Articleshttps://doi.org/10.1038/s41593-019-0486-0
1Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, CA, USA. 2Department of Neurology and Neurological Sciences, Stanford University School of Medicine, Stanford, CA, USA. 3Department of Neurobiology, Stanford University School of Medicine, Stanford, CA, USA. 4Department of Pediatrics Division of Critical Care Medicine, Stanford University School of Medicine, Stanford, CA, USA. 5Center for Pharmacogenomics, Department of Internal Medicine, Washington University School of Medicine, St Louis, MO, USA. 6Present address: Department of Neuroscience and Physiology and Neuroscience Institute, New York University Langone Medical Center, New York, NY, USA. 7Present address: Department of Pharmacology and Therapeutics, The University of Melbourne, Melbourne, Victoria, Australia. *e-mail: [email protected]
A common feature of neurodegenerative diseases, such as Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD), is the accumulation
of neurotoxic proteins in neurons, which leads to neuronal dysfunc-tion and eventually to neuronal death1. Although other cells, includ-ing microglia and astrocytes (glial cells), in the brain also express these proteins and therefore may be directly damaged, the contri-bution of this potential glial damage to neuronal injury is not fully understood. Much research has indicated that the role of glial cells in the brain is neuroprotective in nature; these cells release trophic fac-tors, protect against oxidative stress and excitotoxity, provide essen-tial metabolites and remove damaging agents and cell debris in the milieu of the brain2–4. However, in neurodegenerative diseases, glia are activated to a state that triggers an increased secretion of proin-flammatory factors, which leads to further neurotoxicity through a mechanism collectively termed neuroinflammation5–8. This glial activation and the consequent neuronal damage is attributed to unchecked glial stimulation via exposure to neuronal debris8. In animal models, the activated state of astrocytes, termed the A1 state, is rapidly induced by activated microglia following brain injury, and causes further neuronal injury and cell death9. However, the trigger for this unchecked neuroinflammatory response and whether there are pharmacological agents to prevent A1 astrocyte formation and block neuronal injury have yet to be determined9. Here, we set out to address these questions.
There is a clear role for excessive mitochondrial fragmenta-tion and dysfunction in neurons in neurodegenerative diseases, including in HD, AD, ALS, Parkinson’s disease, dementia, ataxia
and hypertension-induced encephalopathy10–13. Indeed, mitochon-drial fragmentation as a consequence of excessive dynamin-related protein 1 (Drp1)-induced mitochondrial fission is a prototypical feature of these experimental and clinical neurodegenerative dis-eases14–16. Interrupting excessive Drp1-mediated mitochondrial fis-sion can prevent neuronal degeneration17. A heptapeptide (P110) that selectively inhibits the binding of activated Drp1 to one of its mitochondrial receptors, mitochondrial fission 1 (Fis1), without affecting physiological mitochondrial fission16 is neuroprotective in patient-derived cells and in genetic mouse models of AD, HD and ALS12,18–20. We therefore determined whether Drp1–Fis1-mediated excessive mitochondrial fission contributes to glial activation in mouse models of neurodegenerative diseases.
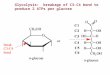
ResultsGlial activation in models of neurodegenerative diseases is inhibited by P110, a selective inhibitor of excessive mitochon-drial fission and fragmentation. Using samples from the same mice described in our previously published studies12,18–20, we found evidence of activation of both microglia and astrocytes in murine models of AD, HD and ALS (Fig. 1) when measured at the end of the study for each cohort (end point in Fig. 1a). We reported that sustained treatment with P110 (the timing and duration of which are indicated by the maroon bars in Fig. 1a) slowed down disease progression in 5XFAD mice18 (an AD model that expresses human APP (which encodes in amyloid-β (Aβ) precursor protein) and PSEN1 (which encodes presenilin 1) transgenes with a total of five AD-linked mutations: the Swedish (K670N/M671L), Florida
Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegenerationAmit U. Joshi 1, Paras S. Minhas2, Shane A. Liddelow 3,6,7, Bereketeab Haileselassie1,4, Katrin I. Andreasson 2, Gerald W. Dorn II5 and Daria Mochly-Rosen 1*
In neurodegenerative diseases, debris of dead neurons are thought to trigger glia-mediated neuroinflammation, thus increas-ing neuronal death. Here we show that the expression of neurotoxic proteins associated with these diseases in microglia alone is sufficient to directly trigger death of naive neurons and to propagate neuronal death through activation of naive astrocytes to the A1 state. Injury propagation is mediated, in great part, by the release of fragmented and dysfunctional microglial mitochon-dria into the neuronal milieu. The amount of damaged mitochondria released from microglia relative to functional mitochon-dria and the consequent neuronal injury are determined by Fis1-mediated mitochondrial fragmentation within the glial cells. The propagation of the inflammatory response and neuronal cell death by extracellular dysfunctional mitochondria suggests a potential new intervention for neurodegeneration—one that inhibits mitochondrial fragmentation in microglia, thus inhibit-ing the release of dysfunctional mitochondria into the extracellular milieu of the brain, without affecting the release of healthy neuroprotective mitochondria.
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience 1635

Articles NATure NeuroscIeNce
(I716V) and London (V717I) mutations in APP, and the M146L and L286V mutations in PSEN1). This treatment also significantly decreased Aβ levels, improved mitochondrial health and short-term and long-term recall in an open-field test18. Sustained P110 treatment also significantly increased the life span of SOD1-G93A mice (an ALS model; survival after paralysis: vehicle = 21 days, P110 = 41.5 days; P = 0.0039)12. Finally, sustained P110 treatment also improved the life span of R6/2 mice (a HD model; dead/survival
at 13 weeks: vehicle = 6/12, P110 = 1/12; P = 0.0006)19,20. Here, we show that this sustained P110 treatment also inhibited neuroinflam-mation in these models, as indicated by decreased levels of activated astrocytes and microglia in histological studies, whereby brain levels of glial fibrillary acidic protein (Gfap) and ionized calcium-binding adapter molecule 1 (Iba-1), respectively, were measured (Fig. 1b–d; Supplementary Fig. 1). The expression of proinflammatory genes (Fig. 1e) and the level of inflammatory cytokine accumulation in the
0
2
4
6
Leve
l (ng
per
µg
prot
ein)
IL-6
– + P110WT
5XFAD G93A
– +WT – +WT
R6/2
<0.0001<0.0001
<0.0001<0.0001
<0.0001<0.0001
0
10
20
30
Fol
d ch
ange
IL-1β
– + P110WT
5XFAD G93A
– +WT – +WT
R6/2
<0.0001<0.0001
<0.0001
<0.00010.0035
0.0403
0
0.05
0.10
0.15
Leve
l (ng
per
µg
prot
ein)
IL-1α
– + P110WT
5XFAD G93A
– +WT – +WT
R6/2
<0.0001<0.0001
<0.0001<0.0001
<0.0001<0.0001
0
1
2
3
4
5
Leve
l (ng
per
µg
prot
ein)
Tnf-α
– + P110WT
5XFAD G93A
– +WT – +WT
R6/2
<0.0001<0.0001
<0.0001<0.0001
<0.0001<0.0001
0
2
4
6
8
10
Fol
d ch
ange
S100β
<0.0001<0.0001
0.00390.0238
<0.00010.0172
0
5
10
15
Fol
d ch
ange
Iba-1
<0.00010.0004
<0.00010.0128
<0.00010.0053
0
2
4
6
8
Fol
d ch
ange
Gfap
– + P110WT
5XFAD G93A
– +WT – +WT
R6/2
– + P110WT
5XFAD G93A
– +WT – +WT
R6/2
– + P110WT
5XFAD G93A
– +WT – +WT
R6/2
<0.0001<0.0001
0.01640.0197
<0.00010.0206
AD HD ALS
Iba-
1G
fap
AD (5XFAD)a
b
c
f g h i j
d e
HD (R6/2) ALS (SOD1-G93A)
0 0 0 1 2 3 4 (Months)1 2 3 (Months)3 6 (Months)
End point (6 months) End point (3 months) End point (4 months)
P110 or Veh (3 mg per kg per day)
WT Veh 5XFAD Veh 5XFAD P110 WT Veh R6/2 Veh R6/2 P110 WT Veh SOD1-G93A Veh SOD1 G93A P110
Tissue gene expression
–2
5XF
AD
R6/
2
WT
G93
A
Veh
P110
WT
Veh
P110
WT
Veh
P110
0
Downregulated
∆∆
Ct (z
-score)
2
Upregulated
Itgam
Itgb2
Itgb3 C3
C1q
a
C1q
b
GFAP
Ccl4
Ttr
Cd8
6
Cd1
4 Il4 Il10
Il13
Il33
Tgf-β
Veg
f
Arg1
Tfam
Nrf2
Pgc
1a
Nox
4
MnS
OD
Cat
alas
e
Proinflammatory Anti-inflammatory Mitochondrial
Fig. 1 | Inhibition of Drp1–Fis1-mediated mitochondrial fission in vivo reduces sustained microglia and astrocyte activation and subsequent proin-flammatory responses in three models of neurodegenerative disease in mice. a, Age of initiation and length of sustained treatment (in months) with P110 or vehicle (Veh; dark red rectangles in the scheme) in mouse models of AD (5XFAD), HD (R6/2) and ALS (SOD1-G93A). b, Representative brain sections immunostained for Gfap and Iba-1 (markers of astrocyte and microglia activation, respectively) in 5XFAD AD mice (hippocampus), R/2 HD mice (striatum) and SOD1-G93A ALS mice (spinal cord) treated with vehicle or P110 (at 3 mg per kg per day) for the time indicated in the red bars in a. Representative of n = 5 mice per group; 2 sections per mouse. Immunohistochemistry of brains of the corresponding WT mice are provided on the left of each mouse model. Scale bars, 50 µm. Note that the tissue samples were from the mice used in our previous publications12,18,19. c,d, Gfap (c) and Iba-1 protein (d) expression levels (determined by quantitating immunostaining) in the brain of each mouse model, presented as a ratio to the corresponding WT mice. e, Expression profiles for select proinflammation-related and anti-inflammation-related genes and mitochondrial-biogenesis-related genes as analyzed by qPCR in the respective tissue samples. Blue and red denote upregulated and downregulated genes, respectively (n = 5 per mice, of 2 technical repeats). f, Plasma concentrations of S100β protein, a peripheral marker of astrocyte activation and blood–brain barrier integrity (n = 5 per mice group). g–j, Protein levels of tissue Tnf-α (g), IL-6 (h), IL-1α (i) and IL-1β (j) in the respective samples (n = 5 per mice group). Probability determined by one-way ANOVA and Benjamini, Krieger and Yekutieli corrections for multiple testing between each treatment group, as above. All data are shown as the mean ± s.d., and P values are indicated.
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience1636

ArticlesNATure NeuroscIeNce
tissue (Fig. 1f–j) also decreased relative to vehicle-treated mice in each of the neurodegenerative disease mouse models, but remained higher than levels of age-matched wild-type (WT) control mice. These data indicate a role for Drp1–Fis1-mediated excessive mito-chondrial fission in neuroinflammation associated with these neu-rodegenerative diseases. Note that at the time of collection of the samples (end point in Fig. 1a) for each model of neurodegenerative diseases, 10% of the P110-treated mice died, none of the matched WT or the 5XFAD mice died in any of the treatment groups, but ~40% of R6/2 and ALS control-treated mice died12,18–20.
Microglia are directly activated by neurotoxic proteins in a mech-anism dependent on Drp1–Fis1-mediated excessive mitochon-drial fission. P110 treatment in vivo may delay neuronal loss, which in turn reduces neuronal debris-induced gliosis. Alternatively, P110 treatment may directly inhibit glial activation. To examine these two possible mechanisms for the anti-inflammatory effect of P110, we performed in vitro studies in which microglia were exposed to neurotoxic proteins in the absence of neurons. Expression of a long track of neurotoxic polyglutamine (Q73) as a model for HD in the microglial cell line BV2 was sufficient to trigger mitochon-drial fragmentation and dysfunction (Fig. 2a,b), and provoked P110-inhibitable neuroinflammatory responses in microglia, as indicated by inflammasome formation, increased cellular reactive oxygen species (ROS) levels and reduced mitochondrial function (Fig. 2c–e) compared with cells expressing the non-pathological polyglutamine (Q23). The levels of the proinflammatory cyto-kines tumor necrosis factor-α (Tnf-α) and interleukin-1β (IL-1β) increased by approximately tenfold in microglia expressing Q73, and P110 treatment blunted the increases by more than 50% (Fig. 2f). We confirmed that P110 treatment of microglia expressing Q73 inhibited Drp1 activation, as indicated by the level of inhibition of Drp1 accumulation in the mitochondrial fraction and the decreased ratio of Drp1 phosphorylation on the activating site (S616) to that on the inactivating site (S637)21 (Fig. 2g,h). To show that these results were not simply due to the transient transfection of BV2, the responses induced by the expression of Q73 in BV2 microglia were similar to those observed in primary microglia isolated from R6/2
mice, a murine model of HD22 (Fig. 2i–k). As expected, mitochon-drial dysfunction and cytokine release were lower in microglia from WT littermates relative to microglia from R6/2 mice (Fig. 2i–k). To confirm that these primary microglia are functional, we stimu-lated them with bacterial-derived lipopolysaccharide (LPS) and found a further decline in ATP production, greater levels of cellular and mitochondrial ROS production and a much greater cytokine release in R6/2-derived microglia relative to microglia derived from WT littermates (Fig. 2i–k). All markers of microglial activation were significantly reduced when excessive mitochondrial fission was inhibited by P110. Stimulation of primary rat microglia with 0.1 μg ml–1 of LPS alone (Supplementary Fig. 2) or with 0.01 μg ml–1 of LPS together with nigericin (an inducer of the NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome23; Fig. 2l–o), in a two-signal model of inflammation in culture, pro-duced similar blunting of microglial activation as that of inhibiting Drp1–Fis1 interactions.
The signature of microglial activation was not unique to models of HD. Expression of the mutant SOD1-G93A in BV2 microglia, as a model of ALS24, or treating microglia with oligomeric amyloid-β peptide-42 (oAβ42), as a model for AD25, also triggered mito-chondrial fragmentation and dysfunction, decreased ATP levels, increased mitochondrial and cell ROS production, and decreased oxidative phosphorylation in the microglia (Fig. 3a–d for SOD1-G93A and Fig. 3f–i for oAβ42). These treatments also increased cyto-kine production and inflammasome formation in the mitochondria of the activated BV2 microglia (Fig. 3e for SOD1-G93A and Fig. 3j for oAβ42), and all of these effects were significantly blunted follow-ing inhibition of the Fis1–Drp1 interaction through treatment of the microglia with P110 (Fig. 3e,j,k). Together, these data show that microglial activation with neurotoxic proteins is different to that induced by the classical activator of inflammatory response LPS, and that this microglial activation depends on Drp1–Fis1-induced excessive mitochondrial fission.
Microglia propagate the proinflammatory response to activate astrocytes. Astrocytes, the other subset of glial cells in the brain, have a wide range of important physiological roles, including
Fig. 2 | Drp1–Fis1-mediated mitochondrial fragmentation and dysfunction are required to induce microglial inflammatory responses in a model of HD (induced by cytotoxic Q73) or LPS activation in culture. a, Top: protocol of treatment for a–h. Bottom: representative photomicrographs of rat BV2 microglia transiently expressing Q23 (control) or Q73 (a model of HD) for 48 h, then treated with vehicle or P110 (1 µM added once, 24 h after transfection, in serum-free and antibiotic-free DMEM) and their mitochondria stained with anti-TOM20 (pseudocolored yellow for microglia) after 24 h. Right: the mitochondrial aspect ratio was quantified using a macro in Fiji (ImageJ)50. b, Intracellular ATP levels and mitochondrial ROS levels quantified after 16 h. c, Levels of inflammasome components (NLRP3 and apoptosis-associated speck-like protein containing a CARD (ASC)) present in mitochondrial fractions of Q23-expressing and Q73-expressing BV2 microglia after 24 h. d, Total cellular ROS levels were quantified after 24 h. e, Oxidative phosphorylation (OXPHOS) and glycolysis rate (using Seahorse) measured in BV2 mouse microglia cells transiently expressing Q23 or Q73 (HD model) for 48 h and treated without or with P110 (1 μM every 24 h). f, Tnf-α and IL-1β levels in cell supernatants from Q23-expressing and Q73-expressing BV2 microglia, treated as in a after 24 h. Con, control. g,h, Mitochondria-associated Drp1 levels in the above BV2 cells (g), as a measure of Drp1 activation, and Drp1 phosphorylation levels at S616-Drp1 (a phosphorylation site correlating with Drp1 activation21) or S637-Drp1 (a site correlating with Drp1 inhibition21) sites (h) in Q23-expressing and Q73-expressing BV2 microglia, treated as in a, after 24 h. i, Top: protocol of treatment for i–k. Bottom: intracellular ATP levels in primary microglia from R6/2 and WT mice that were cultured, treated with vehicle or P110 (1 µM) and stimulated without or with LPS (10 ng ml–1) for 24 h in serum-free and antibiotic-free DMEM. j, Mitochondrial ROS levels and total ROS levels quantified in R6/2 or WT primary mouse microglia treated as in i. k, Levels of released Tnf-α and IL-1β in the culture media of primary mouse microglia cells treated as in i. l, Top: protocol of treatment for l–o. Bottom: representative photomicrographs of primary rat microglia primed with LPS (10 ng ml–1) for 3 h followed by nigericin (1.2 µM) treatment (LPS/Nig) in the presence or absence of P110 (1 µM, added 30 min before LPS) treated for 21 h in serum-free and antibiotic-free DMEM. Right: quantification of the mitochondrial aspect ratio. m, Markers of microglial mitochondrial health, including intracellular ATP levels, mitochondrial ROS levels, OXPHOS, rate of glycolysis, spare respiratory capacity and mitochondrial membrane potential were determined in these control or LPS-primed nigericin-treated microglia. n, Levels of Tnf-α and IL-1α in culture media, and C1q RNA levels in these controls or LPS-primed nigericin-treated primary rat microglia in the absence or presence of P110. o, Nuclear translocation of nuclear factor-κB (Nf-κB) in primary rat microglia, treated as in l. Values underneath are fold change of Nf-κB staining. Probability was calculated by one‐way ANOVA (with Tukey’s post hoc test). All graphs represent the mean ± s.d., and P values are indicated. Scale bars, 5 µm (a and l) or 50 µm (o). All experiments were performed in biologically independent replicates. a, n = 3, each point indicates a single mitochondrion; b, n = 7 (ATP), n = 9 (MitoSOX); c, n = 5 (NLRP3), n = 4 (ASC); d,e, n = 4; f, n = 5; g, n = 8; h, n = 3; i,j, n = 5; k, n = 6 (Tnf-α), n = 5 (IL-1β); l, n = 3, each point indicates a single mitochondrion; m, n = 4 (ATP), n = 12 (MitoSOX), n = 4 (OXHPHOS, glycolysis, spare capacity), n = 20 (TMRM); n, n = 6; o, n = 3.
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience 1637

Articles NATure NeuroscIeNce
neuroprotective roles, when in the A2 state9, and neuroinflamma-tory roles when activated to the A1 state9. Because we noted acti-vation of astrocytes in the murine models of AD, HD and ALS that correlated with the severity of the disease phenotypes (Fig. 1; Supplementary Fig. 1)12,18,19, we assessed possible microglial–astro-cyte cross-talk. We activated cultured microglia in the presence or absence of P110 (Figs. 2 and 3) and expressed Q73 or mutant SOD1-G93A (models of HD and ALS, respectively; see Fig. 4a,g for schemes), or used primary microglia from R6/2 mice and WT litter-mates (see Fig. 4n for the scheme). After culturing for 24 h, we trans-ferred the microglial-conditioned media (MCM) to naive primary astrocytes (Fig. 4a–n) or to R6/2-derived astrocytes and their litter-mate WT astrocytes (Fig. 4o). Transfer of BV2 MCM of Q73 (HD model) or SOD1-A93G (ALS model) cultures to primary astrocytes induced pathological mitochondrial fragmentation in the astrocytes and evoked mitochondrial dysfunction, as evidenced by lower ATP levels, loss of normal inner mitochondrial membrane polarization, increased mitochondrial ROS production and increased cell death (Fig. 4a–e and 4g–k for HD and ALS, respectively). These treat-ments resulted in astrocyte activation to the A1 proinflammatory state (Fig. 4f and 4l for HD and ALS, respectively) and a greatly
increased release of the proinflammatory cytokines Tnf-α and IL-1β (Fig. 4m). All these effects were blunted when the microglia alone were treated with P110 24 h before transferring their media to the naive astrocytes. As the half-life of P110 is ~15 min in culture, the data suggest that Drp1–Fis1 fragmentation in microglia generated a diffusible signal to the astrocytes, which resulted in the activation of astrocytes to the A1 state.
Importantly, to show that propagation of the inflammatory response is not simply due to the genetic manipulation of the BV2 microglial cell line, a twofold stronger propagation of the proin-flammatory signal in astrocytes was induced by transferring con-ditioned media from R6/2 primary microglia to primary microglia from WT mice. These results show that there were increased mito-chondrial and cell ROS levels in naive astrocytes and even higher cellular ROS levels in primary astrocytes isolated from R6/2 mice (Fig. 4n). The propagation of the R6/2 microglia signal in astrocytes was exacerbated by LPS treatment of the donor microglia and was blunted by treatment of the donor microglia with P110 (Fig. 4n). Increased cellular and mitochondrial ROS levels were also found when the proinflammatory microglial medium were transferred to R6/2-derived astrocytes (Fig. 4o), and the increase in cellular
0
0.5
1.0
1.5
2.0
Intr
acel
lula
r A
TP
(fo
ld c
hang
e)
Con Veh P110
LPS/Nig
<0.0001<0.0001
0
0.5
1.0
1.5
2.0
Intr
acel
lula
r A
TP
(fo
ld c
hang
e)
Q23 P110
Q73
Veh
<0.0001
0.0097
0
2
4
6
8
Mito
chon
dria
l asp
ect r
atio
Con Veh P110
LPS/Nig
<0.0001<0.0001
0.01
0.02
0.03
AS
C/V
DA
C r
atio
Q23 Veh P110Q73
<0.0001 0.0038
0
1
2
3
4
5
Fol
d ch
ange
Con Veh P110
LPS/Nig
<0.0001 <0.0001
0
50
100
150
TM
RM
acc
umul
atio
n (%
)
Con Veh P110
LPS/Nig
<0.0001<0.0001
0
2
4
6
8
Mito
SO
X (
fold
cha
nge)
Con Veh P110
LPS/Nig
<0.00010.0043
0
20
40
60
EC
AR
(m
pH m
in–1
)
Con Veh P110
LPS/Nig
<0.0001<0.0001
0.0043
0
100
200
300
400
OC
R (
pmol
min
–1)
Con Veh P110
LPS/Nig
0.0003
Spare respiratory capacity
0
50
100
150
200
OC
R (
pmol
min
–1)
Con Veh P110
LPS/Nig
0.0008
0.0281
0
50
100
150
200
250
EC
AR
(m
pH m
in–1
)
Q23 Veh P110
Q73
<0.0001 0.0021
0
50
100
150
OC
R (
pmol
min
–1)
Q23 Veh P110
Q73
0.0001 0.0033
0
200
400
600
800
1,000
IL-1
β (p
g m
l–1)
Con Veh P110
Q73
<0.0001 <0.0001
0
100
200
300
400
500
TN
F-α
(pg
ml–1
)
TN
F α
(pg
ml–1
)
Tnf-α lL1-α C1q
Con Veh P110
Q73
<0.0001 <0.0001
0
0.01
0.02
0.03
0.04
0.05
Drp
1/V
DA
C r
atio
Q23 Veh P110
Q73
0.0007 0.0079
0
0.01
0.02
0.03
0.04
NLR
P3/
VD
AC
rat
io
Q23 Veh P110
Q73
<0.0001 <0.0001
0.0
0.5
1.0
1.5
Drp
1 ph
osph
oryl
atio
n(f
old
chan
ge)
Q23 Veh P110
Q73
Q23 Veh P110
Q73
Ser 637Ser 616
<0.0001 0.0228
0
10
20
30
Tot
al R
OS
(fo
ld c
hang
e)
Q23 Veh P110Q73
<0.0001 <0.0001
0.0
0.5
1.0
1.5
ng p
er µ
g pr
otei
n
Con Veh P110
LPS/Nig
<0.0001<0.0001
0
2
4
6
Fol
d ch
ange
Con Veh P110
LPS/Nig
<0.0001 <0.0001
0
1
2
3
4
5
Mito
SO
X (
fold
cha
nge)
Q23 Veh P110
Q73
<0.0001 <0.0001
0
2
4
6
8
Mito
chon
dria
l asp
ect r
atio
Q23 Veh P110
Q73
<0.0001<0.0001
0
50
100
150
IL-1
β (p
g m
l–1)
WT R6/2 WT R6/2 WT R6/2– – P110
LPS
P110
<0.0001
<0.0001
<0.0001
0.0012
0
100
200
300
400
500
WT R6/2 WT R6/2 WT R6/2– – P110
LPS
P110
0.0001
0.0001<0.0001
<0.0001
0.0
0.5
1.0
1.5
2.0
Intr
acel
lula
r A
TP
(fo
ld c
hang
e)
WT R6/2 WT R6/2 WT R6/2– – P110
LPS
P110
0.00040.0107
0.0266
0.0189
0.0394
0
2.5
5.0
7.5
10.0
12.5
Tot
al R
OS
(fo
ld c
hang
e)
WT R6/2 WT R6/2 WT R6/2– – P110
LPS
P110
0.0155
<0.0001
0.0022<0.0001
<0.0001
0
2
4
6
Mito
SO
X (
fold
cha
nge)
WT R6/2 WT R6/2 WT R6/2– – P110
LPS
P110
0.0484
0.0110
0.0005
0.0021
0.0168
Q73 Veh P110
Q73
TO
M20
BV2 microglia
Q23/Q73(48 h transfection)
Assaya
e
i
l m
n o
j k
f g h
b c d+/– P110 1 µM
+/– P110 1 µM
Q23 Veh P110N
LRP
3A
SC
VD
AC
Q73
118
25
MW
31
ATP Mitochondrial ROS NLRP3 levels ASC levels
Mitochondrial health Mitochondrial inflammasome formation
HD
microglia
LPS/Nig
+/– Nigericin 1.2 µM,+/– P110 1 µM
LPS 10 ng ml–1
(3 h)Primary rat microglia
Assay
TO
M20
Mitochondrial ROS
Veh P110
ATP
Mitochondrial health
Con
Cellular ROS
Metabolic health
Drp
1V
DA
C
Q23 Veh P110Q73
8181
81
42
31
MWMW
Mitochondrial Drp1 accumulation Drp1 phosphorylation
Q23 Veh P110Q73
s616
s637
Act
in
Primary mouse microgliaR6/2 or WT mice
AssayROS generation Cytokine release
OXPHOS Glycolysis
Cytokine secretion
Mitochondrial membranepotential
OXPHOS Glycolysis
Con Veh P110
LPS/Nig
1 ± 0.2 3.2 ± 0.9 1.52 ± 0.6
NF-κB nuclear translocation
+/– LPS 100 ng
Cytokine release
LPS
/Nig-treated m
icroglia
NF-κB
DAPI
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience1638

ArticlesNATure NeuroscIeNce
ROS production was significantly higher than that of WT litter-mate culture medium transferred to astrocytes (Fig. 4o versus 4n). We also confirmed that the activation of the mouse astrocytes was
due to canonical activation of microglia, as shown by treatment of the microglia with bacterial LPS and transfer of the conditioned media to astrocytes (Fig. 4n,o; Supplementary Fig. 3). Primary
0
1
2
3
4
Tot
al R
OS
(a.
u.)
0.0021<0.0001
0
0.5
1.0
1.5
NLR
P3/
VD
AC
rat
io
0.03550.0061
0
0.5
1.0
Drp
1/V
DA
C r
atio
0.02920.0018
0
1
2
3
4
5
Mito
SO
X (
fold
cha
nge) 0.03680.0012
0
2
4
6
8
Mito
chon
dria
l asp
ect r
atio
Con Veh P110
<0.0001 <0.0001
0
0.5
1.0
1.5
Intr
acel
lula
r A
TP
(fo
ld c
hang
e)
0.0009
0.0140
0
50
100
150
OC
R (
pmol
min
–1)
Con Veh P110
0.0020
0.0170
0
50
100
150
200
250
EC
AR
(m
pH m
in–1
) 0.00030.0156
0
500
1,000
1,500<0.0001
<0.0001
0
20
40
60
80
100<0.0001
0.0003
0
1
2
3
4
Tot
al R
OS
(a.
u.)
0.0024<0.0001
0
50
100
150
OC
R (
pmol
min
–1)
WT Veh P110
SOD1 G93A
WT Veh P110
SOD1 G93A
0.00010.0342
0
50
100
150
200
250
EC
AR
(m
pH m
in–1
) <0.00010.0001
0
20
40
60
80
100
Tnf
-α (
pg m
l–1)
Tnf
-α (
pg m
l–1)
IL-1
β (p
g m
l–1)
IL-1
β (p
g m
l–1)
Con Veh P110
SOD1 G93A
<0.0001<0.0001
0
200
400
600
800
1,000
Con Veh P110
SOD1 G93A
<0.0001<0.0001
0
1
2
3
4
5
Mito
SO
X (
fold
cha
nge) 0.0216
0.0002
0
0.5
1.0
1.5
Intr
acel
lula
r A
TP
(fo
ld c
hang
e)
WT Veh P110
SOD1 G93A
WT Veh P110
SOD1 G93A
WT Veh P110
SOD1 G93A
0.03680.0018
0
2
4
6
8
Mito
chon
dria
l asp
ect r
atio
WT Veh P110
SOD1 G93A
<0.0001 <0.0001WT Veh P110
SOD1 G93A
BV2 microglia
WT or SOD1-G93A(48 h transfection)
a
f
i j k
g h
d e
b cAssay
+/– P110 1 µM
ROS generation
Metabolic health
Control Veh P110
oAβ42 1 µM(2 h)
Assay
+/– P110 1 µM
ROS generation
Metabolic health
OXPHOS Glycolysis
TO
M20
TO
M20
BV2 microglia
)
Cytokine secretion
Cytokine secretion Mitochondrial Drp1 and inflammasome formation
Con Veh P110
NLR
P3
Drp
1V
DA
C
31
118
MW81
AD
microglia m
odelA
LS m
icroglia model
OXPHOS Glycolysis
oAβ42
oAβ42
Con Veh P110oAβ42
oAβ42
Con Veh P110
oAβ42
Con Veh P110
oAβ42
Con Veh P110
oAβ42
Con Veh P110
oAβ42
Con Veh P110
oAβ42
oAβ42
Con Veh P110oAβ42
Con Veh P110oAβ42
Fig. 3 | Drp1–Fis1-mediated mitochondrial fragmentation and dysfunction are required to induce microglial inflammatory response in a model of ALS (by expressing cytotoxic SoD1-G93A) or in a model of AD (induced by treatment with oAβ1-42) in culture. a, Top: protocol of treatment for a–e. Bottom: representative photomicrographs (pseudocolored yellow for microglia) and mitochondrial aspect ratios (right) of BV2 mouse microglia cells transiently expressing human SOD1-WT or mutant SOD1-G93A (a model of ALS) for 48 h and then treated as indicated. b–d, Intracellular ATP (b), ROS levels (c) and mitochondrial OCR and ECAR, measures of mitochondrial OXPHOS capacity and glycolysis, respectively, as determined using Seahorse (d), in BV2 expressing WT or mutant SOD1-G93A after 24 h in serum-free and antibiotic-free DMEM. e, Levels of released Tnf-α and IL-1β the culture media of the BV2 cells treated as in a. f, Top: protocol of treatment for f–k. Bottom: representative photomicrographs (pseudocolored yellow for microglia) and mitochondrial aspect ratios (right) of BV2 microglia treated with or without oAβ42 (1 μM; a model of AD) for 24 h in the presence or absence of P110 (1 μM added once together with oAβ42) in serum-free and antibiotic-free DMEM as a model of AD. g–i, Intracellular ATP (g), ROS levels (h) and metabolic health (i) in BV2 microglia treated with or without oAβ42 (1 μM) for 24 h in defined medium in the presence or absence of P110 (1 µM, added 15 min before oAβ42). j, Levels of released Tnf-α and IL-1β the culture media of cells treated as in f. k, Mitochondrial Drp1 levels and NLRP3 levels in BV2 microglia, treated with or without oligomeric oAβ42 (1 μM) for 24 h in the presence or absence of P110 (1 μM added once together with oAβ42) in serum-free and antibiotic-free DMEM. Probability was calculated by one‐way ANOVA (with Tukey’s post hoc test). All graphs represent the mean ± s.d., and P values are indicated. Scale bars, 5 µm. All experiments were performed in biologically independent replicates. a, n = 3, each point indicates a single mitochondrion; b, n = 3; c, n = 7 (MitoSOX), n = 4 (total ROS); d, n = 4; e, n = 5; f, n = 3, each point indicates a single mitochondrion; g, n = 3; h, n = 4; i, n = 4 (OXPHOS), n = 5 (glycolysis); j, n = 5; k, n = 3.
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience 1639

Articles NATure NeuroscIeNce
rat microglia similarly propagated the inflammatory response in primary rat astrocytes treated with LPS and nigericin (Fig. 5a–f) or by direct treatment of the astrocytes with the proinflammatory cytokines Tnf-α, IL-1α and C1q9 (Fig. 5g–l), which generated similar activation responses in astrocytes, thus serving as positive controls for the general immune activation of astrocytes9. Therefore, propa-gation of the activation signal from all types of microglia to naive astrocytes or to R6/2 primary astrocytes was dependent on Drp1–Fis1-mediated mitochondrial fission in the microglia. Indeed, this effect was inhibited when the microglia were treated with P110 24 h before transfer of their conditioned media to the astrocytes (Figs. 4n,o and 5; Supplementary Fig. 3).
Direct treatment of astrocytes with P110 also inhibited their transition to the proinflammatory A1 state via direct stimulation with Tnf-α, IL-1α and C1q (Fig. 5g–l), thus indicating that Drp1–Fis1-mediated mitochondrial fragmentation also plays a critical role in the activation of astrocytes. Treatment of the astrocytes with a small-molecule inhibitor of Drp1 (Mdivi-1) or with a peptide that inhibits the physiological fission of mitochondria mediated by mito-chondrial fission factor (Mff) and Drp1 (P259)26, exerted similar effects on mitochondrial fragmentation induced with Tnf-α, IL-1α and C1qn (Fig. 5g, quantitated on the right). However, whereas Mdivi-1 improved mitochondrial functions and reduced cell death, P259, the inhibitor of physiological fission was injurious, causing a further reduction in mitochondrial membrane potential and higher mitochondrial ROS production in astrocytes activated by Tnf-α, IL-1α and C1q (Fig. 5i,j). Together, we found that a selective inter-ruption of physiological mitochondrial fission is injurious to the astrocytes. Furthermore, our data indicate that induction of the pro-inflammatory phenotype in microglia or in astrocytes is dependent, at least in part, on Drp1–Fis1-mediated excessive mitochondrial fission and mitochondrial dysfunction, and that the propagation of the proinflammatory signal from activated microglia to naive or primed (R6/2 or WT) is similarly a Drp1–Fis1-dependent process.
Drp1–Fis1-dependent mitochondrial fission increases the ratio of dysfunctional /functional mitochondria released from mouse, rat and human microglia. Mitochondria can be released from mul-tiple cell types2,27. We therefore posited that microglia may release mitochondria or their fragments into the extracellular milieu as physical vessels to propagate the neuroinflammatory signal. In sup-port of this notion, functional mitochondria (with preserved inner membrane potential and ability to generate ATP) were present in
the media of cultured naive primary microglia (Fig. 6b). By con-trast, media of primary microglia activated with LPS and nigericin contained ~50% fewer functional mitochondria (as evidenced by reduced mitochondrial membrane potential and ATP levels in the extracellular mitochondria; Fig. 6b), but more than twofold greater extracellular mitochondrial protein content compared with media from naive primary microglia (as evidenced by western blot analy-ses for the mitochondrial proteins VDAC and TOM20; Fig. 6c, left). P110 treatment of LPS and nigericin-activated primary rat microg-lia did not significantly affect the amount of released mitochondria (Fig. 6c). However, P110 treatment of the microglia greatly reduced the loss of cytochrome c, a small (12 kDa) protein compartmental-ized within the mitochondrial inter-membrane space and a marker for mitochondrial damage28 from the mitochondria released into the culture media (Fig. 6c). Consistent with the improved mito-chondrial structure of the extracellular mitochondria, P110 treat-ment also inhibited the loss of the inner membrane potential and improved ATP production by these extracellular mitochondria, effects that are indicative of improved mitochondrial integrity (Fig. 6b). We can attribute the levels of JC-1 dye (used to assess mito-chondrial membrane potential) and ATP measured in the MCM to the extracellular mitochondria, as filtering the media through a 0.2-μm filter2 (Fig. 6a) resulted in the loss of both the JC-1 signal and ATP (Fig. 6b; MitoΔ).
We also found that microglia from R6/2 mice, BV2 microglia activated by Q73 expression (to model HD) and BV2 microglia expressing mutant SOD1 (to model ALS) released functional mito-chondria into the culture media under basal conditions. However, the integrity of the released mitochondria, evaluated by measuring the mitochondrial membrane potential and ATP production, was lower than their corresponding controls (Fig. 6d,e). The amount of functional extracellular mitochondria was lower if the microglia were activated by expressing mutant huntingtin protein (in microg-lia derived from R6/2 mice), treated without or with LPS (Fig. 6d), or expressing Q73 or mutant SOD protein (Fig. 6e). As before, the extent of the dysfunction of these released mitochondria was greatly blunted if the cells were treated with P110 (Fig. 6d,e). Importantly, we found that a model of human microglia (differentiated from peripheral blood mononuclear cells29) also released functional mitochondria under basal conditions that produced ATP, and that activation of these human microglia with LPS and nigericin greatly reduced the integrity of the expelled mitochondria (Fig. 6f). As before, treatment of the LPS-activated human microglia with P110
Fig. 4 | conditioned media of activated microglia with mitochondrial dysfunction propagate astrocyte activation to the A1 proinflammatory state and dysfunction of mitochondria in cultured astrocytes in multiple models of neurodegenerative diseases. a, Top: protocol of the experiment in a–f. Bottom: representative photomicrographs of mouse primary astrocytes treated with conditioned media from activated BV2 microglia (referred to as MCM from here on) expressing non-pathological Q23 (control) or pathological Q73 (for 48 h; HD model) and treated with vehicle or P110 for 24 h. The MCM was added to astrocytes for 24 h and astrocytes were then stained with anti-TOM20 (pseudocolored green for astrocytes). Right: the mitochondrial aspect ratio was determined as in Fig. 1c. b, Intracellular ATP levels in astrocytes were determined after 24 h of incubation with MCM. c,d, Mitochondrial membrane potential (c) and mitochondrial ROS levels (d) were determined at 6 h after MCM treatments of the astrocytes. e, LDH release from the astrocytes was measured 48 h after MCM treatments. f, Heatmap of RNA transcripts in the astrocytes 24 h after MCM treatments. g, Top: protocol of the experiment in g–l. Bottom: representative photomicrographs of mouse primary astrocytes (pseudocolored green) treated for 12 h with supernatant from BV2 microglia expressing SOD1-WT or SOD1-G93A (MCM). Right: astrocytes were then stained with anti-TOM20 and the mitochondrial aspect was determined as in Fig. 1c. h–j, Intracellular ATP levels at 24 h (h), mitochondrial membrane potential, using TMRM (i), and mitochondrial ROS levels, using MitoSOX (j), were determined 6 h after initiation of treatment with MCM. k, LDH release measured 48 h after MCM treatment. l, Heatmap of RNA transcripts of primary astrocytes primed by treatment with MCM. m, Levels of released Tnf-α and IL-1β into the culture media of cells treated as in a and g. n, Top: protocol of the experiment. Bottom: mitochondrial ROS levels and total ROS levels were measured in primary astrocytes isolated from WT mice treated with supernatant from WT or R6/2 mouse microglia, as in Fig. 2i. o, Top: protocol of the experiment. Bottom: mitochondrial ROS levels and total ROS levels were measured in primary astrocytes isolated from R6/2 mice treated with supernatant from WT or R6/2 microglia treated as in Fig. 2i. Data were evaluated by one-way ANOVA and Holm–Sidak’s multiple comparisons test for multiple testing between each treatment group. All graphs represent the mean ± s.d., and P values are indicated. Scale bars, 20 µm. Note that P110 treatment in the studies shown here and in Figs. 4–6 was restricted to the microglia. As the half-life of the peptide is >30 min, no peptide remained in the MCM to directly affect the astrocytes or the neurons. All experiments were performed in biologically independent replicates. a, n = 3, each point indicates a single mitochondrion; b, n = 3; c,d, n = 5; e, n = 3; f, n = 2; g, n = 3, each point indicates a single mitochondrion; h, n = 4; i,j, n = 4; k, n = 4; l, n = 2; m, n = 5; n, n = 6; o, n = 5.
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience1640

ArticlesNATure NeuroscIeNce
improved the integrity and function of the released mitochondria (Fig. 6f). Together, our data show that inhibition of Drp1–Fis1-mediated mitochondrial fission by P110 reduces the dysfunction
of the mitochondria released from activated microglia, but not the total amount of mitochondria or mitochondrial proteins released into the extracellular media.
0
1
2
3
4
5
Mito
SO
X (
fold
cha
nge)
Q23 Veh P110Q73
0.0097<0.0001
0
25
50
75
100
125
Intr
acel
lula
r A
TP
(%
)
WT Veh P110
G93A
0.0004
<0.0001
0
0.5
1.0
1.5
TM
RM
acc
umul
atio
n(f
old
chan
ge)
Q23 Veh P110Q73
0.02220.0222
0
25
50
75
100
125
Intr
acel
lula
r A
TP
(%
)
Q23 Veh P110Q73
0.00010.0082
0
2
4
6
8
10
Mito
chon
dria
l asp
ect r
atio
Q23 Veh P110Q73
<0.0001
<0.0001
BV2 microglia Mouse primary astrocytes
MCM+/– Q23 or Q73 (48 h)+/– P110 (24 h) Assay
Q73Mitochondrial
membrane potential
Enh
ance
dT
OM
20
Q23
a
d
g
j
m
o
n
k l
h i
e f
b cVeh P110
0
50
100
150
200
250
LDH
rel
ease
(%
)
Q23 Veh P110Q73
0.0004 0.0142
–2
0
2
Cell deathMitochondrial
ROSPan reactive A1 specific A2 specific
Astrocytic activation state
Q23
Veh
P110
Q73
∆∆
Ct (z
-score)∆
∆C
t (z-score)
0
0.5
1.0
1.5
TM
RM
acc
umul
atio
n(f
old
chan
ge)
WT Veh P110
G93A
<0.00010.0172
0
2
4
6
8
Mito
chon
dria
l asp
ect r
atio
WT Veh P110
G93A
<0.0001
<0.0001
BV2 microglia Mouse primary astrocytes
MCM+/– SOD1-G93A (48 h)+/– P110 (24 h) Assay
Mitochondrialmembrane potential
Enh
ance
d
SOD1 G93AWT Veh P110
TO
M20
0
1
2
3
4
5
Mito
SO
X (
fold
cha
nge)
WT Veh P110G93A
WT Veh P110G93A
0.0029<0.0001
0
50
100
150
200
250
LDH
rre
leas
e (%
) 0.02460.0001
Cell deathMitochondrialROS Pan reactive A1 specific A2 specific
Astrocytic activation state
WT
Veh
P110
G93A
–2
0
2
0
20
40
60
80
Tot
al R
OS
(fo
ld c
hang
e)
WT R6/2 WT R6/2 WT R6/2
– – P110
LPS
P110
0.0252
0.0002
<0.0001<0.0001
0
4
8
12
Mito
SO
X (
fold
cha
nge)
WT R6/2 WT R6/2 WT R6/2
– – P110
LPS
P110
0.00740.0241
0.01520.0001
0.0157
0
4
8
12
Mito
SO
X (
fold
cha
nge)
WT R6/2 WT R6/2 WT R6/2– – P110
LPS
P110
<0.0001
<0.0001
<0.0001
0.0010
0.0021
0
20
40
60
80
Tot
al R
OS
(fo
ld c
hang
e)
WT R6/2 WT R6/2 WT R6/2– – P110
LPS
P110
<0.0001
<0.0001
<0.0001
0.0006
0.0397
0
500
1,000
1,500
2,000
2,500
IL-1
β (p
g m
l–1) <0.0001<0.0001
<0.0001 <0.0001
0
200
400
600
800
1,000
Tnf
-α (p
g m
l–1)
Q23 WTVeh P110
Q73
Veh P110
G93A Q23 WTVeh P110
Q73
Veh P110
G93A
<0.0001<0.0001
<0.0001 <0.0001
WT or R6/2 microglia WT astrocytes
MCM+/– LPS (24 h)+/– P110 (24 h)
Assay
WT or R6/2 microglia R6/2 astrocytes
MCM+/– LPS (24 h)+/– P110 (24 h)
Assay
ROS production
ROS production
BV2 microglia Primary mouseastrocytes
MCMQ23, Q73, WT or SOD1-G93A+/– P110 (24 h)
ACM assayt
Lcn2
Steap
4S1p
r3Tim
p1Hsb
p1Cxcl10
Cd4
4Osm
rCp
Serpina
3nAsp
gVim
Gfap
H2-T23
Serping
1H2-D1
Ggta1
Iigp1
Gbp
2Fbln5
Ugt1a
Fkb
p5Psm
b8Srgn
Amigo2
Clcf1
Tgm
1Ptx3
S10
0a10
Sph
k1Cd1
09Ptgs2
Emp1
Slc10
a6Tm4s
f1B3g
nt5
Cd1
4
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience 1641

Articles NATure NeuroscIeNce
Dysfunctional extracellular microglial mitochondria trigger the release of dysfunctional mitochondria from naive astrocytes. As previously reported2, astrocytes also released functional mito-chondria that can produce ATP (Fig. 6g,h, control versus MitoΔ). Moreover, astrocytes activated by conditioned media from LPS and nigericin-treated microglia released more damaged mitochondria, as evidenced by 50% lower mitochondrial membrane integrity and 50% lower ATP levels (Fig. 6g). The released astrocytic mitochon-dria exhibited a substantial loss of cytochrome c (Fig. 6h), which is suggestive of damage to the outer membrane of the extracellular mitochondria. As above, the release of these dysfunctional mito-chondria from the astrocytes was greatly blunted if the microglia were activated in the presence of P110 (Fig. 6g,h). Again, because the half-life of P110 is very short, P110 did not exert its affect directly on the astrocytes. Structural abnormalities of the released astro-cytic mitochondria, as assessed by electron microscopy analyses of the extracellular mitochondria, included reduced cristae struc-tures and breaks in the mitochondrial outer membrane (arrows in Fig. 6i, middle panels versus left panels). By contrast, mitochon-dria isolated from astrocytes treated with conditioned media of LPS and nigericin-treated primary microglial cells co-treated with P110 showed less damage (Fig. 6i, right versus middle panels). These data reveal a role for excessive pathological mitochondrial fission in the genesis and delivery of dysfunctional mitochondria to the extracellular milieu.
To confirm that astrocytes directly release mitochondria, we examined the culture media of astrocytes activated by a mixture of the proinflammatory cytokines Tnf-α, IL1α and C1q. This cytokine activation of the astrocytes reduced the function of the extracellular mitochondria by ~50% relative to those released from naive (control) astrocytes (Fig. 6j). Dysfunction of the extracellu-lar astrocytic mitochondria was greatly blunted by treatment with P110, an inhibitor of Drp1–Fis1-mediated fission, or with Mdivi-1, an inhibitor of the catalytic activity of Drp1 (ref. 30). In contrast, similar to the results above, no benefit was observed after treat-ment with P259, a peptide inhibitor of the Drp1 and Mff interaction and inhibits physiological but not pathological fission26 (Fig. 6j). As the amount of mitochondria in the culture media was unaf-fected when the stressed cells were treated either with the cytopro-tective peptide, P110, or the cell damaging peptide, P259 (see levels of lactate dehydrogenase (LDH) release in Figs. 5k and 6c,h,j and Supplementary Fig. 4c), the mitochondrial content in the media is unlikely to be related to the extent of cell death. Examining the cytochrome c content of the extracellular mitochondria confirmed that the integrity of the mitochondria released from activated astro-cytes treated with P110 or Mdivi-1 was better than those released from activated astrocytes treated with P259 or vehicle (Fig. 6k), thus indicating the selective role of Drp1–Fis1-mediated fission in this process.
Extracellular dysfunctional mitochondria propagate injury from microglia to astrocytes to neurons, whereas extracellular func-tional mitochondria are neuroprotective. As discussed above, much is known about the primary function of all glial cells in pro-tecting and providing essential structural and metabolic support for neurons31. Yet, we found that transfer of conditioned media of microglia-activated astrocytes (Fig. 7) or direct transfer of activated MCM to cultured primary neurons (Supplementary Fig. 4a) was suf-ficient to evoke neuronal damage. ATP production and mitochon-drial inner membrane potential decreased in these neuronal cells, as did the production of neurotoxic ROS (Fig. 7a,b). Propagation of the injurious stimulus from the microglia to astrocytes, and from astro-cytes to neurons, was consistent with whether microglia were acti-vated nonspecifically with LPS (Fig. 7a,b) or by the recapitulation of genetic neurodegenerative diseases by expressing long polygluta-mine (Q73, as a HD model) or SOD1-G93A (ALS model) (Fig. 7c). Extracellular fragmented mitochondria, but not free mitochon-drial DNA32, were the major mediators of the major neurotoxic sig-nal released from activated astrocytes: filtering out mitochondrial particles from activated astrocytic-conditioned media (MitoΔ) attenuated the mitochondrial dysfunction in neurons, as assessed by measuring the level of neuronal mitochondrial respiration (Fig. 7b), and substantially decreased neuronal cell death (Fig. 7d). By contrast, enzymatic degradation of free mitochondrial DNA, a known contributor to cytotoxicity33, using DNase treatment of the conditioned media had only a small effect (Fig. 7d). Finally, the addition of an enriched preparation of extracellular astrocytic mito-chondrial particulates to cultured neurons without soluble material from the conditioned media caused a fivefold increase in neuronal cell death, which was significantly reduced by P110 treatment of the primary microglia that propagated the injurious stimulus to the neurons via the astrocytic mitochondria (Fig. 7e). Therefore, extra-cellular mitochondria alone propagate the injury from the activated microglia to the astrocytes and from the astrocytes to the neurons. We also confirmed results from a previous study2, which showed that intact extracellular astrocytic mitochondria provide neuro-protection. That is, removal of the mitochondria from the naive or P110-protected astrocytic-conditioned media doubled the level of neuronal cell death (Supplementary Fig. 4b; compare the MitoΔ group to the “–” group).
The indirect propagation of the neurotoxic signal from the microglia to the astrocytes and subsequently to the neurons was not due to a transient expression of neurotoxic proteins in the microglia. Microglia isolated from the R6/2 HD mouse model also propagated the mitochondrial dysfunction and neu-ronal death through naive WT astrocytes (Fig. 8a) or through R6/2-derived and likely partially activated astrocytes (Fig. 8b). We observed more than fivefold higher ROS production in the neuronal mitochondria and 50% more neuronal cell death
Fig. 5 | excessive mitochondrial fission and dysfunction occur in activated astrocytes. a, Top: schematic of the experimental design using primary mouse astrocytes treated with conditioned media of LPS/Nig-activated mouse primary microglia (MCM). Bottom: representative photomicrographs of primary rat astrocytes (pseudocolored in green) treated with MCM for 24 h, then stained with anti-TOM20. Right: the mitochondrial aspect ratio was then determined. b–d, Intracellular ATP determined at 24 h (b), and mitochondrial membrane potential (c) and mitochondrial ROS levels (d) determined 12 h after MCM treatment. e, LDH release measured 48 h after MCM treatment. f, Heatmap of RNA transcripts in these astrocytes. g, Top: schematic of the experimental design using primary rat astrocytes treated directly with a combination of Tnf-α, IL-1α and C1q without or with P110 or P259 (1 µM, added once, 15 min before) or 20 µM Mdivi-1 (added once, 15 min before) and then incubated for 24 h in serum-free and antibiotic-free DMEM. Bottom: representative photomicrographs of primary mouse astrocytes (pseudocolored green) as in the scheme and then stained with anti-TOM20. Right: quantification of the mitochondrial aspect ratio. h–l, Intracellular ATP at 24 h (h), mitochondrial membrane potential at 24 h (i), mitochondrial ROS at 24 h (j), LDH release measured at 48 h (k) and heatmap of RNA transcripts of treated rat primary astrocytes at 24 h (l) were determined as above. Data were evaluated by one-way ANOVA and Holm–Sidak’s multiple comparisons tests for multiple testing between each treatment group. All graphs represent the mean ± s.d., and P values are indicated. Scale bars, 20 µm. All experiments were performed in biologically independent replicates. a, n = 3, each point indicates a single mitochondrion; b, n = 3; c,d, n = 5; e, n = 4; f, n = 2; g, n = 3, each point indicates a single mitochondrion; h, n = 5 (Control, Veh, P110), n = 3 (P259, Mdivi-1); i, n = 7 (Control, Veh, P110), n = 4 (P259, Mdivi-1); j, n = 6 (Control, Veh, P110), n = 3 (P259, Mdivi-1); k, n = 5 (Control, Veh, P110), n = 3 (P259, Mdivi-1); l, n = 2.
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience1642

ArticlesNATure NeuroscIeNce
when the injurious stimulus from the microglia was propagated through R6/2-derived astrocytes compared with littermate WT astrocytes (Fig. 8b). Again, a greater propagation of neuro-nal injury was observed when the microglia were also cultured
in the presence of LPS, and P110 treatment of the microglia (R6/2 or WT) was sufficient to greatly blunt the propagation of injury through the astrocytes (R6/2 or WT) to the primary neurons (Fig. 8a,b).
0
50
100
150
200
250
LDH
rel
ease
(%
)
<0.0001
0.0004
0.00910.0507
0
1
2
3
4
5
Mito
SO
X (
fold
cha
nge)
Control Veh P110 P259 Mdivi-1
<0.0001
<0.0001
<0.00010.0011
0
50
100
150
Intr
acel
lula
r A
TP
(%
)
Control Veh P110 P259 Mdivi-1
<0.0001
0.0007
0.0279
0.0199
0
0.5
1.0
1.5
TM
RM
acc
umul
atio
n(f
old
chan
ge)
0.0001
0.01620.0304
0.0366
0
5
10
Mito
chon
dria
l asp
ect r
atio
TNF-α, IL-1α and C1q
TNF-α, IL-1α and C1q
Control Veh P110 P259 Mdivi-1
<0.0001
<0.00010.0012
0.0268
0
50
100
150
200
LDH
rel
ease
(%
)
<0.00010.0071
0
0.5
1.0
1.5
TM
RM
acc
umul
atio
n(f
old
chan
ge)
0.0296
0.0489
0
1
2
3
4
Mito
SO
X (
fold
cha
nge)
Control Veh P110
LPS/Nig
Control Veh P110
LPS/Nig
<0.0001<0.0001
0
50
100
150
Intr
acel
lula
r A
TP
(%
)
0.0001
0.0082
0
2
4
6
8
Mito
chon
dria
l asp
ect r
atio
Control Veh P110
LPS/Nig
Control Veh P110
LPS/Nig
Control Veh P110
LPS/Nig
<0.0001
<0.0001
Lcn2
Steap
4S1p
r3Tim
p1Hsb
p1Cxcl10
Cd4
4Osm
rCp
Serpina
3nAsp
gVim
Gfap
H2-T23
Serping
1H2-D1
Ggta1
Iigp1
Gbp
2Fbln5
Ugt1a
Fkb
p5Psm
b8Srgn
Amigo2
Clcf1
Tgm
1Ptx3
S10
0a10
Sph
k1Cd1
09Ptgs2
Emp1
Slc10
a6Tm4s
f1B3g
nt5
Cd1
4
Lcn2
Steap
4S1p
r3Tim
p1Hsb
p1Cxcl10
Cd4
4Osm
rCp
Serpina
3nAsp
gVim
Gfap
H2-T23
Serping
1H2-D1
Ggta1
Iigp1
Gbp
2Fbln5
Ugt1a
Fkb
p5Psm
b8Srgn
Amigo2
Clcf1
Tgm
1Ptx3
S10
0a10
Sph
k1Cd1
09Ptgs2
Emp1
Slc10
a6Tm4s
f1B3g
nt5
Cd1
4
Primary rat astrocytes
+/– TNF-α, IL-1α and C1q+/– P110, P259 or Mdivi-1 Assay
Cell deathMitochondrial membranepotential
Mitochondrial ROS
Pan reactive A1 specific A2 specific
Astrocytic activation state
–2
2
0
TNF-α, IL-1α and C1q
Control Veh P110 P259 Mdivi-1
TNF-α, IL-1α and C1q TNF-α, IL-1α and C1q
Control Veh P110 P259 Mdivi-1
TNF-α, IL-1α and C1q
Enh
ance
dT
OM
20
Control Veh P110
–2
0
2
Primary ratmicroglia
Primary rat astrocytes
MCM
+/– LPS/Nig
a
d
g
h i
l
j k
e f
b c+/– P110 (24 h)
Assay
Cell death
Mitochondrialmembrane potential
MitochondrialROS Pan reactive A1 specific A2 specific
Astrocytic activation state
LPS/Nig
Control
Veh
P110
LPS
/Nig
∆∆
Ct (z
-score)
∆∆
Ct (z
-score)
Control
Veh
P110
TN
F-α , IL-1α
and C1q
Control Veh P110
Enh
ance
dT
OM
20
P259 Mdivi-1
24 h
24 h
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience 1643

Articles NATure NeuroscIeNce
DiscussionThe relationship between mitochondria and eukaryotes has been critical for the success of metazoan life on Earth. Cell colonization by ancestral α-proteobacteria over a billion years ago conferred advantages in energy production and oxygen utilization. However, host cells had to recognize and protect their increasingly essential endosymbionts while simultaneously identifying and repelling phy-logenetically related pathogenic bacterial invaders. Consequently, mitochondria became immunologically privileged. Nevertheless, misidentification of extracellular mitochondrial DNA, damaged mitochondria or other damage-associated molecular patterns (DAMPs) as bacterial can trigger innate (sterile) immune mecha-nisms that in turn contribute to mitochondrial dysfunction and further propagation of the pathology in acute and chronic inflam-matory diseases32. The mechanisms underlying the mitochondrial loss of the immune-privileged state and their transformation into mediators of pathological entities in neurodegenerative disease are unclear.
Loss of immune-privileged state of mitochondria in models of neurodegenerative diseases. Our results showed that loss of the immune-privileged state of extracellular damaged mitochondria correlates with an increased release of damaged mitochondria from the microglia, and that damaged extracellular mitochondria directly contribute to disease propagation by acting as effectors of the innate immune response by targeting adjacent astrocytes and neurons. We have previously reported that neurons harbor neurotoxic pro-teins12,16,18,20. Our data here showed that the Drp1–Fis1 inhibitory peptide P110 reduces mitochondrial fission and the consequent release of damaged mitochondria from the microglia, thus inhibiting the activation of astrocytes and protecting neurons from innate immune attack. Increased Drp1–Fis1-mediated mitochondrial fission in activated microglia triggers the formation of fragmented and damaged mitochondria that are released from these cells, thus induc-ing the innate immune response. Clinical and experimental studies have identified fragmented mitochondria in biofluids of patients afflicted by subarachnoid hemorrhage and of patients affected by stroke2,34–36, which suggests that their presence in the extracellular space is a biomarker for neurodegeneration and disease severity37,38. Our data showed a causal role of dysfunctional extracellular mito-chondria in propagating neurodegenerative signals from microglia.
Innate immune responses in neurodegenerative diseases begin early in the pathogenesis of these diseases and are associated with little or no infiltration of blood-derived immune cells into the brain39. It is the brain-resident cells, the microglia and astrocytes, that trigger this sterile immune response, thus contributing to neu-ronal dysfunction and degeneration. Although mice exclusively expressing mutant huntingtin in microglia, for example, show increased innate immune responses and increased neuronal cell death22, activated microglia alone are unlikely to cause major neu-rodegenerative disease4,5,40. Rather, our data suggest that a relay of glia-to-neurons-to-glia signaling (Supplementary Fig. 4d) plays an important role in neurodegeneration. Feeding into the vicious cycle, the death of neurons, induced by the neurotoxic proteins, gener-ates further cellular debris and these debris (DAMPs), together with dysfunctional mitochondria released from microglia expressing neurotoxic proteins, exacerbate astrocyte activation and chronic pathogenic inflammation. Thus, neuronal cell death and the final phenotype of the disease occur via innate immune response activation as well as via direct effects of neurotoxic protein-induced neuronal cell death.
Activation of the innate immune response and neurotoxic pro-tein-induced neuronal cell death in models of neurodegenerative diseases are both dependent on Drp1–Fis1-mediated excessive mitochondrial fragmentation. We have previously shown that expression of neurotoxic proteins in cultured neurons and the resulting cell death can be greatly blunted by inhibiting Drp1–Fis1-mediated mitochondrial fragmentation using P110 peptide16,20. Here, we showed that the innate immune response in several mod-els of neurodegenerative diseases is also dependent on pathological Drp1–Fis1-dependent mitochondrial fission in the microglia and the resulting release of dysfunctional mitochondria into the extra-cellular milieu. However, the mechanisms by which mitochondria are released to the extracellular space are still under investigation. For example, mitochondrial transfer may occur via release in extra-cellular vesicles41, structures that are 50–1,000 nm in diameter42, and astrocytes and microglia release extracellular vesicle43–45. But other mechanisms, including direct exocytosis, may contribute to this release46. Also not yet known is the minimal amount of dam-aged mitochondria required for the propagation of neuronal cell death and whether transfer of functional mitochondria between
Fig. 6 | Dysfunctional extracellular mitochondria release from activated primary rat and mouse microglia or astrocytes or human primary monocyte-derived microglial cells occurs in a Drp1–Fis1-specific manner. a, Protocol of the experiment in b and c. b, Levels of mitochondrial membrane potential and ATP in mitochondria-enriched pellets collected from conditioned culture media (CM) of rat primary microglia, as depicted in the scheme. Extracellular mitochondria were removed from the media using 0.2-μm filters (MitoΔ). c, Integrity of the extracellular mitochondria was analyzed for markers of outer membrane proteins (TOM20 and VDAC) and cytochrome c (Cyto c), an inter-membrane space protein that readily leaks out when the integrity of the outer mitochondrial membrane is compromised. β-actin of the cell extracts from the corresponding plates was used as a loading control. d, Top: protocol of the experiment. Bottom: levels of mitochondrial membrane potential and ATP in mitochondria-enriched pellets collected from conditioned culture media of primary microglia isolated from WT and R6/2 mice treated without or with P110 (1 µM, added once 15 min before LPS 0.5 µg ml–1) and for 24 h in serum-free and antibiotic-free DMEM. e, Top: protocol of the experiment. Bottom: levels of mitochondrial membrane potential and ATP in mitochondria-enriched pellet collected from conditioned culture media of transiently expressing Q23 (control) or Q73 (HD model) or human SOD1-WT (control) or SOD1-G93A mutant (ALS model) for 48 h treated without or with P110 (1 µM). f, Top: protocol of the experiment. Bottom: levels of mitochondrial membrane potential and ATP in mitochondrial pellets collected from conditioned culture media of a model of human microglia. Extracellular mitochondria were removed from the media using 0.2-μm filters (MitoΔ), as in a. g, Top: protocol of the experiment. Bottom: membrane potential and ATP levels of the extracellular mitochondria released from rat primary astrocytes treated with rat MCM. Extracellular mitochondria were removed from the media using 0.2-μm filters (MitoΔ). h, The integrity of the extracellular astrocytic mitochondria was analyzed as in c. Extracellular mitochondria were removed from the media using 0.2-μm filters (MitoΔ). i, The morphology and structural organization of the extracellular mitochondria were examined using electron microscopy micrographs of pelleted mitochondria from the conditioned media of astrocytes. Arrows indicate loss of mitochondrial membrane integrity. j, Top: protocol of the experiment. Bottom: membrane potential of the extracellular mitochondria released from primary rat astrocytes treated as in Fig. 5a; in addition to the Drp1–Fis1-selective inhibitor, cells were treated with P259, a Mff–Drp1-selective inhibitor26 (1 μM) or with Mdivi-1 (20 μM), a Drp1 GTPase inhibitor30. k, The integrity of the extracellular mitochondria was analyzed as in c and represented as a ratio to β-actin in total lysate. Extracellular mitochondria were removed from the media using 0.2-μm filters (MitoΔ) as in a. Data were evaluated by one-way ANOVA and Holm–Sidak’s multiple comparisons tests for multiple testing between each treatment group. All graphs represent the mean ± s.d., and P values are indicated (NS, not significant). All experiments were performed in biologically independent replicates. b, n = 12; c, n = 3; d, n = 9; e, n = 7; f, n = 13 (JC-1), n = 7 (ATP); g, n = 13 (JC-1), n = 9 (ATP); h, n = 3; i, n = 2; j,k, n = 3.
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience1644

ArticlesNATure NeuroscIeNce
microglia and astrocytes and from glia to neurons has a role under physiological conditions. What we do know, however, is that extracellular mitochondria are critical in mediating this cell-to-cell propagation of pathological signaling.
The ratio between damaged mitochondria and functional mito-chondria in the extracellular milieu governs the outcome of neu-rons. Although extracellular damaged mitochondria are injurious, transfer of functional mitochondria is protective (Supplementary Fig. 4), as has been previously shown, for example, in a mouse model of acute lung injury47 and in a model of stroke2. Whether the extracellular damaged mitochondria enter the neurons, as has been suggested for functional mitochondria in a previous study2, has not yet been determined. The novel finding of our study is that it is not the amount of the extracellular mitochondria but rather the
ratio between damaged mitochondria and functional mitochondria in the extracellular milieu that governs the outcome of neurons, and is determined by the extent of pathological fission in the donor microglia. The structural and functional integrity of the released glial mitochondria were confirmed by measuring respiration, mito-chondrial membrane potential, ATP production, content of cyto-chrome c (indicating mitochondrial outer membrane integrity) and by electron microscopy analyses. Although the amount of extra-cellular mitochondria was unaltered, all these parameters showed improved structure and function of the extracellular mitochondria released from microglia after treatment with P110.
Our data suggested that selective inhibition of pathological mitochondrial fission in the microglia (mediated by Drp1–Fis1)16 without affecting physiological mitochondrial fission (mediated by Drp1–Mff)26 reduced propagation of neuronal injury by two
0
0.5
1.0
1.5
2.0
JC-1
acc
umul
atio
n(f
old
chan
ge)
WT R6/2 WT R6/2 WT R6/2– – P110
LPS
P110WT R6/2 WT R6/2 WT R6/2
– – P110
LPS
P110
0.0002 <0.0001<0.0001
<0.0001
0.0018
<0.0001
0
0.5
1.0
1.5
2.0
AT
P le
vels
(fo
ld c
hang
e)
0
0.5
1.0
1.5
2.0
AT
P le
vels
(fo
ld c
hang
e)
AT
P le
vels
(fo
ld c
hang
e)
<0.00010.0001
0.0030<0.0001
<0.0001
<0.0001
0
0.5
1.0
1.5
2.0
AT
P le
vels
(fo
ld c
hang
e) <0.0001
0.0019
0
0.5
1.0
1.5
2.0
JC-1
acc
umul
atio
n(f
old
chan
ge)
Con Veh P110
LPS/Nig
MitoΔ Con Veh P110
LPS/Nig
MitoΔ
MitoΔ
<0.00010.0050
0
0.5
1.0
1.5
2.0
JC-1
acc
umul
atio
n(f
old
chan
ge)
Control Veh P110 P259 Mdivi-1
<0.0001
0.0013
0.0004
NS
0
0.5
1.0
1.5
Cyt
o c
/VD
AC
rat
io
Con Veh P110
LPS/Nig
0.0267 0.0321
0
0.5
1.0
1.5
2.0
JC-1
acc
umul
atio
n(f
old
chan
ge)
Con Veh P110
LPS/Nig
<0.0001
<0.0001
<0.0001
0
0.5
1.0
1.5
2.0
AT
P le
vels
(fo
ld c
hang
e)
Con Veh P110
LPS/Nig
<0.0001
<0.0001
<0.0001
0
0.5
1.0
1.5
2.0
Con Veh P110
LPS/Nig
<0.0001
0.0001
0
0.5
1.0
1.5
2.0
JC-1
acc
umul
atio
n(f
old
chan
ge)
Con Veh P110
LPS/Nig
MitoΔ
MitoΔ
MitoΔ
MitoΔ
MitoΔ ΔMito ΔMito
<0.0001<0.0001
<0.0001
0.0078
0.0005
0.0436
0
0.5
1.0
1.5
2.0
JC-1
acc
umul
atio
n(f
old
chan
ge)
Q23 WTVeh P110
Q73
Veh P110
G93A Q23 WTVeh P110
Q73
Veh P110
G93A
<0.0001
0.0123
<0.0001
0.0167
Isolated mitochondrialmembrane potential
Isolated mitochondrial ATP
Primary rat microglia Primary ratastrocytes
MCM (24 h)
LPS +/– Nigericin 1.2 µM or
CM mitochondria isolatedfor mitochondrial
measurement
Cyto c
VDAC
TOM20
MW
31
20
22
10
42
LPS/Nig
Human microglia
CM mitochondria isolated formitochondrial measurement
WT or R6/2 microglia
CM mitochondria isolated formitochondrial measurement
Isolated mitochondrial ATPIsolated mitochondrialmembrane potential
Isolated mitochondrialmembrane potential
Isolated mitochondrial ATP Isolated mitochondrialmembrane potential
Isolated mitochondrial ATP
Q23, Q73, SOD1-WT orSOD1-G93A
(48 h transfection)+/– P110 1 µM (24 h)
P110 1 µM (24 h)
CM mitochondria isolated formitochondrial measurement
BV2 microglia
Primary ratastrocytes
Tnf-α, IL-1α and C1q
Tnf-α, IL-1α and C1q
Tnf-α, IL-1α and C1q Tnf-α, IL-1α and C1q
P110, P259 or Mdivi-1(24 h)
CM mitochondriaisolated by centrifuging
at 13,000 × gfor mitochondrial
measurementCon Veh P11
0P25
9M
divi-1
Isolated mitochondrialmembrane potential
Veh P110
P259
Mdiv
i-1
β-actin
Cyto c
VDAC
TOM20
MW
31
20
22
10
42
Con
500 nm 200 nm 200 nm
500 nm 200 nm 200 nm
200 nm 200 nm 200 nm
200 nm 100 nm 200 nm
Veh P110
LPS/Nig
Collectmedium Supernatant
Cellular debris
Centrifuge1,000 × g for
10 min
Centrifuge13,000 × g for
25 min
Supernatant
MitochondriaATP assayWestern blotting
or
Microglia
Astrocytes
a
d
g
j k
h i
e f
b c
0.2 µm
MitoΔ
JC-1 accumulation
Cells lysed as loading control
+/– Nigericin 1.2 µM orP110 1 µM (24 h)
+/– Nigericin 1.2 µM orP110 1 µM (24 h)
+/– LPS 0.01 µg ml–1
+/– P110 1 µM (24 h)
LPS 0.1 µg ml–1
(3 h)
LPS 0.01 µg ml–1
(3 h)
Primary rat microglia
CM mitochondria isolated formitochondrial measurement
Isolatedmitochondrial ATP
Isolated mitochondrialmembrane potential
Cyto c
VDAC
TOM20
MW31
20
20
10
42
Con Veh P110
Veh P110
Con Veh P110
Veh P110
LPS/Nig
β-actin
Culture media
Total cell lysate
Total cell lysate
Culture media
Culture media
Total cell lysate
0
0.2
0.4
0.6
0.8
1.0
Cyt
o c
/VD
AC
rat
io
Con Veh P110
LPS/Nig
0.0004 0.0025
0
1
2
3
4
5
Cyt
o c
/VD
AC
rat
io
Control Veh P110 P259 Mdivi-1
0.0003
<0.0001
0.0021
NS
β-actin
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience 1645

Articles NATure NeuroscIeNce
mechanisms. First, P110 reduced activation of the innate immune response in microglia and in astrocytes and the cytokine-mediated neuronal cell death induced by extracellular dysfunctional mito-chondria. Second, inhibition of pathological mitochondrial fission by P110 in the donor microglia supported neuronal cell survival by increasing the ratio of healthy to damaged mitochondria released from the donor cells, thus protecting neurons.
Suppression of Drp1–Fis1-mediated mitochondrial fission is a readily translatable approach for interrupting this pathologi-cal microglia-to-astrocyte-to-neuron mitochondrial dysfunction, neuroinflammation and neuronal cell death mechanisms and
supporting transfer of healthy mitochondria to neurons. However, we envision that any means of normalizing the balance between healthy and damaged mitochondria within the neuronal milieu, for example, by clearing damaged and fragmented mito-chondrial with specific antibodies or by introducing healthy mitochondria, could also provide neuronal protection in neuro-degenerative diseases.
online contentAny methods, additional references, Nature Research reporting summaries, source data, statements of code and data availability
0
2
4
6
8
10LD
H r
elea
se (
fold
cha
nge)
NaiveACM
aACM aACM+ P110
<0.0001 <0.0001
0
2
4
6
8
10
LDH
rel
ease
(fo
ld c
hang
e)
NaiveACM
aACM aACM+ P110
aACMMitoΔ
aACM+ DNase
<0.0001<0.0001
0.00020.0208
0
20
40
60
80
100
EC
AR
(m
pH m
in–1
)
Glycolysis
<0.0001
0.0074
<0.0001
0.0083
0
200
400
600
OC
R (
pmol
min
–1)
Q23 Veh P110
Q73
WT Veh P110
G93A
OXPHOS
<0.0001
<0.0001
<0.0001
<0.0001
0
200
400
600
800
1,000
OC
R (
pmol
min
–1)
Maximal respiration
0.0202
<0.0001
0.0049NS
0
100
200
300
400
OC
R (
pmol
min
–1)
Spare respiration capacity
<0.0001<0.0001
<0.0001
0.0323
0
0.2
0.4
0.6
0.8
1.0
TM
RM
acc
umul
atio
n (a
.u.)
Mitochondrial membrane potential
<0.0001 0.0091
0
0.5
1.0
1.5
AT
P le
vels
(fo
ld c
hang
e)
NaiveACM
Veh P110aACM
NaiveACM
Veh P110aACM
NaiveACM
Veh P110aACM
<0.0001 <0.0001
0
1
2
3
4
Mito
SO
X (
fold
cha
nge)
Mitochondrial ROS
0.0053<0.0001
Primary ratmicroglia
Primary ratastrocytes
Primary ratneurons
MCM
MCM
ACM +/– removed mitochondria (MitoΔ)+/– LPS/Nig
a
b
d e
cQ23, Q73, WT or SOD1-G93A
Assay
Primary neuronal mitochondrial health
Rat microglia Rat astrocytes
MCM
LDH assayRat neurons
ACM – isolate mitochondria
Neuronal mitochondrial metabolism
BV2 microglia Primary mouseastrocytes
Primary mouseneurons
MCM
+/– P110 Assay
ACM
Rat microglia Rat astrocytesLDH assay
Rat neurons
ACM +/– removed mitochondria or +/– DNase
Resuspendin DMEM
0
200
400
600
800
OC
R (
pmol
min
–1)
OXPHOS
CM Veh P110
+/– P110
LPS/Nig ACM
MitoΔ MitoΔ CM Veh P110
LPS/Nig ACM
MitoΔ MitoΔ CM Veh P110
LPS/Nig ACM
MitoΔ MitoΔ
<0.0001NS
0.0001<0.0001
0
25
50
75
100
125
OC
R (
pmol
min
–1)
Q23 Veh P110
Q73
WT Veh P110
G93A
Spare respiratory capacity
<0.0001
<0.0001
<0.0001
<0.0001
0
0.5
1.0
1.5
AT
P le
vels
(fo
ld c
hang
e)
Q23 VehP110
Q73
WT VehP110
G93A Q23 VehP110
Q73
WT VehP110
G93A
<0.0001
<0.0001
<0.0001
<0.0001
+/– P110
+/– LPS/Nig +/– LPS/Nig+/– P110
Fig. 7 | Released (extracellular) glial mitochondria propagate mitochondrial dysfunction and cell death from activated microglia to neurons through astrocytes. a, Schematic of the experimental design. b, ATP levels, mitochondrial membrane potential (using TMRM), mitochondrial ROS (using MitoSOX), OXPHOS (OCR measurements), spare respiratory capacity and maximal respiration (using Seahorse) in rat primary cortical neurons treated with activated ACM (aACM; as in Supplementary Fig. 3a) that were activated by media from microglia treated as indicated with LPS/Nig in the presence or absence of P110 (1 μM). Extracellular mitochondria from the MCM were removed before transferring the MCM to the astrocytes using 0.2-μm filters (MitoΔ). OCR, TMRM and MitoSOX were measured at 24 h. c, Top: protocol of the experiment. Bottom: oxygen consumption of mouse primary neurons 24 h after exposure to ACM that were treated with conditioned media of BV2 microglia expressing Q73 or SOD1-G93A or the corresponding controls in the absence or presence of P110. d, Top: scheme of the experimental protocol. Bottom: LDH release (a marker of neuronal cell death) from primary cortical rat neurons treated with rat aACM and the consequence of removal of extracellular mitochondria (filtered through 0.2-μm filters (MitoΔ)) released from the astrocytes or removal of extracellular DNA released from the aACM (using DNase). e, Top: scheme of the experimental protocol. Bottom: rat neuronal cell death (assessed via LDH release) induced by transferring an enriched preparation of extracellular mitochondria isolated from primary rat aACM that were treated by MCM of primary rat microglia treated with LPS/Nig together with or without P110. Data were evaluated by one-way ANOVA and Holm–Sidak’s multiple comparisons tests for multiple testing between each treatment group. All graphs represent the mean ± s.d., and P values are indicated. All experiments were performed in biologically independent replicates. b, n = 7 (naive), n = 10 (aACM, ATP), n = 14 (naive), n = 12 (aACM, TMRM, MitoSOX), n = 9 (Seahorse); c, n = 6; d, n = 13; e, n = 9.
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience1646

ArticlesNATure NeuroscIeNce
and associated accession codes are available at https://doi.org/ 10.1038/s41593-019-0486-0.
Received: 14 August 2018; Accepted: 5 August 2019; Published online: 23 September 2019
References 1. Aguzzi, A. & O’Connor, T. Protein aggregation diseases: pathogenicity and
therapeutic perspectives. Nat. Rev. Drug Discov. 9, 237–248 (2010). 2. Hayakawa, K. et al. Transfer of mitochondria from astrocytes to neurons after
stroke. Nature 535, 551–555 (2016). 3. Belanger, M. & Magistretti, P. J. The role of astroglia in neuroprotection.
Dialogues Clin. Neurosci. 11, 281–295 (2009). 4. Chen, Z. & Trapp, B. D. Microglia and neuroprotection. J. Neurochem. 136,
10–17 (2016). 5. Crotti, A. & Glass, C. K. The choreography of neuroinflammation in
Huntington’s disease. Trends Immunol. 36, 364–373 (2015). 6. Heneka, M. T. et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol.
14, 388–405 (2015).
7. Liu, J. & Wang, F. Role of neuroinflammation in amyotrophic lateral sclerosis: cellular mechanisms and therapeutic implications. Front. Immunol. 8, 1005 (2017).
8. Ransohoff, R. M. How neuroinflammation contributes to neurodegeneration. Science 353, 777–783 (2016).
9. Liddelow, S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017).
10. Itoh, K., Nakamura, K., Iijima, M. & Sesaki, H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 23, 64–71 (2013).
11. Reddy, P. H. Increased mitochondrial fission and neuronal dysfunction in Huntington’s disease: implications for molecular inhibitors of excessive mitochondrial fission. Drug Discov. Today 19, 951–955 (2014).
12. Joshi, A. U. et al. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 10, e8166 (2018).
13. Khalil, B. & Lievens, J. C. Mitochondrial quality control in amyotrophic lateral sclerosis: towards a common pathway? Neural Regen. Res. 12, 1052–1061 (2017).
14. Reddy, P. H. et al. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 67, 103–118 (2011).
0
100
200
300
400
LDH
rel
ease
(%
)
WT R6/2 WT R6/2 WT R6/2– – P110
LPS
P110
0.0004
0.0002
0.0008 <0.0001
0
100
200
300
400
LDH
rel
ease
(%
)
WT R6/2 WT R6/2 WT R6/2
– – P110
LPS
P110
0.0004
0.0001
0.0238 0.0462
0
10
20
30
40
Mito
SO
X (
fold
cha
nge)
WT R6/2 WT R6/2 WT R6/2– – P110
LPS
P110
<0.0001
<0.0001
<0.0001
<0.0001
0
10
20
30
40M
itoS
OX
(fo
ld c
hang
e)
WT R6/2 WT R6/2 WT R6/2
– – P110
LPS
P110
0.0103
<0.0001
0.0015<0.0001
WT or R6/2 microglia WT astrocytes WT neurons
MCM+/– LPS (24 h)a
b
+/– P110 (24 h)
+/– LPS (24 h)
+/– P110 (24 h)
Assay
ACM
WT or R6/2 microglia R6/2 astrocytes
MCM
Assay
ACM
WT neurons
24 h
24 h
Fig. 8 | Wt or R6/2 astrocytes induce Drp1–Fis1-dependent propagation of neuronal mitochondrial dysfunction and neuronal cell death from R6/2 microglia to naive mouse cortical neurons. a, Top: schematic of the experimental design using primary microglia (treated without or with LPS) that were isolated from R6/2 mice or their littermate WT mice. Bottom: mitochondrial ROS and LDH release in mouse primary cortical neurons treated with activated aACM from WT mouse astrocytes. b, Top: the treatment protocol is as in a, except that, in addition to the microglia, the primary astrocytes used were isolated from R6/2 mice or the WT littermates as indicated. Bottom: mitochondrial ROS and LDH release of these cells. All graphs represent the mean ± s.d., and P values are indicated. All experiments were performed in biologically independent replicates. a, n = 5 (MitoSOX), n = 4 (LDH); b, n = 5.
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience 1647

Articles NATure NeuroscIeNce
15. Knott, A. B., Perkins, G., Schwarzenbacher, R. & Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 9, 505–518 (2008).
16. Qi, X., Qvit, N., Su, Y. C. & Mochly-Rosen, D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 126, 789–802 (2013).
17. Grohm, J. et al. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 19, 1446–1458 (2012).
18. Joshi, A. U., Saw, N. L., Shamloo, M. & Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 9, 6128–6143 (2018).
19. Disatnik, M. H. et al. Potential biomarkers to follow the progression and treatment response of Huntington’s disease. J. Exp. Med. 213, 2655–2669 (2016).
20. Guo, X. et al. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Invest. 123, 5371–5388 (2013).
21. Mishra, P. & Chan, D. C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 212, 379–387 (2016).
22. Crotti, A. et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat. Neurosci. 17, 513–521 (2014).
23. Sarkar, S. et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinsons Dis. 3, 30 (2017).
24. Liu, Y., Hao, W., Dawson, A., Liu, S. & Fassbender, K. Expression of amyotrophic lateral sclerosis-linked SOD1 mutant increases the neurotoxic potential of microglia via TLR2. J. Biol. Chem. 284, 3691–3699 (2009).
25. Sondag, C. M., Dhawan, G. & Combs, C. K. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J. Neuroinflamm. 6, 1 (2009).
26. Kornfeld, O. S. et al. Interaction of mitochondrial fission factor with dynamin related protein 1 governs physiological mitochondrial function in vivo. Sci. Rep. 8, 14034 (2018).
27. Falchi, A. M. et al. Astrocytes shed large membrane vesicles that contain mitochondria, lipid droplets and ATP. Histochem. Cell Biol. 139, 221–231 (2013).
28. Gogvadze, V., Orrenius, S. & Zhivotovsky, B. Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim. Biophys. Acta 1757, 639–647 (2006).
29. Ryan, K. J. et al. A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci. Transl Med. 9, eaai7635 (2017).
30. Cassidy-Stone, A. et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204 (2008).
31. Yates, D. Neurodegenerative disease: factoring in astrocytes. Nat. Rev. Neurosci. 16, 67 (2015).
32. West, A. P. et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015).
33. Zhong, Z. et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018).
34. Chou, S. H. et al. Extracellular mitochondria in cerebrospinal fluid and neurological recovery after subarachnoid hemorrhage. Stroke 48, 2231–2237 (2017).
35. Zussman, B., Weiner, G. & Ducruet, A. Mitochondrial transfer into the cerebrospinal fluid in the setting of subarachnoid hemorrhage. Neurosurgery 82, N11–N13 (2018).
36. Hayakawa, K. et al. Protective effects of endothelial progenitor cell-derived extracellular mitochondria in brain endothelium. Stem Cells 36, 1404–1410 (2018).
37. Hayakawa, K. et al. Extracellular mitochondria for therapy and diagnosis in acute central nervous system injury. JAMA Neurol. 75, 119–122 (2018).
38. Wilkins, H. M. et al. Extracellular mitochondria and mitochondrial components act as damage-associated molecular pattern molecules in the mouse brain. J. Neuroimmune Pharm. 11, 622–628 (2016).
39. Labzin, L. I., Heneka, M. T. & Latz, E. Innate immunity and neurodegeneration. Annu. Rev. Med 69, 437–449 (2018).
40. Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C. & Gage, F. H. Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934 (2010).
41. van Niel, G., D’Angelo, G. & Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19, 213–228 (2018).
42. Lai, R. C. et al. MSC secretes at least 3 EV types each with a unique permutation of membrane lipid, protein and RNA. J. Extra. Vesicles 5, 29828 (2016).
43. Taylor, A. R., Robinson, M. B., Gifondorwa, D. J., Tytell, M. & Milligan, C. E. Regulation of heat shock protein 70 release in astrocytes: role of signaling kinases. Dev. Neurobiol. 67, 1815–1829 (2007).
44. Hooper, C. et al. Wnt3a induces exosome secretion from primary cultured rat microglia. BMC Neurosci. 13, 144 (2012).
45. Glebov, K. et al. Serotonin stimulates secretion of exosomes from microglia cells. Glia 63, 626–634 (2015).
46. Hessvik, N. P. & Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 75, 193–208 (2018).
47. Islam, M. N. et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 18, 759–765 (2012).
50. Valente, A. J., Maddalena, L. A., Robb, E. L., Moradi, F. & Stuart, J. A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 119, 315–326 (2017).
AcknowledgementsIn memory of Ben Barres, whose advice and research inspired this study. This work was supported, in part, by a Stanford Discovery Innovation Award and R01 HL52141 to D.M.-R., a Paul & Daisy Soros Fellowship to P.S.M., a postdoctoral fellowship from the Australian National Health and Medical Research Council (GNT1052961) and the Glenn Foundation Glenn Award to S.A.L., RO1AG058047 to K.I.A., and R35 HL135736 to G.W.D. The authors thank N. Raju and S. Avolicino for assistance with immunohistochemistry and J. Perrino for technical support with electron microscopy.
Author contributionsA.U.J. and D.M.-R. generated the hypothesis and the experimental design. A.U.J., D.M.-R. and G.W.D. contributed to the manuscript preparation. K.I.A. contributed to the experimental design. A.U.J. performed isolations of neurons, microglia and astrocytes, collected, analyzed and interpreted the results from earlier mouse studies, and qPCR, ELISA, immunoblots and mitochondrial assays. P.S.M. performed isolations of human monocyte-derived microglia, neurons, microglia and astrocytes, collected data from Seahorse experiments and helped with data analyses. B.H. performed the mitochondrial assays, ELISA and immunoblots and helped with data analyses. S.A.L. isolated microglia and performed microfluidic qPCR and helped with data analyses. All the authors reviewed and edited the manuscript.
competing interestsPatents on P110 and its utility in HD, ALS and other neurodegenerative diseases have been filed by D.M.‐R. and A.U.J. The other authors declare no competing interests.
Additional informationSupplementary information is available for this paper at https://doi.org/10.1038/s41593-019-0486-0.
Reprints and permissions information is available at www.nature.com/reprints.
Correspondence and requests for materials should be addressed to D.M.-R.
Peer review information Nature Neuroscience thanks Robert Friedlander, Krista Spiller and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
© The Author(s), under exclusive licence to Springer Nature America, Inc. 2019
NAtURe NeURoScIeNce | VOL 22 | OCTOBER 2019 | 1635–1648 | www.nature.com/natureneuroscience1648

ArticlesNATure NeuroscIeNce
MethodsData reporting. All procedures were conducted in accordance with the animal care standards of the National Institutes of Health and approved by Stanford University’s Administrative Panel on Laboratory Animal Care. All chemicals were purchased from Sigma‐Aldrich unless stated otherwise.
Peptides. Drp1–Fis1 peptide inhibitor P110 (derived from Drp149–55-GG-TAT47-57), Drp1–Mff peptide inhibitor P259 (derived from Drp122–31-GG-TAT47–57) and control peptide TAT (derived from TAT47–57) were synthesized by Ontores Biotechnologies12. The purity of the peptides was >90% as measured by reverse-phase high-performance liquid chromatography.
Cell lines and transient expression of mutant proteins. BV2 mouse microglial cells were cultured in 75-cm2 flasks or in 6-well or 24-well dishes containing RPMI medium supplemented with 10% fetal bovine serum (FBS), 4 mM glutamine, 100 U ml–1 penicillin, 10 μg ml–1 streptomycin and grown in a 5% CO2 atmosphere at 37 °C. Cultured BV2 microglia were seeded at 5 × 104 cells per ml and allowed to attach before transfection with green fluorescent protein (GFP)-SOD1-WT (control) or mutant SOD1-G93A (ALS-like model) or with Q23 (control) or Q73 (HD-like model) using Lipofectamine 2000 transfection reagent according to the manufacturer’s instructions (Invitrogen) for 48 h. Httex1Q23 and Q73 constructs were obtained from the CHDI Foundation. pF145 pAcGFP1 SOD1-G93A and pF141 pAcGFP1 SOD1-WT were a gift from Elizabeth Fisher (Addgene plasmid nos. 26406 and 26402)48. Transfection efficiency was tested by using a vector containing the GFP gene and usually reached 60%.
Primary neuron cultures. Primary neuron cultures were prepared from cerebral cortices of embryonic day 17 (E17) Sprague–Dawley rat embryos or E17 C57BL/6 mice. In brief, cortices were dissected and dissociated using a papain dissociation system (Worthington Biochemical, cat. no. LK003150). Cells were spread at 20,000 per well of a 96-well plate coated with poly-d-lysine (Sigma, cat. no. P7886) for cell health assays. For Seahorse experiments, 1 × 105 cells per well were seeded in a XF 24-well cell culture microplate and cultured in Neurobasal medium (Invitrogen, cat. no. 21103-049) supplemented with B-27 (Invitrogen, cat. no. 17504044) containing 25 mM glucose, 4 mM glutamine, 1 mM sodium pyruvate and 5% FBS. At 24 h after seeding, the medium was changed to Neurobasal medium supplemented with B-27 and 0.5 mM glutamine. Cells were cultured at 37 °C in a humidified chamber of 95% air and 5% CO2. Cultures were used for experiments from 7 to 10 days after seeding.
Primary astrocyte and microglia cultures from WT rats and mice and from R6/2 mice and their littermates. Primary astrocyte cultures were prepared from cerebral cortices of 2-day-old neonatal Sprague–Dawley rats or postnatal day 1–2 C57BL/6J mice or R6/2 or littermates (B6CBA-TgN (HD exon1)62; JAX stock no. 006494). In brief, dissociated cortical cells were suspended in DMEM/F12 50/50 (Life Technologies, cat. no. 11320-033) containing 25 mM glucose, 4 mM glutamine, 1 mM sodium pyruvate and 10% FBS, and plated on uncoated 25-cm2 flasks at a density of 6 × 105 cells cm–2. Monolayers of type 1 astrocytes were obtained 12–14 days after plating. Cultures were gently shaken, and floating cells (microglia) were collected, resulting in more than 95% pure culture of astrocytes. Astrocytes were dissociated by trypsinization and then reseeded at 3–5 × 105 cells cm–2 or 1.5 × 105 cells per well in 24-well or 1.5 × 106 cells per well in 6-well or 1,200 cells per well in 96-well poly-d-lysine-coated culture vessels in DMEM F12 50/50 containing 15% FBS and 1% penicillin–streptomycin. For Seahorse experiments, astrocytes were seeded at 4 × 104 cells per well in a XF 24-well cell culture microplate. At the start of each experiment, the medium was changed to DMEM without serum and antibiotics. Astrocytic-conditioned media (ACM) were collected 24 h later and cleared of cellular debris by centrifugation at 1,000 × g for 10 min.
Isolated microglia cells collected after shaking, mixed glial cultures and primary microglia were plated at 2.4 × 106 cells per well in 6-well plates or 3 × 105 cells per well in 24-well plates or 2,500 cells per well in 96-well plates and incubated with serum-free and antibiotic free macrophage medium (Invitrogen, cat. no. 12065074) for 24 h before switching to DMEM without serum and antibiotics at the start of various experiments. Collected MCM was then centrifuged at 1,000 × g for 10 min to remove cell debris. For the Seahorse experiments, microglia were seeded at 1.25 × 105 cells per well in a XF 24-well cell culture microplate.
Primary human monocyte-induced microglia. Primary human monocyte-derived microglia-like cells were generated as previously described29,49. In brief, peripheral blood mononuclear cells from de-identified healthy donors (35–65 years old) were obtained from the Stanford Blood Center. Samples were diluted with 20 ml PBS and layered onto 10 ml of Ficoll-Paque (GE Healthcare, cat no. 17144002) using a Pasteur pipette. Tubes were centrifuged at 1,500 r.p.m. for 25 min without brake. The mononuclear cell layer was isolated, resuspended in 50 ml of PBS and then centrifuged at 1,500 r.p.m. for 10 min. Monocytes were then isolated using a Pan Monocyte Human Isolation kit (MACS; Miltenyi Biotech, cat no. 130-096-537) and plated at a confluency of 10 × 106 cells per 10-cm plate. Cells were differentiated for 10 days at 37 °C, 5% CO2 in RPMI medium 1640 GlutaMAX
(ThermoFisher Scientific, cat no. 61870036) and supplemented with 10% FBS, 1% penicillin–streptomycin, 10 ng ml–1 macrophage colony-stimulating factor, 10 ng ml–1 granulocyte–macrophage colony-stimulating factor, 10 ng ml–1 nerve growth factor-β, 100 ng ml–1 chemokine ligand 2 and 100 ng ml–1 IL-34 (all BioLegend CNS). All the experiments were performed in serum-free antibiotic-free RPMI.
oAβ preparation. Synthetic Aβ1–42 was purchased from AnaSpec (cat. no. AS-25381) and prepared according to protocols described elsewhere18. Briefly, lyophilized Aβ1–42 peptides were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP; Sigma Aldrich, cat. no. 325244) and aliquoted into polypropylene microcentrifuge tubes. HFIP was evaporated and the resulting peptide films were stored at −80 °C. Before use, these peptide films were reconstituted at a concentration of 1 mM in dimethylsulfoxide (DMSO; Sigma Aldrich, cat. no. D8418), then subsequently diluted to 100 μM with ice-cold DMEM (phenol-red-free, Invitrogen cat. no. 21063029) and incubated for 24 h at 4 °C to facilitate the formation of oligomers. Resultant peptides were stored at −80 °C until use.
Cell health assays. Cell viability assays. Cell viability was determined by measuring the activity of LDH released using a Cytotoxicity Detection kitPLUS (LDH) (Sigma Aldrich, cat. no. 4744934001) according to the manufacturer’s instructions. Briefly, at the end of the incubation period, supernatants of culture media were collected and centrifuged at 1,000 × g to remove any residual cell debris. The reaction mixture was added to the cell-free supernatant, and color development was measured spectrophotometrically at 492 nm with a reference wavelength at 610 nm. Primary astrocytes and primary neurons were treated with different stimuli for 48 or 24 h, respectively, before analysis.
Mitochondrial ROS production. To determine mitochondrial ROS production, cells were treated with 5 μM MitoSOX Red mitochondrial superoxide indicator (Invitrogen, cat. no. M36008) and 10 μg ml–1 of the nuclear staining dye Hoechst-33342 (Sigma-Aldrich, cat. no. B 2261) at the end of the experiment for 15 min at 37 °C according to the manufacturers’ protocols. Fluorescence was recorded for MitoSOX (excitation (Ex) 510 nm and emission (Em) 580 nm) and Hoechst-33342 (Ex 350 nm and Em 461 nm) in a SpectraMax M2e (Molecular Devices). The MitoSOX fluorescence signal was normalized to the Hoechst reading. Mitochondrial ROS levels in primary microglia were analyzed at 16 h for the HD and ALS models, 21 h for the LPS and nigericin model or 24 h for the AD model after individual stimuli. Mitochondrial ROS levels in primary astrocytes were analyzed either at 6 h for the HD and ALS models or 24 h for all other stimuli. Mitochondrial ROS levels in primary neurons were analyzed at 24 h after stimuli.
ATP measurement. Relative intracellular or extracellular ATP levels were determined using an ATP-based CellTiter-Glo Luminescent Cell Viability kit (Promega, cat. no. G7570), which can perform cell lysis and generate a luminescence signal proportional to the amount of ATP present. In brief, for intracellular ATP levels, opaque-walled 96-well plates with cell lysate (50 μl) were prepared. An equal volume of the single-one-step reagent provided in the kit was added to each well and incubated for 30 min at room temperature. For measuring the ATP content in extracellular mitochondria, cell supernatant was cleared of cellular debris by centrifugation at 1,000 × g for 10 min and then centrifuged at 13,000 × g for 25 min followed by a wash with 1 ml of PBS. The pellet was then resuspended in the 50 µl of serum-free phenol-free DMEM or PBS before an equal volume of the single-one-step reagent provided in the kit with incubation for 30 min at room temperature. ATP content was measured using a luminescence plate reader SpectraMax M2e (Molecular Devices). Mitochondrial ATP levels in primary microglia were analyzed at 16 h for the HD and ALS models, 21 h for the LPS and nigericin model or 24 h for the AD model after individual stimuli. Mitochondrial ATP levels in primary astrocytes were analyzed either at 6 h for the HD and ALS models or 24 h for all other stimuli. Mitochondrial ATP levels in primary neurons were analyzed at 24 h after stimuli. For extracellular mitochondria experiments, ATP levels in the pellet (after 13,000 × g centrifugation) were measured after the end of total stimulation (24 h).
Mitochondria membrane potential measurement. To monitor mitochondrial health, JC-1 dye (Invitrogen, cat. no. T-3168) was used to assess the mitochondrial membrane potential. In brief, cell supernatant was cleared of cellular debris by centrifugation at 1,000 × g for 10 min at the end of the experiment (24 h). The cell supernatant was then loaded with 1 μM JC-1 dye for 30 min at 37 °C. JC-1 dye exhibits potential-dependent accumulation in mitochondria, as indicated by a fluorescence emission shift from green (Ex 485 nm, Em 516 nm) to red (Ex 579 nm, Em 599 nm). Mitochondrial membrane potential was determined by measuring the fluorescent ratio using a SpectraMax M2e (Molecular Devices). Cells were incubated with tetra‐methyl‐rhodamine methyl ester (TMRM; Invitrogen, cat. no. T668) in HBSS (Hank’s balanced salt solution; Invitrogen, cat. no. 14170112) for 30 min at 37 °C, as per the manufacture’s protocol, and the fluorescence was analyzed using a SpectraMax M2e (Molecular Devices, using Ex 360 nm and Em 460 nm). Mitochondrial membrane levels in primary microglia and primary astrocytes were analyzed at 24 h after individual stimuli.
NAtURe NeURoScIeNce | www.nature.com/natureneuroscience

Articles NATure NeuroscIeNce
Bioenergetic profiles. Cells were plated in a Seahorse XF 24-Cell Culture Microplate (Agilent). All Seahorse experiments using microglia, astrocytes and neurons were performed at 24 h after individual stimuli. At the end of treatment, cells were washed twice with Agilent Seahorse XF Media (Agilent) supplemented with 1 mM pyruvate, 2 mM l-glutamine and 2 mM d-glucose. A final volume of 525 μl was placed in each well. Cells were then incubated in a 0% CO2 chamber at 37 °C for 1 h before being placed into a Seahorse XFe24 Analyzer (Agilent). For oxygen consumption rate (OCR) and (extracellular acidification rate) ECAR experiments, cells were treated with 1 μM oligomycin, 2 μM carbonyl cyanide p-trifluoromethoxy phenylhydrazone (FCCP) and 0.5 μM rotenone/antimycin. A total of three OCR and pH measurements were taken after each compound was administered.
Immunocytochemistry and immunohistochemistry. Immunohistochemistry was performed as previously described12. Mitochondrial structure analysis in microglia was performed after 24 h of the initial stimuli. For astrocytes, MCM was added to astrocytes for 24 h before analysis. Cells cultured on 8-well chamber slides were washed with cold PBS, fixed in 4% formaldehyde and permeabilized with 0.1% Triton X-100. After incubation with 2% normal goat serum (to block nonspecific staining), fixed cells were incubated overnight at 4 °C with anti-TOM20 primary antibody (1:500, Santa Cruz). Cells were washed with PBS and incubated for 60 min with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG (1:500 dilution). The cells were then washed gently with PBS and counterstained with Hoechst 33342 (1:10,000 dilution, Molecular Probes) to visualize nuclei. The coverslips were mounted with Slowfade-antifade reagent (Invitrogen). Parameters of mitochondrial morphology were further quantified with Fiji (ImageJ) as previously described50. A detailed workflow is presented in Supplementary Fig. 5.
For the immunohistochemistry studies, mice described in our previous publications were12,18,19 were used. Adult B6SJL Tg (SOD1-G93A) 1 Gur/J male mice with a high copy number of the mutant allele and their WT littermates at 120 days, 5XFAD transgenic male mice and their littermates at 6 months, and hemizygous male R6/2 HD mice and their WT littermates at 12 weeks were analyzed. In brief, mice were killed, tissue dissected and fixed in 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. Tissues were then paraffin‐embedded, and sections were used for immunohistochemical staining of Gfap and Iba-1 (done by Histo-tec Laboratory). The images were acquired using an All‐in‐One Fluorescence Microscope BZ‐X700 (Keyence). The level of Gfap or Iba-1 immunoreactivity was measured by using the mean optical density of diaminobenzidine staining. The white balance of all images was standardized to eliminate color bias.
Isolation of mitochondria-enriched fraction and lysate preparation for western blot analysis. Cells were washed with cold PBS at pH 7.4 and scraped off using mannitol–sucrose buffer containing 210 mM mannitol, 70 mM sucrose, 5 mM MOPS (3-(N-morpholino) propanesulfonic acid), 1 mM EDTA and protease inhibitor cocktail, pH 7.4. The collected cells were passed through a 27-gauge half-inch needle for lysis, followed by centrifugation at 800 × g to pellet nuclei. The post-nuclear supernatant was further centrifuged at 10,000 × g for 20 min to collect a mitochondria-enriched fraction, as previously described12.
Electron microscopy. Pellets from ACM (precleared cell supernatant obtained after centrifugation at 1,000 × g for 10 min followed by 13,000 × g for 15 min) were fixed in 2.0% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4 for 1 h at room temperature on a rocker. They were rinsed in cacodylate buffer, gently scraped and pelleted and post-fixed in 1.0% osmium tetroxide in cacodylate buffer for 1 h on ice. They were then rinsed in buffer and stabilized with a small amount of 2% agarose in PBS to hold them together. Samples were then dehydrated through a graded series of ethanol to 100%, followed by propylene oxide, 100%. Epon resin (Ted Pella) in a 1:1 solution of Epon: propylene oxide was added and then incubated overnight on a rocker at room temperature. The following day, samples were placed in fresh Epon for several hours and then embedded in Epon overnight at 60 °C. Samples were observed in a JEOL 1230 transmission electron microscope at 80 kV, and photographs were taken using a Gatan Multiscan 791 digital camera.
ELISA experiments. Mouse/rat IL-6 (cat. no. 5017218/ 50-183-46), mouse/rat IL-1α (cat. no. 501125107/ 5018351), mouse/rat TNF-α (cat. no. 50−112−8954/ 5017347) and IL-1β (cat. no. 501129749/ 501125471) ELISA kits (eBiosciences) were used to quantify cytokine levels in mouse tissue and cell supernatant according to the manufacturer’s protocols.
RNA isolation and gene expression analysis. RNA isolation was performed using a GenElute Mammalian Total RNA Miniprep kit (Sigma Aldrich, cat. no. RTN350) according to the manufacturer’s protocol. RNA concentration was measured using a Nanodrop instrument (ND −1000; NanoDrop Technologies) and RNA integrity was assessed using a Bioanalyzer (2100; Agilent Technologies). Complementary DNA synthesis was performed using a Quantitect reverse transcription kit (Qiagen, cat. no. 205313) according to the manufacturer’s instructions, with a minimal input of 200 ng total RNA. Quantitative PCR (qPCR)
was performed using a 7300 Real Time PCR system (Applied Biosystems) using the equivalent cDNA amount of 1–2 ng total RNA used in cDNA synthesis. SYBRgreen master mix (Applied Biosystems, cat. no. 43-127-04) and a 2 pmol ml–1 mix of forward and reverse primer sequences were used for 40 cycles of target gene amplification. The following primers were used in this study: 18SrRNA: 5′-CTTAGAGGGACAAGTGGCG, 3′-ACGCTGAGCCAGTCAGTGTA; Arg1: 5′-ATGGAAGAGACCTTCAGCTAC, 3′-GCTGTCTTCCCAAGAGTTGGG; Axl: 5′-ATGGCCGACATTGCCAGTG, 3′-CGGTAGTAATCCCCGTTGTAGA; C1qa: 5′-AAAGGCAATCCAGGCAATATCA, 3′-TGGTTCTGGTATGGACTCTCC; C1qb: 5′-AAGATCCAGAAACACAAGTCCCT, 3′-CCTCCTCACCATCAAAT GTTGG; C3: 5′-CCAGCTCCCCATTAGCTCTG, 3′-GCACTTGCCTCTTTA GGAAGTC; Cat: 5′-TAAGACTGACCAGGGCA, 3′-CAAACCTTGGTGAGAT CGAA; Ccl4: 5′-TTCCTGCTGTTTCTCTTACACCT, 3′-CTGTCTGCCTCTTT TGGTCAG; Cd14: 5′-GGCGCTCCGAGTTGTGACT, 3′-TACCTGCTTCAGC CCAGTGA; Cd86: 5′-TGTTTCCGTGGAGACGCAAG, 3′-TTGAGCCTTTGT AAATGGGCA; Gapdh: 5′-CATCACTGCCACCCAGAAGACTG, 3′-ATGCCA GTGAGCTTCCCGTTCAG; Gas6: 5′-TGCTGGCTTCCGAGTCTTC, 3′-CGGG GTCGTTCTCGAACAC; GFAP: 5′-CCCTGGCTCGTGTGGATTT, 3′-GACCG ATACCACTCCTCTGTC; Il10: 5′-ATTTGAATTCCCTGGGTGAGAAG, 3′-CA CAGGGGAGAAATCGATGACA; Il13: 5′-AGACCAGACTCCCCTGTGCA, 3′-TGGGTCCTGTAGATGGCATTG; Il33: 5′-TGAGACTCCGTTCTGGCC TC, 3′-CTCTTCATGCTTGGTACCCGAT; Il4: 5′-AGATGGATGTGCCAAA CGTCCTCA, 3′-AATATGCGAAGCACCTTGGAAGCC; Itgam: 5′-ATGGAC GCTGATGGCAATACC, 3′-TCCCCATTCACGTCTCCCA; Itgb2: 5′-CACTGT CTCAGTTGTGTACCAAG, 3′-GCTCTGGTGTATCACAGCGAA; Itgb3: 5′GAG CCCATTTTCTTCTCCCG, 3′-GCAACACCATGAATCCATCCC; Megf10: 5′-GA AGACCCCAACGTATGCAG, 3′-CGGTGCAGCTTGTGTAGTAGA; Mertk: 5′-CAGGGCCTTTACCAGGGAGA, 3′-TGTGTGCTGGATGTGATCTTC; Sod2: 5′-ACAGGCCTTATTCCACTGCT, 3′-CAGCATAACGATCGTGGTTT; Nox4: 5′TCTGGCTCT-CCATGAATGTC, 3′-CTGCTTGGAACCTTCTGTGA; Nfe2l2: 5′-TCTCCTCGCTGGAAAAAGAA, 3′-AATGTGCTGGCTGTGCTTTA; Ppargc1a: 5′-TGGAGTGACATCGAGTGTGCT, 3′-GAGTCCACCCAGAAAG CTGT; Tfam: 5′-ATTCCGAAGTGTTTTTCCAGCA, 3′-TCTGAAAGTTTTG CATCTGGGT; Tgfb1: 5′-GGAGAGCCCTGGATACCAAC, 3′-CAACCCAGGT CCTTCCTAAA; Ttr: 5′-TTGCCTCGCTGGACTGGTA, 3′-TTACAGCCACGT CTACAGCAG; Vegfa: 5′-GGAGATCCTTCGAGGAGCACTT, 3′-GGCGATTT AGCAGCAGATATAAGAA; Actb: 5′-TGGAATCCTGTGGCATCCATGAAAC, 3′-TAAAACGCAGCTCAGTAACAGTCCG.
Microfluidic qPCR (pooled-cell samples). Total RNA was extracted from cells using a RNeasy Plus kit (Qiagen, cat. no. 74136) and cDNA synthesis was performed using a SuperScript VILO cDNA synthesis kit (Invitrogen) according to the suppliers’ protocols. Microfluidic qPCR was conducted using 1.25 μl of each cDNA sample pre-amplified using 2.5 μl of 2× Taqman pre-amplification master mix (Applied Biosystems) and 1.25 μl of the primer pool (0.2 pmol each primer per μl, negative primers to rat transcripts for reactive astrocyte subtype were used as previously described9). Pre-amplification was performed (10 min of 95 °C denaturation step and 14 cycles of 15 s at 95 °C and 4 min at 60 °C) and reaction products were diluted 5× in TE buffer (Teknova). Five microliters from a sample mix containing pre-amplified cDNA and amplification Master mix (20 mM MgCl2, 10 mM dNTPs, FastStart Taq polymerase, DNA-binding dye loading reagent, 50× ROX and 20× Evagreen) was loaded into each sample inlet of a 96.96 Dynamic Array chip (Fluidigm) and 5 μl from an assay mix containing DNA-assay loading reagent, as well as forward and reverse primers (10 pmol μl−1) was loaded into each detector inlet. The chip was mixed and loaded in a NanoFlexTM 4-IFC Controller (Fluidigm) and processed in a BioMark Real-Time PCR System (Fluidigm) using a cycling program of 10 min at 95 °C followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s and 72 °C for 30 s. After completion of qPCR, a melting curve of amplified products was determined. Data were collected using BioMark Data Collection Software 2.1.1 build 20090519.0926 (Fluidigm) as the cycle of quantification (Cq), where the fluorescence signal of amplified DNA intersected with background noise. Fluidigm data were corrected for differences in input RNA using the geometric mean of three reference genes (Aldh1l1, Gapdh and Rplp0). Data preprocessing and analysis were completed using Fluidigm Melting Curve Analysis Software 1.1.0 build 20100514.1234 (Fluidigm) and Real-time PCR Analysis Software 2.1.1 build 20090521.1135 (Fluidigm).
Western blotting. Protein concentrations were determined using the Bradford assay (Thermo Fisher Scientific). Proteins were resuspended in Laemmli buffer containing 2‐mercaptoethanol, loaded onto SDS–PAGE gels and transferred onto nitrocellulose membranes, 0.45 μm (Bio‐Rad), as previously described12. For extracellular mitochondria analysis, cell supernatant was cleared of cellular debris by centrifugation at 1,000 × g for 10 min and then centrifuged 13,000 × g for 25 min followed by a wash with 1 ml of PBS. The pellet was then resuspended in the 20 µl of Laemmli buffer containing 2‐mercaptoethanol, loaded onto SDS–PAGE gels and transferred onto nitrocellulose membranes, 0.45 μm (Bio‐Rad), as previously described12. Membranes were cut at appropriate molecular weights and then probed with the indicated antibody and visualized by ECL (0.225 mM p‐coumaric acid; Sigma), 1.25 mM 3‐aminophthalhydrazide (Luminol; Fluka) in 1 M Tris
NAtURe NeURoScIeNce | www.nature.com/natureneuroscience

ArticlesNATure NeuroscIeNce
buffer, pH 8.5. Scanned images of the exposed X‐ray film or images acquired with Azure Biosystems C600 were analyzed using ImageJ to determine the relative band intensity (Supplementary Fig 6). Quantification was performed on samples from independent cultures for each condition. The following antibodies were used in this study: anti-ASC (B3) mouse monoclonal antibody (Santa Cruz Biotechnology, sc-514414; 1:200); anti-COX-2 rabbit polyclonal antibody (Abcam, Ab15191; 1:500); anti-cytochrome c (7H8.2C12) mouse monoclonal antibody (Abcam, ab13575; 1:1,000); anti-Drp1 (clone 8/DLP1 (RUO)) mouse monoclonal antibody (BD Transduction Laboratories, 611113; 1:500); anti-Enolase-1 rabbit polyclonal antibody (Cell Signaling Technology, 3810; 1:500); anti-GFAP rabbit polyclonal antibody (Abcam, ab7260; 1:500); anti-Iba-1 goat polyclonal antibody (Abcam, ab5076; 1:1,000); anti-IL1β rabbit polyclonal antibody (Abcam, ab9722; 1:200); anti-NLRP3 (D4D8T) rabbit monoclonal antibody (Cell Signaling Technology, 15101; 1:200); anti-p20 (D4) mouse monoclonal antibody (Santa Cruz Biotechnology, sc-398715; 1:100); anti-phospho SAPK/JNK (Thr183/Tyr185) rabbit polyclonal antibody (Cell Signaling Technology, 9251; 1:500); anti-phospho NFκB p65 (Ser536) (93H1) rabbit monoclonal antibody (Cell Signaling Technology, 3033; 1:500); anti-phospho p44/42 MAPK (Erk1/2) (Thr202/Tyr204) rabbit polyclonal antibody (Cell Signaling Technology, 9101; 1:500); anti-phospho-Drp1 (Ser616) rabbit polyclonal antibody (Cell Signaling Technology, 3455; 1:200); anti-phospho-Drp1 rabbit polyclonal antibody (Ser637) (Cell Signaling Technology, 4867; 1:200); anti-TOM20 (FL-145) rabbit polyclonal antibody (Santa Cruz Biotechnology, sc-11415; 1:500; discontinued); anti-VDAC1 (20B12AF2) mouse monoclonal antibody (Abcam, 14734; 1:500); and anti-β-actin (8H10D10) mouse monoclonal antibody (Cell Signaling Technology, 3700; 1:500). Secondary antibodies, including anti-mouse IgG (NA931V) and anti-rabbit IgG (NA934V), were obtained from GE Healthcare. Detailed antibody validation profiles are available on the websites of the companies that the antibodies were sourced from.
Tissue total lysate preparation. Samples were processed in the following lysis buffer: 10 mM HEPES-NaOH, pH 7.5, 150 mM NaCl, 1 mM EGTA, 1% Triton X-100, protease inhibitor cocktail and phosphatase inhibitor cocktail. After 20 min of incubation on ice, homogenates were spun at 14,000 r.p.m. for 20 min at 4 °C. The supernatants correspond to the total lysates.
Randomization and blinding. Animal and samples (mice) were randomly assigned to the various experimental groups, and mice were randomly selected for the analyses. In mouse imaging (immunohistochemistry and data acquisition from electron microscopy), the performer(s) was (were) blinded to the experimental design. Additionally, a few biological replicates for mitochondrial transfer experiments, mitochondrial health assays and Seahorse experiments were performed and analyzed by a person blinded to the experimental design.
Statistical analysis. Prism 8.0 (GraphPad Software) was used for the statistical analyses. Data shown are the mean ± s.d., with P < 0.05 considered statistically significant. Group differences were analyzed by one-way analysis of variance (ANOVA) followed by Sidak’s or Benjamini, Krieger and Yekutieli correction multiple comparisons tests or by two-way ANOVA followed by Tukey’s multiple comparisons test for multiple groups. No statistical methods were used to predetermine sample sizes, but our sample sizes were similar to those reported in previous publications9,18,19. Data distribution was assumed to be normal, but this was not formally tested. No data or animals were excluded.
Reporting summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availabilityThe data that support the findings of this study are available from the corresponding author upon reasonable request.
References 48. Stevens, J. C. et al. Modification of superoxide dismutase 1 (SOD1) properties
by a GFP tag—implications for research into amyotrophic lateral sclerosis (ALS). PLoS One 5, e9541 (2010).
49. Minhas, P. S. et al. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol. 20, 50–63 (2019).
NAtURe NeURoScIeNce | www.nature.com/natureneuroscience

1
nature research | reporting summ
aryO
ctober 2018
Corresponding author(s): Daria Mochly-Rosen
Last updated by author(s): Jul 2, 2019
Reporting SummaryNature Research wishes to improve the reproducibility of the work that we publish. This form provides structure for consistency and transparency in reporting. For further information on Nature Research policies, see Authors & Referees and the Editorial Policy Checklist.
StatisticsFor all statistical analyses, confirm that the following items are present in the figure legend, table legend, main text, or Methods section.
n/a Confirmed
The exact sample size (n) for each experimental group/condition, given as a discrete number and unit of measurement
A statement on whether measurements were taken from distinct samples or whether the same sample was measured repeatedly
The statistical test(s) used AND whether they are one- or two-sided Only common tests should be described solely by name; describe more complex techniques in the Methods section.
A description of all covariates tested
A description of any assumptions or corrections, such as tests of normality and adjustment for multiple comparisons
A full description of the statistical parameters including central tendency (e.g. means) or other basic estimates (e.g. regression coefficient) AND variation (e.g. standard deviation) or associated estimates of uncertainty (e.g. confidence intervals)
For null hypothesis testing, the test statistic (e.g. F, t, r) with confidence intervals, effect sizes, degrees of freedom and P value noted Give P values as exact values whenever suitable.
For Bayesian analysis, information on the choice of priors and Markov chain Monte Carlo settings
For hierarchical and complex designs, identification of the appropriate level for tests and full reporting of outcomes
Estimates of effect sizes (e.g. Cohen's d, Pearson's r), indicating how they were calculated
Our web collection on statistics for biologists contains articles on many of the points above.
Software and codePolicy information about availability of computer code
Data collection We used Excel 2015, Prism 6, 7, 8, Fiji ImageJ, BioMark Data Collection Software 2.1.1, Fluidigm Melting Curve Analysis Software 1.1.0, Real-time PCR Analysis Software 2.1
Data analysis We used Prism 7/ 8 for Mac OS X Version 7.0d/ 8.2 for data analysis
For manuscripts utilizing custom algorithms or software that are central to the research but not yet described in published literature, software must be made available to editors/reviewers. We strongly encourage code deposition in a community repository (e.g. GitHub). See the Nature Research guidelines for submitting code & software for further information.
DataPolicy information about availability of data
All manuscripts must include a data availability statement. This statement should provide the following information, where applicable: - Accession codes, unique identifiers, or web links for publicly available datasets - A list of figures that have associated raw data - A description of any restrictions on data availability
Provide your data availability statement here.
Field-specific reportingPlease select the one below that is the best fit for your research. If you are not sure, read the appropriate sections before making your selection.
Life sciences Behavioural & social sciences Ecological, evolutionary & environmental sciences

2
nature research | reporting summ
aryO
ctober 2018For a reference copy of the document with all sections, see nature.com/documents/nr-reporting-summary-flat.pdf
Life sciences study designAll studies must disclose on these points even when the disclosure is negative.
Sample size No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications
Data exclusions No samples were excluded from the study
Replication All attempts were successful.
Randomization Animal/samples (mice) were assigned randomly to the various experimental groups.
Blinding In data collection and analysis (e.g., Seahorse, microfluidic qPCR, as well as imaging and data analysis of IHC and EM), the performer(s) was blinded with experimental design.
Reporting for specific materials, systems and methodsWe require information from authors about some types of materials, experimental systems and methods used in many studies. Here, indicate whether each material, system or method listed is relevant to your study. If you are not sure if a list item applies to your research, read the appropriate section before selecting a response.
Materials & experimental systemsn/a Involved in the study
Antibodies
Eukaryotic cell lines
Palaeontology
Animals and other organisms
Human research participants
Clinical data
Methodsn/a Involved in the study
ChIP-seq
Flow cytometry
MRI-based neuroimaging
AntibodiesAntibodies used Anti-ASC Santa Cruz Biotechnology (Clone: B-3) sc-514414 1:200
Anti-COX-2 Abcam ab15191 1:500 Anti-cytochrome c Abcam [7Clone: H8.2C12] ab13575 1:1000 Anti-Drp1 BD Transduction Laboratories 611113 (Clone: 8/DLP1 (RUO)) (Lot No: 7341636)1:500 Anti-Enolase-1 Cell Signaling Technology (Lot 2) 3810 1:500 Anti-GFAP Abcam ab7260 1:500 Anti-Iba-1 Abcam ab5076 1:1000 Anti-IL1beta Abcam ab9722 1:200 Anti-NLRP3 Cell Signaling Technology (Clone: D4D8T) 15101 1:200 Anti-p20 Santa Cruz Biotechnology (Clone: D-4) sc-398715 1:100 Anti-phospho JNK (Thr183/Tyr185) Cell Signaling Technology (Lot 23) 9251 1:500 Anti-phospho NFkB Cell Signaling Technology (Clone: 93H1) 3033 1:500 Anti-phospho p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Cell Signaling Technology 9101 (Clone: 197G2) 1:500 Anti-phospho-Drp1 (Ser616) Cell Signaling Technology 3455 1:200 Anti-phospho-Drp1 (Ser637) Cell Signaling Technology 4867 1:200 Anti-TOM20 Santa Cruz Biotechnology (Clone: FL-145) sc-11415 1:500; discontinued Anti-VDAC1 Abcam (Clone: 20B12AF2) 14734 1:500 Anti-ebta-actin Cell Signaling Technology 3700 (Clone: 8H10D10, Lot: 16) 1:500 Second antibodies including anti-mouse IgG (#NA931V) and anti- rabbit IgG (NA934V) were from GE Healthcare and used at 1:5000 dilution.
Validation Antibodies specific for the required antigens/epitopes were purchased from commercial sources. Anti-COX2 / Cyclooxygenase 2 antibody (ab15191) Suitable for: IHC-Fr, ICC/IF, Sandwich ELISA, IP, IHC-P, WB Reacts with: Mouse, Rat, Human, Syrian hamster References provided on the vendor datasheet (127 total): 1. Soliman NA et al. The possible ameliorative effect of simvastatin versus sulfasalazine on acetic acid induced ulcerative colitis in adult rats. Chem Biol Interact 298:57-65 (2019). 2. Liu LM et al. Edaravone acts as a potential therapeutic drug against pentylenetetrazole-induced epilepsy in male albino rats by downregulating cyclooxygenase-II. Brain Behav 9:e01156 (2019). 3. Kong Z et al. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed Pharmacother 109:2043-2053 (2019).

3
nature research | reporting summ
aryO
ctober 20184. Li C et al. Correlation of LOX-5 and COX-2 expression with inflammatory pathology and clinical features of adenomyosis. Mol Med Rep 19:727-733 (2019) Enolase-1 Antibody (3810S) Applications: W, IP Species Cross-Reactivity H, M, R, Mk Dai, J., Zhou, Q., et al. (2018), 'Alpha-enolase regulates the malignant phenotype of pulmonary artery smooth muscle cells via the AMPK-Akt pathway.', Nat Commun, 9 (1), pp. 3850 Cha, Y., Han, M. J., et al. (2017), 'Metabolic control of primed human pluripotent stem cell fate and function by the miR-200c- SIRT2 axis.', Nat Cell Biol, 19 (5), pp. 445-456 Anti-Cytochrome C antibody [7H8.2C12] Suitable for: Flow Cyt, IHC-Fr, ICC/IF, WB, IHC-P Reacts with: Mouse, Rat, Horse, Pigeon, Human, Drosophila melanogaster References provided on the vendor datasheet (85 total): 1. Li Z et al. Protective roles of Amanita caesarea polysaccharides against Alzheimer's disease via Nrf2 pathway. Int J Biol Macromol 121:29-37 (2019). 2. Zuo W et al. Hyperglycemia abolished Drp-1-mediated mitophagy at the early stage of cerebral ischemia. Eur J Pharmacol 843:34-44 (2019). NLRP3/NALP3 Antibody NBP2-12446; Reactivity Hu, Mu, Rt, RM Applications WB, ICC/IF, IHC, IHC-P References provided on the vendor datasheet (85 total): 1. Liu B, Lu R, Li H et al. Zhen-wu-tang ameliorates membranous nephropathy rats through inhibiting NF-kB pathway and NLRP3 inflammasome Phytomedicine Apr 1 2019 [PMID: 30991182] (IHC-Fr, Rat) 2. Doyle TM, Chen Z, Durante M, Salvemini D. Activation of sphingosine-1-phosphate receptor 1 in the spinal cord produces mechano-hypersensitivity through the activation of inflammasome and IL-1b pathway J Pain Feb 22 2019 [PMID: 30802544] (WB, Rat) 3. Lyu C, Zhang Y, Gu M et al. IRAK-M Deficiency Exacerbates Ischemic Neurovascular Injuries in Experimental Stroke Mice. Front Cell Neurosci Dec 21 2018 [PMID: 30622459] (WB, Mouse) ASC (B-3): sc-514414 - ASC (B-3) is recommended for detection of ASC of mouse, rat and human origin by Western Blotting. References provided on the vendor datasheet: 1. Zhang, B., et al. 2017. Silybin attenuates LPS-induced lung injury in mice by inhibiting NFκB signaling and NLRP3 activation. Int. J. Mol. Med. 39: 1111-1118. 2. Humphries, F., et al. 2018. The E3 ubiquitin ligase Pellino 2 mediates priming of the NLRP3 inflammasome. Nat. Commun. 9: 1560. Purified Mouse Anti-DLP1 Clone 8/DLP1 (RUO): 611113 - reactivity - Rat (QC Testing) Human, Mouse, Dog (Tested in Development) References provided on the vendor datasheet: 1. Bossya B, Bossy-Wetzela E, Petrillia A, et al. S-Nitrosylation of DRP1 Does Not Affect Enzymatic Activity and is Not Specific to Alzheimer’s Disease. 2010; 20:S513-S526. 2. Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007; 14(6):1086-1094. 3. Jofuku A, Ishihara N, Mihara K. Analysis of functional domains of rat mitochondrial Fis1, the mitochondrial fission-stimulating protein. Biochem Biophys Res Commun. 2005; 333(2):650-659. 4. Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007; 178(1):71-84. 5. Yoon Y, Pitts KR, Dahan S, McNiven MA. A novel dynamin-like protein associates with cytoplasmic vesicles and tubules of the endoplasmic reticulum in mammalian cells. J Cell Biol. 1998; 140(4):779-793. Anti-GFAP antibody (ab7260) Specifically recognizes mammalian GFAP on western blots and immunocytochemically. Detects a band of 55kDa corresponding to GFAP and also a GFAP derived 48kDa band. Some customers have successfully used ab7260 on Zebrafish lysates; however we have conflicting data to suggest that not all batches will be suitable for work on Zebrafish. For further information, please contact Abcam Scientific Support. Reacts with: Mouse, Rat, Cat, Dog, Human, Common marmoset Suitable for: IHC-FoFr, IHC-Fr, IHC-FrFl, ICC/IF, WB, IHC-P, IHC - Wholemount, ICC. References provided on the vendor datasheet: 1. Chang LH et al. Blockade of soluble epoxide hydrolase attenuates post-ischemic neuronal hyperexcitation and confers resilience against stroke with TrkB activation. Sci Rep 8:118 (2018). 2. Yoshimura A et al. TheSox2promoter-driven CD63-GFP transgenic rat model allows tracking of neural stem cell-derived extracellular vesicles. Dis Model Mech 11:N/A (2018). Anti-IL1 beta antibody (ab9722) Suitable for: WB, ELISA, Neutralising, IHC-P, ICC, IHC-Fr, IHC-FoFr, ICC/IFmore details. Reacts with: Mouse, Rat, Human References provided on the vendor datasheet: 1. Hu J et al. Activin A inhibition attenuates sympathetic neural remodeling following myocardial infarction in rats. Mol Med Rep 17:5074-5080 (2018). 2. Tu R et al. Soluble epoxide hydrolase inhibition decreases reperfusion injury after focal cerebral ischemia. Sci Rep 8:5279 (2018). Caspase-1 p20 Antibody (D-4): sc-398715 caspase-1 p20 (D-4) is recommended for detection of caspase-1 precursor and p20 subunit of mouse and rat origin by Western Blotting. References provided on the vendor datasheet: 1. Dolunay, A., et al. 2017. Inhibition of NLRP3 inflammasome prevents LPS-induced inflammatory hyperalgesia in mice: contribution of NFκB, caspase-1/11, ASC, NOX, and NOS isoforms. Inflammation 40: 366-386. 2. Zhang, X., et al. 2017. Resveratrol attenuates early brain injury after experi- mental subarachnoid hemorrhage via inhibition of NLRP3 inflammasome activation. Front. Neurosci. 11: 611. 3. Xiang, Y., et al. 2018. A high concentration of DMSO activates caspase-1 by increasing the cell membrane permeability of potassium. Cytotechnology 70: 313-320. 4. Hou, Y., et al. 2018. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav. Brain Res. 336: 32-39. Phospho-SAPK/JNK (Thr183/Tyr185) Antibody #9251; REACTIVITY; H M R Hm Mk Dm B Sc; Phospho-SAPK/JNK (Thr183/ Tyr185) Antibody detects endogenous levels of p46 and p54 SAPK/JNK dually phosphorylated at threonine 183 and tyrosine 185. This antibody does not recognize unphosphorylated SAPK/JNK. This antibody may slightly cross-react with phospho-Erk1/2 or -p38 phosphorylated at the homologous residues. It will also react with SAPK/JNK singly phosphorylated at Thr183 and singly

4
nature research | reporting summ
aryO
ctober 2018phosphorylated at Tyr185. References provided on the vendor datasheet: (1) Davis, R.J. (1999) Biochem Soc Symp 64, 1-12. (2) Ichijo, H. (1999) Oncogene 18, 6087-93. (3) Kyriakis, J.M. and Avruch, J. (2001) Physiol Rev 81, 807-69. (4) Kyriakis, J.M. (1999) J Biol Chem 274, 5259-62. (5) Leppä, S. and Bohmann, D. (1999) Oncogene 18, 6158-62. (6) Whitmarsh, A.J. and Davis, R.J. (1998) Trends Bio- chem Sci 23, 481-5. Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Antibody #9101; REACTIVITY; H M R Hm Mk Dm B Sc; Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Antibody detects endogenous levels of p44 and p42 MAP Kinase (Erk1 and Erk2) when phosphorylated either individually or dually at Thr202 and Tyr204 of Erk1 (Thr185 and Tyr187 of Erk2). The antibody does not cross-react with the cor- responding phosphorylated residues of either JNK/SAPK or p38 MAP Kinase, and does not cross-react with non- phosphorylated Erk1/2. References provided on the vendor datasheet: (1) Roux, P.P. and Blenis, J. (2004) Microbiol Mol Biol Rev 68, 320-44. (2) Baccarini, M. (2005) FEBS Lett 579, 3271-7. (3) Meloche, S. and Pouysségur, J. (2007) Oncogene 26, 3227-39. (4) Roberts, P.J. and Der, C.J. (2007) Oncogene 26, 3291-310. (5) Rubinfeld, H. and Seger, R. (2005) Mol Biotechnol 31, 151-74. (6) Murphy, L.O. and Blenis, J. (2006) Trends Biochem Sci 31, 268-75. (7) Dalby, K.N. et al. (1998) J Biol Chem 273, 1496-505. (8) Marais, R. et al. (1993) Cell 73, 381-93. (9) Kortenjann, M. et al. (1994) Mol Cell Biol 14, 4815-24. (10) Owens, D.M. and Keyse, S.M. (2007) Oncogene 26, 3203-13. Phospho-DRP1 (Ser616) Antibody #3455; Species Cross-Reactivity* H, (M, R, Mk) Phospho-DRP1 (Ser616) Antibody detects endogenous levels of DRP1 only when phosphorylated at Ser616. References provided on the vendor datasheet: (1) Praefcke, G.J. and McMahon, H.T. (2004) Nat Rev Mol Cell Biol 5, 133-47. (2) Taguchi, N. et al. (2007) J Biol Chem 282, 11521-9. (3) Smirnova, E. et al. (2001) Mol Biol Cell 12, 2245-56. (4) Smirnova, E. et al. (1998) J Cell Biol 143, 351-8. (5) Koch, A. et al. (2003) J Biol Chem 278, 8597-605. (6) Knott, A.B. et al. (2008) Nat Rev Neurosci 9, 505-18. (7) Cereghetti, G.M. et al. (2008) Proc Natl Acad Sci U S A 105, 15803-8. (8) Zunino, R. et al. (2007) J Cell Sci 120, 1178-88. Phospho-DRP1 (Ser637) Antibody #4867 Species Cross-Reactivity* R, (M, Mk) Phospho-DRP1 (Ser637) Antibody detects endogenous levels of DRP1 only when phosphorylated at Ser637. References provided on the vendor datasheet: (1) Praefcke, G.J. and McMahon, H.T. (2004) Nat Rev Mol Cell Biol 5, 133-47. (2) Taguchi, N. et al. (2007) J Biol Chem 282, 11521-9. (3) Smirnova, E. et al. (2001) Mol Biol Cell 12, 2245-56. (4) Smirnova, E. et al. (1998) J Cell Biol 143, 351-8. (5) Koch, A. et al. (2003) J Biol Chem 278, 8597-605. (6) Knott, A.B. et al. (2008) Nat Rev Neurosci 9, 505-18. (7) Cereghetti, G.M. et al. (2008) Proc Natl Acad Sci USA 105, 15803-8. (8) Zunino, R. et al. (2007) J Cell Sci 120, 1178-88. Tom20 Antibody (FL-145): sc-11415; Tom20 (FL-145) is recommended for detection of Tom20 of mouse, rat and human origin by Western Blotting. References provided on the vendor datasheet: 1. Zong, W.X., et al. 2004. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 18: 1272-1282. 2. Usami, Y., et al. 2011. DJ-1 associates with synaptic membranes. Neurobiol. Dis. 43: 651-662. 3. De Marchi, U., et al. 2011. Uncoupling protein 3 (UCP3) modulates the activity of sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) by decreas- ing mitochondrial ATP production. J. Biol. Chem. 286: 32533-32541. 4. Singh, K., et al. 2011. Effect of denervation-induced muscle disuse on mito- chondrial protein import. Am. J. Physiol., Cell Physiol. 300: C138-C145. 5. Pavlov, P.F., et al. 2011. Mitochondrial γ-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein. FASEB J. 25: 78-88. 6. Bénard, G., et al. 2012. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 15: 558-564. 7. Fernandes, R., et al. 2012. NLRP5 mediates mitochondrial function in mouse oocytes and embryos. Biol. Reprod. 86: 138. 8. Selimovic, D., et al. 2012. Apoptosis related protein-1 triggers melanoma cell death via interaction with the juxtamembrane region of p75 neu- rotrophin receptor. J. Cell. Mol. Med. 16: 349-361. Anti-VDAC1 / Porin antibody [20B12AF2] (ab14734); Reacts with: Mouse, Rat, Sheep, Goat, Cat, Dog, Human, Pig, Drosophila melanogaster, Fish, Quail, Common marmoset, Dogfish, Catshark. References provided on the vendor datasheet: 1. Zhang D et al. Increased mitochondrial fission is critical for hypoxia-induced pancreatic beta cell death. PLoS One 13:e0197266 (2018). 2. Vantroys E et al. Severe hepatopathy and neurological deterioration after start of valproate treatment in a 6-year-old child with mitochondrial tryptophanyl-tRNA synthetase deficiency. Orphanet J Rare Dis 13:80 (2018). β-Actin (8H10D10) Mouse mAb #3700; REACTIVITY H M R Hm Mk Dg; β-Actin (8H10D10) Mouse mAb detects endogenous levels of total β-actin protein. References provided on the vendor datasheet: (1) Herman, I.M. (1993) Curr. Opin. Cell Biol. 5, 48–55. (2) Condeelis, J. (2001) Trends Cell Biol. 11, 288–293. (3) Lim, Y.P. et al. (2004) Clin. Cancer Res. 10, 3980–3987. (4) Kayalar, C. et al. (1996) Proc. Natl. Acad. Sci. USA. 93, 2234–2238. (5) Communal, C. et al. (2002) Proc. Natl. Acad. Sci. USA. 99, 6252–6256. (6) Du, J. et al. (2004) J. Clin. Invest. 113, 115–123. Second antibodies including anti-mouse IgG (#NA931V) and anti- rabbit IgG (NA934V) were from GE Healthcare, and anti-goat IgG were from Sigma-Aldrich (#A8919).

5
nature research | reporting summ
aryO
ctober 2018
Eukaryotic cell linesPolicy information about cell lines
Cell line source(s) BV2 microglia were a gift from Dr. Katrin Andreasson, Stanford University
Authentication None of the lines used were authenticated
Mycoplasma contamination Cell lines were not tested for Mycoplasma contamination
Commonly misidentified lines(See ICLAC register)
Cell lines were not tested for Mycoplasma contamination
Animals and other organismsPolicy information about studies involving animals; ARRIVE guidelines recommended for reporting animal research
Laboratory animals Adult B6SJL Tg (SOD1G93A) 1 Gur/J male mice with a high copy number of the mutant allele and their WT littermates at 4-6 weeks, 5XFAD transgenic male mice and their littermates at 3 months, and Hemizygous male R6/2 HD mice and their WT littermates at 4-6 weeks were used.
Wild animals We did not use any wild animals
Field-collected samples The study did not involve field collected samples
Ethics oversight Animal husbandry and procedures were performed according to Stanford guidelines under IACUC approved protocols.
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Human research participantsPolicy information about studies involving human research participants
Population characteristics Peripheral blood mononuclear cells from de-identified healthy donors (35-65 years old) were obtained from the Stanford Blood center
Recruitment N/A
Ethics oversight N/A
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Recommended