Embed Size (px)
Citation preview

ECOLOGICAL CHEMISTRY AND ENGINEERING S
Vol. 15, No. 1 2008
Anna BANEL∗1 i Bogdan ZYGMUNT*
ZASTOSOWANIE POŁĄCZENIA MIKROEKSTRAKCJI DO FAZY STACJONARNEJ I CHROMATOGRAFII GAZOWEJ DO OZNACZANIA LOTNYCH KWASÓW TŁUSZCZOWYCH
W PRÓBKACH ŚRODOWISKOWYCH I POKREWNYCH
APPLICATION OF COMBINATION OF SOLID PHASE MICROEXTRACTION AND GAS CHROMATOGRAPHY FOR DETERMINATION OF VOLATILE FATTY ACIDS
IN ENVIRONMENTAL AND RELATED SAMPLES
Streszczenie: Oznaczanie lotnych kwasów tłuszczowych (LKT) w próbkach środowiskowych wymaga najczęściej izolacji i wzbogacania analitów na etapie przygotowania próbek oraz zastosowania techniki o duŜym potencjale separacyjnym i identyfikacyjnym na etapie oznaczeń końcowych. W pracy dokonano krytycznego przeglądu zastosowań wygodnej i często stosowanej techniki mikroekstrakcji do fazy stacjonarnej (SPME) w połączeniu z wysokosprawną techniką chromatografii gazowej (GC) w oznaczaniu LKT w próbkach środowiskowych i pokrewnych.
Słowa kluczowe: lotne kwasy tłuszczowe, chromatografia gazowa, mikroekstrakcja do fazy stacjonarnej, próbki środowiskowe, ścieki
Wprowadzenie
Monokarboksylowe kwasy tłuszczowe (LKT), zawierające według róŜnych danych literaturowych od 2 do 5 [1, 2], 6 [3], 7 [4-6], a nawet 8 [7] atomów węgla w molekule, naleŜą do grupy związków dość szeroko rozpowszechnionych w środowisku. Powszech-nie związki te występują w ściekach pochodzących z hodowli trzody chlewnej, przemys-łu przetwarzania Ŝywności, w odpadach organicznych, osadach ściekowych oraz odcie-kach ze składowisk odpadów. Głównym źródłem LKT jest proces beztlenowej biodegra-dacji materii organicznej, tj.: węglowodanów, białek i tłuszczów. W środowisku rzadko są one obecne w stanie wolnym, najczęściej występują w postaci estrów, soli lub amidów [8].
* Katedra Chemii Analitycznej, Wydział Chemiczny, Politechnika Gdańska, ul. G. Narutowicza 11/12, 80-952 Gdańsk, tel. 058 347 21 10, fax 058 347 17 83, e-mail: [email protected] 1 Autor do korespondencji: [email protected]

Anna Banel i Bogdan Zygmunt
8
Znajomość właściwości fizykochemicznych danego LKT pozwala dość dobrze prze-widzieć jego zachowanie się w środowisku. Właściwości te determinują równieŜ sposób postępowania podczas oznaczania wybranych analitów z grupy LKT. W tabeli 1 przed-stawiono nomenklaturę, budowę chemiczną oraz wybrane właściwości fizykochemiczne lotnych kwasów tłuszczowych.
Tabela 1 Nomenklatura, budowa chemiczna oraz wybrane właściwości fizykochemiczne lotnych
kwasów tłuszczowych [9-12]
Nazwa systematyczna i zwyczajowa
Wzór chemiczny Temp.
wrzenia [oC]
Rozpuszczalność w wodzie [g/dm3]
pKa M
[g/mol]
Kwas etanowy octowy
CH3COOH 117 duŜa 4,75 60,1
Kwas propionowy propanowy
C2H5COOH 141 duŜa 4,87 74,1
Kwas izobutanowy izomasłowy
(CH3)2CHCOOH 154 210 4,85 88,1
Kwas butanowy masłowy
C3H7COOH 164 średnia 4,81 88,1
Kwas izopentanowy izowalerianowy
(CH3)2CHCH2COOH 177 25 4,78 102,1
Kwas pentanowy walerianowy
C4H9COOH 186 40 4,82 102,1
Kwas heksanowy kapronowy
C5H11COOH 206 10 4,88 116,2
Kwas heptanowy enantowy
C6H13COOH 223 2,6 4,89 130,2
Kwas oktanowy kaprylowy
C7H15COOH 235 0,7 4,89 144,2
Najbardziej odpowiednimi technikami oznaczania LKT są techniki chromatograficz-ne, gdyŜ umoŜliwiają skuteczną separację, a dzięki temu identyfikację i ilościowe ozna-czenie poszczególnych lotnych kwasów tłuszczowych. Wysokosprawna chromatografia cieczowa (HPLC) i chromatografia jonowa (IC) często wymagają skomplikowanej pro-cedury oczyszczania próbki, a często takŜe derywatyzacji, aby zmniejszyć interferencje oraz osiągnąć odpowiednio niską granicę oznaczalności [2]. Natomiast chromatografia gazowa moŜe być z powodzeniem stosowana równieŜ w przypadku małych stęŜeń LKT w próbkach wodnych, np. ściekach. Próbka ścieków, zawierająca lotne kwasy tłuszczo-we, nie nadaje się do bezpośredniego wprowadzenia do kolumny chromatograficznej, groŜąc zanieczyszczeniem lub uszkodzeniem kolumny. Niezgodność matrycy próbki z większością odpowiednich do separacji LKT kolumn do chromatografii gazowej wy-musza jej zmianę. Przed analizą chromatograficzną naleŜy zatem przeprowadzić etap izolacji i wzbogacenia analitów za pomocą odpowiedniej techniki ekstrakcyjnej. Naj-częściej stosowanym sposobem przygotowania próbek wodnych do analizy na zawartość LKT za pomocą techniki GC jest ekstrakcja ciecz-ciecz (LLE) i ekstrakcja do fazy stałej

Zastosowanie połączenia mikroekstrakcji do fazy stacjonarnej i chromatografii gazowej ...
9
(SPE). Techniki te są raczej czasochłonne i Ŝmudne, a ponadto, w szczególności technika LLE, wymagają zastosowania względnie duŜej ilości organicznego rozpuszczalnika o wysokiej czystości. Obserwowane dzisiaj dąŜenie do uproszczenia i miniaturyzacji aparatury do przygotowania próbki i minimalizacji zuŜycia rozpuszczalników spowodo-wały rozwój innych technik, a szczególnie mikroekstrakcji do fazy stacjonarnej (SPME).
Mikroekstrakcja do fazy stacjonarnej (SPME - Solid Phase Microextraction)
SPME jest techniką ekstrakcyjną zaproponowaną w 1990 r. przez Arthura i Pawli-szyna [13]. Metoda ta ciągle cieszy się ogromnym zainteresowaniem, gdyŜ moŜe być stosowana w oznaczaniu szerokiej gamy organicznych związków lotnych i średniolot-nych w próbkach środowiskowych i innych, w tym takŜe w próbkach o skomplikowanych matrycach. Ponadto spełnia ona wymagania aktualnego trendu miniaturyzacji zestawu do przygotowania próbek i prawie całkowitego wyeliminowania rozpuszczalników z tego procesu [14, 15].
Podstawą takiej ekstrakcji jest uŜycie cienkiego włókna ze stopionej krzemionki po-krytego odpowiednim materiałem sorpcyjnym, które przymocowane jest do tłoka mikro-strzykawki do GC. Włókno moŜe być wsuwane bądź wysuwane z igły strzykawki. Kiedy włókno jest eksponowane bezpośrednio w badanej próbce wodnej lub w jej fazie nadpo-wierzchniowej, to następuje podział analitu między fazą wodną (matrycę) a fazą stacjo-narną umieszczoną na włóknie SPME.
Włókno SPME wsunięte do igły strzykawki wprowadza się do dozownika chromato-grafu gazowego, gdzie po wysunięciu anality są desorbowane termicznie i przenoszone za pomocą gazu nośnego do kolumny chromatograficznej w celu ich separacji, a następnie do odpowiedniego detektora [15].
Zalety i wady techniki SPME
Technika SPME jest innowacyjną techniką izolacji i wzbogacania głównie organicz-nych zanieczyszczeń w próbkach środowiskowych [16]. Technika ta jest szybka, prosta, łatwa do automatyzacji oraz moŜliwa do uŜycia w warunkach polowych. Dzięki jej sto-sowaniu moŜna oznaczać organiczne zanieczyszczenia w wodzie na niskim poziomie stęŜeń [16]. Technika SPME jest niewraŜliwa na zawiesiny obecne w próbce, jeśli zasto-suje się ekstrakcję analitów z fazy nadpowierzchniowej. MoŜe być stosowana nie tylko do próbek ciekłych, ale takŜe gazowych oraz stałych (ekstrakcja w fazie nadpowierzch-niowej HS (Head Space) SPME) [17]. Stosowanie rozpuszczalników jest prawie całko-wicie wyeliminowane.
Główną wadą tej techniki ekstrakcyjnej jest względnie duŜy koszt włókna i ograni-czony czas jego uŜytkowania związany z zanieczyszczaniem w czasie ekstrakcji i ewen-tualną degradacją. Częściowa degradacja fazy stacjonarnej włókna, szczególnie jeśli desorpcja prowadzona jest w wysokiej temperaturze, i/lub składników próbki nietrwa-łych termicznie moŜe prowadzić do pojawienia się dodatkowych pików na chromatogra-mie. Pogarszać to moŜe dokładność i precyzję analizy [14].

Anna Banel i Bogdan Zygmunt
10
Dodatkową zaletą tej metody jest moŜliwość jej połączenia z techniką HS. Wokół włókna umieszczonego w fazie gazowej nad ciekłą próbką nie tworzy się stacjonarna warstewka wody, utrudniająca transport analitów do włókna, a duŜa powierzchnia kon-taktu próbki z fazą gazową moŜe być odnawiana w sposób ciągły w wyniku mieszania. W rezultacie transport lotnych analitów z próbki do włókna przez fazę nadpowierzch-niową jest duŜo szybszy niŜ bezpośrednio z próbki do włókna. Równowaga w takim układzie ustala się bardzo szybko [17].
Sposoby stosowania SPME
W zaleŜności od umieszczenia włókna względem próbki mikroekstrakcję do fazy sta-cjonarnej moŜna podzielić na [15, 17, 18]: - bezpośrednią (DI (Direct Immersion) - SPME), - z fazy nadpowierzchniowej (HS-SPME). Te dwa rozwiązania zebrano i opisano w tabeli 2.
Tabela 2 Sposoby prowadzenia mikroekstrakcji do fazy stacjonarnej na włóknie
DI-SPME HS-SPME
Włókno jest bezpośrednio zanurzone w próbce i anality są przenoszone bezpośrednio z matrycy próbki do fazy stacjonarnej.
Włókno SPME wprowadza się do fazy nadpo-wierzchniowej próbki wewnątrz naczynka i poddaje ekspozycji.
Następnie włókno umieszcza się w dozowniku chromatografu gazowego, gdzie następuje termodesorpcja analitów, które są rozdzielane w kolumnie chromatograficznej i oznaczane.
Termodynamiczne aspekty tej techniki przygotowania próbek zostały dokładnie prze-
analizowane i wskazują, Ŝe ilość ekstrahowanych analitów przez włókno jest wprost proporcjonalna do stęŜenia analitów w próbce i niezaleŜna od połoŜenia włókna (w próbce lub w fazie stacjonarnej). Współczynniki podziału analitu między fazą stacjo-narną a fazą nadpowierzchniową (Kfh) oraz analitu między fazą stacjonarną a próbką

Zastosowanie połączenia mikroekstrakcji do fazy stacjonarnej i chromatografii gazowej ...
11
(K fs) mogą być oszacowane na podstawie danych fizykochemicznych i parametrów chromatograficznych [19].
Warunki wpływaj ące na wydajność mikroekstrakcji do fazy stacjonarnej
Z rozwaŜań termodynamicznych wynika, Ŝe takie parametry, jak: temperatura, siła jonowa roztworu, polarność matrycy próbki i rodzaj materiału włókna, wpływają na współczynnik podziału, a tym samym na wydajność ekstrakcji i czułość metody, dlatego powinny być uwzględnione w procesie optymalizacji. Inne parametry, które mogą wpły-wać na czułość oznaczenia, to objętość fazy stacjonarnej, fazy nadpowierzchniowej oraz próbki [17]. Natomiast kinetyka mikroekstrakcji, będąca funkcją temperatury i intensywność mieszania, określa czas ekstrakcji. Wydajność ekstrakcji moŜna równieŜ modyfikować, przeprowadzając anality w pochodne [20].
Pokrycie włókna
Włókno w celu osiągnięcia dobrej selektywności i wydajności ekstrakcji analitów powinno charakteryzować się odpowiednimi właściwościami chemicznymi i fizycznymi [20-22]. W pierwszym etapie wybór fazy stacjonarnej korzysta z zasady „podobne roz-puszcza się w podobnym”, czyli niepolarne anality są skuteczniej ekstrahowane do nie-polarnego pokrycia włókna, najczęściej polidimetylosiloksanu (PDMS), a polarne anality są ekstrahowane do polarnego pokrycia włókna, np. poliakrylanu (PA) [23]. Mieszane fazy (stały sorbent rozproszony w ciekłej fazie stacjonarnej) są stosowane głównie do ekstrakcji bardzo lotnych związków. Wydajność ekstrakcji do takich włókien jest więk-sza w porównaniu z PDMS, ale czas uŜytkowania generalnie krótszy. Proces sorpcji do faz mieszanych jest zazwyczaj w większym stopniu zaleŜny od adsorpcji niŜ absorpcji [21].
Dobór właściwego włókna jest niezmiernie waŜny, poniewaŜ rodzaj i ilość fazy sta-cjonarnej wpływa na czułość i selektywność procedury, korzystającej z mikroekstrakcji do fazy stacjonarnej.
W tabeli 3 przedstawiono krótką charakterystykę handlowo dostępnych włókien do SPME.
Wolne LKT są związkami o silnych właściwościach hydrofilowych i, według wspo-mnianej wyŜej zasady, najbardziej odpowiednie do ich ekstrakcji są włókna mieszane. Typowe fazy mieszane (PDMS-DVB, PDMS-CAR i PDMS-CAR-DVB) uŜyto i porów-nano pod względem moŜliwości oznaczania LKT w próbkach ścieków miejskich za po-mocą HS-SPME-GC-FID [11]. Największe powierzchnie pików uzyskano dla wolnych LKT o większej masie molekularnej (C4-C7). Wszystkie te włókna dały satysfakcjonują-ce powierzchnie pików. Kwasy o mniejszej liczbie węgla w molekule (kwas octowy i propionowy) były ekstrahowane z uŜyciem włókna PDMS-CAR, by uzyskać wystarcza-jąco duŜe pole powierzchni pików. Szczególnie korzystne było jego uŜycie do oznacza-nia kwasu octowego.
W porównaniu z PDMS-DVB, włókno PDMS-CAR-DVB dawało lepszą wartość od-zysku dla kwasu octowego, propionowego i masłowego, podczas gdy dla kwasu waleria-

Anna Banel i Bogdan Zygmunt
12
nowego, heksanowego i heptanowego nie było znaczącej róŜnicy między tymi dwoma włóknami.
Tabela 3 Handlowo dostępne włókna do SPME [19, 22, 24, 26]
Pokrycie włókna Akronim Grubość
filmu [µµµµm]
Oznacze-nie koń-
cowe Zastosowanie
WŁÓKNA NIEPOLARNE
Polidimetylosiloksan PDMS 100 30 7
GC, HPLC GC, HPLC GC, HPLC
Niepolarne związki organiczne: VOCs, PAHs, BTEX
WŁÓKNA POLARNE
Poliakrylan PA 85 GC, HPLC Polarne związki organiczne: triazyny, fosfoorganiczne pestycydy i fenole
WŁÓKNA MIESZANE
Polidimetylosiloksan-polidiwinylobenzen
PDMS-DVB 65 60
GC HPLC
Węglowodory aromatyczne, aromatyczne aminy, VOCs
Polidimetylosiloksan - Carboxen PDMS- CAR
75 GC Gazowe/lotne anality, VOCs, węglowodory
Carbowax - Polidiwinylobenzen CW-DVB 65 GC Polarne związki organiczne, alkohole, ketony, nitroaroma-tyczne związki
Carbowax - Ŝywica z nadrukiem molekularnym
CW-TPR 50 HPLC Anionowe surfaktanty, aroma-tyczne aminy
Polidimetylosiloksan -Polidiwinylobenzen/Carboxen
PDMS-DVB/CAR
50/30 GC Szeroki zakres polarnych analitów
Natomiast do ekstrakcji benzylowych pochodnych LKT z matrycy wodnej zastosowano włókno polarne (PA), które charakteryzuje się stabilnością mechaniczną i zapewniają dobrą powtarzalność ekstrakcji [27]. W przeprowadzonych badaniach porównywano równieŜ wydajność ekstrakcji do włókien PDMS i CW-DVB, uzyskując ponad 10-krotnie mniejszą wydajność dla PDMS. W przypadku włókna CW-DVB wystąpił natomiast problem natury trwałości mechanicznej, wynikający z pęcznienia powierzchni włókna. Dlatego teŜ do dalszej ekstrakcji wykorzystywano jedynie włókno PA.
Włókno pokryte poliakrylanem jest przewaŜnie stosowane do ekstrakcji pochodnych LKT po etapie derywatyzacji lub do jednoczesnej derywatyzacji i sorpcji na włóknie. Derywatyzacja za pomocą takich odczynników, jak np. 1-pentafluorofenylodiazoetan (PFPDE) i pirenylodiazometan (PDAM) w połączeniu z techniką SPME słuŜy do moni-torowania LKT w próbkach wodnych i w powietrzu. Na ogół lepszy odzysk otrzymuje się, prowadząc jednocześnie ekstrakcję i derywatyzację na powierzchni włókna pokryte-go fazą stacjonarną z odczynnikiem derywatyzującym, uzyskując 98% przekształcenie kwasów w estry LKT w fazie stacjonarnej [25].
Grubość warstwy fazy stacjonarnej jest niezmiernie waŜna. Zastosowanie grubej war-stwy fazy stacjonarnej powoduje, Ŝe układ dochodzi do równowagi znacznie dłuŜej.

Zastosowanie połączenia mikroekstrakcji do fazy stacjonarnej i chromatografii gazowej ...
13
NaleŜy wybierać włókno z najmniejszą grubością filmu, które juŜ zapewnia wymaganą czułość [19]. Do oznaczania LKT stosowano handlowo dostępne włókna pokryte filmem fazy stacjonarnej o róŜnych grubościach.
W tabeli 4 przedstawiono właściwości i zakres zastosowania włókien o róŜnej grubo-ści warstwy fazy stacjonarnej [18, 22, 23, 25].
Tabela 4 Porównanie właściwości i zastosowań włókien z grubym i cienkim filmem fazy stacjonarnej
Gruby film Cienki film
Ilo ść sorbowanego analitu większa mniejsza
Właściwości
Skuteczna ekstrakcja związków o wy-sokiej temperaturze wrzenia z matrycy próbki. Szybkość desorpcji jest wówczas prze-dłuŜona i moŜe być niecałkowita (efekt pamięci).
Zapewniona szybka dyfuzja i łatwe uwalnianie związku o wyŜszej tem-peraturze wrzenia podczas termicz-nej desorpcji.
Zastosowanie W szczególności do lotnych związków, gdyŜ zapewniają przeniesienia ich do GC bez znacznych strat.
Do izolacji i wzbogacania substan-cji o wysokiej temperaturze wrze-nia.
Szeroki asortyment włókien do SPME o róŜnej grubości i polarności jest handlowo
dostępny. W tabeli 5 umieszczono parametry (dopuszczalną temperaturę i warunki kon-dycjonowania) zalecane przez producenta dla handlowo dostępnych włókien o róŜnej grubości filmu fazy stacjonarnej.
Tabela 5 Dopuszczalna temperatura pracy i warunki kondycjonowania włókien [19, 26]
Pokrycie włókna Grubość
filmu [µµµµm]
Maksymalna temperatura
desorpcji [oC]
Temperatura kondycjonowania
[oC]
Czas kondycjonowania
[h]
PDMS 100 30 7
280 280 340
250 250 320
1 1
2÷4
PA 85 320 300 2
PDMS-DVB 65 60
270 260 0,5
PDMS-CAR 75 320 280 0,5
CW-DVB 65 265 250 0,5
CW-TPR 50 270 270 4
PDMS-DVB/CAR 50/30 270 270 4
Pierwsze włókna do SPME były opracowane do stosowania w chromatografii gazo-
wej [20]. Natomiast uŜycie techniki SPME w przypadku wysokosprawnej chromatografii cieczowej (HPLC) stwarzało wiele problemów. Wynikały one głównie z rozpuszczania się pokrycia włókna w organicznym rozpuszczalniku fazy ruchomej, pęcznienia pokrycia

Anna Banel i Bogdan Zygmunt
14
włókna, zmian natęŜenia przepływu podczas desorpcji [19]. Obecnie większość włókien moŜe być stosowana równieŜ w HPLC; warunkiem jest odporność pokrycia włókna na organiczny rozpuszczalnik, wchodzący w skład fazy ruchomej [20].
Czas ekstrakcji
Z punktu widzenia powtarzalności i czułości najlepiej, jeśli ekstrakcję (a właściwie sorpcję) prowadzi się do momentu osiągnięcia stanu równowagi pomiędzy próbką a fazą stacjonarną włókna. Czas uzyskania równowagi jest definiowany jak czas, po którym ilość ekstrahowanych analitów pozostaje stała w granicach błędu eksperymentalnego i równa ilości ekstrahowanej w nieskończenie długim czasie. Określając ilość wyekstra-howanego analitu dla stanu równowagi, moŜna obliczyć współczynnik podziału (Kfs) [18], który generalnie wzrasta wraz ze wzrostem długości łańcucha i temperaturą wrzenia analitu [23].
Czas ekspozycji włókna ekstrakcyjnego w fazie nadpowierzchniowej dla wolnych LKT w próbkach ścieków miejskich wynosił 20 min [4, 5], a bezpośrednio z próbek ścieków z farmy trzody chlewnej wynosił równieŜ 20 min [7]. W przypadku ekstrakcji połączonej z derywatyzacją stosowano czas ekspozycji 180÷240 min dla próbek powie-trza, a w fazie nadpowierzchniowej próbek wodnych 60 min [31]. Czas ten jest zaleŜny od współczynnika podziału analitu, współczynnika dyfuzji oraz intensywności mieszania próbki, których wzrost skraca czas potrzebny do osiągnięcia stanu równowagi, poniewaŜ przyspiesza transport analitów w kierunku do włókna [20]. Czas ten jest generalnie krót-szy dla ekstrakcji z fazy nadpowierzchniowej [25].
Temperatura
Temperatura ekstrakcji wpływa na proces SPME w dwojaki sposób [20]. Korzystne i negatywne aspekty podwyŜszenia temperatury procesu ekstrakcji przedstawiono w tabeli 6. Zazwyczaj moŜna ustalić optymalną temperaturę, w której ilość wyekstraho-wanego analitu jest największa. Temperatura ta zaleŜy od składu matrycy i stosowanej fazy stacjonarnej [17]. Najczęściej stosowaną temperaturą w trakcie ekstrakcji LKT było 25°C [11, 12, 14, 26].
Tabela 6
Korzystne i negatywne aspekty podwyŜszonej temperatury ekstrakcji [15, 17, 18, 20, 25]
Korzystne Negatywne
• powoduje wzrost szybkości ekstrakcji przez wzmo-Ŝoną dyfuzję analitów w kierunku włókna
• zmniejsza wartość współczynnika podziału analitów (Kfs), poniewaŜ etap absorpcji jest procesem egzo-termicznym
• pomaga przenieść anality do fazy nadpowierzch-niowej (HS-SPME)
• dla termicznie niestabilnych związków, takich jak np. HMX podwyŜszona temperatura nie jest zaleca-na
• współczynnik dyfuzji w wodzie jest większy i czas ekstrakcji krótszy, ale współczynnik podziału włókno-próbka jest wówczas mniejszy
• zmniejsza wartość współczynnika podziału między fazą stacjonarną a fazą nadpowierzchniową
• ułatwia desorpcję analitów związanych z matrycą i przyspiesza transport analitów o małej lotności

Zastosowanie połączenia mikroekstrakcji do fazy stacjonarnej i chromatografii gazowej ...
15
Mieszanie
Mieszanie odgrywa waŜną rolę w procesie ekstrakcji SPME. Stosowane zazwyczaj sposoby mieszania opisano w tabeli 7 [17, 19, 23, 28].
Tabela 7 Sposoby mieszania stosowane w technice SPME
Metoda mieszania Charakterystyka
magnetyczne
• wymaga zastosowania mieszadełka magnetycznego w naczynku z próbką • jest najczęściej stosowaną techniką w SPME ze względu na łatwość realizacji
w laboratoriach analitycznych, a ponadto moŜe być stosowane w róŜnych wersjach SPME
technika „wiru” • naczynko jest szybko obracane ruchem okręŜnym
stały przepływ próbki
• odnawianie powierzchni włókna i zwiększony transport analitów do włókna
przy uŜyciu ultrad źwięków
• jest najbardziej skutecznym i najszybszym sposobem. Wadą jest ogrzewanie się próbki, a co za tym idzie - moŜliwość degradacji analitów oraz obniŜenie precyzji oznaczeń
Porównując wyniki analiz próbek mieszanych z wykorzystaniem mieszadła magne-tycznego i próbek bez mieszania, większe pola powierzchni pików chromatograficznych LKT uzyskuje się zazwyczaj dla tego pierwszego [4]. Wynika to z faktu, Ŝe skuteczna technika mieszania minimalizuje czas potrzebny do uzyskania stanu równowagi, a tym samym zwiększa odzysk przeprowadzonej ekstrakcji. Czas ten zaleŜy od współczynnika podziału oraz od sposobu i intensywności mieszania. Gdy szybkość mieszania rośnie, to pola powierzchni pików chromatograficznych lotnych kwasów tłuszczowych równieŜ wzrastają [4]. W praktyce trudno jest osiągnąć idealne warunki mieszania dla próbek wodnych, gdyŜ wokół włókna powstaje cienka, nieruchoma warstewka wody, która two-rzy barierę dyfuzyjną. Wpływa to znacząco na wydłuŜenie czasu dochodzenia do stanu równowagi. Najszybszy czas dochodzenia do równowagi w przypadku LKT daje mie-szanie z zastosowaniem ultradźwięków (20 s); natomiast czas w przypadku najbardziej rozpowszechnionego sposobu mieszania za pomocą mieszadła magnetycznego wynosi od 2 do 60 min. Mieszanie wpływa równieŜ korzystnie na dyfuzję składników z fazy ciekłej do fazy nadpowierzchniowej.
Ze względu na stosunkowo duŜą lotność LKT równieŜ bez mieszania moŜna osiągnąć stan równowagi w wersji HS-SPME [4].
Zmiana wartości pH
Przez odpowiedni dobór pH próbki moŜna w znacznym stopniu poprawić czułość procedury analitycznej, korzystając z techniki SPME. ObniŜając wartość pH próbki, moŜna cofnąć dysocjację kwaśnych analitów; przy pH = 2 LKT pozostają praktycznie całkowicie w formie niezdysocjowanej.
Dla analitów kwasowych pH powinno mieć wartość co najmniej o 2 jednostki mniej-szą niŜ pKa analitu, a dla zasadowych analitów o dwie większą niŜ pKb [18]. Jednak nale-

Anna Banel i Bogdan Zygmunt
16
Ŝy pamiętać, Ŝe włókna pokryte fazą PDMS nie mogą być eksponowane na działanie próbki o pH poniŜej 4 i powyŜej 10 [19, 25].
Dodatek soli
Dodatek soli, najczęściej NaCl lub Na2SO4, zwiększa siłę jonową roztworu, co zmniejsza rozpuszczalność wielu substancji organicznych, w tym LKT w wodzie, a tym samym zwiększa ułamek ekstrahowanych analitów do włókna [20, 25, 29]. Zmniejszenie rozpuszczalności wielu związków polarnych moŜe być na tyle duŜe, Ŝe następuje kilka-krotny wzrost współczynnika podziału między fazę stacjonarną a próbkę [17, 19]. Efek-tem towarzyszącym moŜe być spadek selektywności włókna [25]. Włókno po desorpcji musi być bardzo ostroŜnie i dokładnie umyte, poniewaŜ sól zwiększa na ogół jego kru-chość [19].
Wielkość efektu wysolenia próbki zaleŜy od polarności analitu, stęŜenia soli oraz od matrycy próbki [20].
Dodatek rozpuszczalnika
Problem dodatku organicznego rozpuszczalnika do próbki ciekłej nie jest jeszcze gruntownie zbadany. Obecność organicznych rozpuszczalników w próbkach wodnych zazwyczaj zmniejsza ułamek ekstrahowanych analitów. Dodatek rozpuszczalnika orga-nicznego lub wody do próbek stałych i mulistych pozwala na uwolnienie analitów z matrycy i dzięki temu zwiększa skuteczność ekstrakcji [20].
Objętość próbki
WaŜnym parametrem w procesie optymalizacji warunków ekstrakcji LKT jest obję-tość próbki, która ma bezpośredni wpływ na czułość metodyki analitycznej. Objętość próbki jest wielokrotnie większa niŜ objętość fazy stacjonarnej na włóknie i ilość ekstra-howanych analitów jest proporcjonalna do współczynnika podziału, stęŜenia próbki oraz objętości fazy stacjonarnej na włóknie. Związki chemiczne o duŜej wartości współczyn-nika Kfs są bardziej wraŜliwe na zmiany objętości próbki niŜ związki o małym powino-wactwie do włókna. Z tego powodu jednym z kryteriów doboru odpowiedniej objętości próbki jest uwzględnienie wartości współczynnika podziału Kfs. Natomiast dla mikro-ekstracji w fazie nadpowierzchniowej (HS-SPME) objętość fazy gazowej powinna być zminimalizowana, aby osiągnąć jak największe stęŜenie LKT w fazie nadpowierzchnio-wej, a co za tym idzie - uzyskać zwiększoną czułość metodyki [20, 21].
Desorpcja analitów z włókna
Głównymi czynnikami wpływającymi na termiczną desorpcję LKT z włókna SPME są: temperatura, czas desorpcji i połoŜenie igły w dozowniku GC [25]. Wpływ tych czynników został scharakteryzowany w tabeli 8. Skuteczność termicznej desorpcji anali-tów w dozowniku GC jest równieŜ zaleŜna od lotności analitów i grubości pokrycia włókna [30]. Większość dozowników w chromatografach gazowych jest przystosowana do bezpośredniego wprowadzenia włókna. Objętość wkładki wpływa na kształt pików

Zastosowanie połączenia mikroekstrakcji do fazy stacjonarnej i chromatografii gazowej ...
17
chromatograficznych; większa objętość jest powodem poszerzania pasma analizowanych substancji. Dozownik powinien być ustawiony na pracę w układzie bez dzielenia stru-mienia.
Wzrost temperatury powoduje zmniejszenie powinowactwa LKT do włókna, a tym samym zwiększa efektywność ich uwolnienia do dalszej analizy [11, 20], zaś natęŜenie przepływu gazu nośnego determinuje czas przeniesienia LKT z dozownika do kolumny GC.
Tabela 8 Wpływ temperatury, czasu i połoŜenia igły w dozowniku na proces desorpcji termicznej
Temperatura
• zazwyczaj pomiędzy 250÷300°C • zaleŜy od rodzaju pokrycia włókna • optymalna temperatura jest w przybliŜeniu równa temperaturze wrzenia najmniej
lotnego kwasu • początkowa temperatura kolumny GC powinna być:
� utrzymana na niskim poziomie lub kolumna chłodzona, aŜeby zapobiec posze-rzaniu się pasm chromatograficznych
� niŜsza o 80÷100°C od temperatury wrzenia najbardziej lotnego kwasu
Czas • zaleŜy od temperatury desorpcji dla danego kwasu • na ogół pomiędzy 1 s i 1 min [25], 2 min [20], jednakŜe w celu wyeliminowania
efektu pamięci proponuje się zostawić włókno w dozowniku na 5÷10 min
PołoŜenie igły w dozowniku
• waŜne szczególnie dla trudniej lotnych związków chemicznych, gdyŜ dozownik nie jest jednolicie ogrzany
• najlepiej utrzymywać igłę zawsze w tej samej pozycji
Derywatyzacja
Kwasy tłuszczowe są oznaczane za pomocą chromatografii gazowej albo w postaci wolnej, albo zderywatyzowanej [31]. DuŜa polarność i zdolność do odszczepiania proto-nu prowadzi do silnego oddziaływania z próbką wody i tym samym obniŜa wydajność ekstrakcji i czułość procedury. Ponadto silne oddziaływanie wolnych LKT z elementami układu chromatograficznego moŜe w konsekwencji dawać niesymetryczne, silnie ogonu-jące piki. To z kolei utrudnia oznaczanie i pogarsza jakość otrzymanych wyników. Wprowadzenie etapu chemicznej derywatyzacji umoŜliwia rozwiązanie tego problemu. Derywatyzacja, czyli przeprowadzenie analitów w pochodne (w szczególności w estry), poprawia właściwości chromatograficzne poprzez zmianę struktury analitu. Wynikiem tego jest podwyŜszenie lotności oraz obniŜenie polarności i reaktywności analitów, a co za tym idzie - poprawienie jakości pików chromatograficznych [27, 32-34]. Przyczynia się to do zwiększenia czułości i selektywności, a tym samym do obniŜenia granicy ozna-czalności. Krótkołańcuchowe kwasy są bardzo polarne i dobrze rozpuszczalne w wodzie. Ponadto mogą być adsorbowane na ściankach kolumny GC, co jest szczególnie waŜne w przypadku małych stęŜeń (< 1 mmol/dm3) [31]. Dlatego korzystne jest wprowadzenie etapu derywatyzacji do procedur oznaczania LKT. Ze względu na miejsce przeprowa-dzenia tego procesu moŜemy podzielić je na 3 grupy (tab. 9).

Anna Banel i Bogdan Zygmunt
18
Tabela 9 Podział technik derywatyzacji ze względu na miejsce prowadzenia procesu
Derywatyzacja Charakterystyka
Bezpośrednio w matrycy „in situ”
- dodanie do próbki pierwotnej
odczynnika derywatyzującego - po zajściu reakcji pochodne
ekstrahuje się do włókna SPME
Lotne kwasy tłuszczowe Odczynnik derywatyzujący Pochodne LKT
Na włóknie SPME
- pokrycie włókna jest impregno-
wane odczynnikiem derywatyzu-jącym
- wolne kwasy są wyizolowane z matrycy do włókna, gdzie ule-gają derywatyzacji
Lotne kwasy tłuszczowe Odczynnik derywatyzujący Pochodne LKT
W komorze dozownika
chromatografu
- wprowadzenie wyekstrahowane-go analitu do włókna w postaci pary jonowej, a następ-nie do dozownika chromatografu gazowego
- dzięki wysokiej temperaturze panującej wewnątrz dozownika reakcja derywatyzacji zachodzi szybko
- ograniczenie strat analitu w wyniku wyeliminowania etapu ekstrakcji pochodnej
pary jonowe

Zastosowanie połączenia mikroekstrakcji do fazy stacjonarnej i chromatografii gazowej ...
19
Wysoka selektywność i wydajność procesu derywatyzacji moŜe być osiągnięta dzięki właściwemu doborowi odczynnika derywatyzującego oraz techniki derywatyzacyjnej [31].
Większość odczynników derywatyzujących jest wraŜliwa na obecność wody w próbce, dlatego ekstrakcja analitów z matrycy pierwotnej jest często niezbędna przed derywatyzacją. Przykładem jest reakcja metylowania za pomocą np. metanolu z dodat-kiem trifluorku boru jako katalizatora [35], w wyniku której powstają estry metylowe LKT, które są mało stabilne w roztworach wodnych i konieczna jest ich izolacja przed dalszą analizą.
Wprowadzenie grupy pentafluorobenzylowej w procesie derywatyzacji jest szeroko stosowane w oznaczaniu LKT za pomocą techniki GC z detekcją wychwytu elektronów (ECD) lub spektrometrią mas (MS). PFPDE (1-pentafluorofenylodiazoetan) i PFBBr (bromek pentafluorobenzylu) są skutecznymi odczynnikami alkilującymi anality z grupy LKT w matrycy wodnej. PowyŜsze reakcje nie wymagają zmiany środowiska na roz-puszczalnik organiczny [31].
PDAM (pirenylodiazometan) ma stosunkowo wysoką temperaturę wrzenia, dzięki czemu nie interferuje z lotnymi związkami podczas analizy chromatograficznej, umoŜli-wiając tym samym separację i oznaczenie estrów LKT. PDAM łatwo reaguje z LKT w temperaturze pokojowej i daje pochodne trwałe w próbkach wodnych [31]. Ze wzglę-du na małą lotność moŜna łatwo impregnować nim włókna SPME, stosując heksanowy roztwór [36]. Wymagane są łagodne warunki reakcji, uzyskane pochodne są trwałe, a wydajność duŜa.
PFPDE i PDAM są idealnymi odczynnikami derywatyzującymi, które w połączeniu z techniką SPME słuŜą do monitorowania LKT w powietrzu. PFPDE jako odczynnik stosunkowo lotny dodawany jest w nadmiarze do naczynia, w którym znajdują się anali-zowane kwasy. Zachodzi wówczas bezpośrednia derywatyzacja w matrycy próbki. Kolejnym etapem jest mikroekstrakcja do włókna pokrytego poliakrylem (PA) umiesz-czonego nad powierzchnią próbki, zawierającej pochodne LKT.
PDAM jest stosunkowo nielotnym odczynnikiem, dlatego w tym przypadku zaleca się derywatyzację na powierzchni włókna pokrytego PA. Ponad 98% kwasów ulegało przekształceniu w estry LKT w tej fazy stacjonarnej. Wynika z tego, Ŝe lepszy odzysk otrzymano, prowadząc jednocześnie ekstrakcję i derywatyzację na powierzchni włókna pokrytego fazą stacjonarną z odczynnikiem derywatyzującym [31].
Z kolei zastosowanie bromku benzylu umoŜliwia przeprowadzenie derywatyzacji kwasu octowego do octanu benzylu, bezpośrednio w próbkach wodnych [27]. Po etapie derywatyzacji bezpośrednio w matrycy próbki przeprowadza się ekstrakcję HS-SPME na włóknie pokrytym poliakrylem PA.
Jak wyŜej wspomniano, derywatyzację moŜna równieŜ przeprowadzić bezpośrednio w gorącym dozowniku chromatografu gazowego. Taką technikę derywatyzacji zastoso-wano do oznaczania LKT, zawierających powyŜej 4 atomów węgla w molekule, powsta-łych na skutek ozonolizy cyklicznych alkenów. Odczynnikiem derywatyzującym był N,O-bis(trimetylosililo)trifluoroacetamid (BSTFA), wprowadzający grupę trimetylosili-lowej (TMS). Zaletą reakcji sililowania jest jej duŜa szybkość w wysokiej temperaturze dozownika chromatograficznego [37].
Proces derywatyzacji analitów stosuje się w wielu procedurach analitycznych, w któ-rych na etapie oznaczeń końcowych wykorzystuje się chromatografię gazową. Liczne

Anna Banel i Bogdan Zygmunt
20
zalety oraz dostępność szerokiego spektrum odczynników derywatyzujących (tab. 10) umoŜliwiają ciągły rozwój etapu derywatyzacji i wprowadzenia nowych procedur ozna-czania [38].
Tabela 10 Odczynniki stosowane do derywatyzacji LKT ukierunkowanej na analizę chromatograficzną
Odczynnik derywatyzujący Kwas Matryca Oznaczenie
końcowe Litera-
tura PDAM pirenylodiazometan
C2-C5 woda SPME-GC-FID [31]
PFB-Br bromek pentafluorobenzylu PFPDE 1-pentafluorofenylodiazoetan
C2-C5 woda GC-ECD [31]
PDAM pirenylodiazometan PFPDE 1-pentafluorofenylodiazoetan
C2-C5 powietrze GC-FID [31]
C7H7Br bromek benzylu
C2 woda HS-SPME
GC-FID [27]
PDAM pirenylodiazometan
C2-C3, iC4, C4,
iC5, C5-C6 fekalia
In situ/HS-SPME-GC-MS
[36]
BSTFA N,O-bis(trimetlosililo)trifluoroacetamid
C5-C17 powietrze GC-FID [37]
DNPH 2,4-dinitrofenylohydrazyna
C2-C3 woda morska
HPLC-UV/Vis [39]
PFBHA o-2,3,4,5,6-pentafluorobenzylohydroksyloamina
C2-C3 woda HS-GC-MS [39]
DNPH 2,4-dinitrofenylohydrazyna
C1-C4 powietrze HPLC-detektor spektrofotometryczny
[40]
PFB α-2,3,4,5,6-pentafluorotoluen
C2-C5 fekalia GLC-ECD [41]
Metanol z dodatkiem H2SO4 C1-C3,
iC4, C4 kompost HS-GC-FID [42]
Dobór kolumny chromatograficznej
Dobór odpowiedniej kolumny chromatograficznej jest niezwykle waŜny dla separacji analitów oraz ich jakościowego i ilościowego oznaczania. Ogólna zasada wyboru kolum-ny zaleca w pierwszej kolejności odnalezienie w źródłach informacyjnych (publikacje, katalogi firm) odpowiedniej fazy, której charakterystyka jest odpowiednia do rozdziela-nych składników (np. polarną do składników polarnych). Ogólnie wiadomo, Ŝe fazy bardzo polarne są mniej stabilne i słuŜą do analizy związków o węŜszym zakresie tempe-ratur wrzenia oraz mają na ogół gorszą sprawność. Natomiast kolumny z fazą niepolarną są bardziej odporne na utlenianie, łatwiej znoszą niewłaściwe przechowywanie i nie-szczelności układu chromatograficznego.

Zastosowanie połączenia mikroekstrakcji do fazy stacjonarnej i chromatografii gazowej ...
21
Kolumna chromatograficzna powinna mieć duŜą sprawność i odporność na róŜne czynniki [17]. Cechą fazy stacjonarnej powinna być stabilność termiczna i róŜnicowanie retencji poszczególnych składników analizowanej mieszaniny (duŜa selektywność).
Obecnie do oznaczania lotnych kwasów tłuszczowych stosuje się głównie kolumny kapilarne, poniewaŜ zapewniają krótszy czas analizy, duŜą sprawność oraz małe zuŜycie gazu nośnego i fazy stacjonarnej. W analityce wolnych LKT stosuje się głównie polarne fazy stacjonarne; w przypadku prekolumnowej derywatyzacji (np. do estrów LKT) mogą być równieŜ z powodzeniem stosowane niepolarne lub raczej średnio polarne fazy sta-cjonarne.
Do separacji wolnych LKT najbardziej polecaną fazą stacjonarną jest glikol poliety-lenowy (PEG) modyfikowany przez traktowanie kwasem nitrotereftalowym. Faza ta jest powszechnie stosowana w kolumnach kapilarnych i pakowanych. W kolumnach pakowa-nych glikol polietylenowy moŜe być osadzony na róŜnych nośnikach, a takŜe na sadzy grafityzowanej. McGrath i inni [41] do rozdzielania estrów pentafluorobenzylowych LKT wyizolowanych z fekaliów, zastosowali kolumnę pakowaną wypełnioną Chromo-sorbem W, deaktywowanym dimetylochlorosilanem (DCMS) z naniesioną ciekłą fazą 50%metylo-50%fenylopolisiloksanową. Natomiast Manni i Caron [6] do oznaczania wolnych LKT w próbkach ługów ze składowiska odpadów korzystali z kolumny pako-wanej, wypełnionej Chromosorbem WAW (przemyty 1% kwasem ortofosforowym) i pokrytej nośnikiem estrowym. W tabeli 11 przedstawiono krótką charakterystykę naj-częściej stosowanych faz stacjonarnych do oznaczania lotnych kwasów tłuszczowych.
Tabela 11 Ogólna charakterystyka najczęściej stosowanych faz stacjonarnych do oznaczenia LKT
Faza stacjonarna Charakterystyka
fazy stacjonarnej
Postać rozdzielanych LKT
Nazwy zastosowanych
kolumn
Odnośnik literatu-
rowy
Glikol polietylenowy (PEG) polarna wolna Stabilwax, DBWAX
[2, 7, 9]
Glikol politetylenowy modyfiko-wany kwasem nitrotereftalowym
polarna wolna TR-FFAP [4, 5]
95%dimetylo- 5%difenylopolisiloksan
niepolarna róŜne pochodne DB-5 SPB-5
[11, 27]
14%cyjanopropylo- 86%metylosiloksanowa
średniopolarna estry trimetylosililowe RTX-1701, HP-1701
[37, 42]
Detekcja lotnych kwasów tłuszczowych
W celu oznaczania lotnych kwasów tłuszczowych za pomocą GC stosuje się róŜne metody detekcji, zaleŜnie od tego, czy kwasy znajdują się w postaci wolnej czy teŜ po-chodnych (tab. 12).
Najczęściej stosowanymi detektorami w przypadku oznaczania LKT techniką chro-matografii gazowej są: • detektor płomieniowo-jonizacyjny (FID), • detektor wychwytu elektronów (ECD), • spektrometr mas (MS). Do detekcji LKT stosowano równieŜ olfaktometrię [43, 44].

Anna Banel i Bogdan Zygmunt
22
Tabela 12 Zestawienie literaturowe sposobów detekcji stosowanych w oznaczaniu LKT w postaci wolnej i pochodnych
Literatura Detektor
Wolne LKT Pochodne LKT
FID [2, 4, 6, 37] [27, 34, 42]
MS [4, 5, 45, 47, 48] [7, 37, 46]
ECD [41]
Dotychczas najczęściej stosowanymi detektorami w przypadku oznaczania LKT techniką chromatografii gazowej są: detektor płomieniowo-jonizacyjny (FID) i spektro-metr mas (MS). Granice wykrywalności LKT oznaczanych za pomocą GC przedstawio-no w tabeli 13. Wynika z niej, Ŝe chromatografia gazowa sprzęŜona ze spektrometrem mas z ujemną jonizacją chemiczną (GC-CI-MS) daje zdecydowanie niŜsze granice wy-krywalności niŜ pozostałe detektory. Wprowadzając etap derywatyzacji, moŜna uzyskać zwiększoną selektywność i obniŜoną granicę oznaczalności np. poprzez wprowadzanie fluorowanych grup funkcyjnych i zastosowanie detektora ECD.
Tabela 13 Granice wykrywalności LKT
Granica wykrywalności [mg/l]
LKT Detektor
C2 C3 iC4 C4 iC5 C5 C6 C7 Lit.
SPME-GC-FID
3,7 3,3 0,9 0,3 0,7 0,3 - - [2]
HS-SPME-GC-FID
0,675 0,054 - 0,006 0,046 0,019 0,038 [4]
SPME-GC-FID
0,760 0,290 - 0,122 - 0,003 - - [31]
SPME-GC-FID (derywatyzacja PDAM)
- 0,002 - 0,001 - - - - [31]
HS-GC-FID (derywatyzacja: metanol + H2SO4)
5 1 10 1 - 5 - - [42]
HS-SPME-GC-FID
FID
0,0156 - - - - - - - [27]
NCI-MS
0,150 0,005 - 0,002 - 0,002 0,006 0,005 [5]
PCL-MS (CH4) MS
0,115 0,025 - 0,026 - 0,011 0,013 0,010 [5]
SPME-GC-ECD (derywatyzacja PFB-Br)
- - - 0,00005 - 0,00001 - - [31]
SPME-GC-ECD (derywatyzacja PFPDE) ECD
0,00008 0,00006 - 0,00005 - 0,00004 - - [31]

Zastosowanie połączenia mikroekstrakcji do fazy stacjonarnej i chromatografii gazowej ...
23
Literaturowy przegl ąd metodyk oznaczania LKT
Efektywność biologicznego oczyszczania ścieków w duŜym stopniu zaleŜy od zawar-tości LKT, które, jeśli zostaną wprowadzone do środowiska, mogą powodować nieko-rzystne zmiany. Dlatego teŜ naleŜy je monitorować w ściekach surowych i oczyszczo-nych, a takŜe na róŜnych etapach oczyszczania. Znajomość zawartości LKT w ściekach jest pomocna juŜ na etapie projektowania nowoczesnych rozwiązań technologicznych, stosowanych w oczyszczalniach ścieków. Zatem nie moŜe budzić zdziwienia to, Ŝe w ciągu ostatnich lat obserwuje się znaczący wzrost liczby publikacji dotyczących zagadnień oznaczania lotnych kwasów tłuszczo-wych w ściekach i innych próbkach środowiskowych.
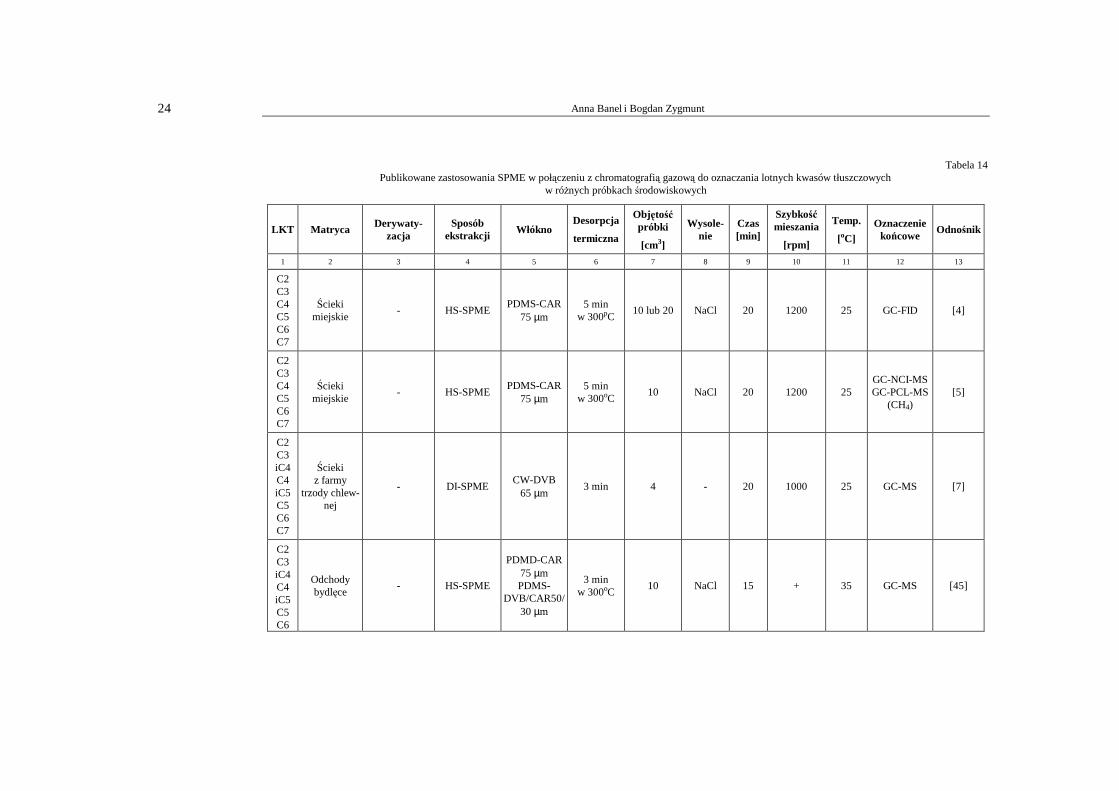
Na podstawie przeprowadzonego studium literaturowego stwierdza się, iŜ najczęstszą techniką oznaczania lotnych kwasów tłuszczowych w próbkach środowiskowych, w tym w ściekach, jest chromatografia gazowa. W tabeli 14 przedstawiono zastosowanie tech-niki GC do oznaczania LKT, w których izolacje i wzbogacanie analitów prowadzi się z wykorzystaniem mikroekstrakcji do fazy stacjonarnej, techniki prostej, wygodnej, sto-sunkowo taniej i dość skutecznej.
Wnioski
Proces beztlenowej biodegradacji materiału organicznego z punktu widzenia jakości środowiska jest zazwyczaj zjawiskiem korzystnym. Powstające wówczas lotne kwasy tłuszczowe, np. w ściekach, zwiększają efektywność biologicznego usuwania związków fosforu i azotu. Jednak LKT mogą mieć pewien niekorzystny wpływ na środowisko; mają bowiem nieprzyjemny zapach i zwiększają mobilność metali cięŜkich i radionukli-dów. Z powyŜszych względów konieczne jest monitorowanie stęŜenia LKT w ściekach na róŜnych etapach oczyszczania biologicznego i w środowisku, w szczególności na składowiskach odpadów. Do oznaczania LKT w próbkach środowiskowych stosuje się na ogół techniki chromatograficzne, w tym najszerzej chromatografię gazową. Przedsta-wiony przegląd literaturowy wskazuje, Ŝe połączenie nowoczesnej techniki przygotowa-nia próbek, tj. mikroekstrakcji do fazy stacjonarnej i chromatografii gazowej z odpowiednim systemem separacyjnym i detekcyjnym, pozwala uzyskać duŜą czułość i selektywność oraz niską granicę oznaczalności.
Dotychczasowe badania pozwalają stwierdzić, Ŝe metodyki, wykorzystujące technikę SPME na etapie przygotowania próbek i chromatografii gazowej sprzęŜonej ze spektro-metrią mas, detektorem płomieniowo-jonizacyjnym lub wychwytu elektronów na etapie oznaczeń końcowych, mogą być z powodzeniem stosowane do identyfikacji i oznaczania ilościowego lotnych kwasów tłuszczowych w ściekach, powietrzu, odcho-dach i innych próbkach.
RóŜnorodność matryc próbek i zwiększające się coraz bardziej rygorystyczne wyma-gania dotyczące jakości wyników zawartości LKT w róŜnych mediach oraz aspekty ochrony środowiska sprawiają, Ŝe powinny być prowadzone dalsze prace nad modyfika-cją juŜ istniejących i opracowaniem nowych procedur analitycznych.
Anna Banel i Bogdan Zygmunt
24
Tabela 14 Publikowane zastosowania SPME w połączeniu z chromatografią gazową do oznaczania lotnych kwasów tłuszczowych
w róŜnych próbkach środowiskowych
LKT Matryca Derywaty-
zacja Sposób
ekstrakcji Włókno
Desorpcja
termiczna
Objętość próbki
[cm3]
Wysole-nie
Czas [min]
Szybkość mieszania
[rpm]
Temp.
[oC] Oznaczenie
końcowe Odnośnik
1 2 3 4 5 6 7 8 9 10 11 12 13
C2 C3 C4 C5 C6 C7
Ścieki miejskie
- HS-SPME PDMS-CAR
75 µm 5 min
w 300pC 10 lub 20 NaCl 20 1200 25 GC-FID [4]
C2 C3 C4 C5 C6 C7
Ścieki miejskie
- HS-SPME PDMS-CAR
75 µm 5 min
w 300oC 10 NaCl 20 1200 25
GC-NCI-MS GC-PCL-MS
(CH4) [5]
C2 C3 iC4 C4 iC5 C5 C6 C7
Ścieki z farmy
trzody chlew-nej
- DI-SPME CW-DVB
65 µm 3 min 4 - 20 1000 25 GC-MS [7]
C2 C3 iC4 C4 iC5 C5 C6
Odchody bydlęce
- HS-SPME
PDMD-CAR 75 µm PDMS-
DVB/CAR50/30 µm
3 min w 300oC
10 NaCl 15 + 35 GC-MS [45]

Zastosowanie połączenia mikroekstrakcji do fazy stacjonarnej i chromatografii gazowej ...
25
C7
C2 C3 iC5 C6
Wydech bydlęcy
- HS-SPME PDMS-DVB/
CAR50/ 30 µm
7 min w 250oC
28 310 - 15 - 30 GC-MS-O [43]
C2 C3 iC4 C4 C5 C6
Pył w chlewni
- HS-SPME PDMS-CAR
85 µm 40 min
w 260oC 40 - 180 - 25 GC-MS-O [44]
C2 C3 iC4 C4 iC5 C5 C6
Pył w chlewni
- HS-SPME PDMS- CAR
75 µm 30÷60 min w 280oC
10 - 30 - 80 GC-MS [47]
C2 C3 iC4 C4 C5 C6 C7 C8
Odchody - HS-SPME PDMS-CAR
75 µm 300oC - - 5 + 20 GC-MS [48]
C2 iC5
Obszary bagienne
- HS-SPME PDMS-CAR
75 µm 5 min
w 300oC 10 NaCl 20 + 25
GC- NCI-MS GC-PCL-MS
(CH4) [49]

Anna Banel i Bogdan Zygmunt
26
C2 C3 C4 C5 C6 C7
Próbki wody
powietrze
In situ/SPME Pirenylodiazo-
metan
DI-SPME
HS-SPME
Wolne kwasy: PA 95 µm Pochodne PDAM:
PA
Wolne kwasy: 3 min
Pochodne PDAM: 4 min
- - - - - GC-FID [11]
C2 Próbki wodne Octan benzylu HS-SPME PA 5 min
w 250°C 3 - 30 - 25 GC-FID [27]
C2 C3 C4 C5
Powietrze
In situ/SPME Pirenylodiazo-
metan (PDAM)
DI-SPME
Wolne kwasy: PA
85 µm Pochodne PDAM:
PA
5 min 40 - 180÷ 240
+ - GC-FID GC-ECD GC-ITMS
[31]
C2 C3 C4 C5
Woda
In situ/SPME Pirenylodiazo-
metan (PDAM)
HS-SPME Pochodne PDAM:
PA 5 min - - 60 - -
GC-FID GC-ECD
[31]
C2 C3 iC4 C4 iC5 C5 C6
Odchody
In situ/SPME Pirenylodiazo-
metan (PDAM)
HS-SPME PA 85 µm 4 min - NaCl - - - GC-MS [36]

Zastosowanie połączenia mikroekstrakcji do fazy stacjonarnej i chromatografii gazowej ...
27
Literatura
[1] śeglin K.: Mat. Seminarium w Polit. Krakow. „Metody oznaczania wskaźników zanieczyszczeń orga-nicznych”. Kraków 1998.
[2] Cruwys J.A., Dnsdale R.M., Hawkes F.R. i Hawkes D.L.: J. Chromatogr. A, 2002, 945, 195-209. [3] Kryłow M. i Lach A.: Mat. Seminarium w Polit. Krakow. „Metody oznaczania wskaźników zanieczysz-
czeń organicznych”. Kraków 1998. [4] Abalos M., Bayona J.M. i Pawliszyn J.: J. Chromatogr. A, 2000, 873, 107-115. [5] Abalos M., Bayona J.M. i Pawliszyn J.: J. Chromatogr. A, 2000, 891, 287-294. [6] Manni G. i Caron F.: J. Chromatogr. A, 1995, 690, 237-242. [7] Shao-Pin Yo: Chemosphere, 1999, 38, 823-834. [8] Światłowska J., Zaborowska A. i Zygmunt B.: Analityka, 2006, 2, 4-6. [9] Kai P. i Schafer A.: Agr. Eng. International: CIGR J. Sci. Res. Develop., 2004, 6, Manuscript BC 04 006. [10] Poradnik fizykochemiczny, praca zbiorowa. WNT, Warszawa 1974. [11] Pan L., Adams M. i Pawliszyn J.: Anal. Chem., 1995, 67, 4396-4403. [12] Wardencki W., Curyło J. i Namieśnik J.: J. Biochem. Biophys. Methods, 2007, 70, 275-288. [13] Psillakis E. i Kalogerakis N.: Trends Anal. Chem., 2002, 21, 53-63. [14] Psillakis E. i Kalogerakis N.: J. Chromatogr. A, 2001, 907, 211-219. [15] Psillakis E. i Kalogerakis N.: J. Chromatogr. A, 2001, 938, 113-120. [16] Psillakis E. i Kalogerakis N.: J. Chromatogr. A, 2003, 999, 145-153. [17] Namieśnik J. i Jamrógiewicz Z.: Fizykochemiczne metody kontroli zanieczyszczeń środowiska. WNT,
Warszawa 1998. [18] Mester Z. i Sturgeon R.: Spectrochim. Acta B, 2005, 60, 1243-1269. [19] de Fa´tima Alpendurada M.: J. Chromatogr. A, 2000, 889, 3-14. [20] Penalver A., Pocurull E., Borrull F. i Marce R.M.: Trends Anal. Chem., 1999, 18, 557-568. [21] Theodoridis G., Koster E.H.M. i de Jong G.J.: J. Chromatogr. B, 2000, 745, 49-82. [22] Zygmunt B., Zaborowska A., Światłowska J. i Namieśnik J.: Curr. Org. Chem., 2007, 11, 241-253. [23] Bulletin 923, Supelco, 1998. [24] Katalog Supelco, 2003/2004, 350-369. [25] Prosen H. i Zupancic-Kralj L.: Trends Anal. Chem., 1999, 18, 272-282. [26] Augusto F. i Pires Valentey A.L.: Trends Anal. Chem., 2002, 21, 428-438. [27] Wittmann Gy., Van Langenhove H. i Dewulf J.: J. Chromatogr. A, 2000, 874, 225-234. [28] Eisert R. i Pawliszyn J.: J. Chromatogr. A, 1997, 776, 293-303. [29] Grebel J.E. i Young C.C., (Mel) Suffet I.H.: J. Chromatogr. A, 2006, 1117, 11-18. [30] Kataoka H., Lord H.L. i Pawliszyn J.: J. Chromatogr. A, 2000, 880, 35-62. [31] Pan L. i Pawliszyn J.: Anal. Chem., 1997, 69, 196-205. [32] Field J.A.: J. Chromatogr. A, 1997, 785, 239-249. [33] Zygmunt B., Jastrzębska A. i Namieśnik J.: Crit. Rev. Anal. Chem., 2001, 31(1), 1-18. [34] Mills G. A. i Walker V.: J. Chromatogr. A, 2000, 902, 267-287. [35] Clark J.T. i Bunch J.E.: J. Chromatogr. Sci., 1997, 35, 206-280. [36] Mills G. A., Walker V. i Mughal H.: J. Chromatogr. B, 1999, 730, 113-122. [37] Docherty K.S. i Ziemann P.J.: J. Chromatogr. A, 2001, 921, 265-275. [38] Wells J.M.: J. Chromatogr. A, 1999, 843, 1-18. [39] Takeda K., Katoh S., Nakatani N. i Sakugawa H.: Anal. Sci., 2006, 22, 1509-1514. [40] Uchiyama S., Matsushima E., Aoyagi S. i Ando M., Anal. Chem., 2004, 76, 5849-5854. [41] McGrath L.T., Weir C.D., Maynard S. i Rowlands B.J.: Anal. Biochem., 1992, 207, 227-230. [42] Himanen M., Latva-Kala K., Itavaara M. i Hanninen K.: J. Environ. Qual., 2006, 35(2), 516-521. [43] Spinhirne J.P., Koziel J.A. i Chirase N.K.: J. Chromatogr. A, 2004, 1025, 63-69. [44] Cai L., Koziel J.A., Lo Y.-C. i Hoff S.J.: J. Chromatogr., 2006, 1102, 60-72. [45] Larreta J., Vallejo A., Bilbao U., Alonso A., Arana G. i Zuloaga O.: J. Chromatogr. A, 2006, 1136, 1-9. [46] Wells J.M. i Zhou You L.: J. Chromatogr. A, 2000, 885, 237-250. [47] Razote E.B., Maghirang R.G., Seitz L.M. i Jeon I.J.: Am. Soc. Agric. Eng., 2004, 47(4), 1231-1238. [48] Miller D.N. i Woodbury B.L.: J. Environ. Qual., 2006, 35(6), 2383-2394. [49] Huang Y., Ortiz L., Aguirre P., Garcia J., Mujeriego R. i Bayona J.M.: Chemosphere, 2005, 59, 769-777.

Anna Banel i Bogdan Zygmunt
28
APPLICATION OF COMBINATION OF SOLID PHASE MICROEXT RACTION AND GAS CHROMATOGRAPHY FOR DETERMINATION OF VOLATIL E FATTY ACIDS
IN ENVIRONMENTAL AND RELATED SAMPLES
Summary: Determination of volatile fatty acids (VFAs) in environmental samples requires an appropriate technique of sample preparation and a technique of final analysis; the latter should enable effective separation and reliable identification of VFAs to be determined. A convenient and frequently used technique of VFA isolation and enrichment is solid phase microextraction (SPME), while gas chromatography (GC) is an effi-cient technique of final analysis. In this work, the combination of these two techniques for determination of VFAs in environmental and related samples has been critically discussed.
Keywords: volatile fatty acids, gas chromatography, solid phase microextraction, environmental sample, waste water







![DOH]DND>HQLH]RVWDMH POSTACIE KLINICZNE … · utratę wody (ok. 1 l/godz.) i elektrolitów. harakterystycznie w próbkach kału nie stwierdza się obecności krwi lub leukocytów](https://img.dokumen.tips/doc/110x75/5c758d4b09d3f220278b78c9/dohdndhqlhrvwdmh-postacie-kliniczne-utrate-wody-ok-1-lgodz-i-elektrolitow.jpg)




![REDOKSOWE PRZEMIANY OSCYLACYJNE DIFTALOCYJANIN …tchie.uni.opole.pl/CDEMfree/Waclawek_redoksowe.pdf · dwa niepodstawione ligandy ftalocyjaninowe [13], do złożonych, ... miały](https://img.dokumen.tips/doc/110x75/5c785cbc09d3f21d538cd111/redoksowe-przemiany-oscylacyjne-diftalocyjanin-tchieuniopoleplcdemfreewaclawek.jpg)
















