Embed Size (px)
Citation preview

rsc.li/chemcomm
ChemCommChemical Communications
rsc.li/chemcomm
ISSN 1359-7345
COMMUNICATIONS. J. Connon, M. O. Senge et al. Conformational control of nonplanar free base porphyrins: towards bifunctional catalysts of tunable basicity
Volume 54Number 14 January 2018Pages 1-112
ChemCommChemical Communications
This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication.
Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available.
You can find more information about Accepted Manuscripts in the Information for Authors.
Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains.
Accepted Manuscript
View Article OnlineView Journal
This article can be cited before page numbers have been issued, to do this please use: B. D. Barve, Y. Kuo
and W. Li, Chem. Commun., 2021, DOI: 10.1039/D1CC04397H.

FEATURE ARTICLE
Please do not adjust margins
Please do not adjust margins
Received 00th January 20xx,Accepted 00th January 20xx
DOI: 10.1039/x0xx00000x
Pd-catalyzed and ligand-enabled alkene difunctionalization via unactivated C–H bond functionalizationBalaji D. Barve,ab Yao-Haur Kuo a and Wen-Tai Li*a
Palladium-catalyzed and ligand-enabled C–H functionalization methods have emerged as a powerful approach for the preparation of therapeutically important motifs and complex natural products. Olefins, owing to their natural abundance, have been extensively employed for the formation of C–C and C–X bonds and the generation of various heterocycles. Traditionally, activated as well as starting materials with preinstalled functional groups, and also halide substrates under transition metal catalysis, have been employed for olefin difunctionalization. However, strategies for employing unactivated C–H bond functionalization to achieve alkene difunctionalization have rarely been explored. A possible solution to this challenge is the application of bulky ligands which enhances the reductive elimination pathway and inhibit β-hydride elimination to selectively yield difunctionalized alkene products. This feature article summarizes the utilization of unreactive C–H bonds in the Pd-catalyzed and ligand-enabled difunctionalization of alkenes.
1 IntroductionIn recent decades, transition metal-catalyzed functionalization of C–H bonds has emerged as a powerful tool for the preparation and diversification of structurally relevant molecules. Among these methods, Pd-catalyzed reactions have emerged as a straightforward and highly efficient technique for the synthesis of natural products and pharmaceutically important motifs.1,2 This strategy is associated with several important features, such as easily accessible starting materials, efficient steps/atom economy, and significant practicality.1 Indeed, there have been pioneering discoveries in this field, such as the Wacker process and the Fujiwara–Moritani reaction.3
The difunctionalization of alkenes, in which two novel functional groups are introduced, one on each side of the carbon–carbon double bond, in a one-pot reaction, is of great interest to researchers in organic synthesis.4 In addition, olefins are readily accessible, abundantly available, and cheap.
The various alkene difunctionalization approaches, including aminocarbonylation,5 carboetherification,6 carboamination,7 diamination,8 carbonylation,9 dioxygenation,10 and dicarbonation,11 are all well documented in the literature. Some of them also utilize the C–H bond functionalization of alkenes to produce a variety of difunctionalized products.8a–b,11a However, the reported strategies have employed traditional alkylating organometallic reagents with intramolecularly substituted sulfonamides or
amides and aryl/pseudo-halides as reaction partners with alkenes. Hence, their applications are limited owing to these expensive starting materials with preinstalled functional groups and the generation of stoichiometric environmentally hazardous byproducts.
Therefore, the Pd-catalyzed functionalization of C–H bonds, by avoiding expensive organometallic reagents and aryl/pseudo-halides to achieve alkene difunctionalization, has received considerable attention and has emerged as an efficient and complementary method for overcoming the disadvantages of traditional methods.12
Traditionally, Pd-catalyzed olefin difunctionalization approaches have always been hampered by challenges to introducing the second functional group via reductive elimination of Pd(II) intermediates and competing β-hydride elimination reactions.13 One strategy to address these difficulties is to employ sterically bulky ligands to promote the reductive elimination pathway and avoid the β-hydride elimination step.10a,14 Additionally, the presence of ligands also increases the rate of reaction and the catalytic reactivity of Pd and significantly improves the stereoselectivity of the reaction.
In this feature article, we have focused on recent advances in the Pd-catalyzed and (organic) ligand-enabled difunctionalization of olefins via unactivated C–H bond functionalization. Generally, C–H bonds having pKa values greater than 30 (pKa > 30) are defined as “unreactive.” However, acidic C−H bonds with a lower pKa may be equally unreactive due to their higher bond dissociation energies (BDE) and vice versa.15 Hence in this feature article, we have used the term “unactivated C–H bond” in most of the descriptions.
In general, most approaches employ the oxidative Fujiwara–Moritani strategy via initial C–H bond functionalization, avoiding the β-hydride elimination process (Heck products), and resulting in alkene difunctionalized derivatives via reductive
a.National Research Institute of Chinese Medicine, Ministry of Health and Welfare, Taipei 11221, Taiwan, R.O.C.
b.Department of Chemistry, National Taiwan Normal University, Taipei 10610, Taiwan, R.O.C.
Page 1 of 13 ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H

ARTICLE ChemComm
2 | Chem. Commun., 2021, 00, 1–12 This journal is © The Royal Society of Chemistry 2021
Please do not adjust margins
Please do not adjust margins
N
R2
R1
R3 R3
Pd(OAc)2(10 mol%)pyridine (L2, 8 mol%)
Phl(OCOt-Bu)2 (2.5 equiv)
AgF (5 equiv)CH3CN, 100 oC, 24 h
N
R2CH2CN
R1
R1 = Ac 90%Piv 61%Boc 80%Me 0%Ph 0%
R3 = NO2 66%F 67%Cl 52%Br 74%I 77%
R3 = F 71%Me 84%
N
CH2CN
R1N
CH2CN
Ac
R3
N
CH2CN
AcR3
R3
N
R2CH2CN
Ac
R2 = Ph 0%CO2Et 44%
N NH2
L2
N
NN O
R3
R2
CNH
R4H
N
N
NO
R2
R3 R4
CNPd(OAc)2 (5 mol %)/L1 (7.5 mol %)
PhI(O2CtBu)2 (1.1 equiv.)
AgF (4 equiv.), MgSO4 (40 mg)R"CH2CN (2.0 mL), 80 oC L1
NO
CN
NO
CN
NO
CN
NO
CN
X
OAc
X = FClBrI
96%98%94%98% 83%90%
92 %
R1R1
NO
Ph
CN
98%
NO
CNNPhth
NO
Ph CN
82%87%
NO
R2
CN
R2 = Ac, Ts 0%
NO
CN
N NO
CNF
F
96% 73%
N O PdII(OAc)2N
N
N O
N O
PdII
NN
OAc
B
CN
+
N N
N N
=
N
NHOAc
N OPdII
N
N
C
+
ESI/MS(m/z): 438
C-H bondactivation
(rds)PhI(OPiv)2
AgF/CH3CN
N OPd
CNXN
ND
X = OPivor F
R R'Pd(II)
[Nu]R'R
Nu
PdIV
[O]reduct.elimin. R'R
Nu
FG[FG]a)
RAr-PdLn R
ArPdLn - H-PdLn
RAr
Heck product
+ H-PdLn
RAr
PdLn
Nu-HRAr
Nu
b)
elimination (Scheme 1a).3,14a Furthermore, Heck intermediates generated during the Pd-catalyzed C–H functionalization process can be turned into the respective alkene difunctionalized products in the presence of appropriate nucleophiles or oxidants (Scheme 1b).12g
Scheme 1. Palladium-catalyzed difunctionalization of alkenes.
2 DicarbonationThe formation of the C–C bond represents a core element of organic chemistry owing to its basic application in the generation of molecular diversity and complexity. The development of efficient strategies for constructing C–C bonds has always been an inspiring goal among researchers in organic synthesis.16 The Pd-catalyzed oxidative Fujiwara–Moritani reaction is an efficient protocol for the generation of C−C bonds via the reaction of arenes and olefins.3
2.1 ArylalkylationLiu et al. reported the arylalkylation of alkenes via the C–H bond functionalization of anilines and unreactive acetonitrile, affording nitrile-bearing indolinones in excellent yields. The protocol avoids strong bases and is performed in the presence of PhI(OPiv)2 and AgF, which acts as a basic additive as well as a promoter of the C(sp3)–H bond cleavage of acetonitrile. The screening of various ligands revealed that bidentate nitrogen-containing ligands improved the reaction outcome, and L1 was selected as the best ligand for this protocol, providing the highest reaction yield (Scheme 2).17
Scheme 2. Pd-catalyzed arylalkylation of alkenes.
The substrate scope indicated that various substituted anilines bearing aryl or alkyl groups on the nitrogen yielded the corresponding products in excellent yields. However, when
electron-withdrawing groups were attached to N, the reaction did not proceed, indicating its limitations. Aryl ring substituents did not show any remarkable effect on reaction outcomes, yielding the respective indolinones in good to excellent yields. However, addition of a substituent on the R4 (R4 = -Et, 54%; -OMe, 32%; -dimethyl, 0%) position reduced the reaction yields substantially.
In 2012, the same group extended the methodology to unactivated alkenes by replacing the previous ligand with pyridine L2, which accelerated the C–H cleavage of aniline during the preparation of nitrile-containing indolines under similar reaction conditions (Scheme 3).18 The ratio of Pd to pyridine (1:0.8) is crucial for this reaction. Investigation of the substrate scope revealed that carbonyl-containing protecting groups on the N yielded the corresponding derivatives in good yields. In contrast, when N was attached to phenyl and methyl groups, the desired products were not generated. The nature of the substituents on the aryl ring had no significant effect on the reaction; electron-donating and -withdrawing groups, as well as halides, afforded the respective products in moderate to good yields. The proposed mechanism for both studies revealed the generation of the Pd-complex C via the coordination of Pd(II) to the olefin and nucleophilic attack on the tethered arene. Then, in the presence of PhI(OPiv)2 and AgF, C(sp3)–H bond activation of CH3CN (rate-determining step) occurred, generating the Pd(IV) complex D, which subsequently underwent reductive elimination to afford the corresponding arylalkylation products (Scheme 4).
Scheme 3. Preparation of nitrile-containing indolines.
Page 2 of 13ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H

ChemComm ARTICLE
This journal is © The Royal Society of Chemistry 2021 Chem. Commun., 2021, 00, 1–12 | 3
Please do not adjust margins
Please do not adjust margins
H HO
N N
O
PdOAc
AcO
-HOAc
H HO
N N
O
PdOAc
HH
ON
N
O
Pd
NO
AcO
A
B
C
NN
O
OPdO
N
CF3
AcO
D
NN
O
OPdII
O
N
PhI(OAc)2+
TMSCF3/CsF
ESI-MS Signals
[A-OAc-] (m/z): 435[D-OAc-] (m/z): 678[D-CF3
-] (m/z): 668
TMS-F
N O
NOCF3
NArF
CO2Et
O
NArF
Et
Et
CO2Et
O
NArF
CO2Et
O
NArF
CO2Et
O
87%Gram scale (86%) 70% 68% 75%
TFAN
NArF
CO2Bn
O
NArF
COEt
O
NArF
CN
O
77% 64% 73%
MeR NArFCONHArF
R
O
Pd(OAc)2 (10 mol %)L4 (40 mol %)
TEMPO (10 mol %)
AgOAc (2 equiv.)K2HPO4 (1.1 equiv.)NaHCO3 (2 equiv.)
O2 (1 atm)C6F5CF3, 120 oC
N O
ButMe
Me
NArF
COPh
O
70%
L4
NArF
CO2Et
O
NArF
CO2Et
O
O
51% (55/45)dr
62% (61/39)dr
ArF = (4-CF3)C6F4
N
R3
NOR2
R1
R2
O
O
N N
OR3 CF3R1
TMSCF3
)-L3
cat. Pd(OAc)2 /L3cat. Yb(OTf)3
PhI(OAc)2 (2 equiv.)CsF, EtOAc, r.t.
NOCF3R
NO
CF3
NPhth
NOCF3X
70%R = OMeMeNO2OCF3
68%76%84%72%
X = FClBrI
83%88%82%89%
NOCF3
R
R
R = Me 61%F 71%
NR
OCF3
R = Ts, 0%
MeMe Me Me
Scheme 4. Proposed mechanism for arylalkylation.
2.2 TrifluoromethylationContinuing their research into C–C bond formation, Liu et al. demonstrated a novel Pd-catalyzed oxidative method for the aryltrifluoromethylation of activated alkenes. The method utilized an inexpensive TMSCF3/CsF combination as the source of the trifluoromethyl group and PhI(OAc)2 as an oxidant to provide the CF3-substituted oxindoles at room temperature (Scheme 5).19 The trifluoromethyl group is enormously important in the agrochemical and pharmaceutical industries. The disadvantages of previous alkene aryltrifluoromethylation methods, such as the use of expensive CF3 sources at higher temperatures, were successfully overcome in this approach.20 The optimization of various nitrogen-containing ligands suggested that L3 afforded the best yield. Various nitrogen-protecting groups, including aryl, alkyl, and silyl moieties, produced the corresponding oxindoles in good yields; in contrast, with the tosyl protecting group, the reaction did not occur. With anilines bearing electron-donating groups, electron-withdrawing groups, and halogens, the reaction occurred easily, affording the corresponding derivatives in excellent yields. The results of detailed control experiments and mechanistic studies suggested that the reaction proceeded via the initial coordination of the alkene to the Pd(II) intermediate B. Nucleophilic attack on the tethered arene generated the Pd intermediate C, which, upon oxidation by PhI(OAc)2/TMSCF3, yielded the C(sp3)−PdIV(CF3) species D, resulting in C(sp3)−CF3 bond formation through reductive elimination, to afford the target products (Scheme 6).
Scheme 5. Synthesis of CF3-substituted oxindoles at room temperature.
Scheme 6. Proposed mechanism of trifluoromethylation.
3 Carboheterofunctionalization of alkenesThe carboheterofunctionalization of alkenes represents a straightforward and atom-economical strategy for preparing therapeutically useful heterocyclic molecules. This approach proceeds via the simultaneous generation of C–X/C–C bonds, resulting in structurally and biologically important oxygen/nitrogen-containing heterocycles.21
3.1 Carboamination via inactivated sp3 C–H bond functionalizationVarious groups have reported the carboamination of alkenes and/or alkynes with aryl compounds substituted with nitrogen-containing or carbonyl groups utilizing ruthenium and rhodium catalysis via C–H bond activation.22 Furthermore, in recent decades, Pd-catalyzed aryl C–H olefination has been extensively studied and reported.23 However, the mechanistically different but challenging Pd-catalyzed olefination of unreactive sp3 C–H bonds has been less well explored. The hurdles in the sp3 C–H activation process are the stability of the sp3 C–H bond and the coordinating ability of alkenes, which may follow a competitive -hydride elimination route, especially in intermolecular reactions. In their pioneering research, Yu et al. demonstrated the Pd-catalyzed difunctionalization of alkenes by employing various ligands and via unreactive sp3 C–H functionalization. They developed a novel approach for -C(sp3)−H olefination and the carbonylation of aliphatic acids by utilizing the quinoline-based ligand L4 and an amide-directing group to generate the respective lactams with all-carbon quaternary carbon centers (Scheme 7).24 The method was successfully applied to prepare -olefinated aliphatic acids. The scope of the reaction indicated that -quaternary center-substituted aliphatic amides, bearing methyl, ethyl, benzyl, or cyclopentyl functional groups provided the target products in moderate to good yields. The -methoxy moiety and -esters successfully afforded the corresponding derivatives in moderate to acceptable yields. Interestingly, cyclic substrates, including tetrahydropyran and piperidine units, provided spirolactams in good yields. Different activated olefins bearing benzyl groups, -unsaturated ketones, and nitrile groups underwent the reaction smoothly, affording the respective lactams in good yields.
Page 3 of 13 ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H

ARTICLE ChemComm
4 | Chem. Commun., 2021, 00, 1–12 This journal is © The Royal Society of Chemistry 2021
Please do not adjust margins
Please do not adjust margins
+
Pd(OAc)2 (10 mol%)L5 (30 mol%)
Ag2CO3, NaOAc, DCMair, 20 h, 105 oC
RNHTf
R1 R2
H
MeO2C NMeO2C
R1 R2
R
Tf N
CF3CF3
L5
NMeO2CCO2Bn
TfNMeO2C
CO2Bn
TfNMeO2C
CO2Bn
Tf73%72% 95%
R1H CO2Bn
NR3CO2Bn
R2
R1
Tf
R2
NHTfR3
Pd(OAc)2 (10 mol %)L6 (30 mol %)
Ag2CO3, Cu(OAc)2, NaOAcDCE, 105 oC, O2, 20 h
NCO2Bn
Tf61%
NCO2Bn
Tf
AcON
CO2Bn
TfAcON
CO2Bn
Tf
MeO2C
77% 75% 65%
N
Ph
L6
HR
NMeO2CR
NsNHNsMeO2C
Pd(OAc)2 (10 mol %)L7 (30 mol %)
Ag2CO3, NaOAc, DCE105 oC, air, 20 h
N
L7
90%
NMeO2CCO2t-Bu
Ns
52%
NMeO2CPh
NsNMeO2CNs
O
NMeO2CNs
Ph
O
75% (10:1)d.r 60% (12:1)
Reactions in the presence of L5-L7
b)
c)
a)
NMeO2CCO2Bn
Tf
77%
NMeO2CP
Tf
77%
O
OEtOEt NMeO2C
Ph
Tf
58%
NMeO2CTf
CF3 NMeO2CTf
Cl
59% 63%
R'
NMeO2CNs
PdII
H XL N
Ns
MeO2C
R'
HR
PdIIHX
L
syn-Addition
NNs
MeO2C
R'R
Pd0LnX2PdIILn
R = CO2R" or ArPathway IIRR'
NHNsMeO2C
Pathway IMichaelAddition
NR
R'
MeO2C
NsR = CN, COMe or COPh
Reoxidation
Ag0 AgI
NSO ONa
ArMeO2C
R'PdII
X
L
R
PdIIR'
NHNsMeO2C
R
X
L
Pd0LnX2PdIILn
Reoxidation
Ag0 AgI
-HydrideElmination-Hydride
Elmination
1,2-MigratoryInsertion
NSO ONa
ArMeO2C
R'PdII
X
L
R
HR'
NHNsMeO2C
C-H Activation
Ar = 4-(NO2)C6H4
A
C
D
E
F
HG
I
B
R
Scheme 7. Carboamination of alkenes via -C−H functionalization of aliphatic acids.
Later, in 2016, the same group investigated the Pd(II)-catalyzed olefination of -C(sp3)–H bonds of amines protected by triflyl (Tf) and 4-nitrobenzenesulfonyl (Ns) groups, yielding a variety of C-2 alkylated pyrrolidines (Scheme 8).25 The series of quinoline-based ligands L5–L7 were developed and applied to appropriate amine substrates for the best results. For example, the 3,4-bis(trifluoromethyl)pyridine ligand was suitable for the transformation of Tf-protected α-amino acid esters in the presence of Ag2CO3 as the sole oxidant. The 3-phenylquinoline ligand was suggested to be superior for the conversion of Tf-protected aliphatic amines, including β-amino alcohols and β-amino acids, to their respective products in the presence of Ag2CO3, Cu(OAc)2, and O2. The application of the Ns protecting group for direct sp3 C–H functionalization of alkyl amines suggests the viability of this method. The reaction fails to proceed in the absence of pyridine- or quinoline-based ligands, suggesting the importance of these ligands for this protocol. The synthetic application of the method was demonstrated by the preparation of natural products and bioactive motifs incorporating the chiral pyrrolidine unit.
Based on these experimental results, an overall mechanism for the generation of pyrrolidines is proposed (Scheme 9). Under basic conditions, the X2PdIILn catalyst initially binds with sulfonamine, resulting in cleavage of the -C(sp3)–H bonds. The five-membered palladacycle species C thus generated coordinates with an alkene and undergoes 1,2-migratory insertion to yield intermediate E; this is followed by β-hydride elimination to generate the olefinated intermediate F and a Pd0Ln species. In the next step, Ag2CO3 re-oxidizes Pd0Ln to X2PdIILn, closing the catalytic cycle. The intermediate F thus formed may undergo two different cyclization reactions to give alkylated or vinylated pyrrolidines, respectively. In the presence of strong electron-withdrawing groups (i.e., CN, COMe, or COPh) conjugated with newly formed double bonds, saturated pyrrolidines are afforded via intramolecular conjugate addition (pathway I). Otherwise, intermediate F undergoes syn-addition followed by β-hydride elimination in the presence of a Pd(II)-catalyst to yield the aza-Wacker E-2-methylene-pyrrolidine products (pathway II).
Scheme 8. Synthesis of pyrrolidines.
3.2 Amino/amidoarylationYang et al. reported the Pd(II)-catalyzed, stereoselective, intramolecular amidoarylation of alkenes utilizing oxygen as a green oxidant and ethyl nicotinate L8 as a ligand for the preparation of an array of indoline derivatives (Scheme 10).26 Ligand screening indicated that ethyl nicotinate was an efficient ligand for this protocol. The substrate scope revealed that anilides substituted at the para-position with electron-deficient and electron-rich groups, as well as halides, provided good to excellent yields of the corresponding indolines.
Scheme 9. Proposed reaction mechanism for generation of pyrrolidines.
Page 4 of 13ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H

ChemComm ARTICLE
This journal is © The Royal Society of Chemistry 2021 Chem. Commun., 2021, 00, 1–12 | 5
Please do not adjust margins
Please do not adjust margins
N
H
NHR2
R1 R1
R2R3
O2
2H2O
Pd(OAc)2 (10 mol %)1, 10-phen (20 mol %)
AcOH (0.06M)130 oC, 24h
NPh
R = H 85%(gram scale, 70%)
Me 71%tBu 66%Cl 62%;OAc 45%CN 56%
R
NPh
61%S
N
Me
PhNPh
Me4NPh
Me3
A'55%
(A/A'= 1.1:1)
53%
NPh
NPh
n
53% n = 1 34%3 52%4 56%
A
R3
symmetric,unsymmetric
terminal,internal,cyclic
NR2
Ph
R2= 4_OMe-Ph, 73%morpholine, 44%py 26%
NAr
Ar1Ar1
NHAr
NAr
Ar1
NAr
PdIILn
Ar1
PdIILnOAcArN
Ar1
NHAr
PdIILnOAc
Ar1
Ar1
NHAr
PdIILnOAc
LnPdII(OAc)2
LnPd0
A
B
C
D
EF
H2O2(1/2 O2 + H2O)
O2
AcOH2 AcOH + O2
H2O2(1/2 O2 + H2O)
AcOH
PhNHAr
G
NN N
O R2
R1 R3HH
XX
R3 R2
R1O
O
Ph
(if R2 = Ph)
orNa2CO3, O2, 70 oC,
5-72 h, dioxane
N
COOEt
Pd(OAc)2, L8
N
O
X
X = HF
ClNO2MeOMe
96%83%93%72%80%77%
N
O
H
n
n = 12
81% ; dr > 24 : 190% ; dr > 24 : 1
N
O
H70% (triple cyclization)
N
63%
L8
Ph
O
NPdII
O
LnX
N
O
LnXPdII
N
PdIILn
O
N
PdIILn
O
H
N
PdII
H X
O
B-1 B-2
C
O2
PdIIX2Ln
Pd0Ln + 2 HX
HX
reductive elimination arene substitution
syn-amidopalladation
HX
N-H deprotonationA
B
-
N
O
HH
X
N
O
1/2 H2O
In the case of meta-substituted anilides, a mixture of regioisomers were formed in moderate to good yields. Disubstituted anilides, naphthyl rings, and α,β-unsaturated group-bearing substrates were compatible with this protocol, affording the target products. The scope of the reaction was extended to prepare the oxidative triply cyclized products. Based on the results of control experiments, the following reaction mechanism was proposed. Anilide, in the presence of a ligated Pd(II) intermediate, generated amidopalladation species A. Upon syn-amidopalladation, this yielded the σ-alkylpalladium intermediate B, and the Pd(II) center thus formed activated the arene via the resonance-stabilized B-2 intermediate (instead of B-1). Based on scope and control experiments, the presence of the Pd(II) species, rather than Pd(IV), is likely to be responsible for activation of the arene. Subsequently, cyclization occurred via C–C bond formation via the reductive elimination of palladacycle C. The regeneration of Pd(II) from Pd(0) by molecular oxygen completed the catalytic cycle (Scheme 11).
Scheme 10. Stereoselective, intramolecular amidoarylation of alkenes.
In 2014, Maiti et al. performed a thorough investigation of the Pd-catalyzed reaction of diarylamines with easily accessible alkenes for the synthesis of N-arylindoles via C–H activation (Scheme 12).27 The generality and viability of the protocol was demonstrated by the preparation of more than 40 novel structurally interesting indole motifs utilizing a variety of symmetrical and unsymmetrical diarylamines bearing different substituents, as well as using terminal/internal/cyclic olefins as coupling partners. Two major advantages found by this study were that the starting materials were readily accessible and the reaction proceeded without directing groups. Based on the results of various control experiments, the proposed mechanism suggested that the initial ortho-palladation of the diarylamine formed species A. The coordination of the alkene with A resulted in intermediate B. Subsequently, -migratory insertion (C) was followed by -hydride elimination to generate 2-(N-arylated)stilbene (D). The formation of the final product G can occur via D and/or an indoline intermediate F. Both routes for the generation of G were supported by the results of a separate study. Furthermore, under standard reaction conditions, F was generated via D in the absence of a stoichiometric oxidant, revealing that F was an intermediate
between D and G. The re-oxidation of Pd(0) to Pd(II) by oxygen (air) via the assumed peroxopalladium(II) complex completed the catalytic cycle. In addition, an alternative Wacker-type mechanism was proposed for this method, involving the Pd(II)-catalyzed activation of the olefin, nucleophilic attack of the amine, followed by intramolecular C–H activation to afford F (Scheme 13).
Scheme 11. Proposed mechanism for amidoarylation of alkenes.
Scheme 12. Pd-catalyzed reaction of diaryl amines with alkenes.
Page 5 of 13 ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H

ARTICLE ChemComm
6 | Chem. Commun., 2021, 00, 1–12 This journal is © The Royal Society of Chemistry 2021
Please do not adjust margins
Please do not adjust margins
NHAr
NHAr
PdIILnOPiv
Ar1
NHAr
PdIILnOPiv
LnPdII(OPiv)2
LnPd0
A
B
C
PivOH2 PivOH + O2
H2O2(1/2 O2 + H2O)
NHAr
H
Ar1
CO2H
HO2C
[O][Cu]
PivOH
Ar1 PdIILnOPiv
CO2H
NHAr
D Ar1
CO2H
ArN
Ar1
O
E
N OPh
RR = H 93%(gram-scale, 78%)
-Me 93%-OMe 83%-F 80%-CO2Me 71%
N O
R1 = Me 99%-OMe 50%
R1
R1
N OPh
n = 1 68%3 89%4 92%
CH3n
N O
S
H3C
CH3
91%
N O
O
H3C
CH3
78%
N OPh
S
35%
Br
NHO O O
R3
R2
R1R1
H
NHR2
R3 Pd(OPiv)2 (10 mol %)1, 10-phenanthroline (20 mol %)
Cu(OAc)2 (0.5 eq.)TFA (4 eq.)
MeOH (2 mL), 110 oC, 24 h(Condition A)
5
Symmetric,unsymmetric
aromatic,aliphatic,heterocyclic
N OPh
OO
N OPh
73% 88%
Scheme 13. Proposed mechanism for N-arylindole synthesis.
In 2015, the same group extended their strategy to the preparation of 4-substituted 2-quinolinones via the Pd-catalyzed dehydrogenative coupling of diarylamines with -unsaturated carboxylic acids. A variety of aryl amines and carboxylic acids containing different functional groups reacted easily to yield the corresponding 4-substituted 2-quinolinones (Scheme 14).28 The use of trifluoroacetic acid (TFA) is crucial for this protocol to suppress the decarboxylation of -unsaturated acids and selectively yield the 4-substituted 2-quinolinones. The substrate scope of this strategy suggests that various substituents, including electron-rich and electron-deficient groups at the ortho-, para-, and meta-positions and halides at the ortho- and para-positions of cinnamic acids, react readily to afford the desired products in acceptable to excellent yields. Electron-rich components gave superior yields to electron-deficient groups. Different aliphatic/heterocyclic acrylic acids as coupling partners afforded the desired products in good yields, while an increase in chain length increased the reactivity of the acrylic acids. Unsymmetrical diarylamines bearing various substituents at different positions were successfully reacted under the same conditions to yield the desired quinolinones. Mechanistically, the protocol follows a mostly similar reaction pathway to that discussed earlier (Scheme 15).
Scheme 14. Pd-catalyzed synthesis of 4-substituted 2-quinolinones.
Scheme 15. Reaction mechanism of Pd-catalyzed synthesis of 4-substituted 2-quinolinones.
Liu et al. demonstrated an enantioselective strategy for the oxidative aminoarylation of olefins via aryl C–H and dual N–H bond cleavage for the efficient preparation of various tetracyclic heterocycles bearing a quaternary carbon center in high yields with excellent enantioselectivity (Scheme 16).29 The treatment of substituted anilines bearing an alkene group with a chiral Qox/Pd(II) (Qox, quinoline–oxazoline; L9) ligand/catalyst, and with Ag2CO3 as the oxidant, afforded a variety of indolines in the presence of a catalytic amount of phenylglyoxylic acid (PGA). The addition of PGA was crucial, promoting the key aminopalladation step and enhancing the enantioselectivity. The screening of a variety of pyridyl–oxazoline (Pox) and oxazoline ligands indicated that Qox L9 afforded a better yield and improved enantioselectivity. Benzanilides substituted with electron-donating, electron-withdrawing, and halide groups at the para- and ortho-positions, as well as disubstituted benzanilides, were compatible with the reaction conditions, affording the target compounds with good to excellent yields and enantioselectivity. An examination of the scope of the reaction for alkenes indicated that the double bond incorporating alkyl/cycloalkyl groups, as well as aromatics bearing electron-donating or electron-withdrawing groups, resulted in good yields and excellent enantioselectivity. In addition, substituents such as allylic ethers, esters, and amine groups were also promising coupling partners, leading to the respective products in moderate to good yields with excellent enantioselectivity. The disadvantage of this protocol is that it can only be applied to terminal olefins, with internal alkenes providing aza-Wacker products instead of the expected aminoarylation derivatives. The gram-scale reaction giving excellent yields and excellent enantioselectivity suggested the viability of this method. Various post-synthetic transformations of the prepared derivatives afforded structurally interesting synthetic derivatives of indolines.
Page 6 of 13ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H

ChemComm ARTICLE
This journal is © The Royal Society of Chemistry 2021 Chem. Commun., 2021, 00, 1–12 | 7
Please do not adjust margins
Please do not adjust margins
NH N
O
HR
O
R
Ar' Ar'
Ar
Ar
Pd(OAc)2 (10 mol %)Qox-L9 (10 mol %)
PhCOCO2H (20-25 mol %)
Ag2CO3 (1.5 equiv.), Li2CO3 (2 equiv.)THF, 50 oC, 72 h
NN
O
Qox-L9
N
O
RR = H
MeClF
OCF3SCF3CF3CNNO2
74%, -94% ee68%, -87% ee70%, -94% ee70%, -88% ee74%, -94% ee73%, -96% ee81%, -96% ee71%, -93% ee98%, -91% ee
N
Et
O
N
O
O
58% (11:1 r.r.), -92% ee
N
O
R
N
O
R
MeO
Cl
R = HCF3CO2Me
75%, -99% ee87%, -97% ee81%, -97% ee
R = H,Ac,SCF3,
66%, -89% ee81%, -95% ee79%, -95% ee
R
R = H 70% -94% eeCO2Me 81% -92% eeAc 75% -94% ee
NAr
O
PdL*OAc
NAr
O
PdL*
NHAr
O
int.IV
int.II
+
Li2CO3
PdL*PGA
int.V
L*PdO
O Ph
O
int.VISN 2
SN 2
L*Pd(PGA)2
PGA
-2HOAc
L*Pd(OAc)2(L* = L9)
A/Li2CO3
HOAc
- OAc-
+
cis-AP step
NAr
O
int.VI
PdL*
PGA
NH
Me PdIIL**
O
int.I
C-H activation Li2CO3
N
Me
PdIIL**
O
int.III
path a
path b
+
NH
PhO
Ar'
A/L2CO3
A
N
O
*Me
Scheme 16. Pd-catalyzed Intramolecular aminoarylation of alkenes.
The interaction of [(L*)Pd(OAc)2] with a benzanilide substrate resulted in the amido-Pd intermediate int. IV, and the face-selective coordination of the olefin to the L*Pd(II) species gave the cis-AP product with high enantioselectivity (83% ee) (Scheme 16, path a). In contrast, the rapid generation of hemilabile bidentate [(L*)Pd(PGA)2] was observed in the presence of PGA, resulting in the [(L*)Pd(PGA)]+ intermediate upon equilibration, which rapidly underwent the predominantly cis-AP (path b). Owing to the bulky steric hindrance of PGA compared with acetate, SN2-type substitution at the alkene coordinated to the L*Pd species in int. VI (path b) showed prominent face selectivity compared with int. IV (path a). Taking into account the high enantioselectivity for trans-AP, alkene interaction with [(L*)Pd(PGA)]+ yielded int. V with high face selectivity (probably due to favorable transition state formation), whereas, in the presence of a base, amidation of the Pd intermediate (int. V) was observed to give int. II. In both pathways, the insertion of an alkene into the amido-Pd bond resulted in the chiral alkyl-Pd(II) int. I. However, it is still difficult to differentiate between these two pathways (Scheme 17).
Scheme 17. Coupling of aliphatic free carboxylic acids with alkenes.
3.3 Carbo-oxygenation3.3.1 sp3 β‑C−H functionalization of free carboxylic acids
The Pd-catalyzed sp3 C−H functionalization of aliphatic acids, including oxygenation, iodination, alkylation, arylation, and fluorination, utilizing various directing groups, has been thoroughly studied in the past decade.30 However, research into the more challenging carboxyl-directed sp3 C−H functionalization leading to alkene difunctionalization has been less comprehensive. Furthermore, it is difficult to prepare structurally interesting -carbonyls via traditional enolate or conjugate addition chemistry. Hence, Pd-catalyzed selective transformation of sp3 -C–H bonds in order to synthesize such carbonyls by utilizing various ligands has emerged as an effective strategy for overcoming these obstacles. The research suggests that identification of suitable ligands and careful reaction conditions are important due weak directing ability of the carboxylate group and competing coordination reactions.
Zhuang et al. employed acetyl-protected aminoethyl phenyl thioether ligand L10 for the Pd(II)-catalyzed sp3 C−H bond activation of free carboxylic acids to afford -lactones and the subsequent synthesis of β-vinylated and -hydroxylated acids (Scheme 18).31 The reported strategies employing directing groups failed to provide these types of synthetic derivatives. In addition, there are no studies of the relevant C−H functionalization or the intramolecular conjugate addition reaction for monoselective -lactone formation in the presence of an olefin. A wide range of alkenes and carboxylic acids were applied in order to prepare the respective -lactones in the presence of Pd(TFA)2, Na2HPO4·7H2O as the base, and Ag2CO3 as the oxidant. The detailed study of L10 indicates that it plays a dual role in this transformation: (1) accelerating the initial rate of the reaction by a factor of 20; and (2) providing greater stability to the Pd catalyst over the course of the reaction, resulting in a higher turnover.
The scope of the reaction indicated that aliphatic acids bearing α-quaternary centers and α-dialkyl substituents afforded good to excellent yields of the corresponding lactones. Furthermore, aliphatic acids substituted at the - or -position with phenyl groups led to the respective lactones in good to excellent yields. Heterocyclic rings on aliphatic rings were readily tolerated in the reaction, resulting in the target compounds. Carboxylic acids bearing an α-hydrogen are challenging for this reaction to succeed due to presence of a labile acidic α-C−H bond and the lack of a suitable Thorpe−Ingold effect. Interestingly, these challenging carboxylic acids could also be reacted using this method. Synthetically important α-amino lactones and highly strained fused bicyclic lactones were also prepared by this strategy. Testing the scope of the reaction with a variety of alkenes revealed that the olefination of pivalic acid with a range of acrylate derivatives afforded the target compounds in excellent yields. Alkenes bearing various electron-withdrawing groups also provided the expected -lactones in excellent yields. The viability and utility of this method was demonstrated by the preparation of γ-lactones at the gram-scale, along with synthetic modifications of the prepared derivatives.
Page 7 of 13 ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H

ARTICLE ChemComm
8 | Chem. Commun., 2021, 00, 1–12 This journal is © The Royal Society of Chemistry 2021
Please do not adjust margins
Please do not adjust margins
O
OMe
Me
CO2Bn
85%
O
OMe
CO2Bn
77% (1.0/0.8)dr
O
O
CO2Et
54%, (1.0/1.0)
O Ph
OOCO2Bn
72%
O
CF3
OCO2Bn
89%
OOCO2Bn
72%
F O
OMe
Me
CO2nPr
70%
O
OMe
Me
85%
O
N
O
OMe
Me
SO2Ph
83%
Me
OMe
HO
OH
R2
R1
OHOH
R
O
OR1
R2
R3OO
R3
R
Pd(OAc)2 (10mol%)L11 (10 mol%)
(2.0 equiv.)Na2HPO4
.7H2O (1.0 equiv.)Ag2CO3 (2.0 equiv.)HFIP, 120 oC, 18 h
AcHNOH
O
Ph
L11
R3
4
Me
O
OMe
CO2Bn
95% (1.0/1.0)dr
Me3
O
OMe
CO2Bn95% (1.0/1.0)
O
O
CO2Bn48% (2.0/1.0)
F
F
O
O
CO2Bn44%
O
O
CO2Bn
52% (1.1/1.0)
O
O
CO2Bn48% (1.3/1.0)
H
OH O
OR1
R2 R
R1
R2
O
R
Pd(TFA)2 (10 mol %)
Na2HPO4.7H2O, Ag2CO3
O
OMe
Me
CONMe2
O
OMe
Me
CN
O
OMe
Me
SO2Ph
O
OMe
Me
P(O)(OEt)2
95% 78% 90% 88%
S
NHAc L10
2
L10 (10 mol %)
HFIP, 120 oC, 12 h
O
O
CO2Et
64%
O
O
CO2Et
54% (1.3:1)d.r.
Cy
O
O
CO2Et
X = CH2, 55%O, 50%
X
O
O
CO2Et
46% (1.6:1)
O
O
COMe51%
O
O
CN40%
O
O
SO2Ph67%
O
O
SO2F51%
O
O
P
40%
OEtOEtO
CO2H
R1
R2
R+O
O
R
R1
R2
Pd(OAc)2 (10 mol%)L12 (20 mol%)
Ag2CO3
Na2HPO4.7H2O (0.2 equiv)
HFIP (2.25 mL), 110 oC
AcHNCO2H
L12
O
Scheme 18. Synthesis of -lactones.
Very recently, three research groups have independently reported the pioneering difunctionalization of alkenes via ligand-enabled sp3 β‑C−H activation of free carboxylic acids.
Yu’s group has contributed significantly in this area and reported Pd-catalyzed, and ligand-influenced remote sp3 -C–H olefination of free carboxylic acids to yield a variety of six-membered lactones (Scheme 19).32 Exploration of ligand reactivities suggested that the mono-N protected amino acid (MPAA) Ac-Phe-OH L11 was an appropriate ligand for this protocol. The substrate scope of this current strategy indicated that carboxylic acids substituted with different alkyl chains, -dialkyl, methoxy, phenoxy phenyl, and 4-methoxyphenyl groups afforded the corresponding lactones in moderate to good yields. Furthermore, 2-methyl benzoic acid substrates bearing benzylic -C(sp3)–H were compatible with this protocol, yielding the bioactive cyclized 3,4-dihydroisocoumarin precursors. Detailed optimization revealed that 2-methylbenzoic acids bearing electron-donating and electron-withdrawing groups at different positions on the aryl rings, as well as disubstituted phenyl and naphthoic acids, afforded the target compounds in acceptable to excellent yields. The alkene substrate scope, including a variety of acrylate derivatives, resulted in the expected -lactone products in good yields. In addition, olefins bearing -unsaturated components, such as Weinreb amide-derived acrylamide, methyl vinyl ketone, and phenyl vinyl sulfone, afforded the target lactones in moderate to good yields. Upon application of pentafluorostyrene as a coupling partner, five-membered lactones were observed to be the major products, which may be attributed to sequential -C(sp3)–H olefination, followed by allylic C–H functionalization.
Scheme 19. Ligand-enabled -C(sp3)–H functionalization of free carboxylic acids.
Gemmeren et al. investigated similar Pd-catalyzed, ligand-enabled C–H activation/olefination of free carboxylic acids via intramolecular Michael addition for the preparation of -lactones (Scheme 20).33 The initial ligand identification suggested that among several amino acid-derived ligands, N-Ac-β-alanine L12 was an efficient ligand for this transformation. The scope of the reaction with respect to acid substrates indicated that the alkyl, cycloalkyl, and aromatic substituents on the acid underwent the reaction smoothly, providing the corresponding lactones in acceptable to good yields with moderate to good diastereoselectivity. Substrates without a quaternary center at the β-position failed to react. The scope of the alkene reaction partner indicated that various olefins incorporating different functional groups were efficiently tolerated under the reaction conditions, affording the respective lactones in good yields. The olefins bearing other electron-withdrawing groups, including vinyl ketone, acrylonitrile, phenyl vinyl sulfone, ethane sulfonyl fluoride, diethyl, and vinylphosphonate groups, were also compatible with these reaction conditions. Based on the results of various control experiments, the following reaction mechanism was proposed for this protocol: the mononuclear Pd(II)-catalyst activated the unreactive -C(sp3)–H bond of the free carboxylic acid, which was the rate-determining step. Then, sequential ligand exchange, carbopalladation, β-H elimination, and de-coordination generated the non-cyclized (open) derivative, which underwent intramolecular Michael addition and cyclization to form the target product (Scheme 21).
Scheme 20. Pd-catalyzed C–H olefination of free carboxylic acids.
Similar to the above studies, Maiti et al. investigated the Pd-catalyzed sp3 -C–H activation of free aliphatic carboxylic acids, which, upon coupling with alkenes in the presence of the N-Ac-Val ligand L13, underwent subsequent C–O cyclization to yield the corresponding -lactones and 3-lactones (Scheme 22).34 Ligand studies suggested that, among the different acetyl-protected amino acids screened for this protocol, N-acetyl valine (L13) was the best ligand, affording the target lactone in 91% yield. The substrate scope of this strategy revealed that -position carboxylic acids bearing ethyl, propyl, and butyl groups,
Page 8 of 13ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H

ChemComm ARTICLE
This journal is © The Royal Society of Chemistry 2021 Chem. Commun., 2021, 00, 1–12 | 9
Please do not adjust margins
Please do not adjust margins
82%
OMeMe
O
CO2Bn
63%,(1.2:1)dr
OMe
O
CO2MeMe
68%
O
O
SO O
Ph
OMe
O
CO2BnMe
67%, (1:1)
OMe
O
CNMe
66%, (1.7:1) 69%
OMeMe
O
O
O
F
71%
OMeMe
O
O
O
NO2
Me OH
R1R2 O
+
Pd(TFA)2 (10 mol%)L13 (20 mol%)
Na2HPO4 (1 equiv.), Ag2CO3 (2 equiv.)HFIP, 12 h, 120 oC
RO
R1
R2
O
R AcHNOH
OL13
O
N
OO
O
MeMeMe
58%
O
N
OO
O
MeMe
O
N
OO
O
MeMe
Me
52% 51%
O
N
OO
O
MeMe
45%OMe
F
terminal,internalmaleimides
Pd(II)
R1R2
Me OH
O
PdO NHAc
OMe
Me
O O
R1
MeR2
A
CH Activationr.d.s.
kH/kD = 2.3Na
PdLn
OR1
R2
ONa
B
olefincoordination
order: 1st w.r.t. acid0.4th w.r.t. olefin
PdLO
R1
R2
ONa
R
Cmigratory insertion
PdLn
O
R
R1
R2
ONa
-hydride elimination
reductive elimination
Ag(I)
Ag(0)
D
E
PdO
HR
R1
R2
ONa
1,4-addition7-endo-trig
1,4-addition6-exo-trig
O
N
O
O
O
R3
R1R2
OO
R
R1R2
PdN
OO
O
Na2HPO4
NaH2PO4
N
OPd
O
O
OO Na
Pd-complex rate limitingC -H activation
N
OPd
O
HO
OO Na
6-memberedpalladacycle
carbopalladation
hydrideelimination
decoordination
reductiveelimination
Pd(0)
2 Ag(I)
2 Ag(0)reoxidation
CO2HCO2Na
O
O
EWG
PdLO
O Na
PdLO
O Na
EWGEWG
PdLO
NaO
EWG
OH
O
EWG
EWG
as well as -diethyl and ethyl-isopropyl functional groups, afforded the target lactones in moderate to good yields.
Scheme 21. Proposed reaction mechanism for -lactone formation.
Examination of the scope of alkenes indicated that different benzyl acrylates, phenethyl acrylates, and aliphatic acrylates, including simple methyl acrylate to bicyclic acrylates, afforded excellent yields under the reaction conditions. Furthermore, long-chain acrylates such as dodecyl, dodecafluoro, and docosyl acrylates were found to be appropriate substrates for this protocol. With the exception of acrylates, various activated alkenes bearing sulfone, nitrile, ketone, and diester groups were readily tolerated under the reaction conditions and afforded the respective -lactones in good to excellent yields. In addition, a variety of maleimides acted as suitable alkene partners, resulting in fused seven-membered lactones under standard reaction conditions. Based on the results of control experiments and the relevant literature, the proposed reaction mechanism indicated that, initially, the carboxylate group binds to the metal catalyst to generate the metal-coordinated complex A, which, when exposed to an alkali metal base, affords the -C–H activated six-membered species B. Next, alkene interaction with B and 1,2-migratory insertion results in an eight-membered cyclo-palladated intermediate D, which gives E upon -hydride elimination; E undergoes reductive elimination followed by Michael addition to yield a six- or seven-membered lactone. Finally, the addition of Ag(I) oxidizes Pd(0) to Pd(II), completing the catalytic cycle (Scheme 23). The further utility of this approach was demonstrated by post-synthetic modifications to the prepared lactones to yield structurally interesting compounds. Presence of the β-quaternary center in the acid is essential for this protocol.
Scheme 22. Synthesis of -lactones and fused seven-membered lactones.
Scheme 23. Proposed reaction mechanism for -C–H activation.
3.3.2 sp2 C−H functionalization of inactivated arenesBuchwald et al. demonstrated the efficient synthesis of tetrahydro-2H-indeno-[2,1-b]furans from acyclic hydroxyalkenes bearing inactivated arenes via mild Pd(II)-catalyzed intramolecular oxidative oxyarylation (Scheme 24).35 The method proceeds via a sequential oxypalladation/C–H functionalization pathway. The screening of various substituted pyridine ligands suggests that ethyl nicotinate L8 was the optimal ligand in the Pd(OAc)2/O2/K2CO3 combination in toluene at 100°C to afford the corresponding tetrahydro-2H-indeno-[2,1-b]furans. The reaction scope encompassed -aryl--hydroxyalkenes with a pyridyl moiety, as well as an array of electron-donating, -withdrawing, -neutral, and halide-substituted aryl groups that were compatible with the reaction conditions. In addition, alkyl and aryl functional groups on the double bond of the alkenes resulted in the corresponding oxyarylated derivatives. The sulindac-derived biologically
Page 9 of 13 ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H

ARTICLE ChemComm
10 | Chem. Commun., 2021, 00, 1–12 This journal is © The Royal Society of Chemistry 2021
Please do not adjust margins
Please do not adjust margins
R2
OOH
R1 R1Pd(OAc)21, 10-phenanthroline
Cu(OAc)2, NaOAcair, ClCH2CH2Cl, 110 oC
O
R1
R2
73%; R1 = CHO, R2 = OMe64%; R1 = COCH3, R2 = H65%; R1 = Br, R2 = Cl42%; R1 = Br, R2 = 2-OMe40%; R1 = R2 = OMe
O
OMe
Cl
58% O Ph89%
Br
O
Br
6
76%
O
CO2Me
5
55%
O
O
Me
OMe
56%
O2N
O
OMe
53%Br
Ph
O2N
OA'
72% A/A'=1:2.3
R2
A
terminal,internalcyclic
O
O2N
63%
O2N 67%; R = H78% Me81% OMe57% Br61% NO2
O O
O
H
Me
74%
O
CF3
H
Me
87%
O
H
ROBr
H
Me
48%
O
N H
Me
R = Me, 81%n-Hex, 58%Ph, 74%
X
XOH
R2
R1 R1
O
R1R1
R3
R3
R2
H
Pd(OAc)2 (5 mol %)L8 (6 mol %)
K2CO3 (0.5 equiv.)toluene, O2 (1 atm)
100 oC, 19 h
73%
N
COOEt
L8
CN
O
50%
O2N
O
45%
S
O2N
O
R2
O2N
O
75%
O
50%Cl
R2 = 4-OCH3 70%4-Cl 69%2-F 42%4-Br 49%
R1H
OHHO2C
Pd(OAc)2 (10mol%)1,10-phen (20 mol%)
Cu(OAc)2.H2O
DCE (4 mL), O2, 130 oC, 24 h
+
O
R2
R1
single regioisomers
Cl
important precursor was successfully prepared using this protocol.
Scheme 24. Pd(II)-catalyzed intramolecular oxidative oxyarylation.
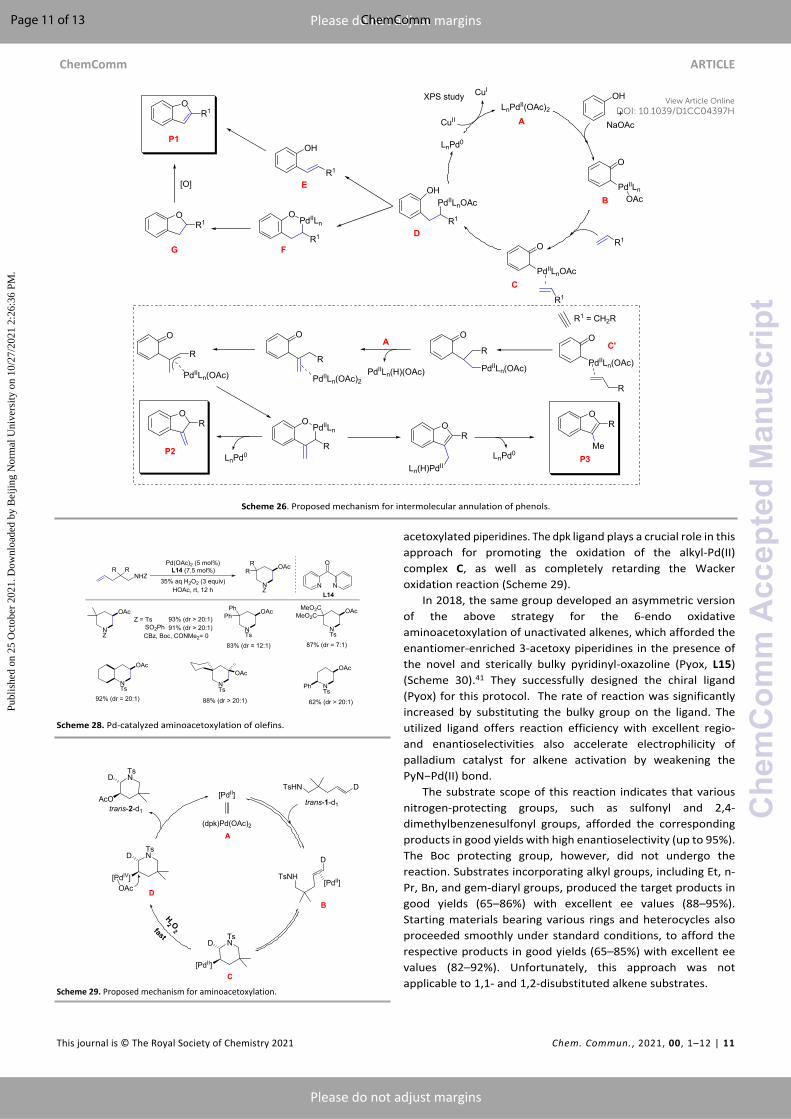
Maiti et al. reported an elegant study of the efficient synthesis of benzofurans/coumarins from phenols and alkenes via Pd-catalyzed intermolecular annulation (Scheme 25).36 The reaction was performed utilizing Pd(OAc)2, 1,10-phenanthroline, Cu(OAc)2, and NaOAc (3 equiv.) at 110°C for 24 h in dichloroethane to yield 2-substituted benzofurans as the carbo-oxygenated difunctionalized products. The substrate scope encompassed phenols, terminal/internal alkenes, and styrenes bearing a variety of substituents, including halogens, to yield the 2-substituted benzofurans. However, substituted coumarins were formed when methyl acrylate was used as the coupling partner. The proposed mechanism began with the ortho-palladation of phenol to form the quinone-type species B. The binding of an alkene with B generated C and, subsequently, D, which could undergo reductive elimination to yield 2-hydroxystilbene (E). Finally, intermediate E was transformed into the target compounds. In addition, the final product may be generated via intermediate F (Scheme 26).
Scheme 25. Synthesis of benzofurans/coumarins from phenols.
Maiti et al. successfully extended the previous strategy for the synthesis of 3-substituted benzofurans by employing cinnamic acids as coupling partners (Scheme 27).37 Carboxylic acid functionality in this protocol served as the traceless directing group for the selective preparation of the target benzofurans. Interestingly, under similar reaction conditions, the relevant 2-arylbenzofurans were not formed. Various
cinnamic acids, including naphthyl, para-tolyl groups, and substituted phenols, afforded the 3-arylated benzofurans in acceptable to excellent yields. Thiophenylacrylic acid generated the expected benzofuran in moderate yield. Phenols bearing nitro, halogen, ester, or cyano groups were compatible with the specified reaction conditions. Electron-rich phenols afforded the expected benzofurans, but in comparatively lower yields. The reaction mechanism for this method mostly follows the previously proposed mechanism, with the release of CO2 leading to the 3-substituted benzofurans. The authors also applied a similar strategy for the preparation of 2,3-disubstituted benzofurans and the relevant deuterium-labeled derivatives.38
3.4 AminoacetoxylationAs mentioned above, Pd-catalyzed amination of alkenes has emerged as an efficient protocol for the preparation of various heterocycles, among which oxygenated piperidines represent an important constituent of various natural products and therapeutically active motifs, such as (−)-cassine, veratramine, and (+)-febrifugine.39 Liu et al. reported the Pd-catalyzed intramolecular aminoacetoxylation of unactivated alkenes to generate different 3-acetoxylated piperidines in high yields with excellent regio- and diastereoselectivity. The reaction was performed under mild reaction conditions utilizing hydrogen peroxide as a green oxidant (Scheme 28).40
Scheme 27. Synthesis of 3-substituted benzofurans employing cinnamic acids.
The screening of various bidentate nitrogen ligands indicated that dipyridinyl ketone (dpk, L14) selectively resulted in the desired product in excellent yield with higher reactivity than other ligands, and was therefore chosen as the best ligand for this protocol. The substrate scope of this reaction suggested that protecting groups such as -Ns, -Ts, and -SO2Ph present on the nitrogen afforded the corresponding products in high yields with greater regioselectivity. In contrast, protecting groups bearing carbonyl moieties were ineffective under this protocol. Substrates incorporating various gem-disubstitutions also reacted efficiently to give the desired products in good to excellent yields. Chain elongation as well as substrates bearing substituents on the carbon adjacent to the nitrogen readily underwent this reaction to produce the target products in good yields. From control experiments, the authors proposed the reaction mechanism for this protocol. Initially, unactivated alkene substrates undergo trans-aminopalladation reaction to generate an alkyl-Pd(II) intermediate C, which upon quick oxidation by H2O2 to form the complex D. Finally, reductive elimination of D provided the target 3-
Page 10 of 13ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H
ChemComm ARTICLE
This journal is © The Royal Society of Chemistry 2021 Chem. Commun., 2021, 00, 1–12 | 11
Please do not adjust margins
Please do not adjust margins
O
OR1
R1
P1
[O]
PdIILnO
R1
F
OH
R1
E
OH
NaOAc+
O
PdIILnOAcB
R1O
PdIILnOAc
R1
C
OH
R1
PdIILnOAc
D
LnPd0
CuII
CuI
LnPdII(OAc)2
A
XPS study
O
R
PdIILn(OAc)
O
R
PdIILn(OAc)2
A
PdIILn(H)(OAc)
O
R
PdIILn(OAc)
O
PdIILn(OAc)
R
C'
R1 = CH2R
OR
P2
PdIILnO
RLnPd0
OR
Ln(H)PdIILnPd0
O
Me
R
P3
G
NZ
OAc
NTs
OAc
NTs
OAc
Ph
93% (dr > 20:1)Z = Ts91% (dr > 20:1) N
Ts
PhPh
OAc
88% (dr > 20:1)
NTs
MeO2CMeO2C
OAc
87% (dr = 7:1)83% (dr = 12:1)
NTs
OAc
62% (dr > 20:1)92% (dr = 20:1)
NHZR R
NZ
RR
OAcPd(OAc)2 (5 mol%)L14 (7.5 mol%)
35% aq H2O2 (3 equiv)HOAc, rt, 12 h
SO2Ph
N N
O
L14
CBz, Boc, CONMe2= 0
TsN
AcO
TsNH
D
[PdII]
TsN
[PdII]
[PdII]
DB
C
fast
H2 O
2
trans-1-d1trans-2-d1
DTsHN
TsN
[PdIV]OAc
D
D
D
(dpk)Pd(OAc)2A
Scheme 26. Proposed mechanism for intermolecular annulation of phenols.
Scheme 28. Pd-catalyzed aminoacetoxylation of olefins.
Scheme 29. Proposed mechanism for aminoacetoxylation.
acetoxylated piperidines. The dpk ligand plays a crucial role in this approach for promoting the oxidation of the alkyl-Pd(II) complex C, as well as completely retarding the Wacker oxidation reaction (Scheme 29).
In 2018, the same group developed an asymmetric version of the above strategy for the 6-endo oxidative aminoacetoxylation of unactivated alkenes, which afforded the enantiomer-enriched 3-acetoxy piperidines in the presence of the novel and sterically bulky pyridinyl-oxazoline (Pyox, L15) (Scheme 30).41 They successfully designed the chiral ligand (Pyox) for this protocol. The rate of reaction was significantly increased by substituting the bulky group on the ligand. The utilized ligand offers reaction efficiency with excellent regio- and enantioselectivities also accelerate electrophilicity of palladium catalyst for alkene activation by weakening the PyN−Pd(II) bond.
The substrate scope of this reaction indicates that various nitrogen-protecting groups, such as sulfonyl and 2,4-dimethylbenzenesulfonyl groups, afforded the corresponding products in good yields with high enantioselectivity (up to 95%). The Boc protecting group, however, did not undergo the reaction. Substrates incorporating alkyl groups, including Et, n-Pr, Bn, and gem-diaryl groups, produced the target products in good yields (65–86%) with excellent ee values (88–95%). Starting materials bearing various rings and heterocycles also proceeded smoothly under standard conditions, to afford the respective products in good yields (65–85%) with excellent ee values (82–92%). Unfortunately, this approach was not applicable to 1,1- and 1,2-disubstituted alkene substrates.
Page 11 of 13 ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H

ARTICLE ChemComm
12 | Chem. Commun., 2021, 00, 1–12 This journal is © The Royal Society of Chemistry 2021
Please do not adjust margins
Please do not adjust margins
Scheme 30. Asymmetric aminoacetoxylation.
4. ConclusionsPd-catalyzed unactivated C–H bond functionalization leading to alkene difunctionalization has emerged as a powerful tool for the synthesis of heterocycles and complex, structurally interesting, frameworks. The approaches so far developed have clear advantages over traditional synthetic methods, including straightforward procedures, atom economy, and redox economy. Furthermore, owing to the various challenges associated with the development of newer versions of Heck coupling reactions, these methods serve as promising alternative pathways for the relevant transformations. Various unactivated C−H bonds, such as sp3 C−H and sp2 C−H bonds, were successfully reacted with olefins in the presence of a Pd catalyst, resulting in alkene difunctionalization. Some of the protocols demonstrated the unique synthesis of important structural motifs, such as -lactones, fused seven-membered lactones, and γ-olefinated aliphatic acids, which are difficult to access via traditional synthetic approaches. This article has also shed light on the reaction mechanisms of the respective protocols. The selection of appropriate ligands is the key factor to ensure the efficiency and desirable outcomes of these methods.
Most of the protocols employ monosubstituted alkenes and styrenes as coupling partners. However, the utilization of challenging and sterically hindered olefins as coupling partners is still difficult to achieve. Furthermore, approaches describing enantioselective C(sp3)–H functionalization leading to alkene difunctionalization are rare. The use of harsh reaction conditions and excessive amounts of particular additives is of concern in some approaches. To overcome these problems, future efforts to develop more generally applicable synthetic protocols, employing mechanistic investigations and novel catalytic systems, are anticipated.
Conflicts of interestThere are no conflicts to declare.
AcknowledgementsWe acknowledge the financial support from the Ministry of Science and Technology of Taiwan (MOST 110-2113-M-077-001-).
Notes and referencesH. 1 (a) P. Kumar, P. J. Nagtilak and M. Kapur, New J. Chem., 2021,
DOI: 10.1039/D1NJ01696B; (b) S. Rana, J. Prasad Biswas, S. Paul, A. Paika and D. Maiti, Chem. Soc. Rev., 2021, 50, 243; (c) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925; (d) S. K. Sinha, G. Zanoni and D. Maiti, Asian J. Org. Chem., 2017, 7, 1178; (e) F. Roudesly, J. Oble, and G. Poli, J. Mol. Catal. A: Chem., 2017, 426, 275; (f) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546; (g) B. D. Barve, Y.-C. Wu, M. El-Shazly, Y.-B. Cheng, M. Korinek, J.-J. Wang and F.-R. Chang, Tetrahedron, 2015,71, 2290; (h) B. D. Barve, Y.-C. Wu, M. El-Shazly, Y.-B. Cheng, M. Korinek, J.-J. Wang and F.-R. Chang, Org. Lett., 2014, 16, 1912; (i) B. D. Barve, Y.-C. Wu, M. El-Shazly, D.-W. Chuang, Y.-B. Cheng, J.-J. Wang and F.-R. Chang, J. Org. Chem., 2014, 79, 3206; (j) B. D. Barve, Y.-C. Wu, M. El-Shazly, D.-W. Chuang, Y.-M. Chung, Y.- H. Tsai, S. -F. Wu, M. Korinek, Y.-C Du, C.-T. Hsieh, J.-J. Wang and F.-R. Chang, Eur. J. Org. Chem. 2012, 6760; (k) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem. Int. Ed., 2012, 36, 8960; (l) L. McMurray, F. O’Hara, J. Matthew and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885.
2 (a) G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis,. P. Verma, N. Chekshin and J. Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571; (b) He, W. G. Whitehurst and M. J. Gaunt, Chem, 2019, 5, 1031; (c) Y. E. Liang, C. L. Lu and W. T. Li, Org. Biomol. Chem., 2019, 17, 7569; (d) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754; (e) S. Agasti, A. Dey and D. Maiti, Chem. Commun., 2017, 53, 6544; (f) N. Thrimurtulu, A. Dey, D. Maiti and C. M. R. Volla, Recent developments in palladium catalyzed natural products synthesis via C-H activation, ch. 9 (Eds.: A. R. Kapdi, D. Maiti), Elsevier, New York. 2017, 453; (g) Y. F. Lin, C. Fong, W. L. Peng, K. C. Tang, Y. E. Liang and W. T. Li, J. Org Chem., 2017, 82, 10855.
3 (a) L. Zhou and W. Lu, Chem. Eur. J., 2014, 20, 634; (b) Y. Fujiwara, I. Moritani, S. Danno, R. Asano and S. Teranishi, J. Am. Chem. Soc., 1969, 91, 7166; (c) I. Moritanl and Y. Fujiwara, Tetrahedron Lett., 1967, 8, 1119; (d) J. Smidt,W. Hafner, R. Jira, R. Sieber, J. Sedlmeier and A. Sabel, Angew. Chem. Int. Ed., 1962, 1, 80.
4 (a) X. Chen, F. Xiao and W.-M. He, Org. Chem. Front., 2021, 8, 5206; (b) Y.-M. Li, J.-F. Fu, L.-Q. He, W.-N. Li and V. Esmail, RSC Adv., 2021, 11, 24474; (c) H.-Q. Ni, C. Phillippa and K. M. Engle, Chem. Commun., 2021, 57, 7610.
5 (a) J. Cheng, X. Qi, M. Li, P. Chen and G. Liu, J. Am. Chem. Soc., 2015, 137, 2480; (b) T. Seidensticker, M. R. L. Furst, R. Frauenlob, J. Vondran, E. Paetzold, U. Kragl, A. J. Vorholt, ChemCatChem., 2015, 7, 4085; (c) H. Liu, N. Yana and P. J. Dyson, Chem. Commun., 2014, 50, 7848; (d) K.-T. Yip, M. Yang, K.-L. Law, N.-Y. Zhu, D. Yang, J. Am. Chem. Soc., 2006, 128, 3130.
6 (a) S. Hayashi, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2009, 131, 6, 2052; (b) J. P. Wolfe, Eur. J. Org. Chem., 2007, 571.
7 (a) K. Pratap and A. Kumar, Org. Lett., 2018, 20, 7451; (b) D. R. White, J. T. Hutt and J. P. Wolfe, J. Am. Chem. Soc., 2015, 137, 11246.
8 (a) A. Iglesias, E. Prez and K. Muniz, Angew. Chem. Int. Ed., 2010, 49, 8109; (b) P. A. Sibbald and F. E. Michael, Org. Lett., 2009, 11, 1147; (c) K. Muniz, C. H. Hovelmann and J. Streuff, J. Am. Chem. Soc. 2008, 130, 763.
9 J.-B. Peng, Adv. Synth. Catal., 2020, 362, 3059.10 (a) B. Tian, P. Chen, X. Leng and G. Liu, Nat. Catal., 2021, 4,
172; (b) J. Huang, J. Li, J. Zheng, W. Wu, W. Hu, L. Ouyang and H. Jiang, Org. Lett., 2017, 19, 3354; (c) A. Wang, H. Jiang and H. Chen, J. Am. Chem. Soc., 2009, 131, 3846; (d) Y. Li, D. Song and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 2962; (e) Y.
NTs
OAc
NTs
OAc
82% (92%)ee
NTs
RR
OAc
NTs
OAc
NHZR R
NZ
RR
OAcPd(OAc)2 (10 mol%)L15 (12 mol%)
PhI(OAc)2 (2.0 equiv.)PhCF3, 0°C
NPh N
O
PhL15
Ph
R= Et, 80% (92%)ee
CH2OAc, 75% (80%)ee
n
n=1 65% (83%)ee
3 85% (90%)ee
X
X= CH2 72% (90%)ee
O 83% (91%)ee
NBOc 70% (91%)ee
Page 12 of 13ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H

ChemComm ARTICLE
This journal is © The Royal Society of Chemistry 2021 Chem. Commun., 2021, 00, 1–12 | 13
Please do not adjust margins
Please do not adjust margins
Zhang and M. S. Sigman, J. Am. Chem. Soc., 2007, 129, 3076 (f) X. Tong, M. Beller and M. K. Tse, J. Am. Chem. Soc., 2007, 129, 4906; (g) L. L. Welbes, T. W. Lyons, K. A. Cychosz and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 5836.
11 (a) J. Jeon, H. Ryu, C. Lee, D. Cho, M. Baik, and S. Hong J. Am. Chem. Soc., 2019, 141, 10048; (b) X. Tong, M. Beller and M. K. Tse, J. Am. Chem. Soc., 2007, 129, 4906. (c) L. L. Welbes, T. W. Lyons, K. A. Cychosz and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 5836.
12 (a) Y. Gao, Y. Gao, W. Wu, X. Yang, W. Liu, X, Yang and C.-J. Li, Chem. Eur. J., 2017, 23, 793; (b) A. Mancinelli, C. Alamillo, J. Albert, X. Ariza, H. Etxabe, J. Farràs, J. Garcia, J. Granell and F. J. Quijada, Organometallics, 2017, 36, 911; (c) M. K. Manna, A. Hossian and R. Jana, Org. Lett., 2015, 17, 672; (d) J.-H. Fan, W.-T. Wei, M.-B. Zhou, R.-J. Song and J.-H. Li, Angew. Chem. Int. Ed., 2014, 53, 6650; (e) M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3680; (f) X. Han and X. Lu, Org. Lett., 2009, 11, 2381; (g) A. Rodriguez and W. J. Moran, Eur. J. Org. Chem., 2009, 1313.
13 E. J. Nigishi, Handbook of Organopalladium Chemistry for Organic Synthesis; Wiley-VCH: Weinheim, 2002, Vol. 2.
14 (a) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413; (b) Q. Zhang, X. Lu and X. Han, J. Org. Chem., 2001, 66, 7676.
15 Y. Qin, L. Zhu, S. Luo, Chem. Rev., 2017, 117, 9433.16 (a) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem.
Rev., 2017, 117, 9333; (b) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170.
17 T. Wu, X. Mu and G. Liu, Angew. Chem. Int. Ed., 2011, 50, 12578.
18 H. Zhang, P. Chen and G. Liu, SynLett, 2012, 23, 2749.19 X. Mu, T. Wu, H.-Y. Wang, Y.-l. Guo and G. Liu, J. Am. Chem.
Soc., 2012, 134, 878.20 X. Wang, L. Truesdale and J.-Q. Yu, J. Am. Chem. Soc., 2010,
132, 3648.21 (a) S. B. Bharate, S. D. Sawant, P. P. Singh and R. A.
Vishwakarma, Chem. Rev., 2013, 113, 6761; (b) J. Alvarez-Builla, J. J. Vaquero, J. Barluenga, Vol. 1, Modern Heterocyclic Chemistry, Wiley-VCH, Weinheim, 2011; (c) M. d’Ischia, A. Napolitano, A. Pezzella, In Comprehensive Heterocyclic Chemistry III, Vol. 3; (Eds.: A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven, R. J. K. Taylor), Elsevier, Oxford, 2008; pp 353−388; (d) L. D. Quin, J. A. Tyrell, Fundamentals of Heterocyclic Chemistry: Importance in Nature and in the Synthesis of Pharmaceuticals, Wiley, Hoboken, NJ, 2007; (e) S.-H. Day, N.-Y. Chiu, L.-T. Tsao, J.-P. Wang and C.-N. Lin, J. Nat. Prod., 2000, 63, 1560.
22 (a) H. Jiang and A. Studer, Chem. Soc. Rev., 2020, 49, 1790; (b) T. Piou and T. Rovis, Nature, 2015, 527, 86; (c) B. M. Trost, K. Imi and I. W. Davies, J. Am. Chem. Soc., 1995, 117, 5371; (d) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529; (e) E. J. Moore, W. R. Pretzer, T. J. O’Connell, J. Harris, L. LaBounty, L. Chou and S. S. Grimmer, J. Am. Chem. Soc., 1992, 114, 5888.
23 (a) W. Ali, G. Prakash and D. Maiti, Chem. Sci., 2021, 12, 2735; (b) A. Deb, A. Hazra, Q. Peng, R. S. Paton and D. Maiti, J. Am. Chem. Soc., 2017, 139, 763; (c) A. Deb, S. Bag, R. Kancherla and D. Maiti, J. Am. Chem. Soc., 2014, 136, 13602; (d) K. J. Stowers, K. C. Fortner and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 6541; (e) K. M. Engle, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137; (f) J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888; (g) J.-J. Li, T.-S. Mei and J.-Q. Yu, Angew. Chem. Int. Ed., 2008, 47, 6452.
24 S. Li, G. Chen, C.-G. Feng, W. Gong and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 5267.
25 H. Jiang, J. He, T. Liu and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 2055.
26 K.-T. Yip and D. Yang, Org. Lett., 2011, 8, 2134.
27 U. Sharma, R. Kancherla, T. Naveen, S. Agasti and D. Maiti, Angew. Chem. Int. Ed., 2014, 53, 11895.
28 R. Kancherla, T. Naveen and D. Maiti, Chem. Eur. J., 2015, 21, 8360.
29 W. Zhang, P. Chen and G. Liu, Angew. Chem. Int. Ed., 2017, 56, 5336.
30 (a) Q. Zhang, X.-S. Yin, K. Chen, S.-Q. Zhang and B.-F. Shi, J. Am. Chem. Soc., 2015, 137, 8219; (b) R.-Y. Zhu, J. He and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 13194; (c) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135; (d) L. D. Tran and O. Daugulis, Angew. Chem. Int. Ed., 2012, 51, 5188; (e) R. Giri, J. Liang, J.-G. Lei, J.-J. Li, D.-H. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Foxman and J.-Q. Yu, Angew. Chem. Int. Ed., 2005, 44, 7420; (f) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem. Int. Ed., 2005, 44, 2112; (g) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154.
31 Z. Zhuang, C.-B. Yu, G. Chen, Q.-F. Wu, Y. Hsiao, C. L. Joe, J. X. Qiao, M. A. Poss and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 10363.
32 H. S. Park, Z. Fan, R.-Y. Zhu and J.-Q. Yu, Angew. Chem. Int. Ed., 2020, 59, 12853.
33 K. K. Ghosh, A. Uttry, A. Mondal, F. Ghiringhelli, P. Wedi and M. van Gemmeren, Angew. Chem. Int. Ed., 2020, 59, 12848.
34 Das, P. Dolui, W. Ali, J. P. Biswas, H. B. Chandrashekar, G. Prakasha and D. Maiti, Chem. Sci., 2020, 11, 9697.
35 R. Zhu and S. L. Buchwald, Angew. Chem. Int. Ed., 2012, 51, 1926.
36 U. Sharma, T. Naveen, A. Maji, S. Manna and D. Maiti, Angew. Chem. Int. Ed., 2013, 52, 12669.
37 S. Agasti, U. Sharma, T. Naveena and D. Maiti, D. Chem. Commun., 2015, 51, 5375.
38 S. Agasti, S. Maity, K. J. Szabo and D. Mait, Adv. Synth. Catal. 2015, 357, 2331.
39 (a) S. Zhu, G. Chandrashekar, L. Meng, K. Robinson and D. Chatterji, Bioorg. Med. Chem., 2012, 20, 927; (b) G. Brunhofer, A. Fallarero, D. Karlsson, A. Batista-Gonzalez, P. Shinde, M. Gopi and P. Vuorela, Bioorg. Med. Chem., 2012, 20, 6669.
40 H. Zhu, P. Chen and G. Liu, Org. Lett., 2015, 17, 1485.41 Q. Xiaoxu, C. Chen, C. Hou, L. Fu, P. Chen and G. Liu, J. Am.
Chem. Soc., 2018, 140, 7415.
Page 13 of 13 ChemComm
Che
mC
omm
Acc
epte
dM
anus
crip
t
Publ
ishe
d on
25
Oct
ober
202
1. D
ownl
oade
d by
Bei
jing
Nor
mal
Uni
vers
ity o
n 10
/27/
2021
2:2
6:36
PM
.
View Article OnlineDOI: 10.1039/D1CC04397H

![ChemComm ViewOnline Dynamic Article Linksszolcsanyi/education/files... · 3. [3,3]-Sigmatropic rearrangements [3,3]-Sigmatropic rearrangements have long been used in organic chemistry,](https://img.dokumen.tips/doc/110x75/5f08617e7e708231d421ba0f/chemcomm-viewonline-dynamic-article-szolcsanyieducationfiles-3-33-sigmatropic.jpg)





![View Article Online ChemComm - Xiamen University · Xiaobo Qu*[a], Tianyu Qiu[a], Di Guo[b], Hengfa Lu[a], Jiaxi Ying[a], Ming Shen[c], Bingwen Hu[c], Vladislav Orekhov[d], and Zhong](https://img.dokumen.tips/doc/110x75/5fbac280afd5c7577578c107/view-article-online-chemcomm-xiamen-university-xiaobo-qua-tianyu-qiua-di.jpg)

![View Article Online ChemComm - Research Explorer · 2017-06-26 · yet despite the existence of sophisticated theoretical treatments [1a] much remains to be done. In recent years](https://img.dokumen.tips/doc/110x75/5f26608f6fa588726d4d7137/view-article-online-chemcomm-research-explorer-2017-06-26-yet-despite-the-existence.jpg)



















