Embed Size (px)
Citation preview

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 1/230
Utviklingsavvik
Genpanel, versjon v02
* Enkelte genomiske regioner har lav eller ingen sekvensdekning ved eksomsekvensering. Detteskyldes at de har stor likhet med andre områder i genomet, slik at spesifikk gjenkjennelse avdisse områdene og påvisning av varianter i disse områdene, blir vanskelig og upålitelig. Dissegenetiske regionene har vi identifisert ved å benytte USCS segmental duplication hvor områderstørre enn 1 kb og ≥90% likhet med andre regioner i genomet, gjenkjennes(https://genome.ucsc.edu).
For noen gener ligger alle ekson i områder med segmentale duplikasjoner: ACTB, ACTG1, ASNS,ATAD3A, CA5A, CFC1, CLCNKB, CYCS, DDX11, GBA, GJA1, MSTO1, PIGC, RBM8A, RPL15, SBDS,SDHA, SHOX, SLC6A8
Vi gjør oppmerksom på at ved identifiseringav ekson oppstrøms for startkodon kaneksonnummereringen endres uten at transkript ID endres.
Avdelingens websider har en full oversikt over områder som er affisert av segmentaleduplikasjoner.
** Transkriptets kodende ekson.
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
AAAS 13666 NM_015665.6 1-16 Achalasia-addisonianism-alacrimiasyndrome, 231550
AARS 20 NM_001605.2 2-21 Epileptic encephalopathy, early infantile,29 616339
AARS2 21022 NM_020745.4 1-22 Combined oxidative phosphorylationdeficiency 8, 614096
AASS 17366 NM_005763.4 2-24 Hyperlysinaemia (Disorders of histidine,tryptophan or lysine metabolism)
ABAT 23 NM_020686.6 2-16 GABA transaminase deficiency(Disorders of neurotransmittermetabolism, gamma-aminobutyrate)
Avdeling for medisinsk genetikk

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 2/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ABCA1 29 NM_005502.4 2-50 Tangier disease (Disorders of highdensity lipoprotein metabolism)
ABCB11 42 NM_003742.4 2-28 Cholestasis, benign recurrentintrahepatic, 2, 605479 Cholestasis, progressive familialintrahepatic 2, 601847
ABCB4 45 NM_000443.4 2-28 Cholestasis, intrahepatic, of pregnancy,3, 614972 Cholestasis, progressive familialintrahepatic 3, 602347
ABCB7 48 NM_004299.6 1-16 Anemia, sideroblastic, with ataxia,301310
ABCC2 53 NM_000392.5 1-32 Dubin-Johnson syndrome, 237500
ABCC6 57 NM_001171.5 1-9 1-31 Pseudoxanthoma elasticum, 264800 Pseudoxanthoma elasticum, formefruste, 177850 Arterial calcification, generalized, ofinfancy, 2, 614473
ABCC9 60 NM_005691.3 1-38 Hypertrichotic osteochondrodysplasia239850 Cardiomyopathy, dilated, 10, 608569
ABCD1 61 NM_000033.4 7-10 1-10 X-linked adrenoleukodystrophy(Disorders of peroxisomal alpha-, betaand omega-oxidation)
ABCD4 68 NM_005050.4 1-19 Methylmalonic aciduria andhomocystinuria, cblJ type, 614857
ABCG5 13886 NM_022436.3 1-13 Sitosterolaemia (Inheritedhypercholesterolaemias)
ABCG8 13887 NM_022437.3 1-13 Sitosterolaemia (Inheritedhypercholesterolaemias)
ABHD12 15868 NM_001042472.3 1-13 Polyneuropathy, Hearing Loss, Ataxia,Retinitis Pigmentosa and Cataract(PHARC)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 3/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ABHD5 21396 NM_016006.6 1-7 Neutral lipid storage disease (Disordersof lipolysis) Chanarin-Dorfman syndrome, 275630
ABL1 76 NM_005157.6 1-11 Congenital heart defects and skeletalmalformations syndrome, 617602
ACAD8 87 NM_014384.2 1-11 Isobutyric aciduria (Organic acidurias)
ACAD9 21497 NM_014049.5 1-18 Mitochondrial complex I deficiency dueto ACAD9 deficiency 611126
ACADM 89 NM_000016.5 1-12 Medium - chain acyl CoA dehydrogenasedeficiency (Disorders of mitochondrialfatty acid oxidation)
ACADS 90 NM_000017.4 1-10 Acyl-CoA dehydrogenase, short-chain,deficiency of 201470
ACADSB 91 NM_001609.4 1-11 2-Methylbutyric aciduria (Organicacidurias)
ACADVL 92 NM_000018.4 1-20 Very long - chain acyl CoAdehydrogenase deficiency (Disorders ofmitochondrial fatty acid oxidation)
ACAN 319 NM_013227.3 2-18 Spondyloepiphyseal dysplasia,Kimberley type 608361 Osteochondritis dissecans, shortstature, and early-onset osteoarthritis,165800
ACAT1 93 NM_000019.4 1-12 Cytosolic acetoacetyl-CoA thiolasedeficiency (Disorders of ketone bodymetabolism)
ACN9 21752 NM_020186.3 1-2 No OMIM phenotype
ACO2 118 NM_001098.3 1-18 Infantile cerebellar-retinal degeneration,614559
ACOX1 119 NM_004035.7 1-14 Peroxisomal acyl-CoA oxidase 1deficiency (Disorders of peroxisomalalpha-, beta and omega-oxidation)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 4/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ACP5 124 NM_001111035.3 4-7 Spondyloenchondrodysplasia, shortstature, SLE, intracranial calcification,spasticity, chilblains, autoimmunehaemolytic anaemia
ACSF3 27288 NM_174917.5 3-11 Combined methylmalonic and malonicaciduria (Organic acidurias)
ACSL4 3571 NM_004458.3 3-16 Mental retardation, X-linked 63, 300387
ACTA1 129 NM_001100.4 2-7 Nemaline myopathy 3, 161800
ACTA2 130 NM_001613.4 2-9 Aortic aneurysm, familial thoracic 6,611788 Moyamoya disease 5, 614042
ACTB 132 NM_001101.5 2-6 2-6 ?Dystonia, juvenile-onset Baraitser Winter syndrome(lissencephaly, pachygyria,polymicrogyria, ptosis, coloboma)
ACTC1 143 NM_005159.5 2-7 Cardiomyopathy, hypertrophic, 11 Cardiomyopathy, dilated, 1R Left ventricular noncompaction 4
ACTG1 144 NM_001614.5 2-6 2-6 Baraitser Winter Syndrome
ACTL6A 24124 NM_004301.5 1-14 developmental delay
ACTL6B 160 NM_016188.5 1-14 Global developmental delay Epileptic encephalopathy, early infantile,76, 618468
ACTN2 164 NM_001103.3 1-21 progressive early-onset muscleweakness
ACVR1 171 NM_001105.5 3-11 Fibrodysplasia ossificans progressiva135100
ACVR2B 174 NM_001106.4 1-11 Heterotaxy syndrome Heterotaxy,visceral, 4, autosomal, 613751

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 5/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ACVRL1 175 NM_000020.3 2-10 Telangiectasia, hereditary hemorrhagic,type 2, 600376 Heritable pulmonary arterialhypertension
ACY1 177 NM_000666.3 2-15 Aminoacylase 1 deficiency (Organicacidurias)
ADA 186 NM_000022.4 1-12 Adenosine deaminase deficiency(Disorders of purine metabolism) Infantile enterocolitis & monogenicinflammatory bowel disease Severe combined immunodeficiencydue to ADA deficiency, 102700
ADAMTS10 13201 NM_030957.4 3-26 Weill-Marchesani syndrome 1, recessive,277600
ADAMTS17 17109 NM_139057.4 1-22 Weill-Marchesani syndrome type 4
ADAMTS9 13202 NM_182920.2 1-39 Nephronophthisis Related Ciliopathy
ADAMTSL2 14631 NM_014694.4 10-19 2-19 Geleophysic dysplasia 1 231050
ADAR 225 NM_001111.5 1-15 Aicardi-Goutieres syndrome 6
ADAT3 25151 NM_138422.4 2 Mental retardation, autosomalrecessive 36, 615286
ADCK3 16812 NM_020247.5 2-15 Coenzyme Q10 deficiency, primary, 4,612016
ADCK4 19041 NM_024876.4 2-15 Nephrotic syndrome, type 9
ADCY5 236 NM_183357.2 1-21 Dyskinesia, familial, with facialmyokymia, 606703
ADK 257 NM_001123.3 1-11 Hypermethioninemia due to adenosinekinase deficiency 614300
ADNP 15766 NM_015339.5 3-5 MENTAL RETARDATION, AUTOSOMALDOMINANT, 28 615873

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 6/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ADPRHL2 21304 NM_017825.3 1-6 Intellectual disability, cerebellaratrophy, ataxia and epilepsy
ADRA2B 282 NM_000682.7 1 Epilepsy, myoclonic, familial adult, 2,607876
ADSL 291 NM_000026.4 1-13 Adenylosuccinate lyase deficiency(Disorders of purine metabolism)
ADSSL1 20093 NM_199165.2 1-13 Myopathy, distal, 5, 617030
AFF2 3776 NM_002025.4 1-21 Mental retardation, X-linked, FRAXE type,309548
AFF3 6473 NM_002285.3 3 3-24 Skeletal dysplasia with severeneurological disease Intellectual disability
AFF4 17869 NM_014423.4 2-21 CORNELIA DE LANGE-LIKE SYNDROME
AFG3L2 315 NM_006796.3 14 1-17 Spinocerebellar ataxia 28
AGA 318 NM_000027.4 1-9 Aspartylglucosaminuria 208400(Patients may be tall for their age, butlack of a growth spurt in pubertytypically causes adults to be short)
AGK 21869 NM_018238.4 16 2-16 Acylglycerol kinase deficiency (Sengersyndrome) (Disorders of complex lipidsynthesis) Sengers syndrome, 212350 Cataract 38, autosomal recessive,614691
AGL 321 NM_000642.3 2-34 Glycogen storage disease type III, Cori(Glycogen storage disorders)
AGO1 3262 NM_012199.5 1-19 Generalized hypotonia Global developmental delay
AGPS 327 NM_003659.4 1-20 Rhizomelic chondrodysplasia punctatatype 3 (Peroxisomal disorders)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 7/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
AGRN 329 NM_198576.4 1-36 Myasthenic syndrome, congenital, 8,with pre- and postsynaptic defects,615120
AGXT 341 NM_000030.3 1-11 Primary hyperoxaluria type I (Disordersof glyoxylate metabolism)
AHDC1 25230 NM_001029882.3 6 XIA-GIBBS SYNDROME
AHI1 21575 NM_017651.4 3-28 Joubert syndrome 3 608629
AIFM1 8768 NM_004208.4 1-16 Cowchock syndrome, 310490 Combined oxidative phosphorylationdeficiency 6, 300816
AIMP1 10648 NM_004757.3 2-7 Leukodystrophy, hypomyelinating, 3,260600
AIMP2 20609 NM_006303.4 1 1-4 Leukodystrophy, hypomyelinating, 17,618006
AIPL1 359 NM_014336.5 1-6 Leber congenital amaurosis 4, 604393 Retinitis pigmentosa, juvenile, 604393 Cone-rod dystrophy, 604393
AIRE 360 NM_000383.4 1-14 Autoimmune polyendocrinopathysyndrome , type I, with orwithout,reversible metaphysealdysplasia, 240300
AK2 362 NM_001625.4 1-6 RETICULAR DYSGENESIS 267500
AKR1D1 388 NM_005989.4 1-9 Neonatal and Adult Cholestasis Bile acid synthesis defect, congenital, 2
AKT1 391 NM_005163.2 2-14 Cowden syndrome 6 OMIM:164730 Proteus syndrome, 176920
AKT3 393 NM_005465.7 2-14 Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome,603387
ALAD 395 NM_000031.6 2-12 Porphyria, acute hepatic, 612740

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 8/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ALAS2 397 NM_000032.5 2-11 X-linked dominant protoporphyria(Porphyrias with acute painfulphotosensitivity)
ALDH18A1 9722 NM_002860.4 2-18 Hypoprolinaemia, Cutis laxa, autosomalrecessive, type IIIa (Disorders ofornithine or proline metabolism) Spastic paraplegia 9A, autosomaldominant, 601162
ALDH1A3 409 NM_000693.4 1-13 ANOPHTHALMIA/MICROPHTHALMIA
ALDH3A2 403 NM_000382.3 1-10 Sjogren-Larsson syndrome, 270200 General Leukodystrophy &Mitochondrial Leukoencephalopathy
ALDH4A1 406 NM_003748.4 1-15 Hyperprolinaemia type II (Disorders ofornithine or proline metabolism)
ALDH5A1 408 NM_001080.3 1-10 Succinic semialdehyde dehydrogenasedeficiency, 271980
ALDH6A1 7179 NM_005589.4 1-12 Methylmalonate semialdehydedehydrogenase deficiency (Organicacidurias)
ALDH7A1 877 NM_001182.5 1-18 Epilepsy, pyridoxine-dependent 266100
ALDOA 414 NM_001243177.3 2-10 Aldolase A deficiency (Glycogen storagedisorders)
ALDOB 417 NM_000035.4 2-9 Neonatal and Adult Cholestasis Hereditary fructose intolerance(Disorders of fructose metabolism)
ALG1 18294 NM_019109.5 6-13 1-13 ALG1-CDG 300141 Mannosyltransferase 1 deficiency(Disorders of protein N-glycosylation)
ALG11 32456 NM_001004127.3 1-4 ALG11-CDG (CDG-IP)
ALG12 19358 NM_024105.4 2-10 Mannosyltransferase 8 deficiency(Disorders of protein N-glycosylation) CONGENITAL DISORDER OFGLYCOSYLATION TYPE 1G (CDG1G)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 9/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ALG13 30881 NM_001099922.3 1-27 Epileptic encephalopathy, early infantile,36 300884 Congenital disorder of glycosylation,type Is
ALG14 28287 NM_144988.4 1-4 ?Myasthenic syndrome, congenital, 15,without tubular aggregates, 616227
ALG2 23159 NM_033087.4 1-2 Myasthenic syndrome, congenital, 14,with tubular aggregates 616228 Mannosyltransferase 2 deficiency(Disorders of protein N-glycosylation)
ALG3 23056 NM_005787.6 1-9 ALG3-CDG (CDG-ID) Mannosyltransferase 6 deficiency ALG3-CDG (Disorders of protein N-glycosylation)
ALG6 23157 NM_013339.4 2-15 Glucosyltransferase 1 deficiency(Disorders of protein N-glycosylation) ALG6-CDG (CDG-IC)
ALG8 23161 NM_024079.5 1-13 ALG8-CDG (CDG-IH) Glucosyltransferase 2 deficiency(Disorders of protein N-glycosylation)
ALG9 15672 NM_024740.2 1-16 Gillessen-Kaesbach-Nishimurasyndrome 263210 Mannosyltransferase 7-9 deficiency(Disorders of protein N-glycosylation) ALG9-CDG 300153
ALKBH8 25189 NM_138775.3 2-12 Intellectual developmental disorder,autosomal recessive 71, 618504
ALMS1 428 NM_015120.4 17-21 1-23 Alstrom Syndrome Bardet-Biedl Syndrome
ALPL 438 NM_000478.6 2-12 Osteogenesis Imperfecta andDecreased Bone Density Hypophosphatasia, infantile, 241500 Hypophosphatasia, childhood, 241510 Odontohypophosphatasia, 146300 Hypophosphatasia, adult, 146300
ALS2 443 NM_020919.4 2-34 Amyotrophic lateral sclerosis 2,autosomal recessive, juvenile, 205100

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 10/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ALX1 1494 NM_006982.3 1-4 Frontonasal dysplasia 3 613456
ALX3 449 NM_006492.3 1-4 Frontonasal dysplasia 1 136760
ALX4 450 NM_021926.4 1-4 Frontonasal dysplasia 2 613451
AMACR 451 NM_014324.6 1-5 Neonatal and Adult Cholestasis Bile acid synthesis defect, congenital, 4214950 Alpha-methylacyl-CoA racemasedeficiency (Disorders of peroxisomalalpha-, beta and omega-oxidation)
AMER1 26837 NM_152424.4 2 Osteopathia striata with cranialsclerosis 300373 Cleft palate
AMN 14604 NM_030943.3 1-12 Intrinsic factor receptor deficiency dueto AMN mutations (Disorders ofcobalamin absorption, transport andmetabolism) Proteinuric renal disease
AMPD2 469 NM_001368809.2 2-19 Pontocerebellar hypoplasia, type 9,615809 ?Spastic paraplegia 63, 615686, AR
AMT 473 NM_000481.4 1-9 Glycine encephalopathy, 605899
ANAPC1 19988 NM_022662.4 2-48 2-48 Rothmund-Thomson Syndrome Type 1
ANKH 15492 NM_054027.6 1-12 Chondrocalcinosis 2 118600 Craniometaphyseal dysplasia 123000
ANKRD11 21316 NM_013275.6 13 3-13 KBG syndrome, 148050 KBG syndrome,148050 (orofacialclefting, intellectual disability, dentalanomalies, dysmorphism)
ANKRD26 29186 NM_014915.2 1-34 THROMBOCYTOPENIA 2 188000

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 11/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ANKS6 26724 NM_173551.5 1-15 Polycystic Kidney Disease,Nephronophthisis And RelatedDisorders 22 Gene Panel Nephronophthisis 16, 615382
ANO10 25519 NM_018075.5 2-13 Spinocerebellar ataxia, autosomalrecessive 10, 613728
ANO3 14004 NM_031418.4 1-27 Dystonia 24, 615034
ANO5 27337 NM_213599.2 1-22 Gnathodiaphyseal dysplasia, 166260 Muscular dystrophy, limb-girdle, type2L, 611307 Miyoshi muscular dystrophy 3, 613319
ANTXR1 21014 NM_032208.2 1-18 GAPO syndrome, 230740
ANTXR2 21732 NM_058172.6 1-17 Hyaline fibromatosis syndrome 228600
AP1B1 554 NM_001127.3 2-3, 6 2-23 Abnormality of copper homeostasis Global developmental delay
AP1S1 559 NM_001283.5 1-5 mental retardation, enteropathy,deafness, peripheral neuropathy,ichthyosis and keratoderma syndrome
AP1S2 560 NM_003916.5 2-5 MENTAL RETARDATION X-LINKED TYPE59 300630 Mental retardation, X-linked syndromic5, 304340
AP2M1 564 NM_001025205.1 2-11 Intellectual developmental disorder 60with seizures, 618587
AP3B1 566 NM_003664.4 1-27 Hermansky-Pudlak syndrome 2 608233
AP3B2 567 NM_004644.5 1-26 Epileptic encephalopathy, early infantile,48 617276
AP4B1 572 NM_006594.5 2-11 Spastic paraplegia 47, autosomalrecessive, 614066
AP4E1 573 NM_007347.5 1-21 Spastic paraplegia 51, autosomalrecessive, 613744

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 12/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
AP4M1 574 NM_004722.4 1-15 Spastic paraplegia 50, autosomalrecessive, 612936
AP4S1 575 NM_007077.4 2-6 Spastic paraplegia 52, autosomalrecessive, 614067
AP5Z1 22197 NM_014855.3 1-17 Spastic paraplegia 48, autosomalrecessive
APC2 24036 NM_005883.2 2-15 Lissencephaly, Subcortical Heterotopia,and Global Developmental Delay
APOA1 600 NM_000039.2 2-4 ApoA-I and apoC-III deficiency, combined
APOA1BP 18453 NM_144772.3 1-6 Encephalopathy, progressive, early-onset, with brain edema and/orleukoencephalopathy 617186
APOA5 17288 NM_052968.5 2-4 Familial hypertriglyceridaemia (Inheritedhypertriglyceridaemias)
APOB 603 NM_000384.3 1-29 Hypercholesterolemia, familial, 2 144010
APOC2 609 NM_000483.5 2-4 Familial apolipoprotein C - II deficiency(Familial chylomicronaemia, Inheritedhypercholesterolaemias)
APOE 613 NM_000041.4 2-4 Familial dysbetalipoproteinaemia(Inherited mixed hyperlipidaemias)
APRT 626 NM_000485.3 1-5 Adenine phosphoribosyl transferasedeficiency (Disorders of purinemetabolism)
APTX 15984 NM_175073.2 3-9 Secondary CoQ10 deficiency(Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly)) Ataxia, early-onset, with oculomotorapraxia and hypoalbuminemia, 208920
AQP1 633 NM_198098.3 1-4 Heritable pulmonary arterialhypertension

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 13/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
AR 644 NM_000044.6 1-8 Androgen insensitivity, 300068Spinaland bulbar muscular atrophy ofKennedy, 313200Androgen insensitivity,partial, with or without breast cancer,312300{Prostate cancer, susceptibilityto}, 176807Hypospadias 1, X-linked,300633
ARCN1 649 NM_001655.5 1-10 Short stature, rhizomelic, withmicrocephaly, micrognathia, anddevelopmental delay, 617164
ARFGEF2 15853 NM_006420.3 1-39 Periventricular heterotopia withmicrocephaly, 608097
ARG1 663 NM_000045.4 1-8 Argininaemia (Urea cycle disorders andinherited hyperammonaemias)
ARHGAP29 30207 NM_004815.4 2-23 Cleft palate
ARHGAP31 29216 NM_020754.4 1-12 Adams-Oliver syndrome 1, 100300
ARHGEF6 685 NM_004840.3 1-22 Mental retardation, X-linked 46, 300436
ARHGEF9 14561 NM_015185.3 1-10 Epileptic encephalopathy, early infantile,8, 300607 Epileptic encephalopathy, early infantile,8 300607
ARID1A 11110 NM_006015.6 1-20 Mental retardation, autosomaldominant 14, 614607 COFFIN-SIRIS SYNDROME 135900
ARID1B 18040 NM_020732.3 1-20 Mental retardation, autosomaldominant 12, 614562 COFFIN SIRIS SYNDROME 135900
ARID2 18037 NM_152641.4 1-21 Coffin-Siris syndrome-like phenotype
ARL13B 25419 NM_182896.3 1-10 Joubert syndrome 8, 612291

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 14/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ARL14EP 26798 NM_152316.3 4 2-4 AUTOSOMAL RECESSIVE MENTALRETARDATION
ARL3 694 NM_004311.4 1-6 JOUBERT SYNDROME, 614615
ARL6 13210 NM_177976.3 3-9 Bardet-Biedl syndrome 3 600151
ARL6IP1 697 NM_015161.3 6 1-6 Spastic paraplegia
ARMC4 25583 NM_018076.5 2-10 2-20 Ciliary dyskinesia, primary, 23, 615451
ARMC9 20730 NM_025139.6 2-21 Joubert syndrome 30, 617622
ARSA 713 NM_000487.6 1-8 Metachromatic leukodystrophy, 250100
ARSB 714 NM_000046.5 1-8 Mucopolysaccharidosis type VI(Maroteaux-Lamy), 253200
ARSE 719 NM_000047.3 9-11 2-11 Chondrodysplasia punctata, X-linkedrecessive, 302950
ARV1 29561 NM_022786.3 1-5 Epileptic encephalopathy, early infantile,38
ARX 18060 NM_139058.3 1-5 Epileptic encephalopathy, early infantile,1, 308350Lissencephaly, X-linked 2,300215Mental retardation, X-linked 29and others, 300419Proud syndrome,300004Partington syndrome,309510Hydranencephaly with abnormalgenitalia, 300215 Hydranencephaly with abnormalgenitalia Lissencephaly, X-linked 2 300215 Mental retardation, X-linked 29 andothers 300419

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 15/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ASAH1 735 NM_177924.5 1-14 Spinal muscular atrophy withprogressive myoclonic epilepsy 159950,Farber lipogranulomatosis 228000, Fetalhydrops, Intellectual disability
ASCC1 24268 NM_001198800.3 2-10 Prenatal Spinal Muscular Atrophy andCongenital Bone Fractures
ASH1L 19088 NM_018489.3 2-28 Mental retardation, autosomaldominant 52, 617796
ASL 746 NM_000048.4 2-17 Argininosuccinic aciduria (Urea cycledisorders and inheritedhyperammonaemias)
ASNS 753 NM_133436.3 3-13 3-13 Asparagine synthetase deficiency,615574 congenital microcephaly, intellectualdisability, progressive cerebral atrophy,intractable seizures
ASPA 756 NM_000049.4 1-6 Canavan disease, 271900 General Leukodystrophy &Mitochondrial Leukoencephalopathy,25655951
ASPH 757 NM_004318.4 1-25 FACIAL DYSMORPHISM, LENSDISLOCATION, ANTERIOR SEGMENTABNORMALITIES, AND SPONTANEOUSFILTERING BLEBS
ASPM 19048 NM_018136.5 1-28 Microcephaly 5, primary, autosomalrecessive, 608716
ASS1 758 NM_000050.4 3-16 intellectual disability Citrullinaemia type1 (Urea cycledisorders and inheritedhyperammonaemias)
ASXL1 18318 NM_015338.6 1-12 Bohring-Opitz syndrome, 605039
ASXL2 23805 NM_018263.6 1-12 Shashi-Pena syndrome 617190 Developmental delay, macrocephaly,and dysmorphic features
2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 16/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
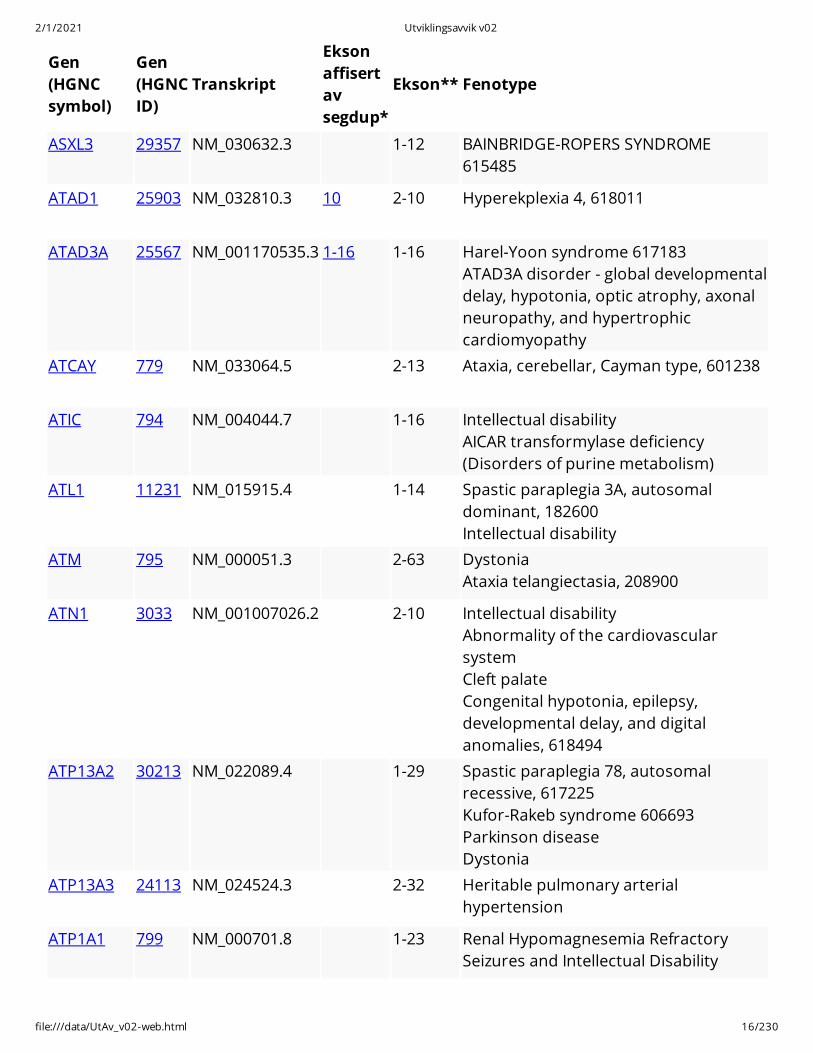
ASXL3 29357 NM_030632.3 1-12 BAINBRIDGE-ROPERS SYNDROME615485
ATAD1 25903 NM_032810.3 10 2-10 Hyperekplexia 4, 618011
ATAD3A 25567 NM_001170535.3 1-16 1-16 Harel-Yoon syndrome 617183 ATAD3A disorder - global developmentaldelay, hypotonia, optic atrophy, axonalneuropathy, and hypertrophiccardiomyopathy
ATCAY 779 NM_033064.5 2-13 Ataxia, cerebellar, Cayman type, 601238
ATIC 794 NM_004044.7 1-16 Intellectual disability AICAR transformylase deficiency(Disorders of purine metabolism)
ATL1 11231 NM_015915.4 1-14 Spastic paraplegia 3A, autosomaldominant, 182600 Intellectual disability
ATM 795 NM_000051.3 2-63 Dystonia Ataxia telangiectasia, 208900
ATN1 3033 NM_001007026.2 2-10 Intellectual disability Abnormality of the cardiovascularsystem Cleft palate Congenital hypotonia, epilepsy,developmental delay, and digitalanomalies, 618494
ATP13A2 30213 NM_022089.4 1-29 Spastic paraplegia 78, autosomalrecessive, 617225 Kufor-Rakeb syndrome 606693 Parkinson disease Dystonia
ATP13A3 24113 NM_024524.3 2-32 Heritable pulmonary arterialhypertension
ATP1A1 799 NM_000701.8 1-23 Renal Hypomagnesemia RefractorySeizures and Intellectual Disability

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 17/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ATP1A2 800 NM_000702.4 1-23 Migraine, familial hemiplegic, 2 602481 Alternating hemiplegia of childhood 1,104290 benign familial infantile convulsions
ATP1A3 801 NM_152296.5 1-23 Alternating Hemiplegia of Childhood(AHC), intellectual disability Cerebellar ataxia, areflexia, pes cavus,optic atrophy and sensorineuralhearing loss (CAPOS, #601338) Catastrophic epilepsy, unusual apneaspells, and postnatal microcephaly CAPOS Syndrome (recurrent mutation)
ATP5A1 823 NM_001001937.1 2-13 Complex V (Mitochondrial respiratorychain disorders (caused by nuclearvariants only), OXPHOS structuralsubunits)
ATP5D 837 NM_001687.5 1-4 ATP5F1D metabolic disorder
ATP5E 838 NM_006886.4 1-2 ?Mitochondrial complex V (ATPsynthase) deficiency, nuclear type 3614053
ATP5F1 840 NM_001688.5 1-7 No OMIM phenotype
ATP5H 845 NM_001003785.2 2-5 No OMIM phenotype
ATP5J2 848 NM_004889.4 1-4 No OMIM phenotype
ATP5L 14247 NM_006476.5 1-3 No OMIM phenotype
ATP5L2 13213 NM_001165877.1 1 No OMIM phenotype
ATP6AP1 868 NM_001183.6 1-10 Immunodeficiency 47 300972
ATP6AP2 18305 NM_005765.3 1-9 Mental retardation, X-linked, syndromic,Hedera type, 300423

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 18/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ATP6V0A2 18481 NM_012463.4 1-20 Cutis laxa, autosomal recessive, type IIA219200
ATP6V1A 851 NM_001690.4 2-15 Epileptic encephalopathy, infantile orearly childhood, 3 618012 Autosomal Recessive Cutis Laxa
ATP6V1B1 853 NM_001692.4 1-14 Renal tubular acidosis with deafness,267300
ATP6V1B2 854 NM_001693.4 1-14 ZIMMERMANN-LABAND SYNDROME
ATP6V1E1 857 NM_001696.4 1-9 Mutations in ATP6V1E1 or ATP6V1ACause Autosomal Recessive Cutis Laxa
ATP7A 869 NM_000052.7 2-23 OCCIPITAL HORN SYNDROME 304150 Menkes disease Spinal muscular atrophy, distal, 300489
ATP7B 870 NM_000053.4 1-21 Wilson disease 277900 Dystonia
ATP8A2 13533 NM_016529.6 1-37 Cerebellar ataxia, mental retardation,and dysequilibrium syndrome 4 615268
ATP8B1 3706 NM_005603.6 2-28 Cholestasis, benign recurrentintrahepatic, 243300 Cholestasis, intrahepatic, of pregnancy,1, 147480 Cholestasis, progressive familialintrahepatic 1, 211600 Byler disease (Disorders of bile acidmetabolism and transport)
ATPAF1 18803 NM_022745.4 1-9 No OMIM phenotype
ATPAF2 18802 NM_145691.4 1-8 Mitochondrial complex V (ATP synthase)deficiency, nuclear type 1, 604273 General Leukodystrophy &Mitochondrial Leukoencephalopathy
ATR 882 NM_001184.4 1-47 Seckel syndrome 1, 210600 MICROCEPHALIC PRIMORDIALDWARFISM I

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 19/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ATRX 886 NM_000489.5 1-35 Alpha-thalassemia/mental retardationsyndrome, 301040 (Microcephaly)
ATXN1 10548 NM_000332.3 8-9 Spinocerebellar ataxia 1, 164400
ATXN10 10549 NM_013236.4 1-12 Spinocerebellar ataxia 10, 603516
ATXN7 10560 NM_000333.3 3-13 Spinocerebellar ataxia 7, 164500
AUH 890 NM_001698.2 1-10 3-methylglutaconic aciduria, type I,250950
AUTS2 14262 NM_015570.4 1-19 SYNDROMIC INTELLECTUAL DISABILITY612100
B3GALNT2 28596 NM_152490.5 1-12 Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies, type A, 11
B3GALT6 17978 NM_080605.4 1 Ehlers-Danlos syndrome, progeroidtype, 2 615349 Spondyloepimetaphyseal dysplasia withjoint laxity, type 1, with or withoutfractures 271640
B3GALTL 20207 NM_194318.4 1-15 O-fucose-specific beta-1,3-N-glucosyltransferase deficiency(Disorders of protein O-glycosylation, O-mannosylglycan synthesis deficiencies) Peters-plus syndrome, 261540
B3GAT3 923 NM_012200.4 3-5 1-5 Multiple joint dislocations, shortstature, craniofacial dysmorphism, withor without congenital heart defects245600 B3GAT3-CDG (Disorders of protein O-glycosylation, O-mannosylglycansynthesis deficiencies)
B3GNT2 15629 NM_006577.6 2 Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies), type A, 13615287

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 20/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
B4GALNT1 4117 NM_001478.5 2-11 Spastic paraplegia 26, autosomalrecessive, 609195
B4GALT1 924 NM_001497.3 1-6 Beta-1,4-galactosyltransferase 1deficiency (Disorders of multipleglycosylation and other glycosylationpathways) Congenital disorder of glycosylation,type IId 607091
B4GALT7 930 NM_007255.3 1-6 Ehlers-Danlos syndrome with shortstature and limb anomalies 130070 B4GALT7-CDG (Disorders of protein O-glycosylation, O-mannosylglycansynthesis deficiencies, Disorders ofprotein O-glycosylation, O-xylosylglycansynthesis deficiencies) Ehlers-Danlos syndrome, progeroidtype, 1, 130070
B9D1 24123 NM_015681.5 1-7 MECKEL SYNDROME 9 614209
B9D2 28636 NM_030578.4 2-4 Joubert syndrome Meckel syndrome 10, 614175
BAAT 932 NM_001701.4 2-4 Neonatal and Adult Cholestasis Hypercholanemia, familial, 607748
BAG3 939 NM_004281.3 1-4 Myopathy, myofibrillar, 6, 612954
BANF1 17397 NM_001143985.1 2-3 NESTOR-GUILLERMO PROGERIASYNDROME 614008
BBS1 966 NM_024649.5 1-17 Polydactyly Bardet-Biedl syndrome 1, 209900
BBS10 26291 NM_024685.4 1-2 Bardet-Biedl syndrome 10, 209900 Polydactyly
BBS12 26648 NM_152618.3 2 Polydactyly Bardet Biedl syndrome 12, 615989
BBS2 967 NM_031885.4 1-17 Bardet-Biedl syndrome 2, 209900 Polydactyly

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 21/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
BBS4 969 NM_033028.5 1-16 Polydactyly Bardet-Biedl syndrome 4, 615982
BBS5 970 NM_152384.3 1-12 Bardet-Biedl syndrome 5, 209900 Polydactyly
BBS7 18758 NM_176824.3 1-19 Polydactyly Bardet-Biedl syndrome 7, 615984
BBS9 30000 NM_198428.3 2-23 Bardet-Biedl syndrome 9, 209900 Polydactyly
BCAP31 16695 NM_001139441.1 5-8 2-8 Deafness, dystonia and cerebellarhypomyelination, 300475
BCAT2 977 NM_001190.4 1-11 Branched-chain amino acid transferase(Disorder of branched-chain amino acidmetabolism not classified as organicaciduria)
BCKDHA 986 NM_000709.4 1-9 Maple syrup urine disease, type Ia BCKD E1 alpha subunit of deficiency(Maple syrup urine disease, disorder ofbranched-chain amino acid metabolismnot classified as organic aciduria)
BCKDHB 987 NM_183050.4 1-10 Maple syrup urine disease, type Ib BCKD E1 beta subunit of deficiency(Maple syrup urine disease, disorder ofbranched-chain amino acid metabolismnot classified as organic aciduria)
BCKDK 16902 NM_005881.4 2-12 Branched-chain ketoaciddehydrogenase kinase deficiency
BCL11A 13221 NM_022893.4 1-4 INTELLECTUAL DISABILITY
BCL11B 13222 NM_138576.4 1-4 Intellectual developmental disorder withdysmorphic facies, speech delay, and T-cell abnormalities, 618092
BCOR 20893 NM_017745.6 2-15 Microphthalmia, syndromic 2, 300166
BCORL1 25657 NM_021946.4 1-12 Intellectual disability, developmentaldelay and dysmorphism

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 22/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
BCS1L 1020 NM_004328.5 3-9 GRACILE syndrome Cholestasis Mitochondrial complex III deficiency,nuclear type 1, 124000 Leigh syndrome, 256000 Bjornstad syndrome, 262000 Mitochondrial Leukoencephalopathy
BFSP2 1041 NM_003571.4 1-7 Cataract 12, multiple types, 611597
BGN 1044 NM_001711.6 2-8 Severe syndromic form of thoracicaortic aneurysm & dissection X-Linked SpondyloepimetaphysealDysplasia
BHLHA9 35126 NM_001164405.1 1 Syndactyly, mesoaxial synostotic, withphalangeal reduction, 609432 Polydactyly SPLIT HAND AND FOOT MALFORMATION220600
BICD2 17208 NM_001003800.2 1-7 Spinal muscular atrophy, lowerextremity-predominant, 2, AD, 615290 arthrogryposis multiplex congenita
BIN1 1052 NM_139343.3 1-19 Myopathy, centronuclear, autosomalrecessive, 255200
BLM 1058 NM_000057.4 2-22 Bloom syndrome, 210900 microcephalic primordial dwarfism
BLOC1S6 8549 NM_012388.3 1-5 HERMANSKY-PUDLAK SYNDROME 9614171
BMP1 1067 NM_006129.5 1-20 Osteogenesis imperfecta, type XIII,614856
BMP2 1069 NM_001200.4 2-3 Brachydactyly, type A2 112600 Short stature, facial dysmorphism, andskeletal anomalies with or withoutcardiac anomalies 617877 Cleft palate

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 23/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
BMP4 1071 NM_001202.6 3-4 Microphthalmia, syndromic 6 607932 Global developmental delay Polydactyly OROFACIAL CLEFT 11 600625
BMPER 24154 NM_133468.5 2-16 DIAPHANOSPONDYLODYSOSTOSIS608022
BMPR1B 1077 NM_001203.3 4-13 Brachydactyly, type A2 112600 Acromesomelic dysplasia, Demirhantype 609441 Chrondrodysplasia, acromesomelic,with genital anomalies, 609441
BMPR2 1078 NM_001204.7 1-13 Pulmonary hypertension, familialprimary, 1, with or without HHT, 178600 Pulmonary venoocclusive disease 1,265450
BNC2 30988 NM_017637.6 1-7 Congenital Lower Urinary TractObstruction
BOLA3 24415 NM_212552.3 1-4 Hyperglycinaemia, non-ketotic (Baker(2014) Brain 137,366) Defective Fe-S/lipoic acid biosynthesis(Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly)) Multiple mitochondrial dysfunctionssyndrome 2, 614299 Disorders of iron homeostasis
BPTF 3581 NM_004459.7 1, 3, 8-9,17-19,27-28,30
1-30 Neurodevelopmental disorder withdysmorphic facies and distal limbanomalies, 617755 Developmental and Speech Delay,Postnatal Microcephaly, andDysmorphic Features
BRAF 1097 NM_004333.6 18 1-18 CARDIOFACIOCUTANEOUS SYNDROME115150 Noonan syndrome 7 613706 LEOPARD syndrome 3 613707

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 24/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
BRAT1 21701 NM_152743.4 2-14 Rigidity and multifocal seizuresyndrome, lethal neonatal 614498
BRCA1 1100 NM_007294.4 2 2-23 Fanconi anemia, complementationgroup S
BRCA2 1101 NM_000059.3 2-27 Fanconi anemia, complementationgroup D1, 605724 Radial Ray abnormality
BRD4 13575 NM_058243.2 2-20 Intellectual disability Microcephaly CORNELIA DE LANGE-LIKE SYNDROME
BRF1 11551 NM_001519.4 1-18 Cerebellofaciodental syndrome, 616202
BRIP1 20473 NM_032043.3 2-20 Radial Ray abnormality Fanconi anemia, complementationgroup J, 609054
BRPF1 14255 NM_001003694.2 2-14 Intellectual developmental disorder withdysmorphic facies and ptosis 617333
BRSK2 11405 NM_001256627.2 1-20 Global developmental delay, Intellectualdisability, Autism, Behavioralabnormality
BRWD3 17342 NM_153252.5 1-41 Mental Retardation, X-linked
BSCL2 15832 NM_032667.6 2-11 Silver spastic paraplegia syndrome,270685 Intractable epilepsy and neurologicalregression Encephalopathy, progressive, with orwithout lipodystrophy 615924 Lipodystrophy, congenital generalized,type 2 269700 Neuropathy, distal hereditary motor,type VA 600794
BSND 16512 NM_057176.3 1-4 BARTTER SYNDROME TYPE 4A 602522

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 25/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
BTD 1122 NM_001281726.1 1-3 Biotinidase deficiency (Disorders ofbiotin metabolism) lactic acidosis with seizures andeczema,immune deficiency
BTRC 1144 NM_033637.4 1-14 Split-hand split-foot malformation 3
BUB1B 1149 NM_001211.5 1-23 MOSAIC VARIEGATED ANEUPLOIDYSYNDROME 1
C10orf2 1160 NM_021830.5 1-5 Progressive external ophthalmoplegia,autosomal dominant, 3, 609286 Mitochondrial DNA depletion syndrome7 (hepatocerebral type), 271245 Spinocerebellar Ataxia, Recessive Perrault syndrome 5, 616138 General Leukodystrophy &Mitochondrial Leukoencephalopathy
C11orf70 28188 NM_032930.3 1-7 PRIMARY CILIARY DYSKINESIA
C11orf83 34399 NM_001085372.3 1-2 ?Mitochondrial complex III deficiency,nuclear type, 616111
C12orf4 1184 NM_020374.4 2-14 Autosomal recessive intellectualdisability
C12orf57 29521 NM_138425.4 1-3 Temtamy syndrome, 218340 COLOBOMA, HYPOPLASTIC CORPUSCALLOSUM AND INTELLECTUALDISABILITY Temtamy syndrome 218340
C12orf65 26784 NM_152269.5 2-3 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Spastic paraplegia 55, autosomalrecessive, 615035 Combined oxidative phosphorylationdeficiency 7, 613559 optic atrophy and spasticity, tibialmuscle weakness and atrophy,peripheral neuropathy

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 26/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
C17orf89 33551 NM_001086521.2 1-3 Mitochondrial complex I deficiency,nuclear type 34
C19orf12 25443 NM_001031726.3 1-3 Neurodegeneration with brain ironaccumulation (NBIA) (Disorder of ironmetabolism) Spastic paraplegia 43, autosomalrecessive, 615043 Dystonia
C19orf70 33702 NM_205767.2 1-4 Combined oxidative phosphorylationdeficiency 37, 618329
C1QBP 1243 NM_001212.4 1-6 Combined oxidative phosphorylationdeficiency 33 617713 Severe Neonatal-, Childhood-, or Later-Onset Cardiomyopathy Associated withCombined Respiratory-ChainDeficiencies
C21orf2 1260 NM_004928.3 1-7 Spondylometaphyseal dysplasia, axial602271 Jeune asphyxiating thoracic dystrophy(JATD) Retinal dystrophy with macularstaphyloma, 617547
C21orf59 1301 NM_021254.4 1-7 PRIMARY CILIARY DYSKINESIA
C2CD3 24564 NM_015531.6 1-31 short-rib polydactyly syndromes (SRPS Orofaciodigital syndrome XIV 615948
C2orf71 34383 NM_001029883.3 1-2 Retinitis pigmentosa 54, 613428
C4orf26 26300 NM_178497.4 1-2 Amelogenesis imperfecta type, IIA4,614832
C5orf42 25801 NM_023073.3 2-52 Oral-facial-digital syndrome type VI Joubert syndrome 17
C6orf57 20957 NM_145267.3 1-3 No OMIM phenotype

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 27/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
C8orf37 27232 NM_177965.4 1-6 Retinitis pigmentosa 64, 614500 Cone-rod dystrophy 16, 614500
C9orf72 28337 NM_018325.5 2-11 Frontotemporal dementia and/oramyotrophic lateral sclerosis 1 105550
C9orf96 28669 NM_153710.5 1-18
CA2 1373 NM_000067.3 1-7 Osteopetrosis, autosomal recessive 3,with renal tubular acidosis, 259730 carbonic anhydrase II deficiency
CA5A 1377 NM_001739.2 1-7 1-7 Hyperammonemia (Urea cycledisorders and inheritedhyperammonaemias)
CA8 1382 NM_004056.6 1-8 Cerebellar ataxia and mentalretardation with or withoutquadrupedal locomotion 3, 613227
CACNA1A 1388 NM_001127221.1 1-47 Epileptic encephalopathy, early infantile,42 617106 Migraine, familial hemiplegic, 1, withprogressive cerebellar ataxia 141500 Spinocerebellar ataxia 6 Episodic ataxia, type 2 Dystonia
CACNA1B 1389 NM_000718.4 1-46 Neurodevelopmental disorder withseizures and nonepileptic hyperkineticmovements, 618497
CACNA1C 1390 NM_000719.7 43-45 1-47 Brugada syndrome 3 611875 Timothy syndrome 601005
CACNA1D 1391 NM_000720.4 1-49 Primary aldosteronism, seizures, andneurologic abnormalities 615474 AD Sinoatrial node dysfunction anddeafness 614896 AR
CACNA1E 1392 NM_000721.4 1-47 Epileptic Encephalopathy withContractures, Macrocephaly, andDyskinesia

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 28/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CACNA1G 1394 NM_018896.5 1-38 Cerebellar atrophy, epilepsy, intellectualdisability Spinocerebellar ataxia 42, early-onset,severe, with neurodevelopmentaldeficits 618087
CACNA1H 1395 NM_021098.3 2-35 Hyperaldosteronism, familial, type IV617027 {Epilepsy, childhood absence,susceptibility to, 6} 611942 {Epilepsy, idiopathic generalized,susceptibility to, 6} 611942
CACNA1S 1397 NM_000069.3 1-44 {Malignant hyperthermia susceptibility5}, 601887 congenital myopathy
CACNA2D2 1400 NM_006030.4 1-38 Cerebellar atrophy with seizures andvariable developmental delay, 618501 Absence epilepsy
CACNB4 1404 NM_000726.4 1-14 JUVENILE MYOCLONIC EPILEPSY 611136 EPISODIC ATAXIA, TYPE 5
CAD 1424 NM_004341.5 1-44 Epileptic encephalopathy, early infantile,50 - MIM 616457
CAMK2A 1460 NM_015981.4 1-19 Intellectual disability
CAMK2B 1461 NM_001220.5 1-23 Mental retardation, autosomaldominant 54 617799
CAMTA1 18806 NM_015215.4 1-23 Cerebellarataxia, nonprogressive, withmental retardation, 614756
CANT1 19721 NM_138793.4 2-4 Desbuquois dysplasia 1 251450 multiple epiphyseal dysplasia type 7,617719.
CAPN1 1476 NM_001198868.2 2-22 Spastic paraplegia 76 autosomalrecessive, 616907
CAPN3 1480 NM_000070.3 1-24 Muscular dystrophy, limb-girdle, type2A, 253600

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 29/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CARKD 25576 NM_001242881.2 1-10 Neurodegenerative disorderexacerbated by febrile illnesses
CARS 1493 NM_001014437.3 1-23 Microcephaly Developmental Delay andBrittle Hair and Nails
CARS2 25695 NM_024537.4 1-15 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Epileptic encephalopathy with complexmovement disorder and regression
CASC5 24054 NM_144508.5 2-26 Microcephaly 4, primary, autosomalrecessive 604321
CASK 1497 NM_003688.3 1-27 Mental retardation, with or withoutnystagmus Mental retardation and microcephalywith pontine and cerebellar hypoplasia FG syndrome 4
CASQ1 1512 NM_001231.5 1-11 Myopathy, vacuolar, with CASQ1aggregates, 616231
CASR 1514 NM_000388.4 2-7 Hypocalciuric hypercalcemia, type I145980 Hyperparathyroidism, neonatal 239200
CAT 1516 NM_001752.4 1-13 Acatalasaemia (Other peroxisomaldisorders)
CAV1 1527 NM_001753.5 1-3 Pulmonary hypertension, primary, 3,615343
CAV3 1529 NM_033337.3 1-2 Muscular dystrophy, limb-girdle, type IC,607801 Rippling muscle disease, 606072 Creatine phosphokinase, elevatedserum, 123320 Myopathy, distal, Tateyama type, 614321Cardiomyopathy, familial hypertrophic,192600
CBL 1541 NM_005188.4 1-16 Noonan syndrome-like disorder with orwithout juvenile myelomonocyticleukemia, 613563

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 30/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CBS 1550 NM_000071.2 3-17 Homocystinuria, B6-responsive andnonresponsive types, 236200 Thrombosis, hyperhomocysteinemic,236200
CC2D1A 30237 NM_017721.5 1-29 Mental retardation, autosomalrecessive 3, 608443
CC2D2A 29253 NM_001080522.2 3-38 COACH syndrome 216360 Meckel syndrome 6 612284 Joubert syndrome 9 612285
CCBE1 29426 NM_133459.4 1-11 Hennekam lymphangiectasia-lymphedema syndrome, 235510
CCDC103 32700 NM_213607.3 2-4 Ciliary dyskinesia, primary, 17, 614679
CCDC11 26530 NM_145020.5 1-8 Heterotaxy, visceral, 6, autosomalrecessive
CCDC114 26560 NM_144577.4 2-14 Ciliary dyskinesia, primary, 20, 615067
CCDC115 28178 NM_032357.4 1-5 Congenital disorder of glycosylation,type IIo 616828
CCDC151 28303 NM_145045.5 1-13 PRIMARY CILLARY DYSKINEASIA 616037
CCDC22 28909 NM_014008.5 1-17 SYNDROMIC X-LINKED INTELLECTUALDISABILITY
CCDC23 29204 NM_199342.4 2-3 Neurodevelopmental disorder withataxia, hypotonia, and microcephaly,618569
CCDC39 25244 NM_181426.2 1-20 CILIARY DYSKINESIA, PRIMARY, 14613807
CCDC40 26090 NM_017950.4 1-20 Ciliary dyskinesia, primary, 15, 613808
CCDC41 17966 NM_016122.3 3-17 Nephronophthisis 18 615862

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 31/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CCDC47 24856 NM_020198.3 2-13 Trichohepatoneurodevelopmentalsyndrome, 618268 Woolly Hair Liver DysfunctionDysmorphic Features and GlobalDevelopmental Delay
CCDC65 29937 NM_033124.5 1-8 Ciliary dyskinesia, primary, 27, 615504
CCDC78 14153 NM_001031737.3 1-14 Myopathy, centronuclear, 4, 614807
CCDC8 25367 NM_032040.5 1 3-M syndrome 3, 614205
CCDC88A 25523 NM_001135597.2 1-32 PEHO syndrome-like, 617507
CCDC88C 19967 NM_001080414.4 1-30 ?Spinocerebellar ataxia 40 616053 AD Hydrocephalus, nonsyndromic,autosomal recessive 236600 AR
CCND2 1583 NM_001759.4 1-5 Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome 3,615938
CCNO 18576 NM_021147.5 1-3 Ciliary diskinesia, primary, 29, 615872
CCT5 1618 NM_012073.5 1-11 autosomal recessive mutilating sensoryneuropathy with spastic paraplegia
CD151 1630 NM_004357.5 3-9 NEPHROPATHY WITH PRETIBIALEPIDERMOLYSIS BULLOSA ANDDEAFNESS 609057
CD96 16892 NM_198196.3 1-15 C syndrome, 211750
CDC42 1736 NM_001791.4 4-6 2-6 Takenouchi-Kosaki syndrome, 616737
CDC45 1739 NM_001178010.2 1-19 Craniosynostosis (Wilkie) (from AnaBeleza) Meier-Gorlin syndrome withcraniosynostosis (from PMID 27374770)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 32/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CDC6 1744 NM_001254.4 2-12 MEIER-GORLIN SYNDROME 5
CDH1 1748 NM_004360.5 1-16 Blepharocheilodontic syndrome 1
CDH11 1750 NM_001797.4 3-13 Elsahy-Waters syndrome
CDH15 1754 NM_004933.3 1-14 Mental retardation, autosomaldominant 3, 612580
CDH2 1759 NM_001792.5 1-16 Syndromic NeurodevelopmentalDisorder with Corpus Collosum, Axon,Cardiac, Ocular, and Genital Defects
CDH23 13733 NM_022124.6 2-68 Usher syndrome, type 1D, 601067 Deafness, autosomal recessive
CDH3 1762 NM_001793.6 1-16 Ectodermal dysplasia, ectrodactyly, andmacular dystrophy 225280 Hypotrichosis, congenital, with juvenilemacular dystrophy, 601553
CDK10 1770 NM_052988.5 1-13 Al Kaissi syndrome, 617694
CDK13 1733 NM_003718.5 1-14 Congenital heart defects, dysmorphicfacial features, and intellectualdevelopment disorder 617360
CDK16 8749 NM_006201.5 2-16
CDK5RAP2 18672 NM_018249.6 1-38 Microcephaly 3, primary, autosomalrecessive, 604804
2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 33/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
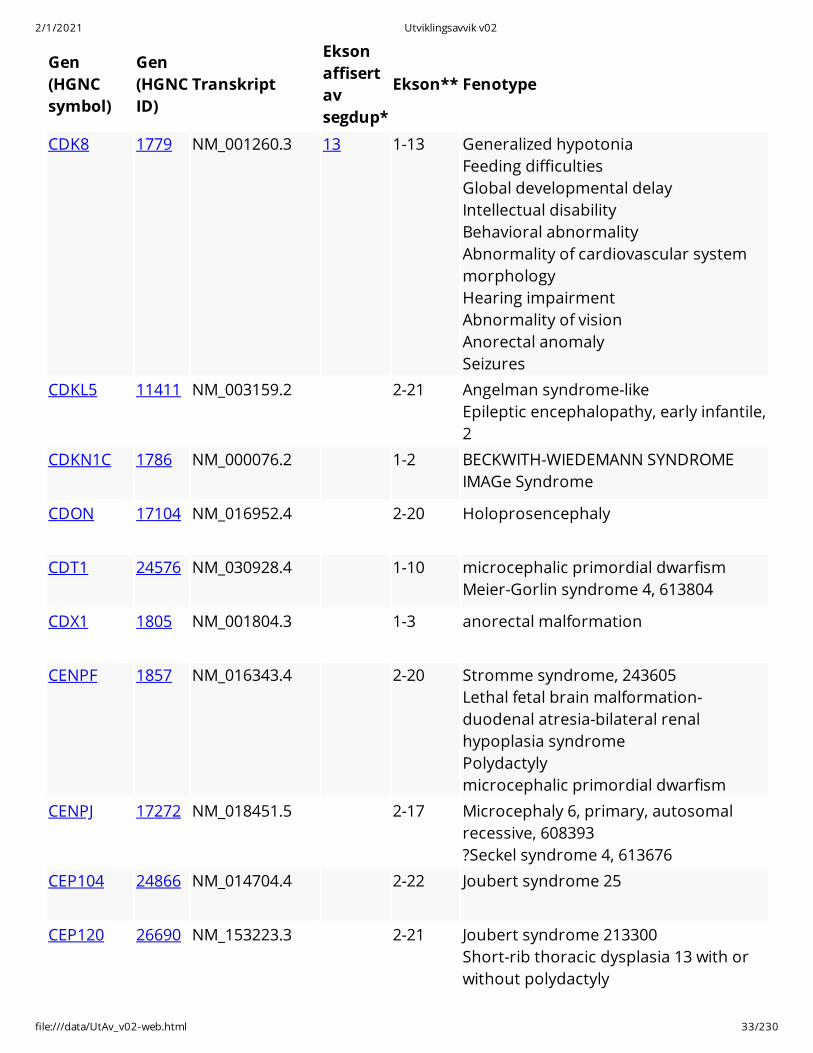
CDK8 1779 NM_001260.3 13 1-13 Generalized hypotonia Feeding difficulties Global developmental delay Intellectual disability Behavioral abnormality Abnormality of cardiovascular systemmorphology Hearing impairment Abnormality of vision Anorectal anomaly Seizures
CDKL5 11411 NM_003159.2 2-21 Angelman syndrome-like Epileptic encephalopathy, early infantile,2
CDKN1C 1786 NM_000076.2 1-2 BECKWITH-WIEDEMANN SYNDROME IMAGe Syndrome
CDON 17104 NM_016952.4 2-20 Holoprosencephaly
CDT1 24576 NM_030928.4 1-10 microcephalic primordial dwarfism Meier-Gorlin syndrome 4, 613804
CDX1 1805 NM_001804.3 1-3 anorectal malformation
CENPF 1857 NM_016343.4 2-20 Stromme syndrome, 243605 Lethal fetal brain malformation-duodenal atresia-bilateral renalhypoplasia syndrome Polydactyly microcephalic primordial dwarfism
CENPJ 17272 NM_018451.5 2-17 Microcephaly 6, primary, autosomalrecessive, 608393 ?Seckel syndrome 4, 613676
CEP104 24866 NM_014704.4 2-22 Joubert syndrome 25
CEP120 26690 NM_153223.3 2-21 Joubert syndrome 213300 Short-rib thoracic dysplasia 13 with orwithout polydactyly

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 34/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CEP135 29086 NM_025009.5 2-25 Microcephaly 8, primary, autosomalrecessive, 614673
CEP152 29298 NM_014985.4 2-26 Microcephaly 9, primary, autosomalrecessive, 614852 Seckel syndrome 5, 613823
CEP164 29182 NM_014956.5 3-33 Nephronophthisis 15 Senior-Loken syndrome
CEP290 29021 NM_025114.4 54 2-54 Meckel syndrome 4 Senior-Loken syndrome Joubert syndrome 5 Bardet-Biedl syndrome 14 615991 Leber congenital amaurosis 10
CEP41 12370 NM_018718.3 1-11 Joubert syndrome 15
CEP55 1161 NM_001127182.2 2-9 microcephaly, delayed development,and bilateral toe syndactyly Meckel-like syndrome autosomal recessive lethal ciliopathy renal dysplasia
CEP57 30794 NM_014679.5 1-11 MOSAIC VARIEGATED ANEUPLOIDYSYNDROME 2
CEP63 25815 NM_025180.4 3-16 Developmental dyslexia ?Seckel syndrome 6, 614728 Microcephaly
CERS1 14253 NM_021267.5 1-7 ?Epilepsy, progressive myoclonic, 8,616230
CFC1 18292 NM_032545.3 1-6 1-6 Heterotaxy, visceral, 2, autosomal605376
CFL2 1875 NM_021914.7 1-4 Nemaline myopathy 7, autosomalrecessive, 610687
CFTR 1884 NM_000492.4 1-27 Neonatal and Adult Cholestasis Cholestasis Cystic fibrosis, 219700 Pancreatitis, 167800

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 35/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CHAMP1 20311 NM_001164144.3 3 INTELLECTUAL DISABILITY
CHAT 1912 NM_020549.4 1-15 Myasthenic syndrome, congenital,associated with episodic apnea, 254210
CHCHD10 15559 NM_213720.3 1-4 Frontotemporal dementia and/oramyotrophic lateral sclerosis 2 ?Myopathy, isolated mitochondrial,autosomal dominant, 616209 Spinal muscular atrophy, Jokela type
CHD2 1917 NM_001271.4 2-39 Epileptic encephalopathy, childhood-onset, 615369
CHD3 1918 NM_001005271.3 1-40 Global developmental delay Intellectual disability Macrocephaly Snijders Blok-Campeau syndrome,618205 Macrocephaly and impaired speech andlanguage
CHD4 1919 NM_001273.5 2-40 Sifrim-Hitz-Weiss syndrome 617159
CHD7 20626 NM_017780.4 2-38 CHARGE SYNDROME 214800 IDIOPATHIC HYPOGONADOTROPICHYPOGONADISM 146110 KALLMANN SYNDROME TYPE 5 612370
CHD8 20153 NM_001170629.2 2-38 Overgrowth with Intellectual disability
CHKB 1938 NM_005198.4 1-11 Muscular dystrophy, congenital,megaconial type, 602541 Choline kinase deficiency (Disorders ofcomplex lipid synthesis)
CHL1 1939 NM_006614.4 3-28 verbal function and developmentaldelay
CHM 1940 NM_000390.4 1-15 Choroideremia, 303100
CHMP1A 8740 NM_002768.5 1-7 Pontocerebellar hypoplasia, type 8,614961

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 36/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CHRDL1 29861 NM_001143981.2 2-12 Megalocornea 1, X-linked 309300
CHRNA1 1955 NM_000079.4 1-9 Myasthenic syndrome, congenital, 1A,slow-channel, 601462 Myasthenic syndrome, congenital, 1B,fast-channel, 608930
CHRNA2 1956 NM_000742.4 2-7 Epilepsy, nocturnal frontal lobe, type 4
CHRNA4 1958 NM_000744.6 1-6 Epilepsy, nocturnal frontal lobe, 1600513
CHRNB1 1961 NM_000747.3 1-11 Myasthenic syndrome, congenital, 2A,slow-channel, 616313
CHRNB2 1962 NM_000748.3 1-6 Epilepsy, nocturnal frontal lobe, 3605375
CHRND 1965 NM_000751.3 1-12 Myasthenic syndrome, congenital, 3A,slow-channel 616321 Multiple pterygium syndrome, lethaltype 253290 Myasthenic syndrome, congenital, 3B,fast-channel 616322
CHRNE 1966 NM_000080.4 1-12 Myasthenic syndrome, congenital, 4A,slow-channel, 605809 Myasthenic syndrome, congenital, 4B,fast-channel, 616324
CHRNG 1967 NM_005199.5 1-12 Escobar syndrome, 265000 Multiple pterygium syndrome, lethaltype, 253290 Myasthenia gravis, neonatal transient fetal akinesia deformation sequencesyndrome/FADS
CHST14 24464 NM_130468.3 1 Ehlers-Danlos syndrome,musculocontractural type 1 601776 CHST14-CDG (Disorders of protein O-glycosylation, O-mannosylglycansynthesis deficiencies)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 37/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CHST3 1971 NM_004273.5 2-3 Spondyloepiphyseal dysplasia withcongenital joint dislocations (recessiveLarsen syndrome) 143095 CHST3-CDG (Disorders of protein O-glycosylation, O-mannosylglycansynthesis deficiencies)
CHST6 6938 NM_021615.5 3 CHST6-CDG (Disorders of protein O-glycosylation, O-mannosylglycansynthesis deficiencies) Macular corneal dystrophy 217800
CHSY1 17198 NM_014918.5 1-3 CHSY1-CDG (Disorders of protein O-glycosylation, O-mannosylglycansynthesis deficiencies) Temtamy preaxial brachydactylysyndrome, 605282
CHUK 1974 NM_001278.5 1-21 Cocoon syndrome, 613630
CIB2 24579 NM_006383.4 1-6 Deafness, autosomal recessive 48,609439 Usher syndrome, type IJ, 614869
CIC 14214 NM_015125.4 1-20 Mental retardation, autosomaldominant 45 617600
CISD2 24212 NM_001008388.5 3 1-3 Wolfram syndrome 2 604928
CIT 1985 NM_001206999.2 2-48 Microcephaly 17, primary, autosomalrecessive, 617090
CIZ1 16744 NM_012127.3 2-17 Dystonia 23, 614860
CKAP2L 26877 NM_152515.5 1-9 Polydactyly Filippi syndrome 272440

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 38/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CLCN2 2020 NM_004366.6 1-24 {Epilepsy, juvenile myoclonic,susceptibility to, 8}, 607628 {Epilepsy, juvenile absence,susceptibility to, 2}, 607628 {Epilepsy, idiopathic generalized,susceptibility to, 11}, 607628 Leukoencephalopathy with ataxia
CLCN4 2022 NM_001830.4 3-13 Raynaud-Claes syndrome 300114 Mental retardation, X-linked 49/15
CLCN5 2023 NM_000084.5 2-12 Proteinuria, low molecular weight, withhypercalciuric nephrocalcinosis 308990 Dent disease 300009 Nephrolithiasis, type I 310468 Hypophosphatemic rickets 300554
CLCN7 2025 NM_001287.6 1-25 Osteopetrosis, autosomal recessive 4611490 Osteopetrosis, autosomal dominant 2166600
CLCNKB 2027 NM_000085.5 2-20 2-20 BARTTER SYNDROME TYPE 4B 613090
CLDN1 2032 NM_021101.5 1-4 Neonatal and Adult Cholestasis Ichthyosis, leukocyte vacuoles, alopeciaand sclerosing cholangitis, 607626
CLDN16 2037 NM_006580.3 1-5 Hypomagnesemia 3, renal 248250
CLDN19 2040 NM_148960.3 1-5 Hypomagnesemia 5, renal, with ocularinvolvement, 248190
CLIC2 2063 NM_001289.6 1-6 ?Mental retardation, X-linked, syndromic32
CLMP 24039 NM_024769.5 1-7 CONGENITAL SHORT BOWEL SYNDROME615237
CLN3 2074 NM_001042432.1 2-16 Ceroid lipofuscinosis, neuronal, 3,204200
CLN5 2076 NM_006493.4 1-4 Ceroid lipofuscinosis, neuronal, 5,256731

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 39/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CLN6 2077 NM_017882.3 1-7 Ceroid lipofuscinosis, neuronal, 6 OMIM
CLN8 2079 NM_018941.4 2-3 Ceroid lipofuscinosis, neuronal, 8
CLP1 16999 NM_006831.3 2-3 PONTOCEREBELLAR HYPOPLASIA, TYPE10
CLPB 30664 NM_030813.6 1-17 3-methylglutaconic aciduria, type VII,with cataracts, neurologic involvementand neutropenia, 616271
CLPP 2084 NM_006012.4 1-6 Perrault syndrome 3, 614129
CLTC 2092 NM_001288653.1 1-32 Mental retardation, autosomaldominant 56, 617854 Overgrowth intellectual disability Epilepsy and intellectual disability
CNBP 13164 NM_003418.5 2-5 Myotonic dystrophy 2, 602668
CNKSR2 19701 NM_014927.5 1-22 Mental retardation, X-linked, syndromic,Houge type 301008
CNNM2 103 NM_017649.5 1-8 Hypomagnesaemia type 6, renal(Disorder of magnesium metabolism)
CNOT1 7877 NM_001265612.2 2-49 Holoprosencephaly 12, with or withoutpancreatic agenesis, 618500
CNOT2 7878 NM_014515.6 2-16 Intellectual developmental disorder withnasal speech, dysmorphic facies, andvariable skeletal anomalies, 618608
CNOT3 7879 NM_014516.4 2-18 CNOT3 syndrome
CNPY3 11968 NM_006586.5 1-6 Epileptic encephalopathy, early infantile,60 617929
CNTN1 2171 NM_001843.4 2-24 ?Myopathy, congenital, Compton-North,612540
CNTNAP1 8011 NM_003632.3 1-24 LETHAL CONGENITAL CONTRACTURESYNDROME 7 616286

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 40/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CNTNAP2 13830 NM_014141.6 1-23 Cortical dysplasia-focal epilepsysyndrome Pitt-Hopkins like syndrome 1
COA3 24990 NM_001040431.3 1-2 Ostergaard et al., 2015, J. Med. Genet.,52, 203-207.
COA4 24604 NM_016565.3 2 No OMIM phenotype
COA6 18025 NM_001012985.2 1-3 Cardioencephalomyopathy, fatalinfantile, due to cytochrome c oxidasedeficiency 4 616501
COA7 25716 NM_023077.3 1-3 Spinocerebellar ataxia, autosomalrecessive, with axonal neuropathy 3OMIM
COASY 29932 NM_025233.7 1-9 Neurodegeneration with brain ironaccumulation 6, 615643 Pontocerebellar hypoplasia, type 12,618266
COG1 6545 NM_018714.3 1-14 Component of COG complex 1deficiency (Disorders of multipleglycosylation and other glycosylationpathways, conserved oligomeric Golgi(COG) complex deficiency) COG1-CDG (CDG-IIG)
COG4 18620 NM_015386.3 1-19 COG4-CDG (CDG-IIJ) Saul-Wilson syndrome 618150 Component of COG complex 4deficiency (Disorders of multipleglycosylation and other glycosylationpathways, conserved oligomeric Golgi(COG) complex deficiency)
COG5 14857 NM_006348.3 1-22 Component of COG complex 5deficiency (Disorders of multipleglycosylation and other glycosylationpathways, conserved oligomeric Golgi(COG) complex deficiency) COG5-CDG

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 41/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
COG6 18621 NM_020751.3 1-19 Shaheen syndrome, 615328 Component of COG complex 6deficiency (Disorders of multipleglycosylation and other glycosylationpathways, conserved oligomeric Golgi(COG) complex deficiency)
COG7 18622 NM_153603.4 1-17 Component of COG complex 7deficiency (Disorders of multipleglycosylation and other glycosylationpathways, conserved oligomeric Golgi(COG) complex deficiency) COG7-CDG (CDG-IIE)
COG8 18623 NM_032382.4 1-5 COG8-CDG (CDG-IIH) Component of COG complex 8deficiency (Disorders of multipleglycosylation and other glycosylationpathways, conserved oligomeric Golgi(COG) complex deficiency)
COL10A1 2185 NM_000493.4 2-3 Metaphyseal chondrodysplasia, Schmidtype 156500
COL11A1 2186 NM_001854.4 1-67 Marshall syndrome 154780 Fibrochondrogenesis 1 228520 Orofacial Clefting with skeletal features Stickler Syndrome Cleft palate
COL11A2 2187 NM_080680.3 1-66 Otospondylomegaepiphyseal dysplasia215150 Fibrochondrogenesis 2 614524 Weissenbacher-Zweymuller syndrome277610 Stickler syndrome, type III Cleft palate
COL12A1 2188 NM_004370.6 2-66 Ullrich congenital muscular dystrophy 2 Bethlem myopathy 2
COL13A1 2190 NM_001130103.2 1-40 Myasthenic syndrome, congenital, 19,616720

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 42/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
COL18A1 2195 NM_130445.4 1-43 Knobloch syndrome, type 1, 267750
COL1A1 2197 NM_000088.3 1-51 Osteogenesis imperfecta, type I 166200 Caffey disease 114000 Osteogenesis imperfecta, type III 259420Osteogenesis imperfecta, type II 166210 Ehlers-Danlos syndrome, type VIIA130060 Ehlers-Danlos syndrome, classic 130000 Osteogenesis imperfecta, type IV 166220Ehlers-Danlos syndrome, type I, 130000 Ehlers-Danlos syndrome, type VIIA,130060 Caffey disease, 114000
COL1A2 2198 NM_000089.4 1-52 Ehlers-Danlos syndrome, cardiacvalvular form 225320 Ehlers-Danlos syndrome, type VIIB130060 Osteogenesis imperfecta, type II 166210 Osteogenesis imperfecta, type III 259420Osteogenesis imperfecta, type IV 166220
COL25A1 18603 NM_198721.4 4 2-37 FIBROSIS OF EXTRAOCULAR MUSCLES,CONGENITAL, 5 616219

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 43/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
COL2A1 2200 NM_001844.5 1-54 Epiphyseal dysplasia, multiple, withmyopia and deafness 132450 Spondyloepiphyseal dysplasia, Stanescutype 616583 Stickler sydrome, type I, nonsyndromicocular 609508 Achondrogenesis, type II orhypochondrogenesis 200610 Kniest dysplasia 156550 Legg-Calve-Perthes disease 150600 Otospondylomegaepiphyseal dysplasia215150 Stickler syndrome, type I 108300 SMED Strudwick type 184250 Spondyloperipheral dysplasia 271700 Platyspondylic skeletal dysplasia,Torrance type 151210 Czech dysplasia 609162 SED congenita 183900 Osteoarthritis with mildchondrodysplasia 604864 Avascular necrosis of the femoral head608805
COL4A1 2202 NM_001845.6 1-52 Brain small vessel disease with orwithout ocular anomalies, 175780 Angiopathy, hereditary, withnephropathy, aneurysms, and musclecramps 611773 Schizencephaly 269160 Variable phenotype - porencephaly,destructive cerebral lesions, eyeanomalies, intracerebral calcification
COL4A2 2203 NM_001846.4 2-48 PORENCEPHALY 2
COL4A3 2204 NM_000091.5 1-52 Alport syndrome, autosomal recessive,203780 Hematuria, benign familial, 141200 Alport syndrome, autosomal dominant,104200

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 44/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
COL4A3BP 2205 NM_001130105.1 1-18 INTELLECTUAL DISABILITY
COL4A4 2206 NM_000092.5 2-48 Alport syndrome, autosomal recessive,203780 Hematuria, familial benign
COL6A1 2211 NM_001848.3 1-35 Bethlem myopathy, 158810 Ullrich congenital muscular dystrophy,254090
COL6A2 2212 NM_001849.4 2-28 Bethlem myopathy, 158810 Ullrich congenital muscular dystrophy,254090
COL6A3 2213 NM_004369.4 2-44 Dystonia 27, 616411 Bethlem myopathy, 158810 Ullrich congenital muscular dystrophy,254090
COL9A1 2217 NM_001851.5 1-38 Epiphyseal dysplasia, multiple, 6 614135 Stickler syndrome, type IV(ophthalmological: myopia, retinaldetachment and cataracts, orofacial:micrognathia, midface hypoplasia andcleft palate, auditory:sensorineuralhearing loss and articular: epiphysealdysplasia) symptoms Cleft palate
COL9A2 2218 NM_001852.4 1-32 Epiphyseal dysplasia, multiple, 2 600204 Stickler syndrome, type V 614284 Cleft palate
COL9A3 2219 NM_001853.4 1-32 Epiphyseal dysplasia, multiple, withmyopathy Stickler syndrome type VI multiple epiphyseal dysplasia 3, with orwithout myopathy - 600969
COLEC10 2220 NM_006438.5 1-6 3MC SYNDROME 3
COLEC11 17213 NM_024027.5 2-7 3MC SYNDROME 2

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 45/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
COLQ 2226 NM_005677.4 1-17 Myasthenic syndrome, congenital, 5,603034
COMP 2227 NM_000095.3 1-19 Epiphyseal dysplasia, multiple, 1 132400 Pseudoachondroplasia 177170
COQ2 25223 NM_015697.8 1-7 Disorders of CoQ10 biosynthesis(Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly)) General Leukodystrophy &Mitochondrial Leukoencephalopathy
COQ4 19693 NM_016035.5 1-7 Disorders of CoQ10 biosynthesis(Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly))
COQ5 28722 NM_032314.4 1-7 No OMIM phenotype
COQ6 20233 NM_182476.3 1-12 Steroid-resistant nephrotic syndrome Disorders of CoQ10 biosynthesis(Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly))
COQ7 2244 NM_016138.5 1-6 ?Coenzyme Q10 deficiency, primary, 8616733
COQ9 25302 NM_020312.4 1-9 Disorders of CoQ10 biosynthesis(Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly)) General Leukodystrophy &Mitochondrial Leukoencephalopathy
COX10 2260 NM_001303.4 6 1-7 Encephalopathy, progressivemitochondrial, with proximal renaltubulopathy due to cytochromecoxidase deficiency Mitochondrial Respiratory ChainComplex IV Deficiency Leigh syndrome due to mitochondrialCOX4 deficiency OMIM

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 46/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
COX11 2261 NM_004375.5 3-4 1-4 No OMIM phenotype
COX14 28216 NM_032901.4 2 Complex IV (Mitochondrial respiratorychain disorders (caused by nuclearvariants only), OXPHOS assemblyfactors)
COX15 2263 NM_004376.7 1-9 Leigh syndrome due to cytochrome coxidase deficiency, 256000 Cardioencephalomyopathy, fatalinfantile, due to cytochrome c oxidasedeficiency 2, 615119
COX16 20213 NM_016468.7 1-4 No OMIM phenotype
COX17 2264 NM_005694.1 1-2 No OMIM phenotype
COX18 26801 NM_173827.4 1-6 No OMIM phenotype
COX19 28074 NM_001031617.3 1-3 No OMIM phenotype
COX20 26970 NM_198076.6 1-4 Mitochondrial complex IV deficiency,220110
COX4I2 16232 NM_032609.3 2-5 Exocrine pancreatic insufficiency,dyserythropoietic anemia, and calvarialhyperostosis 612714
COX6A1 2277 NM_004373.4 1-3 Charcot-Marie-Tooth disease, recessiveintermediate D, 616039
COX6A2 2279 NM_005205.4 1-3 Mitochondrial complex IV deficiency
COX6B1 2280 NM_001863.5 2-4 Mitochondrial Respiratory ChainComplex IV Deficiency
COX6B2 24380 NM_144613.5 2-4 No OMIM phenotype

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 47/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
COX7B 2291 NM_001866.3 1-3 Complex IV (Mitochondrial respiratorychain disorders (caused by nuclearvariants only), OXPHOS structuralsubunits) Linear skin defects with multiplecongenital anomalies Aplasia cutis congenita, reticulolinear,with microcephaly, facial dysmorphismand other congenital anomalies, 300887
CP 2295 NM_000096.4 19 1-19 Cerebellar ataxia, 604290 Hemosiderosis, systemic, due toaceruloplasminemia, 604290
CPA6 17245 NM_020361.5 1-11 Epilepsy, familial temporal lobe, 5614417 AR, AD Febrile seizures, familial, 11 614418
CPAMD8 23228 NM_015692.5 16-17 1-42 Anterior Segment Dysgenesis
CPOX 2321 NM_000097.7 1-7 Harderoporphyria 121300 Coproporphyria 121300
CPS1 2323 NM_001875.5 1-38 Carbamoylphosphate synthetase Ideficiency (Urea cycle disorders andinherited hyperammonaemias)
CPT1A 2328 NM_001876.4 2-19 Carnitine palmitoyltransferase I (CPTI)deficiency (Disorders of carnitinetransport and the carnitine cycle)
CPT1C 18540 NM_001136052.2 3-20 ?Spastic paraplegia 73, autosomaldominant, 616282, AD
CPT2 2330 NM_000098.3 1-5 Carnitine palmitoyltransferase II (CPTII)deficiency (Disorders of carnitinetransport and the carnitine cycle)
CRADD 2340 NM_003805.5 2-3 Mental retardation, autosomalrecessive 34, with variant lissencephaly614499

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 48/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CRB1 2343 NM_201253.3 1-12 Retinitis pigmentosa-12, autosomalrecessive, 600105 Leber congenital amaurosis 8, 613835 Pigmented paravenous chorioretinalatrophy, 172870
CRB2 18688 NM_173689.7 1-13 VENTRICULOMEGALY WITH CYSTICKIDNEY DISEASE
CRBN 30185 NM_016302.3 1-11 Mental retardation, autosomalrecessive 2 607417
CREB3L1 18856 NM_052854.4 1-12 Osteogenesis imperfecta, type XVI616229
CREBBP 2348 NM_004380.3 1-31 CREBBP intellectual disability withouttypical RTS features Rubinstein-Taybi syndrome, 180849
CRELD1 14630 NM_015513.6 1-10 HETEROTAXY SYNDROME 207574
CRIPT 14312 NM_014171.6 1-5 Short stature with microcephaly anddistinctive facies, 615789
CRLF1 2364 NM_004750.5 1-9 Cold-induced sweating syndrome 1272430
CRTAP 2379 NM_006371.5 1-7 Osteogenesis imperfecta, type VII610682
CRX 2383 NM_000554.6 2-4 Cone-rod retinal dystrophy-2, 120970 Leber congenital amaurosis 7, 613829
CRYAA 2388 NM_000394.4 1-3 Cataract 9, multiple types, 604219
CRYAB 2389 NM_001885.3 2-4 Myopathy, myofibrillar 2, 608810 Cataract 16, multiple types, 613763
CRYBA1 2394 NM_005208.4 1-6 Cataract 10, multiple types, 600881
CRYBA4 2396 NM_001886.3 2-6 CATARACT ZONULAR TYPE 2 610425 MICROPHTHALMIA ISOLATED WITHCATARACT TYPE 4 610426
2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 49/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
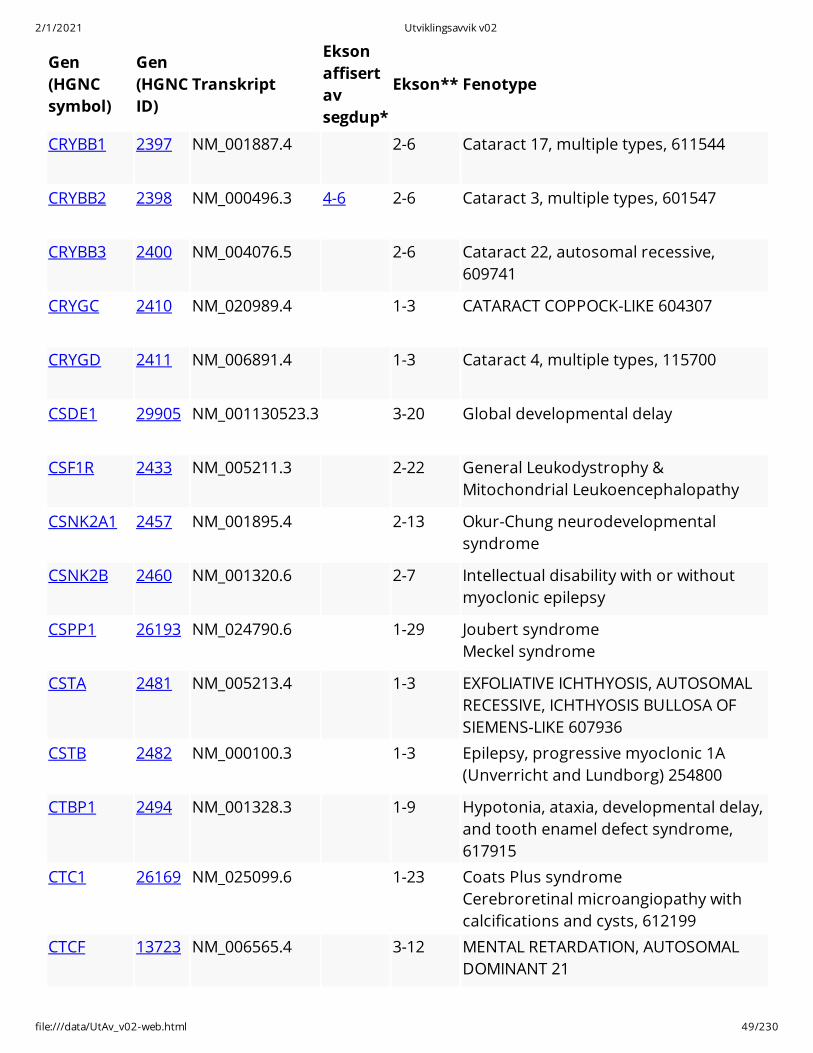
CRYBB1 2397 NM_001887.4 2-6 Cataract 17, multiple types, 611544
CRYBB2 2398 NM_000496.3 4-6 2-6 Cataract 3, multiple types, 601547
CRYBB3 2400 NM_004076.5 2-6 Cataract 22, autosomal recessive,609741
CRYGC 2410 NM_020989.4 1-3 CATARACT COPPOCK-LIKE 604307
CRYGD 2411 NM_006891.4 1-3 Cataract 4, multiple types, 115700
CSDE1 29905 NM_001130523.3 3-20 Global developmental delay
CSF1R 2433 NM_005211.3 2-22 General Leukodystrophy &Mitochondrial Leukoencephalopathy
CSNK2A1 2457 NM_001895.4 2-13 Okur-Chung neurodevelopmentalsyndrome
CSNK2B 2460 NM_001320.6 2-7 Intellectual disability with or withoutmyoclonic epilepsy
CSPP1 26193 NM_024790.6 1-29 Joubert syndrome Meckel syndrome
CSTA 2481 NM_005213.4 1-3 EXFOLIATIVE ICHTHYOSIS, AUTOSOMALRECESSIVE, ICHTHYOSIS BULLOSA OFSIEMENS-LIKE 607936
CSTB 2482 NM_000100.3 1-3 Epilepsy, progressive myoclonic 1A(Unverricht and Lundborg) 254800
CTBP1 2494 NM_001328.3 1-9 Hypotonia, ataxia, developmental delay,and tooth enamel defect syndrome,617915
CTC1 26169 NM_025099.6 1-23 Coats Plus syndrome Cerebroretinal microangiopathy withcalcifications and cysts, 612199
CTCF 13723 NM_006565.4 3-12 MENTAL RETARDATION, AUTOSOMALDOMINANT 21

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 50/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CTDP1 2498 NM_004715.4 1-13 Congenital cataracts, facialdysmorphism, and neuropathy, 604168
CTH 2501 NM_001902.6 1-12 Cystathioninuria, 219500
CTNNA2 2510 NM_001164883.1 2-17 Cortical dysplasia, complex, with otherbrain malformations 9, 618174
CTNNB1 2514 NM_001904.4 2-15 Mental retardation, autosomaldominant 19, 615075
CTNND1 2515 NM_001085458.2 21 3-21 Cleft palate Blepharo-cheiro-dontic syndrome
CTNS 2518 NM_004937.3 3-12 Cystinosis, late-onset juvenile oradolescent nephropathic, 219900
CTSA 9251 NM_000308.4 2-15 Galactosialidosis
CTSC 2528 NM_001814.6 1-7 Haim-Munk syndrome 245010 Papillon-Lefevre syndrome 245000 Periodontitis 1, juvenile 170650
CTSD 2529 NM_001909.5 1-9 Ceroid lipofuscinosis, neuronal, 10OMIM
CTSF 2531 NM_003793.4 1-13 Ceroid lipofuscinosis, neuronal, 13, Kufstype OMIM
CTSK 2536 NM_000396.4 2-8 Pycnodysostosis
CTU2 28005 NM_001012759.3 1-15 Microcephaly, facial dysmorphism, renalagenesis, and ambiguous genitaliasyndrome, 618142
CUBN 2548 NM_001081.4 41-50,61-67
1-67 Intrinsic factor receptor deficiency dueto CUBN mutations (Disorders ofcobalamin absorption, transport andmetabolism) Megaloblastic anemia-1, Finnish type Proteinuric renal disease
CUL3 2553 NM_003590.5 1-16 CUL3-related developmental disorder(monoallelic)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 51/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CUL4B 2555 NM_003588.3 2-22 Mental retardation, X-linked, syndromic15 (Cabezas type), 300354
CUL7 21024 NM_014780.4 2-26 3-M syndrome 1 273750
CUX1 2557 NM_001202543.2 3 1-24 Global developmental delay with orwithout impaired intellectualdevelopment, 618330
CUX2 19347 NM_015267.4 1-22 Epileptic encephalopathy, early infantile,67, 618141
CWC27 10664 NM_005869.4 1-14 Retinitis pigmentosa, skeletal anomaliesand intellectual disability
CWF19L1 25613 NM_018294.6 1-14 Spinocerebellar ataxia, autosomalrecessive 17, 616127 intellectual disability, developmentaldelay
CYB5R3 2873 NM_000398.7 1-9 Methemoglobinemia, type I,250800Methemoglobinemia, type II,250800
CYC1 2579 NM_001916.5 1-7 Mitochondrial complex III deficiency,nuclear type 6, 615453
CYCS 19986 NM_018947.6 2-3 2-3 Thrombocytopenia 4, 612004
CYFIP2 13760 NM_001037333.3 2-31 Epileptic encephalopathy, early infantile65, 618008
CYP1B1 2597 NM_000104.3 2-3 Glaucoma 3A, primary open angle,congenital, juvenile, or adult onset,231300 Peters anomaly, 604229
CYP26B1 20581 NM_019885.3 1-6 Craniosynostosis with radiohumeralfusions and other skeletal andcraniofacial anomalies, 614416
CYP27A1 2605 NM_000784.4 1-9 Severe neonatal cholestasis Cerebrotendinous xanthomatosis General Leukodystrophy &Mitochondrial Leukoencephalopathy

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 52/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
CYP27B1 2606 NM_000785.4 1-9 Vitamin D-dependent rickets, type I264700
CYP2R1 20580 NM_024514.4 1-5 Rickets due to defect in vitamin D 25-hydroxylation, 600081
CYP2U1 20582 NM_183075.3 1-5 Spastic paraplegia 56, autosomalrecessive
CYP7B1 2652 NM_004820.5 1-6 Neonatal and Adult Cholestasis Bile acid synthesis defect, congenital, 3 Spastic paraplegia 5A, autosomalrecessive 270800
D2HGDH 28358 NM_152783.5 2-10 D-2-hydroxyglutaric aciduria
DAG1 2666 NM_004393.6 2-3 Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies), type A, 9616538
DARS 2678 NM_001349.4 1-16 Hypomyelination with brainstem andspinal cord involvement and legspasticity
DARS2 25538 NM_018122.5 1-17 Leukoencephalopathy with brain stemand spinal cord involvement and lactateelevation, 611105 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis)
DBH 2689 NM_000787.4 1-12 Dopamine beta-hydroxylase deficiency
DBT 2698 NM_001918.4 1-11 Dihydrolipoamide branched chaintransacylase deficiency (Maple syrupurine disease, disorder of branched-chain amino acid metabolism notclassified as organic aciduria) Maple syrup urine disease, type II
DCAF17 25784 NM_025000.4 1-14 Woodhouse-Sakati syndrome, 241080

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 53/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
DCC 2701 NM_005215.4 1-29 Gaze palsy, familial horizontal, withprogressive scoliosis, 2 Midline-bridging neuronal commissuredisruption, horizontal gaze palsy,scoliosis, and intellectual disability
DCDC2 18141 NM_016356.5 1-10 Neonatal and Adult Cholestasis Sclerosing cholangitis, neonatal, 617394
DCHS1 13681 NM_003737.4 2-21 PERIVENTRICULAR NEURONALHETEROTOPIA Van Maldergem syndrome 1, 601390
DCPS 29812 NM_014026.6 1-6 Al-Raqad syndrome, 616459
DCX 2714 NM_178153.3 2-7 Lissencephaly, X-linked 300067 Subcortical laminal heterotopia, X-linked300067
DCXR 18985 NM_016286.4 1-8 [Pentosuria] 260800
DDB2 2718 NM_000107.2 1-10 Xeroderma pigmentosum, group E,DDB-negative subtype, 278740
DDC 2719 NM_000790.4 2-14 Aromatic L-amino acid decarboxylasedeficiency Dystonia
DDHD1 19714 NM_001160147.2 1-13 Spastic paraplegia 28, autosomalrecessive, 609340
DDHD2 29106 NM_015214.3 2-17 Spastic paraplegia 54, autosomalrecessive, 615033
DDOST 2728 NM_005216.4 1-11 CONGENITAL DISORDER OFGLYCOSYLATION, TYPE IR
DDR2 2731 NM_006182.4 3-18 Spondylometaepiphyseal dysplasia,short limb-hand type 271665
DDX11 2736 NM_030653.4 2-27 2-27 WARSAW BREAKAGE SYNDROME (WBRS)
DDX3X 2745 NM_001193416.3 1-17 Mental retardation, X-linked 102, 300958

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 54/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
DDX53 20083 NM_182699.4 1
DDX59 25360 NM_001031725.6 2-8 Orofaciodigital syndrome V, 174300
DDX6 2747 NM_004397.6 2-13 Generalized hypotonia Global developmental delay Intellectual disability
DEAF1 14677 NM_021008.3 1-12 ?Dyskinesia, seizures, and intellectualdevelopmental disorder, 617171
DEGS1 13709 NM_003676.4 1-3 Leukodystrophy hypomyelinating 18,MIM 618404) Leukodystrophy hypomyelinating 18,618404
DENND5A 19344 NM_015213.4 1-23 Epileptic encephalopathy, early infantile,49
DEPDC5 18423 NM_001242896.3 2-43 Epilepsy, familial focal, with variable foci1 604364
DES 2770 NM_001927.4 1-9 Muscular dystrophy, limb-girdle, type2R, 615325
DGUOK 2858 NM_080916.3 1-7 Mitochondrial DNA depletion syndrome3 (hepatocerebral type), 251880 Deoxyguanosine kinase deficiency(Disorders of purine metabolism) General Leukodystrophy &Mitochondrial Leukoencephalopathy
DHCR24 2859 NM_014762.4 1-9 Desmosterolosis, 602398
DHCR7 2860 NM_001360.2 3-9 Smith-Lemli-Opitz syndrome, 270400 Cataracts
DHDDS 20603 NM_024887.3 2-9 Developmental delay and seizures withor without movement abnormalities,617836 Retinitis pigmentosa 59 613861 ?Congenital disorder of glycosylation,type 1bb 613861

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 55/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
DHFR 2861 NM_000791.4 6 1-6 Megaloblastic anemia due todihydrofolate reductase deficiency,613839 Dihydrofolate reductase deficiency(Disorders of folate metabolism andtransport)
DHODH 2867 NM_001361.5 1-9 Miller syndrome (postaxial acrofacialdysostosis) 263750
DHPS 2869 NM_001930.4 1-9 Abnormal muscle tone, Globaldevelopmental delay, Intellectualdisability, Seizures, EEG abnormality,Behavioral abnormality, Abnormality ofhead or neck
DHTKD1 23537 NM_018706.7 1-17 Charcot-Marie-Tooth disease, axonal,type 2Q, 615025 2-Oxoadipic aciduria (Disorders ofhistidine, tryptophan or lysinemetabolism)
DHX30 16716 NM_138615.3 3-22 microcephaly, developmental delayintellectual disability, mild cerebralvolume loss, hypotonia, seizures, shortstature, failure to thrive, andgeneralized hirsutism
DIAPH1 2876 NM_005219.5 1-28 Seizures, cortical blindness,microcephaly syndrome, 616632
DIP2B 29284 NM_173602.3 1-38 Mental retardation, FRA12A type,136630
DIS3L2 28648 NM_152383.4 15-21 2-21 PERLMAN SYNDROME 267000
DISP1 19711 NM_032890.5 4-10 Holoprosencephaly
DKC1 2890 NM_001363.5 1-15 DYSKERATOSIS CONGENITA, X-LINKED,305000
DLAT 2896 NM_001931.5 1-14 Dihydrolipoyl transacetylase deficiency(Disorders of pyruvate metabolism) Dystonia

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 56/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
DLD 2898 NM_000108.5 1-14 Dihydrolipoyl dehydrogenase deficiency(Disorders of pyruvate metabolism) Leigh syndrome
DLG1 2900 NM_001098424.1 2-26
DLG2 2901 NM_001142699.1 3-28
DLG3 2902 NM_021120.4 1-19 Mental retardation, X-linked 90, 300850
DLG4 2903 NM_001365.4 1-22 Intellectual disability Marfanoid habitus
DLL1 2908 NM_005618.4 1-11 Neurodevelopmental disorder withnonspecific brain abnormalities andwith or without seizures OMIM
DLL3 2909 NM_016941.4 1-8 Spondylocostal dysostosis 1, autosomalrecessive, 277300
DLL4 2910 NM_019074.4 1-11 Adams-Oliver syndrome 6, 616589
DLX3 2916 NM_005220.3 1-3 Amelogenesis imperfecta, type IV104510 Trichodontoosseous syndrome 190320
DLX4 2917 NM_138281.3 1-3 ?Orofacial cleft 15, 616788
DLX5 2918 NM_005221.6 1-3 Split-hand/foot malformation 1 withsensorineural hearing loss, 220600
DMD 2928 NM_004006.2 1-79 CARDIOMYOPATHY DILATED X-LINKEDTYPE 3B 302045 Duchenne muscular dystrophy, 310200 Becker muscular dystrophy, 300376

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 57/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
DMP1 2932 NM_004407.4 2-6 Multiple synostoses syndrome 2,610017 Chondrodysplasia, Grebe type, 200700 Brachydactyly, type C, 113100 Hypophosphatemic rickets, AR, 241520 Brachydactyly, type A1, C, 615072 Brachydactyly, type A2, 112600 Acromesomelic dysplasia, Hunter-Thompson type, 201250 Du Pan syndrome, 228900 Symphalangism, proximal, 1B, 615298
DMPK 2933 NM_001081563.2 1-14 Myotonic dystrophy 1, 16090
DMXL2 2938 NM_001174116.2 1-43 Epileptic encephalopathy, early infantile,81, 618663 ?Polyendocrine-polyneuropathysyndrome, 616113 Sensorineural Hearing Loss
DNA2 2939 NM_001080449.3 1-21 Progressive external ophthalmoplegiawith mitochondrial DNA deletions,autosomal dominant, 6
DNAAF3 30492 NM_001256714.1 1-12 Ciliary dyskinesia, primary, 2, 606763
DNAH5 2950 NM_001369.2 1-79 CILIARY DYSKINESIA, PRIMARY, 3
DNAH9 2953 NM_001372.4 1-69 Motile Cilia Defects and Situs Inversus
DNAJB6 14888 NM_058246.4 2-10 Muscular dystrophy, limb-girdle, type1E, 603511
DNAJC12 28908 NM_021800.3 1-5 Hyperphenylalaninemia, mild, non-BH4-deficient, 617384
DNAJC19 30528 NM_145261.4 1-6 3-methylglutaconic aciduria, type V
DNAJC5 16235 NM_025219.3 2-5 Ceroid lipofuscinosis, neuronal, 4, Parrytype OMIM
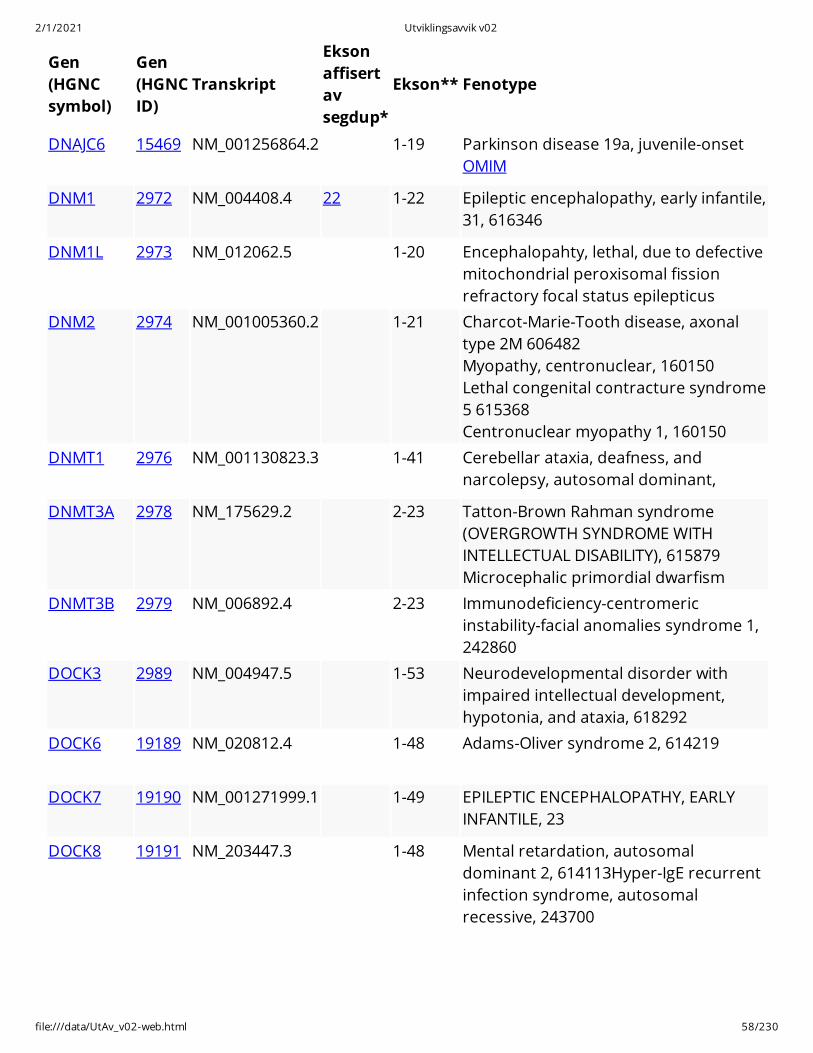
2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 58/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
DNAJC6 15469 NM_001256864.2 1-19 Parkinson disease 19a, juvenile-onsetOMIM
DNM1 2972 NM_004408.4 22 1-22 Epileptic encephalopathy, early infantile,31, 616346
DNM1L 2973 NM_012062.5 1-20 Encephalopahty, lethal, due to defectivemitochondrial peroxisomal fission refractory focal status epilepticus
DNM2 2974 NM_001005360.2 1-21 Charcot-Marie-Tooth disease, axonaltype 2M 606482 Myopathy, centronuclear, 160150 Lethal congenital contracture syndrome5 615368 Centronuclear myopathy 1, 160150
DNMT1 2976 NM_001130823.3 1-41 Cerebellar ataxia, deafness, andnarcolepsy, autosomal dominant,
DNMT3A 2978 NM_175629.2 2-23 Tatton-Brown Rahman syndrome(OVERGROWTH SYNDROME WITHINTELLECTUAL DISABILITY), 615879 Microcephalic primordial dwarfism
DNMT3B 2979 NM_006892.4 2-23 Immunodeficiency-centromericinstability-facial anomalies syndrome 1,242860
DOCK3 2989 NM_004947.5 1-53 Neurodevelopmental disorder withimpaired intellectual development,hypotonia, and ataxia, 618292
DOCK6 19189 NM_020812.4 1-48 Adams-Oliver syndrome 2, 614219
DOCK7 19190 NM_001271999.1 1-49 EPILEPTIC ENCEPHALOPATHY, EARLYINFANTILE, 23
DOCK8 19191 NM_203447.3 1-48 Mental retardation, autosomaldominant 2, 614113Hyper-IgE recurrentinfection syndrome, autosomalrecessive, 243700

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 59/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
DOK7 26594 NM_173660.5 1-7 Limb-girdle muscular dystrophy Fetal akinesia deformation sequence,208150 Myasthenic syndrome, congenital, 10,254300
DOLK 23406 NM_014908.4 1 Congenital disorder of glycosylation,type Im 610768 Dolichol kinase deficiency (Disorders ofmultiple glycosylation and otherglycosylation pathways)
DONSON 2993 NM_017613.4 1-10 Microcephaly, short stature, and limbabnormalities 617604
DPAGT1 2995 NM_001382.4 1-9 Myasthenic syndrome, congenital, 13,with tubular aggregates 614750 UDP-GlcNAc:Dol-P-GlcNac-P transferasedeficiency (Disorders of protein N-glycosylation) DPAGT1-CDG 300129
DPF2 9964 NM_006268.5 11 1-11 Coffin-Siris syndrome 7, 618027
DPH1 3003 NM_001383.5 1-12 Developmental delay with short stature,dysmorphic features, and sparse hair,616901
DPM1 3005 NM_003859.2 1-9 Congenital disorder of glycosylation,type Ie, 608799 GDP-Man:Dol-P mannosyltransferasedeficiency (Disorders of multipleglycosylation and other glycosylationpathways)
DPM2 3006 NM_003863.3 1-4 DPM2-CDG . Musclular dystrophydystroglycanopathy syndrome withsevere epilepsy.
DPM3 3007 NM_153741.2 2 Muscular dystrophy-dystroglycanopathy (limb-girdle), type C,15 612937
DPP6 3010 NM_001936.5 1-26 Mental retardation, autosomaldominant 33, 616311 primary microcephaly

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 60/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
DPYD 3012 NM_000110.4 1-23 Dihydropyrimidine dehydrogenasedeficiency (Disorders of pyrimidinemetabolism) 5-fluorouracil toxicity 274270
DPYS 3013 NM_001385.3 1-9 Dihydropyrimidinase deficiency(Disorders of pyrimidine metabolism)
DRC1 24245 NM_145038.5 1-17 PRIMARY CILARY DYSKINEASIA 244400
DSG1 3048 NM_001942.4 1-15 SEVERE DERMATITIS, MULTIPLEALLERGIES AND METABOLIC WASTING,615508
DSPP 3054 NM_014208.3 2-5 Dentinogenesis imperfecta, Shields typeII, 125490 Deafness, autosomal dominant 36, withdentinogenesis, 605594
DSTYK 29043 NM_015375.3 1-13 Spastic paraplegia 23, 270750 Congenital anomalies of kidney andurinary tract
DVL1 3084 NM_004421.3 1-15 Robinow syndrome, autosomaldominant 2 616331
DVL3 3087 NM_004423.4 1-15 Robinow syndrome, autosomaldominant 3, 616894
DYM 21317 NM_017653.5 2-17 Encephalopahty, lethal, due to defectivemitochondrial peroxisomal fission,614388 Dyggve-Melchior-Clausen disease,223800 Smith-McCort dysplasia, 607326
DYNC1H1 2961 NM_001376.5 1-78 Charcot-Marie-Tooth disease, axonal,type 20, 614228 Mental retardation, autosomaldominant 13, 614563 Spinal muscular atrophy, lowerextremity-predominant, AD, 158600 Early-onset epilepsy Late-onset epilepsy Focal seizures

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 61/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
DYNC1I2 2964 NM_001271788.2 2-16 Abnormality of nervous systemmorphology Abnormality of head or neck Microcephaly Intellectual disability
DYNC2H1 2962 NM_001080463.2 1-90 Short-rib thoracic dysplasia 3 with orwithout polydactyly Thoracic and CranioectodermalDysplasia (Skeletal Ciliopathy) 15 GenePanel Jeune syndrome
DYNC2LI1 24595 NM_016008.4 1-13 Short-rib throacic dysplasia 15 withpolydactyly, 617088
DYRK1A 3091 NM_001396.4 2-12 Mental retardation, autosomaldominant 7, 614104
DYSF 3097 NM_003494.4 1-55 Muscular dystrophy, limb-girdle, type 2B253601 Myopathy, distal, with anterior tibialonset, 606768
DYX1C1 21493 NM_001033560.1 2-9 Ciliary dyskinesia, primary, 25, 615482
EARS2 29419 NM_001083614.2 1-9 Combined oxidative phosphorylationdeficiency 12, 614924 Leukoencephalopathy with thalamusand brainstem involvement and highlactate (LTBL)
EBF3 19087 NM_001005463.3 1-16 Hypotonia, ataxia, and delayeddevelopment syndrome 617330 Intellectual Disability, Ataxia, and FacialDysmorphism
EBP 3133 NM_006579.3 2-5 Chondrodysplasia punctata, X-linkeddominant 302960 MEND syndrome
ECEL1 3147 NM_004826.4 2-18 Arthrogryposis, distal, type 5D, 615065
ECHS1 3151 NM_004092.4 1-8 Mitochondrial short-chain enoyl-CoAhydratase 1 deficiency

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 62/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
EDA 3157 NM_001399.5 1-8 Ectodermal dysplasia 1, hypohidrotic, X-linked, 305100 Tooth agenesis, selective, X-linked 1,313500
EDAR 2895 NM_022336.4 2-12 Ectodermal dysplasia 10B,hypohidrotic/hair/tooth type,autosomal recessive
EDN1 3176 NM_001955.5 1-5 AURICULOCONDYLAR SYNDROME602483
EDNRA 3179 NM_001957.4 2-8 MANDIBULOFACIAL DYSOSTOSIS WITHALOPECIA Cleft palate
EDNRB 3180 NM_000115.5 2-8 ABCD SYNDROME 600501
EED 3188 NM_003797.5 1-12 Cohen-Gibson syndrome, 617561 Overgrowth with Intellectual disability Weaver-like overgrowth syndrome
EEF1A2 3192 NM_001958.5 2-8 Epileptic encephalopathy, early infantile,33 616409
EEF1B2 3208 NM_021121.3 2-7 AUTOSOMAL RECESSIVE MENTALRETARDATION
EFHC1 16406 NM_018100.4 1-11 {Myoclonic epilepsy, juvenile,susceptibility to, 1} 254770
EFNB1 3226 NM_004429.4 1-5 Craniofrontonasal dysplasia 304110
EFTUD2 30858 NM_004247.4 2-28 Mandibulofacial dysostosis, Guion-Almeida type OMIM
EGR2 3239 NM_000399.5 1-2 NEUROPATHY, CONGENITALHYPOMYELINATING, 1 605253
EHMT1 24650 NM_024757.5 1-27 Kleefstra syndrome
EIF2AK3 3255 NM_004836.7 1-17 Wolcott-Rallison syndrome, 226980

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 63/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
EIF2AK4 19687 NM_001013703.4 1-39 Pulmonary venoocclusive disease 2,234810 Heritable pulmonary arterialhypertension
EIF2B1 3257 NM_001414.4 1-9 Childhood ataxia with central nervoussystem hypomyelination/vanishingwhite matter disease General Leukodystrophy &Mitochondrial Leukoencephalopathy
EIF2B2 3258 NM_014239.4 1-8 Childhood ataxia with central nervoussystem hypomyelination/vanishingwhite matter disease General Leukodystrophy &Mitochondrial Leukoencephalopathy Ovarioleukodystrophy, 603896
EIF2B3 3259 NM_020365.5 2-12 Childhood ataxia with central nervoussystem hypomyelination/vanishingwhite matter disease General Leukodystrophy &Mitochondrial Leukoencephalopathy
EIF2B4 3260 NM_015636.3 1-13 Childhood ataxia with central nervoussystem hypomyelination/vanishingwhite matter disease Ovarioleukodystrophy, 603896
EIF2B5 3261 NM_003907.3 1-16 Childhood ataxia with central nervoussystem hypomyelination/vanishingwhite matter disease Ovarioleukodystrophy 603896 General Leukodystrophy &Mitochondrial Leukoencephalopathy
EIF2S3 3267 NM_001415.4 12 1-12 Mental retardation, X-linked, syndromic,Borck type, 300987
EIF3F 3275 NM_003754.3 1-8 Intellectual disability Seizures Behavioral abnormality Sensorineural hearing impairment

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 64/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
EIF4A3 18683 NM_014740.4 1-12 Robin sequence with cleft mandible andlimb anomalies, 268305 Richieri-Costa-Pereira syndrome
ELAC2 14198 NM_018127.7 1-24 infantile hypertrophic cardiomyopathy,lactic acidosis, and isolated complex Ideficiency
ELMO2 17233 NM_133171.5 3-11 3-22 Intraosseous Vascular Malformation
ELN 3327 NM_001278939.1 1-34 Supravalvar aortic stenosis, 185500 Cutis laxa, AD, 123700
ELOVL4 14415 NM_022726.4 1-6 Spinocerebellar ataxia 34 Ichthyosis, spastic quadriplegia, andmental retardation, 614457
ELP2 18248 NM_001242875.3 1-23 Mental retardation, autosomalrecessive 58 617270
EMC1 28957 NM_015047.3 1-23 Global Developmental Delay, Hypotonia,Scoliosis, and Cerebellar Atrophy.
EMD 3331 NM_000117.3 1-6 Emery-Dreifuss muscular dystrophy 1,X-linked 310300
EMG1 16912 NM_006331.8 1-7 Bowen-Conradi syndrome
EML1 3330 NM_004434.3 1-22 congenital hydrocephalus, profoundglobal developmental delay andintractable epilepsy Band heterotopia, 600348
EMX2 3341 NM_004098.4 1-3 Schizencephaly, 269160
ENG 3349 NM_000118.3 1-14 Hereditary hemorrhagic telangiectasia
ENO3 3354 NM_053013.4 2-12 ?Glycogen storage disease XIII

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 65/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ENPP1 3356 NM_006208.3 1-25 Hypophosphatemic rickets, autosomalrecessive, 2 613312 Arterial calcification, generalized, ofinfancy, 1 208000 Cole disease 615522
ENTPD1 3363 NM_001776.6 1-10 Spasticparaplegia 64, 615683
EOGT 28526 NM_001278689.2 4-18 Adams Oliver syndrome 4
EP300 3373 NM_001429.4 1-31 RUBINSTEIN-TAYBI SYNDROME TYPE 2613684
EPB41L1 3378 NM_012156.2 2-22 ?Mental retardation, autosomaldominant 11 614257
EPG5 29331 NM_020964.3 1-44 IMMUNODEFICIENCY WITH CLEFTLIP/PALATE, CATARACT,HYPOPIGMENTATION, AND ABSENTCORPUS CALLOSUM Vici syndrome, 242840
EPHA4 3388 NM_004438.5 1-17
EPM2A 3413 NM_005670.4 1-4 Epilepsy, progressive myoclonic 2A(Lafora)
EPRS 3418 NM_004446.3 1-32 Hypomyelinating Leukodystrophy
EPT1 29361 NM_033505.4 1-10 EPT1-related complex progressivehereditary spastic paraplegia
ERAL1 3424 NM_005702.4 1-10 Perrault syndrome 6, 617565
ERBB3 3431 NM_001982.3 1-28 Hirschprung disease with intestinalpseudo-obstruction Lethal congenital contracturalsyndrome 2 607598

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 66/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ERCC1 3433 NM_202001.3 1-8 FANCONI ANEMIA CEREBROOCULOFACIOSKELETALSYNDROME 4 610758 Xeroderma Pigmentosum
ERCC2 3434 NM_000400.3 1-23 CEREBRO-OCULO-FACIO-SKELETALSYNDROME TYPE 2 (COFS2) Xeroderma pigmentosum, group D,278730 Trichothiodystrophy, 601675
ERCC3 3435 NM_000122.2 1-15 Xeroderma pigmentosum, group B,610651 Trichothiodystrophy, 601675
ERCC4 3436 NM_005236.3 1-11 Radial Ray abnormality PRIMORDIAL DWARFISM 615272 Xeroderma pigmentosum, typeF/Cockayne syndrome, 278760 Fanconi anemia, complementationgroup Q, 61527
ERCC5 3437 NM_000123.3 1-15 Cerebrooculofacioskeletal syndrome 3,616570 Xeroderma pigmentosum, groupG/Cockayne syndrome, 278780
ERCC6 3438 NM_000124.4 2-21 CEREBRO-OCULO-FACIO-SKELETALSYNDROME TYPE 1 214150 De Sanctis-Cacchione syndrome Cockayne syndrome UV-sensitive syndrome Intercranial Calcifications General Leukodystrophy &Mitochondrial Leukoencephalopathy
ERCC6L2 26922 NM_001010895.3 1-14 Bone marrow failure syndrome 2,615715
ERCC8 3439 NM_000082.3 1-12 Cockayne syndrome, type A, 216400(Microcephaly) UV-sensitive syndrome General Leukodystrophy &Mitochondrial Leukoencephalopathy

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 67/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ERF 3444 NM_006494.4 1-4 Craniosynostosis 4 600775 Chitayat syndrome: hyperphalangism,characteristic facies, hallux valgus andbronchomalacia
ERLIN1 16947 NM_006459.4 1-11 Spastic paraplegia 62, 615681
ERLIN2 1356 NM_007175.8 2-12 Spastic paraplegia, autosomal dominantSpastic paraplegia 18, autosomalrecessive, 611225
ERMARD 21056 NM_018341.3 1-18 ?Periventricular nodular heterotopia 6OMIM
ESCO2 27230 NM_001017420.3 2-11 Roberts syndrome, 268300 radial aplasia
ESRP2 26152 NM_024939.3 1-15 cleft lip
ETFA 3481 NM_000126.4 1-12 Electron transfer flavoproteindeficiency, alpha chain (Disorders ofmitochondrial fatty acid oxidation) Glutaric acidemia IIA
ETFB 3482 NM_001985.3 1-6 Glutaric acidemia IIB Electron transfer flavoproteindeficiency, beta chain (Disorders ofmitochondrial fatty acid oxidation)
ETFDH 3483 NM_004453.4 1-13 Glutaric acidemia IIC Disorders of ubiquinone metabolismand biosynthesis ETF-ubiquinone oxidoreductasedeficiency (Disorders of mitochondrialfatty acid oxidation) Mitochondrial Leukoencephalopathy
ETHE1 23287 NM_014297.5 1-7 Ethylmalonic encephalopathy, 602473 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Isolated complex IV deficiency
EVC 3497 NM_153717.3 1-21 Ellis-van Creveld Syndrome Weyers acrodental dysostosis, 193530

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 68/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
EVC2 19747 NM_147127.5 1-22 Ellis-van Creveld syndrome, 225500 Weyers acrofacial dysostosis, 193530
EXOSC3 17944 NM_016042.4 1-4 Pontocerebellar Hypoplasia
EXOSC8 17035 NM_181503.3 1-11 Pontocerebellar hypoplasia, type 1C616081
EXOSC9 9137 NM_001034194.2 1-13 Cerebellar Atrophy with Spinal MotorNeuronopathy
EXPH5 30578 NM_015065.3 1-6 INHERITED SKIN FRAGILITY 615028
EXT1 3512 NM_000127.2 1-11 Multiple exostoses type I (Disorders ofprotein O-glycosylation, O-xylosylglycansynthesis deficiencies) trichorhinophalangeal syndrome type 2-150230
EXT2 3513 NM_207122.1 2-14 Multiple exostoses type II (Disorders ofprotein O-glycosylation, O-xylosylglycansynthesis deficiencies) Seizures, scoliosis, and macrocephalysyndrome, 616682
EXTL3 3518 NM_001440.4 3-7 Immunoskeletal dysplasia withneurodevelopmental abnormalities617425
EYA1 3519 NM_000503.6 3-18 Branchiootorenal syndrome 1, with orwithout cataracts, 113650 Anterior segment anomalies with orwithout cataract, 113650
EZH2 3527 NM_004456.5 2-20 Weaver syndrome

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 69/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
FA2H 21197 NM_024306.5 1-7 Fatty acid 2-hydroxylase deficiency(Disorders of complex lipid synthesis) Early onset dystonia Neurodegeneration with brain ironaccumulation (NBIA) (Disorder of ironmetabolism) Dystonia Spastic paraplegia 35, autosomalrecessive 612319
FAAH2 26440 NM_174912.3 1-11
FAH 3579 NM_000137.3 1-14 Cholestasis Tyrosinemia, type I
FAM105B 25118 NM_138348.6 1-7 Otulin-related auto inflammatorysyndrome (ORAS)
FAM111A 24725 NM_022074.4 3-4 Gracile bone dysplasia 602361 Kenny-Caffey syndrome, type 2 127000
FAM120C 16949 NM_017848.6 1-16
FAM126A 24587 NM_032581.4 2-11 Leukodystrophy, hypomyelinating, 5,610532 Hypomyelination and CongenitalCataract
FAM134B 25964 NM_001034850.2 1-9 Neuropathy, hereditary sensory andautonomic, type IIB, 613115
FAM149B1 29162 NM_173348.2 1-14 Joubert syndrome oral-facial-digital syndrome
FAM161A 25808 NM_001201543.2 1-7 Retinitis pigmentosa 28, 606068
FAM20A 23015 NM_017565.4 1-11 Amelogenesis imperfecta, type IG(enamel-renal syndrome), 204690
FAM20C 22140 NM_020223.4 1-10 Raine syndrome, 259775
FAM46A 18345 NM_017633.3 2-3 Osteogenesis imperfecta, type XVIII617952

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 70/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
FAM58A 28434 NM_152274.5 1-7 STAR syndrome, 300707
FAM92A1 30452 NM_145269.5 8 1-8 postaxial polydactyly type A9
FANCA 3582 NM_000135.4 1-43 Fanconi anemia, complementationgroup A, 227650 Radial Ray abnormality
FANCB 3583 NM_001018113.3 3-10 Vacterl Association, X-Linked, With OrWithout Hydrocephalus Fanconi Anemia, ComplementationGroup B Radial Ray abnormality
FANCC 3584 NM_000136.3 2-15 Fanconi anemia, complementationgroup C, 227645 Radial Ray abnormality
FANCD2 3585 NM_033084.6 12-17,19-28
2-43 Fanconi anemia, complementationgroup D2, 227646 Radial Ray abnormality
FANCE 3586 NM_021922.3 1-10 Fanconi anemia, complementationgroup E, 600901 Radial Ray abnormality
FANCF 3587 NM_022725.4 1 Fanconi anemia, complementationgroup F, 603467 Radial Ray abnormality
FANCG 3588 NM_004629.1 1-14 Radial Ray abnormality Fanconi anemia, complementationgroup G, 614082
FANCI 25568 NM_001113378.1 2-38 Fanconi anemia, complementationgroup I, 609053
FANCL 20748 NM_018062.3 1-14 Radial Ray abnormality Fanconi anemia, complementationgroup L, 614083
FANCM 23168 NM_020937.4 1-23 FANCONI ANEMIA 229154

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 71/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
FAR1 26222 NM_032228.6 12 2-12 SEVERE INTELLECTUAL DISABILITY,EPILEPSY, AND CATARACTS Peroxisomal fatty acyl-CoA reductase 1disorder, 616154
FARS2 21062 NM_006567.5 2-7 Spastic paraplegia 77, autosomalrecessive, 617046 Combined oxidative phosphorylationdeficiency 14, 614946 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis)
FASTKD2 29160 NM_014929.3 2-12 Mitochondrial Respiratory ChainComplex IV Deficiency Combined oxidative phosphorylationdeficiency 44 OMIM
FAT4 23109 NM_024582.4 1-17 PERIVENTRICULAR NEURONALHETEROTOPIA HENNEKAM LYMPHANGIECTASIA-LYMPHEDEMA SYNDROME 2 616006 VAN MALDERGEM SYNDROME 615546
FBLN1 3600 NM_006486.3 1-17 Synpolydactyly, 3/3'4, associated withmetacarpal and metatarsal synostoses608180
FBN1 3603 NM_000138.4 2-66 SHPRINTZEN-GOLDBERGCRANIOSYNOSTOSIS SYNDROME 182212 MARFAN SYNDROME 154700 Geleophysic dysplasia 2 614185 Stiff skin syndrome 184900 Acromicric dysplasia 102370 Weill-Marchesani syndrome 2, dominant608328 MASS syndrome 604308 Marfan lipodystrophy syndrome 616914
FBN2 3604 NM_001999.4 1-65 Contractural arachnodactyly, congenital121050
FBP1 3606 NM_000507.4 1-7 Glycogen Storage Disease Fructose-1,6-bisphosphatase deficiency(Disorders of gluconeogenesis)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 72/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
FBXL3 13599 NM_012158.4 2-5 Intellectual developmental disorder withshort stature, facial anomalies, andspeech defects, 606220
FBXL4 13601 NM_012160.4 3-9 Mitochondrial DNA depletion syndrome13 (encephalomyopathic type), 615471 fatal encephalopathy, lactic acidosis,and severe MTDNA depletion in muscle. Seizures
FBXO11 13590 NM_001190274.2 1-23 cleft lip Intellectual developmental disorder withdysmorphic facies and behavioralabnormalities, 618089
FBXO7 13586 NM_012179.4 1-9 juvenile parkinsonism Dystonia
FBXW11 13607 NM_012300.2 1-12 Global developmental delay SYNDROMIC INTELLECTUAL DISABILITY612100
FBXW4 10847 NM_022039.4 7-9 1-9 SPLIT-HAND/FOOT MALFORMATIONTYPE 3
FDFT1 3629 NM_004462.5 1-8 Profound global developmental delay Intellectual disability Seizures Cortical visual impairment Abnormality of the skin Abnormality of the face
FDX1L 30546 NM_001031734.4 1-5 Mitochondrial myopathy, episodic, withoptic atrophy and reversibleleukoencephalopathy, 251900
FDXR 3642 NM_024417.5 1-12 Auditory neuropathy and optic atrophy617717
FECH 3647 NM_000140.4 1-11 Protoporphyria, erythropoietic, 1177000
FERMT3 23151 NM_031471.6 2-15 Leukocyte adhesion deficiency, type III612840
FEZF1 22788 NM_001024613.4 1-4 HYPOGONADOTROPIC HYPOGONADISMWITH OR WITHOUT ANOSMIA 616030

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 73/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
FGD1 3663 NM_004463.3 1-18 Aarskog-Scott syndrome 305400
FGF10 3666 NM_004465.2 1-3 Polydactyly Limb defects most often involved thethumbs, ranging from total aplasia tohypoplastic, digitalized, triphalangeal,and duplicated thumbs Aplasia of lacrimal and salivary glands,180920
FGF12 3668 NM_021032.4 1-5 Epileptic encephalopathy, early infantile,47, 617166
FGF14 3671 NM_004115.3 1-5 Spinocerebellar ataxia 27
FGF16 3672 NM_003868.3 1-2 Metacarpal 4-5 fusion, 309630
FGF23 3680 NM_020638.3 1-3 Hypophosphatemic rickets, autosomaldominant 193100 Osteomalacia, tumor-induced Tumoral calcinosis,hyperphosphatemic, familial 211900
FGF3 3681 NM_005247.4 1-3 Deafness, congenital with inner earagenesis, microtia, and microdontia,610706
FGF8 3686 NM_033163.4 1-6 Holoprosencephaly
FGF9 3687 NM_002010.3 1-3 Multiple synostoses syndrome 3 612961

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 74/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
FGFR1 3688 NM_023110.3 2-18 Jackson-Weiss syndrome Osteoglophonic dysplasia Pfeiffer syndrome Encephalocraniocutaneous lipomatosis,somatic mosaism, 613001 Hartsfield syndrome, 615465 Hypogonadotropic hypogonadism 2with or without anosmia, 147950 Trigonocephaly 1,190440 Polydactyly Kallmann syndrome 2
FGFR2 3689 NM_000141.4 2-18 Antley-Bixler syndrome without genitalanomalies or disorderedsteroidogenesis 207410 Apert syndrome 101200 Beare-Stevenson cutis gyrata syndrome123790 Pfeiffer syndrome 101600 Craniofacial-skeletal-dermatologicdysplasia 101600 Crouzon syndrome 123500 Jackson-Weiss syndrome 123150 Saethre-Chotzen syndrome 101400 Scaphocephaly, maxillary retrusion, andmental retardation 609579 Bent bone dysplasia syndrome 614592 Craniosynostosis, nonspecific LADD syndrome 149730 Scaphocephaly and Axenfeld-Riegeranomaly Polydactyly Limb defects most often involved thethumbs, ranging from total aplasia tohypoplastic, digitalized, triphalangeal,and duplicated thumbs

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 75/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
FGFR3 3690 NM_000142.4 2-18 Muenke syndrome 602849 Crouzon syndrome with acanthosisnigricans 612247 Muenke syndrome 602849 CATSHL syndrome 610474 SADDAN 616482 Achondroplasia 100800 LADD syndrome 149730 Hypochondroplasia 146000 Polydactyly Limb defects most often involved thethumbs, ranging from total aplasia tohypoplastic, digitalized, triphalangeal,and duplicated thumbs Thanatophoric dysplasia 187600 Focal Epilepsy Epilepsy
FH 3700 NM_000143.3 1-10 Fumarase deficiency (Disorders of thecitric acid cycle) Seizures
FHL1 3702 NM_001449.5 7 3-7 Scapuloperoneal myopathy, X-linkeddominant, 300695 Emery-Dreifuss muscular dystrophy 6,X-linked, 300696 Reducing body myopathy, X-linked 1b,with late childhood or adult onset,300718 Reducing body myopathy, X-linked 1a,severe, infantile or early childhoodonset, 300717
FIG4 16873 NM_014845.5 1-23 Yunis-Varon syndrome 216340 Amyotrophic lateral sclerosis 11 612577 Aplastic/hypoplastic thumbs CHARCOT-MARIE-TOOTH DISEASE, TYPE4J CLEIDOCRANIAL DYSPLASIA WITHMICROGNATHIA, ABSENT THUMBS, ANDDISTAL APHALANGIA YUNIS-VARONSYNDROME 216340

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 76/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
FKBP10 18169 NM_021939.4 1-10 Brucks syndrome Osteogenesis Imperfecta andDecreased Bone Density
FKBP14 18625 NM_017946.4 1-4 Ehlers-Danlos syndrome withprogressive kyphoscoliosis, myopathy,and hearing loss, 614557
FKRP 17997 NM_024301.5 4 Muscular dystrophy-dystroglycanopathy (congenital with orwithout mental retardation), type B, 5606612 Fukutin-related protein deficiency(Disorders of protein O-glycosylation, O-mannosylglycan synthesis deficiencies) Limb-girdle muscular dystrophy Walker-warburg syndrome or muscle-eye-brain disease Muscle- eye- brain disease Warburg syndrome
FKTN 3622 NM_001079802.1 3-11 Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies), type A, 4,253800Muscular dystrophy-dystroglycanopathy (congenital withoutmental retardation), type B, 4,613152Cardiomyopathy, dilated, 1X,611615Muscular dystrophy-dystroglycanopathy (limb-girdle), type C,4, 611588 Fukutin deficiency (Disorders of proteinO-glycosylation, O-mannosylglycansynthesis deficiencies) Cardiomyopathy, dilated, 1X, 611615 Limb-girdle muscular dystrophy
FLAD1 24671 NM_025207.5 1-7 Riboflavin-Responsive and Non-responsive Multiple Acyl-CoADehydrogenase and CombinedRespiratory-Chain Deficiency.

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 77/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
FLNA 3754 NM_001456.3 2-47 Orofacial Clefting with skeletalanomalies Melnick-Needles syndrome, 309350(includes clefting) Terminal osseous dysplasia 300244 Frontometaphyseal dysplasia 305620 X-LINKED CONGENITAL IDIOPATHICINTESTINAL PSEUDOOBSTRUCTION300048 Childhood Interstitial Lung Disease EPILEPTIC ENCEPHALOPATHY Heterotopia, periventricular 300049
FLNB 3755 NM_001457.4 1-46 Skeletal dysplasia with midline cleftpalate Orofacial Clefting with skeletal features Larsen syndrome (includes clefting)MONOALLELIC, autosomal orpseudoautosomal, imprinted statusunknown, 150250 Larsen syndrome 150250 Spondylocarpotarsal synostosissyndrome, 272460 Larsen syndrome, 150250 Boomerang dysplasia, 112310 Atelosteogenesis, type I OMIM
FLNC 3756 NM_001458.4 44-48 1-48 Myopathy, myofibrillar, 5 609524 early-onset restrictive cardiomyopathyand congenital myopathy
FLT4 3767 NM_182925.5 1-30 MILROY DISEASE 153100 Lymphedema, hereditary, IA, 153100 Hemangioma, capillary infantile,somatic, 602089
FLVCR1 24682 NM_014053.4 1-10 Ataxia, posterior column, with retinitispigmentosa,
FLVCR2 20105 NM_017791.3 1-10 Proliferative vasculopathy andhydraencephaly-hydrocephalysyndrome, 225790

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 78/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
FMN2 14074 NM_020066.5 5 1-18 NONSYNDROMIC AUTOSOMAL-RECESSIVE INTELLECTUAL DISABILITY
FMO3 3771 NM_006894.6 2-9 Trimethylaminuria (Disorders andvariants of enzymes that oxidisexenobiotics other than cytochromeP450)
FMR1 3775 NM_002024.5 1-17 FMR1-related disorders include fragile Xsyndrome, fragile X-associatedtremor/ataxia syndrome (FXTAS), andFMR1-related premature ovarianinsufficiency (POI)
FN1 3778 NM_212482.3 1-46 Spondylometaphyseal dysplasia, cornerfracture type 184255
FOLR1 3791 NM_016725.3 2-5 Neurodegeneration due to cerebralfolate transport deficiency, 613068
FOXC1 3800 NM_001453.3 1 Iridogoniodysgenesis, type 1, 601631 Axenfeld-Rieger syndrome, type 3,602482
FOXC2 3801 NM_005251.3 1 Cleft palate Lymphedema-distichiasis syndromewith renal disease and diabetesmellitus, 153400
FOXE1 3806 NM_004473.4 1 Bamforth-Lazarus syndrome, 241850
FOXE3 3808 NM_012186.3 1 Anterior segment mesenchymaldysgenesis, 107250 Aphakia, congenital primary, 610256
FOXF1 3809 NM_001451.3 1-2 Alveolar capillary dysplasia withmisalignment of pulmonary veins,265380
FOXG1 3811 NM_005249.5 1 Rett syndrome, congenital variant
FOXL2 1092 NM_023067.4 1 BLEPHAROPHIMOSIS, PTOSIS, ANDEPICANTHUS INVERSUS SYNDROME110100

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 79/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
FOXN1 12765 NM_003593.2 1-8 T-cell immunodeficiency, congenitalalopecia, and nail dystrophy, 601705
FOXP1 3823 NM_032682.5 6-21 Mental Retardation with LanguageImpairment and Autistic Features
FOXP2 13875 NM_014491.4 2-17 Speech-language disorder-1, 602081
FOXP3 6106 NM_014009.4 2-12 IPEX SYNDROME 304790
FOXRED1 26927 NM_017547.4 1-11 Mitochondrial Respiratory ChainComplex I Deficiency
FRAS1 19185 NM_025074.7 1-74 Fraser syndrome Polydactyly
FREM1 23399 NM_144966.5 3-38 MANITOBA OCULOTRICHOANALSYNDROME 248450
FREM2 25396 NM_207361.6 1-24 Fraser syndrome 2 Polydactyly
FRMD7 8079 NM_194277.2 1-12 NYSTAGMUS 1, CONGENITAL, X-LINKED310700
FRMPD4 29007 NM_014728.3 1-17 Mental retardation, X-linked 104, 300983
FRRS1L 1362 NM_014334.3 1-5 Epileptic encephalopathy, early infantile,37 (MIM 616981)
FTCD 3974 NM_006657.3 1-14 GLUTAMATE FORMIMINOTRANSFERASEDEFICIENCY (FIGLU-URIA)
FTL 3999 NM_000146.4 1-4 Neurodegeneration with brain ironaccumulation 3 606159 Hyperferritinemia-cataract syndrome
FTO 24678 NM_001080432.3 1-9 Growth retardation, developmentaldelay, facial dysmorphism, 612938 Lethal polymalformative syndrome,Boissel type
FTSJ1 13254 NM_012280.4 2-12 Mental retardation, X-linked 9, 309549

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 80/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
FUCA1 4006 NM_000147.4 1-8 Fucosidosis, 230000 seizures General Leukodystrophy &Mitochondrial Leukoencephalopathy
FUK 29500 NM_145059.3 2-24 Seizures Generalized hypotonia Feeding difficulties Intellectual disability Global developmental delay Congenital disorder of glycosylationwith defective fucosylation 2, 618324 Abnormality of vision
FUT8 4019 NM_178155.3 3-11 Intellectual disability Congenital Disorder of Glycosylationwith Defective Fucosylation seizures
FXN 3951 NM_000144.5 5 1-5 Friedreich ataxia with retained reflexes,229300 Hereditary ataxia Defective Fe-S/lipoic acid biosynthesis(Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly))
FXR1 4023 NM_005087.4 17 1-17 Congenital multi-minicore myopathy
FYCO1 14673 NM_024513.4 2-18 Cataract 18, autosomal recessive,610019
FZD2 4040 NM_001466.4 1 Omodysplasia 2, 164745 Robinow syndrome
FZD5 4043 NM_003468.4 2 Autosomal Dominant Coloboma
FZD6 4044 NM_003506.4 2-7 Nail disorder, nonsyndromic congenital,10, (claw-shaped nails), 614157
G6PC 4056 NM_000151.4 1-5 Glycogen storage disease type 1a, vonGierke (Glycogen storage disorders) fasting intolerance with enlarged liver,renal tubular disease

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 81/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
G6PC3 24861 NM_138387.3 1-6 Dursun syndrome
GAA 4065 NM_000152.5 2-20 Glycogen storage disease II, 232300
GABBR2 4507 NM_005458.8 1-19 Rett syndrome Neurodevelopmental disorder withpoor language and loss of hand skills,617903 Epileptic encephalopathy, early infantile,59, 617904
GABRA1 4075 NM_000806.5 3-11 JUVENILE MYOCLONIC EPILEPSY 611136 EPILEPTIC ENCEPHALOPATHY
GABRA2 4076 NM_001114175.2 1-9 Epileptic encephalopathy, early infantile,78, 618557 intellectual disability developmental delay
GABRA5 4079 NM_000810.4 3-11 Epileptic encephalopathy, early infantile,79, 618559 developmental delay
GABRB1 4081 NM_000812.4 1-9 Epileptic encephalopathy, early infantile,45, 617153
GABRB2 4082 NM_021911.2 2-11 Epileptic encephalopathy, infantile orearly childhood, 2, 617829 intellectual disability Epilepsy and intellectual disability
GABRB3 4083 NM_000814.6 1-9 Epilepsy, childhood absence,susceptibility to, 5
GABRG2 4087 NM_000816.3 1-9 Febrile seizures, familial, 8 611277 Epilepsy, generalized, with febrileseizures plus, type 3 611277
GAD1 4092 NM_000817.3 2-17 Developmental and epilepticencephalopathies, cleft palate, jointcontractures and/or omphalocele Cerebral palsy, spastic quadriplegic, 1,603513

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 82/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GALC 4115 NM_000153.4 1-17 Krabbe disease seizures
GALE 4116 NM_000403.4 3-12 Intellectual disability Uridine diphosphate galactose-4-epimerase deficiency (Disorders ofgalactose metabolism)
GALK1 4118 NM_000154.2 1-8 Galactokinase deficiency with cataracts,230200
GALNS 4122 NM_000512.5 1-14 Mucopolysaccharidosis, Type IV (MPS IV,Morquio disease)
GALNT3 4125 NM_004482.4 2-11 Polypeptide N-acetylgalactosaminyltransferase deficiency (Disorders ofprotein O-glycosylation, O-N-acetylgalactosaminylglycan synthesisdeficiencies) Tumoral calcinosis,hyperphosphatemic, familial 211900
GALT 4135 NM_000155.4 1-11 Intellectual disability Classical galactosaemia (Disorders ofgalactose metabolism) Cataracts
GAMT 4136 NM_000156.6 1-6 Cerebral creatine deficiency syndrome2, 612736 Seizures Deficiency of guanidinoacetatemethyltransferase
GARS 4162 NM_002047.4 1-17 Charcot-Marie-Tooth disease, type 2D Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Neuropathy, distal hereditary motor,type VA
GAS2L2 24846 NM_139285.4 1-6 Impaired Cilia Orientation andMucociliary Clearance
GAS8 4166 NM_001481.3 1-11 PRIMARY CILIARY DYSKINESIA

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 83/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GATA1 4170 NM_002049.4 2-6 Radial Ray abnormality
GATA2 4171 NM_032638.5 2-6 Immunodeficiency 21, 614172 Emberger syndrome, 614038
GATA3 4172 NM_001002295.2 2-6 Hypoparathyroidism, sensorineuraldeafness, and renal dysplasia, 146255 Barakat syndrome HDR syndrome
GATA4 4173 NM_002052.5 2-7 Atrioventricular septal defect 4, 614430 ?Testicular anomalies with or withoutcongenital heart disease, 615542
GATA6 4174 NM_005257.5 2-7 Atrioventricular septal defect 5, 614474 Pancreatic agenesis and congenitalheart defects, 600001 Persistent truncus arteriosus, 217095 Tetralogy of Fallot, 187500
GATAD2B 30778 NM_020699.4 2-11 Mental retardation, autosomaldominant 18, 615074
GATC 25068 NM_176818.3 4 1-4 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis)
GATM 4175 NM_001482.3 1-9 Cerebral creatine deficiency syndrome3, 612718 Arginine:glycine amidinotransferasedeficiency (Mitochondrial respiratorychain disorders (caused by nuclearvariants only), disorders of creatininemetabolism)
GBA 4177 NM_001005741.3 2-12 2-12 Gaucher disease (Sphingolipidoses) seizures
GBA2 18986 NM_020944.3 1-17 Spastic paraplegia 46, autosomalrecessive, 614409
GBE1 4180 NM_000158.4 1-16 Glycogen storage disease type IV,Andersen (Glycogen storage disorders) General Leukodystrophy &Mitochondrial Leukoencephalopathy Polyglucosan Body Disease (PGBD)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 84/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GCDH 4189 NM_000159.4 2-12 Dystonia Glutaricaciduria, type I OMIM
GCH1 4193 NM_000161.3 1-6 Dystonia, DOPA-responsive, with orwithout hyperphenylalaninemia progressive spastic paraplegia Spastic paraplegia seizures
GCLC 4311 NM_001498.4 1-16 Gamma-glutamylcysteine synthetasedeficiency (Disorders of the gamma-glutamyl cycle) Hemolytic anemia due to gamma-glutamylcysteine synthetase deficiency230450
GDAP1 15968 NM_018972.4 1-6 Charcot Marie Tooth disease (CMT4A)
GDF1 4214 NM_001492.6 8,7 Right atrial isomerism (Ivemark), 208530Congenital heart defects, multiple types,6, 613854
GDF2 4217 NM_016204.4 1-2 Heritable pulmonary arterialhypertension
GDF5 4220 NM_000557.5 1-2 Brachydactyly, type A1, C 615072 Brachydactyly, type A2 112600 Brachydactyly, type C 113100 Polydactyly Acromesomelic dysplasia, Hunter-Thompson type, 201250 Brachydactyly, type C, 113100 Chondrodysplasia, Grebe type, 200700 Du Pan syndrome, 228900 Brachydactyly, type A2, 112600 Symphalangism, proximal, 1B, 615298 Multiple synostoses syndrome 2,610017

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 85/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GDF6 4221 NM_001001557.4 1-2 Multiple synostoses syndrome type 4 -617898. Klippel-Feil syndrome 1, autosomaldominant, 118100 Microphthalmia, isolated 4, 613094 Microphthalmia with coloboma 6,digenic, 613703 Leber congenital amaurosis 17, 615360
GDI1 4226 NM_001493.3 1-11 Mental retardation, X-linked 41, 300849
GEMIN4 15717 NM_015721.3 1-2 Neurodevelopmental disorder withmicrocephaly, cataracts, and renalabnormalities, 617913
GFAP 4235 NM_002055.5 1-9 Alexander disease, 203450 Autosomal Dominant Ataxia seizures General Leukodystrophy &Mitochondrial Leukoencephalopathy
GFER 4236 NM_005262.3 1-3 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Myopathy, mitochondrial progressive,with congenital cataract, hearing loss,and developmental delay, 613076 Intellectual disability
GFM1 13780 NM_024996.6 1-18 Combined oxidative phosphorylationdeficiency 1, 609060 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) General Leukodystrophy &Mitochondrial Leukoencephalopathy
GFM2 29682 NM_032380.5 2-21 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Early-onset neurological presentationsof mitochondrial disease

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 86/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GFPT1 4241 NM_002056.4 1-19 Myasthenia, congenital, 12, with tubularaggregates(Disorders of protein N-glycosylation) 610542 Limb-girdle congenital myasthenicsyndrome
GHR 4263 NM_000163.5 2-10 Growth hormone insensitivity Short stature, 604271 Increased responsiveness to growthhormone Laron dwarfism, 262500 Proportionate Short Stature/Small forGestational Age
GIF 4268 NM_005142.3 1-9 Intrinsic factor deficiency OMIM
GJA1 4274 NM_000165.5 2 2 Oculodentodigital dysplasia 164200 Syndactyly, type III 186100 Erythrokeratodermia variabilis etprogressiva 133200 Palmoplantar keratoderma withcongenital alopecia 104100 Craniometaphyseal dysplasia,autosomal recessive 218400 Hypoplastic left heart syndrome 1241550
GJA3 4277 NM_021954.4 2 Cataract 14, multiple types, 601885
GJA8 4281 NM_005267.5 2 Cataract 1, multiple types, 116200
GJB1 4283 NM_000166.6 2 Charcot-Marie-Tooth neuropathy, X-linked dominant, 1
GJB2 4284 NM_004004.6 2 Vohwinkel syndrome, 124500 Keratoderma, palmoplantar, withdeafness, 148350 Bart-Pumphrey syndrome, 149200

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 87/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GJB3 4285 NM_024009.3 2 ERYTHROKERATODERMIA VARIABILIS ETPROGRESSIVA 133200 Deafness, autosomal recessive Deafness, autosomal dominant, withperipheral neuropathy Deafness, digenic, GJB2/GJB3, 220290
GJC2 17494 NM_020435.4 2 Autosomal Recessive Ataxia Spastic paraplegia 44, autosomalrecessive, 613206, AR Lymphatic malformation 3, 613480, AD Spastic paraplegia 44, autosomalrecessive, 613206 Leukodystrophy, hypomyelinating, 2,608804 Lymphedema, hereditary, IC, 613480
GK 4289 NM_000167.5 19 1-19 Glycerol kinase deficiency, 307030
GLA 4296 NM_000169.2 1-7 Fabry disease (Sphingolipidoses)
GLB1 4298 NM_000404.4 1-16 Mucopolysaccharidosis type IVB(Morquio), 253010 GM1-gangliosidosis (Sphingolipidoses) seizures
GLDC 4313 NM_000170.2 1-25 Glycine encephalopathy, 605899 seizures
GLDN 29514 NM_181789.4 1-10 Lethal congenital contracture syndrome11 617194
GLE1 4315 NM_001003722.1 1-16 Arthrogryposis, lethal, with anteriorhorn cell disease, 611890
GLI1 4317 NM_005269.3 2-12 Polydactyly, postaxial, type A8, 618123 Polydactyly, preaxial I, 174400
GLI2 4318 NM_005270.5 2-14 Culler-Jones syndrome 615849 Polydactyly Holoprosencephaly

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 88/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GLI3 4319 NM_000168.6 2-15 Greig cephalopolysyndactyly syndrome175700 Polydactyly, postaxial, types A1 and B174200 Polydactyly, preaxial, type IV 174700 Pallister-Hall syndrome 146510
GLIS2 29450 NM_032575.2 1-6 Nephronophthisis
GLIS3 28510 NM_152629.3 2-10 Diabetes mellitus, neonatal, withcongenital hypothyroidism, 610199
GLMN 14373 NM_053274.3 2-19 Glomuvenous malformations, 138000
GLRA1 4326 NM_000171.4 1-9 Hyperekplexia developmental delay infantile spasms and generalized tonic-clonic seizures
GLRB 4329 NM_000824.5 2-10 Hyperekplexia 2, 614619
GLRX5 20134 NM_016417.3 1-2 Defective Fe-S/lipoic acid biosynthesis(Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly)) Anemia, sideroblastic, pyridoxine-refractory, autosomal recessive, 205950 Disorders of iron homeostasis
GLS 4331 NM_014905.5 1-18 Glucosidase 1 deficiency (Disorders ofprotein N-glycosylation) Epileptic encephalopathy, early infantile,71 618328 Global developmental delay,progressive ataxia, and elevatedglutamine 618412
GLUD1 4335 NM_005271.5 2-4, 13 1-13 Hyperinsulinemic hypoglycemia andhyperammonemia (Urea cycle disordersand inherited hyperammonaemias) epilepsy

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 89/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GLUL 4341 NM_002065.6 3-8 Glutamine deficiency, congenital 610015seizures
GLYCTK 24247 NM_145262.4 2-5 D-glyceric aciduria (Disorders of serine,glycine or glycerate metabolism)
GM2A 4367 NM_000405.5 1-4 GM2-gangliosidosis, AB variant, 272750 seizures Hexosaminidase activator deficiency Tay-Sachs disease
GMNN 17493 NM_015895.5 2-7 Autosomal-Dominant PrimordialDwarfism Associated with Meier-GorlinSyndrome
GMPPA 22923 NM_205847.3 2-13 GLYCOSYLATION DISORDERCHARACTERIZED BY INTELLECTUALDISABILITY AND AUTONOMICDYSFUNCTION
GMPPB 22932 NM_013334.3 1-8 Muscular dystrophy-dystroglycanopathy (limb-girdle), type C,14 with features of congenitalmyasthenic syndrome
GNA11 4379 NM_002067.5 1-7 Congenital Hemangioma
GNA14 4382 NM_004297.4 1-7 Congenital vascular tumours
GNAI1 4384 NM_002069.6 1-8 GNAI1 syndrome
GNAI3 4387 NM_006496.4 1-8 Auriculocondylar syndrome 1, 602483
GNAL 4388 NM_001142339.2 2-13 Dystonia 25, 615073
GNAO1 4389 NM_020988.3 1-8 Neurodevelopmental disorder withinvoluntary movements, 617493 Epileptic encephalopathy, early infantile,17
GNAQ 4390 NM_002072.5 7 1-7 Congenital Hemangioma Sturge-Weber syndrome, somatic,mosaic, 185300

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 90/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GNAS 4392 NM_000516.6 1-13 McCune-Albright syndrome Cholestasis Pseudopseudohypoparathyroidism612463 Osseous heteroplasia, progressive166350 ACTH-independent macronodularadrenal hyperplasia 219080 IC
GNB1 4396 NM_002074.5 3-11 Severe Neurodevelopmental Disability,Hypotonia, and Seizures Intellectual disability
GNB5 4401 NM_016194.4 2-13 Language delay and ADHD/cognitiveimpairment with or without cardiacarrhythmia, 617182 early infantile epileptic encephalopathy(EIEE)
GNE 23657 NM_001128227.3 1-12 Nonaka myopathy 605820 Sialuria (Other lysosomal disorders) UDP-GlcNAc epimerase/kinasedeficiency (Disorders of multipleglycosylation and other glycosylationpathways) Limb girdle muscular dystrophy
GNMT 4415 NM_018960.6 1-6 Glycine N-methyltransferase deficiency606664
GNPAT 4416 NM_014236.4 1-16 Rhizomelic chondrodysplasia punctatatype 2 (Peroxisomal disorders)
GNPTAB 29670 NM_024312.5 1-21 Mucolipidosis, Type II Mucolipidosis, Type III Alpha/Beta
GNPTG 23026 NM_032520.5 1-11 Mucolipidosis III, Pseudo-Hurlerpolydystrophy (Other lysosomaldisorders)
GNS 4422 NM_002076.4 1-14 MPS IIID, Sanfilippo D disease(Mucopolysaccharidoses) Mucopolysaccharidosis type IIID 252940
GOLGA2 4425 NM_004486.5 1-26 Secondary dystroglycanopathy

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 91/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GORAB 25676 NM_152281.3 1-5 Geroderma osteodysplasticum
GOSR2 4431 NM_004287.4 3-4 1-6 Epilepsy, progressive myoclonic 6,614018
GOT2 4433 NM_002080.4 10 1-10 Malate-Aspartate Shuttle-RelatedEncephalopathy Global developmental delay Intellectual disability Seizures Increased serum lactate Hyperammonemia Microcephaly Failure to thrive Feeding difficulties Abnormality of nervous systemmorphology
GPAA1 4446 NM_003801.4 1-12 Glycosylphosphatidylinositolbiosynthesis defect 15, 617810 Developmental Delay, Epilepsy,Cerebellar Atrophy, and Osteopenia
GPC3 4451 NM_004484.4 1-8 Simpson-Golabi-Behmel syndrome, type1 312870 Polydactyly
GPC4 4452 NM_001448.3 1-9 KEIPERT SYNDROME 301026
GPC6 4454 NM_005708.5 1-9 Omodysplasia 1 258315
GPD1 4455 NM_005276.4 1-8 Hypertriglyceridemia, transient infantile,614480
GPHN 15465 NM_020806.4 1-23 Mo cofactor deficiency,complementation group C (Disorders ofmolybdenum cofactor metabolism) epileptic encephalopathy
GPR126 13841 NM_020455.5 1-26 LETHAL CONGENITAL CONTRACTURESYNDROME 9 616503

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 92/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GPR179 31371 NM_001004334.4 1-11 Night blindness, congenital stationary(complete), 1E
GPR56 4512 NM_005682.7 3-15 Polymicrogyria, bilateral frontoparietal,606854
GPSM2 29501 NM_013296.5 2-15 DEAFNESS AUTOSOMAL RECESSIVE TYPE82 Chudley-McCullough syndrome 604213
GPT2 18062 NM_133443.4 2-12 Microcephaly Intellectual disability Progressive spasticity
GPX4 4556 NM_001039847.3 1-7 SPONDYLOMETAPHYSEAL DYSPLASIA,SEDAGHATIAN TYPE 250220
GRHL2 2799 NM_024915.4 1-16 ECTODERMAL DYSPLASIA/SHORTSTATURE SYNDROME 616029
GRHL3 25839 NM_198174.3 1-16 Cleft lip Van der Woude syndrome 2, 606713
GRHPR 4570 NM_012203.2 1-9 Primary hyperoxaluria type II (Disordersof glyoxylate metabolism)
GRIA1 4571 NM_001114183.1 1-16 Intellectual disability
GRIA2 4572 NM_001083619.1 1-15 Epileptic encephalopathy intellectualdisability stereotypic hand movements
GRIA3 4573 NM_000828.4 1-15 Mental retardation, X-linked 94, 300699
GRIA4 4574 NM_000829.4 2-17 Neurodevelopmental disorder with orwithout seizures and gait abnormalities,617864
GRID2 4576 NM_001510.4 1-16 Spinocerebellar ataxia, autosomalrecessive 18, 616204
GRIK2 4580 NM_021956.4 1-16 Mental retardation, autosomalrecessive, 6, 611092

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 93/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GRIN1 4584 NM_007327.4 1-20 Neurodevelopmental disorder with orwithout hyperkinetic movements andseizures, autosomal recessive, 617820 involuntary movements severe developmental delay intellectual disability
GRIN2A 4585 NM_000833.5 3-14 Epilepsy with neurodevelopmentaldefects, 613971 LANDAU-KLEFFNER SYNDROME
GRIN2B 4586 NM_000834.4 2-13 Mental retardation, autosomaldominant 6 Epileptic encephalopathy, early infantile,27
GRIN2D 4588 NM_000836.2 2-13 intellectual disability Severe Epileptic EncephalopathyTreatable with NMDA Receptor ChannelBlockers
GRIP1 18708 NM_021150.4 1-24 Fraser syndrome 3 617667 Polydactyly
GRM1 4593 NM_001278064.2 1-8 Spinocerebellar ataxia 44, 617691
GRM6 4598 NM_000843.4 2-11 Night blindness, congenital stationary(complete), 1B
GRN 4601 NM_002087.3 2-13 Aphasia, primary progressive OMIM
GSC 4612 NM_173849.2 1-3 Short stature, auditory canal atresia,mandibular hypoplasia, skeletalabnormalities 602471
GSPT2 4622 NM_018094.5 1 XL INTELLECTUAL DISABILITY
GSS 4624 NM_000178.4 2-13 Glutathione synthetase deficiency withor without 5-oxoprolinuria Pyroglutamic aciduria Glutathione synthetase deficiency(Disorders of the gamma-glutamyl cycle)Fanconi nephropathy

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 94/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GTF2E2 4651 NM_002095.6 2-8 DNA Repair-ProficientTrichothiodystrophy Trichothiodystrophy 6,nonphotosensitive
GTF2H5 21157 NM_207118.3 2-3 Trichothiodystrophy, complementationgroup A, 601675 Photosensitive trichothiodystrophy 3
GTF3C3 4666 NM_012086.5 1-18 Global developmental delay
GTPBP2 4670 NM_019096.5 1-12 Jaberi-Elahi syndrome, 617988 Global developmental delay Intellectual disability Seizures
GTPBP3 14880 NM_133644.4 1-8 Combined oxidative phosphorylationdeficiency 23, 616198 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) mitochondrial translation defectassociated with hypertrophiccardiomyopathy, lactic acidosis andencephalopathy
GUCY2C 4688 NM_004963.4 1-27 Diarrhea 6, 614616 Meconium ileus, 614665
GUF1 25799 NM_021927.3 1-17 ?Epileptic encephalopathy, earlyinfantile, 40, 617065
GUSB 4696 NM_000181.4 11 1-12 MPS VII, Sly disease (MPS IV, Morquiodisease) Mucopolysaccharidosis, Type VII
GYG1 4699 NM_004130.3 1-8 ?Glycogen storage disease XV 613507 Polyglucosan body myopathy 2 616199
GYS1 4706 NM_002103.5 1-16 Glycogen storage disease 0, muscle
GYS2 4707 NM_021957.4 1-16 Glycogen storage disease type 0a, liver(Glycogen storage disorders)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 95/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
GZF1 15808 NM_022482.5 2-6 Larsen syndrome
HAAO 4796 NM_012205.3 1-10 Multiple congenital malformations VACTERL-like phenotype
HACE1 21033 NM_020771.4 1-24 HACE1 related disorder Spastic paraplegia and psychomotorretardation with or without seizures,616756
HADH 4799 NM_005327.6 1-8 3-hydroxyacyl-CoA dehydrogenasedeficiency 231530 Hyperinsulinemic hypoglycemia,familial, 4 609975
HADHA 4801 NM_000182.5 1-20 LCHAD deficiency 609016 Mitochondrial trifunctional proteindeficiency (Disorders of mitochondrialfatty acid oxidation)
HADHB 4803 NM_000183.3 2-16 Mitochondrial trifunctional proteindeficiency (Disorders of mitochondrialfatty acid oxidation)
HAMP 15598 NM_021175.4 1-3 Hereditary haemochromatosis Type 2(Disorder of iron metabolism)
HARS2 4817 NM_012208.4 1-13 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Perrault syndrome 2, 614926
HAX1 16915 NM_006118.4 1-7 Neutropenia, severe congenital 3,autosomal recessive, 610738
HCCS 4837 NM_005333.5 2-7 Linear skin defects with multiplecongenital anomalies 1 Microphthalmia, syndromic 7, 309801
HCFC1 4839 NM_005334.3 1-26 COBALAMIN DISORDER Mental retardation, X-linked 3(methylmalonic acidemia andhomocysteinemia, cblX type ) 309541
HCN1 4845 NM_021072.4 1-8 EPILEPTIC ENCEPHALOPATHY, EARLYINFANTILE, 24

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 96/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
HCN2 4846 NM_001194.4 6-8 1-8 Genetic epilepsy with febrile seizuresplus
HDAC4 14063 NM_006037.3 2-27 Brachydactyly-mental retardationsyndrome, 600430 Albright hereditary osteodystrophy type3, Albright hereditary osteodystrophy-like syndrome, Brachydactyly-intellectual disability, Del(2)(q37) 600430
HDAC6 14064 NM_006044.4 2-29 ?Chondrodysplasia with platyspondyly,distinctive brachydactyly, hydrocephaly,and microphthalmia
HDAC8 13315 NM_018486.3 1-11 Wilson-Turner syndrome 309585 Cornelia de Lange syndrome 5 300882
HEATR2 26013 NM_017802.4 1-13 CILIARY DYSKINESIA, PRIMARY, 18614874
HECW2 29853 NM_020760.4 2-29 Neurodevelopmental disorder withhypotonia, seizures, and absentlanguage
HEPACAM 26361 NM_152722.5 1-7 Megalencephalic leukoencephalopathywith subcortical cysts 2B, remitting, withor without mental retardation (AD),613926 General Leukodystrophy &Mitochondrial Leukoencephalopathy
HERC1 4867 NM_003922.4 2-78 Macrocephaly, dysmorphic facies, andpsychomotor retardation 617011
HES7 15977 NM_032580.4 1-4 Spondylocostal dysostosis 4, autosomalrecessive 613686
HESX1 4877 NM_003865.3 1-4 HESX1-RELATED COMBINED PITUITARYHORMONE DEFICIENCY SEPTOOPTIC DYSPLASIA 256657
HEXA 4878 NM_000520.6 1-14 Tay-Sachs disease, 272800 GM2-GANGLIOSIDOSIS TYPE 1 (GM2G1) Hex A pseudodeficiency, 272800 AR

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 97/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
HEXB 4879 NM_000521.4 1-14 Sandhoff disease, infantile, juvenile, andadult forms, 268800 GM2-GANGLIOSIDOSIS TYPE 2 (GM2G2) seizures myoclonic epilepsy
HFE 4886 NM_000410.3 1-6 Hereditary haemochromatosis Type 1(Disorder of iron metabolism)
HFE2 4887 NM_145277.5 2-3 Hemochromatosis, type 2A, 602390
HGD 4892 NM_000187.4 1-14 Alkaptonuria
HGSNAT 26527 NM_152419.3 1-18 Retinitis Pigmentosa 73 Mucopolysaccharidosis type IIIC(Sanfilippo C) 252930
HIBCH 4908 NM_014362.4 1-14 3-hydroxyisobutryl-CoA hydrolasedeficiency, 250620 Methacrylic aciduria (Organic acidurias)
HINT1 4912 NM_005340.7 1-3 Neuromyotonia and axonal neuropathy,autosomal recessive, 137200
HIST1H1E 4718 NM_005321.2 1 Rahman syndrome, 617537 mild to severe intellectual disability Childhood overgrowth
HIST1H4C 4787 NM_003542.3 1 Growth delay, microcephaly andintellectual disability
HIVEP2 4921 NM_006734.4 5-10 Mental retardation, autosomaldominant 43, 616977 Intellectual disability HIVEP2 associated syndromicdevelopmental delay with intellectualdisability

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 98/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
HK1 4922 NM_000188.2 1-18 Hemolytic anemia due to hexokinasedeficiency, 235700 Neuropathy, hereditary motor andsensory, Russe type, 605285 Abnormal muscle tone Global developmental delay Intellectual disability Visual impairment Neurological speech impairment Ataxia
HLCS 4976 NM_000411.8 4-12 Holocarboxylase synthetase deficiency(Disorders of biotin metabolism)
HMBS 4982 NM_000190.4 1-14 Acute intermittent porphyria (Acuteneuropathic porphyrias)
HMGCL 5005 NM_000191.3 1-9 3-Hydroxy-3-methyl glutaric aciduria(Organic acidurias) Intellectual disability
HMGCS2 5008 NM_005518.4 1-9 HMG-CoA synthase-2 deficiency
HMX1 5017 NM_018942.3 1-2 OCULOAURICULAR SYNDROME 612109
HNF1B 11630 NM_000458.4 1-9 Cholestasis neonatal and adult onset jaundice andcholestasis Renal cysts and diabetes syndrome,137920 Diabetes mellitus, noninsulin-dependent, 125853
HNF4A 5024 NM_175914.4 1-10 Fanconi renotubular syndrome 4, withmaturity-onset diabetes of the young,616026
HNRNPDL 5037 NM_031372.3 1-7 Muscular dystrophy, limb-girdle, type1G 609115
HNRNPH2 5042 NM_019597.5 2 2 Mental retardation, X-linked, syndromic,Bain type, 300986 Neurodevelopmental Disorder inFemales

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 99/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
HNRNPK 5044 NM_002140.4 17 3-17 Au-Kline Syndrome Polydactyly
HNRNPR 5047 NM_001102398.3 10-11 2-11 Intellectual disability Postnatal microcephaly
HNRNPU 5048 NM_031844.3 1-14 Epileptic encephalopathy, early infantile,54, 617391 intellectual disability
HOGA1 25155 NM_138413.4 1-7 Hyperoxaluria, primary, type III 613616
HOXA1 5099 NM_005522.5 1-2 Athabaskan brainstem dysgenesissyndrome, 601536 Bosley-Salih-Alorainy syndrome, 601536
HOXA13 5102 NM_000522.5 1-2 Hand-foot-genital syndrome 140000 Guttmacher syndrome 176305 Polydactyly Hand-foot-uterus syndrome 140000
HOXB1 5111 NM_002144.4 1-2 FACIAL PARESIS, HEREDITARYCONGENITAL, 3 614744
HOXC13 5125 NM_017410.3 1-2 Ectodermal dysplasia 9, hair/nail type,614931
HOXD13 5136 NM_000523.4 1-2 Brachydactyly-syndactyly syndrome610713 Polydactyly ?VACTERL association, 192350
HPCA 5144 NM_002143.3 2-4 Dystonia 2, torsion, autosomalrecessive, 224500
HPD 5147 NM_002150.3 1-14 Hawkinsinuria 140350 Tyrosinemia, type III 276710
HPGD 5154 NM_000860.6 1-7 Cranioosteoarthropathy 259100 Digital clubbing, isolated congenital119900 Hypertrophic osteoarthropathy,primary, autosomal recessive 1 259100
HPRT1 5157 NM_000194.3 1-9 HPRT-related gout Lesch-Nyhan syndrome, 300322

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 100/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
HPS1 5163 NM_000195.5 4-6 3-20 Hermansky-Pudlak syndrome 1 203300
HPSE2 18374 NM_021828.4 1-12 Urofacial syndrome 1, 236730
HR 5172 NM_005144.4 2-19 Alopecia universalis, 203655 Atrichia with papular lesions, 209500 Hypotrichosis 4, 146550
HRAS 5173 NM_005343.4 2-5 CONGENITAL MYOPATHY WITH EXCESSOF MUSCLE SPINDLES 218040 Costello syndrome, 218040 Schimmelpenning-Feuerstein-Mimssyndrome, 218040
HSD17B10 4800 NM_004493.3 1-6 2-METHYL-3-HYDROXYBUTYRYL-COADEHYDROGENASE DEFICIENCY (MHBDDEFICIENCY)
HSD17B4 5213 NM_000414.4 1-24 D-bifunctional protein deficiency,261515 Peroxisomal D-bifunctional proteindeficiency (Disorders of peroxisomalalpha-, beta and omega-oxidation) General Leukodystrophy &Mitochondrial Leukoencephalopathy
HSD3B7 18324 NM_025193.4 2-7 Neonatal and Adult Cholestasis Bile acid sythesis defect, congenital, 1607765 Bile acid synthesis defect, congenital, 1,607765
HSF4 5227 NM_001538.4 3-15 Cataract 5, multiple types, 116800
HSPA9 5244 NM_004134.7 1-17 Even-plus syndrome 616854
HSPB1 5246 NM_001540.5 1-3 Neuropathy, distal hereditary motortype IIB, 608634 Charcot-Marie-Tooth disease, axonal,type 2F OMIM
HSPB8 30171 NM_014365.2 1-3 Neuropathy, distal hereditary motor,type IIA 158590

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 101/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
HSPD1 5261 NM_002156.5 9-12 2-12 Leukodystrophy, hypomyelinating, 4,612233 Spastic paraplegia 13, autosomaldominant, 605280
HSPG2 5273 NM_005529.7 1-97 Dyssegmental dysplasia, Silverman-Handmaker type 224410 Schwartz-Jampel syndrome, type 1255800
HTRA2 14348 NM_013247.4 1-8 3-methylglutaconic aciduria, type VIII
HTT 4851 NM_002111.8 1-67 Huntington disease OMIM Lopes-Maciel-Rodan syndrome OMIM
HUWE1 30892 NM_031407.7 4-84 Mental retardation, X-linked syndromic,Turner type, 300706
HYAL1 5320 NM_153281.1 4-6 MPS IX, Natowicz (MPS IV, Morquiodisease) Mucopolysaccharidosis type IX, 601492
HYDIN 19368 NM_001270974.2 6-84 2-86 CILIARY DYSKINESIA, PRIMARY, 5 608647
HYLS1 26558 NM_145014.2 4 Hydrolethalus syndrome, 236680(includes Cleft palate, Lateral or midlinecleft lip, Lower lip cleft) Joubert syndrome
IARS 5330 NM_002161.5 2-34 Growth retardation, intellectualdevelopmental disorder, hypotonia, andhepatopathy 617093 Microcephaly
IARS2 29685 NM_018060.4 1-23 CAGSSS - Cataracts (CA), growthhormone deficiency (G), sensoryneuropathy (S), sensorineural hearingloss (S), and skeletal dysplasia (S)
IBA57 27302 NM_001010867.4 1-3 Multiple mitochondrial dysfunctionssyndrome 3, 615330 intellectual disability, seizures, loss ofmilestones ?Spastic paraplegia 74, autosomalrecessive, 616451

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 102/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ICK 21219 NM_016513.4 3-15 short-rib thoracic dysplasia withpolydactyly (SRTD) Endocrine-cerebroosteodysplasia,612651 (includes cleft lip, cleft palate)
IDH1 5382 NM_005896.3 3-10 Metaphyseal chondromatosis with D-2-hydroxyglutaric aciduria 614875 Maffucci syndrome 614569 Ollier disease/ Dyschondroplasia166000
IDH2 5383 NM_002168.3 1-11 Mitochondrial isocitrate dehydrogenasedeficiency (Organic acidurias) D-2-hydroxyglutaric aciduria 2 613657
IDH3A 5384 NM_005530.3 1-11 Retinitis pigmentosa with macularpseudocoloboma Infantile encephalopathy
IDH3B 5385 NM_006899.5 1-12 Retinitis pigmentosa 46, 612572
IDS 5389 NM_000202.8 2-3 1-9 MPS II, Hunter disease(Mucopolysaccharidoses) Mucopolysaccharidosis Type II
IDUA 5391 NM_000203.5 1-14 Mucopolysaccharidosis type 1H/S MPS I, Hurler, Scheie disease(Mucopolysaccharidoses)
IER3IP1 18550 NM_016097.5 1-3 Microcephaly, epilepsy, and diabetessyndrome, 614231 Intellectual disability
IFIH1 18873 NM_022168.4 1-16 Singleton-Merten syndrome 1, 182250 seizures Aicardi-Goutieres Syndrome General Leukodystrophy &Mitochondrial Leukoencephalopathy
IFITM5 16644 NM_001025295.3 1-2 Osteogenesis imperfecta, type V, 610967
IFT122 13556 NM_052985.4 15-20 1-31 Cranioectodermal dysplasia

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 103/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
IFT140 29077 NM_014714.4 3-31 Saldino-Mainzer syndrome Jeune syndrome Short-rib thoracic dysplasia 9 with orwithout polydactyly
IFT172 30391 NM_015662.3 1-48 Short-rib thoracic dysplasia 10 with orwithout polydactyly, 615630 Retinitis pigmentosa 71, 616394 Saldino-Mainzer syndrome
IFT27 18626 NM_006860.5 1-7 ?Bardet-Biedl syndrome 19, 615996 Polydactyly
IFT43 29669 NM_052873.3 1-8 Cranioectodermal dysplasia 3, 614099 Sensenbrenner syndrome ?Cranioectodermal dysplasia 3 - 614099 Short-rib thoracic dysplasia 18 withpolydactyly 617866
IFT52 15901 NM_016004.5 2-14 Short-rib thoracic dysplasia 16 with orwithout polydactyly, 617102
IFT80 29262 NM_020800.3 2-20 Short-rib thoracic dysplasia 2 with orwithout polydactyly Jeune syndrome
IFT81 14313 NM_014055.4 2-19 Short-Rib Polydactyly Syndrome
IGF1 5464 NM_000618.5 1-4 Growth retardation with deafness andmental retardation due to IGF1deficiency, 608747 microcephalic primordial dwarfism
IGF1R 5465 NM_000875.5 1-21 Insulin-like growth factor I, resistance to,270450
IGF2 5466 NM_000612.6 2-4 Beckwith-Wiedemann Syndrome Chromosome 11p15.5-Related Russell-Silver Syndrome
IGFBP7 5476 NM_001553.3 1-5 RETINAL ARTERIAL MACROANEURYSMWITH SUPRAVALVULAR PULMONICSTENOSIS 614224
IGHMBP2 5542 NM_002180.2 1-15 Spinal muscular atrophy withrespiratory distress, 604320

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 104/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
IGSF1 5948 NM_001170961.1 2-20 CENTRAL HYPOTHYROIDISM ANDTESTICULAR ENLARGEMENT 300888
IHH 5956 NM_002181.4 1-3 Acrocapitofemoral dysplasia 607778 Brachydactyly, type A1 112500 syndactyly and craniosynostosis
IKBKG 5961 NM_001099857.3 3-10 2-10 Ectodermal, dysplasia, anhidrotic,lymphedema and immunodeficiency300301 Incontinentia pigmenti, 308300
IL11RA 5967 NM_001142784.2 2-13 Craniosynostosis and dental anomalies614188 Crouzon-like craniosynostosis
IL1RAPL1 5996 NM_014271.4 2-11 Mental Retardation, X-linked
IL1RAPL2 5997 NM_017416.2 2-11 X-linked non-syndromic mentalretardation loci
IL1RN 6000 NM_173841.2 1-6 Interleukin 1 receptor antagonistdeficiency 612852
IMPAD1 26019 NM_017813.5 1-5 Chondrodysplasia with jointdislocations, GPAPP type, 614078(includes cleft palate)
INPP5E 21474 NM_019892.6 1-10 Joubert syndrome Mental retardation, truncal obesity,retinal dystrophy, and micropenis,610156
INPP5K 33882 NM_016532.4 1-12 Congenital Muscular DystrophyOverlapping Marinesco-SjogrenSyndrome and Dystroglycanopathy
INPPL1 6080 NM_001567.4 1-28 Opsismodysplasia, 258480
INTS1 24555 NM_001080453.3 2-48 Neurodevelopmental disorder withcataracts, poor growth, and dysmorphicfacies, 618571 Hypotonia Global developmental delay

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 105/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
INVS 17870 NM_014425.5 2-17 Senior-Loken syndrome Nephronophthisis
IQCB1 28949 NM_001023570.4 3-15 Senior-Loken syndrome
IQSEC1 29112 NM_014869.7 1-14 Central hypotonia Global developmental delay Intellectual disability Behavioral abnormality Short stature
IQSEC2 29059 NM_001111125.3 1-15 Rett like phenotype in males Mental retardation, X-linked 1
IRF2BPL 14282 NM_024496.4 1 Neurodevelopmental disorder withregression, abnormal movements, lossof speech, and seizures
IRF6 6121 NM_006147.4 3-9 Orofacial Clefting with skeletal features van der Woude syndrome, 119300 Popliteal pterygium syndrome 1, 119500
IRX5 14361 NM_005853.6 1-3 HYPERTELORISM, SEVERE, WITHMIDFACE PROMINENCE, MYOPIA,MENTAL RETARDATION, AND BONEFRAGILITY
ISCA1 28660 NM_030940.4 4 1-4 MULTIPLE MITOCHONDRIALDYSFUNCTIONS SYNDROME 5, 617613
ISCA2 19857 NM_194279.4 1-4 Multiple mitochondrial dysfunctionssyndrome 4, 616370 infantile neurodegenerativemitochondrial disorder
ISCU 29882 NM_213595.3 1-5 Myopathy with lactic acidosis,hereditary, 255125 Disorders of iron homeostasis Rhabdomyolysis and metabolic muscledisorders
ISG15 4053 NM_005101.4 1-2 Immunodeficiency 38 616126

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 106/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ISPD 37276 NM_001101426.4 1-10 Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies), type A, 7 Congenital Muscular Dystrophy, alpha-dystroglycan related Walker-Warburg syndrome (WWS)
ITCH 13890 NM_031483.7 3-25 AUTOIMMUNE DISEASE, SYNDROMICMULTISYSTEM 613385
ITGA3 6139 NM_002204.4 1-25 INTERSTITIAL LUNG DISEASE,NEPHROTIC SYNDROME, ANDEPIDERMOLYSIS BULLOSA, CONGENITAL614748
ITGA7 6143 NM_002206.3 1-25 Muscular dystrophy, congenital, due toITGA7 deficiency, 613204
ITGA8 6144 NM_003638.3 1-30 RENAL HYPODYSPLASIA/APLASIA 1191830
ITPA 6176 NM_033453.4 1-8 Inosine triphosphatase deficiency(Disorders of purine metabolism) Epileptic encephalopathy, early infantile,35, 616647
ITPR1 6180 NM_002222.6 3-58 Gillespie syndrome 206700 Spinocerebellar ataxia 29, congenitalnonprogressive
IVD 6186 NM_002225.5 1-12 metabolic encephalopathy withhyperammonaemia, hypotonia,recurrent episodes of ketoacidosis, liverimpairment, psychomotor retardation,recurrent infections. Isovaleric aciduria (Organic acidurias)
JAG1 6188 NM_000214.3 1-26 Alagille syndrome Neonatal and Adult Cholestasis Tetralogy of Fallot, 187500 Deafness, congenital heart defects, andposterior embryotoxon
JAGN1 26926 NM_032492.4 1-2 SEVERE CONGENITAL NEUTROPENIA

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 107/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
JAK3 6193 NM_000215.3 2-24 SCID, autosomal recessive, T-negative/B-positive type, 600802
JAM3 15532 NM_032801.5 1-9 Hemorrhagic destruction of the brain,subependymal calcification, andcataracts, 613730
KANSL1 24565 NM_001193466.2 3 2-15 Koolen-De Vries syndrome, 610443 Intellectual Disability Syndrome
KARS 6215 NM_001130089.1 2-15 Deafness, autosomal recessive 89,613916 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Charcot-Marie-Tooth disease, recessiveintermediate, B (Lysyl-tRNA synthetasemutations) (Mitochondrial respiratorychain disorders (caused by nuclearvariants only)) Global developmental delay Intellectual disability Seizures
KAT6A 13013 NM_006766.5 2-17 MENTAL RETARDATION, AUTOSOMALDOMINANT 32
KAT6B 17582 NM_012330.4 3-18 BLEPHAROPHIMOSIS/INTELLECTUALDISABILITY PHENOTYPE WHICH ISNOONAN-LIKE GENITOPATELLAR SYNDROME 606170 SBBYSS syndrome 603736 GTPTS,Ohdo
KATNB1 6217 NM_005886.3 2-20 Lissencephaly 6, with microcephaly,616212 seizures
KBTBD13 37227 NM_001101362.2 1 Nemaline myopathy 6, autosomaldominant, 609273
KCNA1 6218 NM_000217.3 2 Episodic ataxia/myokymia syndrome160120

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 108/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
KCNA2 6220 NM_004974.4 3 hereditary spastic paraplegia and ataxiaEpileptic encephalopathy, early infantile,32
KCNB1 6231 NM_004975.4 1-2 EPILEPTIC ENCEPHALOPATHY, EARLYINFANTILE, 26
KCNC1 6233 NM_001112741.2 1-4 EPILEPSY, PROGRESSIVE MYOCLONIC 7
KCNC3 6235 NM_004977.3 1-4 Spinocerebellar ataxia 13
KCND3 6239 NM_004980.4 2-8 Spinocerebellar ataxia 19, 607346
KCNE1 6240 NM_000219.6 4 JERVELL AND LANGE-NIELSENSYNDROME TYPE 2 612347 LONG QT SYNDROME-5 613695
KCNH1 6250 NM_172362.3 1-11 Hypoplasia of terminal phalanges Temple-Baraitser syndrome, 611816
KCNJ10 6256 NM_002241.5 2 Seizures, Sensorineural Deafness,Ataxia, Mental Retardation, andElectrolyte Imbalance Syndrome SESAME syndrome
KCNJ11 6257 NM_000525.3 1 FAMILIAL HYPERINSULINISM 3272 Diabetes, permanent neonatal, with orwithout neurologic features, 606176 DEND syndrome
KCNJ2 6263 NM_000891.3 2 ANDERSEN CARDIODYSRHYTHMICPERIODIC PARALYSIS Cleft palate
KCNJ6 6267 NM_002240.5 2-4 Keppen-Lubinsky syndrome 614098
KCNJ8 6269 NM_004982.4 2-3 Cantu syndrome
KCNK18 19439 NM_181840.1 1-3 MIGRAINE, WITH OR WITHOUT AURA,SUSCEPTIBILITY TO, 13
KCNK3 6278 NM_002246.3 1-2 Pulmonary arterial hypertension

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 109/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
KCNK4 6279 NM_033310.3 2-7 FHEIG (facial dysmorphism,hypertrichosis, epilepsy, intellectualdisability/developmental delay, andgingival overgrowth)
KCNK9 6283 NM_001282534.2 1-2 Birk-Barel mental retardationdysmorphism syndrome 612292
KCNMA1 6284 NM_002247.4 1-27 Cerebellar atrophy, developmentaldelay, and seizures, 617643 Paroxysmal nonkinesigenic dyskinesia,3, with or without generalized epilepsy,609446
KCNN3 6292 NM_002249.6 1-8 ZIMMERMANN-LABAND SYNDROME
KCNQ1 6294 NM_000218.3 1-16 Long QT syndrome 1, 192500 Jervell and Lange-Nielsen syndrome,220400 Short QT syndrome 2, 609621
KCNQ2 6296 NM_172107.4 1-17 Dystonia Epileptic encephalopathy, early infantile,7 Myokymia Seizures, benign neonatal, 1
KCNQ3 6297 NM_004519.4 1-15 Seizures, benign neonatal, 2 OMIM
KCNQ5 6299 NM_001160133.2 1-15 Intellectual Disability with or withoutEpileptic Encephalopathy
KCNT1 18865 NM_020822.3 1-31 Epileptic encephalopathy, early infantile,14 Epilepsy, nocturnal frontal lobe, 5
KCNT2 18866 NM_198503.5 1-28 ?Epileptic encephalopathy, earlyinfantile, 57
KCTD1 18249 NM_001258221.1 4-5 2-5 Scalp-ear-nipple syndrome, 181270
KCTD17 25705 NM_001282684.1 1-9 Dystonia 26, myoclonic

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 110/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
KCTD3 21305 NM_016121.5 1-18 Epilepsy and global developmentaldelay
KCTD7 21957 NM_153033.4 1-4 NEURONAL CEROID LIPOFUSCINOSIS Epilepsy, progressive myoclonic 3, withor without intracellular inclusions OMIM
KDM1A 29079 NM_001009999.3 1-21 Developmental delay and distinctivefacial features
KDM3B 1337 NM_016604.4 1-24 KDM3B-related intellectual disability,short stature and facial dysmorphism Behavioral abnormality Seizures
KDM5B 18039 NM_006618.5 1-27 Mental retardation, autosomalrecessive 65, 618109 Autism
KDM5C 11114 NM_004187.4 1-26 Mental retardation, X-linked, syndromic,Claes-Jensen type, 300534 -3 epilepsy progressive spasticity hypothyroidism
KDM6A 12637 NM_021140.3 1-29 Kabuki syndrome
KDM6B 29012 NM_001080424.2 4-22 KDM6B-related developmental disorder(monoallelic)
KIAA0195 28983 NM_014738.6 2-32 Global developmental delay Abnormal heart morphology Neurodevelopmental Delay CongenitalHeart Defects and Distinct FacialDysmorphism
KIAA0196 28984 NM_014846.4 2-29 Ritscher-Schinzel syndrome, 220210 Spastic paraplegia 8, autosomaldominant, 603563
KIAA0586 19960 NM_001244189.2 1-34 Joubert syndrome Short-rib thoracic dysplasia 14 withpolydactyly
KIAA0753 29110 NM_014804.3 2-19 Joubert syndrome Short-rib skeletal dysplasia Orofaciodigital syndrome XV 617127

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 111/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
KIAA1109 26953 NM_015312.3 1-84 Brain atrophy, Dandy Walker andContractures Alkuraya-Kucinskas syndrome 617822 seizures
KIAA1279 23419 NM_015634.4 1-7 Goldberg-Shprintzen megacolonsyndrome, 609460 (Microcephaly)
KIAA1715 21610 NM_030650.3 2-13 Neurodevelopmental disorder withepilepsy and hypoplasia of the corpuscallosum 618090
KIAA2022 29433 NM_001008537.3 2-4 Intellectual disability and epilepsy
KIDINS220 29508 NM_020738.4 2-30 Spastic paraplegia, intellectual disability,nystagmus, and obesity.
KIF11 6388 NM_004523.4 1-22 Microcephaly with or withoutchorioretinopathy, lymphedema, ormental retardation, 152950
KIF14 19181 NM_014875.3 2-30 Microcephaly 20, primary, autosomalrecessive, 617914
KIF1A 888 NM_004321.7 2-47 Mental Retardation, Dominant Spastic paraplegia 30, autosomalrecessive, 610357 Mental retardation, autosomaldominant 9, 614255, AD Neuropathy, hereditary sensory, typeIIC, 614213
KIF1C 6317 NM_006612.6 22-23 3-23 Spastic ataxia 2,autosomal recessive
KIF22 6391 NM_007317.3 1-14 Spondyloepimetaphyseal dysplasia withjoint laxity, type 2, 603546
KIF2A 6318 NM_001098511.2 1-21 Cortical dysplasia, complex, with otherbrain malformations 3
KIF5A 6323 NM_004984.4 1-28 Myoclonus, intractable, neonatal,617235 intellectual disability Spastic paraplegia 10, autosomaldominant, 604187
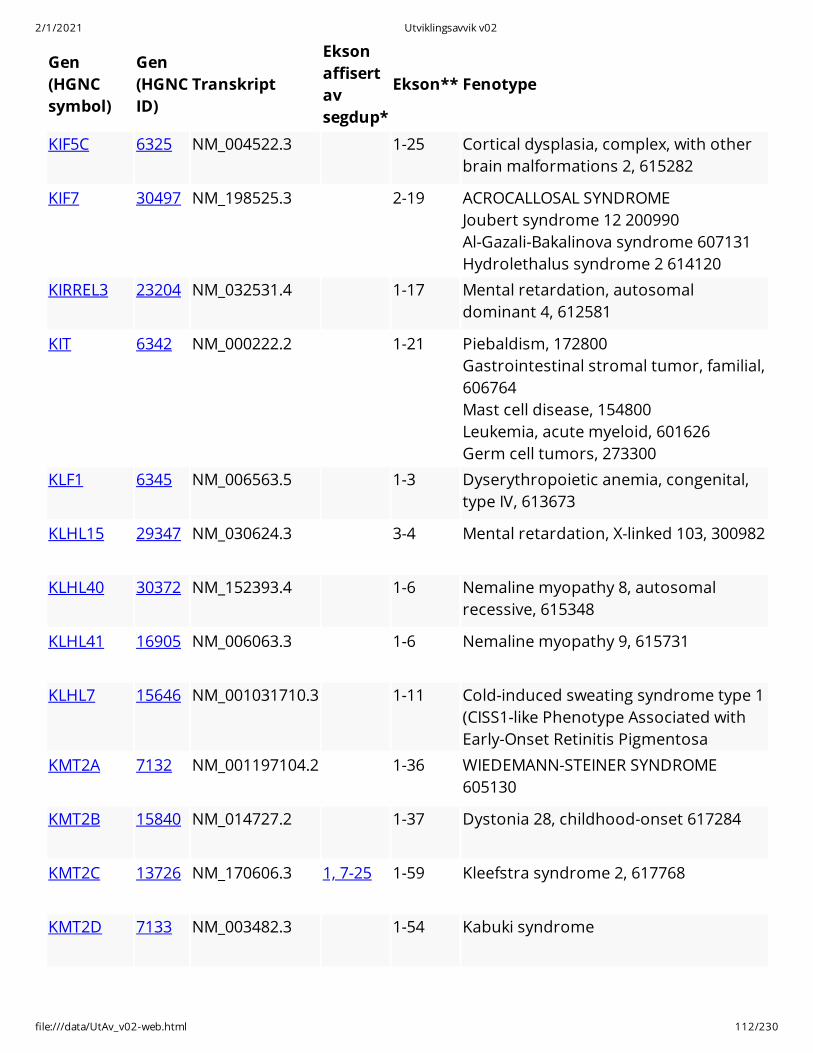
2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 112/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
KIF5C 6325 NM_004522.3 1-25 Cortical dysplasia, complex, with otherbrain malformations 2, 615282
KIF7 30497 NM_198525.3 2-19 ACROCALLOSAL SYNDROME Joubert syndrome 12 200990 Al-Gazali-Bakalinova syndrome 607131 Hydrolethalus syndrome 2 614120
KIRREL3 23204 NM_032531.4 1-17 Mental retardation, autosomaldominant 4, 612581
KIT 6342 NM_000222.2 1-21 Piebaldism, 172800 Gastrointestinal stromal tumor, familial,606764 Mast cell disease, 154800 Leukemia, acute myeloid, 601626 Germ cell tumors, 273300
KLF1 6345 NM_006563.5 1-3 Dyserythropoietic anemia, congenital,type IV, 613673
KLHL15 29347 NM_030624.3 3-4 Mental retardation, X-linked 103, 300982
KLHL40 30372 NM_152393.4 1-6 Nemaline myopathy 8, autosomalrecessive, 615348
KLHL41 16905 NM_006063.3 1-6 Nemaline myopathy 9, 615731
KLHL7 15646 NM_001031710.3 1-11 Cold-induced sweating syndrome type 1(CISS1-like Phenotype Associated withEarly-Onset Retinitis Pigmentosa
KMT2A 7132 NM_001197104.2 1-36 WIEDEMANN-STEINER SYNDROME605130
KMT2B 15840 NM_014727.2 1-37 Dystonia 28, childhood-onset 617284
KMT2C 13726 NM_170606.3 1, 7-25 1-59 Kleefstra syndrome 2, 617768
KMT2D 7133 NM_003482.3 1-54 Kabuki syndrome

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 113/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
KMT2E 18541 NM_182931.3 3-27 Neurodevelopmental disorder andEpilepsy 618512 O'Donnell-Luria-Rodan syndrome,618512
KPTN 6404 NM_007059.4 1-12 Mental retardation, autosomalrecessive 4,1615637
KRAS 6407 NM_004985.5 5 2-5 Cardiofaciocutaneous syndrome 2,615278
KRIT1 1573 NM_194456.1 5-20 CEREBRAL CAVERNOUSMALFORMATIONS TYPE 1 116860
KRT74 28929 NM_175053.4 1-9 HYPOTRICHOSIS SIMPLEX OF THE SCALP2 613981
KY 26576 NM_178554.6 1-11 congenital myopathy
KYNU 6469 NM_003937.3 2-14 Hydroxykynureninuria (Disorders ofhistidine, tryptophan or lysinemetabolism) VACTERL-like phenotype multiple congenital malformations
L1CAM 6470 NM_000425.5 1-28 Hereditary spastic paraplegia X-linked hydrocephalus, MASAsyndrome, 303350 Corpus callosum, partial agenesis of CRASH syndrome Hydrocephalus with congentialidiopathic intestinal pseudoobstruction307000 MASA syndrome
L2HGDH 20499 NM_024884.3 1-10 L-2-hydroxyglutaric aciduria, 236792
LAMA1 6481 NM_005559.4 1-63 CEREBELLAR DYSPLASIA WITH CYSTSWITH OR WITHOUT RETINAL DYSTROPHYAUTOSOMAL RECESSIVE MENTALRETARDATION

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 114/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
LAMA2 6482 NM_000426.3 1-65 Muscular dystrophy, congenital,merosin deficient or partially deficient,607855
LAMB1 6486 NM_002291.3 2-34 COBBLESTONE BRAIN MALFORMATIONWITHOUT MUSCULAR OR OCULARABNORMALITIES Lissencephaly 5
LAMC3 6494 NM_006059.4 1-28 Cortical malformations, occipital,614115
LAMP2 6501 NM_002294.3 1-9 Danon disease
LARGE 6511 NM_004737.6 3-16 Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies), type A, 6,613154 Muscle-eye-brain disease Walker-Warburg syndrome N-acetylglucosaminyltransferase-likeprotein deficiency (Disorders of proteinO-glycosylation, O-mannosylglycansynthesis deficiencies)
LARP7 24912 NM_016648.4 2-13 Alazami syndrome, 615071(Microcephaly and short stature) Primordial dwarfism
LARS 6512 NM_020117.11 1-32 ?Infantile liver failure syndrome 1,615438
LARS2 17095 NM_015340.4 3-22 Perrault syndrome Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis)
LBR 6518 NM_002296.4 2-14 Greenberg skeletal dysplasia 215140 mesomelia rhizomelia Polydactyly

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 115/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
LCAT 6522 NM_000229.2 1-6 Norum disease/LCAT deficiency, 245900 Fish-eye disease, 136120 Lecithin cholesterol acyltransferasedeficiency (Disorders of high densitylipoprotein metabolism)
LCT 6530 NM_002299.4 1-17 Lactose intolerance (Othercarbohydrate disorders) Lactase deficiency, congenital, 223000
LDB3 15710 NM_001080116.1 1-8 Myopathy, myofibrillar, 4, 609452 Left ventricular noncompaction 3, withor without dilated cardiomyopathy,601493
LDHA 6535 NM_005566.4 2-8 Muscle LDH deficiency (Glycogenstorage disorders)
LDLR 6547 NM_000527.5 1-18 Hypercholesterolemia, familial, 1 143890LDL cholesterol level QTL2 143890
LDLRAP1 18640 NM_015627.3 1-9 Hypercholesterolemia, familial, 4 603813
LEMD2 21244 NM_181336.4 1-9 Nuclear Envelopathy with EarlyProgeroid Appearance
LEMD3 28887 NM_014319.5 1-13 Osteopoikilosis, 166700 Buschke-Ollendorff syndrome, 166700 Melorheostosis with osteopoikilosis,155950
LEPRE1 19316 NM_022356.4 1-15 OSTEOGENESIS IMPERFECTA, TYPE VIII610915
LFNG 6560 NM_001040167.2 1-8 ?Spondylocostal dysostosis 3,autosomal recessive, 609813
LGI1 6572 NM_005097.4 1-8 Epilepsy, familial temporal lobe, 1600512
LGI4 18712 NM_139284.3 1-9 Arthrogryposis multiplex congenita,neurogenic, with myelin defect, 617468 Intellectual disability
LHX3 6595 NM_014564.5 1-6 PITUITARY HORMONE DEFICIENCYCOMBINED TYPE 3 221750

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 116/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
LHX4 21734 NM_033343.4 1-6 Pituitary hormone deficiency,combined, 4, 262700
LIAS 16429 NM_006859.4 1-11 Pyruvate dehydrogenase lipoic acidsynthetase deficiency, 614462 Neonatal-onset epilepsy, defectivemitochondrial energy metabolism, andglycine elevation
LIFR 6597 NM_002310.6 2-20 Stuve-Wiedemann syndrome/Schwartz-Jampel type 2 syndrome 601559
LIG4 6601 NM_002312.3 2 SEVERE COMBINED IMMUNODEFICIENCYAUTOSOMAL RECESSIVE T-CELL-NEGATIVE/B-CELL-NEGATIVE/NK-CELL-POSITIVE WITH SENSITIVITY TO IONIZINGRADIATION 602450 microcephalic primordial dwarfism LIG4 syndrome, 606593
LINGO1 21205 NM_032808.7 1-2 LINGO1 related intellectual disabilitywith microcephaly, speech and motordelay
LINS 30922 NM_001040616.3 2-7 Mental retardation, autosomalrecessive 27, 614340
LIPA 6617 NM_000235.4 2-10 Neonatal and Adult Cholestasis lysosomal acid lipase deficiency Wolman disease 278000 Cholesteryl ester storage disease
LIPC 6619 NM_000236.3 1-9 Hepatic lipase deficiency, 614025
LIPN 23452 NM_001102469.1 1-9 ICHTHYOSIS, LAMELLAR, 4 613943
LIPT1 29569 NM_145199.3 2 Lipoyltransferase 1 deficiency Leigh syndrome with secondarydeficiency for pyruvate and alpha-ketoglutarate dehydrogenase.

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 117/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
LIPT2 37216 NM_001144869.3 1-2 Encephalopathy, neonatal severe, withlactic acidosis and brain abnormalities,617668 Mitochondrial Lipoylation DefectAssociated with Severe NeonatalEncephalopathy
LMBR1 13243 NM_022458.4 1-17 Syndactyly, type IV 186200 Laurin-Sandrow syndrome,135750 Acheiropody 200500 Laurin-Sandrow syndrome 135750 Polydactyly, preaxial type II 174500 Hypoplastic or aplastic tibia withpolydactyly 188740 Triphalangeal Thumb-PolysyndactylySyndrome Acheiropody 200500
LMBRD1 23038 NM_018368.4 1-16 Methylmalonic aciduria andhomocystinuria, cblF type, 277380
LMNA 6636 NM_170707.4 1-12 Cardiomyopathy, dilated, 1A 115200 Hutchinson-Gilford progeria 176670 Mandibuloacral dysplasia 248370 Lipodystrophy, familial partial, 2 151660 Restrictive dermopathy, lethal 275210 Cardiomyopathy, dilated, 1A, 115200 Lipodystrophy, familial partial, 2, 151660Emery-Dreifuss muscular dystrophy 3,AR, 181350 Charcot-Marie-Tooth disease, type 2B1,605588 Mandibuloacral dysplasia, 248370 Hutchinson-Gilford progeria, 176670 Restrictive dermopathy, lethal, 275210 Malouf syndrome, 212112 Limb-girdle muscular dystrophy
LMNB1 6637 NM_005573.4 1-11 Adult onset autosomal dominantleukodystrophy (ADLD)
LMOD3 6649 NM_198271.4 1-3 Nemaline myopathy 10 616165

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 118/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
LMX1B 6654 NM_002316.4 1-8 Nail Patella syndrome
LONP1 9479 NM_004793.4 1-18 CODAS (Cerebral, Ocular, Dental,Auricular and Skeletal anomalies)syndrome 600373
LPIN1 13345 NM_145693.4 2-20 Myoglobinuria, acute recurrent,autosomal recessive exercise induced myopathy
LPIN2 14450 NM_014646.2 2-20 Majeed syndrome (Chronic recurrentmultifocal osteomyelitis with congenitaldyserythropoietic anemia) 609628
LPL 6677 NM_000237.3 1-10 Familial lipoprotein lipase deficiency(Familial chylomicronaemia, Inheritedhypercholesterolaemias)
LRAT 6685 NM_004744.5 2-3 LEBER CONGENITAL AMAUROSIS 608553
LRBA 1742 NM_006726.4 2-58 CHILDHOOD-ONSETHYPOGAMMAGLOBULINEMIA 614700
LRIG2 20889 NM_014813.3 1-18 UROFACIAL SYNDROME 236730
LRIT3 24783 NM_198506.5 1-4 AUTOSOMAL-RECESSIVE COMPLETECONGENITAL STATIONARY NIGHTBLINDNESS 615058
LRP2 6694 NM_004525.3 1-79 Donnai-Barrow syndrome, 222448
LRP4 6696 NM_002334.4 1-38 Cenani-Lenz syndactyly Syndactyly type 7 Polydactyly Sclerosteosis 2, 614305 Myasthenic syndrome, congenital, 17,616304

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 119/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
LRP5 6697 NM_002335.4 1, 3-9 1-23 Exudative vitreoretinopathy 4 601813 Osteopetrosis, autosomal dominant 1607634 Osteosclerosis 144750 van Buchem disease, type 2 607636 Osteoporosis-pseudoglioma syndrome259770 Hyperostosis, endosteal 144750
LRPPRC 15714 NM_133259.4 1-38 Leigh syndrome, French-Canadian type,220111 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Isolated complex IV deficiency
LRRC56 25430 NM_198075.4 4-14 Mucociliary Clearance and LateralityDefects
LRRC6 16725 NM_012472.6 1-12 Ciliary dyskinesia, primary, 19, 614935
LSS 6708 NM_001001438.2 1-22 Alopecia Abnormality of the skin Cataract Microcephaly Seizures Abnormality of the genital system Hypotonia Intellectual disability
LTBP2 6715 NM_000428.3 1-36 Glaucoma 3, primary congenital, D,613086 Microspherophakia Weill-Marchesani syndrome 3, recessive,614819
LTBP3 6716 NM_001130144.2 1-28 Geleophysic dysplasia 3 617809 Dental anomalies and short stature610216 Tooth agenesis, selective, 6, 613097
LYRM4 21365 NM_020408.5 1-3 ?Combined oxidative phosphorylationdeficiency 19, 615595

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 120/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
LYRM7 28072 NM_181705.4 1-5 Mitochondrial complex III deficiency,nuclear type 8 severe encephalopathy, lactic acidosisand profound, isolated cIII deficiency inskeletal muscle
LYST 1968 NM_000081.4 3-53 spastic paraplegia Chediak-Higashi syndrome, 214500
LZTFL1 6741 NM_020347.4 1-10 Polydactyly Bardet-Biedl syndrome 17, 615994
LZTR1 6742 NM_006767.4 1-21 Prenatal hydrops increased nuchal translucency Noonan syndrome
MAB21L1 6757 NM_005584.5 1 Intellectual disability Cerebello-Oculo-Facio-Genital syndrome
MAB21L2 6758 NM_006439.5 1 MICROPHTHALMIA, SYNDROMIC 14
MACF1 13664 NM_012090.5 1-93 Intellectual disability Seizures Lissencephaly 9 with complex brainstemmalformation, 618325
MAF 6776 NM_005360.5 1-2 Ayme-Gripp syndrome: CATARACT,DEAFNESS, INTELLECTUAL DISABILITY,SEIZURES, AND A DOWN SYNDROME-LIKE FACIES
MAFB 6408 NM_005461.5 1 Duane Syndrome, Aberrant ExtraocularMuscle Innervation, and Inner-EarDefects
MAG 6783 NM_002361.4 3-11 Spastic paraplegia 75, autosomalrecessive, 616680
MAGEL2 6814 NM_019066.5 1 Schaaf-Yang syndrome ARTHROGRYPOSIS MULTIPLEXCONGENITA Prader-Willi-Like syndrome
MAGI2 18957 NM_012301.4 1-22 Nephrotic syndrome, type 15 617609 Infantile Spasms

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 121/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MAGT1 28880 NM_032121.5 1-10 Immunodeficiency, X-linked, withmagnesium defect, Epstein-Barr virusinfection and neoplasia 300853
MAMLD1 2568 NM_005491.4 1-6 X-LINKED HYPOSPADIAS TYPE 2 300758
MAN1B1 6823 NM_016219.5 1-13 MAN1B1-CDG (Disorders of protein N-glycosylation) Mental retardation, autosomalrecessive 15 614202
MAN2B1 6826 NM_000528.4 1-24 Mannosidosis, alpha-, types I and II
MANBA 6831 NM_005908.4 1-17 Mannosidosis, beta, 248510
MAOA 6833 NM_000240.3 1-15 Brunner syndrome, 300615
MAP1B 6836 NM_005909.5 1-7 Intellectual disability
MAP2K1 6840 NM_002755.3 1-11 Cardiofaciocutaneous syndrome 3,615279
MAP2K2 6842 NM_030662.3 1-11 Cardiofaciocutaneous syndrome 4,615280
MAP3K1 6848 NM_005921.2 1-20 46XY sex reversal 6, 613762
MAP3K7 6859 NM_145331.3 1-17 Frontometaphyseal dysplasia 2, 617137 Cardiospondylocarpofacial syndrome
MAPK10 6872 NM_002753.5 3-14 Epileptic Encephalopathy Lennox-Gastaut type
MAPK8IP3 6884 NM_001040439.2 1-31 Abnormal muscle tone Intellectual Disability with Variable BrainAnomalies
MAPKBP1 29536 NM_001128608.2 2-32 Nephronophthisis 20 617271
MAPRE2 6891 NM_014268.4 1-7 Circumferential Skin Creases KunzeType

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 122/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MARS2 25133 NM_138395.4 1 ?Combined oxidative phosphorylationdeficiency 25 Spastic ataxia 3, autosomal recessive
MASP1 6901 NM_139125.3 1-11 3MC SYNDROME 1
MAST1 19034 NM_014975.3 1-26 Global developmental delay, Intellectualdisability, Abnormality of the corpuscallosum, Cerebellar hypoplasia,Abnormality of the cerebral cortex,Seizures
MAT1A 6903 NM_000429.3 1-9 METHIONINE ADENOSYLTRANSFERASEDEFICIENCY Hypermethioninemia, persistent,autosomal dominant, due tomethionine adenosyltransferase I/IIIdeficiency
MATN3 6909 NM_002381.5 1-8 Multiple Epiphyseal Dysplasia,Dominant Disproportionate Short Stature Epiphyseal dysplasia, multiple, 5, 607078Spondyloepimetaphyseal dysplasia,608728
MATR3 6912 NM_199189.2 5, 18 5-18 Distal Myopathy
MBD5 20444 NM_018328.4 6-15 Mental retardation, autosomaldominant 1
MBOAT7 15505 NM_024298.5 2-8 Intellectual Disability Accompanied byEpilepsy and Autistic Features
MBTPS2 15455 NM_015884.4 1-11 IFAP syndrome with or withoutBRESHECK syndrome,308205 Ichthyosis follicularis, atrichia, andphotophobia with or without brainanomalies, retardation, ectodermaldysplasia, skeletal malformations,hirschsprung disease, ear/eyeanomalies, cleft palate/cryptorchidism,and kidney dysplasia/hypoplasia

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 123/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MC2R 6930 NM_000529.2 2 Glucocorticoid deficiency, due to ACTHunresponsiveness, 202200
MCCC1 6936 NM_020166.5 1-19 3-Methylcrotonyl-CoA carboxylase 1deficiency, 210200
MCCC2 6937 NM_022132.5 1-17 3-Methylcrotonyl-CoA carboxylase 2deficiency, 210210
MCEE 16732 NM_032601.4 1-3 Methylmalonyl-CoA epimerasedeficiency (Organic acidurias) metabolic encephalopathy withhyperammonaemia, hypotonia,recurrent episodes of ketoacidosis, liverimpairment, psychomotor retardation,recurrent infections
MCM3AP 6946 NM_003906.5 1-28 Peripheral neuropathy, autosomalrecessive, with or without impairedintellectual development, 618124
MCOLN1 13356 NM_020533.3 1-14 Mucolipidosis IV (Other lysosomaldisorders)
MCPH1 6954 NM_024596.5 1-14 Microcephaly 1, Primary, AutosomalRecessive
MDH2 6971 NM_005918.4 1-9 Epileptic encephalopathy, early infantile,51
MECOM 3498 NM_001105078.4 3-16 Radioulnar Synostosis withAmegakaryocytic Thrombocytopenia
MECP2 6990 NM_004992.3 2-4 Encephalopathy, neonatal severe Angelman syndrome Mental retardation, X-linked syndromic,Lubs type Rett syndrome
MECR 19691 NM_016011.5 1-10 Dystonia, childhood-onset, with opticatrophy and basal ganglia abnormalities617282
MED12 11957 NM_005120.3 1-45 Opitz-Kaveggia syndrome, 305450 Lujan-Fryns syndrome, 309520 Ohdo syndrome, X-linked, 300895

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 124/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MED12L 16050 NM_053002.5 1-43 Motor delay Delayed speech and languagedevelopment Intellectual disability Behavioral abnormality Abnormality of the abdomen Seizures Abnormality of the corpus callosum
MED13 22474 NM_005121.3 1-30 Delayed speech and languagedevelopment Motor delay Intellectual disability Autistic behavior Attention deficit hyperactivity disorder Abnormality of the eye Constipation
MED13L 22962 NM_015335.4 1-31 Mental retardation and distinctive facialfeatures with or without cardiac defects,616789 Cleft palate
MED17 2375 NM_004268.5 1-12 Microcephaly, postnatal progressive,with seizures and brain atrophy, 613668
MED23 2372 NM_015979.4 1-31 Mental retardation, autosomalrecessive 18, 614249
MED25 28845 NM_030973.3 1-18 Basel-Vanagait-Smirin-Yosef syndrome,616449 Congenital cataract-microcephaly-nevusflammeus simplex-severe intellectualdisability syndrome
MEF2C 6996 NM_002397.5 2-11 Mental retardation, stereotypicmovements, epilepsy, and/or cerebralmalformations
MEGF10 29634 NM_032446.3 3-26 Myopathy, areflexia, respiratorydistress, and dysphagia, early-onset,614399
MEGF8 3233 NM_001410.3 1-41 Polydactyly Carpenter syndrome 2 614976

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 125/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MEIS2 7001 NM_170674.5 1-12 Cleft palate, cardiac defects, and mentalretardation
MEOX1 7013 NM_004527.4 1-3 Klippel-Feil syndrome 2 214300
MESDC2 13520 NM_015154.3 1-3 OSTEOGENESIS IMPERFECTA
MESP2 29659 NM_001039958.2 1-2 Spondylocostal dysostosis 2, autosomalrecessive, 608681
METTL23 26988 NM_001080510.4 2-5 Mental retardation, autosomalrecessive 44, 615942
METTL5 25006 NM_014168.4 1-7 Delayed speech and languagedevelopment Intellectual disability Microcephaly
MFF 24858 NM_020194.5 3-11 Encephalopathy due to defectivemitochondrial and peroxisomal fission 2
MFN2 16877 NM_014874.4 3-19 Charcot-Marie-Tooth disease, type 2A2,609260 Disorders of mitochondrial DNAmaintenance and integrity Hereditary motor and sensoryneuropathy VI, 601152
MFRP 18121 NM_031433.4 1-13 NANOPHTHALMOS 2 609549 MICROPHTHALMIA ISOLATED TYPE 5611040
MFSD2A 25897 NM_001136493.3 1-14 Microcephaly 15, primary, autosomalrecessive, 616486
MFSD8 28486 NM_152778.3 2-13 Ceroid lipofuscinosis, neuronal, 7 OMIM
MGAT2 7045 NM_002408.4 1 Congenital disorder of glycosylation,type IIa, 212066 N-acetylglucosaminyltransferasedeficiency (Disorders of protein N-glycosylation)
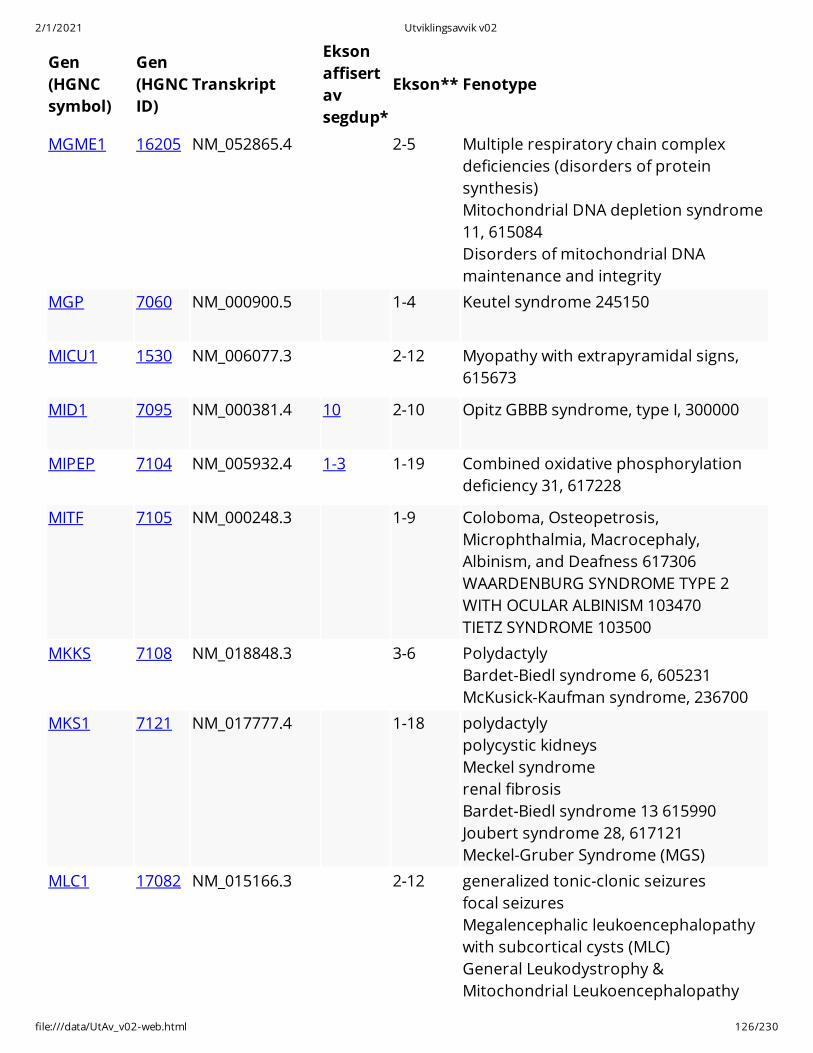
2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 126/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MGME1 16205 NM_052865.4 2-5 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Mitochondrial DNA depletion syndrome11, 615084 Disorders of mitochondrial DNAmaintenance and integrity
MGP 7060 NM_000900.5 1-4 Keutel syndrome 245150
MICU1 1530 NM_006077.3 2-12 Myopathy with extrapyramidal signs,615673
MID1 7095 NM_000381.4 10 2-10 Opitz GBBB syndrome, type I, 300000
MIPEP 7104 NM_005932.4 1-3 1-19 Combined oxidative phosphorylationdeficiency 31, 617228
MITF 7105 NM_000248.3 1-9 Coloboma, Osteopetrosis,Microphthalmia, Macrocephaly,Albinism, and Deafness 617306 WAARDENBURG SYNDROME TYPE 2WITH OCULAR ALBINISM 103470 TIETZ SYNDROME 103500
MKKS 7108 NM_018848.3 3-6 Polydactyly Bardet-Biedl syndrome 6, 605231 McKusick-Kaufman syndrome, 236700
MKS1 7121 NM_017777.4 1-18 polydactyly polycystic kidneys Meckel syndrome renal fibrosis Bardet-Biedl syndrome 13 615990 Joubert syndrome 28, 617121 Meckel-Gruber Syndrome (MGS)
MLC1 17082 NM_015166.3 2-12 generalized tonic-clonic seizures focal seizures Megalencephalic leukoencephalopathywith subcortical cysts (MLC) General Leukodystrophy &Mitochondrial Leukoencephalopathy

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 127/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MLYCD 7150 NM_012213.3 1-5 Malonyl-CoA decarboxylase deficiency(Organic acidurias)
MMAA 18871 NM_172250.3 2-7 Methylmalonic aciduria, vitamin B12-responsive 251100 Defect in adenosylcobalamin synthesis-cbl A (Disorders of cobalaminabsorption, transport and metabolism)
MMAB 19331 NM_052845.4 1-9 Defect in adenosylcobalamin synthesis-cbl B (Disorders of cobalaminabsorption, transport and metabolism) Methylmalonic aciduria, vitamin B12-responsive, due to defect in synthesis ofadenosylcobalamin, cblBcomplementation type 251110
MMACHC 24525 NM_015506.3 1-4 Methylmalonic aciduria andhomocystinuria, cblC type, 277400 Ataxia and hypogonadism
MMADHC 25221 NM_015702.3 2-8 Methylmalonic aciduria andhomocystinuria, cblD type, 277410
MMP13 7159 NM_002427.4 1-10 Spondyloepimetaphyseal dysplasia,Missouri type 602111 Metaphyseal dysplasia, Spahr type -250400
MMP2 7166 NM_004530.6 1-13 Multicentric osteolysis, nodulosis, andarthropathy 259600
MMP21 14357 NM_147191.1 1-7 Heterotaxy, visceral, 7, autosomal OMIM
MMP9 7176 NM_004994.3 1-13 Metaphyseal anadysplasia 2 613073
MN1 7180 NM_002430.3 1-2 MN1 C-terminal truncation syndrome
MNX1 4979 NM_005515.4 1-3 Currarino syndrome 176450
MOCS1 7190 NM_005943.5 1-9 Molybdenum cofactor deficiency, type A,252150

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 128/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MOCS2 7193 NM_176806.4 1-3 Molybdenum cofactor deficiency, type B,252150
MOGS 24862 NM_006302.3 1-4 MOGS-CDG (Disorders of protein N-glycosylation)
MORC2 23573 NM_014941.3 5-27 Axonal type CMT disease type 2Z,616688
MPC1 21606 NM_016098.4 1-5 Mitochondrial pyruvate carrierdeficiency, 614741
MPDU1 7207 NM_004870.4 1-7 Congenital disorder of glycosylation,type If 609180
MPDZ 7208 NM_003829.4 2-46 Hydrocephalus, nonsyndromic,autosomal recessive 2 615219
MPI 7216 NM_002435.3 1-8 Congenital disorder of glycosylation,type Ib 602579 Phosphomannose isomerase deficiency(Disorders of protein N-glycosylation)
MPLKIP 16002 NM_138701.4 1-2 Trichothiodystrophy, nonphotosensitive
MPV17 7224 NM_002437.5 2-8 Mitochondrial DNA depletion syndrome6 (hepatocerebral type) 256810 cholestasis liver failure
MRE11A 7230 NM_005591.3 2-20 Ataxia-telangiectasia-like disorder Nijmegen breakage syndrome-likesevere microcephaly
MRPL3 10379 NM_007208.4 10 1-10 Combined oxidative phosphorylationdeficiency 9, 614582
MRPL44 16650 NM_022915.5 1-4 ?Combined oxidative phosphorylationdeficiency 16, 615395
MRPS14 14049 NM_022100.3 1-3 ?Combined oxidative phosphorylationdeficiency 38, 618378
MRPS16 14048 NM_016065.4 1-3 General Leukodystrophy &Mitochondrial Leukoencephalopathy Combined oxidative phosphorylationdeficiency 2

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 129/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MRPS2 14495 NM_016034.5 1-4 Combined oxidative phosphorylationdeficiency 36 617950 Sensorineural Hearing LossHypoglycemia and Multiple OXPHOSComplex Deficiencies
MRPS22 14508 NM_020191.4 1-8 Combined oxidative phosphorylationdeficiency 5, 611719
MRPS34 16618 NM_023936.2 1-3 Combined oxidativephosphorylationdeficiency 32, 617664 Leigh Syndrome with Instability of theSmall Mitoribosomal Subunit
MRPS7 14499 NM_015971.4 1-5 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis)
MSL3 7370 NM_001282174.1 4-12 Muscular hypotonia Feeding difficulties Neurodevelopmental delay Intellectual disability
MSMO1 10545 NM_006745.5 2-6 Sterol-C4-methyl oxidase deficiency(Disorders of sterol biosynthesis) Microcephaly, congenital cataract, andpsoriasiform dermatitis, 616834
MSTO1 29678 NM_018116.3 1-14 1-14 Myopathy, mitochondrial, and ataxia,617675 Congenital muscular dystrophy withBrain involvment
MSX1 7391 NM_002448.3 1-2 Tooth agenesis, selective, 1, with orwithout orofacial cleft, 106600 Ectodermal dysplasia 3, Witkop type,189500
MSX2 7392 NM_002449.5 2 1-2 Parietal foramina with cleidocranialdysplasia 168550 Craniosynostosis, type 2, 604757
MTFMT 29666 NM_139242.4 1-9 Combined oxidative phosphorylationdeficiency 15 614947 Mitochondrial complex I deficiency,nuclear type 27 618248
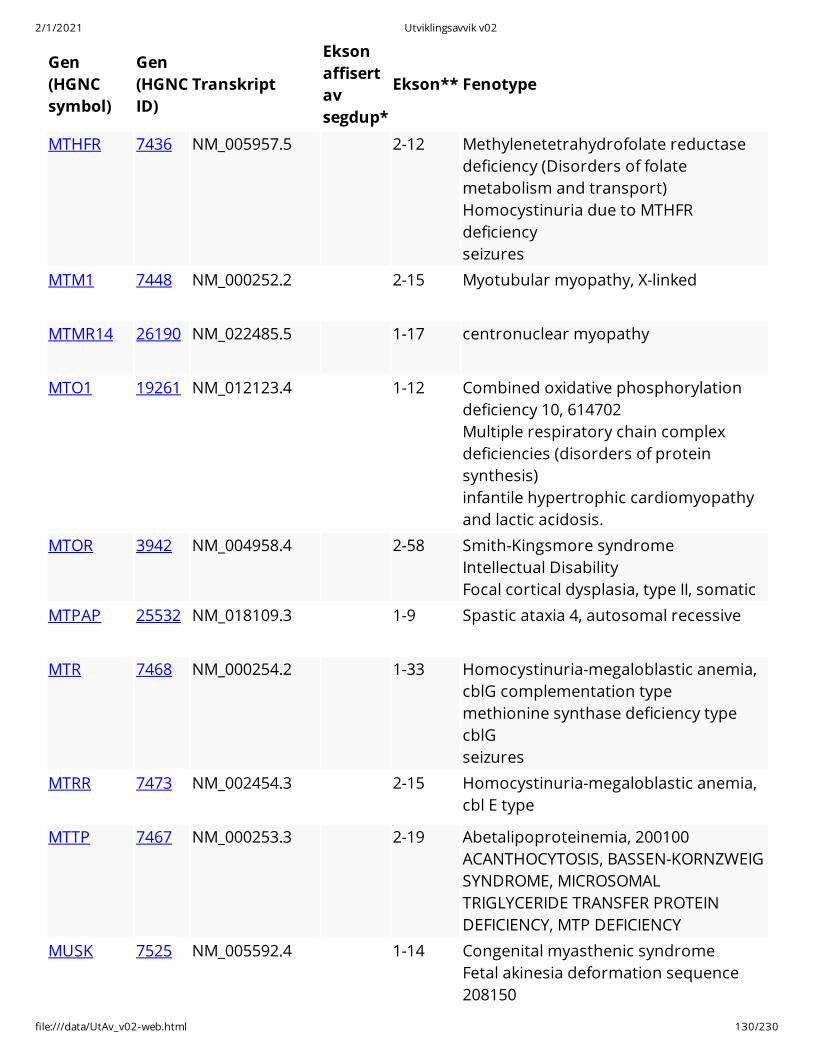
2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 130/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MTHFR 7436 NM_005957.5 2-12 Methylenetetrahydrofolate reductasedeficiency (Disorders of folatemetabolism and transport) Homocystinuria due to MTHFRdeficiency seizures
MTM1 7448 NM_000252.2 2-15 Myotubular myopathy, X-linked
MTMR14 26190 NM_022485.5 1-17 centronuclear myopathy
MTO1 19261 NM_012123.4 1-12 Combined oxidative phosphorylationdeficiency 10, 614702 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) infantile hypertrophic cardiomyopathyand lactic acidosis.
MTOR 3942 NM_004958.4 2-58 Smith-Kingsmore syndrome Intellectual Disability Focal cortical dysplasia, type II, somatic
MTPAP 25532 NM_018109.3 1-9 Spastic ataxia 4, autosomal recessive
MTR 7468 NM_000254.2 1-33 Homocystinuria-megaloblastic anemia,cblG complementation type methionine synthase deficiency typecblG seizures
MTRR 7473 NM_002454.3 2-15 Homocystinuria-megaloblastic anemia,cbl E type
MTTP 7467 NM_000253.3 2-19 Abetalipoproteinemia, 200100 ACANTHOCYTOSIS, BASSEN-KORNZWEIGSYNDROME, MICROSOMALTRIGLYCERIDE TRANSFER PROTEINDEFICIENCY, MTP DEFICIENCY
MUSK 7525 NM_005592.4 1-14 Congenital myasthenic syndrome Fetal akinesia deformation sequence208150

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 131/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MUT 7526 NM_000255.4 2-13 metabolic encephalopathy withhyperammonaemia, hypotonia,recurrent episodes of ketoacidosis, liverimpairment, psychomotor retardation,recurrent infections. Methylmalonyl-CoA mutase deficiency(Organic acidurias)
MVK 7530 NM_000431.4 2-11 Mevalonic aciduria Hyper-IgD syndrome 260920 Porokeratosis 3, multiple types 175900
MYBPC1 7549 NM_002465.4 1-31 Arthrogryposis, distal, type 1B, 614335 Lethal congenital contracture syndrome4, 614915
MYBPC3 7551 NM_000256.3 1-33 Cardiomyopathy, hypertrophic, 4,115197
MYCN 7559 NM_005378.6 2-3 Feingold syndrome (Microcephaly-oculo-digito-esophageal-duodenal)164280
MYF5 7565 NM_005593.3 1-3 External Ophthalmoplegia Rib andVertebral Anomalies
MYH2 7572 NM_017534.6 3-40 Proximal myopathy andophthalmoplegia 605637
MYH3 7573 NM_002470.4 3-41 Arthrogryposis Multiplex Congenita
MYH6 7576 NM_002471.3 3-39 Cardiomyopathy, familial hypertrophic,14, 613251 Atrial septal defect Cardiomyopathy, dilated, 1EE, 613252

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 132/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MYH7 7577 NM_000257.4 3-40 Cardiomyopathy, dilated, 1S Left ventricular noncompaction 5 Cardiomyopathy, hypertrophic, 1 Myopathy, myosin storage, autosomalrecessive Myopathy, myosin storage, autosomaldominant Scapuloperoneal syndrome, myopathictype Laing distal myopathy
MYH8 7578 NM_002472.3 3-40 Carney complex variant, 608837 Trismus-pseudocamptodactylysyndrome (ARTHROGRYPOSIS, DISTAL,TYPE 7
MYH9 7579 NM_002473.5 2-41 May-Hegglin anomaly, 155100 Fechtner syndrome, 153640 Sebastian syndrome, 605249 Deafness, autosomal dominant 17,603622 Epstein syndrome, 153650 Macrothrombocytopenia andprogressive
MYL1 7582 NM_079420.3 1-6 Myopathy, congenital, with fast-twitch(type II) fiber atrophy, 618414
MYLK 7590 NM_053025.4 13-18 4-34 Megacystis Microcolon IntestinalHypoperistalsis Syndrome
MYO18B 18150 NM_032608.7 2-43 KLIPPEL-FEIL SYNDROME 4, AUTOSOMALRECESSIVE, WITH NEMALINE MYOPATHYAND FACIAL DYSMORPHISM
MYO5A 7602 NM_000259.3 1-41 Griscelli syndrome, type 1, 214450 ELEJALDE SYNDROME (ELEJAS)
MYO5B 7603 NM_001080467.3 40 1-40 Microvillus inclusion disease, 251850
MYO7A 7606 NM_000260.4 2-49 USHER SYNDROME TYPE 1B 276900 DEAFNESS AUTOSOMAL RECESSIVE TYPE2 600060

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 133/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
MYO9A 7608 NM_006901.4 2-42 congenital myasthenic syndrome 24,presynaptic 618198
MYOCD 16067 NM_001146312.2 1-14 Congenital megabladder
MYORG 19918 NM_020702.5 2 Basal ganglia calcification, idiopathic, 7,autosomal recessive
MYOT 12399 NM_006790.3 2-10 Muscular dystrophy, limb-girdle, type1A, 159000
MYPN 23246 NM_032578.3 2-20 Childhood-Onset, Slowly ProgressiveNemaline Myopathy
MYRF 1181 NM_001127392.3 1-27 Pulmonary artery and lung hypoplasia,agonadism, omphalocele,diaphragmatic defects, hypoplastic leftheart and scimitar syndrome
MYT1L 7623 NM_015025.4 6-25 Mental retardation, autosomaldominant 39, 616521 obesity
NAA10 18704 NM_003491.4 1-8 N-terminal acetyltransferase deficiency,300855 X-linked anophthalmia syndrome/Lenz OGDEN SYNDROME 300855
NAA15 30782 NM_057175.5 1-20 Mental retardation, autosomaldominant 50, 617787 CONGENITAL HEART DISEASE andNEURODEVELOPMENTAL DISORDER
NACC1 20967 NM_052876.4 2-6 Neurodevelopmental disorder withepilepsy, cataracts, feeding difficulties,and delayed brain myelination, 617393
NADK2 26404 NM_001085411.3 1-12 ?2,4-dienoyl-CoA reductase deficiency616034
NAGA 7631 NM_000262.3 1-9 Kanzaki disease Schindler disease, type I, 609241
NAGLU 7632 NM_000263.4 1-6 MPS IIIB, Sanfilippo B disease(Mucopolysaccharidoses)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 134/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
NAGS 17996 NM_153006.3 1-7 N-Acetylglutamate synthetase deficiency(Urea cycle disorders and inheritedhyperammonaemias)
NALCN 19082 NM_052867.4 2-44 HYPOTONIA, INFANTILE, WITHPSYCHOMOTOR RETARDATION ANDCHARACTERISTIC FACIES 615419 Congenital contractures of the limbsand face, hypotonia, and developmentaldelay 616266
NANS 19237 NM_018946.4 1-6 Spondyloepimetaphyseal dysplasia,Camera-Genevieve type 610442 infantile-onset severe developmentaldelay and skeletal dysplasia
NARS2 26274 NM_024678.6 1-14 Combined oxidative phosphorylationdeficiency 24 seizures
NBAS 15625 NM_015909.4 1-52 Short stature, optic nerve atrophy, andPelger-Huet anomaly, 614800 ACUTE LIVER FAILURE (ALF) IN INFANCYAND CHILDHOOD
NBEA 7648 NM_015678.4 2-17 1-58 NBEA Neurodevelopment disorder withseizures
NBN 7652 NM_002485.4 1-16 NIJMEGEN BREAKAGE SYNDROME251260
NCAPG2 21904 NM_017760.7 2-28 Severe Neurodevelopmental Syndrome Khan-Khan-Katsanis syndrome, 618460
NDE1 17619 NM_001143979.2 3-10 Lissencephaly 4 (with microcephaly),614019
NDP 7678 NM_000266.4 2-3 NORRIE DISEASE Exudative vitreoretinopathy 2, X-linkedOMIM
NDST1 7680 NM_001543.5 2-15 Mental retardation, autosomalrecessive 46, 616116
NDUFA1 7683 NM_004541.4 1-3 Mitochondrial complex I deficiency,252010

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 135/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
NDUFA10 7684 NM_004544.4 1-10 Isolated complex I deficiency Mitochondrial Respiratory ChainComplex I Deficiency Leigh syndrome 256000
NDUFA11 20371 NM_175614.5 1-4 Mitochondrial complex I deficiency,252010
NDUFA12 23987 NM_018838.5 1-4 ?Mitochondrial complex I deficiency,nuclear type 23 618244
NDUFA2 7685 NM_002488.4 1-3 Leigh syndrome due to mitochondrialcomplex I deficiency, 256000
NDUFA4 7687 NM_002489.4 1-4 Isolated complex IV deficiency
NDUFA6 7690 NM_002490.6 1-3 Mitochondrial complex I deficiency,nuclear type 33, 618253
NDUFA9 7693 NM_005002.5 1-11 Leigh syndrome due to mitochondrialcomplex I deficiency, 256000 -3
NDUFAF1 18828 NM_016013.4 2-5 Mitochondrial complex I deficiency,252010 Mitochondrial Leukoencephalopathy
NDUFAF2 28086 NM_174889.5 1-4 Mitochondrial complex I deficiency,252010 Leigh syndrome, 256000 Mitochondrial complex I deficiency252010
NDUFAF3 29918 NM_199069.2 1-5 Mitochondrial complex I deficiency,252010
NDUFAF4 21034 NM_014165.4 3 1-3 Mitochondrial complex I deficiency,252010
NDUFAF5 15899 NM_024120.5 1-11 Mitochondrial complex 1 deficiency,252010
NDUFAF6 28625 NM_152416.4 1-9 Leigh syndrome due to mitochondrialcomplex I deficiency, 256000
NDUFAF7 28816 NM_001083946.2 1-8 No OMIM phenotype

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 136/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
NDUFB11 20372 NM_019056.6 1-3 Linear skin defects with multiplecongenital anomalies 3 Isolated complex I deficiency
NDUFB3 7698 NM_002491.3 2-3 Mitochondrial complex I deficiency,252010
NDUFB8 7703 NM_005004.4 1-5 Mitochondrial complex I deficiency,nuclear type 32, 618252
NDUFB9 7704 NM_005005.3 1-4 ?Mitochondrial complex I deficiency,252010
NDUFS1 7707 NM_005006.7 2-19 Mitochondrial complex I deficiency,252010 LEIGH SYNDROME
NDUFS2 7708 NM_004550.4 2-15 Mitochondrial complex I deficiency,252010 Mitochondrial Leukoencephalopathy Leigh syndrome
NDUFS3 7710 NM_004551.3 1-7 Mitochondrial Respiratory ChainComplex I Deficiency
NDUFS4 7711 NM_002495.4 1-5 Leigh syndrome, 256000 Mitochondrial complex I deficiency,252010
NDUFS6 7713 NM_004553.5 1-4 Mitochondrial complex I deficiency252010
NDUFS7 7714 NM_024407.5 1-8 Mitochondrial complex I deficiency,nuclear type 3 OMIM General Leukodystrophy &Mitochondrial Leukoencephalopathy Leigh syndrome
NDUFS8 7715 NM_002496.4 2-7 Leigh syndrome due to mitochondrialcomplex I deficiency Mitochondrial Leukoencephalopathy
NDUFV1 7716 NM_007103.4 1-10 Mitochondrial Leukoencephalopathy
NDUFV2 7717 NM_021074.5 1-8 Mitochondrial complex I deficiency,252010

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 137/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
NEB 7720 NM_001271208.2 82-105 3-183 nemaline myopathy
NECAP1 24539 NM_015509.4 7-8 1-8 Epileptic encephalopathy, early infantile,21, 615833
NEDD4L 7728 NM_015277.6 1-30 Cleft palate, toe syndactyly,periventricular nodular heterotopia
NEFL 7739 NM_006158.4 1-5 Nemaline Myopathy
NEK1 7744 NM_012224.3 2-34 Short rib thoracic dysplasia 6 with orwithout polydactyly - 263520
NEK8 13387 NM_178170.3 1-15 Renal-hepatic-pancreatic dysplasia Nephronophthisis
NEU1 7758 NM_000434.4 1-6 Sialidosis (Oligosaccharidoses) Mucolipidosis, Type I
NF1 7765 NM_000267.3 9-11, 13-29, 31-35
1-57 Neurofibromatosis, type 1
NFASC 29866 NM_015090.3 3-27 Neurodevelopmental disorder withcentral and peripheral motordysfunction 618356
NFIA 7784 NM_005595.5 1-10 Brain malformations with or withouturinary tract defects, 613735
NFIB 7785 NM_001190737.2 1-11 Macrocephaly, acquired, with impairedintellectual development, 618286
NFIX 7788 NM_001271043.2 1-11 Marshall-Smith syndrome 602535 Sotos syndrome 2 614753
NFU1 16287 NM_001002755.4 1-8 Multiple mitochondrial dysfunctionssyndrome 1
NGLY1 17646 NM_018297.4 1-12 Alacrimia-choreoathetosis-liverdysfunction syndrome Congenital disorder of deglycosylation,615273

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 138/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
NHEJ1 25737 NM_024782.2 2-8 Severe combined immunodeficiencywith microcephaly, growth retardation,and sensitivity to ionizing radiation611291
NHLRC1 21576 NM_198586.3 1 Epilepsy, progressive myoclonic 2B(Lafora)
NHP2 14377 NM_017838.3 1-4 DYSKERATOSIS CONGENITA,AUTOSOMAL RECESSIVE 2 613987
NHS 7820 NM_198270.4 1-8 CATARACT CONGENITAL X-LINKED302200 NANCE-HORAN SYNDROME (NHS)
NIPA1 17043 NM_144599.5 1-5 Spastic paraplegia 6, autosomaldominant, 600363
NIPBL 28862 NM_133433.4 2-47 upper limb anomalies Dislocation of the radial head CORNELIA DE LANGE SYNDROME 1
NKAP 29873 NM_024528.4 1-9 Global developmental delay Intellectual disability
NKX2-1 11825 NM_001079668.3 1-3 Goiter, familial, due to TTF-1 defect(1)Chorea, hereditary benign,118700Choreoathetosis,hypothyroidism, and neonatalrespiratory distress, 610978
NKX2-5 2488 NM_004387.4 1-2 ATRIAL SEPTAL DEFECT WITHATRIOVENTRICULAR CONDUCTIONDEFECTS 108900 CONGENITAL HYPOTHYROIDISM NON-GOITROUS TYPE 5 225250
NKX3-2 951 NM_001189.4 1-2 Spondylo-megaepiphyseal-metaphysealdysplasia, 613330
NKX6-2 19321 NM_177400.3 1-3 Spastic ataxia 8, autosomal recessive,with hypomyelinating leukodystrophy,617560
NLGN3 14289 NM_018977.4 2-7 AUTISM SPECTRUM DISORDERS

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 139/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
NLRP3 16400 NM_004895.4 1-9 Chronic infantile neurologic cutaneousarticular syndrome (CINA) - 607115 CINCA (Infantile-onset multisysteminflammatory disease) 607115
NMNAT1 17877 NM_022787.4 2-5 Leber congenital amaurosis 9, 608553
NNT 7863 NM_012343.4 2-22 Glucocorticoid deficiency 4, with orwithout mineralocorticoid deficiency,614736
NODAL 7865 NM_018055.5 1-3 Heterotaxy, visceral, 5, 270100
NOG 7866 NM_005450.6 1 Brachydactyly, type B2 611377 Multiple synostoses syndrome 1 186500Symphalangism, proximal, 185800 Tarsal-carpal coalition syndrome,186570 Stapes ankylosis with broad thumb andtoes, 184460
NONO 7871 NM_001145408.2 4-13 Mental retardation, X-linked, syndromic34, 300967 Macrocephaly-intellectual disability-leftventricular non compaction syndrome
NOP56 15911 NM_006392.4 1-12 Spinocerebellar ataxia 36, 614153
NOTCH1 7881 NM_017617.5 1-34 Adams-Oliver syndrome 5, 616028 Combination of aplasia cutis congenitaof the scalp vertex and terminaltransverse limb defects (e.g.,amputations, syndactyly, brachydactyly,or oligodactyly)
NOTCH2 7882 NM_024408.4 1-4 1-34 Neonatal and Adult Cholestasis Alagille syndrome 2 Hajdu-Cheney (Serpentine fibulapolycystic kidney) syndrome 102500
NOVA2 7887 NM_002516.4 1-4 Intellectual disability withataxia/spasticity
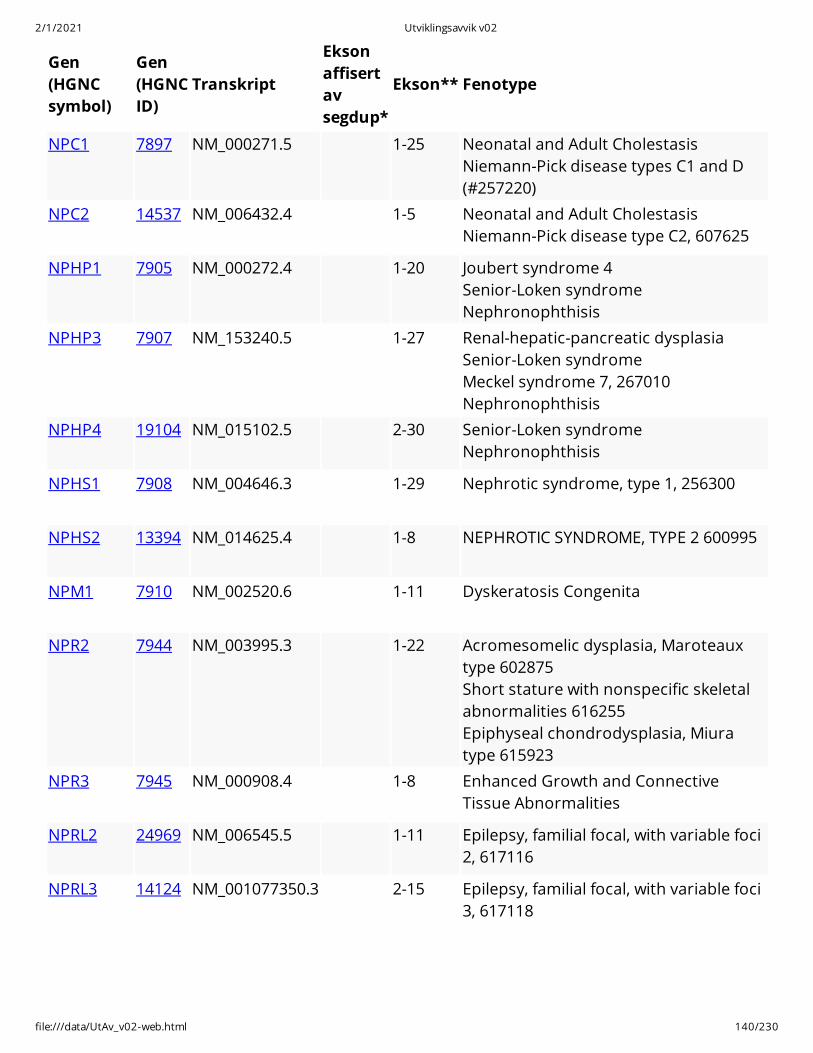
2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 140/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
NPC1 7897 NM_000271.5 1-25 Neonatal and Adult Cholestasis Niemann-Pick disease types C1 and D(#257220)
NPC2 14537 NM_006432.4 1-5 Neonatal and Adult Cholestasis Niemann-Pick disease type C2, 607625
NPHP1 7905 NM_000272.4 1-20 Joubert syndrome 4 Senior-Loken syndrome Nephronophthisis
NPHP3 7907 NM_153240.5 1-27 Renal-hepatic-pancreatic dysplasia Senior-Loken syndrome Meckel syndrome 7, 267010 Nephronophthisis
NPHP4 19104 NM_015102.5 2-30 Senior-Loken syndrome Nephronophthisis
NPHS1 7908 NM_004646.3 1-29 Nephrotic syndrome, type 1, 256300
NPHS2 13394 NM_014625.4 1-8 NEPHROTIC SYNDROME, TYPE 2 600995
NPM1 7910 NM_002520.6 1-11 Dyskeratosis Congenita
NPR2 7944 NM_003995.3 1-22 Acromesomelic dysplasia, Maroteauxtype 602875 Short stature with nonspecific skeletalabnormalities 616255 Epiphyseal chondrodysplasia, Miuratype 615923
NPR3 7945 NM_000908.4 1-8 Enhanced Growth and ConnectiveTissue Abnormalities
NPRL2 24969 NM_006545.5 1-11 Epilepsy, familial focal, with variable foci2, 617116
NPRL3 14124 NM_001077350.3 2-15 Epilepsy, familial focal, with variable foci3, 617118

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 141/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
NR1H4 7967 NM_005123.4 3-11 Neonatal and Adult Cholestasis Cholestasis, Progressive FamilialIntrahepatic 5 ciliopathy
NR2F1 7975 NM_005654.6 1-3 BOSCH-BOONSTRA OPTIC ATROPHYSYNDROME
NR2F2 7976 NM_021005.4 1-3 CONGENITAL HEART DEFECTS and XXsex reversal
NR4A2 7981 NM_006186.4 3-8 Intellectual disability
NR5A1 7983 NM_004959.5 2-7 46XY sex reversal 3, 612965 Premature ovarian failure 7, 612964 Adrenocortical insufficiency Spermatogenic failure 8, 613957
NRAS 7989 NM_002524.5 2-5 NOONAN SYNDROME TYPE 6
NRXN1 8008 NM_001135659.2 2-24 AUTISM Pitt-Hopkins-like syndrome 2, 614325
NRXN2 8009 NM_138732.3 2-20 intellectual disability
NSD1 14234 NM_022455.4 2-23 Sotos syndrome 1 117550
NSDHL 13398 NM_015922.3 2-8 Congenital hemidysplasia withichtyosiform erythroderma and limbdefects (Disorders of sterolbiosynthesis)
NSUN2 25994 NM_017755.6 1-19 Mental Retardation, Recessive
NSUN3 26208 NM_022072.5 1-6 Combined mitochondrial respiratorychain complex deficiency
NT5C2 8022 NM_012229.4 3-19 Spastic paraplegia 45, autosomalrecessive, 613162 Intellectual disability

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 142/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
NT5C3A 17820 NM_016489.13 2-10 Anemia, hemolytic, due to UMPH1deficiency, 266120 Uridine-5 -monophosphate hydrolasesuperactivity (Disorders of pyrimidinemetabolism)
NTNG2 14288 NM_032536.4 2-8 Central hypotonia Global developmental delay Intellectual disability Microcephaly Seizures
NTRK1 8031 NM_001012331.1 1-16 Insensitivity to pain, congenital, withanhidrosis, 256800
NTRK2 8032 NM_006180.4 4-21 Obesity, hyperphagia, anddevelopmental delay, 613886 Epileptic encephalopathy, early infantile,58 (MIM 617830)
NUBPL 20278 NM_025152.3 1-11 Mitochondrial complex I deficiency,252010 Mitochondrial Leukoencephalopathy
NUDT2 8049 NM_001161.5 4-5 Intellectual disability
NUP107 29914 NM_020401.4 1-28 GALLOWAY-MOWAT SYNDROME 7,618348 EARLY-CHILDHOOD-ONSET STEROID-RESISTANT NEPHROTIC SYNDROME
NUP133 18016 NM_018230.3 1-26 GALLOWAY-MOWAT SYNDROME 8,618349
NUP214 8064 NM_005085.4 1-36 Acute Febrile Encephalopathy 618426
NUP62 8066 NM_001193357.2 2 Striatonigral degeneration, infantile,271930 Intellectual disability

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 143/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
NUS1 21042 NM_138459.5 5 1-5 Epilepsy and intellectual disability ?Congenital disorder of glycosylation,type 1aa, 617082 Mental retardation, autosomaldominant 55, with seizures, 617831 Abnormality of extrapyramidal motorfunction
NXN 18008 NM_022463.5 1-8 Robinow syndrome, autosomalrecessive 2, 618529
NYX 8082 NM_022567.2 1-2 Night blindness, congenital stationary(complete), 1A, X-linked, 310500
OAT 8091 NM_000274.4 2-10 Ornithine aminotransferase deficiency(Disorders of ornithine or prolinemetabolism) Gyrate atrophy of choroid and retinawith or without ornithinemia
OBFC1 26200 NM_024928.5 2-10 Cerebroretinal microangiopathy withcalcifications and cysts 2
OBSL1 29092 NM_015311.3 1-21 3-M syndrome 2 612921
OCLN 8104 NM_002538.4 5-9 2-9 Band-like calcification with simplifiedgyration and polymicrogyria Pseudo-TORCH syndrome 1 251290
OCRL 8108 NM_000276.4 1-24 Dent disease 2 300555 Lowe syndrome 309000
ODC1 8109 NM_002539.3 3-12 Global developmental delay Intellectual disability Macrocephaly Alopecia Ectodermal dysplasia
OFD1 2567 NM_003611.3 1-23 Simpson-Golabi-Behmel syndrome, type2 300209 XLR Orofaciodigital syndrome I 311200 XLD Joubert syndrome 10
OGT 8127 NM_181672.3 1-22 Mental retardation, X-linked 106, 300997

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 144/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
OPA1 8140 NM_015560.2 1-28 Optic atrophy plus syndrome, 125250 Progressive External Ophthalmoplegiawith Mitochondrial DNA Deletions
OPA3 8142 NM_025136.4 1-2 Cognitive regression Methylglutaconic aciduria type III,Costeff syndrome (Organic acidurias)
OPHN1 8148 NM_002547.3 2-24 Mental retardation, X-linked, withcerebellar hypoplasia and distinctivefacial appearance, 300486
OPLAH 8149 NM_017570.5 2-28 5-oxoprolinase deficiency 260005
ORAI1 25896 NM_032790.3 1-2 Myopathy, tubular aggregate, 1 160565
ORC1 8487 NM_004153.4 2-17 Meier-Gorlin syndrome 1 microcephalic primordial dwarfism
ORC4 8490 NM_002552.5 2-14 Meier-Gorlin syndrome 2 613800 microcephalic primordial dwarfism
ORC6 17151 NM_014321.4 1-7 Meier-Gorlin syndrome 3, 613803 microcephalic primordial dwarfism
OSGEP 18028 NM_017807.4 1-11 Galloway-Mowat syndrome 3, 617729 Intellectual disability Nephrotic syndrome with primarymicrocephaly
OSTM1 21652 NM_014028.4 1-6 Osteopetrosis, autosomal recessive 5259720
OTC 8512 NM_000531.6 1-10 Ornithine transcarbamylase deficiency(Urea cycle disorders and inheritedhyperammonaemias)
OTOGL 26901 NM_173591.3 1-58 Deafness, autosomal recessive 84B,614944
OTUD6B 24281 NM_016023.5 1-7 Intellectual developmental disorder withdysmorphic facies, seizures, and distallimb anomalies, 617452
OTX2 8522 NM_001270524.2 2-4 MICROPHTHALMIA SYNDROMIC TYPE 5(MCOPS5)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 145/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
OXCT1 8527 NM_000436.4 1-17 Succinyl-CoA:3-Oxoacid-CoA transferase(SCOT) deficiency (Disorders of ketonebody metabolism)
P4HB 8548 NM_000918.4 1-11 Cole-Carpenter syndrome 1
P4HTM 28858 NM_177939.3 1-9 Central hypotonia, Muscular hypotonia,Global developmental delay, Intellectualdisability, Seizures, Abnormality of theeye, Hypoventilation, Sleep apnea,Dysautonomia Hypotonia, hyperventilation, impairedintellectual development,dysautonomia, epilepsy, and eyeabnormalities, 618493
PACS1 30032 NM_018026.4 1-24 Mental retardation, autosomaldominant 17, 615009 Schuurs-Hoeijmakers syndrome, 615009
PACS2 23794 NM_001100913.3 1-25 Epileptic encephalopathy, early infantile,66, 618067 Global developmental delay Intellectual disability Abnormality of the cerebellum
PAFAH1B1 8574 NM_000430.4 2-11 Cerebral Malformation Disorders Lissencephaly/Subcortical BandHeterotopia
PAH 8582 NM_000277.3 1-13 Phenylketonuria
PAK1 8590 NM_001128620.1 2-16 Intellectual developmental disorder withmacrocephaly, seizures, and speechdelay, 618158
PAK3 8592 NM_002578.5 5-18 AGENESIS OF THE CORPUS CALLOSUM Mental Retardation, X-linked
PALB2 26144 NM_024675.4 1-13 Fanconi anemia, complementationgroup N, 610832 Radial Ray abnormality
PAM16 29679 NM_016069.11 1-5 Spondylometaphyseal dysplasia,Megarbane-Dagher-Melike type 613320

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 146/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PANK2 15894 NM_153638.3 1-7 Neurodegeneration with brain ironaccumulation 234200 Dystonia HARP syndrome OMIM
PAPSS2 8604 NM_001015880.2 1-13 Brachyolmia 4 with mild epiphyseal andmetaphyseal changes, 612847
PARK2 8607 NM_004562.3 1-12 Dystonia Parkinson disease, juvenile, type 2,600116
PARN 8609 NM_002582.4 24 1-24 Dyskeratosis congenita, autosomalrecessive 6
PARS2 30563 NM_152268.4 2 Epileptic encephalopathy, early infantile,75, 618437 Alpers syndrome
PAX2 8616 NM_003987.5 1-11 RENAL-COLOBOMA SYNDROME 120330
PAX3 8617 NM_181457.4 1-8 Craniofacial-deafness-hand syndrome,122880 Waardenburg syndrome, type 1, 193500Waardenburg syndrome, type 3, 148820
PAX6 8620 NM_000280.4 4-13 Aniridia, Cerebellar Ataxia, And MentalRetardation COLOBOMA OF OPTIC NERVE 120430 FOVEAL HYPOPLASIA 136520 BILATERAL OPTIC NERVE HYPOPLASIA165550 KERATITIS HEREDITARY 148190 PETERS ANOMALY 604229
PAX7 8621 NM_002584.3 1-8 Hypotonia Ptosis Delayed motor milestones Myopathy, congenital, progressive, withscoliosis, 618578
PAX8 8622 NM_003466.4 2-12 Hypothyroidism, congenital, due tothyroid dysgenesis or hypoplasia,218700

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 147/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PAX9 8623 NM_006194.3 2-5 Tooth agenesis, selective, 3, 604625
PBX1 8632 NM_002585.4 1-9 Congenital anomalies of kidney andurinary tract syndrome with or withouthearing loss, abnormal ears, ordevelopmental delay
PC 8636 NM_000920.4 3-22 Pyruvate carboxylase deficiency(Disorders of gluconeogenesis) lactic acidosis, hypotonia,encephalopathy
PCBD1 8646 NM_000281.4 1-4 Hyperphenylalaninemia, BH4-deficient,D
PCCA 8653 NM_000282.4 1-24 metabolic encephalopathy withhyperammonaemia, hypotonia,recurrent episodes of ketoacidosis, liverimpairment, psychomotor retardation,recurrent infections Propionic aciduria (Organic acidurias)
PCCB 8654 NM_000532.5 1-15 Propionic aciduria (Organic acidurias)
PCDH12 8657 NM_016580.3 1-4 intellectual disability microcephaly epilepsy perithalamic hyperechogenicity periventricular hyperechogenicity midbrain abnormalities hypothalamic abnormalities Microcephaly, seizures, spasticity, andbrain calcification 251280
PCDH19 14270 NM_001184880.2 1-6 Epileptic encephalopathy, early infantile,9
PCGF2 12929 NM_007144.3 3-11 Craniofacial NeurologicalCardiovascular and Skeletal Features Intellectual disability dysmorphic features Global developmental delay

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 148/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PCK1 8724 NM_002591.4 2-10 ?Phosphoenolpyruvate carboxykinasedeficiency, cytosolic 261680
PCLO 13406 NM_033026.6 1-25 ?Pontocerebellar hypoplasia, type 3OMIM
PCNT 16068 NM_006031.6 1-47 Microcephalic OsteodysplasticPrimordial Dwarfism
PCSK9 20001 NM_174936.4 1-12 Hypercholesterolemia, familial, 3 603776
PCYT1A 8754 NM_005017.4 3-10 Spondylometaphyseal dysplasia withcone-rod dystrophy 608940
PCYT2 8756 NM_001256435.3 3-13 Global developmental delay Developmental regression Intellectual disability Spastic paraparesis Seizures Spastic tetraparesis Cerebral atrophy Cerebellar atrophy
PDCD10 8761 NM_145860.1 2-8 CEREBRAL CAVERNOUSMALFORMATIONS TYPE 3 603285
PDE10A 8772 NM_001130690.2 1-22 Childhood-Onset Chorea with BilateralStriatal Lesions Dyskinesia, limb and orofacial, infantile-onset 616921
PDE2A 8777 NM_002599.5 1-31 infantile-onset chorea-predominantmovement disorder
PDE3A 8778 NM_000921.4 1-16 Hypertension and brachydactylysyndrome, 112410
PDE4D 8783 NM_001104631.2 1-15 Acrodysostosis 2, with or withouthormone resistance, 614613
PDE6D 8788 NM_002601.4 1-5 Polydactyly ?Joubert syndrome 22 - MIM 615665
PDE6G 8789 NM_002602.4 2-4 Retinitis pigmentosa 57, 613582

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 149/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PDE6H 8790 NM_006205.3 2-4 RETINAL CONE DYSTROPHY 3 PDE6H610024 ACHROMATOPSIA
PDGFB 8800 NM_002608.4 1-6 Basal ganglia calcification, idiopathic, 5,615483 Fahr syndrome
PDGFRB 8804 NM_002609.4 2-23 PREMATURE AGING SYNDROME,PENTTINEN TYPE 601812 FAMILIAL INFANTILE MYOFIBROMATOSIS228550 Kosaki overgrowth syndrome, 616592 Intellectual disability Basal ganglia calcification, idiopathic, 4615007 Fahr syndrome
PDHA1 8806 NM_000284.4 1-11 Leigh syndrome, X-linked, 308930 Pyruvate dehydrogenase E1-alphadeficiency, 312170 Microcephaly, seizures, very variablephenotype
PDHB 8808 NM_000925.4 1-10 Pyruvate dehydrogenase E1-betadeficiency, 614111
PDHX 21350 NM_003477.3 1-11 Lacticacidemia due to PDX1 deficiency Pyruvate dehydrogenase E3 bindingprotein deficiency (Disorders ofpyruvate metabolism)
PDK3 8811 NM_001142386.3 1-12 ?Charcot-Marie-Tooth disease, X-linkeddominant, 6 300905
PDP1 9279 NM_018444.4 2 Pyruvate dehydrogenase phosphatasedeficiency (Disorders of pyruvatemetabolism)
PDSS1 17759 NM_014317.5 1-12 Coenzyme Q10 deficiency, primary, 2,614651 Disorders of ubiquinone metabolismand biosynthesis

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 150/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PDSS2 23041 NM_020381.4 1-8 Coenzyme Q10 deficiency, primary, 3,614652 Disorders of ubiquinone metabolismand biosynthesis
PDYN 8820 NM_024411.5 3-4 Spinocerebellar ataxia 23
PEPD 8840 NM_000285.4 1-15 Prolidase deficiency, 170100
PET100 40038 NM_001171155.2 1-4 Leigh syndrome Isolated complex IV deficiency Intellectual disability seizures
PET112 8849 NM_004564.3 1-13 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis)
PET117 40045 NM_001164811.2 1-2 lesions in the medulla oblongata
PEX1 8850 NM_000466.3 1-24 Neonatal and Adult Cholestasis INFANTILE REFSUM DISEASE 266510 Adrenoleukodystrophy Peroxisome biogenesis disorder 1A(Zellweger)
PEX10 8851 NM_153818.1 1-6 Peroxisome biogenesis disorder 6A(Zellweger) 614870 ADRENOLEUKODYSTROPHY NEONATAL(NALD)
PEX11B 8853 NM_003846.3 1-4 Intellectual disability Peroxisome biogenesis disorder 14B
PEX12 8854 NM_000286.3 1-3 Peroxisome biogenesis disorder 3A(Zellweger) 614859
PEX13 8855 NM_002618.4 1-4 Peroxisome biogenesis disorder 11A(Zellweger) 614883 ADRENOLEUKODYSTROPHY NEONATAL202370
PEX14 8856 NM_004565.3 1-9 Peroxisome biogenesis disorder 13A(Zellweger) 614887

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 151/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PEX16 8857 NM_004813.3 1-11 Peroxisome biogenesis disorder 8A(Zellweger) 614876
PEX19 9713 NM_002857.3 1-8 Peroxisome biogenesis disorder 12A(Zellweger) 614886
PEX2 9717 NM_000318.3 4 Neonatal and Adult Cholestasis Peroxisome Biogenesis Disorder 5A(Zellweger), 614866 INFANTILE REFSUM DISEASE 266510
PEX26 22965 NM_017929.6 2-6 Peroxisome biogenesis disorder 7A(Zellweger)614872 ADRENOLEUKODYSTROPHY NEONATAL202370 INFANTILE REFSUM DISEASE 266510
PEX3 8858 NM_003630.3 1-12 Peroxisome biogenesis disorder 10A(Zellweger) 614882
PEX5 9719 NM_001131025.1 2-16 ADRENOLEUKODYSTROPHY NEONATAL(NALD) Rhizomelic chondrodysplasia punctata,type 5 616716 ADRENOLEUKODYSTROPHY NEONATAL202370 INFANTILE REFSUM DISEASE 266510 Peroxisome biogenesis disorder 2A(Zellweger)
PEX6 8859 NM_000287.4 1-17 Peroxisome biogenesis disorder 4A(Zellweger) 614862
PEX7 8860 NM_000288.4 1-10 Rhizomelic chondrodysplasia punctata,type 1 Rhizomelic chondrodysplasia punctatatype 1 (Peroxisomal disorders) REFSUM DISEASE 266500 Peroxisome biogenesis disorder 9B,614879
PFKM 8877 NM_000289.6 2-23 Glycogen storage disease VII

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 152/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PGAM2 8889 NM_000290.4 1-3 Glycogen storage disease X 261670
PGAP1 25712 NM_024989.4 1-27 Intellectual disability, encephalopathy,impaired GPI-anchor maturation
PGAP2 17893 NM_001256240.2 2-6 Hyperphosphatasia with mentalretardation syndrome 3 614207 PGAP2-CDG (Disorders ofglycosphingolipid andglycosylphosphatidylinositol anchorglycosylation)
PGAP3 23719 NM_033419.5 1-8 HYPERPHOSPHATASIA WITH MENTALRETARDATION SYNDROME 4
PGK1 8896 NM_000291.4 1-11 Phosphoglycerate kinase 1 deficiency
PGM1 8905 NM_002633.3 1-11 Congenital disorder of glycosylation,type It, 614921 Glycogen Storage Disease Type XIV
PGM3 8907 NM_001199917.2 2-14 Immunodeficiency 23 Severe atopy, autoimmunity, bacterialand viral infections, skeletal anomaliesdysplasia: short stature, brachydactyly,dysmorphic facial features, andintellectual disability cognitiveimpairment, hypomyelination
PHACTR1 20990 NM_030948.4 3-15 Global developmental delay Intellectual disability Seizures
PHEX 8918 NM_000444.6 1-22 Hypophosphatemic rickets, X-linkeddominant 307800
PHF21A 24156 NM_016621.4 3-18 Potocki-Shaffer syndrome, 601224 Intellectual disability
PHF6 18145 NM_032458.3 2-10 Borjeson-Forssman-Lehmannsyndrome, 301900
PHF8 20672 NM_015107.3 2-22 Cleft lip Mental retardation syndrome, X-linked,Siderius type, 300263

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 153/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PHGDH 8923 NM_006623.4 1-12 Neu-Laxova syndrome 1 256520 Phosphoglycerate dehydrogenasedeficiency, 601815
PHIP 15673 NM_017934.7 1-40 Developmental delay, intellectualdisability, obesity, and dysmorphicfeatures, 617991
PHKA1 8925 NM_002637.4 1-32 Muscle glycogenosis, 300559
PHKA2 8926 NM_000292.3 1-33 Glycogen Storage Disorders- Liver hepatomegaly and mild hypoglycaemia
PHKB 8927 NM_000293.3 1-31 Glycogen storage disease type IXHepatic and muscle phosphorylasekinase deficiency (Glycogen storagedisorders)
PHKG2 8931 NM_000294.3 2-10 Glycogen storage disease type IXHepatic phosphorylase kinasedeficiency with cirrhosis (Glycogenstorage disorders)
PHOX2B 9143 NM_003924.4 1-3 CENTRAL HYPOVENTILATIONSYNDROME, CONGENITAL, WITH ORWITHOUT HIRSCHSPRUNG DISEASE209880
PHYH 8940 NM_006214.4 1-9 Refsum disease (Disorders ofperoxisomal alpha-, beta and omega-oxidation)
PIEZO1 28993 NM_001142864.4 1-51 Congenital lymphatic dysplasia withhydrops and/or lymphoedema
PIEZO2 26270 NM_022068.3 4 1-52 Ataxia, dysmetria, contractures &scoliosis with normal cognition but lossof discriminative touch perception ?Marden-Walker syndrome, 248700 Arthrogryposis, distal
PIGA 8957 NM_002641.3 4-6 2-6 PIGA-CDG (Disorders ofglycosphingolipid andglycosylphosphatidylinositol anchorglycosylation)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 154/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PIGB 8959 NM_004855.5 1-12 Epileptic encephalopathy, early infantile,80, 618580 Generalized hypotonia Global developmental delay Intellectual disability Seizures Hearing abnormality Abnormality of vision Elevated alkaline phosphatase Inherited GPI Biosynthesis Defect withan Axonal Neuropathy and MetabolicAbnormality
PIGC 8960 NM_002642.4 2 2 Glycosylphosphatidylinositolbiosynthesis defect 16, 617816
PIGG 25985 NM_001127178.3 1-13 Intellectual Disability with Seizures andHypotonia
PIGH 8964 NM_004569.5 1-4 Glycosylphosphatidylinositolbiosynthesis defect, 17 epilepsy febrile seizures
PIGL 8966 NM_004278.4 1-7 Coloboma, Congenital Heart Disease,Ichthyosiform Dermatosis,MentalRetardation, and Ear AnomaliesSyndrome ZUNICH NEUROECTODERMALSYNDROME PIGL-CDG (Disorders ofglycosphingolipid andglycosylphosphatidylinositol anchorglycosylation) CHIME syndrome 280000
PIGM 18858 NM_145167.3 1 Glycosylphosphatidylinositol deficiency,610293 Hypercoagulability syndrome due toglycosylphosphatidylinositol deficiency Disorders of glycosphingolipid andglycosylphosphatidylinositol anchorglycosylation

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 155/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PIGN 8967 NM_176787.5 23 4-31 PIGN-CDG (Disorders ofglycosphingolipid andglycosylphosphatidylinositol anchorglycosylation) Multiple congenital anomalies-hypotonia-seizures syndrome 1, 614080
PIGO 23215 NM_032634.4 2-11 Hyperphosphatasia with MentalRetardation Syndrome Hyperphosphatasia (Disorders ofglycosphingolipid andglycosylphosphatidylinositol anchorglycosylation)
PIGP 3046 NM_153682.2 2-5 Epileptic encephalopathy, early infantile,55, 617599 Generalized hypotonia Global developmental delay Seizures Intellectual disability Feeding difficulties Cortical visual impairment
PIGQ 14135 NM_004204.4 2-11 Epileptic encephalopathy, early infantile,77, 618548 Intractable seizures developmental delay optic atrophy epilepsy Ohtahara syndrome
PIGS 14937 NM_033198.4 1-12 Neurological Syndrome Fetal Akinesia/Epileptic Encephalopathy
PIGT 14938 NM_015937.6 1-12 Multiple congenital anomalies-hypotonia-seizures syndrome 3

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 156/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PIGU 15791 NM_080476.4 1-12 Glycosylphosphatidylinositolbiosynthesis defect 2, 618590 Global developmental delay Intellectual disability Seizures Cerebral atrophy Cerebellar hypoplasia Scoliosis Intellectual Disability, Central NervousSystem anomalies and Scoliosis
PIGV 26031 NM_017837.3 2-4 Hyperphosphatasia with MentalRetardation Syndrome Hyperphosphatasia (Disorders ofglycosphingolipid andglycosylphosphatidylinositol anchorglycosylation)
PIGW 23213 NM_178517.4 3 Glycosylphosphatidylinositolbiosynthesis defect 11, 616025
PIGY 28213 NM_001042616.2 2 2 Glycosylphosphatidylinositol deficiency
PIH1D3 28570 NM_001169154.1 3-8 X-Linked Primary Ciliary Dyskinesia withOuter and Inner Dynein Arm Defects
PIK3C2A 8971 NM_002645.4 2-33 Oculoskeletodental syndrome 618440
PIK3CA 8975 NM_006218.4 10-14 2-21 Macrodactyly, somatic 155500 Polydactyly CLAPO syndrome, somatic 613089 Megalencephaly-capillary malformation-polymicrogyria syndrome, 602501 Congenital Lipomatous OvergrowthVascular Malformations, Epidermal Neviand Scoliosis/Skeletal/Spinal anomaliessyndrome
PIK3R1 8979 NM_181523.3 2-16 SHORT syndrome 269880 ?Agammaglobulinemia 7, autosomalrecessive, 615214 Immunodeficiency 36, 616005

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 157/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PIK3R2 8980 NM_005027.4 2-16 Polydactyly Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome,603387
PINK1 14581 NM_032409.3 1-8 Parkinson disease 6, early onset, 605909Dystonia
PIP5K1C 8996 NM_012398.3 1-18 Lethal congenital contracturalsyndrome 3 611369
PITRM1 17663 NM_001242309.1 1-24 mental retardation, spinocerebellarataxia, cognitive decline and psychosis
PITX1 9004 NM_002653.5 1-3 Clubfoot, congenital, with or withoutdeficiency of long bones and/or mirror-image polydactyly 119800 Liebenberg syndrome 186550 Polydactyly
PITX2 9005 NM_153427.2 3-5 Axenfeld-Rieger syndrome, type 1,180500 Iridogoniodysgenesis, type 2, 137600 Ring dermoid of cornea, 180550 Peters anomaly, 604229
PITX3 9006 NM_005029.4 2-4 Anterior segment mesenchymaldysgenesis, 107250 Cataract 11, multiple types, 610623 Cataract 11, syndromic, 610623
PKD1 9008 NM_001009944.3 1-33 1-46 Autosomal recessive polycystic kidneydisease (ARPKD) Autosomal dominant polycystic kidneydisease (ADPKD)
PKD1L1 18053 NM_138295.4 1-57 Laterality defects
PKD2 9009 NM_000297.4 1-15 Polycystic kidney disease 2, 613095
PKHD1 9016 NM_138694.4 2-67 Polycystic kidney disease 4, with orwithout hepatic disease 263200

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 158/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PLA2G6 9039 NM_003560.4 2-17 Infantile neuroaxonal dystrophy 1(#256600) Parkinson disease 14 (#612953) Neurodegeneration with brain ironaccumulation 2B (#610217) PLA2G6-associated neurodegeneration(PLAN) Familial cortical myoclonic tremor withepilepsy (FCMTE)
PLAA 9043 NM_001031689.3 1-14 Neurodevelopmental disorder withprogressive microcephaly, spasticity,and brain anomalies, 617527 Lethal Infantile Epileptic Encephalopathy
PLCB1 15917 NM_015192.4 1-32 Epileptic encephalopathy, early infantile,12, 613722
PLCB4 9059 NM_000933.3 1-36 AURICULOCONDYLAR SYNDROME602483
PLCE1 17175 NM_016341.4 2-32 NEPHROTIC SYNDROME, TYPE 3 610725
PLD1 9067 NM_002662.5 2-27 Cardiac valvular defect, developmental
PLEC 9069 NM_000445.5 2-33 Congenital myasthenic syndrome Plectin deficiency Muscular dystrophy with epidermolysisbullosa simplex, 226670
PLEKHA7 27049 NM_175058.5 1-23 cleft lip
PLEKHG2 29515 NM_022835.3 2-19 Leukodystrophy and acquiredmicrocephaly with or without dystonia,616763
PLG 9071 NM_000301.3 1-5 1-19 Plasminogen deficiency, type I
PLK4 11397 NM_014264.5 1-16 Microcephaly and chorioretinopathy,autosomal recessive, 2
PLOD1 9081 NM_000302.4 1-19 Ehlers-Danlos syndrome, type VI 225400

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 159/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PLOD2 9082 NM_182943.3 1-20 Bruck syndrome 2, 609220
PLP1 9086 NM_000533.5 1-7 Spastic paraplegia 2, X-linked, 312920 Pelizaeus-Merzbacher disease, 312080
PLS3 9091 NM_005032.7 2-16 Bone mineral density QTL18,osteoporosis 300910
PMM2 9115 NM_000303.3 1-8 Phosphomannomutase 2 deficiency(Disorders of protein N-glycosylation) Congenital disorder of glycosylation,type Ia 212065
PMPCA 18667 NM_015160.3 1-13 Spinocerebellar ataxia, autosomalrecessive 2 (MIM 213200)
PMPCB 9119 NM_004279.3 1-13 Multiple mitochondrial dysfunctionssyndrome 6, 617954 Neurodegeneration in Early Childhood
PMS2 9122 NM_000535.7 1-5, 9,11-15
1-15 Mismatch repair cancer syndrome,276300
PNKD 9153 NM_015488.5 1-10 Paroxysmal nonkinesigenic dyskinesia,118800
PNKP 9154 NM_007254.4 2-17 Epileptic encephalopathy, early infantile,10, 613402 Ataxia with oculomotor apraxia 4(#616267) Microcephaly, seizures, anddevelopmental delay
PNP 7892 NM_000270.3 1-6 Immunodeficiency due to purinenucleoside phosphorylase deficiency613179
PNPLA1 21246 NM_001145717.1 1-8 CONGENITAL ICHTHYOSIS
PNPLA2 30802 NM_020376.4 2-10 NEUTRAL LIPID STORAGE DISEASE WITHMYOPATHY 610717

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 160/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PNPLA6 16268 NM_006702.5 3-35 Boucher-Neuhauser syndrome, 215470 Oliver-McFarlane syndrome, 275400 Trichomegaly-retina pigmentarydegeneration-dwarfism syndrome Spinocerebellar ataxia,hypogonadotropic hypogonadism andchorioretinal dystrophy (Boucher-Neuhauser syndrome, #215470) Spastic paraplegia 39, autosomalrecessive, 612020
PNPLA8 28900 NM_015723.5 4-12 ?Mitochondrial myopathy with lacticacidosis, 251950
PNPO 30260 NM_018129.4 1-7 Pyridoxamine 5 -oxidase deficiency(Disorders of pyridoxine metabolism) Neonatal epileptic encephalopathy Global developmental delay
PNPT1 23166 NM_033109.5 28 1-28 Deafness, autosomal recessive 70,614934 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Combined oxidative phosphorylationdeficiency 13, 614932
POC1A 24488 NM_015426.5 1-11 Short stature, onychodysplasia, facialdysmorphism, and hypotrichosis,614813 Microcephaly in adulthood primordial dwarfism
POC1B 30836 NM_172240.3 1-12 Joubert Syndrome Senior-Loken Syndrome Cone-rod dystrophy 20, 615973
POGLUT1 22954 NM_152305.3 1-11 Muscular dystrophy, limb-girdle,autosomal recessive 21, 617232
POGZ 18801 NM_015100.4 2-19 INTELLECTUAL DISABILITY

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 161/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
POLA1 9173 NM_016937.4 1-37 Pigmentary disorder, reticulate, withsystemic manifestations, X-linked,301220 X-Linked Intellectual Disabilityassociated with short stature,microcephaly, and hypogonadism VAN ESCH-O'DRISCOLL SYNDROME301030
POLD1 9175 NM_002691.4 2-27 SUBCUTANEOUS LIPODYSTROPHY,DEAFNESS, MANDIBULAR HYPOPLASIAAND MALE HYPOGONADISM Mandibular hypoplasia, deafness,progeroid features, and lipodystrophysyndrome, 615381
POLG 9179 NM_002693.2 2-23 Mitochondrial DNA depletion syndrome4A (Alpers type) 203700 Progressive External Ophthalmoplegiawith Mitochondrial DNA Deletions
POLG2 9180 NM_007215.4 1-8 Disorders of mitochondrial DNAmaintenance and integrity Progressive External Ophthalmoplegiawith Mitochondrial DNA Deletions
POLH 9181 NM_006502.3 4 2-11 Xeroderma pigmentosum, variant type,278750
POLR1A 17264 NM_015425.6 1-34 Acrofacial dysostosis, Cincinnati type616462 cleft palte
POLR1C 20194 NM_203290.4 1-9 Treacher Collins syndrome 3, 248390 Leukodystrophy, hypomyelinating, 11
POLR1D 20422 NM_015972.4 1-2 Treacher Collins syndrome 2, 613717
POLR2A 9187 NM_000937.5 1-29 Global developmental delay Generalized hypotonia Feeding difficulties

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 162/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
POLR3A 30074 NM_007055.4 1-31 Leukodystrophy, hypomyelinating, 7,with or without oligodontia and/orhypogonadotropichypogonadism,607694 Autosomal Recessive Ataxia Leukodystrophy, hypomyelinating, 7,with or without oligodontia and/orhypogonadotropic hypogonadism,607694 Wiedemann-Rautenstrauch syndrome,264090
POLR3B 30348 NM_018082.6 1-28 Leukodystrophy, hypomyelinating, 8,with or without oligodontia and/orhypogonadotropichypogonadism,614381 Pol III-Related Leukodystrophy General Leukodystrophy &Mitochondrial Leukoencephalopathy
POLRMT 9200 NM_005035.4 1-21 No OMIM phenotype
POMGNT1 19139 NM_017739.3 2-22 Protein-O-mannose beta-1,2-N-acetyglucosaminyltransferase deficiency(Disorders of protein O-glycosylation, O-mannosylglycan synthesis deficiencies) Retinitis pigmentosa 76 617123 Congenital Muscular Dystrophy, alpha-dystroglycan related Limb-girdle muscular dystrophy

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 163/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
POMGNT2 25902 NM_032806.6 2 WALKER WARBERG SYNDROME 614830 Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies, type A, 8 OMIMMuscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies, type A, 8 Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies, type A, 8614830 Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies type Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies, type A, 8 OMIMWalker-Warburg syndrome limb girdle muscular dystrophy
POMK 26267 NM_032237.5 4-5 Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies), type A, 12
POMT1 9202 NM_007171.3 2-20 Protein-O-mannosyltransferase 1deficiency (Disorders of protein O-glycosylation, O-mannosylglycansynthesis deficiencies) Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies), type Limb-girdle muscular dystrophy
POMT2 19743 NM_013382.5 1-21 Protein-O-mannosyltransferase 2deficiency (Disorders of protein O-glycosylation, O-mannosylglycansynthesis deficiencies) Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies), type Limb-girdle muscular dystrophy Type 2 lissencephaly

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 164/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
POP1 30129 NM_015029.2 2-16 Anauxetic dysplasia 2, 617396
POR 9208 NM_000941.3 2-16 Antley-Bixler syndrome with genitalanomalies and disorderedsteroidogenesis 201750 Disordered steroidogenesis due tocytochrome P450 oxidoreductase613571
PORCN 17652 NM_203475.3 2-15 Polydactyly Focal dermal hypoplasia, 305600 GOLTZ SYNDROME
POU1F1 9210 NM_000306.4 1-6 POU1F1-RELATED COMBINED PITUITARYHORMONE DEFICIENCY 613038
POU3F3 9216 NM_006236.3 1 Generalized hypotonia Delayed speech and languagedevelopment Global developmental delay Intellectual disability Autistic behavior
PPA2 28883 NM_176869.3 1-12 Sudden arrhythmic cardiac death afterinfectious or alcohol trigger Sudden cardiac failure, infantile, 617222
PPIB 9255 NM_000942.4 1-5 Osteogenesis imperfecta, type IX 259440
PPM1D 9277 NM_003620.4 1-6 Intellectual developmental disorder withgastrointestinal difficulties and highpain threshold, 617450
PPOX 9280 NM_000309.5 2-13 Variegate porphyria (Acute neuropathicporphyrias)
PPP1CB 9282 NM_206876.1 2-9 Rasopathy with developmental delay,short stature and sparse slow-growinghair
PPP1R15B 14951 NM_032833.4 1-2 Microcephaly, short stature, andimpaired glucose metabolism 2, 616817

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 165/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PPP1R21 30595 NM_001135629.3 1-22 Generalized hypotonia, Feedingdifficulties, Profound globaldevelopmental delay, Abnormality ofthe face, Abnormality of vision,Abnormal heart morphology
PPP2CA 9299 NM_002715.4 1-7 Seizures Language impairment Muscular hypotonia Feeding difficulties Intellectual disability Neurodevelopmental disorder andlanguage delay with or withoutstructural brain abnormalities, 618354
PPP2R1A 9302 NM_014225.6 1-15 INTELLECTUAL DISABILITY
PPP2R2B 9305 NM_181678.2 3-11 Spinocerebellar ataxia 12, 604326
PPP2R5D 9312 NM_006245.4 1-16 INTELLECTUAL DISABILITY
PPP3CA 9314 NM_000944.5 1-14 Epileptic encephalopathy, infantile orearly childhood, 1, 617711
PPT1 9325 NM_000310.3 1-9 Ceroid lipofuscinosis, neuronal, 1 OMIM
PQBP1 9330 NM_005710.2 1-6 Renpenning syndrome, 309500
PRDM12 13997 NM_021619.3 1-5 HEREDITARY SENSORY & AUTONOMICNEUROPATHY TYPE VIII 616488
PREPL 30228 NM_006036.4 1-14 HYPOTONIA-CYSTINURIA SYNDROME606407
PRG4 9364 NM_005807.5 2-13 Camptodactyly-arthropathy-coxa vara-pericarditis syndrome 208250
PRICKLE1 17019 NM_153026.3 2-8 Epilepsy, progressive myoclonic 1B612437

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 166/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PRKAG2 9386 NM_016203.4 1-16 Cardiomyopathy, hypertrophic 6,600858 Glycogen storage disease of heart,lethal congenital, 261740 Wolff-Parkinson-White syndrome,194200
PRKAR1A 9388 NM_002734.4 2-11 Acrodysostosis 1, with or withouthormone resistance 101800 Myxoma, intracardiac 255960 Pigmented nodular adrenocorticaldisease, primary, 1 610489
PRKCG 9402 NM_002739.5 1-18 Spinocerebellar ataxia 14
PRKD1 9407 NM_002742.3 1-18 Congenital heart defects andectodermal dysplasia 617364
PRKRA 9438 NM_003690.5 1-8 early-Onset Generalized dystonia-parkinsonism (DYT16), non-responsiveto levo-dopa Dystonia
PRMT7 25557 NM_019023.4 3-19 Short stature, brachydactyly, intellectualdevelopmental disability, and seizures617157 Pseudohypoparathyroidism-likedisorder
PRNP 9449 NM_000311.5 2 Creutzfeldt-Jakob disease Autosomal Dominant Ataxia Gerstmann-Straussler disease Huntington disease-like 1 Insomnia, fatal familial
PRODH 9453 NM_016335.5 2, 6-15 2-15 Hyperprolinaemia type I (Disorders ofornithine or proline metabolism)
PROP1 9455 NM_006261.4 1-3 Pituitary hormone deficiency,combined, 2, 262600
PROSC 9457 NM_007198.4 1-8 Epilepsy, early-onset, vitamin B6-dependent, 617290

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 167/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PRPS1 9462 NM_002764.4 7 1-7 Arts syndrome, 301835 Phosphoribosylpyrophosphatesynthetase superactivity, 300661 Charcot-Marie-Tooth disease, X-linkedrecessive, 5 311070 Deafness, X-linked 1 304500 Gout, PRPS-related 300661
PRR12 29217 NM_020719.3 1-14 Global developmental delay, Intellectualdisability, Abnormality of the iris,Abnormality of vision, Behavioralabnormality
PRRT2 30500 NM_145239.3 2-4 Epilepsy Convulsions, familial infantile, withparoxysmal choreoathetosis, 602066 Seizures, benign familial infantile, 2,605751 Paroxysmal kinesigenic choreoathetosis(PKD1) and infantile convulsions
PRSS12 9477 NM_003619.4 1-13 Mental retardation, autosomalrecessive 1, 249500
PRSS56 39433 NM_001195129.2 1-13 Microphthalmia, isolated 6, 613517
PRUNE 13420 NM_021222.3 1-8 Neurodevelopmental disorder withmicrocephaly, hypotonia, and variablebrain anomalies, 617481
PSAP 9498 NM_002778.4 1-14 Metachromatic leukodystrophy due toSAP-b deficiency, 249900 Combined SAP deficiency Prosaposin deficiency(Sphingolipidoses) Gaucher disease, atypical, 610539 Krabbe disease, atypical, 611722
PSAT1 19129 NM_058179.4 9 1-9 Neu-Laxova syndrome 2 616038 ?Phosphoserine aminotransferasedeficiency 610992
PSMB8 9545 NM_148919.4 1-6 NAKAJO SYNDROME 256040 Autoinflammation, lipodystrophy, anddermatosis syndrome, 256040

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 168/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PSMD12 9557 NM_002816.5 1-11 Stankiewicz-Isidor syndrome, 617516 Syndromic NeurodevelopmentalDisorder
PSPH 9577 NM_004577.4 8 8,4-7 Phosphoserine phosphatase deficiency614023 NEU-LAXOVA 256520
PTCD3 24717 NM_017952.6 1-24 low birth weight, mental retardation,and optic atrophy
PTCH1 9585 NM_000264.5 1-23 BASAL CELL NEVUS SYNDROME Holoprosencephaly
PTCH2 9586 NM_003738.5 1-22 Basal cell nevus syndrome
PTCHD1 26392 NM_173495.3 1-3 AUTISM/ID
PTDSS1 9587 NM_014754.3 1-13 LENZ-MAJEWSKI HYPEROSTOTICDWARFISM
PTEN 9588 NM_000314.8 9 1-9 PROTEUS SYNDROME hemihypertrophy Bannayan-Riley-Ruvalcaba Syndrome Proteus-like syndrome PTEN Hamartoma Tumor Syndrome Macrocephaly and OvergrowthSyndromes Cowden syndrome VATER association with macrocephalyand ventriculomegaly,276950
PTF1A 23734 NM_178161.3 1-2 DIABETES MELLITUS, PERMANENTNEONATAL, WITH CEREBELLAR AGENESIS
PTH 9606 NM_000315.4 2-3 FAMILIAL ISOLATEDHYPOPARATHYROIDISM 146200

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 169/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PTH1R 9608 NM_000316.3 3-16 Failure of tooth eruption, primary125350 Eiken syndrome 600002 Metaphyseal chondrodysplasia, MurkJansen type 156400 Chondrodysplasia, Blomstrand type215045
PTHLH 9607 NM_198965.2 4-6 Humoral hypercalcemia of malignancy Brachydactyly, type E2, 613382
PTPLA 9639 NM_014241.4 1-7 congenital myopathy
PTPN11 9644 NM_002834.4 1-15 NOONAN SYNDROME 1 163950 Metachondromatosis 156250 LEOPARD syndrome 1 151100
PTPN14 9647 NM_005401.5 2-19 CHOANAL ATRESIA AND LYMPHEDEMA613611
PTPN23 14406 NM_015466.4 1-25 Developmental epilepticencephalopathy with hypomyelinationand brain atrophy
PTRH2 24265 NM_016077.4 2 Infantile-onset multisystem neurologic,endocrine, and pancreatic disease,616263
PTRHD1 33782 NM_001013663.1 1-2 Parkinsonism, Intellectual disability
PTS 9689 NM_000317.3 1-6 Intellectual disability 6-Pyruvoyl-tetrahydropterin synthasedeficiency (Disorders of pterinmetabolism) Hyperphenylalaninemia, BH4-deficient,A 261640 Dystonia
PUF60 17042 NM_078480.3 1-12 Verheij syndrome, 615583 Intellectual disability
PUM1 14957 NM_001020658.2 2-22 Spinocerebellar ataxia 47 617931

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 170/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
PURA 9701 NM_005859.5 1 INTELLECTUAL DISABILITY
PUS1 15508 NM_025215.6 1-6 Myopathy, lactic acidosis, andsideroblastic anemia 1 (MLASA) Intellectual disability
PUS3 25461 NM_031307.4 2-4 Global developmental delay Microcephaly
PUS7 26033 NM_019042.5 2-16 Intellectual developmental disorder withabnormal behavior, microcephaly, andshort stature, 618342
PVRL1 9706 NM_203285.2 1-8 Orofacial cleft 7, 225060 Zlotogora-Ogur syndrome CLP, partial syndactyly of digits,intellectual disability, dysmorphism Ectodermal dysplasia, Margarita Islandtype
PVRL4 19688 NM_030916.3 1-9 ECTODERMAL DYSPLASIA-SYNDACTYLYSYNDROME 1 613573
PXDN 14966 NM_012293.3 1-23 CONGENITAL CATARACT, CORNEALOPACITY, AND DEVELOPMENTALGLAUCOMA
PYCR1 9721 NM_006907.4 1-7 Intellectual disability Cutis laxa, autosomal recessive, typeIIb/IIIb (Disorders of ornithine or prolinemetabolism)
PYCR2 30262 NM_013328.4 1-7 Postnatal microcephaly,hypomyelination, and reduced cerebralwhite-matter volume
PYGL 9725 NM_002863.5 1-20 hepatomegaly and mild hypoglycaemia Glycogen storage disease type VI, Hers(Glycogen storage disorders)
PYGM 9726 NM_005609.4 1-20 Glycogen storage disease type V,McArdle (Glycogen storage disorders)
PYROXD1 26162 NM_024854.5 12 1-12 Early-Onset Myopathy with InternalizedNuclei and Myofibrillar Disorganization

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 171/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
QARS 9751 NM_005051.3 1-24 Intellectual disability Microcephaly, progressive, seizures, andcerebral and cerebellar atrophy
QDPR 9752 NM_000320.3 1-7 Hyperphenylalaninemia, BH4-deficient,C Dihydropteridine reductase deficiency Dystonia
QRICH1 24713 NM_017730.3 3-11 Ververi-Brady syndrome, 617982 Intellectual disability QRICH1 syndrome
QRSL1 21020 NM_018292.5 1-11 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis)
RAB11A 9760 NM_004663.5 1-5 Global developmental delay, Intellectualdisability
RAB11B 9761 NM_004218.4 1-5 Neurodevelopmental disorder withataxic gait, absent speech, anddecreased cortical white matter, 617807
RAB18 14244 NM_021252.5 1-7 Warburg micro syndrome 3, 614222
RAB23 14263 NM_183227.2 2-7 Carpenter syndrome 201000 ACROCEPHALOPOLYSYNDACTYLY TYPE 2Polydactyly
RAB33B 16075 NM_031296.3 1-2 Smith-McCort dysplasia 2 615222
RAB39B 16499 NM_171998.4 1-2 MENTAL RETARDATION X-LINKED TYPE72 (MRX72) +/- PARKINSONS Waisman syndrome 311510
RAB3GAP1 17063 NM_012233.3 1-24 Warburg micro syndrome 1 OMIM
RAB3GAP2 17168 NM_012414.4 1-35 Warburg micro syndrome 2 Martsolf syndrome OMIM
RAC1 9801 NM_018890.4 1-7 intellectual disability

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 172/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
RAC3 9803 NM_005052.3 1-6 Neurodevelopmental disorder withstructural brain anomalies anddysmorphic facies, 618577
RAD21 9811 NM_006265.3 14 2-14 Cornelia de Lange syndrome 4, 614701 COHESINOPATHY
RAD51C 9820 NM_058216.3 1-9 Radial Ray abnormality Fanconi anemia, complementationgroup O 613390
RAF1 9829 NM_002880.3 2-17 NOONAN SYNDROME 5
RAI1 9834 NM_030665.4 3-6 Immunodeficiency 9, 612782 SMITH-MAGENIS SYNDROME (SMS)
RALA 9839 NM_005402.4 5 2-5 Global developmental delay, Intellectualdisability, Seizures, Abnormality ofnervous system morphology
RALGAPA1 17770 NM_014990.3 1-40 Neurodevelopmental disorder withhypotonia, neonatal respiratoryinsufficiency, and thermodysregulation618797
RANBP2 9848 NM_006267.5 1-21 1-29 Acute necrotizing encephalopathy(Other metabolic disorders)
RAPSN 9863 NM_005055.5 1-8 Myasthenic syndrome, congenital, 11,associated with acetylcholine receptordeficiency, 616326
RARB 9865 NM_000965.4 1-8 MICROPHTHALMIA, SYNDROMIC 12
RARS 9870 NM_002887.4 1-15 Cerebral hypomyelination Global developmental delay Intellectual disability Seizures Cerebral atrophy Nystagmus Ataxia Feeding difficulties Leukodystrophy, hypomyelinating, 9616140

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 173/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
RARS2 21406 NM_020320.5 1-20 epilepsy Pontocerebellar hypoplasia
RASA1 9871 NM_002890.3 1-25 Parkes Weber syndrome Capillary malformation-arteriovenousmalformation Basal cell carcinoma, somatic, 605462
RASGRP2 9879 NM_153819.1 2-16 Bleeding disorder, platelet-type, 18615888, also with osteopetrosis likebone abnormalities andneurodevelopmental defects
RAX 18662 NM_013435.3 1-3 Microphthalmia, isolated 3, 611038
RBBP8 9891 NM_002894.3 2-19 Jawad syndrome, 251255 Microcephaly with mental retardationand digital anomalies Seckel syndrome 2, 606744 (includesmicrocephaly) Jawad syndrome (microcephaly withmental retardation and digitalanomalies), 251255
RBCK1 15864 NM_031229.4 1-12 Polyglucosan body myopathy 1 with orwithout immunodeficiency 615895
RBM10 9896 NM_005676.5 2-24 Cleft palate TARP syndrome, 311900
RBM8A 9905 NM_005105.4 1-6 1-6 Thrombocytopenia-absent radiussyndrome, 274000
RBP4 9922 NM_006744.4 2-6 Retinol binding protein deficiency(Other disorders of vitamins andcofactors) Posterior segment abnormalities
RBPJ 5724 NM_005349.3 10-12 2-12 Adams-Oliver syndrome 3, 614814
RECQL4 9949 NM_004260.3 1-22 RAPILINO syndrome, 266280 Rothmund-Thomson syndrome, 268400 Baller-Gerold syndrome, 218600

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 174/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
REEP1 25786 NM_022912.3 1-7 Spastic paraplegia 31, autosomaldominant, 610250 ?Neuronopathy, distal hereditarymotor, type VB 614751
REEP2 17975 NM_001271803.2 1-8 Spastic paraplegia
RELN 9957 NM_005045.4 1-65 Lissencephaly 2 (Norman-Roberts type),257320
RERE 9965 NM_012102.4 3-5 3-24 Neurodevelopmental disorder with orwithout anomalies of the brain, eye, orheart 616975
RET 9967 NM_020975.6 1-20 RENAL AGENESIS 191830 MULTIPLE ENDOCRINE NEOPLASIA IIB162300
RFT1 30220 NM_052859.4 1-13 Congenital disorder of glycosylation,type In 612015 Flippase of Man5GlcNAc2-PP-Doldeficiency (Disorders of protein N-glycosylation)
RFX6 21478 NM_173560.4 1-19 MARTINEZ-FRIAS SYNDROME 601346
RHOBTB2 18756 NM_001160036.2 3-12 Epileptic encephalopathy, early infantile,64, 618004 Global developmental delay Intellectual disability Seizures Postnatal microcephaly
RIN2 18750 NM_018993.3 1-11 MACROCEPHALY, ALOPECIA, CUTIS LAXA,AND SCOLIOSIS TALL FOREHEAD, SPARSEHAIR, SKIN HYPEREXTENSIBILITY, ANDSCOLIOSIS
RINT1 21876 NM_021930.6 1-15 Infantile-Onset Recurrent Acute LiverFailure and Skeletal Abnormalities
RIPK4 496 NM_020639.3 1-8 Popliteal pterygium syndrome,Bartsocas-Papas type 263650

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 175/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
RIT1 10023 NM_006912.6 2-6 Noonan syndrome 8, 615355
RLIM 13429 NM_183353.3 5 3-5 Intellectual disability
RMI1 25764 NM_024945.3 3 Bloom Syndrome-like Disorder
RMND1 21176 NM_017909.4 2 2-12 Combined oxidative phosphorylationdeficiency 11, 614922
RNASEH1 18466 NM_002936.5 1-8 Progressive external ophthalmoplegiawith mitochondrial DNA deletions,autosomal recessive 2
RNASEH2A 18518 NM_006397.2 1-8 Aicardi-Goutieres Syndrome
RNASEH2B 25671 NM_024570.4 1-11 Aicardi-Goutieres Syndrome
RNASEH2C 24116 NM_032193.4 1-4 Aicardi-Goutieres Syndrome
RNASET2 21686 NM_003730.6 1-9 Leukoencephalopathy, cystic, withoutmegalencephaly, 612951 Intellectual disability
RNF113A 12974 NM_006978.3 1 Trichothiodystrophy 5,nonphotosensitive, 300953
RNF125 21150 NM_017831.4 1-6 Intellectual disability Tenorio syndrome
RNF13 10057 NM_007282.4 11 3-11 Cortical visual impairment Epileptic encephalopathy, early infantile,73, 618379 Congenital microcephaly Sensorineural hearing impairment Congenital Microcephaly EpilepticEncephalopathy Blindness and Failureto Thrive
RNF170 25358 NM_001160223.1 2-7 Ataxia, sensory, 1, autosomal dominant

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 176/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
RNF216 21698 NM_207111.4 2, 6-8 2-17 Cerebellar ataxia and hypogonadotropichypogonadism, 212840
ROBO3 13433 NM_022370.4 1-28 Gaze palsy, horizontal, with progressivescoliosis, 607313
ROBO4 17985 NM_019055.6 1-18 Bicuspid Aortic Valve and AorticAneurysm 618496
ROGDI 29478 NM_024589.2 1-11 Kohlschutter-Tonz syndrome, 226750
ROR2 10257 NM_004560.4 1-9 Brachydactyly, type B1 113000 Robinow syndrome, autosomalrecessive 268310
RORA 10258 NM_134261.3 1-11 Intellectual developmental disorder withor without epilepsy or cerebellar ataxia,618060
RORB 10259 NM_006914.4 1-10 generalized epilepsies withpredominant absence seizures
RPE65 10294 NM_000329.3 1-14 Leber congenital amaurosis 2, 204100 Retinitis pigmentosa 20, 613794
RPGRIP1 13436 NM_020366.3 1-24 Leber congenital amaurosis 6, 613826 Cone-rod dystrophy 13, 608194
RPGRIP1L 29168 NM_015272.5 2-27 COACH SYNDROME (COACHS) Joubert syndrome Meckel syndrome Meckel-Gruber syndrome
RPIA 10297 NM_144563.3 1-9 Ribose-5-phosphate isomerasedeficiency (Disorders of pentosemetabolism)
RPL10 10298 NM_006013.4 2-7 Mental retardation, X-linked, syndromic,35
RPL11 10301 NM_000975.5 1-6 Diamond-Blackfan anemia with cleftpalate and abnormal thumbs Radial Ray abnormality
RPL13 10303 NM_033251.2 1-5 Spondyloepimetaphyseal Dysplasia withSevere Short Stature

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 177/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
RPL15 10306 NM_002948.4 2-4 2-4 ?Diamond-Blackfan anemia 12 upper limb malformation Triphalangeal thumbs
RPL26 10327 NM_000987.5 2-4 Diamond-Blackfan anemia 11 Hypoplasia or aplasia of radius,unilateral Absent thumb, bilateral
RPL35A 10345 NM_000996.4 2-5 upper limb malformation Diamond-Blackfan anemia 5, 612528 Radial Ray abnormality
RPL5 10360 NM_000969.5 1-8 Cleft palate Diamond-Blackfan anemia 6, 612561 Radial Ray abnormality thumb abnormalities
RPS10 10383 NM_001014.5 2-6 upper limb malformation Diamond-Blackfan anemia 9, 613308 Radial Ray abnormality
RPS19 10402 NM_001022.4 2-6 Mild radial hypoplasia Diamond-Blackfan anemia 1, 105650 Hypoplastic thumbs Absent thumbs Radial Ray abnormality Triphalangeal thumbs
RPS23 10410 NM_001025.5 1-4 Brachycephaly, trichomegaly, anddevelopmental delay 617412 Microcephaly, hearing loss, anddysmorphic features
RPS24 10411 NM_033022.4 1-5 Diamond-Blackfan anemia 3, 610629 Radial Ray abnormality upper limb malformation
RPS26 10414 NM_001029.5 1-4 Cleft palate Diamond-Blackfan anemia 10, 613309 upper limb malformation Radial Ray abnormality
RPS28 10418 NM_001031.5 1-3 DIAMOND-BLACKFAN ANEMIA 15 WITHMANDIBULOFACIAL DYSOSTOSIS DBA15 Cleft palate

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 178/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
RPS29 10419 NM_001032.4 1-3 Diamond-Blackfan anemia 13
RPS6KA3 10432 NM_004586.3 1-22 Coffin-Lowry syndrome
RPS7 10440 NM_001011.4 2-7 Diamond-Blackfan anemia 8, 612563 upper limb malformation Radial Ray abnormality
RRAS 10447 NM_006270.5 1-6 ATYPICAL NOONAN SYNDROME
RRAS2 17271 NM_001177314.1 6 1-6 Noonan syndrome
RRM2B 17296 NM_015713.5 1-9 Mitochondrial depletion syndrome Intellectual disability Progressive external ophthalmoplegiawith mitochondrial DNA deletions,autosomal dominant 5, 613077 Mitochondrial RibonucelotideReductase subunit 2 deficiency(Disorders of purine metabolism)
RSPH1 12371 NM_080860.4 1-9 Ciliary dyskinesia, primary, 24, 615481
RSPH3 21054 NM_031924.6 1-8 PRIMARY CILIARY DYSKINESIA WITHCENTRAL-COMPLEX DEFECTS
RSPO2 28583 NM_178565.5 2-6 Tetra-amelia with lung agenesis
RSPO4 16175 NM_001029871.4 1-5 Anonychia congenita, 206800
RSPRY1 29420 NM_133368.3 2-15 Spondyloepimetaphyseal dysplasia,Faden-Alkuraya type, 616585
RTEL1 15888 NM_032957.5 2-35 DYSKERATOSIS CONGENITA,AUTOSOMAL RECESSIVE 5
RTN2 10468 NM_005619.5 1-11 Spastic paraplegia 12, autosomaldominant, 604805

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 179/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
RTN4IP1 18647 NM_032730.5 1-9 Optic atrophy 10, with or without ataxia,mental retardation, and seizures
RTTN 18654 NM_173630.4 1-49 Intellectual disability Microcephaly, short stature, andpolymicrogyria with seizures
RUNX2 10472 NM_001024630.4 2-9 Metaphyseal dysplasia with maxillaryhypoplasia with or withoutbrachydactyly 156510 Cleidocranial dysplasia, 119600
RUSC2 23625 NM_001135999.1 2-12 Mental retardation, autosomalrecessive 61 OMIM
RYR1 10483 NM_000540.3 1-106 Rhabdomyolysis and metabolic muscledisorders Central core disease, 117000 Minicore myopathy with externalophthalmoplegia, 255320 Neuromuscular disease, congenital,with uniform type 1 fiber 117000 Malignant hyperthermia susceptibility 1145600
RYR2 10484 NM_001035.3 1-105 Ventricular tachycardia,catecholaminergic polymorphic, 1604772
RYR3 10485 NM_001036.5 1-104 childhood-onset nemaline myopathy
SACS 10519 NM_014363.6 2-10 Spastic ataxia, Charlevoix-Saguenay typeIntellectual disability
SALL1 10524 NM_002968.3 2-3 1-3 Radial Ray abnormality Polydactyly Townes Brocks syndrome (Renal-Ear-Anal-Radial syndrome), 107480

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 180/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SALL4 15924 NM_020436.5 1-4 Polydactyly Radial Ray abnormality IVIC syndrome 147750 Duane-radial ray syndrome, 607323 Okihiro (Duane-radial ray) syndrome607323 ACRO-RENAL-OCULAR SYNDROME607323
SAMD9 1348 NM_017654.4 3 MIRAGE - myelodysplasia, infection,restriction of growth, adrenalhypoplasia, genital phenotypes,enteropathy
SAMHD1 15925 NM_015474.3 1-16 Aicardi-Goutieres syndrome-5 (AGS5) seizures
SAR1B 10535 NM_001033503.3 3-8 Anderson disease (Inheritedhypolipidaemias) Chylomicron retention disease
SARS2 17697 NM_017827.4 1-16 Hyperuricemia, pulmonaryhypertension, renal failure, andalkalosis, 613845
SATB2 21637 NM_015265.4 3-12 Glass syndrome Orofacial Clefting with skeletal features Cleft palate, intellectual disability, poor-absent speech, bone fragility- raisedserum alkaline phosphatas
SBDS 19440 NM_016038.4 1-5 1-5 Shwachman-Diamond syndrome260400
SC5D 10547 NM_006918.5 2-5 Lathosterolosis, 607330 Intellectual disability Cataracts
SCAMP5 30386 NM_001178112.2 3-8 Intellectual disability Seizures
SCAPER 13081 NM_020843.4 2-32 Intellectual developmental disorder andretinitis pigmentosa, 618195
SCARB2 1665 NM_005506.4 1-12 Epilepsy, progressive myoclonic 4, withor without renal failure 254900

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 181/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SCARF2 19869 NM_153334.7 1-14 Van den Ende-Gupta syndrome, 600920
SCLT1 26406 NM_144643.4 1-21 Oro-facio-digital syndrome type IX (Adly(2014) Hum Mutat 35,36) Senior-Loken syndrome
SCN11A 10583 NM_014139.3 1-26 Neuropathy, hereditary sensory andautonomic, type VII, 615548 Episodic pain syndrome, familial, 3,615552
SCN1A 10585 NM_001165963.3 4-29 familial hemiplegic migraine 3 Epilepsy, generalized, with febrileseizures plus, type 2 604403 Epileptic encephalopathy, early infantile,6 (Dravet syndrome) 607208 Febrile seizures, familial, 3A 604403 Migraine, familial hemiplegic, 3 609634
SCN1B 10586 NM_001037.5 1-5 Epilepsy, generalized, with febrileseizures plus, type 1 604233 AD Epileptic encephalopathy, early infantile,52 617350 AR
SCN2A 10588 NM_021007.3 2-27 Epileptic encephalopathy, early infantile,11 613721 Seizures, benign familial infantile, 3607745
SCN3A 10590 NM_006922.4 3-28 Epileptic encephalopathy, early infantile,62, 617938 intellectual disability Epilepsy, familial focal, with variable foci4 617935 Epileptic encephalopathy, early infantile,62 617938
SCN4A 10591 NM_000334.4 1-24 Hyperkalemic periodic paralysis, type 2,170500 Myotonia congenita, atypical,acetazolamide-responsive, 608390 Myasthenic syndrome, congenital, 16,614198

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 182/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SCN8A 10596 NM_014191.4 2-27 ?Cognitive impairment with or withoutcerebellar ataxia,614306 Epileptic encephalopathy, earlyinfantile,614558 Seizures, benign familial infantile,617080paroxysmal kinesigenic dyskinesias
SCN9A 10597 NM_002977.3 2-27 Epilepsy, generalized, with febrileseizures plus, type 7 613863 Febrile seizures, familial, 3B 613863
SCO1 10603 NM_004589.4 1-6 Mitochondrial Respiratory ChainComplex IV Deficiency Hepatic failure, early onset, andneurologic disorder
SCO2 10604 NM_005138.2 2 Mitochondrial Respiratory ChainComplex IV Deficiency Myopia 6, 608908 Cardioencephalomyopathy, fatalinfantile, due to cytochrome c oxidasedeficiency 1
SCP2 10606 NM_002979.5 1-16 Sterol carrier protein deficiency(Disorders of peroxisomal alpha-, betaand omega-oxidation) Leukoencephalopathy with dystonia andmotor neuropathy
SCYL1 14372 NM_020680.4 1-18 Episodes of Liver Failure, PeripheralNeuropathy, Cerebellar Atrophy, andAtaxia Spinocerebellar ataxia, autosomalrecessive 21, 616719
SDCCAG8 10671 NM_006642.5 1-18 Intellectual disability SENIOR-LOKEN SYNDROME Bardet-Biedl Syndrome
SDHA 10680 NM_004168.4 1-15 1-15 Leigh syndrome, 256000 Paragangliomas 5, 614165 Mitochondrial respiratory chaincomplex II deficiency, 252011 Cardiomyopathy, dilated, 1GG OMIM

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 183/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SDHAF1 33867 NM_001042631.2 1 Mitochondrial complex II deficiency,252011
SDHAF2 26034 NM_017841.2 1-4 Isolated complex II deficiency Neuro-endocrine Tumours- PCC andPGL
SDHB 10681 NM_003000.3 1-8 Gastrointestinal stromal tumor, 606764 Pheochromocytoma, 171300 Paragangliomas 4, 115310 Isolated complex II deficiency Cowden syndrome 2, 612359 Paraganglioma and gastric stromalsarcoma, 606864 Succinate dehydrogenase-deficientleukoencephalopathy Mitochondrial Leukoencephalopathy
SDHC 10682 NM_003001.3 1-6 Isolated complex II deficiency Neuro-endocrine Tumours- PCC andPGL
SDHD 10683 NM_003002.4 1-4 Mitochondrial respiratory chaincomplex II deficiency 252011
SEC23A 10701 NM_006364.4 2-20 CRANIOLENTICULOSUTURAL DYSPLASIA
SEC23B 10702 NM_006363.6 2-20 COPII component SEC23B (Disorders ofmultiple glycosylation and otherglycosylation pathways, V-ATPasedeficiencies) Dyserythropoietic anemia, congenital,type II, 224100
SEC24D 10706 NM_014822.4 2-23 Osteogenesis Imperfecta, ColeCarpenter syndrome
SECISBP2 30972 NM_024077.5 1-17 THYROID HORMONE METABOLISM,ABNORMAL 609698
SEPN1 15999 NM_020451.3 1-13 Muscular dystrophy, rigid spine, 1,602771 congenital myopathy

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 184/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SEPSECS 30605 NM_016955.4 1-11 Pontocerebellar Hypoplasia
SERAC1 21061 NM_032861.4 2-17 Methylglutaconic aciduria withdeafness, encephalopathy and Leigh-likesyndrome (MEGDEL) (Organic acidurias) Dystonia
SERPINF1 8824 NM_002615.7 2-8 osteogenesis imperfecta
SERPINH1 1546 NM_001235.4 2-5 Osteogenesis Imperfecta andDecreased Bone Density
SERPINI1 8943 NM_005025.4 2-9 Encephalopathy, familial, withneuroserpin inclusion bodies 604218
SET 10760 NM_001122821.2 6-8 1-8 Intellectual disability
SETBP1 15573 NM_015559.3 2-6 Schinzel-Giedion midface retractionsyndrome, 269150
SETD1A 29010 NM_014712.3 2-19 Schizophrenia developmental disorder Intellectual disability Epilepsy
SETD1B 29187 NM_001353345.1 1-16 Epilepsy, developmental delay,intellectual disability, autistic behaviorand craniofacial dysmorphic features
SETD2 18420 NM_014159.6 1-21 Luscan-Lumish syndrome, 616831 intellectual disability SETD2-associated OvergrowthSyndrome
SETD5 25566 NM_001080517.3 3-23 Mental retardation, autosomaldominant 23, 615761

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 185/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SETX 445 NM_015046.7 3-26 Charcot-Marie-Tooth disease Hereditary ataxia Amyotrophic lateral sclerosis/motorneuron disease ataxia with oculomotor apraxia type 2(AOA2), juvenile amyotrophic lateralsclerosis (ALS4) and autosomaldominant ataxia Spinocerebellar ataxia, autosomalrecessive, with axonal neuropathy 2
SF3B4 10771 NM_005850.5 1-6 Acrofacial dysostosis 1, Nager type,154400
SFRP4 10778 NM_003014.4 1-6 Pyle disease, 265900 Metaphyseal dysplasia
SFXN4 16088 NM_213649.2 1-14 Combined oxidative phosphorylationdeficiency 18, 615578
SGCA 10805 NM_000023.4 1-9 Muscular dystrophy, limb-girdle, type2D, 608099
SGCB 10806 NM_000232.4 1-6 Muscular dystrophy, limb-girdle, type2E, 604286
SGCD 10807 NM_000337.5 2-9 Muscular dystrophy, limb-girdle, type2F, 601287
SGCE 10808 NM_003919.3 1-11 Myoclonus-Dystonia maternally imprinted Dystonia-11,myoclonic, 159900
SGCG 10809 NM_000231.2 2-8 Muscular dystrophy, limb-girdle, type2C, 253700
SGPL1 10817 NM_003901.4 2-15 Nephrotic syndrome 14 617575
SGSH 10818 NM_000199.5 1-8 MPS IIIA, Sanfilippo A disease(Mucopolysaccharidoses) seizures
SH3BP2 10825 NM_003023.4 2-13 Cherubism 118400

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 186/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SH3PXD2B 29242 NM_001017995.3 1-13 Frank-ter Haar syndrome, 249420
SHANK1 15474 NM_016148.5 2-24 AUTISM 209850
SHANK2 14295 NM_133266.5 1-11 SUSCEPTIBILITY TO AUTISM TYPE 17(AUTS17)
SHANK3 14294 NM_001372044.1 1-23 PHELAN-MCDERMID SYNDROME
SHH 10848 NM_000193.4 1-3 MICROPHTHALMIA ISOLATED WITHCOLOBOMA TYPE 5 611638 SOLITARY MEDIAN MAXILLARY CENTRALINCISOR 147250 TRIPHALANGEAL THUMB-POLYSYNDACTYLY SYNDROME (TPTPS) Holoprosencephaly
SHOC2 15454 NM_007373.3 2-9 Noonan-like syndrome with looseanagen hair, 607721
SHOX 10853 NM_000451.3 2-6 2-6 Langer mesomelic dysplasia, 249700 Leri-Weill dyschondrosteosis, 127300 Short stature, idiopathic familial 300582
SHROOM3 30422 NM_020859.4 1-11 NEURAL TUBE DEFECT
SHROOM4 29215 NM_020717.3 1-9 Stocco dos Santos X-linked mentalretardation syndrome, 300434
SI 10856 NM_001041.4 2-48 CONGENITAL SUCRASE-ISOMALTASEDEFICIENCY 222900 Disaccharide intolerance 1 (Othercarbohydrate disorders)
SIK1 11142 NM_173354.5 2-14 Epileptic encephalopathy, early infantile,30
SIL1 24624 NM_022464.5 2-10 Marinesco-Sjogren syndrome, 248800
SIM1 10882 NM_005068.2 1-11 Severe obesity with neurobehavioralfeatures

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 187/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SIN3A 19353 NM_001145358.2 17 2-21 Witteveen-Kolk syndrome, 613406 Syndromic intellectual disability
SIX1 10887 NM_005982.4 1-2 Non-syndromic craniosynostosis Brachiootic syndrome 3, 608389 Deafness, autosomal dominant 23,605192
SIX3 10889 NM_005413.4 1-2 Schizencephaly 269160 Holoprosencephaly-2
SIX5 10891 NM_175875.5 1-3 Branchiootorenal syndrome 2, 610896
SKI 10896 NM_003036.4 1-7 Shprintzen-Goldberg syndrome
SKIV2L 10898 NM_006929.5 1-28 Trichohepatoenteric syndrome 2 (Othermetabolic disorders)
SLC10A7 23088 NM_001029998.6 1-12 Chondrodysplasia with multipledislocations and amelogenesisimperfecta
SLC12A3 10912 NM_000339.3 1-26 Gitelman syndrome (Disorder ofmagnesium metabolism) Renal tubular acidosis
SLC12A5 13818 NM_020708.5 1-26 FEBRILE SEIZURES epilepsy of infancy with migrating focalseizures (EIMFS) Epileptic encephalopathy, early infantile,34, 616645
SLC12A6 10914 NM_133647.1 1-25 Agenesis of the corpus callosum withperipheral neuropathy, 218000 -3
SLC13A5 23089 NM_177550.5 1-12 Epileptic encephalopathy, early infantile,25
SLC16A1 10922 NM_003051.3 2-5 Hyperinsulinemic hypoglycemia,familial, 7

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 188/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SLC16A2 10923 NM_006517.5 1-6 Allan-Herndon-Dudley syndrome,300523 General Leukodystrophy &Mitochondrial Leukoencephalopathy Monocarboxylate transporter 8deficiency (MCT8)
SLC17A5 10933 NM_012434.5 1-11 Salla disease, 604369Sialic acid storagedisorder, infantile, 269920 General Leukodystrophy &Mitochondrial Leukoencephalopathy
SLC18A2 10935 NM_003054.6 2-16 Brain Dopamine Serotonin VesicularTransport Disease (Other disorders ofneurotransmitter metabolism)
SLC18A3 10936 NM_003055.3 1 Myasthenic syndrome, congenital, 21,presynaptic, 617239
SLC19A2 10938 NM_006996.3 1-6 Thiamine-responsive megaloblasticanemia syndrome (Disorders ofthiamine metabolism)
SLC19A3 16266 NM_025243.4 2-6 Biotin-responsive basal ganglia disease(Disorders of thiamine metabolism)
SLC1A2 10940 NM_004171.4 1-11 Epileptic encephalopathy, early infantile,41, 617105 Epileptic encephalopathy, early infantile,41, 617105
SLC1A3 10941 NM_004172.5 2-10 Episodic ataxia, type 6,
SLC1A4 10942 NM_003038.5 1-8 Spastic tetraplegia, thin corpuscallosum, and progressivemicrocephaly, 616657
SLC20A2 10947 NM_006749.5 2-11 Basal ganglia calcification, idiopathic, 1213600 Dystonia Fahr syndrome
SLC22A5 10969 NM_003060.4 1-10 Propionicacidemia Carnitine transporter deficiency(Disorders of carnitine transport andthe carnitine cycle)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 189/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SLC24A4 10978 NM_153646.3 1-17 AMELOGENESIS IMPERFECTA.
SLC25A1 10979 NM_005984.5 1-9 Combined D-2- and L-2-hydroxyglutaricaciduria 615182 Disorders of mitochondrial proteintransport Riboflavin transporter deficiency(Disorders of riboflavin transport andmetabolism)
SLC25A12 10982 NM_003705.5 9 1-18 Epileptic encephalopathy, early infantile,39 612949 Disorders of mitochondrial soluteimport (Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly))
SLC25A13 10983 NM_014251.3 1-18 Neonatal and Adult Cholestasis Citrullinemia Type 2 (Urea cycledisorders and inheritedhyperammonaemias)
SLC25A15 10985 NM_014252.4 2, 6-7 2-7 Hyperornithinemia-hyperammonemia-homocitrullinemia syndrome, 238970
SLC25A19 14409 NM_021734.4 3-8 Thiamine metabolism dysfunctionsyndrome 4 (progressivepolyneuropathy type), 613710 Microcephaly, Amish type (Disorders ofthiamine metabolism)
SLC25A20 1421 NM_000387.6 1-9 Carnitine acylcarnitine translocasedeficiency (Disorders of carnitinetransport and the carnitine cycle)
SLC25A21 14411 NM_030631.4 1-10 No OMIM phenotype
SLC25A22 19954 NM_024698.6 2-10 Epileptic encephalopathy, early infantile,3, 609304 Disorders of mitochondrial soluteimport (Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly))

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 190/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SLC25A24 20662 NM_013386.5 1-10 Gorlin-Chaudhry-Moss syndrome(GCMS) Syndrome with Hypertrichosis,Progeroid Appearance, andMitochondrial Dysfunction
SLC25A26 20661 NM_173471.3 2-11 Combined oxidative phosphorylationdeficiency 28 Intra-mitochondrial MethylationDeficiency leading to Clinical findingsranging from neonatal mortalityresulting from respiratory insufficiencyand hydrops to childhood acuteepisodes of cardiopulmonary failureand slowly progressive muscleweakness
SLC25A3 10989 NM_005888.3 2-8 Mitochondrial phosphate carrierdeficiency 610773 Disorders of mitochondrial soluteimport (Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly)) Riboflavin transporter deficiency(Disorders of riboflavin transport andmetabolism)
SLC25A32 29683 NM_030780.5 1-7 ?Exercise intolerance, riboflavin-responsive 616839
SLC25A38 26054 NM_017875.4 1-7 Disorders of mitochondrial soluteimport (Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly)) Anemia, sideroblastic, pyridoxine-refractory, autosomal recessive, 205950

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 191/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SLC25A4 10990 NM_001151.4 1-4 Progressive External Ophthalmoplegiawith Mitochondrial DNADeletions Disorders of mitochondrial DNAmaintenance and integrity Fontaine progeroid syndrome Mitochondrial DNA depletion syndrome12A (cardiomyopathic type) AD 617184 Mitochondrial Leukoencephalopathy
SLC25A42 28380 NM_178526.5 2-8 Metabolic crises, recurrent, withvariable encephalomyopathic featuresand neurologic regression 618416 mitochondrial myopathy
SLC25A46 25198 NM_138773.4 1-8 optic atrophy spectrum disorder Neuropathy, hereditary motor andsensory, type VIB, 616505
SLC26A2 10994 NM_000112.4 2-3 Atelosteogenesis II (includes clefting),256050 De la Chapelle dysplasia (includesclefting), 256050 Diastrophic dysplasia, broadbonehplatyspondylic variant, 222600 Epiphyseal dysplasia, multiple, 4 Achondrogenesis Ib, 600972
SLC27A4 10998 NM_005094.4 2-13 Ichthyosis prematurity syndrome,608649
SLC29A3 23096 NM_018344.6 1-6 Histiocytosis-lymphadenopathy plussyndrome 602782
SLC2A1 11005 NM_006516.3 1-10 Glucose transporter 1 deficiency (blood-brain barrier) (Disorders of glucosetransport) Hereditary ataxia Epileptic encephalopathy GLUT1 deficiency syndrome 1, infantileonset, severe 606777 AD, AR GLUT1 deficiency syndrome 2,childhood onset 612126 AD Stomatin-deficient cryohydrocytosiswith neurologic defects 608885 AD

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 192/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SLC2A10 13444 NM_030777.4 1-5 ARTERIAL TORTUOSITY SYNDROME208050
SLC2A2 11006 NM_000340.2 1-11 Glycogen storage disease type XI(Glycogen storage disorders) Glucose transporter 2 deficiency(Disorders of glucose transport) Fanconi-Bickel Syndrome
SLC30A10 25355 NM_018713.2 1-4 Early onset dystonia Hypermanganesemia with dystonia,polycythemia, and cirrhosis (Disorder ofmagnesium metabolism)
SLC33A1 95 NM_004733.4 6 1-6 Congenital cataracts, hearing loss, andneurodegeneration, 614482
SLC34A1 11019 NM_003052.5 2-13 Nephrolithiasis/osteoporosis,hypophosphatemic, 1, 612286
SLC34A3 20305 NM_080877.2 2-13 Hypophosphatemic rickets withhypercalciuria 241530
SLC35A1 11021 NM_006416.5 1-8 Congenital disorder of glycosylation,type IIf 603585 CMP-sialic acid transporter deficiency(Disorders of multiple glycosylation andother glycosylation pathways) intellectual disability, ataxia, seizures,bleeding diathesis and proteinuria
SLC35A2 11022 NM_001042498.3 1-4 Congenital disorder of glycosylation,type IIm 300896 Epileptic encephalopathy, early infantile,22 (EIEE22)
SLC35A3 11023 NM_012243.3 2-8 ?Arthrogryposis, mental retardation,and seizures (MIM 615553)
SLC35C1 20197 NM_018389.5 1-2 Congenital disorder of glycosylation,type IIc, 266265 GDP-fucose transporter deficiency(Disorders of multiple glycosylation andother glycosylation pathways)

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 193/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SLC35D1 20800 NM_015139.3 1-12 Schneckenbecken dysplasia, 269250
SLC37A4 4061 NM_001164277.1 3-11 Glycogen Storage Disease Ib and Ic heptomgaly, feed intolerance ,inflammatory bowel disease,neutropenia
SLC39A13 20859 NM_152264.4 2-10 Spondylocheirodysplasia, Ehlers-Danlossyndrome-like 612350
SLC39A14 20858 NM_015359.6 2-9 Hypermanganesemia with dystonia 2617013
SLC39A4 17129 NM_130849.4 1-12 Acrodermatitis enteropathica 201100(Disorder of zinc metabolism)
SLC39A8 20862 NM_022154.5 1-8 Congenital disorder of glycosylation,type IIn 616721 Hypomagnesaemia with cerebellaratrophy, hypotonia, strabismus,developmental delay, short stature,mild skeletal dysplasia, and connectivetissue abnormalities (Disorder ofmagnesium metabolism)
SLC3A1 11025 NM_000341.4 1-10 Renal tract calcification (orNephrolithiasis/nephrocalcinosis) Hypotonia-cystinuria syndrome(Disorders of amino acid transport)
SLC40A1 10909 NM_014585.5 1-8 Hemochromatosis, type 4 606069(Disorder of iron metabolism)
SLC45A1 17939 NM_001080397.3 2-9 Intellectual developmental disorder withneuropsychiatric features, 617532
SLC46A1 30521 NM_080669.6 1-6 Hereditary folate malabsorption(Disorders of folate metabolism andtransport)
SLC4A1 11027 NM_000342.4 2-20 Spherocytosis, type 4, 612653 Renal tubular acidosis, distal, AD,179800 Renal tubular acidosis, distal, AR,611590
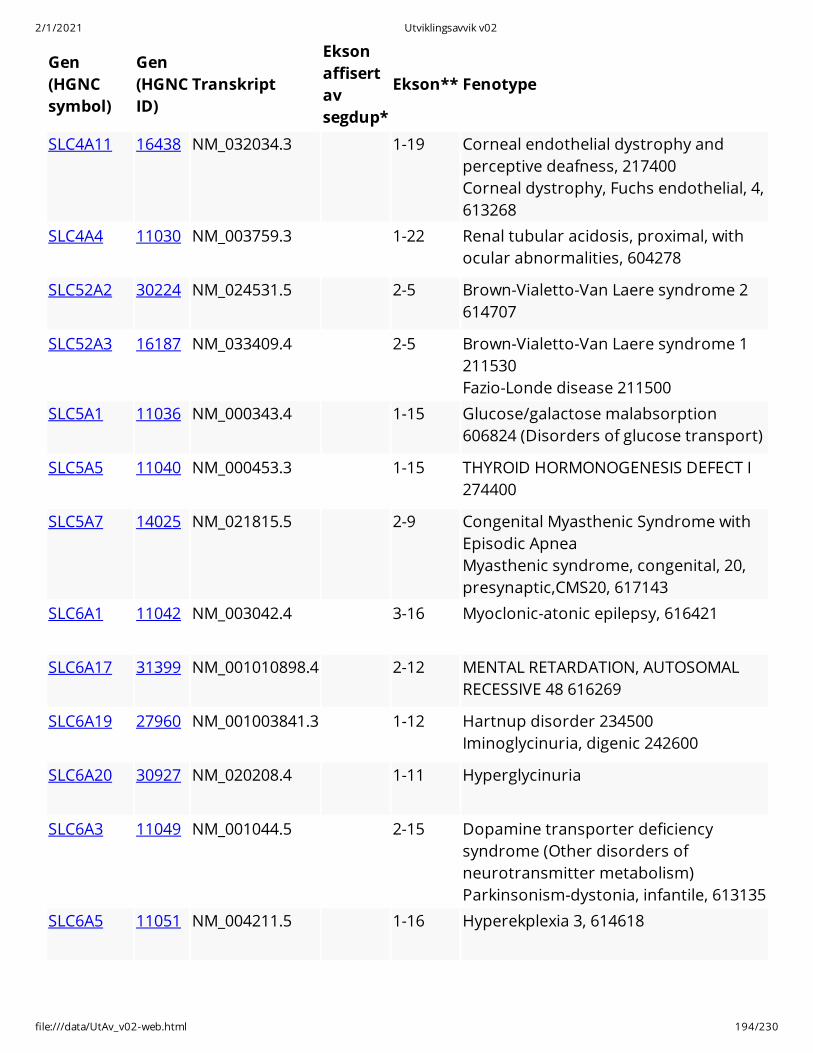
2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 194/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SLC4A11 16438 NM_032034.3 1-19 Corneal endothelial dystrophy andperceptive deafness, 217400 Corneal dystrophy, Fuchs endothelial, 4,613268
SLC4A4 11030 NM_003759.3 1-22 Renal tubular acidosis, proximal, withocular abnormalities, 604278
SLC52A2 30224 NM_024531.5 2-5 Brown-Vialetto-Van Laere syndrome 2614707
SLC52A3 16187 NM_033409.4 2-5 Brown-Vialetto-Van Laere syndrome 1211530 Fazio-Londe disease 211500
SLC5A1 11036 NM_000343.4 1-15 Glucose/galactose malabsorption606824 (Disorders of glucose transport)
SLC5A5 11040 NM_000453.3 1-15 THYROID HORMONOGENESIS DEFECT I274400
SLC5A7 14025 NM_021815.5 2-9 Congenital Myasthenic Syndrome withEpisodic Apnea Myasthenic syndrome, congenital, 20,presynaptic,CMS20, 617143
SLC6A1 11042 NM_003042.4 3-16 Myoclonic-atonic epilepsy, 616421
SLC6A17 31399 NM_001010898.4 2-12 MENTAL RETARDATION, AUTOSOMALRECESSIVE 48 616269
SLC6A19 27960 NM_001003841.3 1-12 Hartnup disorder 234500 Iminoglycinuria, digenic 242600
SLC6A20 30927 NM_020208.4 1-11 Hyperglycinuria
SLC6A3 11049 NM_001044.5 2-15 Dopamine transporter deficiencysyndrome (Other disorders ofneurotransmitter metabolism) Parkinsonism-dystonia, infantile, 613135
SLC6A5 11051 NM_004211.5 1-16 Hyperekplexia 3, 614618

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 195/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SLC6A8 11055 NM_005629.4 1-13 1-13 Creatine transporter deficiency(Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly), disorders of creatininemetabolism) Cerebral creatine deficiency syndrome 1300352
SLC6A9 11056 NM_201649.4 1-14 Glycine encephalopathy with normalserum glycine, 617301
SLC7A7 11065 NM_001126106.4 3-11 Lysinuric protein intolerance (Disordersof amino acid transport)
SLC7A9 11067 NM_014270.5 2-13 Renal tract calcification (orNephrolithiasis/nephrocalcinosis) Cystinuria (Disorders of amino acidtransport)
SLC9A6 11079 NM_006359.3 1-16 Mental retardation, X-linked syndromic,Christianson type, 300243 Angelman-like Syndrome
SLCO2A1 10955 NM_005630.3 1-14 Hypertrophic osteoarthropathy,primary, autosomal recessive 2 614441
SLX4 23845 NM_032444.4 2-15 Fanconi anemia, complementationgroup P, 613951
SMAD3 6769 NM_005902.4 1-9 Loeys-Dietz syndrome 3 613795
SMAD4 6770 NM_005359.6 2-12 JUVENILE POLYPOSIS/HEREDITARYHEMORRHAGIC TELANGIECTASIASYNDROME 175050 Myhre syndrome, 139210
SMAD6 6772 NM_005585.5 1-4 radioulnar synostosis
SMAD9 6774 NM_001127217.2 2-7 Pulmonary arterial hypertension
SMARCA2 11098 NM_003070.5 2-34 COFFIN SIRIS 135900 Nicolaides-Baraitser syndrome, 601358

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 196/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SMARCA4 11100 NM_001128849.2 2-36 COFFIN SIRIS 135900 Rhabdoid tumor predispositionsyndrome 2, 613325Mental retardation,autosomal dominant 16, 614609
SMARCAL1 11102 NM_014140.4 3-18 SCHIMKE IMMUNOOSSEOUS DYSPLASIA242900
SMARCB1 11103 NM_003073.5 1-9 EHMT1-like SYNDROME Rhabdoid tumors, somatic,609322Rhabdoid predispositionsyndrome 1, 609322Mental retardation,autosomal dominant 15, 614608
SMARCC2 11105 NM_003075.5 1-28 Syndromic Intellectual Disability andDevelopmental Delay Coffin-Siris syndrome 8, 618362
SMARCD1 11106 NM_003076.5 1-13 SYNDROMIC INTELLECTUAL DISABILITY612100
SMARCD2 11107 NM_001098426.2 1-13 Specific granule deficiency 2, 617475(includes global developmental delay insome patients)
SMARCE1 11109 NM_003079.5 2-11 COFFIN SIRIS 135900
SMC1A 11111 NM_006306.4 1-25 Cornelia de Lange syndrome 2, 300590
SMC3 2468 NM_005445.3 1-29 Cornelia de Lange syndrome 3, 610759
SMCHD1 29090 NM_015295.2 1-48 Isolated Arhinia/Bosma Arhiniasyndrome Fascioscapulohumeral musculardystrophy 2, digenic 158901
SMG9 25763 NM_019108.4 2-14 Heart and brain malformationsyndrome, 616920
SMO 11119 NM_005631.5 1-12 Cutaneous syndactyly Preaxial polydactyly Curry-Jones syndrome, somatic mosaic601707

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 197/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SMOC1 20318 NM_001034852.3 1-12 Microphthalmia with limb anomalies206920
SMOC2 20323 NM_022138.3 1-13 DENTIN DYSPLASIA, TYPE I, WITHMICRODONTIA AND MISSHAPEN TEETH125400
SMPD1 11120 NM_000543.5 1-6 Niemann-Pick disease, type A, 257200 Niemann-Pick disease, type B, 607616
SMPD4 32949 NM_017951.4 1-20 1-20 Developmental Disorder withMicrocephaly and CongenitalArthrogryposis cerebellar hypoplasia, hypomyelination,microcephaly, arthrogryposis, diabetes
SMS 11123 NM_004595.5 1-11 Mental retardation, X-linked, Snyder-Robinson type, 309583
SNAP25 11132 NM_003081.4 2-8 ?Myasthenic syndrome, congenital 18,616330 Epilepsy and intellectual disability
SNAP29 11133 NM_004782.4 1-5 Cerebral dysgenesis, neuropathy,ichthyosis, and palmoplantarkeratoderma syndrome, 609528 CEDNIK SYNDROME 609528
SNIP1 30587 NM_024700.4 1-4 Psychomotor retardation, epilepsy, andcraniofacial dysmorphism 614501
SNRPB 11153 NM_003091.4 1-7 Cerebrocostomandibular syndrome117650
SNRPE 11161 NM_003094.4 1-5 AUTOSOMAL-DOMINANTHYPOTRICHOSIS SIMPLEX 615059
SNX10 14974 NM_001199835.1 2-7 Osteopetrosis, autosomal recessive 8615085
SNX14 14977 NM_153816.6 1-29 ID, MACROCEPHALY AND CEREBELLARHYPOPLASIA Spinocerebellar ataxia, autosomalrecessive 20, 616354
SON 11183 NM_032195.2 1-7 ZTTK syndrome 617140 Intellectual Disability, CongenitalMalformations, and Failure to Thrive

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 198/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SOS1 11187 NM_005633.3 1-23 Fibromatosis, gingival, 135300Noonansyndrome 4, 610733 NOONAN SYNDROME 4
SOS2 11188 NM_006939.4 1-23 Noonan syndrome 9
SOST 13771 NM_025237.3 1-2 Craniodiaphyseal dysplasia, autosomaldominant 122860
SOX10 11190 NM_006941.4 2-4 KALLMANN SYNDROME WITH DEAFNESS YEMENITE DEAF-BLINDHYPOPIGMENTATION SYNDROME601706 Waardenburg syndrome, type 4C,613266Waardenburg syndrome, type2E, with or without neurologicinvolvement, 611584PCWH syndrome,609136 peripheral demyelinating neuropathy,central dysmyelinating leukodystrophy
SOX11 11191 NM_003108.4 1 Coffin-Siris syndrome 9, 615866 MENTAL RETARDATION, AUTOSOMALDOMINANT, 27 615866
SOX17 18122 NM_022454.4 1-2 Vesicoureteral reflux 3, 613674 Heritable pulmonary arterialhypertension
SOX2 11195 NM_003106.4 1 1 MICROPHTHALMIA SYNDROMIC TYPE 3206900
SOX3 11199 NM_005634.2 1 SEX REVERSAL TYPE 3 300833 Mental retardation, X-linked, withisolated growth hormone deficiency,300123Panhypopituitarism, X-linked,312000
SOX4 11200 NM_003107.3 1 Coffin-Siris syndrome 10, 618506 Syndromic intellectual disability
SOX5 11201 NM_006940.6 1-15 12P12.5 INTRAGENIC DELETIONSASSOCIATED WITH INTELLECTUALDISABILITY

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 199/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SOX9 11204 NM_000346.4 1-3 Campomelic dysplasia with autosomalsex reversal 114290 Cleft palate with skeletal abnormalities Acampomelic campomelic dysplasia,114290
SP7 17321 NM_001173467.3 2-3 Osteogenesis imperfecta, type XII613849
SPAG1 11212 NM_172218.3 2-19 Ciliary dyskinesia, primary, 28, 615505
SPARC 11219 NM_003118.4 2-10 Osteogenesis imperfecta, type XVII616507
SPAST 11233 NM_014946.3 1-17 Spastic paraplegia 4, autosomaldominant, 182601
SPATA5 18119 NM_145207.3 1-16 Epilepsy, hearing loss, and mentalretardation syndrome, 616577
SPECC1L 29022 NM_015330.5 4 3-17 ?Facial clefting, oblique, 1, 600251 Opitz GBBB syndrome, type II (withclefting), 145410
SPEG 16901 NM_005876.5 1-41 Centronuclear myopathy 5 615959
SPG11 11226 NM_025137.4 1-40 Spastic paraplegia 11, autosomalrecessive, 604360 Amyotrophic lateral sclerosis 5, juvenile602099
SPG20 18514 NM_001142294.1 2-9 developmental delay Troyer syndrome, 275900 Spastic paraplegia 20
SPG21 20373 NM_016630.7 2-9 Spastic Paraplegia, Recessive
SPG7 11237 NM_003119.4 1-17 Disorders of mitochondrial DNAmaintenance and integrity Spastic paraplegia 7, 607259
SPINT2 11247 NM_021102.4 1-7 Polydactyly Diarrhea 3, secretory sodium,congenital, syndromic, 270420

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 200/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SPR 11257 NM_003124.5 1-3 Sepiapterin reductase deficiency(Disorders of pterin metabolism) Dopa-Responsive Dystonia Movement disorder, autonomicdysfunction, developmental delay,behavioural difficulties
SPRED1 20249 NM_152594.3 1-7 Legius syndrome, 611431
SPTAN1 11273 NM_001130438.3 2-57 Epileptic encephalopathy, early infantile,5, 613477
SPTBN2 11276 NM_006946.3 2-37 Infantile ataxia with oculomotor andpyramidal signs Spinocerebellar ataxia, autosomalrecessive 14 Spinocerebellar Ataxia, Dominant
SPTLC1 11277 NM_006415.4 3 1-15 Charcot-Marie-Tooth disease Serine palmitoyl transferase deficiency(Disorders of complex lipid synthesis) Familial dysautonomia
SPTLC2 11278 NM_004863.3 1-12 Charcot-Marie-Tooth disease Serine palmitoyl transferase deficiency(Disorders of complex lipid synthesis) Familial dysautonomia
SQSTM1 11280 NM_003900.5 1-8 Dystal Myopathy with rimmed vacuoles,617158
SRCAP 16974 NM_006662.3 3-34 Floating-Harbor syndrome, 136140
SRD5A3 25812 NM_024592.5 4-5 1-5 Congenital disorder of glycosylation,type Iq, 612379Kahrizi syndrome,612713
SRGAP3 19744 NM_014850.4 1-22 3p- syndrome, MIM:613792 (includesintellectual disability)
SRP54 11301 NM_003136.4 2-16 Syndromic neutropenia withShwachman-Diamond-like features
SRPX2 30668 NM_014467.3 2-11 ?Rolandic epilepsy, mental retardation,and speech dyspraxia 300643

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 201/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SRY 11311 NM_003140.3 1 46XY SEX REVERSAL 1 400044 46XY SEX REVERSAL 1
SSBP1 11317 NM_001256513.1 2-7
SSR4 11326 NM_001204526.1 1-7 Congenital disorder of glycosylation,type Iy, 300934
ST14 11344 NM_021978.4 1-19 ICHTHYOSIS AUTOSOMAL RECESSIVEWITH HYPOTRICHOSIS 610765
ST3GAL3 10866 NM_006279.5 2-12 Mental retardation, autosomalrecessive 12, 611090Epilepticencephalopathy, early infantile, 15,615006 Epileptic encephalopathy, early infantile,15
ST3GAL5 10872 NM_003896.4 1-7 Salt and pepper developmentalregression syndrome, 609056 Lactosylceramide alpha-2,3-sialyltransferase deficiency (Disordersof glycosphingolipid andglycosylphosphatidylinositol anchorglycosylation)
STAC3 28423 NM_145064.3 2-12 Myopathy, congenital, Baily-Bloch,255995
STAG1 11354 NM_005862.3 2-34 Mental retardation, autosomaldominant 47, 617635
STAG2 11355 NM_001042749.2 3-35 STAG2-related developmental delay withmicrocephaly and congenital anomalies STAG2-related developmental delay withmicrocephaly and congenital anomalies
STAMBP 16950 NM_006463.5 3-11 Microcephaly-capillary malformationsyndrome 614261
STAR 11359 NM_000349.3 1-7 CHOLESTEROL DESMOLASE-DEFICIENTCONGENITAL ADRENAL HYPERPLASIA201710 Lipoid adrenal hyperplasia, 201710

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 202/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
STAT1 11362 NM_007315.3 3-25 Immunodeficiency 31B, mycobacterialand viral infections, autosomalrecessive, 613796 Immunodeficiency 31C, autosomaldominant, 614162
STAT2 11363 NM_005419.4 2-24 Immunodeficiency 44 616636 elongated mitochondria
STAT5B 11367 NM_012448.4 6-9 2-19 GROWTH HORMONE INSENSITIVITYWITH IMMUNODEFICIENCY 245590
STIL 10879 NM_003035.2 2-17 Microcephaly 7, primary, autosomalrecessive, 612703
STIM1 11386 NM_003156.3 1-12 Myopathy, tubular aggregate, 160565 Stormorken syndrome 185070
STRA6 30650 NM_022369.4 2-19 Microphthalmia, syndromic 9,601186Microphthalmia, isolated, withcoloboma 8, 601186
STRADA 30172 NM_001003787.4 2-13 Polyhydramnios, megalencephaly, andsymptomatic epilepsy, MIM:611087(includes mental retardation)
STS 11425 NM_000351.7 2-10 X-linked ichthyosis (Other disorders inthe metabolism of sterols)
STT3A 6172 NM_001278503.2 3-19 ?Congenital disorder of glycosylation,type Iw, 615596
STUB1 11427 NM_005861.4 1-7 Spinocerebellar ataxia, autosomalrecessive 16, 615768
STX1B 18539 NM_052874.5 1-10 Generalized epilepsy with febrileseizures plus, type 9, 616172
STXBP1 11444 NM_003165.5 1-19 Epileptic encephalopathy, early infantile,4
SUCLA2 11448 NM_003850.2 1-11 Mitochondrial DNA depletion syndrome5 (encephalomyopathic with or withoutmethylmalonicaciduria), 612073

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 203/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SUCLG1 11449 NM_003849.4 1-9 Mitochondrial DNA depletion syndrome9 (encephalomyopathic type withmethylmalonic aciduria),245400 FATAL INFANTILE LACTIC ACIDOSIS308078
SUFU 16466 NM_016169.3 1-12 Joubert Syndrome with Cranio-facial andSkeletal Defects Basal cell nevus syndrome, 109400
SUMF1 20376 NM_182760.4 1-9 Multiple sulfatase deficiency 272200 General Leukodystrophy &Mitochondrial Leukoencephalopathy
SUOX 11460 NM_000456.3 4-6 Sulfite oxidase deficiency, 272300
SURF1 11474 NM_003172.4 1-9 Leigh syndrome, due to COX deficiency,256000 Complex IV (Mitochondrial respiratorychain disorders (caused by nuclearvariants only), OXPHOS assemblyfactors) Charcot-Marie-Tooth disease, type 4K,616684
SUV420H1 24283 NM_017635.5 2-11 Mental retardation, autosomaldominant 51, 617788 KMT5B syndrome
SUZ12 17101 NM_015355.4 1-9 1-16 Weaver-like overgrowth syndrome
SYN1 11494 NM_133499.2 1-13 Epilepsy, X-linked, with variable learningdisabilities and behavior disorders,300491
SYNE1 17089 NM_033071.3 2-146 Spinocerebellar ataxia, autosomalrecessive 8 Emery-Dreifuss muscular dystrophy 4,autosomal dominant 612998
SYNE2 17084 NM_182914.2 2-116 Emery-Dreifuss muscular dystrophy 5,autosomal dominant 612999
SYNGAP1 11497 NM_006772.3 1-19 Mental retardation, autosomaldominant 5, 612621

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 204/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
SYNJ1 11503 NM_003895.3 1-32 Epileptic encephalopathy, early infantile,53, 617389 Parkinson disease 20, early-onset,615530
SYP 11506 NM_003179.2 1-6 Mental retardation, X-linked 96, 300802
SYT1 11509 NM_005639.3 4-11 SYT1-associated neurodevelopmentaldisorder
SYT2 11510 NM_177402.5 2 2-9 Myasthenic syndrome, congenital, 7,presynaptic, 616040
SZT2 29040 NM_015284.4 1-71 Epileptic encephalopathy, early infantile,18, 615476
TAB2 17075 NM_015093.5 4-9 Congenital heart defects,nonsyndromic, 2, 614980
TAC3 11521 NM_013251.4 2-6 HYPOGONADOTROPIC HYPOGONADISM146110
TACO1 24316 NM_016360.4 1-5 Mitochondrial Respiratory ChainComplex IV Deficiency Mitochondrial Leukoencephalopathy
TACR3 11528 NM_001059.3 1-5 HYPOGONADOTROPIC HYPOGONADISM146110
TAF1 11535 NM_004606.5 1-38 Mental retardation, X-linked, syndromic33, 300966 Dystonia-Parkinsonism, X-linked, 314250
TAF13 11546 NM_005645.4 1-4 Mental retardation, autosomalrecessive 60 617432
TAF6 11540 NM_005641.4 2-15 Alazami-Yuan syndrome, 617126 Intellectual disability
TALDO1 11559 NM_006755.2 1-8 Transaldolase deficiency, 606003
TANC2 30212 NM_025185.4 9-10 2-26 Neurodevelopmental Disorder Epilepsy

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 205/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TANGO2 25439 NM_152906.7 2-9 Metabolic encephalomyopathic crises,recurrent, with rhabdomyolysis, cardiacarrhythmias, and neurodegeneration616878
TAOK1 29259 NM_020791.2 2-20 INTELLECTUAL DISABILITY 616579
TAPT1 26887 NM_153365.3 1-14 Osteochondrodysplasia, complex lethal,Symoens-Barnes-Gistelinck type 616897
TARS 11572 NM_152295.4 1-19 Non-photosensitive trichothiodystrophy
TARS2 30740 NM_025150.5 1-18 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis)
TAT 11573 NM_000353.3 2-12 Tyrosinaemia type II (Disorders ofphenylalanine or tyrosine metabolism)
TAZ 11577 NM_000116.5 1-11 Disorders of mitochondrial membranelipids (Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly)) Methylglutaconic aciduria type II, Barthsyndrome (Organic acidurias)
TBC1D20 16133 NM_144628.4 1-8 Warburg micro syndrome 4, 615663
TBC1D23 25622 NM_001199198.3 1-19 Pontocerebellar hypoplasia, type 11,617695
TBC1D24 29203 NM_001199107.2 2-8 Myoclonic epilepsy, infantile, familial,605021Epileptic encephalopathy, earlyinfantile, 16, 615338 NON SYNDROMAL HEARING LOSS
TBC1D7 21066 NM_001143965.3 2-8 Macrocephaly/megalencephalysyndrome, autosomal recessive
TBCD 11581 NM_005993.5 1-39 Encephalopathy, progressive, early-onset, with brain atrophy and thincorpus callosum 617193
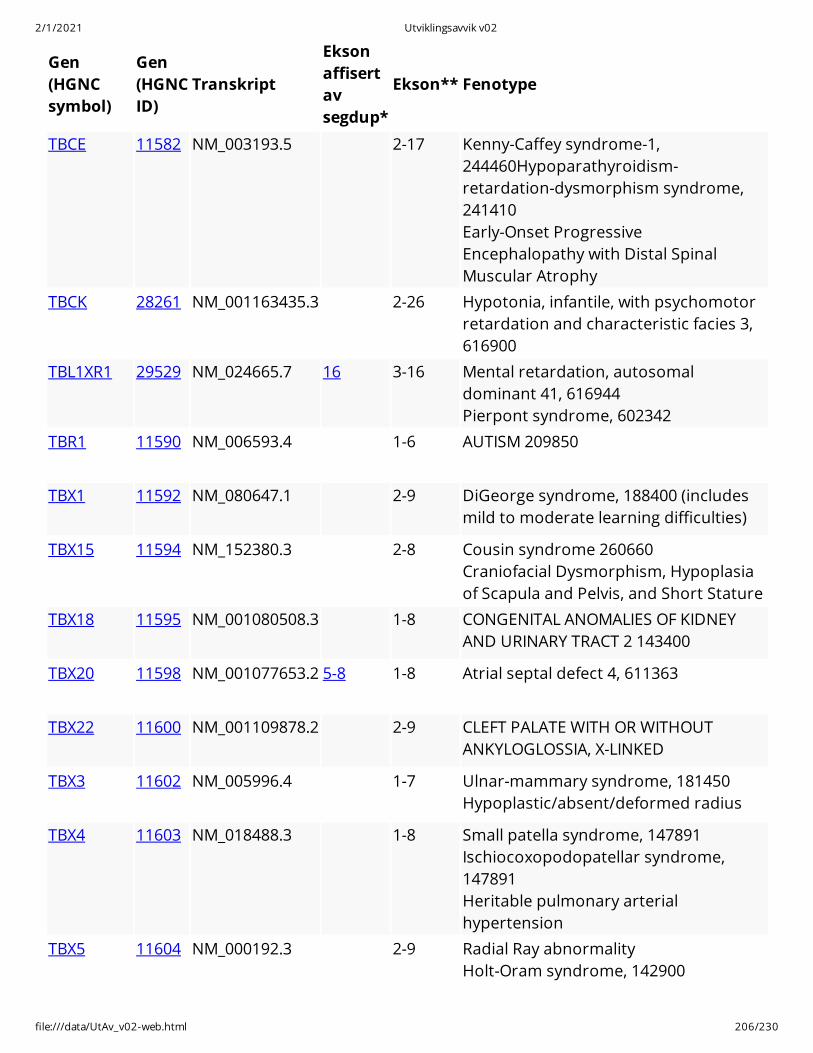
2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 206/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TBCE 11582 NM_003193.5 2-17 Kenny-Caffey syndrome-1,244460Hypoparathyroidism-retardation-dysmorphism syndrome,241410 Early-Onset ProgressiveEncephalopathy with Distal SpinalMuscular Atrophy
TBCK 28261 NM_001163435.3 2-26 Hypotonia, infantile, with psychomotorretardation and characteristic facies 3,616900
TBL1XR1 29529 NM_024665.7 16 3-16 Mental retardation, autosomaldominant 41, 616944 Pierpont syndrome, 602342
TBR1 11590 NM_006593.4 1-6 AUTISM 209850
TBX1 11592 NM_080647.1 2-9 DiGeorge syndrome, 188400 (includesmild to moderate learning difficulties)
TBX15 11594 NM_152380.3 2-8 Cousin syndrome 260660 Craniofacial Dysmorphism, Hypoplasiaof Scapula and Pelvis, and Short Stature
TBX18 11595 NM_001080508.3 1-8 CONGENITAL ANOMALIES OF KIDNEYAND URINARY TRACT 2 143400
TBX20 11598 NM_001077653.2 5-8 1-8 Atrial septal defect 4, 611363
TBX22 11600 NM_001109878.2 2-9 CLEFT PALATE WITH OR WITHOUTANKYLOGLOSSIA, X-LINKED
TBX3 11602 NM_005996.4 1-7 Ulnar-mammary syndrome, 181450 Hypoplastic/absent/deformed radius
TBX4 11603 NM_018488.3 1-8 Small patella syndrome, 147891 Ischiocoxopodopatellar syndrome,147891 Heritable pulmonary arterialhypertension
TBX5 11604 NM_000192.3 2-9 Radial Ray abnormality Holt-Oram syndrome, 142900

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 207/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TBX6 11605 NM_004608.3 2-9 Spondylocostal dysostosis 5 122600
TBXAS1 11609 NM_001061.6 1-13 Ghosal hematodiaphyseal syndrome231095
TCAP 11610 NM_003673.4 1-2 Muscular dystrophy, limb-girdle, type2G, 601954
TCF12 11623 NM_207036.2 2-20 Craniosynostosis 3, 615314
TCF20 11631 NM_005650.3 2-5 Developmental delay with variableintellectual impairment and behavioralabnormalities 618430
TCF4 11634 NM_001083962.2 2-19 Pitt-Hopkins syndrome, 610954
TCIRG1 11647 NM_006019.4 2-20 Osteopetrosis, autosomal recessive 1259700
TCN2 11653 NM_000355.4 1-9 Transcobalamin II deficiency hypotonia, myoclonic like movements,pallor, purpura, anaemia,thrombocytopenia, megaloblastosis,aplastic bone marrow. Intellectual disability SCID
TCOF1 11654 NM_001135243.1 1-26 Treacher Collins syndrome 1 154500
TCTEX1D2 28482 NM_152773.5 1-5 Short-rib thoracic dysplasia 17 with orwithout polydactyly, 617405
TCTN1 26113 NM_001082538.3 1-14 Joubert syndrome
TCTN2 25774 NM_024809.5 1-18 Joubert syndrome, Meckel-Grubersyndrome
TCTN3 24519 NM_015631.6 1-14 Joubert syndrome 18 614815 Meckel-Gruber Mohr-Majewski syndrome Orofaciodigital syndrome IV, 258860

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 208/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TDP2 17768 NM_016614.3 1-7 Spinocerebellar ataxia, autosomalrecessive 23, 616949
TECPR2 19957 NM_014844.5 2-20 Spastic paraplegia 49, autosomalrecessive, 615031
TEK 11724 NM_000459.4 1-23 Venous malformations, multiplecutaneous and mucosal, 600195
TELO2 29099 NM_016111.4 2-21 You-Hoover-Fong syndrome, 616954,syndromic intellectual disability
TERT 11730 NM_198253.3 1-16 Dyskeratosis congenita, autosomaldominant 2 and autosomal recessive 4613989
TFAM 11741 NM_003201.3 7 1-7 ?Mitochondrial DNA depletionsyndrome 15 (hepatocerebral type),617156
TFAP2A 11742 NM_001032280.3 1-7 Branchiooculofacial syndrome, 113620
TFAP2B 11743 NM_003221.4 1-7 Char syndrome, 169100
TFE3 11752 NM_006521.6 1-10 Renal cell carcinoma, papillary, 1 OMIM
TFG 11758 NM_006070.6 2-8 ?Spastic paraplegia 57, autosomalrecessive, 615658, AR Hereditary motor and sensoryneuropathy, Okinawa type, 604484, AD
TFR2 11762 NM_003227.4 1-18 Hereditary haemochromatosis Type 3(Disorder of iron metabolism)
TGDS 20324 NM_014305.4 1-12 CATEL-MANZKE SYNDROME 616145
TGFB1 11766 NM_000660.7 1-7 Camurati-Engelmann disease, 131300
TGFB2 11768 NM_003238.6 1-7 Loeys-Dietz syndrome, type 4, 614816
TGFB3 11769 NM_003239.4 1-7 Loeys-Dietz syndrome 5 615582

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 209/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TGFBR1 11772 NM_004612.4 1-9 Loeys-Dietz syndrome 1 609192
TGFBR2 11773 NM_003242.6 1-7 Loeys-Dietz syndrome 2 610168
TGIF1 11776 NM_173208.2 2-4 Holoprosencephaly 4, 142946
TGM6 16255 NM_198994.3 1-13 Spinocerebellar ataxia 35, 613908
TH 11782 NM_199292.3 1-14 Intellectual disability Early onset dystonia Tyrosine hydroxylase deficiency(Disorders of neurotransmittermetabolism, biogenic amines) Segawa syndrome, recessive, 605407
THAP1 20856 NM_018105.3 1-3 Dystonia 6, torsion, 602629
THOC2 19073 NM_001081550.2 1-38 Mental retardation, X-linked 12/35,300957
THOC6 28369 NM_024339.5 1-13 Beaulieu-Boycott-Innes syndrome,613680 Includes developmental delay andmental retardation
THPO 11795 NM_000460.4 2-6 Thrombocythemia 1, 187950
THRA 11796 NM_199334.4 2-9 HYPOTHYROIDISM, CONGENITAL,NONGOITROUS, 6 614450
THRB 11799 NM_000461.5 3-10 compromised intellectual development
TIA1 11802 NM_022173.4 1-13 Welander distal myopathy, 604454
TIMM22 17317 NM_013337.4 1-4 No OMIM phenotype

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 210/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TIMM50 23656 NM_001001563.5 1-11 3-methylglutaconic aciduria, type IX,617698 intellectual disability and seizure
TIMM8A 11817 NM_004085.4 2 1-2 Mohr-Tranebjaerg syndrome, 304700 Jensen syndrome, 311150 Disorders of mitochondrial proteinimport (Mitochondrial respiratory chaindisorders (caused by nuclear variantsonly))
TIMMDC1 1321 NM_016589.4 1-7 Mitochondrial complex I deficiency,nuclear type 31, 618251
TINF2 11824 NM_001099274.3 1-9 EXUDATIVE RETINOPATHY WITH BONEMARROW FAILURE Dyskeratosis congenita, autosomaldominant 3 613990
TJP2 11828 NM_004817.4 1-23 Cholestasis, progressive familialintrahepatic 4, 615878
TK2 11831 NM_004614.5 1-10 Mitochondrial DNA depletion syndrome2 (myopathic type), 609560 Thymidine kinase 2 deficiency(Disorders of pyrimidine metabolism)
TKT 11834 NM_001135055.2 1-14 Short stature, developmental delay, andcongenital heart defects, 617044
TLK2 11842 NM_006852.4 2-10, 22 2-22 intellectual disability
TMCO1 18188 NM_019026.4 1-7 Craniofacial dysmorphism, skeletalanomalies, and mental retardationsyndrome, 614132
TMEM107 28128 NM_032354.5 1-5 Meckel syndrome 13, 617562 ?Joubert syndrome 29, 617562 Orofaciodigital syndrome XVI, 617563
TMEM126B 30883 NM_018480.5 1-5 Muscle Weakness and Isolated ComplexI Deficiency Isolated complex I deficiency
TMEM138 26944 NM_016464.5 2-5 Joubert syndrome with oculorenaldefect

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 211/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TMEM165 30760 NM_018475.5 1-6 Congenital disorder of glycosylation,type IIk, 614727
TMEM199 18085 NM_152464.3 1-6 Congenital disorder of glycosylation,type IIp 616829 Disorder of Golgi homeostasis
TMEM216 25018 NM_001173990.3 1-5 Neonatal and Adult Cholestasis Joubert syndrome: Meckel-Grubersyndrome
TMEM231 37234 NM_001077416.2 1-6 Meckel syndrome 11, 615397 Joubert syndrome 20, 614970 (includesdevelopmental delay)
TMEM237 14432 NM_001044385.3 1-12 Joubert syndrome 14, 614424
TMEM240 25186 NM_001114748.2 1-4 Spinocerebellar ataxia 21, 607454
TMEM260 20185 NM_017799.4 1-16 Neurodevelopmental, Cardiac, andRenal Syndrome
TMEM38B 25535 NM_018112.3 1-6 Osteogenesis imperfecta, type XIV,615066
TMEM5 13530 NM_014254.3 1-6 SEVERE COBBLESTONE LISSENCEPHALY615041 Muscular dystrophy-dystroglycanopathy (congenital withbrain and eye anomalies), type A, 10615041
TMEM65 25203 NM_194291.2 1-7 TMEM65 related mitochondrialencephalopmyopathy
TMEM67 28396 NM_153704.6 1-28 Meckel syndrome 3, 607361Joubertsyndrome 6, 610688{Bardet-Biedlsyndrome 14, modifier of},209900COACH syndrome,216360Nephronophthisis 11, 613550 nephronophthisis

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 212/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TMEM70 26050 NM_017866.6 1-3 Complex V (Mitochondrial respiratorychain disorders (caused by nuclearvariants only), OXPHOS assemblyfactors)
TMEM8C 33778 NM_001080483.3 1-5 Carey-Fineman-Ziter syndrome, 254940
TMPRSS6 16517 NM_153609.3 1-18 Iron-refractory iron deficiency anemia,206200
TMTC3 26899 NM_181783.4 2-14 Lissencephaly 8, 617255
TMX2 30739 NM_001144012.2 1-7 Intellectual disability Abnormal cortical gyration
TNFRSF11A 11908 NM_003839.4 1-10 Paget disease of bone 2, early-onset602080
TNFRSF11B 11909 NM_002546.4 1-5 Paget disease of bone 5, juvenile-onset239000
TNFRSF13B 18153 NM_012452.3 1-5 IMMUNODEFICIENCY, COMMONVARIABLE, 2 240500
TNFSF11 11926 NM_003701.4 1-5 Osteopetrosis, autosomal recessive 2259710
TNIK 30765 NM_015028.4 1-33 Mental retardation, autosomalrecessive 54, 617028
TNK2 19297 NM_001010938.2 1-15 severe autosomal recessive infantileonset epilepsy EE
TNNI2 11946 NM_003282.4 2-7 Arthrogryposis multiplex congenita,distal, type 2B, 601680
TNNT1 11948 NM_003283.6 2-14 Nemaline Myopathy, Recessive
TNNT3 11950 NM_006757.4 2-16 Arthyrogryposis, distal, type 2B, 601680
TNPO3 17103 NM_012470.3 1-22 Muscular dystrophy, limb-girdle,autosomal dominant 2, 608423

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 213/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TOE1 15954 NM_025077.4 1-8 Pontocerebellar hypoplasia, type 7614969
TOP3A 11992 NM_004618.5 1-19 ?Progressive external ophthalmoplegiawith mitochondrial DNA deletions,autosomal recessive 5, 618098 Bloom Syndrome-like Disorder
TOR1A 3098 NM_000113.3 1-5 Dystonia-1, torsion, 128100 arthrogryposis with developmentaldelay, strabismus and tremor
TOR1AIP1 29456 NM_001267578.1 1-10 Muscular dystrophy, autosomalrecessive, with rigid spine and distaljoint contractures, 617072
TP53RK 16197 NM_033550.4 1-2 GALLOWAY-MOWAT SYNDROME 4,617730
TP63 15979 NM_003722.5 1-14 Rapp-Hodgkin syndrome 129400 Ectrodactyly, ectodermal dysplasia, cleftlip/palate syndrome 3, 604292 Orofacial cleft 8, EEC SYNDROME 3, RappHodgkins syndrome, 129400 Limb-mammary syndrome, 603543
TPK1 17358 NM_022445.4 2-9 Thiamine metabolism dysfunctionsyndrome 5 (episodic encephalopathytype), 614458
TPM2 12011 NM_003289.3 1-9 Arthrogryposis multiplex congenita,distal, type1 108120: Arthrogryposis,distal, type 2B 601680
TPM3 12012 NM_152263.4 1-10 Nemaline myopathy 1, autosomaldominant or recessive, 609284
TPP1 2073 NM_000391.4 1-13 Ceroid lipofuscinosis, neuronal, 2,204500 Hereditary ataxia Spinocerebellar ataxia, autosomalrecessive 7, 609270
TRAF3IP1 17861 NM_015650.4 1-17 Senior-Loken syndrome 9 616629

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 214/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TRAF7 20456 NM_032271.3 2-21 Cardiac, facial, and digital anomalieswith developmental delay, 618164
TRAIP 30764 NM_005879.3 1-15 Seckel syndrome 9, 616777 microcephalic primordial dwarfism
TRAK1 29947 NM_001042646.3 1-16 Epileptic encephalopathy, early infantile,68 618201
TRAP1 16264 NM_016292.3 1-18 VACTERL CAKUT
TRAPPC11 25751 NM_021942.6 2-30 Muscular dystrophy, limb-girdle, type2S, 615356 congenital muscular dystrophy (CMD),progressive fatty liver and infantile-onset cataract
TRAPPC12 24284 NM_016030.6 2-12 Encephalopathy, progressive, early-onset, with brain atrophy and spasticity,617669
TRAPPC2 23068 NM_001011658.4 6 3-6 Spondyloepiphyseal dysplasia tarda,313400
TRAPPC6B 23066 NM_177452.4 1-5 Neurodevelopmental disorder withmicrocephaly, epilepsy, and brainatrophy, 617862
TRAPPC9 30832 NM_031466.7 1-23 Mental retardation, autosomalrecessive 13, 613192
TRDN 12261 NM_006073.4 1-41 Ventricular tachycardia,catecholaminergic polymorphic, 5, withor without muscle weakness, 615441
TREM2 17761 NM_018965.4 1-5 Polycystic lipomembranousosteodysplasia with sclerosingleukoencephalopathy (PLOSL)
TREX1 12269 NM_033629.6 2 Aicardi-Goutieres syndrome 1,dominant and recessive,225750Chilblain lupus,610448Vasculopathy, retinal, withcerebral leukodystrophy,192315{Systemic lupus erythematosus,susceptibility to}, 152700

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 215/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TRIM32 16380 NM_012210.3 2 BARDET-BIEDL SYNDROME TYPE 11209900 Muscular dystrophy, limb-girdle, type2H, 254110
TRIM37 7523 NM_015294.6 1-24 Mulibrey nanism (Other peroxisomaldisorders)
TRIM8 15579 NM_030912.2 1-6 Intellectual disability Early-onset epileptic encephalopathy(EOEE)
TRIO 12303 NM_007118.4 1-57 INTELLECTUAL DISABILITY
TRIP11 12305 NM_004239.4 21 1-21 Achondrogenesis, type IA 200600
TRIP12 12306 NM_004238.3 2-41 Mental retardation, autosomaldominant 49 617752
TRIP13 12307 NM_004237.4 1-13 Mosaic Variegated Aneuploidy andWilms Tumour
TRIP4 12310 NM_016213.5 1-13 Spinal muscular atrophy with congenitalbone fractures 1, 616866
TRIT1 20286 NM_017646.6 1-11 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Combined oxidative phosphorylationdeficiency 35 617873
TRMT1 25980 NM_001136035.3 2-17 Mental retardation, autosomalrecessive 68, 618302
TRMT10A 28403 NM_152292.5 2-8 Microcephaly, short stature, andimpaired glucose metabolism 1, 616033
TRMT10C 26022 NM_017819.4 2 Combined oxidative phosphorylationdeficiency 30, 616974
TRMT5 23141 NM_020810.3 1-5 Combined oxidative phosphorylationdeficiency 26 616539

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 216/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TRMU 25481 NM_018006.5 1-11 {Deafness, mitochondrial, modifier of},580000 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Liver failure, transient infantile, 613070
TRNT1 17341 NM_182916.3 2-8 congenital sideroblastic anemia with Bcell immunodeficiency, fevers, anddevelopmental delay (SIFD) retinitis pigmentosa with erythrocyticmicrocytosis
TRPM1 7146 NM_002420.6 2-27 Night blindness, congenital stationary(complete), 1C, autosomal
TRPM3 17992 NM_020952.6 3-25 Intellectual disability Seizures
TRPM6 17995 NM_017662.5 1-39 Hypomagnesaemia type 1, intestinal(Disorder of magnesium metabolism)
TRPS1 12340 NM_014112.5 2-7 Trichorhinophalangeal syndrome, typeI, 190350 Trichorhinophalangeal syndrome, typeIII, 190351
TRPV3 18084 NM_145068.4 2-18 OLMSTED SYNDROME 614594
TRPV4 18083 NM_021625.5 2-16 Brachyolmia type 3, 113500 Spondylometaphyseal dysplasia,Kozlowski type, 184252 Hereditary motor and sensoryneuropathy, type IIc, 606071 Parastremmatic dwarfism, 168400 SED, Maroteaux type, 184095 Spinal muscular atrophy, distal,congenital nonprogressive, 600175 Digital arthropathy-brachydactyly,familial, 606835
TRPV6 14006 NM_018646.6 1-15 Hyperparathyroidism, transientneonatal, 618188

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 217/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TRRAP 12347 NM_003496.3 2-71 Developmental delay with or withoutdysmorphic facies and autism, 603015
TSC1 12362 NM_000368.5 3-23 Tuberous sclerosis-1,191100Lymphangioleiomyomatosis,606690
TSC2 12363 NM_000548.5 2-42 Tuberous sclerosis-2,613254Lymphangioleiomyomatosis,somatic, 606690
TSEN15 16791 NM_052965.4 1-5 Pontocerebellar hypoplasia, type 2F,617026
TSEN2 28422 NM_025265.4 2-12 Pontocerebellar hypoplasia type 2B,612389
TSEN34 15506 NM_024075.5 2-5 Pontocerebellar Hypoplasia
TSEN54 27561 NM_207346.3 1-11 Pontocerebellar Hypoplasia
TSFM 12367 NM_001172696.2 1-7 Combined oxidative phosphorylationdeficiency 3, 610505
TSHB 12372 NM_000549.5 2-3 HYPOTHRYOIDISM, CONGENITAL,NONGOITROUS 4 275100
TSHR 12373 NM_000369.3 1-10 Thyroid carcinoma with thyrotoxicosis Hyperthyroidism, familial
TSPAN7 11854 NM_004615.3 1-7 Mental retardation, X-linked 58, 300210
TTBK2 19141 NM_173500.4 2-15 Spinocerebellar ataxia 11, 604432
TTC19 26006 NM_017775.4 1-10 Mitochondrial Respiratory ChainComplex III Deficiency
TTC21B 25660 NM_024753.5 1-29 Nephronophthisis 12, 613820 Short-rib thoracic dysplasia 4 with orwithout polydactyly, 613819
TTC25 25280 NM_031421.5 1-11 Primary Ciliary Dyskinesia with Left-Right Body Asymmetry Randomization

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 218/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TTC37 23639 NM_014639.3 4-43 Trichohepatoenteric syndrome 1,222470
TTC7A 19750 NM_020458.4 1-20 INTESTINAL ATRESIA, MULTIPLE 243150
TTC8 20087 NM_198309.3 2-15 Bardet-Biedl syndrome 8,209900Retinitis pigmentosa 51, 613464
TTI2 26262 NM_001102401.4 2-8 Mental retardation, autosomalrecessive 39, 615541
TTN 12403 NM_133378.4 154-155 2-312 Tibial muscular dystrophy, tardive,600334 Muscular dystrophy, limb-girdle, type 2J,608807 Myopathy, early-onset, with fatalcardiomyopathy, 611705
TTPA 12404 NM_000370.3 1-5 Ataxia with Vitamin E Deficiency
TUBA1A 20766 NM_006009.4 2-4 1-4 Lissencephaly 3, 611603
TUBA8 12410 NM_018943.3 1-5 Polymicrogyria with optic nervehypoplasia, 613180
TUBB 20778 NM_178014.4 2-4 1-4 Circumferential Skin Creases KunzeType 156610 Cortical dysplasia, complex, with otherbrain malformations 6, 615771
TUBB2A 12412 NM_001069.3 2-4 1-4 Cortical dysplasia, complex, with otherbrain malformations 5, 615763 infantile-onset epilepsy
TUBB2B 30829 NM_178012.5 4 1-4 Polymicrogyria, symmetric orasymmetric, 610031 Cortical dysplasia, complex, with otherbrain malformations 7, 610031
TUBB3 20772 NM_001197181.2 4 3-4 Cortical dysplasia, complex, with otherbrain malformations 1, 614039

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 219/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TUBB4A 20774 NM_006087.4 4 1-4 Leukodystrophy, hypomyelinating, 6,612438 Dystonia 4, torsion, autosomaldominant, 128101
TUBG1 12417 NM_001070.5 7-10 1-11 Cortical dysplasia, complex, with otherbrain malformations 4, 615412
TUBGCP4 16691 NM_014444.5 1-18 Microcephaly and chorioretinopathy,autosomal recessive, 3, 616335
TUBGCP6 18127 NM_020461.4 1-25 Microcephaly and chorioretinopathywith or without mental retardation,251270
TUFM 12420 NM_003321.5 1-10 Combined oxidative phosphorylationdeficiency 4, 610678
TUSC3 30242 NM_006765.4 1-10 Mental retardation, autosomalrecessive 7, 611093 TUSC3-CDG (Disorders of protein N-glycosylation)
TWIST1 12428 NM_000474.4 1 Craniosynostosis 1 123100 Saethre-Chotzen syndrome with orwithout eyelid anomalies 101400
TWIST2 20670 NM_057179.3 1 ABLEPHARON MACROSTOMIASYNDROME 200110 Barber-Say syndrome, 209885 (includesmental retardation in some patients) Focal facial dermal dysplasia 3, Setleistype, 227260 (can include intellectualdisability and developmental delay)
TXNDC15 20652 NM_024715.3 1-5 Meckel-Gruber syndrome
TXNL4A 30551 NM_006701.5 1-3 Cleft palate Burn-McKeown syndrome, 608572
TXNRD1 12437 NM_182743.3 2-14 genetic generalized epilepsy

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 220/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
TYMP 3148 NM_001953.5 2-10 Thymidine phosphorylase deficiency(Disorders of pyrimidine metabolism) Mitochondrial NeurogastrointestinalEncephalopathy Disease Mitochondrial DNA depletion syndrome1 (MNGIE type), 603041
TYR 12442 NM_000372.5 4-5 1-5 Albinism, oculocutaneous, type IA,203100 Albinism, oculocutaneous, type IB,606952
TYROBP 12449 NM_003332.4 1-5 Nasu-Hakola disease 221770
TYRP1 12450 NM_000550.3 2-8 Albinism, oculocutaneous, type III,203290
UBA1 12469 NM_003334.4 2-26 Spinal muscular atrophy, X-linked 2,infantile, 301830
UBA5 23230 NM_024818.4 9-12 1-12 Epileptic encephalopathy, early infantile,44, 617132
UBAP1 12461 NM_001171201.1 1-6 Hereditary spastic paraplegia
UBE2A 12472 NM_003336.4 1-6 Mental retardation, X-linked syndromic,Nascimento-type, 300860
UBE2T 25009 NM_014176.4 2-7 Fanconi Anemia, ComplementationGroup T, 616435
UBE3A 12496 NM_130838.4 4 2-11 Angelman syndrome, 105830
UBE3B 13478 NM_130466.4 3-28 BLEPHAROPHIMOSIS-MENTALRETARDATION 615057 Kaufman oculocerebrofacial syndrome244450
UBR1 16808 NM_174916.3 1-47 Johanson-Blizzard syndrome, 243800
UBTF 12511 NM_014233.4 2-21 Neurodegeneration, childhood-onset,with brain atrophy, 617672

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 221/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
UCHL1 12513 NM_004181.5 1-9 Spastic paraplegia 79, autosomalrecessive, 615491, AR
UFC1 26941 NM_016406.4 1-6 Severe early-onset encephalopathy withprogressive microcephaly
UFM1 20597 NM_001286704.2 6 2-6 Severe early-onset encephalopathy withprogressive microcephaly, Leukodystrophy hypomyelinating 14,617899
UGT1A1 12530 NM_000463.3 1-5 Neonatal and Adult Cholestasis Crigler-Najjar syndrome, type I 218800 Bilirubin UDP-glucuronosyltransferase 1deficiency (Disorders of bile acidmetabolism and transport)
UMOD 12559 NM_003361.3 2-11 Cystic kidney disease Familial juvenile hyperuricaemicnephropathy (Disorders of purinemetabolism)
UMPS 12563 NM_000373.4 1-6 Intellectual disability Orotic aciduria (Disorders of pyrimidinemetabolism)
UNC80 26582 NM_032504.1 1-64 Hypotonia, infantile, with psychomotorretardation and characteristic facies 2616801
UPB1 16297 NM_016327.3 1-10 Beta-ureidopropionase deficiency(Disorders of pyrimidine metabolism) Beta-ureidopropionase deficiency,613161 (can include mental retardation,developmental delay)
UPF3B 20439 NM_080632.2 1-11 Mental retardation, X-linked, syndromic14, 300676
UQCC1 15891 NM_018244.5 1-10 No OMIM phenotype
UQCC2 21237 NM_032340.4 1-4 Mitochondrial complex III deficiency,nuclear type 7, 615824
UQCR10 30863 NM_001003684.1 1 No OMIM phenotype

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 222/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
UQCR11 30862 NM_006830.4 1-2 No OMIM phenotype
UQCRB 12582 NM_006294.5 3-4 1-4 Mitochondrial complex III deficiency,nuclear type 3, 615158
UQCRC2 12586 NM_003366.4 1-14 Mitochondrial complex III deficiency,nuclear type 5, 615160
UQCRQ 29594 NM_014402.5 2-3 Mitochondrial complex III deficiency,nuclear type 4, 615159
UROC1 26444 NM_144639.3 1-20 Intellectual disability Urocanase deficiency (Disorders ofhistidine, tryptophan or lysinemetabolism)
UROD 12591 NM_000374.5 1-10 Porphyria cutanea tarda (Porphyriaswith erosive photodermatosis)
UROS 12592 NM_000375.3 2-10 Congenital erythropoietic porphyria(Porphyrias with erosivephotodermatosis)
USB1 25792 NM_024598.4 1-7 Poikiloderma with neutropenia
USP18 12616 NM_017414.4 3-11 2-11 Severe pseudo-TORCH syndrome
USP27X 13486 NM_001145073.3 1 Mental retardation 105, 300984
USP7 12630 NM_001286457.2 1-31 USP7-related developmental disorder(monoallelic) Intellectual disability, autism, epilepsy,aggressive behaviour, hypotonia, andhypogonadism
USP9X 12632 NM_001039590.3 2-45 Mental retardation, X-linked 99 300919XLR
UVSSA 29304 NM_020894.4 2-14 UV-sensitive syndrome 3, 614640
VAC14 25507 NM_018052.5 1-19 Striatonigral degeneration, childhood-onset 617054

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 223/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
VAMP1 12642 NM_014231.5 1-5 Spastic ataxia 1, autosomal dominant,108600 Congenital myasthenic syndrome
VAMP2 12643 NM_014232.3 1-5 Generalized hypotonia, Globaldevelopmental delay, Intellectualdisability, Autistic behavior, Stereotypicbehavior, Seizures, Abnormality ofmovement, Cortical visual impairment
VAPB 12649 NM_004738.5 1-6 Spinal muscular atrophy, late-onset,Finkel type 182980 Amyotrophic lateral sclerosis 8 608627
VARS 12651 NM_006295.3 2-30 Neurodevelopmental disorder withmicrocephaly, seizures, and corticalatrophy, 617802
VARS2 21642 NM_001167734.1 1-30 Combined oxidative phosphorylationdeficiency 20, 615917
VCP 12666 NM_007126.5 1-17 Inclusion body myopathy with early-onset Paget disease andfrontotemporal dementia 1, 167320
VDR 12679 NM_001017535.1 4-11 Rickets, vitamin D-resistant, type IIA,277440
VIPAS39 20347 NM_022067.4 3-21 Arthrogryposis, renal dysfunction, andcholestasis 2, 613404 Inherited bleeding disorders
VKORC1 23663 NM_024006.6 1-3 Vitamin K epoxide reductase deficiency(Other disorders of vitamins andcofactors)
VLDLR 12698 NM_003383.5 1-19 Cerebellar hypoplasia and mentalretardation with or withoutquadrupedal locomotion 1, 224050
VMA21 22082 NM_001017980.3 1-3 Myopathy, X-linked, with excessiveautophagy, 310440
VPS11 14583 NM_021729.5 1-17 Leukodystrophy, hypomyelinating, 12,616683
VPS13A 1908 NM_033305.3 1-72 Choreoacanthocytosis 200150

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 224/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
VPS13B 2183 NM_017890.4 2-62 Cohen syndrome, 216550
VPS13D 23595 NM_015378.4 2-70 Spinocerebellar ataxia, autosomalrecessive 4, 607317
VPS33B 12712 NM_018668.4 1-23 Arthrogryposis, renal dysfunction, andcholestasis 1, 208085 CAKUT
VPS37A 24928 NM_152415.3 1-11 Spastic paraplegia 53, 614898, AR
VPS53 25608 NM_001128159.3 1-22 Pontocerebellar hypoplasia, type 2E,615851
VRK1 12718 NM_003384.3 2-13 Pontocerebellar hypoplasia type 1A,607596 Pontocerebellar Hypoplasia withinfantile SMA
VSX2 1975 NM_182894.3 1-5 Microphthalmia with coloboma 3,610092
WAC 17327 NM_016628.5 1-14 INTELLECTUAL DISABILITY WAC syndrome
WARS2 12730 NM_015836.3 1-6 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Neurodevelopmental disorder,mitochondrial, with abnormalmovements and lactic acidosis, with orwithout seizures, 617710
WASF1 12732 NM_003931.3 4-11 ID associated with autistic features,seizures, and developmental delay
WDFY3 20751 NM_014991.5 4-68 Microcephaly 18, primary, autosomaldominant, 617520
WDPCP 28027 NM_015910.7 1-18 ?Bardet-Biedl syndrome 15, 615992 Meckel syndrome ?Congenital heart defects, hamartomasof tongue, and polysyndactyly, 217085

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 225/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
WDR11 13831 NM_018117.12 1-29 KALLMANN SYNDROME
WDR19 18340 NM_025132.4 1-36 Cranioectodermal dysplasia 4, 614378 Short-rib thoracic dysplasia 5 with orwithout polydactyly, 614376 Nephronophthisis 13, 614377 Senior-Loken syndrome 8, 616307 Senior-Loken syndrome
WDR26 21208 NM_025160.6 1-14 Skraban-Deardorff syndrome Intellectual Disability, Seizures,Abnormal Gait, and Distinctive FacialFeatures
WDR34 28296 NM_052844.3 1-9 SEVERE ASPHYXIATING THORACICDYSPLASIA Short-rib thoracic dysplasia 11 with orwithout polydactyly, 615633 Jeune syndrome
WDR35 29250 NM_001006657.2 1-28 Cranioectodermal dysplasia 2, 613610 Short-rib thoracic dysplasia 7 with orwithout polydactyly, 614091
WDR37 31406 NM_014023.4 2-14 Global developmental delay Seizures Abnormality of nervous systemmorphology Abnormality of the cardiovascularsystem Abnormality of the skeletal system Abnormality of the genitourinarysystem
WDR4 12756 NM_033661.4 1-11 Primordial dwarfism global developmental delay.
WDR45 28912 NM_007075.3 3-12 Neurodegeneration with brain ironaccululation 5, 300894
WDR45B 25072 NM_019613.4 9-10 1-10 Neurodevelopmental disorder withspastic quadriplegia and brainabnormalities with or without seizures,617977

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 226/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
WDR48 30914 NM_020839.4 1-19 spastic paraplegia
WDR60 21862 NM_018051.5 1-25 Short-rib thoracic dysplasia 8 with orwithout polydactyly, 615503 Jeune syndrome
WDR62 24502 NM_001083961.2 1-32 Microcephaly 2, primary, autosomalrecessive, with or without corticalmalformations, 604317
WDR73 25928 NM_032856.4 1-8 Galloway Mowat syndrome, whenpatients are ambulant ataxia is arecognised feature
WDR81 26600 NM_001163809.1 1-10 Cerebellar ataxia, mental retardation,and dysequilibrium syndrome 2, 610185
WFS1 12762 NM_006005.3 2-8 Diabetes with additional phenotypessuggestive of a monogenic aetiology Inherited optic neuropathies Mitochondrial respiratory chaindisorders caused by nuclear variantsonly Hereditary ataxia Congenital hearing impairment(profound/severe) Wolfram syndrome 1, 222300
WHSC1 12766 NM_001042424.3 2-22 Intrauterine growth retardation Microcephaly Muscular hypotonia Neurodevelopmental delay
WISP3 12771 NM_198239.2 1-5 Spondyloepiphyseal dysplasia tardawith progressive arthropathy 208230
WNT1 12774 NM_005430.4 1-4 Osteogenesis imperfecta, type XV,615220
WNT10B 12775 NM_003394.4 2-5 Split-hand/foot malformation 6, 225300
WNT3 12782 NM_030753.5 1-4 Tetra-amelia, autosomal recessive,273395

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 227/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
WNT4 12783 NM_030761.5 1-5 MULLERIAN APLASIA ANDHYPERANDROGENISM 158330
WNT5A 12784 NM_003392.4 1-5 Robinow syndrome, autosomaldominant 1 180700
WNT7A 12786 NM_004625.4 1-4 Fuhrmann syndrome, 228930 Ulna and fibula, absence of, with severelimb deficiency, 276820
WRAP53 25522 NM_018081.2 1-10 Dyskeratosis congenita, autosomalrecessive 3, 613988
WT1 12796 NM_024426.6 1-10 DENYS-DRASH SYNDROME 194080 FRASIER SYNDROME FRASIERSYNDROME FRASIER SYNDROME 136680
WWOX 12799 NM_016373.4 1-9 Spinocerebellar ataxia, autosomalrecessive 12, 614322 Epileptic encephalopathy, early infantile,28, 616211
XDH 12805 NM_000379.4 1-36 Xanthinuria type I (Disorders of purinemetabolism)
XPA 12814 NM_000380.3 1-6 progressive intellectual impariment Xeroderma pigmentosum, group A,278700
XPC 12816 NM_004628.4 1-16 Xeroderma pigmentosum, group C,278720
XPNPEP3 28052 NM_022098.4 1-10 nephronophthisis-like nephropathy
XPR1 12827 NM_004736.4 1-15 Basal ganglia calcification (Fahrsyndrome)
XRCC4 12831 NM_022406.4 2-8 Short stature, microcephaly, andendocrine dysfunction, 616541
XYLT1 15516 NM_022166.4 1-12 Desbuquois dysplasia 2, 615777
XYLT2 15517 NM_022167.4 1-11 Spondyloocular syndrome 605822

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 228/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
YAP1 16262 NM_001130145.3 1-9 COLOBOMA, OCULAR, WITH ORWITHOUT HEARING IMPAIRMENT, CLEFTLIP/PALATE, AND/OR MENTALRETARDATION 120433
YARS2 24249 NM_001040436.3 1-5 Multiple respiratory chain complexdeficiencies (disorders of proteinsynthesis) Myopathy, lactic acidosis, andsideroblastic anemia 2, 613561
YME1L1 12843 NM_014263.4 6-8 1-19 ?Optic atrophy 11, 617302
YWHAG 12852 NM_012479.4 1-2 Epileptic encephalopathy, early infantile56, 617665
YY1 12856 NM_003403.5 1-5 Gabriele-de Vries syndrome 617557 INTELLECTUAL DISABILITY
ZBTB11 16740 NM_014415.4 1-11 Intellectual developmental disorder,autosomal recessive 69, 618383
ZBTB18 13030 NM_205768.2 1-2 Mental retardation, autosomaldominant 22, 612337
ZBTB20 13503 NM_001164342.2 2-5 PRIMROSE SYNDROME 259050
ZBTB24 21143 NM_014797.2 2-7 Immunodeficiency-centromericinstability-facial anomalies syndrome 2,614069
ZC3H14 20509 NM_024824.5 1-17 Mental retardation, autosomalrecessive 56, 617125
ZC4H2 24931 NM_018684.4 1-5 ARTHROGRYPOSIS MULTIPLEXCONGENITA AND INTELLECTUALDISABILITY Wieacker-Wolff syndrome 314580
ZDHHC9 18475 NM_016032.4 3-11 Mental retardation, X-linked syndromic,Raymond type, 300799
ZEB2 14881 NM_014795.4 10 2-10 Mowat-Wilson syndrome, 235730

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 229/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ZFP57 18791 NM_001109809.4 2-5 Diabetes mellitus, transient neonatal, 1,601410
ZFYVE26 20761 NM_015346.4 2-42 Spastic paraplegia 15, autosomalrecessive, 270700
ZIC1 12872 NM_003412.4 1-3 CRANIOSYNOSTOSIS 6
ZIC2 12873 NM_007129.5 1-3 Holoprosencephaly 5, 609637
ZIC3 12874 NM_003413.4 1-3 VACTERL ASSOCIATION, X-LINKED, WITHOR WITHOUT HYDROCEPHALUS 319683 Heterotaxy, visceral, 1, X-linked 306955 Congenital heart defects,nonsyndromic, 1, X-linked, 306955
ZMIZ1 16493 NM_020338.4 5-25 Global developmental delay, Intellectualdisability, Feeding difficulties, Growthabnormality, Microcephaly, Abnormalityof the skeletal system, Abnormality ofthe urinary system, Abnormality of thecardiovascular system, Abnormality ofhead or neck
ZMPSTE24 12877 NM_005857.5 1-10 Mandibuloacral dysplasia with type Blipodystrophy, 608612 Restrictive dermopathy, lethal, 275210
ZMYND10 19412 NM_015896.4 1-12 PRIMARY CILIARY DYSKINESIA-22 615444
ZMYND11 16966 NM_006624.5 2-15 INTELLECTUAL DISABILITY
ZNF142 12927 NM_001105537.4 4-10 Neurodevelopmental disorder withimpaired speech and hyperkineticmovements, 618425
ZNF148 12933 NM_021964.2 4-9 Global developmental delay, absent orhypoplastic corpus callosum, anddysmorphic facies, 617260

2/1/2021 Utviklingsavvik v02
file:///data/UtAv_v02-web.html 230/230
Gen(HGNCsymbol)
Gen(HGNCID)
Transkript
Eksonaffisertavsegdup*
Ekson** Fenotype
ZNF292 18410 NM_015021.3 1-8 Intellectual disability Abnormality of the skeletal system Abnormality of the cardiovascularsystem Microcephaly Seizures
ZNF335 15807 NM_022095.4 2-28 Autosomal recessive primarymicrocephaly (MCPH)
ZNF423 16762 NM_015069.4 1-8 Nephronophthisis 14 Joubert syndrome with oculorenaldefect
ZNF462 21684 NM_021224.6 2-13 Ptosis, Prominent metopic ridge,Craniosynostosis, Global developmentaldelay, Intellectual disability, Autisticbehavior
ZNF711 13128 NM_021998.5 3-9 Mental retardation, X-linked 97, 300803
ZNF750 25843 NM_024702.3 2-3 SEBORRHEA-LIKE DERMATITIS WITHPSORIASIFORM ELEMENTS 610227
ZSWIM6 29316 NM_020928.2 1-14 Acromelic frontonasal dysostosis,603671 Neurodevelopmental disorder withmovement abnormalities, abnormalgait, and autistic features, 617865





























