Embed Size (px)
Citation preview

UNIVERSIDADE FEDERAL DO PARÁ
INSTITUTO DE CIÊNCIAS BIOLÓGICAS
PROGRAMA DE PÓS – GRADUAÇÃO EM
BIOLOGIA DE AGENTES INFECCIOSOS E PARASITÁRIOS
ALTERAÇÕES NO PERFIL PROTEICO DE CÉLULAS DE CARCINOMA
HEPATOCELULAR HUMANO HEPG2 EXPERIMENTALMENTE INFECTADAS
COM O VÍRUS DA FEBRE AMARELA, CEPA H111 (FLAVIVIRIDAE: FLAVIVIRUS)
EDUARDO LANA
BELÉM – PARÁ
2014

EDUARDO LANA
ALTERAÇÕES NO PERFIL PROTEICO DE CÉLULAS DE CARCINOMA
HEPATOCELULAR HUMANO HEPG2 EXPERIMENTALMENTE INFECTADAS
COM O VÍRUS DA FEBRE AMARELA, CEPA H111 (FLAVIVIRIDAE: FLAVIVIRUS)
Dissertação apresentada ao Programa de Pós-
Graduação em Biologia de Agentes Infecciosos e
Parasitários do Instituto de Ciências Biológicas da
Universidade Federal do Pará como requisito para a
obtenção do grau de Mestre em Biologia de Agentes
Infecciosos e Parasitários.
Orientador: Prof. Dr. Márcio Roberto Teixeira
Nunes.
BELÉM – PARÁ
2014

EDUARDO LANA
ALTERAÇÕES NO PERFIL PROTEICO DE CÉLULAS DE CARCINOMA
HEPATOCELULAR HUMANO HEPG2 EXPERIMENTALMENTE INFECTADOS
COM VÍRUS DA FEBRE AMARELA, CEPA H111 (FLAVIVIRIDAE: FLAVIVIRUS)
Dissertação apresentada ao Programa de Pós-Graduação em Biologia de Agentes Infecciosos
e Parasitários do Instituto de Ciências Biológicas da Universidade Federal do Pará como
requisito parcial para a obtenção do grau de Mestre em Biologia de Agentes Infecciosos e
Parasitários.
Orientador: Prof. Dr. Márcio Roberto Teixeira Nunes
Seção de Arbovirologia e Febres Hemorrágicas IEC/SVS/MS
Banca examinadora: Prof. Dr. Evonnildo Costa Gonçalves
Instituto de Ciências Biólogicas, UFPA
Dra. Hivana Patrícia Melo Barbosa Dall’Agnol
Instituto Tecnológico Vale – Desenvolvimento Sustentável
Dra. Valéria Lima Carvalho
Seção de Arbovirologia e Febres Hemorrágicas IEC/SVS/MS
Prof. Dr. Pedro Fernando da Costa Vasconcelos
Seção de Arbovirologia e Febres Hemorrágicas IEC/SVS/MS
Belém, 18 de julho de 2014

Dados Internacionais de Catalogação-na-Publicação (CIP)
___________________________________________________________________________
Lana, Eduardo, 1989-
Alterações no perfil proteico de células de carcinoma hepatocelular humano
HepG2 experimentalmente infectadas com o vírus da febre amarela, cepa H111
(flaviviridae:flavivirus) / Eduardo Lana. - 2014.
Orientador: Márcio Roberto Teixeira Nunes.
Dissertação (Mestrado) - Universidade Federal do Pará, Instituto de Ciências
Biológicas, Programa de Pós-Graduação em Biologia de Agentes Infecciosos e
Parasitários, Belém, 2014.
1. Febre amarela. 2. Proteômica. I. Título.
CDD 22. ed. 616.91854
___________________________________________________________________________

EPÍGRAFE
“Somos uma pequena e trêmula
chama que a cada instante ameaça
apagar-se.”
José Saramago
“Deve haver alguma espécie de
sentido, ou o que virá depois?”
Caio Fernando Abreu

DEDICATÓRIA
Dedico este trabalho ao meu avô, Jorge
Lana, e ao meu opa, Werner Radünz, em
respeito a última despedida que nunca
aconteceu.

AGRADECIMENTOS
À Deus, pelo amparo, força e por ser tão bondoso comigo.
À CAPES pelo financiamento da bolsa de estudos mensal a nível de mestrado.
À todo o corpo docente e a turma de mestrado do biênio de 2012-2014 do Programa de
Pós-Graduação em Biologia de Agentes Infecciosos e Parasitários, pelos aprendizados e
discussões enriquecedoras trazidas as salas de aula e por me recepcionarem tão bem.
Aos membros da secretaria da Pós-Graduação BAIP, Raimundo Hosana e Mirna
Tavares, pela presteza e agilidade para resolver problemas e auxiliar nas questões
burocráticas.
À minha família, que no início da possibilidade de sair de casa para ir tão longe foi um
pouco relutante, mas que logo entenderam as minhas causas, sonhos e propósitos me dando
apoio e ajudando incondicionalmente. Eu amo vocês!
À Família Rachid, Eleonora (uma guerreira incrível!), Luiz (um pai exemplar), Thiago
(um irmão atencioso) e Thaís (a irmã mais nova que não tive), por terem me recepcionado de
maneira tão amável e cuidadosa, me ensinando tudo sobre Belém e a gastronomia local.
Ao meu orientador, Dr. Márcio Roberto Teixeira Nunes, por ter aberto um mundo de
possibilidades ao me aceitar como seu aluno. Não sei quem foi mais corajoso de nós dois, mas
obrigado pela confiança depositada e pela força estimulada. Cresci e aprendi muito como
você disse que esperaria de mim em nossa primeira conversa.
Às meninas do Mestrado, que sempre foram grandes mulheres que estiveram ao meu
lado sendo mães, irmãs, tias, avós, professoras e AMIGAS INCRÍVEIS. Que Deus lhes
abençoe como tem me abençoado pela oportunidade em conhecer todas vocês, Andrea, Edina,
Jannyce e Larissa.
Ao Professor Dr. Evonnildo Costa Gonçalves, por ter extrapolado as barreirras
profissionais e se tornado um grande amigo. Obrigado pelos ensinamentos, apoio, audição e
as broncas também, te devo várias.
Ao Jefferson Pereira e Silva, por estes quase dois anos de convivência que me
amadureceram muito pessoalmente e profissionalmente.
À toda a equipe da Seção de Arbovirologia e Febres Hemorrágicas do Instituto
Evandro Chagas, em especial: ao Laboratório de isolamento viral em cultura de células, meu
berço na seção (Creuza Lima, Drª Valéria Lima, Ercília (pela manipulação inicial do H111),
Jamilla (minha irmã que me entende tanto), Maissa (por me emprestar o cupcake amado dela
nos finais de semana), ao Maxwell e a agora Dra Eliana Pinto, por ter me iniciado e ensinado

todas as atividades e rotina do laboratório), ao Laboratório de biologia molecular (Bruno
Tardelli e Alice Queiroz, por todos os empréstimos temporários, Samir Casseb, pelo
tratamento das culturas de células e Natália Vale, por me ensinar a usar o Qubit, não teria sido
tão divertido se não fosse com você), Esterilização (Mari, pela presteza e por estar sempre
alegre e contagiante) e Secretaria (principalmente a Rose, por me ajudar com os problemas
burocráticos e entender a situação final do período das experimentações).
Ao Núcleo de proteômica do Centro de Inovações Tecnológicas – Novamente ao
professor Dr. Evo pelas orientações no laboratório. A Laise por compartilhar experiência e
macetes e ao Sanclayver (San san) por se arriscar junto comigo neste mundo eletrizante do
2D-SDS-PAGE.
A toda a galera da Genômica e Bioinformática do Centro de Inovações Tecnológicas,
pelas experiências, besteiróis e risadas compartilhados na bancada, copa, e sala da Microlins:
Clayton (volta pra terra!), Jedson (não se assustem ele já nasceu assim feio), Poliana (Miss
Haemagogus), Patroca (o guardião oficial do túnel do tempo), Jana (linda linda *-*), Lay
(LayEnne boladona), Daisy (amor em forma de gente) e Rodri (sei que me amas).
À Professora Dra Edina (Seção de Arbovirologia - IEC), por ter acolhido minhas
dúvidas com o teste de viabilidade celular e ajuda com as escalas microscópicas.
À Professora Dra Sílvia Marques (Seção de Bacteriologia/Micologia - IEC), por ter
me acolhido em seu laboratório como a um filho em um momento de desespero e ter me
mostrado com maestria, paciência e carinho os caminhos azuis e tortuosos de Bradford.
Ao Antônio Gregório Dias Júnior, pelos ensinamentos e questionamentos científicos
levantados ao meu projeto. Muito obrigado e muito sucesso na sua caminhada.
Ao Humberto Jácome, pelo apoio, incentivo e por nunca fugir das nossas discussões
científicas valiosas e engraçadas, mesclando nossos conhecimentos com diversão, sempre
instigando ao fortalecimento do pensamento científico e arguidor.
Aos “ICs” Thiago e Mayque, pela oportunidade de ensinar o pouco que aprendi,
ensinar é sempre um caminho de mão dupla.
Aos amigos que ficaram em outros estados, pelo apoio, por ouvirem meus desabafos,
medos e angústias e também pelas visitas.
Aos amigos que por acaso fui conhecendo em Belém e conquistando. Raimundo
Junior, Vinícius Matos e Natacha Malu, obrigado pelo apoio e momentos de descontração.
MUITO OBRIGADO!

SUMÁRIO
1 INTRODUÇÃO ................................................................................................................ 15
1.1 CONSIDERAÇÕES GERAIS SOBRE A FEBRE AMARELA ...................................... 15
1.2 ETIOLOGIA DA FEBRE AMARELA .......................................................................... 17
1.3 EPIDEMIOLOGIA DA FEBRE AMARELA ................................................................. 19
1.4 MANIFESTAÇÕES CLÍNICAS DA FEBRE AMARELA ............................................. 21
1.5 TRATAMENTO, PREVENÇÃO E DIAGNÓSTICO DA FEBRE AMARELA ............. 21
1.6 PROTEÔMICA VIRAL ................................................................................................. 22
2 OBJETIVOS ..................................................................................................................... 25
2.1 OBJETIVO GERAL ...................................................................................................... 25
2.2 OBJETIVOS ESPECÍFICOS ......................................................................................... 25
3 MATERIAL E MÉTODOS ............................................................................................... 26
3.1 ASPECTOS ÉTICOS E DE BIOSSEGURANÇA .......................................................... 26
3.2 CARCINOMA HEPATOCELULAR HUMANO, HEPG2 ............................................ 27
3.2.1 Descongelamento das Células HepG2 ......................................................................... 27
3.2.2 Subcultura das Células HepG2 .................................................................................... 28
3.3 CEPA VIRAL ................................................................................................................ 29
3.4 ESTOQUE VIRAL ........................................................................................................ 29
3.4.1 Estoque Viral em Cultivo de Células de Mosquito Aedes albopictus Clone C6/36 ....... 29
3.5 CONTAGEM DE CÉLULAS E VIABILIDADE CELULAR ........................................ 30
3.6 MULTIPLICIDADE DE INFECÇÃO ............................................................................ 31
3.7 IMUNOFLUORESCÊNCIA INDIRETA ....................................................................... 32
3.8 TITULAÇÃO VIRAL .................................................................................................... 32
3.8.1 Titulação Viral em Células VERO ............................................................................... 33
3.9 INFECÇÃO DAS CÉLULAS HEPG2 PELO VFA ........................................................ 34
3.10 CINÉTICA VIRAL ...................................................................................................... 35
3.11 ANÁLISE PROTEÔMICA .......................................................................................... 35
3.11.1 Extração de Proteínas das Células Cultivadas ............................................................ 35
3.11.2 Clarificação do lisado celular..................................................................................... 35
3.11.3 Quantificação de Proteínas Totais .............................................................................. 36
3.11.4 Eletroforese Bidimensional........................................................................................ 36
3.12 ANÁLISE ESTATÍSTICA ........................................................................................... 37

4 RESULTADOS ................................................................................................................ 38
4.1 Título viral, efeito citopático e teste de viabilidade celular .............................................. 38
4.2 Análise proteômica......................................................................................................... 41
5 DISCUSSÃO .................................................................................................................... 49
5.1 Título viral, efeito citopático e teste de viabilidade celular .............................................. 49
5.2 Análise proteômica......................................................................................................... 49
6 CONCLUSÕES ................................................................................................................ 55
REFERÊNCIAS .................................................................................................................. 56
ANEXO ............................................................................................................................... 63

LISTA DE FIGURAS
Figura 1 - Vírion infeccioso imaturo e maturo da Febre Amarela ......................................... 18
Figura 2 - Organização do genoma e tradução das proteínas virais de flavivírus ................... 19
Figura 3 – Fluxograma de trabalho ....................................................................................... 26
Figura 4 - Curva de cinética da infecção pelo VFA em células HepG2 ................................. 38
Figura 5 - Comportamento da infecção pelo VFA, cepa selvagem Be H111, em cultivo celular
HepG2 ................................................................................................................................. 39
Figura 6 – Resultados de viabilidade celular obtidos para infecção de células HepG2 pelo
VFA ..................................................................................................................................... 40
Figura 7 - Gel bidimensional de proteínas totais de células HepG2 infectadas com o vírus da
Febre Amarela, cepa selvagem Be H111, e corado com solução coloidal de Coomassie. ...... 43
Figura 8 - Gel bidimensional de proteínas totais de células HepG2 do grupo controle corado
com solução coloidal de Coomassie.. ................................................................................... 44
Figura 9 - Representação tridimensional do spot número 1, match ID 244 ............................ 45
Figura 10 - Representação tridimensional do spot número 2, match ID 318 .......................... 46
Figura 11 - Representação tridimensional do spot número 3, match ID 482 .......................... 47
Figura 12 - Representação tridimensional do spot número 4, match ID 484 .......................... 48
LISTA DE TABELAS
Tabela 1 – Características dos géis em relação ao número spots detectados e pareados. ........ 41
Tabela 2 – Spots proteicos diferencialmente expressos. ........................................................ 42
Tabela 3 – Síntese dos resultados encontrados em trabalhos utilizando o método 2-DE
comparativo em infecções experimentais com diferentes vírus. ............................................ 51

LISTA DE ABREVIATURAS, SIGLAS E SÍMBOLOS
2-DE Eletroforese bidimensional
2D-DIGE Gel de eletroforese bidimensional diferencial
% Percentagem
°C Graus centígrados
μg Microgramas
μL Microlitros
ANOVA Análise de variância
AP61 Cultivo celular do invertebrado Aedes pseudoscutellaris
ATCC American type cell collection
BCRJ Banco de células do Rio de Janeiro
BHK-21 Cultivo celular de rim de hamster (Mesocricetus auratus) bebê
BSL2 Nível de biossegurança 2
C Proteína de Capsídeo
C6/36 Cultivo celular do invertebrado Aedes albupictus
Ca2+
Íon de cálcio
CHAPS 3-(cholamidopropyl)dimethylammonio-1-propane sulfonate
cm² Centímetros quadrados
CMC Carboximetilcelulose
CO2 Gás Carbônico
DMEM Meio essencial de Eagle modificado por Dulbecco
D-PBS Solução salina tamponada fofatada incompleta de Dulbecco
dpi Dias pós infecção
DTT Ditiotreitol
E Proteína de Envelope
EM Espectrometria de massas
ECP Efeito citopático
FA Febre amarela
g Gramas
HBsAg Antígeno da Hepatite B
IEC Instituto Evandro Chagas
IFI Imunofluorescência Indireta
IgG Imunoglobulina do tipo G

IgM Imunoglobulina do tipo M
KDa Quilodáltons
Kb Quilobases
KCl Cloreto de potássio
KH2PO4 Fosfato de potássio
KVh Quilovolts hora
L Litro
M Proteína de Membrana
M Molar
MEL Extensão Melanie de arquivo de imagem
Mg2+
Íon de magnésio
mL Mililitros
mm Milímetros
mM Milimolar
mm³ Milimetros cúbicos
MOI Multiplicidade de Infecção
MS Ministério da Saúde
ng Nanograma
NL Não linear
Na2HPO4 Fosfato de sódio
NaCl Cloreto de Sódio
nm Nanômetros
NS Proteína não estrutural
OMS Organização mundial de saúde
pH Potencial hidrogeniônico
PM Peso molecular
PCR Reação em cadeia da polimerase
PFU Unidade formadora de placas
pI Ponto isoelétrico
PrM Proteína Pré-membrana
RPM Rotações por minuto
RT-PCR Transcrição reversa seguida da reação em cadeia da polimerase
SAARB Seção de arbovirologia e febres hemorrágicas
SBF Soro bovino fetal

SDS Dodecil-sulfato de sódio
SVS Secretaria de Vigilância em Saúde
SW13 Cultivo celular de adenocarcinoma humano
TIFF Extensão de arquivo de imagem
UI Unidade internacional
V Volts
VDEN Vírus da Dengue
VERO Cultivo celular de rim de macaco verde africano Cercopithecus aethiops
VFA Vírus da febre amarela
Vh Volts hora
VJE Vírus da Encefalite Japonesa
VNO Vírus do Nilo Ocidental
VROC Vírus Rocio
VSLE Vírus da encefalite de Saint Louis
Vv Volume de estoque viral a ser utilizado para infecção
x Vezes
xg Unidade para força centrífuga relativa

RESUMO
A febre amarela (FA) é uma doença viral endêmica de regiões tropicais da África e das
Américas que afeta principalmente humanos e primatas não humanos, sendo transmitida pela
picada de um mosquito vetor infectado. O agente etiológico, o Vírus da Febre Amarela
(VFA), é capaz de produzir grandes epidemias com alta taxa de mortalidade causada pela
febre hemorrágica pós-infecção. Apesar do controle epidemiológico sobre esta doença e do
estabelecimento de uma vacina eficaz, não existe medicação para tratamento e
frequentemente a FA não é diagnosticada até que o paciente tenha se recuperado ou
sucumbido, isto, se é que o diagnóstico é feito. A falta de informação proteômica sobre o
VFA, amplia a possibilidade da descoberta de biomarcadores proteicos para diagnósticos mais
eficazes e/ou alvos de tratamento, que podem, junto com as ferramentas atuais de controle de
surtos, aumentar a velocidade de resposta sobre possíveis epidemias. Neste trabalho, usamos
técnicas proteômicas para investigar o perfil das proteínas diferenciais de células HepG2
infectadas com o VFA, cepa Be H111. Para isso, cultivos de células HepG2 foram infectadas
com multiplicidade de infecção de 0,01 e avaliados a presença de efeito citopático, título viral
em unidades formadoras de placas (PFU), progressão da infecção através da detecção de
antígenos virais pela imunofluorescência indireta (IFI) e viabilidade celular para determinar o
melhor dia da avaliação da expressão proteica. A cada 24 horas durante 5 dias pós infecção
(dpi) as proteínas foram extraídas com tampão de extração (7M ureia, 2M tioureia e 4% de
CHAPS). As amostras foram analisadas por 2-DE usando fitas IPG pH 3-11 NL para a
focalização isoelétrica seguida de SDS-PAGE vertical para separação da segunda dimensão.
Os mapas 2D das células infectadas e não infectadas (controle) foram visualizados pelo
corante Coomassie G-250. O 4º dpi foi determinado para melhor avaliação das alterações
proteicas, detectando 4 spots de proteínas diferenciais. Estes resultados trazem novas
perspectivas para o aprofundamento nos estudos de biomarcadores celulares para diagnóstico
da FA.
Palavras-chave: Febre Amarela, Vírus da Febre Amarela, Proteômica.

ABSTRACT
Yellow fever (YF) is a viral disease endemic in the tropical parts of Africa and the
Americas that affects mainly human and non-human primates. It is transmitted by the bite of
an infected mosquito vector. The etiologic agent of Yellow Fever Virus (YFV), is the cause of
large epidemics with high mortality caused by post-infection hemorrhagic fever. Despite the
epidemiological control and the establishment of an effective vaccine against this disease,
there is no medication for treatment and YF is often not diagnosed until the patient has
recovered or succumbed, that, if the diagnosis is made. The lack of proteomic information
about YFV increases the possibility of the discovery of protein biomarkers for more effective
targets for diagnostic and/or treatment, which may, with the current tools for outbreak control,
increase the speed of response in possible epidemics. In this work, we used proteomic
approaches to investigate the profile of differentials proteins in infected HepG2 cells with the
YFV, strain Be H111. To do this, cultured HepG2 cells were infected at multiplicity of
infection of 0.01 and it was assessed for the presence of cytopathic effect, virus titers were
calculated by plaque forming units (PFU), the progression of infection by detection of viral
antigens by indirect immunofluorescence (IFI) and cell viability to determine the best day to
assess the protein expression. Every 24 hours for 5 days post infection (dpi) proteins were
extracted with extraction buffer (7M urea, 2M thiourea and 4% CHAPS). The samples were
then analyzed by 2-DE using IPG 3-11 NL pH strips for vertical isoelectric focusing followed
by SDS-PAGE separation in the second dimension. The 2D maps of infected and uninfected
(control) cells were visualized by Coomassie G-250 dye. The 4 dpi was determined to better
elucidate the protein changes, detecting 4 differential protein spots. These results open new
perspectives for the further studies of cellular biomarkers for diagnosis of YF.
Palavras-chave: Yellow Fever, Yellow Fever Virus, Proteomics.

15
1 INTRODUÇÃO
1.1 CONSIDERAÇÕES GERAIS SOBRE A FEBRE AMARELA
Durante as expansões das rotas marítimas com finalidade comercial e o tráfico
negreiro, ocorreu a disseminação de vários patógenos e doenças, dentre estes, a do Vírus da
Febre Amarela (VFA). Acredita-se que o VFA emergiu inicialmente na África, sendo
posteriormente levado para o continente Europeu e Americano (Barrett & Higgs, 2007;
Gardner & Ryman, 2010).
O início da história da febre amarela (FA) é pobremente documentado, no entanto
acredita-se que o termo “febre amarela” foi provavelmente usado pela primeira vez por
Griffin Hughes em seu livro “Natural History of Barbados” de 1750 (Gardner & Ryman,
2010). A primeira descrição de uma provável epidemia de FA foi encontrada em um
manuscrito Maia em Yucatan, no México, em 1648, usando o termo xekik (vômito de sangue
negro ou hematêmese) (Pulendran, 2009; Gardner & Ryman, 2010).
Ao longo do tempo, nos séculos XVIII e XIX, surtos de FA devastaram as Américas
Sul e Central, assim como, cidades portuárias na costa leste da América do Norte, causando
devastação aos colonos americanos, atingindo também, repetidamente várias cidades
européias envolvidas no comércio com o Novo Mundo (Monath, 1994; Staples & Monath,
2008; Gardner & Ryman, 2010).
Ainda por volta do século XIX, reconheceu-se que o contágio da FA não ocorria a
partir do contato inter-humano. No entanto, teorias cientificamente não fundamentadas
atribuíram erroneamente a doença aos miasmas ambientais (Staples & Monath, 2008; Gardner
& Ryman, 2010).
Em 1881, Carlos Finlay, um eminente médico e cientista cubano, avançou de forma
expressiva no campo de estudo da FA quando sugeriu o Culex cubensis (atualmente
conhecido como Aedes aegypti) como o mosquito responsável pela transmissão da doença
(Pulendran, 2009; Gardner & Ryman, 2010). Apesar de várias tentativas, Finlay não foi capaz
de provar sua teoria, que serviria de base para a pesquisa de Walter Reed (Staples & Monath,
2008).
Durante a Guerra Hispano-Americana de 1898, uma grave epidemia de FA eclodiu
entre os camponeses cubanos e os soldados americanos que se encontravam em Havana. Para
cada soldado que morria em batalha, 13 eram acometidos pela FA e evoluíam a óbito. Um
cirurgião geral dos EUA, George Sternberg, enviou Walter Reed e sua equipe para Cuba para
investigar a causa da FA. O trabalho de Reed provou que os mosquitos Ae. aegypti eram o

16
principal modo de transmissão da doença e que a FA era causada por um agente filtrável
encontrado no sangue de pacientes infectados (Staples & Monath, 2008; Pulendran, 2009;
Gardner & Ryman, 2010).
No ano de 1908, programas de controle do mosquito vetor erradicaram a FA dos
principais centros urbanos (Monath, 1994). Embora essas atividades iniciais tenham sido
bem-sucedidas contra FA urbana (transmitida por Ae. aegypti), a meta de erradicação foi
dissipada com a descoberta de que a FA era uma zoonose, mantida por espécies de mosquitos
silvestres e primatas não-humanos na selva amazônica (Staples & Monath, 2008).
Em 1927, mais de um quarto de século após a observação de Reed, Adrian Stokes
isolou o vírus de um homem doente em Gana, África, chamado Asibi (Staples & Monath,
2008; Pulendran, 2009). Max Theiler, em busca de um modelo animal mais barato, mostrou a
viabilidade da infecção em camundongos (Mus musculus). Seguindo os passos de Louis
Pasteur, que tinha atenuado o vírus da raiva por passagem em cérebro de coelho, Theiler
observou que a passagem em série neurotrópica em camundongos resultou em uma perda
progressiva da virulência em macacos rhesus (Macaca mulatta). Isto sugeriu a possibilidade
de desenvolver um vírus atenuado como vacina e, portanto, Theiler escolheu realizar a
passagem do vírus por vários tecidos animais para atenuar sua virulência em humanos. A
maioria das cepas resultou em vírus extremamente letais ou pouco imunogênicos em macacos.
A única exceção foi a cepa YF-17D (do inglês YF, yellow fever), que foi desenvolvida por
176 passagens do vírus primeiramente em cérebro e cultura de tecidos de embrião de
camundongo e posteriormente em embrião e cultura de tecidos de embrião de galinha (Theiler
& Smith, 1937a; 1937b).
Esse trabalho induziu a realização de experimentos em humanos, no qual a cepa YF-
17D foi inoculada pela primeira vez em voluntários humanos, incluindo os autores
desenvolvedores da cepa vacinal, resultando na indução de anticorpos neutralizantes (Theiler
& Smith, 1937b). Em 1945, a Organização Mundial de Saúde permitiu o uso de dois lotes de
sub cepas descendentes da cepa 17D, a cepa 17DD, que tem sido usada na América do Sul, e
17D-204, que tem sido usada no resto do Mundo. Ambas as sub cepas da vacina são
produzidas em ovos de galinha embrionados. Theiler recebeu o Prêmio Nobel em 1951 por
esse trabalho, e este ainda é o único Prêmio Nobel concedido para o desenvolvimento de uma
vacina viral (Pulendran, 2009).
Essas descobertas iniciais serviram como a base para as investigações posteriores
acerca da FA, por conseguinte, gerando avanços nos conhecimentos da epidemiologia,
ecologia, diagnóstico, etiologia e prevenção da febre amarela (Staples & Monath, 2008).

17
1.2 ETIOLOGIA DA FEBRE AMARELA
O VFA é o membro protótipo da família Flaviviridae e gênero Flavivirus, que tem o
seu nome originado da palavra em latim flavus que significa amarelo. O gênero Flavivirus
contém aproximadamente 70 vírus (Monath, 2001; Barrett & Higgs, 2007; Fernandez-Garcia
et al., 2009; Smit et al., 2011), cujo os principais agentes virais associados a doença em
humanos são os Vírus da Febre Amarela, Vírus Dengue (VDEN), Vírus do Nilo Ocidental
(VNO), Vírus da Encefalite Japonesa (VJE), Vírus Rocio (VROC) e o Vírus da Encefalite de
Saint Louis (VSLE) (Vasconcelos, 2002; 2003; Fernandez-Garcia et al., 2009; Smit et al.,
2011).
Em termos ecológicos, os flavivírus são denominados arbovírus, termo derivado da
expressão em inglês Artrophod-borne virus, devido à maioria destes vírus serem transmitidos
por vetores artrópodes (Monath, 2001; Barrett & Higgs, 2007).
O vírion do VFA apresenta morfologia icosaédrica (Smit et al., 2011), nucleocapsídeo
formado por subunidades de proteínas de capsídeo (C), cerca de 25-30 nm de diâmetro,
envoltório bilaminar de natureza lipoprotéica, também conhecido como envelope, que é
originário da célula hospedeira (Vasconcelos, 2003; Gardner & Ryman, 2010) (Figura 1). A
partícula íntegra (vírion mais envelope) mede cerca de 40 nm (Vasconcelos, 2002; King et al.,
2012). O envelope viral é composto por dímeros de glicoproteína do envelope (E) e proteína
de membrana (M) (Monath, 2001; King et al., 2012).
O genoma empacotado no interior do nucleocapsídeo do vírion VFA é constituído de
RNA de fita simples, de polaridade positiva, de aproximadamente 11 kilobases (Kb) de
comprimento (Gardner & Ryman, 2010; King et al., 2012). O genoma completo possui
10.862 nucleotídeos (Vasconcelos, 2003; King et al., 2012) que codifica uma poliproteína de
aproximadamente 3.400 aminoácidos (Fernandez-Garcia et al., 2009), que é posteriormente
processada por clivagem proteolítica (Gardner & Ryman, 2010). Mecanismos de clivagem co-
e pós-traducionais realizadas no polipeptídeo precursor pela ação de enzimas derivadas da
célula hospedeira e viral (enzimas de clivagem) originam três proteínas estruturais (Capsídeo
[C], membrana [M] e envelope [E]) e sete não estruturais (NS1, NS2A, NS2B, NS3, NS4A,
NS4B e NS5) (Fernandez-Garcia et al., 2009; King et al., 2012) (Figura 2).
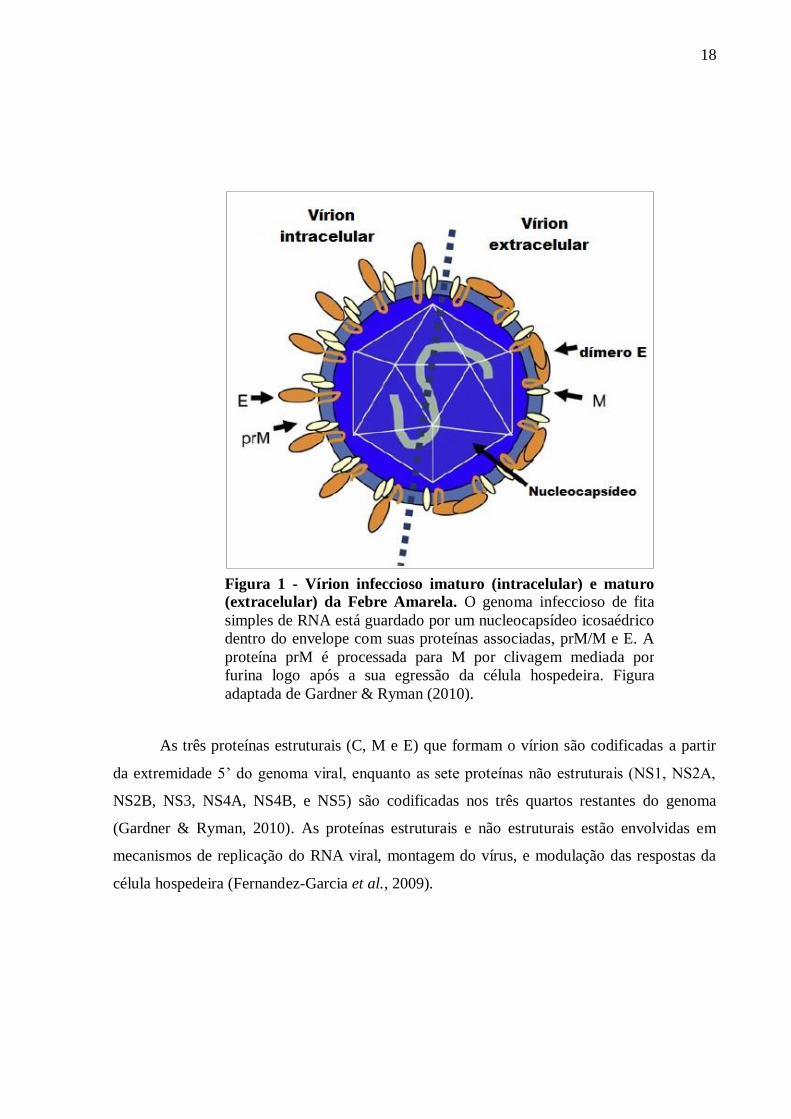
18
As três proteínas estruturais (C, M e E) que formam o vírion são codificadas a partir
da extremidade 5’ do genoma viral, enquanto as sete proteínas não estruturais (NS1, NS2A,
NS2B, NS3, NS4A, NS4B, e NS5) são codificadas nos três quartos restantes do genoma
(Gardner & Ryman, 2010). As proteínas estruturais e não estruturais estão envolvidas em
mecanismos de replicação do RNA viral, montagem do vírus, e modulação das respostas da
célula hospedeira (Fernandez-Garcia et al., 2009).
Figura 1 - Vírion infeccioso imaturo (intracelular) e maturo
(extracelular) da Febre Amarela. O genoma infeccioso de fita
simples de RNA está guardado por um nucleocapsídeo icosaédrico
dentro do envelope com suas proteínas associadas, prM/M e E. A
proteína prM é processada para M por clivagem mediada por
furina logo após a sua egressão da célula hospedeira. Figura
adaptada de Gardner & Ryman (2010).

19
Figura 2 - Organização do genoma e tradução das proteínas virais de flavivírus. A única
região codificante é traduzida em uma poliproteína precursora que é clivada co- e pós-
traducionalmente em três proteínas estruturais (em verde) e sete proteínas não estruturais (em
vermelho). Legenda: UTRs, Regiões não traduzidas 5’ e 3’; nt, nucleotídeos; Kb, quilobases;
aa, aminoácidos; C, proteína do capsídeo; E, proteína de envelope; M, proteína de membrana.
Fonte: Figura adaptada de Fernandez-Garcia et al. (2009).
1.3 EPIDEMIOLOGIA DA FEBRE AMARELA
Notavelmente, a epidemiologia do VFA reflete a distribuição do mosquito vetor
(Barrett & Higgs, 2007), e mesmo depois de uma vacina segura e eficaz (17D) (Vasconcelos
et al.,2004) a doença ainda ocorre em regiões tropicais da África subsariana e das Américas
Central e do Sul (Monath, 2001; Barnett, 2007; Monath, 2008).
A epidemiologia natural da FA em ambos os continentes consiste no ciclo do vírus
entre mosquitos e primatas não humanos (Gardner & Ryman, 2010), sendo que a infecção em
humanos pode ocorrer em duas modalidades epidemiológicas: silvestre e urbana
(Vasconcelos, 2002).
A principal causa de preocupação em nível de saúde pública tanto na África quanto
nas Américas provém do ciclo de transmissão urbana da FA, o qual resulta em uma epidemia
explosiva de alta escala (Vasconcelos, 2002; Gardner & Ryman, 2010) que é iniciada com a
introdução do vírus em áreas com alta densidade populacional, onde o mosquito Ae. aegypti é
o vetor responsável pela disseminação da doença.

20
O ciclo de transmissão silvestre da FA ocorre na mata de áreas endêmicas onde o vírus
é transmitido entre várias espécies de primatas e mosquitos que habitam o dossel da floresta,
sendo que o vírus é “acidentalmente” transmitido para humanos que entram nestas áreas
(Gardner & Ryman, 2010). O ciclo silvestre, além de complexo, não é ainda de inteira
compreensão e varia de acordo com a região onde ocorre (Vasconcelos, 2002). Na África, este
ciclo foi descrito como tendo o Ae. africanus como o principal vetor (Barrett & Higgs, 2007).
Já nas Américas, o ciclo silvestre têm como responsáveis pela transmissão os mosquitos dos
gêneros Haemagogus (Hg. janthinomys, Hg. albomaculatus, etc.) e Sabethes (Sa.
chloropterus, Sa. soperi, etc.), sendo que o Hg. janthinomys é o principal responsável
(Vasconcelos, 2002).
Nas áreas de savana africana há também a transmissão intermediária da FA, que
resulta em epidemias de pequena escala em vilarejos rurais, onde espécies de mosquitos semi-
domésticas (Ae. luteocephalus, Ae. furcifer, Ae. metallicus, Ae. opok, Ae. taylori, Ae. vittatus,
e membros do complexo Ae. simpsoni) (Barrett & Higgs, 2007) infectadas se alimentam de
ambos os hospedeiros, tanto do homem quanto de primatas. Este tipo de surto tem sido
comum na África nas últimas décadas (Gardner & Ryman, 2010).
Segundo estimativa realizada pela Organização Mundial da Saúde (OMS), o VFA
acomete cerca de 200.000 pessoas anualmente (Monath, 2001), incluindo 30.000 mortes
anuais (Barrett & Higgs, 2007). Mais de 90% destes casos ocorrem na África, onde a
transmissão é mantida pela alta densidade de populações do mosquito vetor que estão
próximos a grandes populações humanas não vacinadas em conjunção com um processo de
rápida urbanização, visto que a taxa de aumento da população urbana de 4% ao ano. Já na
América do Sul, a menor taxa de transmissão é em parte devido à alta cobertura vacinal que
ocorre em campanhas de imunização em massa. Contudo, é preocupante a re-infestação de
mosquitos Ae. aegypti nos mais importantes centros urbanos das Américas Central e do Sul,
incluindo cidades onde historicamente ocorreram epidemias de FA urbana e agora estão
habitadas por grandes populações não imunizadas (Gardner & Ryman, 2010).
Com o advento da biologia molecular, mais especificamente de sequenciamento
genômico, caracterização genética e filogenia empregando sequências dos genes de Envelope,
NS5 e regiões não traduzidas 5’ e 3’, sete genótipos do VFA foram descritos, sendo cinco de
ocorrência geográfica na África (Mutebi et al., 2001) e dois nas Américas (Vasconcelos et al.,
2004; Bryant et al., 2005). Nunes et al. (2012) por meio do estudo do genoma completo de
diferentes cepas brasileiras em comparação com cepas Africanas e de Trinidad, bem como,
pela análise filogeográfica, confirmaram dados previamente descritos sobre a heterogeneidade

21
das regiões 3’ não traduzidas (Mutebi et al., 2001; Bryant et al., 2005), bem como, lançaram
as bases para a análise filogeográfica e de dispersão espaço-temporal do VFA nas Américas,
em particular no Brasil.
1.4 MANIFESTAÇÕES CLÍNICAS DA FEBRE AMARELA
Descrito como ''a original febre hemorrágica viral'' a FA é uma sepse viral
pansistêmica que pode apresentar-se clinicamente por febre, prostração, lesão hepática, renal
e miocárdica, hemorragia, choque, e têm 20% a 50% de letalidade (Monath, 2008; Gardner &
Ryman, 2010). A doença clínica varia de não-específica leve, doença abortiva até a febre
hemorrágica fatal (Monath, 2001). Diferenças nas cepas de vírus, bem como fatores imunes
incompletamente compreendidos do hospedeiro, são provavelmente responsáveis pela gama
de sintomas clínicos (Barnett, 2007). Apesar disto, o fígado é o alvo principal da infecção e
pigmentos biliares amarelos são liberados a partir do fígado danificado resultando em
icterícia, por isso o termo "febre amarela" (Pulendran, 2009).
Três fases de FA são descritas. A primeira é durante a viremia que é caracterizada por
febre, mal-estar, mialgia generalizada, náuseas, vômitos, irritabilidade, tontura, e geralmente
uma aparência tóxica. A segunda fase é caracterizada por melhora dos sintomas, incluindo
uma redução de febre, o que pode durar até 48 horas, mas não é observado em todos os casos.
A terceira fase ocorre em aproximadamente 15% dos casos e caracteriza-se pelo retorno da
febre, náuseas, vômitos, icterícia e diátese hemorrágica (Barnett, 2007).
1.5 TRATAMENTO, PREVENÇÃO E DIAGNÓSTICO DA FEBRE AMARELA
Não há medicamento específico para o tratamento da doença (Staples & Monath,
2008). Embora não haja drogas antivirais específicas disponíveis, alguns compostos com
atividade antiviral in vitro foram descritos, incluindo ribavirina e interferon-α (Gardner &
Ryman, 2010), além destes, Monath (2008) revisou trabalhos que relatam o uso de diferentes
dosagens destes compostos e também o uso de anticorpos passivos, interferons,
imunomodulatórios e outras drogas contra a FA. Desta forma, o tratamento medicamentoso
deve se voltar para o combate aos sintomas e os sinais manifestos da doença, como a
medicação a ser prescrita depende das manifestações clínicas, é comum o uso de analgésicos e
antitérmicos, entretanto, se contra-indica o uso de medicamentos que contenham em sua
fórmula o ácido acetil-salicílico ou seus derivados pois eles podem agravar os fenômenos
hemorrágicos (Vasconcelos, 2002; 2003).

22
A vigilância da FA é fundamental para o monitoramento da incidência da doença e
para permitir a detecção precoce de surtos ou sua prevenção, além do acompanhamento de
medidas de controle, a comunicação dos casos é exigida pelo Regulamento Sanitário
Internacional (International Health Regulations) (Gardner & Ryman, 2010). Outra forma
preventiva é o uso da vacina YF17D, como citado anteriormente.
Exames de diagnóstico laboratoriais se baseiam na detecção do vírus ou antígeno viral
no sangue durante a fase pré-ictérica ou por sorologia. Nenhum teste comercial está
disponível e as capacidades de diagnóstico residem unicamente em laboratórios
especializados e de referência (Monath, 2001), pois infelizmente requerem um pessoal de
laboratório altamente treinado, e com acesso a equipamentos e materiais especializados
(Gardner & Ryman, 2010).
O vírus pode ser isolado por inoculação intracerebral em camundongos recém
nascidos ou em cultivos celulares (células VERO, SW13, BHK-21, AP61 e clone C6/36),
particularmente se combinadas com técnica de reação em cadeia da polimerase (PCR), testes
de imunofluorescência indireta usando anticorpos monoclonais ou mediante testes de fixação
do complemento (Monath, 2001; Vasconcelos, 2002; 2003). O diagnóstico sorológico pode
ser feito através da presença de IgM específica para o VFA ou um aumento quatro vezes
maior nos níveis de IgG entre o soro de fase aguda e convalescente na ausência de uma
vacinação recente (Gardner & Ryman, 2010). A reação cruzada entre o VFA e outros
Flavivirus pode dificultar o diagnóstico sorológico, particularmente em indivíduos que vivem
em regiões onde vários Flavivirus são endêmicos (Monath, 2001). Nos casos fatais procura-se
antígenos específicos pela técnica de imunohistoquímica em tecidos hepáticos ou é
evidenciado o genoma viral por RT-PCR do sangue (células e soro) e fígado (Vasconcelos,
2002; 2003). Frequentemente a FA não é diagnosticada até que o paciente tenha se recuperado
ou sucumbido, isto, se é que o diagnóstico é feito (Gardner & Ryman, 2010).
1.6 PROTEÔMICA VIRAL
A proteômica clínica, um subconjunto de atividades da proteômica dentro do campo
da medicina, surgiu com a promessa de acelerar a descoberta de novos alvos de drogas e
proteínas marcadoras de doenças que são úteis para diagnóstico in vitro (Vitzhum et al.,
2005). O recente desenvolvimento das técnicas proteômicas tem estimulado um grande
interesse em aplicar as tecnologias em estudos clínicos translacionais, em particular, pesquisa
por proteínas biomarcadoras (Pan et al., 2011). Um biomarcador é uma substância encontrada

23
em amostras biológicas (Issaq & Veenstra, 2007) que serve como uma característica para o
estado fisiológico da célula em um determinado tempo e pode mudar durante o processo
patológico (Wulfkuhle et al., 2003). Além disso, os biomarcadores são medidas biométricas
que transmitem informações sobre a condição biológica da questão a ser testada, obtendo o
seu valor a partir de sua capacidade de diferenciar entre dois ou mais estados biológicos
(LaBaer, 2005).
Na virologia, a pesquisa proteômica ainda é considerada como um enorme desafio
trazido principalmente por três aspectos; o estudo sobre o proteoma complexo do vírus com
variações inesperadas, o desenvolvimento de técnicas de análise mais precisas, bem como a
compreensão da patogênese viral e dinâmica de interação vírus-hospedeiro. Avanços nessas
áreas serão úteis para o desenvolvimento de vacinas e de descoberta de drogas antivirais
(Zheng et al., 2011). Até agora, um pequeno número de estudos têm utilizado abordagens
proteômicas para mostrar os efeitos de uma infecção viral no proteoma celular e as crescentes
evidências enfatizam a proteômica comparativa como alternativa para o conhecimento das
proteínas diferencialmente expressas em células do hospedeiro que estejam associadas aos
processos fisiopatológicos da infecção por vírus (Maxwell & Frappier, 2007). Além disso,
esta abordagem permite identificar o repertório de fatores do hospedeiro que estão envolvidos
ativamente no ciclo viral infeccioso e caracterizar as respostas hospedeiras, qualitativa e
quantitativamente durante a patogênese viral (Zheng et al., 2012).
De forma geral, as infecções virais podem resultar em modificações pós-traducionais,
tais como a ubiquitinação, fosforilação e glicosilação sem afetar as taxas de transcrição.
Portanto, a análise proteômica das respostas celulares do hospedeiro à infecção por vírus é
mais provável para a avaliação de potenciais fatores celulares envolvidos direta ou
indiretamente na infecção viral e para identificar potenciais alvos de drogas antivirais (Zheng
et al., 2008).
Zheng et al. (2011) relatam que vários vírus de importância clínica e suas infecções no
interior de células hospedeiras foram estudados utilizando diferentes abordagens proteômicas
em conjunção com análise de espectrometria de massas. Somente são relatados em sua
revisão cinco vírus da Família Flaviviridae, o Vírus da Hepatite C, o VDEN, o Vírus da Peste
Suína, o VNO e o Vírus da Diarréia Viral Bovina.
Apesar de ter uma vacina eficaz e a imunização ser o método mais importante de
prevenção contra a FA (Barnett, 2007), esta continua a ser classificada como uma doença
emergente ou re-emergente (Vasconcelos, 2003; Barrett & Higgs, 2007; Gardner & Ryman,
2010) com grande potencial de reurbanização nas Américas. Isto é devido ao atual nível de

24
infestação de Ae. aegypti nos grandes centros urbanos próximos às áreas endêmicas, à
frequente migração de populações entre as áreas endêmicas e não endêmicas, e a baixa
cobertura vacinal contra FA em muitos países (Vasconcelos, 2003; Gardner & Ryman, 2010).
Com a falta de informação proteômica sobre o VFA, estudos como este, podem
ampliar o entendimento da fisiopatologia da doença e também possibilitam a descoberta de
biomarcadores protéicos para diagnósticos mais eficazes e/ou alvos de tratamento, que
podem, junto com as ferramentas atuais de controle de surtos, aumentar a velocidade de
resposta sobre possíveis epidemias. Embora muito se tenha aprendido nos últimos 100 anos,
claramente ainda há muito mais a ser descoberto sobre a FA (Staples & Monath, 2008).

25
2 OBJETIVOS
2.1 OBJETIVO GERAL
Verificar alterações no proteoma de células de carcinoma hepatocelular humano,
HepG2, experimentalmente infectadas pelo Vírus da Febre Amarela, Cepa Be H111
(Flaviviridae: Flavivirus).
2.2 OBJETIVOS ESPECÍFICOS
Observar o provável efeito citopático induzido pelo VFA, a cinética viral
adquirida com MOI de 0,01 e estabelecer o melhor período para análise das
alterações no proteoma celular;
Comparar os padrões dos perfis proteicos de células HepG2 infectadas com
VFA ao grupo controle;

26
3 MATERIAL E MÉTODOS
O material e métodos utilizados estão descritos a seguir, enquanto que a ordem dos
principais experimentos realizados para alcançar os objetivos propostos encontra-se resumida
no fluxograma de trabalho (Figura 3).
3.1 ASPECTOS ÉTICOS E DE BIOSSEGURANÇA
Este projeto para desenvolvimento da dissertação de mestrado não visou a utilização
de humanos, outros animais ou meio ambiente como objetos de pesquisa, sendo utilizada a
Figura 3 – Fluxograma de trabalho. Fluxograma dos principais passos realizados. Caixas
em tons de cinza; representam os experimentos iniciais com cultivos celulares e manuseio
viral. Caixas vermelhas; testes realizados para estabelecer o melhor dia para a análise
diferencial. Caixas verdes; diferenciam os materiais biológicos coletados utilizados. Caixa
roxa; período de análise dos dados gerados a partir dos testes realizados para estabelecer o
melhor dia para a análise diferencial. Caixas azuis; indicam as análises finais partindo da
extração de proteínas até a análise diferencial final. Setas pretas; indicam a ordem do trabalho
realizado. Setas azuis; indicam a ordem do trabalho realizado com as células cultivadas em
placas de seis poços. Setas e conectores laranja; indicam a ordem do trabalho realizado com as
células cultivadas em garrafas.

27
cepa viral já previamente isolada e pertencente ao acervo do Instituto Evandro
Chagas/SVS/MS, Seção de Arbovirologia e Febres Hemorrágicas (SAARB/IEC).
Para a realização dos procedimentos descritos a seguir, foram aplicadas as normas e
critérios pré-estabelecidos e exigidos pelo Comitê Internacional de Biossegurança. Todos os
procedimentos envolvendo a manipulação da cepa viral foram realizados em cabines de
biossegurança e os que envolvam substâncias tóxicas, em capelas de exaustão.
3.2 CARCINOMA HEPATOCELULAR HUMANO, HEPG2
O cultivo epitelial HepG2 (ATCC: HB8065; BCRJ:0103) é considerado nível de
biossegurança 1 e como descrito na patente de criação (Knowles & Aden, 1983), estas células
foram criadas primeiramente com objetivos de produzir uma linhagem de célula hepática
estável aplicáveis à estudos de metabolismo de drogas, em particular, na procura de potencial
carcinógenos e mutagênicos e na produção de componentes vacinais para o Vírus da Hepatite
B, não descartando a cultura de vírus.
A linhagem HepG2 foi derivada de uma biópsia retirada de um adolescente argentino
caucasiano de 15 anos de idade em 1975. Foram caracterizados 55 cromossomos (variando de
50 à 56), tendo como cromossomo marcador o cromossomo 1 rearranjado. Foram testados a
produção de 17 proteínas normais do plasma humano, e dentre elas, apenas a Gc-globulina
estava ausente, e também foi relatado a satisfatória produção do antígeno da hepatite B
(HBsAg) durante a replicação viral do Vírus da Hepatite B (Knowles & Aden, 1983).
3.2.1 Descongelamento das Células HepG2
Para o descongelamento das células HepG2 foi utilizado meio de cultura consistindo
de meio essencial de Eagle modificado por Dulbecco (DMEM – Sigma, EUA), acrescido de
1,5 g/L de bicarbonato de sódio, 10 mM de Hepes (Sigma), 2 mM de L-glutamina (Sigma), e
10% de soro bovino fetal (SBF), sem a adição de antibióticos.
O descongelamento da suspensão celular criopreservada em nitrogênio líquido foi
realizado rapidamente em banho Maria 37°C, porém, antes do total descongelamento a
suspensão celular foi conduzida à cabine de segurança biológica de classe II (B2) para ser
feita uma diluição final de 1:10 em meio de cultura DMEM. A transferência da suspensão
celular para o meio de cultura foi realizada de forma lenta (gota a gota) e seguida por uma
centrifugação de 5 minutos a 800 rotações por minuto (RPM). Após a centrifugação foi
retirado o sobrenadante e o precipitado de células resuspendido em 5 mL de meio e

28
transferido para uma garrafa de 25 cm² de área de crescimento celular a qual foi incubada em
sistema semi aberto em estufa com 5% de CO2 a 37°C.
3.2.2 Subcultura das Células HepG2
Após a formação da monocamada celular foi feito o processo de passagem celular, ou
subcultura para manutenção da linhagem manipulada. De forma geral, existe um padrão
metodológico descrito para culturas celulares de vertebrados (Freshney, 2010; American type
culture collection, 2013) o qual pode ser alterado de forma livre ao manipulador das células,
porém, as mudanças devem ser bem analisadas para não comprometer o cultivo celular. Desta
forma, após a observação da formação da monocamada o meio de cultura foi retirado, e a
monocamada de células lavada três vezes com uma solução salina fosfatada tamponada de
Dulbecco (Dulbecco & Vogt, 1953) incompleta (D-PBS), sem a adição de íons de Ca2+
e
Mg2+
(Knowles & Aden, 1983). A composição do D-PBS é de NaCl (8,0 g), KCl (0,2 g),
Na2HPO4 (1,15 g) e KH2PO4 (0,2 g) diluídos em 1000 mL de água deionizada ultra pura
(Mili-Q) (Milipore, EUA) autoclavada.
Para desprender as células aderidas à parede da garrafa, foi adicionado uma solução
contendo 0,25% de tripsina e 0,02% EDTA em um volume de 0,5 mL/25 cm². Após cobrir
toda a monocamada de células, essas permaneceram em repouso por 30 segundos, quando
logo após, a tripsina foi removida parcialmente, e as células incubadas em estufa 37°C por 2 a
5 minutos, de modo a permitir o desprendimento mais rápido possível das células.
Para inativar a tripsina, logo após o processo descrito acima foi acrescentado meio de
cultura DMEM de crescimento (10% de SBF) com 0,1% de uma formulação de dois
antibióticos (Sigma, EUA) sendo penicilina (100 UI/mL) e estreptomicina (100 μg/mL),
utilizando-se um volume de pelo menos duas vezes ao de tripsina adicionado. A suspensão de
células foi submetida a centrifugação de 1000 RPM por 4 minutos. O sobrenadante foi então
desprezado, e o precipitado de células resuspendido em 3 mL de meio DMEM de
crescimento.
Novas garrafas de cultivo celular foram mantidas em uma divisão split ratio de 1:3
(American type culture collection, 2013) para manutenção da linhagem celular ou para o
experimento de infecção com o VFA.

29
3.3 CEPA VIRAL
Para o presente estudo foi utilizado o isolado Be H111 do VFA pertencente a coleção
de isolamentos da SAARB/IEC com autorização da direção do IEC (Anexo). A cepa em
questão corresponde ao vírus selvagem mantido inoculado em camundongos recém-nascidos
(Mus muscullus).
3.4 ESTOQUE VIRAL
Para alcançar os objetivos deste estudo, foi necessário a síntese de um estoque viral a
partir da recuperação in vitro do isolado do VFA. Esta recuperação foi realizada em duas
passagens em cultivo de células de Aedes albopictus clone C6/36, sendo a segunda passagem
viral utilizando multiplicidade de infecção (MOI) de 0,01.
3.4.1 Estoque Viral em Cultivo de Células de Mosquito Aedes albopictus Clone C6/36
O cultivo de células de mosquito Aedes albopictus, clone C6/36, foi mantido em meio
Leibowitz (L-15 – Sigma) com adição de triptose fosfato (Sigma) a 10%, solução de
aminoácidos não essenciais (fórmula do Dr. Moacir Rebello – Baktron microbiologia, Brasil)
a 1%, bem como uma formulação de dois antibióticos (Gibco, EUA) sendo penicilina e
estreptomicina adicionados ao volume final de 0,1%. O meio de crescimento contém 5% de
SBF, e o de manutenção 2%. A subcultura foi realizada como indicado no sítio eletrônico da
ATCC (American type culture collection, 2013).
Inicialmente, para recuperação do VFA e produção do estoque semente, foi utilizada
uma garrafa grande (175 cm²) com monocamada confluente. O meio de crescimento foi
desprezado e a monocamada lavada delicadamente uma vez com meio de cultivo de
manutenção para retirar células não aderidas à parede da garrafa. Foi realizada a maceração
do cérebro de camundongo recém-nascido infectado em 1,8 mL de uma solução salina
contendo albumina bovina (0,75%), penicilina (100 UI/mL) e estreptomicina (100 µg/mL), o
macerado foi centrifugado por 5 minutos a 5000 RPM, onde o sobrenadante foi utilizado
como inóculo. Logo após, foi inoculado 1 mL do isolado viral em suspensão filtrado em filtro
de seringa sobre a monocamada celular e incubada para adsorção viral por uma hora em
temperatura de 28 a 30°C, com eventuais agitações suaves neste período de tempo. Após a
adsorção, a monocamada foi novamente lavada, e acrescido 50 mL de meio de manutenção
para posterior incubação por 6 a 10 dias em 28 a 30°C.

30
Diariamente foi realizado o acompanhamento em microscópio ótico invertido para
verificar a presença de ECP, ou desagregação das células da parede da garrafa. Quando
notada a presença de um dos dois, foi acrescentado 5 mL (10%) de SBF, com posterior
homogeneização e coleta do líquido do interior da garrafa para centrifugação. Após
centrifugar por 3000 RPM durante 5 minutos, o sobrenadante foi aliquotado em tubos
específicos para congelamento e estes foram armazenados em freezers a -70°C para posterior
titulação viral, enquanto que as células precipitadas foram coletadas em lâminas especiais
para imunofluorescência (Perfecta, Brasil) e testadas por imunofluorescência indireta (IFI)
(subsecção 3.7).
Após a realização do teste de IFI, e com resultado positivo para VFA, foi realizada a
titulação viral (Subsecção 3.8).
Com o título viral do estoque semente já definido, foi realizada uma segunda
passagem do vírus nas células da linhagem C6/36, gerando desta forma o estoque trabalho, só
que desta vez a monocamada foi infectada com diferente MOI, apresentado na subsecção 3.6.
Para realização da segunda passagem foram necessárias duas garrafas grandes com
monocamada confluente. Uma delas foi utilizada para contagem celular (como visto na
subsecção 3.5), e a outra foi inoculada com MOI de 0,01, conforme descrito para a
recuperação viral. Ao final da incubação por 6 a 10 dias pós-infecção (dpi) foi realizado o
mesmo protocolo da primeira inoculação, com acréscimo de SBF, centrifugação e
aliquotamento e armazenagem do sobrenadante, teste de IFI para o precipitado celular e
posterior titulação viral, gerando o estoque viral de trabalho.
3.5 CONTAGEM DE CÉLULAS E VIABILIDADE CELULAR
A contagem de células em câmara de Neubauer foi uma prática frequente no
plaqueamento de células para titulação, teste de viabilidade celular e na infecção utilizando
MOI. Esta prática foi efetuada com o corante vital azul de tripan 0,4% (Sigma), para
diferenciação de células viáveis das não viáveis (Louis & Siegel, 2011).
Após o preparo e homogeneização da suspensão celular foi retirada uma alíquota para
a contagem das células, e a partir desta foi feita uma diluição final de 40 μL de suspensão
celular adicionados a 360 μL de meio de crescimento (1:10), sendo o meio de cultivo
específico para a célula a ser contada. Foram transferidos 300 μL desta diluição celular para
outro microtubo contendo 300 μL de solução de azul de tripan (1:1), onde, após
homogeneização foram transferidos um total de 20 μL, sendo 10 μL em cada borda da câmara

31
de Neubauer, por capilariedade, entre a lamínula e as canaletas em forma de V, então em
seguida foram contadas todas as células.
A contagem foi feita em microscópio ótico, em um aumento de 100 x (aumento de 10
x nas lentes objetivas e 10 x nas lentes oculares), e foram contadas as células presentes nas 8
zonas de canto da câmara. A câmara tem profundidade de 0,1 mm e estas zonas de canto são
de 1 mm x 1 mm de diâmetro e cada zona está dividida em 16 quadrados pequenos, formando
uma matriz de quatro por quatro. Cada zona de canto tem um volume de 0,1 mm³ ou 1 x 10-4
mL (Louis & Siegel, 2011). Gerando desta forma a seguinte fórmula para cálculo, onde Z é a
zona de canto:
Suspensão celular = Média de células x fatores de diluição x volume da zona de canto
Suspensão celular = (Z1+Z2+ Z3+Z4+Z5+Z6+Z7+Z8) x 2 x 10 x 104 células/mL
8
Para calcular o total de células dentro de uma garrafa, parâmetro utilizado para
estabelecer a MOI, foi multiplicada a quantidade de células/mL pela quantidade total de mL
de suspensão celular.
Para determinação do percentual de viabilidade de cada grupo, dividiu-se o número de
células viáveis contadas pelo total de células contadas (viáveis e não viáveis) de ambos os
grupos experimentais. Ou seja:
% de células viáveis = número de células viáveis contadas x 100
total de células contadas
3.6 MULTIPLICIDADE DE INFECÇÃO
A MOI é uma medida quantitativa utilizada para controlar as condições de infecção,
sendo simplesmente a razão do número médio de unidades formadoras de placas (PFU) por
célula utilizada na infecção. Como exemplo, em um ensaio onde a MOI utilizada será de 1,
significa que a inoculação será feita baseada em uma PFU por célula (Wagner et al., 2008).
Para utilização desta medida quantitativa de controle de infecção são necessários dois
parâmetros que são, a concentração de vírus no estoque original (PFU/mL), e a quantidade
total de células a serem infectadas.
Para o cálculo do volume do estoque viral (Vv) titulado necessário para se obter a
MOI desejada, será utilizada a seguinte fórmula:

32
Vv (mL) = MOI x o número total de células a serem infectadas
Título viral (PFU/mL)
3.7 IMUNOFLUORESCÊNCIA INDIRETA
A IFI foi utilizada para confirmar a infecção pelo VFA nas culturas inoculadas. Todos
os testes de IFI foram realizados com o protocolo utilizado na rotina do Laboratório de
Isolamento Viral em Cultura de Células do SAARB/IEC, que segue o protocolo descrito por
Gubler et al. (1984), com adaptações. Os testes, sempre realizados em duplicata, utilizaram
como anticorpos primários os anticorpos, policlonal para o Grupo B e, monoclonal para o
VFA, este último adquirido da Fundação Oswaldo Cruz (Bio-Manguinhos, Rio de Janeiro) e
preparados em uma diluição de 1:20 e 1:900, respectivamente, e como anticorpo secundário o
anticamundongo conjugado com isotiocianato de fluoresceína (FITC) (Cappel, EUA) na
diluição de 1:800.
A leitura das lâminas foi realizada em microscópio de fluorescência (Zeiss) com epi-
iluminação.
3.8 TITULAÇÃO VIRAL
O ECP em células hospedeiras pode ser observado após infecção por muitos vírus que
causam dano ou alterações celulares em decorrência de sua replicação. Mesmo se as células
não são mortas ou lisadas, modificações célulares de uma área devido a uma infecção por
vírus pode ser prontamente observada como uma placa ou foco de infecção (Wagner et al.,
2008).
Trabalhando com diluições e condições apropriadas, uma tal infecção localizada pode
ser o resultado da infecção com um único vírus biologicamente ativo. Uma partícula de vírus
capaz de iniciar uma infecção produtiva é denominado como uma PFU (Wagner et al., 2008).
O processo de formação da placa é iniciado com uma primeira célula infectada que
libera muitos vírus. Se os vírus são mantidos em uma ampla difusão, ocorrerá a infecção na
vizinhança das células infectadas originais que irá infectar as células vizinhas. Este processo
repetido inúmeras vezes, contanto que a interação de células e de vírus é mantida localizada
sob o uso de um meio de cultura celular viscoso, fazem desta área de citopatologia o
resultado de uma única infecção de uma única PFU. As placas resultantes podem ser
facilmente observadas e contadas alguns dias após a infecção (Wagner et al., 2008).

33
3.8.1 Titulação Viral em Células VERO
O cultivo celular VERO (ATCC: CCL-81) é propagado em meio 199 (Gibco)
acrescido de 2,2 g/L de bicarbonato de sódio e 1 mL/L de antibiótico Gentamicina (Nova
Farma, Brasil) e 5% de SBF para o meio de crescimento.
De forma geral, a subcultura de células VERO é semelhante com a descrita para
HepG2 na subsecção 3.2.2, mudando apenas o meio de cultivo utilizado.
Para a titulação do VFA foram utilizadas monocamadas de células VERO preparadas
com concentração celular de 2 x 105 células/mL, confluindo em monocamadas em dois dias
após a sua passagem. A titulação foi realizada em placas de poliestireno (TPP, Suiça) de 6
poços, plaqueadas com 3 mL de meio de crescimento contendo a quantidade de células
citadas.
A técnica utilizada para titulação biológica da cepa em estudo é uma adaptação da
descrita por Malewicz & Jenkin (1979), os quais utilizaram um meio semi-sólido (overlay)
sem SBF.
As placas de poliestireno com monocamada celular confluente tiveram o seu meio de
crescimento retirado e a monocamada foi lavada uma vez com D-PBS. Foram preparadas
diluições em série de 10-1
até 10-8
dos sobrenadantes coletados a serem titulados, utilizando-se
como diluente o meio 199 incompleto com a mesma formulação descrita anteriormente e sem
a adição de SBF.
Foi realizada a inoculação em duplicata de 0,1 mL das diluições a partir de 10-4
em
seus respectivos poços, e no controle negativo foi adicionado apenas o meio de cultura sem
inóculo viral. Logo após, as placas foram incubadas em estufa a 37°C com 5% de CO2 por
uma hora, com agitação a cada 15 minutos para auxiliar na adsorção viral.
Após o período de adsorção viral, as monocamadas foram novamente lavadas com D-
PBS para posterior adição de 3 mL do meio overlay. O meio overlay consiste em meio 199
1X (2,2 g/L de bicarbonato de sódio) e carboximetilcelulose (CMC) de média viscosidade
(Sigma) na concentração final de 1,6%. Após isso, as placas foram incubadas em estufa a
37°C com 5% de CO2 por sete dias.
Após os setes dias de incubação, as monocamadas de células foram fixadas em formol
a 10% durante no mínimo 4 horas, lavadas e posteriormente coradas com cristal violeta a
0,1% durante no mínimo 4 horas. O título viral foi determinado em PFU/mL.

34
As placas formadas nas monocamadas de células foram contadas na primeira diluição
em que se observaram individualizadas. Por conseguinte, foi feita a média de placas e os
números de placas foram multiplicados pelos seus fatores de diluição, e divididos pelo
volume em mL utilizado como inóculo, como apresentado na fórmula a seguir:
Título viral (PFU/mL) = Média de placas x fator de diluição
Volume do inóculo (mL)
3.9 INFECÇÃO DAS CÉLULAS HEPG2 PELO VFA
Neste estudo foram utilizadas 30 garrafas com 25 cm² de área de crescimento celular
contendo monocamadas com confluência de aproximadamente 80% de células HepG2
divididas em dois grupos, grupo controle (sem inóculo viral) e o grupo infectado (inoculados
com VFA). Diariamente foram realizadas extrações de proteínas totais de seis garrafas, três de
cada grupo, durante o período de 1 a 5 dpi. Em paralelo a utilização das garrafas, foram
utilizadas 5 placas (uma pra cada dpi) de seis poços (9,6 cm² de área de crescimento, sendo,
desta forma, um poço equivalente a uma garrafa) mantidas nas mesmas condições que as
garrafas para fazer o teste de viabilidade celular e IFI. Também foram utilizadas garrafas e
uma placa extra, mantidos nas mesmas condições de crescimento celular, as quais foram
tripsinizadas para contagem total de células, e servindo de subsidio para em seguida realizar a
infecção como descrito a seguir.
O meio de crescimento foi removido, e as monocamadas foram lavadas duas vezes
com D-PBS, e inoculadas com MOI 0,01. Para isso, as amostras virais foram diluídas em
meio DMEM sem SBF, padronizando-se o volume final para 3 mL/garrafa e 1 mL/poço. Nos
controles negativos (cultivos não infectados) foram adicionados 3 mL de meio sem inóculo
em cada garrafa ou 1 mL por poço. As garrafas e placas foram incubadas por 1 hora em estufa
37°C a 5% de CO2 e homogeneizadas cuidadosamente a cada 15 minutos para o espalhamento
da suspensão viral na monocamada.
Após a adsorção viral, os inóculos virais foram removidos e as monocamadas lavadas
uma vez com D-PBS. Foi adicionado um volume de 10 mL (garrafa) e 3 mL (poço) de meio
DMEM de manutenção (3% de SBF). Seguindo-se, então, a incubação em estufa 37°C a 5%
CO2 por até 5 dias. As células foram observadas nesse intervalo de tempo objetivando a
pesquisa de provável ECP em microscópio ótico invertido (Zeiss).

35
3.10 CINÉTICA VIRAL
Para a curva da cinética viral, durante o preparo das garrafas para extração de
proteínas, foram coletados os sobrenadantes das garrafas do grupo infectado. Em sequência da
adição de 10% de SBF, e foram centrifugados a 2000 RPM, alíquotados e estocados em
freezers a -70°C, para posterior titulação viral pelo método descrito na subsecção 3.8. Os
sobrenadantes do grupo controle também foram coletados para posterior uso como controle
negativo no teste de titulação.
3.11 ANÁLISE PROTEÔMICA
3.11.1 Extração de Proteínas das Células Cultivadas
As extrações de proteínas foram realizadas em ambos os grupos, diariamente, entre os
dias 1 a 5 após a inoculação, sendo os sobrenadantes celulares coletados conforme descrito na
subsecção 3.10.
Posteriormente, as monocamadas de ambos os grupos foram lavadas duas vezes com
10 mL de D-PBS gelado. Foi, então, adicionado 1 mL de uma solução de tampão de extração
(7 M de Uréia, 2 M de Tiuréia e 4% de CHAPS (3-(cholamidopropyl)dimethylammonio-1-
propane sulfonate)) com 1 μL de inibidor de protease Protease Inhibitor Mix (GE Helthcare)
sobre as monocamadas, agitando levemente para que esta solução cubra totalmente a
monocamada celular, submetendo, então, as garrafas a um banho de gelo por 5 minutos.
Após o banho de gelo, as monocamadas foram rapidamente coletadas com pipeta
sorológica. O lisado celular foi transferido para microtubos do tipo eppendorf, e mantidos em
gelo/refrigeração durante a ultrassonicação, realizada por 30 segundos divididos em dois
períodos de 15 segundos cada, em uma frequência de onda de 60% do ultrassonicador. Após
isto, permanecendo em banho de gelo/refrigeração durante todo o período de clarificação e
quantificação.
3.11.2 Clarificação do lisado celular
Após a extração, os tubos com os lisados celulares permaneceram em banho de gelo, e
foram centrifugados a 8000 xg por 10 minutos a 4°C para a precipitação dos “debris”
celulares.

36
Os sobrenadantes foram cuidadosamente transferidos para tubos tipo eppendorf e
imediatamente quantificados. Durante esta etapa, os tubos permaneceram sob refrigeração
constante, isto é, em um recipiente com gelo.
3.11.3 Quantificação de Proteínas Totais
A quantificação de proteínas totais foi realizada com auxílio do 2-D Quant kit (GE
Healthcare), de acordo com as recomendações do fabricante. Após a quantificação, as
amostras foram aliquotadas e então armazenadas em freezers a -80°C até posterior uso.
3.11.4 Eletroforese Bidimensional
Os extratos proteicos foram aplicados em uma quantia de 125 µg de proteínas em
IPG-strip, 7 cm, pH 3-11NL (GE Healthcare) pelo método de re-hidratação em gel por no
mínimo 16 horas para a realização da primeira dimensão. A focalização isoelétrica foi
realizada na unidade IPGphor III (GE Healthcare) com o seguinte perfil: 100 V por 30
minutos, 300 V por 200 Vh, gradualmente até 1000 V por 300 Vh, gradualmente até 5000 V
por 4000 Vh, permanecendo em 5000 V por 1500 Vh, com uma focagem total de 6 kV. Antes
da segunda dimensão, as strips foram passadas por um processo de redução em tampão de
equilíbrio (6 M uréia, 0,075 M TrisHCl pH 8,8; glicerol 29%; SDS 2%; azul de bromofenol
0,02%) contendo 10 mg/mL de DTT (ditiotreitol) durante 15 minutos e em seguida,
alquilados por 15 minutos na mesma solução de equilíbrio mencionada anteriormente,
contendo iodacetamida (25 mg/mL). Na segunda dimensão os spots protéicos foram
separados em gel de SDS-poliacrilamida 12,5% com auxilio do sistema MiniVE (GE
Healthcare), em uma voltagem constante de 100V por aproximadamente 2 horas.
Os spots de proteínas foram visualizados após coloração em solução coloidal de
Comassie Blue G 250, overnight sob agitação, seguido da captura de imagem através do
ImageScanner III (GE Healthcare), software Labscan (GE Healthcare), no qual a imagem é
importada em formato TIFF e MEL. Após aquisição das imagens do gel estas foram
analisadas e os mapas diferenciais das proteínas gerados usando o programa ImageMaster 2D
Platinum 7.0 (GE Healthcare).

37
3.12 ANÁLISE ESTATÍSTICA
Para a análise estatística dos dados obtidos nos testes de viabilidade celular, foi
utilizado o programa BioEstat 5.0 (Instituto Mamirauá), os dados foram submetidos ao teste
de análise de variância (ANOVA) com um critério seguido do teste Tukey, ambos assumindo
valor de p < 0,05 para consideração de diferença estatística significante. Os resultados de
ANOVA obtidos para os valores dos spots diferenciais foram obtidos automaticamente pelo
programa ImageMaster 2D Platinum 7.
As tabelas e figuras com representações gráficas foram geradas no Microsoft Office
Excel 2007 (Microsoft).

38
4. RESULTADOS
4.1 TÍTULO VIRAL, EFEITO CITOPÁTICO E TESTE DE VIABILIDADE CELULAR
Com o intuito de fazer uma observação mais extensa do comportamento e progressão
da infecção viral da cepa selvagem Be H111 do VFA em células HepG2, optou-se por uma
inoculação contendo uma MOI de 0,01. Desta forma, foi obtida uma curva de cinética viral
(Figura 3), em que os maiores títulos virais observados foram a partir do 3º dpi. O ECP foi
caracterizado pela presença de células não aderidas/mortas na monocamada celular. No grupo
infectado, foram observados ECP bem caracterizados no quarto e quinto dpi em comparação
com o grupo controle, que apresentou leves características de morte celular no quinto dia,
semelhante ao terceiro dia do grupo infectado (Figura 4 A e B). Estes resultados de ECP
acompanham as características da curva viral, mostrando que a alta taxa de replicação viral
contribui para a formação do ECP em cultivo celular.
Figura 4 - Curva de cinética da infecção pelo VFA em células HepG2. Título viral de
acordo com os dias pós-infecção. Os títulos virais em PFU/mL estão indicados em escala
logarítmica de base 10 (log10).
Também foram observados com os resultados obtidos da IFI o aumento crescente da
progressão da infecção viral através da expressão de antígenos virais detectados tanto por
anticorpos policlonais (Figura 4 C) quanto por monoclonais (dados não mostrados).
4,00
4,50
5,00
5,50
6,00
6,50
7,00
0 1 2 3 4 5 6
Título viral (Log
10)
Dias pós-infecção

39
Figura 5 - Comportamento da infecção pelo VFA, cepa selvagem Be H111, em cultivo celular HepG2. A e B, acompanhamento do ECP durante o período de
cinco dias (aumento de 100x). C, resultado dos testes de IFI durante o período de cinco dias com anticorpo policlonal para o grupo B dos arbovírus (aumento de 100x).

40
Os resultados de ECP foram reproduzíveis em todos os experimentos realizados.
Para quantificar este achado foi realizado o experimento de viabilidade celular através da
contagem celular com Azul de Tripan 0,4%.
Nos ensaios para verificar a viabilidade celular relacionada à infecção viral, os
valores das médias das contagens de células viáveis foram comparados entre os dias por
grupo e também entre os grupos por dia. Os resultados obtidos nos testes de viabilidade
celular estão representados na Figura 5. Na comparação entre os dias por grupo, o grupo
controle não apresentou diferença estatística (p > 0,05), demonstrando que a condição de
cultivo em meio de manutenção (3% SBF) não afeta estatisticamente a viabilidade celular.
Já no grupo infectado os dias 1 e 2 tiveram diferenças significativas (p < 0,05 e p < 0,01)
quando comparados com os dias 4 e 5, enquanto no dia 3 apenas foi observada diferença
significativa (p < 0,05) quando comparado ao dia 5, demonstrando que a atividade de
replicação viral está envolvida na diminuição da viabilidade celular. Entre as comparações
dos grupos por dia, apenas os dias 4 e 5 obtiveram diferença estatística (p < 0,05) entre os
grupos, demonstrando quantitativamente o já notado ECP.
Figura 6 – Resultados de viabilidade celular obtidos para infecção de células HepG2
pelo VFA. Os sinais e letras estão incluídos para delimitar os grupos e dias que tiveram
diferenças estatísticas no teste de ANOVA um critério seguido de Tukey. Na comparação
entre grupos por dias, “*” representa p < 0,05. Na comparação entre os dias por grupo, “a”,
*
*
a;d b;e
c
a;b
c;d;e
40,00
50,00
60,00
70,00
80,00
90,00
100,00
1 2 3 4 5
Via
bil
idad
e c
elu
lar
(%)
Dias pós-infecção
Controle Infectado

41
“b” e “c” representam diferença onde p < 0,05 e “d” e “e” diferença onde o valor de p <
0,01.
4.2 ANÁLISE PROTEÔMICA
Na tentativa de definir o melhor dpi para fazer a análise proteômica diferencial da
infecção viral, foram levados em consideração todos os dados descritos anteriormente.
Inicialmente optou-se por fazer a análise com o 3º dpi, devido ao alto título viral obtido, a
presença de expressão de antígenos virais detectados pela IFI, a ausência de ECP entre o
grupo controle e infectado, levando em consideração que a infecção viral pelo VFA induz o
mecanismo de morte celular por apoptose (Mariannaeu et al., 1998; Xiao et al., 2001), e
entre as características bioquímicas da apoptose há a ativação da cascata de proteases
(Elmore, 2007), o que evidentemente atrapalharia os objetivos do trabalho. Apesar dos
critérios adotados, a análise dos géis pelo programa ImageMaster 2D Platinum 7 não
identificou nenhum spot diferencial.
Em uma reavaliação dos dados obtidos, considerando principalmente o aumento de
células expressando os antígenos virais no teste de IFI e que nas comparações estatísticas de
viabilidade celular entre os dias por grupo não houve diferença entre os dias 3 e 4, optou-se
pela tentativa de avaliação do 4º dpi.
Para a detecção dos spots proteicos nos géis corados por Comassie foram utilizados
os parâmetros de Smooth 2, saliência 100 e área mínima 10. Desta forma, foram detectados
no mínimo 521 e no máximo 612 spots nos géis de ambos os grupos, e como resultado do
matching (pareamento dos géis), foram encontrados uma média de 85% de spots pareados.
Os dados resultantes da detecção e processo de matching dos géis estão apresentados na
tabela 1.
Tabela 1 – Características dos géis em relação ao número spots detectados e pareados.
Amostra Grupo Spots Matches Matches (%)
C1D4 Controle 521 450 86
C2D4 Controle 612 463 76
C3D4 Controle 559 505 90
H1D4 Infectado 545 453 83
H2D4 Infectado 589 476 81
H3D4 Infectado 547 512 94
Para a análise de seleção dos spots diferenciais foram levados em consideração a
presença do mesmo spot nos três géis do mesmo grupo, valor da análise estatística por
ANOVA com p < 0,05 e, quando presente nos dois grupos, tiver uma diferença de expressão

42
de pelo menos 1,5 vezes entre os grupos. Seguindo estas considerações foram obtidos 4
spots proteicos diferenciais (Figura 6 e 7), onde dois foram caracterizados como spots
diferenciais (por estarem presente em apenas um dos grupos) e dois como expressamente
diferenciais (por estarem presentes nos dois grupos e terem diferença de expressão > que 1,5
vezes) (Tabela 2) podendo serem visualizados em representação gráfica tridimensional nas
figuras 8, 9, 10 e 11.
Tabela 2 – Spots proteicos diferencialmente expressos.
Legenda: Match ID é o número de identificação do pareamento feito automaticamente pelo
software ImageMaster 2D Platinum 7. pI é o ponto isoelétrico, o pH encontrado da proteína
separada. PM, peso molecular da proteína encontrada. “+” representa a presença do spot
proteico no grupo, enquanto que “–” é a ausência e “++” é a maior expressão da proteína no
grupo.
Nº do spot Match
ID pI PM
Diferença em
Vezes Controle Infectado
ANOVA
(valor de p)
1 224 5,70 45738 2,86362 + ++ 0,00011383
2 318 6,97 67299 2,26962 + ++ 0,0113139
3 482 7,16 67299
– + 0,00067245
4 484 6,97 70341 – + 0,00423055

43
Figura 7 - Gel bidimensional de proteínas totais de células HepG2 infectadas com o
vírus da Febre Amarela, cepa selvagem Be H111, e corado com solução coloidal de
Coomassie. Os spots identificados com o número 1 e 2 foram expressamente diferenciais
e os spots 3 e 4 diferenciais. Os valores de pH e peso molecular dos spots estão na Tabela
2.

44
Figura 8 - Gel bidimensional de proteínas totais de células HepG2 do grupo controle
corado com solução coloidal de Coomassie. Os spots identificados com o número 1 e 2
foram os expressamente diferenciais. Os valores de pH e peso molecular dos spots estão na
Tabela 2.

45
Figura 9 - Representação tridimensional do spot número 1, match ID 224. Do lado
esquerdo estão os géis do grupo controle, na direita os infectados. A linha verde faz a
marcação do spot selecionado, a vermelha são outros spots detectados.
Grupo Controle Grupo Infectado
46
Figura 10 - Representação tridimensional do spot número 2, match ID 318. Do lado
esquerdo estão os géis do grupo controle, na direita os infectados. A linha verde faz a
marcação do spot selecionado, a vermelha são outros spots detectados
Grupo Controle Grupo Infectado

47
Figura 11 - Representação tridimensional do spot número 3, match ID 482. Do lado
esquerdo estão os géis do grupo controle, na direita os infectados. A linha verde faz a
marcação do spot selecionado apenas no grupo infectado devido à ausência deste spot no
grupo controle, e a vermelha são outros spots detectados.
Grupo Controle Grupo Infectado

48
Figura 12 - Representação tridimensional do spot número 4, match ID 484. Do lado
esquerdo estão os géis do grupo controle, na direita os infectados. A linha verde faz a
marcação do spot selecionado apenas no grupo infectado devido à ausência deste spot no
grupo controle, e a vermelha são outros spots detectados.
Grupo Controle Grupo Infectado

49
5 DISCUSSÃO
5.1 TÍTULO VIRAL, EFEITO CITOPÁTICO E TESTE DE VIABILIDADE CELULAR
A caracterização da infecção viral realizada com MOI 0,01 para um
acompanhamento lento e progressivo da infecção viral mostrou resultados satisfatórios com
a progressão evidente e bem definida. Marianneau et al. (1998) demonstraram em apenas
algumas células HepG2 infectadas o aparecimento de ECP e apoptose no 3º dpi, o que é
visualizado também nos resultados deste trabalho, onde há pouco aparecimento de ECP que
é visualizado pelo teste de viabilidade celular (provável apoptose) com uma queda de 14%,
o que não foi significante estatisticamente, no 3º dpi. Esta semelhança foi encontrada apesar
dos MOIs diferentes utilizados, pois Marianneau et al.(1998) utilizaram MOIs de 1, 10 e 20,
e neste trabalho o único MOI utilizado foi o de 0,01, o que sugere um padrão
comportamental da replicação viral na indução de apoptose celular.
A técnica para titulação viral sem SBF de Malewicz & Jenkin (1979) utilizada para
titulação do Vírus da Dengue, foi adaptada para células VERO (CCL-81) e apresentou
formação nítida de placas, apesar de não produzirem resultados antes de 7 dias de incubação
como descrito pelos autores. Os títulos virais obtidos com o MOI de 0,01 se apresentaram
diferentes dos alcançados por Mariannaeu et al. (1998), devido as diferenças de MOIs e
talvez ao método de titulação. O maior título viral visto foi de 9 x 105 pfu/mL com MOI 20
após 2 dpi em um método utilizando células AP61 (Mariannaeu et al., 1998), enquanto que
neste trabalho no mesmo dpi o título foi de 1 x 105 pfu/mL (Figura 3) utilizando células
VERO.
5.2 ANÁLISE PROTEÔMICA
Rachas et al. (2014) reafirmam a necessidade de métodos de diagnósticos precoces
para respostas de controle mais rápidas durante a ocorrência de possíveis surtos de FA. Já no
relato de Okamoto et al. (2010), a detecção e os esforços de diagnóstico de patógenos
tipicamente utilizam de métodos de identificação patógeno-específica que são baseados em
técnicas imunológicas ou de amplificação de genes, o que também é visualizado na grande
área da arbovirologia, tanto no campo da pesquisa científica quanto em vigilância (Lanciotti,
2003; Parida et al., 2004, 2005, 2006, 2007; Peyrefitte et al., 2008; Toriniwa & Komiya,
2006) incluindo estudos específicos com o VFA (Drosten et al., 2002; Bae et al., 2003;
Weidmann et al. 2010; Nunes et al., 2011; Dash et al., 2012; Domingo et al., 2012; Kwallah
et al., 2013; Escadafal et al., 2014).

50
Desta forma, as ferramentas proteômicas são uma nova metodologia para a detecção
de componentes das interações proteicas tanto quanto dos processos fisiopatológicos que
ocorrem durante a infecção viral (Maxwell & Frappier, 2007; Zheng et al., 2008). A
ausência de estudos proteômicos com o VFA aumenta a importância dos resultados deste
trabalho, e dificulta a comparação dos seus achados. No presente estudo foram visualizados
4 spots proteicos diferenciais, sendo que em outros estudos com diferentes vírus conseguiu-
se identificar através do mesmo método de separação por gel de eletroforese bidimensional
(2-DE) (O’Farrell, 1975) de 17 a 102 spots diferenciais (Tabela 3). Estas diferenças podem
ser explicadas devido aos vírus estudados, células utilizadas como hospedeiras, tipo de
extração das proteínas, tratamento e manipulação da amostra, tamanho e intervalo de pH das
strips e aos métodos de detecção de spots proteicos.
De fato, diferentes vírus infectam diferentes tipos celulares devido as afinidades de
interações entre receptores celulares e proteínas virais, além de induzirem diferentes padrões
proteicos como resposta à infecção. Quanto ao processo de extração de proteínas celulares,
foi observado que diferentes tratamentos (uso de ultrassonicador ou congelamento rápido) e
tampões utilizados para lisar as células e solubilizar as proteínas, geram diferentes
quantidades de proteínas disponíveis na amostra, afetando também os padrões apresentados,
como visto durante as experimentações de padronização deste trabalho (dados não
mostrados). Sobre os tratamentos e manipulação da amostra, há diferentes protocolos
caseiros de precipitação (Englard & Seifter, 1990) e kits comerciais disponíveis para a
eliminação de impurezas e interferentes (excesso de sais, material genético e lipídeos) das
amostras, estes prometem melhorar a focalização isoelétrica das proteínas, mas podem
ocasionar perdas proteicas durante a sua aplicação.
O tamanho e intervalo de pH da strip trazem o melhoramento da resolução dos spots
proteicos separados na segunda dimensão, quanto maior o tamanho da strip e menor seu
intervalo de pH mais separados os spots ficarão entre si, evitando sobreposições de pontos
isoelétricos das proteínas devido a maior cobertura de faixa de pH alcançada. Na tabela 3 se
torna evidente que quanto maior o tamanho da strip maior a quantidade de spots detectados
maior o número de spots diferenciais detectados. Os estudos que utilizaram géis com
tamanho igual ou superior a 17 cm (Sun et al., 2008; Zheng et al., 2008; Zhang et al., 2009)
obtiveram detecção superior a 1300 spots por gel e os maiores números de spots diferenciais
detectados. Porém, os métodos de detecção tem grande importância para a revelação dos
spots proteicos, como visto na detecção de até 200 spots a mais (Pattanakitsakul et al., 2007)
em comparação com os resultados obtidos neste trabalho.

51
Tabela 3 – Síntese dos resultados encontrados em trabalhos utilizando o método 2-DE comparativo em infecções experimentais com diferentes vírus.
Referência Vírus
família:gênero
Célula
hospedeira
Tamanho
da strip
Faixa
de pH Método de detecção
Spots totais
detectados
Spots
diferenciais
Spots
identificados
Horas pós infecção
analisadas
Alfonso et al., 2004 Vírus da Peste Suína Africana
Asfarviridae
Vero
(CCL-81)
17 cm
3-10
Prata compatível com
EM
não relata
90
68 10 e 24
Pattanakitsakul et al., 2007
Vírus Dengue sorotipo2
Flaviviridae:Flavivirus
HepG2
7 cm
3-10
NL
SYPRO Ruby
até 800
17
17 24
Sun et al., 2008
Vírus Clássico da Peste Suína
Flaviviridae:Pestivirus
PK-15
17 cm
3-10
Prata/Coomassie G250
mais de 1300
35
21 48
Zheng et al., 2008
Vírus da Doença de Gumboro
Birnaviridae
Fibroblasto
de embrião
de galinha
24 cm
5-8
Prata compatível com
EM
1400-1650
102
81
12, 48 e
96
Zhang et al., 2009
Circovírus Suíno tipo 2 Circoviridae
PK-15
24 cm
5-8 L
Prata compatível com EM
1450-1600
61
37 12, 48 e 96
Presente estudo
Vírus da Febre Amarela
Flaviviridae:Flavivirus
HepG2
7 cm
3-11
NL
Coomassie G250
521-612
4
0 72 e 96
cm centímetros;
EM Espectrometria de massas;
NL Não linear;
L Linear;

52
O que se espera das técnicas de detecção de spots proteicos é que: sejam sensíveis
suficientes para poucas cópias de proteínas; permitam análises quantitativas; tenham grande
linearidade; tenham um grande alcance dinâmico; sejam compatíveis com EM; não sejam
tóxicos; que causem baixo impacto ambiental; e de custo acessível. Mas infelizmente
nenhuma das técnicas disponíveis possui todas estas características juntas (Westermeier &
Naven, 2002).
As colorações com Coomassie ganharam a popularidade devido a seu baixo custo,
praticidade de uso e ótima compatibilidade com os métodos de caracterização proteica como
através da EM, apesar das limitações de pouca sensibilidade e alcance dinâmico. Porém, a
sensibilidade varia para os diferentes tipos de coloração por Coomassie (Neuhoff et al.,1988).
O Coomassie coloidal tem uma sensibilidade de 8-10 ng de proteínas enquanto que nos outros
tipos desta coloração a sensibilidade é de 30-100 ng de proteínas (Patton, 2002).
A coloração por prata tem maior sensibilidade, chegando a 0,5 ng de proteínas
(Heukeshoven & Dernick, 1985), mas tem um alcance dinâmico inferior ao do Coomassie,
além de ser um tanto complexo, com vários passos e processos que devem ser parados em
tempo arbitrário para evitar o excesso de reações, e a modificação da técnica para utilização
compatível com a EM diminui a sua sensibilidade (Patton, 2002).
Existem alguns corantes fluorescentes disponíveis, eles possuem um grande alcance
dinâmico e por isso tem boa empregabilidade para quantificação de proteínas (Westermeier &
Naven, 2002). A literatura relata que o SYPRO Ruby apresenta melhor sensibilidade que as
colorações por prata (Lopez et al., 2000; Berggren et al., 2000). Além disto, o SYPRO Ruby é
um método de coloração de fácil aplicação (Patton, 2002) e tem melhor reprodutibilidade na
quantificação de proteínas de baixa intensidade nos géis de 2-DE e geram grande
convergência da sequência aminoacídica na identificação através do perfilamento de massas
dos pepitídeos destas proteínas (Lopez et al., 2000), demonstrando assim que este corante é
superior a coloração por prata para aplicações em proteômica (Patton, 2002).
Hoving et al. (2000) relataram as vantagens da utilização de géis com ultrazoom
(vários géis com diferentes faixas reduzidas de pH das strips para caracterização de uma
amostra) para o aumento da resolução dos padrões proteicos apresentados em géis 2-DE
ressaltando também as desvantagens no aumento da quantidade de géis e de amostras
utilizados, dando importância aos métodos de detecção e descrevendo o SYPRO Ruby como o
possível melhor corante para uso em estudos proteômicos.
A 2-DE é uma técnica poderosa que oferece, por uma perspectiva, alta eficiência para
separação de misturas proteicas complexas, mas, em contrapartida, é caracterizado por sua

53
baixa reprodutibilidade (Robotti et al., 2011) devido as variações de gel para gel, como a
localização dos spots e o baixo alcance dinâmico das técnicas de coloração normalmente
utilizadas (Ye et al., 2010). É provável que estas variações intrínsecas de gel para gel
mascaram as diferenças biológicas entre as amostras e comprometem as comparações
qualitativas em nível de expressão de proteínas. Estas variações parecem surgir da absorção
problemática das proteínas pelas strips de focalização isoelétrica, transferência incompleta de
proteínas da primeira para a segunda dimensão e locais não homogênios na composição do
gel, campo de força ou gradiente de pH (Bergh & Arckens, 2004). Estas limitações podem ser
superadas quando deixado de lado o clichê ‘um gel, uma amostra’, e for implementado uma
abordagem multiplex, utilizando um único gel para separação de múltiplas amostras de
proteínas, seguido pela visualização independente destas amostras, como acontece na técnica
de gel de eletroforese bidimensional diferencial (2D-DIGE) (Bergh & Arckens, 2004; Ye et
al., 2010).
No estudo de Ye et al. (2010), onde o foco era a identificação de proteínas virais do
Vírus Vaccinia, a técnica de 2D-DIGE mostrou ter maior sensibilidade, reprodutibilidade e
eficiência que o 2-DE comparativo, apresentando 5 proteínas virais por 2D-DIGE e apenas 1
proteína viral utilizando o 2-DE; observou-se também mais proteínas diferenciais, mas como
o estudo tinha foco específico as outras proteínas não foram relatadas.
Nos estudos com vírus da família Flaviviridae, empregando a técnica 2D-DIGE,
Pastorino et al. (2009) utilizaram strips de 18 cm em combinação com dois espectros de pH
diferentes e conseguiram fazer o match de 1950 spots proteicos na faixa de pH 4-7 e 1674 na
faixa de pH 6-11, alcançando a quantidade de 127 spots diferenciais, dos quais 93 foram
completamente identificados para elucidar a infecção do VNO em células VERO. Enquanto
que Li et al. (2010), utilizando strips de um única faixa de pH (3-10) com tamanho de 24 cm
conseguiu um total de aproximadamente 1400 spots proteicos, sendo 15 diferenciais e 8
proteínas completamente caracterizadas durante a infecção de cultura de células endoteliais
primárias de suíno infectadas experimentalmente pelo Vírus clássico da Peste Suína. Em
comparação com o estudo de Li et al., (2010) a diferença na quantidade de spots detectados é
de quase três vezes menor nos resultados obtidos no presente estudo, esta diferença
acompanha a mesma proporção em relação ao número de spots diferenciais, mostrando certa
linearidade dos achados. Comparando com as observações de Pastorino et al., (2009), a
diferença é muito maior devido a utilização de menores faixas de pH em géis de grandes
dimensões, demonstrando novamente que o emprego destes materiais amplia o número de
resultados para análises proteômicas mais profundas.

54
Os resultados adquiridos com a técnica utilizada mostraram novas informações quanto
às alterações no perfil proteico e biologia da célula hospedeira induzidas pela infecção do
VFA. Contudo o emprego de outras técnicas (tanto de detecção de spots como também a
utilização de géis de ultrazoom) podem aumentar a quantidade e relevância destas
informações. Desta forma, é importante enfatizar que diferentes técnicas utilizadas em estudos
proteômicos, detectam diferentemente as proteínas (Westermeier & Naven, 2002), como
exemplo, a coloração por prata que normalmente produz padrões diferentes dos padrões
adquiridos com Coomassie e outros procedimentos de coloração como com SYPRO Ruby
(Görg et al., 2000). Logo, os resultados obtidos no presente trabalho sugerem a necessidade
de maiores explorações nesta temática, ampliando os conhecimentos sobre o VFA e também
trazem novas perspectivas para o aprofundamento nos estudos de biomarcadores celulares
para diagnóstico e tratamento da FA.

55
6 CONCLUSÕES
Observou-se efeito citopático induzido pelo VFA no 4º dpi, identificado pela
microscopia óptica e quantificado através do teste de viabilidade celular.
A cinética viral adquirida com MOI de 0,01 apresentou o maior título no 5º dpi,
apresentando discreta variação de valores a partir do 3º dpi.
O melhor período para análise das alterações no proteoma celular foi o 3º dpi, no
entanto, em comparação ao grupo controle não foram observadas diferenças de expressão
proteica pela técnica de coloração com Coomassie coloidal.
O período do 4º dpi, apresentou 4 spots diferenciais pela técnica de coloração com
Coomassie coloidal;

56
REFERÊNCIAS
ALFONSO, P., RIVERA, J., HERNÁEZ, B., ALONSO, C., ESCRIBANO, J. M.
Identification of cellular proteins modified in response to African swine fever virus
infection by proteomics. Proteomics 4: 2037-2046, 2004.
AMERICAN TYPE CELL COLLECTION (ATCC). Cell lines and Hybridomas, Estados
Unidos, Disponível em: <http://www.atcc.org/>, acesso em março de 2013.
BAE, H. G., NITSCHE, A., TEICHMANN, A., BIEL, S. S., NIEDRIG, M. Detection of
yellow fever virus: a comparison of quantitative real-time PCR and plaque assay.
Journal of Virological Methods 110 (2): 185-191, 2003.
BARNETT, E. D. Yellow fever: Epidemiology and prevention. Clinical Infectious Diseases
44: 850–856, 2007.
BARRETT, A. D. T., HIGGS, S. Yellow fever: A disease that has yet to be conquered.
Annual Review of Entomology 52: 209–229, 2007.
BERGGREN, K., CHERNOKALSKAYA, E., STEINBERG, T. H., KEMPER, C., LOPEZ,
M. F., DIWU, Z., HAUGLAND, R. P., PATTON, W. F. Background-free, high
sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-
polyacrylamide gels using a luminescent ruthenium complex. Electrophoresis 21: 2509-
2521, 2000.
BERGH, G. V. D. & ARCKENS, L. Fluorescent two-dimensional difference gel
electrophoresis unveils the potential of gel-based proteomics. Current Opinion in
Biotechnology 15: 38-43, 2004.
BRYANT, J. E., VASCONCELOS, P. F. C., RIJNBRAND, R. C. A., MUTEBI, J. P.,
HIGGS, S., BARRETT, A. D. T. Size Heterogeneity in the 3’ Noncoding Region of
South American Isolates of Yellow Fever Virus. Journal of virology 79 (6):3807–3821,
2005.
DASH, P. K., BOUTONNIER, A., PRINA, E., SHARMA, S., REITER, P. Development of a
SYBR green I based RT-PCR assay for yellow fever virus: application in assessment of
YFV infection in Aedes aegypti. Virology Journal 9: 27, 2012.
DOMINGO, C., PATEL, P., YILLAH, J., WEIDMANN, M., MENDEZ, J. A. NAKOUNÉ,
E. R., NIEDRIG, M. Advanced yellow fever virus genome detection in point-of-care
facilities and reference laboratories. Journal of Clinical Microbiology 50 (12): 4054–
4060, 2012.
DROSTEN, C., GÖTTIG, S., SCHILLING, S., ASPER, M., PANNING, M., SCHMITZ, H.,
GÜNTHER, S. Rapid detection and quantification of RNA of ebola and marburg viruses,

57
lassa virus, crimean-congo hemorrhagic fever virus, rift valley fever virus, dengue virus,
and yellow fever virus by real-time reverse transcription-PCR. Journal of Clinical
Microbiology 40 (7): 2323–2330, 2002.
DULBECCO, R., VOGT, M. Plaque formation and isolation of pure lines with poliomyelitis
viruses. The Journal of experimental medicine 99 (2): 167-182, 1954.
ELMORE, S. Apoptosis: a review of programmed cell death. Toxicologic Pathology 35 (4):
495-516, 2007.
ENGLARD, S. & SEIFTER, S. Precipitation techniques. Methods in Enzymology 182:
285–300, 1990.
ESCADAFAL, C.; FAYE, O., SALL, A. A., FAYE, O., WEIDMANN, M., STROHMEIER,
O., VON STETTEN, F., DREXLER, J., EBERHARD, M., NIEDRIG, M., PATEL, P.
Rapid molecular assays for the detection of yellow fever virus in low-resource settings.
PLoS Neglected Tropical Diseases 8 (3): e2730, 2014.
FERNANDEZ-GARCIA, M. D., MAZZON, M., JACOBS, M., AMARA, A. Pathogenesis of
Flavivirus infections: Using and abusing the host cell. Cell Host & Microbe 5:318-328,
2009.
FRESHNEY, R. I. Culture of animal cells: a manual of basic technique and specialized
applications. Wiley-BLACKWELL: Nova Jersey, EUA, 6 ed. ISBN 978-0-470-52812-9,
2010, p 732.
GARDNER, C. G., RYMAN, K. D. Yellow fever: A reemerging threat. Clinical Laboratory
Medicine 30: 237-260, 2010.
GUBLER, D. J., KUNO, G., SATHER, G. E., VELEZ, M., OLIVER, A. Mosquito cell
cultures and specific monoclonal antibodies in surveillance for dengue viruses. American
Journal of tropical medicine and hygiene 33 (1): 158-165, 1984.
GÖRG, A., OBERMAIER, C., BOGUTH, G., HARDER, A., SCHEIBE, B.,
WILDGRUBER, R., WEISS, W. The current state of two-dimensional electrophoresis
with immobilized pH gradients. Electrophoresis 21: 1037-1053, 2000.
HEUKESHOVEN, J. & DERNICK, R. Simplified method for silver staining of proteins in
polyacrylamide gels and the mechanism of silver staining. Electrophoresis 6: 103-112,
1985.
HOVING, S., VOSHOL, H., OOSTRUM, J. V. Towards high performance two-dimensional
gel electrophoresis using ultrazoom gels. Electrophoresis 21: 2617-2621, 2000.

58
ISSAQ, H. J., VEENSTRA, T. D. The role of electrophoresis in disease biomarker discovery.
Electrophoresis 28: 1980-1988, 2007.
KING, A. M. Q., ADAMS, M. J., CARSTENS, E. B., LEFKOWITZ, E. J. Virus Taxonomy:
Cassification and Nomenclature of Viruses. Ninth Report of the International
Committee on Taxonomy of viruses. Elsevier, ISBN: 978-0-12-384684-6, 2012.
KNOWLES, B. B., ADEN, D. P. Human hepatoma derived cell line, process for preparation
thereof, and uses therefor. Patente, EUA, 4.393.133, 1983.
KWALLAH, A. O., INOUE, S., MUIGAI, A. W. T., KUBO, T., SANG, R., MORITA, K.,
MWAU, M. A real-time reverse transcription loop-mediated isothermal amplification
assay for the rapid detection of yellow fever virus. Journal of Virological Methods 193
(1): 23–27, 2013.
LABAER, J. So, you want to look for biomarkers (introduction to the special biomarkers
issue). Journal of Proteome Research 4: 1053-1059, 2005.
LANCIOTTI, R. S. Molecular amplification assays for the detection of flaviviruses.
Advances in Virus Research 61: 67–99, 2003.
LI, S., QU, H., HAO, J., SUN, J., GUO, H., GUO, C., SUN, B., TU, C. Proteomic analysis of
primary porcine endothelial cells after infection by classical swine fever virus.
Biochimica et Biophysica Acta 1804: 1882-1888, 2010.
LOPEZ, M. F., BERGGREN, K., CHERNOKALSKAYA, E., LAZAREV, A., ROBINSON,
M., PATTON, W. F. A comparsion of silver stain and SYPRO Ruby protein gel stain
with respect to protein detection in two-dimensional gels and identification by peptide
mass profiling. Electrophoresis 21: 3673-3683, 2000.
LOUIS, K. S., SIEGEL, A. C. Cell viability analysis using trypan blue: manual and
automated methods. In: Mammalian cell viability: methods and protocols, Methods in
molecular biology 740, M. J. Stoddart (ed.), DOI 10.1007/978-1-61779-108-6_2, ©
Springer Science+Business Media, LLC, 2011.
MALEWICZ, B., JENKIN, H. M. Development of dengue virus plaques under serum-free
overlay medium. Journal of clinical microbiology 9 (5): 609-614, 1979.
MARIANNEAU, P., STEFFAN, A. M., ROYER, C., DROUET, M. T., KIRN, A., DEUBEL,
V. Differing infection patterns of dengue and yellow fever viruses in a human hepatoma
cell line. The Journal of Infectious Diseases 178: 1270-1278, 1998.
MAXWELL, K.L., FRAPPIER, L. Viral proteomics. Microbiology and Molecular Biology
Reviews 71: 398–411, 2007.

59
MONATH, T. P. Treatment of yellow fever. Antiviral Research 78: 116–124, 2008.
MONATH, T. P. Yellow fever and dengue - the interactions of virus, vector and host in the
re-emergence of epidemic disease. Seminars in Virology 5: 133-145, 1994.
MONATH, T. P. Yellow fever: An update. Lancet Infectious Diseases 1:11-20, 2001.
MUTEBI, J. P., WANG, H., LI, L., BRYANT, J.E., BARRETT, A. D. T. Phylogenetic and
evolutionary relationships along yellow fever virus isolates in Africa. Journal of
Virology 85: 6999-7008, 2001.
NEUHOFF, V., ARNOLD, N., TAUBE, D., EHRHARDT, W. Improved staining of proteins
in polyacrylamide gels including isoelectric focusing gels with clear background at
nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis
9: 255-262, 1988.
NUNES, M. R. T., PALACIOS, G., CARDOSO, J. F., MARTINS, L. C., SOUSA JR., E. C.,
LIMA, C. P. S., MEDEIROS, D. B. A., SAVJI, N., DESAI, A., RODRIGUES, S. G.,
CARVALHO, V. L., LIPKIN, W. I., VASCONCELOS, P. F. C. Genomic and
Phylogenetic Characterization of Brazilian Yellow Fever Virus Strains. Journal of
Virology 86 (24): 13263–13271, 2012.
NUNES, M. R. T., PALACIOS, G., NUNES, K. N. B., CASSEB, S. M. M., MARTINS,
LÍVIA C., QUARESMA, J. A. S., SAVJI, N., LIPKIN, W. I., VASCONCELOS, P. F. C.
Evaluation of two molecular methods for the detection of yellow fever virus genome.
Journal of Virological Methods 174 (1): 29-34, 2011.
O’FARRELL, P. H. High resolution two-dimensional electrophoresis of proteins. The
Journal of Biological Chemistry 250 (10): 4007–4021, 1975.
OKAMOTO, K., ENDO, Y., INOUE, S.,, NABESHIMA, T., NGA, P. T., GUILLERMO, P.
H., YU, F., LOAN, D. P., TRANG, B. M., NATIVIDAD, F. F., HASEBE, F., MORITA,
K. Development of a rapid and comprehensive proteomics-based arboviruses detection
system. Journal of Virological Methods 167 (1): 31-36, 2010.
PAN, S., CHEN, R., AEBERSOLD, R. BRENTNALL, T. A. Mass spectrometry based
glycoproteomics-From a proteomics perspective. Molecular & Cellular Proteomics 10
(1), 2011.
PARIDA, M. M., SANTHOSH, S. R., DASH, P. K., TRIPATHI, N. K., SAXENA, P.,
AMBUJ, S., SAHNI, A. K., LAKSHMANA RAO, P. V., MORITA, K. Development and
evaluation of reverse transcription-loop-mediated isothermal amplification assay for rapid
and real-time detection of Japanese encephalitis virus. Journaul of Clinical
Microbiology 44 (11): 4172–4178, 2006.

60
PARIDA, M. M., SANTHOSH, S.R., DASH, P.K., TRIPATHI, N.K., LAKSHMI, V.,
MAMIDI, N., SHRIVASTVA, A., GUPTA, N., SAXENA, P., BABU, J. P.,
LAKSHMANA RAO, P. V., MORITA, K. Rapid and real-time detection of
Chikungunya virus by reverse transcription loop-mediated isothermal amplification
assay. Journaul of Clinical Microbiology 45 (2): 351–357, 2007.
PARIDA, M., HORIOKE, K., ISHIDA, H., DASH, P. K., SAXENA, P., JANA, A. M.,
ISLAM, M. A., INOUE, S., HOSAKA, N., MORITA, K. Rapid detection and
differentiation of dengue virus serotypes by a real-time reverse transcription-loop-
mediated isothermal amplification assay. Journaul of Clinical Microbiology 43 (6):
2895–2903, 2005.
PARIDA, M., POSADAS, G., INOUE, S., HASEBE, F., MORITA, K. Real-time reverse
transcription loop-mediated isothermal amplification for rapid detection of West Nile
virus. Journaul of Clinical Microbiology 42 (1): 257–263, 2004.
PARKER C., WARREN M., LOISELLE D., DICHEVA N., SCARLETT C., BORCHERS C.
Identification of components of protein complexes. In: Methods in Molecular Biology.
(Totowa, NJ Ed.). Humana Press, 2005.
PASTORINO, B., BOUCOMONT-CHAPEAUBLANC, E., PEYREFITTE, C. N.,
BELGHAZI, M. FUSAÏ, T., ROGIER, C., TOLOU, H. J., ALMERAS, L. Identification
of cellular proteome modifications in response to west nile virus infection. Molecular &
Cellular Proteomics 8 (7): 1623-1637, 2009.
PATTANAKITSAKUL, N., RUNGROJCHAROENKIT, K., KANLAYA, R.,
SINCHAIKUL, S., NOISAKRAN, S., CHEN, S., MALASIT, P., THONGBOONKERD,
V. Proteomic analysis of host responses in HepG2 cells during Dengue virus infection.
Journal of Proteome Research 6, 4592–4600, 2007.
PATTON, W. F. Detection technologies in proteome analysis. Journal of Chromatography
B 771: 3-31, 2002.
PEYREFITTE, C. N., BOUBIS, L., COUDRIER, D., BOULOY, M., GRANDADAM, M.,
TOLOU, H. J., PLUMET, S. Real-time reverse-transcription loop-mediated isothermal
amplification for rapid detection of rift valley Fever virus. Journaul of Clinical
Microbiology 46 (11), 3653–3659.
PULENDRAN, B. Learning immunology from the yellow fever vaccine: innate immunity to
systems vaccinology. Nature Reviews Immunology 9: 741-747, 2009.
ROBOTTI, E., DEMARTINI, M., GOSETTI, F., CALABRESE, G., MARENGO, E.
Development of a classification and ranking method for the identification of possible

61
biomarkers in two-dimensional gel-electrophoresis based on principal component
analysis and variable selection procedures. Molecular BioSystems 7: 677-686, 2011.
SMIT, J. M., MOESKER, B., RODENHUIS-ZYBERT, I., WILSCHUT, J. Flavivirus cell
entry and membrane fusion. Viruses 3 (2): 160-171, 2011.
STAPLES, J. E., MONATH, T. P. Yellow fever: 100 years of discovery. JAMA 300 (8):
960-962, 2008.
SUN, J., JIANG, Y., SHI, Z., YAN, Y., GUO, H., HE, F., TU, C. Proteomic alteration of PK-
15 cells after infection by classical swine fever virus. Journal of Proteome Research 7:
5263-5269, 2008.
THEILER, M. & SMITH, H. H. The effect of prolonged cultivation in vitro upon the
pathogenicity of yellow fever virus. Journal of Experimental Medicine 65: 767-786,
1937a.
THEILER, M. & SMITH, H. H. The use of yellow fever virus modified by in vitro cultivation
for human immunization. Journal of Experimental Medicine 65: 787-800, 1937b.
TORINIWA, H. & KOMIYA, T. Rapid detection and quantification of Japanese encephalitis
virus by real-time reverse transcription loop-mediated isothermal amplification.
Microbiology and Immunology 50 (5): 379–387, 2006.
VASCONCELOS, P. F. C. Febre amarela. Revista da Sociedade Brasileira de Medicina
Tropical 36 (2): 275-293, 2003.
VASCONCELOS, P. F. C. Febre amarela: Reflexões sobre a doença, as perspectivas para o
século XXI e o risco da reurbanização. Revista Brasileira de Epidemiologia 5 (3): 244-
258, 2002.
VASCONCELOS, P. F. C., BRYANT, J. E., TRAVASSOS DA ROSA, A. P. A., TESH, R.
B., RODRIGUES, S. G., BARRETT, A. D.T. Genetic divergence and dispersal of
Yellow Fever Virus, Brazil. Emerging Infectious Diseases 10 (9): 1578-1584, 2004.
VITZHUM, F., BEHRENS, F., ANDERSON, N. L., SHAW, J. H. Proteomics: From basic
research to diagnostic application. A Review of Requirements & Needs. Journal of
Proteome Research 4: 1086-1097, 2005.
WAGNER, E. L., HEWLETT, M. J., BLOOM, D. C., CAMERINI, D. Basic Virology. 3 ed.
Blackwell Publishing, 2008. 581p.
WEIDMANN, M., FAYE, O., FAYE, O., KRANASTER, R., MARX, A., NUNES, M. R. T.,
VASCONCELOS, P. F. C., HUFERT, F. T., SALL, A. A. Improved LNA probe-based

62
assay for the detection of African and south American yellow fever virus strains. Journal
of Clinical Virology 48 (3): 187-192, 2010.
WESTERMEIER, R. & NAVEN, T. Proteomics in practice: A laboratory manual of proteome
analysis. Weinheim, Wiley-VCH Verlag GmbH, 2002. 311p.
WULFKUHLE, J. D., LIOTTA, L. A., PETRICOIN, E. F. Early detection: Proteomic
applications for the early detection of cancer. Nature Reviews Cancer 3: 267-275, 2003.
XIAO, S. Y., ZHANG, H., GUZMAN, H., TESH, R. B. Experimental yellow fever virus
infection in the golden hamster (Mesocricetus auratus). II. Pathology. The Journaul of
Infectious Diseases 183 (10): 1437-1444, 2001.
YE, Y., MAR, E. C., TONG, S., SAMMONS, S., FANG, S., ANDERSON, L. J., WANG, D.
Application of proteomics methods for pathogen discovery. Journal of Virological
Methods 163: 87-95, 2010.
ZHANG, X., ZHOU, J., WU, Y., ZHENG, X., MA, G., WANG, Z., JIN, Y., HE, J., YAN, Y.
Differential proteome analysis of host cells infected with porcine circovirus type 2.
Journal of Proteome Research 8: 5111-5119, 2009.
ZHENG, J., SUGRUE, R. J., TANG, K. Mass spectrometry based proteomic studies on
viruses and hosts – A review. Analytica Chimica Acta 702: 149-159, 2011.
ZHENG, J.,TAN, B. H., SUGRUE, R., TANG, K. Current approaches on viral infection:
proteomics and functional validations. Frontiers in Microbiology 3, Artigo 393, 2012.
ZHENG, X., HONG, L., SHI, L., GUO, J., SUN, Z., ZHOU, J. Proteomics analysis of host
cells infected with infectious bursal disease virus. Molecular & Cellular Proteomics 7
(3): 612-625, 2008.

63
ANEXO





























