Embed Size (px)
Citation preview

http://tpx.sagepub.com/
Toxicologic Pathology
http://tpx.sagepub.com/content/35/1/116The online version of this article can be found at:
DOI: 10.1080/01926230601060025
2007 35: 116Toxicol PatholAlessandra Livraghi and Scott H. Randell
Cystic Fibrosis and Other Respiratory Diseases of Impaired Mucus Clearance
Published by:
http://www.sagepublications.com
On behalf of:
Society of Toxicologic Pathology
can be found at:Toxicologic PathologyAdditional services and information for
http://tpx.sagepub.com/cgi/alertsEmail Alerts:
http://tpx.sagepub.com/subscriptionsSubscriptions:
http://www.sagepub.com/journalsReprints.navReprints:
http://www.sagepub.com/journalsPermissions.navPermissions:
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from

Toxicologic Pathology, 35:116–129, 2007Copyright C© by the Society of Toxicologic PathologyISSN: 0192-6233 print / 1533-1601 onlineDOI: 10.1080/01926230601060025
Cystic Fibrosis and Other Respiratory Diseases of Impaired
Mucus Clearance
ALESSANDRA LIVRAGHI1 AND SCOTT H. RANDELL1,2
1Cystic Fibrosis/Pulmonary Research and Treatment Center, Department of Medicine, The University of North Carolina at Chapel Hill2Department of Cell and Molecular Physiology, The University of North Carolina at Chapel Hill
ABSTRACT
Exposed to a diverse array of potentially noxious agents, the respiratory tract is protected by a highly developed innate defense system. Physiologicallyregulated epithelial ion and water transport coordinated with mucin secretion, beating cilia, and cough results in continuous flow of fluid and mucusover airway surfaces toward the larynx. This cleansing action is the initial and perhaps most quantitatively important innate defense mechanism.Repeated lung infections and eventual respiratory insufficiency characteristic of human cystic fibrosis (CF) and primary ciliary dyskinesia (PCD)illustrate the consequences of impaired mucus clearance. Altered mucus clearance likely contributes to the initiation, progression, and chronicity ofother airway diseases characterized by inflammation and mucous secretory cell hyper/metaplasia that afflict millions worldwide, including chronicobstructive pulmonary disease (COPD). This review concisely discusses the pathophysiology of human diseases characterized by genetic defects thatimpair mucus clearance. It then explores animal models in which components of the mucus clearance system have been disrupted. These modelsfirmly establish the importance of mucus clearance for respiratory health, and will help elucidate disease mechanisms and therapeutic strategies inCF, PCD and COPD.
Keywords. Chronic obstructive pulmonary disease; cilia; mucin; mucociliary clearance; cystic fibrosis; primary ciliary dyskinesia.
INTRODUCTION
Respiratory tract diseases such as asthma, chronic obstruc-tive pulmonary disease (COPD) and lung cancer typically re-sult from complex interactions between the environment andhost genetics. Constantly exposed to the environment, theairways and lungs have evolved to efficiently deliver inhaledair to the alveoli, while protecting gas exchange structuresfrom potentially harmful airborne chemicals, particles, andpathogens. A layer of fluid and mucus, propelled by ciliabeat and cough, continuously flows over conducting airwaysurfaces towards the pharynx. Mucus clearance is a critical,physiologically regulated, protective function of the airwaysand lungs.
The genetic defects in cystic fibrosis (CF) and primary cil-iary dyskinesia (PCD) impair mucus clearance, and study ofthese diseases has provided key insights regarding the patho-biology that occurs when constant protection of the respira-tory tract by effective mucus clearance is lost. In this review,we will concisely discuss the structural components and phys-iology of mucus clearance and the pathophysiology of CF andPCD in humans. We will then summarize animal models inwhich genes involved in the mucus clearance system havebeen disrupted or modified to model the human diseases.
Address correspondence to: Scott H. Randell, UNC CF Center, CB 7248,Room 4011 Thurston-Bowles Building, Chapel Hill, NC 27599; e-mail:[email protected]
Abbreviations: CF, cystic fibrosis; PCD, primary ciliary dyskinesia;COPD, chronic obstructive pulmonary disease; PCL, peri-ciliary layer;ASL, airway surface liquid; CFTR, cystic fibrosis transmembrane con-ductance regulator; ENaC, epithelial sodium channel; OMIM, OnlineMendelian Inheritance in Man; TEM, transmission electron microscopy;PD, potential difference; Isc, short circuit current.
Respiratory Tract Physical Structure, Innate and AcquiredHost Defense
Integrated airway and lung defense is the product of acomplex network, acting at distinct levels of organization(Figure 1). The nasal passages, nasopharynx and pharynxconstitute the upper airways and the trachea to ∼6th genera-tion bronchi are the proximal, large lower airways. Portionsof the nasal cavity and all the large airways are normally linedby a pseudostratified mucociliary epithelium consisting prin-cipally of basal, intermediate, ciliated and mucous secretory(goblet) cells. The proximal portion of the small, distal air-ways (bronchioles) is also lined by a columnar pseudostrati-fied epithelium, which transitions in more distal airways to ashorter, columnar to cuboidal epithelium in which Clara cellsreplace mucous secretory cells.
Finally, the alveolar region is composed mainly of thin typeI, and cuboidal type II alveolar epithelial cells. Thus, a con-tinuous epithelial layer forms a vital physical barrier betweenthe outside environment and the underlying lung parenchymaand circulatory system. Just as the relative abundance of celltypes changes along the respiratory tract axis, each segmenthas specialized functions that are key to host defense. Theepithelial physical barrier, in combination with secreted an-timicrobial factors, broad specificity soluble binding proteins(opsonins), mucus, mucociliary clearance, cough, phagocyticand natural killer cells constitutes the innate immune system(Martin and Frevert, 2005; Zaas and Schwartz, 2005). Thissystem is constantly on guard and requires no previous ex-posure to elicit a protective response.
To eliminate noxious stimuli, enhanced mucus and fluid se-cretion, cilia beat, antimicrobials, cytokines, chemokines andinflammatory cells are induced when pathogen associatedpatterns or other chemical motifs are recognized by innateimmune system receptors. The cell and cytokine/chemokine
116
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from
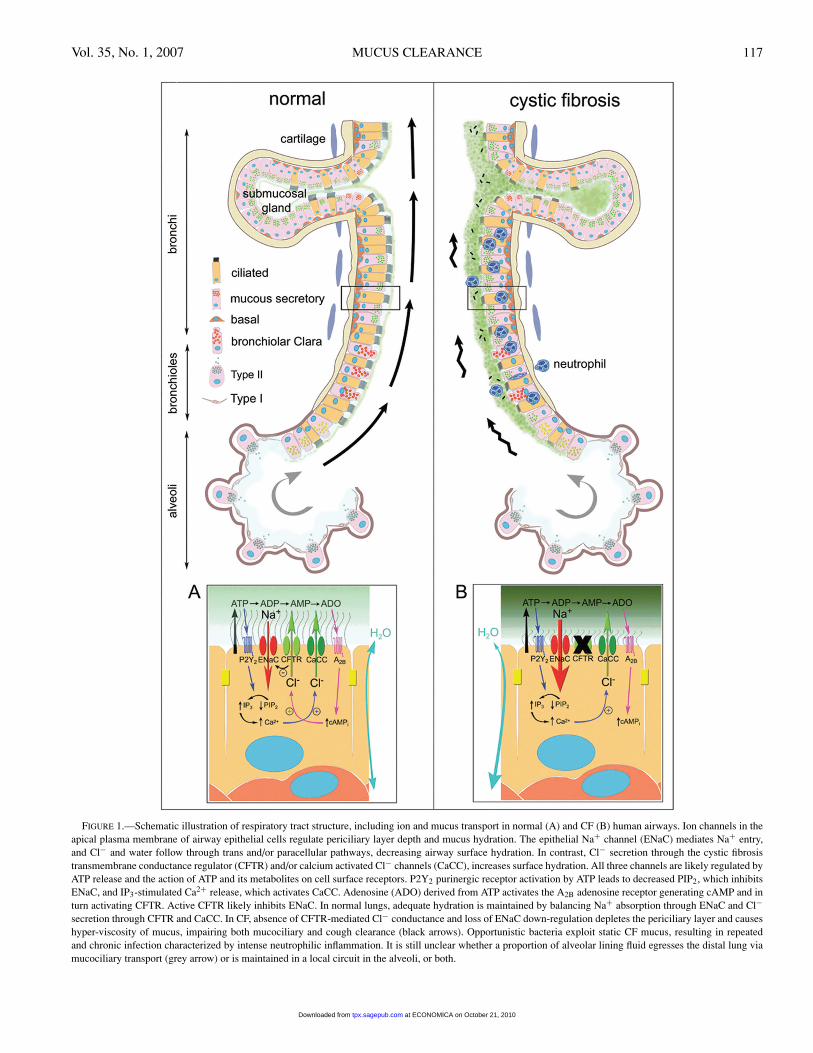
Vol. 35, No. 1, 2007 MUCUS CLEARANCE 117
FIGURE 1.—Schematic illustration of respiratory tract structure, including ion and mucus transport in normal (A) and CF (B) human airways. Ion channels in theapical plasma membrane of airway epithelial cells regulate periciliary layer depth and mucus hydration. The epithelial Na+ channel (ENaC) mediates Na+ entry,and Cl− and water follow through trans and/or paracellular pathways, decreasing airway surface hydration. In contrast, Cl− secretion through the cystic fibrosistransmembrane conductance regulator (CFTR) and/or calcium activated Cl− channels (CaCC), increases surface hydration. All three channels are likely regulated byATP release and the action of ATP and its metabolites on cell surface receptors. P2Y2 purinergic receptor activation by ATP leads to decreased PIP2, which inhibitsENaC, and IP3-stimulated Ca2+ release, which activates CaCC. Adenosine (ADO) derived from ATP activates the A2B adenosine receptor generating cAMP and inturn activating CFTR. Active CFTR likely inhibits ENaC. In normal lungs, adequate hydration is maintained by balancing Na+ absorption through ENaC and Cl−secretion through CFTR and CaCC. In CF, absence of CFTR-mediated Cl− conductance and loss of ENaC down-regulation depletes the periciliary layer and causeshyper-viscosity of mucus, impairing both mucociliary and cough clearance (black arrows). Opportunistic bacteria exploit static CF mucus, resulting in repeatedand chronic infection characterized by intense neutrophilic inflammation. It is still unclear whether a proportion of alveolar lining fluid egresses the distal lung viamucociliary transport (grey arrow) or is maintained in a local circuit in the alveoli, or both.
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from

118 LIVRAGHI AND RANDELL TOXICOLOGIC PATHOLOGY
milieu induced by the innate immune system is critical for theinvolvement and development of the more specialized adap-tive immune response; for example, specific memory and ef-fector T and B cells. Mucous secretory and ciliated cells playkey roles in mucociliary clearance, which in conjunction withcough, provides mucus clearance, perhaps the quantitativelymost important airway innate defense mechanism (Knowlesand Boucher, 2002; Randell and Boucher, 2006).
Mucus, Mucous Secretory Cells, and MucinsMucus is the collective term for mucins, ions, water and
other substances normally present on mucosal surfaces. Byvirtue of highly glycosylated secreted mucins, lipids, andsoluble proteins, mucus binds and entraps a broad array ofinhaled particles. Since increased airway secretions charac-terize obstructive lung diseases, including asthma, COPD andCF, particular attention has been paid to the regulation ofmucous secretory cell number, mucin gene expression andglycoprotein production, and the secretory pathways that me-diate mucin release (Thornton and Sheehan, 2004; Rose andVoynow, 2006; Williams et al., 2006). Beyond obstruction ofairflow, excessive mucus that fails to clear may form plaquesand plugs that can serve as a nidus for infection by oppor-tunistic pathogens, especially facultative anaerobic bacteriasuch as P. aeruginosa, able to adapt to relatively hypoxicconditions.
Often, single cell, free-living bacteria (planktonic) evolveinto alginate-producing colonies or biofilms that are muchmore resistant to attack from both neutrophils and antibi-otics, thus becoming difficult to eradicate (Worlitzsch et al.,2002; Matsui et al., 2005). The robust inflammatory responsetriggered by recurrent, or chronic infection can lead to severeremodeling of the airways perpetuating the vicious cycle ini-tiated by mucus accumulation.
FIGURE 2.—Cilia structure in respiratory tract epithelial cells. (A) Air-liquid interface cultures of normal human tracheobronchial epithelial cells display mucoussecretory and ciliated cell differentiation similar to the in vivo morphology. The porous culture support is visible below the cells. Formalin-fixation and paraffinsection, H&E stain. (B) Low power TEM view across the apical plasma membrane of a ciliated cell demonstrates cilia axonemes and basal bodies (BB). (C and D)Cross-section views of cilia axonemes at low and high power, respectively, reveal the 9 + 2 arrangement of microtubules and inner (IDA) and outer dynein arms(ODA), and radial spoke apparatus (RSA) structures. B, C, and D were prepared using conventional TEM procedures.
Ciliated Cells. At least 8 categories of cilia, or cilia de-rived organelles, with different functions have been describedin the human body (Afzelius, 2004). In the respiratory tract,ciliated cells have ∼200 cilia per cell, each with a diameterof 250 nm and a length of ∼6 μm. There are an estimated 109
cilia/cm2of upper and large airway surface. Cilia in the upperand large airways are denser and longer than in the bronchi-oles. Each respiratory cilia axoneme has a typical 9 + 2 mi-crotubule structure (9 outer doublets and 2 central single mi-crotubules) with characteristic inner and outer dynein arms,nexin links, and radial spoke apparatus. Each axoneme insertsinto the ciliated cell at a specialized structure that terminatesin a cytoplasmic basal body that resembles the centriole ofthe mitotic apparatus (Figure 2). It is estimated that >200unique proteins constitute the cilia and basal body structures(Fliegauf and Omran, 2006). In normal lungs, cilia beat incoordinated, directional, metachronal waves at a frequencyestimated to be ∼10–20 Hz, and mucus and the embeddedparticles are constantly transported towards the pharynx to beswallowed or expectorated, thereby promoting airway steril-ity (Meeks and Bush, 2000).
The Peri-Ciliary Layer (PCL), Glands, and Cough. To-gether with cilia and mucus, an adequate peri-ciliary layer(PCL) is required for effective mucociliary clearance. ThePCL lines the airway surface and provides a low viscositymilieu in which cilia can freely beat to propel overlying mu-cus. The existence of the PCL has been demonstrated in vivoby osmium/perfluorocarbon fixation techniques followed bytransmission electron microscopy (Sims et al., 1991) or invitro by confocal microscopy (Matsui et al., 1998; Tarranand Boucher, 2002) (Figure 3). The ability of the airway ep-ithelium to regulate PCL volume to maintain an ∼7 μm-thicklayer required for cilia to beat and effectively clear mucus is
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from

Vol. 35, No. 1, 2007 MUCUS CLEARANCE 119
FIGURE 3.—An adequate periciliary liquid (PCL) layer is essential for mucustransport. (A) Perflurocarbon-OsO4 fixation preserves the morphology of thePCL and overlying mucus. The PCL is visible as a clear layer in a thin sectionof a well-differentiatied air-liquid interface culture of human tracheobronchialepithelial cells, Epon-embedding, Richardson’s stain. (B) TEM of an equivalentarea as depicted in the box in panel A. Dense mucus is visible above the clearPCL. Two polystyrene beads, which were added to track mucus movement, arevisible at the mucus-PCL border (asterisks). (C) The PCL as viewed in a livecell preparation by X-Z plane confocal microscopy. Texas red dextran-labeledairway surface liquid (ASL) is on the apical surface (red) and the cells werelabeled with calcein (green).
critical for lung innate defense (Matsui et al., 1998; Tarranet al., 2001, 2005).
As discussed in the legend of Figure 1, recent studies sug-gest that PCL volume homeostasis represents the coordinatedbalancing of Cl− secretion and Na+ absorption and is sensi-tive to changes in the concentration of soluble mediators (nu-cleotides and nucleosides) present in the PCL (Lazarowskiet al., 2004; Tarran, 2004, 2006a, 2006b). Upper and proxi-mal airways also contain submucosal glands that contribute toairway secretions and amplify the ability of airway epithelialcells to produce antimicrobial factors and mucus (Wine andJoo, 2004; Inglis and Wilson, 2005). Finally, cough is an im-portant innate defense mechanism that prevents pulmonaryaspiration, promotes ciliary activity and clears airway debris(Chang, 2006; McCool, 2006). The combination of mucocil-iary and cough clearance constitutes “mucus clearance.”
Cystic Fibrosis (CF). In the following section, we high-light the seminal characteristics of CF. For a more a com-
prehensive treatise on the history, genetics, diagnosis, patho-genesis, microbiology, treatment, or controversies in this in-tensely studied disorder, the reader is directed to an excellentrecent textbook chapter (Boucher, 2005) and review articles(Davis, 2006; Rowe et al., 2005). CF is the most common life-threatening genetic disease of Caucasians, affecting ∼1/2500and ∼1/17,000 live births in the White and Black populationsof the United States, respectively. It has been recognized as adistinct disease entity since the 1930s. It is an autosomal re-cessive, monogenic disorder resulting from mutations in thecystic fibrosis transmembrane conductance regulator (CFTR)gene, which was discovered in 1989 (Online Mendelian In-heritance in Man (OMIM) # 602421, National Center forBiotechnology Information).
The CFTR protein is a cAMP-activated transmembraneanion channel expressed at a variety of epithelial surfaces,where it is thought to principally conduct Cl− ions. How-ever, CFTR also coordinately regulates the function of otherepithelial ion channels located in the apical membrane. Thereare over 1400 different recognized CFTR mutations but themajority, from ∼80% to >95%, depending on the patient eth-nicity, can be detected in a screen of 97 of the most commonmutations. CFTR mutations include deletions (the deletion of3 base pairs resulting in the loss of phenylalanine at position508, known as �F508, is the most common CF mutation),missense, frameshift, nonsense, and also mutations causingalterations in RNA splicing.
Acting through a variety of molecular and cellular mech-anisms, severe mutations result in an almost total absence offunctional CFTR protein at the cell surface. Milder mutationsresulting in partial CFTR function may produce abnormal-ities in other organs but respiratory disease may be mild oreven absent. CF is a multi-system disorder with a childhoodonset. The defining feature is abnormal ion and water trans-port across a variety of epithelial surfaces, which leads tothe characteristic salty sweat and elevated nasal and rectaltrans-epithelial electrical potential differences (PD) that canbe measured for diagnostic purposes. Relatively dehydrated,thickened secretions and mucus accumulation/obstruction ofseveral tubular duct structures are characteristic of CF.
Significant morbidities include obstructive azoospermia inmales due to in utero blockage, subsequent fibrotic oblitera-tion and total loss of the vas deferens, epididymis and seminalvesicles. In the female, the uterine cervix can be inappropri-ately obstructed with thickened mucus. Other morbidities areprincipally associated with the gastrointestinal tract. Meco-nium ileus in the newborn is highly suggestive of CF andrecurrent bouts of intestinal obstruction are common. Gas-trointestinal changes and frequent coughing increases thefrequency of rectal prolapse. Biliary tract obstruction vari-ably results in liver dysfunction and fibrosis. Pancreatic in-volvement is highly prevalent and obstruction begins in utero,which serves as the basis for the blood trypsinogen neonatalscreening test.
Exocrine pancreatic insufficiency and duct blockage de-crease digestive enzyme secretion into the intestine and canresult in significant nutritional deficiencies and steatorrheaif not properly treated. Ultimately, postobstructive atrophyresults in complete replacement of the exocrine pancreas byfibrosis and fatty tissue, with eventual disruption of the islets,which can cause insulin dependent hyperglycemia. Progres-sive pancreatic lesions are illustrated in Figure 4.
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from

120 LIVRAGHI AND RANDELL TOXICOLOGIC PATHOLOGY
FIGURE 4.—Progressive obstructive pancreatic pathology in CF. Low (A) and higher power (B) views of the pancreas of a 9-year-old pancreatic insufficient CFmale that died of a non-CF related cause exhibits eosinophilic material in dilated ducts, residual exocrine pancreatic acini and numerous islets, whereas the pancreasof a 47-year-old pancreatic insufficient CF female (C) contains mainly fat and islets with minimal fibrosis in this particular view. Routine formalin-fixed paraffinsections, H&E stain.
Neonatal respiratory distress is not specifically associatedwith CF and, except for dilated mucus gland ducts, the lungsare apparently normal at birth. Postnatal respiratory tract le-sions are, by far, the leading cause of morbidity and mor-tality in CF. In the upper airways, rhinitis and sinusitis dueto infection are highly prevalent and development of nasalpolyposis is common. Recurrent bacterial bronchopneumo-nia, often initially due to H. influenzae or S. aureus and thenP. aeruginosa, is frequent in newborns and young childrenwith CF. Evidence for lung infection with P. aeruginosa oc-curs in ∼60% of CF individuals by age 3, which is likely tobe an underestimate (Burns et al., 2001; West et al., 2002).Repeated and ultimately chronic infection with mucoidP. aeruginosa, or other characteristic Gram-negative bacte-ria, including Burkholderia sp. (previously known as Pseu-domonas cepacia), S. maltophilia, A. xylosoxidans, as wellas variable involvement of Aspergillus and nontuberculousmycobacteria, leads to progressive bronchiectasis, bronchi-olectasis and respiratory insufficiency that ultimately causesrespiratory failure and death of >95% of CF patients,currently with a median survival age of ∼37 years.
The sequence of events predisposing to airway infectionin CF has been debated through the years, but clarified morerecently. Generalized immune deficiency and specific ab-normalities in acquired immunity are highly unlikely, sincesystemic infection is not characteristic of CF. In fact, sep-sis due to P. aeruginosa, even after decades of lung infec-tion, is rare, presumably due to effective humoral immu-nity. More likely, the disease represents a failure of localairway defense. It has been suggested that bacteria adheremore readily to CF airway epithelial cells due to enhancedexpression of the cell surface ganglioside asialoGM1, pro-moting infection (Saiman and Prince, 1993). Paradoxically,it has also been proposed that CFTR serves as a bacterial re-ceptor and that its absence leads to failure to internalize andkill bacteria (Pier, 2000). Defects in anti-microbial activityof airway fluid (Ganz, 2002) or in neutrophil phagocytosis(Berger et al., 1989) likely occur in the inflamed CF airway
environment but are unlikely to be the primary defect. Thepresence of an intrinsic hyper-inflammatory phenotype in CFcells, even in the absence of infection, is debated (Machen,2006). A consensus now exists that respiratory tract patho-physiology in CF principally results from the inability to se-crete Cl− and regulate Na+ absorption, which causes relativedehydration of the airway surface, depleting the PCL andcausing accumulation of hyper-viscous mucus that cannotbe cleared by mucociliary clearance or cough (Matsui et al.,1997).
As discussed previously and as illustrated in Figure 5,mucus plaques and plugs serve as a nidus for intra-luminalinfection. Interestingly, hypoxic microenvironments, whichare exploited by characteristic CF pathogens, develop inmacroscopic mucus accumulations, even within ventilatedairways (Worlitzsch et al., 2002). Bacteria resident withinthe thickened luminal mucus may evade chemical antimi-crobial factors and phagocytes (Matsui et al., 2005). Com-plex bacterial evolution and host adaptation occurs in thechronically infected airway, which is likely unique in CFdue to the constant and severe degree of mucus dehydrationand impaired mucus clearance. Bacterial colonies exhibitingbiofilm-like properties may develop, which are difficult orimpossible to eradicate. The continuous presence of bacte-ria and the accompanying intense inflammation ultimatelyremodel the airway wall, causing the ubiquitous mucous se-cretory cell hyperplasia and metaplasia, submucosal glandenlargement, hypertrophy of the bronchial circulation, ecta-sis of bronchi and bronchioles, and variable parenchymal cystformation, sometimes progressing to cavitary disease, withadjacent fibrosis and pleural involvement.
Primary Ciliary Dyskinesia (PCD). PCD illustrates theimportance of normally beating cilia to maintain healthy air-ways. In recent years, there has been impressive progressto elucidate the role of cilia in diverse biological processes.It has been demonstrated that motile, rotating, 9 + 0 ciliaat the embryonic node function to break bilateral symmetry
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from
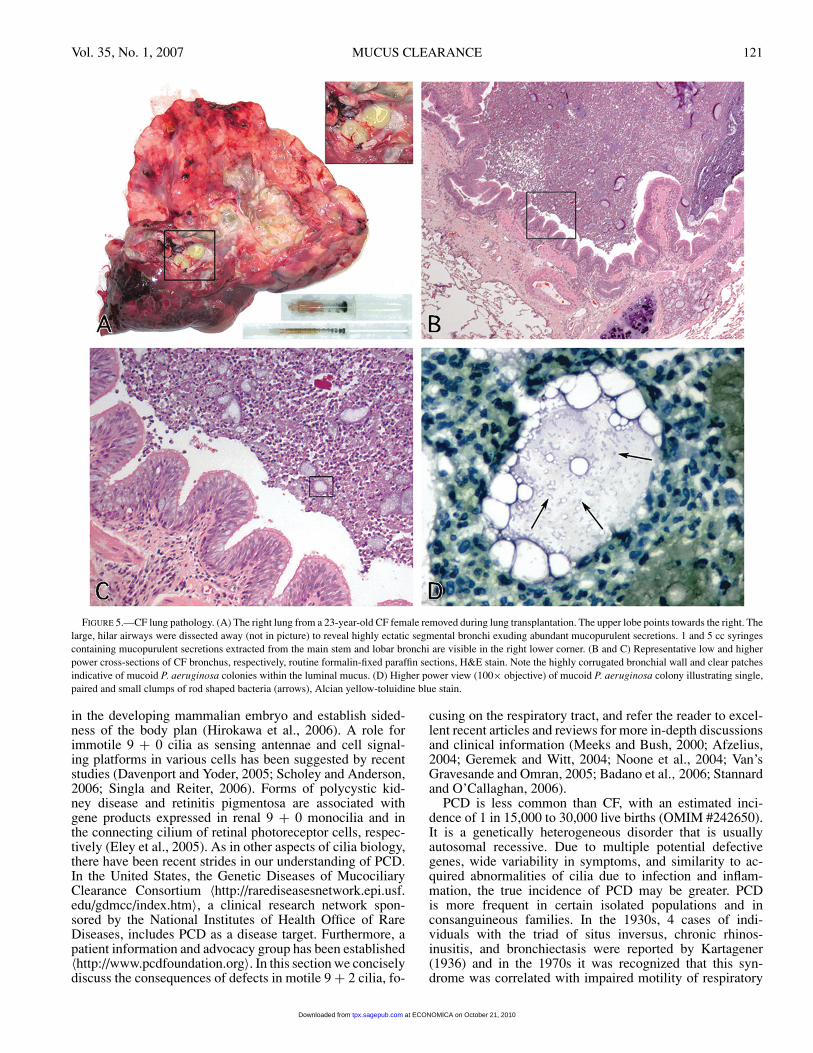
Vol. 35, No. 1, 2007 MUCUS CLEARANCE 121
FIGURE 5.—CF lung pathology. (A) The right lung from a 23-year-old CF female removed during lung transplantation. The upper lobe points towards the right. Thelarge, hilar airways were dissected away (not in picture) to reveal highly ectatic segmental bronchi exuding abundant mucopurulent secretions. 1 and 5 cc syringescontaining mucopurulent secretions extracted from the main stem and lobar bronchi are visible in the right lower corner. (B and C) Representative low and higherpower cross-sections of CF bronchus, respectively, routine formalin-fixed paraffin sections, H&E stain. Note the highly corrugated bronchial wall and clear patchesindicative of mucoid P. aeruginosa colonies within the luminal mucus. (D) Higher power view (100× objective) of mucoid P. aeruginosa colony illustrating single,paired and small clumps of rod shaped bacteria (arrows), Alcian yellow-toluidine blue stain.
in the developing mammalian embryo and establish sided-ness of the body plan (Hirokawa et al., 2006). A role forimmotile 9 + 0 cilia as sensing antennae and cell signal-ing platforms in various cells has been suggested by recentstudies (Davenport and Yoder, 2005; Scholey and Anderson,2006; Singla and Reiter, 2006). Forms of polycystic kid-ney disease and retinitis pigmentosa are associated withgene products expressed in renal 9 + 0 monocilia and inthe connecting cilium of retinal photoreceptor cells, respec-tively (Eley et al., 2005). As in other aspects of cilia biology,there have been recent strides in our understanding of PCD.In the United States, the Genetic Diseases of MucociliaryClearance Consortium 〈http://rarediseasesnetwork.epi.usf.edu/gdmcc/index.htm〉, a clinical research network spon-sored by the National Institutes of Health Office of RareDiseases, includes PCD as a disease target. Furthermore, apatient information and advocacy group has been established〈http://www.pcdfoundation.org〉. In this section we conciselydiscuss the consequences of defects in motile 9 + 2 cilia, fo-
cusing on the respiratory tract, and refer the reader to excel-lent recent articles and reviews for more in-depth discussionsand clinical information (Meeks and Bush, 2000; Afzelius,2004; Geremek and Witt, 2004; Noone et al., 2004; Van’sGravesande and Omran, 2005; Badano et al., 2006; Stannardand O’Callaghan, 2006).
PCD is less common than CF, with an estimated inci-dence of 1 in 15,000 to 30,000 live births (OMIM #242650).It is a genetically heterogeneous disorder that is usuallyautosomal recessive. Due to multiple potential defectivegenes, wide variability in symptoms, and similarity to ac-quired abnormalities of cilia due to infection and inflam-mation, the true incidence of PCD may be greater. PCDis more frequent in certain isolated populations and inconsanguineous families. In the 1930s, 4 cases of indi-viduals with the triad of situs inversus, chronic rhinos-inusitis, and bronchiectasis were reported by Kartagener(1936) and in the 1970s it was recognized that this syn-drome was correlated with impaired motility of respiratory
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from
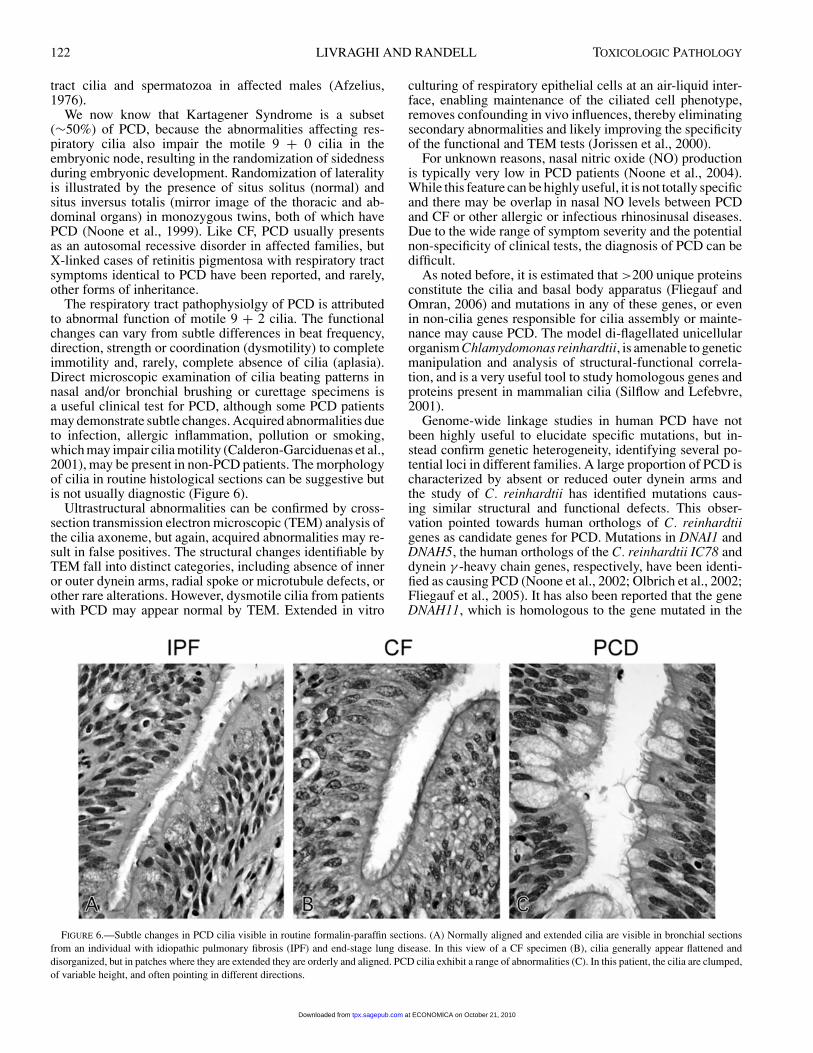
122 LIVRAGHI AND RANDELL TOXICOLOGIC PATHOLOGY
tract cilia and spermatozoa in affected males (Afzelius,1976).
We now know that Kartagener Syndrome is a subset(∼50%) of PCD, because the abnormalities affecting res-piratory cilia also impair the motile 9 + 0 cilia in theembryonic node, resulting in the randomization of sidednessduring embryonic development. Randomization of lateralityis illustrated by the presence of situs solitus (normal) andsitus inversus totalis (mirror image of the thoracic and ab-dominal organs) in monozygous twins, both of which havePCD (Noone et al., 1999). Like CF, PCD usually presentsas an autosomal recessive disorder in affected families, butX-linked cases of retinitis pigmentosa with respiratory tractsymptoms identical to PCD have been reported, and rarely,other forms of inheritance.
The respiratory tract pathophysiolgy of PCD is attributedto abnormal function of motile 9 + 2 cilia. The functionalchanges can vary from subtle differences in beat frequency,direction, strength or coordination (dysmotility) to completeimmotility and, rarely, complete absence of cilia (aplasia).Direct microscopic examination of cilia beating patterns innasal and/or bronchial brushing or curettage specimens isa useful clinical test for PCD, although some PCD patientsmay demonstrate subtle changes. Acquired abnormalities dueto infection, allergic inflammation, pollution or smoking,which may impair cilia motility (Calderon-Garciduenas et al.,2001), may be present in non-PCD patients. The morphologyof cilia in routine histological sections can be suggestive butis not usually diagnostic (Figure 6).
Ultrastructural abnormalities can be confirmed by cross-section transmission electron microscopic (TEM) analysis ofthe cilia axoneme, but again, acquired abnormalities may re-sult in false positives. The structural changes identifiable byTEM fall into distinct categories, including absence of inneror outer dynein arms, radial spoke or microtubule defects, orother rare alterations. However, dysmotile cilia from patientswith PCD may appear normal by TEM. Extended in vitro
FIGURE 6.—Subtle changes in PCD cilia visible in routine formalin-paraffin sections. (A) Normally aligned and extended cilia are visible in bronchial sectionsfrom an individual with idiopathic pulmonary fibrosis (IPF) and end-stage lung disease. In this view of a CF specimen (B), cilia generally appear flattened anddisorganized, but in patches where they are extended they are orderly and aligned. PCD cilia exhibit a range of abnormalities (C). In this patient, the cilia are clumped,of variable height, and often pointing in different directions.
culturing of respiratory epithelial cells at an air-liquid inter-face, enabling maintenance of the ciliated cell phenotype,removes confounding in vivo influences, thereby eliminatingsecondary abnormalities and likely improving the specificityof the functional and TEM tests (Jorissen et al., 2000).
For unknown reasons, nasal nitric oxide (NO) productionis typically very low in PCD patients (Noone et al., 2004).While this feature can be highly useful, it is not totally specificand there may be overlap in nasal NO levels between PCDand CF or other allergic or infectious rhinosinusal diseases.Due to the wide range of symptom severity and the potentialnon-specificity of clinical tests, the diagnosis of PCD can bedifficult.
As noted before, it is estimated that >200 unique proteinsconstitute the cilia and basal body apparatus (Fliegauf andOmran, 2006) and mutations in any of these genes, or evenin non-cilia genes responsible for cilia assembly or mainte-nance may cause PCD. The model di-flagellated unicellularorganism Chlamydomonas reinhardtii, is amenable to geneticmanipulation and analysis of structural-functional correla-tion, and is a very useful tool to study homologous genes andproteins present in mammalian cilia (Silflow and Lefebvre,2001).
Genome-wide linkage studies in human PCD have notbeen highly useful to elucidate specific mutations, but in-stead confirm genetic heterogeneity, identifying several po-tential loci in different families. A large proportion of PCD ischaracterized by absent or reduced outer dynein arms andthe study of C. reinhardtii has identified mutations caus-ing similar structural and functional defects. This obser-vation pointed towards human orthologs of C. reinhardtiigenes as candidate genes for PCD. Mutations in DNAI1 andDNAH5, the human orthologs of the C. reinhardtii IC78 anddynein γ -heavy chain genes, respectively, have been identi-fied as causing PCD (Noone et al., 2002; Olbrich et al., 2002;Fliegauf et al., 2005). It has also been reported that the geneDNAH11, which is homologous to the gene mutated in the
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from
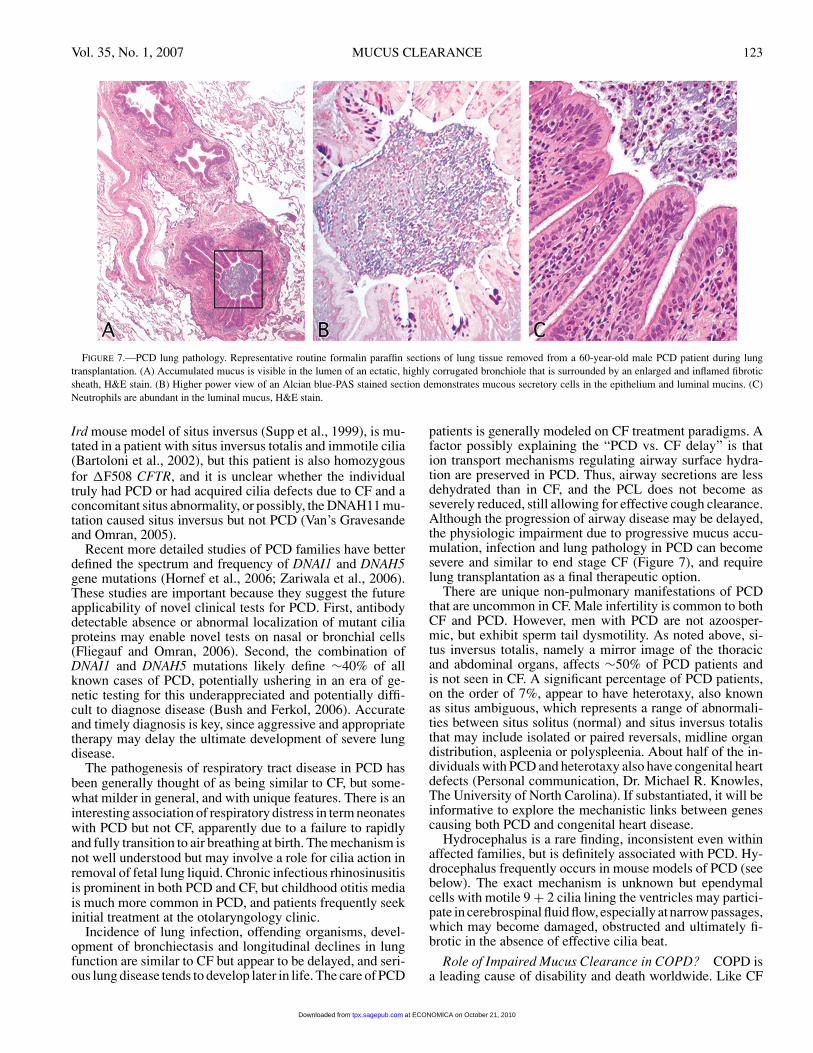
Vol. 35, No. 1, 2007 MUCUS CLEARANCE 123
FIGURE 7.—PCD lung pathology. Representative routine formalin paraffin sections of lung tissue removed from a 60-year-old male PCD patient during lungtransplantation. (A) Accumulated mucus is visible in the lumen of an ectatic, highly corrugated bronchiole that is surrounded by an enlarged and inflamed fibroticsheath, H&E stain. (B) Higher power view of an Alcian blue-PAS stained section demonstrates mucous secretory cells in the epithelium and luminal mucins. (C)Neutrophils are abundant in the luminal mucus, H&E stain.
Ird mouse model of situs inversus (Supp et al., 1999), is mu-tated in a patient with situs inversus totalis and immotile cilia(Bartoloni et al., 2002), but this patient is also homozygousfor �F508 CFTR, and it is unclear whether the individualtruly had PCD or had acquired cilia defects due to CF and aconcomitant situs abnormality, or possibly, the DNAH11 mu-tation caused situs inversus but not PCD (Van’s Gravesandeand Omran, 2005).
Recent more detailed studies of PCD families have betterdefined the spectrum and frequency of DNAI1 and DNAH5gene mutations (Hornef et al., 2006; Zariwala et al., 2006).These studies are important because they suggest the futureapplicability of novel clinical tests for PCD. First, antibodydetectable absence or abnormal localization of mutant ciliaproteins may enable novel tests on nasal or bronchial cells(Fliegauf and Omran, 2006). Second, the combination ofDNAI1 and DNAH5 mutations likely define ∼40% of allknown cases of PCD, potentially ushering in an era of ge-netic testing for this underappreciated and potentially diffi-cult to diagnose disease (Bush and Ferkol, 2006). Accurateand timely diagnosis is key, since aggressive and appropriatetherapy may delay the ultimate development of severe lungdisease.
The pathogenesis of respiratory tract disease in PCD hasbeen generally thought of as being similar to CF, but some-what milder in general, and with unique features. There is aninteresting association of respiratory distress in term neonateswith PCD but not CF, apparently due to a failure to rapidlyand fully transition to air breathing at birth. The mechanism isnot well understood but may involve a role for cilia action inremoval of fetal lung liquid. Chronic infectious rhinosinusitisis prominent in both PCD and CF, but childhood otitis mediais much more common in PCD, and patients frequently seekinitial treatment at the otolaryngology clinic.
Incidence of lung infection, offending organisms, devel-opment of bronchiectasis and longitudinal declines in lungfunction are similar to CF but appear to be delayed, and seri-ous lung disease tends to develop later in life. The care of PCD
patients is generally modeled on CF treatment paradigms. Afactor possibly explaining the “PCD vs. CF delay” is thation transport mechanisms regulating airway surface hydra-tion are preserved in PCD. Thus, airway secretions are lessdehydrated than in CF, and the PCL does not become asseverely reduced, still allowing for effective cough clearance.Although the progression of airway disease may be delayed,the physiologic impairment due to progressive mucus accu-mulation, infection and lung pathology in PCD can becomesevere and similar to end stage CF (Figure 7), and requirelung transplantation as a final therapeutic option.
There are unique non-pulmonary manifestations of PCDthat are uncommon in CF. Male infertility is common to bothCF and PCD. However, men with PCD are not azoosper-mic, but exhibit sperm tail dysmotility. As noted above, si-tus inversus totalis, namely a mirror image of the thoracicand abdominal organs, affects ∼50% of PCD patients andis not seen in CF. A significant percentage of PCD patients,on the order of 7%, appear to have heterotaxy, also knownas situs ambiguous, which represents a range of abnormali-ties between situs solitus (normal) and situs inversus totalisthat may include isolated or paired reversals, midline organdistribution, aspleenia or polyspleenia. About half of the in-dividuals with PCD and heterotaxy also have congenital heartdefects (Personal communication, Dr. Michael R. Knowles,The University of North Carolina). If substantiated, it will beinformative to explore the mechanistic links between genescausing both PCD and congenital heart disease.
Hydrocephalus is a rare finding, inconsistent even withinaffected families, but is definitely associated with PCD. Hy-drocephalus frequently occurs in mouse models of PCD (seebelow). The exact mechanism is unknown but ependymalcells with motile 9 + 2 cilia lining the ventricles may partici-pate in cerebrospinal fluid flow, especially at narrow passages,which may become damaged, obstructed and ultimately fi-brotic in the absence of effective cilia beat.
Role of Impaired Mucus Clearance in COPD? COPD isa leading cause of disability and death worldwide. Like CF
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from

124 LIVRAGHI AND RANDELL TOXICOLOGIC PATHOLOGY
and PCD, submucosal gland enlargement, mucous secretorycell hyperplasia in the large airways and metaplasia in thesmall airways and sputum production are common featuresof COPD (Szilasi et al., 2006). Thickening of the bronchi-olar wall due to fibrosis, an increased volume of epithelialcells and the presence of luminal inflammatory mucus exu-dates are correlated with the severity of COPD (Hogg et al.,2004). Toxic particles and gases, the main cause of COPD,inhibit cilia function and Cl− secretion (Sisson et al., 1994;Kreindler et al., 2005; Cantin et al., 2006), and emphysemaand loss of small airways tethers may distort and/or promotecollapse of the airway wall, all of which would decreasethe efficiency of mucociliary and cough clearance. Bacte-rial infection and exacerbation by viral illness are prominentfeatures of COPD, and this sequence is also important inCF and PCD. Thus, reduced efficiency of mucus clearancelikely plays an important role in the pathogenesis of COPD.A more detailed comparison of mucus clearance in CF andCOPD is available in a recent review (Randell and Boucher,2006).
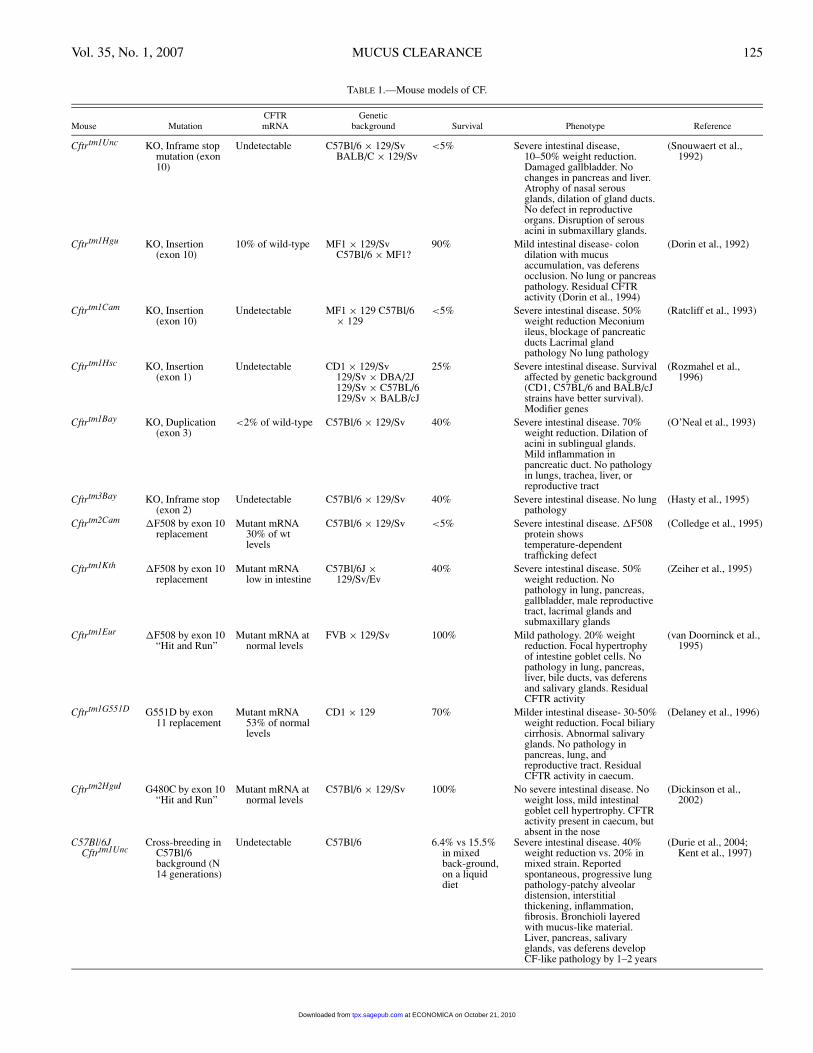
Animal Models of CF. The cloning of the human CFTRgene pointed the CF research community towards generationof an animal model, in which the gene could be either deletedor mutated, to enable study of the pathophysiology and po-tential treatments. To date, both knockout mice and thoseharboring specific mutations in murine Cftr, notably �F508,have been created. A brief summary of CF mouse genotypeand phenotype, adapted from the European working groupon CFTR expression web site 〈http://pen2.igc.gulbenkian.pt/cftr/vr/f/scholte mouse models table.pdf〉, is given in Ta-ble 1. Detailed reviews of the pathophysiological featuresof CF mice have been published (Grubb and Boucher, 1999;Guilbault et al., 2006). Briefly, a consistent phenotype is ex-hibited in the gastrointestinal tract of CF mice. Untreatedmice typically die shortly after birth due to meconium ileusor after weaning from intestinal obstruction/rupture causedby impaired luminal fluid secretion, which correlates withthe absence of functional CFTR protein in the gut epitheliumand resembles the human CF intestinal phenotype. The inci-dence of death due to intestinal obstruction can be reducedby a liquid diet or by supplementing the drinking water witha laxative (Colyte).
An unexpected finding was the lack of an overt pul-monary phenotype. All of the CF mice generated exhib-ited inconsistent or minor pathological alterations in thelungs. In Cftrtm1Unc (knockout) mice, the nasal septal ep-ithelium exhibited hyperplastic and hypertrophic mucous se-cretory cells (Snouwaert et al., 1992) in conjunction withreduced PCL volume, in comparison to wild-type littermates(Tarran et al., 2001). When Cftrtm1Unc mice were backcrossedonto the C57BL/6 strain, patchy alveolar distension, intersti-tial thickening, early mild neutrophilic infiltrate at day 30, anincrease in monocytes at 3 months, and fibrosis were reported(Kent et al., 1997; Durie et al., 2004;). G551D CF mice ex-hibited inspissated eosinophilic material in the lumen of thepharyngeal submucosal glands (Delaney et al., 1996).
A number of hypotheses have been formulated to explainminimal pulmonary pathology in CF mice. Except for theproximal trachea, murine airways are devoid of submucosalglands (Pack et al., 1981), which may be an important dif-ference from humans, but is not consistent with the presence
of disease in gland-free human distal bronchioles. As dis-cussed previously, human CF is characterized by both theabsence of cAMP-stimulated, CFTR-mediated Cl− secretionand Na+hyperabsorption, due to unregulated ENaC activityin the upper and lower airways. Short circuit current (Isc), asmeasured using Ussing chambers, is an index of ion transportacross the epithelium.
In excised mouse nasal epithelium virtually all of thebasal Isc is inhibitable by amiloride (ENaC-mediated) andin Cftrtm1Unc mice amiloride-sensitive Isc is elevated indi-cating sodium hyperabsorption (Clarke et al., 1992). Thismimics the higher nasal PD found in CF humans, whichis also thought to be due to up-regulation of ENaC activ-ity. However, in the mouse tracheal and bronchial epithe-lium, the amiloride-sensitive Isc accounts for only part ofthe basal Isc. Tracheas from both wild-type and Cftrtm1Unc
mice respond with significant Cl− secretion when stimulatedwith forskolin, typically used to activate adenyl cyclase, in-crease cellular cAMP and activate CFTR. However, in freshlyexcised mouse tracheas, forskolin also elevates intracellu-lar Ca2+, activating an alternative (non-CFTR,) Cl− channel(Grubb et al., 1994).
Therefore, CFTR seems to have a small role in the mouselower airways where it is replaced by prominent activity of al-ternative, Ca2+-activated Cl− channels. Thus, minimal spon-taneous lung pathology in CF mice is most likely due to re-gional, species-specific differences in ion transport betweenmice and humans. There is an active research effort to cre-ate CF in larger animals that may be physiologically moresimilar to humans, including ferrets, pigs and sheep (Scholteet al., 2004; Li et al., 2006).
Scnn1b Transgenic Mice. Based on the hypothesis thathydration of the airway surface represents the balance be-tween Cl− secretion and Na+ absorption, mice that hyper-absorb Na+ in the lower airways were created by overex-pressing the β subunit of ENaC (encoded by the Scnn1bgene) using the airway-specific Clara cell secretory protein(CCSP) promoter (Mall et al., 2004). The tracheas of neona-tal and adult Scnn1b-transgenic mice exhibited elevated basaland amiloride-sensitive Isc, compared to nontransgenic litter-mates. Accordingly, tracheal and bronchial PCL height wassignificantly reduced, the percent solids content of lower air-way mucus was significantly increased and mucus transport,measured in vivo, was significantly reduced in comparisonto wild-type littermates.
Scnn1b overexpression did not have adverse effects on fe-tal survival, but Scnn1b mice showed a significant postnatalmortality that began in the first days of life and continued upto 4 weeks, with an overall mortality of ∼50%. Scnn1b miceexhibited postnatal mucus accumulation and obstruction inthe large and small airways associated with goblet cell meta-plasia and neutrophil infiltration (Figure 8). The neutrophil-attracting chemokines macrophage inflammatory protein-2(MIP-2) and KC (the two murine analogues of human IL-8)were significantly increased. Lungs were free of cultureablebacteria, indicating that the neutrophilic inflammation oc-curred in the absence of active infection. Interestingly, theupregulation of MIP-2 and KC was not a direct effect ofScnn1b overexpression in the epithelial cells, since their se-cretion was not increased in epithelial cultures derived fromScnn1b airways.
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from
Vol. 35, No. 1, 2007 MUCUS CLEARANCE 125
TABLE 1.—Mouse models of CF.
CFTR GeneticMouse Mutation mRNA background Survival Phenotype Reference
Cftrtm1Unc KO, Inframe stopmutation (exon10)
Undetectable C57Bl/6 × 129/SvBALB/C × 129/Sv
<5% Severe intestinal disease,10–50% weight reduction.Damaged gallbladder. Nochanges in pancreas and liver.Atrophy of nasal serousglands, dilation of gland ducts.No defect in reproductiveorgans. Disruption of serousacini in submaxillary glands.
(Snouwaert et al.,1992)
Cftrtm1Hgu KO, Insertion(exon 10)
10% of wild-type MF1 × 129/SvC57Bl/6 × MF1?
90% Mild intestinal disease- colondilation with mucusaccumulation, vas deferensocclusion. No lung or pancreaspathology. Residual CFTRactivity (Dorin et al., 1994)
(Dorin et al., 1992)
Cftrtm1Cam KO, Insertion(exon 10)
Undetectable MF1 × 129 C57Bl/6× 129
<5% Severe intestinal disease. 50%weight reduction Meconiumileus, blockage of pancreaticducts Lacrimal glandpathology No lung pathology
(Ratcliff et al., 1993)
Cftrtm1Hsc KO, Insertion(exon 1)
Undetectable CD1 × 129/Sv129/Sv × DBA/2J129/Sv × C57BL/6129/Sv × BALB/cJ
25% Severe intestinal disease. Survivalaffected by genetic background(CD1, C57BL/6 and BALB/cJstrains have better survival).Modifier genes
(Rozmahel et al.,1996)
Cftrtm1Bay KO, Duplication(exon 3)
<2% of wild-type C57Bl/6 × 129/Sv 40% Severe intestinal disease. 70%weight reduction. Dilation ofacini in sublingual glands.Mild inflammation inpancreatic duct. No pathologyin lungs, trachea, liver, orreproductive tract
(O’Neal et al., 1993)
Cftrtm3Bay KO, Inframe stop(exon 2)
Undetectable C57Bl/6 × 129/Sv 40% Severe intestinal disease. No lungpathology
(Hasty et al., 1995)
Cftrtm2Cam �F508 by exon 10replacement
Mutant mRNA30% of wtlevels
C57Bl/6 × 129/Sv <5% Severe intestinal disease. �F508protein showstemperature-dependenttrafficking defect
(Colledge et al., 1995)
Cftrtm1Kth �F508 by exon 10replacement
Mutant mRNAlow in intestine
C57Bl/6J ×129/Sv/Ev
40% Severe intestinal disease. 50%weight reduction. Nopathology in lung, pancreas,gallbladder, male reproductivetract, lacrimal glands andsubmaxillary glands
(Zeiher et al., 1995)
Cftrtm1Eur �F508 by exon 10“Hit and Run”
Mutant mRNA atnormal levels
FVB × 129/Sv 100% Mild pathology. 20% weightreduction. Focal hypertrophyof intestine goblet cells. Nopathology in lung, pancreas,liver, bile ducts, vas deferensand salivary glands. ResidualCFTR activity
(van Doorninck et al.,1995)
Cftrtm1G551D G551D by exon11 replacement
Mutant mRNA53% of normallevels
CD1 × 129 70% Milder intestinal disease- 30-50%weight reduction. Focal biliarycirrhosis. Abnormal salivaryglands. No pathology inpancreas, lung, andreproductive tract. ResidualCFTR activity in caecum.
(Delaney et al., 1996)
Cftrtm2HguI G480C by exon 10“Hit and Run”
Mutant mRNA atnormal levels
C57Bl/6 × 129/Sv 100% No severe intestinal disease. Noweight loss, mild intestinalgoblet cell hypertrophy. CFTRactivity present in caecum, butabsent in the nose
(Dickinson et al.,2002)
C57Bl/6JCftrtm1Unc
Cross-breeding inC57Bl/6background (N14 generations)
Undetectable C57Bl/6 6.4% vs 15.5%in mixedback-ground,on a liquiddiet
Severe intestinal disease. 40%weight reduction vs. 20% inmixed strain. Reportedspontaneous, progressive lungpathology-patchy alveolardistension, interstitialthickening, inflammation,fibrosis. Bronchioli layeredwith mucus-like material.Liver, pancreas, salivaryglands, vas deferens developCF-like pathology by 1–2 years
(Durie et al., 2004;Kent et al., 1997)
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from

126 LIVRAGHI AND RANDELL TOXICOLOGIC PATHOLOGY
FIGURE 8.—Lung pathology in Scnn1b (βENaC-overexpressing) mice. (A and B) Low power view of lung tissue sections from an adult Scnn1b mouse, H&E andAlcian blue-PAS stain, respectively. (C–H) Higher magnification views of boxed areas in panels A and B, H&E (C–E) and Alcian blue-PAS stain (F–H). Note thevariable degree of highly adherent, accumulated mucus in different airways and the presence of neutrophils in luminal mucus (inset in panel C).
It was proposed that inflammation occurs because en-vironmental inflammatory stimuli accumulate in the lungsof Scnn1b mice due to poor mucus clearance. Finally, H.influenzae or P. aeruginosa instilled into the trachea persistedlonger in Scnn1b mice, indicating impaired airway clearance.In summary, the Scnn1b mice exhibit airway Na+ hyper-absorption, a hallmark of human CF, and recapitulate manyfeatures of the human disease, including mucus accumula-tion and neutrophilic airway inflammation. This mouse isthus a useful in vivo tool for investigating pathophysiologicmechanisms relevant to CF and other diseases.
Animal Models of PCD. A phenotype similar to PCD oc-curs naturally in both dogs and pigs, which appears identicalto that seen in humans, except that hydrocephalus is a promi-nent feature (Edwards et al., 1989; Roperto et al., 1993). Thegenetic basis of canine and porcine PCD-like airway diseaseremains unknown. Numerous mouse models with left-rightsymmetry defects have been identified and are the subject ofvarious reviews (Ibanez-Tallon et al., 2003; Eley et al., 2005),but due to the lack of respiratory tract symptoms these are notclassified as PCD models (Geremek and Witt, 2004). Table2 summarizes the mouse models with known genetic defectsaffecting 9 + 2 motile cilia function.
Mouse axonemal dynein heavy chain 5, encoded by theMdnah5 gene, is the homologue of human DNAH5. Mdnah5-
deficient mice exhibit early mortality (∼4–5 weeks), hydro-cephalus, respiratory cilia immotility due to the completelack of outer dynein arms, randomization of sidedness, and aPCD-like respiratory phenotype characterized by early devel-opment of chronic rhinitis (Ibanez-Tallon et al., 2002). Tar-geted mutation of the mouse homologue of human DNAI1,Mdhc7, which encodes for an inner dynein arm heavy chainresults in reduced sperm motility and male infertility. Thesemice show a 50% reduction of the respiratory ciliary beatfrequency but apparently this does not cause airway disease(Neesen et al., 2001; Vernon et al. ,2005; Woolley et al.,2005). Other candidate genes for PCD include transcriptionfactors implicated in ciliated cell differentiation. Hfh4 (alsoknown as Foxj1) is a transcription factor that regulates dyneingene expression and Hfh4-deficient mice completely lack ep-ithelial cell cilia and may be a model for cilia aplasia. Thesemice exhibit hydrocephalus and randomization of sidedness,but still have monocilia in the embryonic nodal pole (Chenet al., 1998; Brody et al., 2000). However, mutations in theFOXJ1 gene have not been found in PCD patients, generallyexcluding it as a candidate gene for PCD (Maiti et al., 2000).
It was reported that deletion of DNA polymerase λ gene(Poll) in mice produced inner dynein arm defects in ependy-mal and respiratory cilia and a phenotype characteristic ofPCD, including hydrocephalus, situs inversus, chronic sup-purative sinusitis, and male sterility (Kobayashi et al., 2002).
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from

Vol. 35, No. 1, 2007 MUCUS CLEARANCE 127
TABLE 2.—Mouse models of PCD.
MutationsHuman Protein found in
Mouse Gene homolog Mutation function PCD patients Phenotype Reference
Mdnah5−/− Mdnah5 DNAH5 Insertional KO Axonemaldynein heavychain
yes Perinatal mortality, growthfailure, mucusaccumulation/inflammationin nasal sinuses, middle earinfection, situs inversus,ciliary immotility,hydrocephalus, deficiencyin outer dynein arms
(Ibanez-Tallonet al., 2002)
Mdhc7−/− Mdhc7 DNAH1 Insertional KO Axonemaldynein heavychain
no Reduced sperm motility,reduced ciliary beatfrequency, no airwayphenotype
(Neesen et al.,2001)
Hfh4−/− hfh4 aliasFoxj1
HFH4 FOXJ1 Insertional KO Transcriptionfactor,regulation ofciliogenesis
no Perinatal mortality, growthfailure situs inversus,absence of cilia in theairways, partial penetranceof hydrocephalus, nodalcilia are present
(Brody et al.,2000; Chenet al,. 1998)
Poll−/− Poll Dpcd POLL and DPCD Insertional KO Unknown no Perinatal mortality,hydrocephalus, situsinversus, chronic sinusitis,infertility. Defective innerdynein arms of ependimaland respiratory cilia
(Kobayashi et al,.2002; Zariwalaet al., 2004a)
However, it was difficult to reconcile a mutation in a DNApolymerase with an axonemal defect and there was evidencethat mice with deletion of the sequence encoding for the cat-alytic domain of Poll were viable and fertile (Bertocci et al.,2002). Thus, the targeting construct used to generate Poll−/−mice was examined in further detail.
The region of chromosome 19 that was deleted to generatethe Poll−/− mouse also included the first exon and the startcodon of a newly identified gene, Dpcd, which is transcribedfrom the opposite strand relative to Poll. Dpcd is predictedto encode for a 23 KDa protein of unknown function. SinceDpcd mRNA is expressed in airway epithelial cells and itsexpression is upregulated during ciliogenesis, it has been sug-gested that mutation in Dpcd can cause PCD, although thusfar no mutations in the human homolog of Dpcd have beenfound in 51 unrelated PCD patients (Zariwala et al., 2004).
Collectively, the existing animal models of PCD are char-acterized by a high incidence of hydrocephalus and perina-tal mortality. These pathologic features hamper the estab-lishment of models suitable for long-term studies of chronicinfection characteristic of the human pulmonary PCD phe-notype. Nevertheless, mouse models where a homolog ofa candidate human gene has been targeted can confirm thelikely role of that gene in PCD, and novel animal modelsmay also reveal important differences in the physiology ofthe mucociliary clearance system between mice and humans.It is possible to genetically manipulate mice to alter candidatePCD genes only in the trachea and lungs, and future develop-ment of such mice may avoid hydrocephalus, enabling morein-depth study of the lower respiratory tract.
SUMMARY AND CONCLUSION
Mucus clearance, the continuous flow of fluid and mucusover airway surfaces towards the larynx, is a vital protective,innate defense mechanism. Failure of this system, due to ge-netic defects in specific components, occurs in CF and PCD
and results in repeated lung infections and eventual respira-tory insufficiency. These diseases illustrate key principles un-derlying efficient mucus clearance and the pathophysiologicconsequences of its impairment. Altered mucus clearancelikely plays a role in several common acquired respiratorytract diseases, including COPD. A greater understanding ofthe basic science of mucus clearance and new animal modelswill promote advances in the diagnosis and treatment of thisimportant class of diseases.
ACKNOWLEDGMENTS
The authors thank Dr. Michael R. Knowles for critical as-sessment of the manuscript.
REFERENCES
Afzelius, B. A. (1976). A human syndrome caused by immotile cilia. Science193, 317–9.
Afzelius, B. A. (2004). Cilia-related diseases. J Pathol 204, 470–7.Badano, J. L., Mitsuma, N., Beales, P. L., and Katsanis, N. (2006). The cil-
iopathies: an emerging class of human genetic disorders. Annu Rev Ge-nomics Hum Genet 7, 125–48.
Bartoloni, L., Blouin, J. L., Pan, Y., Gehrig, C., Maiti, A. K., Scamuffa, N.,Rossier, C., Jorissen, M., Armengot, M., Meeks, M., Mitchison, H. M.,Chung, E. M., Delozier-Blanchet, C. D., Craigen, W. J., and Antonarakis,S. E. (2002). Mutations in the DNAH11 (axonemal heavy chain dyneintype 11) gene cause one form of situs inversus totalis and most likelyprimary ciliary dyskinesia. Proc Natl Acad Sci USA 99, 10282–6.
Berger, M., Sorensen, R. U., Tosi, M. F., Dearborn, D. G., and Doring, G. (1989).Complement receptor expression on neutrophils at an inflammatory site,the Pseudomonas-infected lung in cystic fibrosis. J Clin Invest 84, 1302–13.
Bertocci, B., De Smet, A., Flatter, E., Dahan, A., Bories, J.-C., Landreau, C.,Weill, J.-C., and Reynaud, C.-A. (2002). Cutting edge: DNA polymerasesmicro and lambda are dispensable for Ig gene hypermutation. J Immunol168, 3702–6.
Boucher, R. C., Knowles M. R., and Jankaskas, J. R. (2005). Cystic fibrosis.In: Textbook of Respiratory Medicine (M. A. Nadels, ed., Vol. 1), pp.1217–51. Elsevier, Philadelphia.
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from

128 LIVRAGHI AND RANDELL TOXICOLOGIC PATHOLOGY
Brody, S. L., Yan, X. H., Wuerffel, M. K., Song, S.-K., and Shapiro, S. D. (2000).Ciliogenesis and left-right axis defects in forkhead factor HFH-4-null mice.Am J Respir Cell Mol Biol 23, 45–51.
Burns, J. L., Gibson, R. L., McNamara, S., Yim, D., Emerson, J., Rosenfeld, M.,Hiatt, P., McCoy, K., Castile, R., Smith, A. L., and Ramsey, B. W. (2001).Longitudinal assessment of Pseudomonas aeruginosa in young childrenwith cystic fibrosis. J Infect Dis 183, 444–52.
Bush, A., and Ferkol, T. (2006). Movement: the emerging genetics of primaryciliary dyskinesia. Am J Respir Crit Care Med 174, 109–10.
Calderon-Garciduenas, L., Valencia-Salazar, G., Rodriguez-Alcaraz, A.,Gambling, T. M., Garcia, R., Osnaya, N., Villarreal-Calderon, A., Devlin,R. B., and Carson, J. L. (2001). Ultrastructural nasal pathology in childrenchronically and sequentially exposed to air pollutants. Am J Respir CellMol Biol 24, 132–8.
Cantin, A. M., Hanrahan, J. W., Bilodeau, G., Ellis, L., Dupuis, A., Liao, J.,Zielenski, J., and Durie, P. (2006). Cystic fibrosis transmembrane conduc-tance regulator function is suppressed in cigarette smokers. Am J RespirCrit Care Med 173, 1139–44.
Chang, A. B. (2006). The physiology of cough. Paediatr Respir Rev 7, 2–8.Chen, J., Knowles, H. J., Hebert, J. L., and Hackett, B. P. (1998). Mutation of
the mouse hepatocyte nuclear factor/forkhead homologue 4 gene resultsin an absence of cilia and random left-right asymmetry. J Clin Invest 102,1077–82.
Clarke, L. L., Grubb, B. R., Gabriel, S. E., Smithies, O., Koller, B. H., andBoucher, R. C. (1992). Defective epithelial chloride transport in a gene-targeted mouse model of cystic fibrosis. Science 257, 1125–8.
Davenport, J. R., and Yoder, B. K. (2005). An incredible decade for the primarycilium: a look at a once-forgotten organelle. Am J Physiol Renal Physiol289, F1159–69.
Davis, P. B. (2006). Cystic fibrosis since 1938. Am J Respir Crit Care Med 173,475–82.
Delaney, S. J., Alton, E. W., Smith, S. N., Lunn, D. P., Farley, R., Lovelock, P.K., Thomson, S. A., Hume, D. A., Lamb, D., Porteous, D. J., Dorin, J. R.,and Wainwright, B. J. (1996). Cystic fibrosis mice carrying the missensemutation G551D replicate human genotype-phenotype correlations. EmboJ 15, 955–63.
Durie, P. R., Kent, G., Phillips, M. J., and Ackerley, C. A. (2004). Characteristicmultiorgan pathology of cystic fibrosis in a long-living cystic fibrosis trans-membrane regulator knockout murine model. Am J Pathol 164, 1481–93.
Edwards, D. F., Kennedy, J. R., Patton, C. S., Toal, R. L., Daniel, G. B.,and Lothrop, C. D. (1989). Familial immotile-cilia syndrome in Englishspringer spaniel dogs. Am J Med Genet 33, 290–8.
Eley, L., Yates, L. M., and Goodship, J. A. (2005). Cilia and disease. Curr OpinGenet Develop Genet Dis 15, 308–14.
Fliegauf, M., Olbrich, H., Horvath, J., Wildhaber, J. H., Zariwala, M. A.,Kennedy, M., Knowles, M. R., and Omran, H. (2005). Mislocalizationof DNAH5 and DNAH9 in respiratory cells from patients with primaryciliary dyskinesia. Am J Respir Crit Care Med 171, 1343–9.
Fliegauf, M., and Omran, H. (2006). Novel tools to unravel molecular mecha-nisms in cilia-related disorders. Trends Genet 22, 241–5.
Ganz, T. (2002). Antimicrobial polypeptides in host defense of the respiratorytract. J Clin Invest 109, 693–7.
Geremek, M., and Witt, M. (2004). Primary ciliary dyskinesia: genes, candidategenes and chromosomal regions. J Appl Genet 45, 347–61.
Grubb, B. R., and Boucher, R. C. (1999). Pathophysiology of gene-targetedmouse models for cystic fibrosis. Physiol Rev 79, S193–214.
Grubb, B. R., Paradiso, A. M., and Boucher, R. C. (1994). Anomalies in iontransport in CF mouse tracheal epithelium. Am J Physiol 267, C293–300.
Guilbault, C., Saeed, Z., Downey, G. P., and Radzioch, D. (2006). Cystic fibrosismouse models. Am J Respir Cell Mol Biol, in press.
Hirokawa, N., Tanaka, Y., Okada, Y., and Takeda, S. (2006). Nodal flow and thegeneration of left-right asymmetry. Cell 125, 33–45.
Hogg, J. C., Chu, F., Utokaparch, S., Woods, R., Elliott, W. M., Buzatu, L.,Cherniack, R. M., Rogers, R. M., Sciurba, F. C., Coxson, H. O., and Pare,P. D. (2004). The nature of small-airway obstruction in chronic obstructivepulmonary disease. N Engl J Med 350, 2645–53.
Hornef, N., Olbrich, H., Horvath, J., Zariwala, M. A., Fliegauf, M., Loges, N. T.,Wildhaber, J., Noone, P. G., Kennedy, M., Antonarakis, S. E., Blouin, J. L.,Bartoloni, L., Nublein, T., Ahrens, P., Griese, M., Kuhl, H., Sudbrak, R.,Knowles, M. R., Reinhardt, R., and Omran, H. (2006). DNAH5 mutationsare a common cause of primary ciliary dyskinesia with outer Dynein armdefects. Am J Respir Crit Care Med.
Ibanez-Tallon, I., Gorokhova, S., and Heintz, N. (2002). Loss of function ofaxonemal dynein Mdnah5 causes primary ciliary dyskinesia and hydro-cephalus. Hum Mol Genet 11, 715–21.
Ibanez-Tallon, I., Heintz, N., and Omran, H. (2003). To beat or not to beat:roles of cilia in development and disease. Hum Mol Genet 12 (Spec No 1),R27–35.
Inglis, S. K., and Wilson, S. M. (2005). Cystic fibrosis and airway submucosalglands. Pediatr Pulmonol 40, 279–84.
Jorissen, M., Willems, T., and Van der Schueren, B. (2000). Ciliary functionanalysis for the diagnosis of primary ciliary dyskinesia: advantages ofciliogenesis in culture. Acta Otolaryngol 120, 291–5.
Kartagener, M. H. A. (1936). Situs viscerum inversus und Polyposis nasi ineinem Falle familiaerer Bronchiektasien. Beitr. Klin. Tuberk. 331–3.
Kent, G., Iles, R., Bear, C. E., Huan, L.-J., Griesenbach, U., McKerlie, C.,Frndova, H., Ackerley, C., Gosselin, D., Radzioch, D., O’Brodovich, H.,Tsui, L.-C., Buchwald, M., and Tanswell, A. K. (1997). Lung disease inmice with cystic fibrosis. J Clin Invest 100, 3060–9.
Knowles, M. R., and Boucher, R. C. (2002). Mucus clearance as a primary innatedefense mechanism for mammalian airways. J Clin Invest 109, 571–7.
Kobayashi, Y., Watanabe, M., Okada, Y., Sawa, H., Takai, H., Nakanishi, M.,Kawase, Y., Suzuki, H., Nagashima, K., Ikeda, K., and Motoyama, N.(2002). Hydrocephalus, situs inversus, chronic sinusitis, and male infertil-ity in dna polymerase lambda-deficient mice: possible implication for thepathogenesis of immotile cilia syndrome. Mol Cell Biol 22, 2769–76.
Kreindler, J. L., Jackson, A. D., Kemp, P. A., Bridges, R. J., and Danahay,H. (2005). Inhibition of chloride secretion in human bronchial epithelialcells by cigarette smoke extract. Am J Physiol Lung Cell Mol Physiol 288,L894–902.
Lazarowski, E. R., Tarran, R., Grubb, B. R., Van Heusden, C. A., Okada, S.,and Boucher, R. C. (2004). Nucleotide release provides a mechanism forairway surface liquid homeostasis. J Biol Chem 279, 36855–64.
Li, Z., Sun, X., Chen, J., Liu, X., Wisely, S. M., Zhou, Q., Renard, J. P., Leno,G. H., and Engelhardt, J. F. (2006). Cloned ferrets produced by somaticcell nuclear transfer. Dev Biol 293, 439–48.
Machen, T. E. (2006). Innate immune response in CF airway epithelia: hyper-inflammatory? Am J Physiol Cell Physiol 291, C218–30.
Maiti, A. K., Bartoloni, L., Mitchison, H. M., Meeks, M., Chung, E., Spiden, S.,Gehrig, C., Rossier, C., DeLozier-Blanchet, C. D., Blouin, J., Gardiner, R.M., and Antonarakis, S. E. (2000). No deleterious mutations in the FOXJ1(alias HFH-4) gene in patients with primary ciliary dyskinesia (PCD).Cytogenet Cell Genet 90, 119–22.
Mall, M., Grubb, B. R., Harkema, J. R., O’Neal, W. K., and Boucher, R.C. (2004). Increased airway epithelial Na+ absorption produces cysticfibrosis-like lung disease in mice. Nat Med 10, 487–93.
Martin, T. R., and Frevert, C. W. (2005). Innate immunity in the lungs. Proc AmThorac Soc 2, 403–11.
Matsui, H., Grubb, B. R., Tarran, R., Randell, S. H., Gatzy, J. T., Davis, C. W., andBoucher, R. C. (1998). Evidence for periciliary liquid layer depletion, notabnormal ion composition, in the pathogenesis of cystic fibrosis airwaysdisease. Cell 95, 1005–15.
Matsui, H., Johnson, L. G., Randell, S. H., and Boucher, R. C. (1997). Lossof binding and entry of liposome-DNA complexes decreases transfectionefficiency in differentiated airway epithelial cells. J Biol Chem 272, 1117–26.
Matsui, H., Verghese, M. W., Kesimer, M., Schwab, U. E., Randell, S. H.,Sheehan, J. K., Grubb, B. R., and Boucher, R. C. (2005). Reduced three-dimensional motility in dehydrated airway mucus prevents neutrophil cap-ture and killing bacteria on airway epithelial surfaces. J Immunol 175,1090–9.
McCool, F. D. (2006). Global physiology and pathophysiology of cough: ACCPevidence-based clinical practice guidelines. Chest 129, 48S–53S.
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from

Vol. 35, No. 1, 2007 MUCUS CLEARANCE 129
Meeks, M., and Bush, A. (2000). Primary ciliary dyskinesia (PCD). PediatrPulmonol 29, 307–16.
Neesen, J., Kirschner, R., Ochs, M., Schmiedl, A., Habermann, B., Mueller, C.,Holstein, A. F., Nuesslein, T., Adham, I., and Engel, W. (2001). Disruptionof an inner arm dynein heavy chain gene results in asthenozoospermia andreduced ciliary beat frequency. Hum Mol Genet 10, 1117–28.
Noone, P. G., Bali, D., Carson, J. L., Sannuti, A., Gipson, C. L., Ostrowski, L. E.,Bromberg, P. A., Boucher, R. C., and Knowles, M. R. (1999). Discordantorgan laterality in monozygotic twins with primary ciliary dyskinesia. AmJ Med Genet 82, 155–60.
Noone, P. G., Leigh, M. W., Sannuti, A., Minnix, S. L., Carson, J. L., Hazucha,M., Zariwala, M. A., and Knowles, M. R. (2004). Primary ciliary dyskine-sia: diagnostic and phenotypic features. Am J Respir Crit Care Med 169,459–67.
Noone, P. G., Zariwala, M., Sannuti, A., Minnix, S., Leigh, M. W., Carson, J.,and Knowles, M. R. (2002). Mutations in DNAI1 (IC78) cause primaryciliary dyskinesia. Chest 121, 97S.
Olbrich, H., Haffner, K., Kispert, A., Volkel, A., Volz, A., Sasmaz, G., Reinhardt,R., Hennig, S., Lehrach, H., Konietzko, N., Zariwala, M., Noone, P. G.,Knowles, M., Mitchison, H. M., Meeks, M., Chung, E. M., Hildebrandt, F.,Sudbrak, R., and Omran, H. (2002). Mutations in DNAH5 cause primaryciliary dyskinesia and randomization of left-right asymmetry. Nat Genet30, 143–4.
Pack, R. J., Al-Ugaily, L. H., and Morris, G. (1981). The cells of the tracheo-bronchial epithelium of the mouse: a quantitative light and electron mi-croscope study. J Anat 132, 71–84.
Pier, G. B. (2000). Role of the cystic fibrosis transmembrane conductance regu-lator in innate immunity to Pseudomonas aeruginosa infections. Proc NatlAcad Sci USA 97, 8822–8.
Randell, S. H., and Boucher, R. C. (2006). Effective mucus clearance is essentialfor respiratory health. Am J Respir Cell Mol Biol 35, 20–8.
Roperto, F., Galati, P., and Rossacco, P. (1993). Immotile cilia syndrome in pigs.A model for human disease. Am J Pathol 143, 643–7.
Rose, M. C., and Voynow, J. A. (2006). Respiratory tract mucin genes and mucinglycoproteins in health and disease. Physiol Rev 86, 245–78.
Rowe, S. M., Miller, S., and Sorscher, E. J. (2005). Cystic fibrosis. N Engl JMed 352, 1992–2001.
Saiman, L., and Prince, A. (1993). Pseudomonas aeruginosa pili bind toasialoGM1 which is increased on the surface of cystic fibrosis epithelialcells. J Clin Invest 92, 1875–80.
Scholey, J. M., and Anderson, K. V. (2006). Intraflagellar transport and cilium-based signaling. Cell 125, 439–42.
Scholte, B. J., Davidson, D. J., Wilke, M., and De Jonge, H. R. (2004). Animalmodels of cystic fibrosis. J Cyst Fibros 3 Suppl 2, 183–90.
Silflow, C. D., and Lefebvre, P. A. (2001). Assembly and motility of eukaryoticcilia and flagella. Lessons from Chlamydomonas reinhardtii. Plant Physiol127, 1500–7.
Sims, D. E., Westfall, J. A., Kiorpes, A. L., and Horne, M. M. (1991). Preser-vation of tracheal mucus by nonaqueous fixative. Biotech Histochem 66,173–80.
Singla, V., and Reiter, J. F. (2006). The primary cilium as the cell’s antenna:signaling at a sensory organelle. Science 313, 629–33.
Sisson, J. H., Papi, A., Beckmann, J. D., Leise, K. L., Wisecarver, J., Brodersen,B. W., Kelling, C. L., Spurzem, J. R., and Rennard, S. I. (1994). Smokeand viral infection cause cilia loss detectable by bronchoalveolar lavagecytology and dynein ELISA. Am J Respir Crit Care Med 149, 205–13.
Snouwaert, J. N., Brigman, K. K., Latour, A. M., Malouf, N. N., Boucher, R. C.,Smithies, O., and Koller, B. H. (1992). An animal model for cystic fibrosismade by gene targeting. Science 257, 1083–8.
Stannard, W., and O’Callaghan, C. (2006). Ciliary function and the role of ciliain clearance. J Aerosol Med 19, 110–5.
Supp, D. M., Brueckner, M., Kuehn, M. R., Witte, D. P., Lowe, L. A., McGrath, J.,Corrales, J., and Potter, S. S. (1999). Targeted deletion of the ATP binding
domain of left-right dynein confirms its role in specifying development ofleft-right asymmetries. Development 126, 5495–504.
Szilasi, M., Dolinay, T., Nemes, Z., and Strausz, J. (2006). Pathology of chronicobstructive pulmonary disease. Pathol Oncol Res 12, 52–60.
Tarran, R. (2004). Regulation of airway surface liquid volume and mucus trans-port by active ion transport. Proc Am Thorac Soc 1, 42–6.
Tarran, R., and Boucher, R. C. (2002). Thin-film measurements of airway surfaceliquid volume/composition and mucus transport rates in vitro. MethodsMol Med 70, 479–92.
Tarran, R., Button, B., and Boucher, R. C. (2006a). Regulation of normal andcystic fibrosis airway surface liquid volume by phasic shear stress. AnnuRev Physiol 68, 543–61.
Tarran, R., Button, B., Picher, M., Paradiso, A. M., Ribeiro, C. M., Lazarowski,E. R., Zhang, L., Collins, P. L., Pickles, R. J., Fredburg, J. J., and Boucher,R. C. (2005). Normal and cystic fibrosis airway surface liquid homeostasis:the effects of phasic shear stress and viral infections. J Biol Chem 280,35751–9.
Tarran, R., Grubb, B. R., Parsons, D., Picher, M., Hirsh, A. J., Davis, C. W., andBoucher, R. C. (2001). The CF salt controversy: in vivo observations andtherapeutic approaches. Mol Cell 8, 149–58.
Tarran, R., Trout, L., Donaldson, S. H., and Boucher, R. C. (2006b). Solublemediators, not cilia, determine airway surface liquid volume in normaland cystic fibrosis superficial airway epithelia. J Gen Physiol 127, 591–604.
Thornton, D. J., and Sheehan, J. K. (2004). From mucins to mucus: toward a morecoherent understanding of this essential barrier. 10.1513/pats.2306016.Proc Am Thorac Soc 1, 54–61.
Van’s Gravesande, K. S., and Omran, H. (2005). Primary ciliary dyskinesia:clinical presentation, diagnosis and genetics. Ann Med 37, 439–49.
Vernon, G. G., Neesen, J., and Woolley, D. M. (2005). Further studies on knock-out mice lacking a functional dynein heavy chain (MDHC7). 1. Evidencefor a structural deficit in the axoneme. Cell Motil Cytoskeleton 61, 65–73.
West, S. E., Zeng, L., Lee, B. L., Kosorok, M. R., Laxova, A., Rock, M. J.,Splaingard, M. J., and Farrell, P. M. (2002). Respiratory infections withPseudomonas aeruginosa in children with cystic fibrosis: early detectionby serology and assessment of risk factors. JAMA 287, 2958–67.
Williams, O. W., Sharafkhaneh, A., Kim, V., Dickey, B. F., and Evans, C. M.(2006). Airway mucus: from production to secretion. Am J Respir CellMol Biol 34, 527–36.
Wine, J. J., and Joo, N. S. (2004). Submucosal glands and airway defense. ProcAm Thorac Soc 1, 47–53.
Woolley, D. M., Neesen, J., and Vernon, G. G. (2005). Further studies on knock-out mice lacking a functional dynein heavy chain (MDHC7). 2. A devel-opmental explanation for the asthenozoospermia. Cell Motil Cytoskeleton61, 74–82.
Worlitzsch, D., Tarran, R., Ulrich, M., Schwab, U., Cekici, A., Meyer, K. C.,Birrer, P., Bellon, G., Berger, J., Weiss, T., Botzenhart, K., Yankaskas, J.R., Randell, S., Boucher, R. C., and Doring, G. (2002). Effects of reducedmucus oxygen concentration in airway Pseudomonas infections of cysticfibrosis patients. J Clin Invest 109, 317–25.
Zaas, A. K., and Schwartz, D. A. (2005). Innate immunity and the lung: defenseat the interface between host and environment. Trends Cardiovasc Med15, 195–202.
Zariwala, M. A., Leigh, M. W., Ceppa, F., Kennedy, M. P., Horvath, J., Olbrich,H., Loges, N. T., Duriez, B., Escudier, E., Mitchison, H. M., Chodhari,R., Chung, E. M., Morgan, L. M., de Iongh, R. U., Rutland, J., Pradal, U.,Omran, H., Amselem, S., and Knowles, M. R. (2006). Mutations of DNAI1in primary ciliary dyskinesia: evidence of founder effect in a commonmutation. Am J Respir Crit Care Med 174, 858–66.
Zariwala, M., O’Neal, W. K., Noone, P. G., Leigh, M. W., Knowles, M. R.,and Ostrowski, L. E. (2004). Investigation of the possible role of a novelgene, DPCD, in primary ciliary dyskinesia. Am J Respir Cell Mol Biol 30,428–34.
at ECONOMICA on October 21, 2010tpx.sagepub.comDownloaded from





























