Embed Size (px)
Citation preview

Tolerance of a Phage Element by Streptococcus pneumoniae Leads to aFitness Defect during Colonization
Hilary K. DeBardeleben,a Elena S. Lysenko,a Ankur B. Dalia,c Jeffrey N. Weisera,b
Departments of Microbiologya and Pediatrics,b University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA; Department of Molecular Biology andMicrobiology, Tufts University School of Medicine, Boston, Massachusetts, USAc
The pathogenesis of the disease caused by Streptococcus pneumoniae begins with colonization of the upper respiratory tract.Temperate phages have been identified in the genomes of up to 70% of clinical isolates. How these phages affect the bacterialhost during colonization is unknown. Here, we examined a clinical isolate that carries a novel prophage element, designatedSpn1, which was detected in both integrated and episomal forms. Surprisingly, both lytic and lysogenic Spn1 genes were ex-pressed under routine growth conditions. Using a mouse model of asymptomatic colonization, we demonstrate that the Spn1�
strain outcompeted the Spn1� strain >70-fold. To determine if Spn1 causes a fitness defect through a trans-acting factor, weconstructed an Spn1� mutant that does not become an episome or express phage genes. This mutant competed equally with theSpn1� strain, indicating that expression of phage genes or phage lytic activity is required to confer this fitness defect. In vitro, wedemonstrate that the presence of Spn1 correlated with a defect in LytA-mediated autolysis. Furthermore, the Spn1� strain dis-played increased chain length and resistance to lysis by penicillin compared to the Spn� strain, indicating that Spn1 alters thecell wall physiology of its host strain. We posit that these changes in cell wall physiology allow for tolerance of phage gene prod-ucts and are responsible for the relative defect of the Spn1� strain during colonization. This study provides new insight into howbacteria and prophages interact and affect bacterial fitness in vivo.
Streptococcus pneumoniae, or the pneumococcus, is a Gram-positive opportunistic pathogen responsible for �1 million
deaths worldwide each year (http://www.who.int/immunization/monitoring_surveillance/burden/estimates/Pneumo_hib/en/). Al-though it is a leading source of infection, the pneumococcus is mostcommonly found in a commensal relationship with its human host(1). Pneumococcal colonization is characterized by sequential andoverlapping episodes that each last for days to months. Colonizingpneumococci provide the reservoir of organisms causing disease andare the main source of bacteria for host-to-host transmission (2).Therefore, colonizing pneumococci are likely to be the major focus ofselective pressure and adaptation for the organism. During coloniza-tion, pneumococci encounter various challenges, one of which is pre-dation by bacteriophages. Up to 70% of clinical isolates of S. pneu-moniae harbor genetic elements resembling prophages based onthe presence of the phage-encoded lysin, which is a homologue ofthe chromosomally encoded cell wall amidase, LytA (3). Howthese prophage elements affect the physiology of their pneumo-coccal host during colonization has not been fully explored.
The predator-prey dynamic between phages and the bacteriathey infect has been an important factor driving bacterial evolu-tion (4). This arms race has resulted in the evolution of phages thatpromote the fitness of their bacterial host, including its survivalwithin a mammalian host and ability to cause disease (5). Forexample, prophages may encode toxins or other virulence factorsthat increase the ability of their bacterial host to persist duringinfection. Phages may also impact genetic diversity within a spe-cies through lysogenic conversion or horizontal gene transfer,which facilitates bacterial adaptation to the host environment(e.g., antibiotic resistance) (6).
In contrast to the positive effects of phages on bacterial patho-genesis, few studies have assessed the negative effect of phages onbacterial survival during infection. Aspects of the temperate phagelife cycle that could have a negative impact are phage lysis, in-
creased burden of DNA and protein synthesis, and cis-acting ef-fects on genes flanking the phage attachment site. The variety ofphage resistance mechanisms employed by bacteria indicates thatnegative effects of carrying prophage are a significant factor inbacterial fitness (7).
In the case of the pneumococcus, prophage SV-1 has beenshown to enhance biofilm formation by increasing amounts ofextracellular DNA (8). Similarly, pneumococcal prophage MM1has been shown to increase bacterial adherence to human epithe-lial cells in culture (9). However, the contribution of these or otherpneumococcal phages to positive or negative effects on bacterialfitness in vivo has not been demonstrated.
Using a newly identified prophage, Spn1, found in a highlysuccessful pneumococcal lineage (10), and a well-establishedmouse model (11), we examined how the prophage affects S.pneumoniae during colonization. The bacterial isolate harboringSpn1 was obtained from a human challenge study and shown to befit for colonizing the natural host for up to 122 days (10) and themurine nasopharynx for 21 days (11). We find that the Spn1 ele-ment is stably present and transcriptionally active in this isolate.However, the presence of Spn1 correlates with a negative impacton bacterial fitness during colonization. We also provide evidencethat Spn1 is associated with changes in the cell wall physiology ofits host. We posit that this change is a mechanism for tolerance ofphage products, which may be the underlying cause of this fitnessdefect.
Received 10 February 2014 Accepted 5 May 2014
Published ahead of print 9 May 2014
Address correspondence to Jeffrey N. Weiser, [email protected].
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JB.01556-14
2670 jb.asm.org Journal of Bacteriology p. 2670 –2680 July 2014 Volume 196 Number 14
on June 19, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

MATERIALS AND METHODSBacterial growth conditions. Unless noted otherwise, bacteria weregrown in C�Y medium, pH �6.8 (12), without shaking in sealed tubesat 37°C. Growth was monitored via absorbance readings at 620 nm.Bacteria were plated on blood agar plates or tryptic soy medium (TS)plus catalase (4,741 U/plate) (Worthington Chemicals). C�Y was sup-plemented with streptomycin (200 �g/ml), kanamycin (500 �g/ml),erythromycin (1 �g/ml), neomycin (5 �g/ml), or spectinomycin (200�g/ml) where indicated.
Sequencing of Spn1. P1121, a bacterial isolate from a human chal-lenge study (Table 1), was sequenced on an Illumina HiSeq and de novoassembled using CLC’s Main Workbench (CLC bio). Two assembled con-tigs containing prophage coding regions were identified by NCBI’s BLASTprogram. The gap between the two contigs was sequenced using Sangersequencing technology. Once the sequence was complete, the genome wassubmitted to RAST for annotation (13).
RNA isolation and qRT-PCR. RNA was isolated using the TRIzolreagent and protocol (Life Technologies), with modifications. Bacteriawere harvested at mid-log phase, and a 2:1 volume of RNAprotect bacte-rial reagent (Qiagen) was added to the culture. Bacteria were then resus-pended in TRIzol, frozen, and then disrupted by bead beating for 5 min.RNA was isolated by phenol-chloroform extraction as written in theTRIzol protocol. After resuspension of the RNA in RNase-free water,samples were treated with DNase (Qiagen) for 30 min at 37°C. DNasewas inactivated at 65°C for 10 min before RNA was used to make cDNA.Quantitative reverse transcription-PCR (qRT-PCR) was carried out usinga high-capacity cDNA reverse transcription kit (Applied Biosystems).Quantitative PCR was performed with the Power SYBR green PCR mastermix (Applied Biosystems). mRNA levels were quantified using �CT
(threshold cycle) values and compared to an internal control, gyrA. Prim-ers are listed in Table 2.
DNase protection assay. Supernatants from cultures left untreated ortreated with 5 �g/ml mitomycin C (Sigma-Aldrich) were filtered througha 0.22-�m-syringe filter. Samples were divided and either left untreated ortreated with DNase (final concentration, roughly 270 Kunitz units/ml)(Qiagen) and incubated for 30 min at 37°C. DNase was then inactivated at65°C for 10 min. DNA was quantified via quantitative PCR using thePower SYBR green PCR master mix (Applied Biosystems).
Generation of mutants. Bacterial mutants were created in the P1121background by first amplifying regions upstream and downstream of the geneof interest via PCR. For each mutant, a set of primers 1 to 6 was designed.Primers amplifying the flanking regions are designated 1 and 2 (upstream)and 5 and 6 (downstream). Antibiotic cassettes were amplified using primers
3 and 4. PCR was carried out using standard techniques with either Taq DNApolymerase (Invitrogen) and related reagents for non-cloning-related PCRassays or Platinum Pfx polymerase (Invitrogen) and related reagents for PCRrelated to cloning. Primers used are listed in Table 2. Primers 2 to 5 weretagged with an overlapping sequence. A DNA fragment was then createdusing overlap-extension PCR (14) with the amplified regions flanking theantibiotic resistance cassette of choice. Antibiotic resistance markers usedwere erythromycin resistance, Emr (pCR2.1-TOPO plasmid with Emr inser-tion from pMU1328 [15]), kanamycin resistance, Knr (Janus cassette [16]),and spectinomycin resistance, Spr (pCR2.1-TOPO with Spr insertion frompBI143 [aad9; GenBank accession number U30830] [17]). Each of the mark-ers used contains its own promoters. This PCR fragment was transformeddirectly into bacteria using a transformation protocol as previously described(18). Mutants with an unmarked deletion were made via the Janus cassette aspreviously described (16). Primers and antibiotic cassettes used for each mu-tant strain are listed in Table 2. Constructs were confirmed by sequencingacross junctions.
Mitomycin C assays. Bacteria were grown to mid-log phase and thendiluted to an optical density at 620 nm (OD620) of 0.1 in C�Y mediumwith or without 0.5 �g/ml of mitomycin C (Sigma-Aldrich). Growth at37°C without shaking was recorded as the OD620 more than 4 h aftertreatment. Samples were collected for RNA isolation 2 h after addition ofmitomycin C. Control strains TIGR4 (MM1�) and TIGR4M (MM1�)were obtained from V. Fischetti, Rockefeller University (Table 1).
Mouse model of colonization. Bacteria were grown to mid-log phase(OD620 of 0.5) in C�Y medium. One ml of culture was pelleted andresuspended in 100 �l of phosphate-buffered saline (PBS). For competi-tive assays, bacteria were combined in a 1:1 ratio after resuspension. Ten�l of resuspended bacteria was dropped onto the nostrils of nonanesthe-tized wild-type female C57BL/6 mice (6 to 8 weeks of age). Inoculumcounts were �1 � 106 CFU/ml. For competitive assays, at least 50 coloniesof the inoculum were plated to determine streptomycin sensitivity orresistance and, thereby, the input ratio. Seven days after inoculation, nasallavages were taken as previously described (19) and plated on TS withneomycin (5 �g/ml) to determine CFU counts. For competitive assays, atleast 50 colonies from each mouse were tested for streptomycin resistanceor sensitivity to determine an output ratio. Competitive indices (CIs) werecalculated as (output ratio)/(input ratio). Statistics were calculated on logvalues of all competitive indices.
In vitro competitive growth assay. Bacterial strains were grown to anOD620 of 0.5 in C�Y and then diluted to an OD620 of 0.1. One ml of eachstrain and 2 ml of fresh culture, for a total of 4 ml, were combined to start
TABLE 1 Bacterial strains
Strain no. Designation Background Descriptiona Source or reference
P1121 Spn1� Human challenge study isolate, serotype 23F 11P1397 Spn1� Smr P1121 Smr spontaneous mutation This studyP2197 MM1� TIGR4 10P2198 MM1� TIGR4 10P2282 Spn1� �lytA P1121 lytA::Spr This studyP2308 Spn1� �mml P1121 mml::Kmr This studyP2345 Spn1� �SP_1563 P1121 SP_1563::Spr This studyP2348 Spn1� �SP_1564 P1121 SP_1564::Emr This studyP2352 Spn1� Smr P1397 Unmarked in-frame Spn1 deletion, Smr This studyP2373 Spn1� �pblB P1121 pblB::Emr This studyP2379 Spn1� �lytA P2385 lytA::Spr transformed with P2282 This studyP2380 Spn1� �mml �lytA P2308 mml::Kmr lytA::Spr transformed with P2282 This studyP2385 Spn1� P2352 Transformed with P1121 to remove Smr This studyP2386 Spn1� �int �mml P2385 mml::Kmr int::Emr transformed with P2387 This studyP2387 Spn1� �int �mml P2308 mml::Kmr int::Emr transformed with P2388 This studyP2388 Spn1� �int P1121 int::Emr This studya Smr, streptomycin resistant; Spr, spectinomycin resistant; Kmr, kanamycin resistant; Emr, erythromycin resistant.
Pneumococcal Phage Affects Colonization Fitness
July 2014 Volume 196 Number 14 jb.asm.org 2671
on June 19, 2018 by guesthttp://jb.asm
.org/D
ownloaded from
the assay. Cultures were incubated, without shaking, at 37°C and platedfor bacterial counts at 0 h and 5 h on selective media for an input andoutput ratio, respectively, to calculate the competitive index.
Growth and autolysis assays. Bacteria were diluted from mid-log-phase culture to a low starting OD620 (�0.05), and then they were grownfor 24 h in a 96-well plate in an incubating plate reader set at 37°C in 180
�l C�Y medium with 10 �l of catalase (30,000 U/ml) (WorthingtonChemicals). Penicillin sensitivity was determined across a range of con-centrations (50 ng/ml exceeded the MIC for both strains, defined as totalinhibition of growth) under these conditions. Absorbance at 600 nm wasmeasured every 15 min with 5 s of shaking on a low setting before everyreading. To maximize autolysis, cocultures were carried out in brain heart
TABLE 2 Primers for mutant construction and qRT-PCR
Primer Gene Direction Sequence Source
AttF SP_1563 Forward TGTGGGTGGTGGTCCTGTCG This studyAttR SP_1564 Reverse ATGCCAAACTGGCCCGTCAC This studyEGP4 int Forward GAAGATAGGAGGATAAACTGG 22EGP9 mml Reverse AACTGCAGAAATTGTTCTTTCACCGCAGG 22Spn1-2 mml Reverse CATTATCCATTAAAAATCAAACAACTGCAGAAATTGTTCTTTCACCGCAGG This studySpn1-3 Janus Forward CCTGCGGTGAAAGAACAATTTCTGCAGTTGTTTGATTTTTAATGGATAATG This studySpn1-4 Janus Reverse CCAGTTTATCCTCCTATCTTCCTTTCCTTATGCTTTTGGAC This studySpn1-5 int Forward GTCCAAAAGCATAAGGAAAGGAAGATAGGAGGATAAACTGG This study1563-1 SP_1561 Forward TATAGCCGCCCGGTGTCTGG This study1563-2 SP_1563 Reverse CACTTTATTAATTTGTTCGTATGTATTCAGTTTCTCCTTTGTTTTTTCTAGTCAGTTTAT This study1563-3 SpecR Forward ATAAACTGACTAGAAAAAACAAAGGAGAAACTGAATACATACGAACAAATTAATAAAGTG This study1563-4 SpecR Reverse CGAATCCTATGTGACTCGTGGTTCTTTTTTCCCGAGCTCGAATTGACGCGGATCC This study1563-5 SP_1563 Forward GGATCCGCGTCAATTCGAGCTCGGGAAAAAAGAACCACGAGTCACATAGGATTCG This study1563-6 mml Reverse CGCTGCAACAGGCTGGCAGA This study1564-1 int Forward TGGAGAAGAAAACTCCCCAGGCA This study1564-2 SP_1564 Reverse GGTGCAAGTCAGCACGAACGATATAAAGCAACCCCTTGAATTATCAA This study1564-3 ermR Forward ATAATTCAAGGGGTTGCTTTATATCGTTCGTGCTGACTTGCACC This study1564-4 ermR Reverse GATGCGCATTATACAGGTGAAAAATGAGTAACGTGTAACTTTCCAAAT This study1564-5 SP_1564 Forward ATTTGGAAAGTTACACGTTACTCATTTTTTCACCTGTATAATGCGCATC This study1564-6 SP_1565 Reverse GGCCAGCCGTCGAGTAGTGC This studymml-1 SP_1563 Forward GCAGCCTTTTATGCCCACCTACGCC This studymml-2 mml Reverse CTCTGGAATAGGCATAGACACTATCCACCGCAGGCTCAGGCTTGCGG This studymml-3 kanR Forward CCGCAAGCCTGAGCCCTGCGGTGGATAGTGTCTATGCCTATTCCAGAG This studymml-4 kanR Reverse GACGCATGGAAAGGACGATAGGGACACGTTTTTGTGGTGAGAAAC This studymml-5 mml Forward GTTTCTTCACCACAAAAACGTGTCCCTATCGTCCTTTCCATGCGTC This studymml-6 pblB Reverse CTGAGCATAAGGAATAGGAGGTGTG This studyint-1 int Forward ATGTGGATGGAAGAACTTTCCAAC This studyint-2 int Reverse GGTGCAAGTCAGCACGAAACAAGTGCTTTTATTTCTTGCATGGT This studyint-3 ermR Forward ACCATGCAAGAAATAAAAGCACTTGTTTCGTGCTGACTTGCACC This studyint-4 ermR Reverse GTACTTTTGGTGATATTCTCGACGATTAAGAGTAACGTGTAACTTTCCAAAT This studyint-5 int Forward ATTTGGAAAGTTACACGTTACTCTTAATCGTCGAGAATATCACCAAAAGTAC This studyint-6 int Reverse AGTATCTAATTTATTGACCAGTTTCTCCTCC This studyPblB-1 ORF61 Forward GAAGCTGGTAAAGAGGCGGT This studyPblB-2 pblB Reverse ATGGTGCAAGTCAGCACGAATCCTTGGCAAACATGGCTCT This studyPblB-3 ermR Forward AGAGCCATGTTTGCCAAGGATTCGTGCTGACTTGCACCAT This studyPblB-4 ermR Reverse ATGGTTTCAGCCTCTTGAGCTAGTAACGTGTAACTTTCCAA This studyPblB-5 pblB Forward TTGGAAAGTTACACGTTACTAGCTCAAGAGGCTGAAACCAT This studyPblB-6 ORF55 Reverse GTCAGGATTGCCCTCTGCAT This studyLytA-1 dinF Forward TTGGCTAGTTCGACAGATGGTTAC This studyLytA-2 lytA Reverse CACTTTATTAATTTGTTCGTATGTATTCTTCTACTCCTTATCAATTAAAACAACTCA This studyLytA-3 specR Forward TGAGTTGTTTTAATTGATAAGGAGTAGAAGAATACATACGAACAAATTAATAAAGTG This studyLytA-4 specR Reverse ATGCGCTGTTCTGATTTGAAAGACATTCCCCCGAGCTCGAATTGACGCGGATCC This studyLytA-5 lytA Forward GGATCCGCGTCAATTCGAGCTCGGGGGAATGTCTTTCAAATCAGAACAGCGCAT This studyLytA-6 SP_1935 Reverse TGAGTTCTATTGGCATTTTCTCTG This studyRTmmlF mml Forward CCACTCAACAGGCAACCGTA This studyRTmmlR mml Reverse CTTCCGTTGTTCACAGGACC This studyRTholinF holin Forward TTGCGTAAAGGCGGAGAGAA This studyRTholinR holin Reverse AGCTACTTGCTCAACGGCAT This studyRTPblBF pblB Forward TATCAAGGCCGAATCGGTGG This studyRTPblBR pblB Reverse GGCACCATTCCCCATACCAA This studyRTORF42F ORF42 Forward GGACGCAGACTACAGCAAGT This studyRTORF42R ORF42 Reverse CATATCGGCAGGCACGTACT This studyRTORF20F ORF20 Forward AAGAGTCAAATCGGTGGCGT This studyRTORF20R ORF20 Reverse GCTCCTTTGTCAGCAATCGAC This studyRTC1RepF C1Rep Forward TCGCACAGATAATCCTACCATCGCT This studyRTC1RepR C1Rep Reverse TGAAGCGCATAGCAGTAGAGGCA This studyRTintF int Forward CGACTTAGCGAACGTTTCCAGCCA This studyRTintR int Reverse ACGTGGGGGTCGTGGTGTCC This studyRT1563F SP_1563 Forward CGACCTCAACCGTCACAAGA This studyRT1563R SP_1563 Reverse TAGCAGCAGTCACCGATAGC This studyRT1564F SP_1564 Forward AGGTCGTTACGGGTTTCTCG This studyRT1564R SP_1564 Reverse ACCTATCTCCAGCGAGCAGA This studyRTgyrAF gyrA Forward GCCCTTTGGCAGTCCGACCA This studyRTgyrAR gyrA Reverse ACGTGGGGGTCGTGGTGTCC This study
DeBardeleben et al.
2672 jb.asm.org Journal of Bacteriology
on June 19, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

infusion medium (BHI), and 50 �l of each of the strains was grown tomid-log phase and diluted to a low OD620 (�0.05).
Western blotting. Whole-cell lysates were created by resuspendingbacteria (harvested at mid-log phase) in loading dye (30 mM Tris-HCl,pH 6.8, 2% SDS, 10% glycerol, and 2% �-mercaptoethanol) and incubat-ing them at 95°C for 5 min. Lysates were loaded onto a 10% glycine gel(Mini-Protean TGX gel; Bio-Rad) and run under standard conditions.Protein was transferred to a polyvinylidene difluoride (PVDF) membranevia a semidry transfer. After transfer, the membrane was allowed to dry,blocked with 2% bovine serum albumin (BSA) solution in phosphate-buffered saline, and incubated with a 1:100 dilution of anti-LytA rabbitIgG (obtained from R. Lopez, Biological Research Center, Madrid, Spain)or a 1:40 dilution of anti-pneumolysin mouse IgG (Novacastra Laborato-ries Ltd., Buffalo Grove, IL) for 2 h. This was followed by incubation withan appropriate secondary antibody conjugated to alkaline phosphatase(anti-rabbit IgG or anti-mouse IgG, respectively, diluted 1:5,000[Sigma]). Proteins were visualized with a 5-bromo-4-chloro-3=-indolyl-phosphate and nitroblue tetrazolium reaction.
Microscopy. Bacteria were grown to mid-log phase in C�Y medium.Samples were blinded before images at �400 magnification were taken ona Nikon E600 bright-field microscope. ImageJ analysis was performedusing the “threshold” function followed by the “analyze particles” func-tion to quantify particle size. For transmission electron microscopy, sam-ples were grown to mid-log phase and then pelleted. Pelleted bacteria werehigh-pressure frozen, freeze substituted into acetone with 2% OsO4 and0.1% uranyl acetate, embedded in EPON resin, cut into 70-nm sections,and imaged on a JEOL 1010 microscope on an AMT camera.
Nucleotide sequence accession number. The sequence for the phageelement Spn1 was deposited in GenBank under accession numberKJ417497.
RESULTSIdentification of phage element Spn1. Phage element Spn1(GenBank accession no. KJ417497) was first identified in a humancolonization isolate of Streptococcus pneumoniae via PCR usingthe prophage typing system described by Romero et al. (20). Sub-sequent sequencing of the entire locus revealed a novel phage el-ement, of �42 kb, most closely related to phage 040922 (GenBankaccession no. FR671406.1). However, the integrase (int) andlysin (mml) of Spn1 shared 99% and 95% identity, respectively,with the previously characterized pneumococcal phage MM1(GenBank accession no. AJ302074.2). The entirety of Spn1 wasnot identified in any previously published pneumococcal genome;therefore, it is not likely to be common. The sequence of phageelement Spn1 was annotated using RAST, which predicted 63open reading frames. Similar to lambda phage, the genome wasorganized into lytic (lysis, structural, and replication genes) andlysogenic functional clusters (Fig. 1A). The Spn1 phage elementintegrates between two convergently transcribed genes, annotatedas SP_1563 (a pyridine-nucleotide-disulfide-oxidoreductase) andSP_1564 (conserved hypothetical protein) in the TIGR4 genome(GenBank accession no. AE005672.3). The attachment site islikely to be the same attachment site for MM1, a 15-bp sequencelocated at the 3= end of the coding sequence for SP_1564 (Fig. 1B)(21). Neither flanking gene’s coding region is interrupted by inte-gration of Spn1. Both flanking genes are highly conserved amongS. pneumoniae genomes.
Spn1 activity. We designed PCR primers to amplify across theattachment site of both the prophage and the bacteria to deter-mine if Spn1 is capable of excising (Table 2). Both sets of primersamplified a product in noninduced genomic DNA samples, indi-cating a background level of spontaneous excision and circular-ization of Spn1 from the P1121 genome (Fig. 1C). Using qRT-
PCR on bacterial RNA isolated from noninduced log-phasecultures, we determined that Spn1 expresses its lysogenic genes asexpected but also expresses lytic genes, including those encodingits lysin (Mml) and holins (Fig. 1D). Despite the expression of lyticgenes, we were unable to identify plaques on any indicator strainusing a plaque assay that was previously described for S. pneu-moniae (22). Additionally, there was no evidence of phage parti-cles using a DNase protection assay or in transmission electronmicrographs of strain P1121. A selectable marker was insertedwithin Spn1 to determine whether the prophage could be cured, asindicated by the loss of the marker during passage. No Spn1�
revertants were identified during in vitro growth (spontaneouscure rate of 0.002 per generation). Spn1 was then removed fromstrain P1121 using Janus cassette technology to generate an un-marked, in-frame deletion, Spn1� (16).
Spn1 induction by mitomycin C. The P1121 lysogen wastreated with the DNA cross-linking agent mitomycin C (5 �g/ml)to trigger the SOS response and activate the Spn1 prophage. Mi-tomycin C treatment of strain TIGR4 with or without phage MM1served as controls. There was inhibition of growth correlating withthe presence of phage MM1 but no effect of mitomycin C on thegrowth of strain P1121 carrying Spn1 compared to the isogenicSpn1� construct (Fig. 2A). To confirm that the mitomycin Ctreatment was activating the Spn1 prophage under these condi-tions, phage gene expression was assessed. Gene expression wasgreatly increased in the presence of mitomycin C for lytic andlysogenic genes across Spn1 (Fig. 2B). Thus, Spn1 is transcription-ally activated by mitomycin C, but this treatment is not sufficientto induce lysis of its bacterial host.
Spn1 causes a defect in fitness in vivo. To determine the effectof Spn1 on its bacterial host in vivo, we used a murine nasopha-ryngeal colonization model. When tested individually, the Spn1�
and Spn1� strains each colonized mice at similar densities at 7days postinoculation (Fig. 3A). This time point was chosen be-cause it is the extent of a period of stable colonization beforeclearance becomes prominent (23). To compare their relative fit-nesses, we competed Spn1� and Spn1� strains 1:1 in the samemodel of colonization. At 7 days postinoculation, the Spn1�
strain significantly outcompeted the Spn1� strain (CI, 70) (Fig.3B). Competitive growth in vitro did not correlate with competi-tive effect in vivo, indicating that Spn1 confers a fitness disadvan-tage that is specific to the in vivo environment (Fig. 3B). In order todifferentiate strains postcolonization, one strain was marked withan rpsL mutation conferring resistance to streptomycin. Datashown is a combination of experiments with either the Spn1� orSpn1� strain having been marked to ensure that the streptomycinresistance phenotype was not the cause of the fitness effect in vitroor in vivo.
A trans-acting factor from Spn1 affects in vivo fitness. Toassess whether the effect of Spn1 on in vivo fitness was caused bycis-acting effects on the flanking genes, we generated mutants inSP_1563 and SP_1564 in strain P1121 and tested them in the col-onization model. The loss of SP_1563 resulted in a defect in colo-nization (Fig. 4A). To determine if Spn1 was affecting transcrip-tion of the flanking genes, we compared mRNA levels of Spn1�
and Spn1� strains using qRT-PCR. Expression of SP_1563 andSP_1564 was unaffected by the presence of Spn1 (Fig. 4B). There-fore, an effect of Spn1 on either flanking gene was unlikely to bethe cause of the fitness defect in vivo.
We attempted to reinsert the Spn1 prophage in the Spn1�
Pneumococcal Phage Affects Colonization Fitness
July 2014 Volume 196 Number 14 jb.asm.org 2673
on June 19, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

strain but were unable to identify transformants acquiring thislarge genetic element. By placing selectable markers within boththe int and mml genes at opposite poles of the prophage, it waspossible to obtain transformants of the Spn1� strain in which theentire locus was restored. However, interruption of both the intand mml genes resulted in a strain in which Spn1 no longer excisedspontaneously (Fig. 1C) or transcribed genes across the entireprophage (Fig. 4C). Theoretically, this mutant should maintainany cis-acting effects on the flanking genes but lack trans-actingeffects due to phage gene expression. The competitive index of theSpn1� �int �mml strain versus the Spn1� strain was not signifi-cantly different from 1, indicating that the two strains competeequally (Fig. 4D). This result suggests that a trans-acting factor
requiring phage gene expression is required for the effect of Spn1on in vivo fitness. This result also eliminated the possibility that theburden of carrying an additional �42 kb of DNA was responsiblefor our observations on fitness during colonization.
Three candidate genes were identified as potential trans-actingfactors in the fitness effect of Spn1. First, the phage lysin, Mml, wasinterrupted to create an Spn1� �mml strain. The competitive in-dex of Spn1� versus Spn1� �mml was significantly greater than 1,indicating that the Spn1-mediated fitness effect was not due toMml (Fig. 4E). The second candidate gene, PblB, a large phage tailprotein, was removed to create the Spn1� �pblB strain. Compe-tition between Spn1� and Spn1� �pblB resulted in a competitiveindex not significantly different from 1, indicating no competitive
FIG 1 Genetic map and activity of Spn1. (A) The strain P1121, including the genetic element Spn1, was sequenced on an Illumina Hi-Seq and annotated usingRAST. Sixty-three open reading frames were identified and are indicated as block arrows showing the direction of transcription. Genes were arranged in lytic orlysogenic clusters by putative function: lysis genes (black), structural genes (dark gray), replication genes (white), and lysogeny genes (light gray). (B) Schematicof Spn1 (circle) integration between genes SP_1563 and SP_1564 (dashed arrows show the direction of transcription). The attachment site, 15 bp at the 3= endof the coding region of SP_1564, is indicated by a black bar. The light gray bar indicates a noncoding intergenic region. Small arrows represent the location ofprimers. (C) PCR products showing the presence of a circularized element (primers a and b), integration at the attachment site (primers a and d), or theattachment site lacking Spn1 (primers c and d) in the indicated strains. Size markers are indicated in base pairs. (D) Levels of mRNA relative to the housekeepinggene gyrA as determined by qRT-PCR for genes across Spn1. The position of specific genes is designated in panel A and shaded to match the functional clusterin the genetic map. Data are shown as means � standard errors of the means (SEM).
DeBardeleben et al.
2674 jb.asm.org Journal of Bacteriology
on June 19, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

advantage for either strain (Fig. 4E). Lastly, the integrase gene(int), a DNA binding protein that mediates integration of thephage (24), was interrupted to create Spn1� �int. The competitiveindex of Spn1� versus Spn1� �int is significantly greater than 1,indicating that Spn1� has the fitness advantage. This leads us toconclude that Mml, PblB, or integrase alone does not confer thefitness defect to the Spn1� strain in vivo.
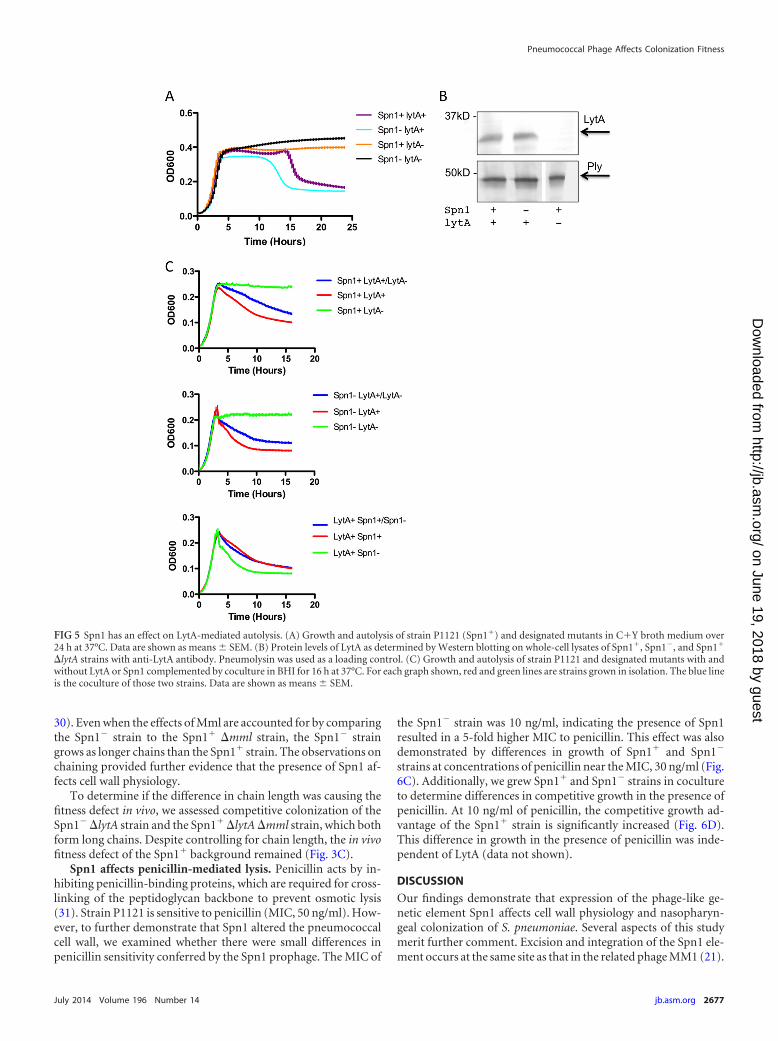
The presence of Spn1 correlates with delay in autolysis. Thephage lysin, Mml, and the major autolysin of S. pneumoniae, LytA,are homologous and thought to be functionally redundant cellwall amidases (22). In order to determine if autolysis is affected bythe presence of Spn1, we measured bacterial growth in nutrientbroth medium over a period of 24 h. Autolysis was delayed in the
Spn1� strain by �5 h (Fig. 5A). As a control, lytA was interruptedto ensure autolysis was LytA dependent in both the Spn1� andSpn1� strains. No autolysis was observed in the absence of LytA.In agreement with in vitro competition experiments (Fig. 3B), theSpn1� strain outgrew the Spn1� strain; however, this effect wasreversed in the absence of LytA (Fig. 5A). Genetic mutations inMml had no effect on growth or autolysis (data not shown). Wechecked levels of LytA expression by Western blotting and de-tected no difference in expression of LytA between Spn1� andSpn1� strains (Fig. 5B).
To determine if Spn1 affected LytA activity, we complementedLytA activity by comparing growth of LytA� and LytA� bacteriasingly and in a coculture. If active, the LytA secreted from the
FIG 2 Activation of Spn1 by mitomycin C. (A) Growth of strain P1121 with or without Spn1 and strain TIGR4 with or without MM1 in C�Y medium.Mitomycin C (5 �g/ml) was added at time zero. (B) Relative transcript abundances as determined by qRT-PCR for the Spn1 gene indicated relative to the internalcontrol, gyrA, is shown relative to treatment without mitomycin C. Data are shown as means � SEM. One-sample t test was used to determine if the relativeincrease in gene expression was greater than 1. *, P 0.05.
FIG 3 Spn1 in colonization. (A) Density of Spn1� and Spn1� in nasal lavages obtained 7 days after inoculation. The dashed line indicates the limit of detection.Comparisons between strains were made using the Mann-Whitney test. (B) Competitive indexes (CI; output ratio/input ratio) of Spn1�/Spn1� following growthfor 5 h in C�Y medium (in vitro) or 7 days of colonization (in vivo). (C) Competitive indexes for Spn1� �lytA/Spn1� �lytA and Spn1� �lytA/Spn1� �lytA�mmlto determine the competitive advantage without autolysis and with equivalent chain length, respectively. Each symbol is from a single mouse or culture. The CIis based on patching �50 colonies on selective medium to distinguish strains. One-sample t test was used to determine if CIs were significantly different from ahypothetical value of 1, which would indicate no competitive advantage or disadvantage. *, P 0.05; ***, P 0.001; ns, nonsignificant.
Pneumococcal Phage Affects Colonization Fitness
July 2014 Volume 196 Number 14 jb.asm.org 2675
on June 19, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

LytA� strain should be able to lyse the LytA� strain in coculture,resulting in a growth curve similar to that of an LytA� straingrown in isolation. We measured the growth of a coculture ofLytA� and LytA� strains in both Spn1� and Spn1� backgrounds.As expected, the growth pattern of the cocultured strains mim-icked that of an LytA� strain grown in isolation, indicating LytA isactive and able to lyse LytA� strains in both the Spn1� and theSpn1� backgrounds (Fig. 5C). This was confirmed by plating bac-teria and colony counts at 0, 5, and 8 h (data not shown). To test ifLytA’s target, the cell wall, is altered in the Spn1� strain, we cocul-tured the Spn1� and Spn1� strains where both strains containedLytA. The growth curve mimicked the curve of Spn1� strainsgrown in isolation (Fig. 5C). This led us to conclude that Spn1confers resistance to LytA-mediated autolysis.
LytA has previously been associated with virulence (25). Todetermine if the delay in autolysis was the cause of the fitnessdefect in vivo, we competed Spn1� �lytA and Spn1� �lytA strainsin a 1:1 ratio in the murine model of colonization. The competi-tive advantage of the Spn1� strain was lytA independent (Fig. 3C).
Thus, the altered cell wall physiology conferred by Spn1 may resultin a fitness defect in vivo independent of its effects on promotingresistance to LytA-mediated autolysis.
Spn1 affects chain length. LytA is also involved in cleavage ofthe cell wall to allow for cell separation during division. Deletionof lytA leads to the formation of long chains in some strains ofpneumococci (26). Additionally, chain length has been shown tohave an important role during both colonization and invasivedisease (27, 28). Using bright-field microscopy, we observed thatthe mean chain length of the Spn1� strain was greater than that ofthe Spn1� strain (Fig. 6A and B). The role of the phage-encodedhomologue Mml in septation and chain length has not been elu-cidated. Surprisingly, deletion of lytA or mml did not lead to longchains in Spn1� bacteria. However, Spn1� �lytA �mml bacteriadid form long chains, a result consistent with the functional re-dundancy of these amidases. An alignment of the amino acid se-quences of Mml and LytA showed that all catalytically active res-idues and those involved in dimerization are conserved, which isconsistent with their functional redundancy (data not shown) (29,
FIG 4 Spn1 gene expression is required for the fitness defect in vivo. (A) Density of Spn1�(WT), Spn1� �SP_1563, and Spn1� �SP_1564 in nasal lavagesobtained 7 days after inoculation. Comparisons between mutant and WT strains were made by Mann-Whitney test. (B) Levels of mRNA relative to the internalcontrol, gyrA, as determined by qRT-PCR of genes SP_1563 and SP_1564 in the presence (black bars) or absence (white bars) of Spn1. Data are shown asmeans � SEM. (C) Levels of mRNA compared to the internal control, gyrA, as determined by qRT-PCR for sample phage genes in either the Spn1� strain or theSpn1� �int �mml strain. Expression was undetected (ND) in the Spn1� �int �mml strain. Data are shown as means � SEM. (D) Competitive indexes ofSpn1�/Spn1� �int �mml following growth for 5 h in C�Y medium or 7 days of colonization. (E) Competitive indexes of Spn1� �pblB/Spn1�, Spn1�/Spn1�
�mml, and Spn1�/Spn1� �int following 7 days of colonization. One-sample t test was used to determine significant differences from a hypothetical value of 1,which would indicate no competitive advantage. *, P 0.05; **, P 0.01; ns, nonsignificant.
DeBardeleben et al.
2676 jb.asm.org Journal of Bacteriology
on June 19, 2018 by guesthttp://jb.asm
.org/D
ownloaded from
30). Even when the effects of Mml are accounted for by comparingthe Spn1� strain to the Spn1� �mml strain, the Spn1� straingrows as longer chains than the Spn1� strain. The observations onchaining provided further evidence that the presence of Spn1 af-fects cell wall physiology.
To determine if the difference in chain length was causing thefitness defect in vivo, we assessed competitive colonization of theSpn1� �lytA strain and the Spn1� �lytA �mml strain, which bothform long chains. Despite controlling for chain length, the in vivofitness defect of the Spn1� background remained (Fig. 3C).
Spn1 affects penicillin-mediated lysis. Penicillin acts by in-hibiting penicillin-binding proteins, which are required for cross-linking of the peptidoglycan backbone to prevent osmotic lysis(31). Strain P1121 is sensitive to penicillin (MIC, 50 ng/ml). How-ever, to further demonstrate that Spn1 altered the pneumococcalcell wall, we examined whether there were small differences inpenicillin sensitivity conferred by the Spn1 prophage. The MIC of
the Spn1� strain was 10 ng/ml, indicating the presence of Spn1resulted in a 5-fold higher MIC to penicillin. This effect was alsodemonstrated by differences in growth of Spn1� and Spn1�
strains at concentrations of penicillin near the MIC, 30 ng/ml (Fig.6C). Additionally, we grew Spn1� and Spn1� strains in cocultureto determine differences in competitive growth in the presence ofpenicillin. At 10 ng/ml of penicillin, the competitive growth ad-vantage of the Spn1� strain is significantly increased (Fig. 6D).This difference in growth in the presence of penicillin was inde-pendent of LytA (data not shown).
DISCUSSION
Our findings demonstrate that expression of the phage-like ge-netic element Spn1 affects cell wall physiology and nasopharyn-geal colonization of S. pneumoniae. Several aspects of this studymerit further comment. Excision and integration of the Spn1 ele-ment occurs at the same site as that in the related phage MM1 (21).
FIG 5 Spn1 has an effect on LytA-mediated autolysis. (A) Growth and autolysis of strain P1121 (Spn1�) and designated mutants in C�Y broth medium over24 h at 37°C. Data are shown as means � SEM. (B) Protein levels of LytA as determined by Western blotting on whole-cell lysates of Spn1�, Spn1�, and Spn1�
�lytA strains with anti-LytA antibody. Pneumolysin was used as a loading control. (C) Growth and autolysis of strain P1121 and designated mutants with andwithout LytA or Spn1 complemented by coculture in BHI for 16 h at 37°C. For each graph shown, red and green lines are strains grown in isolation. The blue lineis the coculture of those two strains. Data are shown as means � SEM.
Pneumococcal Phage Affects Colonization Fitness
July 2014 Volume 196 Number 14 jb.asm.org 2677
on June 19, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

Through PCR analysis, it appears that a circularized form of theprophage occurs during normal bacterial growth, although wewere unable to detect packaged phage by transmission electronmicroscopy or DNase protection assays. We were also unable todemonstrate phage infectivity by plaque assays (even after induc-tion by mitomycin C), transduction, or detection of lysogenicconversion using a marked phage after coculture, in vitro or invivo. Furthermore, we were unable to transfer phage Spn1 to otherpneumococcal backgrounds, including TIGR4 and R36A. It re-mains possible that under different growth conditions or in itsnatural environment, Spn1 forms infectious phage particles. Spn1could have become defective after infecting this strain, or it couldhave been obtained through transformation rather than infection.It was unexpected that the lytic genes of Spn1 were expressedduring normal growth, yet we did not detect increased bacteriallysis compared to that of an isogenic strain lacking Spn1. Thecircularized form may be a precursor to Spn1 gene expression(and allow for amplification of gene expression), since mutationsat both ends of the element in Spn1� �int �mml prevented bothcircularization and expression of lytic and lysogenic genes. Theinability of Spn1 to silence expression of lytic genes, which is nottypical of lambdoid phages, appears to have required adaptationby the host bacterium (32).
The tolerance of prophage gene expression, even when furtherinduced with mitomycin C, suggests that the phage lysins, whichusually target the cell wall, are ineffective against strain P1121. It ispossible that the lysins are inactive or only a small portion of thepopulation is being activated by mitomycin C. However, our ob-servations suggest that strain P1121 modifies its cell wall so that it
is less susceptible to lysis. Several lines of evidence point to analteration in cell wall physiology when this strain carries Spn1. TheSpn1� strain has a delayed autolysis phenotype not due to anyalteration in amounts or activity of the major amidase responsiblefor autolysis, LytA. This suggests that LytA’s target, the cell wall, ismore resistant to its enzymatic activity. Moreover, the Spn1�
strain showed an increased resistance to penicillin. However, wedid not detect an increased amount of peptidoglycan when puri-fied from the Spn1� strain or a thicker cell wall in electron micro-graphs of the Spn1� strain (data not shown). The effect of peni-cillin suggests that alteration of the cell wall is due to increasedcross-linking, since this is the step inhibited by the drug. Increasedintegrity through more extensive cross-linking within the cell wallcould also explain the increased resistance to LytA and delayedautolysis, phenotypes previously linked to altered penicillin resis-tance (33, 34). Finally, alterations in cell wall physiology may beseen as differences in cell division/separation resulting in abnor-mal chain formation. The presence of Spn1 correlated with de-creased average chain length.
Studies have shown that lytA mutants do not autolyze and insome strains display increased chain length (26). However, in thisstudy, the Spn1� strain demonstrated delayed autolysis, indicat-ing resistance to LytA, but had shorter chains, which is usuallylinked to an increase in susceptibility to LytA. This apparent dis-crepancy could be indicative of differences in secretion and local-ization of amidases on the bacterial cell surface. A recent study onMml and LytA showed that phosphorylcholine is required forsecretion of both of these lysins. The authors of that study pro-posed that LytA binds the phosphorylcholine as it is added to the
FIG 6 Spn1 has an effect on the cell wall. (A) Images of strain P1121 and the mutant indicated in mid-log-phase culture using a bright-field microscope. The barindicates 10 �m. (B) Average pixels per particle as determined by ImageJ analysis to quantify chain length. Student t test was used to compare groups. ***, P 0.001. ns, nonsignificant. Data are shown as means � SEM. (C) Growth of Spn1� and Spn1� strains in 30 ng/ml of penicillin in tryptic soy broth for 5 h at 37°C.Data are shown as means � SEM. (D) Competitive indexes (CI; output ratio/input ratio) of Spn1�/Spn1� following growth for 5 h with increasing concentra-tions of penicillin. Each symbol is from a single culture. A Student t test was used to determine significant differences between groups. ***, P 0.001.
DeBardeleben et al.
2678 jb.asm.org Journal of Bacteriology
on June 19, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

cell wall during division (35). This is supported by another studydemonstrating that LytA is localized to the nascent peptidoglycan(33). This suggests that the change in the cell wall that leads toresistance to LytA in the Spn1� strain occurs during maturation ofthe peptidoglycan, leaving the nascent peptidoglycan formed dur-ing division susceptible to LytA-mediated cleavage, resulting inreduced chain length. In contrast, despite conservation of catalyticamino acid residues between Mml and LytA, Mml was function-ally redundant only in the chain length phenotype. This also maybe due to localization of Mml at the nascent peptidoglycan andindicative of separate regulatory machinery for LytA secretionduring autolysis.
The precise nature of the alteration in the pneumococcal cellwall required for tolerance of Spn1 is the subject of further inves-tigation. Factors regulating cell wall synthesis, quantity of pepti-doglycan per cell, and level of cross-linking are incompletely un-derstood. These differences could be the direct action of anunknown protein on the prophage. However, it is possible that theoverproduction of prophage products, including lysin and holins,leads to a general stress response in the host bacterium. Stressresponses in bacteria often induce changes in the cell wall. Forexample, in Staphylococcus aureus there is evidence of cell wallthickening in the presence of vancomycin (36). Also, the CiaRHand WalRK two-component systems in S. pneumoniae arethought to maintain cell wall integrity in the presence of variousstressors (37, 38).
The presence of Spn1 was associated with reduced fitness dur-ing competitive colonization, an effect that may be related to thechanges in its cell wall. There are several phenotypes associatedwith the presence of Spn1 that may affect fitness during coloniza-tion. Most notably, greater chain length increases adherence toepithelial cells (28), and the presence and action of LytA is impor-tant during pathogenesis, although the exact mechanism is un-clear (25). However, when we eliminate differences in chainlength and the effects of LytA but maintain the presence of Spn1,we still observe a fitness defect in the Spn1� strain during com-petitive colonization. This implies that the inherent differences inthe cell wall, not these related phenotypes, are responsible for thefitness defect in vivo. Theoretically, alteration of the cell wall inresponse to the prophage could be a metabolic burden on theSpn1� strain, which gives the Spn1� strain the edge in competitivecolonization. The in vitro competition data support this concept,since the competitive effect is lost in a nutrient-rich environment.Additionally, the cell wall anchors many factors linked to fitnessfor colonization, which also could be playing a role (39).
The competitive fitness defect and alteration of the cell wall areboth dependent on prophage gene expression. This indicates thata trans-acting factor encoded on the prophage is responsible forthese phenotypes. We eliminated the potential contribution ofthree candidate trans-acting factors, Mml, PblB, and integrase.Mml has previously been shown to be redundant with LytA inphage progeny release, indicating it contributes to bacterial lysis(22). Integrase is a DNA binding protein involved in the excisionand integration of the prophage (24). PblB was originally anno-tated on phages of Streptococcus mitis and is thought to mediateadherence to platelets (40). We saw no differences in adherence ofthe Spn1� and Spn1� bacteria to A549 human epithelial cells inculture (data not shown). We also investigated the role of theputative holins encoded upstream of Mml. We found that dele-tion of the holins did not alter the phenotype of the Spn1� strain
in any in vitro assays and did not improve the fitness of the Spn1�
strain in vivo (data not shown). While we eliminated several po-tential trans-acting factors, there are �63 open reading framesannotated on Spn1, many of which have an unknown functionand could be affecting bacterial fitness. The increased protein syn-thesis burden of carrying Spn1 also could account for the effect onfitness in vivo.
The question remains as to why the bacterium does not spon-taneously cure Spn1 if it is associated with a fitness cost. S. pneu-moniae is highly proficient at recombination and it should readilyeliminate any disadvantageous genetic elements, yet Spn1 remainsstably integrated (41). Moreover, in a study of human experimen-tal pneumococcal colonization, Spn1 was present in the initialclinical isolate used to inoculate several volunteers and also in allof the isolates retrieved from participants up to 122 days later (10and data not shown). The success of this lineage may be due to afactor found in the human host, such as exposure to penicillin, anatural population of phage, or competition with other pneumo-cocci. The human host from which P1121 or its predecessor wereisolated may have been exposed to penicillin, thereby selecting forthe presence of Spn1. Also, colonization by multiple strains ofpneumococci commonly occurs in children (42). Studies done inE. faecalis, a close relative of S. pneumoniae, show that lysogens arebetter able to compete against strains of the same species by way ofphage lysis (43). Furthermore, prophages are known to protectagainst superinfection by other related phages. Therefore, it ispossible that selective pressure from penicillin, other pneumo-cocci, or other phages during colonization of a human host wassufficient to maintain this element.
In conclusion, we have determined that a novel prophage,Spn1, is associated with altering the bacterial cell wall and a fitnessdefect during competitive colonization. This type of interactionbetween a prophage and its bacterial host, where the bacteria tol-erate the phage despite its negative effects, has, to our knowledge,not been described previously.
ACKNOWLEDGMENTS
This work was supported by grants from The National Institutes of Healthto J.N.W. (R01 AI078538 and T32 A1060516) and the Morphology Coreof the Center for Molecular Studies in Digestive and Liver Diseases (NIHP30 DK050306).
We also thank the Electron Microscopy Resource Laboratory.
REFERENCES1. Bogaert D, De Groot R, Hermans PWM. 2004. Streptococcus pneumoniae
colonisation: the key to pneumococcal disease. Lancet Infect. Dis. 4:144 –154. http://dx.doi.org/10.1016/S1473-3099(04)00938-7.
2. Weiser JN. 2010. The pneumococcus: why a commensal misbehaves. J.Mol. Med. 88:97–102. http://dx.doi.org/10.1007/s00109-009-0557-x.
3. Ramirez M, Severina E, Tomasz A. 1999. A high incidence of prophagecarriage among natural isolates of Streptococcus pneumoniae. J. Bacteriol.181:3618 –3625.
4. Weinbauer MG, Rassoulzadegan F. 2004. Are viruses driving microbialdiversification and diversity? Environ. Microbiol. 6:1–11. http://dx.doi.org/10.1046/j.1462-2920.2003.00539.x.
5. Brüssow H, Canchaya C, Hardt W-D. 2004. Phages and the evolution ofbacterial pathogens: from genomic rearrangements to lysogenic conver-sion. Microbiol. Mol. Biol. Rev. 68:560 – 602. http://dx.doi.org/10.1128/MMBR.68.3.560-602.2004.
6. Ubukata K, Konno M, Fujii R. 1975. Transduction of drug resistance totetracycline, chloramphenicol, macrolides, lincomycin and clindamycinwith phages induced from Streptococcus pyogenes. J. Antibiot. 28:681– 688.http://dx.doi.org/10.7164/antibiotics.28.681.
Pneumococcal Phage Affects Colonization Fitness
July 2014 Volume 196 Number 14 jb.asm.org 2679
on June 19, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

7. Labrie SJ, Samson JE, Moineau S. 2010. Bacteriophage resistance mechanisms.Nat. Rev. Microbiol. 8:317–327. http://dx.doi.org/10.1038/nrmicro2315.
8. Carrolo M, Frias MJ, Pinto FR, Melo-Cristino J, Ramirez M. 2010.Prophage spontaneous activation promotes DNA release enhancing bio-film formation in Streptococcus pneumoniae. PLoS One 5:e15678. http://dx.doi.org/10.1371/journal.pone.0015678.
9. Loeffler JM, Fischetti VA. 2006. Lysogeny of Streptococcus pneumoniaewith MM1 phage: improved adherence and other phenotypic changes.Infect. Immun. 74:4486 – 4495. http://dx.doi.org/10.1128/IAI.00020-06.
10. McCool TL, Cate TR, Moy G, Weiser JN. 2002. The immune response topneumococcal proteins during experimental human carriage. J. Exp. Med.195:359 –365. http://dx.doi.org/10.1084/jem.20011576.
11. Wu HY, Virolainen A, Mathews B, King J, Russell MW, Briles DE. 1997.Establishment of a Streptococcus pneumoniae nasopharyngeal colonizationmodel in adult mice. Microb. Pathog. 23:127–137. http://dx.doi.org/10.1006/mpat.1997.0142.
12. Lacks S, Hotchkiss RD. 1960. A study of the genetic material determiningan enzyme in pneumococcus. Biochim. Biophys. Acta 39:508 –518. http://dx.doi.org/10.1016/0006-3002(60)90205-5.
13. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA,Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, OlsonR, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T,Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V,Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations usingsubsystems technology. BMC Genomics 9:75. http://dx.doi.org/10.1186/1471-2164-9-75.
14. Higuchi R, Krummel B, Saiki RK. 1988. A general method of in vitropreparation and specific mutagenesis of DNA fragments: study of proteinand DNA interactions. Nucleic Acids Res. 16:7351–7367. http://dx.doi.org/10.1093/nar/16.15.7351.
15. Achen MG, Davidson BE, Hillier AJ. 1986. Construction of plasmidvectors for the detection of streptococcal promoters. Gene 45:45– 49. http://dx.doi.org/10.1016/0378-1119(86)90130-7.
16. Sung CK, Li H, Claverys JP, Morrison DA. 2001. An rpsL cassette, janus,for gene replacement through negative selection in Streptococcus pneu-moniae. Appl. Environ. Microbiol. 67:5190 –5196. http://dx.doi.org/10.1128/AEM.67.11.5190-5196.2001.
17. Smith CJ, Rollins LA, Parker AC. 1995. Nucleotide sequence determi-nation and genetic analysis of the Bacteroides plasmid, pBI143. Plasmid34:211–222. http://dx.doi.org/10.1006/plas.1995.0007.
18. Hsieh Y-C, Wang J-T, Lee W-S, Hsueh P-R, Shao P-L, Chang L-Y, LuC-Y, Lee C-Y, Huang F-Y, Huang L-M. 2006. Serotype competence andpenicillin resistance in Streptococcus pneumoniae. Emerg. Infect. Dis. 12:1709 –1714. http://dx.doi.org/10.3201/eid1211.060414.
19. McCool TL, Weiser JN. 2004. Limited role of antibody in clearance of Strep-tococcus pneumoniae in a murine model of colonization. Infect. Immun.72:5807–5813. http://dx.doi.org/10.1128/IAI.72.10.5807-5813.2004.
20. Romero P, García E, Mitchell TJ. 2009. Development of a prophagetyping system and analysis of prophage carriage in Streptococcus pneu-moniae. Appl. Environ. Microbiol. 75:1642–1649. http://dx.doi.org/10.1128/AEM.02155-08.
21. Gindreau E, López R, García P. 2000. MM1, a temperate bacteriophageof the type 23F Spanish/U. S. A. multiresistant epidemic clone of Strepto-coccus pneumoniae: structural analysis of the site-specific integration sys-tem. J. Virol. 74:7803–7813. http://dx.doi.org/10.1128/JVI.74.17.7803-7813.2000.
22. Frias MJ, Melo-Cristino J, Ramirez M. 2009. The autolysin LytAcontributes to efficient bacteriophage progeny release in Streptococcuspneumoniae. J. Bacteriol. 191:5428 –5440. http://dx.doi.org/10.1128/JB.00477-09.
23. Van Rossum AMC, Lysenko ES, Weiser JN. 2005. Host and bacterialfactors contributing to the clearance of colonization by Streptococcuspneumoniae in a murine model. Infect. Immun. 73:7718 –7726. http://dx.doi.org/10.1128/IAI.73.11.7718-7726.2005.
24. Obregón V, García P, López R, García JL. 2003. Molecular and biochem-ical analysis of the system regulating the lytic/lysogenic cycle in the pneu-mococcal temperate phage MM1. FEMS Microbiol. Lett. 222:193–197.http://dx.doi.org/10.1016/S0378-1097(03)00281-7.
25. Orihuela CJ, Gao G, Francis KP, Yu J, Tuomanen EI. 2004. Tissue-specific contributions of pneumococcal virulence factors to pathogenesis.J. Infect. Dis. 190:1661–1669. http://dx.doi.org/10.1086/424596.
26. Sanchez-Puelles JM, Ronda C, Garcia JL, Garcia P, Lopez R, Garcia E.
1986. Searching for autolysin functions. Characterization of a pneumo-coccal mutant deleted in the lytA gene. Eur. J. Biochem. 158:289 –293.
27. Dalia AB, Weiser JN. 2011. Minimization of bacterial size allows forcomplement evasion and is overcome by the agglutinating effect of anti-body. Cell Host Microbe 10:486 – 496. http://dx.doi.org/10.1016/j.chom.2011.09.009.
28. Rodriguez JL, Dalia AB, Weiser JN. 2012. Increased chain length pro-motes pneumococcal adherence and colonization. Infect. Immun. 80:3454 –3459. http://dx.doi.org/10.1128/IAI.00587-12.
29. Mellroth P, Sandalova T, Kikhney A, Vilaplana F, Hesek D, Lee M,Mobashery S, Normark S, Svergun D, Henriques-Normark B, AchourA. 2014. Structural and functional insights into peptidoglycan access forthe lytic amidase LytA of Streptococcus pneumoniae. mBio 5:e01120 –13.http://dx.doi.org/10.1128/mBio.01120-13.
30. Romero P, López R, García E. 2007. Key role of amino acid residues in thedimerization and catalytic activation of the autolysin LytA, an importantvirulence factor in Streptococcus pneumoniae. J. Biol. Chem. 282:17729 –17737. http://dx.doi.org/10.1074/jbc.M611795200.
31. Waxman DJ, Strominger JL. 1983. Penicillin-binding proteins and themechanism of action of beta-lactam antibiotics. Annu. Rev. Biochem.52:825– 869. http://dx.doi.org/10.1146/annurev.bi.52.070183.004141.
32. Ptashne M. 2004. A genetic switch: phage lambda revisited. Cold SpringHarbor Laboratory Press, Cold Spring Harbor, NY.
33. Mellroth P, Daniels R, Eberhardt A, Rönnlund D, Blom H, Widengren J,Normark S, Henriques-Normark B. 2012. LytA, major autolysin of Strepto-coccus pneumoniae, requires access to nascent peptidoglycan. J. Biol.Chem. 287:11018 –11029. http://dx.doi.org/10.1074/jbc.M111.318584.
34. Novak R, Braun JS, Charpentier E, Tuomanen E. 1998. Penicillin tol-erance genes of Streptococcus pneumoniae: the ABC-type manganese per-mease complex Psa. Mol. Microbiol. 29:1285–1296. http://dx.doi.org/10.1046/j.1365-2958.1998.01016.x.
35. Frias MJ, Melo-Cristino J, Ramirez M. 2013. Export of the pneumococ-cal phage SV1 lysin requires choline-containing teichoic acids and is ho-lin-independent. Mol. Microbiol. 87:430 – 445. http://dx.doi.org/10.1111/mmi.12108.
36. Cui L, Ma X, Sato K, Okuma K, Tenover FC, Mamizuka EM, GemmellCG, Kim M-N, Ploy M-C, El-Solh N, Ferraz V, Hiramatsu K. 2003. Cellwall thickening is a common feature of vancomycin resistance in Staphy-lococcus aureus. J. Clin. Microbiol. 41:5–14. http://dx.doi.org/10.1128/JCM.41.1.5-14.2003.
37. Mascher T, Heintz M, Zähner D, Merai M, Hakenbeck R. 2006. TheCiaRH system of Streptococcus pneumoniae prevents lysis during stressinduced by treatment with cell wall inhibitors and by mutations in pbp2xinvolved in beta-lactam resistance. J. Bacteriol. 188:1959 –1968. http://dx.doi.org/10.1128/JB.188.5.1959-1968.2006.
38. Dubrac S, Bisicchia P, Devine KM, Msadek T. 2008. A matter of life anddeath: cell wall homeostasis and the WalKR (YycGF) essential signal trans-duction pathway. Mol. Microbiol. 70:1307–1322. http://dx.doi.org/10.1111/j.1365-2958.2008.06483.x.
39. Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role ofStreptococcus pneumoniae virulence factors in host respiratory coloniza-tion and disease. Nat. Rev. Microbiol. 6:288 –301. http://dx.doi.org/10.1038/nrmicro1871.
40. Bensing BA, Siboo IR, Sullam PM. 2001. Proteins PblA and PblB ofStreptococcus mitis, which promote binding to human platelets, are en-coded within a lysogenic bacteriophage. Infect. Immun. 69:6186 – 6192.http://dx.doi.org/10.1128/IAI.69.10.6186-6192.2001.
41. Hiller NL, Ahmed A, Powell E, Martin DP, Eutsey R, Earl J, Janto B,Boissy RJ, Hogg J, Barbadora K, Sampath R, Lonergan S, Post JC, HuFZ, Ehrlich GD. 2010. Generation of genic diversity among Streptococcuspneumoniae strains via horizontal gene transfer during a chronic poly-clonal pediatric infection. PLoS Pathog. 6:e1001108. http://dx.doi.org/10.1371/journal.ppat.1001108.
42. Brugger SD, Frey P, Aebi S, Hinds J, Mühlemann K. 2010. Multiplecolonization with S. pneumoniae before and after introduction of the sev-en-valent conjugated pneumococcal polysaccharide vaccine. PLoS One5:e11638. http://dx.doi.org/10.1371/journal.pone.0011638.
43. Duerkop BA, Clements CV, Rollins D, Rodrigues JLM, Hooper LV.2012. A composite bacteriophage alters colonization by an intestinal com-mensal bacterium. Proc. Natl. Acad. Sci. U. S. A. 109:17621–17626. http://dx.doi.org/10.1073/pnas.1206136109.
DeBardeleben et al.
2680 jb.asm.org Journal of Bacteriology
on June 19, 2018 by guesthttp://jb.asm
.org/D
ownloaded from





























