Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1994 by The American Society for Biochemistry and Molecular Biology, Inc
Vol. 269, No. 35, Issue of September 2, pp. 22282-22294, 1994 Printed in U.S.A.
GreA-induced Transcript Cleavage in Transcription Complexes Containing Escherichia coli RNA Polymerase Is Controlled by Multiple Factors, Including Nascent Transcript Location and Structure*
(Received for publication, January 21, 1994, and in revised form, May 12, 1994)
GuoHua FengS, Donna N. Lee+§, Daguang WangS, Cathleen L. ChanS, and Robert Landickall From the Departments of $Biology and IBiochemistry and Molecular Biophysics, Washington University, St. Louis, Missouri 63130
The Escherichia coli GreA and GreB proteins induce cleavage of 3’ fragments from nascent transcripts in halted transcription complexes. We have overproduced and purified the GreA protein and tested how it affects initiation, pausing, and termination by E. coli RNA po- lymerase. Recombinant GreA induced cleavage of two to three nucleotide fragments in two promoter-proximal complexes, whereas an apparently endogenous cleavage removed a single larger fragment. Both types of cleav- age stopped once the transcript was shortened to -10 nucleotides. However, during initiation, GreA induced cleavage of transcripts as short as four nucleotides, in- hibiting their release as abortive products and stimulat- ing both productive initiation and “primer-shifting” at a weak promoter. GreA induced repetitive cleavage over a long distance in complexes containing a long G-less nas- cent transcript. However, reverse translocation was in- hibited in transcription complexes that contained a G- rich, C-less nascent transcript. Substituting IMP for GMP in the transcript relieved inhibition. Finally, GreA had little effect on transcription through the his and trp leader pause sites or on termination at nine different p-independent terminators. We propose that transcript cleavage and reverse translocation are controlled in part by backsliding of the nascent transcript through an RNA-binding site.
Until recently, transcription was considered a simple, repeat- ing process of nucleotide addition to the 3’-end of the growing transcript and translocation of RNA polymerase on the DNA template. This view was challenged by the discovery in 1989 that 2-10-ntl fragments can be hydrolyzed off the 3’-end of the nascent transcript and that elongation resumes from the newly generated 3”OH on the upstream transcript fragment upon addition of NTPs (Surratt et al., 1991). Subsequent studies soon
GM38660 and National Science Foundation Grant DMB-8957331 (to R. * This work was supported by National Institutes of Health Grant
L.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisernent” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Center, Durham, NC 27710. 5 Present address: Dept. of Biochemistry, Duke University Medical
11 To whom corresuondence should be addressed, Tel.: 314-935-6029: ,, ~~ ~~.
Fax: 314-935-4432. 1
The abbreviations used are: nt, nucleotide; bp, base paifis); dNTPs, deoxynucleoside triphosphates; HPLC, high-pressure liquid chromatog- raphy; IPTG, isopropylthiogalactoside; nt, nucleotide(s1; NTPs, nucleo- side triphosphates; PCR, polymerase chain reaction; pol 11, RNA polym- erase 11; BSA, bovine serum albumin.
revealed that either of two -17-kDa cellular polypeptides, GreA and GreB, greatly stimulate the cleavage reaction (Borukhov et al., 1992, 1993a).
GreA and GreB possess similar amino acid sequences but stimulate different cleavage activities. Whereas GreA induces removal of two to three nucleotide fragments, GreB may cause removal of up to a 12-nt fragment. Transcript cleavage in ar- rested complexes reactivates transcription (Borukhov et al., 1993a; Krummel and Chamberlin, 1992). Both GreA and GreB prevent formation of arrested complexes on an rrnB template, but only GreB rescues arrested complexes once they form (Borukhov et al., 1993a). RNA polymerase itself may be capable of transcript cleavage, which is merely stimulated by GreA or GreB.
The mechanism of action and biological role of these proteins are not known. However, transcript cleavage appears to be an evolutionarily conserved function. The eukaryotic elongation factor TFIIS (S-11) induces a cleavage reaction in pol I1 tran- scription complexes similar to the effect of GreA and GreB on Escherichia coli complexes (reviewed in Kassavetis and Gei- duschek, 1993). TFIIS-induced cleavage allows read-through of several different pol I1 arrest sites i n vitro (Izban and Luse, 1992a, 1992b, 1993a, 1993b; Reines, 1992; Gu et al., 1993; Guo et al., 1993; Wang and Hawley, 1993). Transcript cleavage also occurs in purified transcription complexes containing vaccinia virus RNA polymerase, one subunit of which (Rpo30) is similar in sequence to TFIIS (Hagler and Shuman, 1993).
Although no sequence similarity is evident between the pro- karyotic and eukaryotic cleavage factors, their ubiquity sug- gests that transcript cleavage must play an important role in transcription or its regulation. Two possibilities are (i) prevent- ing accumulation of arrested complexes on the DNA (Reines, 1992) and (ii) increasing transcriptional fidelity by removal of misincorporated nucleotides (Erie et al., 1993; Kassavetis and Geiduschek, 1993; Wang and Hawley, 1993).
Several important questions about the effects of transcript cleavage on RNA synthesis remain to be answered. Although GreA and GreB stimulate transcript cleavage in several differ- ent promoter-proximal transcription complexes i n vitro, their effect on transcription complexes well removed from the pro- moter region has not been tested. The discovery of transcript cleavage raised the question of whether previous studies of pausing, termination, and antitermination could have been compromised by the presence of cleavage factors in the RNA polymerase preparations used (Borukhov et al., 1992). Tran- script cleavage could be a component of these regulatory mechanisms; it also might affect initiation or elongation of RNA chains.
To address these questions, we developed a simple purifica- tion procedure for GreA, which relied on overexpression of re-
22282

GreA-induced Dunscript Cleavage 22283
combinant GreAin E. coli and its resistance to irreversible heat denaturation. Using well-developed methods for in vitro tran- scription with purified E. coli RNA polymerase, we tested the effects of GreA on several different steps in the transcription cycle. We report here results that, together with other studies of transcript cleavage, are most easily explained by a model in which the rate of transcript cleavage is governed by backsliding of the nascent transcript through the active site. This process probably is controlled by different interactions between RNA polymerase and the 3'-proximal region of transcript, an up- stream segment of transcript held in a tight RNA-binding site, and the DNA template.
MATERIALS AND METHODS Enzymes and Reagent~-[cr-~~P]GTP and [a-32PlCTP were purchased
from Amersham Corp. HPLC-purified NTPs were obtained from Phar- macia LKB Biotechnology Inc. 3"dNTPs were obtained from Boeh- ringer Mannheim. ApU dinucleotide, Sephadex G-50, and heparin were obtained from Sigma. Restriction endonucleases and other DNA-modi- fying enzymes were obtained from New England Biolabs or United State Biochemicals Inc. (Cleveland, OH) and used according to the manufacturer's instructions.
RNA polymerase was purified from E. coli strain MRE6OO (Grain Processing Co. Muscatine, I A ) by the method of Burgess and Jendrisak (1975) with the modifications described by Hager et al. (1990). GreB was undetectable in this preparation by quantitative Western blotting with anti-GreB polyclonal antisera (ECLTM detection method, Amersham), which had a limit of detection of 0.1 ng GreB/pg RNA polymerase (less than -1:375 molar ratio; data not shown). Anti-GreA antisera revealed 0.5 ng GreNpg RNA polymerase (-1:75 molar ratio).
Isolation of the GreA Gene and Construction of an Overproducing Plasmid-Based on the DNA sequence of greA reported by Sparkowski and Das (19901, we synthesized two DNA oligonucleotides: 5'-CAC- GAGCTCAAGCTATTCCGATGACCTT and B'-CACAAGCTTGAGTATT- GGGTAATTCTTA (underlined nucleotides define the restriction sites SacI and HindIII, respectively; italicized nucleotides correspond to the 5'- and 3'-ends of the greA gene, respectively). We used the oligonucle- otides to amplifygreA by PCR from chromosomal DNA obtained from E. coli strain JM109 (Yanisch-Perron et al., 1985). The PCR-amplified DNA fragment was digested with SacI, treated with T4 DNA polymer- ase, and then digested with HindIII. The expression plasmid pTrc99c (Amann et al., 1988) was digested with NcoI, repaired with the Klenow fragment of DNA polymerase I and then digested with HindIII. Ligation of these fragments yielded pDNL278, which contained an intact greA gene under the control of the inducible trc promoter. We verified the complete DNA sequence of greA in pDNL278 by dideoxy sequencing of the double-stranded plasmid DNA.
Purification of GreA Protein-Overproduction and purification of GreA was followed by SDS-polyacrylamide gel electrophoresis (Fig. 1). Plasmid pDNL278 was transformed into E. coli strain JM109. 2.4 liters of 2 x LB medium with 100 pg ampicillidml was inoculated with an overnight culture of the transformed cells. Expression of GreA was induced by addition of IPTG to 1 mM when the culture reached early log phase (-20 Klett units). Growth was continued for another 3 h, and the cells were harvested at Klett -90 by centrifugation (if not stated oth- erwise, all centrifugation was at 10,000 x g for 15 min, and all purifi- cation steps were conducted at 4 "C). The cell pellet was suspended in 150 ml of lysis buffer (0.1 M Tris-HCI, pH 8.0,O.l M NaCl, 10 mM EDTA, 34 p~ phenylmethylsulfonyl fluoride, and 3 mg lysozyme/ml) and blended at low speed in a Waring Blender for 2 min. After 20 min a t 4 "C, 50 ml of 2% sodium deoxycholate was added, and the mixture was blended at high speed for 30 s. Cell debris was removed by centrifuga- tion, and 6 ml of 10% dialyzed polyethyleneimine was added to the supernatant (0.3% final concentration). The precipitate was removed by centrifugation, and the supernatant was heated at 68-70 "C for 20 min. Heat-insoluble material was removed by centrifugation, and powdered ammonium sulfate was added to the supernatant to a concentration of 1.5 M. After 30 min at 4 "C, the precipitate was removed by centrifuga- tion, more ammonium sulfate was added to a final concentration of 3.0 M, and the mixture was left at 4 "C overnight. The 1.5-3.0 M ammonium sulfate fraction was collected by centrifugation at 10,000 x g for 30 min, and the protein pellet was dissolved in 40 ml of TE (50 mM Tris-HC1, pH 8.0, 5 mM EDTA). After filtration through a 0.22-pm filter, the solution was loaded on a Mono Q fast protein liquid chromatography column previously equilibrated with TE buffer and eluted with a gradient of
NaCl in TE (Borukhov et al., 1992; Fig. 1B). DNA lkmplates-All templates used in this study were amplified
from plasmid DNA by PCR (Higuchi et al., 1988) using oligonucleotides that hybridized upstream from the promoter and downstream from the region of interest. All but one (amplified from pDW135, see below) contained the wild-type T7 A1 promoter or a derivative with an different initially transcribed region, and form either A20 or G16 complexes when incubated with RNA polymerase, ApU dinucleotide, ATP, CTP, and GTP (see below).
Plasmids pAR1707 (T7 Te terminator; Studier and Rosenberg, 19811, pCPG-T3, pCPG-P14, pCPG-tonB, pCPG-rrnB T1, and pCPG-AtR2 con- tained the wild-type T7 A1 promoter and were obtained from M. Cham- berlin (Reynolds et al., 1992) (wild-type initial transcript sequence is 5'-AUCGAGAGGGACACGGCGAAU). Plasmid pRL407 encodes a slightly different initial transcript (5'-AUCGAGAGGGACACGGGGGG- GAUCC, underlined sequence corresponds to BamHI site) followed by the E. coli trp leader region pause and termination signals (Lee et al., 1990). Plasmids pRL522, which contains the E. coli trp leader region terminator, and pRL537, which contains the S. marcesens trp leader region terminator (Landick et al., 1990a), were derived from pRL418, which contains a short polylinker between the T7 A1 promoter and the rrnB T1T2 terminators (Chan and Landick, 1989). pRL522 contains an EcoRI-BamHI fragment from pRL448 (Landick et al., 1990b), repaired with the Klenow fragment of DNA polymerase I, and then ligated into the HincII site of pRL418.
Plasmid pCL185 contains Salmonella typhimurium his pause site and was derived from pCL208 (Chan and Landick, 1993) by a sponta- neous transversion of G to T at +17 of the T7 A1 transcription unit, so that it encodes the initial transcript 5'-AUCGAGAGGGACACGGUG- GAUCC and generates G16 complexes rather than A20 complexes in the absence of UTP. pDNL279 contains E. coli trp leader pause signal and was constructed by replacing the BamHI-Hind111 fragment of pCL185 with a BamHI-Hind111 fragment from pRL458 (Lee et al., 1990) so that it also generates G16 complexes in the absence of UTP.
Two other plasmids that were derived from pRL418 by PCR mu- tagenesis served as the starting point for several other plasmids. pRL550 specifies the initial transcript 5'-AUCCCACACCAAACACAC- CAUGGUACGUAGGAUCC (underlined sequence corresponds to re- striction endonuclease recognition sites for NcoI, SnaBI, and BamHI) and pRL551 specifies the initial transcript 5-AUCGAGAGCGACACGA- CACAUAUGUACGUACGGAUC (underlined sequence corresponds to restriction endonuclease recognition sites for NdeI, SnaBI, and BamHI). Both plasmids contain polylinker and other sequences down- stream from the BamHI site identical to those in pRL418 (Chan and Landick, 1989). pGF104 contains the terminator from the S. typhi- murium his leader region and was derived from pRL551 by insertion between the BamHI and SphI sites of a BglII-SphI DNA fragment with
AGAGAAAGCCCCCGGAAGATGCATCTI'CCGGGGGCTTTTTTMTG- the top-strand sequence: GATCTI'CCAGTGGTGCTAGCACGCT-
GCGCGCGATACAGACCGGTTCGAACAGGATAAAGAGGAACGCAG- AATGTTAGACAACACCCGCTTACGCATAGCTATTCAGAAATCAG- GCCGTTTAAGCGATGATTCACGAGAATTGCTGCTGGCCCGCTGCGGC- ATAAAAATTAATTTACACACTCAGCGCCTGATTGCGATGGCGGAA- AACATGCCGATTGATGACTCTAGACCTGCAGGCATG.
The G-less template plasmids pRL596 and pRL597 were derived from pRL550 and contain 147 and 392 bp G-less cassettes, respectively. pRL550 was digested with NcoI, treated with Mung Bean nuclease, and then digested with BamHI. The large fragment from this digestion was ligated to an ApaI-BamHI fragment from pRL542 (London et al., 1991) to create pRL596 and to a SacI to BamHI fragment from pC&T (Sawa- dogo and Roeder, 1985) to create pRL597. The ApaI and SacI ends of these fragments were made blunt with T4 DNA polymerase prior to cleavage with the second enzyme.
The C-less template plasmid, pDW135, was obtained from pRL418 by digestion with SacI, treatment with T4 DNApolymerase, digestion with XbaI to remove the T7 A1 promoter, and then ligation to an Eco47III- XbaI fragment from pDE13 (Erie et al., 1993) that contained the A P r promoter. The resulting plasmid, pDW1, was digested with XbaI and then ligated to an XbaI-NheI fragment from pRL542 (London et al., 1991). This plasmid, pDW3, was linearized with XbaI, and 1 pg was treated with 5 units of exonuclease I11 for 1 min in 15 pl of exonuclease buffer. The exonuclease-treated DNA was digested with Mung Bean nuclease and then recircularized by ligation. One plasmid recovered from this procedure, pDW135, contained a 135-bp C-less sequence downstream of the APr promoter.
In Vitro Dunscription Assays-Unless otherwise stated, all tran- scription reactions were performed in buffer A (20 mM Tris acetate, pH 8.0, 20 mM NaCI, 20 mM MgCl,, 14 mM P-mercaptoethanol, 2% (vh)

22284 GreA-induced Dunscript Cleavage glycerol, and 20 pg of acetylated BSNml. Transcription complexes halted in the promoter-proximal region (G16 for pCL185 and pDNL279, and A20 for all other templates) were formed by incubation of RNA polymerase and DNA template with 200 PM ApU, 2.5 p~ ATP and CTP, and 2.5 p~ [a-32PlGTP at 37 "C for 5-20 min.
To obtain G16 and A20 complexes free of NTPs, 10 pmol of RNA polymerase and 5 pmol of DNA template were first incubated at 37 "C for 5 min in 50 pl of bufferA and NTPs, then mixed with 50 pl of ice-cold dilution buffer (Mg2"free buffer A with 40 pg heparidml and 20 mM EDTA) and immediately loaded on a Sephadex G-50 spin column equili- brated with the dilution buffer.
The effect of GreA on transcription initiation was studied using the pRL522 and pRL550 templates. On the pRL522 template, RNA polym- erase, DNA template, and dinucleotide ApU were incubated at 37 "C for 10 min in 12 pl of buffer A. To start transcription, 8 p1 of nucleotide mix was added, producing final concentrations of 200 p~ ApU, 5 PM each of GTP, CTP, and ATP or 10 PM 3'-dATP (see figure legends). GreA, when added, was included in the NTP mix. Transcripts were labeled with either [a-32PlGTP or [a-32P]CTP (see Fig. 3 legend). On the pRL550 template, open complexes were first formed by incubating RNA polym- erase with DNA template at 37 "C for 10 min in buffer A. Initiation was then started by adding rifampicin to 10 pg/ml and NTPs to 10 p~ ATP, UTP, and 5 p~ [a-32PlCTP with or without GreA.
To obtain transcription complexes halted after transcription of the G-less regions of the pRL596 and pRL597 templates, open complexes were first formed by incubation of 10 pmol of RNA polymerase and 5 pmol of DNA in 25 p1 of buffer A at 37 "C for 10 min. Transcription was started by addition of 25 pl of initiation mix that yielded final concen- trations of 50 p ATP and UTP, 10 p~ [O~-~~PICTP (-200 CUmmol), and 10 pg rifampicidml. After incubation at 37 "C for 10 min, the reaction mixtures were diluted with 50 pl of 1 x PC buffer (20 mM Tris acetate, pH 8.0, 150 mM KCl, 4 mM magnesium acetate, 0.1 mM EDTA, 0.1 mM dithiothreitol, 20 pg acetylated BSNml, 8 pg heparidml, and 4% (vh) glycerol) and filtered through Sepharose CL-GB columns (0.8 x 15 cm; 7.5-ml bed volume) as described previously (Landick and Yanofsky, 1987).
To obtain transcription complexes halted after transcription of the C-less region of the pDW135 template, we used the same method as for the G-less templates but with either 50 p~ ATP, UTP, and 10 p~ [a-32PlGTP (200 CUmmol) or 160 PM ATP, 200 p~ ITP, and 15 p~ [a-32PlUTP (290 Ci/mmol) as final NTP concentrations (see figure leg- ends). The isolated transcription complexes were adjusted to the com- position of buffer A and then incubated with either BSA, GreA, or 2 mM PP, to measure transcript cleavage.
To obtain transcription complexes at either trp or his pause site, G16 complexes were formed on DNA templates from pDNL279 (for trp) or pCL185 (for his) and then elongated with 20 p~ GTP, 150 p each of ATP, CTP, and UTP, and 160 pg heparidml at 37 "C for 15 s, and then chilled rapidly in an ice-water slurry. The chilled reactions were imme- diately passed through Sepharose CL-GB gel filtration columns as de- scribed above.
To obtain trp A92 complexes (Fig. SA), the isolated trp paused tran- scription complexes were incubated for 15 min at 37 "C with 10 p~ each GTP, UTP, and CTP and then centrifuged through a Sephadex G-50 column (Levin et al., 1987). This yielded trp G84 complexes which were then incubated for 5 min at 37 "C with 10 p each ATP, GTP, and CTP and again centrifuged through Sephadex G-50. To obtain his All0 com- plexes, the isolated his paused transcription complexes were incubated for 15 min at 37 "C with 10 p~ each GTP, ATP, and CTP and then centrifuged through Sephadex G-50.
For termination, pausing, and transcript elongation experiments, the A20 or G16 complexes were elongated directly in the presence either BSA or GreA as indicated in figure and table legends. To generate a sequence ladder of RNA transcripts, open complexes or promoter-proxi- mal transcription complexes (G16 or A20) were elongated separately with 25 p~ NTPs and 25 PM of one of the 3'-deoxynucleotides at 37 "C for 10 min. All transcription reactions were stopped by addition of an equal volume of stop buffer (89 mM Tris-borate, pH 8.3, 2.5 mM EDTA, 10 M
urea, and 0.5% (wh) each bromphenol blue and xylene cyanol). The samples were heated at 65 "C for 2 min and electrophoresed through 7 M urea, 0.5 x TBE polyacrylamide gels (19:l acrylamidehis(acry1a- mide), if not stated otherwise).
Identification of Abortive Tkanscripts and Cleavage Products-The sequence of the wild-type T7 A1 promoter transcript is 5'-AUC- GAGAGGG... . To identify the RNA bands corresponding to abortive transcripts, we synthesized ApUpC, ApUpCpG, or ApUpCpGpdA using the following combinations of nucleotides: ApU+[a-32PlCTP, ApU+ CTP+[CY-~~P]GTP, and ApU+CTP+[a-32PlGTP+3'-dATP. Because these
short RNAs lack 5'- or 3'-phosphates, they migrate anomalously in a high percentage polyacrylamide, 7 M urea gel, in the order slowest to fastest of 3-, 4-, and 5-mer. Anomalous migration of short oligoribonucle- otides lacking terminal phosphates also is reported by Krummel(1989), who observed increasing mobility from 3- to 6-mer, then decreasing so 3- equals 9-mer. To confirm the identity of abortive RNAs that comigrated with these standards, we isolated them from the gel and treated them with either polynucleotide kinase or alkaline phosphatase. Kinase treatment increased the mobilities of these RNAs, whereas phosphatase treatment had no effect.
We assigned the GreA-induced cleavage products as 5'-phosphoryl dimers and trimers by three criteria: (i) their mobilities were un- changed by kinase treatment; (ii) their mobilities decreased to positions near ApUpC after treatment with alkaline phosphatase; (iii) in a 20% polyacrylamide, 7 M urea gel, they comigrated with pApU and pApUpC generated by kinase treatment of ApU or ApUpC with [y-32PlATP.
Quantitation of Proteins and RNA Tkanscripts-SDS-gel electro- phoresis was performed as described by Laemmli (1970). The amount of GreA in fractions during purification was estimated by scanning Coo- massie Blue-stained SDS-polyacrylamide gels with a computing densi- tometer (Molecular Dynamics), using purified GreA as standard. The total amount of protein in each fraction was determined with the Brad- ford assay (Bradford, 1976), using BSA as standard. Radioactivity in RNA transcripts was quantitated using an AMBIS radioanalytic imag- ing system or a Molecular Dynamics PhosphorImager. Termination ef- ficiencies and pause half-lives were calculated as described previously (Landick et al., 1990a; Lee et al., 1990).
RESULTS
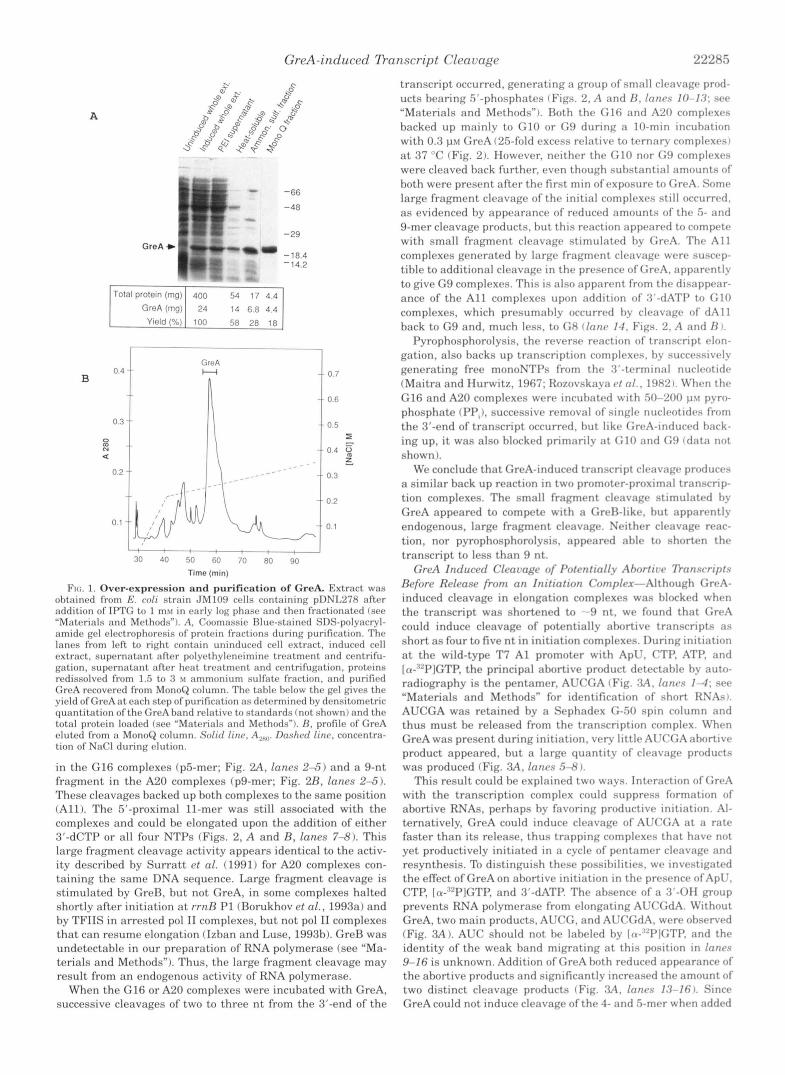
Overexpression and Purification of GreA Protein-We con- structed a GreA-overproducing plasmid, pDNL278, on which expression of GreA was driven by the trc promoter and could be induced with IPTG (see "Materials and Methods"). Exposure of cells containing pDNL278 to IPTG in early log phase caused overexpression of GreA to above 6% of the total soluble protein (Fig. L4). The overproduced GreAremained soluble and had no effect on cell growth when compared with the cells transformed with a control plasmid containing the greA gene in orientation opposite to the trc promoter (data not shown). While trying to simplify the published purification scheme (Borukhov et al., 19921, we found that GreA was heat stable. Thus, we purified GreA by heating the soluble cell extract to 68-70 "C for 20 min followed by differential ammonium sulfate precipitation and anion-exchange chromatography (Fig. 1; see "Materials and Methods"). Approximately 4.4 mg of purified GreA protein was obtained from 2.4 liters of cell culture (about 5 g of cell paste). The purified GreA protein had no nuclease activity when incu- bated with 32P-labeled RNA (data not shown) and gave the same transcript cleavage activity as the endogenous GreA pu- rified directly from E. coli strain MRE6OO by Borukhov et al. (1992).
GreA-induced Dunscript Cleavage in Tho Promoter-proxi- mal Halted Elongation Complexes Was Blocked a t Dunscript Lengths of 8-10 nt and Was Distinct from Endogenous Dan- script Cleavage-To characterize the activity of recombinant GreA protein, we first tested its effect on two promoter-proxi- mal halted elongation complexes, the G16 and A20 complexes. These transcription complexes were prepared on templates that contained a T7 A1 promoter followed by slightly different initially transcribed regions and isolated by centrifugal gel fil- tration (see "Materials and Methods"). GreA induced a similar pattern of transcript cleavage in both complexes (Fig. 2). A fraction of the complexes inactivated during the gel filtration, as evidenced by a portion of the G16 or A20 transcripts that subsequently could be neither cleaved nor elongated (Fig. 2). Control experiments suggested that at least some of the refrac- tory RNAs were released from the transcription complexes (data not shown).
When the G16 and A20 complexes were incubated without GreA, a one-step cleavage reaction occurred that removed a five-nt fragment from the 3"proximal portion of the transcript
GreA-induced Danscript C l c n u n g ~ 22285
A
- - 66 - -48 - -29
Yield (%) 58 28 18
B 0 4 1
0.3 1 7 0.2 .. "
-
-~ 0 7
- - 0 6
- 0.5 s
- - 0.4 $
- z
- - 0.3
- ~ 0.2
- - 0.1
I . I 30 40 50 60 70 BO 90
Time (min)
Ftc:. 1. Over-expression and purification of GreA. Extract was ohtainrd from E. colr strain J M l O I ) crlls containing pDN127X af t r r nddition of 11°K: to 1 mxt in rarly log phnsr and thrn fractionatrd (ser "Materials and Mrthotls"). A, Coomassic I3lur-stainrtl SDS-polvacryl- amitlr grl rlectrophorrsis of protrin fractions during purification. Thr l a w s from I r f t to right contain uninducrtl cell extract. intlucrd crll rxtract. suprrnntant nftrr polyrthylrneiminr trrntmrnt and crntrifu- gation, supc.rnal.ant aftrr h r a t trrntmrnt and centrifugation, proteins rrdissolvrd from 1.5 to 3 51 ammonium sulfntr fraction, and purifirtl GrrA recoverrd from MonoQ column. Thc, tahle hrlow t h r grl givrs the yield ofGrcAat rach strp o f purification as tlrtrrminrd hy drnsitometric quantitation ofthr GrrA hand relative- to stnndards (not shown) and the total protrin loadrd l s r r "Materinls and Methods"). R , profilr of GrrA rlutrtl from a MonoQ column. Solid linr, 1)nshvtl l i n r , concrntra- tion o f NaCI during rlution.
in the G16 complexes (p5-mer; Fig. 2 A , lanrs 2-5) and a 9-nt fragment in the A20 complexes (p9-mer; Fig. 2B, Innas 2-55). These cleavages backed up hoth complexes to the same position (All). The 5'-proximal 11-mer was still associated with the complexes and could be elongated upon the addition of either 3'-dCTP or a11 four NTPs (Figs. 2, A and B , lanes 7-8). This large fragment cleavage activity appears identical to the activ- ity described by Surrat t at nl. (1991) for A20 complexes con- taining the same DNA sequence. Large fragment cleavage is stimulated hy GreR, but not GreA, in some complexes halted shortly after initiation at rrnR P1 (Rorukhov at nl., 1993a) and by TFIIS in arrested pol I1 complexes, but not pol I1 complexes that can resume elongation (Izhan and Luse, 199%). GreR was undetectable in our preparation of RNA polymerase (see "Ma- terials and Methods"). Thus, the large fragment cleavage may result from a n endogenous activity of RNA polymerase.
When the G16 or A20 complexes were incuhated with GreA, successive cleavages of two to three nt from the 3'-end of the
transcript occurred, generating a group of small c l r a \ ~ ~ g r prod- ucts hearing 5'-phosphates (Figs. 2, A and /3, lnnc7.s lO-l.'l: s w "Materials and Methods"). Both the G l f i and A20 complrxrs backed up mainly to G I 0 or G9 during a 10-min incuhatinn with 0.3 par GreA (25-fnld ~ X C C S S relnt ivr t.o trrna? complrxrs I
at 37 "C (Fig. 2). However. neither the G10 nor G9 complrxvs were cleaved hack further, even though suhstnntial amounts of hoth were present after tho first min ofrxposurc to GrrA. Somr large fragment cleavage of the initial complexrs still occurrrd, as evidenced by appearance of reduced amounts of the. 5- and %mer cleavage products, hut this rraction appc.:lrrd to comprtr with small fragment clcavagr stimulated hy Grc-A. Thc All complexes generatrd hy Iargv fragment clenvagr wrrr suscc.p- tihle to additional cleavage in thr prrsrncr o f (;rrA. apparrntly to give G9 complexes. This is also apparvnt from thc. dis:lppc.nr- ance of the Al l complc.xcs upon addition of :1'-tiATP to ( ; l o complexes, which presumahly occurrrd hy cl(~~v:tgc. of dAl1 hack to G9 and, much less. to G8 r l n r r c - 1 4 , Figs. 2. A and /{ 1.
Pyrophosphorolysis, t h r revrrsc rt.action of transcript (*Ion- gation, also hacks up transcription comp1cxc.s. hy succrssivcLly generating free monoNTPs from the :l '-trrminnl nurlwticir (Maitra and Hurwitz. 1967; Rozovsknya vt o l . . 19H2 ). Whvn thtb G l 6 and A20 complexes wcrr incuhntc4 \\fith 50-200 p \ f pyro- phosphate (PI',), succrssive rt.moval of singlr nuclw)tidw from the 3'-end of transcript occurrcvi, hut likr (;rc.A-intluccd hack- ing up, i t was also hlocked primarily at (;10 and (;$) ( d a t a n o t shown 1.
We conclude that GrrA-induced transcript cl~:~v:~cr protiurw a similar back up reaction in two promotrr-proximal transcrip- tion complexes. The small f r a m c n t cle:~vage stimulatrd hy GreA appeared to compete with a Grt.B-likv. hut apparently endogenous, large f r a p e n t clcavagr. Nrither clr:~vag~ r e x - tion, nor pyrophosphorolysis, appeared ahlr to shnrtrn thc transcript to less than 9 nt.
GrrA Inducari Clcovngr of Potrrttinl1,v Ahortirv 7hnscr ip t . s Brfora Rrlrnsr from nn Initiation Complrx-Although GrrA- induced cleavage in elongation complexrs was hlocked whrn the transcript was shortrned to 9 nt. we found that GrrA could induce cleavage of potentially ahortivr transcripts as short as four to five nt in initiation complexes. During initiation a t t he wild-type T7 A1 promoter with ApC:, CTP, ATP, and Itr-T'IGTP, the principal abortive product d(.trctahle hy a u t n - radiography is the pentamer, AUC(;A (Fig. :<A, Innvs 1 - 4 ; s w "Materials and Methods" for identification of short RNAsr. AUCGA was retained by a Sephadcx G-50 spin column and thus must be released from the transcription complrx. Whrn GreAwas present during initiation, v e y littlrAUC(;A ahortivr product appeared. hut a large quantity of clravagc products was produced (Fig. 3A. loncs .5+ ).
This result could he explained two ways. Intwaction of GrrA with the transcription complex could suppress fnrmation of abortive RNAs, perhaps by favoring productivr initiation. AI- ternatively, GreA could induce clravngc of AUCGA a t a rntc faster than its release, thus trapping complexes that h:rw not yet productively initiated in a cycle of pc.ntamrr clravagr and resynthesis. To distinguish thwe possihi1itit.s. wv invrstigatcci the effect of GreA on ahortive initiation in the prrsrncr o f Apt'. CTP, [n-:"P]GTP, and 3'-dATP. The ahscncc. of a .7 ' -011 ~~oro r~p prevents RNA polymerase from elongating AUCGdA. LVithout GreA, two main products. AUCG, and AU(:GdA, werr ohsrned (Fig. 3A ). AUC should not he lahrlcd hy [n-"PIGTP, and thr identity of the weak hand mimating at this position in Ionrs 9-16 is unknown. Addition of (;reA hoth rcviucrd npprarnncc of the ahortive products and simificantlv incrrascd the amount of two distinct cleavage products (Fig. 3.4, Innrs 1.'-161. Sincr GreAcould not induce cleavage of the 4- anti 5-mrr whrn addrd

22286 GreA-induced Danscript Cleavage
I Control GreA
FIG. 2. GreA-induced cleavage in two promoter-proximal ternary com- plexes, G16 and A20. GI6 ( A ) and A20 ( R ) transcription complexes were formed on templates pCL185 and pCPG-T3 (see “Materials and Methods”). The complex- es were separated from unincorporated NTPs by centrifugation through Seph- adex G-50. About 0.6 pmol of isolated com- plexes were adjusted to buffer A and in- cubated with either acetylated DSA as a control (lanes 1-5) or with 10 pmol of GreA (lanrs 9-13) at 37 “C for the times indicated. Portions ( 5 pi) were removed from the reaction mixtures a t the times indicated and mixed with 5 pl of stop buffer. Lanes 6 4 and 14-16 in each panel contain samples incubated with the indi- cated NTPs after the 10-min cleavage re- action. The RNA transcripts were electro- phoresed through a 20% (20:2. acrylamidd his(acry1amidel) 7 h! urea 0.5 x TRE gel. The sequences of the G16 and A20 tran- scripts are shown below each panel. p5- and p9-mer indicate the large fragment cleavage products hearing 5‘-phosphates.
“A:: *
p5mer -+
Lanes : 2 3 L 5 6 7 8 9 10 11 :213141516 +I
AUCGAGAGGGACACGG U
pCL185 transcript A
r 15
G16
A
to reactions after they were released, nor cleave purified RNAs (data not shown), both potentially abortive RNAs must have been cleaved prior to release from the transcription complexes.
Since one cleavage product should be produced in each cycle of cleavage and resynthesis, the rate of short RNA synthesis in the absence or presence of GreA could be compared using the sum of the abortive and cleaved products. Addition of GreA increased this rate about 5-fold (Fig. 3R). Presumably, GreA- induced cleavage of 4- and 5-mer prevented their release, thus allowing repeated resynthesis and cleavage of short initiation intermediates. Curiously, addition of GreA also caused forma- tion of some A20 complexes in these experiments (Fig. 3A, lanes 13-16). Apparently, by repeatedly inducing removal of 3’-dA, GreA increased the likelihood that trace amounts ofATP could be incorporated and yield A20 complexes.
To map the site of GreA-induced cleavage of the abortive RNAs, we tested initiation with ApU, GTP, and 3’-dATP, but using [a-”PICTP as label instead. AUCG and AUCGdA, but not AUC, were significantly reduced by addition of GreA. However, the 3”cleavage fragments were not labeled, indicating they did not contain CMP (Fig. 3A lunes 17-24 ). GreA-induced cleavage must have occurred after C3, with release of unlabeled pG from AUCG or pGdA from AUCGdA. Thus, although GreA could not induce cleavage of RNAs shorter than -9 n t after backing up from a n elongation complex (Fig. 2). i t could induce cleavage of potentially abortive RNAs as short as 4 nt.
GreA Stimulated Productive Initiation at a Weak Promoter- The finding that GreA induced cleavage of potentially abortive transcripts and stimulated A20 complex formation under con- ditions where productive initiation was inhibited raises the possibility t h a t GreA may affect normal transcription initiation by increasing the number of chances for the complex to initiate productively. Although no increase in productive transcript (A20) was observed upon GreA addition to reactions on a wild- type T7 A1 promoter (Fig. 3A ), productive initiation could be so efficient t ha t an effect of GreA was undetectable. Therefore, we examined the effect of GreA in a single-round initiation experi- ment at a weak derivative of the T7 A1 promoter (pRL550; see “Materials and Methods”). On the pRL550 template, which lacks GMP in the first 20 nt of transcript and initiates poorly,
f A 1 1 fG lO f G9
C p9rnnr
AUCGAGAGGGACACGGCGAA U r‘ 5 10 15 20
pCPG T3Te transcript A70
B
addition of GreA increased the amount of productive transcript about 3-fold (Fig. 4).
Interestingly, GreA stimulated formation not only U2 1 RNA. hut also of two other transcripts four to five nt larger than U21 (Fig. 4). To identify these larger RNAs. they were !i’-end-la- beled by transcription in the presence of Iy-~”TIATP. isolated, and then sequenced enzymatically. Both transcripts contained additional non-templated nucleotides at the 5’-end transcripts: 5’-AUCCAUCCC for the smaller and !i‘-AUCCCAf/C,’CC for the larger (data not shown; underlined italics indicate normal 5’ sequence). Apparently, these RNAs arose by “primer slippage,“ a reaction also observed at the rrnR PI (Borukhov rt ul., 1993b) and A P,: promoters. Initially, synthesized AUCC or AUCCC must shift from its normal position in the initiation complex into a site where it both stabilizes the complex and allows extension in a new register from its 3’-end. Thus, GreA-induced cleavage of pentamer RNA in an unstable initiation intcrme- diate not only increased the probability of transcribing past the pentamer stage, but also allowed more time for primer slippage, to occur.
GreA Induced Reverse Danslocution for olvr 200 hp i n Pro- moter-Distal Danscription Complcxcs Formrd on a G-lcss Templates-To test whether GreA could induce transcript cleavage in promoter-distal transcription complexes. we iso- lated several different types of defined transcription complexes: complexes halted at the end of a 392- or a 147-nt G-less tract in the absence of GTP, complexes halted at the end of a 135-nt C-less tract in the absence of CTP, and complexes paused a t the trp or his leader pause sites.
When C392 complexes were incubated with 500 nv GreA ( a 30-fold molar excess), after removal of NTPs by gel-filtration, successive rounds of transcript cleavage caused reverse trans- location of RNA polymerase. After 60 min. the complexes were distributed over an -200 bp region Fig. 5A 1. Addition of NTPs allowed all the reverse translocated complexes to resume elon- gation. Thus, GreA was able to act on transcription complexes far removed from a promoter. Interestingly. the backward movement of the complexes during transcript cleavage was
~~~ ~~
’ K. Severinov and A. Goldfarh, manuscript suhmittcd.

GreA-induced Danscript Cleavage 22287
A
1
111 .- "
+ I
-]&G;GAGGGACACGGGGGA.UCCU 10 15 20 Cleavage [ Ip
Products 2 4 6 R 1 0
Tmn Imm)
0 2 4 6 8 10 12 14 16 18 20 22
Time (min)
FIG. 3. GreA-induced cleavage of abortive transcripts formed at the T7 A1 promoter. Open complexes were formed at the T7 A 1 promoter by incuhating 2.5 pmol of RNA polymerase with 0.4 pmol of pRL522 DNA templntc. in 12 pI of huffer A at 37 "C for 5 min. Tran- scription was initiated by adding 8 pI of NTP mix to achieve final concentrations of 200 p~ ApU and either 5 p~ each of ATP, CTP, and In-:'*PlGTP (Innrs 1-8) . or 10 PSI 3'-dATP, 5 pal CTP, and In-:"PlGTP (Innrs 9-16), or 10 p~ 3'-dATP, 5 p~ [n-."PICTP, a n d G T P ( 1 n n ~ s 17-24). GreA (20-fold molar excess) was added together with NTPs. Portions (4 pl) were removed from the reaction mixtures at the times indicated and mixed with 4 pl of stop huffer. A, RNA transcripts were separated in a 15% polyacrylamide, 7 M urea, 0.5 x TRE gel (19:1.5, acrylamidd his(acry1amide)). The positions of productively initiated transcript (A20). abortive transcripts (ApUpC, ApUpCpC, and AplJpCpCpA) and the cleavage products are marked on the r ight. B . comparison of ahor- tive initiation rate in the presence and ahsence of GreA (quantitated from Innvs 9 to 16 in Fig. 3A ).
punctuated by prominent pauses where the cleavage reaction was relatively slow. These pause sites were spaced 10-30 bp apart and were approximately equal in strength, a result sim- ilar to that reported for TFIIS-induced reverse translocation by pol I1 transcription complexes (Wang and Hawley, 1993).
We observed formation of a similar pattern of paused inter- mediates after incuhation of purified C147 complexes with GreA (Fig. 5 R ) . One species, formed near position 140, per- sisted even when most complexes had backed up very far and could not be chased upon addition of NTPs (Fig. 5B ); i t probably entered an arrested configuration during reverse translocation since, although i t also formed in GreR reactions, those com- plexes resumed elongation upon addition of NTPs (data not shown). Three important observations emerge by comparing the reverse translocation pause sites to those occurring during pyrophosphorolysis or during transcript elongation: ( i ) the rate of reverse translocation was much slower than that of elonga- tion; ( i i ) forward and reverse pausing occurred at different sites; and (iii) pausing during pyrophosphorolysis occurred at sites near but not always identical to those for GreA-induced
pRL550 RNA lranscnpl AUCCCACACCAAACACACCAUGGUACGU + 1 r 1s 2o
non-lranscnbed slrand ACAGCCATCCCACACCAAACACACC AT GGTACGTAG r u2 1
Flc:. 4. GreA stimulated productive initiation at a weak T7 A1 promoter. Open transcription complexrs wrrr fnrmrd hy tncuhating 1 pmol of RNA polymerase with 0.4 pmol of pR1,550 trmpl;ltr In 15 pI of huffer A a t 37 'C for 10 min. 10 pI of NTP rnlx was thrn addrd to klvc. final concentrations of 10 p~ ATP, UTI'. 5 11\1 lm- '~ ' l ' l~ 'Tl '~lK~ ( : l 'mmol~ . and 10 pg rifampicinlml. GrrA r20-fnld molar excrss) was add14 wlth the NTPs. Portions 15 p l ) wrrr r rmovrd from the rwtrtlon rnlxturr9 at the times indicated and mixed wlth 15 pI of stop huffrr. A. I'hospho- rImager print of RNA transcripts srparatcd In :I 15'; pnly:tcr?;lam~de. 7 Y urra . 0.5 x TRE gel. 1,nnc.s ( ' cnntaln samplrs incuhrltrd with I 0 0 pv NTPs after 10 min of incuhation with or without G w A . N . comparison of the total amounts of productivr transcripts in t h r a h w n r r 1 1 or presence (01 of GrrA and thr prrcrnt of prnductivr transcripts from primer slippage in the ahsrncr (...I or prrrrncr (AI of GreA as quantl- t a t rd from Fig. 4A.
reverse translocation (Fig. 5B ). Finally, when we examined the short RNAs produced during these reactions on a high percent- age gel, we observed only 2- and %mer cleavage products (Fig. 5 C ) . However, some transcription complexes had hacked up to the promoter-proximal region. as evidenced by the presence of 9-10 n t RNA that could be re-elongated upon addition of NTPs (Fig. 5C).
The Presencr of GMP in thr Nascrnt 7hnsrript.s of 7hnsrrrp. tion Complexrs Formed on a C-lrss Trmplatr Inhihitrd GrrA- a n d P~rophosphorolysis-inducrd Rrorrsr 7hnslocntion"The absence of Gs in the C392 and C147 transcripts might contrih- U t e to extensive reverse translocation (Wang and Hawley, 1993; Izban and Luse, 1992a). To test this idea. we prepared C13.5 complexes halted on the C-less pDW135 template (see "Mate- rials and Methods"). When the G135 complexes were incuhatcd with either 500 nu GreA or 2 mu PP,, hoth cleavage anti pyro- phosphorolysis were significantly inhihited after removal of 4-11 nt (Fig. 6A) . Even after 60 min, very few GreA-treated complexes escaped these early blocks, and those that did stopped at prominent sites 10-20 hp further upstream. On thc C-less template, hoth GreA and PP, produced pausing. mostly at the same sites (confirmed by coelectrophorrsis of products; data not shown). PP, produced more efficient reverse translo- cation and also induced pausing at s i tes + I and -1 relative to the GreA-induced pause sites (Fig. 6A ).
To test whether G in the transcript was responsible for the slower reverse translocation on the C-less template. we re- placed GTP with ITP during formation of the "G1.75" complexps. When these IMP-containing complexes were incuhated with GreA, reverse translocation was no longer hlocked at the early

22288 GreA-induced Dunscript Cleavage
-309
-240
-217
- 201
- rw -180
- I M I
lanes 1 7 3 4 5 6 7 8 9 1011171314
I
r' T7A1 promoler G.less (392 bp)
C147+
'-I! "c140
~ t r A l 3 0
I "c120
L "VllO
^. rClOO
. "cw
I "lo - -. - - 1
r!n UCUCAUAUAUCCVUAUCCUCUCCUCA$CVCUCCCUCCUC~
10 100 110
170 AUCUCCCCCC CUCACACUCA UUUCUCAUUC CACUCCC GGG
$30 140 I50
\ t C147
- 9-70 "I
Innes 1 .' I .I g, / , i A 't 1 0
FIG. 5. GreA-induced reverse translocation, pyrophosphoroly- ais, and transcription on G-less templates. C392 and C147 tran- scription complexes were formed on DNA tvmplate pRL597 and pRL596,
sites (Fig. 6R ). However, pausing still occurred at thc same si tes observed for the GMP-containing complexrs (Fig. 6 B ) . From this, we conclude that the presence of G in the nascent tran- script slows cleavage at reverse translocation pausr sites, hut that the locations of the sites appear to he determined by other interactions in transcription complrx, perhaps betwc.cn RNA polymerase and the DNA template. Gs in transcript might slow cleavage and reverse translocation by affrcting clravagr di- rectly or by slowing reverse movement of thc. transcript through the transcription complex (see "Discussion").
GreA Had Littlr or No Effrct on Pnusinx n t t h r his nnd t rp Lender Pause Sitrs or on 7ivninntion Efficiency nt Nirw Diffir- ent p-Indeprndrnf Trrminntors-Since GreA induced transcript cleavage in promoter-distal transcription complrxrs. wr next tested whether it altered either pausing or termination by RNA polymerase. We tested the effect of GreA on pausing in the leader regions of the S. typhimurium his and E . C 0 / 1 t rp oper- ons. We saw no significant change in the half-lifr of pausing in the presence of GreA (Table I) . We also measured termination eficiency at nine different pindependrnt terminators and found GreA had no significant effect on the eflicirncy of termi- nation at any of these termination sites {Tahlr 11) (scc "Discus- sion").
Effect of GreA on 7hnscription Complr.rc~s nt nntl Around the his a n d trp lnndrr Pnusr Sitrs-The finding that pausing a t the his and f r p leader sites was unaffected by GreA even though some cleavage events could be rapid raised several questions for us: ( i ) does cleavage occur at these sites; ( i i , are they also pause sites for reverse translocation; and C i i i ) if cl(.;lvagc. dors occur, what effect does a relatively well-defined nascent-tran- script secondary structure, the pause RNA hairpin. have on reverse translocation? To address thew questions, we isolated transcription complexes paused a t the his and trp leader pausr sites by gel filtration and incuhated them with GreA or PP,. Interestingly, cleavage of the trp paused complexes was slow relative to most other transcription complexes examined to date (Fig. 7A, lanes 44)). At a saturating concentration of CreA (-10 ~ M J , the rate of the URI complex cleavage was 5 x 10 ' s" (data not shown). The his paused complexes were evrn mom
. "
respectively, and isolated by filtration through Sc.pharosc. ('1,-613 IHW
"Materials and Methods"). A, - 1.6 pmol of ('392 romplrxw containing some free RNA polymerase (still present aRrr gc.1 liltr;rtinn) w w r incu- bated with either I3SA as control f lnnrs 2-5 I or 50 pmol nf (;rrA ~ l r r n r s 6-14) in 100 pI of huffrr A at 37 C. Portinns 1 1 0 pI I of thr rractinn mixtures were removed at the times indicatrd and mixed wlth I O pI nf stop buffer. IMnp I C C ' ) contained complc.xrs incuh;~trcl with 100 p v NTPs prior to the cleavage reaction. IL1nr.s .i and 14 (('I containrd complexes incuhated with 100 psc NTPs aRer trratmrnt with ( ;rrA for 60 min. Size standards are marked on the right. R . 0.6 pmol of C1.17 complexes containing some free RNA polymrrnsr wrrv incuhatrd with either RSA as control l lnnrs 1 4 1 . 100 pmol of (;rrA f I r r n v s S-l21. or 2 m!.j PP, ( lanes 13-20) in 100 pI of buffer A at .'17 ' C , Trzlnscript elonga- tion ( /ones 21-25) was conducted in 50 p51 ATP, UTI'. 5 p \ ~ Icr-"l'I('Tl' (200 Ci/mmol). and 10 pg rifampicin/ml. I'ortions I 1 0 1 1 1 1 w r r r rwnoved from the reaction mixtures at the times indicated and mixcd with 10 111
of stop huffer, except for the pyrophosphorolysis rwctions. which werv instead precipitated by addition of cold trichloroncc*tic acid t o 6';. washed three times with 7OC4 ethanol. and then dissnlvetl in 20 pI of stop buffer. The faint ladder of hands presrnt in / o n r s 13-20 rc-sult.3 from acid degradation; the positions of the I'l',-shnrtvnc.d RNAs c m hr identified as strong hands ahnve this hackpnund. Thr srquence of the. C147 transcript is shown helow thc. gel. The .?'-tc.rminnl h m r s of RNAs present at major pause sites during elongatinn f . ' . ) and rrversr trans- location ( * ) and of an apparent nucleolvtic depadatinn product f I are marked on both sides of the gel and on the sequencr h e . 1 1 ~ thca ~ r l . The sequence ladder ( lanes 27-29) was generatrd with 3'-droxynuclrotidrs as described under "Materials and Methods." C', the samr samplrs shown in Fig. 5R were loaded on a 2 0 4 polyacrylamidr urra grl to show the small cleavage produch and RNAs in complc~xrs hnckrtl to the promoter-proximal region.

GreA-induced Ranscript Cleavage 22289
G135-
120- '
110--1
100 -i
90-1
80 -
I
I
. "135 2
0- 120
"110
- 1 0 0
- 90
- 80
. , .> .: 5 G ; f ! :$ ;o ' 1 1 ; ) l : ! ; < ; ; r ~ . r ) ~ / 1 ~ 1 ~ 1 7 n : " : v : w r . z 1- 70 . , , .; 7, ,. ., , . , ,
+ l r I hPr promoter ~AUGUAGGAGU GGAAUGAGAA AUGAGUGUGA GGGGGGGAGA
UAGAGGAGGGAGAGGUGAGGAGAGGAUAA~G~UAUAUGAGAUGAGUAGGG 50
20 30 40
60 70 83 '(0
AG~AUUGG&'$ GAGUAGGGGG UGA~AGGGAU GAGGAUGAUA GGGGG cccc 110 120 130
0 0
- \ 0 0 0 0 6 , t
G135
plexes were formed on a C-less template derivrd from pDW135 hy incubation with RNA polymerase,ATP. UTI', and Irr-'"PIGTI'as drscrihrd unrlrr FIG. 6. GreA-induced transcript cleavage, pyrophosphorolysis, and transcription on a C-lenn template. A. GI35 trnnwription rom-
"Material and Mrthods." -0.X pmol of G135 complex and frer RNA polymerase were incuhabd with either M A a s control rlnnrn X-IO I , 50 p m d ofGreA(lanrs 11-17) or 2 mM PP, ( lanes 18-2.5) in 100 pI of buffer A. Transcript elongation tlanrs .7-71 and srqurnce laddrr prrparation rlnnr-s 1 and 2 ) were performed as described in the legend to Fig. 5, Portions (10 pl ) werr removed from the reaction mlxture9 a t th r t imrs ind lca t rd and processed as described in the lrgend to Fig. 5. The sequence of '2135 transcript is shown hrlow the grl. Thr 3 ' - t r rmina l haws of RNAs prrsrnt a t major pause sites during elongation (1) and reverse translocation ( * ) are marked on the r ight and on thr srqurncr hrlow the gel. I < . 11.35 transcription complrxes with IMP-containing transcripts were prepared by incubation of the pl)W135 C-Irss trrnplate with RNA pnlymrrase. 50 p~ ATP, 200 p~ ITP. and 12 phl 1w"PIUTP as described under "Matrrial and Methods." ~ 2.5 pmol of I135 cornplrx and frrr RNA polymrrasr wrrr incuhnted with either RSA as control (lanes 1-7) or 50 pmol of GreA (lanrs 4-11 in 45 pI of huffer A at 37 .C', Portions (5 p l ~ wrrr removrd from the reaction mixtures at the t imes indicated and mixed with 15 PI of stop buffer.
T A I M I Effcrt of GrrA on tmnscriptional pausing
~~ ~ - -~
I'nunr hnlf-lifrcslh
4 r r A +GrrA Sitr" . - ""
""
E. coli trp leader pause 133 ? 2 125 * 5 S. !.vphimurium his leader pause 266 9 2x0 ? 6
~ ~
" For comparison see previous studies by Chan and Landick (19x9, 1993) for the his pause site and Lee rt nl. 11990) for the trp pause site.
'I Pausing was measured on templates from pCL185 (his pause) and pRIA07 ( t rp pause). Promoter-proximal complexes (G16 for his and A20 for trp) were formed by incubating 2.5 pmol of RNA polymerase with 0.5 pmol of DNA template in 20 pI of buffer A with ApU. ATP, CTP, and la-:"PIGTP as descrihed under "Material and Methods." To start elon- gation. 30 pl of chase mix was added to produce a final concentration of 100 pg hepar idml , 100 phl each ofATP, CTP, UTP, 5 pu GTP, and 1 p~ rither hovine serum albumin or GrrA 120-fold molar excess to RNA polymerase). Samples werr removed at various times thereafter, added to a stop mix, electrophoresed, and quantitated to calculate pause half- livrs as described under "Materials and Methods." Errors given arr 2 S.D. in the regression analyses.
resistant to the action of GreA, showing almost no cleavage even after 30 min (Fig. 7B, lanes 20-23 ). When cleavage of the trp complexes did occur, several additional di- or trinucleotides were removed rapidly, but further backing up was strongly inhibited when they reached G71. Interestingly, this is pre- cisely the position of the first base pair in the putative Irp pause RNA hairpin (Chan and Landick, 1993).
To determine if similar hehavior occurred when RNA polym- erase reached the pause sites hy reverse translocation, we walked the paused complexes forward hy incubation with a limited set of NTPs, by 11 nt to A92 for t rp and hy eight nt to A l l 0 for his. Incubation of these complexes with either GreA or PP, backed up both to the pause regions, although more rapidly for his than for trp (Fig. 7A, lanes l l - l .? and 2.'1-2.5; Fig. 7 8 . lanes 1-6 and 11-15). The his complexes stopped at three cnn- tiguous sites of which the transcription pausct site was the central and most prominent. The trp complexes stopped pri- manly one nt 5' to the transcription pause site. A fraction of the trp complexes backed up past the pause region, hut stopped at G71, the same position at which complexes hacked up from the pause site stopped (compare Fig. 7A Inn(, I,? with Lanr 6 I.
We conclude from these experiments that the paused config- uration is relatively resistant to GreA-induced transcript clew- age, consistent with the lack of effect of GreA on pausing (Tahlr I ) . Further, complexes that approach the pause site in the re- verse direction exhibit a reduced rate of movement in the pause region, but often one nt before or after the pause site ohserved during chain elongation. This could reflect the constraints of di- and trinucleotide steps. However. the same hehavior is evident during pyrophosphorolysis, which suggests instead that tran- scription complexes may enter a paused configuration at slightly different positions during the forward and rewrse movement.

22290 GreA-induced Danscript Cleavage TABLE I1
Effect of GreA on p-independent termination
Site Plasmid used for efficiency (%Ib Termination
terndate‘
Phage T7 early terminator E. coli rrnB T1
pAR1707 79 f 1 pCPG rrnB T1 76 t 1
S. typhimurium his leader terminator pGF104 70 -c 1 E. coli trp leader terminator pRL407 6 5 f 1
E. coli P14 pCPG P14 43 -c 2 E. coli tonB pCPG tonB 25 -c 2 Phage T3 early terminator pCPG T3 14 -c 1 S. marcesens trp leader terminator pRL522 8 -c 1
A t R Z pCPG A h2 56 f 3
79 f 1 76 -c 2 69 f 1 60 f 2 57 f 3 42 f 2 26 f 2 19 f 1 9 f 1
Rosenberg, 1981); pCPG rrnBT1, pCPG A t,,, pCPG P14, pCPG tonB, a References for the terminator plasmids are: pAR1707 (Studier and
and pCPG T3 (Reynolds et al., 1992); pRL407 (Landick et al., 1990a); pGF104 and pRL522 (this work, see “Materials and Methods”).
pmol of RNApolymerase with 0.2 pmol of DNAtemplate at 37 “C for 10 * Promoter-proximal A20 complexes were formed by incubating 1
min in 10 pl of buffer A with 200 p~ ApU, 2.5 p~ ATP, 2.5 p~ CTP, and 3.75 p~ [w3’P1GTP (270 CUmmol) as described under “Material and Methods.” A 3-pl portion of the A20 complexes was mixed with 7 p1 of chase mix with or without GreA to give final concentrations of 100 p~ NTPs, 100 pg heparidml, and 1 p GreA (molar ratio to RNA polym- erase, 25:l) and incubated at 37 “C for 10 min. RNA products were electrophoresed and quantitated to determine termination efficiencies as described under “Materials and Methods.” Errors are 1 S.D. in trip- licate repetitions.
DISCUSSION
Our aims in this study were to test the possible involvement of GreA in regulation of transcript elongation and to character- ize its effects on defined transcription complexes. By construct- ing a GreA-overproducing plasmid and devising an efficient method for purification, we obtained large quantities of GreA which made these experiments possible and which will allow detailed study of its structure and function in the future. We found that GreA could induce transcript cleavage in initiation complexes, halted elongation complexes, both near and well removed from a promoter, and naturally paused transcription complexes. Repetitive cleavage caused reverse translocation of RNA polymerase over 200 bp on G-less templates but was in- hibited when the nascent transcript was reduced to - 10 nt, contained GMP or formed secondary structures near the com- plex. Interestingly, we have recently found that recombinant GreB protein at high concentration yields virtually identical result^.^ The large fragment cleavage reported for some com- plexes treated with GreB (Borukhov et al., 1993a) and which we observed in the absence of GreA (Fig. 2) is stimulated only at low GreB concentrations (5-10 nm), whereas higher concen- trations of GreB act similarly to GreA.
Biological Function of GreA-Two aspects of our results bear on the biological function of transcript cleavage. First, overpro- duction of GreA (or GreB3) to -6% of total cell protein had minimal effect on cell growth. Based on comparison of purifi- cation yields (Fig. 1 and Borukhov et al., 19921, its normal level must be <0.03% or at most equimolar with RNA polymerase (Bremer and Dennis, 1987). Since saturating the rate of tran- script cleavage in trp paused transcription complexes required more than 200-fold molar excess of GreA, it is unlikely that an increased rate of transcript cleavage alters RNA metabolism significantly in cells grown in rich medium. Rapid resynthesis of excised RNA fragments must compensate for any increase in the cleavage rate. Of course, cleavage factor overproduction may affect growth in more restrictive conditions and deletion or alteration of the cleavage factor genes may affect RNA metab- olism.
G. Feng, unpublished results.
Second, micromolar levels of GreA (and GreB, results not shown) had little if any effect on transcriptional pausing or on the efficiency of p-independent termination (Tables I and 11). A preliminary experiment also revealed no dramatic effect on the pattern of p-dependent termination at A tR1 (data not shown). Thus, it is unlikely that transcript cleavage plays a significant role in these mechanisms, answering, at least for these sites, the concern raised by Borukhov et al. (1992) (see Introduction). Our results suggest that cleavage factors do not exert major allosteric effects on nucleotide addition by RNA polymerase or alter the rate-determining step at terminators. However, it is possible that transcript release occurs slowly after commitment to a termination pathway, in which case stimulation of release by GreA would have escaped detection in our experiments.
Our results are consistent with the idea that the major bio- logical role of transcript cleavage is to recycle transcription complexes back to active form when they enter an unactivated or arrested state upon nucleotide misincorporation or other malfunction of the transcriptional apparatus, rather than regu- lation of normal transcript elongation.
GreA-induced Dunscript Cleavage Stimulates Productive Initiation-During initiation, however, transcript cleavage may play some special function. We found that potentially abor- tive RNA transcripts as short as four nt were cleaved readily. Cleavage inhibited abortive transcript release and stimulated both productive initiation and “primer shifting” at a weak pro- moter derived from T7 A1 (Figs. 3 and 4).
Two explanations are apparent to us. First, GreA may di- rectly suppress release of short transcripts. Second, cleavage may indirectly increase productive initiation if it occurs in an altered conformation of the transcription complex that is in equilibrium with the elongation competent form. Such an equi- librium between activated (elongation competent) and unacti- vated (cleavage competent) transcription complexes was postu- lated by Erie et al. (1993) to explain inhibition of nucleotide misincorporation by GreA.
To understand the second model, consider two possible path- ways for abortive transcript cleavage (depicted for the G-less transcript-producing template from pRL550 where stimulation of productive initiation was observed; the abortive intermedi- ates are inferred from experiments on the wild-type T7 A1 promoter, see Figs. 3 and 4; * denotes the activated state which is competent for nucleotide addition, see Erie et al., 1993) (Schemes 1 and 2):
Cleavage J I
Lt AU* - AUC* AUCCC* + AUCCCA* + Initiation
faat Productive
AUC c AUCCC Released oligoribonucleotides
SCHEME 1
AU* + AUC* - + AUCCC* + AUCCCA initiation fast Productive
JT Cleavage 0
8 1 AUC - AUCCC Cleavage-susceptible conformation
AUC AUCCC Released oligoribonucleotides
SCHEME 2
In pathway 1 productive initiation and abortive release are in direct competition. Cleavage and resynthesis ofAUCCC* can slow transcription past the 5-mer, but cannot change the ratio of productive to abortive synthesis or increase the rate of pro- ductive initiation. In pathway 2, only cleavage and release

GreA-induced Danscript Cleavage 22291
FIG. 7. GreA-induced cleavage and pyrophosphorolysis of transcripts in paused transcription complexes. The paused transcription complexes, trp-U81 (A) and his-U1O2 (B), were formed on templates amplified from pDNL279 and pCL185, respectively, and isolated by fil- tration through Sepharose CL-GB (see "Materials and Methods"). Numbering corresponds to actual template positions for the trp complexes and to the corre- sponding positions in the wild-type his leader transcript for the his complexes, in both cases to facilitate comparison to re- cently published results (Lee and Land- ick, 1992; Chan and Landick, 1993). To prepare trp A92 complexes (A) and his AllO complexes (B), portions of the iso- lated pause complexes were incubated with limited sets of NTPs a t 37 "C for 15 min as described under "Materials and Methods." -1 pmol of transcription com- plexes and free RNA polymerase were in- cubated with either 1 pg of BSA as con- trol, 50 pmol of GreA, or 2 mM PP, in 45 1.11 of buffer A a t 37 "C. Portions (5 pl) were removed from the reaction mixtures a t the times indicated and mixed with 15 p1 of stop buffer. Pyrophosphorolysis samples were precipitated with trichloro- acetic acid and washed as described in the legend of Fig. 5 prior to dissolution in stop buffer. The RNA sequence ladders were generated with 3'-deoxynucleotides as de- scribed under "Materials and Methods." The trp and his RNA hairpin structures are shown below the gels.
Complexes
0 3 0 C 1 5 3 0 C Time (min) PPI Control I GreA IControl I GreA Ladder PPi A92 UBI trppause I A92 equence U81
r=-"q I. . r 3 0 C I l 5 3 O C I O 3 0 C I l 5 3 0 C I A U G C I 1 - 5 - -1 .- -
A92 -4
U81-4
A -1
G71 -+ * -
--a - 5
lanes 1 2 3 4 5 6 7 8 9 10 11 1213 1415 16 17 18 19 20 21 22 23 242526
6oA U A U
G C U G
55 C E G C i G 6 5
f rppause hairpin U= A u- c C E G A= U70
UGGUUGGCGCEGUAUUCACCAUGCGUAAAGCAA
50 4 8o f Y G7l Pause (U81) A92
Complexes A1 10 complex U102 his-pause Sequence
Time(mWo2 6102030C~O30C~26102030CO30C~6102030C~l 5 1 0 1 5 2 0 3 0 C A U G C PPI (Controll GreA Control) GreA I PPI Ladder
A1 10.
u102\
_. -?
r -
I
his pause hairpin 75 C E G
65 CACCAgCAU CGAUGUGUGCU GGAAGACA UU
95 100 4 , l o+
Pause(UlO2) A l l O
compete directly and repeated resynthesis of the AUCCC* in- termediate can increase the fraction of initiation events that lead to productive initiation. Thus, GreA either suppresses abortive transcript release by a steric or allosteric effect on the transcription complex or transcript cleavage proceeds by path- way 2. Either model could account for stimulation of primer- shifted initiation (Fig. 4; Borukhov et al., 1993bI3 by increasing the lifetime of 3- to 5-mer oligoribonucleotides in the initiation complex and thus the effective concentration of precursor in the primer-shift pathway. How Are the Rates of Cleavage and Reverse Danslocation
Controlled3Come transcription complexes are susceptible to rapid transcript cleavage (see Figs. 3, 4, and 7 and Surratt et al., 1991; Borukhov, 1992, 1993a) whereas others are highly resistant (see Figs. 2 and 8). Extensive reverse translocation
occurs only when the transcript lacks GMP (Figs. 5 and 6). Similar backing up has been reported for TFIIS and pol I1 on other templates (Reines, 1992; Gu et al., 1993; Guo and Price, 1993; Wang and Hawley, 1993; Izban and Luse, 1992a).
Like pausing during chain elongation (Chan and Landick, 1993, 1994), variation in the rate of transcript cleavage prob- ably results from the combined effects of different interactions between RNA polymerase and the nucleic acid chains. Possible participants include single-stranded segments of transcript, RNA secondary structures, the single-stranded transcription bubble, and double-stranded DNA outside the bubble. These contacts could either directly influence the cleavage reaction or could affect backward translocation of DNA or RNA through the enzyme.
One clear difference is the requirement for proper alignment

22292
FIG. 8. Model for GreA-induced transcript cleavage and reverse translocation. A, three potential RNA- binding sites in the transcription complex (see text). The transcript in site I may be positioned either by contacts to RNA po- lymerase or by base pairing to the DNA template strand (see Chan and Landick, 1994; Lee and Landick, 1992). B, tran- script slippage model. Only the sites and the relative position of the nascent tran- script (0) are indicated. The two configu- rations of transcription complex with re- positioned active sites may be stabilized by both GreA and GreB for the smaller slippage interval and by GreB alone for the larger slippage interval (see text). C, active site slippage model. D, possible configurations of various transcription complex intermediates discussed in text.
GreA-induced Danscript Cleavage
A Open Complex
I Product Site
III Hairpin-binding Site
D Different Transcription Complex Conformations
( 1) Q (3 (4) (5) Initiation Early Elongation Complex Complex
Elongation Structure Inside Structure Outside Complex Complex Complex
of the relevant phosphodiester bond in the cleavage site rather than alignment of the NTP and transcript 3’-OH, which strongly influences the elongation rate (see Chan and Landick, 1993; Aivazashvili, et al. 1982). This may account for the some- what more uniform strength and regular distribution of pause sites observed during processive transcript cleavage (Figs. 5 and 6 ; Wang and Hawley, 1993; Guo and Price, 1993) and for the many cases where forward and reverse pause sites differ (Figs. 5 and 6). Where these pause sites exactly or nearly co- incide, such as at the his and trp leader pause sites (Fig. 7), it is likely that interactions other than in the catalytic site pre- dominate. The slight positional differences in forward and re- verse pausing at these sites may reflect the absence of nucle- otide addition as a component of the reaction, rather than any fundamental difference in the conformation of the paused con- figurations. Indeed, substitutions of the 3’4erminal nucleotide at the his pause site produce slight shifts in the position of pausing (Chan and Landick, 1993).
Unfortunately we do not know the structures of the DNA and RNA in contact with RNA polymerase. We assume that during cleavage the transcription bubble resembles the 12-22-nt structure reported for elongation complexes (Krummel, 1992; Lee and Landick, 1992; Kainz and Roberts, 1992; see accom- panying paper). The configuration of the transcript is problem- atic. Recent results (reviewed in Das, 1993; Chamberlin, 1994; Chan and Landick, 1994) suggest it may occupy two separate transcript-binding sites (Fig. a): an upstream transcript exit site and a 3‘-proximal site of either RNA-DNA hybrid or single- stranded RNA in which up to - 10 rounds of nucleotide addition may occur prior to a translocation event (see Yager and von Hippel, 1992; Lee and Landick, 1992; Borukhov et al., 1993; Chan and Landick, 1994; Chamberlin, 1994). Borukhov et al. (1993a) suggest that only transcript located in site I is suscep- tible to cleavage. Work in our laboratory (see Chan and Land- ick, 1993, 1994) and that of Chamberlin (Amdt and Chamber-
lin, 1990) is consistent with the additional existence of an RNA hairpin-binding site (Fig. 8A ). As refined below, this model can explain our findings.
l tvo Models for l).anscript Cleavage-The key revision is that transcript cleavage probably is catalyzed by the same amino acids that catalyze chain elongation. Izban and Luse (1994) recently found that pyrophosphate cleaves the nascent RNAin an arrested pol I1 transcription complex by nucleophilic attack on the same internal phosphodiester bond that is at- tacked by water during TFIIS-stimulated transcript cleavage. Since pyrophosphorolysis is the chemical reverse of nucleotide addition, this result strongly suggests that cleavage occurs in the RNApolymerase active site. The similarity of reverse trans- location driven by pyrophosphate or by GreA (Figs. 5-7) is also consistent with this view. Further, cleavage of RNA, stimulated by TFIIS for pol I1 and by GreA for E. coli RNA polymerase, occurs in binary complexes formed between RNA polymerase and RNA (Johnson and Chamberlin, 1994; Altmann et al., 1994). After cleavage, these complexes catalyze non-templated reaction of NTPs with the newly generated 3’-hydroxyl, further supporting the idea that cleavage is catalyzed by the active site.
This view suggests that binding of GreA, GreB, or TFIIS stimulates cleavage by stabilizing the positioning of the active site over an internal phosphodiester bond, so that hydrolysis, rather than nucleotide addition, is catalyzed. In principle, this could involve slippage of either the RNA transcript through the active site (transcript slippage model, Fig. 8B) or of the active site along the transcript (active site slippage model, Fig. 8C). In either model, the different positions of active site along the transcript correspond to different, interconvertible, energetic states of the transcription complex. Normally, active site posi- tioning at the 3’-end of the transcript or over the first phos- phodiester bond must predominate (the two states of the mi- croscopic translocation cycle of NTP addition, see Erie and von Hippel, 1992; Chan and Landick, 1994). That transcript short-

GreA-induced Dunscript Cleavage 22293
ening occurs 500-1000 times slower than elongation may re- flect the thermodynamic favorability of an unoccupied NTP- binding site (leftmost conformation, Fig. 8, B and C ) relative to the slipped conformations. Cleavage factors may stabilize the slipped configurations and also could accelerate cleavage by lowering the activation barrier to hydrolysis once the active site is repositioned. These slipped conformations also may explain both the unactivated kinetic states described by Erie et al. (1993) and arrested transcription complexes (Chamberlin, 1994; see accompanying article).
The observations of both small and large cleavage intervals (Surratt et al., 1991; Borukhov, 1993a; Fig. 21, which are dif- ferentially favored by GreA, GreB, and RNA polymerase in the absence of cleavage factors, are consistent with these models and suggest that two different minor configurations of the tran- scription complex in which the active site is repositioned over different phosphodiester bonds (Fig. 8, B and C ) predominate among the slipped conformations. The inverse concentration dependence of GreB-induced large fragment cleavage may be explained if GreB stabilizes the larger slippage step with high affinity, but induces slow cleavage, and stabilizes the smaller slippage step with low affinity, but induces rapid cleavage. Arrested complexes may form from an initial intermediate with a large slippage interval, since GreA-induced small fragment cleavage can compete with large fragment cleavage (Fig. 2), even though GreA cannot reactivate arrested complexes (Borukhov et al., 1993a).
Comparison of the Models to Results-Cleavage of poten- tially abortive transcripts as short as four nt may occur because the nascent transcript remains in site I until the transition to elongation mode occurs (Fig. 8 0 , panel 1 ) . Thus, the steps described above for cleavage-induced enhancement of produc- tive initiation could occur either by transcript slippage or active site slippage in this configuration of the complex.
Extensive transcript shortening (Figs. 5 and 6) requires that the transcript move backwards from site I1 to site I (Fig. 8 0 , panel 3 ). Thus transcript slippage must occur at least occasion- ally. To explain inhibition of cleavage when the transcript is reduced to -10 nt (Fig. 2; Izban and Luse, 1992a) or when secondary structure is present, we suggest this slippage occurs efficiently only if single-stranded RNA is available to reoccupy site I1 (Fig. 80,paneZ 2). This suggests that site I1 contacts -10 nt, which is consistent with other estimates of transcript-RNA contacts (Lee and Landick, 1992; Rice et al., 1992, see Chan and Landick, 1994).
However, several other results strongly suggest that the ac- tive site can move within the transcription complex. Mustaev et al. (1993) found that RNA chains of up to nine nt can be syn- thesized when the initiating nucleotide is cross-linked to RNA polymerase. Further, the increasing size of cleavage fragments released by complexes treated with GreB at successive tem- plate positions (Borukhov et al., 1993a; see also Fig. 2) is most easily explained by active site slippage back to the same posi- tion. Therefore, we favor the view that either transcript slip- page back through site I or active site slippage along the tran- script in site I1 or both may occur depending on the template position.
Secondary structures may block reverse slippage either by preventing transcript re-entry into site I1 (Fig. 8 0 , panel 4 ) or by occupying an internal site so that slippage is not possible (Fig. 8 0 , panel 5). Inhibition of reverse translocation in com- plexes containing GMP in the nascent RNA may result from such structures, since even in the C-less transcript G-U, G-A, and G-G base pairs can form (Wyatt and Tinoco, 1993). Alter- natively, GMP may inhibit cleavage either by hydrogen bonding with unusual stability to DNA or RNA polymerase, slowing
transcript slippage, or by positioning adjacent phosphodiester bonds unfavorably for cleavage. Regardless of the explanation for the effects of GMP and IMP, RNA structure must play some role in determining the rates of cleavage and reverse translo- cation. We have recently found that the cleavage becomes facile in his paused transcription complexes when base substitutions that destabilize the pause hairpin are present and is again inhibited when compensatory substitutions that restore the structure are intr~duced.~ However, since IMP-containing tran- scription complexes exhibit some of the same reverse pause sites as GMP-containing complexes (Fig. 61, the overall rate of reverse translocation almost certainly is determined by the combined influences of reverse passage of both DNA and RNA strands through the transcription complex. How these RNA polymerase-DNA contacts change upon transcript cleavage is addressed in the accompanying paper.
ments on the manuscript and S. Borukhov, M. Chamberlin, S. Darst, A. Acknowledgments-We thank members of our laboratory for com-
Goldfarb, M. Izban, D. Luse, and K. Severinov for stimulating discus- sions and for sharing results prior to publication. The initial cloning of greA in our laboratory was accomplished with help from a University City High School student, Heather Williams, during a summer research internship supported by the National Science Foundation. Her contri- bution to the project is happily acknowledged.
REFERENCES
Aivazashvili, V. A,, Bibilashvili, R. S., Vartikyan, R. M., and Kutateladze, T. A.
Altmann, C. R., Solow-Cordero, D. E., and Chamberlin, M. J. (1994) Proc. Natl.
Amann, E., Ochs, B., and Abel, K.-J. (1988) Gene (Amst.) 6 9 , 3 0 1 3 1 5 Amdt, K. M., and Chamberlin, M. J. (1990) J. Mol. Biol. 213, 79-108 Borukhov, S., Polyakov, A., Nikiforov, V., and Goldfarb, A. (1992) Proc. Natl. Acad.
Borukhov, S., Sagitov, V., and Goldfarb, A. (1993a) Cell 72, 4 5 9 4 6 6 Borukhov, S., Sagitov, V., Josaitis, C. A., Gourse, R. L., and Goldfarb, A. (1993b) J.
Bradford, M. M. (1976) Anal. Biochem. 72,248-254 Bremer, H., and Dennis, P. P. (1987) in Escherichia coli and Salmonella typhi-
murium: Cellular and Molecular Biology (Niedhardt, F. C., Ingraham, J. L., Low, K. B., Magasanik, B., Schaecbter, M. and Umbarger, H. E., eds) pp. 1527- 1542, American Society for Microbiology, Washington, D. C.
(1982) Mol. Biol. 16, 711-722
Acad. Sci. U. S. A. 91,3784-3788
Sci. U. S. A. 89,8899-8902
Biol. Chem. 268,23477-23482
Burgess, R. R., and Jendrisak, J. J. (1975) Biochemistry 14,4634-4638 Chamberlin, M. J. (1994) Haruey Lect. 8 8 , 1-21 Chan, C. L., and Landick, R. (1989) J. Biol. Chem. 264,20796-20804 Chan, C. L., and Landick, R. (1993) J. Mol. Biol. 233, 2 5 4 2 Chan, C. L., and Landick, R. (1994) in Danscription: Mechanisms and Regulation
Das, A. (1993) Annu. Reo. Biochem. 62, 893-930 (Conaway, R., and Conaway, J., eds) pp, 297-320. Raven Press, New York
Erie, D., Hajiseyedjavadi, O., Young, M., and van Hippel, P. (1993) Science 262,
Erie, D. A,, Yager, T. D., and von Hippel, P. H. (1992) Annu. Rev. Biophys. Biomol.
Gu, W., Powell, W., Mote, J., Jr., and Reines, D. (1993) J. Biol. Chem. 268,25604-
Guo, H., and Price, D. H. (1993) J. Biol. Chem. 268,18762-18770 Hager, D. A,, Jin, D. J., and Burgess, R. R. (1990) Biochemistry 29, 7890-7894 Hagler, J., and Shuman, S. (1993) J. Biol. Chem. 268, 216G2173 Heumann, H., Metzger, W., and Niehorster, M. (1986) Eur. J. Biochem. 168,575-
Higuchi, R., Krummel, B., and Saiki, R. K. (1988) NucleicAcidsRes. 16,7351-7367 Izban, M. G., and Luse, D. S. (1992a) Genes & Deu. 6, 1342-1356 Izban, M. G., and Luse, D. S. (1992b) J. Biol. Chem. 267,13647-13655 Izban, M. G., and Luse, D. S. (1993a) J. Biol. Chem. 268, 12864-12873
Johnson, T., and Chamberlin, M. J. (1994) Cell 77, 212-224 Izban, M. G., and Luse, D. S. (1993b) J. Biol. Chem. 268, 12874-12885
Kassavetis, G. A,, and Geiduschek, E. P. (1993) Science 269,944-945 Krummel, B. (1990) Structural Dansitions of E. coli: RNA Polymerase during
Danscription Initiation and Elongation, Ph. D. Thesis. University of California, Berkeley
867-873
Struct. 21, 379415
25616
579
Krummel, B., and Chamberlin, M. J. (1992) J. Mol. Biol. 226, 239-250 Laemmli, U. K. (1970) Nature 227, 6 8 0 4 8 5 Landick, R., and Yanofsky, C. (1987) J. Mol. Biol. 196, 363-377 Landick, R., Stewart, J., and Lee, D. N. (1990a) Genes & Deu. 4, 1623-1636 Landick, R., Yanofsky, C., Phung, L., and Choo., K. (1990b) J. Mol. Biol. 216,25-37 Lee, D. N., and Landick, R. (1992) J. Mol. Biol. 228, 759-777 Lee, D. N., Phung, L., Stewart, J., and Landick, R. (1990) J. Biol. Chem. 266,
Levin, J. R., Krummel, B., and Chamberlin, M. J. (1987) J. Mol. Biol. 196,85-100 London, L., Keene, R. G., and Landick, R. (1991) Mol. Cell. Biol. 11, 4599-4615 Maitra, V, and Hurwitz, J. (1967) J. Biol. Chem. 242, 48974907
15145-15153
D. Wang and R. Landick, manuscript in preparation.

22294 GreA-induced llanscript Cleavage Mustaev, A., Kashlev, M., Zaychikov, E., Grachev, M., and Goldfarb, A. (1993) J. 80574061
Reines, D. (1992) J. Biol. Chem. 267, 37953800 Reynolds, R., and Chamberlin, M. J. (1992) J. Mol. Bid. 224, 53-63 Sparkowski, J., and Das, A. (1990) Nucleic Acids Res. 18, 6443 Reynolds, R., Bermudez-Cruz, R. M., and Chamberlin, M. J. (1992) J. Mol. Biol. Studier, w., and &senberg, A. (lgS1) ” 153* 503-527
Rice, G. A., &ne, c. M., and Chamberlin, M. J. (1991) Proc. Natl. Acad. Sei. wang, D., and ~ ~ ~ l ~ ~ , D, K, (1993) pro,.. ~ ~ ~ 1 , ~ ~ ~ d , sei, U, 8. A. 90, 843447
Rozovskaya, T. A.3 Chencbik, A. A., and Beabealashvih R. s. (1982) Lett. Atkins, J. F., eds) pp. 465-496, Cold Spring Harbor Laboratory Press, Cold Wyatt, J. R., and Tinoco, I., Jr. (1993) in The RNA World (Gesteland, R. F., and
Rudd, M. D, Izban, M. G., and Luse, D. S. (1994) Proc. Natl. Acad. Sci. U. S. A. 91, Yanisch-Perron, C., Vlera, J., and Messing, J. (1985) Gene (Amst.) 33, 103-119
Biol. Chem. 268,19185-19187 Sawadogo, M., and Roeder, R. G. (1985) Proc. Natl. Acad. Sci. U. S. A. 82, 4394- 4398
224,31-51
U. S. A. 88,1245-1249
137,100-104
Surratt, C. K., Milan, S. C., and Chamberlin, M. J. (1991) Proc. Natl. Acad. Sci. U. S. A. 88, 7983-7987
Spring Harbor, NY





























