Embed Size (px)
Citation preview

Complex Regulation of Human Neuronal Nitric-oxide Synthase Exon1c Gene TranscriptionESSENTIAL ROLE OF Sp AND ZNF FAMILY MEMBERS OF TRANSCRIPTION FACTORS*
Received for publication, October 10, 2001, and in revised form, April 2, 2002Published, JBC Papers in Press, April 17, 2002, DOI 10.1074/jbc.M109802200
Dieter Saur‡, Barbara Seidler, Heidi Paehge, Volker Schusdziarra, and Hans-Dieter Allescher
From the Department of Internal Medicine II, Technische Universitat Munchen, Munich 81675, Germany
Neuronal nitric-oxide synthase (nNOS) is expressed ina variety of human tissues and shows a complex tran-scriptional regulation with the presence of nine alterna-tive first exons (1a–1i) resulting in nNOS transcriptswith differing 5�-untranslated regions. We previouslydemonstrated that nNOS exon 1c, one of the predomi-nant transcripts in the human gastrointestinal tract, isdriven by a separate promoter (Saur, D., Paehge, H.,Schusdziarra, V., and Allescher, H. D. (2000) Gastroen-terology 118, 849–858). The present study focused on thequantitative expression of nNOS first exon variants indifferent human tissues and the characterization of thebasal nNOS exon 1c promoter. In human brain, skeletalmuscle, colon, and TGW-nu-I neuroblastoma cells, firstexon expression patterns were analyzed by quantitativereal-time reverse transcription-PCR. In these tissues/cells exon 1c was one of the most abundant first exons ofnNOS. By transient transfections of TGW-nu-I and HeLacells with reporter plasmids containing a series of 5� and3� deletions in the exon 1c regulatory region, the mini-mal TATA-less promoter was localized within 44 basepairs. Gel mobility shift assays of this cis-regulatoryregion revealed a high complexity of the basal promoterwith a cooperative binding of several transcription fac-tors, like Sp and ZNF family members. When the Spbinding site of the minimal promoter construct was mu-tated, promoter activity was completely abolished inboth cell lines, whereas mutation of the common bind-ing site of ZNF76 and ZNF143 resulted in a decrease of53% in TGW-nu-I and 37% in HeLa cells. In DrosophilaSchneider cells expression of Sp1, the long Sp3 isoform,ZNF76 and ZNF143 potently transactivated the nNOSexon 1c promoter. These results identify the critical reg-ulatory region for the nNOS exon 1c basal promoter andstress the functional importance of multiple proteincomplexes involving Sp and ZNF families of transcrip-tion factors in regulating nNOS exon 1c transcription.
Nitric oxide (NO),1 a ubiquitous multifunctional mediator, issynthesized by nitric-oxide synthases (NOS) during the oxida-
tion of L-arginine to L-citrulline. In the central and peripheralnervous system, skeletal muscle, the macula densa of the kid-ney, testis, and neutrophils, neuronal NOS (nNOS) is the pre-dominant enzyme for the generation of nitric oxide. NO, syn-thesized by nNOS, acts as neurotransmitter, neuromodulator,or intracellular signaling molecule. It is involved in synapticplasticity, regulation of gene expression, development, differ-entiation, and regeneration and plays an important role inneurodegenerative disorders and stroke as a mediator of neu-rotoxicity (for review see Refs. 1–4). In the gastrointestinaltract NO generated by nNOS acts as an important mediator ofthe non-adrenergic non-cholinergic inhibitory innervation ofintestinal smooth muscle (5) and as a neuromodulator withinthe enteric nervous system (6).
Although the transcriptional regulation of the other twoNOS enzymes, the calcium-dependent endothelial NOS and thecalcium-independent inducible NOS, are extensively studied(for review see Refs. 3, 7, 8), little is known about the transcrip-tional regulation of the nNOS gene (3, 4, 9, 10), which isconsidered to be responsible for the largest proportion of NO inthe body (1). Although usually named constitutive, recent ob-servations suggest a tightly regulated gene expression of nNOSin response to different physiological and pathophysiologicalstimuli, resulting in an up- or down-regulation of nNOS mRNA(3, 9).
Recently nine distinct first exons, called exons 1a–1i, ofnNOS mRNA have been identified, leading to nNOS mRNAvariants with different 5�-untranslated regions and transla-tional efficiencies (11). The nNOS gene is therefore believed tobe one of the most complex genes known in terms of first exonusage and alternative splicing (11–13). It has been shown thatnNOS exons 1c (12) and 1f and 1g (13) (former called exons 15�3,15�2, and 15�1, respectively), which show high abundant expres-sion in the human gastrointestinal tract (12), are driven byseparate promoters in HeLa cells. The use of multiple alterna-tive promoters allows a cell-, tissue-, and site-specific transcrip-tional regulation of nNOS in different physiological and patho-physiological stages.
An altered expression or biological activity of nNOS has beenlinked to several physiological conditions, like aging and preg-nancy, as well as different pathophysiological conditions anddiseases such as ischemia/hypoxia and injuries of the centralnervous system, inherited diabetes insipidus, heart failure,arteriosclerosis, achalasia, diabetic gastroparesis, and hyper-trophic pyloric stenosis (1–4, 7, 14–17). nNOS� mutant mice,
* This work was supported by Deutsche ForschungsgemeinschaftSonderforschungsbereich 391 C5, KKF TU Munich F71-98 and F47-01.Preliminary results were presented at the annual meeting of the Amer-ican Gastroenterological Association in Atlanta (2001). The costs ofpublication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The nucleotide sequence(s) reported in this paper has been submittedto the GenBankTM/EBI Data Bank with accession number(s) AJ308545.
‡ To whom correspondence should be addressed: II. MedizinischeKlinik und Poliklinik, Technische Universitat Munchen, Ismaninger-strasse 22, Munchen D-81675, Germany. Tel.: 49-89-4140-4377; Fax:49-89-4140-4932; E-mail: [email protected].
1 The abbreviations used are: NO, nitric oxide; NOS, nitric-oxide
synthase; nNOS, neuronal nitric-oxide synthase; EMSA, electro-phoretic mobility shift assay; FAM, 6-carboxy-fluorescein; FBS, fetalbovine serum; GSP, gene-specific primer; nt, nucleotide(s); RAGE, rapidamplification of genomic ends; RT, reverse transcriptase; SL2 cells,Drosophila Schneider cells; PBS, phosphate-buffered saline; GAPDH,glyceraldehyde-3-phosphate dehydrogenase.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 277, No. 28, Issue of July 12, pp. 25798–25814, 2002© 2002 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org25798
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

generated by targeted disruption of the nNOS gene by homol-ogous recombination, showed a gastrointestinal phenotype re-sembling hypertrophic pyloric stenosis with delayed gastricemptying of solids and fluids (18, 19). In addition, these micehave a hypertensive lower esophageal sphincter with impairedrelaxation (20). A recent study comprising 27 families withinherited infantile pyloric stenosis identified nNOS as a sus-ceptibility gene for this disorder (15), and expression of nNOSexon 1c mRNA is significantly reduced in the pyloric sphincterof patients with infantile hypertrophic pyloric stenosis.2
Therefore it is of physiological and pathophysiological inter-est to investigate the molecular basis of nNOS exon 1c tran-scription. In addition, the distribution and quantitative expres-sion of alternative first exon nNOS variants in different humantissues was determined.
Here we present evidence for a tissue-specific expression ofnNOS first exon variants, with exon 1c being one of the pre-
dominant forms in human brain, skeletal muscle, gastrointes-tinal tissue, and neuroblastoma cells. We furthermore charac-terize the basal promoter of exon 1c, showing a high complexitywith a cooperative binding of several Sp and ZNF family mem-bers of transcription factors.
EXPERIMENTAL PROCEDURES
Materials—The cell lines TGW-nu-I (human neuroblastoma) andME-180 (human cervix carcinoma) were kindly provided to our labora-tory by Dr. Esumi (21) and Dr. E. R. Werner (22), respectively. Themammalian expression plasmids pcDNA3 ZNF76 and pcDNA3 143were a gift from Dr. P. Carbon (23), and CB6-MZF-1 (alternative nameCB6-ZNF42), under the control of the cytomegalovirus early promoter,was generously provided by Dr. R. Hromas (24). The plasmid pPac-Sp1,which expresses Sp1 from the Drosophila actin promoter, and the“empty” control plasmid pPac0, containing only the Drosophila actinpromoter, were generously provided by Dr. R. Tjian (25). The expressionvector for Sp2 (pPac-Sp2) was a gift from Dr. J. D. Noti (26), and theexpression plasmids for the short isoforms of Sp3 (pPac-Sp3), the longisoform of Sp3 (pPacUSp3), Sp4 (pPac-Sp4), and the �-galactosidaseexpression plasmid p97b were generously provided by Dr. G. Suske(27–30). All cell culture reagents were obtained from Invitrogen (Gron-
2 D. Saur, J. M. Vanderwinden, M. H. De-Laet, and H.-D. Allescher,manuscript in preparation.
TABLE ISense (S) and antisense strand (AS) primers and TaqMan probes
Sequences of primers and probes for RT-PCR, real-time RT-PCR, 5�-RAGE-PCR, constructions of pPac-ZNF42, pPac-ZNF76, and pPac-ZNF143,generation of 5� deletions of pGL3–5891/�49, point mutations of pGL3–90/�49, and site-directed mutagenesis of pGL3–5891/�49 and pGL3–332/�49. Boldface letters indicate mutated bases.
Name Sequence (5� to 3�)
ZNF42 (S) CGCAGGTCCAGGTAGTGTAAGCZNF42 (AS) CTCTCCGATGCTCTTCCAGGZNF76 (S) CAGGTGACGGTACAGAAAGAAGCZNF76 (AS) TGATGAGCGGTGGTGTAGAGACZNF143 (S) TCATAGATGGCCAGGTCATTCAGZNF143 (AS) CCAATCATTCCAGTACCTGCTACACZNF42 XhoI (S) CGGCTCGAGGCCGCCACCATGAATGGTCCCCTTGTGTAZNF42 XhoI (AS) CGGCTCGAGCTACTCGGCGCTGTGGACZNF76 BamHI (S) CGGGATCCGCCGCCACCATGGAGAGZNF76 BamHI (AS) CGGGATCCTCAGCAGCCACTCTCCGZNF143 BamHI (S) CGGGATCCGCCGCCACCATGGCAGAGTTTCCTGGAGGAGGGZNF143 BamHI (AS) CGGGATCCTTAATCATCCAACCCTGnNOS exon 1a (S) CCCATGCTCTAGCTTGGAGCnNOS exon 1b (S) GCTGCTGCCACCTCTAAATGAnNOS exon 1c (S) ATGCCAGGGTGAGGCCTTnNOS exon 1d (S) GCTGAGAGATGGAAGAACTGGCnNOS exon 1e (S) GGAGGAGCCTTGGAGGGAnNOS exon 1f (S) CGCTGGGTGAGGAGCTACTTAGnNOS exon 1g (S) GGGAGGAAGAGGAGGAGTACGAnNOS exon 1h (S) TTCCTCGGTGCCCGACTnNOS exon 1i (S) CACCTGGAGCCTCTTAACTTTCAGnNOS exon 2 (AS) GTGACGCATGATAGATGTGAACTATTCnNOS exon 2 Probe AGATCTGATCCGCATCTAACAGGCTGGCnNOS exon 6 (S) CAGTGGTCCAAGCTGCAGGTAnNOS exon 7 (AS) GGTGGCATACTTGACATGGTTACAnNOS exon 7 Probe TCGATGCCCGTGACTGCACCACP-5891 (S) AAACATAAATTCCATTGCATTCCAP-2774 (S) ACTTTGGGAGGCTGAGGCAGP-1938 (S) GTACCTGGCACATAATAGGTP-1520 (S) AGCTCCTGTCCTCAGGTGATCTP-332 (S) GCTGGGGGGAAACCTGAGP-279 (AS) CAGCAGTGGCAGCGTCTGP-83 (S) ACCTCCCAGCCTGCCCCTGP�49 (AS) GTCACCCCCTCTCAGACAGTGP-90 Sp1/ZNF42-M1 (S) AGAGCTCGAGCCACCTCCCAGCCTGCCCCTGGTTAGGP-90 Sp1-M2 (S) AGAGCTCGAGCCACCTCCCAGCCTGCCCCTGGGGATTGGCP-90 Ap2/Olf-1-M (S) AGAGCTCGAGCCACCTCCCAGCCTGCTTCTGP-90 Staf-M (S) AGAGCTCGAGCCACCTCTTAGTCTGCCCCTGSDM-Sp1/ZNF42-M1 (S) CTCCCAGCCTGCCCCTGGTTAGGGGCCACCTGGTGTCSDM-Sp1/ZNF42-del (S) CCAGCCTGCCCCTCACCTGGTGTCSDM-Staf-M (S) CAGCAGAGCCACCTCTTAGTCTGCCCCTGGGGAGGGSDM-Staf-del (S) CAGCAGAGCCACCCTGGGGAGGGSDM-Staf/Sp1/ZNF42-M1 (S) CAGCAGAGCCACCTCTTAGTCTGCCCCTGGTTAGGGGCCACCTGGTGTCSDM-Staf/Sp1/ZNF42-del (S) CAGCAGAGCCACCACCTGGTGTCGSP1-AS/ex 1c (AS) CCTGGCATGGTGGCGGTCACGSP2-AS/ex 1c (AS) GTCACCCCCTCTCAGACAGTGCTCAP1 GTAATACGACTCACTATAGGGCAP2 ACTATAGGGCACGCGTGGT
nNOS Exon 1c Basal Promoter Regulation 25799
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

ingen, Netherlands). Polyclonal antibodies against Sp1 (PEP 2 X), Sp2(K-20 X), Sp3 (D-20 X), Sp4 (V-20 X), NF�B p65 (C-20 X), NF-�B p50(C-19 X), Ap-2a (C-17 X) were purchased from Santa Cruz Biotechnol-ogies (Heidelberg, Germany). Consensus and mutant consensus oligo-nucleotides used in electrophoretic mobility shift assays (EMSAs) wereobtained from Santa Cruz Biotechnologies (Ap2, Myc-Max, NF-I, YY1,NF�B, p53, Sp1, USF-1), or synthesized (Staf, Olf-1, ZNF42, MAZ) byMWG (Ebersberg, Germany). Primers were made by MWG and Taq-Man-probes by Applied Biosystems (Weiterstadt, Germany). Restric-tion endonucleases were obtained from New England BioLabs (Mann-heim, Germany). [�-32P]ATP was supplied from Amersham Biosciences,Inc. (Freiburg, Germany). The Escherichia coli strain TOP10 (Invitro-gen) was used for transformation and amplification of DNA-containingplasmids.
Tissue Preparation—Tissues from human rectum were obtainedfrom surgical resections for malignant disease and prepared as previ-ously described (12).
RNA Isolation—Liquid nitrogen-frozen rectum muscle layer prepa-rations and fresh TGW-nu-I, ME-180, and HeLa cells were homoge-nized with a Polytron homogenizer (Kinematica, Switzerland), and totalRNA was isolated using the guanidine isothiocyanate/phenol/chloro-form extraction method as previously described (12, 31). Pooled totalRNA from human adult brain and skeletal muscle was purchased fromCLONTECH (Heidelberg, Germany).
RT-PCR—Reverse transcription and PCR amplification were carriedout exactly as described before (12, 31). RT-PCR was performed withspecific primer pairs for ZNF42, ZNF76, and ZNF143 (for all primerssee Table I) for 35 cycles using random hexamer-primed cDNA fromhuman brain, skeletal muscle, rectum, TGW-nu-I cells, ME-180 cells,and HeLa cells (annealing 58 °C, 30 s; extension 72 °C, 60 s; denatur-ation 94 °C, 30 s). Amplification products were cloned into pCRII plas-mid (Invitrogen) and subjected to DNA sequence analysis (GATC, Kon-stanz, Germany).
Quantitative Real-time RT-PCR—Real-time quantitative RT-PCRanalyses for the alternative first exons 1a–1i and exon 6/7 of humannNOS mRNA were performed using an ABI Prism 7700 SequenceDetection System instrument (Applied Biosystems). cDNAs for quanti-tative analysis were generated with the TaqMan reverse transcriptionreagents (Applied Biosystems) as recommended by the manufacturer,using murine leukemia virus reverse transcriptase, random hexamerprimers, and 5 �g of total RNA from human rectum and from TGW-nu-I, ME-180, and HeLa cells. In addition, total RNA obtained fromCLONTECH and isolated from pooled human brain and skeletal musclewas used. nNOS transcripts were amplified with intron-spanning, iso-form-specific primers and probes complementary to the alternative firstexons 1a–1i (forward primers) and the common exon 2 (reverse primer,probe). As a parameter for total nNOS mRNA expression, an intron-spanning pair of exon 6- and exon 7-specific primers present in allknown human nNOS cDNAs was used with an exon 7-specific internalprobe. Primers and TaqMan probes were designed to meet specificcriteria by using the Primer Express software (Applied Biosystems).The 5�-end of the probe was labeled with a fluorescent reporter (6-carboxy-fluorescein (FAM)) and the 3�-end with a quencher dye (6-carboxyltetramethylrhodamine). Sequences of primers and fluorescentprobes used in the study are shown in Table I. GAPDH, HPRT, andTF2D primers and probes were purchased from Applied Biosystems.
The principle of real-time RT-PCR has been described in detailelsewhere (32, 33). Briefly, real-time PCR is based on a sequence-specific probe labeled with a 5� reporter and 3� quencher dye. When theprobe is intact, reporter dye emission is quenched, but during theextension phase of the PCR, the nucleolytic activity of the Taq-DNApolymerase cleaves the hybridized probe, and due to the separation ofreporter and quencher dye a fluorescence signal is released that ismonitored by the sequence detector. A computer algorithm normalizesthe signal to an internal reference (�Rn) and calculates the thresholdcycle number (Ct), when �Rn becomes equal to 10 standard deviationsof the baseline. Ct is used for quantification of the input mRNA number.For each amplicon, the amount of target and endogenous reference(glyceraldehyde-3-phosphate dehydrogenase, HPRT, TF2D) was deter-mined from a standard curve generated by serial 5-fold dilutions(25,000 to 8 copies) of plasmids containing the respective target se-quence. The standard curve was amplified in triplicate during everyexperiment, and the amount of target gene was normalized by theendogenous reference. Quantitative PCR was performed using the Taq-Man Universal PCR Master Mix (Applied Biosystems), with cDNAscorresponding to 100 ng of total RNA, 200 nM probe, and 900 nM primersin a 25-�l final reaction mix (1 PCR cycle 50 °C, 2 min; 95 °C, 10 min;50 PCR cycles 60 °C, 1 min; 95 °C, 15 s). Signals were analyzed by the
ABI Prism Sequence Detection System software version 1.7 (AppliedBiosystems).
Cloning of the 5�-Flanking Region of Exon 1c—The 5�-flanking regionof human nNOS exon 1c was determined by rapid amplification ofgenomic ends (RAGE) using the human GenomeWalker kit (CLONTECH)as previously described (12). Digested and adapter-ligated genomic DNAfragments obtained from CLONTECH were amplified in a first round ofPCR (seven cycles 94 °C, 25 s; 72 °C, 7 min and 32 cycles 94 °C, 25 s; 67 °C,7 min adding 5 s each cycle) using the antisense gene-specific primerGSP1-AS/ex 1c (10 pmol) (for all primers see Table I) and the sensegeneric primer AP1 from CLONTECH specific for the ligated adapters.The second round of PCR was performed with nested antisense gene-specific primer GSP2-AS/ex 1c (10 pmol) and sense generic primer AP2 (5cycles 94 °C, 25 s; 72 °C, 7 min; 20 cycles 94 °C, 25 s; 67 °C 7 min adding5 s each cycle). PCR products were cloned into pCRII plasmid and sub-jected to commercial sequence analysis (GATC). The BLASTn programwas employed to search for sequence identity. Potential cis-acting DNAsequences and putative consensus elements were identified by analysiswith the MatInspector professional software (Genomatix Software GmbH,Munich, Germany) using the Transfac data base.
Plasmid Constructions—The human genomic 5�-flanking region ofnNOS exon 1c was obtained by 5�-RAGE PCR. To create a 5940-bpfragment containing the promoter of exon 1c (nt �5891 to �49), 100 ng ofgenomic DNA was PCR-amplified (30 cycles at 94 °C, 20 s; 60 °C, 30 s;72 °C, 7 min) using a proofreading polymerase (Pwo, Roche MolecularBiochemicals, Mannheim, Germany) and the primers P-5891 (sense) andP�49 (antisense) (see Table I for primer sequences). The gel-purified PCRproduct was blunt end-cloned into the SmaI site of the promoter-/enhanc-erless firefly luciferase reporter gene vector pGL3-basic (Promega, Mann-heim, Germany) in the forward (pGL3�5891/�49) and reverse(pGL3�49/�5891) orientation. Reporter gene constructs containing 5�and 3� deletions of the promoter region of nNOS exon 1c were generatedby PCR, exonuclease III/S1 nuclease digestion, or restriction endonucle-ase digestion. pGL3�2774/�49, pGL3�1938/�49, pGL3�1520/�49 andpGL3�332/�49 were prepared by PCR and blunt end cloning similar tothe preparation of pGL3�5891/�49, using the sense primers P-2774,P-1938, P-1520, and P-332 combined with the common antisense primerP�49. For construction of pGL3�5891/�279, sense primer P-5891 wasused with antisense primer P-279. pGL3�226/�49 was prepared by restric-tion endonuclease digestion of pGL3�332/�49 with PflmI and MluI, fol-lowed by blunting and religation. Additional 5� deletions of pGL3�332/�49(pGL3�278/�49, pGL3�241/�49, pGL3�174/�49, pGL3�131/�49,pGL3�90/�49, pGL3�83/�49, pGL3�63/�49, pGL3�48/�49, pGL3�34/�49, pGL3�24/�49, pGL3�14/�49, pGL3�4/�49, pGL3 � 18/�49), wereprepared by successive exonuclease III and S1 nuclease digestions andre-ligations. pGL3�90/�49 Sp1/ZNF42-M1, containing a point muta-tion of the common Sp1 and ZNF42 binding site, pGL3�90/�49 Sp1-M2, containing only a mutated Sp1 element, pGL3�90/�49 Ap2/Olf-1-M, containing a mutation of the common Ap2 and Olf-1 binding site,and pGL3�90/�49 Staf-M, containing a mutated Staf consensus se-quence (common binding site for ZNF76 and ZNF143), were preparedby PCR using the common antisense primer P�49 combined with thefollowing sense primers: P-90 Sp1/ZNF42-M1, P-90 Sp1-M2, P-90 Ap2/Olf-1-M, and P-90 Staf-M. Gel-purified PCR products were blunt end-cloned into pGL3-basic in the forward orientation. Mutations and de-letions of the common Sp1/ZNF42 binding site, the Staf binding site,and both the Sp1/ZNF42 and Staf binding sites in the longer promoterconstructs pGL3�5981/�49 and pGL3�332/�49 were generated byusing the QuikChange XL site-directed mutagenesis kit (Stratagene,Heidelberg, Germany) exactly as described by the manufacturer withthe following primers: SDM-Sp1/ZNF42-M1 (mutation of the Sp1/ZNF42 binding site), SDM-Sp1/ZNF42-del (deletion of the Sp1/ZNF42binding site), SDM-Staf-M (mutation of the Staf binding site), SDM-Staf-del (deletion of the Staf binding site), SDM-Staf/Sp1/ZNF42-M1(mutation of the Staf and Sp1/ZNF42 binding sites), and SDM-Staf/Sp1/ZNF42-del (deletion of the Staf and Sp1/ZNF42 binding sites). To con-struct plasmids that express the transcription factors ZNF42, ZNF76,and ZNF143 from the Drosophila actin promoter, the eukaryotic ex-pression vectors CB6-ZNF42 (alternative name CB6-MZF-1), pcDNA3ZNF76, and pcDNA3 ZNF143 were used as templates for PCR to gen-erate DNAs containing the complete coding sequences of these tran-scription factors. Primers for ZNF42 contained XhoI, and primers forZNF76 and ZNF143 contained BamHI restriction sites and a Kozakconsensus sequence upstream of the ATG start codon. The XhoI andBamHI restriction sites were used to clone the coding sequences ofZNF42, ZNF76, and ZNF143 into the respective XhoI or BamHI sites ofthe expression plasmid pPac0, which contains only the Drosophila actinpromoter. Integrity of all cloned sequences was confirmed by automated
nNOS Exon 1c Basal Promoter Regulation25800
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

DNA sequencing (GATC) using an ABI Prism 377 DNA sequencer(Applied Biosystems).
Cell Culture, Transient Expression, and Reporter Gene Assays—HeLa cells were cultured and transiently cotransfected with the differ-ent nNOS exon 1c-pGL3 promoter gene constructs and the herpessimplex virus thymidine kinase promoter-driven Renilla luciferase ex-pression vector pRL-TK (Promega) to normalize for transfection effi-ciency and cell number essentially as described previously (12). TGW-nu-I cells were cultured in minimum Eagle’s medium containing 10%
fetal bovine serum (FBS), 25 mM L-glutamine, 100 units/ml penicillin,and 100 �g/ml streptomycin sulfate (37 °C/5%CO2). 50–60% confluentcells were transiently cotransfected by lipid-mediated transfer using0.95 �g of plasmid DNA of each pGL3 construct and 0.05 �g of pRL-TKDNA with 6 �l of Plus reagent and 4 �l of LipofectAMINE (Invitrogen)per 3.5-cm dish. Cells were incubated in FBS and antibiotic-free mini-mum Eagle’s medium containing the lipid-coated DNA complexes for3 h and in complete medium for an additional 48 h. ME-180 cells werecultured in McCoy�s 5A medium as described before (22). About 60%
FIG. 1. A, schematic exon structure of alternatively spliced mRNA transcripts of human nNOS (adapted from Wang et al. (11)). Nine distinct firstexons (exon 1a–1i) driven by separate promoters and spliced to the common exon 2 of nNOS are shown. Arrows indicate the positions of sense andantisense strand primers used in quantitative real-time RT-PCR (Fig. 2). The position of the internal 6-carboxy-fluorescein (FAM)-labeled TaqManProbe is marked above exon 2 by a line, and the translational initiation codon is noted by a vertical arrow. B, genomic organization of the alternativefirst exons 1a, 1b, and 1c of the nNOS gene. Exons 1a and 1b are located in the 5�-upstream genomic DNA region of exon 1c. The arrows below thenucleotide sequence depict the 5� deletion sites of the different nNOS pGL3 promoter constructs used in reporter gene assays. The nucleotidesequence reported in this study has been deposited in the EMBL/GenBankTM data base with accession number AJ308545. C, nucleotide sequenceof human nNOS exon 1c (in boldface letters) and in part the 5�-flanking region including the minimal promoter. The transcription start site isindicated as �1 and underlined. Cis-acting elements as determined by gel shift assays (Fig. 4, A–F) are indicated by lines above the sequence.Arrows below the nucleotide sequence depict the 5� deletion sites of the different nNOS pGL3 promoter constructs used in reporter gene assays.
nNOS Exon 1c Basal Promoter Regulation 25801
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

confluent cells were transfected with 0.38 �g of each pGL3 test plasmidDNA and 0.02 �g of pRL-TK DNA using 4 �l of Effectene and 3.2 �l ofEnhancer (Qiagen) per 3.5-cm dish.
Approximately 2 � 106 Drosophila Schneider cells (SL2 cells) wereplated the day prior to transfection onto six-well plates and cultured at25 °C in DES Expression Media containing 10% FBS. After washingand addition of fresh medium without FBS, cells were transfected using9 �l of Cellfectin (Invitrogen) per well along with 1 �g of each pGL3construct, 0.5 �g of the �-galactosidase expression plasmid p97b fornormalization of transfection efficiencies, and variable amounts of theexpression plasmids pPac-Sp1, pPac-Sp2, pPac-USp3, pPac-Sp3, pPac-Sp4, pPac-ZNF42, pPac-ZNF76, and pPac-ZNF143. Variations in theamount of the expression plasmids were compensated with the emptyplasmid pPac0 to adjust the total DNA content of the transfection mixto 2 �g per well. After 24 h of transfection, medium was removed andcells were incubated for an additional 24 h in complete medium con-taining FBS. SV40 promoter/enhancer-directed pGL3-control and pro-moter-/enhancerless pGL3-basic plasmids (both Promega) were used aspositive and negative controls in all experiments, respectively.
48 h after transfection, cells were harvested by treatment with lysisbuffer (Promega). Total cellular protein was determined by Bio-Rad IIassay and �-galactosidase activity was assayed using the �-Galactosid-ase Enzyme Assay System (Promega) essentially as recommended bythe manufacturer. Firefly and Renilla luciferase activities were meas-ured in a luminometer (EG&G Berthold, Bad Wildbad, Germany) byusing the Dual Luciferase Reporter Assay System (Promega) as previ-ously described (12). All values were determined from three independ-ent transfection experiments, each done in triplicate, and are expressedas mean values � S.D. Data of TGW-nu-I and HeLa cell transfectionsare presented as relative luciferase activity of firefly/Renilla luciferase.Values for SL2 cells are expressed as -fold induction of normalizedfirefly luciferase activity relative to that obtained following cotransfec-tion of the pGL3 reporter plasmids with empty pPac0, which does notexpress Sp/ZNF proteins. In SL2 cells firefly luciferase activity wasnormalized in all samples for total protein content, because the tran-scription of the �-galactosidase expression vector p97b, intended asinternal control, was highly inducible by cotransfection with pPac-ZNF76 and pPac-ZNF143 (but not with pPac-Sp1–4 and pPac-ZNF42).
Electrophoretic Mobility Shift Assays—Nuclear extracts were pre-pared from cultured untransfected TGW-nu-I and HeLa cells and fromDrosophila Schneider cells transiently transfected with the expressionplasmids pPac-Sp1, pPac-Sp2, pPac-USp3, pPac-Sp3, pPac-Sp4, pPac-ZNF42, pPac-ZNF76, and pPac-ZNF143. Cells were harvested from10-cm dishes by scraping, washed once with ice-cold PBS, centrifuged,resuspended in 400 �l of ice-cold buffer A (10 mM HEPES (pH 7.9), 10mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 1 mM phenyl-methylsulfonyl fluoride) and incubated for 20 min at 4 °C. After addi-tion of 25 �l of Nonidet P-40 (10%), the probes were vortexed for 10 s attop speed, followed by a centrifugation step (10,000 � g, 5 min, 4 °C).The pelleted nuclei were resuspended in ice-cold high salt buffer C (20mM HEPES (pH 7.9), 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM
dithiothreitol, 1 mM phenylmethylsulfonyl fluoride) and vigorouslymixed on a shaker for 30 min at 4 °C to extract nuclear proteins. Thesamples were subjected to centrifugation (10,000 � g, 5 min, 4 °C) andthe supernatant was stored at �80 °C. For EMSAs 10 �g of the appro-priate nuclear extract was incubated in 20 �l of binding buffer (25 mM
Tris-HCl (pH 8.0), 50 mM KCl, 6.25 mM MgCl2, 10% glycerol, 50 �g/mlbovine serum albumin, 1 �g of poly(dI-dC) (Amersham Biosciences,Inc.)) with 20 fmol of the indicated 32P-labeled double-stranded oligo-nucleotide probe and in certain experiments with unlabeled competitoroligonucleotides (see Table II and under “Materials”). 32P-Labeledprobes were prepared by 5�-end labeling using T4 polynucleotide kinaseand [�-32P]ATP. Reactions were incubated for 30 min at 22 °C andloaded onto a non-denaturing, 5% polyacrylamide gel (37:1 ratio ofacrylamide to N,N�-methylenebisacrylamide) in 0.5 � Tris borateEDTA (TBE) buffer. Electrophoresis was performed at 250 V for 3 h in0.5� TBE buffer with buffer recirculation at 4 °C, followed by autora-diography at �80 °C with intensifying screens. For supershift assays,antibodies (2 �l) were added 30 min after addition of the labeled probeand incubated for an additional 30 min.
Western Blot Analysis—Nuclear extracts (60 �g of protein per lane)from transfected SL2 cells and untransfected TGW-nu-I and HeLa cellswere separated by 7.5% SDS-PAGE and transferred to polyvinylidenedifluoride membranes (Bio-Rad) as previously described (12, 31). Blotswere probed with Sp1, Sp2, and Sp3 antibodies diluted 1:1000. Signaldetection of the immunoreactive bands was facilitated by enhancedchemiluminescence using the ECL system (Amersham Biosciences,Inc.).
Data Analysis—Unless otherwise indicated, all data were deter-mined from three independent experiments, each done in triplicate, andare expressed as mean values � S.D. Comparisons among data setswere made with analysis of variance, followed by Student’s t test.Values of p � 0.05 or less were considered to be statistically significant.
RESULTS
Expression Pattern of nNOS First Exon mRNA Vari-ants—We analyzed mRNA expression of the nine alternativefirst exon variants of human nNOS (Fig. 1A) by real-timequantitative RT-PCR (5� nuclease assay) in human brain, skel-etal muscle, rectum, TGW-nu-I neuroblastoma cells, ME-180cervix carcinoma cells, and HeLa cells. In addition, as a param-eter for total nNOS mRNA expression, a sequence betweenexon 6 and exon 7, encoding parts of the oxygenase domain ofnNOS (hem binding site that is essential for NOS activity (34)),was amplified. As internal controls GAPDH, HPRT, and TF2DmRNA expression were quantified. Real-time RT-PCR of thedifferent first exons revealed high expression of nNOS exon 1cin the investigated human tissues (Fig. 2, A–C) and the TGW-nu-I cell line (Fig. 2D), whereas ME-180 cells were nNOS exon1c-negative (Fig. 2E). The other first exons showed varyingexpression patterns with a cell- and tissue-specific expression(see Fig. 2, A–F). Exon 1f and exon 1g are highly abundant inthe brain and rectum and very lowly abundant in skeletalmuscle (Fig. 2, A–C, F; where bars are not evident despitepositive RT-PCR results in Fig. 2F, values are less than theresolution shown in the figure). In contrast exon 1a is highlyexpressed in skeletal muscle (Fig. 2B) but missing in TGW-nu-Icells (Fig. 2D) and lowly abundant in the rectum (Fig. 2C).HeLa cells were nNOS mRNA-negative but showed high ex-pression of the investigated housekeeping genes (data notshown). Fig. 2F summarizes the distribution of alternative firstexons of nNOS in the investigated tissues and cell lines. nNOSfirst exon expression was normalized against GAPDH (Fig. 2,A–E) and showed no significant difference when other house-keeping genes (HPRT, TF2D) were used as internal controls(data not shown).
Cloning of the 5�-Flanking Region of nNOS Exon 1c—Tofurther determine the partially known promoter sequence ofnNOS exon 1c (formerly called exon 15�3) (12), we used rapidamplification of genomic ends (RAGE) to obtain a 5939-bp DNAfragment of the 5�-flanking regulatory region of exon 1c span-ning nucleotides (nt) �5891 to �49 (Fig. 1B). This nucleotidesequence has been deposited in the EMBL/GenBankTM database with accession number AJ308545. The transcriptionalstart site of exon 1c was identified previously to be located 84bp upstream of the exon 1c/intron 1 splice junction at an ade-nine (�1) (Fig. 1, B and C) (12). Computer-based analysis of thesequence immediately upstream of this region using the Mat-Inspector professional software, revealed no canonical TATA orCAAT boxes as initiators of transcription, but a putative tran-scription initiation site with overlapping consensus sequencesfor Staf (common binding site for ZNF76 and ZNF143 (23)), p53half site, Olf-1, Ap2, Myc/Max, ZNF42 (alternative title MZF-1(24)), Sp1 and MAZ between nt �90 and �47 bp relative to thetranscription start site. The obtained 5�-flanking region of exon1c contains in addition the sequence of nNOS exon 1a (nt3656–3755) derived from EMBL accession number AF049712(11) and exon 1b (nt 5567–5635) derived from EMBL accessionnumber AF049713 (11) (see Fig. 1B). Exon 1c is located at nt5892–5975 of the submitted sequence with EMBL accessionnumber AJ308545. Therefore exons 1a, 1b, and 1c are locatedin close proximity within 2400 bp, similar to the genomic struc-ture of exons 1f and 1g (former called exons 15�2 and 15�1) ofhuman (13) and exons 1b and 1c of rat nNOS (35). Further-more, the BLASTn search revealed that the putative transcrip-tion initiation site of exon 1c is highly conserved between rat
nNOS Exon 1c Basal Promoter Regulation25802
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

FIG. 2. Quantitative analysis of human nNOS mRNA expression by real-time RT-PCR in human brain (A), skeletal muscle (B),rectum (C), TGW-nu-I neuroblastoma cells (D), and ME-180 cervix carcinoma cells (E). A set of nine forward primers, specific for the ninealternative first exons of nNOS (exon 1a–1i) were used with a common exon 2-specific reverse primer and an internal exon 2-specific 6-carboxy-fluorescein (FAM)-labeled TaqMan probe. For primer locations see arrows in Fig. 1A. As a parameter for total nNOS mRNA expression, a pair ofexon 6- and exon 7-specific primers present in all known nNOS cDNAs were used with an exon 7-specific internal FAM-labeled probe. Relativeamounts of transcripts were calculated using standard curves and dividing the expression levels of the different nNOS variants by the expressionlevels of the GAPDH housekeeping gene measured in the same RNA preparation. Results shown are the mean � S.D. of one (pooled RNA obtainedfrom CLONTECH; A and B) and three independent RNA isolations (C–E). Individual cDNA samples were analyzed in triplicate with a given pairof primers. F, distribution of alternative first exon variants of human nNOS in the brain, skeletal muscle, rectum, TGW-nu-I cells, ME-180 cells,and HeLa cells as determined by real-time RT-PCR. ��� indicates a high, �� a moderate, and � a low expression level; � indicates negativeRT-PCR results (where bars are not evident (A–E) despite positive RT-PCR results, values are less than the resolution of the figure).
nNOS Exon 1c Basal Promoter Regulation 25803
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

FIG. 3. Functional analysis of the human nNOS exon 1c promoter in TGW-nu-I (A), HeLa (B), and ME-180 (C) cells. 5� and 3� deletionswere introduced into the nNOS exon 1c 5�-flanking region, and the indicated DNA fragments were ligated into the promoter-/enhancerless firefly
nNOS Exon 1c Basal Promoter Regulation25804
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

and human with a sequence homology of 100% between nt �84and �41 relative to the transcription start site of human exon1c and nt 320 and 363 of rat nNOS exon 1b (EMBL accessionnumber AF008911) (35). Note that the nomenclature of alter-native first exons of nNOS is different in rat and human withhuman exon 1c corresponding to rat exon 1b (11, 35).
Identification of the Basal Promoter Region of nNOS Exon1c—Previously, we showed that exon 1c (designated exon 15�3)of nNOS is driven by a separate promoter within 332 bp up-stream of the transcription start site in HeLa cells (12). How-ever, neither the minimal promoter, nor the major transcrip-tion factors that regulate basal transcriptional activation wereelucidated. To examine the sequence that is necessary for basaltranscription of nNOS exon 1c, we analyzed various 5� and 3�deletions of the 5�-flanking region of exon 1c by reporter geneassays. The 5.9-kb promoter fragment of exon 1c (see Fig. 1B),obtained by 5�-RAGE, was cloned 5� to the firefly luciferasereporter gene of the pGL3-basic plasmid, resulting inpGL3�5891/�49. Different 5� and 3� deletions of the exon 1cpromoter were generated by PCR, or restriction endonucleaseand exonuclease III digestion of the pGL3�5891/�49 andpGL3�332/�49 plasmids (Fig. 1, B and C). These constructswere transiently transfected into nNOS exon 1c-positive TGW-nu-I neuroblastoma cells (see Fig. 2, D and F) and nNOS-negative HeLa cells (see Fig. 2F), as determined by quantita-tive real-time RT-PCR. After transfections of TGW-nu-I andHeLa cells with pGL3�5891/�49, we obtained a 13.7- and9.1-fold increase of the normalized promoter activity comparedwith that of promoter-/enhancerless pGL3-basic, respectively(Fig. 3, A and B). After deletion of 3117 bp from full-lengthpGL3�5891/�49, a decrease of 57% (p � 0.008) in promoteractivity could be observed in TGW-nu-I cells (Fig. 3A). In con-trast, an increase of 67% (p � 0.007) was evident in HeLa cells(Fig. 3B). For both cell lines, progressive deletions from �2774/�49 to �1520/�49 did not show a significant change of tran-scriptional activity. Further deletion to �332/�49 resulted in adecrease of 44% (p � 0.012) in TWG-nu-I cells, whereaspromoter activity showed no significant change in HeLa cells.These observations suggest the presence of cis regulatory se-quences exhibiting positive effects on the promoter activitybetween �5891 and �2774 and between �1520 and �332specifically in TGW-nu-I cells, whereas sequences between�5891 and �2774 negatively affect promoter activity in HeLa
cells. This argues for a cell-specific regulation of nNOS exon 1cpromoter activity by cis acting elements in the upstream 5�-flanking region of exon 1c.
Up to 242 bp (to position �90) could be further deleted inHeLa cells without a significant change of transcriptional ac-tivity. In contrast, in TGW-nu-I cells, we observed a drop inactivity of 33% (not significant) between �278 and �241, anincrease between �241 and �131 of 72% (p � 0.01), and anadditional profound rise of 108% (p � 0.001) after deletion ofsequences between �131 and �90. However, deletion of anadditional 27 bp (to position �63) abolished promoter activitycompletely in both cell lines (p � 0.001). Values seen for this�63/�49 construct, as well as for further deletions up to �18/�49, essentially reflected those of pGL3-basic. The antisenseconstruct of pGL3�5891/�49, named pGL3 � 49/�5891,showed only background activity. As positive control the SV40promoter/enhancer-directed pGL3-control vector was used inTGW-nu-I (Fig. 3A) and HeLa cells (data not shown).
These data demonstrate that the basal promoter for exon 1cis disrupted by deletion to �63 relative to the exon 1c tran-scription start site, and therefore, they strongly suggest thatthe promoter resides within the predicted region between �90and �47. Interestingly, adjacent upstream 5� regions regulatethis basal promoter activity differentially in nNOS-positiveTGW-nu-I cells and nNOS-negative HeLa cells, demonstratingdistinct mechanisms of activation in these two cell lines.
When 350 bp was removed from the 3�-end of pGL3�5891/�49, a �5891/�279 construct resulted that lacks the minimalpromoter and the transcription start site of exon 1c but con-tains the putative promoters and transcription start sites ofexon 1a and exon 1b. After transient transfection of this con-struct, normalized promoter activity showed just backgroundactivity in exon 1a and exon 1b mRNA-negative TGW-nu-I cellsand in nNOS mRNA-negative HeLa cells (Fig. 3, A and B).These results demonstrate that the upstream 5�-flanking re-gion of exon 1c, containing exons 1a and 1b and their putativepromoters, can influence transcription of exon 1c but does notactivate transcription of exon 1a and/or exon 1b in both celllines under the investigated conditions.
Because exon 1c promoter constructs are active in endoge-nous nNOS-negative HeLa cells, we further investigated thisdiscrepancy by reporter gene assays using nNOS-positive butexon 1c-negative ME-180 cells. After transient transfection
TABLE IIDouble-stranded oligonucleotides used for electrophoretic mobility shift assays
DNA sequences for double-stranded oligonucleotides used as probes and/or competitors in gel shift assays. Boldface letters indicate mutatedbases. Note: Sequences of oligonucleotides purchased from Santa Cruz Biotechnologies (see “Materials”) are not listed.
Name Sequence (5� to 3�; sense strand) Reference
�90/�47 AGAGCCACCTCCCAGCCTGCCCCTGGGGAGGGGCCACCTGGTGTC�90/�63 AGAGCCACCTCCCAGCCTGCCCCTGGGGA�90/�47 Sp1 Mut AGAGCCACCTCCCAGCCTGCCCCTGGTTAGGGGCCACCTGGTGTCStaf consensus ATTACCCATAATGCATTGCGGT (23, 38, 57)Staf Mut ATTAGTTCTAATGCATTGCGGT (23, 38, 57)Olf-1 consensus CACAATTCCCCAGGGAGAAGGA (59)Olf-1 Mut CACAATGGTCCAGGGAGAAGGA (59)ZNF42 consensus CGCCAGGCCTCCCCCTCCCGAGGATGTAAC (47)ZNF42 Mut CGCCAGGCCTCCTTTTCCCGAGGATGTAAC (47)MAZ consensus GAAAAAGAAGGGAGGGGAGGGATC (60)MAZ Mut GAAAAAGAAGGGAATTCAGGGATC (60)
luciferase expression vector pGL3-basic. As positive control an SV40 promoter/enhancer-directed firefly luciferase control vector (pGL3-control)was used (data not shown for HeLa and ME-180 cells in B and C, respectively). pGL3-basic and an antisense construct of pGL3�5891/�49 namedpGL3 � 49/�5891 were used as a negative control. Constructs were transiently cotransfected into TGW-nu-I (A), HeLa (B), and ME-180 (C) cellswith the herpes simplex virus thymidine kinase promoter driven Renilla luciferase expression vector pRL-TK as internal control as describedunder “Experimental Procedures.” To control for transfection efficiency, the firefly luciferase activity of the test plasmids was corrected for Renillaluciferase activity of pRL-TK. Data are expressed as means � S.D. of three independent experiments in triplicate. Where bars and/or error barsare not evident, values are less than the resolution of the figure.
nNOS Exon 1c Basal Promoter Regulation 25805
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

nNOS Exon 1c Basal Promoter Regulation25806
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

with different nNOS exon 1c pGL3 promoter constructs, weobserved a moderate, but significant increase in relative lucif-erase activity over that of promoter-/enhancerless pGL3-basic(Fig. 3C). Therefore, endogenous nNOS-negative HeLa cellsand nNOS exon 1c-negative ME-180 cells are able to transac-tivate nNOS exon 1c reporter constructs.
Identification of Transcription Factors Binding to the BasalnNOS Exon 1c Promoter—Computer-based sequence inspec-tion of the GC-rich (66%) and TATA-less basal promoter regionbetween �90 and �47 of exon 1c indicated a variety of putativecis-regulatory elements, like Staf (consensus binding site forZNF143 and ZNF 76 (23)) (�85 to �64), p53 half-site (�79 to�70), Olf-1 (�78 to �57), Ap2 (�73 to �62), Myc/Max (�61 to�48), ZNF42 (alternative title MZF-1) (�70 to �63), and a lowaffinity GC box with binding sites for Sp1 (�69 to �57) andMAZ (�67 to �59). To assess transcription factor binding, adouble-stranded 44-bp 32P-labeled oligonucleotide (�90/�47)(see Table II for all oligonucleotides used in gel shift experi-ments, except consensus and mutant consensus oligonucleo-tides for Ap2, Myc-Max, NF-I, YY1, NF�B, p53, Sp1, USF-1,which were purchased from Santa Cruz Biotechnologies), in-cluding these elements, was used in electrophoretic mobilityshift assays (EMSAs). A series of five shifted protein-DNAcomplexes were observed after incubation of TGW-nu-I nuclearextracts with the labeled probe (Fig. 4A, lane 2), whereas onlytwo protein-DNA complexes were evident using HeLa nuclearextracts (Fig. 4B, lane 2). These complexes were competed witha 100-fold molar excess of unlabeled �90/�47 probe, establish-ing binding specificity (Fig. 4, A and B, lane 3). Shifted bandswithout competition after addition of the unlabeled �90/�47probe were considered as nonspecific binding and were markedby an asterisk in Fig. 4 (A–D). Using a 100-fold molar excess ofan unlabelled Sp1 consensus oligonucleotide (containing thebinding site for the transcription factors Sp1, Sp3, and Sp4)complexes I/III (TGW-nu-I cells, Fig. 4A, lane 7) and complex I(HeLa cells, Fig. 4B, lane 7) were completely competed, andcomplex II (both cell lines) was partially competed. There wasno further competition of complex II, when a 200- and 500-foldmolar excess of unlabeled Sp1 consensus oligonucleotides wasused (both cell lines, data not shown). No competition of thecomplexes was observed with a 100- and 200-fold molar excessof a commercial mutant Sp1 oligonucleotide (Fig. 4, A and B,lane 8). Using TGW-nu-I nuclear extracts a slight reduction inthe protein-DNA complexes I, II, and III was seen with a�90/�63 oligonucleotide where the GC box is disrupted (Fig.4A, lane 4) and with a �90/�47 oligonucleotide in which theSp1 binding site was mutated (Fig. 4A, lane 5), whereas com-plexes IV and V were completely competed by these two oligo-nucleotides. With HeLa cell nuclear extracts complex I wasslightly and complex II was completely competed using the�90/�63 and �90/�47 Sp1-Mut competitors (Fig. 4B, lanes 4and 5). Furthermore, no competition of any complex was ob-
served using an unrelated YY1 consensus oligonucleotide(TGW-nu-I cells, Fig. 4A, lane 23; HeLa cells, Fig. 4B, lane 21).When protein-DNA complexes were incubated with antibodiesagainst Sp1, Sp2, Sp3, and Sp4 or combinations of these anti-bodies, supershifts of different complexes were observed withTGW-nu-I and HeLa cell nuclear extracts (Fig. 4, C and D). Forthe Sp1 antibody a supershift of complex I/III (TGW-nu-I cells)and complex I (HeLa cells), for the Sp2 antibody a supershift ofcomplex I (TGW-nu-I and HeLa cells), for the Sp3 antibody asupershift of complex I/II/III (TGW-nu-I cells) and complex II(HeLa cells) was observed whereas for the Sp4 antibody nosupershift was seen. A combination of Sp1, Sp2, and Sp3 anti-bodies led to different supershifts as shown in Fig. 4C forTGW-nu-I cells and Fig. 4D for HeLa cells. When all threeantibodies were added to the binding reaction, complexes I/II/III (TGW-nu-I cells) and complexes I/II (HeLa cells) were su-pershifted with a nearly complete abrogation. Using antibodiesagainst goat and rabbit IgG, no supershift of any complex wasevident (data not shown). Taken together these results demon-strate that Sp1, Sp2, and Sp3 can interact with the 44-bpminimal promoter of exon 1c. To further characterize the re-tarded complex II obtained with TGW-nu-I and HeLa cells,which was not completely competed by an Sp1 consensus oli-gonucleotide, and the unaffected complexes IV and V (TGW-nu-I cells), consensus oligonucleotides for the transcription fac-tors Staf (ZNF76 and ZNF143), p53, Olf-1, Ap2, Myc/Max, ZNF42, MAZ, NF-�, NF-1, YY1, and USF were used. Myc/Max,NF-1, USF, p53 (data not shown), and YY1 (Fig. 4A, lane 23;Fig. 4B, lane 21) had no effects on nucleoprotein complex for-mation. Unspecific competition of the retarded protein-DNAcomplexes V (MAZ, Fig. 4A, lane 20) and IV/V (Olf-1, Fig. 4A,lanes 11 and 12) was evident using TGW-nu-I cell nuclearextracts and a 100-fold excess of unlabelled consensus andmutated consensus oligonucleotides for MAZ and Olf-1. In con-trast, oligonucleotides containing Staf, ZNF42, Ap2, and NF-�binding sites were specific in competition studies. An oligonu-cleotide with a Staf binding site (consensus sequence for ZNF76and ZNF143) competed partially complex II (both cell lines,Fig. 4, A and B, lane 9), whereas ZNF42 consensus oligonucleo-tides competed complexes I/III/IV/V (TGW-nu-I cells, Fig. 4A,lane 15) and complex I (HeLa cells, Fig. 4B, lane 15). Additionof oligonucleotides containing mutations in the Staf and ZNF42binding sites failed to compete protein-DNA complexes (Fig. 4,A and B, lane 16). A combination of Sp1 and Staf consensusoligonucleotides resulted in a complete competition of complexII (both cell lines, Fig. 4, A and B, lane 13), whereas Sp1combined with other consensus oligonucleotides like Olf-1 (Fig.4, A and B, lane 14) and ZNF42 (data not shown) showed thesame degree of competition as Sp1 alone. This observationsuggests a cooperative binding of Sp and ZNF76/143 transcrip-tion factors to the basal nNOS exon 1c promoter.
Consensus oligonucleotides for Ap2 and NF-�B showed a
FIG. 4. Identification of multiple DNA-protein complexes in the nNOS exon 1c basal promoter. Electromobility shift assays (EMSAs)were performed using nuclear extracts from TGW-nu-I (A, C, E) and HeLa (B, D, F) cells as described under “Experimental Procedures.” A and B,32P-labeled oligonucleotide probes spanning the �90 to �47 region of the basal nNOS exon 1c promoter were incubated with 10 �g of TGW-nu-I(A) and HeLa (B) cell nuclear extracts resulting in five (panel A) and two (panel B) specific shifted protein-DNA complexes, respectively. Nonspecificbands are indicated by an asterisk. In lanes 3, 4, and 5 a 100-fold molar excess of unlabeled �90/�47, �90/�63, and �90/�47 Sp1 Mutoligonucleotides were used as competitors. To characterize shifted protein-DNA complexes, unlabeled consensus and mutated consensus oligonu-cleotides for the indicated transcription factors were used, as well as combinations of these oligonucleotides (panel A: lanes 7–16, 20–23; panel B:lanes 7–21). Lane 1 represents labeled probe alone. C and D, detection of protein-DNA complexes using antibodies against Sp1, Sp2, Sp3, Sp4, andcombinations of these antibodies. EMSAs were done as described above using TGW-nu-I (C) and HeLa (D) cell nuclear extracts and the 32P-labeled�90/�47 spanning probe of the basal nNOS exon 1c promoter. Supershift assays were performed by incubation of the binding reaction with thelabeled probe for 30 min followed by the addition of 2 �l of each antibody and an additional incubation for 30 min at room temperature. Nonspecificbands are indicated by an asterisk. E and F, EMSAs of TGW-nu-I (E) and HeLa (F) cell nuclear extracts binding to the �90/�63 region of the nNOSexon 1c promoter. Gel shift assays were performed as described above (A and B), except that a 32P-labeled oligonucleotide probe spanning the �90to �63 region of the minimal exon 1c promoter was used. Lane 1 represents probe alone, and 10 �g of the respective nuclear extracts was addedto the binding reaction in lane 2, resulting in four (panel E) and six (panel F) specific shifted protein-DNA complexes, respectively. A 100-fold molarexcess of various unlabeled oligonucleotides were added as competitors in lanes 3–10 as indicated.
nNOS Exon 1c Basal Promoter Regulation 25807
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

FIG. 5. Mutagenesis of transcription factor binding sites decreases transcription from the nNOS exon 1c minimal promoter.TGW-nu-I (A, C, E) and HeLa (B, D, F) cells were transiently cotransfected with pRL-TK and different wild type or mutant reporter gene constructscontaining targeted substitutions or deletions in transcription factor binding sites as described under “Experimental Procedures.” To adjust fortransfection efficiency, the firefly luciferase activity of the test plasmids was corrected for Renilla luciferase activity (pRL-TK). Data are expressedas percent luciferase activity relative to the respective “full-length” wild type pGL3 construct and represent the means � S.D. of three independentexperiments in triplicate. A and B, the percent luciferase activities of wild type pGL3�90/�49, 5�-deleted pGL3�83/�49, and pGL3�48/�49, thepromoter-/enhancerless pGL3-basic vector, or mutant pGL3�90/�49 reporter gene constructs containing the indicated targeted substitutions inthe binding site of Sp1 (pGL3�90/�49 Sp1-M2), the common binding sites of Sp1 and ZNF42 (pGL3�90/�49 Sp1/ZNF42-M1), Ap2 and Olf1(pGL3�90/�49 Ap2/Olf1-M), ZNF76 and ZNF143 (called Staf binding site; pGL3�90/�49 Staf-M) are plotted for TGW-nu-I (A) and HeLa (B) cells.C and D, the percent luciferase activities of wild type pGL3�5891/�49, 5�-deleted pGL3�48/�49, pGL3-basic vector, and the indicated mutantpGL3�5891/�49 constructs containing targeted substitutions or deletions in the Sp1/ZNF42, Staf, and both the Sp1/ZNF42 and Staf bindingelement are plotted for TGW-nu-I (C) and HeLa (D) cells. E and F, the percent luciferase activities of wild type pGL3�332/�49, 5�-deletedpGL3�48/�49, pGL3-basic vector, and the indicated mutant pGL3�332/�49 constructs containing targeted substitutions or deletions in theSp1/ZNF42, Staf, and both the Sp1/ZNF42 and Staf binding element are plotted for TGW-nu-I (E) and HeLa (F) cells.
nNOS Exon 1c Basal Promoter Regulation25808
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

clear reduction in the protein-DNA complexes I/II/III (TGW-nu-I cells, Fig. 4A, lanes 21 and 22) and complex I (HeLa cells,Fig. 4B, lanes 17 and 20), whereas mutated oligonucleotidesfailed to compete (Fig. 4B, lane 18 and data not shown). How-ever, using Ap2 and NF-�B (subunits p50 and p65) antibodies,we were unable to observe a supershift or abrogation of anycomplex (data not shown). The causal mechanism of this ob-servation is unclear. Ap2 and NF-� may not be able to bind tothe minimal promoter of exon 1c autonomously and thus do notplay a direct role in exon 1c basal promoter activation. There-fore, competition of the retarded bands in the gel shift assayscould be due to protein-protein interactions of Ap2/NF-�B withother nuclear factors whose DNA binding affinity and specific-
ity could be increased by the presence of Ap2 or NF-�B. Suchmechanisms have been demonstrated recently (36, 37), andtherefore Ap2 and NF-�B could participate in the formation ofmultiple protein complexes that enhance or repress the trans-activation potential of other transcription factors like Sp1.
To determine transcription factor binding in the absence ofSp and GC box activity, a 22-bp 32P-labeled �90/�63 oligonu-cleotide with a disrupted GC box and Sp1 binding site was usedin EMSAs. A series of four and six shifted protein-DNA com-plexes was observed after incubation with TGW-nu-I (Fig. 4E,lane 2) and HeLa (Fig. 4F, lane 2) cell nuclear extracts, respec-tively. These complexes were completely competed with a 100-fold molar excess of unlabeled �90/�63 probe (Fig. 4, E and F,
FIG. 5—continued
nNOS Exon 1c Basal Promoter Regulation 25809
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from
lane 3). Using a 100-fold molar excess of an unlabelled Sp1consensus oligonucleotide, the signal intensity of the protein-DNA complexes II and III were amplified, and the signal in-tensity of complex IV was reduced with TGW-nu-I cell nuclearextracts (Fig. 4E, lane 4). In contrast, complete competition ofcomplex I was observed using HeLa cell nuclear extracts (Fig.4F, lane 4). When a ZNF42 consensus oligonucleotide was used,complex IV was completely competed and complexes II and IIIwere shifted or competed resulting in a retarded band betweencomplex II and III in TGW-nu-I cells (Fig. 4E, lane 5), whereasno competition was observed with a mutated ZNF42 consensusoligonucleotide (Fig. 4E, lane 6). The significance of the de-scribed observation remains unclear but demonstrates thatZNF42 is able to modulate transcription factor binding to the�90/�63 sequence. In HeLa cells ZNF42 consensus oligonu-cleotides resulted in only an unspecific competition of complexIII (Fig. 4F, lanes 5 and 6). A Staf consensus oligonucleotidecompeted complex I/II/III (TGW-nu-I cells, Fig. 4E, lane 9) andcomplex II/III/IV/V (HeLa cells, Fig. 4F, lane 9). However,complex VI of Fig. 4F (HeLa cells) could not specifically becompeted by any of the used consensus oligonucleotides, indi-cating that an as yet unknown transcription factor present inHeLa cells is able to bind to the �90 to �63 sequence of theexon 1c promoter. No competition was observed with mutantStaf (Fig. 4, E and F, lane 10), mutant Sp1 (data not shown),and unrelated YY1 (Fig. 4E, lane 7, Fig. 4F, lane 8) consensusoligonucleotides using nuclear extracts from both cell lines.
Collectively, these results identify a multiplicity of protein-DNA complexes within the nNOS exon 1c basal promoter in-volving Sp1, Sp2, Sp3, and members of the ZNF family oftranscription factors. These factors have differential effects onnucleoprotein complex formation. Some of them increase,whereas others decrease, band intensities, suggesting modula-tory effects on protein-DNA interactions.
Confirmation of Transcription Elements and PromoterTransactivation by Mutagenesis and Transient Transfec-tions—To further clarify the role of the different cis regulatoryelements in the basal nNOS exon 1c promoter, reporter geneconstructs for pGL3�90/�49 containing point mutations in theSp1/ZNF42 (pGL3�90/�49 Sp1/ZNF42-M1), Sp1 (pGL3�90/�49 Sp1-M2), Ap2/Olf-1 (pGL3�90/�49 Ap2/Olf-1-M), and
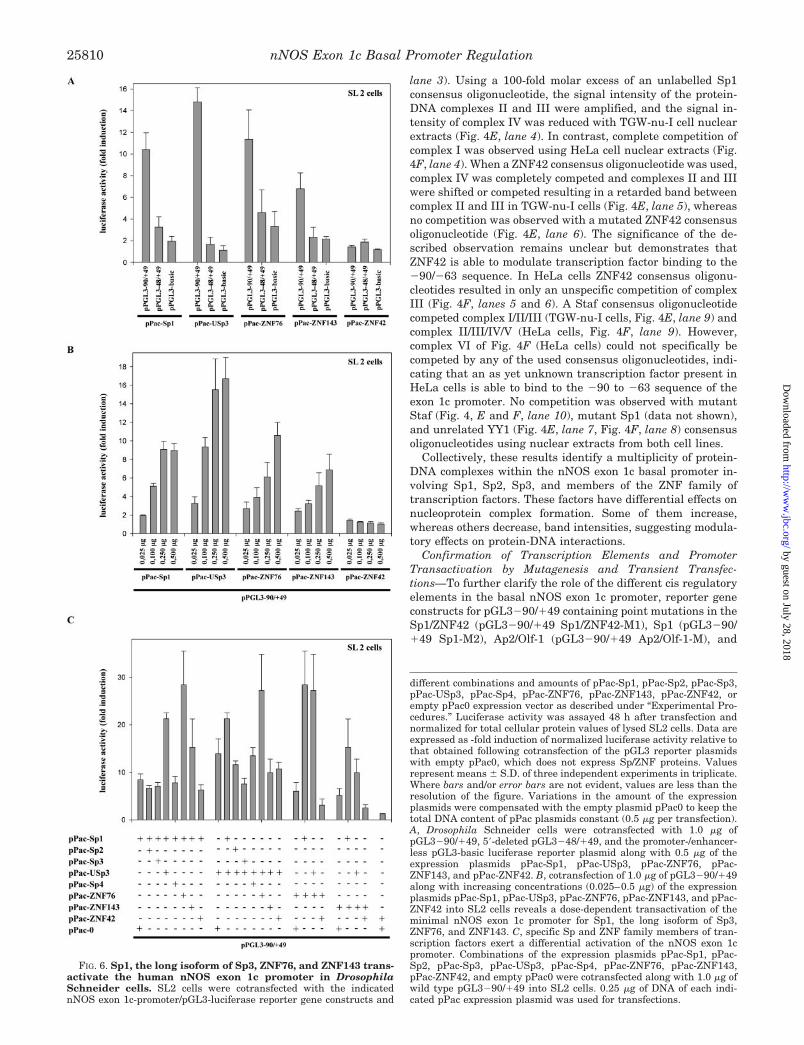
FIG. 6. Sp1, the long isoform of Sp3, ZNF76, and ZNF143 trans-activate the human nNOS exon 1c promoter in DrosophilaSchneider cells. SL2 cells were cotransfected with the indicatednNOS exon 1c-promoter/pGL3-luciferase reporter gene constructs and
different combinations and amounts of pPac-Sp1, pPac-Sp2, pPac-Sp3,pPac-USp3, pPac-Sp4, pPac-ZNF76, pPac-ZNF143, pPac-ZNF42, orempty pPac0 expression vector as described under “Experimental Pro-cedures.” Luciferase activity was assayed 48 h after transfection andnormalized for total cellular protein values of lysed SL2 cells. Data areexpressed as -fold induction of normalized luciferase activity relative tothat obtained following cotransfection of the pGL3 reporter plasmidswith empty pPac0, which does not express Sp/ZNF proteins. Valuesrepresent means � S.D. of three independent experiments in triplicate.Where bars and/or error bars are not evident, values are less than theresolution of the figure. Variations in the amount of the expressionplasmids were compensated with the empty plasmid pPac0 to keep thetotal DNA content of pPac plasmids constant (0.5 �g per transfection).A, Drosophila Schneider cells were cotransfected with 1.0 �g ofpGL3�90/�49, 5�-deleted pGL3�48/�49, and the promoter-/enhancer-less pGL3-basic luciferase reporter plasmid along with 0.5 �g of theexpression plasmids pPac-Sp1, pPac-USp3, pPac-ZNF76, pPac-ZNF143, and pPac-ZNF42. B, cotransfection of 1.0 �g of pGL3�90/�49along with increasing concentrations (0.025–0.5 �g) of the expressionplasmids pPac-Sp1, pPac-USp3, pPac-ZNF76, pPac-ZNF143, and pPac-ZNF42 into SL2 cells reveals a dose-dependent transactivation of theminimal nNOS exon 1c promoter for Sp1, the long isoform of Sp3,ZNF76, and ZNF143. C, specific Sp and ZNF family members of tran-scription factors exert a differential activation of the nNOS exon 1cpromoter. Combinations of the expression plasmids pPac-Sp1, pPac-Sp2, pPac-Sp3, pPac-USp3, pPac-Sp4, pPac-ZNF76, pPac-ZNF143,pPac-ZNF42, and empty pPac0 were cotransfected along with 1.0 �g ofwild type pGL3�90/�49 into SL2 cells. 0.25 �g of DNA of each indi-cated pPac expression plasmid was used for transfections.
nNOS Exon 1c Basal Promoter Regulation25810
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

Staf (pGL3�90/�49 Staf-M) binding sites were constructed(see Fig. 5, A and B). These constructs were transiently ex-pressed in TGW-nu-I (Fig. 5A) and HeLa cells (Fig. 5B), andpromoter activities were compared with that of the wild typeplasmid pGL3�90/�49. Furthermore, 5�-deleted pGL3�83/�49, pGL3�48/�49, and promoter-/enhancerless pGL3-basicwere used as controls. Mutation of the common Sp1/ZNF42 bind-ing site, as well as the Sp1 binding site, alone abolished promoteractivity of nNOS exon 1c to that of pGL3-basic in both cell lines(Fig. 5, A and B). In TGW-nu-I cells, the pGL3�90/�49 promoterconstruct containing a mutated Staf element resulted in a 53%decrease in luciferase activity compared with the pGL3�90/�49wild type construct (Fig. 5A), whereas a decrease of 37% wasdetected in HeLa cells (Fig. 5B). In TGW-nu-I and HeLa cells,constructs of pGL3�90/�49 with mutations in the Ap2/Olf-1binding site were 94% and 98.5% as active as wild typepGL3�90/�49, respectively, indicating that the Ap2 and Olf-1consensus binding sites in the exon 1c minimal promoter play norole for basal transcription (Fig. 5, A and B). Taken together,these data clearly demonstrate the critical cis-acting roles for Spand ZNF binding motifs in the exon 1c minimal promoter.
To investigate the importance of this basal promoter regionfor the transactivation of the longer reporter constructspGL3�5891/�49 and pGL3�332/�49, site-specific mutationsand deletions in the Sp and Staf binding sites were introducedinto the wild type plasmids. After transient expression inTGW-nu-I cells, promoter activities of all mutated/deletedpGL3�5891/�49 plasmids reflected that of pGL3-basic (Fig.5C). In HeLa cells, the construct pGL3�5891/�49 Staf/Sp1/ZNF42-M1, which contains a double mutation of the Staf andSp1/ZNF42 binding sites, abolished promoter activity com-pletely, and the construct pGL3�5891/�49 Staf/Sp1/ZNF42-del, with a deletion of both sites, was 11% as active as wild typepGL3�5891/�49 (Fig. 5D). Plasmids containing a mutation(pGL3�5891/�49 Sp1/ZNF42-M1) or deletion (pGL3�5891/�49 Sp1/ZNF42-del) of the Sp1/ZNF42 site displayed a drop of87 and 79% in functional promoter activity relative to the wildtype construct, respectively, and mutation (pGL3�5891/�49Staf-M) and deletion (pGL3�5891/�49 Staf-del) of the Stafelement resulted in a decrease of 68 and 71%, respectively (Fig.5D). The same mutations and deletions were incorporated in tothe pGL3�332/�49 construct. After transient expression inTGW-nu-I and HeLa cells, relative functional reporter activi-ties revealed no important differences compared with that ofthe mutations/deletions in the setting of the full-length �5891/�49 construct (Fig. 5, E and F).
Collectively, these data clearly indicate the major impor-tance of the GC-rich region between �90 and �47 relative tothe exon 1c transcription start site to promote nNOS exon 1cexpression.
Activation of the nNOS Exon 1c Promoter in DrosophilaSchneider Cells—To further determine whether members ofthe Sp and ZNF families of transcription factors functionallyinteract with the basal promoter of exon 1c, transient transfec-tion experiments were performed with Drosophila Schneidercells (SL2 cells), which lack many mammalian transcriptionfactors, like Sp-related proteins (25, 26), ZNF76 and ZNF143(23, 38). Expression constructs under the control of an SL2-specific promoter for Sp1 (pPac-Sp1), Sp2 (pPac-Sp2), the long(pPac-USp3) and short isoforms of Sp3 (pPac-Sp3), Sp4 (pPac-Sp4), ZNF42 (pPac-ZNF42), ZNF76 (pPac-ZNF76), ZNF 143(pPac-ZNF143) and empty pPac0 were cotransfected along withreporter vectors (pGL3) under the control of different nNOSexon 1c promoters. Normalized luciferase activities for thepGL3 reporter constructs were compared with that in cotrans-fections with the empty SL2 expression vector pPac0. As shown
in Fig. 6A cotransfection of pGL3�90/�49 with pPac-Sp1 in-duced a 10.4-fold increase in relative luciferase activity overcotransfection with pPac0. In contrast, values slightly abovethose of pGL3-basic (representing basal luciferase activity)were observed after cotransfection of SL2 cells with pPac-Sp1and pGL3�48/�49, which lacks the GC box of the basal exon 1cpromoter (Fig. 6A). A 14.8-fold increase was seen, when thelong isoform of Sp3 (pPac-USp3) was cotransfected withpGL3�90/�49, whereas cotransfection experiments withpGL3�48/�49 again showed only background activity (Fig.6A). Cotransfection of pPac-ZNF76 and pPac-ZNF143 alongwith pGL3�90/�49 resulted in a 11.6- and 6.8-fold transacti-vation of pGL3�90/�49, respectively, whereas induction ofpGL3�48/�49 by ZNF76 and ZNF143 reflected that of pGL3basic (Fig. 6A). No transactivation of pGL3�90/�49 andpGL3�48/�49 promoter activity was seen after cotransfectionwith pPac-ZNF42, indicating that ZNF42 is not able to inducethe basal nNOS exon 1c promoter autonomously (Fig. 6A).Likewise, there was no significant stimulation of normalizedluciferase activity over that of pGL3-basic, when either pPac-Sp2, pPac-Sp3 encoding the short isoforms of Sp3, or pPac-Sp4was cotransfected with pGL3�90/�49 or pGL3�48/�49 (datanot shown). Fig. 6B shows the effects of increasing amounts ofpPac-Sp1, pPac-USp3, pPac-ZNF76, pPac-ZNF143, and pPac-ZNF42 on pGL3�90/�49 luciferase activity. Sp1, the long iso-form of Sp3, ZNF76, and ZNF143 exhibited a dose-dependenttransactivation of pGL3�90/�49, whereas pGL3�48/�49 wasagain not activated. Increasing amounts of pPac-ZNF42 had noeffect on transactivation of pGL3�90/�49 (Fig. 6B).
In combination experiments (Fig. 6C), the stimulatory effectsof Sp1 and the long Sp3 isoform (21.3-fold induction), as well asSp1 and ZNF143 (15.3-fold induction) were additive, whereasSp1 combined with ZNF76 (28.4-fold induction) and full-lengthSp3 combined with ZNF76 (27.2-fold induction) resulted in apotentiation of nNOS exon 1c basal promoter transactivation.Cotransfection of pPac-Sp1 with pPac-Sp3 or pPac-Sp4, as wellas cotransfection of pPac-USp3 with Sp2, Sp4, ZNF143, orZNF42, had no effect on promoter activation of pGL3�90/�49(Fig. 6C). However, combining pPac-Sp1 with pPac-Sp2 orpPac-ZNF42, pPac-USp3 with pPac-Sp3, pPac-ZNF76 withpPac-ZNF42, and pPac-ZNF143 with pPac-ZNF42 resulted in aslight, but statistically significant decrease of nNOS exon 1cpromoter activity (Fig. 6C). Thus ZNF and Sp family membersare able to exert positive and negative effects on the transac-tivation of the nNOS exon 1c minimal promoter.
Expression of Sp1, Sp2, isoforms of Sp3, and Sp4 in SL2 cellsafter transient transfection with the different pPac plasmidswas verified by Western blot analysis of SL2 cell nuclear ex-tracts using selective antibodies against Sp1, Sp2, Sp3, andSp4 (data not shown). Where no antibodies were available(ZNF42, ZNF76, and ZNF143), expression in SL2 cells aftertransient transfection was confirmed using EMSAs. Binding ofexpressed ZNF42, ZNF76, and ZNF143, as well as Sp1 and theshort and long isoforms of Sp3 to the minimal exon 1c promoter(�90/�47) and the respective consensus oligonucleotides(ZNF42, Staf, Sp1) was seen in gel shift assays using nuclearextracts from SL2 cells transiently transfected with the differ-ent expression plasmids (pPac-Sp1, pPac-USp3, pPac-Sp3,pPac-ZNF42, pPac-ZNF76, pPac-ZNF143) (data not shown). Incontrast to Sp1 and Sp3 isoforms, expressed Sp2 and Sp4proteins showed no binding to the exon 1c minimal promoter.In addition, there was no binding of expressed Sp2 to the Sp1consensus oligonucleotide as previously described (26, 27, 30,39) (data not shown). Control experiments showed that Sp1,Sp2, and Sp3 proteins and ZNF42, ZNF76, and ZNF143mRNAs are expressed in TGW-nu-I and HeLa cells, enabling
nNOS Exon 1c Basal Promoter Regulation 25811
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

the regulation of nNOS exon 1c promoter activity in mamma-lian cells (data not shown). In contrast to Sp1, Sp2, Sp3,ZNF76, and ZNF143, mRNA for ZNF42 was not detectable inhuman brain, skeletal muscle, and rectum by RT-PCR, indicat-ing that ZNF42 plays no role in the transcriptional regulationof nNOS exon 1c in these tissues under the investigated con-ditions. This is in agreement with previous findings, demon-strating that ZNF42 is a specific transcriptional regulator ofmyeloid differentiation (24).
In summary, these results identify Sp1, full-length Sp3,ZNF76, and ZNF143 as potent transcriptional activators of thenNOS exon 1c promoter, whereas the short isoforms of Sp3 andZNF42 exhibit a specific repressive effect on Sp/ZNF-mediatedtranscriptional activation.
DISCUSSION
A variety of human nNOS mRNA variants have been de-scribed recently (3, 4, 11–13, 40). Among these, transcriptswith different untranslated first exons are generated by alter-native promoter usage (12, 13). Real-time quantitative RT-PCRwas used to determine the expression patterns and the quan-titative distribution of nine alternative first exons of nNOS(exons 1a–1i) (11) and revealed a cell- and tissue-specific ex-pression with exon 1c being one of the predominant variants inhuman brain, skeletal muscle, rectum, and TGW-nu-I neuro-blastoma cells. Because exon 1c is highly expressed in humanbrain and skeletal muscle, it seems responsible for the largestproportion of nNOS mRNA in the body. Furthermore, nNOSexon 1c mRNA expression is significantly reduced in the pyloricsphincter of patients with infantile hypertrophic pyloric steno-sis.2 Therefore the transcriptional regulation of this variant isof special interest.
To characterize the structure and the expressional regula-tion of nNOS exon 1c, we cloned its genomic 5�-flanking regionand analyzed the basal promoter. A 5939-bp genomic 5�-flank-ing DNA fragment of exon 1c, which contains in addition nNOSexon 1a and exon 1b, was obtained by 5�-RAGE PCR. By 5� and3� deletion analysis of reporter plasmids, using nNOS exon 1cmRNA-positive TGW-nu-I, nNOS-negative HeLa and nNOSmRNA-positive, but exon 1c-negative ME-180 cells, the mini-mal promoter was localized to position �90 to �47, relative tothe transcription start site of exon 1c. This region is highlyconserved between different species with a sequence homologyof 100% between rat (nt 320 to 363 of EMBL accession numberAF008911) and man (nt �84 to �41 of exon 1c). Such a highdegree of conservation argues for an important, biologicallyconserved function of this transcriptional control region (41).
Interestingly, nNOS exon 1c reporter plasmids were active inHeLa and ME-180 cells that lack endogenous nNOS exon 1cmRNA. This indicates that cell-specific expression of nNOS exon1c is mediated by distinct mechanisms that cannot influencetranscription of the investigated reporter gene constructs. Suchcell-specific transcriptional control mechanisms can be due torepressive cis-acting elements that inhibit transcription in dis-tinct cell types. They are often localized within the upstream5�-flanking region or within the first intron of a gene, as shownfor the growth-associated protein 43 (42). Therefore additionalsequences in the far upstream region or in the first intron maymediate cell-specific expression of nNOS exon 1c. In addition thechromatin structure and acetylation state that predicts DNAsequence accessibility plays an important role in cell-specificgene regulation (for review see Ref. 43). Therefore condensationand thus silencing of the nNOS gene could also be responsible forthe lack of endogenous nNOS expression in HeLa cells.
To detect transcription factors binding to the basal nNOSexon 1c promoter, EMSAs, including competition and super-shift analysis were performed. They identified Sp and ZNF
family members of transcription factors as the critical factorsregulating the exon 1c �90/�47 basal promoter region. Forexample, Sp1 and isoforms of Sp3 are binding to the low affin-ity GC box (GGGAGGGG). Although this motif contains an A inplace of the consensus C, it has been demonstrated to bindtranscription factors of the Sp family, which activate or represstranscription substantially (30, 41, 44). Because the minimalpromoter of nNOS exon 1c is GC-rich, TATA-less, and Sp-regulated, it resembles those for constitutively expressedgenes, like dihydrofolate reductase, endothelial NOS, or theserotonin 1a receptor (41, 45–47). The regulation of such pro-moters is poorly understood. It has been shown that G/C-richpromoter regions, lacking a canonical TATA box, can bind Sp1molecules that interact with multiple components of the tran-scriptional machinery (for review see Ref. 48), and thereforeSp1 plays a critical role in the assembly of the transcriptioninitiation complex (45). A number of transcription factors hasbeen documented as acting in combination with Sp1 or promot-ing its displacement from the same or an overlapping site (28,30, 37, 49). Among these, Sp2, Sp3, and Sp4 belong to the sametranscription factor family (27, 28, 30). They share a highlyhomologous DNA binding domain, but only Sp1, Sp3, and Sp4recognize the classic Sp1 GC-box in vitro with similar affinities(27, 30, 39). Sp3 mRNA encodes for different isoforms, arisingfrom alternative translation initiation sites (50). These iso-forms of Sp3 have been found to exert both activating andinhibiting effects on gene transcription (26, 28, 30, 49–53). Incontrast, Sp1 is typically an activator of transcription (30, 48).Because Sp1, Sp2, and isoforms of Sp3 are ubiquitous ex-pressed (28), this could explain why the investigated reporterplasmids under the control of the nNOS exon 1c minimal pro-moter are expressed in nNOS exon 1c-negative HeLa andME-180 cells. When the effects of Sp transcription factors onthe exon 1c promoter were examined in Drosophila Schneidercells that lack Sp, ZNF76, and ZNF143 binding activity (23, 25,26, 38), Sp1 and the long isoform of Sp3 potently stimulatedtranscription, whereas Sp2 and the short isoforms of Sp3 andSp4 were transcriptionally inactive on their own. ExpressedSp1 and the short and long isoforms of Sp3 were able to bind tothe exon 1c minimal promoter region in gel shift assays,whereas the Sp2 and Sp4 proteins did not form protein-DNAcomplexes. However, an Sp2-specific antibody resulted in asupershift of complex I in EMSAs using TGW-nu-I and HeLacell nuclear extracts. This suggests that Sp2 is not able to bindto the minimal promoter of exon 1c autonomously. The super-shift of the retarded band I in the gel shift assays could be dueto protein-protein interactions of Sp2 with other nuclear fac-tors, not present in SL2 cells, resulting in an increase in DNAbinding affinity and specificity of Sp2 (52). Such mechanismshave been described for a variety of transcription factors likeGATA members and the p65 subunit of NF-�B (37, 54). Incotransfection experiments the transactivating effects of ex-pressed Sp1 and full-length Sp3 were completely additive,whereas Sp2 selectively repressed Sp1-mediated transcrip-tional activation of the nNOS exon 1c basal promoter. This wasa moderate effect with a decrease from a 8.4- to a 6.7-foldinduction of luciferase activity. However, this observation dem-onstrates a specific and differential modulation of promotertransactivation by different members of the Sp family of tran-scription factors. A similar effect was observed for the combi-nation of short and long isoforms of Sp3, where the shortisoform selectively repressed transactivation of the exon 1cbasal promoter by full-length Sp3 (decrease from a 13.9- to a7.5-fold induction). The mode of action could be due to forma-tion or disruption of protein-protein (self-)interactions andmultimerization that increase or decrease the DNA binding
nNOS Exon 1c Basal Promoter Regulation25812
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

affinity of the respective transcription factors or to competitionfor common DNA recognition sites (30, 37, 49, 50, 52, 54). Incontrast to Sp2 and the short isoforms of Sp3, Sp4 did not alterpromoter transactivation by Sp1 and the long isoform of Sp3. Inaddition to Sp transcription factors, we demonstrated that exon1c promoter activity is regulated by different members of theZNF family of transcription factors. ZNF42, ZNF76, andZNF143 specifically bind to the minimal exon 1c promoter ingel shift experiments, and cotransfection of ZNF76 or ZNF143resulted in a strong induction of the exon 1c promoter in Dro-sophila Schneider cells, whereas ZNF42 by itself was transcrip-tionally inert. Combining ZNF and Sp transcription factors inSL2 cells resulted in differential effects on nNOS exon 1c pro-moter activity. ZNF76 potentiated the stimulatory effects ofSp1 and the long isoform of Sp3, whereas ZNF143 combinedwith Sp1 showed an additive effect. In contrast, no effect wasseen when ZNF143 was coexpressed with the long isoform ofSp3. ZNF42 significantly repressed exon 1c promoter transac-tivation of ZNF76, ZNF143, and Sp1. These findings demon-strate a differential regulation of the nNOS exon 1c promoterby Sp and ZNF family members of transcription factors andsuggest highly coordinated protein-protein interactions andprotein-DNA binding to overlapping cis-acting elements. Thisobservation was confirmed by the results of gel shift competi-tion studies. Complete competition of the retarded complex IIobtained with TGW-nu-I cell nuclear extracts could be demon-strated only by using a combination of a 100-fold molar excessof unlabelled Sp1 and Staf (ZNF76 and ZNF143 binding site)oligonucleotides, whereas Sp1 or Staf alone were not able tocompete this band completely.
The involvement of members of the Sp and ZNF families oftranscription factors on transactivation of the basal humannNOS exon 1c promoter was verified by mutating the consen-sus sequences and transfecting different reporter constructsinto HeLa and TGW-nu-I cells. The mutated GC box completelyabolished promoter activity of all tested exon 1c reporter plas-mids in TGW-nu-I cells and reduced promoter activity by 78%or more in HeLa cells. Mutation of the Staf (ZNF76 andZNF143) binding site decreased relative luciferase activity ofthe minimal promoter construct (pGL3�90/�49) by 53% inTGW-nu-I and 37% in HeLa cells. This effect was even morepronounced in the longer promoter constructs pGL3�332/�49and pGL3�5891/�49 with a drop of 76 and 90%, respec-tively, in TGW-nu-I cells and 68 and 60%, respectively, inHeLa cells. Collectively, these data stress the essential role forSp and Staf binding motifs for the initiation of transcriptionfrom the TATA less human nNOS exon 1c promoter.
Little is known about the role of the ZNF transcription factorfamily members ZNF76 (55) and ZNF143 (56) in transactiva-tion of mRNA promoters. Both transcription factors resemblethe Drosophila Kruppel segmentation gene product due to thepresence of repeated Cys2-His2 zinc finger domains that areconnected by conserved sequences. They contain seven tan-demly repeated zinc fingers, showing high homology to theXenopus laevis transcriptional activator Staf (23), originallyidentified in Xenopus as the transactivator of the tRNASec gene(38). In addition Xenopus Staf possesses the capacity to stim-ulate expression from TATA-box containing RNA polymerase IImRNA promoters (23, 57). ZNF76 and 143 are highly identicaland functionally equivalent to the X. laevis Staf and, therefore,believed to be the human homologues (23). Both transcriptionfactors are expressed in a broad range of tissues (23), includinghuman brain, skeletal muscle, rectum, TGW-nu-I, and HeLacells, and are able to bind tightly to Staf consensus-responsiveelements present in the X. laevis tRNASec and the human U6promoter with identical affinities (23). When expressed in Dro-
sophila Schneider cells, lacking ZNF76 and ZNF143 activity(23, 38), both factors activated transcription from the TATA-box containing thymidine kinase mRNA promoter through theStaf binding site (23). Here we demonstrate for the first timethat the human homologues of Staf can stimulate transcriptionfrom a TATA-less promoter in Drosophila Schneider cells ontheir own. In addition, we demonstrate a synergistic activationof transcription by combinations of Sp1, full-length Sp3,ZNF76, and ZNF143, whereas ZNF42, Sp2, and the short iso-forms of Sp3 repressed transcriptional activation in certaincases. It has been suggested that the cell content of distincttranscription factors is a critical factor influencing the tran-scription of TATA-less promoters (49). Therefore, differences inthe cellular levels of Sp1, Sp2, Sp3 isoforms, ZNF42, ZNF76,and ZNF143 in different cell and tissue types or changes of thecellular levels in response to physiological and pathologicalconditions (30, 51, 58) could conceivably exert profound effectson the expression of nNOS exon 1c. This adds another level ofcomplexity and cell specificity to the already intricate regula-tion of nNOS gene expression resulting from the use of multiplepromoters (3, 4, 10, 12, 13, 40).
In conclusion, our results demonstrate that nNOS exon 1c isthe predominant nNOS mRNA variant in the body. The mini-mal exon 1c promoter could be localized to 44 bp, with a coop-erative binding of several transcription factors of the Sp andZNF families. This portends many possibilities for tissue- andcell-specific control in response to cellular requirements. Re-cent findings support a complex and tightly regulated geneexpression of nNOS, showing profound changes in differentphysiological and pathophysiological states (1–4, 14, 16, 17, 21,35). However, the causal mechanisms are unknown, and there-fore the results of this study could provide insight into the rolesof changes of nNOS gene transcription, including possible ther-apeutic strategies for increasing nNOS gene expression.
Acknowledgments—We thank Dr. R. Tjian (University of California,Berkeley, CA), Dr. J. D. Noti (Guthrie Research Institute, Sayre, PA),Dr. G. Suske (University of Marburg, Germany), Dr. P. Carbon (Uni-versity of Strasbourg, France), and Dr. R. Hromas (University of Indi-ana, Indianapolis, IN) for providing the expression plasmids pPac0,pPac-Sp1, pPac-Sp2, pPac-Sp3, pPac-Usp3, pPac-Sp4, p97b, pcDNA3ZNF76, pcDNA3 ZNF143, and CB6-MZF-1 used in this study. We thankDr. E. R. Werner (University of Innsbruck, Austria) and Dr. H. Esumi(National Cancer Center Research Institute East, Chiba, Japan) forproviding the ME-180 and TGW-nu-I cell lines, respectively. We thankU. Goetz for excellent technical assistance and P. Ruth for his sugges-tions during the preparation of the manuscript.
REFERENCES
1. Bredt, D. S. (1999) Free Radic. Res. 31, 577–5962. Dawson, V. L., and Dawson, T. M. (1998) Prog. Brain Res. 118, 215–2293. Forstermann, U., Boissel, J. P., and Kleinert, H. (1998) FASEB J. 12, 773–7904. Wang, Y., Newton, D. C., and Marsden, P. A. (1999) Crit. Rev. Neurobiol. 13,
21–435. Stark, M. E., and Szurszeswski, J. H. (1992) Gastroenterology 103, 1928–19496. Allescher, H. D., Kurjak, M., Huber, A., Trudrung, P., and Schusdziarra, V.
(1996) Am. J. Physiol. 271, G568–G5747. Papapetropoulos, A., Rudic, R. D., and Sessa, W. C. (1999) Cardiovasc. Res. 43,
509–5208. Geller, D. A., and Billiar, T. R. (1998) Cancer Metastasis Rev. 17, 7–239. Sasaki, M., Gonzalez-Zulueta, M., Huang, H., Herring, W. J., Ahn, S., Ginty,
D. D., Dawson, V. L., and Dawson, T. M. (2000) Proc. Natl. Acad. Sci.U. S. A. 97, 8617–8622
10. Deans, Z., Dawson, S. J., Xie, J., Young, A. P., Wallace, D., and Latchman,D. S. (1996) J. Biol. Chem. 271, 32153–32158
11. Wang, Y., Newton, D. C., Robb, G. B., Kau, C. L., Miller, T. L., Cheung, A. H.,Hall, A. V., VanDamme, S., Wilcox, J. N., and Marsden, P. A. (1999) Proc.Natl. Acad. Sci. U. S. A. 96, 12150–12155
12. Saur, D., Paehge, H., Schusdziarra, V., and Allescher, H. D. (2000) Gastroen-terology 118, 849–858
13. Xie, J., Roddy, P., Rife, T., Murad, F., and Young, A. P. (1995) Proc. Natl. Acad.Sci. U. S. A. 92, 1242–1246
14. Vanderwinden, J. M., Mailleux, P., Schiffmann, S. N., Vanderhaeghen, J. J.,and De-Laet, M. H. (1992) N. Engl. J. Med. 327, 511–515
15. Chung, E., Curtis, D., Chen, G., Marsden, P. A., Twells, R., Xu, W., andGardiner, M. (1996) Am. J. Hum. Genet. 58, 363–370
16. Mearin, F., Mourelle, M., Guarner, F., Salas, A., Riveros, M., V, Moncada, S.,and Malagelada, J. R. (1993) Eur J. Clin. Invest. 23, 724–728
nNOS Exon 1c Basal Promoter Regulation 25813
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

17. Watkins, C. C., Sawa, A., Jaffrey, S., Blackshaw, S., Barrow, R. K., Snyder,S. H., and Ferris, C. D. (2000) J. Clin. Invest. 106, 373–384
18. Huang, P. L., Dawson, T. M., Bredt, D. S., Snyder, S. H., and Fishman, M. C.(1993) Cell 75, 1273–1286
19. Mashimo, H., Kjellin, A., and Goyal, R. K. (2000) Gastroenterology 119,766–773
20. Sivarao, D. V., Mashimo, H. L., Thatte, H. S., and Goyal, R. K. (2001) Gastro-enterology 121, 34–42
21. Ogura, T., Nakayama, K., Fujisawa, H., and Esumi, H. (1996) Neurosci. Lett.204, 89–92
22. Werner-Felmayer, G., Werner, E. R., Fuchs, D., Hausen, A., Mayer, B., Reibnegger,G., Weiss, G., and Wachter, H. (1993) Biochem. J. 289, 357–361
23. Myslinski, E., Krol, A., and Carbon, P. (1998) J. Biol. Chem. 273, 21998–2200624. Hromas, R., Collins, S. J., Hickstein, D., Raskind, W., Deaven, L. L., O’Hara,
P., Hagen, F. S., and Kaushansky, K. (1991) J. Biol. Chem. 266,14183–14187
25. Courey, A. J., and Tjian, R. (1988) Cell 55, 887–89826. Noti, J. D. (1997) J. Biol. Chem. 272, 24038–2404527. Hagen, G., Muller, S., Beato, M., and Suske, G. (1992) Nucleic Acids Res. 20,
5519–552528. Hagen, G., Muller, S., Beato, M., and Suske, G. (1994) EMBO J. 13, 3843–385129. Hagen, G., Dennig, J., Preiss, A., Beato, M., and Suske, G. (1995) J. Biol.
Chem. 270, 24989–2499430. Philipsen, S., and Suske, G. (1999) Nucleic Acids Res. 27, 2991–300031. Huber, A., Saur, D., Kurjak, M., Schusdziarra, V., and Allescher, H. D. (1998)
Am. J. Physiol. 275, G1146–G115632. Gibson, U. E., Heid, C. A., and Williams, P. M. (1996) Genome Res. 6, 995–100133. Bustin, S. A. (2000) J. Mol. Endocrinol. 25, 169–19334. Stuehr, D. J. (1999) Biochim. Biophys. Acta 1411, 217–23035. Lee, M. A., Cai, L., Hubner, N., Lee, Y. A., and Lindpaintner, K. (1997) J. Clin.
Invest. 100, 1507–151236. Terzano, S., Flora, A., Clementi, F., and Fornasari, D. (2000) J. Biol. Chem.
275, 41495–4150337. Perkins, N. D., Agranoff, A. B., Pascal, E., and Nabel, G. J. (1994) Mol. Cell.
Biol. 14, 6570–658338. Schuster, C., Myslinski, E., Krol, A., and Carbon, P. (1995) EMBO J. 14,
3777–378739. Kingsley, C., and Winoto, A. (1992) Mol. Cell. Biol. 12, 4251–426140. Wang, Y., Goligorsky, M. S., Lin, M., Wilcox, J. N., and Marsden, P. A. (1997)
J. Biol. Chem. 272, 11392–1140141. Parks, C. L., and Shenk, T. (1996) J. Biol. Chem. 271, 4417–443042. Weber, J. R., and Skene, J. H. (1997) J. Neurosci. 17, 7583–759343. Lemon, B., and Tjian, R. (2000) Genes Dev. 14, 2551–256944. Song, J., Ugai, H., Kanazawa, I., Sun, K., and Yokoyama, K. K. (2001) J. Biol.
Chem. 276, 19897–1990445. Azizkhan, J. C., Jensen, D. E., Pierce, A. J., and Wade, M. (1993) Crit. Rev.
Eukaryot. Gene Expr. 3, 229–25446. Zhang, R., Min, W., and Sessa, W. C. (1995) J. Biol. Chem. 270, 15320–1532647. Laumonnier, Y., Nadaud, S., Agrapart, M., and Soubrier, F. (2000) J. Biol.
Chem. 275, 40732–4074148. Naar, A. M., Ryu, S., and Tjian, R. (1998) Cold Spring Harbor Symp. Quant.
Biol. 63, 189–19949. Kwon, H. S., Kim, M. S., Edenberg, H. J., and Hur, M. W. (1999) J. Biol. Chem.
274, 20–2850. Kennett, S. B., Udvadia, A. J., and Horowitz, J. M. (1997) Nucleic Acids Res.
25, 3110–311751. Rajakumar, R. A., Thamotharan, S., Menon, R. K., and Devaskar, S. U. (1998)
J. Biol. Chem. 273, 27474–2748352. Bakovic, M., Waite, K. A., and Vance, D. E. (2000) J. Lipid Res. 41, 583–59453. Majello, B., De Luca, P., and Lania, L. (1997) J. Biol. Chem. 272, 4021–402654. Merika, M., and Orkin, S. H. (1995) Mol. Cell. Biol. 15, 2437–244755. Ragoussis, J., Senger, G., Mockridge, I., Sanseau, P., Ruddy, S., Dudley, K.,
Sheer, D., and Trowsdale, J. (1992) Genomics 14, 673–67956. Tommerup, N., and Vissing, H. (1995) Genomics 27, 259–26457. Schaub, M., Myslinski, E., Schuster, C., Krol, A., and Carbon, P. (1997) EMBO
J. 16, 173–18158. Ammendola, R., Mesuraca, M., Russo, T., and Cimino, F. (1992) J. Biol. Chem.
267, 17944–1794859. Kudrycki, K., Stein-Izsak, C., Behn, C., Grillo, M., Akeson, R., and Margolis,
F. L. (1993) Mol. Cell. Biol. 13, 3002–301460. Bossone, S. A., Asselin, C., Patel, A. J., and Marcu, K. B. (1992) Proc. Natl.
Acad. Sci. U. S. A. 89, 7452–7456
nNOS Exon 1c Basal Promoter Regulation25814
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from

AllescherDieter Saur, Barbara Seidler, Heidi Paehge, Volker Schusdziarra and Hans-Dieter
TRANSCRIPTION FACTORSTranscription: ESSENTIAL ROLE OF Sp AND ZNF FAMILY MEMBERS OF
Complex Regulation of Human Neuronal Nitric-oxide Synthase Exon 1c Gene
doi: 10.1074/jbc.M109802200 originally published online April 17, 20022002, 277:25798-25814.J. Biol. Chem.
10.1074/jbc.M109802200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/277/28/25798.full.html#ref-list-1
This article cites 60 references, 31 of which can be accessed free at
by guest on July 28, 2018http://w
ww
.jbc.org/D
ownloaded from





![· Gefitinib Gefitinib 1. Non-small cell lung cancer EGFR DNA EGF-R exon 19 deletion, exon 21 [1.858R] substitution mutations, L861Q G719X EGFR exon 20](https://img.dokumen.tips/doc/110x75/5e51ddba1b664701f40175b0/gefitinib-gefitinib-1-non-small-cell-lung-cancer-egfr-dna-egf-r-exon-19-deletion.jpg)

![CRISPR/Cas9-mediated genome editing induces exon skipping ... · HeLa cells can cause skipping of exon 3, exon 4, or exons 3, 4, and 5 [18]. We also detected infrequent exon skipping](https://img.dokumen.tips/doc/110x75/60db8f117fb86d112c69c947/crisprcas9-mediated-genome-editing-induces-exon-skipping-hela-cells-can-cause.jpg)
















![Nant - Final[1]](https://img.dokumen.tips/doc/110x75/577cdab71a28ab9e78a658a6/nant-final1.jpg)




