Embed Size (px)
Citation preview

The Domain Structure of Proteins: Prediction and
Organization.
Golan YonaDept. of Computer Science
Cornell University(joint work with Niranjan Nagarajan)
Golan Yona, Cornell University
Golan Yona, Cornell University
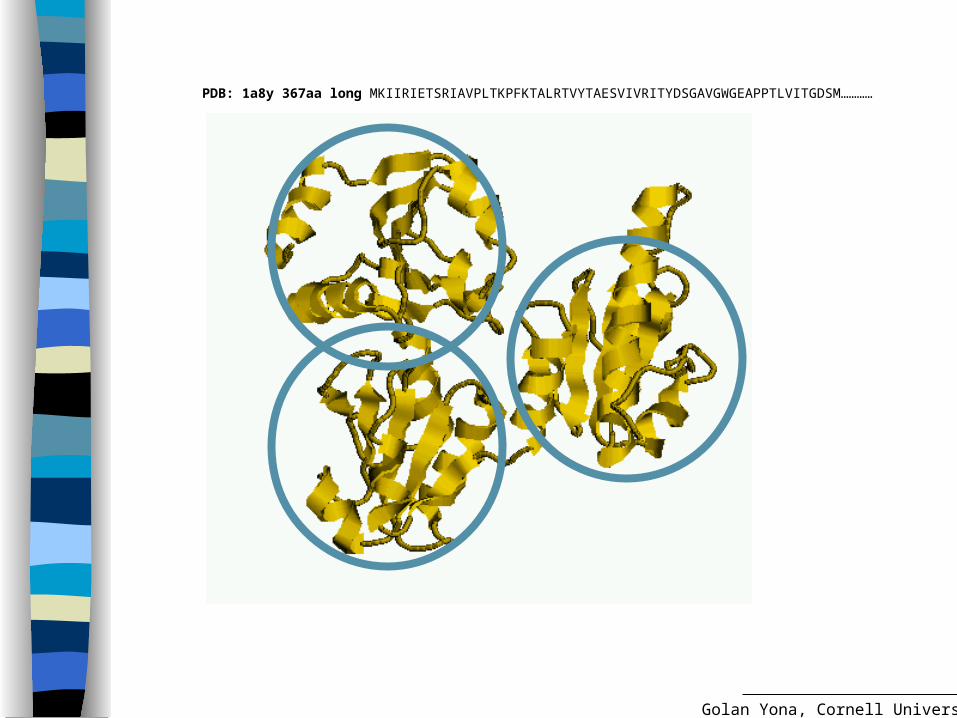
PDB: 1a8y 367aa long MKIIRIETSRIAVPLTKPFKTALRTVYTAESVIVRITYDSGAVGWGEAPPTLVITGDSM…………

Golan Yona, Cornell University
The domain structure of a protein
A domain is considered the fundamental unit of protein structure, folding, function, evolution and design.
Compact Stable Folds independently? Has a specific function
Golan Yona, Cornell University
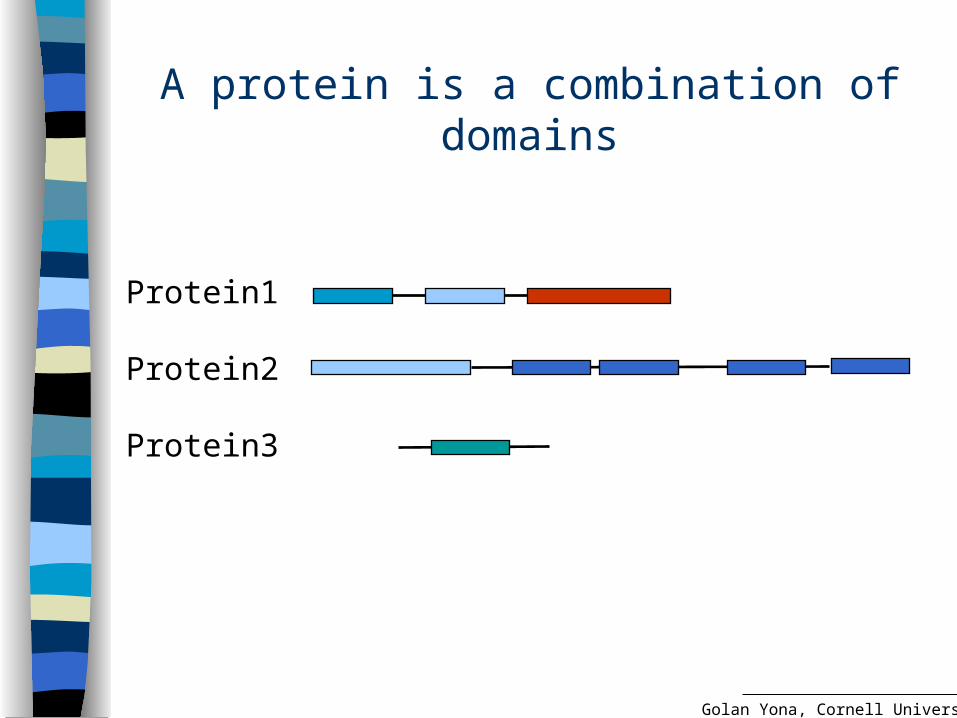
A protein is a combination of domains
Protein1
Protein2
Protein3

Golan Yona, Cornell University
Any signals that might indicate domain boundaries?
A very weak signal if any in the sequence
Usually domain delineation is done based on structure
Best methods available – manual! But structural information is sparse..

Golan Yona, Cornell University
Definitions and assumptions
Domain: continuous sequence that corresponds to an elemental building block of protein folds.
A subsequence that is likely to be stable as an independent folding unit.
Was formed as an independent unit, and later was combined with others – more complex functions.
There are traces of the autonomous units..

Golan Yona, Cornell University
First step..
Gather data – database search Histogram of matches is informative but
noisy
Mutations, insertions, deletions, conflicting evidence
sequence

Golan Yona, Cornell University
Previous methods
Methods based on the use of similarity searches and knowledge of sequence termini to delineate domain boundaries using heuristics/rules (MKDOM, Domainer, DIVCLUS, DOMO).
Methods that rely on expert knowledge of protein families to construct models like HMMs to identify other members of the family (Pfam, TigrFam, SMART).
Methods that try to infer domain boundaries by using sequence information to predict tertiary structure first (SnapDragon. Rigden’s covariance analysis)
Methods that use multiple alignments to predict domain boundaries (PASS, Domination).
Others..(e.g. CSA and DGS = guess based on size)

Golan Yona, Cornell University
How do you evaluate the different methods?
No universal measures A variety of qualitative and quantitative
evaluation criteria, external resources and manual analysis are used to verify domain boundaries

Golan Yona, Cornell University
Method outline
Source/test data – SCOP Processed data - alignments Learning system:
– Domain-information-content scores– NN– Probabilistic model
Evaluation
“A Multi-Expert System for the Automatic Detection of Protein Domains from Sequence Information” Niranjan Nagaragan and Golan Yona, in the proceedings of RECOMB2003

Golan Yona, Cornell University
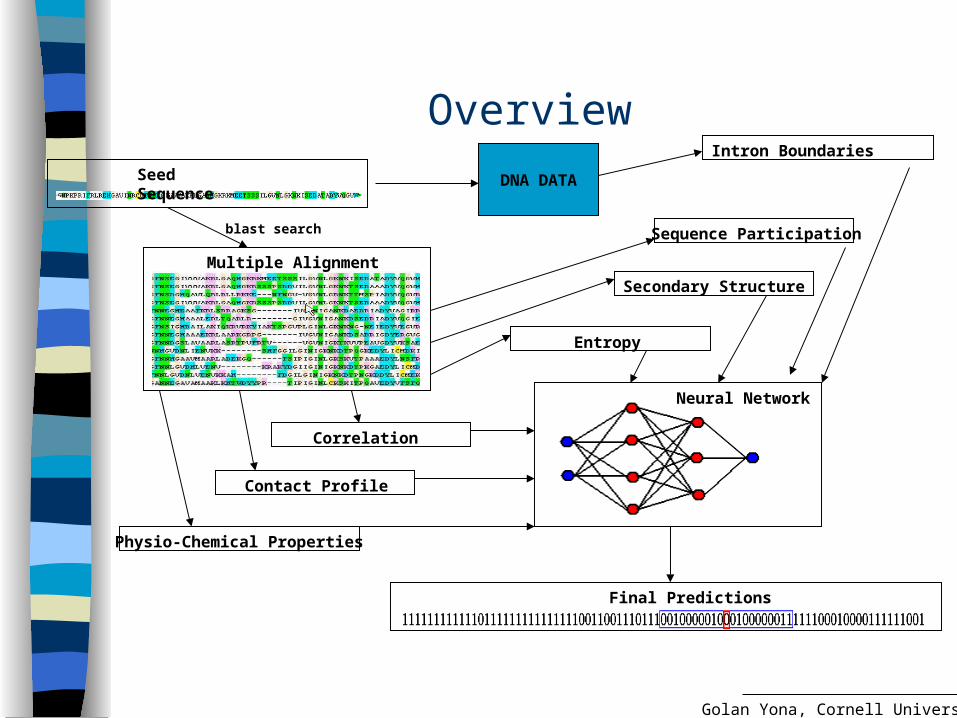
OverviewSeed Sequence
Multiple Alignment
blast search
Neural Network
Correlation
Entropy
Sequence Participation
Contact Profile
Secondary Structure
Physio-Chemical Properties
Final Predictions
DNA DATA
Intron Boundaries

Golan Yona, Cornell University
The source/test data set
PDB structures with their partitions into domains as defined in SCOP:– 1ctf: domain1 1-76 domain2 77-123
Remove sequences shorter than 40 aa and almost identical entries

Golan Yona, Cornell University
Alignments
Search each query against a database of ~1 million non-redundant sequences
Remove fragments first Two phase alignment procedure
– First phase: blast– Second phase: multiple iteration psi-blast
Select one representative from each group of similar proteins
Remove proteins that are less than 90% covered (missing information)
Number of domains ranging from 1-7 Final set: 605 multi-domain proteins and 576 single
domain proteins (1/4)

Golan Yona, Cornell University
The domain-information-content of an alignment column
Measures that (are believed) to reflect structural properties of proteins
A total of 20 measures– Conservation measures – Consistency and correlation measures – Measures of structural flexibility – Residue type based measures – Predicted secondary structure information– Intron-exon data

Golan Yona, Cornell University
Conservation measures
Entropy: some positions are more conserved than others
Class entropy: some positions have preference towards a class of amino-acids (similar physio-chemical properties)
Evolutionary pressure (span): sum of pairwise similarities
Motivation: consider the mutual similarity of amino acids

Golan Yona, Cornell University
Consistency and correlation measures
All domain appearances should maintain its integrity Consistency: difference in sequence counts
Asymmetric correlation: consistency of individual sequences.
Symmetric correlation: reinforcement by missing sequences
Measures are averaged over a window

Golan Yona, Cornell University
Consistency and correlation measures – cont.
Sequence termination: strong but elusive– Fragments– Premature halt in alignment– Loosely aligned
Product of left and right termination scores: given c sequences that terminate at a position, with evalues e1,e2,e3,…ec
Golan Yona, Cornell University

Golan Yona, Cornell University
Measures of structural flexibility
Indel entropy: variability indicates structural flexibility (likely to occur near domain boundaries)
Correlated mutations: indicative of contacts Contact profiles

Golan Yona, Cornell University
Contact profile

Golan Yona, Cornell University
Residue type based measures
hydrophobic vs. hydrophilic cystines and prolines Classes of amino acids
Predicted secondary structures Helices and strands are rigid Loops are more abundant near domain
boundaries

Golan Yona, Cornell University
Intron-exon data
Exon boundaries are expected to coincide with domain boundaries
1
1 2
3
2
1 3
2
Protein1
Protein2
Protein3

Golan Yona, Cornell University
Score refinement and normalization
Smoothing using a window w (optimized)
Unification to a single scale – zscore over all positions

Golan Yona, Cornell University
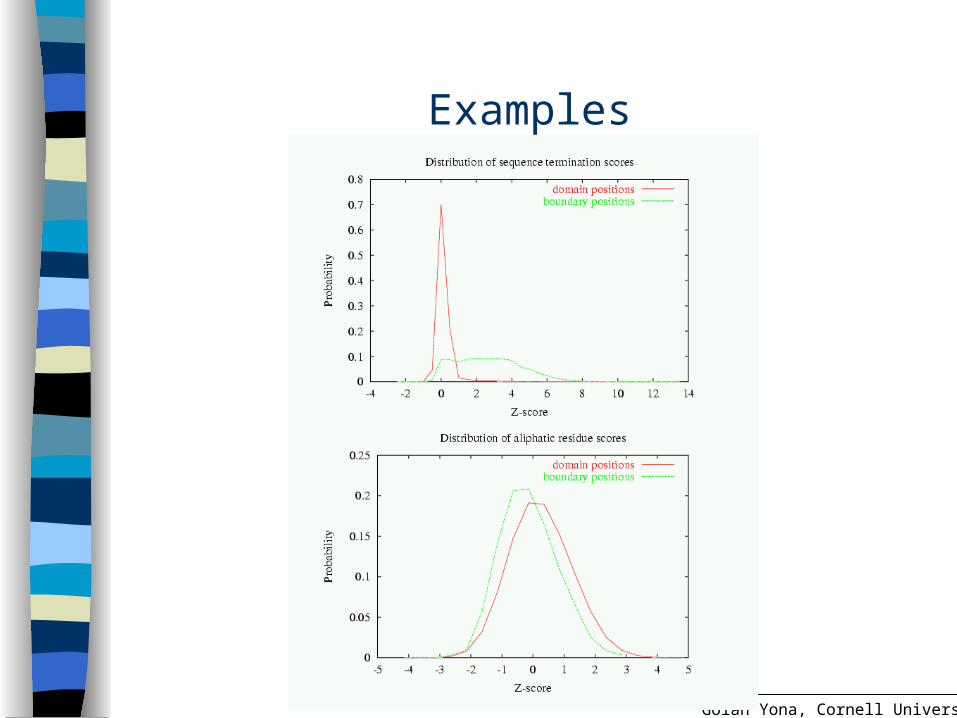
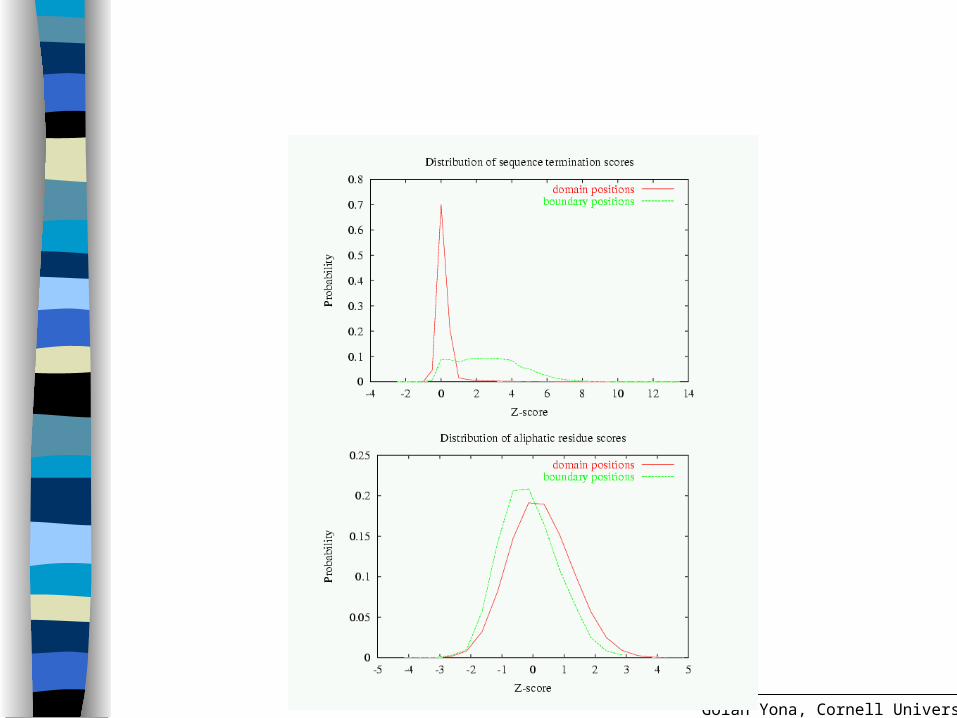
Maximizing the information content of scores
Opt for the most distinct distributions of domain positions vs. boundary positions
Affected by the parameters (w smoothing factor) and x (boundary window size)
Use the Jensen-Shannon divergence measure
Golan Yona, Cornell University
Examples
Golan Yona, Cornell University
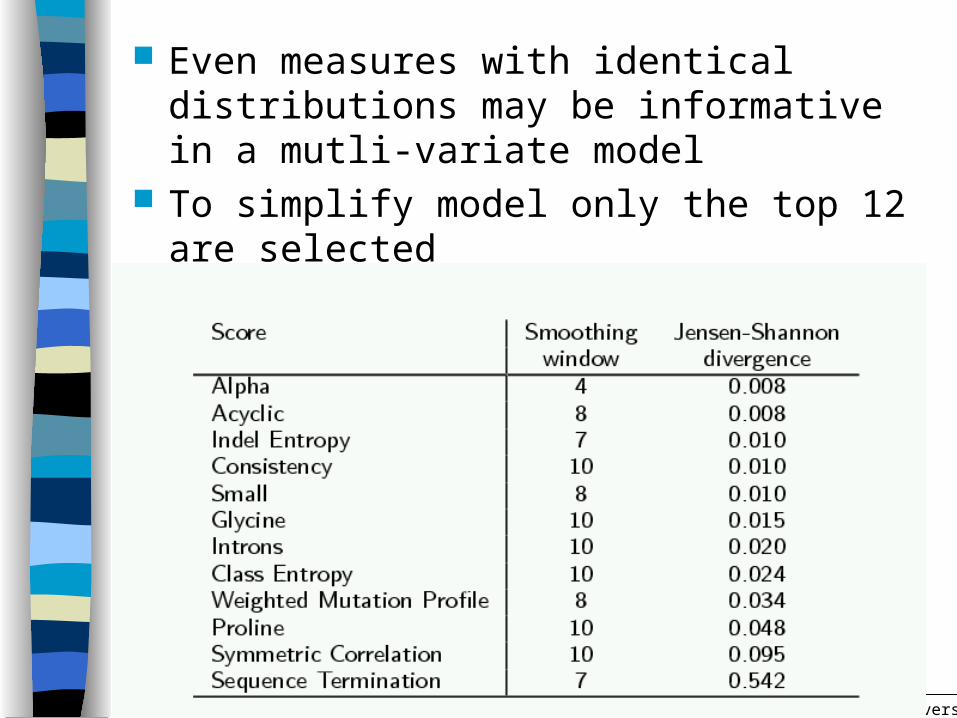
Even measures with identical distributions may be informative in a mutli-variate model
To simplify model only the top 12 are selected

Golan Yona, Cornell University
The learning system
A neural network is trained to model effectively the complex decision boundary surface
Predicts correctly 94% of domain positions and 88% of the transitions in the test set
Also tried mapping from multiple positions (local input neighborhood) to single/multiple output
Golan Yona, Cornell University
OverviewSeed Sequence
Multiple Alignment
blast search
Neural Network
Correlation
Entropy
Sequence Participation
Contact Profile
Secondary Structure
Physio-Chemical Properties
Final Predictions
DNA DATA
Intron Boundaries
Golan Yona, Cornell University
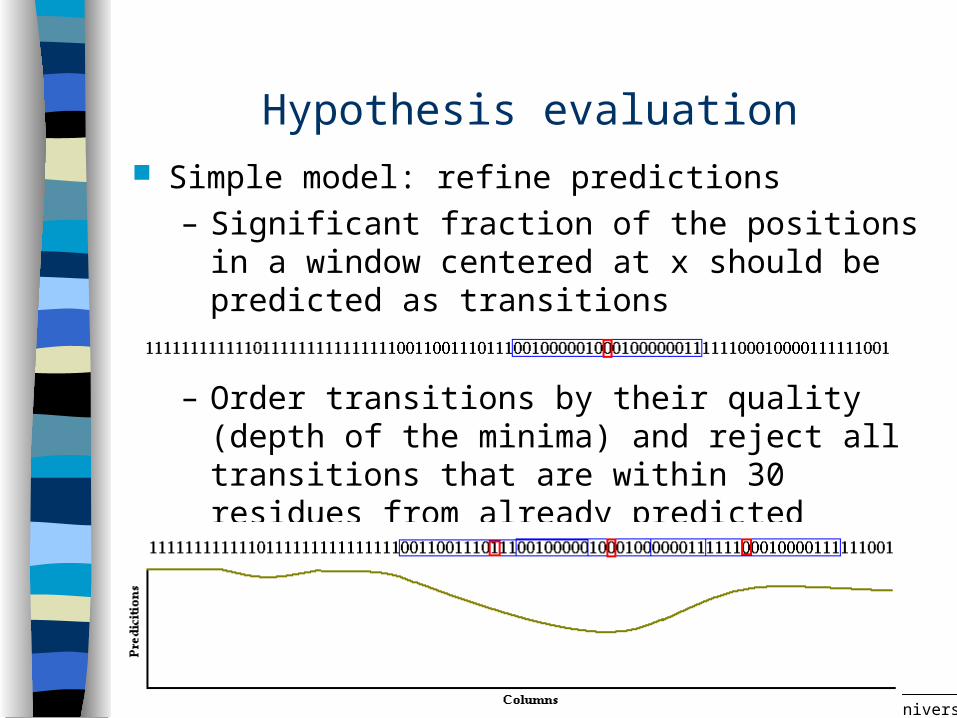
Hypothesis evaluation Simple model: refine predictions
– Significant fraction of the positions in a window centered at x should be predicted as transitions
– Order transitions by their quality (depth of the minima) and reject all transitions that are within 30 residues from already predicted transitions

Golan Yona, Cornell University
The domain generator model
Multiple hypotheses – find the “best one”
Assume a model: random generator that moves repeatedly between a domain state and a linker state and emits one domain or transition at a time according to different source probability distributions.
Total probability is the product

Golan Yona, Cornell University
Formally..S = D1 D2 Dn We are given a sequence S (multiple
alignment) of length L and a possible partition into n domains D=D1,D2,..Dn of lengths l1,l2,..,ln (NN output)
Find the partition that will maximize the posterior probability P(D/S)
Maximize the product of the likelihood and the prior

Golan Yona, Cornell University
Calculating the prior P(D)
For an arbitrary protein of length L what is the probability to observe D
Approximate using a simplified model: given the length of the protein, the generator selects the number of domains first and then selects the length of one domain at a time, considering the domains that were already generated.

Golan Yona, Cornell University
The prior probabilities
Approximate P0(li/L) by P0(li) normalized to the relevant range.
P0(li/L) is derived based on experimental data

Golan Yona, Cornell University
The prior probabilities (cont.)
Calculate Prob(n/L) = Prob(n,L)/P(L)
1
2

Golan Yona, Cornell University
The likelihood
Use probabilities of observed scores considering the two different sources
The model D partitions the sequence S into n domains and n-1 transitions: D1,T1,D2,T2,…,Tn-
1,Dn that correspond to the subsequences s1,t1,s2,t2,..,tn-1,sn
Assume domains are independent of each other (additional test can be used)

Golan Yona, Cornell University
…likelihood
Each term P(si/Di) and P(tj/Tj) is a product over the probabilities of the individual positions, each one is estimated by the joint probability distribution of the 12 features
How to estimate this probability? (independence assumption does not hold)
Golan Yona, Cornell University

Golan Yona, Cornell University
Likelihood of individual position
Given k random variables X1,X2,..,Xk their joint prob. Distribution
Use first order dependencies For each pair, calculate the distance between
the joint prob. Distribution and the product of the marginal distributions

Golan Yona, Cornell University
Sort all pairs based on their dependency, and pick the most dependent one (denoted by Y1, Y2) and start the expansion
Select the next one based on the strongest dependency with variables that are already in the expansion

Golan Yona, Cornell University
Denote by Z=PILLAR(Y) the random variable that Y is most dependent on
Of all possible dependencies involving Y3 pick P(Y3/Z) and add it to the expansion
Proceed until you exhaust all variables Maximize support, minimize error The expansion is different for domain and
transition regions

Golan Yona, Cornell University
Finally..
Enumerate all possible hypotheses, calculate the posterior probability for each one, and output the one that maximizes the prob.
Golan Yona, Cornell University
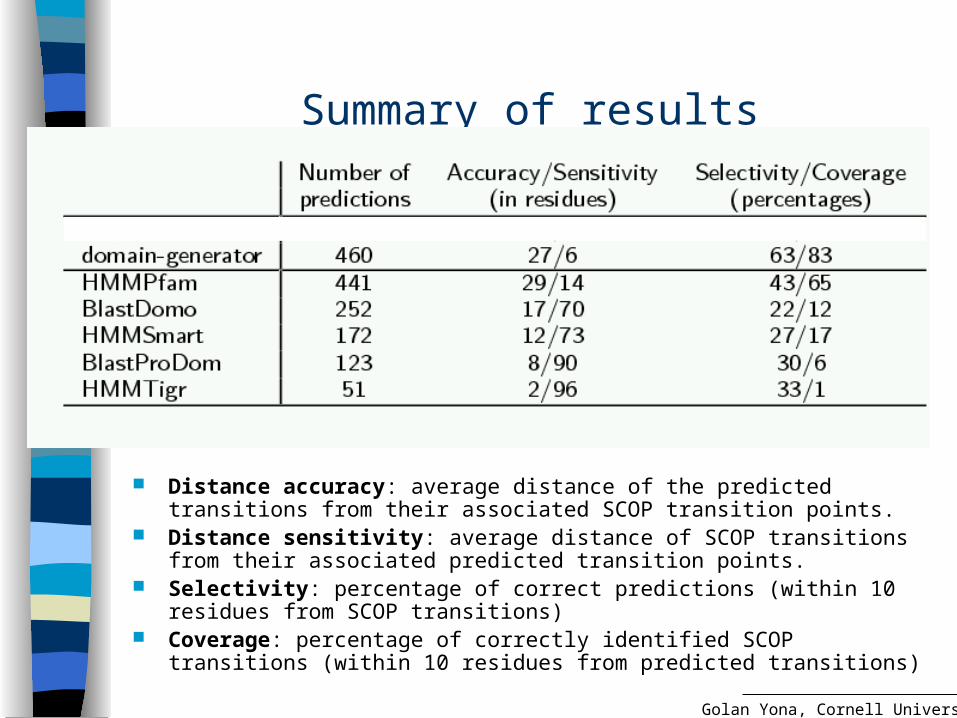
Summary of results
Distance accuracy: average distance of the predicted transitions from their associated SCOP transition points.
Distance sensitivity: average distance of SCOP transitions from their associated predicted transition points.
Selectivity: percentage of correct predictions (within 10 residues from SCOP transitions)
Coverage: percentage of correctly identified SCOP transitions (within 10 residues from predicted transitions)

Golan Yona, Cornell University
Examples
PDB ID: 2gep Domain Definition:
8-72, 73-272, 273-352, 353-497 Predicted Domains:
1-75, 76-270, 271-352, 353-497 PFam Definition:
1-67, 273-345, 356-425

Golan Yona, Cornell University
Examples
PDB ID: 1b6s chain D Domain Definition: 1-78, 79-276, 277-355 Predicted Domains: 1-73, 74-271, 272-355 PFam Definition: 30-167

Golan Yona, Cornell University
Examples
PDB ID: 1acc Domain Definition:
14-735 Predicted Domains:
1-158, 159-583, 584-735 PFam Definition:
103-544

Golan Yona, Cornell University
Conclusions
A method for predicting the domain structure of a protein from sequence information alone
Protein/DNA data, multiple features, optimization based on information theory principles, learning system and final prediction using the domain-generator model (with confidence values).
Exhaustive hypothesis evaluation Fully automatic and fast Perform very well even compared to the best manual and
semi-manual methods out there (also on CATH data) Dare to say …can be used to verify domain assignments
based on structural data Improvements: other learning systems, more features

Golan Yona, Cornell University
Acknowledgments
Niranjan Nagarajan
SCOP CATH PSI-BLAST Pfam InterPro
NSF





























