Embed Size (px)
Citation preview

Gemeinsames Praktikum
des Fachbereichs Bio- und Chemieingenieurwesen
Der Versuch gehört zum Praktikumsbereich Technische Chemie A
Versuch TC4
Flüssig-Flüssig-Extraktion
Versuchsinhalt:
• Darstellung ternärer Gemische im Dreiecksdiagramm
• Flüssig-Flüssig-Gleichgewicht des Stoffsystems Essigsäure / n-Butylacetat / Wasser
• Binodalkurve als Grenzlinie der Löslichkeit
• Konoden als Verbindungslinien zweier im Gleichgewicht stehender Phasen
• Konjugatlinie zur graphischen Interpolation der Gleichgewichtsdaten
• Experimentelle Überprüfung einiger Gleichgewichtsdaten
• Bilanzierung einer vierstufigen Gegenstromextraktion durch Stufenkonstruktion im Dreiecks- und im Verteilungsdiagramm
• Experimentelle Durchführung der vierstufigen Gegenstromextraktion in einer kontinuierlich betriebenen Mischer-Scheider-Laborapparatur
• Vergleich der theoretischen Stufenkonstruktion mit den Versuchsergebnissen
• Ermittlung der Stufenwirkungsgrade der Laborapparatur

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 2 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Inhaltsverzeichnis
Inhaltsverzeichnis .........................................................................................................................................2
1. Einleitung .................................................................................................................................................3
2. Grundlagen ..............................................................................................................................................4
2.1 Die Flüssig-Flüssig-Extraktion..............................................................................................................4 2.2 Das Dreieckskoordinatensystem und seine Eigenschaften....................................................................5 2.3 Binodalkurve und Konoden...................................................................................................................6 2.4 Mess- und Interpolationsverfahren sowie Modellgleichungen..............................................................8 für flüssig-flüssig-Phasengleichgewichte 2.5 Das Verteilungsdiagramm .....................................................................................................................9 2.6 Extraktionsverfahren ...........................................................................................................................10 2.7 Darstellung der Gegenstromextraktion in Dreieckskoordinaten .........................................................12 2.8 Ausführung der Stufenkonstruktion bei unterschiedlichen Vorgaben.................................................13 2.9 Übertragung der Stufenkonstruktion in das Verteilungsdiagramm .....................................................14 2.10 Die praktische Extraktionsstufe und der Wirkungsgrad......................................................................15
3. Versuchsbeschreibung ..........................................................................................................................16
3.1 Stoffdaten ..............................................................................................................................................16 3.2 Methoden zur Bestimmung von Flüssig-Flüssig-Phasengleichgewichten ............................................17 3.3 Laboranlage zur kontinuierlichen vierstufigen Gegenstromextraktion .................................................18
4. Aufgabenstellung und Versuchsdurchführung...................................................................................19
Nomenklatur ...............................................................................................................................................21
Literatur:.....................................................................................................................................................21

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 3 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
1. Einleitung Unter Flüssig-Flüssig-Extraktion versteht man die Trennung einer Lösung mit Hilfe einer weiteren flüssi-gen Komponente (Lösungsmittel), die mit der Ausgangslösung eine Mischungslücke bildet und ein aus-reichendes Lösungsvermögen für den abzutrennenden Stoff besitzt.
Dieses Trennverfahren sollte als Alternative bzw. Ergänzung zu destillativen Verfahren insbesondere im-mer dann in Erwägung gezogen werden, wenn in dem zu trennenden Gemisch:
a) die Siedepunkte der einzelnen Komponenten dicht zusammenliegen, wenn also eine Rektifikation nur mit großer Trennstufenzahl und hohem Rücklaufverhältnis, d.h. großem Energieaufwand möglich wäre,
b) einzelne Komponenten beim Verdampfen Azeotrope bilden, wenn also als destillative Verfahren nur aufwendige Methoden wie die Zweidruck-Rektifikation sowie die Azeotrop- oder Extraktiv-Rektifika-tion in Frage kommen,
c) die abzutrennende Substanz nur in geringen Mengen vorhanden ist und eine höhere Siedetemperatur aufweist als die restlichen Komponenten, d.h. dass diese Hauptkomponenten mit großem Energieauf-wand abdestilliert werden müssten,
d) temperaturempfindliche Substanzen enthalten sind, die auf diesem Wege in ein Lösemittel überführt werden können, aus dem sie sich dann durch schonende Destillation oder auch ein anderes Verfahren wie z.B. Kristallisation abtrennen lassen.
Selbstverständlich muss für das eingesetzte Lösungsmittel im Normalfall ein geschlossener Stoffkreislauf realisiert werden. Dies geschieht meistens durch Rektifikation – u.U. aber auch durch eine erneute Ex-traktion – und erfordert häufig einen großen Aufwand. Ob ein Trennverfahren unter Anwendung der Flüs-sig-Flüssig-Extraktion sinnvoll ist, kann letztendlich nur anhand der Wirtschaftlichkeit des Gesamtverfah-rens entschieden werden.
Ein Beispiel dafür, dass die Anwendung der Flüssig-Flüssig-Extraktion trotz großen Aufwandes wirtschaft-lich günstiger sein kann als die direkte Rektifikation, ist die Aufkonzentrierung wässriger Essigsäure unterhalb 60 Gew-%. Die destillative Trennung ist hier problematisch und wird in der Technik zusätzlich durch Nebenprodukte aus der Essigsäure-Herstellung erschwert. Als Extraktionsmittel eignet sich z.B. n-Butylacetat, wie es in diesem Versuch verwendet wird. Obwohl sich Essigsäure in derartigen Lösungs-mitteln zu geringeren Gewichtsanteilen als in Wasser löst, und das Lösungsmittel deshalb in großen Men-gen eingesetzt und durch Rektifikation wieder gereinigt werden muss, sind solche Verfahren wirtschaft-lich interessant.
Um die Flüssig-Flüssig-Extraktion in diesem Praktikumsversuch anschaulich zu machen, wurde das be-schriebene Stoffsystem aus der Technik gewählt. Es wird allerdings hier in umgekehrter Richtung einge-setzt, also zur Extraktion von Essigsäure aus n-Butylacetat mit Wasser als Lösungsmittel. Verglichen mit dem technischen Original-Verfahren lässt sich diese Extraktion mit kleineren Stoffmengen und leichter er-fassbaren Konzentrationen zur Demonstration der Zusammenhänge durchführen.
Das erste Ziel dieses Versuches ist es, die Darstellung ternärer Gemische in Dreiecks-Koordinaten und die graphischen Möglichkeiten zur Durchführung von Bilanzierungen in einem solchen Diagramm ken-nenzulernen. An einigen Beispielen wird die Messung von Gleichgewichtsdaten mit den einfachen in die-sem Versuch zur Verfügung stehenden Methoden gezeigt. Anschließend wird eine vierstufige Gegen-stromextraktion zunächst durch eine graphische Stufenkonstruktion ausgelegt und das Verfahren dann mit den dabei ermittelten Betriebsdaten in einer Laborapparatur nachvollzogen. Die theoretisch erhaltenen und die im praktischen Betrieb gewonnenen Werte werden miteinander verglichen und der Wirkungsgrad der Laborapparatur wird bestimmt.
Informieren Sie sich über technische Ausführungsformen von Extraktionsapparaten sowie ihre besonderen Eigenschaften und Anwendungsfälle in der Literatur (Überblick in [1], ausführlicher z.B. in [2 – 4]).

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 4 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
2. Grundlagen
2.1 Die Flüssig-Flüssig-Extraktion
Ausgangsgemisch F Extrakt E
Raffinat R Lösungsmittel L
Extraktionsstufe
Abb. 1 Schematische Darstellung einer Extraktionsstufe
Man bringt die zu trennende Lösung (Ausgangsgemisch oder Feed F), die aus dem Trägerstoff T und dem zu extrahierenden Stoff C (Extraktstoff) besteht, mit einer bestimmten Menge des Lösungsmittels L in innigen Kontakt, z.B. durch Dispergieren. Da das ternäre System T, L und C eine Mischungslücke bildet, in die bei genügender Lösungsmittelzufuhr das Gemisch aus Zulauf und Lösungsmittel fällt, entstehen zwei ineinander unlösliche flüssige ternäre Phasen, zwischen denen ein Stoffaustausch stattfindet: Der zu extrahierende Stoff C verteilt sich zwischen dem Trägerstoff T und dem Lösungsmittel L, d.h. er wandert von der einen Phase durch die Phasengrenzfläche in die andere Phase. Dort ändert sich seine Konzentra-tion, bis ein Gleichgewichtszustand zwischen den beiden Phasen erreicht ist.
Nach Trennung der Phasen wird die Raffinatphase R abgezogen. Diese hat sich aus dem Ausgangsge-misch gebildet, besteht also im wesentlichen aus dem Trägerstoff T sowie der Restmenge des Stoffes C und im Trägerstoff gelöstem Lösungsmittel L. Die ebenfalls abgezogene Extraktphase E – entstanden aus dem Lösungsmittel – besteht aus Lösungsmittel L sowie der extrahierten Menge des Stoffes C und im Lösungsmittel gelöstem Trägerstoff T.
Was Trägerstoff T und Raffinat R bzw. was Lösungsmittel L und Extrakt E ist, ergibt sich aus den Vor-gaben des Verfahrens gemäß Abb. 1 und nicht zwangsläufig aus dem Stoffsystem. Wie in der Einleitung erwähnt, können für das gleiche Stoffsystem durchaus zwei verschiedene Verfahren mit wechselseitigem Austausch von Trägerstoff T und Lösungsmittel L angewandt werden.
Die in Abb. 1 schematisch dargestellte Extraktionsstufe besteht aus einer Mischer-Scheider-Kombination (Abb. 2). Im Mischer wird – meist mechanisch, z.B. durch Rühren – das Ausgangsgemisch F innig mit dem Lösungsmittel L vermischt. Die Trennung der beiden im Gleichgewicht stehenden Phasen in Raffinat R und Extrakt E geschieht im auf den Mischer folgenden Abscheider (oft einfach Scheider genannt).
Ausgangs- Lösungsmittel L Raffinat Rgemisch F
Phasen- grenzfläche
Extrakt E
Mischer Scheider
Abb. 2: Aufbau einer Extraktionsstufe aus Mischer und Scheider Jedes einzelne Mischen und Trennen der Phasen wird eine praktische Extraktionsstufe genannt. Das Ex-traktionsverfahren besteht aus einer oder mehreren aufeinander folgenden Stufen. Mehrstufenverfahren sind die am häufigsten in der Technik anzutreffenden Ausführungsformen, weil mit zunehmender Stufen-zahl – einen gleichbleibenden Extraktionseffekt vorausgesetzt – die Lösungsmittelmenge verringert wer-den kann. Die optimalen Bedingungen lassen sich bei Kenntnis des Phasengleichgewichtes für die meisten Extraktionsprobleme rechnerisch – oder wie hier im Versuch graphisch – ermitteln. Extraktionsversuche sind nur für nicht eindeutig definierte Ausgangsgemische oder Lösungsmittelmischungen notwendig.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 5 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Für die Extraktion lassen sich also drei charakteristische Vorgänge unterscheiden:
• Vermischung des zu extrahierenden Gemisches mit dem Extraktionsmittel
Durch innige Vermischung der beiden Flüssigkeiten wird die Einstellung des Verteilungsgleichgewich-tes beschleunigt. Bis zu welchem Grad das Gleichgewicht tatsächlich erreicht wird, hängt von der Durchmischung und der Mischzeit ab. Es gibt Mischer, die eine so feine Verteilung herbeiführen, dass das Gleichgewicht praktisch sofort erreicht wird.
• Trennung der Phasen in Schichten
Nach dem Mischen werden die beiden Flüssigkeiten in den Scheider geführt, wo eine Schichtung der Phasen erfolgt. Dieser Vorgang ist für die Wirksamkeit einer Extraktionsstufe genauso wichtig wie die Gleichgewichtseinstellung. Schwierigkeiten bei der Phasentrennung – z.B. Emulsionsbildung – können sowohl durch Eigenschaften des Stoffsystems, wie z.B. zu geringe Unterschiede zwischen den Phasen in der Dichte und / oder der Oberflächenspannung, aber auch durch eine zu heftige vorherige Vermi-schung verursacht werden.
• Entfernung und Wiedergewinnung des Extraktionsmittels
Die Extraktion ist fast immer mit anderen Trennoperationen, vorzugsweise mit einer Destillation ver-bunden, um das Lösungsmittel zurückzugewinnen und im Kreislauf wieder in die Extraktion zurück-zuführen. Die Wiedergewinnung des Lösungsmittels ist oft ebenso wichtig wie die Extraktion selbst.
Eine apparative Einrichtung, in der Gleichgewicht zwischen beiden Phasen erreicht wird, entspricht einer theoretischen Extraktionsstufe *). Der Grad der Annäherung an das Gleichgewicht ist ein Maß für die Wirksamkeit der betreffenden Apparatestufe. Hierbei ist in vielen Fällen der Wirkungsgrad, der angibt, wie weit die praktischen Verhältnisse den theoretischen nahekommen, empirisch zu bestimmen.
Zur graphischen Darstellung der Vorgänge in einer Extraktionsanlage und der Gleichgewichtsdaten des zu betrachtenden Dreistoffsystems bedient man sich unterschiedlicher Diagramme, die im folgenden erläutert werden. 2.2 Das Dreieckskoordinatensystem und seine Eigenschaften
Abb. 3: Konzentrationsangaben für ternäre Gemische im Dreieckskoordinatensystem
(hier z.B. als Molenbrüche)
Abb. 4: Mischungsregel und Hebelgesetz im Dreieckskoordinatensystem
*) Analog zu anderen thermischen Trennverfahren wie der Rektifikation oder Absorption, gibt es auch für die
Extraktion Ausführungsformen, bei denen ein kontinuierlicher Stoffübergang ohne eine zeitlich getrennte Durch-mischung und Phasenabscheidung erfolgt, z.B. in der Füllkörper-Packung einer Extraktions-Kolonne. Hier defi-niert man einen bestimmten Abschnitt der Packung , der in der Wirkung einer einmaligen Gleichgewichts-einstellung entspricht, als Höhen-Äquivalent für eine theoretische Stufe.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 6 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Daten ternärer Stoffsysteme, also auch die Phasengleichgewichte für flüssige ternäre Systeme bei kon-stanter Temperatur und konstantem Druck lassen sich vorteilhaft in Dreieckskoordinaten darstellen. Im folgenden wird die besonders anschauliche Form des gleichseitigen Dreiecks genutzt. In der Literatur (z.B. [5]) erfolgt die Darstellung häufig in einem rechtwinkligen Dreieck, wodurch sich an den Zusam-menhängen aber nichts ändert.
Die Ecken des Dreiecks (siehe Abb. 3) stellen die reinen Komponenten T, L und C dar. Punkte auf den drei Seiten geben die Zusammensetzung der drei binären Gemische TL, LC und TC wieder. Jede ternäre Mischung aus den drei Stoffen T, L und C wird durch einen Punkt innerhalb des Dreiecks repräsentiert.
Im Dreiecksdiagramm können Konzentrationsangaben x i benutzt werden, die die Bedingung *)
x i
i
=∑ 11
bzw. x i
i
=∑ 1001
% (1)
erfüllen, z.B. Molenbruch (siehe Abb. 3) oder Mol-% sowie Gewichtsanteile oder Gew-%, wie für die folgende Versuchsdurchführung **). Die Konzentrationsangabe muss für die Gesamtmenge des entstande-nen Gemisches gelten, unabhängig davon, ob es homogen ist oder in mehrere Phasen zerfällt (eine Kon-zentrationsangabe z.B. in Mol / l wäre für ein zweiphasiges Gemisch wenig sinnvoll).
Im Dreiecksdiagramm gelten folgende Gesetzmäßigkeiten (siehe Abb. 4):
1. Jede von einer Ecke ausgehende Gerade teilt nach dem 2. Strahlensatz die Gegenseite und alle Paral-lelen zu ihr im gleichen Verhältnis.
Fügt man also zu dem Gemisch M (bestehend aus L und C im Verhältnis 3/7) eine dritte Komponente T hinzu, dann liegen alle möglichen ternären Gemische auf der Linie TM, da sich an dem Verhältnis von L : C = 3 : 7 durch diese Zugabe von T nichts ändert.
2. Wenn z.B. die Konzentration in Molenbrüchen angegeben ist, ergibt sich für das Molverhältnis der zugefügten Komponente T zur Molzahl des Ausgangsgemisches M im Mischpunkt N aus dem Verhält-nis der Abstände zwischen den Punkten:
MoleMole
MoleMole Mole
TM
TL C
MNTN
= + = (2)
Diese Beziehung lässt sich in das sogenannte „Hebelgesetz“ umformen:
( ) ( ) ( ) ( )Mole MoleT TN M MN⋅ = ⋅ (3)
3. Die Zusammensetzung irgendeiner neuen Mischung R, die dadurch gebildet wird, dass man zwei ter-näre Mischungen F und S miteinander mischt, liegt, entsprechend der oberen Gesetzmäßigkeit, auf der Verbindungslinie FS der beiden Ausgangsgemische. Es gilt wiederum das Hebelgesetz. Machen Sie sich klar, dass das Hebelgesetz die Gesamt- und Teil-Massenbilanzen darstellt (F und S sind im Ver-hältnis 1 : 2 gemischt worden).
2.3 Binodalkurve und Konoden
Für Extraktionen sind nur Dreistoffsysteme mit Mischungslücken geeignet. Besonders wichtig ist dabei der Verlauf der Grenze zwischen dem flüssigen Einphasen- und dem Zweiphasengebiet.
Ist in einem Dreistoffsystem C sowohl mit T als auch mit L in beliebigen Verhältnissen mischbar, wäh-rend T und L nur teilweise miteinander mischbar sind (Auftreten einer Mischungslücke), dann erhält man die in Abb. 5 schematisch gezeigten Verhältnisse.
*) Durch die Festlegung der Summe ist das ternäre System bereits durch zwei Konzentrations-Angaben bestimmt und kann deshalb in einem zweidimensionalen Diagramm eindeutig dargestellt werden.
**) Volumenanteile oder Vol-% sind häufig ungeeignet, weil sich die Volumina in Mischungen nicht immer additiv nach Bedingung (1) verhalten (z.B. Wasser und Ethanol).

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 7 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Abb. 5 Darstellung von Phasengleichgewichten ternärer Gemische in Dreieckskoordinaten
Der Abstand TX stellt in diesem Dreistoffsystem die Löslichkeit von L in T dar. LY entspricht der Lös-lichkeit von T in L.
Die Kurve X-P-Y ist eine Sättigungsisotherme bei konstantem Druck. Man nennt sie auch Phasengrenz-kurve, Binode oder Binodalkurve. Sie stellt die Grenze zwischen dem homogenen einphasigen Gebiet und dem heterogenen zweiphasigen Gebiet dar. Der Einfluss der Temperatur ist meistens stark (im allgemeinen Verkleinerung der Mischungslücke mit zunehmender Temperatur), während sich der Druck auf die flüssi-gen Phasen weniger auswirkt.
Jeder Punkt der Binodalkurve steht mit einem anderen Binodenpunkt im Gleichgewicht. Die Linien, die im Gleichgewicht stehende Punkte miteinander verbinden, nennt man Konoden (unendlich viele).
Alle Gemische, die einem Punkt im Zweiphasengebiet entsprechen (z.B. K), zerfallen entlang einer Kono-de in zwei ternäre Phasen. Die Zusammensetzung solcher Phasen, die miteinander im Gleichgewicht ste-hen, ist durch die Endpunkte der jeweiligen Konoden auf der Binodalkurve gegeben. Wenn T der Träger-stoff und L das Lösungsmittel ist, dann ist nach den Definitionen in Abb. 1 R das Raffinat und E das Ex-trakt (aus dem Gemisch K in Abb. 5 scheiden sich also R 1 als Raffinat und E 1 als Extrakt ab).
Die Mengen der beiden Gleichgewichtsphasen, die man auch konjugierte Phasen nennt, berechnet sich über das Hebelgesetz:
Extraktmenge
Raffinatmenge
Menge
Menge
Abstand
Abstand= =
E
R
KR
KE (für die Konode durch K) (4)
Die Komponente C verteilt sich auf beide flüssige Phasen. Im dem Maße, wie die Konzentration an C zu-nimmt, werden die Konoden kürzer. Die Zusammensetzungen der jeweiligen Gleichgewichtsphasen nä-hern sich einander und werden schließlich identisch. Sie verschmelzen an einem bestimmten Punkt P, dem „kritischen Punkt“, der die Extraktseite („Extraktast“) der Binodalkurve von der Raffinatseite („Raffinat-ast“) trennt. Außerhalb der Binodalkurve existiert in Systemen mit einer einzigen Mischlücke nur eine homogene Phase (es gibt auch sehr komplizierte Stoffsysteme, z.B. mit mehreren Mischungslücken [5]).
Üblicherweise – wie auch in Abb. 5 dargestellt – verlaufen die Konoden nicht parallel. Sie können ihre Neigung stark aufwärts oder abwärts variieren, wenn sich die Konzentration von C in der Mischung än-dert. Im allgemeinen fällt weder der kritische Punkt P auf das Maximum der Binodalkurve noch liegt die-ses Maximum in der Mitte des Diagramms. Wenn sich die Neigung der Konoden in ihrer Tendenz ändert, tritt mindestens eine waagerechte Konode auf (solutropes System), d.h. die Konzentration des Extrakt-stoffes C ist in diesem Falle für Extrakt und Raffinat gleich. Machen Sie sich klar, dass die Konsequenzen nicht die gleichen sind, wie bei einem azeotropen Punkt in der Destillationstechnik.
Grundsätzlich ist es für ein wirtschaftliches Extraktionsverfahren günstig, wenn sich der Extraktstoff im Extrakt besser löst als im Raffinat (entsprechend den ansteigenden Konoden in Abb. 5). Dies ist aber nicht Voraussetzung für die Extraktion, da man mit entsprechend großen Mengen an Lösemittel den Extraktstoff auch aus dem Trägerstoff extrahieren kann, wenn die Löslichkeit im Extrakt geringer ist als im Raffinat (in Abb. 5 würden die Konoden dann abfallen, siehe auch das in der Einleitung angesprochene Verfahren zur Extraktion von Essigsäure aus verdünnten wässrigen Lösungen).

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 8 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
2.4 Mess- und Interpolationsverfahren sowie Modellgleichungen für flüssig-flüssig-Phasengleichgewichte
Um das Phasengleichgewicht darstellen zu können, genügt es, eine begrenzte Anzahl von Konoden durch Versuche zu ermitteln und die Binodalkurve als Ausgleichskurve durch die jeweiligen Konodenendpunkte E und R (siehe Abb. 5) zu legen.
Um eine maximale Redundanz der Messergebnisse zu erreichen, sollte man nach Möglichkeit alle erreich-baren Informationen nutzen (siehe Abb. 5): • Zusammensetzung des Mischpunktes (K in Abb. 5), z.B. durch genaues Einwiegen, • Zusammensetzung der Extrakt- und Raffinat-Phase durch Analyse, am besten aller Komponenten, • Menge der Extrakt- und Raffinat-Phase, um nach dem Hebelgesetz die Bilanz kontrollieren zu können.
Wenn nicht alle diese Messmöglichkeiten zur Verfügung stehen – wie hier im Versuch – kann man auch mit einfacheren Methoden, die nur einige der o.g. Daten benötigen, brauchbare Messwerte erzielen Eine zusätzliche, anschauliche Methode zur Messung von Punkten auf der Binodalkurve ist die „Trübungs-Titration“, die unmittelbar den Übergang vom homogenen zum heterogenen Gebiet oder umgekehrt erfasst.
Für die Auslegung eines Extraktionsverfahrens (siehe Kapitel 2.7) muss man jeden beliebigen Punkt der Binodalkurve und die durch diesen Punkt verlaufende Konode ermitteln können. Alle Messdaten sind mehr oder weniger fehlerbehaftet, selbst wenn man alle o.g. Möglichkeiten zur Kontrolle und Absicherung der Werte nutzen kann. Es ist also immer mit einer Streuung der Messpunkte zu rechnen. Wenn man zahl-reiche Konoden vermessen hat, könnte man in erster Näherung einfach durch eine lineare Interpolation Zwischenwerte bestimmen. Dabei würde man jedoch voraussetzen – ohne es begründen zu können –, dass die beiden verwendeten Konoden richtig sind, und dass zwischen ihnen ein linearer Zusammenhang besteht.
Abb. 6 Interpolationsverfahren
mit Hilfe der Konjugatlinien
Sinnvoller ist es, den ganzen Datensatz als Einheit zu be-trachten und gemeinsam auszuwerten und für den Verlauf sowohl der Binodalkurve als auch der Konodensteigung ein Ausgleichsverfahren einzusetzen.
Zur graphischen Interpolation der Konoden konstruiert man eine sog. „Konjugatlinie“ (d.h. Verbindungslinie), nach den zwei Methoden in Abb. 6.
Die effektivere Art benutzt ein angehängtes zweites Drei-ecksdiagramm T-L-G. Jeder Punkt P-X-Y-Z der Konju-gatlinie entsteht aus den beiden Endpunkten R i und E i ei-ner Konode, indem zu den Außenseiten des Dreiecks T-G und L-G Parallelen gezogen werden (z.B. Punkt Y aus R 2 und E 2 ; zahlenmäßig kann man die Punkte mit den Ko-ordinaten der Konzentration an L in E i und an T in R i eintragen). Die Konjugatlinie verläuft vom Punkt Z, der die gegenseitige Löslichkeit von T und L wiedergibt bis zum kritischen Punkt P. Ohne ein zweites Diagramm kommt die zweite Konjugatlinie P-U-V-W aus, die Paral-lelen zu T-L und T-C zieht. Wegen der flacheren Schnitt-winkel ist diese Methode jedoch weniger genau.
Umgekehrt kann man mit Hilfe der Konjugatlinien aus je-dem Punkt der Binodalkurve den konjugierten Punkt kon-struieren, z.B. kommt man von E 3 über X oder U nach R 3.
Der wesentliche Vorteil dieses graphischen Verfahrens ist, dass man für Bionodalkurve und Konjugatlinie Aus-gleichskurven einzeichnen kann, die alle Messdaten gleichwertig wiedergeben und offensichtlich herausfal-lende Einzelwerte erkennen lassen.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 9 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Neben der hier vorgestellten graphischen Methode verwendet man heute überwiegend Modellgleichungen, die vor allem auch für die Anwendung in Computerprogrammen erforderlich sind. Sie basieren haupt-sächlich auf den für nicht-ideale Flüssigkeits-Dampf-Gleichgewichte (siehe Versuch TC1) entwickelten Ansätzen NRTL-Gleichung (Non Random Two-Liquid Equation) und UNIQUAC-Gleichung (Universal Quasi-Chemical Equation) [5]. Ein Flüssigkeitsgemisch mit Mischungslücke ist ein Beispiel für eine sehr große Abweichung vom Verhalten eines idealen Flüssigkeitsgemisches, in dem sich die verschiedenen Komponenten gegenseitig nicht beeinflussen würden. Die genannten Gleichungen sind in der Lage, die Flüssigkeits-Dampf-Gleichgewichte auch derartiger Mehrkomponenten-Mischungen richtig wiederzuge-ben. Der Zusammenhang mit den beiden flüssigen Phasen besteht darin, dass diese im Gleichgewicht die gleiche Dampfphase haben müssen, wodurch ein Berechnungs-Ansatz gegeben ist.
Mit Hilfe dieser Gleichungssysteme kann man durch Parameter-Anpassung eine optimale Wiedergabe der Messwerte erreichen und erkennen, ob alle Daten zusammenpassen. Die in der Literatur veröffentlichten flüssig-flüssig Gleichgewichtsdaten sind in [5] zusammen mit diesen Parametern verfügbar. Intensiv wird auch an Gleichungssystemen gearbeitet, die eine Vorhersage von flüssig-flüssig Gleichgewichtsdaten aus der Molekülstruktur ohne aufwendige Messungen erlauben. Ein Ansatzpunkt ist die aus der UNIQUAC-Gleichung entwickelte UNIFAC-Methode (Universal Functional Group Activity Coefficients), die für die Vorhersage von Flüssigkeits-Dampf-Gleichgewichten bereits erfolgreich zum Einsatz kommt [6]. 2.5 Das Verteilungsdiagramm
Außer den bisher beschriebenen Dreiecksdiagrammen benutzt man als Darstellungsart im rechtwinkligen Koordinatensystem die sogenannten Verteilunsdiagramme (analog zum McCabe-Thiele-Diagramm in der Destillationstechnik), die allerdings nur den Verlauf der Konoden wiedergeben, d.h. nur die Konzentrati-onsverteilung einer Komponente zwischen zwei heterogenen Phasen wird dargestellt. Löslichkeitsdaten, wie sie das Dreieckskoordinatensystem vermittelt, können aus Verteilungsdiagrammen nicht abgelesen werden. Sie sind jedoch leichter zu handhaben und für viele Zwecke ausreichend. Das Verteilungsdia-gramm ist vor allem wertvoll, wenn verschiedene Extraktionsmittel miteinander verglichen werden sollen.
Hier trägt man die Konzentration des zu extrahierenden Stoffes C in den beiden Phasen y = Extraktphase und x = Raffinatphase gegeneinander auf. Eine Möglichkeit zur Konstruktion solcher Verteilungsdia-gramme mit Hilfe von Dreieckskoordinaten und den Zusammenhang zwischen den beiden Darstellungs-arten zeigt Abb. 7.
Abb. 7 Zusammenhang der Darstellungen
im Dreiecksdiagramm und im Verteilungsdiagramm
Durch Übertragen der gegenseitigen Löslichkeiten eines ternären Systems mit wenigen gemessenen Kono-den vom Dreiecksdiagramm in das Verteilungsdiagramm lassen sich Konoden gut interpolieren (Abb. 7). Wenn man aus dem Verteilungsdiagramm die gesuchte (d.h. noch nicht vorhandene) Konode durch einen Punkt M im Dreiecksdiagramm erhalten will, muss man dem Verteilungsdiagramm durch mehrmaliges Probieren diejenigen im Gleichgewicht stehenden Konzentrationen xR und yE entnehmen, deren Verbin-dungslinie im Dreiecksdiagramm durch M verläuft.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 10 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
2.6 Extraktionsverfahren Bei der Flüssig-Flüssig-Extraktion sind folgende Verfahren gebräuchlich:
a) das einstufige Extraktionsverfahren
b) die mehrstufigen Extraktionsverfahren • Kreuzstromextraktion • Gegenstromextraktion • Gegenstromextraktion mit Rücklauf
2.6.1 Die einstufige Extraktion
Die einstufige Extraktion findet fast nur im Labormaßstab Anwendung (z.B. durch Verwendung eines Schüttelrichters); in der Technik spielt sie nur eine untergeordnete Rolle.
Das Verfahren wurde bereits in Abb. 1 schematisch dargestellt: Eine gewisse Menge der Mischung F aus Trägerstoff T und zu extrahierendem Stoff C wird mit einer entsprechenden Menge des Lösungsmittels L im Mischer gut in Berührung gebracht, damit sich möglichst rasch das Gleichgewicht einstellt. Nach dem Entmischen werden die Phasen getrennt, d.h. die Raffinatphase R und die Extraktphase E abgezogen. Die einstufige Extraktion kann sowohl satzweise als auch kontinuierlich durchgeführt werden. 2.6.2 Die mehrstufigen Extraktionsverfahren 2.6.2.1 Die Kreuzstromextraktion
Abb. 8 Extraktion im Kreuzstrom von Feed / Raffinat und Lösungsmittel Man kann die Extraktion effektiver gestalten, wenn man die Gesamtmenge des Lösungsmittels aufteilt und die Extraktion stufenweise durchführt, also mehrere Mischer-Scheider-Einheiten hintereinander anordnet und jeder Stufe immer wieder frisches Lösungsmittel zuführt. Die Raffinatphase einer Stufe wird zum Zulauf der nächsten Stufe. Man spricht dann von einer mehrstufigen Kreuzstromextraktion, wie sie schematisch in Abb. 8 dargestellt ist. Die mehrstufige Kreuzstromextraktion kann entweder gemäß Bild 8 kontinuierlich durchgeführt oder auch satzweise betrieben werden. Bei der kontinuierlichen Durchführung strömt die zu extrahierende Mischung von einer Extraktionsstufe zur anderen und wird dabei immer wieder mit frischem Lösungsmittel

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 11 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
behandelt, bis sie schließlich als Raffinat aus der letzten Stufe austritt. In jeder Stufe wird ein Extrakt gewonnen, dessen Gehalt an Extraktstoff C von Stufe zu Stufe abnimmt. Genau das gleiche Verfahren kann satzweise durchgeführt werden, indem man die Mischung F in einer Extraktionsapparatur immer wieder mit frischem Lösungsmittel behandelt, nachdem man den jeweils anfallenden Extrakt nach Phasentrennung abgezogen hat.
Ein Unterschied zwischen der kontinuierlichen und der satzweisen Durchführung der einstufigen und der mehrstufigen Extraktion besteht darin, dass die Konzentrationsänderungen durch Stoffaustausch bei der satzweisen Durchführung innerhalb des Gefäßes mit der Zeit stattfindet, während bei der kontinuierlichen Arbeitsweise der Konzentrationsverlauf innerhalb der Anlage ortsabhängig wird. 2.6.2.2 Die Gegenstromextraktion
Abb. 9 Gegenstromextraktion
Die mehrstufige Gegenstromextraktion gemäß Abb. 9 wird technisch kontinuierlich durchgeführt. Hierbei wird das zu trennende Gemisch F und das Lösungsmittel L an den entgegengesetzten Enden der Extrak-tionsapparatur zugeführt und beide durchströmen diese kontinuierlich im Gegenstrom: Die Raffinatphase einer Stufe wird zum Zulauf der nächsten Stufe und die Extraktphase einer Stufe wird zum Lösungsmittel der vorigen Stufe.
Die Gegenstromextraktion ist die am weitesten verbreitete Extraktionsmethode. Durch das auch bei ande-ren technischen Prozessen häufig genutzte Gegenstromprinzip wird in jeder Stufe eine hohe Konzentrati-onsdifferenz für einen schnellen Stofftransport erreicht. Das Raffinat kommt mit dem frischen Lösungs-mittel in Kontakt und wird dadurch optimal abgereichert. Das Extrakt erreicht im Kontakt mit dem Zulauf-gemisch die größtmögliche Konzentration an Extraktstoff. Im allgemeinen kann man davon ausgehen, dass mit der Gegenstromextraktion das beste Ergebnis mit dem geringsten Lösungsmitteleinsatz erreichbar ist.
Durch einen Produktrücklauf erreicht man bei dem typischen Anwendungsfall der Rektifikation die An-reicherung des Produktes in der flüssigen Phase, so dass man in einen Bereich des Flüssigkeits-Dampf-Gleichgewichtes gelangt, in dem man hohe Produktkonzentration im Dampf erzielbar ist (siehe Versuch TV4). Ähnlich ist es in speziellen Fällen bei der Extraktion sinnvoll, einen Rücklauf einzusetzen. Man kann so ggf. durch Konzentrations-Verschiebungen eine effektivere Trennung erreichen, z.B. weil in die-sem Bereich die Konoden günstiger verlaufen oder weil sich die Mischungslücke vergrößert oder auch weil eine Emulsionsgefahr reduziert wird.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 12 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
2.7 Darstellung der Gegenstromextraktion in Dreieckskoordinaten In Abb. 9 ist eine mehrstufige Gegenstromanlage, bestehend aus 4 Mischer-Scheider-Batterien gezeigt worden. Eine Gegenstromextraktion lässt sich nicht wie die Kreuzstromextraktion einfach Stufe für Stufe im Dreiecksdiagramm verfolgen, da die jeweilige Raffinatkonzentration, die der nächsten Stufe zufließt, zunächst noch unbekannt ist.
Eine Mengenbilanz um die erste, erste + zweite bis um die erste bis n-te Stufe ergibt nach Abb. 10:
Abb. 10 Mengenbilanz für die Gegenstromextraktion 1. Stufe m F + m E 2 = m R 1 + m E 1 m R 1 – m E 2 = m F – m E 1 = Q (5)
1. bis 2. Stufe m R 2 – m E 3 = m F – m E 1 = Q (6) • • •
1. bis n-te Stufe m R n – m L = m F – m E 1 = Q (7)
Diese Gleichungen sagen aus, dass die Differenzmenge zwischen der Raffinatphase einer beliebigen Stufe und der Extraktphase der nachfolgenden Stufe für jeden Mengenbilanzquerschnitt einen konstanten Diffe-renzmengenstrom Q ergibt, der der Differenz zwischen dem Feed und der Extraktphase der ersten Stufe entspricht.
Abb. 11 Graphische Konstruktion zur Ermittlung der Stufenzahl für die Gegenstromextraktion Jede der Bilanzgleichungen (5) bis (7) wird nach dem Mischungsgesetz im Dreiecksdiagramm durch eine Gerade dargestellt, die für eine beliebige Stufe i durch den Punkt für das diese Stufe verlassende Raffinat R i und den Punkt für das in diese Stufe eintretenden Extrakt aus der nächsten Stufe E i + 1 (für die n-te Stufe das Lösungsmittel L) geht. Die Geraden für alle n Stufen schneiden sich außerdem in einem festen Punkt Q, der daher auch Pol oder Arbeitspunkt genannt wird (siehe Abb. 11, für die Zulaufseite der Anlage kommt noch eine Gerade Q–F–E 1 hinzu).

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 13 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Der Pol Q liegt (meist) außerhalb des Dreiecks (rechts oder links, je nach der Lage von F und E 1) und stellt somit kein wirkliches Gemisch dar, da es negative Mengen und negative Konzentrationen nicht gibt. Er kennzeichnet die Zusammensetzung und die Menge eines fiktiven Gemisches und hat lediglich eine konstruktive Bedeutung, deren mathematische Begründung oben angeführt ist. Eine konkrete Vorstellung sollte man mit diesem Pol nicht verbinden.
Die Lage des Pols Q lässt sich einfach festlegen, wenn die Konzentrationen bekannt bzw. gefordert sind:
• des in der 1. Stufe zulaufenden Gemisches F, • des von der 1. Stufe ablaufenden Extraktes E 1, • des von der letzten (n-ten) Stufe ablaufenden und durch die Problemstellung definierten Raffinates R n , • des der letzten Stufe zulaufenden Extraktionsmittels (meist weitgehend reines Lösungsmittel).
Das Vorgehen ist gemäß Abb. 11 folgendermaßen:
Man fängt an bei einem beliebigen Ende, z.B. bei Stufe 1, wo das zu extrahierende Gemisch, der Zulauf F, eintritt und das endgültige Extrakt E1 die Anlage verlässt. Gemäß Gleichung (7) verbindet man zuerst die Zustandspunkte R n mit L sowie F mit E1 durch je eine Gerade, deren Schnittpunkt Q ergibt. Mit Hilfe der Konode E1–R1 wird dann die Zusammensetzung der Raffinatphase, die die erste Stufe verlässt, bestimmt. Eine neue Arbeitslinie von Punkt R 1 zum Pol Q gemäß Gleichung (5) legt in der Verlängerung auf dem Extraktast der Binodalkurve die Konzentration von E 2 fest. E 2 stellt die Zusammensetzung der Extraktphase dar, die die 2. Stufe verlässt und in die 1. Stufe eintritt. Die Punkte Q , R 1 und E 2 liegen dann auf einer Geraden, wie dies Gleichung (5) fordert. Den zum Extrakt E 2 im Gleichgewicht stehenden Raffinatablauf R 2 erhält man wiederum über die von E 2 ausgehende Konode.
Das Konstruktionsverfahren wechselt also zwischen der Massenbilanz der Stoffe, die in den Querschnitten außerhalb der Extraktionsstufen fließen – dargestellt mit Hilfe der Geraden durch den Polpunkt Q – und dem Phasengleichgewicht in den Extraktionsstufen – dargestellt durch eine Konode, die ausgehend von einem Punkt auf der Binodalkurve mittels der Konjugatlinie gezeichnet werden kann.
Dieses Verfahren wird solange fortgesetzt, bis eine Raffinat-Zusammensetzung erreicht bzw. überschritten ist, die der verlangten Raffinat-Endkonzentration R n entspricht. Die Zahl der benötigten Schritte (Gleichgewichtseinstellungen) zur Erzielung der gewünschten Raffinat-Endkonzentration entspricht der Anzahl der erforderlichen theoretischen Extraktionsstufen. Es ist dabei durchaus sinnvoll, auch Bruchteile von theoretischen Extraktionsstufen abzuschätzen, da die Zahl der benötigten praktischen Trennstufen oh-nehin höher liegt (praktische Stufenzahl = theoretische Stufenzahl / Wirkungsgrad, siehe Kapitel 2.10). 2.8 Ausführung der Stufenkonstruktion bei unterschiedlichen Vorgaben
Abb. 12 Gegenstromextraktion: Zusammenhang zwischen dem Lösungsmittel- Feed-Verhältnis und den Endkonzentrationen
von Extrakt und Raffinat
Aus technischen und / oder wirtschaftlichen Grün-den können für ein Extraktionsverfahren drei der folgenden Betriebsparameter festgelegt werden, die beiden anderen ergeben sich daraus: • Feed-Konzentration (liegt nahezu immer fest), • Raffinat-Endkonzentration R n , • Extrakt-Endkonzentration E 1 , • Lösungsmittel-Feed-Verhältnis, • Stufenzahl (einschließlich Wirkungsgrad)
Der Mischungspunkt M, der sich aus der Lö-sungsmittelmenge L und der Zulaufmenge F er-rechnen lässt, liegt auf der Verbindungslinie FL, die er entsprechend dem Hebelgesetz im folgen-den Verhältnis teilt:
Menge F
Menge L=
LM
FM (8)

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 14 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Durch Umstellung der Gleichung (7) m m m mR n L F E− = − 1 (7)
erhält man m m m m MF L E R n+ = + =1 (9)
d.h., dass auch E1 und Rn auf einer Geraden liegen, die ebenfalls durch M gehen muss. Wenn die Zusam-mensetzungen E1 und Rn vorgegeben sind, lässt sich das Lösungsmittel-Feed-Verhältnis L / F ermitteln. Sind dagegen L / F und damit M sowie E 1 oder R n bekannt ergibt sich die Zusammensetzung R n bzw. E 1 als Verlängerung der Geraden E 1–M bzw. R n–M bis zum jeweiligen Schnittpunkt mit der Binodalkurve.
Die weitere Bestimmung des Polpunktes Q und der Stufenzahl n erfolgt dann wie zuvor beschrieben.
Wenn man die Stufenzahl n fest vorgeben will, ist der Vorgang zwar der gleiche wie bisher beschrieben, aber in diesem Fall muss die Lösung durch Ausprobieren gefunden werden. Wird z.B. bei bekanntem Feed die Endkonzentration im Raffinat R n oder im Extrakt E 1 festgelegt, müssen das Lösungsmittel-Feed-Ver-hältnis = L / F und dementsprechend E 1 bzw. R n variiert werden. Man nimmt zunächst ein Verhältnis L / F an und erhält daraus nach der beschriebenen Konstruktion gemäß Abb. 12 die zweite Zusammensetzung E1 bzw. Rn . Dann führt man – z.B. ausgehend von E1 – das graphische Verfahren mit der gegebenen Stu-fenzahl n durch. Ist die erhaltene Raffinat-Endkonzentration R n größer (kleiner) als der geforderte Wert, so ist das Verhältnis L / F zu niedrig (zu hoch) gewählt worden und muss solange verändert werden, bis die gewünschte Endkonzentration im Raffinat erreicht ist. Dabei ändern sich jeweils selbstverständlich auch die zweite Zusammensetzung E1 bzw. Rn sowie der Polpunkt Q und damit die gesamte Konstruktion. 2.9 Übertragung der Stufenkonstruktion in das Verteilungsdiagramm
In dem im Kapitel 2.5 vorgestellten Verteilungsdiagramm werden auf der Ordinate die Konzentration y des Extraktstoffes in der Extraktphase und auf der Abszisse die Konzentration des Extraktstoffes x in der Raffinatphase aufgetragen. Die Verteilungskurve stellt die Gleichgewichtszustände zwischen Extrakt- und Raffinatphase dar.
Abb. 13 Übertragung der Stufenkonstruktion vom Dreiecks- in das Verteilungs-Diagramm Zur Aufstellung der Arbeitslinie *) wird ein beliebiger Querschnitt der schematischen Extraktionsappara-tur in Abb. 13 oben betrachtet. Durch diesen Querschnitt strömen z.B. der Zulauf F mit der Konzentration x F und das Extrakt der ersten Stufe E 1 mit der Konzentration y E 1 . Beide Konzentrationen bilden den ei-nen Endpunkt der Arbeitslinie im Verteilungsdiagramm. Analog wird für alle Querschnitte vorgegangen.
*) Die Arbeitslinie ist bei der Extraktion nicht zwangsläufig eine Gerade, da sich die Mengenströme zwischen Raffinat- und Extrakt-Phase von Stufe zu Stufe verschieben können.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 15 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Der andere Endpunkt der Arbeitslinie liegt durch die Konzentration y L des Lösungsmittels L (bei reinem Lösungsmittel ist y L = 0) und der Konzentration x R n des austretenden Raffinates im entsprechenden Bilanzquerschnitt fest.
Beginnend mit der Feed-Konzentration wird zwischen Arbeitslinie und Verteilungskurve die Stufenkon-struktion eingezeichnet. Die einzuzeichnende Stufenkonstruktion muss natürlich ebenso viele Extraktions-stufen liefern, wie die Konstruktion im Dreiecksdiagramm ergeben hatte. 2.10 Die praktische Extraktionsstufe und der Wirkungsgrad
Die Stufenkonstruktion in Abb. 13 liefert – wie in allen bisher besprochenen Fällen – die theoretische Stufenzahl, da in den einzelnen Extrakt- und Raffinat-Phasen die Gleichgewichtskonzentrationen ange-nommen werden. In einem technischen Extraktionsapparat, also einer praktischen Extraktionsstufe wird das Gleichgewicht nie ganz erreicht. Um die Wirksamkeit einer praktischen Extraktionsstufe zu beschrei-ben, definiert man den Wirkungsgrad η wie folgt (bezogen auf den Extraktstoff C in der Extraktphase):
η = =− +
′ − +≤
praktisch erzielte Anreicherung
theoretisch mögliche Anreicherungim Gleichgewichtszustand
yE i yE i
yE i yE i
1
11 (10)
y´E i : theoretische Gleichge- wichts-Konzentration an Extraktstoff C, die maxi- mal im Extrakt der i-ten Stufe erreichbar ist
yE i : praktische (gemessene) Konzentration an Extrakt- stoff C im Extrakt der i-ten Stufe
yE i + 1 : praktische (gemessene) Konzentration an Extrakt- stoff C im Extrakt der (i + 1)-ten Stufe, d.h. im Extraktionsmittel- zulauf der i-ten Stufe
Abb. 14 Ermittlung des Stufenwirkungsgrades
Der Wirkungsgrad lässt sich besonders anschaulich im Verteilungsdiagramm (Abb. 14) demonstrieren. Gemäß Gleichung (10) ist in Abb. 14 ist der Stufenwirkungsgrad der i-ten Stufe gegeben durch das Ver-hältnis der senkrechten Strecke i – i´´, der praktisch in der Stufe erreichten Konzentrationsänderung im Extrakt, zum senkrechten Abstand i´ – i´´ der Arbeitslinie von der Gleichgewichtskurve, die die maximale Konzentration angibt, wenn das Gleichgewicht wirklich vollständig erreicht wird. Umgekehrt kann die Stufenkonstruktion nach Abb. 14 für eine reale Extraktionsanlage durchgeführt werden, indem man als Stufenhöhe für eine Extraktionsstufe von der Arbeitslinie (i´´) ausgehend nur den dem Wirkungsgrad ent-sprechenden Anteil (i´´ bis i) des vollen Abstandes bis zur Gleichgewichtskurve (i´´ bis i´) verwendet.
Als wesentliche Gründe dafür, dass der Wirkungsgrad einer Extraktionsstufe unter 1 liegt, sind zu nennen: • der Gleichgewichtszustand wird im Mischer nicht erreicht weil
• die Durchmischung und damit die Phasengrenzfläche für den Stofftransport nicht ausreicht • die Kontaktzeit zu kurz ist (in Anbetracht der erreichten Phasengrenzfläche)
• die Phasentrennung reicht nicht aus, so dass Teile der jeweils anderen Phase mitgeschleppt werden.
Der Wirkungsgrad hängt infolgedessen nicht nur von der Konstruktion des Extraktionsapparates ab, son-dern auch vom eingesetzten Stoffsystem und den Betriebsbedingungen, insbesondere vom Stoffdurchsatz.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 16 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
3. Versuchsbeschreibung
3.1 Stoffdaten
Trägerstoff T n-Butylacetat (Essigsäure-n-butylester, „BuAc“), Molmasse 116,16 kg/kMol Extraktstoff C Essigsäure („HAc“), Molmasse 60,05 kg/kMol Lösungsmittel L Wasser, Molmasse 18,02 kg/kMol
Die eingesetzten Stoffe werden als 100 % rein angenommen.
In der Literatur [5] sind die folgenden Daten für das Flüssig-Flüssig-Phasengleichgewicht bei 25 °C ange-geben, einschließlich der angepassten Parameter für Modellgleichungen (siehe Kapitel 2.4). Die Modell-gleichungen basieren auf den Wechselwirkungen zwischen den Molekülen in der Flüssigkeit und verwen-den molare Konzentrationen (Molenbruch oder Mol-%). Mit Hilfe der in diesem Versuch zur Verfügung stehenden vereinfachten Analysenmethoden (siehe Kapitel 4.6) lassen sich die Molenbrüche jedoch nicht unmittelbar bestimmen. Deshalb wird in diesem Versuch mit Massenbrüchen (Gew-%) gearbeitet, und die Daten aus [5] sind in Gew-% umgerechnet worden. An den im Kapitel 2 beschriebenen Methoden ändert sich dadurch grundsätzlich nichts *). In der Tabelle sind die Endpunkte von 15 Konoden sowie der kriti-sche Punkt aufgeführt (die Zuordnung Raffinat- und Extrakt-Phase entspricht den Vorgaben dieses Ver-suches).
Konode Gew-% in der Raffinatphase Gew-% in der Extraktphase Nr. BuAc HAc Wasser BuAc HAc Wasser
1 97,1 1,3 1,6 0,8 3,7 95,5 2 94,8 3,5 1,7 1,0 6,7 92,3 3 93,0 5,0 2,0 1,0 10,1 88,9 4 90,8 6,4 2,8 1,2 13,2 85,6 5 88,0 8,9 3,1 1,3 16,1 82,6 6 85,1 10,9 4,0 1,6 19,3 79,1 7 81,4 13,6 5,0 1,7 21,5 76,8 8 79,8 14,6 5,6 1,9 25,3 72,8 9 77,0 16,7 6,3 2,3 27,8 69,9 10 72,1 20,4 7,5 2,9 30,7 66,4 11 70,2 21,4 8,4 3,9 32,8 63,3 12 64,8 25,2 10,0 4,7 35,3 60,0 13 60,5 27,8 11,7 6,1 37,6 56,3 14 54,2 31,4 14,4 8,9 39,4 51,7 15 45,5 35,8 18,7 12,8 42,1 45,1
Als Koordinaten für den kritischen Punkt wurden mit Hilfe der an die Daten angepassten Modellgleichung berechnet:
P 26,1 43,3 30,6
Tabelle 1 Flüssig-Flüssig-Phasengleichgewicht bei 25 °C für das Stoffsystem Essigsäure – n-Butylacetat – Wasser [5]
*) Infolge des großen Unterschiedes in der Molmasse zwischen n-Butylacetat und Wasser sind die Zahlenwerte der Gleichgewichtsdaten in Mol-% und Gew-% sehr unterschiedlich. Verwirrend ist zunächst die Tatsache, dass die Konodensteigung für beide Konzentrationsmaße unterschiedlich ist. In molaren Konzentrationen ist die Löslich-keit von Essigsäure in n-Butylacetat höher als in Wasser, in Massenanteilen ist es umgekehrt. Das Ergebnis der Auslegung einer Extraktionsanlage ist insgesamt natürlich unabhängig von der Wahl des Konzentrationsmaßes.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 17 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
3.2 Methoden zur Bestimmung von Flüssig-Flüssig-Phasengleichgewichten
Wie im Kapitel 2.4 erwähnt, bietet die genaue Einwaage eines Gemisches aus dem heterogenen Gebiet, die Trennung der beiden Phasen und die Erfassung ihrer Menge und aller Konzentrationen die besten Voraussetzungen zur Erfassung möglichst zuverlässiger Flüssig-Flüssig-Phasengleichgewichtsdaten. Da dies hier in diesem Versuch nicht möglich ist, sollen einige vereinfachte Methoden vorgestellt und experimentell überprüft werden. 3.2.1 Trübungs-Titration zur Ermittlung der Binodalkurve
Durch tropfenweise Zugabe eines Stoffes zu einem bekannten Gemisch kann man die Binodalkurve ge-zielt so langsam überschreiten, dass sich der Wechsel vom homogenen zum heterogenen Gebiet oder um-gekehrt durch die Trübung bzw. Klärung der stark gerührten Lösung gut erkennen lässt. Man erhält durch diese sehr einfache Methode, die zur Auswertung das Hebelgesetz nach Abb. 4 nutzt, jeweils einzelne Punkte auf der Binodalkurve. Diese Methode liefert die vollständige Zusammensetzung der Messpunkte, die sich somit in Massen- oder Mol-Anteilen (Gew-% oder Mol-%) angeben lassen.
Zur Aufstellung eines geschlossenen Kurvenzugs ist es ratsam, die Ausgangsgemische so zu wählen, dass die sich ergebenden Punkte entlang der gesuchten Binodalkurve möglichst gleiche Abstände voneinander aufweisen. Ein Versuchspunkt sollte möglichst in der Nähe des (geschätzten) Kurvenmaximums liegen. 3.2.2 Untersuchung der beiden Phasen eines heterogenen Gemisches
HAc C
Wasser L
mER
E
BuAc T
MmR
Konode
Abb. 15 Auswertung der beiden Phasen aus einer heterogenen
Mischung M nach dem Hebelgesetz
Ausgehend von einem bekannten (am besten genau eingewogenen) heterogenen Gemisch (Punkt M in Abb. 15) weiß man, daß die bei-den gebildeten Phasen E und R im Dreiecksdiagramm mit dem Mischpunkt M auf einer Geraden liegen müssen. Man kann die Menge der beiden Phasen durch Phasentrennung und Auswiegen oder durch Bestimmung beider Volumina gleichzeitig in einem Messzylinder und anschließende Messung der Dichte beider Phasen ermitteln. Nach dem Hebelgesetz Gleichung (2) und (3) ist dadurch das Verhältnis der Massen *) der beiden Phasen m R zu m E und so-mit auch das Verhältnis der Abstände E-M zu M-R zugänglich.
Um aus dem Verhältnis der Abstände die konkrete Lage im Dreiecksdiagramm bestimmen zu können sind aber noch weitere Informationen erforderlich.
3.2.2.1 Einpassung in die Binodalkurve
Wenn die Binodalkurve bereits bekannt ist – z.B. aus den o.g. Trübungs-Titrationen – kann man die Tatsa-che nutzen, dass E und R auf der Binodalkurve liegen müssen. Durch Probieren kann man die Konode so um den Punkt M drehen (Lineal), dass sowohl die Punkte E und R auf der Binodalkurve liegen, als auch das ermittelte Massenverhältnis der Phasen erfüllt wird. Damit ist die Konodenlage eindeutig bestimmt. 3.2.2.2 Analyse einer Komponente in den Phasen
Als einfache Analysenmethode steht die Titration der Essigsäure mit 0,1-n-Natronlauge **) zur Verfü-gung. Dadurch lassen sich zwei waagerechte Linien im Dreiecksdiagramm nach Abb. 15 für die Essigsäu-rekonzentrationen C in der Phase E und R festlegen. Auch hier kann die Lage der Konode eindeutig defi-niert werden, indem man sie durch Probieren so um den Punkt M dreht, dass sowohl Punkt E und R den analysierten Essigsäurekonzentrationen entsprechen, als auch das ermittelte Massenverhältnis der Phasen erfüllt wird.
*) Auf diesem Wege ist nur das Verhältnis der Massen nicht jedoch der Molzahlen zugänglich. Die Ergebnisse können deshalb nur für ein Phasendiagramm mit Konzentrationsangaben in Gew-% verwendet werden.
**) Die Titration erfolgt mit Phenolphthalein als Indikator für einen leicht alkalischen Umschlagsbereich, wie er für Essigsäure benötigt wird. Eine Verseifung des n-Butylacetats ist unter diesen Bedingungen vernachlässigbar.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 18 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
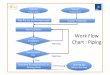
3.3 Laboranlage zur kontinuierlichen vierstufigen Gegenstromextraktion
Abb. 16 Fließbild der kontinuierlichen vierstufigen Gegenstromextraktion
Die Versuchsapparatur zur kontinuierlich arbeitenden Gegenstromextraktion ist schematisch in Abb. 16 wiedergegeben. Das Betreiben derartiger Laborapparaturen ergibt sich aus der Notwendigkeit, besonders bei Neuentwicklungen, Aussagen über den Wirkungsgrad der Mischer-Scheider-Kombination zu erhalten, da bei alleiniger Kenntnis der Gleichgewichtsdaten des zu untersuchenden Systems lediglich die theoreti-sche Trennstufenzahl berechenbar ist. Der Apparatur werden über zwei Dosierpumpen aus den Vorrats-gefäßen Feed und Lösungsmittel kontinuierlich zugeführt.
Abb. 17 Mischer-Scheider-Einheit der Laborapparatur
Jede Extraktionsstufe setzt sich aus einer Mischer- und einer Scheider-Einheit zu-sammen, wie sie in Abb. 17 dargestellt sind (es stehen zwei etwas unterschied-liche Apparaturen zur Verfügung). Der Mischer stellt nach seinem Funktions-prinzip eine Kreiselpumpe dar. Ihm flie-ßen Raffinat- und Extrakt-Phase getrennt zu, steigen in der hohlen Welle hoch und werden durch die hohlen Rührbalken in den Mischraum geschleudert, in dem eine intensive Durchmischung erfolgt. Aus dem Mischraum gelangt das Zweiphasen-gemisch in den Scheider und trennt sich unter dem Einfluss der Schwerkraft. Die leichtere Phase tritt oben aus dem Über-lauf aus, die schwerere Phase von dringt unten durch die Öffnungen in das äußere Rohr des Scheiders ein, steigt darin empor und verlässt den Scheider durch das kleinere, konzentrisch im großen Rohr angebrachte Fallrohr.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 19 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
4. Aufgabenstellung und Versuchsdurchführung
Die Stoffe Essigsäure (ätzend) und n-Butylacetat (Geruchsbelästigung) müssen mit äußerster Sorgfalt und Vorsicht gehandhabt werden !
Die ausgehängten Sicherheitsvorschriften sind unbedingt einzuhalten !
Auf die Pflicht zum Tragen einer Schutzbrille wird ausdrücklich hingewiesen !
Das Feed-Gemisch darf nur im Abzug angesetzt und in den vorgesehenen bruchsicheren Behältern transportiert werden !
Gemische mit n-Butylacetat dürfen nicht ins Abwasser gelangen ! (auch die titrierten Lösungen nicht)
Das Raffinat darf nicht in den Abfallbehälter geschüttet werden, sondern ist im vorgesehenen Behälter im Abzug zu verwahren !
1. Tragen Sie die im Kapitel 3.1 aufgeführten flüssig-flüssig Phasengleichgewichts-Daten auf dem vom
Assistenten erhaltenen Dreieckskoordinaten-Papier ein. • Zeichnen Sie die Binodalkurve, die Konoden und die Konjugatlinie als Ausgleichskurven ein.
Diese Vorarbeiten sind Voraussetzung für ein erfolgreiches Versuchsergebnis und sollten dem-entsprechend sehr sorgfältig erfolgen.
2. Bestimmen Sie die gegenseitige Löslichkeit von n-Butylacetat und Wasser durch Trübungs-Titration (siehe Kapitel 3.2.1) • Vorlage von ca. 100 g n-Butylacetat (genau auswiegen) und titrieren mit Wasser (Dichte 1 kg/l), • Vorlage von ca. 100 g Wasser (genau auswiegen) und titrieren mit n-Butylacetat
(Dichte des n-Butylacetats mit Hilfe der Mohr-Westphal´schen Waage bestimmen *) ), • rechnen Sie die Ergebnisse in Gew-% aus, tragen Sie sie in das Dreiecksdiagramm ein und dis-
kutieren Sie evtl. vorhandene Abweichungen von den Literaturdaten.
3. Bestimmen Sie die Lage von zwei Konoden durch Trennung der Phasen (siehe Kapitel 3.2.2), • setzen Sie jeweils ca. 100 g der beiden vom Assistenten angegebenen Gemische in einem ver-
schließbaren Messzylinder an (die Konzentration ist nur ungefähr einzuhalten, muss aber exakt ausgewogen werden), stellen Sie das Phasengleichgewicht durch Schütteln her und lesen Sie nach der Phasentrennung die Volumina ab.
• Einpassung in die Binodalkurve, • bestimmen Sie die Dichte der Phasen mit Hilfe der Mohr-Westphal´schen Waage *), • ermitteln Sie das Ergebnis nach Kapitel 3.2.2.1, zeichnen Sie es in das Dreiecksdia-
gramm ein und diskutieren Sie evtl. vorhandene Abweichungen von den Literaturdaten.
• Analyse der Essigsäure in beiden Phasen, • titrieren Sie genau eingewogene Proben beider Phasen mit 0,1n-Natronlauge **), • ermitteln Sie das Ergebnis nach Kapitel 3.2.2.2, zeichnen Sie es in das Dreiecksdia-
gramm ein und diskutieren Sie evtl. vorhandene Abweichungen von den Literaturdaten.
*) Die Gebrauchsanweisung für die Mohr-Westphal´sche Waage hängt im Praktikumsraum aus.
**) Die Probemenge für die Titrationen ist so zu wählen (Vorabschätzung der Konzentration z.B. aus dem Gleich-gewichtsdiagramm), dass etwa 10 ml an 0,1 n-Natronlauge verbraucht werden (Titration notfalls wiederholen)

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 20 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
4. Bestimmen Sie in dem noch vorhandenen Raffinat aus der letzten Versuchsdurchführung den Essig-säuregehalt durch Titrieren einer genau eingewogenen Probe mit 0,1n-Natronlauge, • ermitteln Sie daraus mit Hilfe der Binodalkurve die Gesamtzusammensetzung , • entnehmen Sie am Dreiwegehahn des Feedbehälters den Rest der Zulauflösung , bestimmen Sie
dessen Masse und titrieren Sie dessen Essigsäure-Konzentration mit 0,1 n-Natronlauge (der Wassergehalt der Rest-Feedmenge soll vernachlässigt werden).
5. Setzen Sie aus dem alten Raffinat und der Rest-Feedmenge unter Verwendung der in Punkt 4 gewon-nenen Analysendaten durch Zuwiegen von Essigsäure und n-Butylacetat insgesamt ca. 3 kg neues Feed-Gemisch (genau auswiegen) mit einem Konzentrationsverhältnis Essigsäure : n-Butylacetat = 40 : 60 Gew-% an, kontrollieren Sie die Essigsäure-Konzentration durch Titration und füllen Sie das Gemisch in den Zulaufbehälter ein, • zeichnen Sie den Punkt für das Feed-Gemisch in das Dreiecksdiagramm ein.
6. Lassen Sie den Zulaufbehälter für das Lösungsmittel Wasser am Dreiwegehahn ab und füllen Sie eine genau bekannte Menge Wasser ein; leeren Sie die Raffinat- und Extrakt-Sammelbehälter.
7. Führen Sie die Stufenkonstruktion für vier theoretische Extraktionsstufen gemäß Kapitel 2.8 durch, um das erforderliche Lösungsmittel / Feed-Verhältnisses und die Extrakt-Endkonzentration E 1 zur Erzielung der vom Assistenten angegebenen Raffinat-Endkonzentration R n zu ermitteln
(Feed-Gemisch siehe Punkt 5, einen Startwert für das L / F-Verhältnis erhalten Sie vom Assistenten).
8. Nehmen Sie nach Absprache mit dem Assistenten die vierstufige Labor-Extrationsanlage mit dem ermittelten Lösungsmittel / Feed-Verhältnis in Betrieb, • überprüfen und korrigieren Sie zunächst die Dosierpumpen *) • schreiben Sie die Uhrzeit der Anlageninbetriebnahme und aller weiteren Aktionen auf.
9. Nach ca. 2 Stunden wird die erste Raffinatprobe (Raffinat 4) mit Hilfe einer Injektionsspritze mit PTFE-Schlauch aus dem dazu vorgesehenen Gefäß gezogen. 2 - 3 g der Probe werden auf 0,001 g genau in einen Erlenmeyerkolben eingewogen, mit Wasser verdünnt und mit 0,1 n NaOH gegen Phenolphthalein titriert. Die nächsten R4-Proben werden im Abstand von 20 Minuten gezogen.
Weichen zwei nacheinander gezogene Proben um weniger als 5% (relativ) in ihrer Essigsäure-Konzentration voneinander ab, so gilt das Gleichgewicht als erreicht.
10. Stellen Sie die Anlage ab, damit sich durch die folgenden Probenahmen keine Konzentrationsver-schiebungen mehr ergeben können (Zeit aufschreiben).
11. Das tatsächlich erreichte Verhältnis von Lösungsmittel / Feed, das für die Konstruktion im Dreiecks-diagramm maßgebend ist, wird aus den verbrauchten Mengen berechnet. Dazu wird der in beiden Vorratsbehältern (Feed und Lösungsmittel) verbliebene Rest in ein Gefäß abgelassen, genau ausge-wogen und aus der Differenz zur eingefüllten Menge der Verbrauch bestimmt (Feed wieder in den Vorratsbehälter geben ! ).
12. Bestimmen Sie die insgesamt entstandenen Mengen an Raffinat und Extrakt durch Wiegen.
Das Raffinat muss im dafür vorgesehenen Behälter im Abzug für den nächsten Versuch aufbewahrt werden. Das Extrakt wird in den Abfall-Sammelbehälter gegeben.
*) Zunächst ist darauf zu achten, dass alle Leitungen luftfrei sind (ggf. entlüften). Wenn Luft in eine Pumpe geraten
ist, kann die Förderung zusammenbrechen. In diesem Fall muss das Druckhalteventil oben auf der Pumpe, das durch einen konstanten Gegendruck die Fördergenauigkeit erhöht, kurz entlastet werden (Assistenten fragen). Die Förderleistung der eingesetzten Magnet-Membranpumpen ist durch die Taktfrequenz (beide Zulaufpumpen sind gekoppelt) und die Hubhöhe gegeben. Die Fördermenge soll jeweils bei ca. 1000 g/h liegen. Das L / F-Verhältnis lässt sich über die Hubhöhe variieren (großer Drehknopf hinter der Klappe). Zur Kontrolle der Fördermenge wird der Weg zur Anlage geschlossen und der seitliche Ablauf geöffnet, wo die Flüssigkeit aufgefangen und gewogen und somit der Förderstrom bestimmt werden kann (sinnvolle Messzeit wählen). Es ist unbedingt darauf zu achten, dass bei laufenden Pumpen weder beide Wege verschlossen werden (Pumpen werden beschädigt) noch beide We-ge geöffnet werden (Anlage läuft leer). Beachten Sie: eine zu starke Änderung der Betriebsbedingungen kann zu solchen Störungen in der Anlage führen (Verschiebung aller Phasen), dass die Versuchszeit nicht mehr ausreicht.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 4 Seite: 21 Chemieingenieurwesen Flüssig-Flüssig-Extraktion Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
13. Aus jedem Scheider werden durch die dazu vorgesehenen Probeöffnungen mit Hilfe von (trockenen !) Injektionsspritzen mit PTFE-Schlauch Proben zur Konzentrationsbestimmung von Raffinat (1 - 4) und Extrakt (1 - 4) gezogen. Diese 8 Proben werden titrimetrisch auf ihre Essigsäurekonzentration untersucht. Es sind Doppelbestimmungen mit Mittelwertbildung auszuführen, wobei die Titrationen bei mangelhafter Übereinstimmung (schlechter als 5 % relativ) zu wiederholen sind.
14. Führen Sie die theoretische Stufenkonstruktion mit dem tatsächlich erreichten Lösungsmittel / Feed-Verhältnis (siehe Punkt 11) erneut durch, • tragen Sie die gemessenen Raffinat- und Extrakt-Konzentrationen in das Dreiecksdiagramm ein, • bilanzieren Sie die kontinuierliche Gegenstromextraktion anhand aller gemessenen Mengen-
ströme und Analysenergebnisse (L, F, R 4, E 1); die theoretischen und praktischen Verhältnisse L / F und R 4 / E 1 sind zu vergleichen.
Diese Ergebnisse (Punkt 14) sind bereits am Ende des Versuches vorzulegen.
15. Erstellen Sie ein Verteilungsdiagramm und zeichnen Sie die Gleichgewichtskurve ein, • übertragen Sie sowohl die Daten der theoretischen Stufenkonstruktion wie auch die gemessenen
Daten in das Verteilungsdiagramm und zeichnen Sie die sich daraus ergebenden Arbeitslinien ein,
• berechnen Sie die Wirkungsgrade der einzelnen Stufen für die kontinuierliche Gegenstrom-extraktion.
16. Zur Versuchsdiskussion sind alle gezeichneten Diagramme mit qualitativen Angaben zu den Kurven-verläufen zu diskutieren. Weiterhin sind die berechneten Wirkungsgrade zu diskutieren.
Nomenklatur
BuAc = n-Butylacetat HAc = Essigsäure
C = Extraktstoff E = Extrakt F = Zulauf, Feed L = Lösungsmittel M = Mischpunkt
P = kritischer Punkt R = Raffinat T = Trägerstoff
m = Masse [kg , g] n = Anzahl der Extraktionsstufen
η = Wirkungsgrad ρ = Dichte [g / ml]
Literatur:
[1] Ullmann's Encyclopedia of Industrial Chemistry, Chapter: „Liquid-liquid extraction“, aktuelle elektronische Ausgabe über die Homepage der Universitätsbibliothek zugänglich.
letzte deutsche Ausgabe in Buchform: Ullmanns Encyklopädie der technischen Chemie, 4. Auflage, Band 2 „Verfahrenstechnik I, Grundoperationen“, Kapitel „Flüssig-flüssig-Extraktion“, Verlag Chemie, Weinheim, 1972
[2] Gmehling, J.; Brehm, A.: „Lehrbuch der Technischen Chemie“, Band 2 „Grundoperationen“, Georg Thieme Verlag, Stuttgart, 1996
[3] Sattler, K.: „Thermische Trennverfahren“, VCH Verlagsgesellschaft mbH, Weinheim, 1995
[4] Schlünder, E., Thurner, F., „Destillation, Absorption, Extraktion“, Georg Thieme Verlag, Stuttgart, 1986
[5] Sørensen, J. M.; Arlt, W.: „Liquid-liquid equilibrium data collection“, Chemistry Data Series, Vol. V, DECHEMA Frankfurt am Main, ab 1979
[6] Lohmann, J.; Joh, R.; Nienhaus, B,; Gmehling, J.: „Verbesserung der Vorhersagequalität und Erwei-terung der Anwendungsgebiete der Gruppenbeitragsmethode Modified UNIFAC (Dortmund)“,
Chem. Ing. Tech. 70 (1998) 138-142
[7] Welzenbacher, U., „Neue Datenblätter für gefährliche Arbeitsstoffe nach der Gefahrstoffverordnung“, WEKA Verlag, Augsburg 1994