Embed Size (px)
DESCRIPTION
Emerging evidence suggests that immunotherapy may play an important role in treating prostate cancer.
Citation preview

Editor-in-Chief, James O. Armitage, MD | ASCOPost.com
Our Aging Population: Challenges in Caring for Older Patients With CancerA Conversation With Hyman Muss, MDBy Ronald Piana
Managing older-aged cancer patients represents one of the major challenges to our health-care
system. Caring for older cancer patients, with their fre-quent multiple morbidities and a variable health status, requires special integration of an oncologic and geriat-ric approach. Moreover, our aging population will pose serious challenges to an already overtaxed cancer deliv-
ery system and will intensify the need for oncologists to be optimally trained to manage their older patients. Over the past 20 years, the oncology community has seen the development of geriatric programs and a fo-cus on research in older cancer patients. To shed light on the state of geriatric oncology, The ASCO Post spoke with Hyman Muss, MD, a leader in the field.
Practicing Like ‘Mini-Geriatricians’
During your career in geri-atric oncology, what has been the most significant advance?
Older patients, at least in affluent Western countries, are the majority of patients who have cancer, and the numbers are continuing to
Genitourinary Cancers Symposium
H ealth-care fraud is a long-standing prob-lem in the United States, accounting for
$75 billion in government expenses per year,1 while total spending on government health-care programs is over $1 trillion. Two decades ago, the Department of Justice increased its ef-forts to combat health-care fraud. This change was stimulated by the Federal False Claims Act, a 1986 legislation that allows qui tam re-lators (commonly termed “whistle-blowers”) to receive up to 30% of financial recoveries from successfully concluded False Claims Act investigations.
In 1996, the Federal Health Care Fraud and
Health-Care Fraud and Abuse: Implications for
Oncology
By Z. Kevin Lu, PhD, Brian Chen, JD, PhD, Zaina Qureshi, PhD, MPH, Oliver Sartor, MD,
and Charles Bennett, MD, PhD, MPP
Oncology Meetings CoverageGenitourinary Cancers Symposium �����������������������������1, 3–6, 8–9GI Cancers Symposium ������������ 16, 18–19
Richard ‘Buz’ Cooper, MD, on Health-Care Resources �������������������������33Inside the Black Box ���������������������������������42Direct From ASCO ���������������������������47–50Five Questions to Guide Myeloma Care ��������������������������������������������59Issues With Herbal Supplements ������������76Patient’s Corner: A Perspective ��������������89David Hui, MD, MSc, on End-of-Life Care ������������������������������������93
MORE IN THIS ISSUE
continued on page 37
Geriatrics for the Oncologist
Intensified Immunotherapy Encouraging in Chemotherapy-Naive Metastatic Prostate CancerBy Alice Goodman
Emerging evidence suggests that immunotherapy may play an important role in treating prostate
cancer. In particular, preliminary results have shown that combining a new vaccine with ipilimumab (Yervoy) boosts overall survival in men with castra-tion-resistant prostate cancer.1 A study comparing data from three independent trials of the vaccine won a Merit Award at the 2015 Genitourinary Cancers Symposium.
Rilimogene galvacirepvec/rilimogene glafolivec (Prostvac) is a poxvirus-based, prostate-specific vaccine being developed by Bavarian Nordic in partnership with the National Cancer Institute (NCI). Ipilimumab, an immune checkpoint inhibitor, is approved for the treat-ment of melanoma and is being evaluated in a phase III trial in metastatic castration-resistant prostate cancer.
“On the heels of the success of immunotherapy in metastatic melanoma, immunotherapy is an active area of study in prostate and other cancers. The approach in
this phase I study was to prime the immune cells with [the vaccine] and then intensify immunotherapy with ipilimumab to further enhance the immune re-sponse,” said lead author Harpreet Singh, MD, of the National Cancer Institute.
Overall Survival Benefit
Two previous phase II trials by the same group looked at the vaccine alone and found an overall sur-vival benefit in men with metastatic castration-resistant prostate cancer. The first trial, which included 125 men, found a median overall survival of 25.1 months. The second trial, which included 32 men, showed a median
Harpreet Singh, MD
Managing Bladder Cancer 6, 8–9 | Hypofractionated Breast Irradiation 20, 23, 24 | FDA Update 14, 66–67 VOLUME 6, ISSUE 5MARCH 25, 2015
continued on page 4 continued on page 97
The most important advance in geriatric oncology over the course of my career has been the recognition that older cancer patients need to be managed differently than their younger counterparts.
—Hyman Muss, MD
Author affiliations on page 97.
Disclaimer: This commentary represents the views of the authors and may not necessarily reflect the views of ASCO.
A Harborside Press® PublicationSend your comments to [email protected]
Charles Bennett, MD, PhD, MPP

PAGE 2 The ASCO Post | MARCH 25, 2015
Disclaimer: The ideas and opinions expressed in The ASCO Post™ do not necessarily reflect those of Harborside Press®, LLC, HSP News Service, LLC, or the American Society of Clinical Oncology, Inc. (ASCO®). The mention of any product, service, or therapy in this publication should not be construed as an endorsement of the products mentioned. It is the responsibility of the treating physician or other health-care provider, relying on independent experience and knowledge of the patient, to determine the appropri-ate treatment for the patient. Readers are advised to check the appropriate medical literature and the product information currently provided by the manufacturer of each product or therapy to be administered to verify the dosage, method, and duration of administration, or contraindications. Readers are also encouraged to contact the manufacturer with questions about the features or limitations of any products. Harborside Press®, HSP News Service, LLC, and ASCO® assume no responsibility for any injury or damage to persons or property arising out of or related to any use of material contained in this publication or to any errors or omissions.
James O. Armitage, MD Editor-in-Chief
Elizabeth Reed, MD Deputy Editor University of Nebraska Medical Center
Associate EditorsJame Abraham, MD Cleveland Clinic
Manmeet Ahluwalia, MD, FACP Cleveland Clinic
Joseph S. Bailes, MD Texas Oncology
Laurence H. Baker, DO University of Michigan Health System
Richard R. Barakat, MD Memorial Sloan Kettering Cancer Center
Charles L. Bennett, MD, PhD, MPP University of South Carolina, Columbia
Douglas W. Blayney, MD Stanford University Medical Center
Philip D. Bonomi, MD Rush University Medical Center
Richard Boxer, MD University of Wisconsin School of Medicine
Harold J. Burstein, MD Dana-Farber Cancer Institute
Robert W. Carlson, MD National Comprehensive Cancer Network
Barrie R. Cassileth, PhD Memorial Sloan Kettering Cancer Center
Jay S. Cooper, MD Maimonides Medical Center
John Cox, DO Texas Oncology
E. David Crawford, MD University of Colorado
Nancy E. Davidson, MD University of Pittsburgh Cancer Institute
George D. Demetri, MD Dana-Farber Cancer Institute
Paul F. Engstrom, MD Fox Chase Cancer Center
David S. Ettinger, MD Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Bishoy Morris Faltas, MD Weill Cornell Medical College
John A. Fracchia, MD New York Urological Associates
Alison Freifeld, MD University of Nebraska Medical Center
Louis B. Harrison, MD Moffitt Cancer Center
Jimmie C. Holland, MD Memorial Sloan Kettering Cancer Center
Clifford A. Hudis, MD, FACP Memorial Sloan Kettering Cancer Center
Nora Janjan, MD, MPSA, MBA National Center for Policy Analysis
Hagop M. Kantarjian, MD MD Anderson Cancer Center
Mario E. Lacouture, MD Memorial Sloan Kettering Cancer Center
Theodore S. Lawrence, MD, PhD University of Michigan Comprehensive Cancer Center
Stephen J. Lemon, MD, MPH Oncology Associates, PC, Omaha
Michael P. Link, MD Stanford University Medical Center
John L. Marshall, MD Ruesch Center for the Cure of GI Cancer at Georgetown University
Mary S. McCabe, RN, MA Memorial Sloan Kettering Cancer Center
William T. McGivney, PhD Philadelphia, Pennsylvania
James L. Mulshine, MD Rush University Medical Center
Derek Raghavan, MD, PhD Levine Cancer Institute Carolinas HealthCare System
Steven T. Rosen, MD City of Hope National Medical Center
Lee S. Schwartzberg, MD University of Tennessee Health Science Center
Andrew D. Seidman, MD Memorial Sloan Kettering Cancer Center
Samuel Silver, MD, PhD University of Michigan Health System
George W. Sledge, MD Indiana University
Thomas J. Smith, MD Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Jamie Von Roenn, MD American Society of Clinical Oncology
Lynn D. Wilson, MD Yale University School of Medicine
Stanley H. Winokur, MD Singer Island, Florida
William C. Wood, MD Winship Cancer Institute, Emory University
International EditorsClement Adebamowo, BM, ChB (Hons), ScD University of Ibadan, Nigeria
Eduardo Cazap, MD, PhD International Union Against Cancer (UICC) Buenos Aires, Argentina
Rakesh Chopra, MD Artemis Health Institute Gurgaon, India
Nagi El-Saghir, MD American University of Beirut, Lebanon
Mary Gospodarowicz, MD Princess Margaret Hospital Toronto, Ontario, Canada
Jacek Jassem, MD Medical University of Gdansk, Poland
David Khayat, MD Pitie-Salpetriere Hospital, Paris, France
Tony Mok, MD The Chinese University of Hong Kong Shatin, Hong Kong
Eliezer Robinson, MD National Council for Oncology Israeli Cancer Association, Haifa, Israel
Nagahiro Saijo, MD, PhD Kinki University School of Medicine Osaka, Japan
John F. Smyth, MD University of Edinburgh Edinburgh, Scotland
Daniel A. Vorobiof, MD Sandton Oncology Centre Johannesburg, South Africa
Harborside Press® Publishing Staff Conor Lynch, Executive Editor [email protected]
Cara H. Glynn, Director of Editorial [email protected]
Andrew Nash, Associate Director of Editorial [email protected]
Jo Cavallo, Senior Editor and Correspondent [email protected]
Randi Londer Gould and Susan Reckling, Senior Editors [email protected] [email protected]
Sarah McGullam, Web Editor [email protected]
Michael Buckley, Art Director [email protected]
Regine M. Lombardo, Senior Graphic Designer [email protected]
Terri Caivano, Layout Artist [email protected]
Gail van Koot, Editorial Coordinator [email protected]
Elizabeth Janetschek, Editorial Assistant [email protected]
Norman Virtue, Production Manager [email protected]
Shannon Meserve, Circulation Manager [email protected]
Jeannine Coronna, Vice President, Director of Operations [email protected]
Frank Buchner, Chief Technology Officer [email protected]
Leslie Dubin, Vice-President, Director of Sales [email protected]
Anthony Cutrone, President [email protected]
John A. Gentile, Jr, Chairman [email protected]
Contributing Writers:Charlotte Bath, Margot Fromer, Alice Goodman, Caroline Helwick, Susan London, Caroline McNeil, Eileen O’Gara-Kurtis, Ronald Piana, Matthew Stenger, Marian Wiseman
Contributing Artists: Portraits by Keith Witmer, Keith Witmer Illustrations.
Disclosure information available at ASCOPost.com.
Editorial Board
The ASCO Post (ISSN 2154-3283), USPS Publicaton Number 6885, is published semi-monthly, except monthly in January by Harborside Press®, LLC, 37 Main Street, Cold Spring Harbor, NY 11724, under a license arrangement with the American Society of Clinical Oncology, Inc. (ASCO®). Periodicals Postage paid at Cold Spring Harbor, NY, and additional mailing offices.
Change of Address: Postmaster send address changes to The ASCO Post, c/o Harborside Press®, LLC, 37 Main Street, Cold Spring Harbor, NY 11724. ASCO Members: If you would like to cancel your subscription to The ASCO Post or need to update your mailing address, please visit your personalized page on ASCO.org. For personal-ized service, please contact ASCO Member Services at (888) 282-2552, (703) 299-0158, or via email at [email protected]. Non ASCO Members: To initiate or cancel a subscription or to update your mailing address, please email [email protected] or fax (631) 692-0805.
Copyright ©2015 by Harborside Press®, LLC. All rights reserved. Reproduction in whole or in part, in any form, without prior written permission of the publisher is pro-
hibited. For permission inquiries, contact [email protected].
Editorial Mission: The ASCO Post communicates timely in-formation to a broad audience of oncology specialists, help-ing to advance the highest quality multidisciplinary cancer care. The ASCO Post publishes highly validated coverage of cancer research and policy news, patient care and clinical practice issues, and thoughtful commentary from leaders in the field and others with an interest in clinical oncology.
Circulation: The ASCO Post is sent free of charge to ap-proximately 27,000 physicians and nurses, including all US-based ASCO members. Medical, surgical, pediatric, and gynecologic oncologists, hematologists, and he-matologist/oncologists in the United States who are not members of ASCO will be eligible for a complimentary subscription. ASCO members outside of the United States receive complimentary access to The ASCO Post online at www.ASCOPost.com.
Paid subscriptions to The ASCO Post are available for all other interested individuals. Individual Domestic: $300;
Canada: $436; Individual International: $575; Institutional Domestic: $370; Canada: $507; Institutional International: $645. Single Copy Domestic: $57; Canada: $65; Interna-tional: $72. Contact [email protected].
Correspondence: Address general inquiries to Harbor-side Press®, LLC, 37 Main Street, Cold Spring Harbor, NY 11724. Phone: 631.692.0800; Fax: 631.692.0805. Address editorial correspondence to James O. Armitage, MD, Edi-tor-in-Chief, c/o Cara Glynn, phone: 631.935.7654; e-mail: [email protected].
Advertising: For information on advertising rates, re-prints, or supplements, contact Leslie Dubin, phone: 631.935.7660; e-mail: [email protected].
Notice to Advertisers: Advertiser and advertising agency recognize and accept that the following language ap-pears within the publication: “All statements, including product claims, are those of the person or organization making the statement or claim. Neither the publisher nor ASCO adopts any such statement or claim as its own, and any such statement or claim does not necessarily reflect the opinion of the publisher or ASCO.”
Advertiser and advertising agency accept and assume li-ability for all content (including text, representations, il-lustrations, opinions, and facts) of advertisements print-ed, and also assume responsibility for any claims made against the publisher or ASCO arising from or related to such advertisements. In the event that legal action or a claim is made against the publisher or ASCO arising from or related to such advertisements, advertiser and advertising agency agree to fully defend, indemnify, and hold harmless the publisher and ASCO, and to pay any judgment, expenses, and legal fees incurred by the publisher and by ASCO as a result of said legal action or claim. The publisher reserves the right to reject any advertising that it believes is not in keeping with the publication’s standards.
The publisher is not liable for delays in delivery and/or non-delivery in the event of Act of God, action by any government or quasi-governmental entity, fire, flood, in-surrection, riot, explosion, embargo, strikes (whether legal or illegal), labor or material shortage, transportation inter-ruption of any kind, work slow-down, or any condition be-yond the control of the publisher affecting production or delivery in any manner.

ASCOPost.com | MARCH 25, 2015 PAGE 3
Genitourinary Cancers Symposium
No Survival Benefit Reported With Docetaxel Added to Hormone Therapy in Metastatic Prostate Cancer By Alice Goodman
Docetaxel added to androgen-de-privation therapy did not improve
overall survival over androgen-depriva-tion therapy alone in hormone-naive met-astatic prostate cancer, according to an updated analysis of the GETUG-AFU 15 trial presented at the 2015 Genitourinary Cancers Symposium.1 In a retrospective analysis component of the trial, docetaxel
provided an additional 14 months of sur-vival overall and a 4-month difference in patients with high-volume disease, but neither was statistically significant.
“We wanted to see whether docetaxel in an earlier setting could improve survival in metastatic cancer,” said presenting au-thor Gwenaelle Gravis, MD, of the Insti-tut Paoli-Calmettes, Marseille, France.
Study DetailsGETUG-AFU 15 enrolled 385 pa-
tients between October 2004 and Decem-ber 2008 and randomized them to receive either androgen-deprivation therapy plus docetaxel or androgen-deprivation thera-py alone (luteinizing hormone-releasing hormone agonist or maximum androgen blockade or bilateral orchiectomy). The study was conducted at 30 centers (29 in France and 1 in Belgium).
The patients’ median age was 63 years. Fifty-eight percent had a Gleason score of ≥ 8. Most cases were metastatic at diagnosis: 76% in the docetaxel arm and 67% in the androgen-deprivation therapy alone arm. The median follow-up was 50 months.
In the original analysis, with a me-dian follow-up of 50 months, the time to disease progression was prolonged in the docetaxel arm, but median survival was not significantly different, Dr. Gra-vis noted. In the updated analysis, with a median follow-up of 80+ months, me-dian overall survival was 46.5 months (range = 39.1–60.6 months) for andro-gen-deprivation therapy alone vs 60.9 months (range = 46.1–71.4 months;
hazard ratio = 0.9 (0.7–1.2) for andro-gen-deprivation therapy plus docetaxel.
Results Differ From the CHAARTED Study
These results differed from those of the E2805 CHAARTED study present-ed by Christopher Sweeney, MD, at the 2014 ASCO Annual Meeting.2 That
study showed a 17-month overall sur-vival improvement with docetaxel added to androgen-deprivation therapy in men with high-volume disease, from 32.2 months to 49.2 months (P = .0006).
“In this retrospective analysis of GETUG-AFU 15, the majority of pa-tients were low volume, around 53%, and
80% of patients in the androgen-depriva-tion therapy–alone arm received docetax-el after castration resistance. As distinct from the CHAARTED study, we found that the median overall survival was not significantly different,” she said.
When patients in GETUG-AFU 15 were analyzed retrospectively according to disease volume, no significant differ-ence was observed for low-volume dis-ease between the two arms at a median follow-up of 81 months. Median overall survival was not reached with androgen-deprivation therapy alone and was 83.1 months for androgen-deprivation ther-apy plus docetaxel. In the high-volume
disease group, median overall survival was 35.1 months vs 39 months, respec-tively. This represented a nonsignificant 4-month difference favoring docetaxel at a median follow-up of 84 months.
In a multivariate analysis, prognostic factors included the extent of metasta-ses (high or low volume) and the level of alkaline phosphatase at baseline were independant prognostic factors for sur-vival. Treatment arm was not significant.
There was no difference in overall sur-vival in unselected patients with metastatic prostate cancer. In a retrospective analysis, no improvement was observed in patients
EXPERT POINT OF VIEW
“GETUG-AFU 15 sought to improve outcomes in meta-
static hormone-naive prostate cancer, but the study failed its primary objec-tive,” noted formal discussant Eric J. Small, MD, of the University of Cali-fornia, San Francisco.
In the overall analysis of this pre-viously published trial, with no en-richment for any group, there was a 14-month difference in overall sur-vival, which did not reach statistical significance, he reminded listeners.
However, the CHAARTED study
showed a highly statistically significant 13.6-month difference in survival with the addition of docetaxel to hormone therapy. This trial had more patients, and the benefit of early docetaxel was driven by high-volume patients, with no benefit in low-volume patients.
Potential reasons for the overall survival difference between the two trials include one-third fewer high-volume patients in the GETUG-AFU 15 trial and no enrichment for high-volume patients, Dr. Small said.
“The GETUG-AFU 15 trial is
underpowered, but results are direc-tionally consistent with the ECOG CHAARTED trial, and the discrep-ancy between these trials can be ex-plained,” he stated.
“In summary, high-volume disease does worse than low-volume disease and should be treated with androgen-deprivation therapy plus docetaxel. At this time, low-volume disease in hormone-sensitive prostate cancer should not be treated with docetaxel. Identification of biomarkers to pre-dict which patients may benefit from docetaxel is a research priority,” Dr. Small said.
Dr. Small looks forward to the results of the CALGB 90203 trial, which is comparing prostatectomy vs neoadjuvant androgen-deprivation therapy plus docetaxel followed by prostatectomy and includes correla-tive analysis of molecular tissue with clinical outcomes. n
Disclosure: Dr. Small reported no potential conflicts of interest.
High-volume disease does worse than low-volume disease and should be treated with androgen-deprivation therapy plus docetaxel.
—Eric J. Small, MD
Genitourinary Oncology
In a retrospective analysis, no improvement [in overall survival] was observed in patients with low-volume disease with the addition of docetaxel.
—Gwenaelle Gravis, MD
Updated Analysis of the GETUG-AFU 15 Trial
■ The results of the GETUG-AFU 15 study diverge from those of the E3805 CHAARTED study, which found a significant survival difference in high-volume metastatic prostate cancer when docetaxel was added to hormone therapy.
■ Several factors may be responsible for this difference, including the fact that GETUF-AFU 15 was underpowered to detect a significant difference in survival in men with high-volume disease.
■ Results of yet another trial in this setting are awaited, and then all three studies can be considered in determining the best course of treatment for hormone-sensitive chemotherapy-naive metastatic prostate cancer.
continued on page 4

PAGE 4 The ASCO Post | MARCH 25, 2015
Genitourinary Cancers Symposium
overall survival of 26.6 months. Both tri-als demonstrated improved overall sur-vival over that predicted by the Halabi nomogram (a model that uses histori-cal data to estimate survival of prostate cancer patients after castration): The first study showed an 8.5-month im-
provement in median overall survival, and the second had a similar 9.1-month improvement in median overall survival compared with predicted survival, Dr. Singh told The ASCO Post.
These studies led to an international phase III trial of the vaccine in men with metastatic castration-resistant prostate cancer and minimal symptoms who are chemotherapy-naive. That trial has just finished accrual.
“It is clear that vaccine therapy alone improves overall survival,” Dr. Singh stated.
Phase I TrialShe also presented updated phase I
results of a study of 30 patients with metastatic castration-resistant prostate cancer (and baseline characteristics similar to those in the phase II trials)
treated with the combination of the vaccine plus escalating doses of ipilim-umab. Follow-up was about 80 months.
The predicted survival of chemo-therapy-naive patients with metastatic castration-resistant prostate cancer is 18.5 months on the Halabi nomogram, she said. The updated overall survival analysis showed a median overall sur-
vival of 31.3 months using the inten-sified immunotherapy approach. The most impressive results were seen in the group receiving the highest dose of ipil-imumab (10 mg/kg) plus the vaccine: a
median overall survival of 37.2 months.“There is a tail in the curve. About
20% of these patients on the high-est dose of ipilimumab are alive at 80 months,” Dr. Singh said.
Hypothesis-Generating Data“These hypothesis-generating data
suggest it is rational to combine vac-cine and immunotherapy. Anti–PD-1 or anti–PD-L1 immunotherapies [anti-bodies against programmed cell death protein 1 or its ligand] are of interest in this regard. They appear to have less toxicity than ipilimumab. If we com-bine the vaccine with one of these
agents, we may improve outcomes,” she stated.
Many others in the field of prostate cancer are excited about the promise of immunotherapy. In the Genitouri-nary Cancers Symposium Daily News, L. Michael Glodé, MD, University of Colorado at Denver, wrote: “The re-markable advances recently reported in melanoma [with immunotherapies] … provide hope that we may be only in the earliest phase of truly effective treat-ment of metastatic prostate cancer, with
the ultimate goal of long-term remis-sions or even immunologically driven ‘cure.’”2
Dr. Glodé called the trial results reported by Dr. Singh “promising” and suggested that using the new vac-cine could be more cost-effective than sipuleucel-T (Provenge), a vaccine ap-proved in the predocetaxel setting. n
Disclosure: Dr. Singh reported no potential conflicts of interest. Dr. Glodé and colleagues received research funding for the phase II trial of Prostvac from Bavarian Nordic.
References1. Singh H, Madan RA, Dahut WL, et al:
Combining active immunotherapy and im-mune checkpoint inhibitors in prostate can-cer. 2015 Genitourinary Cancers Symposium. Abstract 172. Presented February 26, 2015.
2. Glodé LM: Immunotherapy for pros-tate cancer: Past, present, and future. 2015 Genitourinary Cancers Symposium Daily News, February 27, 2015.
Intensified Immunotherapycontinued from page 1 EXPERT POINT OF VIEW
ASCO Expert and GU News Planning Team Member, Charles Ryan, MD, of the Univer-
sity of California, San Franciso, Helen Diller Fam-ily Comprehensive Cancer Center, said the results presented by Singh et al at the 2015 Genitourinary Cancers Symposium were “encouraging” and that it makes sense to exploit two complementary arms of the immune system: The vaccine initiates or “jump starts” response to the tumor, and then active im-munotherapy enhances the durability of response.
Dr. Ryan noted that the Halabi nomogram was developed before the availability of newer therapies like abiraterone (Zytiga) and enzalutamide (Xtandi), so projected survival using that nomogram is probably not appli-cable in the modern era.
The rilimogene galvacirepvec/rilimogene glafolivec (Prostvac) vaccine has several potential advantages over sipuleucel-T (Provenge), Dr. Ryan commented. “Prostvac is off the shelf, whereas sipuleucel-T relies on har-vesting enough of the patient’s own immune cells. Thus, there is greater po-tential for a uniform product with Prostvac,” he said. “It is also possible that Prostvac will be less expensive, but that [remains to be seen].”
Regarding the approach of combining the new vaccine with anti–PD-L1, Dr. Ryan said it is possible that an anti–PD-L1 agent would be safer than ipilimumab (Yervoy), “but this would require further validation in prostate cancer. Ipilimumab is better studied in prostate cancer than anti–PD-L1.” n
Disclosure: Dr. Ryan reported no potential conflicts of interest.
Charles Ryan, MD These hypothesis-generating data suggest it is rational to combine vaccine and immunotherapy. Anti–PD-1
or anti–PD-L1 immunotherapies are of interest in this regard…. If we combine the vaccine with one of these
agents, we may improve outcomes. —Harpreet Singh, MD
Immunotherapy in Advanced Prostate Cancer
■ A new prostate-specific cancer vaccine (rilimogene galvacirepvec/rilimogene glafolivec [Prostvac]) has been shown to improve survival in men with chemotherapy-naive metastatic prostate cancer.
■ The new vaccine is undergoing phase III testing.
■ Preliminary evidence suggests that combining the vaccine with an immune checkpoint inhibitor can further extend survival in chemotherapy-naive metastatic prostate cancer.
with low-volume disease with the addi-tion of docetaxel, and a nonsignificant dif-ference of 4 months was observed in the high-volume disease group.
Possible explanations for the differ-ence between the current results and those in the CHAARTED study include
the increased use of salvage docetaxel in GETUG-AFU 15 and that the study was underpowered to detect a difference in the 183 patients with high-volume disease (91 on androgen-deprivation therapy alone and 92 on androgen-deprivation therapy plus docetaxel).
When available, results of the STAMPEDE trial will be considered in the context of both CHAARTED and
GETUG-AFU 15 to determine the best course of treatment, she said. n
Disclosure: Dr. Gravis has served as a consultant or advisor to and traveled on behalf of Sanofi.
References1. Gravis G, Boher JM, Joly F, et al: An-
drogen deprivation therapy plus docetaxel versus ADT alone for hormone-naive meta-static prostate cancer: Long-term analysis
of the GETUG-AFU 15 phase III trial. 2015 Genitourinary Cancers Symposium. Abstract 140. Presented February 26, 2015.
2. Sweeney C, Chen, Y-H, Carducci MA, et al: Impact on overall survival with chemohormonal therapy versus hormonal therapy for hormone-sensitive newly meta-static prostate cancer: An ECOG-led phase III randomized trial. 2014 ASCO Annual Meeting. Abstract LBA2.
Docetaxel Plus Hormone Therapycontinued from page 3
L. Michael Glodé, MD

ASCOPost.com | MARCH 25, 2015 PAGE 5
Genitourinary Cancers Symposium
High-Dose Radiotherapy Provides Meaningful Improvements in Localized Prostate Cancer—But Not Increased Survival YetBy Alice Goodman
High-dose radiotherapy failed to improve overall survival at 7 years
compared with standard-dose radio-therapy in men with stage II localized prostate cancer. However, the high-dose schedule had several advantages, includ-ing improved rates of local tumor control and distant metastasis, according to the
results of the phase III Radiation Ther-apy Oncology Group (RTOG) 0126 study1 presented at the 2015 Genitouri-nary Cancers Symposium.
“We were disappointed that we failed to see an improvement in over-all survival,” said lead author Jeff M. Michalski, MD, of Washington Uni-versity School of Medicine, St. Louis. “Dose escalation did not significantly decrease the rate of death from prostate cancer. However, dose escalation does offer some valuable outcomes, just not improvement in survival,” he added.
Study DetailsAt a median follow-up of 7 years, the
5-year overall survival rates were 88% and 89% in the high-dose and standard radiotherapy arms, respectively, and 10-year overall survival rates were 67% and 66% in the high-dose and standard arms, respectively. These differences were not statistically significant.
RTOG 0126 evaluated whether a
79.2-Gy dose of three-dimensional conformal radiotherapy (3D-CRT) or intensity-modulated radiotherapy improved overall survival compared with the standard 3D-CRT or inten-sity-modulated radiotherapy dose of 70.2 Gy. The 79.2-Gy dose was chosen for the trial because it had been previ-
ously identified as feasible and associ-ated with fewer side effects than other dose schedules, Dr. Michalski said.
A total of 1,532 patients were ran-domized 1:1 to receive either 44 frac-tions of high-dose radiotherapy or 39 fractions of standard radiotherapy. The median age of patients was 69 years, and 66% received 3D-CRT. No patient received hormone therapy.
Meaningful ImprovementsHigh-dose radiotherapy did show
improvements over standard therapy regarding the rates of biochemical failure, distant metastasis, and time to local tumor progression. Ten-year rates of biochemical failure were 30% and 45% in the high-dose and stan-dard radiotherapy arms, respectively (P < .0001).
Ten-year rates of time to local tu-mor progression were 4% and 8% in the high-dose and standard radiotherapy arms, respectively (P = .0059), and the
10-year rates of distant metastasis were 5% and 8% in the high-dose and stan-dard arms, respectively (P = .026).
Less salvage therapy was used in the high-dose radiotherapy arm (13.5% vs 20.6%, P = .0002).
High-dose radiotherapy incurred greater late toxicity, including grade 2 or higher gastrointestinal and genito-urinary adverse events. Dr. Michalski said there are strategies that can reduce these side effects such as intensity-mod-ulated radiotherapy and careful atten-tion to rectal dosimetry.
Further study is planned to test
dose-escalated therapy. The RTOG 0815 trial, completing accrual, will eval-uate dose-escalated radiotherapy with or without a short course of androgen-deprivation therapy. n
Disclosure: Dr. Michalski reported no potential conflicts of interest.
Reference1. Michalski JM, Moughan J, Purdy J,
et al: A randomized trial of 79.2 Gy ver-sus 70.2 Gy radiation therapy for localized prostate cancer. 2015 Genitourinary Can-cers Symposium. Abstract 4. Presented February 26, 2015.
EXPERT POINT OF VIEW
Commenting on this study, formal discussant D. Andrew Loblaw, MD, a radiation oncologist at the Odette Cancer Centre, Sunnybrook
Health Sciences Centre, Toronto, Canada, said: “The trial showed that after a median follow-up of 7.0 years, there were no differences in overall survival between the two groups. This, however, is not surprising given the follow-up; one wouldn’t expect to see a difference in survival until at least a median of 13 years of follow-up. There was a clinically significant difference in biochemical disease-free survival, local tumor control, and a trend toward improvement in time to metastatic disease, all favoring the higher-dose group.”
Dr. Loblaw acknowledged the higher risk of moderate gastrointestinal and genitourinary toxicities associated with the higher-dose group. “The chance
of serious side effects in the high-dose group was only nominally higher in the gastrointestinal realm,” he said.
“Overall, this study is consistent with the five other randomized dose-es-calated external-beam studies—data that have already changed practice. Al-though very well conducted, we’ll have to wait another 7 years to see whether this study can address the question of whether five additional external-beam treatments improve overall survival,” Dr. Loblaw stated.
Dose escalation has already entered the realm of clinical practice. Accord-ing to Dr. Loblaw, in both Canada and Europe, doses higher than 74 Gy are standard, as per guidelines. “In the United States, the National Comprehen-sive Cancer Network Guidelines don’t specify dose, but all of the physicians I have interacted with offer dose escalation,” he said.
Disclosure: Dr. Loblaw reported no potential conflicts of interest.
High-Dose Radiotherapy in Localized Prostate Cancer
■ At a median follow-up of 7 years, high-dose radiotherapy did not improve survival over standard-dose radiotherapy in stage II localized prostate cancer, according to a phase III RTOG trial.
■ High-dose radiotherapy did improve the rates of local tumor control and distant metastasis.
■ Experts say that with longer follow-up, a survival difference may emerge.
■ Dose escalation has become common practice now in this setting.
Dose escalation did not significantly decrease the rate of death from prostate cancer. However, dose escalation does offer some valuable outcomes, just not improvement in survival.
—Jeff M. Michalski, MD
We’ll have to wait another 7 years to see whether this study can address the question of whether five additional external-beam treatments improve overall survival.
—D. Andrew Loblaw, MD
Genitourinary Oncology

PAGE 6 The ASCO Post | MARCH 25, 2015
Genitourinary Cancers Symposium
Antiangiogenesis Plus Chemotherapy Pursued in Advanced Bladder Cancer By Alice Goodman
Two separate phase II studies lend support to the concept of antiangio-
genesis in advanced bladder cancer. The combination of an antiangiogenic agent and chemotherapy may fulfill an unmet need in this disease, the studies suggest. Both studies were presented at the 2015 Genitourinary Cancers Symposium.
The first study offered proof of con-cept for using the combination of pazo-panib (Votrient) and weekly paclitaxel.1 However, for now GlaxoSmithKline has halted the development of pazopanib for bladder cancer. The second study was positive for the combination of ramu-cirumab (Cyramza) and docetaxel, and a phase III study comparing that com-bination with docetaxel alone has been mounted in the second-line setting.
“We are moving forward with the antiangiogenic approach [in bladder cancer]. These studies provide proof of concept that the target is worthy of pursuit,” said Matthew Galsky, MD, Director of Genitourinary Medical Oncology at Icahn School of Medicine at Mount Sinai, New York, who was not involved in either of these trials.
Dr. Galsky said that the first ran-domized phase III trial of antiangio-
genic therapies in this setting, CALGB 90601, has completed accrual and is ongoing. That trial is comparing gem-citabine/cisplatin plus or minus beva-cizumab (Avastin). “We are waiting for results of this trial. In addition, there may be other antiangiogenic drugs that will be found useful,” he added.
Paclitaxel Plus Pazopanib“Although platinum-based combi-
nation chemotherapy achieves good responses in metastatic bladder cancer, patients will recur, and effective treat-ment is an unmet need,” stated lead author of the phase II study Sandy Srinivas, MD, Stanford University School of Medicine, Palo Alto, California.
“Many drugs have been tested in metastatic disease, but none is ap-proved in the United States,” she added.
The combination of pazopanib plus weekly paclitaxel achieved high response
rates and was tolerable. Based on the pos-itive results, a phase III trial was mount-ed. In August 2014, GlaxoSmithKline halted development of pazopanib, leav-ing the combination treatment in limbo. It is possible that another drug company will pick this up and go forward, but for now, that is unknown.
The phase II study combined pazo-panib with weekly paclitaxel, based on data suggesting that this combination would be effective.
Patients with pretreated recurrent urothelial cancer were included in the study (n = 32). The median age was 67 years; patients were predominately male; 34% had upper tract tumors; 50% had two prior chemotherapy regimens; 28% had liver metastases. The median time from the last chemotherapy was less than 3 months in more than 50% of patients.
A two-stage design called for stop-ping the trial if no response was ob-served in the first nine patients, but
that did not happen. The most common adverse events
included fatigue, diarrhea, nausea/vom-iting, and neuropathy. There were few grade 3 and 4 adverse events. Febrile neutropenia occurred in 6%, and 44% of patients required growth factor support.
The overall response rate was 50%; there were 3 complete responses, 12
unconfirmed partial responses, and 11 confirmed partial responses. These re-sponse rates are encouraging compared with expected response rates in this set-ting, Dr. Srinivas said. One patient is still alive 55 months later, she added.
Responses were seen in both high- and low-risk patients. Dose modifica-tions of pazopanib were employed in the majority of patients.
Ramucirumab Plus DocetaxelInterim results of a randomized
phase II trial showed significant im-
provement in progression-free survival with ramucirumab plus docetaxel as second-line therapy for advanced meta-static urothelial carcinoma,2 supporting further evaluation of this combination.
“These patients have limited thera-peutic options,” said lead author Daniel Petrylak, MD, Yale University Cancer Center, New Haven, Connecticut.
“Interim results of the phase II trial support the initiation of a phase III registration trial called RANGE. This study will compare docetaxel and ramucirumab vs docetaxel plus place-bo in locally advanced or unresectable or metastatic urothelial cancer that has progressed on first-line platinum ther-apy,” he told listeners.
The open-label, multicenter, phase II trial conducted in the United States and Canada randomized 139 patients to one of three arms: docetaxel with ramu-
EXPERT POINT OF VIEW
Formal discussant of both trials, Jonathan Rosenberg, MD, of the Genitourinary Di-
vision at Memorial Sloan Kettering Cancer Cen-ter in New York, agreed that both studies move the field of antiangiogenesis in advanced bladder cancer forward.
“The very high overall response rates in the pazopanib plus paclitaxel study have not been seen in the second-line setting. The progression-free survival is also quite good. This study shows
that the combination of chemotherapy and antiangiogenesis inhibition may play a role in bladder cancer. Tolerability is an issue, since the majority of patients required dose reductions and 44% needed growth factor support,” Dr. Rosenberg said.
The second study had positive results, and the combination of ramuci-rumab plus docetaxel will be studied in phase III. “In this study, the median progression-free survival was very respectable,” Dr. Rosenberg continued.
“Both studies were done in previously treated metastatic bladder cancer. There is reason to be hopeful that antiangiogenesis plus chemotherapy will turn out to be a valid option in a disease with no approved therapies,” con-cluded Dr. Rosenberg. n
Disclosure: Dr. Rosenberg has served as a consultant to Eli Lilly.
Genitourinary Oncology
Jonathan Rosenberg, MD
Ramucirumab reduced the risk of disease progression by 61% compared with docetaxel.
—Daniel Petrylak, MD
Treating Advanced Bladder Cancer
■ In the United States, there are no approved drugs for metastatic bladder cancer that has progressed on first-line platinum-based chemotherapy, and thus it represents an unmet need.
■ Phase II studies in this setting showed encouraging response rates and progression-free survival for separate combinations of an antiangiogenic agent plus chemotherapy.
■ Both trials support further study of antiangiogenic therapies and chemotherapy.
■ The phase III RANGE study should shed some light on this issue.
Matthew Galsky, MD
Sandy Srinivas, MD
continued on page 8
Residual disease in myeloma goes deep.1-5
shouldn’t we stRive to go deepeR?
Growing evidence supports using a long-term treatment approach for continuous disease suppression and improved outcomes.6-8
Proteasomes regulate intracellular protein degradation. Their inhibition induces endoplasmic reticulum stress within myeloma cells and impacts support mechanisms within the bone marrow microenvironment.9-11
At Takeda, we’re committed to achieving a deeper understanding of multiple myeloma and helping to address the challenges patients face today and tomorrow.
Even after achieving complete response, myeloma cells can persist.1-5 It’s time to reconsider the way we approach myeloma:
Takeda Oncology and are registered trademarks of Takeda Pharmaceutical Company Limited.Other trademarks are the property of their respective owners.
Copyright © 2015, Millennium Pharmaceuticals, Inc. All rights reserved. Printed in the USA USO/NON/15/0002 January/15
ReFeRenCes: 1. Rawstron AC, Child JA, de Tute RM, et al. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the Medical Research Council Myeloma IX Study. J Clin Oncol. 2013;31(20):2540-2547. 2. Paiva B, Vidriales MB, Cerveró J, et al. Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood. 2008;112(10):4017-4023. 3. Biran N, Ely S, Chari A. Controversies in the assessment of minimal residual disease in multiple myeloma: clinical significance of minimal residual disease negativity using highly sensitive techniques [published online ahead of print September 16, 2014]. Curr Hematol Malig Rep. doi:10.1007/s11899-014-0237-y. 4. Munshi NC, Anderson KC. Minimal residual disease in multiple myeloma. J Clin Oncol. 2013;31(20):2523-2526. 5. Martinez-Lopez J, Lahuerta JJ, Pepin F, et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood. 2014;123(20):3073-3079. 6. Palumbo A, Niesvizky R. Sustained disease control in transplant-ineligible patients: the role of continuous therapy. Leuk Res. 2012;36(suppl 1):S19-S26. 7. Girnius S, Munshi NC. Challenges in multiple myeloma diagnosis and treatment. Leuk Suppl. 2013;2(suppl):S3-S9. 8. Palumbo A, Gay F, Musto P, et al. Continuous treatment (CT) versus fixed duration of therapy (FDT) in newly diagnosed myeloma patients: PFS1, PFS2, OS endpoints. J Clin Oncol. 32:5s, 2014 (suppl; abstr 8515). 9. Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4(5):349-360. 10. Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther. 2011;10(11):2034-2042. 11. Borrello I. Can we change the disease biology of multiple myeloma? Leuk Res. 2012;36(suppl 1):S3-S12.
IXAZ14CDNY7406_A_EMC_JrnlAd_KingSize_r6.indd 1 1/29/15 1:02 PM

Residual disease in myeloma goes deep.1-5
shouldn’t we stRive to go deepeR?
Growing evidence supports using a long-term treatment approach for continuous disease suppression and improved outcomes.6-8
Proteasomes regulate intracellular protein degradation. Their inhibition induces endoplasmic reticulum stress within myeloma cells and impacts support mechanisms within the bone marrow microenvironment.9-11
At Takeda, we’re committed to achieving a deeper understanding of multiple myeloma and helping to address the challenges patients face today and tomorrow.
Even after achieving complete response, myeloma cells can persist.1-5 It’s time to reconsider the way we approach myeloma:
Takeda Oncology and are registered trademarks of Takeda Pharmaceutical Company Limited.Other trademarks are the property of their respective owners.
Copyright © 2015, Millennium Pharmaceuticals, Inc. All rights reserved. Printed in the USA USO/NON/15/0002 January/15
ReFeRenCes: 1. Rawstron AC, Child JA, de Tute RM, et al. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the Medical Research Council Myeloma IX Study. J Clin Oncol. 2013;31(20):2540-2547. 2. Paiva B, Vidriales MB, Cerveró J, et al. Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood. 2008;112(10):4017-4023. 3. Biran N, Ely S, Chari A. Controversies in the assessment of minimal residual disease in multiple myeloma: clinical significance of minimal residual disease negativity using highly sensitive techniques [published online ahead of print September 16, 2014]. Curr Hematol Malig Rep. doi:10.1007/s11899-014-0237-y. 4. Munshi NC, Anderson KC. Minimal residual disease in multiple myeloma. J Clin Oncol. 2013;31(20):2523-2526. 5. Martinez-Lopez J, Lahuerta JJ, Pepin F, et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood. 2014;123(20):3073-3079. 6. Palumbo A, Niesvizky R. Sustained disease control in transplant-ineligible patients: the role of continuous therapy. Leuk Res. 2012;36(suppl 1):S19-S26. 7. Girnius S, Munshi NC. Challenges in multiple myeloma diagnosis and treatment. Leuk Suppl. 2013;2(suppl):S3-S9. 8. Palumbo A, Gay F, Musto P, et al. Continuous treatment (CT) versus fixed duration of therapy (FDT) in newly diagnosed myeloma patients: PFS1, PFS2, OS endpoints. J Clin Oncol. 32:5s, 2014 (suppl; abstr 8515). 9. Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4(5):349-360. 10. Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther. 2011;10(11):2034-2042. 11. Borrello I. Can we change the disease biology of multiple myeloma? Leuk Res. 2012;36(suppl 1):S3-S12.
IXAZ14CDNY7406_A_EMC_JrnlAd_KingSize_r6.indd 1 1/29/15 1:02 PM

PAGE 8 The ASCO Post | MARCH 25, 2015
Genitourinary Cancers Symposium
cirumab, docetaxel with icrucumab, or docetaxel alone. Patients were treated until disease progression. The icrucu-mab arm failed to show any meaningful benefit, and that drug is not expected to be pursued further in bladder cancer.
At an interim analysis, median pro-gression-free survival was 22 weeks with ramucirumab plus docetaxel and 10.4 weeks with docetaxel alone. Sub-
group analysis showed that all groups benefited from the combination.
Overall survival in the ramucirumab plus docetaxel arm was more than 11 months at the time of the analysis, but survival data are not yet mature. Again, this is considered to be better than that expected from historic controls.
“Ramucirumab reduced the risk of disease progression by 61% compared with docetaxel,” Dr. Petrylak said.
Toxicities that occurred in more than 5% of patients on the combination
arm were fatigue, febrile neutropenia, pneumonia, diarrhea, and stomatitis. n
Disclosure: Dr. Galsky has received research funding from Bristol-Myers Squibb, Novartis, Dendreon, Celgene, and Janssen; is on the adisory board of Merck, Lilly, Novartis, and Astellas; has served as a consultant for BioMotiv; and has stock in Dual Therapeutics. Dr. Srinivas reported no potential conflicts of interest. Dr. Petrylak has served as a consultant or advisor to and received research funding from Eli Lilly and Co, Genentech, and Sanofi and has travelled on behalf of Genentech and Sanofi.
References1. Srinivas S, Narayanan S, Harshman LC,
et al: Phase II study of pazopanib with weekly paclitaxel in refractory urothelial cancer. 2015 Genitourinary Cancers Symposium. Abstract 294. Presented February 27, 2015.
2. Petrylak DP, Tagawa ST, Kohli M, et al: Interim results of a randomized phase 2 study of docetaxel with ramucirumab ver-sus docetaxel in second-line advanced or metastatic urothelial carcinoma. 2015 Gen-itourinary Cancers Symposium. Abstract 295. Presented February 27, 2015.
Antiangiogenesis Plus Chemotherapycontinued from page 6
Adjuvant Chemotherapy Favored in Locally Advanced Bladder Cancer Not Treated With Neoadjuvant TherapyBy Alice Goodman
A large observational study presented at the 2015 Genitourinary Cancers
Symposium in Orlando, Florida, found that adjuvant chemotherapy extended the likelihood of survival in locally ad-vanced bladder cancer compared with observation alone.1 Using three different approaches to propensity scores adjust-ment for confounders, the authors found that adjuvant chemotherapy was associ-ated with a 28% to 31% higher probabil-ity of survival in patients with locally ad-vanced bladder cancer, postcystectomy, compared to observation alone.
“In a real-world population, adju-
vant chemotherapy provided a survival benefit in patients with locally advanced bladder cancer who had not been treated
with neoadjuvant chemotherapy. This level 2 evidence lends support to the use of adjuvant chemotherapy in locally
advanced bladder cancer patients who have not received neoadjuvant chemo-therapy—the current standard of care,” said lead author Matthew D. Galsky, MD, Associate Professor of Medicine and Urology at The Tisch Cancer Insti-tute, Icahn School of Medicine at Mount Sinai, New York. “These data can help inform shared decision-making.”
Poor AccrualDr. Galsky told the The ASCO Post
that three recent randomized trials of adjuvant chemotherapy in this setting have had poor rates of accrual. Of a to-tal of 1,610 planned patients, only 620 (39%) enrolled in these trials.
“I think accrual was poor, at least in part, because doctors think they know the answer. However, doctors are split on this issue, and so are trial results, so doctors may be biased toward favoring the results of one trial or another,” Dr. Galsky said.
In addition, the guidelines are split on this issue. The National Comprehen-sive Cancer Network Guidelines advo-cate for adjuvant chemotherapy (cat-egory 2B recommendation) for patients who have not received neoadjuvant therapy, whereas the European Associa-tion of Urology recommends against it.
“There are no sufficiently powered randomized trials to answer this ques-tion, and historical experience suggests such a trial cannot be done. We con-ducted our population-based observa-tional study to provide the next level of evidence,” Dr. Galsky said.
Study DetailsThe study was based on 5,653 pa-
tients with locally advanced urothelial bladder cancer enrolled on the National
Genitourinary Oncology
EXPERT POINT OF VIEW
Commenting on the study pre-sented by Galsky et al at the
Genitourinary Cancers Symposium, Matthew I. Milowsky, MD, Section Chief, Genitourinary Cancer, at the
Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, agreed with Dr. Galsky that it is unlikely that we will ever have an appropriately powered, prospective clinical trial to define the role of adju-vant therapy in bladder cancer.
“The study by Galsky et al is an ex-
tremely important contribution to the existing literature,” he stated. “Com-parative effectiveness studies are one mechanism by which to answer clini-cally important questions that we have
been unable to adequately address with randomized clinical trials.”
Further ConsiderationsAddressing some of the study’s
limitations, Dr. Milowsky said: “Al-though the National Cancer Data-base has limitations, it is a robust
source of hospital registry data, repre-senting approximately 70% of newly diagnosed cancer cases nationwide. The authors’ use of propensity score matching to determine the treatment effect while accounting for variables that predict for receiving the treat-ment and multiple imputation analy-sis to account for incomplete data sig-nificantly strengthens their findings.”
He added, “The conclusion that adjuvant chemotherapy was associ-ated with an improvement in survival in patients with ≥ pT3 and/or pN-positive bladder cancer with a hazard ratio of 0.72 is strikingly similar to the benefit that has been seen in meta-analyses and retrospective series. It is likely the right time to stop asking for another randomized clinical trial and recommend the use of adjuvant cisplatin-based chemotherapy in ap-propriately selected patients.”
Disclosure: Dr. Milowsky reported no potential conflicts of interest.
It is likely the right time to stop asking for another randomized clinical trial and recommend the use of adjuvant cisplatin-based chemotherapy in appropriately selected patients.
—Matthew I. Milowsky, MD
continued on page 9
The best available evidence supports use of neoadjuvant therapy in this setting. These data lend more support to consider adjuvant chemotherapy in appropriate patients who have not received neoadjuvant chemotherapy.
—Matthew D. Galsky, MD

ASCOPost.com | MARCH 25, 2015 PAGE 9
Genitourinary Cancers Symposium
Cancer Database who were treated with radical cystectomy between 2003 and 2007: 4,360 were treated with observa-tion after surgery, and 1,293 received adjuvant chemotherapy. These patients had not received neoadjuvant chemo-therapy or radiation to the primary tumor. Patients in the adjuvant chemo-therapy group received multiagent che-motherapy within 90 days of surgery.
The authors found that patients with positive lymph nodes were about twice as likely to receive adjuvant chemother-apy, and those with private insurance were 1.3 times more likely to be treated with adjuvant chemotherapy.
In addition to the main analysis, a subset analysis found that adjuvant chemotherapy was favored over ob-servation for all subsets, including age, gender, nodal status, and number of involved nodes. Study limitations included its retrospective design, the lack of details on the type of chemo-therapy, and no details on cancer-spe-cific survival.
Closing Thoughts“The best available evidence supports
use of neoadjuvant therapy in this setting. Our data lend more support to consider adjuvant chemotherapy in appropriate patients who have not received neoadju-vant chemotherapy,” Dr. Galsky stated.
Whether adjuvant chemotherapy should be used to treat a patient who
has had neoadjuvant chemotherapy and has residual cancer in the tumor speci-men is an issue not addressed by the current study. n
Disclosure: Dr. Galsky has received research funding from Bristol Myers, Novartis, Dendreon, Celgene, and Janssen; is on the adisory board of Merck, Lilly, Novartis, and Astellas; has served as a consultant for
BioMotiv; and has stock in Dual Therapeutics.
Reference1. Galsky MD, Stensland K, Moshier EL,
et al: Comparative effectiveness of adjuvant chemotherapy versus observation in patients with ≥ pT3 and/or pN+ bladder cancer. 2015 Genitourinary Cancers Symposium. Abstract 292. Presented February 27, 2015.
Locally Advanced Bladder Cancercontinued from page 8
Role of Adjuvant Chemotherapy in
Bladder Cancer
■ Guidelines differ on the appropriate use of adjuvant chemotherapy in locally advanced bladder cancer patients who have not received neoadjuvant chemotherapy—the current standard of care.
■ Randomized trials to address this issue have had poor accrual and are underpowered.
■ In the absence of level 1 data, a large, real-world, observational study tackled this issue and found that adjuvant therapy is likely to extend survival over observation alone in this setting by at least 28%.
■ This study provides level 2 evidence that can be used in shared decision-making between patients and physicians.
The ASCO Post Wants to Hear
From YouWrite to The ASCO Post at [email protected]
Advertisement not displayed in digital edition at advertiser’s request

PAGE 10 The ASCO Post | MARCH 25, 2015
Announcements
AACR Names Nancy E. Davidson, MD, President-Elect for 2015–2016By Jo Cavallo
The American Association for Cancer Research (AACR) has announced
the election of Nancy E. Davidson, MD, Director of the University of Pittsburgh Cancer Institute and UPMC Cancer-
Center in Pittsburgh, as its President-Elect for 2015–2016. Dr. Davidson will officially become President-Elect at the AACR Annual Meeting 2015 in Phila-delphia, April 18–22. She will start her
presidential term in April 2016.Dr. Davidson has focused her career
on clinical and translational breast cancer research, cancer biology and treatment, and the role of apoptosis and mecha-
nisms of epigenetic regulation of gene expression of the estrogen receptor alpha (ESR1) gene in breast cancer treatment.
“We are delighted that Dr. Davidson has been elected to serve as the next
Advertisement not displayed in digital edition at advertiser’s request

ASCOPost.com | MARCH 25, 2015 PAGE 11
Announcements
continued on page 12
AACR President-Elect,” said Margaret Foti, PhD, MD (hc), Chief Executive Officer of the AACR, in a statement. “She is an acknowledged expert in breast cancer research whose clinical and translational work has had a pro-found impact on the lives of patients. Dr. Davidson will lead the association with much energy and dedication, and
it will be an honor to work with her to make further strides in our mission to prevent and cure all cancers.”
Exciting Time in Cancer Research
“With deaths from cancer declining and the number of cancer survivors on the rise, this is an exciting time in cancer
research and care,” said Dr. Davidson in a statement. “I am honored to be given the opportunity to work with AACR and its members on our singular focus to advance scientific discoveries that can translate to exceptional patient care.” Dr. Davidson is also Distinguished Pro-fessor of Medicine and Pharmacology and Chemical Biology, Associate Vice
Chancellor for Cancer Research, Hill-man Professor of Oncology, and Pro-fessor at the Clinical and Translational Science Institute at the University of Pittsburgh and Adjunct Professor of Oncology at Johns Hopkins University School of Medicine in Baltimore.
Dr. Davidson is known for her stud-ies involving the role of hormones and the estrogen receptor in breast carcino-gens that have defined the molecular mechanisms driving the disease, as well as for her efforts to establish novel ther-apeutic approaches for patients who fail to respond to common treatment mo-dalities. Dr. Davidson has led clinical trials investigating chemotherapy and endocrine-related therapies for treating premenopausal breast cancer and has increased the understanding of the po-tential of angiogenesis inhibitors, such as bevacizumab (Avastin), in the treat-ment of metastatic breast cancer.
A Formidable CareerDr. Davidson has been recognized
for her work in breast cancer research with many awards throughout her career, including ASCO’s Gianni Bo-nadonna Breast Cancer Award, the AACR Women in Cancer Research Charlotte Friend Memorial Lecture-ship, the Potamkin Award from the Pennsylvania Breast Cancer Coalition, the Distinguished Alumna Award from Johns Hopkins University Alumni As-sociation, and the Rosaline E. Franklin Award for Women in Science from the National Cancer Institute.
Dr. Davidson is an elected member of the Institute of Medicine, the Asso-
I am honored to be given the opportunity to work with
AACR and its members on our singular focus to advance
scientific discoveries that can translate to exceptional
patient care. —Nancy E. Davidson, MD
Advertisement not displayed in digital edition at advertiser’s request

PAGE 12 The ASCO Post | MARCH 25, 2015
Announcements
ciation of American Physicians, and the American College of Physicians. Dr. Davidson is a Past President of ASCO and currently serves as a member of the scientific advisory committee of Break-
through Breast Cancer and the scientif-ic advisory board of the V Foundation for Cancer Research.
The AACR, whose mission is to pre-vent and cure cancer through research, education, communication, and col-laboration, has more than 35,000 mem-bers in 101 countries. n
AACR Names Presidentcontinued from page 11
Dr. Davidson will lead the association with much
energy and dedication, and it will be an honor to work with her to make further strides in
our mission to prevent and cure all cancers.
—Margaret Foti, PhD, MD (hc)
Winship Cancer Institute of Emory University Names New Chief Medical Officer and Chief Quality Officer
Sagar Lonial, MD, has been named Chief Medical Officer at Winship
Cancer Institute of Emory University and Charles A. Staley, MD, has been named Chief Quality Officer, accord-ing to an announcement recently re-leased by the Cancer Institute. Both physicians join Winship’s senior lead-ership team and will advance Win-ship’s clinical programs and services within all of its facilities.
Dr. Lonial, Professor and Executive Vice Chair of Emory’s Department of Hematology and Medical Oncology, is an internationally recognized authority in the management and research related to B-cell malignancies, including multi-ple myeloma. As Chief Medical Officer of Winship, Dr. Lonial will oversee all
clinical care initiatives impacting both clinicians and patients.
Dr. Staley, Professor and Director of Emory’s Division of Surgical Oncol-ogy, specializes in the management of
patients with gastrointestinal cancers. He previously served as Winship’s Chief Medical Officer and in this new role will assume responsibility for the institute’s quality improvement processes across
all disciplines and campuses.“I can’t think of two physicians who
are better suited to take on the criti-cal leadership roles of Winship’s Chief Medical Officer and Chief Quality Of-
JOB#
: 483
34
CLIE
NT: E
xelix
is DE
SC: J
ourn
al Ad
A si
ze
FILE
NAM
E: E
XL_X
LX_Q
4833
4_JA
_D01
.indd
DA
TE: 5
-12-
2014
6:1
1 PM
RO
UND:
1
1 PG: P
ared
esA/
Cruz
, Feli
x AD
: L B
udre
au
PM: N
one
AE: K
Ruf
f CW
: S G
uest
La
st S
aved
: 5-1
2-20
14 6
:11
PMTR
IM: 1
5” x
10.
25”
BLEE
D: 1
7.5”
x 1
1.25
” SA
FETY
: 6.7
5” x
9.7
5”
PROD
: M H
aight
IN
K Sp
ec: 4
c PR
INT
SCAL
E: 7
5%FO
NTS:
Neo
San
s Pr
o (B
old),
DIN
Pro
(Bold
, Med
ium, R
egula
r, Bl
ack,
Italic
), He
lvet
ica
Neue
LT
Std
(65
Mediu
m)
IMAG
ES: 4
8334
_JA_W
hale
s_fn
.tif (
CMYK
; 300
ppi;
100
%),
4833
4_KM
Cha
rt_O
L3_v
2a_A
LT S
IZE_
2415
.eps
(100
%),
4833
4_JA
_Sol
o_W
hale
s_fn
.psd
(CMY
K; 3
00 p
pi; 1
00%
), Ex
elix
is_b
lk.e
ps (2
0.6%
, 19.
01%
), Co
met
riq_
gene
ric_®
_80m
g_4C
.eps
(24.
69%
)IN
KS:
Cya
n,
Mag
enta
, Y
ellow
, B
lack
DOC
PATH
: Mac
intos
h HD
:Use
rs:p
ared
esa:
Desk
top:
EXL_
XLX_
Q483
34_JA
_D01
.indd
NOTE
S: N
one
EXL_
XLX_
Q483
34_JA
_D01
.indd
Ga
lley:
1
S&H
Phar
maG
raph
ics
Disk
DATE
SIGN
OFF
PGQC
TCAD
CDCW
AE/A
SED
PROD
4.0 11.2months months
1.00.90.80.70.60.50.40.30.20.10.0
0 3 6 9 12 15 18 21
No. of patients at risk:COMETRIQ® 219 121 78 55 31 12 2 1 Placebo 111 35 11 6 3 2 0 0
Months
Prob
abili
ty o
f pat
ient
s w
ho a
re p
rogr
essi
on fr
ee
4.0 11.2months
median
months
median
COMETRIQ® (n=219) Placebo (n=111)
HR=0.2895% CI: 0.19, 0.40P<0.0001
PFS1.00.90.80.70.60.50.40.30.20.10.0
0 3 6 9 12 15 18 21
No. of patientsat risk:COMETRIQ® 219 121 78 55 31 12 2 1Placebo 111 35 11 6 3 2 0 0
Months
Prob
abili
ty o
f pat
ient
s w
ho a
re p
rogr
essi
on fr
ee
4.0 11.2months
median
months
median
COMETRIQ® (n=219)Placebo (n=111)
HR=0.2895% CI: 0.19, 0.40P<0.0001
PFS
Attack from multiple anglesCOMETRIQ® has been shown to inhibit the activity of MET; VEGFR-1, -2, and -3; RET; and other receptor tyrosine kinases, in vitro• These tyrosine kinases are involved in both normal cellular function and pathologic
processes such as oncogenesis, metastasis, tumor angiogenesis, and maintenance of the tumor microenvironment
MET=hepatocyte growth factor receptor; VEGFR=vascular endothelial cell growth factor receptor; RET=rearranged during transfection.
COMETRIQ® (cabozantinib) is indicated for the treatment of patients with progressive, metastatic medullary thyroid cancer (MTC)
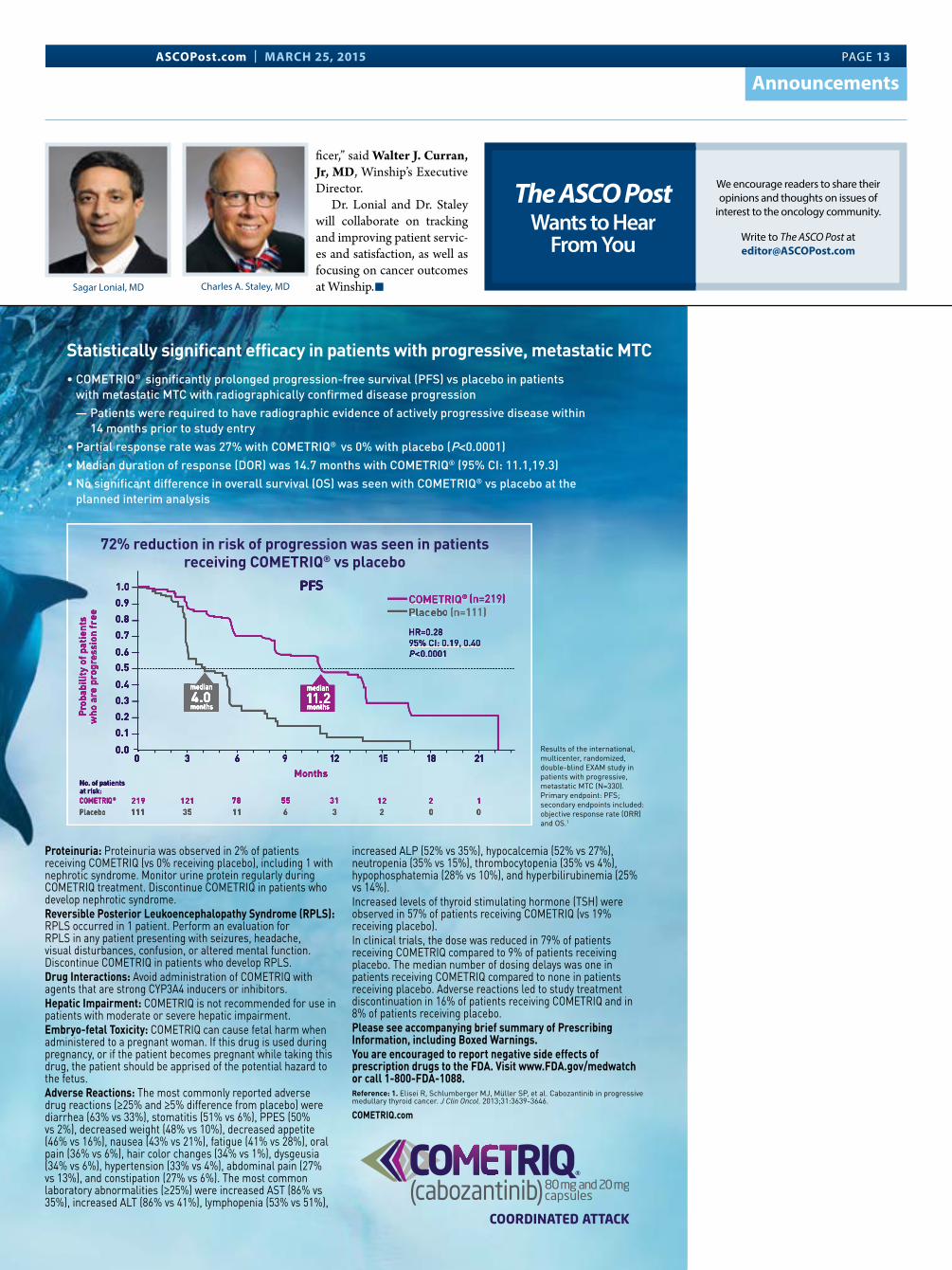
Statistically significant efficacy in patients with progressive, metastatic MTC• COMETRIQ® significantly prolonged progression-free survival (PFS) vs placebo in patients
with metastatic MTC with radiographically confirmed disease progression — Patients were required to have radiographic evidence of actively progressive disease within
14 months prior to study entry• Partial response rate was 27% with COMETRIQ® vs 0% with placebo (P<0.0001)• Median duration of response (DOR) was 14.7 months with COMETRIQ® (95% CI: 11.1,19.3)• No significant difference in overall survival (OS) was seen with COMETRIQ® vs placebo at the
planned interim analysis
72% reduction in risk of progression was seen in patients receiving COMETRIQ® vs placebo
© 2014 Exelixis, Inc. 210 East Grand Avenue, So. San Francisco, CA 94080 Printed in USA 05/14 [COM-0086]COORDINATED ATTACK
Important Safety InformationWARNING: PERFORATIONS AND FISTULAS, and HEMORRHAGE• Perforations and Fistulas: Gastrointestinal perforations
occurred in 3% and fistula formation in 1% of COMETRIQ®-treated patients. Discontinue COMETRIQ in patients with perforation or fistula.
• Hemorrhage: Severe, sometimes fatal, hemorrhage including hemoptysis and gastrointestinal hemorrhage occurred in 3% of COMETRIQ-treated patients. Monitor patients for signs and symptoms of bleeding. Do not administer COMETRIQ to patients with severe hemorrhage.
Perforations and Fistulas: Serious gastrointestinal (GI) perforations and fistulas were reported, of which one GI fistula was fatal. Non-GI fistulas including tracheal/esophageal were reported in 4% of COMETRIQ-treated patients. Two of these were fatal. Monitor patients for symptoms of perforations and fistulas.Hemorrhage: Serious and sometimes fatal hemorrhage occurred with COMETRIQ. Events ≥ Grade 3 occurred in 3% of COMETRIQ patients vs 1% receiving placebo. Do not administer COMETRIQ to patients with a recent history of hemorrhage or hemoptysis.Thrombotic Events: COMETRIQ treatment results in an increased incidence vs placebo of venous thromboembolism (6% vs 3%) and arterial thromboembolism (2% vs 0%). Discontinue COMETRIQ in patients who develop an acute myocardial infarction or any other clinically significant arterial thromboembolic complication.
Wound Complications: Wound complications have been reported with COMETRIQ. Stop treatment with COMETRIQ at least 28 days prior to scheduled surgery. Resume COMETRIQ therapy after surgery based on clinical judgment of adequate wound healing. Withhold COMETRIQ in patients with dehiscence or wound healing complications requiring medical intervention.Hypertension: COMETRIQ treatment results in an increased incidence of treatment-emergent hypertension vs placebo (61% vs 30%). Monitor blood pressure prior to initiation and regularly during COMETRIQ treatment. Withhold COMETRIQ for hypertension that is not adequately controlled with medical management; when controlled, resume COMETRIQ at a reduced dose. Discontinue COMETRIQ for severe hypertension that cannot be controlled with anti-hypertensive therapy.Osteonecrosis of the Jaw (ONJ): ONJ occurred in 1% of COMETRIQ-treated patients. ONJ can manifest as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration or erosion, persistent jaw pain or slow healing of the mouth or jaw after dental surgery. Perform an oral examination prior to initiation of COMETRIQ and periodically during COMETRIQ therapy. Advise patients regarding good oral hygiene practices. For invasive dental procedures, withhold COMETRIQ treatment for at least 28 days prior to scheduled surgery, if possible.Palmar- Plantar Erythrodysesthesia Syndrome (PPES): PPES occurred in 50% of patients treated with COMETRIQ and was severe in 13% of patients. Withhold COMETRIQ in patients who develop intolerable Grade 2 PPES or Grade 3-4 PPES until improvement to Grade 1; resume COMETRIQ at a reduced dose.
Proteinuria: Proteinuria was observed in 2% of patients receiving COMETRIQ (vs 0% receiving placebo), including 1 with nephrotic syndrome. Monitor urine protein regularly during COMETRIQ treatment. Discontinue COMETRIQ in patients who develop nephrotic syndrome.Reversible Posterior Leukoencephalopathy Syndrome (RPLS): RPLS occurred in 1 patient. Perform an evaluation for RPLS in any patient presenting with seizures, headache, visual disturbances, confusion, or altered mental function. Discontinue COMETRIQ in patients who develop RPLS.Drug Interactions: Avoid administration of COMETRIQ with agents that are strong CYP3A4 inducers or inhibitors.Hepatic Impairment: COMETRIQ is not recommended for use in patients with moderate or severe hepatic impairment.Embryo-fetal Toxicity: COMETRIQ can cause fetal harm when administered to a pregnant woman. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.Adverse Reactions: The most commonly reported adverse drug reactions (≥25% and ≥5% difference from placebo) were diarrhea (63% vs 33%), stomatitis (51% vs 6%), PPES (50% vs 2%), decreased weight (48% vs 10%), decreased appetite (46% vs 16%), nausea (43% vs 21%), fatigue (41% vs 28%), oral pain (36% vs 6%), hair color changes (34% vs 1%), dysgeusia (34% vs 6%), hypertension (33% vs 4%), abdominal pain (27% vs 13%), and constipation (27% vs 6%). The most common laboratory abnormalities (≥25%) were increased AST (86% vs 35%), increased ALT (86% vs 41%), lymphopenia (53% vs 51%),
increased ALP (52% vs 35%), hypocalcemia (52% vs 27%), neutropenia (35% vs 15%), thrombocytopenia (35% vs 4%), hypophosphatemia (28% vs 10%), and hyperbilirubinemia (25% vs 14%).Increased levels of thyroid stimulating hormone (TSH) were observed in 57% of patients receiving COMETRIQ (vs 19% receiving placebo).In clinical trials, the dose was reduced in 79% of patients receiving COMETRIQ compared to 9% of patients receiving placebo. The median number of dosing delays was one in patients receiving COMETRIQ compared to none in patients receiving placebo. Adverse reactions led to study treatment discontinuation in 16% of patients receiving COMETRIQ and in 8% of patients receiving placebo.Please see accompanying brief summary of Prescribing Information, including Boxed Warnings.You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.FDA.gov/medwatch or call 1-800-FDA-1088.Reference: 1. Elisei R, Schlumberger MJ, Müller SP, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31:3639-3646.
COMETRIQ.com
Results of the international, multicenter, randomized, double-blind EXAM study in patients with progressive, metastatic MTC (N=330). Primary endpoint: PFS; secondary endpoints included: objective response rate (ORR) and OS.1
C M Y KCosmos Communications 1
1ej
27902a 05.12.14 133
Q1 Q2
S:6.75” S:6.75”
S:9.75”
T:15”
T:10.25”
B:17.5”
B:11.25”
ASCOPost.com | MARCH 25, 2015 PAGE 13
Announcements
ficer,” said Walter J. Curran, Jr, MD, Winship’s Executive Director.
Dr. Lonial and Dr. Staley will collaborate on tracking and improving patient servic-es and satisfaction, as well as focusing on cancer outcomes at Winship.nSagar Lonial, MD Charles A. Staley, MD
The ASCO Post Wants to Hear
From You
We encourage readers to share their opinions and thoughts on issues of
interest to the oncology community.
Write to The ASCO Post at [email protected]
JOB#
: 483
34
CLIE
NT: E
xelix
is DE
SC: J
ourn
al Ad
A si
ze
FILE
NAM
E: E
XL_X
LX_Q
4833
4_JA
_D01
.indd
DA
TE: 5
-12-
2014
6:1
1 PM
RO
UND:
1
1 PG: P
ared
esA/
Cruz
, Feli
x AD
: L B
udre
au
PM: N
one
AE: K
Ruf
f CW
: S G
uest
La
st S
aved
: 5-1
2-20
14 6
:11
PMTR
IM: 1
5” x
10.
25”
BLEE
D: 1
7.5”
x 1
1.25
” SA
FETY
: 6.7
5” x
9.7
5”
PROD
: M H
aight
IN
K Sp
ec: 4
c PR
INT
SCAL
E: 7
5%FO
NTS:
Neo
San
s Pr
o (B
old),
DIN
Pro
(Bold
, Med
ium, R
egula
r, Bl
ack,
Italic
), He
lvet
ica
Neue
LT
Std
(65
Mediu
m)
IMAG
ES: 4
8334
_JA_W
hale
s_fn
.tif (
CMYK
; 300
ppi;
100
%),
4833
4_KM
Cha
rt_O
L3_v
2a_A
LT S
IZE_
2415
.eps
(100
%),
4833
4_JA
_Sol
o_W
hale
s_fn
.psd
(CMY
K; 3
00 p
pi; 1
00%
), Ex
elix
is_b
lk.e
ps (2
0.6%
, 19.
01%
), Co
met
riq_
gene
ric_®
_80m
g_4C
.eps
(24.
69%
)IN
KS:
Cya
n,
Mag
enta
, Y
ellow
, B
lack
DOC
PATH
: Mac
intos
h HD
:Use
rs:p
ared
esa:
Desk
top:
EXL_
XLX_
Q483
34_JA
_D01
.indd
NOTE
S: N
one
EXL_
XLX_
Q483
34_JA
_D01
.indd
Ga
lley:
1
S&H
Phar
maG
raph
ics
Disk
DATE
SIGN
OFF
PGQC
TCAD
CDCW
AE/A
SED
PROD
4.0 11.2months months
1.00.90.80.70.60.50.40.30.20.10.0
0 3 6 9 12 15 18 21
No. of patients at risk:COMETRIQ® 219 121 78 55 31 12 2 1 Placebo 111 35 11 6 3 2 0 0
Months
Prob
abili
ty o
f pat
ient
s w
ho a
re p
rogr
essi
on fr
ee
4.0 11.2months
median
months
median
COMETRIQ® (n=219) Placebo (n=111)
HR=0.2895% CI: 0.19, 0.40P<0.0001
PFS1.00.90.80.70.60.50.40.30.20.10.0
0 3 6 9 12 15 18 21
No. of patientsat risk:COMETRIQ® 219 121 78 55 31 12 2 1Placebo 111 35 11 6 3 2 0 0
Months
Prob
abili
ty o
f pat
ient
s w
ho a
re p
rogr
essi
on fr
ee
4.0 11.2months
median
months
median
COMETRIQ® (n=219)Placebo (n=111)
HR=0.2895% CI: 0.19, 0.40P<0.0001
PFS
Attack from multiple anglesCOMETRIQ® has been shown to inhibit the activity of MET; VEGFR-1, -2, and -3; RET; and other receptor tyrosine kinases, in vitro• These tyrosine kinases are involved in both normal cellular function and pathologic
processes such as oncogenesis, metastasis, tumor angiogenesis, and maintenance of the tumor microenvironment
MET=hepatocyte growth factor receptor; VEGFR=vascular endothelial cell growth factor receptor; RET=rearranged during transfection.
COMETRIQ® (cabozantinib) is indicated for the treatment of patients with progressive, metastatic medullary thyroid cancer (MTC)
Statistically significant efficacy in patients with progressive, metastatic MTC• COMETRIQ® significantly prolonged progression-free survival (PFS) vs placebo in patients
with metastatic MTC with radiographically confirmed disease progression — Patients were required to have radiographic evidence of actively progressive disease within
14 months prior to study entry• Partial response rate was 27% with COMETRIQ® vs 0% with placebo (P<0.0001)• Median duration of response (DOR) was 14.7 months with COMETRIQ® (95% CI: 11.1,19.3)• No significant difference in overall survival (OS) was seen with COMETRIQ® vs placebo at the
planned interim analysis
72% reduction in risk of progression was seen in patients receiving COMETRIQ® vs placebo
© 2014 Exelixis, Inc. 210 East Grand Avenue, So. San Francisco, CA 94080 Printed in USA 05/14 [COM-0086]COORDINATED ATTACK
Important Safety InformationWARNING: PERFORATIONS AND FISTULAS, and HEMORRHAGE• Perforations and Fistulas: Gastrointestinal perforations
occurred in 3% and fistula formation in 1% of COMETRIQ®-treated patients. Discontinue COMETRIQ in patients with perforation or fistula.
• Hemorrhage: Severe, sometimes fatal, hemorrhage including hemoptysis and gastrointestinal hemorrhage occurred in 3% of COMETRIQ-treated patients. Monitor patients for signs and symptoms of bleeding. Do not administer COMETRIQ to patients with severe hemorrhage.
Perforations and Fistulas: Serious gastrointestinal (GI) perforations and fistulas were reported, of which one GI fistula was fatal. Non-GI fistulas including tracheal/esophageal were reported in 4% of COMETRIQ-treated patients. Two of these were fatal. Monitor patients for symptoms of perforations and fistulas.Hemorrhage: Serious and sometimes fatal hemorrhage occurred with COMETRIQ. Events ≥ Grade 3 occurred in 3% of COMETRIQ patients vs 1% receiving placebo. Do not administer COMETRIQ to patients with a recent history of hemorrhage or hemoptysis.Thrombotic Events: COMETRIQ treatment results in an increased incidence vs placebo of venous thromboembolism (6% vs 3%) and arterial thromboembolism (2% vs 0%). Discontinue COMETRIQ in patients who develop an acute myocardial infarction or any other clinically significant arterial thromboembolic complication.
Wound Complications: Wound complications have been reported with COMETRIQ. Stop treatment with COMETRIQ at least 28 days prior to scheduled surgery. Resume COMETRIQ therapy after surgery based on clinical judgment of adequate wound healing. Withhold COMETRIQ in patients with dehiscence or wound healing complications requiring medical intervention.Hypertension: COMETRIQ treatment results in an increased incidence of treatment-emergent hypertension vs placebo (61% vs 30%). Monitor blood pressure prior to initiation and regularly during COMETRIQ treatment. Withhold COMETRIQ for hypertension that is not adequately controlled with medical management; when controlled, resume COMETRIQ at a reduced dose. Discontinue COMETRIQ for severe hypertension that cannot be controlled with anti-hypertensive therapy.Osteonecrosis of the Jaw (ONJ): ONJ occurred in 1% of COMETRIQ-treated patients. ONJ can manifest as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration or erosion, persistent jaw pain or slow healing of the mouth or jaw after dental surgery. Perform an oral examination prior to initiation of COMETRIQ and periodically during COMETRIQ therapy. Advise patients regarding good oral hygiene practices. For invasive dental procedures, withhold COMETRIQ treatment for at least 28 days prior to scheduled surgery, if possible.Palmar- Plantar Erythrodysesthesia Syndrome (PPES): PPES occurred in 50% of patients treated with COMETRIQ and was severe in 13% of patients. Withhold COMETRIQ in patients who develop intolerable Grade 2 PPES or Grade 3-4 PPES until improvement to Grade 1; resume COMETRIQ at a reduced dose.
Proteinuria: Proteinuria was observed in 2% of patients receiving COMETRIQ (vs 0% receiving placebo), including 1 with nephrotic syndrome. Monitor urine protein regularly during COMETRIQ treatment. Discontinue COMETRIQ in patients who develop nephrotic syndrome.Reversible Posterior Leukoencephalopathy Syndrome (RPLS): RPLS occurred in 1 patient. Perform an evaluation for RPLS in any patient presenting with seizures, headache, visual disturbances, confusion, or altered mental function. Discontinue COMETRIQ in patients who develop RPLS.Drug Interactions: Avoid administration of COMETRIQ with agents that are strong CYP3A4 inducers or inhibitors.Hepatic Impairment: COMETRIQ is not recommended for use in patients with moderate or severe hepatic impairment.Embryo-fetal Toxicity: COMETRIQ can cause fetal harm when administered to a pregnant woman. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.Adverse Reactions: The most commonly reported adverse drug reactions (≥25% and ≥5% difference from placebo) were diarrhea (63% vs 33%), stomatitis (51% vs 6%), PPES (50% vs 2%), decreased weight (48% vs 10%), decreased appetite (46% vs 16%), nausea (43% vs 21%), fatigue (41% vs 28%), oral pain (36% vs 6%), hair color changes (34% vs 1%), dysgeusia (34% vs 6%), hypertension (33% vs 4%), abdominal pain (27% vs 13%), and constipation (27% vs 6%). The most common laboratory abnormalities (≥25%) were increased AST (86% vs 35%), increased ALT (86% vs 41%), lymphopenia (53% vs 51%),
increased ALP (52% vs 35%), hypocalcemia (52% vs 27%), neutropenia (35% vs 15%), thrombocytopenia (35% vs 4%), hypophosphatemia (28% vs 10%), and hyperbilirubinemia (25% vs 14%).Increased levels of thyroid stimulating hormone (TSH) were observed in 57% of patients receiving COMETRIQ (vs 19% receiving placebo).In clinical trials, the dose was reduced in 79% of patients receiving COMETRIQ compared to 9% of patients receiving placebo. The median number of dosing delays was one in patients receiving COMETRIQ compared to none in patients receiving placebo. Adverse reactions led to study treatment discontinuation in 16% of patients receiving COMETRIQ and in 8% of patients receiving placebo.Please see accompanying brief summary of Prescribing Information, including Boxed Warnings.You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.FDA.gov/medwatch or call 1-800-FDA-1088.Reference: 1. Elisei R, Schlumberger MJ, Müller SP, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31:3639-3646.
COMETRIQ.com
Results of the international, multicenter, randomized, double-blind EXAM study in patients with progressive, metastatic MTC (N=330). Primary endpoint: PFS; secondary endpoints included: objective response rate (ORR) and OS.1
C M Y KCosmos Communications 1
1ej
27902a 05.12.14 133
Q1 Q2
S:6.75” S:6.75”
S:9.75”
T:15”
T:10.25”
B:17.5”
B:11.25”

PAGE 14 The ASCO Post | MARCH 25, 2015
FDA Update
Priority Review Granted for Trabectedin in Advanced Soft-Tissue Sarcoma
The U.S. Food and Drug Adminis-tration (FDA) has granted Prior-
ity Review for the New Drug Application (NDA) for trabectedin to treat patients with advanced soft-tissue sarcoma, in-cluding liposarcoma and leiomyosarcoma subtypes, who have received prior chemo-
therapy including an anthracycline. The NDA for trabectedin was submitted to the FDA on November 24, 2014.
Trabectedin is a novel, multimod-al, synthetically produced antitumor agent—originally derived from the sea squirt Ecteinascidia turbinata—that pre-
vents tumor cells from multiplying.The filing is based on the phase III
randomized, open-label study ET743-SAR-3007. This trial is evaluating the safety and efficacy of trabectedin vs da-carbazine for the treatment of patients with advanced liposarcoma and leio-myosarcoma. Results will be presented at a later date. n
JOB#
: 483
34
CLIE
NT: E
xelix
is DE
SC: J
ourn
al Ad
A si
ze
FILE
NAM
E: E
XL_X
LX_Q
4833
4_JA
_D01
.indd
DA
TE: 5
-12-
2014
6:1
1 PM
RO
UND:
1
1 PG: P
ared
esA/
Cruz
, Feli
x AD
: L B
udre
au
PM: N
one
AE: K
Ruf
f CW
: S G
uest
La
st S
aved
: 5-1
2-20
14 6
:11
PMTR
IM: 1
5” x
10.
25”
BLEE
D: 1
7.5”
x 1
1.25
” SA
FETY
: 6.7
5” x
9.7
5”
PROD
: M H
aight
IN
K Sp
ec: 4
c PR
INT
SCAL
E: 7
5%FO
NTS:
Non
e
IMAG
ES: C
omet
riq_7
x10_
Jan1
4_BS
_p1.
pdf (
100%
), Co
met
riq_7
x10_
Jan1
4_BS
_p2.
pdf (
100%
)IN
KS:
Blac
kDO
C PA
TH: M
acint
osh
HD:U
sers
:par
edes
a:De
skto
p:EX
L_XL
X_Q4
8334
_JA_D
01.in
ddNO
TES:
Non
e
EXL_
XLX_
Q483
34_JA
_D01
.indd
Ga
lley:
2
S&H
Phar
maG
raph
ics
Disk
DATE
SIGN
OFF
PGQC
TCAD
CDCW
AE/A
SED
PROD
COMETRIQ® (cabozantinib) capsules BRIEF SUMMARY OF PRESCRIBING INFORMATION Initial U.S. Approval: 2012
1. INDICATIONS AND USAGECOMETRIQ is indicated for the treatment of patients with progressive, metastatic medullary thyroid cancer (MTC).
2. DOSAGE AND ADMINISTRATION 2.1 Recommended Dose: The recommended daily dose of COMETRIQ is 140 mg (one 80-mg and three 20-mg capsules). Do not administer COMETRIQ with food. Instruct patients not to eat for at least 2 hours before and at least 1 hour after taking COMETRIQ. Continue treatment until disease progression or unacceptable toxicity occurs. Swallow COMETRIQ capsules whole. Do not open COMETRIQ capsules. Do not take a missed dose within 12 hours of the next dose. Do not ingest foods (e.g., grapefruit, grapefruit juice) or nutritional supplements that are known to inhibit cytochrome P450 during COMETRIQ.2.2 Dosage Adjustments: For Adverse Reactions : Withhold COMETRIQ for NCI CTCAE Grade 4 hematologic adverse reactions, Grade 3 or greater non-hematologic adverse reactions or intolerable Grade 2 adverse reactions. Upon resolution/improvement of the adverse reaction (i.e., return to baseline or resolution to Grade 1), reduce the dose as follows:• If previously receiving 140-mg daily dose, resume treatment at 100 mg
daily (one 80-mg and one 20-mg capsule)• If previously receiving 100-mg daily dose, resume treatment at 60 mg daily
(three 20-mg capsules)• If previously receiving 60-mg daily dose, resume at 60 mg if tolerated,
otherwise, discontinue COMETRIQPermanently discontinue COMETRIQ for any of the following: development of visceral perforation or fistula formation; severe hemorrhage; serious arterial thromboembolic event (e.g., myocardial infarction, cerebral infarction); nephrotic syndrome; malignant hypertension, hypertensive crisis, persistent uncontrolled hypertension despite optimal medical management; osteonecrosis of the jaw; or reversible posterior leukoencephalopathy syndrome. In Patients With Hepatic Impairment : COMETRIQ is not recommended for use in patients with moderate and severe hepatic impairment. In Patients Taking CYP3A4 Inhibitors : Avoid the use of concomitant strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, nefazodone, saquinavir, telithromycin, ritonavir, indinavir, nelfinavir, voriconazole) in patients receiving COMETRIQ. For patients who require treatment with a strong CYP3A4 inhibitor, reduce the daily COMETRIQ dose by 40 mg (for example, from 140 mg to 100 mg daily or from 100 mg to 60 mg daily). Resume the dose that was used prior to initiating the CYP3A4 inhibitor 2 to 3 days after discontinuation of the strong inhibitor. In Patients Taking Strong CYP3A4 Inducers: Avoid the chronic use of concomitant strong CYP3A4 inducers (e.g., phenytoin, carbamazepine, rifampin, rifabutin, rifapentine, phenobarbital) if alternative therapy is available. Do not ingest foods or nutritional supplements (e.g., St. John’s Wort [Hypericum perforatum]) that are known to induce cytochrome P450 activity. For patients who require treatment with a strong CYP3A4 inducer, increase the daily COMETRIQ dose by 40 mg (for example, from 140 mg to 180 mg daily or from 100 mg to 140 mg daily) as tolerated. Resume the dose that was used prior to initiating the CYP3A4 inducer 2 to 3 days after discontinuation of the strong inducer. The daily dose of COMETRIQ should not exceed 180 mg.
4. CONTRAINDICATIONS None.
5. WARNINGS AND PRECAUTIONS5.1 Perforations and Fistulas: Gastrointestinal (GI) perforations and fistulas were reported in 3% and 1% of COMETRIQ-treated patients, respectively. All were serious and one GI fistula was fatal (<1%). Non-GI fistulas including tracheal/esophageal were reported in 4% of COMETRIQ-treated patients. Two (1%) of these were fatal. Monitor patients for symptoms of perforations and fistulas. Discontinue COMETRIQ in patients who experience a perforation or a fistula. 5.2 Hemorrhage: Serious and sometimes fatal hemorrhage occurred with COMETRIQ. The incidence of Grade ≥3 hemorrhagic events was higher in COMETRIQ-treated patients compared with placebo (3% vs. 1%). Do not administer COMETRIQ to patients with a recent history of hemorrhage or hemoptysis. 5.3 Thrombotic Events: COMETRIQ treatment results in an increased incidence of thrombotic events (venous thromboembolism: 6% vs. 3% and arterial thromboembolism: 2% vs. 0% in COMETRIQ-treated and placebo-treated patients, respectively). Discontinue COMETRIQ in patients who develop an acute myocardial infarction or any other clinically significant arterial thromboembolic complication.
5.4 Wound Complications: Wound complications have been reported with COMETRIQ. Stop treatment with COMETRIQ at least 28 days prior to scheduled surgery. Resume COMETRIQ therapy after surgery based on clinical judgment of adequate wound healing. Withhold COMETRIQ in patients with dehiscence or wound healing complications requiring medical intervention. 5.5 Hypertension: COMETRIQ treatment results in an increased incidence of treatment-emergent hypertension with Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (modified JNC criteria) stage 1 or 2 hypertension identified in 61% of COMETRIQ-treated patients compared with 30% of placebo-treated patients in the randomized trial. Monitor blood pressure prior to initiation and regularly during COMETRIQ treatment. Withhold COMETRIQ for hypertension that is not adequately controlled with medical management; when controlled, resume COMETRIQ at a reduced dose. Discontinue COMETRIQ for severe hypertension that cannot be controlled with anti-hypertensive therapy.5.6 Osteonecrosis of the Jaw (ONJ): Osteonecrosis of the jaw (ONJ) occurred in 1% of COMETRIQ-treated patients. ONJ can manifest as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration or erosion, persistent jaw pain or slow healing of the mouth or jaw after dental surgery. Perform an oral examination prior to initiation of COMETRIQ and periodically during COMETRIQ therapy. Advise patients regarding good oral hygiene practices. For invasive dental procedures, withhold COMETRIQ treatment for at least 28 days prior to scheduled surgery, if possible. 5.7 Palmar-Plantar Erythrodysesthesia Syndrome: Palmar-plantar erythrodysesthesia syndrome (PPES) occurred in 50% of patients treated with cabozantinib and was severe (≥Grade 3) in 13% of patients. Withhold COMETRIQ in patients who develop intolerable Grade 2 PPES or Grade 3-4 PPES until improvement to Grade 1; resume COMETRIQ at a reduced dose.5.8 Proteinuria: Proteinuria was observed in 4 (2%) patients receiving COMETRIQ, including one with nephrotic syndrome, as compared to none of the patients receiving placebo. Monitor urine protein regularly during COMETRIQ treatment. Discontinue COMETRIQ in patients who develop nephrotic syndrome. 5.9 Reversible Posterior Leukoencephalopathy Syndrome: Reversible Posterior Leukoencephalopathy Syndrome (RPLS), a syndrome of subcortical vasogenic edema diagnosed by characteristic finding on MRI, occurred in one (<1%) patient. Perform an evaluation for RPLS in any patient presenting with seizures, headache, visual disturbances, confusion or altered mental function. Discontinue COMETRIQ in patients who develop RPLS. 5.10 Drug Interactions: Avoid administration of COMETRIQ with agents that are strong CYP3A4 inducers or inhibitors. 5.11 Hepatic Impairment: COMETRIQ is not recommended for use in patients with moderate or severe hepatic impairment. 5.12 Embryo-Fetal Toxicity: COMETRIQ can cause fetal harm when administered to a pregnant woman. Cabozantinib was embryolethal in rats at exposures below the recommended human dose, with increased incidences of skeletal variations in rats and visceral variations and malformations in rabbits. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
6. ADVERSE REACTIONS6.1 Clinical Trial Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The safety of COMETRIQ was evaluated in 330 patients with progressive, metastatic medullary thyroid cancer randomized to receive 140 mg of COMETRIQ (n=214) or placebo (n=109) administered daily until disease progression or intolerable toxicity occurred in a randomized, double-blind, controlled trial. The data described below reflect a median exposure to COMETRIQ for 204 days. The population exposed to COMETRIQ was 70% male, 90% white, and had a median age of 55 years. Adverse reactions which occurred in ≥25% of COMETRIQ-treated patients occurring more frequently in the COMETRIQ arm with a between-arm difference of ≥5% included, in order of decreasing frequency: diarrhea, stomatitis, palmar-plantar erythrodysesthesia syndrome (PPES), decreased weight, decreased appetite, nausea, fatigue, oral pain, hair color changes, dysgeusia, hypertension, abdominal pain, and constipation. The most common laboratory abnormalities (>25%) were increased AST, increased ALT, lymphopenia, increased alkaline phosphatase, hypocalcemia, neutropenia, thrombocytopenia, hypophosphatemia, and hyperbilirubinemia. Grade 3-4 adverse reactions and laboratory abnormalities which occurred in ≥5% of COMETRIQ-treated patients occurring more frequently in the COMETRIQ arm with a between-arm difference of ≥2% included, in order of decreasing frequency: diarrhea, PPES, lymphopenia, hypocalcemia, fatigue, hypertension, asthenia, increased ALT, decreased weight, stomatitis, and decreased appetite (see Table 1, Table 2). Fatal adverse reactions occurred in 6% of patients receiving COMETRIQ and resulted from hemorrhage, pneumonia, septicemia, fistulas, cardiac arrest, respiratory failure, and unspecified death. Fatal adverse reactions occurred in 5% of patients receiving placebo and resulted from septicemia, pneumonia, and general deterioration. The dose was reduced in 79% of patients receiving COMETRIQ compared to 9% of patients receiving placebo. The median number of dosing delays was one in patients receiving COMETRIQ compared to none in patients receiving placebo. Adverse reactions led to study treatment discontinuation in 16% of patients receiving COMETRIQ and in 8% of patients receiving placebo. The most frequent adverse reactions leading to permanent discontinuation in patients treated with COMETRIQ were: hypocalcemia, increased lipase, PPES, diarrhea, fatigue, hypertension, nausea, pancreatitis, tracheal fistula formation, and vomiting. Increased levels of thyroid stimulating hormone (TSH) were observed in 57% of patients receiving COMETRIQ after the first dose compared to 19% of patients receiving placebo (regardless of baseline value). Ninety-two percent (92%) of patients on the COMETRIQ arm had a prior thyroidectomy, and 89% were taking thyroid hormone replacement prior to the first dose.
Table 1. Per-Patient Incidence of Selected Adverse Reactions in Protocol XL184-301 Occurring at a Higher Incidence in COMETRIQ-Treated Patients [Between-Arm Difference of ≥5% (All Grades)1 or ≥2% (Grades 3-4)]
MedDRA System Organ Class/ Preferred Terms
Cabozantinib (n=214)
Placebo (n=109)
All Grades
Grades 3-4
All Grades
Grades 3-4
GASTROINTESTINAL DISORDERSDIARRHEA 63 16 33 2
STOMATITIS2 51 5 6 0
NAUSEA 43 1 21 0
ORAL PAIN3 36 2 6 0
CONSTIPATION 27 0 6 0
ABDOMINAL PAIN4 27 3 13 1
VOMITING 24 2 2 1
DYSPHAGIA 13 4 6 1
DYSPEPSIA 11 0 0 0
HEMORRHOIDS 9 0 3 0
GENERAL DISORDERS AND ADMINISTRATION SITE CONDITIONSFATIGUE 41 9 28 3
ASTHENIA 21 6 15 1
INVESTIGATIONSDECREASED WEIGHT 48 5 10 0
METABOLISM AND NUTRITION DISORDERSDECREASED APPETITE 46 5 16 1
DEHYDRATION 7 2 2 1
MUSCULOSKELETAL AND CONNECTIVE TISSUE DISORDERSARTHRALGIA 14 1 7 0
MUSCLE SPASMS 12 0 5 0
MUSCULOSKELETAL CHEST PAIN 9 1 4 0
NERVOUS SYSTEM DISORDERSDYSGEUSIA 34 0 6 0
HEADACHE 18 0 8 0
DIZZINESS 14 0 7 0
PARESTHESIA 7 0 2 0
PERIPHERAL SENSORY NEUROPATHY
7 0 0 0
PERIPHERAL NEUROPATHY 5 0 0 0
PSYCHIATRIC DISORDERSANXIETY 9 0 2 0
RESPIRATORY, THORACIC, AND MEDIASTINAL DISORDERSDYSPHONIA 20 0 9 0
SKIN AND SUBCUTANEOUS TISSUE DISORDERSPPES5 50 13 2 0
HAIR COLOR CHANGES/ DEPIGMENTATION, GRAYING
34 0 1 0
RASH 19 1 10 0
DRY SKIN 19 0 3 0
ALOPECIA 16 0 2 0
ERYTHEMA 11 1 2 0
HYPERKERATOSIS 7 0 0 0
VASCULAR DISORDERSHYPERTENSION 33 8 4 0
HYPOTENSION 7 1 0 01 National Cancer Institute Common Terminology Criteria for Adverse Events Version 3.0. 2 Includes the following terms: stomatitis, aphthous stomatitis, mouth ulceration, mucosal inflammation.
3 Includes the following terms: oral pain, oropharyngeal pain, glossitis, burning mouth syndrome, glossodynia.
4 Includes the following terms: abdominal pain, abdominal pain lower, abdominal pain upper, abdominal rigidity, abdominal tenderness, esophageal pain.
5Palmar-plantar erythrodysesthesia syndrome.
WARNING: PERFORATIONS AND FISTULAS, and HEMORRHAGESee full prescribing information for complete boxed warning. Perforations and Fistulas: Gastrointestinal perforations occurred in 3% and fistula formation in 1% of COMETRIQ-treated patients. Discontinue COMETRIQ in patients with perforation or fistula. (5.1) Hemorrhage: Severe, sometimes fatal, hemorrhage including hemoptysis and gastrointestinal hemorrhage occurred in 3% of COMETRIQ-treated patients. Monitor patients for signs and symptoms of bleeding. Do not administer COMETRIQ to patients with severe hemorrhage. (5.2)
of therapy. Infertility: There are no data on the effect of COMETRIQ on human fertility. Cabozantinib impaired male and female fertility in animal studies. 8.6 Hepatic Impairment: Cabozantinib pharmacokinetics has not been studied in patients with hepatic impairment. There are limited data in patients with liver impairment (serum bilirubin greater than 1.5 times the upper limit of normal). COMETRIQ is not recommended for use in patients with moderate or severe hepatic impairment, as safety and efficacy have not been established. 8.7 Renal Impairment: No dose adjustment is recommended for patients with mild or moderate renal impairment. There is no experience with COMETRIQ in patients with severe renal impairment.10. OVERDOSAGE One case of overdosage was reported in a patient who inadvertently took twice the intended dose (200 mg daily) for nine days. The patient suffered Grade 3 memory impairment, Grade 3 mental status changes, Grade 3 cognitive disturbance, Grade 2 weight loss, and Grade 1 increase in BUN. The extent of recovery was not documented.17. PATIENT COUNSELING INFORMATIONSee FDA-approved patient labeling (Patient Information and Instructions for Use). Inform patients of the following: • COMETRIQ often causes diarrhea which may be severe in some cases. Inform patients
of the need to contact their healthcare provider if severe diarrhea occurs during treatment with COMETRIQ.
• COMETRIQ often causes palmar-plantar erythrodysesthesia syndrome. Advise patients to contact their healthcare provider for progressive or intolerable rash.
• COMETRIQ often causes sores in the mouth, oral pain, changes in taste, nausea or vomiting. Advise patients to contact their healthcare provider if any of these symptoms are severe or prevent patients from eating and drinking.
• COMETRIQ often causes weight loss which may be significant in some cases. Advise patients to report significant weight loss.
• To contact their healthcare provider before any planned surgeries, including dental procedures.
• COMETRIQ may interact with other drugs; advise patients to inform their healthcare provider of all prescription or nonprescription medication or herbal products that they are taking.
• Patients of childbearing potential must use effective contraception during therapy and for at least four months following their last dose of COMETRIQ.
• Breast-feeding mothers must discontinue nursing while receiving COMETRIQ therapy. • COMETRIQ should not be taken with food. Instruct patients not to eat for at least
2 hours before and at least 1 hour after taking COMETRIQ. COMETRIQ capsules should not be opened or crushed but should be taken with a full glass (at least 8 ounces) of water.
• Patients should not consume grapefruits or grapefruit juice while taking COMETRIQ treatment.
Reference ID: 3223542
Distributed by Exelixis, Inc.11/2012
© 2012 Exelixis, Inc. 210 East Grand Avenue, So. San Francisco, CA 94080 Printed in USA 11/12 [24523]
Table 2. Percent-Patient Incidence of Laboratory Abnormalities Occurring at a Higher Incidence in COMETRIQ-Treated Patients in Protocol XL184-301 [Between- Arm Difference of ≥5% (All Grades) or ≥2% (Grades 3-4)]
ADVERSE EVENT COMETRIQ (n=214) Placebo (n=109)
All Grades
Grade 3-4 All Grades Grade 3-4
CHEMISTRIES
INCREASED AST 86 3 35 2
INCREASED ALT 86 6 41 2
INCREASED ALP 52 3 35 3
HYPOCALCEMIA 52 12 27 3
HYPOPHOSPHATEMIA 28 3 10 1
HYPERBILIRUBINEMIA 25 2 14 5
HYPOMAGNESEMIA 19 1 4 0
HYPOKALEMIA 18 4 9 3
HYPONATREMIA 10 2 5 0
HEMATOLOGIC
LYMPHOPENIA 53 16 51 11
NEUTROPENIA 35 3 15 2
THROMBOCYTOPENIA 35 0 4 3
ALT, alanine aminotransferase; ALP, alkaline phosphatase; AST, aspartate aminotransferase
Nearly all COMETRIQ-treated patients (96% vs. 84% placebo) experienced elevated blood pressure and there was a doubling in the incidence of overt hypertension in COMETRIQ-treated patients over placebo-treated patients (61% vs. 30%) according to modified Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC) staging criteria. No patients developed malignant hypertension.
Table 3. Per-Patient Incidence of Hypertension in Protocol XL184-301
HYPERTENSION, JNC1 STAGE COMETRIQ N=2113 (%)
Placebo N=1073 (%)
Normal: Grade 0: Systolic <120 mmHg and Diastolic <80 mmHg
4
15
Pre-hypertension: Systolic ≥120 mmHg or Diastolic ≥80 mmHg
34
54
Stage 1: Systolic ≥140 mmHg or Diastolic ≥90 mmHg
46
25
Stage 2: Systolic ≥160 mmHg or Diastolic ≥100 mmHg
15 5
Malignant: Diastolic ≥120 mmHg
0
0
1 Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure, JAMA 2003:289:2560. Criteria applied were modified, as multiple readings were not available per timepoint, and therefore not averaged.
2 Subjects classified by highest category based on all recorded blood pressure readings beginning after the first dose through 30 days after last dose.
3Subjects with at least two blood pressure measurements after the first dose.
7. DRUG INTERACTIONS 7.1 Effect of CYP3A4 Inhibitors: Administration of a strong CYP3A4 inhibitor, ketoconazole (400 mg daily for 27 days) to healthy subjects increased single-dose plasma cabozantinib exposure (AUC0-inf) by 38%. Avoid taking a strong CYP3A4 inhibitor (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, voriconazole) when taking COMETRIQ. 7.2 Effect of CYP3A4 Inducers: Administration of a strong CYP3A4 inducer, rifampin (600 mg daily for 31 days) to healthy subjects decreased single-dose plasma cabozantinib exposure (AUC0-inf) by 77%. Avoid chronic coadministration of strong CYP3A4 inducers (e.g., dexamethasone, phenytoin, carbamazepine, rifampin, rifabutine, rifapentine, phenobarbital, St. John’s Wort) with COMETRIQ.8. USE IN SPECIFIC POPULATIONS 8.1 Pregnancy: Pregnancy Category D.Risk Summary: Based on its mechanism of action, COMETRIQ can cause fetal harm when administered to a pregnant woman. Cabozantinib was embryolethal in rats at exposures below the recommended human dose, with increased incidences of skeletal variations in rats and visceral variations and malformations in rabbits. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Animal Data: In an embryo-fetal development study in which pregnant rats were administered daily doses of cabozantinib during organogenesis, increased loss of pregnancy compared to controls was observed at doses as low as 0.03 mg/kg (less than 1% of the human exposure by AUC at the recommended dose). Findings included delayed ossifications and skeletal variations at doses equal to or greater than 0.01 mg/kg/day (approximately 0.03% of the human exposure by AUC at the recommended dose). In pregnant rabbits administered cabozantinib daily during organogenesis, there were findings of visceral malformations and variations including reduced splenic size and missing lung lobe at 3 mg/kg (approximately 11% of the human exposure by AUC at the recommended dose). 8.2 Nursing Mothers: It is unknown whether cabozantinib or its metabolites are excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from COMETRIQ, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. 8.3 Pediatric Use: The safety and effectiveness of COMETRIQ in pediatric patients have not been studied. 8.4 Geriatric Use: Clinical studies of COMETRIQ did not include sufficient numbers of patients aged 65 years and over to determine whether they respond differently from younger patients.8.5 Females and Males of Reproductive Potential: Contraception: Use effective contraception during treatment with COMETRIQ and up to 4 months after completion
KCosmos Communications 1
1ej
27902a 05.12.14 133
Q1 Q2
S:6.75” S:6.75”
S:9.75”
T:15”
T:10.25”
B:17.5”
B:11.25”

ASCOPost.com | MARCH 25, 2015 PAGE 15
Journal Spotlight
Gastric, Breast Cancer Risk in Carriers of CDH1 Gene Mutations
In a new study,1 more precise estimates of age-associated risks of gastric and
breast cancer were derived for carriers of the CDH1 gene mutation, a cancer-pre-disposing gene that is abnormal in families meeting criteria for clinically defined he-reditary diffuse gastric cancer (HDGC).
David G. Huntsman, MD, of the British Columbia Cancer Agency, and colleagues tested for CDH1 germline mutations in 183 new families with HDGC. Penetrance (the proportion of people with a gene mutation who will show clinical disease) was derived from
75 mutation-positive families from this and other study groups, comprising 3,858 individuals. Germline DNA from 144 HDGC families without the CDH1 mutations also was screened for 55 cancer-associated genes, to determine if other genes are associated with HDGC.
The authors identified 31 distinct CDH1 mutations in 34 of 183 families (19%). They estimate that by age 80, the cumulative incidence of gastric can-cer is 70% for men and 56% for women, with a risk of breast cancer for women of 42%. They also identified candidate mutations in 16 of 144 probands (the person who is the starting point in a family being studied).
“These data should assist in the ge-netic counseling and management of at-risk individuals from CDH1-positive HDGC families,” the study concludes.
‘Major Advance’In a related editorial,2 James M. Ford,
MD, of the Stanford University School of Medicine, said, “The study by Dr. Hunts-man and colleagues assembles the larg-est group of genetically defined HDGC families to date (75 families, comprising 3,858 individuals) to determine age-specific penetrance of gastric and breast cancer. These updated risk assessments
should be considered the new standard for genetic counseling, and will be includ-ed in the next International Gastric Can-cer Linkage Consortium guidelines.… The study provides a major advance. Fur-ther clinical and genetic research is nec-essary to identify more biomarkers and better methods of screening individuals at high risk,” he concluded. n
References1. Hansford S, Kaurah P, Li-Chang H,
et al: Hereditary diffuse gastric cancer syn-drome. JAMA Oncol. February 12, 2015 (early release online).
2. Ford JM: Hereditary gastric cancer. JAMA Oncol. February 12, 2015 (early re-lease online).
David G. Huntsman, MD
James M. Ford, MD
Genetics/Genomics
JOB#
: 483
34
CLIE
NT: E
xelix
is DE
SC: J
ourn
al Ad
A si
ze
FILE
NAM
E: E
XL_X
LX_Q
4833
4_JA
_D01
.indd
DA
TE: 5
-12-
2014
6:1
1 PM
RO
UND:
1
1 PG: P
ared
esA/
Cruz
, Feli
x AD
: L B
udre
au
PM: N
one
AE: K
Ruf
f CW
: S G
uest
La
st S
aved
: 5-1
2-20
14 6
:11
PMTR
IM: 1
5” x
10.
25”
BLEE
D: 1
7.5”
x 1
1.25
” SA
FETY
: 6.7
5” x
9.7
5”
PROD
: M H
aight
IN
K Sp
ec: 4
c PR
INT
SCAL
E: 7
5%FO
NTS:
Non
e
IMAG
ES: C
omet
riq_7
x10_
Jan1
4_BS
_p1.
pdf (
100%
), Co
met
riq_7
x10_
Jan1
4_BS
_p2.
pdf (
100%
)IN
KS:
Blac
kDO
C PA
TH: M
acint
osh
HD:U
sers
:par
edes
a:De
skto
p:EX
L_XL
X_Q4
8334
_JA_D
01.in
ddNO
TES:
Non
e
EXL_
XLX_
Q483
34_JA
_D01
.indd
Ga
lley:
2
S&H
Phar
maG
raph
ics
Disk
DATE
SIGN
OFF
PGQC
TCAD
CDCW
AE/A
SED
PROD
COMETRIQ® (cabozantinib) capsules BRIEF SUMMARY OF PRESCRIBING INFORMATION Initial U.S. Approval: 2012
1. INDICATIONS AND USAGECOMETRIQ is indicated for the treatment of patients with progressive, metastatic medullary thyroid cancer (MTC).
2. DOSAGE AND ADMINISTRATION 2.1 Recommended Dose: The recommended daily dose of COMETRIQ is 140 mg (one 80-mg and three 20-mg capsules). Do not administer COMETRIQ with food. Instruct patients not to eat for at least 2 hours before and at least 1 hour after taking COMETRIQ. Continue treatment until disease progression or unacceptable toxicity occurs. Swallow COMETRIQ capsules whole. Do not open COMETRIQ capsules. Do not take a missed dose within 12 hours of the next dose. Do not ingest foods (e.g., grapefruit, grapefruit juice) or nutritional supplements that are known to inhibit cytochrome P450 during COMETRIQ.2.2 Dosage Adjustments: For Adverse Reactions : Withhold COMETRIQ for NCI CTCAE Grade 4 hematologic adverse reactions, Grade 3 or greater non-hematologic adverse reactions or intolerable Grade 2 adverse reactions. Upon resolution/improvement of the adverse reaction (i.e., return to baseline or resolution to Grade 1), reduce the dose as follows:• If previously receiving 140-mg daily dose, resume treatment at 100 mg
daily (one 80-mg and one 20-mg capsule)• If previously receiving 100-mg daily dose, resume treatment at 60 mg daily
(three 20-mg capsules)• If previously receiving 60-mg daily dose, resume at 60 mg if tolerated,
otherwise, discontinue COMETRIQPermanently discontinue COMETRIQ for any of the following: development of visceral perforation or fistula formation; severe hemorrhage; serious arterial thromboembolic event (e.g., myocardial infarction, cerebral infarction); nephrotic syndrome; malignant hypertension, hypertensive crisis, persistent uncontrolled hypertension despite optimal medical management; osteonecrosis of the jaw; or reversible posterior leukoencephalopathy syndrome. In Patients With Hepatic Impairment : COMETRIQ is not recommended for use in patients with moderate and severe hepatic impairment. In Patients Taking CYP3A4 Inhibitors : Avoid the use of concomitant strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, nefazodone, saquinavir, telithromycin, ritonavir, indinavir, nelfinavir, voriconazole) in patients receiving COMETRIQ. For patients who require treatment with a strong CYP3A4 inhibitor, reduce the daily COMETRIQ dose by 40 mg (for example, from 140 mg to 100 mg daily or from 100 mg to 60 mg daily). Resume the dose that was used prior to initiating the CYP3A4 inhibitor 2 to 3 days after discontinuation of the strong inhibitor. In Patients Taking Strong CYP3A4 Inducers: Avoid the chronic use of concomitant strong CYP3A4 inducers (e.g., phenytoin, carbamazepine, rifampin, rifabutin, rifapentine, phenobarbital) if alternative therapy is available. Do not ingest foods or nutritional supplements (e.g., St. John’s Wort [Hypericum perforatum]) that are known to induce cytochrome P450 activity. For patients who require treatment with a strong CYP3A4 inducer, increase the daily COMETRIQ dose by 40 mg (for example, from 140 mg to 180 mg daily or from 100 mg to 140 mg daily) as tolerated. Resume the dose that was used prior to initiating the CYP3A4 inducer 2 to 3 days after discontinuation of the strong inducer. The daily dose of COMETRIQ should not exceed 180 mg.
4. CONTRAINDICATIONS None.
5. WARNINGS AND PRECAUTIONS5.1 Perforations and Fistulas: Gastrointestinal (GI) perforations and fistulas were reported in 3% and 1% of COMETRIQ-treated patients, respectively. All were serious and one GI fistula was fatal (<1%). Non-GI fistulas including tracheal/esophageal were reported in 4% of COMETRIQ-treated patients. Two (1%) of these were fatal. Monitor patients for symptoms of perforations and fistulas. Discontinue COMETRIQ in patients who experience a perforation or a fistula. 5.2 Hemorrhage: Serious and sometimes fatal hemorrhage occurred with COMETRIQ. The incidence of Grade ≥3 hemorrhagic events was higher in COMETRIQ-treated patients compared with placebo (3% vs. 1%). Do not administer COMETRIQ to patients with a recent history of hemorrhage or hemoptysis. 5.3 Thrombotic Events: COMETRIQ treatment results in an increased incidence of thrombotic events (venous thromboembolism: 6% vs. 3% and arterial thromboembolism: 2% vs. 0% in COMETRIQ-treated and placebo-treated patients, respectively). Discontinue COMETRIQ in patients who develop an acute myocardial infarction or any other clinically significant arterial thromboembolic complication.
5.4 Wound Complications: Wound complications have been reported with COMETRIQ. Stop treatment with COMETRIQ at least 28 days prior to scheduled surgery. Resume COMETRIQ therapy after surgery based on clinical judgment of adequate wound healing. Withhold COMETRIQ in patients with dehiscence or wound healing complications requiring medical intervention. 5.5 Hypertension: COMETRIQ treatment results in an increased incidence of treatment-emergent hypertension with Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (modified JNC criteria) stage 1 or 2 hypertension identified in 61% of COMETRIQ-treated patients compared with 30% of placebo-treated patients in the randomized trial. Monitor blood pressure prior to initiation and regularly during COMETRIQ treatment. Withhold COMETRIQ for hypertension that is not adequately controlled with medical management; when controlled, resume COMETRIQ at a reduced dose. Discontinue COMETRIQ for severe hypertension that cannot be controlled with anti-hypertensive therapy.5.6 Osteonecrosis of the Jaw (ONJ): Osteonecrosis of the jaw (ONJ) occurred in 1% of COMETRIQ-treated patients. ONJ can manifest as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration or erosion, persistent jaw pain or slow healing of the mouth or jaw after dental surgery. Perform an oral examination prior to initiation of COMETRIQ and periodically during COMETRIQ therapy. Advise patients regarding good oral hygiene practices. For invasive dental procedures, withhold COMETRIQ treatment for at least 28 days prior to scheduled surgery, if possible. 5.7 Palmar-Plantar Erythrodysesthesia Syndrome: Palmar-plantar erythrodysesthesia syndrome (PPES) occurred in 50% of patients treated with cabozantinib and was severe (≥Grade 3) in 13% of patients. Withhold COMETRIQ in patients who develop intolerable Grade 2 PPES or Grade 3-4 PPES until improvement to Grade 1; resume COMETRIQ at a reduced dose.5.8 Proteinuria: Proteinuria was observed in 4 (2%) patients receiving COMETRIQ, including one with nephrotic syndrome, as compared to none of the patients receiving placebo. Monitor urine protein regularly during COMETRIQ treatment. Discontinue COMETRIQ in patients who develop nephrotic syndrome. 5.9 Reversible Posterior Leukoencephalopathy Syndrome: Reversible Posterior Leukoencephalopathy Syndrome (RPLS), a syndrome of subcortical vasogenic edema diagnosed by characteristic finding on MRI, occurred in one (<1%) patient. Perform an evaluation for RPLS in any patient presenting with seizures, headache, visual disturbances, confusion or altered mental function. Discontinue COMETRIQ in patients who develop RPLS. 5.10 Drug Interactions: Avoid administration of COMETRIQ with agents that are strong CYP3A4 inducers or inhibitors. 5.11 Hepatic Impairment: COMETRIQ is not recommended for use in patients with moderate or severe hepatic impairment. 5.12 Embryo-Fetal Toxicity: COMETRIQ can cause fetal harm when administered to a pregnant woman. Cabozantinib was embryolethal in rats at exposures below the recommended human dose, with increased incidences of skeletal variations in rats and visceral variations and malformations in rabbits. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
6. ADVERSE REACTIONS6.1 Clinical Trial Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The safety of COMETRIQ was evaluated in 330 patients with progressive, metastatic medullary thyroid cancer randomized to receive 140 mg of COMETRIQ (n=214) or placebo (n=109) administered daily until disease progression or intolerable toxicity occurred in a randomized, double-blind, controlled trial. The data described below reflect a median exposure to COMETRIQ for 204 days. The population exposed to COMETRIQ was 70% male, 90% white, and had a median age of 55 years. Adverse reactions which occurred in ≥25% of COMETRIQ-treated patients occurring more frequently in the COMETRIQ arm with a between-arm difference of ≥5% included, in order of decreasing frequency: diarrhea, stomatitis, palmar-plantar erythrodysesthesia syndrome (PPES), decreased weight, decreased appetite, nausea, fatigue, oral pain, hair color changes, dysgeusia, hypertension, abdominal pain, and constipation. The most common laboratory abnormalities (>25%) were increased AST, increased ALT, lymphopenia, increased alkaline phosphatase, hypocalcemia, neutropenia, thrombocytopenia, hypophosphatemia, and hyperbilirubinemia. Grade 3-4 adverse reactions and laboratory abnormalities which occurred in ≥5% of COMETRIQ-treated patients occurring more frequently in the COMETRIQ arm with a between-arm difference of ≥2% included, in order of decreasing frequency: diarrhea, PPES, lymphopenia, hypocalcemia, fatigue, hypertension, asthenia, increased ALT, decreased weight, stomatitis, and decreased appetite (see Table 1, Table 2). Fatal adverse reactions occurred in 6% of patients receiving COMETRIQ and resulted from hemorrhage, pneumonia, septicemia, fistulas, cardiac arrest, respiratory failure, and unspecified death. Fatal adverse reactions occurred in 5% of patients receiving placebo and resulted from septicemia, pneumonia, and general deterioration. The dose was reduced in 79% of patients receiving COMETRIQ compared to 9% of patients receiving placebo. The median number of dosing delays was one in patients receiving COMETRIQ compared to none in patients receiving placebo. Adverse reactions led to study treatment discontinuation in 16% of patients receiving COMETRIQ and in 8% of patients receiving placebo. The most frequent adverse reactions leading to permanent discontinuation in patients treated with COMETRIQ were: hypocalcemia, increased lipase, PPES, diarrhea, fatigue, hypertension, nausea, pancreatitis, tracheal fistula formation, and vomiting. Increased levels of thyroid stimulating hormone (TSH) were observed in 57% of patients receiving COMETRIQ after the first dose compared to 19% of patients receiving placebo (regardless of baseline value). Ninety-two percent (92%) of patients on the COMETRIQ arm had a prior thyroidectomy, and 89% were taking thyroid hormone replacement prior to the first dose.
Table 1. Per-Patient Incidence of Selected Adverse Reactions in Protocol XL184-301 Occurring at a Higher Incidence in COMETRIQ-Treated Patients [Between-Arm Difference of ≥5% (All Grades)1 or ≥2% (Grades 3-4)]
MedDRA System Organ Class/ Preferred Terms
Cabozantinib (n=214)
Placebo (n=109)
All Grades
Grades 3-4
All Grades
Grades 3-4
GASTROINTESTINAL DISORDERSDIARRHEA 63 16 33 2
STOMATITIS2 51 5 6 0
NAUSEA 43 1 21 0
ORAL PAIN3 36 2 6 0
CONSTIPATION 27 0 6 0
ABDOMINAL PAIN4 27 3 13 1
VOMITING 24 2 2 1
DYSPHAGIA 13 4 6 1
DYSPEPSIA 11 0 0 0
HEMORRHOIDS 9 0 3 0
GENERAL DISORDERS AND ADMINISTRATION SITE CONDITIONSFATIGUE 41 9 28 3
ASTHENIA 21 6 15 1
INVESTIGATIONSDECREASED WEIGHT 48 5 10 0
METABOLISM AND NUTRITION DISORDERSDECREASED APPETITE 46 5 16 1
DEHYDRATION 7 2 2 1
MUSCULOSKELETAL AND CONNECTIVE TISSUE DISORDERSARTHRALGIA 14 1 7 0
MUSCLE SPASMS 12 0 5 0
MUSCULOSKELETAL CHEST PAIN 9 1 4 0
NERVOUS SYSTEM DISORDERSDYSGEUSIA 34 0 6 0
HEADACHE 18 0 8 0
DIZZINESS 14 0 7 0
PARESTHESIA 7 0 2 0
PERIPHERAL SENSORY NEUROPATHY
7 0 0 0
PERIPHERAL NEUROPATHY 5 0 0 0
PSYCHIATRIC DISORDERSANXIETY 9 0 2 0
RESPIRATORY, THORACIC, AND MEDIASTINAL DISORDERSDYSPHONIA 20 0 9 0
SKIN AND SUBCUTANEOUS TISSUE DISORDERSPPES5 50 13 2 0
HAIR COLOR CHANGES/ DEPIGMENTATION, GRAYING
34 0 1 0
RASH 19 1 10 0
DRY SKIN 19 0 3 0
ALOPECIA 16 0 2 0
ERYTHEMA 11 1 2 0
HYPERKERATOSIS 7 0 0 0
VASCULAR DISORDERSHYPERTENSION 33 8 4 0
HYPOTENSION 7 1 0 01 National Cancer Institute Common Terminology Criteria for Adverse Events Version 3.0. 2 Includes the following terms: stomatitis, aphthous stomatitis, mouth ulceration, mucosal inflammation.
3 Includes the following terms: oral pain, oropharyngeal pain, glossitis, burning mouth syndrome, glossodynia.
4 Includes the following terms: abdominal pain, abdominal pain lower, abdominal pain upper, abdominal rigidity, abdominal tenderness, esophageal pain.
5Palmar-plantar erythrodysesthesia syndrome.
WARNING: PERFORATIONS AND FISTULAS, and HEMORRHAGESee full prescribing information for complete boxed warning. Perforations and Fistulas: Gastrointestinal perforations occurred in 3% and fistula formation in 1% of COMETRIQ-treated patients. Discontinue COMETRIQ in patients with perforation or fistula. (5.1) Hemorrhage: Severe, sometimes fatal, hemorrhage including hemoptysis and gastrointestinal hemorrhage occurred in 3% of COMETRIQ-treated patients. Monitor patients for signs and symptoms of bleeding. Do not administer COMETRIQ to patients with severe hemorrhage. (5.2)
of therapy. Infertility: There are no data on the effect of COMETRIQ on human fertility. Cabozantinib impaired male and female fertility in animal studies. 8.6 Hepatic Impairment: Cabozantinib pharmacokinetics has not been studied in patients with hepatic impairment. There are limited data in patients with liver impairment (serum bilirubin greater than 1.5 times the upper limit of normal). COMETRIQ is not recommended for use in patients with moderate or severe hepatic impairment, as safety and efficacy have not been established. 8.7 Renal Impairment: No dose adjustment is recommended for patients with mild or moderate renal impairment. There is no experience with COMETRIQ in patients with severe renal impairment.10. OVERDOSAGE One case of overdosage was reported in a patient who inadvertently took twice the intended dose (200 mg daily) for nine days. The patient suffered Grade 3 memory impairment, Grade 3 mental status changes, Grade 3 cognitive disturbance, Grade 2 weight loss, and Grade 1 increase in BUN. The extent of recovery was not documented.17. PATIENT COUNSELING INFORMATIONSee FDA-approved patient labeling (Patient Information and Instructions for Use). Inform patients of the following: • COMETRIQ often causes diarrhea which may be severe in some cases. Inform patients
of the need to contact their healthcare provider if severe diarrhea occurs during treatment with COMETRIQ.
• COMETRIQ often causes palmar-plantar erythrodysesthesia syndrome. Advise patients to contact their healthcare provider for progressive or intolerable rash.
• COMETRIQ often causes sores in the mouth, oral pain, changes in taste, nausea or vomiting. Advise patients to contact their healthcare provider if any of these symptoms are severe or prevent patients from eating and drinking.
• COMETRIQ often causes weight loss which may be significant in some cases. Advise patients to report significant weight loss.
• To contact their healthcare provider before any planned surgeries, including dental procedures.
• COMETRIQ may interact with other drugs; advise patients to inform their healthcare provider of all prescription or nonprescription medication or herbal products that they are taking.
• Patients of childbearing potential must use effective contraception during therapy and for at least four months following their last dose of COMETRIQ.
• Breast-feeding mothers must discontinue nursing while receiving COMETRIQ therapy. • COMETRIQ should not be taken with food. Instruct patients not to eat for at least
2 hours before and at least 1 hour after taking COMETRIQ. COMETRIQ capsules should not be opened or crushed but should be taken with a full glass (at least 8 ounces) of water.
• Patients should not consume grapefruits or grapefruit juice while taking COMETRIQ treatment.
Reference ID: 3223542
Distributed by Exelixis, Inc.11/2012
© 2012 Exelixis, Inc. 210 East Grand Avenue, So. San Francisco, CA 94080 Printed in USA 11/12 [24523]
Table 2. Percent-Patient Incidence of Laboratory Abnormalities Occurring at a Higher Incidence in COMETRIQ-Treated Patients in Protocol XL184-301 [Between- Arm Difference of ≥5% (All Grades) or ≥2% (Grades 3-4)]
ADVERSE EVENT COMETRIQ (n=214) Placebo (n=109)
All Grades
Grade 3-4 All Grades Grade 3-4
CHEMISTRIES
INCREASED AST 86 3 35 2
INCREASED ALT 86 6 41 2
INCREASED ALP 52 3 35 3
HYPOCALCEMIA 52 12 27 3
HYPOPHOSPHATEMIA 28 3 10 1
HYPERBILIRUBINEMIA 25 2 14 5
HYPOMAGNESEMIA 19 1 4 0
HYPOKALEMIA 18 4 9 3
HYPONATREMIA 10 2 5 0
HEMATOLOGIC
LYMPHOPENIA 53 16 51 11
NEUTROPENIA 35 3 15 2
THROMBOCYTOPENIA 35 0 4 3
ALT, alanine aminotransferase; ALP, alkaline phosphatase; AST, aspartate aminotransferase
Nearly all COMETRIQ-treated patients (96% vs. 84% placebo) experienced elevated blood pressure and there was a doubling in the incidence of overt hypertension in COMETRIQ-treated patients over placebo-treated patients (61% vs. 30%) according to modified Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC) staging criteria. No patients developed malignant hypertension.
Table 3. Per-Patient Incidence of Hypertension in Protocol XL184-301
HYPERTENSION, JNC1 STAGE COMETRIQ N=2113 (%)
Placebo N=1073 (%)
Normal: Grade 0: Systolic <120 mmHg and Diastolic <80 mmHg
4
15
Pre-hypertension: Systolic ≥120 mmHg or Diastolic ≥80 mmHg
34
54
Stage 1: Systolic ≥140 mmHg or Diastolic ≥90 mmHg
46
25
Stage 2: Systolic ≥160 mmHg or Diastolic ≥100 mmHg
15 5
Malignant: Diastolic ≥120 mmHg
0
0
1 Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure, JAMA 2003:289:2560. Criteria applied were modified, as multiple readings were not available per timepoint, and therefore not averaged.
2 Subjects classified by highest category based on all recorded blood pressure readings beginning after the first dose through 30 days after last dose.
3Subjects with at least two blood pressure measurements after the first dose.
7. DRUG INTERACTIONS 7.1 Effect of CYP3A4 Inhibitors: Administration of a strong CYP3A4 inhibitor, ketoconazole (400 mg daily for 27 days) to healthy subjects increased single-dose plasma cabozantinib exposure (AUC0-inf) by 38%. Avoid taking a strong CYP3A4 inhibitor (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, voriconazole) when taking COMETRIQ. 7.2 Effect of CYP3A4 Inducers: Administration of a strong CYP3A4 inducer, rifampin (600 mg daily for 31 days) to healthy subjects decreased single-dose plasma cabozantinib exposure (AUC0-inf) by 77%. Avoid chronic coadministration of strong CYP3A4 inducers (e.g., dexamethasone, phenytoin, carbamazepine, rifampin, rifabutine, rifapentine, phenobarbital, St. John’s Wort) with COMETRIQ.8. USE IN SPECIFIC POPULATIONS 8.1 Pregnancy: Pregnancy Category D.Risk Summary: Based on its mechanism of action, COMETRIQ can cause fetal harm when administered to a pregnant woman. Cabozantinib was embryolethal in rats at exposures below the recommended human dose, with increased incidences of skeletal variations in rats and visceral variations and malformations in rabbits. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Animal Data: In an embryo-fetal development study in which pregnant rats were administered daily doses of cabozantinib during organogenesis, increased loss of pregnancy compared to controls was observed at doses as low as 0.03 mg/kg (less than 1% of the human exposure by AUC at the recommended dose). Findings included delayed ossifications and skeletal variations at doses equal to or greater than 0.01 mg/kg/day (approximately 0.03% of the human exposure by AUC at the recommended dose). In pregnant rabbits administered cabozantinib daily during organogenesis, there were findings of visceral malformations and variations including reduced splenic size and missing lung lobe at 3 mg/kg (approximately 11% of the human exposure by AUC at the recommended dose). 8.2 Nursing Mothers: It is unknown whether cabozantinib or its metabolites are excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from COMETRIQ, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. 8.3 Pediatric Use: The safety and effectiveness of COMETRIQ in pediatric patients have not been studied. 8.4 Geriatric Use: Clinical studies of COMETRIQ did not include sufficient numbers of patients aged 65 years and over to determine whether they respond differently from younger patients.8.5 Females and Males of Reproductive Potential: Contraception: Use effective contraception during treatment with COMETRIQ and up to 4 months after completion
KCosmos Communications 1
1ej
27902a 05.12.14 133
Q1 Q2
S:6.75” S:6.75”
S:9.75”
T:15”
T:10.25”
B:17.5”
B:11.25”

PAGE 16 The ASCO Post | MARCH 25, 2015
Gastrointestinal Cancers Symposium
Many Cancer Patients at Risk for Hepatitis B Virus Reactivation By Caroline Helwick
“In 2015, no cancer patients should be cured of their malignancy
only to die of reactivation of hepatitis B virus (HBV),” according to Anna S. Lok, MD, the Alice Lohrman Andrews Research Professor in Hepatology and Director of Clinical Hepatology at the University of Michigan, Ann Arbor.
“I recommend screening all patients before starting on chemotherapy. As-sess the risk of HBV reactivation, and if the risk is moderate or high, start pro-phylaxis as soon as possible,” Dr. Lok told attendees at the 2015 Gastrointes-tinal Cancers Symposium.
“Some oncologists argue that not all patients with reactivation will have a bad outcome and ask whether we can just monitor patients and start antivirals on demand [ie, upon signs of reactiva-tion],” she said. “But we don’t always catch things in time, and in multiple studies, outcomes for prophylaxis are better than for treatment on demand,” she explained.
Gregory J. Gores, MD, Professor of Medicine at the Mayo Clinic, Roches-ter, Minnesota, moderated the session and commented on Dr. Lok’s presenta-
tion. “There are probably a dozen differ-ent guidelines from multiple societies on the issue of HBV reactivation. The program committee of this meeting se-lected Dr. Lok to give her perspective because of her depth of knowledge in this topic and her pragmatic approach.”
Understanding HBV Reactivation
The reactivation of HBV is an abrupt increase in viral replication in a person with chronic or past HBV infection. Reactivation can occur in persons who are surface antigen–positive (HBsAg+) or surface antigen–negative (HBsAg–) and core antibody–positive (anti-HBc+) with or without surface anti-body. Many patients have normal levels
of liver enzymes and undetectable HBV DNA at baseline, but chemotherapy leads to immunosuppression and its sequelae: increases in HBV DNA, el-evations in liver enzymes, and in some cases development of acute liver failure, and even death.
Control of HBV by the host im-mune response becomes weakened in the setting of immunosuppressive therapy, allowing HBV to replicate at a faster rate. It has been most ex-tensively studied in lymphoma (with the use of the anti-CD20 antibody rituximab [Rituxan]), but it also oc-curs with other chemotherapy agents, corticosteroids alone (in high enough doses), biologics (antitumor necrosis factor-alpha [TNF] agents, interleu-kin-17, anti-CTLA4), and more clas-sic immunosuppressive agents such as methotrexate. According to case reports, even some molecularly tar-geted agents, including tyrosine kinase inhibitors, the mTOR inhibitor evero-
limus (Afinitor), and transarterial che-moembolization with cytotoxic drugs, can cause HBV reactivation.
Special ConcernsRituximab creates the most concern,
as it carries a more than fivefold in-creased risk for HBV reactivation vs oth-er regimens among HBsAg–/anti-HBc+ patients. Also concerning is the potential for delayed reactivation, up to 2 years fol-
lowing discontinuation of rituximab. Reports of 109 fatalities from HBV
reactivation associated with rituximab led the U.S. Food and Drug Administra-tion (FDA) to issue a warning in 2013 and a recommendation that all patients receiving this drug be screened at base-line for HBsAg and anti-HBc, that those testing positive be monitored during and after treatment, and that antiviral prophylaxis be considered. Patients should remain on prophylaxis for 1 year after treatment ends.
Another special group includes pa-tients with hepatocellular carcinoma, who should definitely receive an antivi-ral, according to Dr. Lok, “because they have underlying liver disease that led to the cancer and the data have shown that hepatocellular carcinoma patients on an antiviral have less recurrence and better survival. In this case, there are other rationales for the antiviral, not just for prevention of reactivation.”
Male gender and elevated HBV
DNA at baseline indicate greater sus-ceptibility to reactivation. An increased risk is also associated with high-inten-sity immunosuppressive therapy and conditioning regimens prior to bone marrow transplant, immunosuppres-sive therapy after solid organ transplant, high-dose steroids, and anthracyclines. HBsAg+ patients are more at risk than HBsAg–/anti-HBc+ patients.
Reactivation Seen in at Least 30% of HBsAg+ Patients
Determining the incidence of treat-ment-associated HBV reactivation is diffi-cult, because clinical trials tend to exclude patients with HBV infection. “But when drugs are used in the real world, problems arise, and we have little data to guide our management,” Dr. Lok indicated.
In lymphoma patients, from whom most of the data come, Dr. Lok docu-mented HBV-related hepatitis in almost half of HBsAg+ patients and in 4% of HBsAg–/anti-HBc+ patients. This “seemingly trivial” increase in enzymes triggered nonfatal liver failure in 4% and death in 4% of HBsAg+ patients.1
In a systematic review of patients receiving chemotherapy for various malignancies, HBV reactivation was observed in 38.7%, leading to hepatitis in 33%, liver failure in 13%, and death in 5.5% of patients who did not receive antiviral prophylaxis.2 Another study of HBsAg+ patients observed reactivation in 30%, including 58% with lymphoma and 25% with other malignancies.3
Optimizing Prophylaxis Among the five FDA-approved
nucleoside/nucleotide analogs (dosed once daily) are lamivudine (Epivir), adefovir (Hepsera), entecavir (Bara-clude), telbivudine (Tyzeka), and te-nofovir (Viread). Most of the data on prophylaxis are on lamivudine, the first of these agents approved.
A systematic review,2 involving 275 treated patients and 475 controls, showed that lamivudine reduced HBV reactivation and HBV-related hepatitis by 79% to 100%, reduced HBV-related liver failure by 100%, and reduced HBV-related deaths by 80% to 100%. Lamivu-dine-treated patients also were less likely to die of cancer, since chemotherapy in-terruption is less likely when reactivation is prevented, Dr. Lok noted.
The preferred agents are now en-tecavir and tenofovir, as they are the
Infectious Disease
Table: Risk-Based Prevention of HBV Reactivation
Risk HBsAg+ HBsAg–, anti-HBc+ Antiviral Therapy
High Chemotherapy, anti-CD20 or anti-CD56 treatment; IST for transplan-tation; steroids in combination with other IST
Chemotherapy for hematologic malignancies, anti-CD20 or anti-CD56 treatment
Prophylaxis
Moderate Anti-TNF treatment, maintenance low-dose steroids; other IST without steroid
Chemotherapy for solid tumors; IST for transplantation; steroids in combination with other IST
Prophylaxis or on- demand (monitor)
Low Steroids alone for a few days Anti-TNF, maintenance low-dose steroid, other IST without steroid
No prophylaxis
HBc = hepatitis B core antibody; HBsAg = hepatitis B surface antigen; IST = immunosuppressive therapy.
I recommend screening all patients before starting on chemotherapy. Assess the risk of HBV reactivation, and if the risk is moderate or high, start prophylaxis as soon as possible.
—Anna S. Lok, MD
Gregory J. Gores, MD
continued on page 18

MULTIPLE MYELOMA NEVER GIVES UP. BUT NEITHER DO WE.™
© 2014 Celgene Corporation 02/14 US-CELG140017
Celgene.com/NeverGiveUp
#mmaware@Celgene_Myeloma
to raise awareness of multiple myeloma.
Join us this March on Twitter for
31 ACTS31 FACTSJoin us this March on Twitter for
31 ACTS31 FACTS
MARCH ISMYELOMAAWARENESSMONTH
C M Y K
Cosmos Communications 1
9ej
27127a 02.19.14 133
Q1 Q2
S:9.5”S
:13
”
T:10.5”T:1
4”
B:11”B
:14
.5”
Celgene MM-Pink Ribon_US-CELG140017.indd 1 2/27/15 5:14 PM

PAGE 18 The ASCO Post | MARCH 25, 2015
Gastrointestinal Cancers Symposium
most potent viral suppressors and the least likely to cause drug resistance. In a recent randomized, controlled trial of rituximab-treated patients, entecavir was significantly more effective than la-mivudine in HBsAg+ patients in terms of HBV reactivation (6.6% vs 30%, P = .001), HBV-related hepatitis (0% vs 13.3%, P = .003), and chemotherapy disruption (1.6% vs 18.3%, P = .002).4
Algorithm to Guide Prophylaxis
An algorithm recently coauthored by Dr. Lok can guide clinicians in the prevention of HBV reactivation.5 It calls for all cancer patients to be screened before starting treatment and classifies patients as (1) HBsAg+; (2) HBsAg–, anti-HBc+; and (3) HB-sAg–, anti-HBc–.
Risk within the first two groups is further stratified by HBV DNA levels.
For HBsAg+ and HBsAg–/anti-HBc+ patients, those considered at high-risk (see table on page 16) should receive a prophylactic antiviral; moderate-risk patients can receive prophylaxis or be monitored and receive on-demand an-tivirals; low-risk patients can receive usual care, as can the third group.
For patients with undetectable or very low levels of HBV DNA, Dr. Lok does not advocate delaying chemother-apy. However, this step may be wise for patients with high levels, “because once patients are immunosuppressed, the re-sponse to the antiviral appears to be less robust,” she noted. n
Disclosure: Dr. Lok has held consulting or advisory roles for Gilead Sciences and GlaxoSmithKline and has received research funding from Gilead Sciences and Bristol-Myers Squibb. Dr. Gore reported no potential conflicts of interest.
References1. Lok AS, Liang RH, Chiu EK, et al:
Reactivation of hepatitis B virus replica-
tion in patients receiving cytotoxic thera-py: Report of a prospective study. Gastro-enterology 100:182-188, 1991.
2. Loomba R, Rowley A, Wesley R, et al: Systematic review: The effect of preven-tive lamivudine on hepatitis B reactivation during chemotherapy. Ann Intern Med 148:519-528, 2008.
3. Yeo W, Zee B, Zhong S, et al: Com-prehensive analysis of risk factors associ-ating with hepatitis B virus reactivation in cancer patients undergoing cytotoxic chemotherapy. Br J Cancer 90:1306-1311, 2004.
4. Huang H, Li X, Zhu J, et al: Entecavir vs lamivudine for prevention of hepatitis B virus reactivation among patients with untreated diffuse large B-cell lymphoma receiving R-CHOP chemotherapy: A ran-domized clinical trial. JAMA 312:2521-2530, 2014.
5. Hwang JP, Lok AS: Management of patients with hepatitis B who require im-munosuppressive therapy. Nat Rev Gas-troenterol Hepatol 11:209-219, 2014.
Hepatitis B Viruscontinued from page 16
Reactivation of Hepatitis B Virus in Patients With Cancer
■ The immunosuppressive effects of anticancer agents put patients at risk for reactivation of hepatitis B virus (HBV).
■ All cancer patients should be screened for HBV infection, and risk for reactivation of HBV should be assessed.
■ Patients at moderate to high risk of HBV reactivation should receive antiviral prophylaxis, preferably with entecavir and tenofovir, before starting treatment for cancer.
■ Rituximab is associated with the highest risk of HBV reactivation among chemotherapy treatments.
Optimal Timing of Rectal Surgery: 60 Days or Less Post Chemoradiation TherapyBy Caroline Helwick
For the treatment of locally ad-vanced rectal cancer, the optimal
timing between the end of neoadjuvant chemoradiation therapy and surgical re-section appears to be 60 days, according to an analysis of the National Cancer Database presented at the 2015 Gastro-intestinal Cancers Symposium.1
An interval of 60 days or less was associated with a higher likelihood of pathologic complete response. A lon-ger interval did not further increase re-sponse and, in fact, was associated with higher risks of positive margins and mortality, reported Jonathan C. Salo, MD, a surgical oncologist at the Levine Cancer Institute at Carolinas Medical Center, Charlotte, North Carolina.
“Several retrospective studies have shown that pathologic complete re-sponse rates increase with increasing ra-diation-surgery interval. Most retrospec-
tive studies have shown no difference in overall mortality with longer radiation-surgery interval, compared with a short-er interval, and some have even shown
improved survival in patients who had a longer interval,” Dr. Salo said.
Dr. Salo and colleagues assessed pa-tients in the National Cancer Database, which represents 70% of newly diagnosed cancer patients in the United States; con-
tains 30 million records; and provides staging, treatment, and survival data. They examined data from 6,805 patients diagnosed with rectal adenocarcinoma
between 2004 and 2006 and who un-derwent preoperative radiation therapy (most with chemotherapy), followed by radical surgical resection (excluding trans-anal excision). Surgery was done within 6 months of diagnosis and within 4 months of radiation therapy. The median follow-up for these patients was 5.6 years.
Pathologic Complete Response Optimized at 60 Days
“Increasing the radiation-surgery in-terval was associated with an increase in the rate of pathologic complete re-sponse up to 60 days (P = .0003), but the rate did not appear to increase thereafter,” Dr. Salo reported.
A longer radiation-surgery interval beyond 60 days was associated with an increase in the rate of positive surgi-cal margins. Positive margin rates were 4.1% for an interval up to 30 days; 5.4% for an interval between 30 and 44 days; 4.8% for an interval between 45 and 59 days; 6.7% for an interval between 60 and 74 days; and 7.7% for an interval more than 75 days (P = .0067).
An interval beyond 60 days was also associated with shorter overall survival. When stratified by intervals of up to 60 days vs more than 60 days, the longer interval was associated with a 25% increased mortality risk, which was highly significant (P < .0001), he reported.
Dr. Salo proposed two explanations for the relationship between longer radiation-surgery interval and worse overall survival. It is possible, he said, that an increasing interval directly affects cancer outcomes, possibly through tumor regrowth, as suggested by the increase in positive margins shown in his study.
Other Prognostic FactorsPatients with longer intervals in
the database may also have been af-fected by other factors, such as comor-
National Cancer Database Analysis on Timing of Rectal Surgery
■ Analysis of the National Cancer Database (2004–2006) found the optimal time between neoadjuvant chemoradiation therapy and surgery is around 60 days for most patients.
■ Much shorter intervals between neoadjuvant chemoradiation therapy and surgery were associated with lower rates of pathologic complete response, whereas intervals beyond 60 days were associated with increased rates of positive surgical margins and a 25% increase in mortality.
Surgical margins were a highly powerful prognostic indicator, as was postoperative length of stay.
—Jonathan C. Salo, MD
continued on page 19

THE 1ST FDA-APPROVED ANTI–PD-1 THERAPY
KEYTRUDA:
TO HELP FIGHT TUMORS
In appropriate patients with advanced melanoma
ANTI–PD-1 EFFICACY
SELECTED SAFETY INFORMATION• Immune-mediated adverse reactions
occurred with KEYTRUDA, including pneumonitis, colitis, hepatitis, hypophysitis, nephritis, hyperthyroidism, and hypothyroidism. Based on the severity of the adverse reaction, KEYTRUDA should be withheld or discontinued and corticosteroids administered. For more information regarding immune-mediated adverse reactions, please read the additional Selected Safety Information on the next page.
Please see additional Selected Safety Information on the next page and the Brief Summary of Prescribing Information on the adjacent pages.
24% overall response rate in the 2 mg/kg arm (95% confidence interval [CI], 15–34; n=89); 1 complete response (1%) and 20 partial responses (23%).
86% of patients with an objective response (n=18/21) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
Releases programmed death receptor-1 (PD-1) pathway–mediated inhibition of the immune response.
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use
INDICATIONS AND USAGE
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Immune-Mediated Pneumonitis: Pneumonitis occurred in 12 (2.9%) of 411 melanoma patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to development of pneumonitis was 5 months (range 0.3 weeks–9.9 months). The median duration was 4.9 months (range 1 week–14.4 months). Five of eight patients with Grade 2 and the one patient with Grade 3 pneumonitis required initial treatment with high-dose systemic corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. The median initial dose of high-dose corticosteroid treatment was 63.4 mg/day of prednisone or equivalent with a median duration of treatment of 3 days (range 1–34) followed by a corticosteroid taper. Pneumonitis led to discontinuation of KEYTRUDA in 3 (0.7%) patients. Pneumonitis completely resolved in seven of the nine patients with Grade 2–3 pneumonitis.
Monitor patients for signs and symptoms of pneumonitis. Evaluate patients with suspected pneumonitis with radiographic imaging and administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for moderate (Grade 2) pneumonitis, and permanently discontinue KEYTRUDA for severe (Grade 3) or life-threatening (Grade 4) pneumonitis.
Immune-Mediated Colitis: Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset of colitis was 6.5 months (range 2.3–9.8). The median duration was 2.6 months (range 0.6 weeks–3.6 months). All three patients with Grade 2 or 3 colitis were treated with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) with a median initial dose of 70 mg/day of prednisone or equivalent; the median duration of initial treatment was 7 days (range 4–41), followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to colitis. All four patients with colitis experienced complete resolution of the event.
Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for moderate (Grade 2) or severe (Grade 3) colitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) colitis.
Immune-Mediated Hepatitis: Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The time to onset was 22 days for the case of Grade 4 hepatitis which lasted 1.1 months. The patient with Grade 4 hepatitis permanently discontinued KEYTRUDA and was treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) systemic corticosteroids followed by a corticosteroid taper. Both patients with hepatitis experienced complete resolution of the event.
Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
Immune-Mediated Hypophysitis: Hypophysitis occurred in 2 (0.5%) of 411 patients, consisting of one Grade 2 and one Grade 4 case (0.2% each), in patients receiving KEYTRUDA in Trial 1. The time to onset was 1.7 months for the patient with Grade 4 hypophysitis and 1.3 months for the patient with Grade 2 hypophysitis. Both patients were treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) corticosteroids followed by a corticosteroid taper and remained on a physiologic replacement dose.
Monitor for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for moderate (Grade 2) hypophysitis, withhold or discontinue KEYTRUDA for severe (Grade 3) hypophysitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hypophysitis.
Renal Failure and Immune-Mediated Nephritis: Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. The time to onset of autoimmune nephritis was 11.6 months after the first dose of KEYTRUDA (5 months after the last dose) and lasted 3.2 months; this patient did not have a biopsy. Acute interstitial nephritis was confirmed by renal biopsy in two patients with Grades 3–4 renal failure. All three patients fully recovered renal function with treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper.
Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for moderate (Grade 2) nephritis, and permanently discontinue KEYTRUDA for severe (Grade 3), or life-threatening (Grade 4) nephritis.
Immune-Mediated Hyperthyroidism and Hypothyroidism: Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset was 1.5 months (range 0.5–2.1). The median duration was 2.8 months (range 0.9 to 6.1). One of two patients with Grade 2 and the one patient with Grade 3 hyperthyroidism required initial treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to hyperthyroidism. All five patients with hyperthyroidism experienced complete resolution of the event.
Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The median time to onset of hypothyroidism was 3.5 months (range 0.7 weeks–19 months). All but two of the patients with hypothyroidism were
treated with long-term thyroid hormone replacement therapy. The other two patients only required short-term thyroid hormone replacement therapy. No patient received corticosteroids or discontinued KEYTRUDA for management of hypothyroidism.
Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders.
Administer corticosteroids for Grade 3 or greater hyperthyroidism, withhold KEYTRUDA for severe (Grade 3) hyperthyroidism, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
Other Immune-Mediated Adverse Reactions: Other clinically important immune-mediated adverse reactions can occur.
The following clinically significant, immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA in Trial 1: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, and adrenal insufficiency.
Across clinical studies with KEYTRUDA in approximately 2000 patients, the following additional clinically significant, immune-mediated adverse reactions were reported in less than 1% of patients: myasthenic syndrome, optic neuritis, and rhabdomyolysis.
For suspected immune-mediated adverse reactions, ensure adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
Embryofetal Toxicity: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for 4 months after the last dose of KEYTRUDA.
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail above.
• Immune-mediated pneumonitis.
• Immune-mediated colitis.
• Immune-mediated hepatitis.
• Immune-mediated hypophysitis.
• Renal failure and immune-mediated nephritis.
• Immune-mediated hyperthyroidism and hypothyroidism.
• Immune-mediated adverse reactions.
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS and PRECAUTIONS section reflect exposure to KEYTRUDA in Trial 1, an uncontrolled, open-label, multiple cohort trial in which 411 patients with unresectable or metastatic melanoma received KEYTRUDA at either 2 mg/kg every 3 weeks or 10 mg/kg every 2 or 3 weeks. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 24.6 months) with a median of 10 doses (range 1 to 51). The study population characteristics were: median age of 61 years (range 18–94), 39% age 65 years or older, 60% male, 97% white, 73% with M1c disease, 8% with brain metastases, 35% with elevated LDH, 54% with prior exposure to ipilimumab, and 47% with two or more prior systemic therapies for advanced or metastatic disease.
KEYTRUDA was discontinued for adverse reactions in 9% of the 411 patients. Adverse reactions, reported in at least two patients, that led to discontinuation of KEYTRUDA were: pneumonitis, renal failure, and pain. Serious adverse reactions occurred in 36% of patients receiving KEYTRUDA. The most frequent serious adverse drug reactions reported in 2% or more of patients in Trial 1 were renal failure, dyspnea, pneumonia, and cellulitis.
Table 1 presents adverse reactions identified from analyses of the 89 patients with unresectable or metastatic melanoma who received KEYTRUDA 2 mg/kg every three weeks in one cohort of Trial 1. Patients had documented disease progression following treatment with ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This cohort of Trial 1 excluded patients with severe immune-related toxicity related to ipilimumab, defined as any Grade 4 toxicity requiring treatment with corticosteroids or Grade 3 toxicity requiring corticosteroid treatment (greater than 10 mg/day prednisone or equivalent dose) for greater than 12 weeks; a medical condition that required systemic corticosteroids or other immunosuppressive medication; a history of pneumonitis or interstitial lung disease; or any active infection requiring therapy, including HIV or hepatitis B or C. Of the 89 patients in this cohort, the median age was 59 years (range 18–88), 33% were age 65 years or older, 53% were male, 98% were white, 44% had an elevated LDH, 84% had Stage M1c disease, 8% had brain metastases, and 70% received two or more prior therapies for advanced or metastatic disease. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 15.3 months) with a median of nine doses (range 1 to 23). Fifty-one percent of patients were exposed to KEYTRUDA for greater than 6 months and 21% for greater than 1 year.
KEYTRUDA was discontinued for adverse reactions in 6% of the 89 patients. The most common adverse reactions (reported in at least 20% of patients) were fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, arthralgia, and diarrhea.
32946_BUILD.indd 1 12/9/14 4:21 PM32946_01_p.indd 5 12/10/14 12:57 PM

(% of patients)
1%
23% Partial Response (PR) (n=20)
Complete Response (CR) (n=1) 86% Of responses were ongoing in patients who responded to KEYTRUDA
(n=18/21)
Study design: A multicenter, open-label, randomized, dose-comparative study cohort of the ongoing KEYNOTE-001 Phase 1b trial in patients with unresectable or metastatic melanoma and progression of disease. Key eligibility criteria included prior treatment with ipilimumab (2 or more doses at 3 mg/kg or higher) and a BRAF or MEK inhibitor, if BRAF V600 mutation–positive; and disease progression within 24 weeks following the last dose of ipilimumab. Patients were randomized to receive 2 mg/kg (n=89) or 10 mg/kg (n=84) of KEYTRUDA every 3 weeks until unacceptable toxicity or disease progression. The major efficacy outcome measures were confirmed overall response rate, as assessed by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and duration of response. Tumor response was assessed every 12 weeks.
KEYTRUDA provided ongoing responses in patients who responded
Scan here or visit keytruda.com to learn more.
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14 keytruda.com
Questions about access for KEYTRUDA? Call The Merck Access Program at 855-257-3932.
• Among the 21 patients with an objective response, 3 (14%) had progression of disease 2.8, 2.9, and 8.2 months after initial response.
• The remaining 18 patients (86%) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
• One additional patient developed 2 new asymptomatic lesions at the first tumor assessment concurrent with a 75% decrease in overall tumor burden.
—KEYTRUDA was continued and this reduction in tumor burden was durable for 5+ months.
• There were objective responses in patients with and without BRAF V600 mutation–positive melanoma.
• Similar overall response rate results were observed in the 10-mg/kg arm.
• Patients continued treatment with KEYTRUDA until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging.
For patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor
KEYTRUDA:DURABILITY OF RESPONSE24% overall response rate (complete response+partial response)
with single-agent KEYTRUDA (95% CI, 15–34)
• Pneumonitis occurred in 12 (2.9%) of 411 patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
• Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
• Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
• Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor patients for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
• Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
• Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
• Other clinically important immune-mediated adverse reactions can occur. The following clinically significant immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, adrenal insufficiency, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
SELECTED SAFETY INFORMATION
SELECTED SAFETY INFORMATION (CONTINUED)• For suspected immune-mediated adverse reactions, ensure
adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
• Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
• The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
• It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
• Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Please see the Brief Summary of the Prescribing Information on the adjacent pages.
Data for 2 mg/kg every 3 weeks (n=89)
32946_BUILD.indd 2 12/9/14 4:21 PM32946_01_p.indd 6 12/10/14 12:57 PM
(% of patients)
1%
23% Partial Response (PR) (n=20)
Complete Response (CR) (n=1) 86% Of responses were ongoing in patients who responded to KEYTRUDA
(n=18/21)
Study design: A multicenter, open-label, randomized, dose-comparative study cohort of the ongoing KEYNOTE-001 Phase 1b trial in patients with unresectable or metastatic melanoma and progression of disease. Key eligibility criteria included prior treatment with ipilimumab (2 or more doses at 3 mg/kg or higher) and a BRAF or MEK inhibitor, if BRAF V600 mutation–positive; and disease progression within 24 weeks following the last dose of ipilimumab. Patients were randomized to receive 2 mg/kg (n=89) or 10 mg/kg (n=84) of KEYTRUDA every 3 weeks until unacceptable toxicity or disease progression. The major efficacy outcome measures were confirmed overall response rate, as assessed by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and duration of response. Tumor response was assessed every 12 weeks.
KEYTRUDA provided ongoing responses in patients who responded
Scan here or visit keytruda.com to learn more.
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14 keytruda.com
Questions about access for KEYTRUDA? Call The Merck Access Program at 855-257-3932.
• Among the 21 patients with an objective response, 3 (14%) had progression of disease 2.8, 2.9, and 8.2 months after initial response.
• The remaining 18 patients (86%) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
• One additional patient developed 2 new asymptomatic lesions at the first tumor assessment concurrent with a 75% decrease in overall tumor burden.
—KEYTRUDA was continued and this reduction in tumor burden was durable for 5+ months.
• There were objective responses in patients with and without BRAF V600 mutation–positive melanoma.
• Similar overall response rate results were observed in the 10-mg/kg arm.
• Patients continued treatment with KEYTRUDA until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging.
For patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor
KEYTRUDA:DURABILITY OF RESPONSE24% overall response rate (complete response+partial response)
with single-agent KEYTRUDA (95% CI, 15–34)
• Pneumonitis occurred in 12 (2.9%) of 411 patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
• Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
• Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
• Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor patients for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
• Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
• Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
• Other clinically important immune-mediated adverse reactions can occur. The following clinically significant immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, adrenal insufficiency, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
SELECTED SAFETY INFORMATION
SELECTED SAFETY INFORMATION (CONTINUED)• For suspected immune-mediated adverse reactions, ensure
adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
• Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
• The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
• It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
• Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Please see the Brief Summary of the Prescribing Information on the adjacent pages.
Data for 2 mg/kg every 3 weeks (n=89)
32946_BUILD.indd 2 12/9/14 4:21 PM32946_01_p.indd 2 12/10/14 12:57 PM

(% of patients)
1%
23% Partial Response (PR) (n=20)
Complete Response (CR) (n=1) 86% Of responses were ongoing in patients who responded to KEYTRUDA
(n=18/21)
Study design: A multicenter, open-label, randomized, dose-comparative study cohort of the ongoing KEYNOTE-001 Phase 1b trial in patients with unresectable or metastatic melanoma and progression of disease. Key eligibility criteria included prior treatment with ipilimumab (2 or more doses at 3 mg/kg or higher) and a BRAF or MEK inhibitor, if BRAF V600 mutation–positive; and disease progression within 24 weeks following the last dose of ipilimumab. Patients were randomized to receive 2 mg/kg (n=89) or 10 mg/kg (n=84) of KEYTRUDA every 3 weeks until unacceptable toxicity or disease progression. The major efficacy outcome measures were confirmed overall response rate, as assessed by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and duration of response. Tumor response was assessed every 12 weeks.
KEYTRUDA provided ongoing responses in patients who responded
Scan here or visit keytruda.com to learn more.
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14 keytruda.com
Questions about access for KEYTRUDA? Call The Merck Access Program at 855-257-3932.
• Among the 21 patients with an objective response, 3 (14%) had progression of disease 2.8, 2.9, and 8.2 months after initial response.
• The remaining 18 patients (86%) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
• One additional patient developed 2 new asymptomatic lesions at the first tumor assessment concurrent with a 75% decrease in overall tumor burden.
—KEYTRUDA was continued and this reduction in tumor burden was durable for 5+ months.
• There were objective responses in patients with and without BRAF V600 mutation–positive melanoma.
• Similar overall response rate results were observed in the 10-mg/kg arm.
• Patients continued treatment with KEYTRUDA until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging.
For patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor
KEYTRUDA:DURABILITY OF RESPONSE24% overall response rate (complete response+partial response)
with single-agent KEYTRUDA (95% CI, 15–34)
• Pneumonitis occurred in 12 (2.9%) of 411 patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
• Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
• Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
• Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor patients for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
• Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
• Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
• Other clinically important immune-mediated adverse reactions can occur. The following clinically significant immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, adrenal insufficiency, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
SELECTED SAFETY INFORMATION
SELECTED SAFETY INFORMATION (CONTINUED)• For suspected immune-mediated adverse reactions, ensure
adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
• Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
• The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
• It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
• Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Please see the Brief Summary of the Prescribing Information on the adjacent pages.
Data for 2 mg/kg every 3 weeks (n=89)
32946_BUILD.indd 2 12/9/14 4:21 PM32946_01_p.indd 6 12/10/14 12:57 PM
(% of patients)
1%
23% Partial Response (PR) (n=20)
Complete Response (CR) (n=1) 86% Of responses were ongoing in patients who responded to KEYTRUDA
(n=18/21)
Study design: A multicenter, open-label, randomized, dose-comparative study cohort of the ongoing KEYNOTE-001 Phase 1b trial in patients with unresectable or metastatic melanoma and progression of disease. Key eligibility criteria included prior treatment with ipilimumab (2 or more doses at 3 mg/kg or higher) and a BRAF or MEK inhibitor, if BRAF V600 mutation–positive; and disease progression within 24 weeks following the last dose of ipilimumab. Patients were randomized to receive 2 mg/kg (n=89) or 10 mg/kg (n=84) of KEYTRUDA every 3 weeks until unacceptable toxicity or disease progression. The major efficacy outcome measures were confirmed overall response rate, as assessed by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and duration of response. Tumor response was assessed every 12 weeks.
KEYTRUDA provided ongoing responses in patients who responded
Scan here or visit keytruda.com to learn more.
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14 keytruda.com
Questions about access for KEYTRUDA? Call The Merck Access Program at 855-257-3932.
• Among the 21 patients with an objective response, 3 (14%) had progression of disease 2.8, 2.9, and 8.2 months after initial response.
• The remaining 18 patients (86%) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
• One additional patient developed 2 new asymptomatic lesions at the first tumor assessment concurrent with a 75% decrease in overall tumor burden.
—KEYTRUDA was continued and this reduction in tumor burden was durable for 5+ months.
• There were objective responses in patients with and without BRAF V600 mutation–positive melanoma.
• Similar overall response rate results were observed in the 10-mg/kg arm.
• Patients continued treatment with KEYTRUDA until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging.
For patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor
KEYTRUDA:DURABILITY OF RESPONSE24% overall response rate (complete response+partial response)
with single-agent KEYTRUDA (95% CI, 15–34)
• Pneumonitis occurred in 12 (2.9%) of 411 patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
• Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
• Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
• Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor patients for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
• Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
• Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
• Other clinically important immune-mediated adverse reactions can occur. The following clinically significant immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, adrenal insufficiency, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
SELECTED SAFETY INFORMATION
SELECTED SAFETY INFORMATION (CONTINUED)• For suspected immune-mediated adverse reactions, ensure
adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
• Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
• The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
• It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
• Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Please see the Brief Summary of the Prescribing Information on the adjacent pages.
Data for 2 mg/kg every 3 weeks (n=89)
32946_BUILD.indd 2 12/9/14 4:21 PM32946_01_p.indd 2 12/10/14 12:57 PM

THE 1ST FDA-APPROVED ANTI–PD-1 THERAPY
KEYTRUDA:
TO HELP FIGHT TUMORS
In appropriate patients with advanced melanoma
ANTI–PD-1 EFFICACY
SELECTED SAFETY INFORMATION• Immune-mediated adverse reactions
occurred with KEYTRUDA, including pneumonitis, colitis, hepatitis, hypophysitis, nephritis, hyperthyroidism, and hypothyroidism. Based on the severity of the adverse reaction, KEYTRUDA should be withheld or discontinued and corticosteroids administered. For more information regarding immune-mediated adverse reactions, please read the additional Selected Safety Information on the next page.
Please see additional Selected Safety Information on the next page and the Brief Summary of Prescribing Information on the adjacent pages.
24% overall response rate in the 2 mg/kg arm (95% confidence interval [CI], 15–34; n=89); 1 complete response (1%) and 20 partial responses (23%).
86% of patients with an objective response (n=18/21) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
Releases programmed death receptor-1 (PD-1) pathway–mediated inhibition of the immune response.
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use
INDICATIONS AND USAGE
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Immune-Mediated Pneumonitis: Pneumonitis occurred in 12 (2.9%) of 411 melanoma patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to development of pneumonitis was 5 months (range 0.3 weeks–9.9 months). The median duration was 4.9 months (range 1 week–14.4 months). Five of eight patients with Grade 2 and the one patient with Grade 3 pneumonitis required initial treatment with high-dose systemic corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. The median initial dose of high-dose corticosteroid treatment was 63.4 mg/day of prednisone or equivalent with a median duration of treatment of 3 days (range 1–34) followed by a corticosteroid taper. Pneumonitis led to discontinuation of KEYTRUDA in 3 (0.7%) patients. Pneumonitis completely resolved in seven of the nine patients with Grade 2–3 pneumonitis.
Monitor patients for signs and symptoms of pneumonitis. Evaluate patients with suspected pneumonitis with radiographic imaging and administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for moderate (Grade 2) pneumonitis, and permanently discontinue KEYTRUDA for severe (Grade 3) or life-threatening (Grade 4) pneumonitis.
Immune-Mediated Colitis: Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset of colitis was 6.5 months (range 2.3–9.8). The median duration was 2.6 months (range 0.6 weeks–3.6 months). All three patients with Grade 2 or 3 colitis were treated with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) with a median initial dose of 70 mg/day of prednisone or equivalent; the median duration of initial treatment was 7 days (range 4–41), followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to colitis. All four patients with colitis experienced complete resolution of the event.
Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for moderate (Grade 2) or severe (Grade 3) colitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) colitis.
Immune-Mediated Hepatitis: Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The time to onset was 22 days for the case of Grade 4 hepatitis which lasted 1.1 months. The patient with Grade 4 hepatitis permanently discontinued KEYTRUDA and was treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) systemic corticosteroids followed by a corticosteroid taper. Both patients with hepatitis experienced complete resolution of the event.
Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
Immune-Mediated Hypophysitis: Hypophysitis occurred in 2 (0.5%) of 411 patients, consisting of one Grade 2 and one Grade 4 case (0.2% each), in patients receiving KEYTRUDA in Trial 1. The time to onset was 1.7 months for the patient with Grade 4 hypophysitis and 1.3 months for the patient with Grade 2 hypophysitis. Both patients were treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) corticosteroids followed by a corticosteroid taper and remained on a physiologic replacement dose.
Monitor for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for moderate (Grade 2) hypophysitis, withhold or discontinue KEYTRUDA for severe (Grade 3) hypophysitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hypophysitis.
Renal Failure and Immune-Mediated Nephritis: Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. The time to onset of autoimmune nephritis was 11.6 months after the first dose of KEYTRUDA (5 months after the last dose) and lasted 3.2 months; this patient did not have a biopsy. Acute interstitial nephritis was confirmed by renal biopsy in two patients with Grades 3–4 renal failure. All three patients fully recovered renal function with treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper.
Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for moderate (Grade 2) nephritis, and permanently discontinue KEYTRUDA for severe (Grade 3), or life-threatening (Grade 4) nephritis.
Immune-Mediated Hyperthyroidism and Hypothyroidism: Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset was 1.5 months (range 0.5–2.1). The median duration was 2.8 months (range 0.9 to 6.1). One of two patients with Grade 2 and the one patient with Grade 3 hyperthyroidism required initial treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to hyperthyroidism. All five patients with hyperthyroidism experienced complete resolution of the event.
Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The median time to onset of hypothyroidism was 3.5 months (range 0.7 weeks–19 months). All but two of the patients with hypothyroidism were
treated with long-term thyroid hormone replacement therapy. The other two patients only required short-term thyroid hormone replacement therapy. No patient received corticosteroids or discontinued KEYTRUDA for management of hypothyroidism.
Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders.
Administer corticosteroids for Grade 3 or greater hyperthyroidism, withhold KEYTRUDA for severe (Grade 3) hyperthyroidism, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
Other Immune-Mediated Adverse Reactions: Other clinically important immune-mediated adverse reactions can occur.
The following clinically significant, immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA in Trial 1: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, and adrenal insufficiency.
Across clinical studies with KEYTRUDA in approximately 2000 patients, the following additional clinically significant, immune-mediated adverse reactions were reported in less than 1% of patients: myasthenic syndrome, optic neuritis, and rhabdomyolysis.
For suspected immune-mediated adverse reactions, ensure adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
Embryofetal Toxicity: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for 4 months after the last dose of KEYTRUDA.
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail above.
• Immune-mediated pneumonitis.
• Immune-mediated colitis.
• Immune-mediated hepatitis.
• Immune-mediated hypophysitis.
• Renal failure and immune-mediated nephritis.
• Immune-mediated hyperthyroidism and hypothyroidism.
• Immune-mediated adverse reactions.
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS and PRECAUTIONS section reflect exposure to KEYTRUDA in Trial 1, an uncontrolled, open-label, multiple cohort trial in which 411 patients with unresectable or metastatic melanoma received KEYTRUDA at either 2 mg/kg every 3 weeks or 10 mg/kg every 2 or 3 weeks. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 24.6 months) with a median of 10 doses (range 1 to 51). The study population characteristics were: median age of 61 years (range 18–94), 39% age 65 years or older, 60% male, 97% white, 73% with M1c disease, 8% with brain metastases, 35% with elevated LDH, 54% with prior exposure to ipilimumab, and 47% with two or more prior systemic therapies for advanced or metastatic disease.
KEYTRUDA was discontinued for adverse reactions in 9% of the 411 patients. Adverse reactions, reported in at least two patients, that led to discontinuation of KEYTRUDA were: pneumonitis, renal failure, and pain. Serious adverse reactions occurred in 36% of patients receiving KEYTRUDA. The most frequent serious adverse drug reactions reported in 2% or more of patients in Trial 1 were renal failure, dyspnea, pneumonia, and cellulitis.
Table 1 presents adverse reactions identified from analyses of the 89 patients with unresectable or metastatic melanoma who received KEYTRUDA 2 mg/kg every three weeks in one cohort of Trial 1. Patients had documented disease progression following treatment with ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This cohort of Trial 1 excluded patients with severe immune-related toxicity related to ipilimumab, defined as any Grade 4 toxicity requiring treatment with corticosteroids or Grade 3 toxicity requiring corticosteroid treatment (greater than 10 mg/day prednisone or equivalent dose) for greater than 12 weeks; a medical condition that required systemic corticosteroids or other immunosuppressive medication; a history of pneumonitis or interstitial lung disease; or any active infection requiring therapy, including HIV or hepatitis B or C. Of the 89 patients in this cohort, the median age was 59 years (range 18–88), 33% were age 65 years or older, 53% were male, 98% were white, 44% had an elevated LDH, 84% had Stage M1c disease, 8% had brain metastases, and 70% received two or more prior therapies for advanced or metastatic disease. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 15.3 months) with a median of nine doses (range 1 to 23). Fifty-one percent of patients were exposed to KEYTRUDA for greater than 6 months and 21% for greater than 1 year.
KEYTRUDA was discontinued for adverse reactions in 6% of the 89 patients. The most common adverse reactions (reported in at least 20% of patients) were fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, arthralgia, and diarrhea.
32946_BUILD.indd 1 12/9/14 4:21 PM32946_01_p.indd 1 12/10/14 12:57 PM
Immunogenicity: As with all therapeutic proteins, there is the potential for immunogenicity. Because trough levels of pembrolizumab interfere with the electrochemiluminescent (ECL) assay results, a subset analysis was performed in the patients with a concentration of pembrolizumab below the drug tolerance level of the anti-product antibody assay. In this analysis, none of the 97 patients who were treated with 2 mg/kg every 3 weeks tested positive for treatment-emergent anti-pembrolizumab antibodies.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to KEYTRUDA with the incidences of antibodies to other products may be misleading.
Table 2: Laboratory Abnormalities Increased from Baseline in ≥20% of Patients with Unresectable or Metastatic Melanoma
KEYTRUDA 2 mg/kg every 3 weeks
N=89
Laboratory Test All Grades (%)
Grades 3–4 (%)
ChemistryHyperglycemia 40 2*Hyponatremia 35 9Hypoalbuminemia 34 0Hypertriglyceridemia 25 0Increased Aspartate Aminotransferase 24 2*Hypocalcemia 24 1
HematologyAnemia 55 8*
* Grade 4 abnormalities in this table limited to hyperglycemia, increased aspartate aminotransferase, and anemia (one patient each).
DRUG INTERACTIONS
No formal pharmacokinetic drug interaction studies have been conducted with KEYTRUDA.
USE IN SPECIFIC POPULATIONS
Pregnancy: Pregnancy Category D.
Risk Summary : Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus.
Animal Data: Animal reproduction studies have not been conducted with KEYTRUDA to evaluate its effect on reproduction and fetal development, but an assessment of the effects on reproduction was provided. A central function of the PD-1/PD-L1 pathway is to preserve pregnancy by maintaining maternal immune tolerance to the fetus. Blockade of PD-L1 signaling has been shown in murine models of pregnancy to disrupt tolerance to the fetus and to result in an increase in fetal loss; therefore, potential risks of administering KEYTRUDA during pregnancy include increased rates of abortion or stillbirth. As reported in the literature, there were no malformations related to the blockade of PD-1 signaling in the offspring of these animals; however, immune-mediated disorders occurred in PD-1 knockout mice.
Human IgG4 (immunoglobulins) are known to cross the placenta; therefore, pembrolizumab has the potential to be transmitted from the mother to the developing fetus. Based on its mechanism of action, fetal exposure to pembrolizumab may increase the risk of developing immune-mediated disorders or of altering the normal immune response.
Nursing Mothers: It is not known whether KEYTRUDA is excreted in human milk. No studies have been conducted to assess the impact of KEYTRUDA on milk production or its presence in breast milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
Pediatric Use: Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Geriatric Use: Of the 411 patients treated with KEYTRUDA, 39% were 65 years and over. No overall differences in safety or efficacy were reported between elderly patients and younger patients.
Renal Impairment: Based on a population pharmacokinetic analysis, no dose adjustment is needed for patients with renal impairment.
Hepatic Impairment: Based on a population pharmacokinetic analysis, no dose adjustment is needed for patients with mild hepatic impairment [total bilirubin (TB) less than or equal to ULN and AST greater than ULN or TB greater than 1 to 1.5 times ULN and any AST]. KEYTRUDA has not been studied in patients with moderate (TB greater than 1.5 to 3 times ULN and any AST) or severe (TB greater than 3 times ULN and any AST) hepatic impairment.
Females and Males of Reproductive Potential: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for at least 4 months following the last dose of pembrolizumab.
OVERDOSAGE
There is no information on overdosage with KEYTRUDA.
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
• Inform patients of the risk of immune-mediated adverse reactions that may require corticosteroid treatment and interruption or discontinuation of KEYTRUDA, including:
— Pneumonitis: Advise patients to contact their healthcare provider immediately for new or worsening cough, chest pain, or shortness of breath.
— Colitis: Advise patients to contact their healthcare provider immediately for diarrhea or severe abdominal pain.
— Hepatitis: Advise patients to contact their healthcare provider immediately for jaundice, severe nausea or vomiting, or easy bruising or bleeding.
— Hypophysitis: Advise patients to contact their healthcare provider immediately for persistent or unusual headache, extreme weakness, dizziness or fainting, or vision changes.
— Nephritis: Advise patients to contact their healthcare provider immediately for signs or symptoms of nephritis.
— Hyperthyroidism and Hypothyroidism: Advise patients to contact their healthcare provider immediately for signs or symptoms of hyperthyroidism and hypothyroidism.
• Advise patients of the importance of keeping scheduled appointments for blood work or other laboratory tests.
• Advise women that KEYTRUDA may cause fetal harm. Instruct women of reproductive potential to use highly effective contraception during and for 4 months after the last dose of KEYTRUDA.
• Advise nursing mothers not to breastfeed while taking KEYTRUDA.
For more detailed information, please read the Prescribing Information.
uspi-mk3475-iv-1409r000
Revised: 09/2014
Copyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved.
ONCO-1116177-0000 11/14
Table 1: Adverse Reactions in ≥10% of Patients with Unresectable or Metastatic Melanoma
KEYTRUDA 2 mg/kg every 3 weeks
N=89
Adverse Reaction All Grades (%)
Grade 3* (%)
General Disorders and Administration Site ConditionsFatigue 47 7Peripheral edema 17 1Chills 14 0Pyrexia 11 0
Gastrointestinal DisordersNausea 30 0Constipation 21 0Diarrhea 20 0Vomiting 16 0Abdominal pain 12 0
Respiratory, Thoracic And Mediastinal DisordersCough 30 1Dyspnea 18 2
Skin And Subcutaneous Tissue DisordersPruritus 30 0Rash 29 0Vitiligo 11 0
Metabolism and Nutrition DisordersDecreased appetite 26 0
Musculoskeletal and Connective Tissue DisordersArthralgia 20 0Pain in extremity 18 1Myalgia 14 1Back pain 12 1
Nervous System DisordersHeadache 16 0Dizziness 11 0
Blood and Lymphatic System DisordersAnemia 14 5
Psychiatric DisordersInsomnia 14 0
Infections and InfestationsUpper respiratory tract infection 11 1
*There were no Grade 5 adverse reactions reported. Of the ≥10% adverse reactions, none was reported as Grade 4.
Other clinically important adverse reactions observed in up to 10% of patients treated with KEYTRUDA were: Infections and infestations: sepsis
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use (continued)
32946_BUILD.indd 3 12/9/14 4:21 PM32946_01_p.indd 4 12/10/14 12:57 PM

THE 1ST FDA-APPROVED ANTI–PD-1 THERAPY
KEYTRUDA:
TO HELP FIGHT TUMORS
In appropriate patients with advanced melanoma
ANTI–PD-1 EFFICACY
SELECTED SAFETY INFORMATION• Immune-mediated adverse reactions
occurred with KEYTRUDA, including pneumonitis, colitis, hepatitis, hypophysitis, nephritis, hyperthyroidism, and hypothyroidism. Based on the severity of the adverse reaction, KEYTRUDA should be withheld or discontinued and corticosteroids administered. For more information regarding immune-mediated adverse reactions, please read the additional Selected Safety Information on the next page.
Please see additional Selected Safety Information on the next page and the Brief Summary of Prescribing Information on the adjacent pages.
24% overall response rate in the 2 mg/kg arm (95% confidence interval [CI], 15–34; n=89); 1 complete response (1%) and 20 partial responses (23%).
86% of patients with an objective response (n=18/21) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
Releases programmed death receptor-1 (PD-1) pathway–mediated inhibition of the immune response.
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use
INDICATIONS AND USAGE
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Immune-Mediated Pneumonitis: Pneumonitis occurred in 12 (2.9%) of 411 melanoma patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to development of pneumonitis was 5 months (range 0.3 weeks–9.9 months). The median duration was 4.9 months (range 1 week–14.4 months). Five of eight patients with Grade 2 and the one patient with Grade 3 pneumonitis required initial treatment with high-dose systemic corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. The median initial dose of high-dose corticosteroid treatment was 63.4 mg/day of prednisone or equivalent with a median duration of treatment of 3 days (range 1–34) followed by a corticosteroid taper. Pneumonitis led to discontinuation of KEYTRUDA in 3 (0.7%) patients. Pneumonitis completely resolved in seven of the nine patients with Grade 2–3 pneumonitis.
Monitor patients for signs and symptoms of pneumonitis. Evaluate patients with suspected pneumonitis with radiographic imaging and administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for moderate (Grade 2) pneumonitis, and permanently discontinue KEYTRUDA for severe (Grade 3) or life-threatening (Grade 4) pneumonitis.
Immune-Mediated Colitis: Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset of colitis was 6.5 months (range 2.3–9.8). The median duration was 2.6 months (range 0.6 weeks–3.6 months). All three patients with Grade 2 or 3 colitis were treated with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) with a median initial dose of 70 mg/day of prednisone or equivalent; the median duration of initial treatment was 7 days (range 4–41), followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to colitis. All four patients with colitis experienced complete resolution of the event.
Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for moderate (Grade 2) or severe (Grade 3) colitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) colitis.
Immune-Mediated Hepatitis: Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The time to onset was 22 days for the case of Grade 4 hepatitis which lasted 1.1 months. The patient with Grade 4 hepatitis permanently discontinued KEYTRUDA and was treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) systemic corticosteroids followed by a corticosteroid taper. Both patients with hepatitis experienced complete resolution of the event.
Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
Immune-Mediated Hypophysitis: Hypophysitis occurred in 2 (0.5%) of 411 patients, consisting of one Grade 2 and one Grade 4 case (0.2% each), in patients receiving KEYTRUDA in Trial 1. The time to onset was 1.7 months for the patient with Grade 4 hypophysitis and 1.3 months for the patient with Grade 2 hypophysitis. Both patients were treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) corticosteroids followed by a corticosteroid taper and remained on a physiologic replacement dose.
Monitor for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for moderate (Grade 2) hypophysitis, withhold or discontinue KEYTRUDA for severe (Grade 3) hypophysitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hypophysitis.
Renal Failure and Immune-Mediated Nephritis: Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. The time to onset of autoimmune nephritis was 11.6 months after the first dose of KEYTRUDA (5 months after the last dose) and lasted 3.2 months; this patient did not have a biopsy. Acute interstitial nephritis was confirmed by renal biopsy in two patients with Grades 3–4 renal failure. All three patients fully recovered renal function with treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper.
Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for moderate (Grade 2) nephritis, and permanently discontinue KEYTRUDA for severe (Grade 3), or life-threatening (Grade 4) nephritis.
Immune-Mediated Hyperthyroidism and Hypothyroidism: Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset was 1.5 months (range 0.5–2.1). The median duration was 2.8 months (range 0.9 to 6.1). One of two patients with Grade 2 and the one patient with Grade 3 hyperthyroidism required initial treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to hyperthyroidism. All five patients with hyperthyroidism experienced complete resolution of the event.
Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The median time to onset of hypothyroidism was 3.5 months (range 0.7 weeks–19 months). All but two of the patients with hypothyroidism were
treated with long-term thyroid hormone replacement therapy. The other two patients only required short-term thyroid hormone replacement therapy. No patient received corticosteroids or discontinued KEYTRUDA for management of hypothyroidism.
Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders.
Administer corticosteroids for Grade 3 or greater hyperthyroidism, withhold KEYTRUDA for severe (Grade 3) hyperthyroidism, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
Other Immune-Mediated Adverse Reactions: Other clinically important immune-mediated adverse reactions can occur.
The following clinically significant, immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA in Trial 1: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, and adrenal insufficiency.
Across clinical studies with KEYTRUDA in approximately 2000 patients, the following additional clinically significant, immune-mediated adverse reactions were reported in less than 1% of patients: myasthenic syndrome, optic neuritis, and rhabdomyolysis.
For suspected immune-mediated adverse reactions, ensure adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
Embryofetal Toxicity: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for 4 months after the last dose of KEYTRUDA.
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail above.
• Immune-mediated pneumonitis.
• Immune-mediated colitis.
• Immune-mediated hepatitis.
• Immune-mediated hypophysitis.
• Renal failure and immune-mediated nephritis.
• Immune-mediated hyperthyroidism and hypothyroidism.
• Immune-mediated adverse reactions.
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS and PRECAUTIONS section reflect exposure to KEYTRUDA in Trial 1, an uncontrolled, open-label, multiple cohort trial in which 411 patients with unresectable or metastatic melanoma received KEYTRUDA at either 2 mg/kg every 3 weeks or 10 mg/kg every 2 or 3 weeks. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 24.6 months) with a median of 10 doses (range 1 to 51). The study population characteristics were: median age of 61 years (range 18–94), 39% age 65 years or older, 60% male, 97% white, 73% with M1c disease, 8% with brain metastases, 35% with elevated LDH, 54% with prior exposure to ipilimumab, and 47% with two or more prior systemic therapies for advanced or metastatic disease.
KEYTRUDA was discontinued for adverse reactions in 9% of the 411 patients. Adverse reactions, reported in at least two patients, that led to discontinuation of KEYTRUDA were: pneumonitis, renal failure, and pain. Serious adverse reactions occurred in 36% of patients receiving KEYTRUDA. The most frequent serious adverse drug reactions reported in 2% or more of patients in Trial 1 were renal failure, dyspnea, pneumonia, and cellulitis.
Table 1 presents adverse reactions identified from analyses of the 89 patients with unresectable or metastatic melanoma who received KEYTRUDA 2 mg/kg every three weeks in one cohort of Trial 1. Patients had documented disease progression following treatment with ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This cohort of Trial 1 excluded patients with severe immune-related toxicity related to ipilimumab, defined as any Grade 4 toxicity requiring treatment with corticosteroids or Grade 3 toxicity requiring corticosteroid treatment (greater than 10 mg/day prednisone or equivalent dose) for greater than 12 weeks; a medical condition that required systemic corticosteroids or other immunosuppressive medication; a history of pneumonitis or interstitial lung disease; or any active infection requiring therapy, including HIV or hepatitis B or C. Of the 89 patients in this cohort, the median age was 59 years (range 18–88), 33% were age 65 years or older, 53% were male, 98% were white, 44% had an elevated LDH, 84% had Stage M1c disease, 8% had brain metastases, and 70% received two or more prior therapies for advanced or metastatic disease. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 15.3 months) with a median of nine doses (range 1 to 23). Fifty-one percent of patients were exposed to KEYTRUDA for greater than 6 months and 21% for greater than 1 year.
KEYTRUDA was discontinued for adverse reactions in 6% of the 89 patients. The most common adverse reactions (reported in at least 20% of patients) were fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, arthralgia, and diarrhea.
32946_BUILD.indd 1 12/9/14 4:21 PM32946_01_p.indd 1 12/10/14 12:57 PM
Immunogenicity: As with all therapeutic proteins, there is the potential for immunogenicity. Because trough levels of pembrolizumab interfere with the electrochemiluminescent (ECL) assay results, a subset analysis was performed in the patients with a concentration of pembrolizumab below the drug tolerance level of the anti-product antibody assay. In this analysis, none of the 97 patients who were treated with 2 mg/kg every 3 weeks tested positive for treatment-emergent anti-pembrolizumab antibodies.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to KEYTRUDA with the incidences of antibodies to other products may be misleading.
Table 2: Laboratory Abnormalities Increased from Baseline in ≥20% of Patients with Unresectable or Metastatic Melanoma
KEYTRUDA 2 mg/kg every 3 weeks
N=89
Laboratory Test All Grades (%)
Grades 3–4 (%)
ChemistryHyperglycemia 40 2*Hyponatremia 35 9Hypoalbuminemia 34 0Hypertriglyceridemia 25 0Increased Aspartate Aminotransferase 24 2*Hypocalcemia 24 1
HematologyAnemia 55 8*
* Grade 4 abnormalities in this table limited to hyperglycemia, increased aspartate aminotransferase, and anemia (one patient each).
DRUG INTERACTIONS
No formal pharmacokinetic drug interaction studies have been conducted with KEYTRUDA.
USE IN SPECIFIC POPULATIONS
Pregnancy: Pregnancy Category D.
Risk Summary : Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus.
Animal Data: Animal reproduction studies have not been conducted with KEYTRUDA to evaluate its effect on reproduction and fetal development, but an assessment of the effects on reproduction was provided. A central function of the PD-1/PD-L1 pathway is to preserve pregnancy by maintaining maternal immune tolerance to the fetus. Blockade of PD-L1 signaling has been shown in murine models of pregnancy to disrupt tolerance to the fetus and to result in an increase in fetal loss; therefore, potential risks of administering KEYTRUDA during pregnancy include increased rates of abortion or stillbirth. As reported in the literature, there were no malformations related to the blockade of PD-1 signaling in the offspring of these animals; however, immune-mediated disorders occurred in PD-1 knockout mice.
Human IgG4 (immunoglobulins) are known to cross the placenta; therefore, pembrolizumab has the potential to be transmitted from the mother to the developing fetus. Based on its mechanism of action, fetal exposure to pembrolizumab may increase the risk of developing immune-mediated disorders or of altering the normal immune response.
Nursing Mothers: It is not known whether KEYTRUDA is excreted in human milk. No studies have been conducted to assess the impact of KEYTRUDA on milk production or its presence in breast milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
Pediatric Use: Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Geriatric Use: Of the 411 patients treated with KEYTRUDA, 39% were 65 years and over. No overall differences in safety or efficacy were reported between elderly patients and younger patients.
Renal Impairment: Based on a population pharmacokinetic analysis, no dose adjustment is needed for patients with renal impairment.
Hepatic Impairment: Based on a population pharmacokinetic analysis, no dose adjustment is needed for patients with mild hepatic impairment [total bilirubin (TB) less than or equal to ULN and AST greater than ULN or TB greater than 1 to 1.5 times ULN and any AST]. KEYTRUDA has not been studied in patients with moderate (TB greater than 1.5 to 3 times ULN and any AST) or severe (TB greater than 3 times ULN and any AST) hepatic impairment.
Females and Males of Reproductive Potential: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for at least 4 months following the last dose of pembrolizumab.
OVERDOSAGE
There is no information on overdosage with KEYTRUDA.
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
• Inform patients of the risk of immune-mediated adverse reactions that may require corticosteroid treatment and interruption or discontinuation of KEYTRUDA, including:
— Pneumonitis: Advise patients to contact their healthcare provider immediately for new or worsening cough, chest pain, or shortness of breath.
— Colitis: Advise patients to contact their healthcare provider immediately for diarrhea or severe abdominal pain.
— Hepatitis: Advise patients to contact their healthcare provider immediately for jaundice, severe nausea or vomiting, or easy bruising or bleeding.
— Hypophysitis: Advise patients to contact their healthcare provider immediately for persistent or unusual headache, extreme weakness, dizziness or fainting, or vision changes.
— Nephritis: Advise patients to contact their healthcare provider immediately for signs or symptoms of nephritis.
— Hyperthyroidism and Hypothyroidism: Advise patients to contact their healthcare provider immediately for signs or symptoms of hyperthyroidism and hypothyroidism.
• Advise patients of the importance of keeping scheduled appointments for blood work or other laboratory tests.
• Advise women that KEYTRUDA may cause fetal harm. Instruct women of reproductive potential to use highly effective contraception during and for 4 months after the last dose of KEYTRUDA.
• Advise nursing mothers not to breastfeed while taking KEYTRUDA.
For more detailed information, please read the Prescribing Information.
uspi-mk3475-iv-1409r000
Revised: 09/2014
Copyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved.
ONCO-1116177-0000 11/14
Table 1: Adverse Reactions in ≥10% of Patients with Unresectable or Metastatic Melanoma
KEYTRUDA 2 mg/kg every 3 weeks
N=89
Adverse Reaction All Grades (%)
Grade 3* (%)
General Disorders and Administration Site ConditionsFatigue 47 7Peripheral edema 17 1Chills 14 0Pyrexia 11 0
Gastrointestinal DisordersNausea 30 0Constipation 21 0Diarrhea 20 0Vomiting 16 0Abdominal pain 12 0
Respiratory, Thoracic And Mediastinal DisordersCough 30 1Dyspnea 18 2
Skin And Subcutaneous Tissue DisordersPruritus 30 0Rash 29 0Vitiligo 11 0
Metabolism and Nutrition DisordersDecreased appetite 26 0
Musculoskeletal and Connective Tissue DisordersArthralgia 20 0Pain in extremity 18 1Myalgia 14 1Back pain 12 1
Nervous System DisordersHeadache 16 0Dizziness 11 0
Blood and Lymphatic System DisordersAnemia 14 5
Psychiatric DisordersInsomnia 14 0
Infections and InfestationsUpper respiratory tract infection 11 1
*There were no Grade 5 adverse reactions reported. Of the ≥10% adverse reactions, none was reported as Grade 4.
Other clinically important adverse reactions observed in up to 10% of patients treated with KEYTRUDA were: Infections and infestations: sepsis
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use (continued)
32946_BUILD.indd 3 12/9/14 4:21 PM32946_01_p.indd 4 12/10/14 12:57 PM

The Merck Access Program for KEYTRUDA for Injection
Visitmerckaccessprogram-keytruda.com
Call855-257-3932
• Speak with a dedicated representative Monday to Friday between 8 AM and 8 PM ET.
• Ask to be contacted by a field reimbursement associate.
OR
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14
32946_BUILD.indd 4 12/9/14 4:21 PM32946_01_p.indd 3 12/10/14 12:57 PM

ASCOPost.com | MARCH 25, 2015 PAGE 19
Gastrointestinal Cancers Symposium
bidities, poor access to care, and older age, which can adversely affect cancer outcomes. Multivariate Cox model-ing showed a number of factors asso-ciated with overall survival, including age (P < .0001), gender (P = .0034), insurance status (P = .0029), Charlson comorbidity score (P < .0001), tumor grade (P = .0004), clinical nodal status (P = .0006), pathologic tumor grade (P < .0001), pathologic nodal status (P < .0001), surgical margins (P < .0001),
length of hospital stay (P < .0001), and radiation-surgery interval (P < .0001). On the other hand, the type of resec-tion did not seem to affect long-term survival.
“Surgical margins were a highly powerful prognostic indicator, as was postoperative length of stay,” Dr. Salo noted. “Radiation dose, radiation boost, and elapsed radiation therapy time did not make a difference, but the interval between radiation therapy and surgery was important, even in the multivariate analysis, and was an
important prognosticator for overall survival.”
The study found the delay between radiation therapy and surgery was longer for patients who were older; had more comorbidities; and were Hispanic, black, or uninsured. Longer intervals related to comorbidities or poor access to care could be related to worse survival but would not explain an increase in positive surgical margins with longer intervals, he suggested. He hypothesized that the interval be-tween radiation therapy and surgery
might directly be detrimental to over-all survival, perhaps through regrowth of tumor. n
Disclosure: Dr. Salo reported no potential conflicts of interest.
Reference1. Huntington CR, Boselli D, Hill JS,
Salo JC: Optimal timing of surgical resec-tion after radiation therapy in locally ad-vanced rectal adenocarcinoma: An analy-sis of the National Cancer Database. 2015 Gastrointestinal Cancers Symposium. Ab-stract 510. Presented January 17, 2015.
EXPERT POINT OF VIEW
Nancy Kemeny, MD, Attending Physician at Memorial Sloan
Kettering Cancer Center and Professor of Medicine at Weill Cornell Medical College, New York, discussed the study at the session and noted several limita-tions: The data could be outdated (che-motherapy and surgery could be better now), some patient- and treatment-related specifics were lacking or not reported, and the pathologic complete response rate of 6.8% is lower than most current reports (12%–28%).
However, she said that this large retrospective study “has given support to the concept” that a longer interval (more than 60 days) from the end of chemoradiation therapy to surgery is necessary to increase the chance of complete response, but waiting more than 60 days decreases overall survival.”
Discussing what is already known about rectal cancer treatment and outcomes, she pointed out that the attainment of a complete response is predictive of outcomes “and may allow for more tailored treatment.”
For example, she indicated that many such patients are now avoiding sur-gery at Memorial Sloan Kettering, and results for this nonoperative management approach were report-ed at the Symposium.1 (These results
were also reported in the February 10 issue of The ASCO Post. Visit ASCOPost.com to view them.)
Elaborating on the importance of achieving a complete response, she cited a study that assessed patients 6 weeks after neoadjuvant treatment
and found that poor responders (by maximum standardized uptake val-ues on PET/CT) were less likely to achieve a complete response (7% vs 38%) and less likely to demonstrate tumor regression (16% vs 45%).2
She added that large, bulky tumors need more time to achieve a complete response, but longer intervals may not be necessary for all tumors. “High” rectal tumors may need less time; however, “low” rectal tumors may need longer intervals when sphincter
preservation is sought, she said.Dr. Kemeny indicated that al-
though it may take time to develop a complete response, in the absence of a tumor response, these longer inter-vals actually may be harmful. A longer wait time could facilitate local tumor growth and metastasis, enhance fibro-sis, and delay the resumption of post-operative chemotherapy. n
Disclosure: Dr. Kemeny reported no potential conflicts of interest.
References1. Smith JJ, Chow OS, Eaton A, et al:
Organ preservation in patients with rectal cancer with clinical complete response after neoadjuvant therapy. 2015 Gastro-intestinal Cancers Symposium. Abstract 509. Presented January 17, 2015.
2. Perez RO, Habr-Gama A, São Julião GP, et al: Optimal timing for assessment of tumor response to neoadjuvant chemo-radiation in patients with rectal cancer: Do all patients benefit from waiting lon-ger than 6 weeks? Int J Radiat Oncol Biol Phys 84:1159-1165, 2012.
Gastrointestinal Oncology
Rectal Surgerycontinued from page 18
This study has given support to the concept that a longer interval from the end of chemoradiation therapy to surgery is necessary to increase the chance of complete response, but waiting more than 60 days decreases overall survival.
—Nancy Kemeny, MD
Don’t Miss These Important Reports in This Issue of The ASCO Post
Sandra Sanchez-Reilly, MD, AGSF, FAAHPM, on Avoiding Burnout see page 54
Leah Christl, PhD, and Albert Deisseroth, MD, PhD, on Biosimilar Products see page 42
Joseph Mikhael, MD, MEd, on ‘the Big 5’ in Relapsed Myeloma see page 59
Patricia Ganz, MD, on New Breast Cancer Survivors’ Mobile App see page 78
David Hui, MD, MSc, on Physical Signs of Impending Death see page 93
Visit The ASCO Post online at ASCOPost.com

PAGE 20 The ASCO Post | MARCH 25, 2015
Journal Spotlight
Hypofractionated Whole-Breast Irradiation After Breast-Conserving Surgery Used in Up to One-Third of Eligible PatientsBy Matthew Stenger
In a study reported in JAMA, Justin E. Bekelman, MD, of the Univer-
sity of Pennsylvania Perelman School of Medicine, and colleagues found that approximately two-thirds of pa-tients with early-stage breast cancer for whom hypofractionated whole-breast irradiation (for 3–5 weeks) was en-dorsed received conventional whole-breast irradiation (for 5–7 weeks) after breast-conserving surgery.1 Health-care expenditures were significantly lower for patients receiving hypofrac-tionated whole-breast irradiation.
In 2011, the American Society for Radiation Oncology (ASTRO) prac-tice guidelines endorsed hypofraction-ated whole-breast irradiation as being of equal effectiveness for in-breast tumor control and comparable with regard to long-term adverse effects compared with conventional whole-breast irradiation in patients with early-stage breast cancer who are at least 50 years old, have patho-logic stage T1 or T2N0 disease, have no prior chemotherapy, and have radiation dose heterogeneity higher or lower than 7% of prescription dose.2 The guidelines also permitted hypofractionated whole-breast irradiation for other patients with early-stage breast cancer, including those younger than age 50.
Study DetailsIn this retrospective observational
cohort study, administrative claims data from 14 commercial health-care plans covering 7.4% of U.S. adult women in 2013 were used to classify patients with incident early-stage breast cancer treated with lumpectomy and whole-breast ir-radiation from 2008 and 2013 into a hypofractionation-endorsed cohort (n = 8,924) and a hypofractionation-per-mitted cohort (n = 6,719). The hypo-fractionation-endorsed cohort included patients 50 years of age and older without prior chemotherapy or axillary lymph
node involvement, and the hypofraction-ation-permitted cohort included patients younger than 50 years of age or those with prior chemotherapy or axillary lymph node involvement.
Hypofractionated whole-breast irra-diation was defined as 11 to 24 fractions (3–5 weeks of whole-breast irradiation), and conventional whole-breast irradia-tion was defined as 25 to 40 fractions (5–7 weeks of whole-breast irradiation). Uptake of and costs associated with hy-pofractionated whole-breast irradiation were assessed for 2008 through 2013,
covering the years before and after the re-lease of the ASTRO guidelines.
Use of hypofractionated vs conven-tional whole-breast irradiation was ana-lyzed using multivariable logistic regres-sion, with adjustment for patient, clinical, demographic, and contextual variables, including year of treatment, age, comor-bid disease, history of chemotherapy before and after lumpectomy, axillary lymph node involvement, practice set-ting, use of intensity-modulated radio-therapy, density of radiation oncologists by hospital service area, population density of county of residence, median household income, and level of educa-tional attainment.
In the hypofractionation-endorsed cohort, the mean age was 62.7 years, and 66.0% of patients were younger than age 65. In the hypofractionation-permitted cohort, the mean age was 52.7 years, 47.0% were younger than age 50, 20.4% had axillary lymph node involvement,
10.6% received chemotherapy prior to surgery, and 59.6% received chemothera-py prior to whole-breast irradiation.
Increasing Use Use of hypofractionated whole-breast
irradiation increased from 10.6% (95% confidence interval [CI] = 8.8%–12.5%) in 2008 to 34.5% (95% CI = 32.2%–36.8%) in 2013 in the hypofractionation-endorsed cohort (adjusted odds ratio [OR] per year = 1.4, P < .001) and from 8.1% (95% CI = 6.0%–10.2%) in 2008 to 21.2% (95% CI = 18.9%–23.6%) in 2013 (adjusted OR per year = 1.3, P < .001) in the hypofractionation-permitted cohort.
In adjusted analyses, younger wom-en were less likely to receive hypofrac-tionated whole-breast irradiation in the hypofractionation-endorsed cohort, with the modality being used in 15.4% of those aged 50 to 54 years, 23.5% of those aged 70 to 74 years (P < .001), and 29.4% of those older than age 75 (P < .001). Age was not significantly as-sociated with use of hypofractionated whole-breast irradiation in the hypo-fractionation-permitted cohort.
In the hypofractionation-endorsed cohort, use of hypofractionated whole-breast irradiation was more likely in the hospital outpatient setting vs freestanding facilities (21.6% vs 17.5%, adjusted OR = 1.4, P < .001). Use of intensity-modulated radiotherapy vs no intensity-modulated radiotherapy was associated with a sig-nificantly greater likelihood of use of hy-pofractionated whole-breast irradiation in both the hypofractionation-endorsed co-hort (27.1% vs 19.5%, adjusted OR = 1.5, P < .001) and the hypofractionation-per-mitted cohort (18.2% vs 12.2%, adjusted OR = 1.6, P < .001).
CostsAdjusted mean total health-care ex-
penditures (adjusted for inflation to 2013) in the 1 year after diagnosis were $28,747 for hypofractionated whole-breast irradiation and $31,641 for con-
ventional whole-breast irradiation in the hypofractionation-endorsed cohort (dif-ference = $2,894, P < .001) and $64,273 for hypofractionated whole-breast ir-radiation and $72,860 for conventional whole-breast irradiation in the hypofrac-tionation-permitted cohort (difference = $8,587, P < .001). Estimated savings with hypofractionated whole-breast irradia-tion were 9.1% in the hypofractionation-endorsed cohort and 11.8% in the hypo-fractionation-permitted cohort. There were no differences in mean total patient out-of-pocket expenses between hypo-fractionated and conventional whole-breast irradiation in either cohort.
Adjusted mean radiotherapy-related expenditures within the 3.5-month ra-diation period were significantly lower for hypofractionated whole-breast ir-radiation than for conventional whole-breast irradiation in both the hypofrac-tionation-endorsed cohort ($12,622 vs $16,961, difference = $4,338, P < .001) and the hypofractionation-permitted co-hort ($14,974 vs $19,762, difference = $4,785, P < .001). Patient out-of-pocket radiotherapy-related expenses were low-er with hypofractionated whole-breast ir-radiation in both cohorts ($617 vs $746, P < .001; $519 vs $619, P = .04).
The investigators concluded: “Hy-pofractionated whole-breast irradiation after breast-conserving surgery increased among women with early-stage breast cancer in 14 U.S. commercial health-care plans between 2008 and 2013. How-ever, only 34.5% of patients with hypo-fractionation-endorsed and 21.2% with hypofractionation-permitted early-stage breast cancer received hypofractionated whole-breast irradiation in 2013.” n
Disclosure: For full disclosures of the study authors, visit jama.jamanetwork.com.
References1. Bekelman JE, Sylwestrzak G, Barron J,
et al: Uptake and costs of hypofractionated vs conventional whole breast irradiation after breast conserving surgery in the United States, 2008-2013. JAMA 312:2542-2550, 2014.
2. Smith BD, Bentzen SM, Correa CR, et al: Fractionation for whole breast irradiation: An American Society for Radiation Oncol-ogy (ASTRO) evidence-based guideline. Int J Radiat Oncol 81:59-68, 2011.
See commentary by Lori J. Pierce, MD, on page 23.
Breast Cancer
Hypofractionated whole-breast irradiation after breast-conserving surgery increased among women with early-stage breast cancer in 14 U.S. commercial health-care plans between 2008 and 2013.
—Justin E. Bekelman, MD, and colleagues
Hypofractionated Whole-Breast Irradiation After Breast-Conserving Surgery
■ In 2013, 34.5% of “hypofractionation-endorsed” and 21.2% of “hypofractionation-permitted” patients received hypofractionated whole-breast irradiation (ie, 3–5 weeks of radiotherapy as opposed to 5–7 weeks).
■ Hypofractionated whole-breast irradiation was associated with cost savings.

Advertisement not displayed in digital edition at advertiser’s request

Advertisement not displayed in digital edition at advertiser’s request

ASCOPost.com | MARCH 25, 2015 PAGE 23
Perspective
Increasing the Use of Hypofractionated Radiation in Early-Stage Breast Cancer: The Way ForwardBy Lori J. Pierce, MD
B ekelman and colleagues are to be congratulated on the publication
of an important paper—reviewed in this issue of The ASCO Post—alerting us all to the underutilization of hypo-fractionated whole-breast irradiation in the treatment of early-stage breast cancer.1
As background, recent randomized radiation trials have proven what pre-clinical radiation biology studies and biologic models previously predicted. Specifically, these trials demonstrated that delivering fewer large daily ra-diation doses (hypofractionation) to a moderate total dose over approxi-mately 4 weeks resulted in clinical outcomes comparable to those with conventional fractionation, which uses smaller daily fractions to higher total doses over 5 to 6 weeks.2-4
Preclinical laboratory studies in conjunction with linear-quadratic modeling suggested that short-course hypofractionation would result in high rates of tumor control while min-imizing normal-tissue toxicity.5,6 This was indeed demonstrated in four re-cent randomized trials. These results were unexpected, since early studies using hypofractionation to high total doses resulted in increased rates of late normal-tissue toxicity and mor-bidity. This, in turn, led to the aban-donment of using large daily doses and the adoption of smaller fractions as standard practice in most radia-tion centers. However, an important difference between the earlier hypo-fractionation schedules and the more recent hypofractionation trials was the reduction in the total dose in the recent trials, which, as predicted by biologic models, resulted in low rates of normal-tissue toxicity.
ASTRO Task ForceIn 2009, the American Society for
Radiation Oncology (ASTRO) con-vened a task force to review the data from the randomized hypofraction-ation trials and generate an evidence-based guideline for treatment rec-ommendations for early-stage breast
cancer.7 There was clear consensus among task force members that the data demonstrated no significant dif-ference in the rates of tumor control in the breast, cosmesis, or complications between hypofractionated radiation and conventional fractionation in women 50 years and older with patho-logic T1,2 N0 cancers who did not re-ceive systemic chemotherapy and in whom dose homogeneity was within 7% of the prescribed dose. These pa-tients represented the majority of the patients entered in the randomized trials. A consensus was not reached for other patient subgroups underrep-resented in the trials. This guideline was published in 2011.
A Closer Look at the Bekelman Study
Bekelman et al now report the rates of usage of hypofractionated radiotherapy using claims data from
14 commercial health-care plans in the United States in patients treated with breast-conserving surgery and radiotherapy from 2008 to 2013.1 They show usage in 34.5% of the hypofractionation-“endorsed” co-hort and 21.2% in those women for whom a consensus by the ASTRO task force was not reached (ie, the hypofractionation-“permitted” co-hort). These rates of hypofraction-ated whole-breast irradiation were low despite equivalent results dem-onstrated in the clinical trials and the reduction in the adjusted mean total health-care expenditures associated with the shorter course.
The methodology in their study was rigorous, but the data were ret-rospective and based on claims thus subject to misclassification bias. The authors were unable to ascertain many
of the clinical and pathologic factors (such as tumor size, nodal status, and dose homogeneity) important in the selection of patients in some of the randomized trials and important in the development of the ASTRO guideline. As the authors noted, mis-classification would have biased the estimates of the use of hypofraction-ated radiotherapy downward.
The MROQC InitiativeThe utilization of hypofraction-
ated whole-breast irradiation in early-stage breast cancer patients treated with breast conservation was also recently reported by Jagsi et al using data prospectively collected in the state of Michigan from radiation on-cology practices participating in the Michigan Radiation Oncology Qual-ity Consortium (MROQC).8 This is a quality initiative funded by Blue Cross Blue Shield of Michigan.
Of MROQC participants regis-tered between October 2011 and December 2013 with T1-2 N0 can-cers, 31% were identified as having re-ceived hypofractionated whole-breast irradiation. When patients were fur-ther selected using factors consistent with the ASTRO-endorsed cohort (ie, age 50 years and older, no receipt of chemotherapy, and breast separa-tion < 25 cm [a metric used to assess dose homogeneity]), the rate of hy-pofractionation use was increased to 43%. Although this rate was higher than that in the unselected cohort, these results demonstrated a lower rate of hypofractionated radiotherapy utilization than expected in patients who were excellent candidates for this fractionation schedule.
The reasons behind the low utiliza-tion rate of hypofractionated whole-
breast irradiation are unclear. The financial differential for radiation on-cology practices has been suggested as a contributing factor. Providers could also have been hesitant to use large fractions because of persistent con-cerns over placing patients at risk for the long-term toxicities observed in the hypofractionation studies of the past. Even in the ASTRO evidence-based guideline (published before the publication of the 10-year re-sults of the START trials), task force members had “lingering uncertainty regarding late effects of hypofrac-tionated whole-breast irradiation on cardiac function.”7 Thus, the relative “newness” of the hypofractionation schedules in the randomized trials may have tempered enthusiasm. Per-haps the low uptake was due in part to patient decisions not to choose the more-recent fractionation schemes with 10-year follow-up, when 20+ years of outcomes data were available for conventional whole-breast frac-tionated therapy.
Increasing AwarenessAt present, we don’t understand
the reason(s) leading to the under-utilization of the short-course hy-pofractionated regimens: this must be carefully studied. However, if the increased trajectory for use observed among the hypofractionation-en-dorsed group in the last year of the Bekelman et al study is sustained, it suggests that large-fraction radiother-apy is being integrated into routine American practice. Continued edu-cation regarding the effectiveness of the shorter 4-week course, which of-fers greater patient convenience and health-care savings, will hopefully result in the increased use of this frac-tionation schedule.
ASTRO’s Choosing Wisely cam-paign encourages discussion of whole-breast hypofractionation in women 50 years and older diagnosed with early-stage disease.9 An update of the ASTRO guidelines is also an-ticipated in the coming months, and this will increase awareness of the clinical utility and practicality of the shorter course. Finally, prospective analyses from clinical trials and ob-servational studies such as MROQC,
Breast Cancer
Dr. Pierce is Professor of Radiation Oncol-ogy at the University of Michigan School of Medicine in Ann Arbor and Director of the Michigan Radiation Oncology Quality Consortium.
Continued education regarding the effectiveness of the shorter 4-week course, which offers greater patient convenience and health-care savings, will hopefully result in the increased use of this fractionation schedule.
—Lori J. Pierce, MD
continued on page 24

PAGE 24 The ASCO Post | MARCH 25, 2015
Perspective
as well as retrospective analyses from studies such as that by Bekelman et al, will also help to educate health-care providers and the patients we serve. n
Disclosure: Dr. Pierce reported no potential conflicts of interest.
References1. Bekelman JE, Sylwestrzak G, Bar-
ron J, et al: Uptake and costs of hypofrac-tionated vs conventional whole breast ir-radiation after breast conserving surgery
in the United States, 2008-2013. JAMA 312:2542-2550, 2014.
2. Whelan TJ, Pignol JP, Levine MN, et al: Long-term results of hypofraction-ated radiation therapy for breast cancer. N Engl J Med 362:513-520, 2010.
3. Owen JR, Ashton A, Bliss JM, et al: Effect of radiotherapy fraction size on tu-mour control in patients with early-stage breast cancer after local tumor excision: Long-term results of a randomised trial. Lancet Oncol 7:467-471, 2006.
4. Haviland JS, Owen JR, Dewar JA, et al: The UK Standardisation of Breast Radiotherapy (START) trials of radio-
therapy hypofractionation for treatment of early breast cancer: 10-year follow-up results of two randomised controlled tri-als. Lancet Oncol 14:1086-1094, 2013.
5. Fowler JF: The linear-quadratic for-mula and progress in fractionated radio-therapy. Br J Radiol 62:679-694, 1989.
6. Whelan TJ, Kim DH, Sussman J: Clinical experience using hypofraction-ated radiation schedules in breast cancer. Semin Radiat Oncol 18:257-264, 2008.
7. Smith BD, Bentzen SM, Correa CR, et al: Fractionation for whole breast irra-diation: An American Society for Radia-tion Oncology (ASTRO) evidence-based
guideline. Int J Radiat Oncol Biol Phys 81:59-68, 2011.
8. Jagsi R, Griffith KA, Heimburger D, et al: Choosing wisely? Patterns and correlates of the use of hypofractionated whole-breast radiation therapy in the state of Michigan. Int J Radiat Oncol Biol Phys 90:1010-1016, 2014.
9. American Society for Radiation Oncology: Five things physicians and patients should question. Available at: http://www.choosing wisely.org/doc-tor-patient-lists/american-society-for-radiation-oncology. Accessed January 29, 2015.
Lori J. Pierce, MDcontinued from page 23
New Analysis Reports Increased Risk of Thyroid Cancer After Breast Cancer Diagnosis
Breast cancer survivors are at increased risk of developing thyroid cancer, es-
pecially within 5 years of their breast can-cer diagnosis, according to a new analysis of a large national database. The study results were presented at the Endocrine Society’s 97th Annual Meeting.1
“Recognition of this association be-tween breast and thyroid cancer should prompt vigilant screening for thyroid can-cer among breast cancer survivors,” said lead investigator Jennifer Hong Kuo, MD, Assistant Professor of Surgery at Co-lumbia University.
Until now, Dr. Kuo said, the relation-
ship between breast and thyroid cancer has been controversial, largely based on single-institution studies that have sug-gested a possible increase in thyroid can-cer incidence after breast cancer.
Study DetailsThe researchers used the National
Cancer Institute’s (NCI) Surveillance, Epidemiology, and End Results (SEER) 9 database to identify the number of indi-viduals with a diagnosis of breast and/or thyroid cancer between 1973 and 2011. They found 704,402 patients with only breast cancer, 49,663 patients with only
thyroid cancer, and 1,526 patients who de-veloped thyroid cancer after breast cancer.
Compared with patients with breast cancer alone, women who had breast cancer followed by thyroid cancer were younger on average when diagnosed with their breast cancer. They also were more likely to have had invasive ductal carcino-ma and to have received radiation therapy as part of their breast cancer treatment.
There was no difference in risk based on whether the breast cancer was hor-mone receptor–positive or had spread to lymph nodes.
Compared with patients who had only thyroid cancer, breast cancer survi-vors who developed thyroid cancer were more likely to have a more aggressive type of thyroid cancer, but the cancers were smaller in size and fewer patients required additional radioactive iodine treatment. Because thyroid cancer tends to occur at younger ages than breast cancer does, breast cancer survivors who then devel-oped thyroid cancer were older on average than those with only thyroid cancer: 62 vs 45 years, respectively, Dr. Kuo reported.
Next Steps in Screening The study findings showed that breast
cancer survivors developed thyroid cancer at a median of 5 years. Therefore, Dr. Kuo recommended that every year for the first 5 years after a breast cancer diagnosis, pa-tients should undergo a dedicated thyroid exam—especially survivors who received radiation therapy. Radiation therapy to the head, neck, or chest is a known risk factor for thyroid cancer, according to the Endo-
crine Society’s Hormone Health Network.Dr. Kuo said she plans to study wheth-
er tamoxifen treatment, typically given for 5 years after a breast cancer diagnosis, may play a role in increasing the risk of thyroid cancer. n
Disclosure: The study authors reported no potential conflicts of interest.
Reference1. Kuo JH, Chabot J, Terry MB, et al: In-
creased incidence of thyroid cancer among breast cancer survivors: An analysis of the SEER 9-database. Poster THR-049. Pre-sented at the Endocrine Society’s 97th An-nual Meeting, March 5–8, 2015, San Diego, California.
Journal Spotlight
Thyroid Cancer Risk
Women who had breast can-cer followed by thyroid
cancer were younger on average when diagnosed with their breast cancer than those with breast cancer alone.
They also were more likely to have had invasive ductal carcino-ma and to have received radiation therapy as part of their breast cancer treatment.
Researchers plan to study whether tamoxifen treatment, typically given for 5 years after a breast cancer diagnosis, may play a role in increasing the risk of thy-roid cancer. n
Apoptotic HeLa Cell—Scanning electron micrograph of an apoptotic HeLa cell. Source: National Institutes of Health.
Send your high-resolution image and caption to [email protected]. Please include your name and a caption for the photo.
Images in Oncology

CLIENT: Lilly FINISH SIZE: 21.75” wide x 14” high
JOB#: ELRALU 27020 (Tabloid) ARTIST: MC
LOCATION: Mechanicals COLLECT DATE:
R|O|U|T|I|N|G| |S|T|A|G|E M8ED CW AD CPM ACD AE inserv
SINCE THE APPROVAL OF DOCETAXEL IN 1999, NO SECOND-LINE REGIMEN HAS EXTENDED OVERALL SURVIVAL VERSUS DOCETAXEL ACROSS A BROAD POPULATION OF METASTATIC NSCLC PATIENTS1-3
NSCLC=non-small cell lung cancer.
DID YOU KNOW?
B:14
.25
in
B:11 inT:
14 in
T:10.875 inS:
13 in
S:9.5 in
27020_ELRALU_NowApproved_LUNG_JournAd_Sprd_SnglPg_TAB_M8.indd 2 1/9/15 6:29 PM

OS
PR
OB
AB
ILIT
Y
1.0
0.8
0.6
0.4
0.2
0.0
0 3 6 9 12 15 18 21 24 27 30 33 36
TIME FROM RANDOMIZATION (MONTHS)
527 415 329 231 156 103628
625
CYRAMZA+ docetaxelPlacebo+ docetaxel 501 386 306 197 129 86
Number at Risk
70 45 23 11 2 0
56 36 23 9 0 0
CYRAMZA+ docetaxel
Placebo+ docetaxel
Hazard Ratio (95% CI)=0.86 (0.75, 0.98); P=0.024
10.5MONTHS
CYRAMZA+ docetaxel
(n=628)
Placebo+ docetaxel(n=625)
15% INCREASEIN MEDIAN OS
(9.5, 11.2)
MONTHS9.1
(8.4, 10.0)
OVERALL SURVIVAL: MEDIAN - MONTHS (95% CI) MAJOR OUTCOME MEASURE
VISIT www.CYRAMZAHCP.com
ADVANCING THE SECOND-LINE TREATMENT OF METASTATIC NSCLC4
• The percentage of deaths at the time of analysis was 68% (428 patients) and 73% (456 patients) in the CYRAMZA plus docetaxel and placebo plus docetaxel arms, respectively4
Demonstrated improvements across all three efficacy outcomes (OS, PFS, ORR)4
• Median PFS with CYRAMZA plus docetaxel was 4.5 months (95% CI: 4.2, 5.4) vs 3.0 months (95% CI: 2.8, 3.9) with placebo plus docetaxel (hazard ratio 0.76 [95% CI: 0.68, 0.86]; P<0.001)
— The percentage of events at the time of analysis was 89% (558 patients) and 93% (583 patients) in the CYRAMZA plus docetaxel and placebo plus docetaxel arms, respectively
• ORR with CYRAMZA plus docetaxel was 23% (95% CI: 20, 26) vs 14% (95% CI: 11, 17) with placebo plus docetaxel (P<0.001)*
CI=confidence interval; OS=overall survival; PFS=progression-free survival; ORR=objective response rate.
*ITT population. Disease progression and tumor response were assessed by investigators in accordance with Response Evaluation Criteria in Solid Tumors (RECIST) 1.1.5 ORR is defined as complete plus partial response.
REVEL TRIAL DESIGN (N=1253)The phase III REVEL trial evaluated the efficacy and safety of CYRAMZA plus docetaxel vs placebo plus docetaxel in patients with metastatic NSCLC with disease progression on or after platinum-based chemotherapy. Major efficacy outcome measure was OS. Supportive efficacy outcome measures were PFS and ORR. All patients were required to have Eastern Cooperative Oncology Group performance status 0 or 1. Patients were randomized 1:1 (N=1253) to receive either CYRAMZA 10 mg/kg or placebo, in combination with docetaxel at 75 mg/m2 every 21 days.4
CYRAMZA PLUS DOCETAXEL DEMONSTRATED A STATISTICALLYSIGNIFICANT IMPROVEMENT IN OVERALL SURVIVAL VS DOCETAXEL4
CYRAMZA is the first antiangiogenic agent FDA approvedin combination with docetaxel for the second-line treatment of metastatic NSCLC, including nonsquamous and squamous histologies.4
WARNING: HEMORRHAGECYRAMZA increased the risk of hemorrhage, including severe and sometimes fatal hemorrhagic events. Permanently discontinue CYRAMZA in patients who experience severe bleeding.
Warnings and PrecautionsHemorrhage • CYRAMZA increased the risk of hemorrhage and gastrointestinal
hemorrhage, including severe and sometimes fatal hemorrhagic events. In Study 3, which evaluated CYRAMZA plus docetaxel in metastatic non-small cell lung cancer (NSCLC), the incidence of severe bleeding was 2.4% for CYRAMZA plus docetaxel and 2.3% for placebo plus docetaxel. Patients with NSCLC receiving therapeutic anticoagulation or chronic therapy with NSAIDs or other antiplatelet therapy other than once-daily aspirin or with radiographic evidence of major airway or blood vessel invasion or intratumor cavitation were excluded from Study 3; therefore, the risk of pulmonary hemorrhage in these groups of patients is unknown. Permanently discontinue CYRAMZA in patients who experience severe bleeding.
Arterial Thromboembolic Events • Serious, sometimes fatal, arterial thromboembolic events (ATEs)
including myocardial infarction, cardiac arrest, cerebrovascular accident, and cerebral ischemia occurred in clinical trials including 1.7% of 236 patients who received CYRAMZA as a single agent for gastric cancer in Study 1. Permanently discontinue CYRAMZA in patients who experience a severe ATE.
Hypertension• An increased incidence of severe hypertension occurred in
patients receiving CYRAMZA plus docetaxel (6%) as compared to placebo plus docetaxel (2%). Control hypertension prior to initiating treatment with CYRAMZA. Monitor blood pressure every 2 weeks or more frequently as indicated during treatment. Temporarily suspend CYRAMZA for severe hypertension until medically controlled. Permanently discontinue CYRAMZA if medically significant hypertension cannot be controlled with
antihypertensive therapy or in patients with hypertensive crisis or hypertensive encephalopathy.
Infusion-Related Reactions • Prior to the institution of premedication recommendations across
clinical trials of CYRAMZA, infusion-related reactions (IRRs) occurred in 6 out of 37 patients (16%), including 2 severe events. The majority of IRRs across trials occurred during or following a first or second CYRAMZA infusion. Symptoms of IRRs included rigors/tremors, back pain/spasms, chest pain and/or tightness, chills, flushing, dyspnea, wheezing, hypoxia, and paresthesia. In severe cases, symptoms included bronchospasm, supraventricular tachycardia, and hypotension. Monitor patients during the infusion for signs and symptoms of IRRs in a setting with available resuscitation equipment. Immediately and permanently discontinue CYRAMZA for Grade 3 or 4 IRRs.
Gastrointestinal Perforations• CYRAMZA is an antiangiogenic therapy that can increase the
risk of gastrointestinal perforation, a potentially fatal event. In Study 3, the incidence of gastrointestinal perforation was 1% for CYRAMZA plus docetaxel versus 0.3% for placebo plus docetaxel. Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation.
Impaired Wound Healing• CYRAMZA has not been studied in patients with serious or
nonhealing wounds. CYRAMZA is an antiangiogenic therapy with the potential to adversely affect wound healing. Withhold CYRAMZA prior to surgery. Resume CYRAMZA following the surgical intervention based on clinical judgment of adequate wound healing. If a patient develops wound healing complications during therapy, discontinue CYRAMZA until the wound is fully healed.
Clinical Deterioration in Child-Pugh B or C Cirrhosis• Clinical deterioration, manifested by new onset or worsening
encephalopathy, ascites, or hepatorenal syndrome, was reported in patients with Child-Pugh B or C cirrhosis who received single-agent CYRAMZA. Use CYRAMZA in patients with Child-Pugh B or C cirrhosis only if the potential benefits of treatment are judged to outweigh the risks of clinical deterioration.
Reversible Posterior Leukoencephalopathy Syndrome (RPLS)• RPLS has been reported at a rate of <0.1% in clinical studies
with CYRAMZA. Confirm the diagnosis of RPLS with MRI and discontinue CYRAMZA in patients who develop RPLS. Symptoms may resolve or improve within days, although some patients with RPLS can experience ongoing neurologic sequelae or death.
Most Common Adverse Reactions• The most commonly reported adverse reactions (all grades; Grade
3/4) occurring in ≥5% of patients receiving CYRAMZA plus docetaxel and ≥2% higher than placebo plus docetaxel in Study 3 were neutropenia (55% vs 46%; 49% vs 40%), fatigue/asthenia (55% vs 50%; 14% vs 11%), stomatitis/mucosal inflammation (37% vs 19%; 7% vs 2%), epistaxis (19% vs 7%; <1% vs <1%), febrile neutropenia (16% vs 10%; 16% vs 10%), peripheral edema (16% vs 9%; 0% vs <1%), thrombocytopenia (13% vs 5%; 3% vs <1%), lacrimation increased (13% vs 5%; <1% vs 0%), and hypertension (11% vs 5%; 6% vs 2%).
• The most common serious adverse events with CYRAMZA plus docetaxel in Study 3 were febrile neutropenia (14%), pneumonia (6%), and neutropenia (5%). The use of granulocyte colony-stimulating factors was 42% in CYRAMZA plus docetaxel-treated patients versus 37% in patients who received placebo plus docetaxel.
• Treatment discontinuation due to adverse reactions occurred more frequently in CYRAMZA plus docetaxel-treated patients (9%) than in placebo plus docetaxel-treated patients (5%). The most common adverse events leading to treatment discontinuation of CYRAMZA were infusion-related reaction (0.5%) and epistaxis (0.3%).
• Clinically relevant adverse reactions reported in ≥1% and <5% of CYRAMZA plus docetaxel-treated patients in Study 3 were hyponatremia (4.8% CYRAMZA plus docetaxel versus 2.4% for placebo plus docetaxel) and proteinuria (3.3% CYRAMZA plus docetaxel versus 0.8% placebo plus docetaxel).
Drug Interactions• No pharmacokinetic interactions were observed between
ramucirumab and docetaxel.
Use in Specific Populations• Pregnancy Category C: Based on its mechanism of action,
CYRAMZA may cause fetal harm. Advise females of reproductive potential to avoid getting pregnant, including use of adequate contraception, while receiving CYRAMZA and for at least 3 months after the last dose of CYRAMZA. Animal models link angiogenesis, VEGF and VEGF Receptor 2 to critical aspects of female reproduction, embryofetal development, and postnatal development. There are no adequate or well-controlled studies of ramucirumab in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus.
• Nursing Mothers: It is recommended to discontinue nursing or discontinue CYRAMZA due to the potential risks to the nursing infant.
• Females of Reproductive Potential: Advise females of reproductive potential that CYRAMZA may impair fertility.
Please see Brief Summary of Prescribing Information for CYRAMZA, including Boxed Warning for hemorrhage, on the next page.
RB-L HCP ISI 17DEC2014
References: 1. Reck M, Kaiser R, Mellemgaard A, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomized controlled trial. Lancet Oncol. 2014;15:143-155. 2. Supplement to: Reck M, Kaiser R, Mellemgaard A, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomized controlled trial. Lancet Oncol. 2014;15:143-155. 3. National Cancer Institute. Cancer drug information. FDA approval for docetaxel. http://www.cancer.gov/cancertopics/druginfo/fda-docetaxel/print. Accessed August 26, 2014. 4. CYRAMZA (ramucirumab) [package insert]. Indianapolis, IN: Eli Lilly and Company; 2014. 5. Garon EB, Ciuleanu T-E, Arrieta O, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet. 2014;384(9944):665-673.
RB93740 12/2014 PRINTED IN USA © Lilly USA, LLC 2014. ALL RIGHTS RESERVED.CYRAMZA® is a registered trademark of Eli Lilly and Company.
IMPORTANT SAFETY INFORMATION FOR CYRAMZA
CYRAMZA® (ramucirumab), in combination with docetaxel, is indicated for the treatment of patients with metastatic NSCLC with disease progression on or after platinum-based chemotherapy. Patients with epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving CYRAMZA.
NEW FDA APPROVAL
B:14.25 inB:14
.25
in
B:11 in B:11 in
T:14 inT:14
in
T:10.875 in T:10.875 in
S:13 inS:13
in
S:9.5 in S:9.5 in
27020_ELRALU_NowApproved_LUNG_JournAd_Sprd_SnglPg_TAB_M8.indd 3-4 1/9/15 6:29 PM

OS
PR
OB
AB
ILIT
Y
1.0
0.8
0.6
0.4
0.2
0.0
0 3 6 9 12 15 18 21 24 27 30 33 36
TIME FROM RANDOMIZATION (MONTHS)
527 415 329 231 156 103628
625
CYRAMZA+ docetaxelPlacebo+ docetaxel 501 386 306 197 129 86
Number at Risk
70 45 23 11 2 0
56 36 23 9 0 0
CYRAMZA+ docetaxel
Placebo+ docetaxel
Hazard Ratio (95% CI)=0.86 (0.75, 0.98); P=0.024
10.5MONTHS
CYRAMZA+ docetaxel
(n=628)
Placebo+ docetaxel(n=625)
15% INCREASEIN MEDIAN OS
(9.5, 11.2)
MONTHS9.1
(8.4, 10.0)
OVERALL SURVIVAL: MEDIAN - MONTHS (95% CI) MAJOR OUTCOME MEASURE
VISIT www.CYRAMZAHCP.com
ADVANCING THE SECOND-LINE TREATMENT OF METASTATIC NSCLC4
• The percentage of deaths at the time of analysis was 68% (428 patients) and 73% (456 patients) in the CYRAMZA plus docetaxel and placebo plus docetaxel arms, respectively4
Demonstrated improvements across all three efficacy outcomes (OS, PFS, ORR)4
• Median PFS with CYRAMZA plus docetaxel was 4.5 months (95% CI: 4.2, 5.4) vs 3.0 months (95% CI: 2.8, 3.9) with placebo plus docetaxel (hazard ratio 0.76 [95% CI: 0.68, 0.86]; P<0.001)
— The percentage of events at the time of analysis was 89% (558 patients) and 93% (583 patients) in the CYRAMZA plus docetaxel and placebo plus docetaxel arms, respectively
• ORR with CYRAMZA plus docetaxel was 23% (95% CI: 20, 26) vs 14% (95% CI: 11, 17) with placebo plus docetaxel (P<0.001)*
CI=confidence interval; OS=overall survival; PFS=progression-free survival; ORR=objective response rate.
*ITT population. Disease progression and tumor response were assessed by investigators in accordance with Response Evaluation Criteria in Solid Tumors (RECIST) 1.1.5 ORR is defined as complete plus partial response.
REVEL TRIAL DESIGN (N=1253)The phase III REVEL trial evaluated the efficacy and safety of CYRAMZA plus docetaxel vs placebo plus docetaxel in patients with metastatic NSCLC with disease progression on or after platinum-based chemotherapy. Major efficacy outcome measure was OS. Supportive efficacy outcome measures were PFS and ORR. All patients were required to have Eastern Cooperative Oncology Group performance status 0 or 1. Patients were randomized 1:1 (N=1253) to receive either CYRAMZA 10 mg/kg or placebo, in combination with docetaxel at 75 mg/m2 every 21 days.4
CYRAMZA PLUS DOCETAXEL DEMONSTRATED A STATISTICALLYSIGNIFICANT IMPROVEMENT IN OVERALL SURVIVAL VS DOCETAXEL4
CYRAMZA is the first antiangiogenic agent FDA approvedin combination with docetaxel for the second-line treatment of metastatic NSCLC, including nonsquamous and squamous histologies.4
WARNING: HEMORRHAGECYRAMZA increased the risk of hemorrhage, including severe and sometimes fatal hemorrhagic events. Permanently discontinue CYRAMZA in patients who experience severe bleeding.
Warnings and PrecautionsHemorrhage • CYRAMZA increased the risk of hemorrhage and gastrointestinal
hemorrhage, including severe and sometimes fatal hemorrhagic events. In Study 3, which evaluated CYRAMZA plus docetaxel in metastatic non-small cell lung cancer (NSCLC), the incidence of severe bleeding was 2.4% for CYRAMZA plus docetaxel and 2.3% for placebo plus docetaxel. Patients with NSCLC receiving therapeutic anticoagulation or chronic therapy with NSAIDs or other antiplatelet therapy other than once-daily aspirin or with radiographic evidence of major airway or blood vessel invasion or intratumor cavitation were excluded from Study 3; therefore, the risk of pulmonary hemorrhage in these groups of patients is unknown. Permanently discontinue CYRAMZA in patients who experience severe bleeding.
Arterial Thromboembolic Events • Serious, sometimes fatal, arterial thromboembolic events (ATEs)
including myocardial infarction, cardiac arrest, cerebrovascular accident, and cerebral ischemia occurred in clinical trials including 1.7% of 236 patients who received CYRAMZA as a single agent for gastric cancer in Study 1. Permanently discontinue CYRAMZA in patients who experience a severe ATE.
Hypertension• An increased incidence of severe hypertension occurred in
patients receiving CYRAMZA plus docetaxel (6%) as compared to placebo plus docetaxel (2%). Control hypertension prior to initiating treatment with CYRAMZA. Monitor blood pressure every 2 weeks or more frequently as indicated during treatment. Temporarily suspend CYRAMZA for severe hypertension until medically controlled. Permanently discontinue CYRAMZA if medically significant hypertension cannot be controlled with
antihypertensive therapy or in patients with hypertensive crisis or hypertensive encephalopathy.
Infusion-Related Reactions • Prior to the institution of premedication recommendations across
clinical trials of CYRAMZA, infusion-related reactions (IRRs) occurred in 6 out of 37 patients (16%), including 2 severe events. The majority of IRRs across trials occurred during or following a first or second CYRAMZA infusion. Symptoms of IRRs included rigors/tremors, back pain/spasms, chest pain and/or tightness, chills, flushing, dyspnea, wheezing, hypoxia, and paresthesia. In severe cases, symptoms included bronchospasm, supraventricular tachycardia, and hypotension. Monitor patients during the infusion for signs and symptoms of IRRs in a setting with available resuscitation equipment. Immediately and permanently discontinue CYRAMZA for Grade 3 or 4 IRRs.
Gastrointestinal Perforations• CYRAMZA is an antiangiogenic therapy that can increase the
risk of gastrointestinal perforation, a potentially fatal event. In Study 3, the incidence of gastrointestinal perforation was 1% for CYRAMZA plus docetaxel versus 0.3% for placebo plus docetaxel. Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation.
Impaired Wound Healing• CYRAMZA has not been studied in patients with serious or
nonhealing wounds. CYRAMZA is an antiangiogenic therapy with the potential to adversely affect wound healing. Withhold CYRAMZA prior to surgery. Resume CYRAMZA following the surgical intervention based on clinical judgment of adequate wound healing. If a patient develops wound healing complications during therapy, discontinue CYRAMZA until the wound is fully healed.
Clinical Deterioration in Child-Pugh B or C Cirrhosis• Clinical deterioration, manifested by new onset or worsening
encephalopathy, ascites, or hepatorenal syndrome, was reported in patients with Child-Pugh B or C cirrhosis who received single-agent CYRAMZA. Use CYRAMZA in patients with Child-Pugh B or C cirrhosis only if the potential benefits of treatment are judged to outweigh the risks of clinical deterioration.
Reversible Posterior Leukoencephalopathy Syndrome (RPLS)• RPLS has been reported at a rate of <0.1% in clinical studies
with CYRAMZA. Confirm the diagnosis of RPLS with MRI and discontinue CYRAMZA in patients who develop RPLS. Symptoms may resolve or improve within days, although some patients with RPLS can experience ongoing neurologic sequelae or death.
Most Common Adverse Reactions• The most commonly reported adverse reactions (all grades; Grade
3/4) occurring in ≥5% of patients receiving CYRAMZA plus docetaxel and ≥2% higher than placebo plus docetaxel in Study 3 were neutropenia (55% vs 46%; 49% vs 40%), fatigue/asthenia (55% vs 50%; 14% vs 11%), stomatitis/mucosal inflammation (37% vs 19%; 7% vs 2%), epistaxis (19% vs 7%; <1% vs <1%), febrile neutropenia (16% vs 10%; 16% vs 10%), peripheral edema (16% vs 9%; 0% vs <1%), thrombocytopenia (13% vs 5%; 3% vs <1%), lacrimation increased (13% vs 5%; <1% vs 0%), and hypertension (11% vs 5%; 6% vs 2%).
• The most common serious adverse events with CYRAMZA plus docetaxel in Study 3 were febrile neutropenia (14%), pneumonia (6%), and neutropenia (5%). The use of granulocyte colony-stimulating factors was 42% in CYRAMZA plus docetaxel-treated patients versus 37% in patients who received placebo plus docetaxel.
• Treatment discontinuation due to adverse reactions occurred more frequently in CYRAMZA plus docetaxel-treated patients (9%) than in placebo plus docetaxel-treated patients (5%). The most common adverse events leading to treatment discontinuation of CYRAMZA were infusion-related reaction (0.5%) and epistaxis (0.3%).
• Clinically relevant adverse reactions reported in ≥1% and <5% of CYRAMZA plus docetaxel-treated patients in Study 3 were hyponatremia (4.8% CYRAMZA plus docetaxel versus 2.4% for placebo plus docetaxel) and proteinuria (3.3% CYRAMZA plus docetaxel versus 0.8% placebo plus docetaxel).
Drug Interactions• No pharmacokinetic interactions were observed between
ramucirumab and docetaxel.
Use in Specific Populations• Pregnancy Category C: Based on its mechanism of action,
CYRAMZA may cause fetal harm. Advise females of reproductive potential to avoid getting pregnant, including use of adequate contraception, while receiving CYRAMZA and for at least 3 months after the last dose of CYRAMZA. Animal models link angiogenesis, VEGF and VEGF Receptor 2 to critical aspects of female reproduction, embryofetal development, and postnatal development. There are no adequate or well-controlled studies of ramucirumab in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus.
• Nursing Mothers: It is recommended to discontinue nursing or discontinue CYRAMZA due to the potential risks to the nursing infant.
• Females of Reproductive Potential: Advise females of reproductive potential that CYRAMZA may impair fertility.
Please see Brief Summary of Prescribing Information for CYRAMZA, including Boxed Warning for hemorrhage, on the next page.
RB-L HCP ISI 17DEC2014
References: 1. Reck M, Kaiser R, Mellemgaard A, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomized controlled trial. Lancet Oncol. 2014;15:143-155. 2. Supplement to: Reck M, Kaiser R, Mellemgaard A, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomized controlled trial. Lancet Oncol. 2014;15:143-155. 3. National Cancer Institute. Cancer drug information. FDA approval for docetaxel. http://www.cancer.gov/cancertopics/druginfo/fda-docetaxel/print. Accessed August 26, 2014. 4. CYRAMZA (ramucirumab) [package insert]. Indianapolis, IN: Eli Lilly and Company; 2014. 5. Garon EB, Ciuleanu T-E, Arrieta O, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet. 2014;384(9944):665-673.
RB93740 12/2014 PRINTED IN USA © Lilly USA, LLC 2014. ALL RIGHTS RESERVED.CYRAMZA® is a registered trademark of Eli Lilly and Company.
IMPORTANT SAFETY INFORMATION FOR CYRAMZA
CYRAMZA® (ramucirumab), in combination with docetaxel, is indicated for the treatment of patients with metastatic NSCLC with disease progression on or after platinum-based chemotherapy. Patients with epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving CYRAMZA.
NEW FDA APPROVAL
B:14.25 inB:14
.25
in
B:11 in B:11 in
T:14 inT:14
in
T:10.875 in T:10.875 in
S:13 inS:13
in
S:9.5 in S:9.5 in
27020_ELRALU_NowApproved_LUNG_JournAd_Sprd_SnglPg_TAB_M8.indd 3-4 1/9/15 6:29 PM

CYRAMZA® (ramucirumab) injection RB-L HCP BS 17Dec2014 CYRAMZA® (ramucirumab) injection RB-L HCP BS 17Dec2014
CYRAMZA RB-L HCP BS 17Dec2014 Brief Summary 9.5 x 13 PRINTER VERSION 1 OF 2
CYRAMZA® (ramucirumab) injection BRIEF SUMMARY: For complete safety, please consult the full Prescribing Information.
WARNING: HEMORRHAGECYRAMZA increased the risk of hemorrhage, including severe and sometimes fatal hemorrhagic events. Permanently discontinue CYRAMZA in patients who experience severe bleeding.
INDICATIONS AND USAGE Non-Small Cell Lung Cancer: CYRAMZA, in combination with docetaxel, is indicated for the treatment of patients with metastatic non-small cell lung cancer (NSCLC) with disease progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving CYRAMZA.
CONTRAINDICATIONS None.
WARNINGS AND PRECAUTIONS Hemorrhage CYRAMZA increased the risk of hemorrhage and gastrointestinal hemorrhage, including severe and sometimes fatal hemorrhagic events. In Study 1, the incidence of severe bleeding was 3.4% for CYRAMZA and 2.6% for placebo. In Study 2, the incidence of severe bleeding was 4.3% for CYRAMZA plus paclitaxel and 2.4% for placebo plus paclitaxel. Patients with gastric cancer receiving nonsteroidal anti-inflammatory drugs (NSAIDs) were excluded from enrollment in Studies 1 and 2; therefore, the risk of gastric hemorrhage in CYRAMZA-treated patients with gastric tumors receiving NSAIDs is unknown. In Study 3, the incidence of severe bleeding was 2.4% for CYRAMZA plus docetaxel and 2.3% for placebo plus docetaxel. Patients with NSCLC receiving therapeutic anticoagulation or chronic therapy with NSAIDS or other anti-platelet therapy other than once daily aspirin or with radiographic evidence of major airway or blood vessel invasion or intratumor cavitation were excluded from Study 3; therefore, the risk of pulmonary hemorrhage in these groups of patients is unknown. Permanently discontinue CYRAMZA in patients who experience severe bleeding. Arterial Thromboembolic Events Serious, sometimes fatal, arterial thromboembolic events (ATEs) including myocardial infarction, cardiac arrest, cerebrovascular accident, and cerebral ischemia occurred in clinical trials including 1.7% of 236 patients who received CYRAMZA as a single agent for gastric cancer in Study 1. Permanently discontinue CYRAMZA in patients who experience a severe ATE. Hypertension An increased incidence of severe hypertension occurred in patients receiving CYRAMZA as a single agent (8%) as compared to placebo (3%) and in patients receiving CYRAMZA plus paclitaxel (15%) as compared to placebo plus paclitaxel (3%) and in patients receiving CYRAMZA plus docetaxel (6%) as compared to placebo plus docetaxel (2%). Control hypertension prior to initiating treatment with CYRAMZA. Monitor blood pressure every two weeks or more frequently as indicated during treatment. Temporarily suspend CYRAMZA for severe hypertension until medically controlled. Permanently discontinue CYRAMZA if medically significant hypertension cannot be controlled with antihypertensive therapy or in patients with hypertensive crisis or hypertensive encephalopathy. Infusion-Related Reactions Prior to the institution of premedication recommendations across clinical trials of CYRAMZA, infusion-related reactions (IRRs) occurred in 6 out of 37 patients (16%), including two severe events. The majority of IRRs across trials occurred during or following a first or second CYRAMZA infusion. Symptoms of IRRs included rigors/tremors, back pain/spasms, chest pain and/or tightness, chills, flushing, dyspnea, wheezing, hypoxia, and paresthesia. In severe cases, symptoms included bronchospasm, supraventricular tachycardia, and hypotension. Monitor patients during the infusion for signs and symptoms of IRRs in a setting with available resuscitation equipment. Immediately and permanently discontinue CYRAMZA for Grade 3 or 4 IRRs. Gastrointestinal Perforations CYRAMZA is an antiangiogenic therapy that can increase the risk of gastrointestinal perforation, a potentially fatal event. Four of 570 patients (0.7%) who received CYRAMZA as a single agent in clinical trials experienced gastrointestinal perforation. In Study 2, the incidence of gastrointestinal perforations was also increased in patients that received CYRAMZA plus paclitaxel (1.2%) as compared to patients receiving placebo plus paclitaxel (0.3%). In Study 3, the incidence of gastrointestinal perforation was 1% for CYRAMZA plus docetaxel and 0.3% for placebo plus docetaxel. Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation. Impaired Wound Healing CYRAMZA has not been studied in patients with serious or non-healing wounds. CYRAMZA is an antiangiogenic therapy with the potential to adversely affect wound healing. Withhold CYRAMZA prior to surgery. Resume following the surgical intervention based on clinical judgment of adequate wound healing. If a patient develops wound healing complications during therapy, discontinue CYRAMZA until the wound is fully healed. Clinical Deterioration in Patients with Child-Pugh B or C Cirrhosis Clinical deterioration, manifested by new onset or worsening encephalopathy, ascites, or hepatorenal syndrome was reported in patients with Child-Pugh B or C cirrhosis who received single-agent CYRAMZA. Use CYRAMZA in patients with Child-Pugh B or C cirrhosis only if the potential benefits of treatment are judged to outweigh the risks of clinical deterioration. Reversible Posterior Leukoencephalopathy Syndrome (RPLS) RPLS has been reported with a rate of <0.1% in clinical studies with CYRAMZA. Confirm the diagnosis of RPLS with MRI and discontinue CYRAMZA in patients who develop RPLS. Symptoms may resolve or improve within days, although some patients with RPLS can experience ongoing neurologic sequelae or death.
ADVERSE REACTIONS Clinical Trials Experience Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
CYRAMZA Administered in Combination with DocetaxelStudy 3 was a multinational, randomized, double-blind study conducted in patients with NSCLC with disease progression on or after one platinum-based therapy for locally advanced or metastatic disease. Patients received either CYRAMZA 10 mg/kg intravenously plus docetaxel 75 mg/m2 intravenously every 3 weeks or placebo plus docetaxel 75 mg/m2 intravenously every 3 weeks. Due to an increased incidence of neutropenia and febrile neutropenia in patients enrolled in East Asian sites, Study 3 was amended and 24 patients (11 CYRAMZA plus docetaxel, 13 placebo plus docetaxel) at East Asian sites received a starting dose of docetaxel at 60 mg/m2 every 3 weeks. Study 3 excluded patients with an ECOG PS of 2 or greater, bilirubin greater than the upper limit of normal (ULN), uncontrolled hypertension, major surgery within 28 days, radiographic evidence of major airway or blood vessel invasion by cancer, radiographic evidence of intra-tumor cavitation, or gross hemoptysis within the preceding 2 months, and patients receiving therapeutic anticoagulation or chronic anti-platelet therapy other than once daily aspirin. The study also excluded patients whose only prior treatment for advanced NSCLC was a tyrosine kinase (epidermal growth factor receptor [EGFR] or anaplastic lymphoma kinase [ALK]) inhibitor. The data described below reflect exposure to CYRAMZA plus docetaxel in 627 patients in Study 3. Demographics and baseline characteristics were similar between treatment arms. Median age was 62 years; 67% of patients were men; 84% were White and 12% were Asian; 33% had ECOG PS 0; 74% had non-squamous histology and 25% had squamous histology. Patients received a median of 4.5 doses of CYRAMZA; the median duration of exposure was 3.5 months, and 195 (31% of 627) patients received CYRAMZA for at least six months. In Study 3, the most common adverse reactions (all grades) observed in CYRAMZA plus docetaxel-treated patients at a rate of ≥30% and ≥2% higher than placebo plus docetaxel were neutropenia, fatigue/asthenia, and stomatitis/mucosal inflammation. Treatment discontinuation due to adverse reactions occurred more frequently in CYRAMZA plus docetaxel-treated patients (9%) than
in placebo plus docetaxel-treated patients (5%). The most common adverse events leading to treatment discontinuation of CYRAMZA were infusion-related reaction (0.5%) and epistaxis (0.3%). For patients with non-squamous histology, the overall incidence of pulmonary hemorrhage was 7% and the incidence of ≥Grade 3 pulmonary hemorrhage was 1% for CYRAMZA plus docetaxel compared to 6% overall incidence and 1% for ≥Grade 3 pulmonary hemorrhage for placebo plus docetaxel. For patients with squamous histology, the overall incidence of pulmonary hemorrhage was 10% and the incidence of ≥Grade 3 pulmonary hemorrhage was 2% for CYRAMZA plus docetaxel compared to 12% overall incidence and 2% for ≥Grade 3 pulmonary hemorrhage for placebo plus docetaxel. The most common serious adverse events with CYRAMZA plus docetaxel were febrile neutropenia (14%), pneumonia (6%), and neutropenia (5%). The use of granulocyte colony-stimulating factors was 42% in CYRAMZA plus docetaxel-treated patients versus 37% in patients who received placebo plus docetaxel. In patients ≥65 years, there were 18 (8%) deaths on treatment or within 30 days of discontinuation for CYRAMZA plus docetaxel and 9 (4%) deaths for placebo plus docetaxel. In patients <65 years, there were 13 (3%) deaths on treatment or within 30 days of discontinuation for CYRAMZA plus docetaxel and 26 (6%) deaths for placebo plus docetaxel. Table 4 provides the frequency and severity of adverse reactions in Study 3.
Table 4: Adverse Reactions Occurring at Incidence Rate ≥5% and a ≥2% Difference Between Arms in Patients Receiving CYRAMZA in Study 3
Adverse Reactions (MedDRA) System Organ Class
CYRAMZA plus docetaxel (N=627) Placebo plus docetaxel (N=618)All Grades
(Frequency %) Grade 3-4
(Frequency %) All Grades
(Frequency %) Grade 3-4
(Frequency %) Blood and Lymphatic System Disorders Febrile neutropenia 16 16 10 10
Neutropenia 55 49 46 40
Thrombocytopenia 13 3 5 <1
Gastrointestinal Disorders Stomatitis/Mucosal inflammation
37 7 19 2
Eye Disorders Lacrimation increased 13 <1 5 0
General Disorders and Administration Site Disorders Fatigue/Asthenia 55 14 50 11
Peripheral edema 16 0 9 <1
Respiratory, Thoracic, and Mediastinal Disorders Epistaxis 19 <1 7 <1
Vascular Disorders Hypertension 11 6 5 2
Clinically relevant adverse drug reactions reported in ≥1% and <5% of the CYRAMZA plus docetaxel-treated patients in Study 3 were hyponatremia (4.8% CYRAMZA plus docetaxel versus 2.4% for placebo plus docetaxel) and proteinuria (3.3% CYRAMZA plus docetaxel versus 0.8% placebo plus docetaxel).
Immunogenicity As with all therapeutic proteins, there is the potential for immunogenicity. In 19 clinical trials, 70/2131 (3.3%) of CYRAMZA-treated patients with post baseline serum samples tested positive for treatment-emergent anti-ramucirumab antibodies by an enzyme-linked immunosorbent assay (ELISA). Neutralizing antibodies were detected in 12 of the 70 patients who tested positive for treatment-emergent anti-ramucirumab antibodies. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to CYRAMZA with the incidences of antibodies to other products may be misleading.
DRUG INTERACTIONS No pharmacokinetic (PK) interactions were observed between ramucirumab and docetaxel.
USE IN SPECIFIC POPULATIONS Pregnancy Pregnancy Category CRisk Summary Based on its mechanism of action, CYRAMZA may cause fetal harm. Animal models link angiogenesis, VEGF and VEGF Receptor 2 (VEGFR2) to critical aspects of female reproduction, embryofetal development, and postnatal development. There are no adequate or well-controlled studies of ramucirumab in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus. Animal Data No animal studies have been specifically conducted to evaluate the effect of ramucirumab on reproduction and fetal development. In mice, loss of the VEGFR2 gene resulted in embryofetal death and these fetuses lacked organized blood vessels and blood islands in the yolk sac. In other models, VEGFR2 signaling was associated with development and maintenance of endometrial and placental vascular function, successful blastocyst implantation, maternal and feto-placental vascular differentiation, and development during early pregnancy in rodents and non-human primates. Disruption of VEGF signaling has also been associated with developmental anomalies including poor development of the cranial region, forelimbs, forebrain, heart, and blood vessels. Nursing Mothers It is not known whether CYRAMZA is excreted in human milk. No studies have been conducted to assess CYRAMZA’s impact on milk production or its presence in breast milk. Human IgG is excreted in human milk, but published data suggests that breast milk antibodies do not enter the neonatal and infant circulation in substantial amounts. Because many drugs are excreted in human milk and because of the potential risk for serious adverse reactions in nursing infants from ramucirumab, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother. Pediatric Use The safety and effectiveness of CYRAMZA in pediatric patients have not been established. In animal studies, effects on epiphyseal growth plates were identified. In cynomolgus monkeys, anatomical pathology revealed adverse effects on the epiphyseal growth plate (thickening and osteochondropathy) at all doses tested (5-50 mg/kg). Ramucirumab exposure at the lowest weekly dose tested in the cynomolgus monkey was 0.2 times the exposure in humans at the recommended dose of ramucirumab as a single agent. Geriatric Use Of the 563 CYRAMZA-treated patients in two randomized gastric cancer clinical studies, 36% were 65 and over, while 7% were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects. Of the 1253 patients in Study 3, 455 (36%) were 65 and over and 84 (7%) were 75 and over. Of the 627 patients who received CYRAMZA plus docetaxel in Study 3, 237 (38%) were 65 and over, while 45 (7%) were 75 and over. In an exploratory subgroup analysis of Study 3, the hazard ratio for overall survival in patients less than 65 years old was 0.74 (95% CI: 0.62, 0.87) and in patients 65 years or older was 1.10 (95% CI: 0.89, 1.36). Renal Impairment No dose adjustment is recommended for patients with renal impairment based on population PK analysis. Hepatic Impairment No dose adjustment is recommended for patients with mild hepatic impairment (total bilirubin within upper limit of normal [ULN] and aspartate aminotransferase [AST] >ULN or total bilirubin >1.0-1.5 times ULN and any AST)
CYRAMZA RB-L HCP BS 17Dec2014 Brief Summary 9.5 x 13 PRINTER VERSION 2 OF 2
CYRAMZA® (ramucirumab) injection RB-L HCP BS 17Dec2014
based on population PK analysis. Clinical deterioration was reported in patients with Child-Pugh B or C cirrhosis who received single-agent CYRAMZA. Females and Males of Reproductive Potential FertilityAdvise females of reproductive potential that CYRAMZA may impair fertility. ContraceptionBased on its mechanism of action, CYRAMZA may cause fetal harm. Advise females of reproductive potential to avoid getting pregnant while receiving CYRAMZA and for at least 3 months after the last dose of CYRAMZA.
DOSAGE AND ADMINISTRATION
Do not administer CYRAMZA as an intravenous push or bolus.
Recommended Dose and Schedule The recommended dose of CYRAMZA is 10 mg/kg administered by intravenous infusion over approximately 60 minutes on day 1 of a 21-day cycle prior to docetaxel infusion. Continue CYRAMZA until disease progression or unacceptable toxicity Premedication Prior to each CYRAMZA infusion, premedicate all patients with an intravenous histamine H1 antagonist (e.g., diphenhydramine hydrochloride). For patients who have experienced a Grade 1 or 2 infusion reaction, also premedicate with dexamethasone (or equivalent) and acetaminophen prior to each CYRAMZA infusion. Dose Modifications Infusion-Related Reactions (IRR)• Reduce the infusion rate of CYRAMZA by 50% for Grade 1 or 2 IRRs. • Permanently discontinue CYRAMZA for Grade 3 or 4 IRRs. Hypertension• Interrupt CYRAMZA for severe hypertension until controlled with medical management. • Permanently discontinue CYRAMZA for severe hypertension that cannot be controlled with antihypertensive therapy. Proteinuria• Interrupt CYRAMZA for urine protein levels ≥2 g/24 hours. Reinitiate treatment at a reduced dose of 8 mg/kg every 2 weeks once the urine protein level returns to <2 g/24 hours. If the protein level ≥2 g/24 hours reoccurs, interrupt CYRAMZA and reduce the dose to 6 mg/kg every 2 weeks once the urine protein level returns to <2 g/24 hours. • Permanently discontinue CYRAMZA for urine protein level >3 g/24 hours or in the setting of nephrotic syndrome. Wound Healing Complications• Interrupt CYRAMZA prior to scheduled surgery until the wound is fully healed. Arterial Thromboembolic Events, Gastrointestinal Perforation, or Grade 3 or 4 Bleeding• Permanently discontinue CYRAMZA.
For toxicities related to docetaxel, refer to the current respective prescribing information.
PATIENT COUNSELING INFORMATION Advise patients: • That CYRAMZA can cause severe bleeding. Advise patients to contact their health care provider for bleeding or symptoms of bleeding including lightheadedness. • Of increased risk of an arterial thromboembolic event. • To undergo routine blood pressure monitoring and to contact their health care provider if blood pressure is elevated or if symptoms from hypertension occur including severe headache, lightheadedness, or neurologic symptoms. • To notify their health care provider for severe diarrhea, vomiting, or severe abdominal pain. • That CYRAMZA has the potential to impair wound healing. Instruct patients not to undergo surgery without first discussing this potential risk with their health care provider. • Of the potential risk for maintaining pregnancy, risk to the fetus, or risk to postnatal development during and following treatment with CYRAMZA and the need to avoid getting pregnant, including use of adequate contraception, for at least 3 months following the last dose of CYRAMZA. • To discontinue nursing during CYRAMZA treatment.
Additional information can be found at www.CYRAMZAhcp.com.
Eli Lilly and Company, Indianapolis, IN 46285, USA
Copyright © 2014, Eli Lilly and Company. All rights reserved.
RB-L HCP BS 17Dec2014

ASCOPost.com | MARCH 25, 2015 PAGE 29
Journal Spotlight
Adjuvant Ovarian Suppression May Benefit Women With Premenopausal Breast Cancer Who Received Prior ChemotherapyBy Matthew Stenger
In a phase III trial (SOFT) reported in The New England Journal of Medi-
cine, Prudence A. Francis, MD, of Peter MacCallum Cancer Centre, Mel-bourne, Australia, Meredith M. Regan, ScD, of IBCSG Statistical Centre at Dana-Farber Cancer Institute, Boston, and colleagues found that the addition of ovarian suppression to tamoxifen did not improve disease-free survival among all women with premenopausal hormone receptor–positive breast can-cer but appeared to have a beneficial ef-fect in those who had received adjuvant
chemotherapy and remained premeno-pausal.1 Treatment with exemestane plus ovarian suppression vs tamoxifen alone was associated with even greater benefit in this higher-risk group.
Study DetailsIn the study, 3,066 premenopausal
women with breast cancer were ran-domized to receive 5 years of tamoxi-fen (n = 1,021), tamoxifen plus ovarian suppression (n = 1,024), or exemes-tane plus ovarian suppression (n = 1,021) between December 2003 and January 2011. Randomization was stratified by receipt or nonreceipt of adjuvant chemotherapy. Ovarian sup-pression was achieved by triptorelin (Trelstar depot in the U.S.) treatment, bilateral oophorectomy, or bilateral ovarian irradiation.
Patients had a median age of 43
years, 46.7% of the patients had not re-ceived prior chemotherapy and 53.3% had received prior chemotherapy and remained premenopausal, and 34.9% of patients had node-positive disease.
The primary analysis tested the hy-pothesis that tamoxifen plus ovarian suppression would improve disease-free survival vs tamoxifen alone. The study was originally designed to com-pare disease-free survival between the three treatment groups with three pairwise comparisons. However, en-rolled patients were older and had low-
er-risk characteristics than anticipated, and the rate of disease-free survival was higher than expected. A protocol amendment to the statistical analysis plan was adopted in 2011; it designat-ed the test of superiority of tamoxifen/ovarian suppression over tamoxifen alone as the primary analysis and the comparison of exemestane/ovarian suppression vs tamoxifen alone as a secondary analysis.
Outcomes in Total Population After a median follow-up of 67
months, 14.7% of patients had recur-rent disease or a second invasive can-cer or had died. The rate of disease-free survival at 5 years was 86.6% in the tamoxifen/ovarian suppression group vs 84.7% in the tamoxifen group (hazard ratio [HR] = 0.83, P = .10).
Breast Cancer
continued on page 30
Adding ovarian suppression to tamoxifen did not provide a significant benefit in the overall study population.
—Prudence A. Francis, MD, Meredith M. Regan, ScD, and colleagues
CYRAMZA® (ramucirumab) injection RB-L HCP BS 17Dec2014 CYRAMZA® (ramucirumab) injection RB-L HCP BS 17Dec2014
CYRAMZA RB-L HCP BS 17Dec2014 Brief Summary 9.5 x 13 PRINTER VERSION 1 OF 2
CYRAMZA® (ramucirumab) injection BRIEF SUMMARY: For complete safety, please consult the full Prescribing Information.
WARNING: HEMORRHAGECYRAMZA increased the risk of hemorrhage, including severe and sometimes fatal hemorrhagic events. Permanently discontinue CYRAMZA in patients who experience severe bleeding.
INDICATIONS AND USAGE Non-Small Cell Lung Cancer: CYRAMZA, in combination with docetaxel, is indicated for the treatment of patients with metastatic non-small cell lung cancer (NSCLC) with disease progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving CYRAMZA.
CONTRAINDICATIONS None.
WARNINGS AND PRECAUTIONS Hemorrhage CYRAMZA increased the risk of hemorrhage and gastrointestinal hemorrhage, including severe and sometimes fatal hemorrhagic events. In Study 1, the incidence of severe bleeding was 3.4% for CYRAMZA and 2.6% for placebo. In Study 2, the incidence of severe bleeding was 4.3% for CYRAMZA plus paclitaxel and 2.4% for placebo plus paclitaxel. Patients with gastric cancer receiving nonsteroidal anti-inflammatory drugs (NSAIDs) were excluded from enrollment in Studies 1 and 2; therefore, the risk of gastric hemorrhage in CYRAMZA-treated patients with gastric tumors receiving NSAIDs is unknown. In Study 3, the incidence of severe bleeding was 2.4% for CYRAMZA plus docetaxel and 2.3% for placebo plus docetaxel. Patients with NSCLC receiving therapeutic anticoagulation or chronic therapy with NSAIDS or other anti-platelet therapy other than once daily aspirin or with radiographic evidence of major airway or blood vessel invasion or intratumor cavitation were excluded from Study 3; therefore, the risk of pulmonary hemorrhage in these groups of patients is unknown. Permanently discontinue CYRAMZA in patients who experience severe bleeding. Arterial Thromboembolic Events Serious, sometimes fatal, arterial thromboembolic events (ATEs) including myocardial infarction, cardiac arrest, cerebrovascular accident, and cerebral ischemia occurred in clinical trials including 1.7% of 236 patients who received CYRAMZA as a single agent for gastric cancer in Study 1. Permanently discontinue CYRAMZA in patients who experience a severe ATE. Hypertension An increased incidence of severe hypertension occurred in patients receiving CYRAMZA as a single agent (8%) as compared to placebo (3%) and in patients receiving CYRAMZA plus paclitaxel (15%) as compared to placebo plus paclitaxel (3%) and in patients receiving CYRAMZA plus docetaxel (6%) as compared to placebo plus docetaxel (2%). Control hypertension prior to initiating treatment with CYRAMZA. Monitor blood pressure every two weeks or more frequently as indicated during treatment. Temporarily suspend CYRAMZA for severe hypertension until medically controlled. Permanently discontinue CYRAMZA if medically significant hypertension cannot be controlled with antihypertensive therapy or in patients with hypertensive crisis or hypertensive encephalopathy. Infusion-Related Reactions Prior to the institution of premedication recommendations across clinical trials of CYRAMZA, infusion-related reactions (IRRs) occurred in 6 out of 37 patients (16%), including two severe events. The majority of IRRs across trials occurred during or following a first or second CYRAMZA infusion. Symptoms of IRRs included rigors/tremors, back pain/spasms, chest pain and/or tightness, chills, flushing, dyspnea, wheezing, hypoxia, and paresthesia. In severe cases, symptoms included bronchospasm, supraventricular tachycardia, and hypotension. Monitor patients during the infusion for signs and symptoms of IRRs in a setting with available resuscitation equipment. Immediately and permanently discontinue CYRAMZA for Grade 3 or 4 IRRs. Gastrointestinal Perforations CYRAMZA is an antiangiogenic therapy that can increase the risk of gastrointestinal perforation, a potentially fatal event. Four of 570 patients (0.7%) who received CYRAMZA as a single agent in clinical trials experienced gastrointestinal perforation. In Study 2, the incidence of gastrointestinal perforations was also increased in patients that received CYRAMZA plus paclitaxel (1.2%) as compared to patients receiving placebo plus paclitaxel (0.3%). In Study 3, the incidence of gastrointestinal perforation was 1% for CYRAMZA plus docetaxel and 0.3% for placebo plus docetaxel. Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation. Impaired Wound Healing CYRAMZA has not been studied in patients with serious or non-healing wounds. CYRAMZA is an antiangiogenic therapy with the potential to adversely affect wound healing. Withhold CYRAMZA prior to surgery. Resume following the surgical intervention based on clinical judgment of adequate wound healing. If a patient develops wound healing complications during therapy, discontinue CYRAMZA until the wound is fully healed. Clinical Deterioration in Patients with Child-Pugh B or C Cirrhosis Clinical deterioration, manifested by new onset or worsening encephalopathy, ascites, or hepatorenal syndrome was reported in patients with Child-Pugh B or C cirrhosis who received single-agent CYRAMZA. Use CYRAMZA in patients with Child-Pugh B or C cirrhosis only if the potential benefits of treatment are judged to outweigh the risks of clinical deterioration. Reversible Posterior Leukoencephalopathy Syndrome (RPLS) RPLS has been reported with a rate of <0.1% in clinical studies with CYRAMZA. Confirm the diagnosis of RPLS with MRI and discontinue CYRAMZA in patients who develop RPLS. Symptoms may resolve or improve within days, although some patients with RPLS can experience ongoing neurologic sequelae or death.
ADVERSE REACTIONS Clinical Trials Experience Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
CYRAMZA Administered in Combination with DocetaxelStudy 3 was a multinational, randomized, double-blind study conducted in patients with NSCLC with disease progression on or after one platinum-based therapy for locally advanced or metastatic disease. Patients received either CYRAMZA 10 mg/kg intravenously plus docetaxel 75 mg/m2 intravenously every 3 weeks or placebo plus docetaxel 75 mg/m2 intravenously every 3 weeks. Due to an increased incidence of neutropenia and febrile neutropenia in patients enrolled in East Asian sites, Study 3 was amended and 24 patients (11 CYRAMZA plus docetaxel, 13 placebo plus docetaxel) at East Asian sites received a starting dose of docetaxel at 60 mg/m2 every 3 weeks. Study 3 excluded patients with an ECOG PS of 2 or greater, bilirubin greater than the upper limit of normal (ULN), uncontrolled hypertension, major surgery within 28 days, radiographic evidence of major airway or blood vessel invasion by cancer, radiographic evidence of intra-tumor cavitation, or gross hemoptysis within the preceding 2 months, and patients receiving therapeutic anticoagulation or chronic anti-platelet therapy other than once daily aspirin. The study also excluded patients whose only prior treatment for advanced NSCLC was a tyrosine kinase (epidermal growth factor receptor [EGFR] or anaplastic lymphoma kinase [ALK]) inhibitor. The data described below reflect exposure to CYRAMZA plus docetaxel in 627 patients in Study 3. Demographics and baseline characteristics were similar between treatment arms. Median age was 62 years; 67% of patients were men; 84% were White and 12% were Asian; 33% had ECOG PS 0; 74% had non-squamous histology and 25% had squamous histology. Patients received a median of 4.5 doses of CYRAMZA; the median duration of exposure was 3.5 months, and 195 (31% of 627) patients received CYRAMZA for at least six months. In Study 3, the most common adverse reactions (all grades) observed in CYRAMZA plus docetaxel-treated patients at a rate of ≥30% and ≥2% higher than placebo plus docetaxel were neutropenia, fatigue/asthenia, and stomatitis/mucosal inflammation. Treatment discontinuation due to adverse reactions occurred more frequently in CYRAMZA plus docetaxel-treated patients (9%) than
in placebo plus docetaxel-treated patients (5%). The most common adverse events leading to treatment discontinuation of CYRAMZA were infusion-related reaction (0.5%) and epistaxis (0.3%). For patients with non-squamous histology, the overall incidence of pulmonary hemorrhage was 7% and the incidence of ≥Grade 3 pulmonary hemorrhage was 1% for CYRAMZA plus docetaxel compared to 6% overall incidence and 1% for ≥Grade 3 pulmonary hemorrhage for placebo plus docetaxel. For patients with squamous histology, the overall incidence of pulmonary hemorrhage was 10% and the incidence of ≥Grade 3 pulmonary hemorrhage was 2% for CYRAMZA plus docetaxel compared to 12% overall incidence and 2% for ≥Grade 3 pulmonary hemorrhage for placebo plus docetaxel. The most common serious adverse events with CYRAMZA plus docetaxel were febrile neutropenia (14%), pneumonia (6%), and neutropenia (5%). The use of granulocyte colony-stimulating factors was 42% in CYRAMZA plus docetaxel-treated patients versus 37% in patients who received placebo plus docetaxel. In patients ≥65 years, there were 18 (8%) deaths on treatment or within 30 days of discontinuation for CYRAMZA plus docetaxel and 9 (4%) deaths for placebo plus docetaxel. In patients <65 years, there were 13 (3%) deaths on treatment or within 30 days of discontinuation for CYRAMZA plus docetaxel and 26 (6%) deaths for placebo plus docetaxel. Table 4 provides the frequency and severity of adverse reactions in Study 3.
Table 4: Adverse Reactions Occurring at Incidence Rate ≥5% and a ≥2% Difference Between Arms in Patients Receiving CYRAMZA in Study 3
Adverse Reactions (MedDRA) System Organ Class
CYRAMZA plus docetaxel (N=627) Placebo plus docetaxel (N=618)All Grades
(Frequency %) Grade 3-4
(Frequency %) All Grades
(Frequency %) Grade 3-4
(Frequency %) Blood and Lymphatic System Disorders Febrile neutropenia 16 16 10 10
Neutropenia 55 49 46 40
Thrombocytopenia 13 3 5 <1
Gastrointestinal Disorders Stomatitis/Mucosal inflammation
37 7 19 2
Eye Disorders Lacrimation increased 13 <1 5 0
General Disorders and Administration Site Disorders Fatigue/Asthenia 55 14 50 11
Peripheral edema 16 0 9 <1
Respiratory, Thoracic, and Mediastinal Disorders Epistaxis 19 <1 7 <1
Vascular Disorders Hypertension 11 6 5 2
Clinically relevant adverse drug reactions reported in ≥1% and <5% of the CYRAMZA plus docetaxel-treated patients in Study 3 were hyponatremia (4.8% CYRAMZA plus docetaxel versus 2.4% for placebo plus docetaxel) and proteinuria (3.3% CYRAMZA plus docetaxel versus 0.8% placebo plus docetaxel).
Immunogenicity As with all therapeutic proteins, there is the potential for immunogenicity. In 19 clinical trials, 70/2131 (3.3%) of CYRAMZA-treated patients with post baseline serum samples tested positive for treatment-emergent anti-ramucirumab antibodies by an enzyme-linked immunosorbent assay (ELISA). Neutralizing antibodies were detected in 12 of the 70 patients who tested positive for treatment-emergent anti-ramucirumab antibodies. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to CYRAMZA with the incidences of antibodies to other products may be misleading.
DRUG INTERACTIONS No pharmacokinetic (PK) interactions were observed between ramucirumab and docetaxel.
USE IN SPECIFIC POPULATIONS Pregnancy Pregnancy Category CRisk Summary Based on its mechanism of action, CYRAMZA may cause fetal harm. Animal models link angiogenesis, VEGF and VEGF Receptor 2 (VEGFR2) to critical aspects of female reproduction, embryofetal development, and postnatal development. There are no adequate or well-controlled studies of ramucirumab in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus. Animal Data No animal studies have been specifically conducted to evaluate the effect of ramucirumab on reproduction and fetal development. In mice, loss of the VEGFR2 gene resulted in embryofetal death and these fetuses lacked organized blood vessels and blood islands in the yolk sac. In other models, VEGFR2 signaling was associated with development and maintenance of endometrial and placental vascular function, successful blastocyst implantation, maternal and feto-placental vascular differentiation, and development during early pregnancy in rodents and non-human primates. Disruption of VEGF signaling has also been associated with developmental anomalies including poor development of the cranial region, forelimbs, forebrain, heart, and blood vessels. Nursing Mothers It is not known whether CYRAMZA is excreted in human milk. No studies have been conducted to assess CYRAMZA’s impact on milk production or its presence in breast milk. Human IgG is excreted in human milk, but published data suggests that breast milk antibodies do not enter the neonatal and infant circulation in substantial amounts. Because many drugs are excreted in human milk and because of the potential risk for serious adverse reactions in nursing infants from ramucirumab, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother. Pediatric Use The safety and effectiveness of CYRAMZA in pediatric patients have not been established. In animal studies, effects on epiphyseal growth plates were identified. In cynomolgus monkeys, anatomical pathology revealed adverse effects on the epiphyseal growth plate (thickening and osteochondropathy) at all doses tested (5-50 mg/kg). Ramucirumab exposure at the lowest weekly dose tested in the cynomolgus monkey was 0.2 times the exposure in humans at the recommended dose of ramucirumab as a single agent. Geriatric Use Of the 563 CYRAMZA-treated patients in two randomized gastric cancer clinical studies, 36% were 65 and over, while 7% were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects. Of the 1253 patients in Study 3, 455 (36%) were 65 and over and 84 (7%) were 75 and over. Of the 627 patients who received CYRAMZA plus docetaxel in Study 3, 237 (38%) were 65 and over, while 45 (7%) were 75 and over. In an exploratory subgroup analysis of Study 3, the hazard ratio for overall survival in patients less than 65 years old was 0.74 (95% CI: 0.62, 0.87) and in patients 65 years or older was 1.10 (95% CI: 0.89, 1.36). Renal Impairment No dose adjustment is recommended for patients with renal impairment based on population PK analysis. Hepatic Impairment No dose adjustment is recommended for patients with mild hepatic impairment (total bilirubin within upper limit of normal [ULN] and aspartate aminotransferase [AST] >ULN or total bilirubin >1.0-1.5 times ULN and any AST)
CYRAMZA RB-L HCP BS 17Dec2014 Brief Summary 9.5 x 13 PRINTER VERSION 2 OF 2
CYRAMZA® (ramucirumab) injection RB-L HCP BS 17Dec2014
based on population PK analysis. Clinical deterioration was reported in patients with Child-Pugh B or C cirrhosis who received single-agent CYRAMZA. Females and Males of Reproductive Potential FertilityAdvise females of reproductive potential that CYRAMZA may impair fertility. ContraceptionBased on its mechanism of action, CYRAMZA may cause fetal harm. Advise females of reproductive potential to avoid getting pregnant while receiving CYRAMZA and for at least 3 months after the last dose of CYRAMZA.
DOSAGE AND ADMINISTRATION
Do not administer CYRAMZA as an intravenous push or bolus.
Recommended Dose and Schedule The recommended dose of CYRAMZA is 10 mg/kg administered by intravenous infusion over approximately 60 minutes on day 1 of a 21-day cycle prior to docetaxel infusion. Continue CYRAMZA until disease progression or unacceptable toxicity Premedication Prior to each CYRAMZA infusion, premedicate all patients with an intravenous histamine H1 antagonist (e.g., diphenhydramine hydrochloride). For patients who have experienced a Grade 1 or 2 infusion reaction, also premedicate with dexamethasone (or equivalent) and acetaminophen prior to each CYRAMZA infusion. Dose Modifications Infusion-Related Reactions (IRR)• Reduce the infusion rate of CYRAMZA by 50% for Grade 1 or 2 IRRs. • Permanently discontinue CYRAMZA for Grade 3 or 4 IRRs. Hypertension• Interrupt CYRAMZA for severe hypertension until controlled with medical management. • Permanently discontinue CYRAMZA for severe hypertension that cannot be controlled with antihypertensive therapy. Proteinuria• Interrupt CYRAMZA for urine protein levels ≥2 g/24 hours. Reinitiate treatment at a reduced dose of 8 mg/kg every 2 weeks once the urine protein level returns to <2 g/24 hours. If the protein level ≥2 g/24 hours reoccurs, interrupt CYRAMZA and reduce the dose to 6 mg/kg every 2 weeks once the urine protein level returns to <2 g/24 hours. • Permanently discontinue CYRAMZA for urine protein level >3 g/24 hours or in the setting of nephrotic syndrome. Wound Healing Complications• Interrupt CYRAMZA prior to scheduled surgery until the wound is fully healed. Arterial Thromboembolic Events, Gastrointestinal Perforation, or Grade 3 or 4 Bleeding• Permanently discontinue CYRAMZA.
For toxicities related to docetaxel, refer to the current respective prescribing information.
PATIENT COUNSELING INFORMATION Advise patients: • That CYRAMZA can cause severe bleeding. Advise patients to contact their health care provider for bleeding or symptoms of bleeding including lightheadedness. • Of increased risk of an arterial thromboembolic event. • To undergo routine blood pressure monitoring and to contact their health care provider if blood pressure is elevated or if symptoms from hypertension occur including severe headache, lightheadedness, or neurologic symptoms. • To notify their health care provider for severe diarrhea, vomiting, or severe abdominal pain. • That CYRAMZA has the potential to impair wound healing. Instruct patients not to undergo surgery without first discussing this potential risk with their health care provider. • Of the potential risk for maintaining pregnancy, risk to the fetus, or risk to postnatal development during and following treatment with CYRAMZA and the need to avoid getting pregnant, including use of adequate contraception, for at least 3 months following the last dose of CYRAMZA. • To discontinue nursing during CYRAMZA treatment.
Additional information can be found at www.CYRAMZAhcp.com.
Eli Lilly and Company, Indianapolis, IN 46285, USA
Copyright © 2014, Eli Lilly and Company. All rights reserved.
RB-L HCP BS 17Dec2014

PAGE 30 The ASCO Post | MARCH 25, 2015
Perspective
On multivariate analysis adjusting for prognostic factors, the HR was 0.78, P = .03. The 5-year rate of disease-free survival with exemestane/ovarian sup-pression was 89.0% (HR = 0.68, 95% confidence interval [CI] = 0.53–0.86, vs tamoxifen).
The 5-year rate of freedom from breast cancer was 88.4% with tamoxi-fen/ovarian suppression vs 86.4% with tamoxifen (HR = 0.81, P = 0.09; ad-justed HR = 0.75, P = 0.02) and 90.9% with exemestane/ovarian suppression (HR = 0.64, 95% CI = 0.49–0.83). The 5-year rate of freedom from distant tu-mor recurrence was 91.3% with tamox-ifen/ovarian suppression vs 90.7% with tamoxifen (HR = 0.88, 95% CI = 0.66–1.18) and 93.0% with exemes-tane/ovarian suppression (HR = 0.71, 95% CI = 0.52–0.96, vs tamoxifen). Overall survival at 5 years was 96.7% with tamoxifen/ovarian suppression vs 95.1% with tamoxifen (HR = 0.74, P = 0.13) and 95.3% with exemestane/ovarian suppression (HR = 0.97, 95% CI = 0.68–1.40, vs tamoxifen).
Patients Who Had No Prior Chemotherapy
Among patients who did not receive chemotherapy (median age, 46), more than 95% remained free from breast cancer at 5 years in all treatment groups. The 5-year rates for the tamoxifen/ovar-ian suppression, tamoxifen, and exemes-tane/ovarian suppression groups were 93.4%, 93.3%, and 95.2% for disease-free survival; 95.1%, 95.8%, and 97.1% for freedom from breast cancer; 98.7%, 98.6%, and 99.3% for freedom from distant tumor recurrence; and 99.2%, 99.8%, and 98.8% for overall survival. The treatment groups weren’t statisti-cally compared (ie, no statistical test and P value) within the subgroup of patients who didn’t receive prior chemotherapy.
Patients Who Had Prior Chemotherapy
Among patients who had received prior chemotherapy and remained pre-menopausal (median age, 40), 5-year disease-free survival rates in the tamoxi-fen/ovarian suppression, tamoxifen, and exemestane/ovarian suppression groups were 80.7% (HR = 0.82, 95% CI
= 0.64–1.07, vs tamoxifen), 77.1%, and 83.8% (HR = 0.70, 95% CI = 0.53–0.92, vs tamoxifen). Five-year rates of freedom from breast cancer were 82.5% (HR = 0.78, 95% CI = 0.60–1.02), 78.0%, and 85.7% (HR = 0.65, 95% CI = 0.49–0.87). Five-year rates of freedom from distant tumor recurrence were 84.8% (HR = 0.87, 95% CI = 0.64–1.17), 83.6%, and 87.8% (HR = 0.72, 95% CI = 0.52–0.98). Five-year overall survival rates were 94.5% (HR = 0.64, 95% CI = 0.42–0.96), 90.9%, and 92.3% (HR = 0.87, 95% CI = 0.59–1.27).
The investigators concluded: “Add-ing ovarian suppression to tamoxifen did not provide a significant benefit in the overall study population. How-
ever, for women who were at sufficient risk for recurrence to warrant adju-vant chemotherapy and who remained premenopausal, the addition of ovar-ian suppression improved disease out-comes. Further improvement was seen with the use of exemestane plus ovarian suppression.” n
Disclosure: This study was supported by the International Breast Cancer Study Group, Ipsen, the National Cancer Institute, and Pfizer. For full disclosures of the study authors, visit www.nejm.org.
Reference1. Francis PA, Regan MM, Fleming GF,
et al: Adjuvant ovarian suppression in pre-menopausal breast cancer. N Engl J Med. December 11, 2014 (early release online).
Adjuvant Ovarian Suppressioncontinued from page 29
Adjuvant Ovarian Suppression in Premenopausal Breast Cancer
■ The addition of ovarian suppression to tamoxifen did not appear to improve outcomes among all patients.
■ Improvements in outcomes were observed with the addition of ovarian suppression to tamoxifen (and further improvement with ovarian suppression plus exemestane) compared with tamoxifen alone in higher-risk patients who had received prior chemotherapy and remained premenopausal.
SOFT Trial Results Inconclusive: Further Study Needed By Edith A. Perez, MD
The results of the SOFT trial—presented at the 2014 San An-
tonio Breast Cancer Symposium, re-ported recently by Francis et al in The New England Journal of Medicine,1 and reviewed in this issue of The ASCO Post—were not as conclusive as we had
hoped. In essence, the study enrolled women with resected early-stage hor-
mone receptor–positive breast cancer who were randomized to receive one of three antiestrogen approaches. The re-sults demonstrated that most of these patients had a very good outcome in response to the physician’s decisions (in consultation with the patient) of
surgery, chemotherapy, radiation ther-apy, plus one of the three hormonal ap-proaches tested.
This is good news, although it does not imply that all women may not have had side effects of these treatments nor did not develop tumor recurrence or will not develop tumor recurrence
in the years to come. However, at least at the reported follow-up, survival was greater than 95%, and about 85% of patients were alive without tumor recurrence.
Deciding Who Needs Chemotherapy
In terms of the main hypothesis of the trial, the results demonstrated that we cannot recommend ovarian func-tion suppression as standard therapy for all patients with hormone recep-tor–positive breast cancer diagnosed in the premenopausal stage. This statement is based on the lack of sta-tistical difference between tamoxifen and tamoxifen with ovarian function suppression. However, the important caveat to this overall “negative result” of this study is that for the subset of pa-tients whose physicians thought they should receive chemotherapy, ovarian function suppression appeared to add to tamoxifen (or exemestane).
The challenge here is that the deci-sion for chemotherapy was left to phy-sician discretion, so the study does not
provide guidelines on how to “exactly” assign need for chemotherapy. There-fore, we need to await further under-standing of molecular profiling added to clinicopathologic characteristics to eventually make more informed and accurate decisions than those we can make today regarding who “needs” chemotherapy to optimize disease-free and overall survival.
Thus, we are still left with a situa-tion where we need to continue dis-cussing options with each patient, sharing the results of this trial (effi-cacy as well as toxicities observed), while awaiting further study follow-up related to long-term breast-related events to determine whether there are clinically relevant negative effects of ovarian function suppression. n
Disclosure: Dr. Perez reported no potential conflicts of interest.
Reference1. Francis PA, Regan MM, Fleming
GF, et al: Adjuvant ovarian suppression in premenopausal breast cancer. N Engl J Med 372:436-446, 2015.
Dr. Perez is Deputy Director at Large, Mayo Clinic Cancer Center; Group Vice Chair, Alliance for Clinical Trials in On-cology; and Director, Mayo Clinic Breast Cancer Translational Genomics Program.
The challenge here is that the decision for chemotherapy was left to physician discretion, so the study does not provide guidelines on how to exactly assign need for chemotherapy.
—Edith A. Perez, MD

when it’s time for first-line Cll
treAtment
indicationGAZYVA® (obinutuzumab), in combination with chlorambucil, is indicated for the treatment of patients with previously untreated chronic lymphocytic leukemia (CLL).
Boxed WArninGs: HePAtitis B VirUs reACtiVAtiOn AnD PrOGressiVe MUltifOCAl leUKOenCePHAlOPAtHY• Hepatitis B Virus (HBV) reactivation, in some cases resulting in fulminant hepatitis, hepatic failure,
and death, can occur in patients receiving CD20-directed cytolytic antibodies, including GAZYVA. screen all patients for HBV infection before treatment initiation. Monitor HBV positive patients during and after treatment with GAZYVA. Discontinue GAZYVA and concomitant medications in the event of HBV reactivation
• Progressive Multifocal leukoencephalopathy (PMl) including fatal PMl, can occur in patients receiving GAZYVA
147311_L01_INSRT.indd 1 10/13/14 4:42 PM

iMPOrtAnt sAfetY infOrMAtiOn
stArt with GAZYVA®
Hepatitis B Virus reactivation • Hepatitis B virus (HBV) reactivation, in some cases
resulting in fulminant hepatitis, hepatic failure, and death, can occur in patients treated with anti-CD20 antibodies including GAZYVA. HBV reactivation has been reported in patients who are hepatitis B surface antigen (HBsAg) positive and also in patients who are HBsAg negative but are hepatitis B core antibody (anti-HBc) positive. Reactivation has also occurred in patients who appear to have resolved hepatitis B infection (i.e., HBsAg negative, anti-HBc positive, and hepatitis B surface antibody [anti-HBs] positive)
• HBV reactivation is defined as an abrupt increase in HBV replication manifesting as a rapid increase in serum HBV DNA level, or detection of HBsAg in a person who was previously HBsAg negative and anti-HBc positive. Reactivation of HBV replication is often followed by hepatitis, i.e., increase in transaminase levels and, in severe cases, increase in bilirubin levels, liver failure, and death
• Screen all patients for HBV infection by measuring HBsAg and anti-HBc before initiating treatment with GAZYVA. For patients who show evidence of hepatitis B infection (HBsAg positive [regardless of antibody status] or HBsAg negative but anti-HBc positive), consult physicians with expertise in managing hepatitis B regarding monitoring and consideration for HBV antiviral therapy
• Monitor patients with evidence of current or prior HBV infection for clinical and laboratory signs of hepatitis or HBV reactivation during and for several months following treatment with GAZYVA
• In patients who develop reactivation of HBV while receiving GAZYVA, immediately discontinue GAZYVA and any concomitant chemotherapy, and institute appropriate treatment. Resumption of GAZYVA in patients whose HBV reactivation resolves should be discussed with physicians with expertise in managing hepatitis B. Insufficient data exist regarding the safety of resuming GAZYVA in patients who develop HBV reactivation
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) include obinutuzumab (GAZYVA®) + chlorambucil as a preferred first-line regimen for CLL patients with comorbidities, without del(17p)2,a,b
For the first-line treatment of CLL in combination with chlorambucil1
a NCCN treatment suggestions for patients with del(17p) are not segmented by age or comorbidities. Obinutuzumab (GAZYVA) + chlorambucil is included as a suggested treatment for this patient population. Suggested treatment regimens are listed in alphabetical order.b Obinutuzumab (GAZYVA) + chlorambucil is listed first as a suggested treatment regimen for this patient population. Treatment regimens are listed in order of preference. Note: All recommendations are category 2A unless otherwise indicated.
147311_L01_INSRT.indd 2 10/13/14 4:42 PM
Progressive Multifocal leukoencephalopathy (PMl) • JC virus infection resulting in PML, which can be fatal, was
observed in patients treated with GAZYVA. Consider the diagnosis of PML in any patient presenting with new onset or changes to preexisting neurologic manifestations. Evaluation of PML includes, but is not limited to, consultation with a neurologist, brain MRI, and lumbar puncture. Discontinue GAZYVA therapy and consider discontinuation or reduction of any concomitant chemotherapy or immunosuppressive therapy in patients who develop PML
infusion reactions • GAZYVA can cause severe and life-threatening infusion
reactions. Two-thirds of patients experienced a reaction to the first 1000 mgs infused of GAZYVA. Infusion reactions can also occur with subsequent infusions. Symptoms may include hypotension, tachycardia, dyspnea, and respiratory symptoms (eg, bronchospasm, larynx and throat irritation,
wheezing, laryngeal edema). Other common symptoms include nausea, vomiting, diarrhea, hypertension, flushing, headache, pyrexia, and chills
• Premedicate patients with acetaminophen, antihistamine, and a glucocorticoid. Institute medical management for infusion reactions as needed. Closely monitor patients during the entire infusion. Infusion reactions within 24 hours of receiving GAZYVA have occurred
• For patients with any Grade 4 infusion reactions, including but not limited to anaphylaxis, acute life-threatening respiratory symptoms, or other life-threatening infusion reaction: Stop the GAZYVA infusion. Permanently discontinue GAZYVA therapy
Please see the following pages for additional important safety information and brief summary of full Prescribing information, including Boxed WArninGs.
neArLY A 1-YeAr imProVement in meDiAn Pfs (23.0 months Vs 11.1 months; hr=0.16; 95% Ci, 0.11-0.24; P<0.0001)1
iMPOrtAnt sAfetY infOrMAtiOn (COnt’D)
enhanced response rates1
• GAZYVA + Clb more than doubled response rates vs Clb monotherapy (75.9% vs 32.1%, respectively) • More than 1 in 4 patients receiving GAZYVA + Clb achieved a complete response (27.8% vs 0.9%, respectively)
the most common Grade 3-4 adverse reactions were infusion reactions, neutropenia, and thrombocytopenia1
• The most common adverse reactions were infusion reactions, neutropenia, thrombocytopenia, anemia, pyrexia, cough, and musculoskeletal disorders
02621467691118
16
0
2
0
03969111146201208238
n at risk
Chlorambucil
GAZYVA+ chlorambucil
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
0 24 2721181512963
Pfs
time (months)
Chlorambucil (n=118)GAZYVA + chlorambucil (n=238)
11.1 23.0
Hr=0.16; 95% CI, 0.11-0.24; P<0.0001
84%risk
reduction
siGnifiCAntlY sUPeriOr Pfs: GAZYVA in combination with Clb more than doubled median Pfs vs Clb monotherapy1
PFS, progression-free survival; Clb, chlorambucil; HR, hazard ratio; CI, confidence interval. Cll-11 trial Design1: GAZYVA, in combination with chlorambucil, was evaluated in a Phase III, open-label, multicenter, 3-arm, randomized, parallel-group comparative study in patients with previously untreated CD20+ chronic lymphocytic leukemia and coexisting medical conditions and/or reduced renal function. Patients with creatinine clearance <30 mL/min or inadequate liver function were excluded. The primary endpoint was progression-free survival. Overall response rate and complete response rate were secondary endpoints.
147311_L01_INSRT.indd 3 10/13/14 4:42 PM

iMPOrtAnt sAfetY infOrMAtiOn
stArt with GAZYVA®
Hepatitis B Virus reactivation • Hepatitis B virus (HBV) reactivation, in some cases
resulting in fulminant hepatitis, hepatic failure, and death, can occur in patients treated with anti-CD20 antibodies including GAZYVA. HBV reactivation has been reported in patients who are hepatitis B surface antigen (HBsAg) positive and also in patients who are HBsAg negative but are hepatitis B core antibody (anti-HBc) positive. Reactivation has also occurred in patients who appear to have resolved hepatitis B infection (i.e., HBsAg negative, anti-HBc positive, and hepatitis B surface antibody [anti-HBs] positive)
• HBV reactivation is defined as an abrupt increase in HBV replication manifesting as a rapid increase in serum HBV DNA level, or detection of HBsAg in a person who was previously HBsAg negative and anti-HBc positive. Reactivation of HBV replication is often followed by hepatitis, i.e., increase in transaminase levels and, in severe cases, increase in bilirubin levels, liver failure, and death
• Screen all patients for HBV infection by measuring HBsAg and anti-HBc before initiating treatment with GAZYVA. For patients who show evidence of hepatitis B infection (HBsAg positive [regardless of antibody status] or HBsAg negative but anti-HBc positive), consult physicians with expertise in managing hepatitis B regarding monitoring and consideration for HBV antiviral therapy
• Monitor patients with evidence of current or prior HBV infection for clinical and laboratory signs of hepatitis or HBV reactivation during and for several months following treatment with GAZYVA
• In patients who develop reactivation of HBV while receiving GAZYVA, immediately discontinue GAZYVA and any concomitant chemotherapy, and institute appropriate treatment. Resumption of GAZYVA in patients whose HBV reactivation resolves should be discussed with physicians with expertise in managing hepatitis B. Insufficient data exist regarding the safety of resuming GAZYVA in patients who develop HBV reactivation
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) include obinutuzumab (GAZYVA®) + chlorambucil as a preferred first-line regimen for CLL patients with comorbidities, without del(17p)2,a,b
For the first-line treatment of CLL in combination with chlorambucil1
a NCCN treatment suggestions for patients with del(17p) are not segmented by age or comorbidities. Obinutuzumab (GAZYVA) + chlorambucil is included as a suggested treatment for this patient population. Suggested treatment regimens are listed in alphabetical order.b Obinutuzumab (GAZYVA) + chlorambucil is listed first as a suggested treatment regimen for this patient population. Treatment regimens are listed in order of preference. Note: All recommendations are category 2A unless otherwise indicated.
147311_L01_INSRT.indd 2 10/13/14 4:42 PM
Progressive Multifocal leukoencephalopathy (PMl) • JC virus infection resulting in PML, which can be fatal, was
observed in patients treated with GAZYVA. Consider the diagnosis of PML in any patient presenting with new onset or changes to preexisting neurologic manifestations. Evaluation of PML includes, but is not limited to, consultation with a neurologist, brain MRI, and lumbar puncture. Discontinue GAZYVA therapy and consider discontinuation or reduction of any concomitant chemotherapy or immunosuppressive therapy in patients who develop PML
infusion reactions • GAZYVA can cause severe and life-threatening infusion
reactions. Two-thirds of patients experienced a reaction to the first 1000 mgs infused of GAZYVA. Infusion reactions can also occur with subsequent infusions. Symptoms may include hypotension, tachycardia, dyspnea, and respiratory symptoms (eg, bronchospasm, larynx and throat irritation,
wheezing, laryngeal edema). Other common symptoms include nausea, vomiting, diarrhea, hypertension, flushing, headache, pyrexia, and chills
• Premedicate patients with acetaminophen, antihistamine, and a glucocorticoid. Institute medical management for infusion reactions as needed. Closely monitor patients during the entire infusion. Infusion reactions within 24 hours of receiving GAZYVA have occurred
• For patients with any Grade 4 infusion reactions, including but not limited to anaphylaxis, acute life-threatening respiratory symptoms, or other life-threatening infusion reaction: Stop the GAZYVA infusion. Permanently discontinue GAZYVA therapy
Please see the following pages for additional important safety information and brief summary of full Prescribing information, including Boxed WArninGs.
neArLY A 1-YeAr imProVement in meDiAn Pfs (23.0 months Vs 11.1 months; hr=0.16; 95% Ci, 0.11-0.24; P<0.0001)1
iMPOrtAnt sAfetY infOrMAtiOn (COnt’D)
enhanced response rates1
• GAZYVA + Clb more than doubled response rates vs Clb monotherapy (75.9% vs 32.1%, respectively) • More than 1 in 4 patients receiving GAZYVA + Clb achieved a complete response (27.8% vs 0.9%, respectively)
the most common Grade 3-4 adverse reactions were infusion reactions, neutropenia, and thrombocytopenia1
• The most common adverse reactions were infusion reactions, neutropenia, thrombocytopenia, anemia, pyrexia, cough, and musculoskeletal disorders
02621467691118
16
0
2
0
03969111146201208238
n at risk
Chlorambucil
GAZYVA+ chlorambucil
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
0 24 2721181512963
Pfs
time (months)
Chlorambucil (n=118)GAZYVA + chlorambucil (n=238)
11.1 23.0
Hr=0.16; 95% CI, 0.11-0.24; P<0.0001
84%risk
reduction
siGnifiCAntlY sUPeriOr Pfs: GAZYVA in combination with Clb more than doubled median Pfs vs Clb monotherapy1
PFS, progression-free survival; Clb, chlorambucil; HR, hazard ratio; CI, confidence interval. Cll-11 trial Design1: GAZYVA, in combination with chlorambucil, was evaluated in a Phase III, open-label, multicenter, 3-arm, randomized, parallel-group comparative study in patients with previously untreated CD20+ chronic lymphocytic leukemia and coexisting medical conditions and/or reduced renal function. Patients with creatinine clearance <30 mL/min or inadequate liver function were excluded. The primary endpoint was progression-free survival. Overall response rate and complete response rate were secondary endpoints.
147311_L01_INSRT.indd 3 10/13/14 4:42 PM

iMPOrtAnt sAfetY infOrMAtiOn (COnt’D)
© 2014 Genentech USA, Inc. All rights reserved. GAZ0002401103 Printed in USA. August 2014
infusion reactions (cont’d) • For patients with Grade 1, 2, or 3 infusion reactions:
Interrupt GAZYVA for Grade 3 reactions until resolution of symptoms. Interrupt or reduce the rate of the infusion for Grade 1 or 2 reactions and manage symptoms
• For patients with preexisting cardiac or pulmonary conditions, monitor more frequently throughout the infusion and the post-infusion period since they may be at greater risk of experiencing more severe reactions. Hypotension may occur as part of the GAZYVA infusion reaction. Consider withholding antihypertensive treatments for 12 hours prior to and during each GAZYVA infusion, and for the first hour after administration until blood pressure is stable. For patients at increased risk of hypertensive crisis, consider the benefits versus the risks of withholding their hypertensive medication
tumor lysis syndrome (tls) • Acute renal failure, hyperkalemia, hypocalcemia,
hyperuricemia, and/or hyperphosphatemia from TLS can occur within 12-24 hours after the first infusion. Patients with high tumor burden and/or high circulating lymphocyte count (>25 x 109/L) are at greater risk for TLS and should receive appropriate tumor lysis prophylaxis with anti-hyperuricemics (e.g., allopurinol) and hydration beginning 12-24 hours prior to the infusion of GAZYVA. For treatment of TLS, correct electrolyte abnormalities, monitor renal function and fluid balance, and administer supportive care, including dialysis, as indicated
infections • Serious bacterial, fungal, and new or reactivated viral
infections can occur during and following GAZYVA therapy. Do not administer GAZYVA to patients with an active infection. Patients with a history of recurring or chronic infections may be at increased risk of infection
neutropenia • GAZYVA, in combination with chlorambucil, caused Grade
3 or 4 neutropenia in 34% of patients. Patients with Grade 3 to 4 neutropenia should be monitored frequently with regular laboratory tests until resolution. Anticipate, evaluate, and treat any symptoms or signs of developing infection
• Neutropenia can also be of late onset (occurring more than 28 days after completion of treatment) and/or prolonged (lasting longer than 28 days)
• Patients with neutropenia are strongly recommended to receive antimicrobial prophylaxis throughout the treatment period. Antiviral and antifungal prophylaxis should be considered
thrombocytopenia • GAZYVA, in combination with chlorambucil, caused Grade 3 or 4 thrombocytopenia in 11% of patients in the trial. In 5% of patients, GAZYVA caused acute thrombocytopenia occurring within 24 hours after the GAZYVA infusion.
thrombocytopenia (cont’d) Fatal hemorrhagic events during Cycle 1 have also
been reported. Monitor all patients frequently for thrombocytopenia and hemorrhagic events, especially during the first cycle. In patients with Grade 3 or 4 thrombocytopenia, monitor platelet counts more frequently until resolution and consider subsequent dose delays of GAZYVA and chlorambucil or dose reductions of chlorambucil. Transfusion of blood products (ie, platelet transfusion) may be necessary. Consider withholding concomitant medications which may increase bleeding risk (platelet inhibitors, anticoagulants), especially during the first cycle
immunization • The safety and efficacy of immunization with live or
attenuated viral vaccines during or following GAZYVA therapy has not been studied. Immunization with live virus vaccines is not recommended during treatment and until B-cell recovery
Pregnancy: Category C • Women of childbearing potential should use effective
contraception while receiving GAZYVA and for 12 months following treatment. GAZYVA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus
Geriatric Use • Two hundred and forty previously untreated CLL patients
received GAZYVA in combination with chlorambucil. Of the 109 patients ≥75 years of age, 49 (45%) experienced serious adverse events and 5 (5%) experienced adverse events leading to death. For 131 patients <75 years of age, 39 (30%) experienced a serious adverse event and 3 (2%) experienced an adverse event leading to death. Similar rates were observed in the comparator arm
Additional important safety information • The most common adverse reactions (≥10%) were: infusion
reactions (69%), neutropenia (40%), thrombocytopenia (15%), anemia (12%), pyrexia (10%), cough (10%), and musculoskeletal disorders (17%)
• Grade 3/4 adverse reactions were: infusion reactions (21%), neutropenia (34%), thrombocytopenia (11%), anemia (4%), leukopenia (5%), and pyrexia (<1%)
You are encouraged to report side effects to Genentech and the fDA. You may contact Genentech by calling 1-888-835-2555. You may contact the fDA by visiting www.fda.gov/medwatch, or calling 1-800-fDA-1088.
references: 1. GAZYVA full Prescribing information. south san francisco, CA: Genentech UsA, inc.; June 2014. 2. referenced with permission from the nCCn Clinical Practice Guidelines in oncology (nCCn Guidelines®) for non-hodgkin’s Lymphomas V.1.2014. © national Comprehensive Cancer network, inc. 2014. All rights reserved. Accessed march 31, 2014. to view the most recent and complete version of the guideline, go online to www.nccn.org. nAtionAL ComPrehensiVe CAnCer networK®, nCCn®, nCCn GUiDeLines®, and all other nCCn Content are trademarks owned by the national Comprehensive Cancer network, inc.
Please see the following pages for brief summary of full Prescribing information, including Boxed WArninGs.
Visit GAZYVA.com for more information
147311_L01_INSRT.indd 4 10/13/14 4:42 PM

ASCOPost.com | MARCH 25, 2015 PAGE 31
Journal Spotlight
continued on page 32
Physician-Controlled Decisions in Cancer Care Linked to Lower Quality Rating
P atients who described physician-controlled decisions about their
cancer care vs shared decision-making were less likely to report receiving ex-cellent quality of care, according to a study published by JAMA Oncology.1
The Institute of Medicine has called
for shared decision-making and accom-modation of patient preferences to im-prove the overall quality of health care. Although metrics of quality are contro-versial, patients’ reports about the qual-ity of their care are increasingly impor-tant health -care performance measures.
Nancy L. Keating, MD, MPH, of Harvard Medical School, and col-leagues surveyed patients in the Cancer Care Outcomes Research and Surveil-lance Consortium (CanCORS) study who were diagnosed with lung and/or colorectal cancer. The authors analyzed
responses from 5,315 patients who re-ported decision roles for 10,817 treat-ment decisions. They then assessed the association between patients’ roles in their decisions and their reported quality of care and physician communication.
WARNING: HEPATITIS B VIRUS REACTIVATION and PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
• Hepatitis B Virus (HBV) reactivation, in some cases resulting in fulminant hepatitis, hepatic failure, and death, can occur in patients receiving CD20-directed cytolytic antibodies, including GAZYVA. Screen all patients for HBV infection before treatment initiation. Monitor HBV-positive patients during and after treatment with GAZYVA. Discontinue GAZYVA and concomitant medications in the event of HBV reactivation [see Warnings and Precautions (5.1)].
• Progressive Multifocal Leukoencephalopathy (PML) including fatal PML, can occur in patients receiving GAZYVA [see Warnings and Precautions (5.2)].
1 INDICATIONS AND USAGEGAZYVA, in combination with chlorambucil, is indicated for the treatment of patients with previously untreated chronic lymphocytic leukemia (CLL) [see Clinical Studies (14.1)].
4 CONTRAINDICATIONSNone.
5 WARNINGS AND PRECAUTIONS
5.1 Hepatitis B Virus Reactivation Hepatitis B virus (HBV) reactivation, in some cases resulting in fulminant hepatitis, hepatic failure, and death, can occur in patients treated with anti-CD20 antibodies such as GAZYVA. HBV reactivation has been reported in patients who are hepatitis B surface antigen (HBsAg) positive and also in patients who are HBsAg negative but are hepatitis B core antibody (anti-HBc) positive. Reactivation has also occurred in patients who appear to have resolved hepatitis B infection (i.e., HBsAg negative, anti-HBc positive, and hepatitis B surface antibody [anti-HBs] positive).
HBV reactivation is defined as an abrupt increase in HBV replication manifesting as a rapid increase in serum HBV DNA level or detection of HBsAg in a person who was previously HBsAg negative and anti-HBc positive. Reactivation of HBV replication is often followed by hepatitis, i.e., increase in transaminase levels and, in severe cases, increase in bilirubin levels, liver failure, and death.
Screen all patients for HBV infection by measuring HBsAg and anti-HBc before initiating treatment with GAZYVA. For patients who show evidence of hepatitis B infection (HBsAg positive [regardless of antibody status] or HBsAg negative but anti-HBc positive), consult physicians with expertise in managing hepatitis B regarding monitoring and consideration for HBV antiviral therapy.
Monitor patients with evidence of current or prior HBV infection for clinical and laboratory signs of hepatitis or HBV reactivation during and for several months following treatment with GAZYVA. HBV reactivation has been reported for other CD20-directed cytolytic antibodies following completion of therapy.
In patients who develop reactivation of HBV while receiving GAZYVA, immediately discontinue GAZYVA and any concomitant chemotherapy, and institute appropriate treatment. Resumption of GAZYVA in patients whose HBV reactivation resolves should be discussed with physicians with expertise in managing hepatitis B. Insufficient data exist regarding the safety of resuming GAZYVA in patients who develop HBV reactivation.
5.2 Progressive Multifocal LeukoencephalopathyJC virus infection resulting in progressive multifocal leukoencephalopathy (PML), which can be fatal, was observed in patients treated with GAZYVA. Consider the diagnosis of PML in any patient presenting with new onset or changes to preexisting neurologic manifestations. Evaluation of PML includes, but is not limited to, consultation with a neurologist, brain MRI, and lumbar puncture. Discontinue GAZYVA therapy and consider discontinuation or reduction of any concomitant chemotherapy or immunosuppressive therapy in patients who develop PML.
5.3 Infusion Reactions GAZYVA can cause severe and life-threatening infusion reactions. Two thirds of patients experienced a reaction to the first 1000 mg infused of GAZYVA. Infusion reactions can also occur with subsequent infusions. Symptoms may include hypotension, tachycardia, dyspnea, and respiratory symptoms (e.g., bronchospasm, larynx and throat irritation, wheezing, laryngeal edema). Other common symptoms include nausea, vomiting, diarrhea, hypertension, flushing, headache, pyrexia, and chills [see Adverse Reactions (6.1)].
Premedicate patients with acetaminophen, antihistamine and a glucocorticoid. Institute medical management (e.g., glucocorticoids, epinephrine, bronchodilators, and/or oxygen) for infusion reactions as needed. Closely monitor patients during the entire infusion. Infusion reactions within 24 hours of receiving GAZYVA have occurred [see Dosage and Administration (2)].
For patients with any Grade 4 infusion reactions, including but not limited to anaphylaxis, acute life-threatening respiratory symptoms, or other life-threatening infusion reaction: Stop the GAZYVA infusion. Permanently discontinue GAZYVA therapy.
For patients with Grade 1, 2 or 3 infusion reactions: Interrupt GAZYVA for Grade 3 reactions until resolution of symptoms. Interrupt or reduce the rate of the infusion for Grade 1 or 2 reactions and manage symptoms [see Dosage and Administration (2)].
For patients with preexisting cardiac or pulmonary conditions, monitor more frequently throughout the infusion and the post-infusion period since they may be at greater risk of experiencing more severe reactions. Hypotension may occur as part of the GAZYVA infusion reaction. Consider withholding antihypertensive treatments for 12 hours prior to, during each GAZYVA infusion, and for the first hour after administration until blood pressure is stable. For patients at increased risk of hypertensive crisis, consider the benefits versus the risks of withholding their antihypertensive medication.
5.4 Tumor Lysis SyndromeAcute renal failure, hyperkalemia, hypocalcemia, hyperuricemia, and/or hyperphosphatemia from Tumor Lysis Syndrome (TLS) can occur within 12–24 hours after the first infusion. Patients with high tumor burden and/or high circulating lymphocyte count (> 25 x 109/L) are at greater risk for TLS and should receive appropriate tumor lysis prophylaxis with anti-hyperuricemics (e.g., allopurinol) and hydration beginning 12–24 hours prior to the infusion of GAZYVA [see Dosage and Administration (2.2)]. For treatment of TLS, correct electrolyte abnormalities, monitor renal function and fluid balance, and administer supportive care, including dialysis as indicated.
5.5 InfectionsSerious bacterial, fungal, and new or reactivated viral infections can occur during and following GAZYVA therapy. Fatal infections have been reported with GAZYVA. Do not administer GAZYVA to patients with an active infection. Patients with a history of recurring or chronic infections may be at increased risk of infection.
5.6 Neutropenia GAZYVA in combination with chlorambucil caused Grade 3 or 4 neutropenia in 33% of patients in the trial. Patients with Grade 3 to 4 neutropenia should be monitored frequently with regular laboratory tests until resolution. Anticipate, evaluate, and treat any symptoms or signs of developing infection.
Neutropenia can also be of late onset (occurring more than 28 days after completion of treatment) and/or prolonged (lasting longer than 28 days).
Patients with neutropenia are strongly recommended to receive antimicrobial prophylaxis throughout the treatment period. Antiviral and antifungal prophylaxis should be considered.
5.7 ThrombocytopeniaGAZYVA in combination with chlorambucil caused Grade 3 or 4 thrombocytopenia in 10% of patients in the trial. In 4% of patients, GAZYVA caused acute thrombocytopenia occurring within 24 hours after the GAZYVA infusion. Fatal hemorrhagic events during Cycle 1 have also been reported in patients treated with GAZYVA.
Monitor all patients frequently for thrombocytopenia and hemorrhagic events, especially during the first cycle. In patients with Grade 3 or 4 thrombocytopenia, monitor platelet counts more frequently until resolution and consider subsequent dose delays of GAZYVA and chlorambucil or dose reductions of chlorambucil. Transfusion of blood products (i.e., platelet transfusion) may be necessary. Consider withholding concomitant medications which may increase bleeding risk (platelet inhibitors, anticoagulants), especially during the first cycle.
5.8 ImmunizationThe safety and efficacy of immunization with live or attenuated viral vaccines during or following GAZYVA therapy has not been studied. Immunization with live virus vaccines is not recommended during treatment and until B-cell recovery.
6 ADVERSE REACTIONSThe following adverse reactions are discussed in greater detail in other sections of the label: • Hepatitis B reactivation [see Warnings and Precautions (5.1)] • Progressive multifocal leukoencephalopathy [see Warnings
and Precautions (5.2)] • Infusion reactions [see Warnings and Precautions (5.3)] • Tumor lysis syndrome [see Warnings and Precautions (5.4)] • Infections [see Warnings and Precautions (5.5)] • Neutropenia [see Warnings and Precautions (5.6)] • Thrombocytopenia [see Warnings and Precautions (5.7)]
The most common adverse reactions (incidence ≥ 10%) were infusion reactions, neutropenia, thrombocytopenia, anemia, pyrexia, cough, nausea, and diarrhea.
6.1 Clinical Trial Experience Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in Tables 3–6 below are based on a safety population of 773 previously untreated patients with CLL. Patients were treated with chlorambucil alone, GAZYVA in combination with chlorambucil, or rituximab in combination with chlorambucil. The Stage 1 analysis compared GAZYVA in combination with chlorambucil vs. chlorambucil alone, and Stage 2 compared GAZYVA in combination with chlorambucil vs. rituximab in combination with chlorambucil. Patients received three 1000 mg doses of GAZYVA on the first cycle and a single dose of 1000 mg once every 28 days for 5 additional cycles in combination with chlorambucil (6 cycles of 28 days each in total). In the last 140 patients enrolled, the first dose of GAZYVA was split between day 1 (100 mg) and day 2 (900 mg) [see Dosage and Administration (2.1)]. In total, 81% of patients received all 6 cycles (of 28 days each) of GAZYVA-based therapy.
Table 3 Summary of Adverse Reactions Reported in ≥ 5% of Patients and at Least 2% Greater in the GAZYVA Treated Arm (Stage 1)
GAZYVA® (obinutuzumab) Injection, for intravenous infusion Initial U.S. Approval: 2013 This is a brief summary of information about GAZYVA. Before prescribing, please see full Prescribing Information.
Injury, poisoning, and procedural complicationsInfusion 69 21 0 0reactions
Blood and lymphatic system disordersb
Neutropenia 41 35 18 16
Thrombocytopenia 15 11 8 4
Anemia 12 5 10 4
Leukopenia 7 5 0 0
General disorders and administration site conditionsPyrexia 10 < 1 7 0
Respiratory, thoracic, and mediastinal disordersCough 10 0 7 < 1
Infections and infestationsUrinary tract 6 2 3 < 1infection
Musculoskeletal and connective tissue disorderBack pain 5 < 1 2 0
Adverse Reactions (MedDRAa) System Organ Class
GAZYVA + Chlorambucil
n = 336
Rituximab + Chlorambucil
n = 321
All Grades %
Grades 3–4 %
All Grades %
Grades 3–4 %
Table 4 Summary of Adverse Reactions Reported in ≥ 5% of Patients and at Least 2% Greater in the GAZYVA Treated Arm (Stage 2)
Injury, poisoning and procedural complicationsInfusion 66 20 38 4reactions
Blood and lymphatic system disordersb
Neutropenia 38 33 32 28
Thrombocytopenia 14 10 7 3
Leukopenia 6 4 2 < 1
General disorders and administration site conditionsPyrexia 9 < 1 7 < 1
Gastrointestinal disordersDiarrhea 10 2 8 < 1
Constipation 8 0 5 0
Infections and infestationsNasopharyngitis 6 < 1 3 0
Urinary tract 5 1 2 < 1infection
a MedDRA coded adverse reactions as reported by investigators. b Adverse events reported under “Blood and lymphatic
system disorders” reflect those reported by investigator as clinically significant.
Adverse Reactions (MedDRAa) System Organ Class
GAZYVA + Chlorambucil
n = 241
Chlorambucil n = 116
All Grades %
Grades 3–4 %
All Grades %
Grades 3–4 %
02-12478_R01_GAUS_BriefSummary_A_size.indd 1 1/29/15 3:00 PM

PAGE 32 The ASCO Post | MARCH 25, 2015
Journal Spotlight
A total of 58% of patients preferred shared roles in decision-making about their cancer, 36% preferred patient-controlled decisions, and 6% preferred physician-controlled decisions. Of the treatment decisions made by patients, 42% were regarding surgery, 36% regard-
ing chemotherapy, and 22% regarding ra-diation therapy. The patients in the study reported their actual decision-making pro-cess was patient-controlled in 39% of deci-sions, shared in 44% of decisions, and con-trolled by physicians in 17% of decisions.
Patients reported their care by the physician performing the treatment as excellent in 67.8% of cases. While a pa-
tient’s preferred role in decision-making was not associated with quality ratings, patient reports that treatment deci-sions were controlled by physicians (vs shared) were associated with lower odds of excellent patient-reported quality.
Overall, 55.8% of patients gave their physicians the highest possible rating of communication. However, patients who preferred physician-controlled to shared decisions were less likely to give top ratings to their physicians, as were patients who reported actually experi-encing physician-controlled vs shared decisions, the results show.
“Given the increasing emphasis on patient experiences and ratings in health care, these results highlight the benefits of promoting shared decision making among all patients with cancer, even those who express preferences for less active roles,” the study concludes.
Shared Decision-MakingIn a related commentary,2 Sarah
T. Hawley, PhD, MPH, and Reshma Jagsi, MD, DPhil, both of the Univer-sity of Michigan, wrote, “We find it un-surprising that even patients who pre-ferred a physician-controlled decision rated the physician communication outcomes highest when the actual deci-sion-making process was more shared, as the individual items that constitute the communication measure described elements most likely to be absent when the actual decision is not shared.”
They continued, “More compelling is the association found between shared decision-making and patient appraisal of excellent quality of care. It is intriguing that this association remained, even when controlling for preferred role. Dr. Kehl and colleagues conclude from this finding that it is important to promote shared de-cision-making, even among patients who may seek less active roles. Yet these results are in some contrast to prior work that has suggested that it is the match between pa-tients’ preferred and actual involvement that contributes to greater satisfaction with care. These conflicting results under-score the need for further work to better quantify and link measures of shared deci-sion making to patient appraisal of care,” they concluded. n
Disclaimer: This work of the CanCORS Consortium was supported by grants from the National Cancer Institute and the Department of Veterans Affairs.
References1. Kehl KL, Landrum MB, Arora NK, et
al: JAMA Oncol. February 12, 2015 (early release online).
2. Hawley ST, Jagsi R: JAMA Oncol. February 12, 2015 (early release online).
Physician-Controlled Decisionscontinued from page 31
Nancy L. Keating, MD, MPH
Infusion Reactions: The incidence of infusion reactions was 65% with the first infusion of GAZYVA. The incidence of Grade 3 or 4 infusion reactions was 20% with 7% of patients discontinuing therapy. The incidence of reactions with subsequent infusions was 3% with the second 1000 mg and < 1% thereafter. No Grade 3 or 4 infusion reactions were reported beyond the first 1000 mg infused.
Of the first 53 patients receiving GAZYVA on the trial, 47 (89%) experienced an infusion reaction. After this experience, study protocol modifications were made to require pre-medication with a corticosteroid, antihistamine, and acetaminophen. The first dose was also divided into two infusions (100 mg on day 1 and 900 mg on day 2). For the 140 patients for whom these mitigation measures were implemented, 74 patients (53%) experienced a reaction with the first 1000 mg (64 patients on day 1, 3 patients on day 2, and 7 patients on both days) and < 3% thereafter [see Dosage and Administration (2)].
Neutropenia: The incidence of neutropenia reported as an adverse reaction was 38% in the GAZYVA treated arm and 32% in the rituximab treated arm, with the incidence of serious adverse events being 1% and < 1%, respectively (Table 4). Cases of late-onset neutropenia (occurring 28 days after completion of treatment or later) were 16% in the GAZYVA treated arm and 12% in the rituximab treated arm.
Infection: The incidence of infections was similar between GAZYVA and rituximab treated arms. Thirty-eight percent of patients in the GAZYVA treated arm and 37% in the rituximab treated arm experienced an infection, with Grade 3–4 rates being 11% and 13%, respectively. Fatal events were reported in 1% of patients in both arms.
Thrombocytopenia: The overall incidence of thrombocytopenia reported as an adverse reaction was higher in the GAZYVA treated arm (14%) compared to the rituximab treated arm
(7%), with the incidence of Grade 3–4 events being 10% and 3%, respectively (Table 4). The difference in incidences between the treatment arms is driven by events occurring during the first cycle. The incidence of thrombocytopenia (all grades) in the first cycle were 11% in the GAZYVA and 3% in the rituximab treated arms, with Grade 3–4 rates being 8% and 2%, respectively. Four percent of patients in the GAZYVA treated arm experienced acute thrombocytopenia (occurring within 24 hours after the GAZYVA infusion).
The overall incidence of hemorrhagic events and the number of fatal hemorrhagic events were similar between the treatment arms, with 3 in the rituximab and 4 in the GAZYVA treated arms. However, all fatal hemorrhagic events in patients treated with GAZYVA occurred in Cycle 1.
Tumor Lysis Syndrome: The incidence of Grade 3 or 4 tumor lysis syndrome was 2% in the GAZYVA treated arm versus 0% in the rituximab treated arm.
Musculoskeletal Disorders: Adverse events related to musculoskeletal disorders (all events from the System Organ Class), including pain, have been reported in the GAZYVA treated arm with higher incidence than in the rituximab treated arm (18% vs. 15%).
Liver Enzyme Elevations: Hepatic enzyme elevations have occurred in patients who received GAZYVA in clinical trials and had normal baseline hepatic enzyme levels (AST, ALT, and ALP). The events occurred most frequently within 24-48 hours of the first infusion. In some patients, elevations in liver enzymes were observed concurrently with infusion reactions or tumor lysis syndrome. In the pivotal study, there was no clinically meaningful difference in overall hepatotoxicity adverse events between all arms (4% of patients in the GAZYVA treated arm). Medications commonly used to prevent infusion reactions (e.g., acetaminophen) may also be implicated in these events. Monitor liver function tests during treatment, especially during the first cycle. Consider treatment interruption or discontinuation for hepatotoxicity.
6.2 ImmunogenicitySerum samples from patients with previously untreated CLL were tested during and after treatment for antibodies to GAZYVA. Of the GAZYVA treated patients, 7% (18/271) tested positive for anti-GAZYVA antibodies at one or more time points. Neutralizing activity of anti-GAZYVA antibodies has not been assessed.
Immunogenicity data are highly dependent on the sensitivity and specificity of the test methods used. Additionally, the observed incidence of a positive result in a test method may be influenced by several factors, including sample handling, timing of sample collection, drug interference, concomitant medication, and the underlying disease. Therefore, comparison of the incidence of antibodies to GAZYVA with the incidence of antibodies to other products may be misleading. Clinical significance of anti-GAZYVA antibodies is not known.
6.3 Additional Clinical Trial ExperienceWorsening of Pre-existing Cardiac Conditions: Fatal cardiac events have been reported in patients treated with GAZYVA.
7 DRUG INTERACTIONSNo formal drug interaction studies have been conducted with GAZYVA.
8 USE IN SPECIFIC POPULATIONS8.1 PregnancyPregnancy Category CRisk SummaryThere are no adequate and well-controlled studies of GAZYVA in pregnant women. Women of childbearing potential should use effective contraception while receiving GAZYVA and for 12 months following treatment. GAZYVA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Mothers who have been exposed to GAZYVA during pregnancy should discuss the safety and timing of live virus vaccinations for their infants with their child’s healthcare providers.
Animal DataIn a pre- and post-natal development study, pregnant cynomolgus monkeys received weekly intravenous doses of 25 or 50 mg/kg obinutuzumab from day 20 of pregnancy until parturition. There were no teratogenic effects in animals. The high dose results in an exposure (AUC) that is 2.4 times the exposure in patients with CLL at the recommended label dose. When first measured on day 28 postpartum, obinutuzumab was detected in offspring, and B cells were completely depleted. The B-cell counts returned to normal levels, and immunologic function was restored within 6 months after birth.
8.3 Nursing MothersIt is not known whether obinutuzumab is excreted in human milk. However, obinutuzumab is excreted in the milk of lactating cynomolgus monkeys and human IgG is known to be excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from obinutuzumab, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric UseThe safety and effectiveness of GAZYVA in pediatric patients has not been established.
GAZYVA® [obinutuzumab]
Manufactured by: Genentech, Inc.
A Member of the Roche Group South San Francisco, CA 94080-4990
U.S. License No: 1048
Initial US Approval: 2013
Code Revision Date: December 2014
GAZYVA is a registered trademark of Genentech, Inc.
GAZ/011615/0009 1/15
© 2015 Genentech, Inc.
Table 5 Post-Baseline Laboratory Abnormalities by CTCAE Grade in ≥ 5% of Patients and at Least 2% Greater in the GAZYVA Treated Arm (Stage 1)
Table 6 Post-Baseline Laboratory Abnormalities by CTCAE Grade in ≥ 5% of Patients and at Least 2% Greater in the GAZYVA Treated Arm (Stage 2)
HematologyNeutropenia 78 48 53 27
Lymphopenia 80 40 9 3
Leukopenia 84 37 12 < 1
ChemistryHypocalcemia 38 3 33 2
Hyperkalemia 33 5 18 3
Hyponatremia 30 8 12 3
AST 29 1 16 0(SGOT increased)
Creatinine 30 < 1 20 2increased
ALT 27 2 16 0 (SGPT increased)
Hypoalbuminemia 23 < 1 15 < 1
Alkaline phosphatase 18 0 11 0increased
Hypokalemia 15 1 5 < 1
HematologyNeutropenia 76 46 69 41
Lymphopenia 80 39 50 16
Leukopenia 84 35 62 16
Thrombocytopenia 48 13 40 8
Anemia 39 10 37 10
ChemistryHypocalcemia 37 3 32 <1
Hyperkalemia 14 1 10 <1
Hyponatremia 26 7 18 2
AST 27 2 21 <1(SGOT increased)
ALT 28 2 21 1 (SGPT increased)
Hypoalbuminemia 23 <1 16 <1
8.5 Geriatric UseOf 336 previously untreated CLL patients who received GAZYVA in combination with chlorambucil, 273 patients (81%) were ≥ 65 years of age and 156 patients (46%) were ≥ 75 years of age. The median age was 74 years. Of the 156 patients ≥ 75 years of age, 72 (46%) experienced serious adverse events and 11 (7%) experienced adverse events leading to death. For 180 patients < 75 years of age, 59 (33%) experienced a serious adverse event and 4 (2%) an adverse event leading to death. No significant differences in efficacy were observed between patients ≥ 75 years of age and those < 75 years of age [see Clinical Studies (14.1)].
8.6 Renal ImpairmentBased on population pharmacokinetic analysis, a baseline creatinine clearance (CrCl) ≥ 30 mL/min does not affect the pharmacokinetics of GAZYVA. GAZYVA has not been studied in patients with a baseline CrCl < 30 mL/min [see Clinical Pharmacology (12.3)].
8.7 Hepatic ImpairmentGAZYVA has not been studied in patients with hepatic impairment.
10 OVERDOSAGEThere has been no experience with overdose in human clinical trials. Doses ranging from 50 mg up to and including 2000 mg per infusion have been administered in clinical trials. For patients who experience overdose, treatment should consist of immediate interruption or reduction of GAZYVA and supportive therapy.
17 PATIENT COUNSELING INFORMATIONAdvise patients to seek immediate medical attention for any of the following:
• Signs and symptoms of infusion reactions including dizziness, nausea, chills, fever, vomiting, diarrhea, breathing problems, or chest pain [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
• Symptoms of tumor lysis syndrome such as nausea, vomiting, diarrhea, and lethargy [see Warnings and Precautions (5.4) and Adverse Reactions (6.1)].
• Signs of infections including fever and cough [see Warnings and Precautions (5.5) and Adverse Reactions (6.1)].
• Symptoms of hepatitis including worsening fatigue or yellow discoloration of skin or eyes [see Warnings and Precautions (5.1)].
• New or changes in neurological symptoms such as confusion, dizziness or loss of balance, difficulty talking or walking, or vision problems [see Warnings and Precautions (5.2)].
Advise patients of the need for:
• Periodic monitoring of blood counts [see Warnings and Precautions (5.6 and 5.7) and Adverse Reactions (6.1)].
• Avoid vaccinations with live viral vaccines [see Warnings and Precautions (5.8)].
• Patients with a history of hepatitis B infection (based on the blood test) should be monitored and sometimes treated for their hepatitis [see Warnings and Precautions (5.1)].
InvestigationsGAZYVA
+ Chlorambucil n = 241
Chlorambucil n = 116
All Grades %
Grades 3–4 %
All Grades %
Grades 3–4 %
InvestigationsGAZYVA
+ Chlorambucil n = 336
Rituximab + Chlorambucil
n = 321
All Grades %
Grades 3–4 %
All Grades %
Grades 3–4 %
02-12478_R01_GAUS_BriefSummary_A_size.indd 2 1/29/15 3:00 PM

ASCOPost.com | MARCH 25, 2015 PAGE 33
Expert’s Corner
Ongoing Controversies in Allocating Our Health-Care ResourcesA Conversation With Richard ‘Buz’ Cooper, MDBy Ronald Piana
Disparities of care that result in poor-er outcomes among certain popu-
lations have long been an issue addressed by the cancer community and its major organizations such as ASCO. While ethnicity and race play key roles in this ongoing debate over equitable allocation of our precious health-care resources, another persistent issue frames the larger story of the haves and have-nots in can-cer care outcomes: poverty and the poli-tics of health-care distribution.
To bring clarity to the issue of how we apportion health-care services and the doctors who deliver them, The ASCO Post visited with Richard “Buz” Cooper, MD, Director of the Center for the Future of the Healthcare Workforce and a Senior Fellow in the Leonard Da-vis Institute of Health Economics at the University of Pennsylvania. At the Upper East Side apartment he shares with his wife, Barrie R. Cassileth, MS, PhD, the Laurance S. Rockefeller Chair in Integrative Medicine at Memo-rial Sloan Kettering Cancer Center, Dr. Cooper shared his experiences in hema-tology and oncology, as well as his cur-rent work in public health.
Early CareerWith the Manhattan skyline stretch-
ing out from the 32nd-floor apartment, Dr. Cooper, a tall man with a profes-sorial bearing, explained that his early career as a hematologist was influenced by his work at the National Cancer In-stitute (NCI) in 1963, when Drs. Emil Frei and Emil J. Freireich first began us-ing combination chemotherapy to treat childhood acute leukemia. “It was an extraordinary experience to see these children with a heretofore fatal disease respond to this first-time-ever treat-ment. It was truly a remarkable time in oncology, and I wanted to be part of it,” said Dr. Cooper.
After his stint at the NCI came to a close, Dr. Cooper finished his hematol-ogy fellowship at Boston City Hospi-tal. In 1971, he went to the University of Pennsylvania to start a hematology/oncology program. “Interest was build-ing to expand the nation’s cancer cen-ters, so we gathered our resources and developed a cancer center attached to the university, which broke the mold of the freestanding centers that the NCI preferred,” said Dr. Cooper. He added, “Dr. Peter Nowell, who discovered the Philadelphia chromosome, assumed the director’s mantle for the first years; then I took over, and we developed the center into a nationally recognized multidisci-plinary cancer institute.”
The next chapter in Dr. Cooper’s ca-reer took him back to his hometown of Milwaukee, where he became Dean of the Medical College of Wisconsin. “It
was a small school that struggled finan-cially. Much like the Penn cancer cen-ter, which we really made out of whole cloth, the Medical College, in a sense, had to be built from the ground up. It was a 9-year project of love, and when I left the College it was on the cusp of being a powerhouse,” said Dr. Cooper.
Physician Supply and DemandThe next period of Dr. Cooper’s ca-
reer was transformative and, in part, in-forms his current work in public health. During the Clinton administration’s health-reform initiative, a startling ob-servation emerged: We were heading for a surplus of 100,000 doctors by 2000. According to Dr. Cooper, this projection would reduce the recruit-ing of students into medical school and drastically cut residency programs, causing long-term reductions in Ameri-ca’s physician force.
“This doctor surplus dogma was held by many experts. A friend of mine at the [American Medical Association] asked if I could address this issue. I did,
and I soon found a glaring error in the surplus projection: Their conclusion was based on outdated census data, which assumed that the U.S. population was not going to increase. So, I called the Census Bureau and had them send me the current data,” said Dr. Cooper.
Dr. Cooper applied the Census Bu-reau’s updated projection of population growth with the existing projections of physician growth based on the numbers coming out of medical schools, current-ly working, and those retiring.
“The administration’s projection of a surplus of doctors by 2000 was wrong. Instead, we were on track for a danger-ous shortage of doctors,” said Dr. Coo-per. “My findings caused an outrage. But there were nuances that the ad-ministration was missing. For instance, at one point, lung cancer was virtually untreatable so of course you didn’t need
lung cancer specialists. But advances in detection and treatment fostered a need for more oncologists to treat that and other diseases. We needed more doc-tors, not fewer.”
In 2009, Dr. Cooper was called to testify at a Senate hearing on the need for innovative models in health care, which was, in part, a subtle call for a reduction in physician services. During the hearing, Dr. Cooper made his opin-ion clear: “My analysis of our need to expand physician supply and encourage innovation in physician practices stands against a set of beliefs that more phy-sicians and more health care may not be good for the nation…. In fact, the preponderance of data do not support these conclusions.”
To this day, Dr. Cooper laments that after all his work on the issue of physician supply and demand, there was still a core of influential policymakers who favored a reduction in the number of physicians. “I have an upcoming debate at the National Resident Matching Program, but the fact that I’m arguing whether or not we need
to expand physician supply is sad. There is an entrenched body of opinion in cer-tain sectors of health care that refuses to recognize, for instance, that we are facing a workforce shortage crisis in oncology,” he said.
Health-Care Disparities Dr. Cooper further commented that
along with the erroneous statistics used to project a surplus of physicians, the government-driven agenda was claim-ing that addressing waste and inefficien-cy would ameliorate the need for more doctors and services.
“The fundamental underpinning of this grand assumption is derived from The Dartmouth Atlas of Health Care, which contends that regional variations in cost are driven by variations in how physicians practice,” he said. Although Dr. Cooper agrees that there is sub-stantial waste and inefficiency in the health-care system, he contends that it is economic disparity in care, and not physician practice patterns, that drive overutilization of health-care services.
He stressed that poorer people, for a number of socioeconomic reasons, are less healthy than economically stable or affluent people and therefore cost more to treat. “For example,” he said, “Mississippi, which is the poorest state in the country, has very high per capita Medicare spending, simply because the population presents with serious health issues that are costly to treat.”
Dr. Cooper emphasized his point: “Health-care costs on the affluent Up-per East Side of New York are about 80% of the national average. But walk a few blocks up to Harlem, for instance, and the per capita costs are about three times the national average. The same health-care providers are serving pa-tients from both areas, but the patients from Harlem simply cost more to care for because of the negative health con-sequences of living in poverty. And if you merge the data from the two eco-nomically divergent areas, you get a very distorted view of costs relative to physician services in Manhattan.”
In Dr. Cooper’s opinion, projects like The Dartmouth Atlas and other cost-ef-fectiveness initiatives that ostensibly seek to decrease waste and inefficiency in our health-care system have an unintended consequence—they erode physician au-tonomy and move the locus of control
Health-Care Policy
Richard ‘Buz’ Cooper, MD
continued on page 34
All of these academic conclusions about overutilization of physician services being the main driver of our so-
called fiscal crisis in health-care point at specialties, such as oncology, as the main cause. However, it is just the
opposite; our specialists drive innovation and excellent, cost-effective care in our system.
—Richard “Buz” Cooper, MD
Infusion Reactions: The incidence of infusion reactions was 65% with the first infusion of GAZYVA. The incidence of Grade 3 or 4 infusion reactions was 20% with 7% of patients discontinuing therapy. The incidence of reactions with subsequent infusions was 3% with the second 1000 mg and < 1% thereafter. No Grade 3 or 4 infusion reactions were reported beyond the first 1000 mg infused.
Of the first 53 patients receiving GAZYVA on the trial, 47 (89%) experienced an infusion reaction. After this experience, study protocol modifications were made to require pre-medication with a corticosteroid, antihistamine, and acetaminophen. The first dose was also divided into two infusions (100 mg on day 1 and 900 mg on day 2). For the 140 patients for whom these mitigation measures were implemented, 74 patients (53%) experienced a reaction with the first 1000 mg (64 patients on day 1, 3 patients on day 2, and 7 patients on both days) and < 3% thereafter [see Dosage and Administration (2)].
Neutropenia: The incidence of neutropenia reported as an adverse reaction was 38% in the GAZYVA treated arm and 32% in the rituximab treated arm, with the incidence of serious adverse events being 1% and < 1%, respectively (Table 4). Cases of late-onset neutropenia (occurring 28 days after completion of treatment or later) were 16% in the GAZYVA treated arm and 12% in the rituximab treated arm.
Infection: The incidence of infections was similar between GAZYVA and rituximab treated arms. Thirty-eight percent of patients in the GAZYVA treated arm and 37% in the rituximab treated arm experienced an infection, with Grade 3–4 rates being 11% and 13%, respectively. Fatal events were reported in 1% of patients in both arms.
Thrombocytopenia: The overall incidence of thrombocytopenia reported as an adverse reaction was higher in the GAZYVA treated arm (14%) compared to the rituximab treated arm
(7%), with the incidence of Grade 3–4 events being 10% and 3%, respectively (Table 4). The difference in incidences between the treatment arms is driven by events occurring during the first cycle. The incidence of thrombocytopenia (all grades) in the first cycle were 11% in the GAZYVA and 3% in the rituximab treated arms, with Grade 3–4 rates being 8% and 2%, respectively. Four percent of patients in the GAZYVA treated arm experienced acute thrombocytopenia (occurring within 24 hours after the GAZYVA infusion).
The overall incidence of hemorrhagic events and the number of fatal hemorrhagic events were similar between the treatment arms, with 3 in the rituximab and 4 in the GAZYVA treated arms. However, all fatal hemorrhagic events in patients treated with GAZYVA occurred in Cycle 1.
Tumor Lysis Syndrome: The incidence of Grade 3 or 4 tumor lysis syndrome was 2% in the GAZYVA treated arm versus 0% in the rituximab treated arm.
Musculoskeletal Disorders: Adverse events related to musculoskeletal disorders (all events from the System Organ Class), including pain, have been reported in the GAZYVA treated arm with higher incidence than in the rituximab treated arm (18% vs. 15%).
Liver Enzyme Elevations: Hepatic enzyme elevations have occurred in patients who received GAZYVA in clinical trials and had normal baseline hepatic enzyme levels (AST, ALT, and ALP). The events occurred most frequently within 24-48 hours of the first infusion. In some patients, elevations in liver enzymes were observed concurrently with infusion reactions or tumor lysis syndrome. In the pivotal study, there was no clinically meaningful difference in overall hepatotoxicity adverse events between all arms (4% of patients in the GAZYVA treated arm). Medications commonly used to prevent infusion reactions (e.g., acetaminophen) may also be implicated in these events. Monitor liver function tests during treatment, especially during the first cycle. Consider treatment interruption or discontinuation for hepatotoxicity.
6.2 ImmunogenicitySerum samples from patients with previously untreated CLL were tested during and after treatment for antibodies to GAZYVA. Of the GAZYVA treated patients, 7% (18/271) tested positive for anti-GAZYVA antibodies at one or more time points. Neutralizing activity of anti-GAZYVA antibodies has not been assessed.
Immunogenicity data are highly dependent on the sensitivity and specificity of the test methods used. Additionally, the observed incidence of a positive result in a test method may be influenced by several factors, including sample handling, timing of sample collection, drug interference, concomitant medication, and the underlying disease. Therefore, comparison of the incidence of antibodies to GAZYVA with the incidence of antibodies to other products may be misleading. Clinical significance of anti-GAZYVA antibodies is not known.
6.3 Additional Clinical Trial ExperienceWorsening of Pre-existing Cardiac Conditions: Fatal cardiac events have been reported in patients treated with GAZYVA.
7 DRUG INTERACTIONSNo formal drug interaction studies have been conducted with GAZYVA.
8 USE IN SPECIFIC POPULATIONS8.1 PregnancyPregnancy Category CRisk SummaryThere are no adequate and well-controlled studies of GAZYVA in pregnant women. Women of childbearing potential should use effective contraception while receiving GAZYVA and for 12 months following treatment. GAZYVA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Mothers who have been exposed to GAZYVA during pregnancy should discuss the safety and timing of live virus vaccinations for their infants with their child’s healthcare providers.
Animal DataIn a pre- and post-natal development study, pregnant cynomolgus monkeys received weekly intravenous doses of 25 or 50 mg/kg obinutuzumab from day 20 of pregnancy until parturition. There were no teratogenic effects in animals. The high dose results in an exposure (AUC) that is 2.4 times the exposure in patients with CLL at the recommended label dose. When first measured on day 28 postpartum, obinutuzumab was detected in offspring, and B cells were completely depleted. The B-cell counts returned to normal levels, and immunologic function was restored within 6 months after birth.
8.3 Nursing MothersIt is not known whether obinutuzumab is excreted in human milk. However, obinutuzumab is excreted in the milk of lactating cynomolgus monkeys and human IgG is known to be excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from obinutuzumab, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric UseThe safety and effectiveness of GAZYVA in pediatric patients has not been established.
GAZYVA® [obinutuzumab]
Manufactured by: Genentech, Inc.
A Member of the Roche Group South San Francisco, CA 94080-4990
U.S. License No: 1048
Initial US Approval: 2013
Code Revision Date: December 2014
GAZYVA is a registered trademark of Genentech, Inc.
GAZ/011615/0009 1/15
© 2015 Genentech, Inc.
Table 5 Post-Baseline Laboratory Abnormalities by CTCAE Grade in ≥ 5% of Patients and at Least 2% Greater in the GAZYVA Treated Arm (Stage 1)
Table 6 Post-Baseline Laboratory Abnormalities by CTCAE Grade in ≥ 5% of Patients and at Least 2% Greater in the GAZYVA Treated Arm (Stage 2)
HematologyNeutropenia 78 48 53 27
Lymphopenia 80 40 9 3
Leukopenia 84 37 12 < 1
ChemistryHypocalcemia 38 3 33 2
Hyperkalemia 33 5 18 3
Hyponatremia 30 8 12 3
AST 29 1 16 0(SGOT increased)
Creatinine 30 < 1 20 2increased
ALT 27 2 16 0 (SGPT increased)
Hypoalbuminemia 23 < 1 15 < 1
Alkaline phosphatase 18 0 11 0increased
Hypokalemia 15 1 5 < 1
HematologyNeutropenia 76 46 69 41
Lymphopenia 80 39 50 16
Leukopenia 84 35 62 16
Thrombocytopenia 48 13 40 8
Anemia 39 10 37 10
ChemistryHypocalcemia 37 3 32 <1
Hyperkalemia 14 1 10 <1
Hyponatremia 26 7 18 2
AST 27 2 21 <1(SGOT increased)
ALT 28 2 21 1 (SGPT increased)
Hypoalbuminemia 23 <1 16 <1
8.5 Geriatric UseOf 336 previously untreated CLL patients who received GAZYVA in combination with chlorambucil, 273 patients (81%) were ≥ 65 years of age and 156 patients (46%) were ≥ 75 years of age. The median age was 74 years. Of the 156 patients ≥ 75 years of age, 72 (46%) experienced serious adverse events and 11 (7%) experienced adverse events leading to death. For 180 patients < 75 years of age, 59 (33%) experienced a serious adverse event and 4 (2%) an adverse event leading to death. No significant differences in efficacy were observed between patients ≥ 75 years of age and those < 75 years of age [see Clinical Studies (14.1)].
8.6 Renal ImpairmentBased on population pharmacokinetic analysis, a baseline creatinine clearance (CrCl) ≥ 30 mL/min does not affect the pharmacokinetics of GAZYVA. GAZYVA has not been studied in patients with a baseline CrCl < 30 mL/min [see Clinical Pharmacology (12.3)].
8.7 Hepatic ImpairmentGAZYVA has not been studied in patients with hepatic impairment.
10 OVERDOSAGEThere has been no experience with overdose in human clinical trials. Doses ranging from 50 mg up to and including 2000 mg per infusion have been administered in clinical trials. For patients who experience overdose, treatment should consist of immediate interruption or reduction of GAZYVA and supportive therapy.
17 PATIENT COUNSELING INFORMATIONAdvise patients to seek immediate medical attention for any of the following:
• Signs and symptoms of infusion reactions including dizziness, nausea, chills, fever, vomiting, diarrhea, breathing problems, or chest pain [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
• Symptoms of tumor lysis syndrome such as nausea, vomiting, diarrhea, and lethargy [see Warnings and Precautions (5.4) and Adverse Reactions (6.1)].
• Signs of infections including fever and cough [see Warnings and Precautions (5.5) and Adverse Reactions (6.1)].
• Symptoms of hepatitis including worsening fatigue or yellow discoloration of skin or eyes [see Warnings and Precautions (5.1)].
• New or changes in neurological symptoms such as confusion, dizziness or loss of balance, difficulty talking or walking, or vision problems [see Warnings and Precautions (5.2)].
Advise patients of the need for:
• Periodic monitoring of blood counts [see Warnings and Precautions (5.6 and 5.7) and Adverse Reactions (6.1)].
• Avoid vaccinations with live viral vaccines [see Warnings and Precautions (5.8)].
• Patients with a history of hepatitis B infection (based on the blood test) should be monitored and sometimes treated for their hepatitis [see Warnings and Precautions (5.1)].
InvestigationsGAZYVA
+ Chlorambucil n = 241
Chlorambucil n = 116
All Grades %
Grades 3–4 %
All Grades %
Grades 3–4 %
InvestigationsGAZYVA
+ Chlorambucil n = 336
Rituximab + Chlorambucil
n = 321
All Grades %
Grades 3–4 %
All Grades %
Grades 3–4 %
02-12478_R01_GAUS_BriefSummary_A_size.indd 2 1/29/15 3:00 PM

PAGE 34 The ASCO Post | MARCH 25, 2015
Expert’s Corner
from the medical professional to the reg-ulators and health-care administrators.
“All of these academic conclusions about overutilization of physician ser-vices being the main driver of our so-called fiscal crisis in health-care point at specialties, such as oncology, as the main cause. However, it is just the op-posite; specialists drive innovation and excellent, cost-effective care.”
He continued, “In fact, when all of the states across the country are criti-cally examined, more total spending and more specialists are associated with better-quality health outcomes—just
the opposite of The Dartmouth Atlas message, but just what you would ex-pect if you analyze how the delivery of health-care services truly affects popula-tions of vastly different socioeconomic structures. Furthermore, more spend-ing at the high end improves outcomes because the care consists of a broader spectrum of beneficial services.”
Poverty and Health-Care Costs“There is a small group of researchers
who generally get lumped into the “social justice world” of medicine. They’re a good bunch of professionals who have col-lected abundant evidence showing that poverty is strongly associated with poor
health status, greater per capita spending, more hospital readmissions, and poorer outcomes. It is the single strongest factor in geographic variations in health care and the single greatest contributor to excess spending,” said Dr. Cooper.
He stressed that in order to fully understand how different populations access health care and utilize services, we must develop a risk-assessment methodology that captures all of the so-cial and demographic variables. “A new report by the [Institute of Medicine] has recommended such an approach. It will be difficult to implement over time, given the complexity of these variables, but at least it points us in the right di-
rection and blows the whistle on those policymakers who slice and dice the data to arrive at politically expedient conclusions,” said Dr. Cooper.
Dr. Cooper is a firm believer that more doctors—especially oncologists and other specialists—are ultimately needed to deliver high-quality cost-ef-fective medical care as our aging popu-lation grows. He also is firm about an-other subject: Poverty is a huge driver of health-care costs. To that end, Dr. Cooper is working on the last chapter of a soon-to-be-published book titled Cancer Through the Lens of Poverty. n
Disclosure: Dr. Cooper reported no potential conflicts of interest.
Richard ‘Buz’ Cooper, MDcontinued from page 33
Apply as individuals or teams at www.ASCO.org/TeamsQuestions: [email protected]
NCI-ASCO Teams in Cancer Care Project:Call for Participants
The National Cancer Institute (NCI) and American Society of Clinical Oncology (ASCO) are collaborating to illustrate models of team-based care at a workshop and in JOP content. Teamwork involves delineating roles and responsibilities for multidisciplinary care across many settings. At a time of increasing expectations and pressure to demonstrate value in oncology care, this project will explore how oncology care can provide an effective model.
Goal: Illustrate in presentations and manuscripts effective approaches to delivering coordinated, patient-centered cancer care and apply the evidence for effective cancer care teams
Who: Approximately 10 writing groups will be formed with the following participants:
n Clinicians (physicians, nurse practitioners, physician assistants, and nurses)
n Patient advocates
n Researchers studying team effectiveness in health care (not only oncology) in the United States or Canada
What: Groups including clinicians, patient advocates, and team researchers will develop a presentation and manuscript applying principles of team-based care to a case within the cancer care continuum (prevention, screening, diagnosis, active treatment of curable and incurable disease, survivorship, end of life).
Important Dates:n Applications due: June 15, 2015n Selection of teams: July 17, 2015n In-person meeting at ASCO offices, Alexandria, VA: August 28, 2015n Presentations at NCI-ASCO Workshop: February 25, 2016
Visit The ASCO Post website at ASCOPost.com

ASCOPost.com | MARCH 25, 2015 PAGE 35
Announcements
UPMC Whitfield Cancer Centre Receives Joint Commission International Accreditation
For the third time since 2008, the University of Pittsburgh Medi-
cal Center (UPMC) Whitfield Cancer Centre, operated by UPMC in Water-ford, Ireland, has successfully achieved
accreditation from the Joint Commis-sion International ( JCI). This recogni-tion is based on an extensive review of the center’s patient safety, quality stan-dards, and processes.
The JCI accreditation process fo-cuses on determining whether a health care facility has the right systems and processes in place to support high-quality and safe patient care and has
the culture and capacity to continu-ously improve care. JCI’s surveyors examine crucial issues such as patient and family education, access to care,
continued on page 36
Erbitux® (cetuximab)injection, for intravenous infusionBrief Summary of Prescribing Information. For complete prescribing information consult official package insert.
WArNiNG: SEriOuS iNFuSiON rEACtiONS and CArDiOPuLMONArY ArrEStinfusion reactions: Serious infusion reactions occurred with the administration of Erbitux in approximately 3% of patients in clinical trials, with fatal outcome reported in less than 1 in 1000. [See Warnings and Precautions, Adverse Reactions.] immediately interrupt and permanently discontinue Erbitux infusion for serious infusion reactions. [See Dosage and Administration (2.4) in Full Prescribing information, Warnings and Precautions.]Cardiopulmonary Arrest: Cardiopulmonary arrest and/or sudden death occurred in 2% of patients with squamous cell carcinoma of the head and neck treated with Erbitux and radiation therapy in Study 1 and in 3% of patients with squamous cell carcinoma of the head and neck treated with European union (Eu)-approved cetuximab in combination with platinum-based therapy with 5-fluorouracil (5-Fu) in Study 2. Closely monitor serum electrolytes, including serum magnesium, potassium, and calcium, during and after Erbitux administration. [See Warnings and Precautions, Clinical Studies (14.1) in Full Prescribing information.]
iNDiCAtiONS AND uSAGESquamous Cell Carcinoma of the Head and Neck (SCCHN): Erbitux® (cetuximab) is indicated in combination with radiation therapy for the initial treatment of locally or regionally advanced squamous cell carcinoma of the head and neck. [See Clinical Studies (14.1) in Full Prescribing Information.]Erbitux is indicated in combination with platinum-based therapy with 5-FU for the first-line treatment of patients with recurrent locoregional disease or metastatic squamous cell carcinoma of the head and neck. [See Clinical Studies (14.1) in Full Prescribing Information.]Erbitux, as a single agent, is indicated for the treatment of patients with recurrent or metastatic squamous cell carcinoma of the head and neck for whom prior platinum-based therapy has failed. [See Clinical Studies (14.1) in Full Prescribing Information.]K-Ras Mutation-negative, EGFr-expressing Colorectal Cancer: Erbitux is indicated for the treatment of K-Ras mutation-negative (wild-type), epidermal growth factor receptor (EGFR)-expressing, metastatic colorectal cancer (mCRC) as determined by FDA-approved tests for this use [see Dosage and Administration (2.2) in Full Prescribing Information, Warnings and Precautions, Clinical Studies (14.2) in Full Prescribing Information] • in combination with FOLFIRI (irinotecan, 5-fluorouracil, leucovorin) for first-line treatment, • in combination with irinotecan in patients who are refractory to irinotecan-based chemotherapy, • as a single agent in patients who have failed oxaliplatin- and irinotecan-based chemotherapy or who are
intolerant to irinotecan. [See Warnings and Precautions, Clinical Pharmacology (12.1) in Full Prescribing Information, Clinical Studies (14.2) in Full Prescribing Information.]
Limitation of Use: Erbitux is not indicated for treatment of K-Ras mutation-positive colorectal cancer [see Warnings and Precautions, Clinical Studies (14.2) in Full Prescribing Information].CONtrAiNDiCAtiONSNoneWArNiNGS AND PrECAutiONSinfusion reactions: Serious infusion reactions, requiring medical intervention and immediate, permanent discontinuation of Erbitux included rapid onset of airway obstruction (bronchospasm, stridor, hoarseness), hypotension, shock, loss of consciousness, myocardial infarction, and/or cardiac arrest. Severe (NCI CTC Grades 3 and 4) infusion reactions occurred in 2–5% of 1373 patients in Studies 1, 3, 5, and 6 receiving Erbitux, with fatal outcome in 1 patient. [See Clinical Studies (14.1, 14.2) in Full Prescribing Information.]Approximately 90% of severe infusion reactions occurred with the first infusion despite premedication with antihistamines. Monitor patients for 1 hour following Erbitux infusions in a setting with resuscitation equipment and other agents necessary to treat anaphylaxis (eg, epinephrine, corticosteroids, intravenous antihistamines, bronchodilators, and oxygen). Monitor longer to confirm resolution of the event in patients requiring treatment for infusion reactions. Immediately and permanently discontinue Erbitux in patients with serious infusion reactions. [See Boxed Warning, Dosage and Administration (2.4) in Full Prescribing Information.]Cardiopulmonary Arrest: Cardiopulmonary arrest and/or sudden death occurred in 4 (2%) of 208 patients treated with radiation therapy and Erbitux as compared to none of 212 patients treated with radiation therapy alone in Study 1. Three patients with prior history of coronary artery disease died at home, with myocardial infarction as the presumed cause of death. One of these patients had arrhythmia and one had congestive heart failure. Death occurred 27, 32, and 43 days after the last dose of Erbitux. One patient with no prior history of coronary artery disease died one day after the last dose of Erbitux. In Study 2, fatal cardiac disorders and/or sudden death occurred in 7 (3%) of 219 patients treated with EU-approved cetuximab and platinum-based therapy with 5-FU as compared to 4 (2%) of 215 patients treated with chemotherapy alone. Five of these 7 patients in the chemotherapy plus cetuximab arm received concomitant cisplatin and 2 patients received concomitant carboplatin. All 4 patients in the chemotherapy-alone arm received cisplatin. Carefully consider use of Erbitux in combination with radiation therapy or platinum-based therapy with 5-FU in head and neck cancer patients with a history of coronary artery disease, congestive heart failure, or arrhythmias in light of these risks. Closely monitor serum electrolytes, including serum magnesium, potassium, and calcium, during and after Erbitux. [See Boxed Warning, Warnings and Precautions.]Pulmonary toxicity: Interstitial lung disease (ILD), including 1 fatality, occurred in 4 of 1570 (<0.5%) patients receiving Erbitux in Studies 1, 3, and 6, as well as other studies, in colorectal cancer and head and neck cancer. Interrupt Erbitux for acute onset or worsening of pulmonary symptoms. Permanently discontinue Erbitux for confirmed ILD.Dermatologic toxicity: Dermatologic toxicities, including acneiform rash, skin drying and fissuring, paronychial inflammation, infectious sequelae (for example, S. aureus sepsis, abscess formation, cellulitis, blepharitis, conjunctivitis, keratitis/ulcerative keratitis with decreased visual acuity, cheilitis), and hypertrichosis occurred in patients receiving Erbitux therapy. Acneiform rash occurred in 76–88% of 1373 patients receiving Erbitux in Studies 1, 3, 5, and 6. Severe acneiform rash occurred in 1–17% of patients.Acneiform rash usually developed within the first two weeks of therapy and resolved in a majority of the patients after cessation of treatment, although in nearly half, the event continued beyond 28 days. Monitor patients receiving Erbitux for dermatologic toxicities and infectious sequelae. Instruct patients to limit sun exposure during Erbitux therapy. [See Dosage and Administration (2.4) in Full Prescribing Information.]use of Erbitux in Combination With radiation and Cisplatin: In a controlled study, 940 patients with locally advanced SCCHN were randomized 1:1 to receive either Erbitux in combination with radiation therapy and cisplatin or radiation therapy and cisplatin alone. The addition of Erbitux resulted in an increase in the incidence of Grade 3–4 mucositis, radiation recall syndrome, acneiform rash, cardiac events, and electrolyte disturbances compared to radiation and cisplatin alone. Adverse reactions with fatal outcome were reported in 20 patients (4.4%) in the Erbitux combination arm and 14 patients (3.0%) in the control arm. Nine patients in the Erbitux arm (2.0%) experienced myocardial ischemia compared to 4 patients (0.9%) in the control arm. The main efficacy outcome of the study was progression-free survival (PFS). The addition of Erbitux to radiation and cisplatin did not improve PFS.Hypomagnesemia and Electrolyte Abnormalities: In patients evaluated during clinical trials, hypomagne- semia occurred in 55% of 365 patients receiving Erbitux in Study 5 and two other clinical trials in colorectal cancer and head and neck cancer, respectively, and was severe (NCI CTC Grades 3 and 4) in 6–17%.In Study 2, where EU-approved cetuximab was administered in combination with platinum-based therapy, the addition of cetuximab to cisplatin and 5-FU resulted in an increased incidence of hypomagnesemia (14% vs. 6%) and of Grade 3–4 hypomagnesemia (7% vs. 2%) compared to cisplatin and 5-FU alone. In contrast, the incidences of hypomagnesemia were similar for those who received cetuximab, carboplatin, and 5-FU compared to carboplatin and 5-FU (4% vs. 4%). No patient experienced Grade 3–4 hypomagnesemia in either arm in the carboplatin subgroup.
The onset of hypomagnesemia and accompanying electrolyte abnormalities occurred days to months after initiation of Erbitux (cetuximab). Periodically monitor patients for hypomagnesemia, hypocalcemia, and hypokalemia, during and for at least 8 weeks following the completion of Erbitux. Replete electrolytes as necessary.K-Ras testing in Metastatic or Advanced Colorectal Cancer Patients: Determination of K-Ras mutational status in colorectal tumors using an FDA-approved test indicated for this use is necessary for selection of patients for treatment with Erbitux. Erbitux is indicated only for patients with EGFR-expressing K-Ras mutation-negative (wild-type) mCRC. Erbitux is not an effective treatment for patients with colorectal cancer that harbor somatic mutations in codons 12 and 13 (exon 2). Studies 4 and 5, conducted in patients with colorectal cancer, demonstrated a benefit with Erbitux treatment only in the subset of patients whose tumors were K-Ras mutation-negative (wild-type). Erbitux is not effective for the treatment of K-Ras mutation-positive colorectal cancer as determined by an FDA-approved test for this use. [See Indications and Usage (1.2) in Full Prescribing Information, Clinical Pharmacology (12.1) in Full Prescribing Information, Clinical Studies (14.2) in Full Prescribing Information].Perform the assessment for K-Ras mutation status in colorectal cancer in laboratories with demonstrated proficiency in the specific technology being utilized. Improper assay performance can lead to unreliable test results.Refer to an FDA-approved test’s package insert for instructions on the identification of patients eligible for the treatment of Erbitux.Epidermal Growth Factor receptor (EGFr) Expression and response: Because expression of EGFR has been detected in nearly all SCCHN tumor specimens, patients enrolled in the head and neck cancer clinical studies were not required to have immunohistochemical evidence of EGFR tumor expression prior to study entry.Patients enrolled in the colorectal cancer clinical studies were required to have immunohistochemical evidence of EGFR tumor expression. Primary tumor or tumor from a metastatic site was tested with the DakoCytomation EGFR pharmDx™ test kit. Specimens were scored based on the percentage of cells expressing EGFR and intensity (barely/faint, weak-to-moderate, and strong). Response rate did not correlate with either the percentage of positive cells or the intensity of EGFR expression. ADvErSE rEACtiONSThe following adverse reactions are discussed in greater detail in other sections of the label:• Infusion reactions [See Boxed Warning, Warnings and Precautions.]• Cardiopulmonary arrest [See Boxed Warning, Warnings and Precautions.]• Pulmonary toxicity [See Warnings and Precautions.]• Dermatologic toxicity [See Warnings and Precautions.]• Hypomagnesemia and Electrolyte Abnormalities [See Warnings and Precautions.]The most common adverse reactions in Erbitux clinical trials (incidence ≥25%) include cutaneous adverse reactions (including rash, pruritus, and nail changes), headache, diarrhea, and infection. The most serious adverse reactions with Erbitux are infusion reactions, cardiopulmonary arrest, dermatologic toxicity and radiation dermatitis, sepsis, renal failure, interstitial lung disease, and pulmonary embolus. Across Studies 1, 3, 5, and 6, Erbitux was discontinued in 3–10% of patients because of adverse reactions. Clinical trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The data below reflect exposure to Erbitux in 1373 patients with SCCHN or colorectal cancer in randomized Phase 3 (Studies 1 and 5) or Phase 2 (Studies 3 and 6) trials treated at the recommended dose and schedule for medians of 7 to 14 weeks. [See Clinical Studies (14) in Full Prescribing Information.]infusion reactions: Infusion reactions, which included pyrexia, chills, rigors, dyspnea, bronchospasm, angioedema, urticaria, hypertension, and hypotension occurred in 15–21% of patients across studies. Grades 3 and 4 infusion reactions occurred in 2–5% of patients; infusion reactions were fatal in 1 patient.infections: The incidence of infection was variable across studies, ranging from 13–35%. Sepsis occurred in 1–4% of patients. renal: Renal failure occurred in 1% of patients with colorectal cancer. Squamous Cell Carcinoma of the Head and Neck Erbitux in Combination with Radiation Therapy — Table 1 contains selected adverse reactions in 420 patients receiving radiation therapy either alone or with Erbitux for locally or regionally advanced SCCHN in Study 1. Erbitux was administered at the recommended dose and schedule (400 mg/m2 initial dose, followed by 250 mg/m2 weekly). Patients received a median of 8 infusions (range 1–11).
table 1: incidence of Selected Adverse reactions (≥10%) in Patients with Locoregionally Advanced SCCHN
Erbitux plus radiation (n=208)
radiation therapy Alone (n=212)
body System Preferred Term
Grades 1–4
Grades 3 and 4
Grades 1–4
Grades 3 and 4
% of Patientsbody as a WholeAsthenia 56 4 49 5Fevera 29 1 13 1Headache 19 <1 8 <1Infusion Reactionb 15 3 2 0Infection 13 1 9 1Chillsa 16 0 5 0DigestiveNausea 49 2 37 2Emesis 29 2 23 4Diarrhea 19 2 13 1Dyspepsia 14 0 9 1Metabolic/NutritionalWeight Loss 84 11 72 7Dehydration 25 6 19 8Alanine Transaminase, highc 43 2 21 1Aspartate Transaminase, highc 38 1 24 1Alkaline Phosphatase, highc 33 <1 24 0respiratoryPharyngitis 26 3 19 4Skin/AppendagesAcneiform Rashd 87 17 10 1Radiation Dermatitis 86 23 90 18Application Site Reaction 18 0 12 1Pruritus 16 0 4 0a Includes cases also reported as infusion reaction. b Infusion reaction is defined as any event described at any time during the clinical study as “allergic reaction” or “anaphylactoid reaction”, or any event occurring on the first day of dosing described as “allergic reaction”, “anaphylactoid reaction”, “fever”, “chills”, “chills and fever”, or “dyspnea”. c Based on laboratory measurements, not on reported adverse reactions, the number of subjects with tested samples varied from 205–206 for Erbitux plus Radiation arm; 209–210 for Radiation alone. d Acneiform rash is defined as any event described as “acne”, “rash”, “maculopapular rash”, “pustular rash”, “dry skin”, or “exfoliative dermatitis”.
The incidence and severity of mucositis, stomatitis, and xerostomia were similar in both arms of the study. Late Radiation Toxicity — The overall incidence of late radiation toxicities (any grade) was higher in Erbitux in combination with radiation therapy compared with radiation therapy alone. The following sites were affected: salivary glands (65% versus 56%), larynx (52% versus 36%), subcutaneous tissue (49% versus 45%), mucous membrane (48% versus 39%), esophagus (44% versus 35%), skin (42% versus 33%). The incidence of Grade 3 or 4 late radiation toxicities was similar between the radiation therapy alone and the Erbitux plus radiation treatment groups.
Erb0813PBS_693US13PBS02201_7x9wip3.indd 1 8/19/13 2:01 PM
14220302_0124301_CrossTumor_ROB_v4_M.indd 1 5/16/14 12:40 PM

PAGE 36 The ASCO Post | MARCH 25, 2015
Announcements
and medication management. The pro-cess requires hospitals to demonstrate a track record of standards compliance and relies on candid interviews with patients, nurses, and physicians about care practices.
“The success of this third accredita-
tion survey reflects the ongoing efforts of the whole Cancer Centre team and their dedication to continuing to pro-vide a quality service to all cancer pa-tients,” said Catriona McDonald, Di-rector of Operations and Radiotherapy Services Manager.
“Demonstrating compliance with JCI standards serves as validation of an
organization’s commitment to an inter-nationally recognized, time-tested, and comprehensive level of quality,” said Cheryl Brill, UPMC’s Vice President of International Clinical Operations and Quality.
UPMC Whitfield Cancer Centre offers the most advanced radiation therapy, including intensity-modulated
radiation therapy and image-guided radiation therapy, to residents of the southeast region of Ireland. One of only four ambulatory care centers to be JCI-accredited in Ireland, the center is committed to delivering the highest standard of radiation therapy and sup-portive care for patients with all types of cancer. n
UPMC Whitfield Cancer Centrecontinued from page 35
The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to Erbitux (cetuximab) with the incidence of antibodies to other products may be misleading.Postmarketing Experience: The following adverse reactions have been identified during post-approval use of Erbitux. Because these reactions are reported from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.• Aseptic meningitis • Mucosal inflammation DruG iNtErACtiONSA drug interaction study was performed in which Erbitux was administered in combination with irinotecan. There was no evidence of any pharmacokinetic interactions between Erbitux and irinotecan.uSE iN SPECiFiC POPuLAtiONSPregnancy: Pregnancy Category C — There are no adequate and well-controlled studies of Erbitux in pregnant women. Based on animal models, EGFR has been implicated in the control of prenatal development and may be essential for normal organogenesis, proliferation, and differentiation in the developing embryo. Human IgG is known to cross the placental barrier; therefore, Erbitux may be transmitted from the mother to the developing fetus, and has the potential to cause fetal harm when administered to pregnant women. Erbitux should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.Pregnant cynomolgus monkeys were treated weekly with 0.4 to 4 times the recommended human dose of cetuximab (based on body surface area) during the period of organogenesis (gestation day [GD] 20–48). Cetuximab was detected in the amniotic fluid and in the serum of embryos from treated dams at GD 49. No fetal malformations or other teratogenic effects occurred in offspring. However, significant increases in embryolethality and abortions occurred at doses of approximately 1.6 to 4 times the recommended human dose of cetuximab (based on total body surface area).Nursing Mothers: It is not known whether Erbitux is secreted in human milk. IgG antibodies, such as Erbitux, can be excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Erbitux, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. If nursing is interrupted, based on the mean half-life of cetuximab [see Clinical Pharmacology (12.3) in Full Prescribing Information], nursing should not be resumed earlier than 60 days following the last dose of Erbitux.Pediatric use: The safety and effectiveness of Erbitux in pediatric patients have not been established. The pharmacokinetics of cetuximab, in combination with irinotecan, were evaluated in pediatric patients with refractory solid tumors in an open-label, single-arm, dose-finding study. Erbitux was administered once-weekly, at doses up to 250 mg/m2, to 27 patients ranging from 1 to 12 years old; and in 19 patients ranging from 13 to 18 years old. No new safety signals were identified in pediatric patients. The pharmacokinetic profiles of cetuximab between the two age groups were similar at the 75 and 150 mg/m2 single dose levels. The volume of the distribution appeared to be independent of dose and approximated the vascular space of 2–3 L/m2. Following a single dose of 250 mg/m2, the geometric mean AUC0-inf (CV%) value was 17.7 mg•h/mL (34%) in the younger age group (1–12 years, n=9) and 13.4 mg•h/mL (38%) in the adolescent group (13–18 years, n=6). The mean half-life of cetuximab was 110 hours (range 69 to 188 hours) for the younger age group, and 82 hours (range 55 to 117 hours) for the adolescent age group.
Geriatric use: Of the 1662 patients who received Erbitux (cetuximab) with irinotecan, FOLFIRI or Erbitux monotherapy in six studies of advanced colorectal cancer, 588 patients were 65 years of age or older. No overall differences in safety or efficacy were observed between these patients and younger patients.Clinical studies of Erbitux conducted in patients with head and neck cancer did not include sufficient number of subjects aged 65 and over to determine whether they respond differently from younger subjects.OvErDOSAGEThe maximum single dose of Erbitux administered is 1000 mg/m2 in one patient. No adverse events were reported for this patient. NONCLiNiCAL tOxiCOLOGYCarcinogenesis, Mutagenesis, impairment of Fertility: Long-term animal studies have not been performed to test cetuximab for carcinogenic potential, and no mutagenic or clastogenic potential of cetuximab was observed in the Salmonella-Escherichia coli (Ames) assay or in the in vivo rat micronucleus test. Menstrual cyclicity was impaired in female cynomolgus monkeys receiving weekly doses of 0.4 to 4 times the human dose of cetuximab (based on total body surface area). Cetuximab-treated animals exhibited increased incidences of irregular or absent cycles, as compared to control animals. These effects were initially noted beginning week 25 of cetuximab treatment and continued through the 6-week recovery period. In this same study, there were no effects of cetuximab treatment on measured male fertility parameters (ie, serum testosterone levels and analysis of sperm counts, viability, and motility) as compared to control male monkeys. It is not known if cetuximab can impair fertility in humans.Animal Pharmacology and/or toxicology: In cynomolgus monkeys, cetuximab, when administered at doses of approximately 0.4 to 4 times the weekly human exposure (based on total body surface area), resulted in dermatologic findings, including inflammation at the injection site and desquamation of the external integument. At the highest dose level, the epithelial mucosa of the nasal passage, esophagus, and tongue were similarly affected, and degenerative changes in the renal tubular epithelium occurred. Deaths due to sepsis were observed in 50% (5/10) of the animals at the highest dose level beginning after approximately 13 weeks of treatment.PAtiENt COuNSELiNG iNFOrMAtiONAdvise patients:• To report signs and symptoms of infusion reactions such as fever, chills, or breathing problems.• Of the potential risks of using Erbitux during pregnancy or nursing and of the need to use adequate
contraception in both males and females during and for 6 months following the last dose of Erbitux therapy. • That nursing is not recommended during, and for 2 months following the last dose of Erbitux therapy. • To limit sun exposure (use sunscreen, wear hats) while receiving and for 2 months following the last dose
of Erbitux.
Erbitux® is a registered trademark of ImClone LLC a wholly-owned subsidiary of Eli Lilly and Company.Manufactured by ImClone LLC a wholly-owned subsidiary of Eli Lilly and Company, Branchburg, NJ 08876 USADistributed and marketed by Bristol-Myers Squibb Company, Princeton, NJ 08543 USACo-marketed by Eli Lilly and Company, Indianapolis, IN 46285 USA
Copyright © 2004–2013 ImClone LLC a wholly-owned subsidiary of Eli Lilly and Company, and Bristol-Myers Squibb Company. All rights reserved.
1236886B3 Rev August 2013
693US13PBS02201
Erb0813PBS_693US13PBS02201_7x9wip3.indd 3 8/19/13 2:01 PM
14220302_0124301_CrossTumor_ROB_v4_M.indd 3 5/16/14 12:40 PM
Study 2: EU-Approved Cetuximab in Combination with Platinum-based Therapy with 5-Fluorouracil — Study 2 used EU-approved cetuximab. Since U.S.-licensed Erbitux (cetuximab) provides approximately 22% higher exposure relative to the EU-approved cetuximab, the data provided below may underestimate the incidence and severity of adverse reactions anticipated with Erbitux for this indication. However, the tolerability of the recommended dose is supported by safety data from additional studies of Erbitux [see Clinical Pharmacology (12.3) in Full Prescribing Information].Table 2 contains selected adverse reactions in 434 patients with recurrent locoregional disease or metastatic SCCHN receiving EU-approved cetuximab in combination with platinum-based therapy with 5-FU or platinum-based therapy with 5-FU alone in Study 2. Cetuximab was administered at 400 mg/m2 for the initial dose, followed by 250 mg/m2 weekly. Patients received a median of 17 infusions (range 1–89).
table 2: incidence of Selected Adverse reactions (≥10%) in Patients with recurrent Locoregional Disease or Metastatic SCCHN
Eu-Approved Cetuximab plus Platinum-based therapy with 5-Fu
(n=219)
Platinum-based therapy with 5-Fu Alone
(n=215)System Organ Class Preferred Term
Grades 1–4
Grades 3 and 4
Grades 1–4
Grades 3 and 4
% of PatientsEye DisordersConjunctivitis 10 0 0 0Gastrointestinal DisordersNausea 54 4 47 4Diarrhea 26 5 16 1General Disorders and Administration Site ConditionsPyrexia 22 0 13 1Infusion Reactiona 10 2 <1 0infections and infestationsInfectionb 44 11 27 8Metabolism and Nutrition DisordersAnorexia 25 5 14 1Hypocalcemia 12 4 5 1Hypokalemia 12 7 7 5Hypomagnesemia 11 5 5 1Skin and Subcutaneous tissue DisordersAcneiform Rashc 70 9 2 0Rash 28 5 2 0Acne 22 2 0 0Dermatitis Acneiform 15 2 0 0Dry Skin 14 0 <1 0Alopecia 12 0 7 0a Infusion reaction defined as any event of “anaphylactic reaction”, “hypersensitivity”, “fever and/or chills”, “dyspnea”, or “pyrexia” on the first day of dosing. b Infection – this term excludes sepsis-related events which are presented separately. c Acneiform rash defined as any event described as “acne”, “dermatitis acneiform”, “dry skin”, “exfoliative rash”, “rash”, “rash erythematous”, “rash macular”, “rash papular”, or “rash pustular”. Chemotherapy = cisplatin + 5-fluorouracil or carboplatin + 5-fluorouracil
For cardiac disorders, approximately 9% of subjects in both the EU-approved cetuximab plus chemotherapy and chemotherapy-only treatment arms in Study 2 experienced a cardiac event. The majority of these events occurred in patients who received cisplatin/5-FU, with or without cetuximab as follows: 11% and 12% in patients who received cisplatin/5-FU with or without cetuximab, respectively, and 6% or 4% in patients who received carboplatin/5-FU with or without cetuximab, respectively. In both arms, the incidence of cardiovascular events was higher in the cisplatin with 5-FU containing subgroup. Death attributed to cardiovascular event or sudden death was reported in 3% of the patients in the cetuximab plus platinum-based therapy with 5-FU arm and 2% in the platinum-based chemotherapy with 5-FU alone arm.Colorectal CancerStudy 4: EU-Approved Cetuximab in Combination with FOLFIRI — Study 4 used EU-approved cetuximab. U.S.-licensed Erbitux provides approximately 22% higher exposure to cetuximab relative to the EU-approved cetuximab. The data provided below for Study 4 is consistent in incidence and severity of adverse reactions with those seen for Erbitux in this indication. The tolerability of the recommended dose is supported by safety data from additional studies of Erbitux [see Clinical Pharmacology (12.3) in Full Prescribing Information].Table 3 contains selected adverse reactions in 667 patients with K-Ras mutation-negative (wild-type), EGFR-expressing, metastatic colorectal cancer receiving EU-approved cetuximab plus FOLFIRI or FOLFIRI alone in Study 4 [see Warnings and Precautions]. Cetuximab was administered at the recommended dose and schedule (400 mg/m2 initial dose, followed by 250 mg/m2 weekly). Patients received a median of 26 infusions (range 1–224).
table 3: incidence of Selected Adverse reactions Occurring in ≥10% of Patients with K-Ras Mutation-negative (Wild-type) and EGFr-expressing, Metastatic Colorectal Cancera
Eu-Approved Cetuximab plus FOLFiri
(n=317)
FOLFiri Alone
(n=350)body System Preferred Term
Grades
1–4bGrades 3 and 4
Grades
1–4Grades 3 and 4
% of Patientsblood and Lymphatic System DisordersNeutropenia 49 31 42 24Eye DisordersConjunctivitis 18 <1 3 0Gastrointestinal DisordersDiarrhea 66 16 60 10Stomatitis 31 3 19 1Dyspepsia 16 0 9 0General Disorders and Administration Site ConditionsInfusion-related Reactionc 14 2 <1 0Pyrexia 26 1 14 1infections and infestationsParonychia 20 4 <1 0investigationsWeight Decreased 15 1 9 1Metabolism and Nutrition DisordersAnorexia 30 3 23 2
(Continued)
table 3: incidence of Selected Adverse reactions Occurring in ≥10% of Patients with K-Ras Mutation-negative (Wild-type) and EGFr-expressing, Metastatic Colorectal Cancera
Eu-Approved Cetuximab plus FOLFiri
(n=317)
FOLFiri Alone
(n=350)body System Preferred Term
Grades
1–4bGrades 3 and 4
Grades
1–4Grades 3 and 4
% of PatientsSkin and Subcutaneous tissue DisordersAcne-like Rashd 86 18 13 <1 Rash 44 9 4 0 Dermatitis Acneiform 26 5 <1 0 Dry Skin 22 0 4 0 Acne 14 2 0 0 Pruritus 14 0 3 0Palmar-plantar Erythrodysesthesia Syndrome 19 4 4 <1Skin Fissures 19 2 1 0a Adverse reactions occurring in at least 10% of Erbitux (cetuximab) combination arm with a frequency at least 5% greater than that seen in the FOLFIRI arm. b Adverse reactions were graded using the NCI CTC, V 2.0. c Infusion related reaction is defined as any event meeting the medical concepts of allergy/anaphylaxis at any time during the clinical study or any event occurring on the first day of dosing and meeting the medical concepts of dyspnea and fever or by the following events using MedDRA preferred terms: “acute myocardial infarction”, “angina pectoris”, “angioedema”, “autonomic seizure”, “blood pressure abnormal”, “blood pressure decreased”, “blood pressure increased”, “cardiac failure”, “cardiopulmonary failure”, “cardiovascular insufficiency”, “clonus”, “convulsion”, “coronary no-reflow phenomenon”, “epilepsy”, “hypertension”, “hypertensive crisis”, “hypertensive emergency”, “hypotension”, “infusion related reaction”, “loss of consciousness”, “myocardial infarction”, “myocardial ischaemia”, “prinzmetal angina”, “shock”, “sudden death”, “syncope”, or “systolic hypertension”. d Acne-like rash is defined by the events using MedDRA preferred terms and included “acne”, “acne pustular”, “butterfly rash”, “dermatitis acneiform”, “drug rash with eosinophilia and systemic symptoms”, “dry skin”, “erythema”, “exfoliative rash”, “folliculitis”, “genital rash”, “mucocutaneous rash”, “pruritus”, “rash”, “rash erythematous”, “rash follicular”, “rash generalized”, “rash macular”, “rash maculopapular”, “rash maculovesicular”, “rash morbilliform”, “rash papular”, “rash papulosquamous”, “rash pruritic”, “rash pustular”, “rash rubelliform”, “rash scarlatiniform”, “rash vesicular”, “skin exfoliation”, “skin hyperpigmentation”, “skin plaque”, “telangiectasia”, or “xerosis”.
Erbitux Monotherapy — Table 4 contains selected adverse reactions in 242 patients with K-Ras mutation-negative (wild-type), EGFR-expressing, metastatic colorectal cancer who received best supportive care (BSC) alone or with Erbitux in Study 5 [see Warnings and Precautions]. Erbitux was administered at the recommended dose and schedule (400 mg/m2 initial dose, followed by 250 mg/m2 weekly). Patients received a median of 17 infusions (range 1–51).
table 4: incidence of Selected Adverse reactions Occurring in ≥10% of Patients with K-Ras Mutation-negative (Wild-type), EGFr-expressing, Metastatic Colorectal Cancer treated with Erbitux Monotherapya
Erbitux plus bSC (n=118)
bSC alone (n=124)
body System Preferred Term
Grades 1–4b
Grades 3 and 4
Grades 1–4
Grades 3 and 4
% of PatientsDermatology/SkinRash/Desquamation 95 16 21 1Dry Skin 57 0 15 0Pruritus 47 2 11 0Other-Dermatology 35 0 7 2Nail Changes 31 0 4 0Constitutional SymptomsFatigue 91 31 79 29Fever 25 3 16 0Infusion Reactionsc 18 3 0 0Rigors, Chills 16 1 3 0PainPain-Other 59 18 37 10Headache 38 2 11 0Bone Pain 15 4 8 2PulmonaryDyspnea 49 16 44 13Cough 30 2 19 2GastrointestinalNausea 64 6 50 6Constipation 53 3 38 3Diarrhea 42 2 23 2Vomiting 40 5 26 5Stomatitis 32 1 10 0Other-Gastrointestinal 22 12 16 5Dehydration 13 5 3 0Mouth Dryness 12 0 6 0Taste Disturbance 10 0 5 0infectionInfection without neutropenia 38 11 19 5MusculoskeletalArthralgia 14 3 6 0NeurologyNeuropathy-sensory 45 1 38 2Insomnia 27 0 13 0Confusion 18 6 10 2Anxiety 14 1 5 1Depression 14 0 5 0a Adverse reactions occurring in at least 10% of Erbitux plus BSC arm with a frequency at least 5% greater than that seen in the BSC alone arm. b Adverse reactions were graded using the NCI CTC, V 2.0. c Infusion reaction is defined as any event (chills, rigors, dyspnea, tachycardia, bronchospasm, chest tightness, swelling, urticaria, hypotension, flushing, rash, hypertension, nausea, angioedema, pain, sweating, tremors, shaking, drug fever, or other hypersensitivity reaction) recorded by the investigator as infusion-related.
Erbitux in Combination with Irinotecan — The most frequently reported adverse reactions in 354 patients treated with Erbitux plus irinotecan in clinical trials were acneiform rash (88%), asthenia/malaise (73%), diarrhea (72%), and nausea (55%). The most common Grades 3–4 adverse reactions included diarrhea (22%), leukopenia (17%), asthenia/malaise (16%), and acneiform rash (14%).immunogenicity: As with all therapeutic proteins, there is potential for immunogenicity. Immunogenic responses to cetuximab were assessed using either a double antigen radiometric assay or an ELISA assay. Due to limitations in assay performance and sampling timing, the incidence of antibody development in patients receiving Erbitux has not been adequately determined. Non-neutralizing anti-cetuximab antibodies were detected in 5% (49 of 1001) of evaluable patients without apparent effect on the safety or antitumor activity of Erbitux.
(Continued)
Erb0813PBS_693US13PBS02201_7x9wip3.indd 2 8/19/13 2:01 PM
14220302_0124301_CrossTumor_ROB_v4_M.indd 2 5/16/14 12:40 PM

ASCOPost.com | MARCH 25, 2015 PAGE 37
Geriatrics for the Oncologist
grow at an explosively high rate. This has led me to conclude that the most impor-tant advance in geriatric oncology over the course of my career has been the rec-ognition that older cancer patients need to be managed differently than their younger counterparts. To that end, we all need to
learn to do routine geriatric assessments, and as oncologists, learn to practice like mini-geriatricians, factoring all the other health-related baggage common in older patients into our treatment plans.
Now that we’ve gained more knowl-edge in this area, it’s important that a proper assessment be done so older pa-tients do not get low-balled in treatment
decisions. The danger using age in rec-ommending a treatment plan is to deny state-of-the-art treatment to those who deserve it and may benefit from it.
Functional, Not Chronologic AgeBesides a chronologic measure, how
do we determine who is a geriatric cancer patient?
When we see an older patient, the primary question we should be asking is not what the patient’s age is, but what is the patient’s life expectancy and how might treatment affect function and quality of life. There are some very good online tools to help in this assessment process. One, ePrognosis (eprognosis.ucsf.edu), is a tool for estimating the life expectancy of elders. It is easy to use, accurate, and a great help for busy clinicians.
I show how ePrognosis works in all my lectures at ASCO meetings. There are several tools that can be used de-pending upon the status of the patient. The tools require that a few extra ques-tions be asked of the older patient, and then the calculators allow you to get a fairly good picture of your patient’s 5- and 10-year life expectancy—which is essential in determining an appropriate treatment program. Our guesses on life expectancy, even by the most seasoned clinician, are notoriously bad.
There is also an emerging group of predictive models for chemotherapy-related toxicity in which you can input clinical and geriatric information and get an idea of the risk of your patient having grade 3 and higher chemo-therapy toxicity. Martine Extermann, MD, PhD, has developed a wonderful model called the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH). It is specifically designed to score laboratory test values and geri-atric assessment parameters besides age, such as functional and nutritional status, comorbidity, cognition, psycho-logical state, and social support, to pre-dict the risk of severe toxicity from che-
Hyman Muss, MDcontinued from page 1 GUEST EDITOR
Dr. Lichtman is an Attending Physician at Memorial Sloan
Kettering Cancer Center, Commack, New York, and Professor of Medi-cine, Weill Cornell Medical College, New York. He is also President Elect of the International Society of Geriat-ric Oncology (www.siog.org).
Stuart M. Lichtman, MD
continued on page 38
The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to Erbitux (cetuximab) with the incidence of antibodies to other products may be misleading.Postmarketing Experience: The following adverse reactions have been identified during post-approval use of Erbitux. Because these reactions are reported from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.• Aseptic meningitis • Mucosal inflammation DruG iNtErACtiONSA drug interaction study was performed in which Erbitux was administered in combination with irinotecan. There was no evidence of any pharmacokinetic interactions between Erbitux and irinotecan.uSE iN SPECiFiC POPuLAtiONSPregnancy: Pregnancy Category C — There are no adequate and well-controlled studies of Erbitux in pregnant women. Based on animal models, EGFR has been implicated in the control of prenatal development and may be essential for normal organogenesis, proliferation, and differentiation in the developing embryo. Human IgG is known to cross the placental barrier; therefore, Erbitux may be transmitted from the mother to the developing fetus, and has the potential to cause fetal harm when administered to pregnant women. Erbitux should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.Pregnant cynomolgus monkeys were treated weekly with 0.4 to 4 times the recommended human dose of cetuximab (based on body surface area) during the period of organogenesis (gestation day [GD] 20–48). Cetuximab was detected in the amniotic fluid and in the serum of embryos from treated dams at GD 49. No fetal malformations or other teratogenic effects occurred in offspring. However, significant increases in embryolethality and abortions occurred at doses of approximately 1.6 to 4 times the recommended human dose of cetuximab (based on total body surface area).Nursing Mothers: It is not known whether Erbitux is secreted in human milk. IgG antibodies, such as Erbitux, can be excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Erbitux, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. If nursing is interrupted, based on the mean half-life of cetuximab [see Clinical Pharmacology (12.3) in Full Prescribing Information], nursing should not be resumed earlier than 60 days following the last dose of Erbitux.Pediatric use: The safety and effectiveness of Erbitux in pediatric patients have not been established. The pharmacokinetics of cetuximab, in combination with irinotecan, were evaluated in pediatric patients with refractory solid tumors in an open-label, single-arm, dose-finding study. Erbitux was administered once-weekly, at doses up to 250 mg/m2, to 27 patients ranging from 1 to 12 years old; and in 19 patients ranging from 13 to 18 years old. No new safety signals were identified in pediatric patients. The pharmacokinetic profiles of cetuximab between the two age groups were similar at the 75 and 150 mg/m2 single dose levels. The volume of the distribution appeared to be independent of dose and approximated the vascular space of 2–3 L/m2. Following a single dose of 250 mg/m2, the geometric mean AUC0-inf (CV%) value was 17.7 mg•h/mL (34%) in the younger age group (1–12 years, n=9) and 13.4 mg•h/mL (38%) in the adolescent group (13–18 years, n=6). The mean half-life of cetuximab was 110 hours (range 69 to 188 hours) for the younger age group, and 82 hours (range 55 to 117 hours) for the adolescent age group.
Geriatric use: Of the 1662 patients who received Erbitux (cetuximab) with irinotecan, FOLFIRI or Erbitux monotherapy in six studies of advanced colorectal cancer, 588 patients were 65 years of age or older. No overall differences in safety or efficacy were observed between these patients and younger patients.Clinical studies of Erbitux conducted in patients with head and neck cancer did not include sufficient number of subjects aged 65 and over to determine whether they respond differently from younger subjects.OvErDOSAGEThe maximum single dose of Erbitux administered is 1000 mg/m2 in one patient. No adverse events were reported for this patient. NONCLiNiCAL tOxiCOLOGYCarcinogenesis, Mutagenesis, impairment of Fertility: Long-term animal studies have not been performed to test cetuximab for carcinogenic potential, and no mutagenic or clastogenic potential of cetuximab was observed in the Salmonella-Escherichia coli (Ames) assay or in the in vivo rat micronucleus test. Menstrual cyclicity was impaired in female cynomolgus monkeys receiving weekly doses of 0.4 to 4 times the human dose of cetuximab (based on total body surface area). Cetuximab-treated animals exhibited increased incidences of irregular or absent cycles, as compared to control animals. These effects were initially noted beginning week 25 of cetuximab treatment and continued through the 6-week recovery period. In this same study, there were no effects of cetuximab treatment on measured male fertility parameters (ie, serum testosterone levels and analysis of sperm counts, viability, and motility) as compared to control male monkeys. It is not known if cetuximab can impair fertility in humans.Animal Pharmacology and/or toxicology: In cynomolgus monkeys, cetuximab, when administered at doses of approximately 0.4 to 4 times the weekly human exposure (based on total body surface area), resulted in dermatologic findings, including inflammation at the injection site and desquamation of the external integument. At the highest dose level, the epithelial mucosa of the nasal passage, esophagus, and tongue were similarly affected, and degenerative changes in the renal tubular epithelium occurred. Deaths due to sepsis were observed in 50% (5/10) of the animals at the highest dose level beginning after approximately 13 weeks of treatment.PAtiENt COuNSELiNG iNFOrMAtiONAdvise patients:• To report signs and symptoms of infusion reactions such as fever, chills, or breathing problems.• Of the potential risks of using Erbitux during pregnancy or nursing and of the need to use adequate
contraception in both males and females during and for 6 months following the last dose of Erbitux therapy. • That nursing is not recommended during, and for 2 months following the last dose of Erbitux therapy. • To limit sun exposure (use sunscreen, wear hats) while receiving and for 2 months following the last dose
of Erbitux.
Erbitux® is a registered trademark of ImClone LLC a wholly-owned subsidiary of Eli Lilly and Company.Manufactured by ImClone LLC a wholly-owned subsidiary of Eli Lilly and Company, Branchburg, NJ 08876 USADistributed and marketed by Bristol-Myers Squibb Company, Princeton, NJ 08543 USACo-marketed by Eli Lilly and Company, Indianapolis, IN 46285 USA
Copyright © 2004–2013 ImClone LLC a wholly-owned subsidiary of Eli Lilly and Company, and Bristol-Myers Squibb Company. All rights reserved.
1236886B3 Rev August 2013
693US13PBS02201
Erb0813PBS_693US13PBS02201_7x9wip3.indd 3 8/19/13 2:01 PM
14220302_0124301_CrossTumor_ROB_v4_M.indd 3 5/16/14 12:40 PM
Study 2: EU-Approved Cetuximab in Combination with Platinum-based Therapy with 5-Fluorouracil — Study 2 used EU-approved cetuximab. Since U.S.-licensed Erbitux (cetuximab) provides approximately 22% higher exposure relative to the EU-approved cetuximab, the data provided below may underestimate the incidence and severity of adverse reactions anticipated with Erbitux for this indication. However, the tolerability of the recommended dose is supported by safety data from additional studies of Erbitux [see Clinical Pharmacology (12.3) in Full Prescribing Information].Table 2 contains selected adverse reactions in 434 patients with recurrent locoregional disease or metastatic SCCHN receiving EU-approved cetuximab in combination with platinum-based therapy with 5-FU or platinum-based therapy with 5-FU alone in Study 2. Cetuximab was administered at 400 mg/m2 for the initial dose, followed by 250 mg/m2 weekly. Patients received a median of 17 infusions (range 1–89).
table 2: incidence of Selected Adverse reactions (≥10%) in Patients with recurrent Locoregional Disease or Metastatic SCCHN
Eu-Approved Cetuximab plus Platinum-based therapy with 5-Fu
(n=219)
Platinum-based therapy with 5-Fu Alone
(n=215)System Organ Class Preferred Term
Grades 1–4
Grades 3 and 4
Grades 1–4
Grades 3 and 4
% of PatientsEye DisordersConjunctivitis 10 0 0 0Gastrointestinal DisordersNausea 54 4 47 4Diarrhea 26 5 16 1General Disorders and Administration Site ConditionsPyrexia 22 0 13 1Infusion Reactiona 10 2 <1 0infections and infestationsInfectionb 44 11 27 8Metabolism and Nutrition DisordersAnorexia 25 5 14 1Hypocalcemia 12 4 5 1Hypokalemia 12 7 7 5Hypomagnesemia 11 5 5 1Skin and Subcutaneous tissue DisordersAcneiform Rashc 70 9 2 0Rash 28 5 2 0Acne 22 2 0 0Dermatitis Acneiform 15 2 0 0Dry Skin 14 0 <1 0Alopecia 12 0 7 0a Infusion reaction defined as any event of “anaphylactic reaction”, “hypersensitivity”, “fever and/or chills”, “dyspnea”, or “pyrexia” on the first day of dosing. b Infection – this term excludes sepsis-related events which are presented separately. c Acneiform rash defined as any event described as “acne”, “dermatitis acneiform”, “dry skin”, “exfoliative rash”, “rash”, “rash erythematous”, “rash macular”, “rash papular”, or “rash pustular”. Chemotherapy = cisplatin + 5-fluorouracil or carboplatin + 5-fluorouracil
For cardiac disorders, approximately 9% of subjects in both the EU-approved cetuximab plus chemotherapy and chemotherapy-only treatment arms in Study 2 experienced a cardiac event. The majority of these events occurred in patients who received cisplatin/5-FU, with or without cetuximab as follows: 11% and 12% in patients who received cisplatin/5-FU with or without cetuximab, respectively, and 6% or 4% in patients who received carboplatin/5-FU with or without cetuximab, respectively. In both arms, the incidence of cardiovascular events was higher in the cisplatin with 5-FU containing subgroup. Death attributed to cardiovascular event or sudden death was reported in 3% of the patients in the cetuximab plus platinum-based therapy with 5-FU arm and 2% in the platinum-based chemotherapy with 5-FU alone arm.Colorectal CancerStudy 4: EU-Approved Cetuximab in Combination with FOLFIRI — Study 4 used EU-approved cetuximab. U.S.-licensed Erbitux provides approximately 22% higher exposure to cetuximab relative to the EU-approved cetuximab. The data provided below for Study 4 is consistent in incidence and severity of adverse reactions with those seen for Erbitux in this indication. The tolerability of the recommended dose is supported by safety data from additional studies of Erbitux [see Clinical Pharmacology (12.3) in Full Prescribing Information].Table 3 contains selected adverse reactions in 667 patients with K-Ras mutation-negative (wild-type), EGFR-expressing, metastatic colorectal cancer receiving EU-approved cetuximab plus FOLFIRI or FOLFIRI alone in Study 4 [see Warnings and Precautions]. Cetuximab was administered at the recommended dose and schedule (400 mg/m2 initial dose, followed by 250 mg/m2 weekly). Patients received a median of 26 infusions (range 1–224).
table 3: incidence of Selected Adverse reactions Occurring in ≥10% of Patients with K-Ras Mutation-negative (Wild-type) and EGFr-expressing, Metastatic Colorectal Cancera
Eu-Approved Cetuximab plus FOLFiri
(n=317)
FOLFiri Alone
(n=350)body System Preferred Term
Grades
1–4bGrades 3 and 4
Grades
1–4Grades 3 and 4
% of Patientsblood and Lymphatic System DisordersNeutropenia 49 31 42 24Eye DisordersConjunctivitis 18 <1 3 0Gastrointestinal DisordersDiarrhea 66 16 60 10Stomatitis 31 3 19 1Dyspepsia 16 0 9 0General Disorders and Administration Site ConditionsInfusion-related Reactionc 14 2 <1 0Pyrexia 26 1 14 1infections and infestationsParonychia 20 4 <1 0investigationsWeight Decreased 15 1 9 1Metabolism and Nutrition DisordersAnorexia 30 3 23 2
(Continued)
table 3: incidence of Selected Adverse reactions Occurring in ≥10% of Patients with K-Ras Mutation-negative (Wild-type) and EGFr-expressing, Metastatic Colorectal Cancera
Eu-Approved Cetuximab plus FOLFiri
(n=317)
FOLFiri Alone
(n=350)body System Preferred Term
Grades
1–4bGrades 3 and 4
Grades
1–4Grades 3 and 4
% of PatientsSkin and Subcutaneous tissue DisordersAcne-like Rashd 86 18 13 <1 Rash 44 9 4 0 Dermatitis Acneiform 26 5 <1 0 Dry Skin 22 0 4 0 Acne 14 2 0 0 Pruritus 14 0 3 0Palmar-plantar Erythrodysesthesia Syndrome 19 4 4 <1Skin Fissures 19 2 1 0a Adverse reactions occurring in at least 10% of Erbitux (cetuximab) combination arm with a frequency at least 5% greater than that seen in the FOLFIRI arm. b Adverse reactions were graded using the NCI CTC, V 2.0. c Infusion related reaction is defined as any event meeting the medical concepts of allergy/anaphylaxis at any time during the clinical study or any event occurring on the first day of dosing and meeting the medical concepts of dyspnea and fever or by the following events using MedDRA preferred terms: “acute myocardial infarction”, “angina pectoris”, “angioedema”, “autonomic seizure”, “blood pressure abnormal”, “blood pressure decreased”, “blood pressure increased”, “cardiac failure”, “cardiopulmonary failure”, “cardiovascular insufficiency”, “clonus”, “convulsion”, “coronary no-reflow phenomenon”, “epilepsy”, “hypertension”, “hypertensive crisis”, “hypertensive emergency”, “hypotension”, “infusion related reaction”, “loss of consciousness”, “myocardial infarction”, “myocardial ischaemia”, “prinzmetal angina”, “shock”, “sudden death”, “syncope”, or “systolic hypertension”. d Acne-like rash is defined by the events using MedDRA preferred terms and included “acne”, “acne pustular”, “butterfly rash”, “dermatitis acneiform”, “drug rash with eosinophilia and systemic symptoms”, “dry skin”, “erythema”, “exfoliative rash”, “folliculitis”, “genital rash”, “mucocutaneous rash”, “pruritus”, “rash”, “rash erythematous”, “rash follicular”, “rash generalized”, “rash macular”, “rash maculopapular”, “rash maculovesicular”, “rash morbilliform”, “rash papular”, “rash papulosquamous”, “rash pruritic”, “rash pustular”, “rash rubelliform”, “rash scarlatiniform”, “rash vesicular”, “skin exfoliation”, “skin hyperpigmentation”, “skin plaque”, “telangiectasia”, or “xerosis”.
Erbitux Monotherapy — Table 4 contains selected adverse reactions in 242 patients with K-Ras mutation-negative (wild-type), EGFR-expressing, metastatic colorectal cancer who received best supportive care (BSC) alone or with Erbitux in Study 5 [see Warnings and Precautions]. Erbitux was administered at the recommended dose and schedule (400 mg/m2 initial dose, followed by 250 mg/m2 weekly). Patients received a median of 17 infusions (range 1–51).
table 4: incidence of Selected Adverse reactions Occurring in ≥10% of Patients with K-Ras Mutation-negative (Wild-type), EGFr-expressing, Metastatic Colorectal Cancer treated with Erbitux Monotherapya
Erbitux plus bSC (n=118)
bSC alone (n=124)
body System Preferred Term
Grades 1–4b
Grades 3 and 4
Grades 1–4
Grades 3 and 4
% of PatientsDermatology/SkinRash/Desquamation 95 16 21 1Dry Skin 57 0 15 0Pruritus 47 2 11 0Other-Dermatology 35 0 7 2Nail Changes 31 0 4 0Constitutional SymptomsFatigue 91 31 79 29Fever 25 3 16 0Infusion Reactionsc 18 3 0 0Rigors, Chills 16 1 3 0PainPain-Other 59 18 37 10Headache 38 2 11 0Bone Pain 15 4 8 2PulmonaryDyspnea 49 16 44 13Cough 30 2 19 2GastrointestinalNausea 64 6 50 6Constipation 53 3 38 3Diarrhea 42 2 23 2Vomiting 40 5 26 5Stomatitis 32 1 10 0Other-Gastrointestinal 22 12 16 5Dehydration 13 5 3 0Mouth Dryness 12 0 6 0Taste Disturbance 10 0 5 0infectionInfection without neutropenia 38 11 19 5MusculoskeletalArthralgia 14 3 6 0NeurologyNeuropathy-sensory 45 1 38 2Insomnia 27 0 13 0Confusion 18 6 10 2Anxiety 14 1 5 1Depression 14 0 5 0a Adverse reactions occurring in at least 10% of Erbitux plus BSC arm with a frequency at least 5% greater than that seen in the BSC alone arm. b Adverse reactions were graded using the NCI CTC, V 2.0. c Infusion reaction is defined as any event (chills, rigors, dyspnea, tachycardia, bronchospasm, chest tightness, swelling, urticaria, hypotension, flushing, rash, hypertension, nausea, angioedema, pain, sweating, tremors, shaking, drug fever, or other hypersensitivity reaction) recorded by the investigator as infusion-related.
Erbitux in Combination with Irinotecan — The most frequently reported adverse reactions in 354 patients treated with Erbitux plus irinotecan in clinical trials were acneiform rash (88%), asthenia/malaise (73%), diarrhea (72%), and nausea (55%). The most common Grades 3–4 adverse reactions included diarrhea (22%), leukopenia (17%), asthenia/malaise (16%), and acneiform rash (14%).immunogenicity: As with all therapeutic proteins, there is potential for immunogenicity. Immunogenic responses to cetuximab were assessed using either a double antigen radiometric assay or an ELISA assay. Due to limitations in assay performance and sampling timing, the incidence of antibody development in patients receiving Erbitux has not been adequately determined. Non-neutralizing anti-cetuximab antibodies were detected in 5% (49 of 1001) of evaluable patients without apparent effect on the safety or antitumor activity of Erbitux.
(Continued)
Erb0813PBS_693US13PBS02201_7x9wip3.indd 2 8/19/13 2:01 PM
14220302_0124301_CrossTumor_ROB_v4_M.indd 2 5/16/14 12:40 PM

PAGE 38 The ASCO Post | MARCH 25, 2015
Geriatric Oncology
Just like in younger patients, keeping fit and eating a healthy diet is the best advice we can give to prevent cancer and other diseases that increase with aging.
—Hyman Muss, MD
Geriatrics for the Oncologist
Geriatrics for the Oncologist is guest edited by Stuart Lichtman, MD, and developed in collaboration with the International Society of Geriatric Oncology (SIOG). Visit SIOG.org for more on geriatric oncology.
motherapy in older patients. A similar model, the Cancer and Aging Research Group (CARG) model, has been devel-oped by Arti Hurria, MD.
More Data Needed on Chemoprevention
Is chemoprevention a management tool in geriatric oncology?
Chemoprevention in the elderly cancer patient has little clinical application. And there aren’t enough trials in this area to understand what, if any, cancer chemopre-vention strategies may benefit the geriatric population. In elderly patients with a life expectancy of 5 years or more, screening for breast and colorectal cancers may have benefit, but we need more information.
In terms of chemoprevention, what can we really prevent?
For instance, most of the chemopreven-tion studies with tamoxifen do not show a significant survival benefit, so I think there is no reason to put an older cancer patient on tamoxifen or an aromatase inhibitor, especially considering the side effects. In short, there are no large data sets to prove the benefit of chemoprevention strategies in the elderly, and I certainly wouldn’t go in that direction with my patients. Just like in younger patients, keeping fit and eating a healthy diet is the best advice we can give to prevent cancer and other diseases that increase with aging.
An Educational OpportunitySince community doctors deliver the
majority of cancer care, is there a knowl-edge gap in the busy community setting about managing the more complex needs
of their geriatric patients?I don’t think either academic or com-
munity doctors are up to speed with the special assessment and care of geriatric cancer patients. For instance, we looked within our own institution at the Uni-versity of North Carolina Lineberger Comprehensive Cancer Center at data we collected about patient falls; we found that almost none of the patients who had a history of falls were referred for physical or occupational therapy evalution, even though these interven-tions in older patients with falls lower morbidity and mortality. This shows that most doctors think that because most cancer patients are older, we intui-tively know how to care for them, but at least in this instance, we didn’t deliver the kind of specialized care needed in this more challenging population.
There are many educational oppor-tunities for doctors to learn more about their older patients. For instance, ASCO
has a terrific series of slides on gen-eral geriatric care at ASCO University; there are other equally good websites, and I have a series of slides on geriatric oncology on our Lineberger Compre-hensive Cancer Center website that can be downloaded. I am always available to colleagues who want to get hold of this valuable information. But the truth is that clinicians simply don’t have the time for extra educational activity. With all the new demands of electronic medi-cal records, meaningful use, other data collection, and paperwork, who wants to go home after a long day in the clinic and begin studying slides on how to do a geriatric assessment? We need to fig-ure out how to get this information into all training programs and make it part of lifelong learning.
Not One-Size-Fits-All Treatment Decisions
One common misconception about older patients that needs changing is that they should always be spared the rigors of aggressive therapy. This attitude is be-ing proved false, but it may be difficult to convince oncologists, because there are so few clinical trials that include the elderly. What’s your opinion on this?
I think the word on the issue of under-treating elderly cancer patients is finally getting out, and both community and academic clinicians are becoming more aware that otherwise healthy older cancer patients deserve state-of-the-art therapies. That said, treating elderly patients, as I’ve mentioned, depends largely on doing thorough and accurate assessments.
For instance, the small potential ben-efit of chemotherapy is usually unwar-ranted in an 80-year-old patient with a few positive nodes who has hormone receptor–positive cancer. However, a
75-year-old woman with triple-negative breast cancer who is in otherwise good shape should be treated with the same state-of-the-art therapies as her younger counterpart. Treatment decisions in the elderly are not one-size-fits-all; it is all about patient-by-patient assessment.
Don’t Overlook Early DementiaDementia in the elderly patient with
cancer is a big challenge. Are we doing a good job meeting that challenge?
No. A clinician must do a cognitive function test in the office to identify pa-tients with dementia. I’ve learned that very few doctors do these tests routinely, quite frankly because it is a time-con-suming practice, even though a screen can be done in a few minutes. For ex-ample, a well-dressed older person who
is articulate when answering yes-or-no questions frequently may not know what she had for breakfast. Early dementia is frequently overlooked. So, you have to probe, and that takes time, but it’s part of geriatric best practices and worth it.
Defining Value and QualityAny thoughts on how the movement
in oncology to value over quantity might affect geriatric oncology, especially in an environment that is growing increasingly sensitive over health-care costs?
The terms “value” and “quality” are variable across the whole spectrum of cancer care. If I can define what value is for my patient, and then we’ll deter-mine whether the cost is worth it for one quality-adjusted year of life. Do you think a 75-year-old retired person who has led a productive life has less value as a human being than a 15-year-old who is incarcerated for drugs? The examples are endless, so definitions of value and cost can be fuzzy and unfair. Quality is another tough one. Is it that I follow national treatment guidelines or have free parking?
Closing ThoughtsWould you like to share any final
thoughts?I am very pleased that so many great
cancer organizations are now paying at-tention to the issues of elderly patients. ASCO, in particular, has been a leader in this field, providing valuable venues for training and education for the oncology community and the younger doctors en-tering oncology. This is the way forward, which will finally integrate geriatric on-cology into our daily practice. n
Disclosure: Dr. Muss reported no potential conflicts of interest.
Hyman Muss, MDcontinued from page 37
For more information, visit http://www.siog.org/
Who will crack the cancer code?It’s the question that millions of people are asking. Pushing us to explore every idea, continually refining our approach, and collaborating with innovators across the globe to explore cancer genomes as never before. Leading us to identify cancer mutations and mechanisms, like PD-1 interactions and EGFR, discoveries that help all of us develop more targeted therapies. Together, we can find solutions to the toughest problems, because the more answers we find, the more lives we save.
Videos, whitepapers and more at DiscoverCareBelieve.org/code.
© 2015 Dana-Farber Cancer Institute

Who will crack the cancer code?It’s the question that millions of people are asking. Pushing us to explore every idea, continually refining our approach, and collaborating with innovators across the globe to explore cancer genomes as never before. Leading us to identify cancer mutations and mechanisms, like PD-1 interactions and EGFR, discoveries that help all of us develop more targeted therapies. Together, we can find solutions to the toughest problems, because the more answers we find, the more lives we save.
Videos, whitepapers and more at DiscoverCareBelieve.org/code.
© 2015 Dana-Farber Cancer Institute

PAGE 40 The ASCO Post | MARCH 25, 2015
In the Clinic
Lenalidomide in Combination With Dexamethasone in Newly Diagnosed Multiple MyelomaBy Matthew Stenger
On February 18, 2015, the indica-tion for lenalidomide (Revlimid)
in combination with dexamethasone was expanded to include patients with newly diagnosed multiple myeloma.1 Lenalidomide in combination with dexamethasone was previously ap-proved for use in patients with multiple myeloma who had received at least one prior treatment.
Supporting TrialThe expanded indication is based on
findings in an open-label phase III trial in which 1,623 newly diagnosed patients who were not candidates for stem cell transplant were randomized to receive dexamethasone 40 mg/d (20 mg/d for age > 75 years) on days 1, 8, 15, and 22 of 28-day cycles plus lenalidomide 25 mg/d on days 1 to 21 of 28-day cycles either continuously until progressive disease (continuous group, n = 535) or for up to 18 cycles (18-cycle group, n = 541) or melphalan, prednisone, and thalidomide (MPT, n = 547).1,2 All pa-tients received prophylactic anticoagu-lation, usually with aspirin. The primary endpoint was progression-free survival.
Overall, patients had a median age of 73 years (35% > 75 years); 59% had International Staging System stage I/II and 41% had stage III disease; 9% had severe renal impairment and 23% had moderate renal impairment; and East-ern Cooperative Oncology Group per-formance status was 0 in 29%, 1 in 49%, and 2 in 21%.
Median progression-free survival was significantly longer in the continu-ous lenalidomide group vs the MPT group (25.5 vs 21.2 months, hazard ratio [HR] = 0.72, P < .0001) as well as vs the 18-cycle lenalidomide group (20.7 months, HR = 0.70, 95% con-fidence interval [CI] = 0.60–0.82). No difference was observed between the 18-cycle lenalidomide group and the MPT group (HR = 1.03, 95% CI = 0.89–1.20). Overall response rates were 75.1% vs 62.3% and 73.4%. Me-
dian overall survival was 58.9 months with continuous lenalidomide vs 48.5 months with MPT (HR = 0.75, 95% CI = 0.62–0.90) and 56.7 months in the 18-cycle lenalidomide group (HR = 0.91, 95% CI = 0.75–1.09; HR = 0.83, 95% CI = 0.69–0.99, vs MPT).
How It WorksLenalidomide is a thalidomide ana-
log with immunomodulatory, antian-giogenic, and antineoplastic properties. It inhibits cell proliferation; induces apoptosis of certain hematopoietic tu-mor cells, including mantle cell lym-phoma, multiple myeloma, and del (5q) myelodysplastic syndromes in vitro; and delays tumor growth in he-matopoietic tumor models, including multiple myeloma. The immunomodu-latory properties of the agent include activation of T cells and natural killer (NK) cells, increased numbers of NK T cells, and inhibition of proinflammatory cytokines (eg, tumor necrosis factor-α and interleukin 6) by monocytes. The
combination of lenalidomide and dexa-methasone synergizes the inhibition of cell proliferation and the induction of apoptosis in multiple myeloma cells.
How It Is GivenIn multiple myeloma, the recom-
mended starting dose of lenalidomide is 25 mg/d on days 1 to 21 of repeated 28-day cycles in combination with dexa-methasone, with treatment continued until disease progression or unaccept-able toxicity. In patients who are eligible for autologous stem cell transplantation, hematopoietic stem cell mobilization should occur within 4 cycles of lenalido-
mide-containing therapy. Starting doses should be 10 mg once daily for moderate renal impairment, 15 mg every 48 hours for severe renal impairment, and 5 mg once daily following dialysis in patients requiring dialysis.
Treatment should be interrupted in patients developing grade 3 or 4 thrombocytopenia or neutropenia, and complete blood cell counts should be followed weekly; upon recovery, treat-ment should be resumed at a lower dose. Treatment should be interrupted for other grade 3 or 4 adverse events and resumed at a lower dose upon reso-lution to grade ≤ 2.
Safety ProfileIn the phase III trial in newly diag-
nosed myeloma, the most common adverse events of any grade in the con-tinuous lenalidomide group included diarrhea (46%), anemia (44%), neutro-penia (35%), fatigue (33%), and back pain (32%), and the most common grade 3 or 4 events were neutropenia
(28%), anemia (18%), and pneumo-nia (11%). Infections were observed in 75% of patients in the continuous le-nalidomide group vs 56% of patients in the MPT group.
In the continuous lenalidomide group, the most common adverse events leading to dose interruption were infections (29%), the most com-mon adverse events leading to dose reduction were hematologic events (11%), and the most common adverse events leading to treatment discontinu-ation were infections (3.4%). In both lenalidomide groups, the frequency of adverse events was generally highest in the first 6 months of treatment, except with regard to cataracts; the frequency of cataracts increased from 0.7% during the first 6 months of treatment to 9.6% by the second year of treatment with continuous lenalidomide.
Lenalidomide carries boxed warn-
ings for embryo-fetal toxicity, hemato-logic toxicity, and venous and arterial thromboembolism. To avoid embryo-fetal exposure, lenalidomide is available only through a restricted distribution program, the Revlimid REMSTM pro-gram, formerly known as the “RevAs-sist®” program (information available at www.celgeneriskmanagement.com or 1-888-423-5436).
Patients with del (5q) myelodysplas-tic syndromes should have complete blood cell counts monitored weekly for the first 8 weeks and monthly there-after. A significantly increased risk of deep vein thrombosis and pulmonary embolism and a risk of myocardial in-farction and stroke have been observed in patients with multiple myeloma; antithrombotic prophylaxis is recom-mended.
Lenalidomide also carries warnings/precautions for serious and fatal cardiac adverse events (observed in patients with chronic lymphocytic leukemia), second primary malignancies, hepatic failure including fatalities, allergic re-actions including fatalities, tumor lysis syndrome including fatalities, tumor flare reactions (observed in chronic lymphocytic leukemia and lymphoma), and impaired stem cell mobilization.
Lenalidomide is contraindicated during pregnancy and in patients with demonstrated hypersensitivity to the agent. n
References1. Revlimid (lenalidomide) capsules
prescribing information, Celgene Cor-poration, February 2015. Available at http://www.revlimid.com/wp-content/ uploads/2013/11/PI.pdf. Accessed March 3, 2015.
2. Benboubker L, Dimopoulos MA, Dispenzieri A, et al: Lenalido-mide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med 371:906-917, 2014.
In the Clinic provides overviews of novel oncology agents, addressing indications, mechanisms, admin-istration recommendations, safety profiles, and other essential infor-mation needed for the appropriate clinical use of these drugs.
Hematology
OF NOTELenalidomide is a thalidomide analog with immunomodulatory, antiangiogenic, and antineoplas-tic properties. The combination of lenalidomide and dexamethasone synergizes the inhibition of cell pro-liferation and the induction of apop-tosis in multiple myeloma cells.
OF NOTELenalidomide carries boxed warn-ings for embryo-fetal toxicity, he-matologic toxicity, and venous and arterial thromboembolism.
Expanded Indication for Lenalidomide in Combination With Dexamethasone
■ Lenalidomide (Revlimid) in combination with dexamethasone is now indicated in the treatment of patients with newly diagnosed multiple myeloma.
■ In multiple myeloma, the recommended starting dose of lenalidomide is 25 mg/d on days 1 to 21 of repeated 28-day cycles in combination with dexamethasone.

ASCOPost.com | MARCH 25, 2015 PAGE 41
Announcements
New Lurie Cancer Center Program Combines Oncology With Genomics to Provide More Personalized Cancer Care
The Robert H. Lurie Comprehen-sive Cancer Center of North-
western University, in collaboration with the Northwestern Medicine De-velopmental Therapeutics Institute and Northwestern Memorial Hospital, has launched a new research program, Northwestern Onco-SET (Sequence, Evaluate, Treat). The program’s goal is to provide a more personalized, precision medicine option for cancer patients by combining oncology with genomics. This program will initially focus on pa-tients with any type of cancer that is not responsive to traditional therapies.
Molecularly Defined Targets“Northwestern Onco-SET will help
establish Chicago as a national and in-ternational leader in precision medicine for cancer,” said Leonidas Platanias, MD, PhD, Director of the Lurie Can-cer Center. “This is the first time cancer treatment in Chicago will be offered in a
comprehensive, multidisciplinary pro-gram. Onco-SET will use molecularly defined genomic targets as a basis for determining treatment options, includ-ing novel, early-phase clinical trials.”
Onco-SET personalizes cancer care for patients by sequencing the indi-vidual genetic profile of their tumors and evaluating the results to provide the treatments or clinical trials that will benefit them most. Some of these approaches include site-agnostic, pathway-driven treatments, which use therapies developed to target the spe-cific genetic abnormalities of one type of cancer and apply them to treating a different kind of cancer if it shares the same genetic abnormalities.
“As part of our work with Onco-SET, we are also planning to initiate a pilot program of site-agnostic, pathway-driv-en tumor clinics,” added Dr. Platanias.
Wide Spectrum of SpecialistsTo evaluate and discuss the best
treatment options for each patient, Onco-SET created the Lurie Cancer
Center’s Molecular Tumor Board, which brings together a group of experts to re-view every tumor’s genomic profile. The board comprises a wide spectrum of can-cer specialists, including pathologists;
medical, surgical, and radiation oncolo-gists; cancer geneticists; genome biolo-gists; molecular scientists; bioethicists; and bioinformaticists.
By offering cancer patients care
within Onco-SET, the program is also expanding the Lurie Cancer Center’s preclinical research by collecting and analyzing detailed data from each pa-tient’s tumor genomic profiles. n
Leonidas Platanias, MD, PhD Advertisement not displayed in digital edition at advertiser’s request

PAGE 42 The ASCO Post | MARCH 25, 2015
Inside the Black BoxInside the Black Box
Development and Approval of Biosimilar ProductsBy Leah Christl, PhD, and Albert Deisseroth, MD, PhD
The Biologics Price Competi-tion and Innovation Act of 2009
(BPCI Act) was passed as part of health reform (the Affordable Care Act) and was signed into law on March 23, 2010. The BPCI Act created an abbreviated licensure pathway for biologic products shown to be bio-similar to or interchangeable with an FDA-licensed reference product. The ASCO Post spoke with Drs. Christl and Deisseroth about biosimilars and the BPCI Act.
Understanding BiosimilarsFirst, can you tell us what a biosimilar
product is?Dr. Christl: To understand bio-
similar products, it is important first to understand how biologic products are defined and regulated. Many of today’s important medications are biologic products, generally derived from a liv-ing organism. This material can come from many sources, including humans, animals, microorganisms, or yeast. Bio-logic products are licensed under the Public Health Service Act (PHS Act), unlike drugs that are approved under the Food, Drug, and Cosmetic Act (FD&C Act).
In addition to creating an abbrevi-ated pathway for licensure of biosimi-lar biologic products, the BPCI Act revised the definition of “biologic product” to include proteins, except for chemically synthesized polypep-tides. The term “biologic product” is defined under the PHS Act as “a virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or
derivative, allergenic product, protein (except any chemically synthesized polypeptide), or analogous product … applicable to the prevention, treat-ment, or cure of a disease or condi-tion of human beings.…” Historically, some proteins, such as human growth hormone, have been approved as drugs under the FD&C Act, whereas other proteins, such as blood factors, have been licensed as biologics under the PHS Act.
A biosimilar is a biologic prod-uct that is highly similar to an FDA-licensed reference product, notwith-standing minor differences in clinically inactive components, and has no clini-cally meaningful differences in terms of safety, purity, and potency (ie, safety and effectiveness) from the reference product. The term “reference product” here refers to the single biologic prod-uct licensed under the PHS Act against which a biosimilar is evaluated. A bio-similar product can only be licensed if it has the same mechanism(s) of action, route of administration, dosage form, and strength as the reference biologic product and only for the indications and condition(s) of use that have been approved for the reference biologic product.
What is the abbreviated license path-way for biologic products?
Dr. Christl: A biologic product that is demonstrated to be “biosimilar” to the reference product may rely for licensure on, among other things, publicly avail-able information regarding FDA’s pre-vious determination that the reference product is safe, pure, and potent. This licensure pathway under section 351(k) of the PHS Act permits a biosimilar bio-logic product to be licensed based on less than a full complement of product-spe-cific preclinical and clinical data.
Can you clarify the differences between “stand-alone” and biosimilar product de-velopment?
Dr. Christl: As previously noted, the reference product is the single bio-logic product licensed under the PHS Act against which a biosimilar is evalu-ated. The reference product is licensed by FDA under section 351(a) of the PHS Act based on a full complement of product-specific preclinical and clinical data. The reference product can be con-
sidered a “stand-alone” application.The goal of “stand-alone” develop-
ment is to independently demonstrate that the product is safe and effective for the intended conditions of use, and drug development starts with preclinical re-search and progresses through phase I, II, and III trials. The goal of biosimilar development is to demonstrate biosimi-larity between the proposed product and a reference product and not to indepen-dently establish the safety and effective-ness of the proposed product.
What is required to demonstrate biosimilarity?
Dr. Christl: The PHS Act requires that a 351(k) application include in-formation demonstrating biosimilar-ity based upon data derived from (1)
analytic studies demonstrating that the biologic product is “highly simi-lar” to the reference product, notwith-standing minor differences in clinically inactive components; (2) animal stud-ies (including the assessment of toxic-ity); and (3) a clinical study or studies (including the assessment of immu-nogenicity and pharmacokinetics or pharmacodynamics) sufficient to dem-onstrate safety, purity, and potency in one or more appropriate conditions of use for which the reference prod-uct is licensed and for which licensure is sought for the biosimilar product. FDA may determine, in its discretion, that an element described above is unnecessary in a 351(k) application. FDA has articulated in guidance to in-dustry that it will use a totality of the ev-idence approach to review applications for biosimilar products, consistent with a longstanding Agency approach to evaluation of scientific evidence.
What is analytic similarity?Dr. Christl: Analytic similarity
studies generate data to characterize the reference product variability and prod-uct quality characteristics, characterize
There is no single study that will demonstrate biosimilarity. That is why FDA recommends a stepwise
approach to data generation and evaluation of residual uncertainty at each step.
—Leah Christl, PhD
FDA Clinical Reviewers
Albert Deisseroth, MD, PhD
Leah Christl, PhD
INSIDE THE BLACK BOX is an occasional column provid-ing insight into the U.S. Food and Drug Administration (FDA) and its policies and procedures. In this installment, Leah Christl, PhD, and Albert Deisseroth, MD, PhD, an-swer questions about biosimilar products. Dr. Christl is the Associate Director for Therapeutic Biologics and the team leader for the Office of New Drugs’ Therapeutic Biologics and Biosimilars Team in the Center for Drug Evaluation and Research. Dr. Deisseroth is a team leader in the Divi-sion of Hematologic Products.

ASCOPost.com | MARCH 25, 2015 PAGE 43
Inside the Black Box
the proposed biosimilar product qual-ity characteristics and variability, and identify and evaluate the potential im-pact of observed differences between the proposed biosimilar product and the reference product. The sponsor must demonstrate that the products are highly similar. The potential effect of differences on safety, purity, and poten-cy should be addressed and supported by appropriate data.
Analytic similarity data are the foun-dation of a biosimilar development program and provide the necessary ex-tensive structural and functional char-acterization of the molecule (critical quality attributes and clinically active components and the impact of manu-facturing changes that occur during product development) and an under-standing of the relationship between quality attributes and the clinical safety and efficacy profile. This aids in deter-mining the residual uncertainty about biosimilarity and in predicting expected clinical similarity.
Role of Clinical StudiesWhat clinical studies are expected?Dr. Christl: As a scientific mat-
ter, FDA expects an adequate clinical pharmacokinetic and pharmacody-namic (if relevant, eg, the endpoint reflects the biologic effect(s) of the product) comparison between the proposed biosimilar product and the reference product. Also, as a scientific matter, at least one clinical study that includes a comparison of the immu-nogenicity of the proposed and refer-ence product generally will be expect-ed. A comparative clinical study will be necessary to support demonstra-tion of biosimilarity if there are resid-ual uncertainties about whether there are clinically meaningful differences between the proposed and reference products based on structural and functional characterization, animal testing, human pharmacokinetic and pharmacodynamic data, and clinical immunogenicity assessment.
Can you tell us more about the clinical studies that may be necessary?
Dr. Christl: If analytic differences have been observed, then any clinical study should be designed to best evalu-ate their potential impact.
A comparative clinical pharmacoki-netic study is generally expected, and pharmacodynamic data are desirable on a case-by-case basis. The goal is to demonstrate similarity by assessing whether there are clinically meaning-ful differences in pharmacokinetics
(and pharmacodynamics) between the proposed biosimilar and the reference product. The assumption is that simi-lar exposure (and pharmacodynamic response) will provide similar efficacy and safety (ie, an exposure-response relationship exists). Depending on the product, the study may be conducted in healthy volunteers or patients.
A comparative clinical study for a bi-osimilar development program should be designed to investigate whether there are clinically meaningful differ-ences between the proposed product and the reference product. Important determinants of the study design can include the complexity of the product; limitations of analytic testing; the ex-tent of knowledge about critical quality attributes; the degree of understanding of the mechanism of action of the refer-ence product; the extent of knowledge about how differences in analytic, ani-mal, and clinical pharmacokinetics or pharmacodynamics predict differences in clinical outcome; and the sensitiv-ity of the study design (eg, population, dose, endpoint[s]).
An adequate scientific justification must be provided for the choice of study design, study population, study endpoint(s), estimated effect size for the reference product, and study margin(s). The potential exists for a biosimilar product to be approved for one or more conditions of use for which the U.S.-licensed reference product is licensed based on extrapola-tion of clinical data intended to dem-onstrate biosimilarity in one condition of use. Sufficient scientific justification for extrapolation is necessary.
You mentioned a “totality-of-the-evidence” approach. Can you elaborate further?
Dr. Christl: There is no single study that will demonstrate biosimilarity. That is why FDA recommends a step-wise approach to data generation and evaluation of residual uncertainty at each step. If analytic differences are ob-served, their potential impact(s) should be evaluated before proceeding to the next step in development.
For example, the results of analytic studies would provide information on the animal studies that would be war-ranted as the next step in evaluating residual uncertainty, and the results of the analytic and animal studies would provide information needed for the design of clinical pharmacokinetic and pharmacodynamics studies and the clinical immunogenicity assessment. All of these data together would be
used to determine the residual uncer-tainty about the biosimilarity of the two products and the type of addition-al clinical data that would be needed.
First Biosimilar Marketing Application
FDA held a meeting of the Oncology Drug Advisory Committee (ODAC) on January 7, 2015, which was the first bio-similar marketing application discussed at a public advisory committee meeting. Could you summarize what kind of mol-ecule was under review and its history?
Dr. Deisseroth: Sandoz submitted a 351(k) biologics license application requesting licensure of a granulocyte colony-stimulating factor (G-CSF) product, referred to during develop-ment as EP2006, as a biosimilar to U.S.-licensed filgrastim (Neupogen). Filgrastim is an analog of G-CSF, a hematopoietic growth factor that in-duces proliferation and differentiation of neutrophil-committed progenitor cells into neutrophils. Filgrastim also induces the release of neutrophils as well as CD34-positive hematopoietic progenitor cells from the marrow into the peripheral blood. Filgrastim is made up of 175 amino acids without glycosylation, so, from a molecular perspective, this product may be one of the easier ones to replicate in terms of manufacturing.
In its application, Sandoz requested that EP2006 be designated as a bio-similar to filgrastim for all of its five currently approved indications; they include decreasing the incidence of in-fections in patients with nonmyeloid malignancies receiving myelosuppres-sive anticancer drugs; reducing the du-ration of neutropenia in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by marrow transplantation; reducing the incidence and duration of sequelae of neutropenia in symptomatic patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutrope-nia; mobilizing hematopoietic progeni-tor cells into the peripheral blood for collection by leukapheresis; and reduc-ing the time to neutrophil recovery and the duration of fever, following induc-tion or consolidation chemotherapy treatment of adults with acute myeloid leuekmia.
Since 2009, Sandoz has marketed Zarzio, a European Union (EU)–ap-proved biosimilar product to EU-ap-proved filgrastim. Marketing experi-ence with biosimilar filgrastim outside of the United States includes in excess of 7.5 million days of patient exposure.
What did Sandoz present at the advi-sory committee meeting, and what was the reaction of FDA to this presentation?
Dr. Deisseroth: The foundation of biosimilar applications and a key hurdle to surmount is whether the biosimilar product is “highly similar” to the reference product from an ana-lytic perspective. FDA agreed with the conclusions of Sandoz that EP2006 is “highly similar” to U.S.-licensed filgras-tim based on its extensive studies of the physicochemical and functional prop-erties of the two products. FDA also agreed with Sandoz’s conclusion that its nonclinical studies supported a demon-stration of biosimilarity of its product, EP2006, to U.S.-licensed filgrastim.
Sandoz then summarized its exten-sive clinical pharmacokinetic and phar-macodynamic studies in normal human subjects comparing biosimilar filgras-tim with both U.S.-licensed filgrastim and EU-approved filgrastim at a wide range of doses. It is important to note that it was necessary for Sandoz to build a scientific bridge between biosimilar filgrastim, EU-approved filgrastim, and U.S.-licensed filgrastim to justify the relevance of the data generated using EU-approved filgrastim as part of the basis demonstrating biosimilarity to U.S.-licensed filgrastim.
Sandoz also summarized immuno-genicity and safety evaluations, which had been carried out in normal human subjects and in patients with breast cancer. FDA agreed with Sandoz that these studies did not show any clinical-ly meaningful differences between the products, which is the second criterion for demonstrating biosimilarity of two products.
Finally, Sandoz presented safety, ef-ficacy, and immunogenicity data from a
GUEST EDITOR
Inside the Black Box is Guest Edited by Richard Pazdur, MD, Director of the FDA’s Office of Hematology and Oncology Products.
Richard Pazdur, MD
continued on page 44

PAGE 44 The ASCO Post | MARCH 25, 2015
Inside the Black Box
comparative clinical study that random-ized patients with breast cancer receiv-ing chemotherapy between biosimilar filgrastim and U.S.-licensed filgrastim to support its conclusion that there were no clinically meaningful differences between biosimilar filgrastim and U.S.- licensed filgrastim. FDA commented that although it agreed with Sandoz’s conclusions, a sponsor generally will not need to conduct a comparative clinical study to evaluate efficacy and safety to support demonstration of biosimilarity unless there are residual uncertainties from the analytic, animal, and/or clinical pharmacokinetic and pharmacodynamic studies that need to be evaluated further.
The final issue addressed by the advi-sory committee was whether biosimilar filgrastim should be licensed for all of the five indications for which filgras-tim is currently approved. FDA recom-mended that biosimilar filgrastim be licensed for all five of the filgrastim indi-cations due to the common mechanism of action for all of these indications and the totality of the evidence presented by Sandoz. The advisory committee voted unanimously that biosimilar filgrastim should receive licensure as a biosimilar to U.S.-licensed filgrastim for all of the indications for which filgrastim is cur-rently approved.
Final CommentsWhat are your final comments about
biosimilar development?Dr. Christl: There is no “one-size-
fits-all” development plan for a biosimi-lar. The principles discussed herein and as outlined in FDA guidances (www.
fda.gov/Drugs/GuidanceComplian-ceRegulatoryInformation/Guidances/ucm290967.htm) should be used. FDA scientists are available to meet with companies at each step of development and when an application is submit-ted to evaluate the applicant’s integra-tion of various types of information to determine whether the applicant’s
product is biosimilar to the reference product. Additional information can be found at www.fda.gov/Drugs/De-velopmentApprovalProcess/How-DrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBi-ologicApplications/Biosimilars/de-fault.htm.
Note: On March 6, 2015, subse-
quent to this interview, FDA licensed the product discussed in this article as Zarxio (filgrastim-sndz), a biosimilar to U.S.-licensed filgrastim. More informa-tion about the approval of this biosimi-lar can be found at www.ascopost.com/ViewNews.aspx?nid=23634. n
Disclosure: Drs. Christl and Deisseroth reported no potential conflicts of interest.
Biosimilar Productscontinued from page 43
Did You Know?Sales of herbal products reached $6 billion in 2013.
See page 76 for more on increasing problems in herbal supplements.
LIVE: 14.15”
LIVE: 9.575”
TRIM: 15.15”
TRIM: 10.575”
BLEED: 15.4”
BLEED: 10.825”
FOLD: 7.575”
SAFETY: 6.575”
FOLD: 7.575”
SAFETY: 6.575”
THIS ADVERTISEMENT PREPARED BY NeONClient: TaihoProduct: Taiho Oncology Job#: 10249957Job Name: The ASCO Post - King Island Size Ad Code: TOI-2014-US-002
Publications:The ASCO Post
Colors: 4CSizes: Bleed: 15.4" W x 10.825" H
Sm Trim: 15.15" W x 10.575" H Live: 14.15" W x 9.575" H
10249957 ASCO SPREAD AD KING ISLAND SIZE
It’s all in the deliveryCaring for cancer differently...
Taiho Oncology, Inc., is focused on bringing novel technology to cornerstone chemotherapies for a wide range of tumor types—including colorectal cancer and a variety of solid tumors. By developing oral oncolytic therapies, we are aspiring to deliver more meaningful moments to patients, and to redefine the way the world treats cancer.
To learn more, please visit www.TaihoOncology.com.
Introducing
Making the human connection
© TAIHO ONCOLOGY, INC. 11/2014 All rights reserved. TOI-2014-US-002

ASCOPost.com | MARCH 25, 2015 PAGE 45
Announcements
FDA Names Geoffrey Kim, MD, Director of the Division of Oncology Products
The U.S. Food and Drug Admin-istration (FDA) recently an-
nounced the appointment of Geoffrey Kim, MD, as Director of the Division of Oncology Products 1 (DOP1) in the Office of Hematology Oncology Products. Dr. Kim assumed this posi-
tion effective March 22, 2015.Dr. Kim previously served as the
Acting Deputy Director in Division of Oncology Products 1. He was involved with numerous cross-center working groups, developing policies pertain-ing to in-vitro companion diagnostics,
combination products, and dose find-ing optimization strategies for oncol-ogy products.
Dr. Kim is also the gynecologic ma-lignancies scientific liaison for Office of Hematology Oncology Products, and he is active with outreach to the ovarian
cancer community through the Society for Gynecologic Oncologists, Gyneco-logic Oncology Group, and the Ovar-ian Cancer National Alliance. Dr. Kim began working at the FDA in 2010 as a Medical Officer in Division of Oncol-ogy Products 1.
He received his bachelor’s degree at UCLA, his medical degree at the New York Medical College, and completed his residency in internal medicine at the Montefiore Medical Center. He per-formed laboratory research at these in-stitutions with a particular focus on cell adhesion molecules and cellular signal-ing. Dr. Kim completed his medical on-cology fellowship at the National Can-cer Institute (NCI) where he was active in both laboratory and clinical research in the NCI molecular signaling section and the ovarian cancer clinic.
Amna Ibrahim, MD, served as Act-ing Division Director of Division of Oncology Products 1 during the tran-sition period. Dr. Ibrahim will assume her previous role as the Deputy Divi-sion Director of Division of Oncology Products 1. n
Editorial CorrespondenceJames O. Armitage, MD
Editor-in-Chief e-mail: [email protected]
Cara H. Glynn Director of Editorial
e-mail: [email protected]: 631.935.7654
Andrew Nash Assoc. Director of Editorial
e-mail: [email protected] Phone: 631.935.7657
Editorial OfficeHarborside Press
37 Main Street Cold Spring Harbor, NY 11724
Phone: 631.692.0800 Fax: 631.692.0805 ASCOPost.com
HarborsidePress.com
Contact The ASCO Post
LIVE: 14.15”
LIVE: 9.575”
TRIM: 15.15”
TRIM: 10.575”
BLEED: 15.4”
BLEED: 10.825”
FOLD: 7.575”
SAFETY: 6.575”
FOLD: 7.575”
SAFETY: 6.575”
THIS ADVERTISEMENT PREPARED BY NeONClient: TaihoProduct: Taiho Oncology Job#: 10249957Job Name: The ASCO Post - King Island Size Ad Code: TOI-2014-US-002
Publications:The ASCO Post
Colors: 4CSizes: Bleed: 15.4" W x 10.825" H
Sm Trim: 15.15" W x 10.575" H Live: 14.15" W x 9.575" H
10249957 ASCO SPREAD AD KING ISLAND SIZE
It’s all in the deliveryCaring for cancer differently...
Taiho Oncology, Inc., is focused on bringing novel technology to cornerstone chemotherapies for a wide range of tumor types—including colorectal cancer and a variety of solid tumors. By developing oral oncolytic therapies, we are aspiring to deliver more meaningful moments to patients, and to redefine the way the world treats cancer.
To learn more, please visit www.TaihoOncology.com.
Introducing
Making the human connection
© TAIHO ONCOLOGY, INC. 11/2014 All rights reserved. TOI-2014-US-002

PAGE 46 The ASCO Post | MARCH 25, 2015
In the Clinic
Panobinostat in Combination With Bortezomib-Dexamethasone in Previously Treated Multiple Myeloma By Matthew Stenger
On February 23, 2015, panobinostat (Farydak) was granted accelerated
approval for use in combination with bort-ezomib (Velcade) and dexamethasone in the treatment of patients with multiple my-eloma who have received at least two prior regimens including bortezomib and an im-munomodulatory agent.1,2 As a condition of accelerated approval, the sponsor is re-quired to conduct a trial to verify and de-scribe the clinical benefit of panobinostat in patients with multiple myeloma.
Supporting Study Approval was based on outcomes in a
double-blind phase III trial in which 758 patients with relapsed multiple myeloma received oral panobinostat or placebo in combination with bortezomib and dexa-methasone.2,3 Data supporting efficacy in the current indication were from a pre-specified subgroup analysis of progres-sion-free survival involving 193 patients (94 in the panobinostat group and 99 in the control group) who had received prior treatment with both bortezomib and an immunomodulatory agent and a median of two prior therapies. In this subgroup, the median age was 60 years (range, 28–79 years), and 76% had received at least two previous lines of treatment.
In the subgroup analysis, median pro-gression-free survival was 10.6 months (95% confidence interval [CI] = 7.6–13.8 months) in the panobinostat group vs 5.8 months (95% CI = 4.4–7.1 months) in the control group (hazard ratio = 0.52, 95% CI = 0.36–0.76). Objective response rates were 58.5% vs 41.4%, including complete response in 8.5% vs 2.0% and near-complete response in 13.8% vs 7.1%.
How It WorksPanobinostat is a histone deacetylase
(HDAC) inhibitor. These enzymes cata-lyze the removal of acetyl groups from lysine residues of histones and some non-histone proteins. Inhibition of HDAC enzymatic activity results in increased acetylation of histone proteins, an epi-genetic alteration that results in relaxing
of chromatin and leads to transcriptional activation. In in vitro studies, panobino-stat resulted in accumulation of acetylated histones and other proteins and induced cell-cycle arrest or apoptosis of some transformed cells. Panobinostat treat-ment resulted in increased levels of acety-lated histones in mouse xenografts and exhibited greater cytotoxicity in tumor cells vs normal cells.
How It Is GivenThe recommended starting dose of
panobinostat is 20 mg once every other day for three doses per week in weeks 1 and 2 of each 21-day cycle for up to 8 cy-cles. In patients with clinical benefit who do not have unresolved severe or medi-cally significant toxicity, continuation of treatment for an additional 8 cycles should be considered (total duration of 16 cycles over 48 weeks). The recommended dose of bortezomib is 1.3 mg/m2, and the recommended dose of dexamethasone is 20 mg. In cycles 1 through 8, the 21-day cycle consists of panobinostat on days 1, 3, and 5; bortezomib on days 1 and 4; and dexamethasone on days 1, 2, 4, and 5 of weeks 1 and 2, with no treatment during week 3. In cycles 9 through 16, the 21-day cycle consists of panobinostat on days 1, 3, and 5; bortezomib on day 1; and dexa-methasone on days 1 and 2 of weeks 1 and 2, with no treatment during week 3.
Dose reduction of panobinostat for toxicity should occur in 5-mg increments; if a reduction to less than 10 mg three times per week is required, panobinostat should be discontinued. Panobinostat la-beling provides specific instructions for dose modification for panobinostat and bortezomib for thrombocytopenia, neu-tropenia, and diarrhea and for panobino-stat for anemia and nausea/vomiting.
The starting dose of panobinostat should be reduced to 15 mg in patients with mild hepatic impairment and to 10 mg in those with moderate hepatic impairment; panobinostat should be
avoided in patients with severe hepatic impairment. The starting dose of panobi-nostat should be reduced to 10 mg when it is coadministered with strong CYP3A inhibitors (eg, boceprevir, clarithromycin, conivaptan, indinavir, itraconazole, keto-conazole, and lopinavir-ritonavir).
Safety ProfileIn the total population of the phase III
trial, the most common adverse events of any grade occurring at an incidence of at least 5% higher in the panobinostat vs con-trol groups were diarrhea (68% vs 42%), fatigue (60% vs 42%), and nausea (36% vs 21%), and the most common grade 3 or 4 adverse events were diarrhea (25% vs 8%), fatigue (25% vs 12%), vomiting (7% vs 1%), and nausea (6% vs 1%). The most common grade 3 or 4 laboratory abnor-malities were thrombocytopenia (67% vs 31%), lymphopenia (53% vs 40%), neu-tropenia (34% vs 11%), leukopenia (23% vs 8%), hypophosphatemia (20% vs 12%), anemia (18% vs 19%), hypokalemia (18% vs 7%), and hyponatremia (13% vs 7%).
Serious adverse events occurred in 60% vs 42%, with the most frequent in the pan-obinostat group being pneumonia (18%), diarrhea (11%), thrombocytopenia (7%), fatigue (6%), and sepsis (6%). Adverse events led to discontinuation of panobino-stat in 36% of patients, with the most com-mon causes being diarrhea, fatigue, and pneumonia. Electrocardiographic changes, including new T-wave changes and ST-segment depression, occurred in 64% vs 42% of patients, with arrhythmia occurring in 12% vs 5%. The incidence of death not due to progressive disease was 7% vs 3.2%.
Panobinostat carries boxed warnings for severe diarrhea and severe and fatal car-diac ischemic events, severe arrhythmia, and electrocardiographic changes. It also carries warnings/precautions for hemor-rhage, including fatal and serious gastroin-testinal and pulmonary hemorrhage, hepa-totoxicity, and embryo-fetal toxicity.
Monitoring should include complete
blood cell count; electrocardiography; measurement of serum electrolytes, in-cluding potassium and magnesium; and liver function tests prior to and during therapy. Prior to treatment, the platelet count should be ≥ 100 × 109/L, and the neutrophil count should be ≥ 1.5 × 109/L. Patients should receive transfusion for low platelet counts as needed.
Complete blood cell count should be monitored weekly or more often as clini-cally indicated. QT correction formula Fridericia (QTcF) should be < 450 msec prior to initiation of treatment; treatment should be interrupted for QTcF increases to ≥ 480 msec, with electrolyte abnormal-ities being corrected. Treatment should be permanently discontinued if QT pro-longation does not resolve.
In the phase III trial, electrocardiogra-phy was performed at baseline and prior to initiation of each cycle for the first 8 cycles. Abnormal electrolyte values should be cor-rected before treatment. In the phase III tri-al, electrolyte monitoring was performed prior to the start of each cycle, at day 11 of cycles 1 through 8, and at the start of each cycle for cycles 9 through 16. The panobi-nostat dose should be adjusted if abnormal liver function tests are observed.
Women should be advised of potential hazard to the fetus and to avoid pregnancy while taking panobinostat. n
References 1. U.S. Food and Drug Administration:
Approved drugs. Panobinostat. Available at http://www.fda.gov/Drugs/Information-OnDrugs/ApprovedDrugs/ucm435339.htm. Accessed March 4, 2015.
2. Farydak (panobinostat) capsules pre-scribing information, Novartis Pharmaceu-ticals, February 2015. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/205353s000lbl.pdf. Accessed March 4, 2015.
3. San-Miguel JF, Hungria VT, Yoon SS, et al: Panobinostat plus bortezomib and dexa-methasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma. Lancet Oncol 15:1195-1206, 2014.
In the Clinic provides overviews of novel oncology agents, addressing indications, mechanisms, admin-istration recommendations, safety profiles, and other essential infor-mation needed for the appropriate clinical use of these drugs.
Hematology
OF NOTEPanobinostat carries boxed warn-ings for severe diarrhea and severe and fatal cardiac ischemic events, severe arrhythmia, and electrocar-diographic changes.
Panobinostat in Previously Treated Multiple Myeloma
■ Panobinostat (Farydak) was granted accelerated approval for use in combination with bortezomib (Velcade) and dexamethasone in the treatment of patients with multiple myeloma who have received at least two prior regimens, including bortezomib and an immunomodulatory agent.
■ The recommended starting dose of panobinostat is 20 mg once every other day for three doses per week in weeks 1 and 2 of each 21-day cycle for up to 8 cycles.

THE 1ST FDA-APPROVED ANTI–PD-1 THERAPY
KEYTRUDA:
TO HELP FIGHT TUMORS
In appropriate patients with advanced melanoma
ANTI–PD-1 EFFICACY
SELECTED SAFETY INFORMATION• Immune-mediated adverse reactions
occurred with KEYTRUDA, including pneumonitis, colitis, hepatitis, hypophysitis, nephritis, hyperthyroidism, and hypothyroidism. Based on the severity of the adverse reaction, KEYTRUDA should be withheld or discontinued and corticosteroids administered. For more information regarding immune-mediated adverse reactions, please read the additional Selected Safety Information on the next page.
Please see additional Selected Safety Information on the next page and the Brief Summary of Prescribing Information on the adjacent pages.
24% overall response rate in the 2 mg/kg arm (95% confidence interval [CI], 15–34; n=89); 1 complete response (1%) and 20 partial responses (23%).
86% of patients with an objective response (n=18/21) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
Releases programmed death receptor-1 (PD-1) pathway–mediated inhibition of the immune response.
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use
INDICATIONS AND USAGE
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Immune-Mediated Pneumonitis: Pneumonitis occurred in 12 (2.9%) of 411 melanoma patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to development of pneumonitis was 5 months (range 0.3 weeks–9.9 months). The median duration was 4.9 months (range 1 week–14.4 months). Five of eight patients with Grade 2 and the one patient with Grade 3 pneumonitis required initial treatment with high-dose systemic corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. The median initial dose of high-dose corticosteroid treatment was 63.4 mg/day of prednisone or equivalent with a median duration of treatment of 3 days (range 1–34) followed by a corticosteroid taper. Pneumonitis led to discontinuation of KEYTRUDA in 3 (0.7%) patients. Pneumonitis completely resolved in seven of the nine patients with Grade 2–3 pneumonitis.
Monitor patients for signs and symptoms of pneumonitis. Evaluate patients with suspected pneumonitis with radiographic imaging and administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for moderate (Grade 2) pneumonitis, and permanently discontinue KEYTRUDA for severe (Grade 3) or life-threatening (Grade 4) pneumonitis.
Immune-Mediated Colitis: Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset of colitis was 6.5 months (range 2.3–9.8). The median duration was 2.6 months (range 0.6 weeks–3.6 months). All three patients with Grade 2 or 3 colitis were treated with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) with a median initial dose of 70 mg/day of prednisone or equivalent; the median duration of initial treatment was 7 days (range 4–41), followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to colitis. All four patients with colitis experienced complete resolution of the event.
Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for moderate (Grade 2) or severe (Grade 3) colitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) colitis.
Immune-Mediated Hepatitis: Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The time to onset was 22 days for the case of Grade 4 hepatitis which lasted 1.1 months. The patient with Grade 4 hepatitis permanently discontinued KEYTRUDA and was treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) systemic corticosteroids followed by a corticosteroid taper. Both patients with hepatitis experienced complete resolution of the event.
Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
Immune-Mediated Hypophysitis: Hypophysitis occurred in 2 (0.5%) of 411 patients, consisting of one Grade 2 and one Grade 4 case (0.2% each), in patients receiving KEYTRUDA in Trial 1. The time to onset was 1.7 months for the patient with Grade 4 hypophysitis and 1.3 months for the patient with Grade 2 hypophysitis. Both patients were treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) corticosteroids followed by a corticosteroid taper and remained on a physiologic replacement dose.
Monitor for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for moderate (Grade 2) hypophysitis, withhold or discontinue KEYTRUDA for severe (Grade 3) hypophysitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hypophysitis.
Renal Failure and Immune-Mediated Nephritis: Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. The time to onset of autoimmune nephritis was 11.6 months after the first dose of KEYTRUDA (5 months after the last dose) and lasted 3.2 months; this patient did not have a biopsy. Acute interstitial nephritis was confirmed by renal biopsy in two patients with Grades 3–4 renal failure. All three patients fully recovered renal function with treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper.
Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for moderate (Grade 2) nephritis, and permanently discontinue KEYTRUDA for severe (Grade 3), or life-threatening (Grade 4) nephritis.
Immune-Mediated Hyperthyroidism and Hypothyroidism: Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset was 1.5 months (range 0.5–2.1). The median duration was 2.8 months (range 0.9 to 6.1). One of two patients with Grade 2 and the one patient with Grade 3 hyperthyroidism required initial treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to hyperthyroidism. All five patients with hyperthyroidism experienced complete resolution of the event.
Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The median time to onset of hypothyroidism was 3.5 months (range 0.7 weeks–19 months). All but two of the patients with hypothyroidism were
treated with long-term thyroid hormone replacement therapy. The other two patients only required short-term thyroid hormone replacement therapy. No patient received corticosteroids or discontinued KEYTRUDA for management of hypothyroidism.
Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders.
Administer corticosteroids for Grade 3 or greater hyperthyroidism, withhold KEYTRUDA for severe (Grade 3) hyperthyroidism, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
Other Immune-Mediated Adverse Reactions: Other clinically important immune-mediated adverse reactions can occur.
The following clinically significant, immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA in Trial 1: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, and adrenal insufficiency.
Across clinical studies with KEYTRUDA in approximately 2000 patients, the following additional clinically significant, immune-mediated adverse reactions were reported in less than 1% of patients: myasthenic syndrome, optic neuritis, and rhabdomyolysis.
For suspected immune-mediated adverse reactions, ensure adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
Embryofetal Toxicity: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for 4 months after the last dose of KEYTRUDA.
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail above.
• Immune-mediated pneumonitis.
• Immune-mediated colitis.
• Immune-mediated hepatitis.
• Immune-mediated hypophysitis.
• Renal failure and immune-mediated nephritis.
• Immune-mediated hyperthyroidism and hypothyroidism.
• Immune-mediated adverse reactions.
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS and PRECAUTIONS section reflect exposure to KEYTRUDA in Trial 1, an uncontrolled, open-label, multiple cohort trial in which 411 patients with unresectable or metastatic melanoma received KEYTRUDA at either 2 mg/kg every 3 weeks or 10 mg/kg every 2 or 3 weeks. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 24.6 months) with a median of 10 doses (range 1 to 51). The study population characteristics were: median age of 61 years (range 18–94), 39% age 65 years or older, 60% male, 97% white, 73% with M1c disease, 8% with brain metastases, 35% with elevated LDH, 54% with prior exposure to ipilimumab, and 47% with two or more prior systemic therapies for advanced or metastatic disease.
KEYTRUDA was discontinued for adverse reactions in 9% of the 411 patients. Adverse reactions, reported in at least two patients, that led to discontinuation of KEYTRUDA were: pneumonitis, renal failure, and pain. Serious adverse reactions occurred in 36% of patients receiving KEYTRUDA. The most frequent serious adverse drug reactions reported in 2% or more of patients in Trial 1 were renal failure, dyspnea, pneumonia, and cellulitis.
Table 1 presents adverse reactions identified from analyses of the 89 patients with unresectable or metastatic melanoma who received KEYTRUDA 2 mg/kg every three weeks in one cohort of Trial 1. Patients had documented disease progression following treatment with ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This cohort of Trial 1 excluded patients with severe immune-related toxicity related to ipilimumab, defined as any Grade 4 toxicity requiring treatment with corticosteroids or Grade 3 toxicity requiring corticosteroid treatment (greater than 10 mg/day prednisone or equivalent dose) for greater than 12 weeks; a medical condition that required systemic corticosteroids or other immunosuppressive medication; a history of pneumonitis or interstitial lung disease; or any active infection requiring therapy, including HIV or hepatitis B or C. Of the 89 patients in this cohort, the median age was 59 years (range 18–88), 33% were age 65 years or older, 53% were male, 98% were white, 44% had an elevated LDH, 84% had Stage M1c disease, 8% had brain metastases, and 70% received two or more prior therapies for advanced or metastatic disease. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 15.3 months) with a median of nine doses (range 1 to 23). Fifty-one percent of patients were exposed to KEYTRUDA for greater than 6 months and 21% for greater than 1 year.
KEYTRUDA was discontinued for adverse reactions in 6% of the 89 patients. The most common adverse reactions (reported in at least 20% of patients) were fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, arthralgia, and diarrhea.
32946_BUILD.indd 1 12/9/14 4:21 PM32946_01_p.indd 5 12/10/14 12:57 PM

(% of patients)
1%
23% Partial Response (PR) (n=20)
Complete Response (CR) (n=1) 86% Of responses were ongoing in patients who responded to KEYTRUDA
(n=18/21)
Study design: A multicenter, open-label, randomized, dose-comparative study cohort of the ongoing KEYNOTE-001 Phase 1b trial in patients with unresectable or metastatic melanoma and progression of disease. Key eligibility criteria included prior treatment with ipilimumab (2 or more doses at 3 mg/kg or higher) and a BRAF or MEK inhibitor, if BRAF V600 mutation–positive; and disease progression within 24 weeks following the last dose of ipilimumab. Patients were randomized to receive 2 mg/kg (n=89) or 10 mg/kg (n=84) of KEYTRUDA every 3 weeks until unacceptable toxicity or disease progression. The major efficacy outcome measures were confirmed overall response rate, as assessed by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and duration of response. Tumor response was assessed every 12 weeks.
KEYTRUDA provided ongoing responses in patients who responded
Scan here or visit keytruda.com to learn more.
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14 keytruda.com
Questions about access for KEYTRUDA? Call The Merck Access Program at 855-257-3932.
• Among the 21 patients with an objective response, 3 (14%) had progression of disease 2.8, 2.9, and 8.2 months after initial response.
• The remaining 18 patients (86%) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
• One additional patient developed 2 new asymptomatic lesions at the first tumor assessment concurrent with a 75% decrease in overall tumor burden.
—KEYTRUDA was continued and this reduction in tumor burden was durable for 5+ months.
• There were objective responses in patients with and without BRAF V600 mutation–positive melanoma.
• Similar overall response rate results were observed in the 10-mg/kg arm.
• Patients continued treatment with KEYTRUDA until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging.
For patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor
KEYTRUDA:DURABILITY OF RESPONSE24% overall response rate (complete response+partial response)
with single-agent KEYTRUDA (95% CI, 15–34)
• Pneumonitis occurred in 12 (2.9%) of 411 patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
• Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
• Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
• Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor patients for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
• Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
• Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
• Other clinically important immune-mediated adverse reactions can occur. The following clinically significant immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, adrenal insufficiency, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
SELECTED SAFETY INFORMATION
SELECTED SAFETY INFORMATION (CONTINUED)• For suspected immune-mediated adverse reactions, ensure
adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
• Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
• The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
• It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
• Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Please see the Brief Summary of the Prescribing Information on the adjacent pages.
Data for 2 mg/kg every 3 weeks (n=89)
32946_BUILD.indd 2 12/9/14 4:21 PM32946_01_p.indd 6 12/10/14 12:57 PM
(% of patients)
1%
23% Partial Response (PR) (n=20)
Complete Response (CR) (n=1) 86% Of responses were ongoing in patients who responded to KEYTRUDA
(n=18/21)
Study design: A multicenter, open-label, randomized, dose-comparative study cohort of the ongoing KEYNOTE-001 Phase 1b trial in patients with unresectable or metastatic melanoma and progression of disease. Key eligibility criteria included prior treatment with ipilimumab (2 or more doses at 3 mg/kg or higher) and a BRAF or MEK inhibitor, if BRAF V600 mutation–positive; and disease progression within 24 weeks following the last dose of ipilimumab. Patients were randomized to receive 2 mg/kg (n=89) or 10 mg/kg (n=84) of KEYTRUDA every 3 weeks until unacceptable toxicity or disease progression. The major efficacy outcome measures were confirmed overall response rate, as assessed by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and duration of response. Tumor response was assessed every 12 weeks.
KEYTRUDA provided ongoing responses in patients who responded
Scan here or visit keytruda.com to learn more.
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14 keytruda.com
Questions about access for KEYTRUDA? Call The Merck Access Program at 855-257-3932.
• Among the 21 patients with an objective response, 3 (14%) had progression of disease 2.8, 2.9, and 8.2 months after initial response.
• The remaining 18 patients (86%) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
• One additional patient developed 2 new asymptomatic lesions at the first tumor assessment concurrent with a 75% decrease in overall tumor burden.
—KEYTRUDA was continued and this reduction in tumor burden was durable for 5+ months.
• There were objective responses in patients with and without BRAF V600 mutation–positive melanoma.
• Similar overall response rate results were observed in the 10-mg/kg arm.
• Patients continued treatment with KEYTRUDA until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging.
For patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor
KEYTRUDA:DURABILITY OF RESPONSE24% overall response rate (complete response+partial response)
with single-agent KEYTRUDA (95% CI, 15–34)
• Pneumonitis occurred in 12 (2.9%) of 411 patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
• Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
• Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
• Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor patients for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
• Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
• Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
• Other clinically important immune-mediated adverse reactions can occur. The following clinically significant immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, adrenal insufficiency, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
SELECTED SAFETY INFORMATION
SELECTED SAFETY INFORMATION (CONTINUED)• For suspected immune-mediated adverse reactions, ensure
adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
• Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
• The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
• It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
• Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Please see the Brief Summary of the Prescribing Information on the adjacent pages.
Data for 2 mg/kg every 3 weeks (n=89)
32946_BUILD.indd 2 12/9/14 4:21 PM32946_01_p.indd 2 12/10/14 12:57 PM

(% of patients)
1%
23% Partial Response (PR) (n=20)
Complete Response (CR) (n=1) 86% Of responses were ongoing in patients who responded to KEYTRUDA
(n=18/21)
Study design: A multicenter, open-label, randomized, dose-comparative study cohort of the ongoing KEYNOTE-001 Phase 1b trial in patients with unresectable or metastatic melanoma and progression of disease. Key eligibility criteria included prior treatment with ipilimumab (2 or more doses at 3 mg/kg or higher) and a BRAF or MEK inhibitor, if BRAF V600 mutation–positive; and disease progression within 24 weeks following the last dose of ipilimumab. Patients were randomized to receive 2 mg/kg (n=89) or 10 mg/kg (n=84) of KEYTRUDA every 3 weeks until unacceptable toxicity or disease progression. The major efficacy outcome measures were confirmed overall response rate, as assessed by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and duration of response. Tumor response was assessed every 12 weeks.
KEYTRUDA provided ongoing responses in patients who responded
Scan here or visit keytruda.com to learn more.
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14 keytruda.com
Questions about access for KEYTRUDA? Call The Merck Access Program at 855-257-3932.
• Among the 21 patients with an objective response, 3 (14%) had progression of disease 2.8, 2.9, and 8.2 months after initial response.
• The remaining 18 patients (86%) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
• One additional patient developed 2 new asymptomatic lesions at the first tumor assessment concurrent with a 75% decrease in overall tumor burden.
—KEYTRUDA was continued and this reduction in tumor burden was durable for 5+ months.
• There were objective responses in patients with and without BRAF V600 mutation–positive melanoma.
• Similar overall response rate results were observed in the 10-mg/kg arm.
• Patients continued treatment with KEYTRUDA until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging.
For patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor
KEYTRUDA:DURABILITY OF RESPONSE24% overall response rate (complete response+partial response)
with single-agent KEYTRUDA (95% CI, 15–34)
• Pneumonitis occurred in 12 (2.9%) of 411 patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
• Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
• Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
• Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor patients for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
• Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
• Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
• Other clinically important immune-mediated adverse reactions can occur. The following clinically significant immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, adrenal insufficiency, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
SELECTED SAFETY INFORMATION
SELECTED SAFETY INFORMATION (CONTINUED)• For suspected immune-mediated adverse reactions, ensure
adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
• Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
• The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
• It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
• Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Please see the Brief Summary of the Prescribing Information on the adjacent pages.
Data for 2 mg/kg every 3 weeks (n=89)
32946_BUILD.indd 2 12/9/14 4:21 PM32946_01_p.indd 6 12/10/14 12:57 PM
(% of patients)
1%
23% Partial Response (PR) (n=20)
Complete Response (CR) (n=1) 86% Of responses were ongoing in patients who responded to KEYTRUDA
(n=18/21)
Study design: A multicenter, open-label, randomized, dose-comparative study cohort of the ongoing KEYNOTE-001 Phase 1b trial in patients with unresectable or metastatic melanoma and progression of disease. Key eligibility criteria included prior treatment with ipilimumab (2 or more doses at 3 mg/kg or higher) and a BRAF or MEK inhibitor, if BRAF V600 mutation–positive; and disease progression within 24 weeks following the last dose of ipilimumab. Patients were randomized to receive 2 mg/kg (n=89) or 10 mg/kg (n=84) of KEYTRUDA every 3 weeks until unacceptable toxicity or disease progression. The major efficacy outcome measures were confirmed overall response rate, as assessed by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and duration of response. Tumor response was assessed every 12 weeks.
KEYTRUDA provided ongoing responses in patients who responded
Scan here or visit keytruda.com to learn more.
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14 keytruda.com
Questions about access for KEYTRUDA? Call The Merck Access Program at 855-257-3932.
• Among the 21 patients with an objective response, 3 (14%) had progression of disease 2.8, 2.9, and 8.2 months after initial response.
• The remaining 18 patients (86%) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
• One additional patient developed 2 new asymptomatic lesions at the first tumor assessment concurrent with a 75% decrease in overall tumor burden.
—KEYTRUDA was continued and this reduction in tumor burden was durable for 5+ months.
• There were objective responses in patients with and without BRAF V600 mutation–positive melanoma.
• Similar overall response rate results were observed in the 10-mg/kg arm.
• Patients continued treatment with KEYTRUDA until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging.
For patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor
KEYTRUDA:DURABILITY OF RESPONSE24% overall response rate (complete response+partial response)
with single-agent KEYTRUDA (95% CI, 15–34)
• Pneumonitis occurred in 12 (2.9%) of 411 patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
• Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
• Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
• Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor patients for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
• Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
• Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
• Other clinically important immune-mediated adverse reactions can occur. The following clinically significant immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, adrenal insufficiency, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
SELECTED SAFETY INFORMATION
SELECTED SAFETY INFORMATION (CONTINUED)• For suspected immune-mediated adverse reactions, ensure
adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
• Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
• The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
• It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
• Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Please see the Brief Summary of the Prescribing Information on the adjacent pages.
Data for 2 mg/kg every 3 weeks (n=89)
32946_BUILD.indd 2 12/9/14 4:21 PM32946_01_p.indd 2 12/10/14 12:57 PM

THE 1ST FDA-APPROVED ANTI–PD-1 THERAPY
KEYTRUDA:
TO HELP FIGHT TUMORS
In appropriate patients with advanced melanoma
ANTI–PD-1 EFFICACY
SELECTED SAFETY INFORMATION• Immune-mediated adverse reactions
occurred with KEYTRUDA, including pneumonitis, colitis, hepatitis, hypophysitis, nephritis, hyperthyroidism, and hypothyroidism. Based on the severity of the adverse reaction, KEYTRUDA should be withheld or discontinued and corticosteroids administered. For more information regarding immune-mediated adverse reactions, please read the additional Selected Safety Information on the next page.
Please see additional Selected Safety Information on the next page and the Brief Summary of Prescribing Information on the adjacent pages.
24% overall response rate in the 2 mg/kg arm (95% confidence interval [CI], 15–34; n=89); 1 complete response (1%) and 20 partial responses (23%).
86% of patients with an objective response (n=18/21) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
Releases programmed death receptor-1 (PD-1) pathway–mediated inhibition of the immune response.
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use
INDICATIONS AND USAGE
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Immune-Mediated Pneumonitis: Pneumonitis occurred in 12 (2.9%) of 411 melanoma patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to development of pneumonitis was 5 months (range 0.3 weeks–9.9 months). The median duration was 4.9 months (range 1 week–14.4 months). Five of eight patients with Grade 2 and the one patient with Grade 3 pneumonitis required initial treatment with high-dose systemic corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. The median initial dose of high-dose corticosteroid treatment was 63.4 mg/day of prednisone or equivalent with a median duration of treatment of 3 days (range 1–34) followed by a corticosteroid taper. Pneumonitis led to discontinuation of KEYTRUDA in 3 (0.7%) patients. Pneumonitis completely resolved in seven of the nine patients with Grade 2–3 pneumonitis.
Monitor patients for signs and symptoms of pneumonitis. Evaluate patients with suspected pneumonitis with radiographic imaging and administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for moderate (Grade 2) pneumonitis, and permanently discontinue KEYTRUDA for severe (Grade 3) or life-threatening (Grade 4) pneumonitis.
Immune-Mediated Colitis: Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset of colitis was 6.5 months (range 2.3–9.8). The median duration was 2.6 months (range 0.6 weeks–3.6 months). All three patients with Grade 2 or 3 colitis were treated with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) with a median initial dose of 70 mg/day of prednisone or equivalent; the median duration of initial treatment was 7 days (range 4–41), followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to colitis. All four patients with colitis experienced complete resolution of the event.
Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for moderate (Grade 2) or severe (Grade 3) colitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) colitis.
Immune-Mediated Hepatitis: Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The time to onset was 22 days for the case of Grade 4 hepatitis which lasted 1.1 months. The patient with Grade 4 hepatitis permanently discontinued KEYTRUDA and was treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) systemic corticosteroids followed by a corticosteroid taper. Both patients with hepatitis experienced complete resolution of the event.
Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
Immune-Mediated Hypophysitis: Hypophysitis occurred in 2 (0.5%) of 411 patients, consisting of one Grade 2 and one Grade 4 case (0.2% each), in patients receiving KEYTRUDA in Trial 1. The time to onset was 1.7 months for the patient with Grade 4 hypophysitis and 1.3 months for the patient with Grade 2 hypophysitis. Both patients were treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) corticosteroids followed by a corticosteroid taper and remained on a physiologic replacement dose.
Monitor for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for moderate (Grade 2) hypophysitis, withhold or discontinue KEYTRUDA for severe (Grade 3) hypophysitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hypophysitis.
Renal Failure and Immune-Mediated Nephritis: Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. The time to onset of autoimmune nephritis was 11.6 months after the first dose of KEYTRUDA (5 months after the last dose) and lasted 3.2 months; this patient did not have a biopsy. Acute interstitial nephritis was confirmed by renal biopsy in two patients with Grades 3–4 renal failure. All three patients fully recovered renal function with treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper.
Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for moderate (Grade 2) nephritis, and permanently discontinue KEYTRUDA for severe (Grade 3), or life-threatening (Grade 4) nephritis.
Immune-Mediated Hyperthyroidism and Hypothyroidism: Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset was 1.5 months (range 0.5–2.1). The median duration was 2.8 months (range 0.9 to 6.1). One of two patients with Grade 2 and the one patient with Grade 3 hyperthyroidism required initial treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to hyperthyroidism. All five patients with hyperthyroidism experienced complete resolution of the event.
Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The median time to onset of hypothyroidism was 3.5 months (range 0.7 weeks–19 months). All but two of the patients with hypothyroidism were
treated with long-term thyroid hormone replacement therapy. The other two patients only required short-term thyroid hormone replacement therapy. No patient received corticosteroids or discontinued KEYTRUDA for management of hypothyroidism.
Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders.
Administer corticosteroids for Grade 3 or greater hyperthyroidism, withhold KEYTRUDA for severe (Grade 3) hyperthyroidism, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
Other Immune-Mediated Adverse Reactions: Other clinically important immune-mediated adverse reactions can occur.
The following clinically significant, immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA in Trial 1: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, and adrenal insufficiency.
Across clinical studies with KEYTRUDA in approximately 2000 patients, the following additional clinically significant, immune-mediated adverse reactions were reported in less than 1% of patients: myasthenic syndrome, optic neuritis, and rhabdomyolysis.
For suspected immune-mediated adverse reactions, ensure adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
Embryofetal Toxicity: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for 4 months after the last dose of KEYTRUDA.
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail above.
• Immune-mediated pneumonitis.
• Immune-mediated colitis.
• Immune-mediated hepatitis.
• Immune-mediated hypophysitis.
• Renal failure and immune-mediated nephritis.
• Immune-mediated hyperthyroidism and hypothyroidism.
• Immune-mediated adverse reactions.
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS and PRECAUTIONS section reflect exposure to KEYTRUDA in Trial 1, an uncontrolled, open-label, multiple cohort trial in which 411 patients with unresectable or metastatic melanoma received KEYTRUDA at either 2 mg/kg every 3 weeks or 10 mg/kg every 2 or 3 weeks. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 24.6 months) with a median of 10 doses (range 1 to 51). The study population characteristics were: median age of 61 years (range 18–94), 39% age 65 years or older, 60% male, 97% white, 73% with M1c disease, 8% with brain metastases, 35% with elevated LDH, 54% with prior exposure to ipilimumab, and 47% with two or more prior systemic therapies for advanced or metastatic disease.
KEYTRUDA was discontinued for adverse reactions in 9% of the 411 patients. Adverse reactions, reported in at least two patients, that led to discontinuation of KEYTRUDA were: pneumonitis, renal failure, and pain. Serious adverse reactions occurred in 36% of patients receiving KEYTRUDA. The most frequent serious adverse drug reactions reported in 2% or more of patients in Trial 1 were renal failure, dyspnea, pneumonia, and cellulitis.
Table 1 presents adverse reactions identified from analyses of the 89 patients with unresectable or metastatic melanoma who received KEYTRUDA 2 mg/kg every three weeks in one cohort of Trial 1. Patients had documented disease progression following treatment with ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This cohort of Trial 1 excluded patients with severe immune-related toxicity related to ipilimumab, defined as any Grade 4 toxicity requiring treatment with corticosteroids or Grade 3 toxicity requiring corticosteroid treatment (greater than 10 mg/day prednisone or equivalent dose) for greater than 12 weeks; a medical condition that required systemic corticosteroids or other immunosuppressive medication; a history of pneumonitis or interstitial lung disease; or any active infection requiring therapy, including HIV or hepatitis B or C. Of the 89 patients in this cohort, the median age was 59 years (range 18–88), 33% were age 65 years or older, 53% were male, 98% were white, 44% had an elevated LDH, 84% had Stage M1c disease, 8% had brain metastases, and 70% received two or more prior therapies for advanced or metastatic disease. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 15.3 months) with a median of nine doses (range 1 to 23). Fifty-one percent of patients were exposed to KEYTRUDA for greater than 6 months and 21% for greater than 1 year.
KEYTRUDA was discontinued for adverse reactions in 6% of the 89 patients. The most common adverse reactions (reported in at least 20% of patients) were fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, arthralgia, and diarrhea.
32946_BUILD.indd 1 12/9/14 4:21 PM32946_01_p.indd 1 12/10/14 12:57 PM
Immunogenicity: As with all therapeutic proteins, there is the potential for immunogenicity. Because trough levels of pembrolizumab interfere with the electrochemiluminescent (ECL) assay results, a subset analysis was performed in the patients with a concentration of pembrolizumab below the drug tolerance level of the anti-product antibody assay. In this analysis, none of the 97 patients who were treated with 2 mg/kg every 3 weeks tested positive for treatment-emergent anti-pembrolizumab antibodies.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to KEYTRUDA with the incidences of antibodies to other products may be misleading.
Table 2: Laboratory Abnormalities Increased from Baseline in ≥20% of Patients with Unresectable or Metastatic Melanoma
KEYTRUDA 2 mg/kg every 3 weeks
N=89
Laboratory Test All Grades (%)
Grades 3–4 (%)
ChemistryHyperglycemia 40 2*Hyponatremia 35 9Hypoalbuminemia 34 0Hypertriglyceridemia 25 0Increased Aspartate Aminotransferase 24 2*Hypocalcemia 24 1
HematologyAnemia 55 8*
* Grade 4 abnormalities in this table limited to hyperglycemia, increased aspartate aminotransferase, and anemia (one patient each).
DRUG INTERACTIONS
No formal pharmacokinetic drug interaction studies have been conducted with KEYTRUDA.
USE IN SPECIFIC POPULATIONS
Pregnancy: Pregnancy Category D.
Risk Summary : Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus.
Animal Data: Animal reproduction studies have not been conducted with KEYTRUDA to evaluate its effect on reproduction and fetal development, but an assessment of the effects on reproduction was provided. A central function of the PD-1/PD-L1 pathway is to preserve pregnancy by maintaining maternal immune tolerance to the fetus. Blockade of PD-L1 signaling has been shown in murine models of pregnancy to disrupt tolerance to the fetus and to result in an increase in fetal loss; therefore, potential risks of administering KEYTRUDA during pregnancy include increased rates of abortion or stillbirth. As reported in the literature, there were no malformations related to the blockade of PD-1 signaling in the offspring of these animals; however, immune-mediated disorders occurred in PD-1 knockout mice.
Human IgG4 (immunoglobulins) are known to cross the placenta; therefore, pembrolizumab has the potential to be transmitted from the mother to the developing fetus. Based on its mechanism of action, fetal exposure to pembrolizumab may increase the risk of developing immune-mediated disorders or of altering the normal immune response.
Nursing Mothers: It is not known whether KEYTRUDA is excreted in human milk. No studies have been conducted to assess the impact of KEYTRUDA on milk production or its presence in breast milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
Pediatric Use: Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Geriatric Use: Of the 411 patients treated with KEYTRUDA, 39% were 65 years and over. No overall differences in safety or efficacy were reported between elderly patients and younger patients.
Renal Impairment: Based on a population pharmacokinetic analysis, no dose adjustment is needed for patients with renal impairment.
Hepatic Impairment: Based on a population pharmacokinetic analysis, no dose adjustment is needed for patients with mild hepatic impairment [total bilirubin (TB) less than or equal to ULN and AST greater than ULN or TB greater than 1 to 1.5 times ULN and any AST]. KEYTRUDA has not been studied in patients with moderate (TB greater than 1.5 to 3 times ULN and any AST) or severe (TB greater than 3 times ULN and any AST) hepatic impairment.
Females and Males of Reproductive Potential: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for at least 4 months following the last dose of pembrolizumab.
OVERDOSAGE
There is no information on overdosage with KEYTRUDA.
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
• Inform patients of the risk of immune-mediated adverse reactions that may require corticosteroid treatment and interruption or discontinuation of KEYTRUDA, including:
— Pneumonitis: Advise patients to contact their healthcare provider immediately for new or worsening cough, chest pain, or shortness of breath.
— Colitis: Advise patients to contact their healthcare provider immediately for diarrhea or severe abdominal pain.
— Hepatitis: Advise patients to contact their healthcare provider immediately for jaundice, severe nausea or vomiting, or easy bruising or bleeding.
— Hypophysitis: Advise patients to contact their healthcare provider immediately for persistent or unusual headache, extreme weakness, dizziness or fainting, or vision changes.
— Nephritis: Advise patients to contact their healthcare provider immediately for signs or symptoms of nephritis.
— Hyperthyroidism and Hypothyroidism: Advise patients to contact their healthcare provider immediately for signs or symptoms of hyperthyroidism and hypothyroidism.
• Advise patients of the importance of keeping scheduled appointments for blood work or other laboratory tests.
• Advise women that KEYTRUDA may cause fetal harm. Instruct women of reproductive potential to use highly effective contraception during and for 4 months after the last dose of KEYTRUDA.
• Advise nursing mothers not to breastfeed while taking KEYTRUDA.
For more detailed information, please read the Prescribing Information.
uspi-mk3475-iv-1409r000
Revised: 09/2014
Copyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved.
ONCO-1116177-0000 11/14
Table 1: Adverse Reactions in ≥10% of Patients with Unresectable or Metastatic Melanoma
KEYTRUDA 2 mg/kg every 3 weeks
N=89
Adverse Reaction All Grades (%)
Grade 3* (%)
General Disorders and Administration Site ConditionsFatigue 47 7Peripheral edema 17 1Chills 14 0Pyrexia 11 0
Gastrointestinal DisordersNausea 30 0Constipation 21 0Diarrhea 20 0Vomiting 16 0Abdominal pain 12 0
Respiratory, Thoracic And Mediastinal DisordersCough 30 1Dyspnea 18 2
Skin And Subcutaneous Tissue DisordersPruritus 30 0Rash 29 0Vitiligo 11 0
Metabolism and Nutrition DisordersDecreased appetite 26 0
Musculoskeletal and Connective Tissue DisordersArthralgia 20 0Pain in extremity 18 1Myalgia 14 1Back pain 12 1
Nervous System DisordersHeadache 16 0Dizziness 11 0
Blood and Lymphatic System DisordersAnemia 14 5
Psychiatric DisordersInsomnia 14 0
Infections and InfestationsUpper respiratory tract infection 11 1
*There were no Grade 5 adverse reactions reported. Of the ≥10% adverse reactions, none was reported as Grade 4.
Other clinically important adverse reactions observed in up to 10% of patients treated with KEYTRUDA were: Infections and infestations: sepsis
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use (continued)
32946_BUILD.indd 3 12/9/14 4:21 PM32946_01_p.indd 4 12/10/14 12:57 PM

THE 1ST FDA-APPROVED ANTI–PD-1 THERAPY
KEYTRUDA:
TO HELP FIGHT TUMORS
In appropriate patients with advanced melanoma
ANTI–PD-1 EFFICACY
SELECTED SAFETY INFORMATION• Immune-mediated adverse reactions
occurred with KEYTRUDA, including pneumonitis, colitis, hepatitis, hypophysitis, nephritis, hyperthyroidism, and hypothyroidism. Based on the severity of the adverse reaction, KEYTRUDA should be withheld or discontinued and corticosteroids administered. For more information regarding immune-mediated adverse reactions, please read the additional Selected Safety Information on the next page.
Please see additional Selected Safety Information on the next page and the Brief Summary of Prescribing Information on the adjacent pages.
24% overall response rate in the 2 mg/kg arm (95% confidence interval [CI], 15–34; n=89); 1 complete response (1%) and 20 partial responses (23%).
86% of patients with an objective response (n=18/21) had ongoing responses with durations ranging from 1.4+ to 8.5+ months, which included 8 patients with ongoing responses of 6 months or longer.
Releases programmed death receptor-1 (PD-1) pathway–mediated inhibition of the immune response.
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use
INDICATIONS AND USAGE
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Immune-Mediated Pneumonitis: Pneumonitis occurred in 12 (2.9%) of 411 melanoma patients, including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to development of pneumonitis was 5 months (range 0.3 weeks–9.9 months). The median duration was 4.9 months (range 1 week–14.4 months). Five of eight patients with Grade 2 and the one patient with Grade 3 pneumonitis required initial treatment with high-dose systemic corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. The median initial dose of high-dose corticosteroid treatment was 63.4 mg/day of prednisone or equivalent with a median duration of treatment of 3 days (range 1–34) followed by a corticosteroid taper. Pneumonitis led to discontinuation of KEYTRUDA in 3 (0.7%) patients. Pneumonitis completely resolved in seven of the nine patients with Grade 2–3 pneumonitis.
Monitor patients for signs and symptoms of pneumonitis. Evaluate patients with suspected pneumonitis with radiographic imaging and administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for moderate (Grade 2) pneumonitis, and permanently discontinue KEYTRUDA for severe (Grade 3) or life-threatening (Grade 4) pneumonitis.
Immune-Mediated Colitis: Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset of colitis was 6.5 months (range 2.3–9.8). The median duration was 2.6 months (range 0.6 weeks–3.6 months). All three patients with Grade 2 or 3 colitis were treated with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) with a median initial dose of 70 mg/day of prednisone or equivalent; the median duration of initial treatment was 7 days (range 4–41), followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to colitis. All four patients with colitis experienced complete resolution of the event.
Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for moderate (Grade 2) or severe (Grade 3) colitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) colitis.
Immune-Mediated Hepatitis: Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The time to onset was 22 days for the case of Grade 4 hepatitis which lasted 1.1 months. The patient with Grade 4 hepatitis permanently discontinued KEYTRUDA and was treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) systemic corticosteroids followed by a corticosteroid taper. Both patients with hepatitis experienced complete resolution of the event.
Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
Immune-Mediated Hypophysitis: Hypophysitis occurred in 2 (0.5%) of 411 patients, consisting of one Grade 2 and one Grade 4 case (0.2% each), in patients receiving KEYTRUDA in Trial 1. The time to onset was 1.7 months for the patient with Grade 4 hypophysitis and 1.3 months for the patient with Grade 2 hypophysitis. Both patients were treated with high-dose (greater than or equal to 40 mg prednisone or equivalent per day) corticosteroids followed by a corticosteroid taper and remained on a physiologic replacement dose.
Monitor for signs and symptoms of hypophysitis. Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for moderate (Grade 2) hypophysitis, withhold or discontinue KEYTRUDA for severe (Grade 3) hypophysitis, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hypophysitis.
Renal Failure and Immune-Mediated Nephritis: Nephritis occurred in 3 (0.7%) patients, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. The time to onset of autoimmune nephritis was 11.6 months after the first dose of KEYTRUDA (5 months after the last dose) and lasted 3.2 months; this patient did not have a biopsy. Acute interstitial nephritis was confirmed by renal biopsy in two patients with Grades 3–4 renal failure. All three patients fully recovered renal function with treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper.
Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for moderate (Grade 2) nephritis, and permanently discontinue KEYTRUDA for severe (Grade 3), or life-threatening (Grade 4) nephritis.
Immune-Mediated Hyperthyroidism and Hypothyroidism: Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA in Trial 1. The median time to onset was 1.5 months (range 0.5–2.1). The median duration was 2.8 months (range 0.9 to 6.1). One of two patients with Grade 2 and the one patient with Grade 3 hyperthyroidism required initial treatment with high-dose corticosteroids (greater than or equal to 40 mg prednisone or equivalent per day) followed by a corticosteroid taper. One patient (0.2%) required permanent discontinuation of KEYTRUDA due to hyperthyroidism. All five patients with hyperthyroidism experienced complete resolution of the event.
Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA in Trial 1. The median time to onset of hypothyroidism was 3.5 months (range 0.7 weeks–19 months). All but two of the patients with hypothyroidism were
treated with long-term thyroid hormone replacement therapy. The other two patients only required short-term thyroid hormone replacement therapy. No patient received corticosteroids or discontinued KEYTRUDA for management of hypothyroidism.
Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders.
Administer corticosteroids for Grade 3 or greater hyperthyroidism, withhold KEYTRUDA for severe (Grade 3) hyperthyroidism, and permanently discontinue KEYTRUDA for life-threatening (Grade 4) hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
Other Immune-Mediated Adverse Reactions: Other clinically important immune-mediated adverse reactions can occur.
The following clinically significant, immune-mediated adverse reactions occurred in less than 1% of patients treated with KEYTRUDA in Trial 1: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, and adrenal insufficiency.
Across clinical studies with KEYTRUDA in approximately 2000 patients, the following additional clinically significant, immune-mediated adverse reactions were reported in less than 1% of patients: myasthenic syndrome, optic neuritis, and rhabdomyolysis.
For suspected immune-mediated adverse reactions, ensure adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
Embryofetal Toxicity: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for 4 months after the last dose of KEYTRUDA.
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail above.
• Immune-mediated pneumonitis.
• Immune-mediated colitis.
• Immune-mediated hepatitis.
• Immune-mediated hypophysitis.
• Renal failure and immune-mediated nephritis.
• Immune-mediated hyperthyroidism and hypothyroidism.
• Immune-mediated adverse reactions.
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS and PRECAUTIONS section reflect exposure to KEYTRUDA in Trial 1, an uncontrolled, open-label, multiple cohort trial in which 411 patients with unresectable or metastatic melanoma received KEYTRUDA at either 2 mg/kg every 3 weeks or 10 mg/kg every 2 or 3 weeks. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 24.6 months) with a median of 10 doses (range 1 to 51). The study population characteristics were: median age of 61 years (range 18–94), 39% age 65 years or older, 60% male, 97% white, 73% with M1c disease, 8% with brain metastases, 35% with elevated LDH, 54% with prior exposure to ipilimumab, and 47% with two or more prior systemic therapies for advanced or metastatic disease.
KEYTRUDA was discontinued for adverse reactions in 9% of the 411 patients. Adverse reactions, reported in at least two patients, that led to discontinuation of KEYTRUDA were: pneumonitis, renal failure, and pain. Serious adverse reactions occurred in 36% of patients receiving KEYTRUDA. The most frequent serious adverse drug reactions reported in 2% or more of patients in Trial 1 were renal failure, dyspnea, pneumonia, and cellulitis.
Table 1 presents adverse reactions identified from analyses of the 89 patients with unresectable or metastatic melanoma who received KEYTRUDA 2 mg/kg every three weeks in one cohort of Trial 1. Patients had documented disease progression following treatment with ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This cohort of Trial 1 excluded patients with severe immune-related toxicity related to ipilimumab, defined as any Grade 4 toxicity requiring treatment with corticosteroids or Grade 3 toxicity requiring corticosteroid treatment (greater than 10 mg/day prednisone or equivalent dose) for greater than 12 weeks; a medical condition that required systemic corticosteroids or other immunosuppressive medication; a history of pneumonitis or interstitial lung disease; or any active infection requiring therapy, including HIV or hepatitis B or C. Of the 89 patients in this cohort, the median age was 59 years (range 18–88), 33% were age 65 years or older, 53% were male, 98% were white, 44% had an elevated LDH, 84% had Stage M1c disease, 8% had brain metastases, and 70% received two or more prior therapies for advanced or metastatic disease. The median duration of exposure to KEYTRUDA was 6.2 months (range 1 day to 15.3 months) with a median of nine doses (range 1 to 23). Fifty-one percent of patients were exposed to KEYTRUDA for greater than 6 months and 21% for greater than 1 year.
KEYTRUDA was discontinued for adverse reactions in 6% of the 89 patients. The most common adverse reactions (reported in at least 20% of patients) were fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, arthralgia, and diarrhea.
32946_BUILD.indd 1 12/9/14 4:21 PM32946_01_p.indd 1 12/10/14 12:57 PM
Immunogenicity: As with all therapeutic proteins, there is the potential for immunogenicity. Because trough levels of pembrolizumab interfere with the electrochemiluminescent (ECL) assay results, a subset analysis was performed in the patients with a concentration of pembrolizumab below the drug tolerance level of the anti-product antibody assay. In this analysis, none of the 97 patients who were treated with 2 mg/kg every 3 weeks tested positive for treatment-emergent anti-pembrolizumab antibodies.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to KEYTRUDA with the incidences of antibodies to other products may be misleading.
Table 2: Laboratory Abnormalities Increased from Baseline in ≥20% of Patients with Unresectable or Metastatic Melanoma
KEYTRUDA 2 mg/kg every 3 weeks
N=89
Laboratory Test All Grades (%)
Grades 3–4 (%)
ChemistryHyperglycemia 40 2*Hyponatremia 35 9Hypoalbuminemia 34 0Hypertriglyceridemia 25 0Increased Aspartate Aminotransferase 24 2*Hypocalcemia 24 1
HematologyAnemia 55 8*
* Grade 4 abnormalities in this table limited to hyperglycemia, increased aspartate aminotransferase, and anemia (one patient each).
DRUG INTERACTIONS
No formal pharmacokinetic drug interaction studies have been conducted with KEYTRUDA.
USE IN SPECIFIC POPULATIONS
Pregnancy: Pregnancy Category D.
Risk Summary : Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PDL-1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus.
Animal Data: Animal reproduction studies have not been conducted with KEYTRUDA to evaluate its effect on reproduction and fetal development, but an assessment of the effects on reproduction was provided. A central function of the PD-1/PD-L1 pathway is to preserve pregnancy by maintaining maternal immune tolerance to the fetus. Blockade of PD-L1 signaling has been shown in murine models of pregnancy to disrupt tolerance to the fetus and to result in an increase in fetal loss; therefore, potential risks of administering KEYTRUDA during pregnancy include increased rates of abortion or stillbirth. As reported in the literature, there were no malformations related to the blockade of PD-1 signaling in the offspring of these animals; however, immune-mediated disorders occurred in PD-1 knockout mice.
Human IgG4 (immunoglobulins) are known to cross the placenta; therefore, pembrolizumab has the potential to be transmitted from the mother to the developing fetus. Based on its mechanism of action, fetal exposure to pembrolizumab may increase the risk of developing immune-mediated disorders or of altering the normal immune response.
Nursing Mothers: It is not known whether KEYTRUDA is excreted in human milk. No studies have been conducted to assess the impact of KEYTRUDA on milk production or its presence in breast milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA.
Pediatric Use: Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
Geriatric Use: Of the 411 patients treated with KEYTRUDA, 39% were 65 years and over. No overall differences in safety or efficacy were reported between elderly patients and younger patients.
Renal Impairment: Based on a population pharmacokinetic analysis, no dose adjustment is needed for patients with renal impairment.
Hepatic Impairment: Based on a population pharmacokinetic analysis, no dose adjustment is needed for patients with mild hepatic impairment [total bilirubin (TB) less than or equal to ULN and AST greater than ULN or TB greater than 1 to 1.5 times ULN and any AST]. KEYTRUDA has not been studied in patients with moderate (TB greater than 1.5 to 3 times ULN and any AST) or severe (TB greater than 3 times ULN and any AST) hepatic impairment.
Females and Males of Reproductive Potential: Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use highly effective contraception during treatment with KEYTRUDA and for at least 4 months following the last dose of pembrolizumab.
OVERDOSAGE
There is no information on overdosage with KEYTRUDA.
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
• Inform patients of the risk of immune-mediated adverse reactions that may require corticosteroid treatment and interruption or discontinuation of KEYTRUDA, including:
— Pneumonitis: Advise patients to contact their healthcare provider immediately for new or worsening cough, chest pain, or shortness of breath.
— Colitis: Advise patients to contact their healthcare provider immediately for diarrhea or severe abdominal pain.
— Hepatitis: Advise patients to contact their healthcare provider immediately for jaundice, severe nausea or vomiting, or easy bruising or bleeding.
— Hypophysitis: Advise patients to contact their healthcare provider immediately for persistent or unusual headache, extreme weakness, dizziness or fainting, or vision changes.
— Nephritis: Advise patients to contact their healthcare provider immediately for signs or symptoms of nephritis.
— Hyperthyroidism and Hypothyroidism: Advise patients to contact their healthcare provider immediately for signs or symptoms of hyperthyroidism and hypothyroidism.
• Advise patients of the importance of keeping scheduled appointments for blood work or other laboratory tests.
• Advise women that KEYTRUDA may cause fetal harm. Instruct women of reproductive potential to use highly effective contraception during and for 4 months after the last dose of KEYTRUDA.
• Advise nursing mothers not to breastfeed while taking KEYTRUDA.
For more detailed information, please read the Prescribing Information.
uspi-mk3475-iv-1409r000
Revised: 09/2014
Copyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved.
ONCO-1116177-0000 11/14
Table 1: Adverse Reactions in ≥10% of Patients with Unresectable or Metastatic Melanoma
KEYTRUDA 2 mg/kg every 3 weeks
N=89
Adverse Reaction All Grades (%)
Grade 3* (%)
General Disorders and Administration Site ConditionsFatigue 47 7Peripheral edema 17 1Chills 14 0Pyrexia 11 0
Gastrointestinal DisordersNausea 30 0Constipation 21 0Diarrhea 20 0Vomiting 16 0Abdominal pain 12 0
Respiratory, Thoracic And Mediastinal DisordersCough 30 1Dyspnea 18 2
Skin And Subcutaneous Tissue DisordersPruritus 30 0Rash 29 0Vitiligo 11 0
Metabolism and Nutrition DisordersDecreased appetite 26 0
Musculoskeletal and Connective Tissue DisordersArthralgia 20 0Pain in extremity 18 1Myalgia 14 1Back pain 12 1
Nervous System DisordersHeadache 16 0Dizziness 11 0
Blood and Lymphatic System DisordersAnemia 14 5
Psychiatric DisordersInsomnia 14 0
Infections and InfestationsUpper respiratory tract infection 11 1
*There were no Grade 5 adverse reactions reported. Of the ≥10% adverse reactions, none was reported as Grade 4.
Other clinically important adverse reactions observed in up to 10% of patients treated with KEYTRUDA were: Infections and infestations: sepsis
Brief Summary of the Prescribing Information for KEYTRUDA® (pembrolizumab) for injection, for intravenous use (continued)
32946_BUILD.indd 3 12/9/14 4:21 PM32946_01_p.indd 4 12/10/14 12:57 PM

The Merck Access Program for KEYTRUDA for Injection
Visitmerckaccessprogram-keytruda.com
Call855-257-3932
• Speak with a dedicated representative Monday to Friday between 8 AM and 8 PM ET.
• Ask to be contacted by a field reimbursement associate.
OR
Merck OncologyCopyright © 2014 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. ONCO-1116177-0000 11/14
32946_BUILD.indd 4 12/9/14 4:21 PM32946_01_p.indd 3 12/10/14 12:57 PM

ASCOPost.com | MARCH 25, 2015 PAGE 47
Direct From ASCO
2015 Pre–Annual Meeting Seminars Feature Important Topics for Modern Cancer Care Providers
A SCO will once again be offering a series of Pre–Annual Meeting
Seminars ahead of its 2015 Annual Meeting in Chicago. First offered in 2012, the Pre–Annual Meeting Semi-nars are a series of in-depth educational opportunities dedicated to topics of in-terest in the oncology community.
The seminars are conveniently scheduled ahead of the Annual Meet-ing in Chicago for attendees looking to maximize their learning and network-ing opportunities. All seminars are held onsite at McCormick Place in Chicago from 12:30 PM on Thursday, May 28, until 11:00 AM on Friday, May 29.
Each year, ASCO members and volunteers propose topics for the Pre–Annual Meeting Seminars, which are then reviewed and discussed by ASCO leadership. Once the topics are selected,
planning committees select faculty and finalize the programs.
The 2015 program will feature two new seminar topics: “Faculty Develop-ment for Medical Educators: Advancing My Career and Teaching Repertoire” and “The Economics of Cancer Care.” In addition, three of the popular topics fea-
tured at the 2014 Pre–Annual Meeting Seminars are returning in 2015: “New Drugs in Oncology,” “Genetics and Ge-nomics for the Practicing Clinician,” and “Hematology for the Oncologist,” which is cosponsored by the American Society of Hematology (ASH).
Faculty Development for Medical Educators
The first new seminar added to the 2015 Pre–Annual Meeting Seminars is “Faculty Development for Medical Edu-
cators: Advancing My Career and Teach-ing Repertoire.” This seminar will feature an interactive workshop discussing per-sonal career development, the challeng-es and opportunities for medical educa-tors, development of strong educational portfolios, and how to maximize the use of technology for teaching.
“Traditionally, the focus of profes-sional development within oncology has centered on the development of the clinical or laboratory-based inves-tigator,” said seminar Co-Chair Jill Gilbert, MD, Associate Professor of Medicine and Director of the Hema-tology/Oncology Fellowship Program at the Vanderbilt University School of Medicine. “However, ASCO rec-ognizes that we are entering a new era of medical education—a renaissance of sorts—and that landmark changes in the health-care landscape demand that established providers and trainees are prepared to take on the challenges of delivering high-quality, compassion-ate care, with attention to fiscal respon-sibility and patient involvement.”
In this changing environment, it is anticipated that medical educators will serve as the catalyst for such change both on an institutional and a national professional organization level.
“ASCO recognizes the current gap in development opportunities for the
medical educator, and with the Pre–Annual Meeting Seminars, now begins its own journey as a national leader in medical education professional devel-opment,” Dr. Gilbert said. “As session Co-Chair, I strongly encourage medical educators at all stages of their careers to take advantage of this first-of-its-kind, exciting opportunity.”
The Economics of Cancer CareThe second new 2015 seminar will
discuss the dramatically shifting land-scape of financing and organization in cancer care. ASCO recognizes that many stakeholders, including cancer care providers, are likely affected by the rapid pace of change occurring in re-imbursement models, treatment path-ways, and how best to provide quality care. “The Economics of Cancer Care” will provide participants sufficient background knowledge to make better-informed decisions in their relations with patients and payers.
This seminar will cover current and innovative strategies for rewarding re-search and development in cancer ther-apeutics and diagnostics, trends in trial costs, drug pricing, financial burdens placed on patients, the basics of reim-bursement and coverage decision-mak-ing, lessons from innovative reimburse-
continued on page 49
With the new Pre–Annual Meeting Seminars, [ASCO] now begins its own journey as a national leader in medical education professional development.
—Jill Gilbert, MD
FDA Takes Steps to Simplify Compassionate Use Process, Invites Comment
The U.S. Food and Drug Adminis-tration (FDA) introduced a more
streamlined form for requesting per-mission for patient access to investiga-tional drugs outside of clinical trials.
The new form is available for com-ment in a draft guidance for industry entitled “Individual Patient Expand-ed Access Applications: Form FDA 3926.” This draft guidance introduces and describes draft Form FDA 3926: Individual Patient Expanded Access-Investigational New Drug Applica-tion (IND). When finalized, the form will be available for physicians to use for expanded access requests for indi-
vidual patient INDs. In a post on the FDA Voice Blog,
Peter Lurie, MD, MPH, Associ-ate FDA Commissioner for Public Health Strategy and Analysis, wrote that the agency developed the new form in response to concerns from physicians and patients that the cur-rent form was too difficult. It re-quired 26 types of information and seven attachments, taking physicians about 100 hours to fill out. The new form requires only eight elements of information and a single attachment, and should take physicians about 45 minutes to complete. n
ASCO Supports FDA’s Proposed Regulation of Laboratory Developed Tests
In comments submitted to the U.S. Food and Drug Administration
(FDA), ASCO expressed its support for the agency’s draft guidance, “Frame-work for Regulatory Oversight of Labo-ratory Developed Tests (LDTs).”
ASCO also strongly recommends that the agency proceed with regulatory authority in a way that helps ensure on-going innovation in the field of molecu-lar testing, as well as timely patient access to validated molecular diagnostics that help improve use of precision medicine. Increasingly, multiplex genomic and proteomic tests are being used to guide therapy selection for people with cancer.
“ASCO believes that the tests used to detect those abnormalities must be of the highest quality and thoroughly validated before being offered to doc-tors and patients,” ASCO President Peter Paul Yu, MD, FACP, FASCO, wrote. “Our patients depend on high- quality tests as much as they depend on carefully studied, safe, and effective drugs to achieve the best possible out-comes.”
Read the FDA’s draft guidance on LDT regulation at www.fda.gov/down-loads/MedicalDevices/DeviceRegu-lationandGuidance/GuidanceDocu-ments/UCM416685.pdf. n

PAGE 48 The ASCO Post | MARCH 25, 2015
Direct From ASCO
Beating the Odds in The Big Casino
More than 35 ASCO members contributed personal essays to
a recently published collection of sto-ries about humanism in medicine, in-cluding ASCO Past Presidents Paul A. Bunn, Jr, MD, FASCO, and Emil J. Freireich, MD, FASCO, and cur-rent President-Elect Julie M. Vose, MD, MBA, FASCO. The Big Casino: America’s Best Cancer Doctors Share Their Most Powerful Stories is a collabo-ration between Stanley H. Winokur, MD, Medical Director of Axess Oncol-ogy and ASCO member, and Vincent Coppola, a journalist and author who was diagnosed with cancer in 2012.
In the interview that follows, Dr. Winokur discusses the book’s con-tributors, themes, and his own essays. “Young doctors, especially, are going to be inspired by the stories in the book,”
he said. “The authors are their mentors, their heroes, being completely real.”
Humanizing PhysiciansThe author list is impressive. Were you
surprised by the response when you asked for contributors?
The doctors who answered the call are some of the busiest doctors in the world. And without fail, they all re-sponded that they were grateful for the opportunity. I believe there are hun-dreds more doctors who would have contributed. It’s therapeutic to share these stories. I’m looking at creating a website where people can continue to submit their stories as a vehicle to share the gifts we’ve been given.
What guidance did you give to the au-thors for their contributions?
I personally talked with every au-thor I invited, and I said, “You’re a rock star in the world of medicine. Everyone knows who you are as a scientist and a clinician. I’d like you to write about who you are as a human being.” Patients talk about how grateful they are to their doctors. This book is about our grati-tude for our patients, and the precious
gifts they’ve given us—gifts of courage, strength, hope, humor, determination, and joy. So the question I asked the au-thors was, “What have you gotten from your patients?”
Humor and HopeYou contributed two stories to the
Young doctors, especially, are going to be inspired by the stories in the book. The authors are their mentors, their heroes, being completely real.
—Stanley H. Winokur, MD
Helping Patients Understand Palliative Care
The booklet Palliative Care: Improv-ing Quality of Life for Patients and
Families is one of the latest additions to the ASCO Answers collection of pa-tient education materials developed by ASCO for people with cancer and their caregivers. These materials provide on-cologist-approved information in a con-versational tone, to give patients and their caregivers the tools and resources
they need to become active participants in their cancer care.
In the Palliative Care booklet, pa-tients and their families learn about this specialized area of medicine and how it differs from hospice care. It explains that palliative care is more than receiving a drug to ease physical symptoms, but rather, involves sup-porting a patient’s physical, social,
spiritual, and emotional health, from diagnosis into recovery.
Additionally, patients and caregiv-ers can find a list of questions to ask the health-care team, a list of national or-ganizations that can provide additional information and support, and a diction-ary of terms related to palliative care that they might hear during diagnosis, treatment, and recovery.
Talking about palliative care soon after a cancer diagnosis helps patients better understand their prognosis and goals of treatment, clarifies their expec-tations, and helps them maintain the best quality of life. By using this book-let, patients and their families are bet-ter able to talk with members of their health-care team about their concerns,
continued on page 50
Accelerating Breakthroughs
Launching Careers
Improving Cancer Care
ConquerCancerFoundation.org
continued on page 49

ASCOPost.com | MARCH 25, 2015 PAGE 49
Direct From ASCO
Top 10 most-accessed articles published in 2011 in Journal of Clinical Oncology
Top 5 articles recently published in
Journal of Oncology Practice
What’s Hot in
JOPPhase II Study of Paclitaxel Given Once per Week Along With
Trastuzumab and Pertuzumab in Patients With Human Epidermal
Growth Factor Receptor 2–Positive Metastatic Breast Cancer
by Chau Dang, et al
Duration of Androgen Suppression Before Radiotherapy for
Localized Prostate Cancer: Radiation Therapy Oncology Group
Randomized Clinical Trial 9910
by Thomas M. Pisansky, et al
Reproducible and Sustained Efficacy of Targeted Therapy With
Vemurafenib in Patients With BRAFV600E-Mutated Erdheim-
Chester Disease
by Julien Haroche, et al
Relationship Between Male Pattern Baldness and the Risk of
Aggressive Prostate Cancer: An Analysis of the Prostate, Lung,
Colorectal, and Ovarian Cancer Screening Trial
by Cindy Ke Zhou, et al
Risk of Marrow Neoplasms After Adjuvant Breast Cancer
Therapy: The National Comprehensive Cancer Network
Experience
by Antonio C. Wolff, et al
JOP.ascopubs.org
Volume 7, Issue 3 May 2011
The Authoritative Resource for Oncology Practices
Report on the ASCO 2010 Provider-Payer Initiative Meeting By Michael N. Neuss, MD, et al
Subspecialization in Community Oncology: Option or Necessity? By Dean H. Gesme, MD, et al
Current Hepatitis B Screening Practices and Clinical Experience of Reactivation in Patients Undergoing Chemotherapy for Solid Tumors: A Nationwide Survey of Medical Oncologists By Fiona L. Day, FRACP, et al
Barriers to Recruitment of Rural Patients in Cancer Clinical Trials By Shamsuddin Virani, MB, BS, et al
Partners and Partnerships: Trends in Private Oncology Practice By Thomas A. Paivanas, MHSA
Journal of oncology Practice
www.jop.ascopubs.org
ment, coverage and treatment models, options for reimbursement reform, and quality in the outpatient setting.
New Drugs in OncologyReturning in 2015 is the seminar
“New Drugs in Oncology.” This semi-nar’s focus will be the theoretical and
practical aspects of both recently ap-proved drugs and those on their way to approval, including information on mechanisms of action, administration, toxicity, side effect management, and use in the clinic.
The seminar will be co-chaired by George Sledge, MD, FASCO, Profes-sor of Medicine at the Stanford Uni-versity Medical Center, and Anthony Tolcher, MD, Director of Clinical Re-search at South Texas Accelerated Re-search Therapeutics.
Genetics and Genomics for the Practicing Clinician
Another of the returning seminars is “Genetics and Genomics for the Practicing Clinician.” Oncology is ben-efiting from revolutionary advances in DNA-sequencing technology and genomic analysis, and, as a result, the field is changing very rapidly, accord-ing to seminar Co-Chair, Michael F. Berger, PhD, Associate Director of the Marie-Josée and Henry R. Kravis Cen-ter for Molecular Oncology at Memo-rial Sloan Kettering Cancer Center.
Tumor genomic profiling is now routinely performed in academic and—increasingly—community settings.
“This practice can provide impor-tant diagnostic and prognostic infor-mation and enable the selection of therapies tailored to a patient’s molecu-lar profile,” Dr. Berger said. “However, it can also reveal unexpected inherited susceptibilities to cancer and other dis-eases, with major consequences for pa-tients and their families.”
Practicing oncologists and other cancer providers must work to un-derstand the variety of tests available, when they should be administered, and what to consider when interpret-ing the results.
This Pre–Annual Meeting Seminar will cover both tumor and inherited genetic variation, as well as explain the variety of tests available to the clinician and when they should be administered.
“We’ve put together a really exciting program covering both tumor and germ-line analysis, focusing on case studies illustrative of scenarios frequently en-countered by clinicians,” Dr. Berger said.
Hematology for the OncologistIn 2014, ASCO teamed up with ASH
to offer a seminar titled, “Hematology for the Oncologist,” and this popular seminar has been brought back for the 2015 Pre–Annual Meeting Seminars.
“Medical oncologists are constantly being asked to consult on benign hema-tology issues, whether or not they have done additional formal training in he-matology,” said seminar Co-Chair Gary I. Cohen, MD, Director of the Sandra and Malcolm Berman Cancer Institute at the Greater Baltimore Medical Center and Associate Professor of Oncology at Johns Hopkins School of Medicine.
For that reason, this cosponsored seminar will focus on hematologic is-sues that medical oncologists com-monly encounter in a consultative
practice, such as the etiology and treat-ment of disorders of red cells or plate-lets, myeloproliferative syndromes,
clotting disorders, and more. “As oncologists in general practice,
we have worked diligently with our ASH colleagues to provide current up-dates on both the common and com-plex hematologic problems encoun-tered in a typical oncology consultative
practice,” Dr. Cohen said. “I encourage attendance by all oncologists to get a better understanding of these critical clinical care issues that we all face in ev-eryday practice.”
Register NowAnyone interested in attending the
Pre–Annual Meeting Seminars can register at the ASCO Annual Meeting Registration Site. Registration includes a boxed lunch on Thursday and a conti-nental breakfast on Friday. If registered on or before April 22, 2015, registra-tion is $100 for ASCO members and $200 for nonmembers. These live ac-tivities have been approved for AMA PRA Category 1 Credit™. n
© 2015. American Society of Clinical Oncology. All rights reserved.
George Sledge, MD, FASCO
Gary I. Cohen, MD
Pre–Annual Meeting Seminarscontinued from page 47
and work with them to manage the challenges that come with cancer.
The new Palliative Care booklet can be purchased in packs of 125 from the ASCO University Bookstore at cancer.net/estore. ASCO members receive a 20% discount, and all pa-tient education materials ship for free. It is also available to download at cancer.net/palliativecare.
For more information on quality-of-life issues, your patients can also turn
to Cancer.Net’s symptom management resources. At cancer.net/sideeffects, patients can find articles on more than 45 side effects of cancer and can-cer treatments. These articles explain symptoms that should be reported to their health-care team and how each side effect can be managed. n
Originally printed in ASCO Connection. © American Society of Clinical Oncology. “Helping Patients Understand Palliative Care.” ASCO Connection, January 2015: 35. All rights reserved.
Palliative Care Bookletcontinued from page 48

PAGE 50 The ASCO Post | MARCH 25, 2015
Direct From ASCO
ASCO’s Patient Resources for Colorectal Cancer Awareness Month
S tock your practice with Cancer.Net resources. Cancer.Net has a
comprehensive guide to colorectal can-cer at www.cancer.net/colorectal and a shorter, one-page colorectal cancer fact sheet. You will also find specialized re-sources for survivorship, palliative care, and managing the cost of cancer care. Copies can be purchased from the ASCO University Bookstore at cancer.net/ eStore. Shipping is free, and ASCO members save 20%. n
© 2015. American Society of Clinical Oncology. All rights reserved.
book. The first, “How to Tell Patients They Have Cancer,” demonstrates a sense of gallows humor. Is that an essential trait when working in oncology?
Yes, absolutely. That story hap-pened when I was in medical school, about 40 years ago, and it shows the intense fear that doctors had of being real. We talked about “neoplasms” and “lesions” because we didn’t know how to tell patients that they had cancer. We didn’t want to use that word. It was our armor, to avoid telling patients they might die. There was a dark humor in the way we discussed it. That dark hu-mor exists today in other ways. As doc-tors, we very rarely share those kinds of stories with the public because we don’t want to be seen as minimizing someone’s suffering or to be perceived as insincere. The humor is really about our own fear.
If readers take away only one message from the book, what do you hope they un-derstand?
Hope is a precious gift. We should never say to our patients, “There’s nothing else I can do for you.” There might not be any more drugs or thera-pies we can give them, but we can give them our compassion and our gratitude. We can stand next to them
and hold their hands. There is always something we can do.
Dr. Winokur’s hope is that every oncologist and patient will have the opportunity to read The Big Casino. The book is being sold at cost, and no editor or contributor is profiting from its sales. Print and ebook edi-
tions are available on Amazon.com; visit thebigcasino.org for a full list of contributors and information about discounted bulk orders for your training program, cancer center, or practice. n
Originally printed in ASCO Connection. © American Society of Clinical Oncology.
“Beating the Odds in The Big Casino.” ASCO Connection, January 2015: 32-34. All rights reserved.
The Big Casinocontinued from page 48
See page 86 to read an excerpt from The Big Casino,
or visit http://bit.ly/1wk9nsm for past essays published in
The ASCO Post.
NOW APPROVED
Visit LENVIMAinfo.com
LENVIMATM is a trademark used by Eisai Inc. under license from Eisai R&D Management Co., Ltd. © 2015 Eisai Inc. All rights reserved. Printed in USA/February 2015 LENV0185
Important Safety Information
Warnings and Precautions
Hypertension was reported in 73% of LENVIMA-treated patients (of which 44% were ≥ Grade 3) and 16% of patients in the placebo group. Control blood pressure prior to treatment and monitor blood pressure after 1 week, then every 2 weeks for the fi rst 2 months, and then at least monthly during treatment. Withhold LENVIMA for Grade 3 hypertension; resume at a reduced dose when hypertension is controlled at ≤ Grade 2. Discontinue LENVIMA for life-threatening hypertension.
Cardiac dysfunction was reported in 7% of LENVIMA-treated patients (2% Grade 3 or greater). Monitor patients for clinical symptoms or signs of cardiac decompensation. Withhold LENVIMA for development of Grade 3 cardiac dysfunction until improved to Grade 0 or 1 or baseline. Resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of cardiac dysfunction. Discontinue LENVIMA for Grade 4 cardiac dysfunction.
Arterial thromboembolic events were reported in 5% of LENVIMA-treated patients; events of Grade 3 or greater were 3%. Discontinue LENVIMA following an arterial thrombotic event. LENVIMA has not been studied in patients who have had an arterial thromboembolic event within the previous 6 months.
4% of LENVIMA-treated patients experienced an increase in ALT and 5% experienced an increase in AST that was Grade 3 or greater. Monitor liver function before initiation and during treatment with LENVIMA. Withhold LENVIMA for the development of ≥ Grade 3 liver impairment until resolved to Grade 0 to 1 or baseline. Resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of hepatotoxicity. Discontinue LENVIMA for hepatic failure.
Proteinuria was reported in 34% of LENVIMA-treated patients (of which 11% were Grade 3). Monitor for proteinuria before initiation of, and periodically during treatment. Obtain a 24 hour urine protein if urine dipstick proteinuria ≥2+ is detected. Withhold LENVIMA for ≥ 2 grams of proteinuria/24 hours and resume at a reduced dose when proteinuria is <2 gm/24 hours. Discontinue LENVIMA for nephrotic syndrome.
LENVIMATM (lenvatinib) is indicated for the treatment of patients with
locally recurrent or metastatic, progressive, radioactive iodine-refractory
di erentiated thyroid cancer (DTC).
Events of renal impairment were reported in 14% of LENVIMA-treated patients. Renal failure or impairment ≥ Grade 3 was 3% in LENVIMA-treated patients. Withhold LENVIMA for development of Grade 3 or 4 renal failure / impairment until resolved to Grade 0 to 1 or baseline. Resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of renal impairment.
Events of gastrointestinal perforation or fi stula were reported in 2% of LENVIMA-treated patients. Discontinue LENVIMA in patients who develop gastrointestinal perforation or life-threatening fi stula.
QT/QTc interval prolongation was reported in 9% of LENVIMA-treated patients (2% Grade 3 or greater). Monitor ECG in patients with congenital long QT syndrome, CHF, bradyarrhythmias, or patients taking drugs known to prolong the QT interval. Monitor and correct electrolyte abnormalities in all patients. Withhold LENVIMA for the development of ≥ Grade 3 QT interval prolongation. Resume LENVIMA at a reduced dose when QT prolongation resolves to Grade 0 or 1 or baseline.
Hypocalcemia ≥ Grade 3 was reported in 9% of LENVIMA-treated patients. Monitor blood calcium levels at least monthly and replace calcium as necessary during LENVIMA treatment. Interrupt and adjust LENVIMA dosing as necessary depending on severity, presence of ECG changes, and persistence of hypocalcemia.
Reversible posterior leukoencephalopathy syndrome (RPLS) was reported in 3 patients across clinical studies in which 1108 patients received LENVIMA. Confi rm the diagnosis of RPLS with MRI. Withhold LENVIMA for RPLS until fully resolved. Resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of neurologic symptoms.
Hemorrhagic events occurred in 35% of LENVIMA-treated patients and in 18% of the placebo group. The incidence of Grade 3-5 hemorrhage was similar between arms at 2% and 3%, respectively. The most frequently reported hemorrhagic event was epistaxis (11% Grade 1 and 1% Grade 2). Discontinuation due to hemorrhagic events occurred in 1% of LENVIMA-treated patients. There was one case of fatal intracranial hemorrhage among 16 patients who received LENVIMA and had CNS metastases at baseline. Withhold LENVIMA for the development of Grade 3 hemorrhage until resolved to Grade 0 to 1. Resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of hemorrhage. Discontinue LENVIMA in patients who experience Grade 4 hemorrhage.
LENVIMA impairs exogenous thyroid suppression. Elevation of TSH level above 0.5 mU/L was observed post baseline in 57% of LENVIMA-treated patients. Monitor TSH levels monthly and adjust thyroid replacement medication as needed.
LENVIMA can cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use e¤ ective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy.
Advise women not to breastfeed during treatment with LENVIMA.
Adverse Reactions
The most common adverse reactions observed in LENVIMA-treated patients vs. placebo treated patients respectively were hypertension (73% vs 16%), fatigue (67% vs 35%), diarrhea (67% vs 17%), arthralgia/myalgia (62% vs 28%), decreased appetite (54% vs 18%), weight decreased (51% vs 15%), nausea (47% vs 25%), stomatitis (41% vs 8%), headache (38% vs 11%), vomiting (36% vs 15%), proteinuria (34% vs 3%), palmar-plantar erythrodysesthesia syndrome (32% vs 1%), abdominal pain (31% vs 11%), and dysphonia (31% vs 5%).
Please see Brief Summary of Prescribing Information on the following pages.
78618ha_b.indd 1-2 2/23/15 4:50 PM

ASCOPost.com | MARCH 25, 2015 PAGE 51
Announcements
New Oncology Care Model: Encouraging Better Coordination for Cancer Care
On February 12, 2015, the U.S. Department of Health and Hu-
man Services (HHS) announced its new Oncology Care Model, a multi-payer payment and care delivery model intended to support better coordina-tion for cancer care. The initiative will
include 24-hour access to practitioners for beneficiaries undergoing cancer treatment and is based on coordinated, person-centered care, aimed at reward-ing value of care, rather than volume.
“Based on feedback from the medi-cal, consumer, and business communi-
ties, we are launching this new model of care to support clinicians’ work with their patients,” said Patrick Conway, MD, Centers for Medicare & Medicaid Services (CMS) Chief Medical Officer and Deputy Administrator for Innova-tion and Quality. “We aim to provide
Medicare beneficiaries struggling with cancer with high-quality care around the clock and to reward doctors for the value, not volume, of care they provide.”
As part of the Department’s approach to improving health delivery, the Oncol-
Health-Care Policy
continued on page 52
NOW APPROVED
Visit LENVIMAinfo.com
LENVIMATM is a trademark used by Eisai Inc. under license from Eisai R&D Management Co., Ltd. © 2015 Eisai Inc. All rights reserved. Printed in USA/February 2015 LENV0185
Important Safety Information
Warnings and Precautions
Hypertension was reported in 73% of LENVIMA-treated patients (of which 44% were ≥ Grade 3) and 16% of patients in the placebo group. Control blood pressure prior to treatment and monitor blood pressure after 1 week, then every 2 weeks for the fi rst 2 months, and then at least monthly during treatment. Withhold LENVIMA for Grade 3 hypertension; resume at a reduced dose when hypertension is controlled at ≤ Grade 2. Discontinue LENVIMA for life-threatening hypertension.
Cardiac dysfunction was reported in 7% of LENVIMA-treated patients (2% Grade 3 or greater). Monitor patients for clinical symptoms or signs of cardiac decompensation. Withhold LENVIMA for development of Grade 3 cardiac dysfunction until improved to Grade 0 or 1 or baseline. Resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of cardiac dysfunction. Discontinue LENVIMA for Grade 4 cardiac dysfunction.
Arterial thromboembolic events were reported in 5% of LENVIMA-treated patients; events of Grade 3 or greater were 3%. Discontinue LENVIMA following an arterial thrombotic event. LENVIMA has not been studied in patients who have had an arterial thromboembolic event within the previous 6 months.
4% of LENVIMA-treated patients experienced an increase in ALT and 5% experienced an increase in AST that was Grade 3 or greater. Monitor liver function before initiation and during treatment with LENVIMA. Withhold LENVIMA for the development of ≥ Grade 3 liver impairment until resolved to Grade 0 to 1 or baseline. Resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of hepatotoxicity. Discontinue LENVIMA for hepatic failure.
Proteinuria was reported in 34% of LENVIMA-treated patients (of which 11% were Grade 3). Monitor for proteinuria before initiation of, and periodically during treatment. Obtain a 24 hour urine protein if urine dipstick proteinuria ≥2+ is detected. Withhold LENVIMA for ≥ 2 grams of proteinuria/24 hours and resume at a reduced dose when proteinuria is <2 gm/24 hours. Discontinue LENVIMA for nephrotic syndrome.
LENVIMATM (lenvatinib) is indicated for the treatment of patients with
locally recurrent or metastatic, progressive, radioactive iodine-refractory
di erentiated thyroid cancer (DTC).
Events of renal impairment were reported in 14% of LENVIMA-treated patients. Renal failure or impairment ≥ Grade 3 was 3% in LENVIMA-treated patients. Withhold LENVIMA for development of Grade 3 or 4 renal failure / impairment until resolved to Grade 0 to 1 or baseline. Resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of renal impairment.
Events of gastrointestinal perforation or fi stula were reported in 2% of LENVIMA-treated patients. Discontinue LENVIMA in patients who develop gastrointestinal perforation or life-threatening fi stula.
QT/QTc interval prolongation was reported in 9% of LENVIMA-treated patients (2% Grade 3 or greater). Monitor ECG in patients with congenital long QT syndrome, CHF, bradyarrhythmias, or patients taking drugs known to prolong the QT interval. Monitor and correct electrolyte abnormalities in all patients. Withhold LENVIMA for the development of ≥ Grade 3 QT interval prolongation. Resume LENVIMA at a reduced dose when QT prolongation resolves to Grade 0 or 1 or baseline.
Hypocalcemia ≥ Grade 3 was reported in 9% of LENVIMA-treated patients. Monitor blood calcium levels at least monthly and replace calcium as necessary during LENVIMA treatment. Interrupt and adjust LENVIMA dosing as necessary depending on severity, presence of ECG changes, and persistence of hypocalcemia.
Reversible posterior leukoencephalopathy syndrome (RPLS) was reported in 3 patients across clinical studies in which 1108 patients received LENVIMA. Confi rm the diagnosis of RPLS with MRI. Withhold LENVIMA for RPLS until fully resolved. Resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of neurologic symptoms.
Hemorrhagic events occurred in 35% of LENVIMA-treated patients and in 18% of the placebo group. The incidence of Grade 3-5 hemorrhage was similar between arms at 2% and 3%, respectively. The most frequently reported hemorrhagic event was epistaxis (11% Grade 1 and 1% Grade 2). Discontinuation due to hemorrhagic events occurred in 1% of LENVIMA-treated patients. There was one case of fatal intracranial hemorrhage among 16 patients who received LENVIMA and had CNS metastases at baseline. Withhold LENVIMA for the development of Grade 3 hemorrhage until resolved to Grade 0 to 1. Resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of hemorrhage. Discontinue LENVIMA in patients who experience Grade 4 hemorrhage.
LENVIMA impairs exogenous thyroid suppression. Elevation of TSH level above 0.5 mU/L was observed post baseline in 57% of LENVIMA-treated patients. Monitor TSH levels monthly and adjust thyroid replacement medication as needed.
LENVIMA can cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use e¤ ective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy.
Advise women not to breastfeed during treatment with LENVIMA.
Adverse Reactions
The most common adverse reactions observed in LENVIMA-treated patients vs. placebo treated patients respectively were hypertension (73% vs 16%), fatigue (67% vs 35%), diarrhea (67% vs 17%), arthralgia/myalgia (62% vs 28%), decreased appetite (54% vs 18%), weight decreased (51% vs 15%), nausea (47% vs 25%), stomatitis (41% vs 8%), headache (38% vs 11%), vomiting (36% vs 15%), proteinuria (34% vs 3%), palmar-plantar erythrodysesthesia syndrome (32% vs 1%), abdominal pain (31% vs 11%), and dysphonia (31% vs 5%).
Please see Brief Summary of Prescribing Information on the following pages.
78618ha_b.indd 1-2 2/23/15 4:50 PM

PAGE 52 The ASCO Post | MARCH 25, 2015
Announcements
ogy Care Model is one of many innova-tive payment and care delivery models developed by the CMS Innovation Cen-ter and advanced by the Affordable Care Act. The model was created in response to feedback from the oncology community, patient advocates, and the private sector
that a new way of paying for and deliver-ing oncology care is needed. This model will invest in physician-led practices, al-lowing the practices to innovate and de-liver higher-quality care to their patients.
A Closer Look at the ModelThe model focuses on three key ar-
eas: (1) linking payment to quality of
care, (2) improving and innovating in care delivery, and (3) sharing informa-tion more broadly to providers, con-sumers, and others to support better decisions while maintaining privacy.
The Oncology Care Model encourag-es participating practices to improve care and lower costs through episode-based, performance-based payments that finan-
Oncology Care Modelcontinued from page 51
Patrick Conway, MD
LENV-55860_M2_BriefSum_Islnd.indd2-19-2015 6:47 PM Vincent Jeffrey / Suke Yawata
Client CodeClient
LiveOverall TrimBleed
# of Colors
LENV0176 Eisai/LENVIMA
7.0625” x 9.5”7.3125” x 10”None
Black
Colors Black
FontsUnivers LT Std (67 Bold Condensed, 47 Light Con-densed, 47 Light Condensed Oblique)
Job info Fonts & ColorsImages
Saved at
None
from hsvjeffrey5552 by
Printed At
Eisai_K.ai (3.6%)
Notes Brief Summary - Island Spread
LENVIMA™ (lenvatinib) BRIEF SUMMARY – See package insert for full prescribing information.1 INDICATIONS AND USAGELENVIMA is indicated for the treatment of patients with locally recurrent or metastatic, progressive, radioactive iodine-refractory differentiated thyroid cancer (DTC).2 DOSAGE AND ADMINISTRATION2.1 Recommended DoseThe recommended daily dose of LENVIMA is 24 mg (two 10 mg capsules and one 4 mg capsule) orally taken once daily with or without food. Continue LENVIMA until disease progression or until unacceptable toxicity occurs.Take LENVIMA at the same time each day. If a dose is missed and cannot be taken within 12 hours, skip that dose and take the next dose at the usual time of administration.Severe Renal or Hepatic ImpairmentThe recommended dose of LENVIMA is 14 mg taken orally once daily in patients with severe renal impairment (creatinine clearance [CLcr] less than 30 mL/min calculated by the Cockroft-Gault equation) or severe hepatic impairment (Child-Pugh C).2.2 Dose ModificationsHypertension• Assess blood pressure prior to and periodically during treatment. Initiate or adjust medical management to
control blood pressure prior to and during treatment.• Withhold LENVIMA for Grade 3 hypertension that persists despite optimal antihypertensive therapy; resume
at a reduced dose (see Table 1) when hypertension is controlled at less than or equal to Grade 2.• Discontinue LENVIMA for life-threatening hypertension.Cardiac dysfunction or hemorrhage • Discontinue for a Grade 4 event.• Withhold LENVIMA for development of Grade 3 event until improved to Grade 0 or 1 or baseline.• Either resume at a reduced dose (see Table 1) or discontinue LENVIMA depending on the severity and
persistence of the adverse event.Arterial thrombotic event • Discontinue LENVIMA following an arterial thrombotic event.Renal failure and impairment or hepatotoxicity• Withhold LENVIMA for development of Grade 3 or 4 renal failure/impairment or hepatotoxicity until resolved
to Grade 0 to 1 or baseline.• Either resume at a reduced dose (see Table 1) or discontinue LENVIMA depending on the severity and
persistence of renal impairment or hepatotoxicity.• Discontinue LENVIMA for hepatic failure.Proteinuria• Withhold LENVIMA for ≥2 grams of proteinuria/24 hours.• Resume at a reduced dose (see Table 1) when proteinuria is <2 gm/24 hours.• Discontinue LENVIMA for nephrotic syndrome.Gastrointestinal perforation or fistula formation• Discontinue LENVIMA in patients who develop gastrointestinal perforation or life-threatening fistula.QT prolongation• Withhold LENVIMA for the development of Grade 3 or greater QT interval prolongation.• Resume LENVIMA at a reduced dose (see Table 1) when QT prolongation resolves to Grade 0 or 1 or baseline.Reversible posterior leukoencephalopathy syndrome (RPLS)• Withhold for RPLS until fully resolved.• Upon resolution, resume at a reduced dose or discontinue LENVIMA depending on the severity and
persistence of neurologic symptoms.Manage other adverse reactions according to the instructions in Table 1. Based on the absence of clinical experience, there are no recommendations on resumption of dosing in patients with Grade 4 clinical adverse reactions that resolve.
Table 1 Recommended Dose Modifications for Persistent and Intolerable Grade 2 or Grade 3 Adverse Reactions or Grade 4 Laboratory Abnormalitiesa
Adverse Reaction Modification Adjusted Doseb
First occurrence Interrupt until resolved to Grade 0-1 or baseline
20 mg (two 10 mg capsules) orally once daily
Second occurrencec Interrupt until resolved to Grade 0-1 or baseline
14 mg (one 10 mg capsule plus one 4 mg capsule)
orally once daily
Third occurrencec Interrupt until resolved to Grade 0-1 or baseline
10 mg (one 10 mg capsule) orally once daily
a Initiate medical management for nausea, vomiting, or diarrhea prior to interruption or dose reduction of LENVIMA
b Reduce dose in succession based on the previous dose level (24 mg, 20 mg, or 14 mg per day)c Refers to the same or a different adverse reaction that requires dose modification
4 CONTRAINDICATIONSNone.5 WARNINGS AND PRECAUTIONS5.1 HypertensionIn Study 1 hypertension was reported in 73% of LENVIMA-treated patients and 16% of patients in the placebo group. The median time to onset of new or worsening hypertension was 16 days for LENVIMA-treated patients. The incidence of Grade 3 hypertension was 44% as compared to 4% for placebo, and the incidence of Grade 4 hypertension was less than 1% in LENVIMA-treated patients and none in the placebo group.Control blood pressure prior to treatment with LENVIMA. Monitor blood pressure after 1 week, then every 2 weeks for the first 2 months, and then at least monthly thereafter during treatment with LENVIMA. Withhold LENVIMA for Grade 3 hypertension despite optimal antihypertensive therapy; resume at a reduced dose when hypertension is controlled at less than or equal to Grade 2. Discontinue LENVIMA for life-threatening hypertension.5.2 Cardiac DysfunctionIn Study 1, cardiac dysfunction, defined as decreased left or right ventricular function, cardiac failure, or pulmonary edema, was reported in 7% of LENVIMA-treated patients (2% Grade 3 or greater) and 2% (no Grade 3 or greater) of patients in the placebo group. The majority of these cases in LENVIMA-treated patients (14 of 17 cases) were based on findings of decreased ejection fraction as assessed by echocardiography. Six of 261 (2%) LENVIMA-treated patients in Study 1 had greater than 20% reduction in ejection fraction as measured by echocardiography compared to no patients who received placebo.Monitor patients for clinical symptoms or signs of cardiac decompensation. Withhold LENVIMA for development of Grade 3 cardiac dysfunction until improved to Grade 0 or 1 or baseline. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of cardiac dysfunction. Discontinue LENVIMA for Grade 4 cardiac dysfunction.5.3 Arterial Thromboembolic EventsIn Study 1, arterial thromboembolic events were reported in 5% of LENVIMA-treated patients and 2% of patients in the placebo group. The incidence of arterial thromboembolic events of Grade 3 or greater was 3% in LENVIMA-treated patients and 1% in the placebo group.Discontinue LENVIMA following an arterial thrombotic event. The safety of resuming LENVIMA after an arterial thromboembolic event has not been established and LENVIMA has not been studied in patients who have had an arterial thromboembolic event within the previous 6 months.5.4 HepatotoxicityIn Study 1, 4% of LENVIMA-treated patients experienced an increase in alanine aminotransferase (ALT) and 5% experienced an increase in aspartate aminotransferase (AST) that was Grade 3 or greater. No patients in the placebo group experienced Grade 3 or greater increases in ALT or AST. Across clinical studies in which 1108 patients received LENVIMA, hepatic failure (including fatal events) was reported in 3 patients and acute hepatitis was reported in 1 patient.Monitor liver function before initiation of LENVIMA, then every 2 weeks for the first 2 months, and at least monthly thereafter during treatment. Withhold LENVIMA for the development of Grade 3 or greater liver impairment until
resolved to Grade 0 to 1 or baseline. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of hepatotoxicity. Discontinue LENVIMA for hepatic failure.5.5 ProteinuriaIn Study 1, proteinuria was reported in 34% of LENVIMA-treated patients and 3% of patients in the placebo group. The incidence of Grade 3 proteinuria in LENVIMA-treated patients was 11% compared to none in the placebo group.Monitor for proteinuria before initiation of, and periodically throughout treatment. If urine dipstick proteinuria greater than or equal to 2+ is detected, obtain a 24 hour urine protein. Withhold LENVIMA for ≥2 grams of proteinuria/24 hours and resume at a reduced dose when proteinuria is <2 gm/24 hours. Discontinue LENVIMA for nephrotic syndrome.5.6 Renal Failure and ImpairmentIn Study 1, events of renal impairment were reported in 14% of LENVIMA-treated patients compared to 2% of patients in the placebo group. The incidence of Grade 3 or greater renal failure or impairment was 3% in LENVIMA-treated patients and 1% in the placebo group. The primary risk factor for severe renal impairment in LENVIMA-treated patients was dehydration/hypovolemia due to diarrhea and vomiting.Withhold LENVIMA for development of Grade 3 or 4 renal failure/impairment until resolved to Grade 0 to 1 or baseline. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of renal impairment.5.7 Gastrointestinal Perforation and Fistula FormationIn Study 1, events of gastrointestinal perforation or fistula were reported in 2% of LENVIMA-treated patients and 0.8% of patients in the placebo group.Discontinue LENVIMA in patients who develop gastrointestinal perforation or life-threatening fistula.5.8 QT Interval ProlongationIn Study 1, QT/QTc interval prolongation was reported in 9% of LENVIMA-treated patients and 2% of patients in the placebo group. The incidence of QT interval prolongation of Grade 3 or greater was 2% in LENVIMA-treated patients compared to no reports in the placebo group. Monitor electrocardiograms in patients with congenital long QT syndrome, congestive heart failure, bradyarrhythmias, or those who are taking drugs known to prolong the QT interval, including Class Ia and III antiarrhythmics.Monitor and correct electrolyte abnormalities in all patients. Withhold LENVIMA for the development of Grade 3 or greater QT interval prolongation. Resume LENVIMA at a reduced dose when QT prolongation resolves to Grade 0 or 1 or baseline.5.9 HypocalcemiaIn Study 1, 9% of LENVIMA-treated patients experienced Grade 3 or greater hypocalcemia compared to 2% in the placebo group. In most cases hypocalcemia responded to replacement and dose interruption/dose reduction.Monitor blood calcium levels at least monthly and replace calcium as necessary during LENVIMA treatment. Interrupt and adjust LENVIMA dosing as necessary depending on severity, presence of ECG changes, and persistence of hypocalcemia.5.10 Reversible Posterior Leukoencephalopathy SyndromeAcross clinical studies in which 1108 patients received LENVIMA, there were 3 reported events of reversible posterior leukoencephalopathy syndrome (RPLS). Confirm the diagnosis of RPLS with MRI. Withhold for RPLS until fully resolved. Upon resolution, resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of neurologic symptoms.5.11 Hemorrhagic EventsIn Study 1, hemorrhagic events occurred in 35% of LENVIMA-treated patients and in 18% of the placebo group. However, the incidence of Grade 3-5 hemorrhage was similar between arms at 2% and 3%, respectively. The most frequently reported hemorrhagic event was epistaxis (11% Grade 1 and 1% Grade 2). Discontinuation due to hemorrhagic events occurred in 1% of LENVIMA-treated patients.Across clinical studies in which 1108 patients received LENVIMA, Grade 3 or greater hemorrhage was reported in 2% of patients. In Study 1, there was 1 case of fatal intracranial hemorrhage among 16 patients who received lenvatinib and had CNS metastases at baseline.Withhold LENVIMA for the development of Grade 3 hemorrhage until resolved to Grade 0 to 1. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of hemorrhage. Discontinue LENVIMA in patients who experience Grade 4 hemorrhage.5.12 Impairment of Thyroid Stimulating Hormone SuppressionLENVIMA impairs exogenous thyroid suppression. In Study 1, 88% of all patients had a baseline thyroid stimulating hormone (TSH) level less than or equal to 0.5 mU/L. In those patients with a normal TSH at baseline, elevation of TSH level above 0.5 mU/L was observed post baseline in 57% of LENVIMA-treated patients as compared with 14% of patients receiving placebo.Monitor TSH levels monthly and adjust thyroid replacement medication as needed in patients with DTC.5.13 Embryofetal ToxicityBased on its mechanism of action and data from animal reproduction studies, LENVIMA can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of lenvatinib during organogenesis at doses below the recommended human dose resulted in embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy.6 ADVERSE REACTIONSThe following adverse reactions are discussed elsewhere in the label. Please see the Warnings and Precautions sections in the full prescribing information.• Hypertension• Cardiac Dysfunction• Arterial Thromboembolic Events• Hepatotoxicity• Proteinuria• Renal Failure and Impairment• Gastrointestinal Perforation and Fistula Formation• QT Interval Prolongation• Hypocalcemia• Reversible Posterior Leukoencephalopathy Syndrome• Hemorrhagic Events• Impairment of Thyroid Stimulating Hormone Suppression6.1 Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Safety data obtained in 1108 patients with advanced solid tumors who received LENVIMA as a single agent across multiple clinical studies was used to further characterize risks of serious adverse drug reactions. The median age was 60 years (range 21-89 years). The dose range was 0.2 mg to 32 mg. The median duration of exposure in the entire population was 5.5 months.The safety data described below are derived from Study 1 which randomized (2:1) patients with radioactive iodine-refractory differentiated thyroid cancer (RAI-refractory DTC) to LENVIMA (n=261) or placebo (n=131). The median treatment duration was 16.1 months for LENVIMA and 3.9 months for placebo. Among 261 patients who received LENVIMA in Study 1, median age was 64 years, 52% were women, 80% were White, 18% were Asian, and 2% were Black; 4% identified themselves as having Hispanic or Latino ethnicity.In Study 1, the most common adverse reactions observed in LENVIMA-treated patients (greater than or equal to 30%) were, in order of decreasing frequency, hypertension, fatigue, diarrhea, arthralgia/myalgia, decreased appetite, weight decreased, nausea, stomatitis, headache, vomiting, proteinuria, palmar-plantar erythrodysesthesia (PPE) syndrome, abdominal pain, and dysphonia. The most common serious adverse reactions (at least 2%) were pneumonia (4%), hypertension (3%), and dehydration (3%).Adverse reactions led to dose reductions in 68% of patients receiving LENVIMA and 5% of patients receiving placebo; 18% of patients discontinued LENVIMA and 5% discontinued placebo for adverse reactions. The most common adverse reactions (at least 10%) resulting in dose reductions of LENVIMA were hypertension (13%), proteinuria (11%), decreased appetite (10%), and diarrhea (10%); the most common adverse reactions (at least 1%) resulting in discontinuation of LENVIMA were hypertension (1%) and asthenia (1%).Table 2 presents the percentage of patients in Study 1 experiencing adverse reactions at a higher rate in LENVIMA-treated patients than patients receiving placebo in the double-blind phase of the DTC study.
Table 2 Adverse Reactions Occurring in Patients with a Between-Group Difference of Greater than or Equal to 5% All Grades or Greater than or Equal to 2% Grades 3 and 4
Adverse Reaction
LENVIMA 24 mg N=261
Placebo N=131
All Grades (%)
Grades 3-4 (%)
All Grades (%)
Grades 3-4 (%)
Vascular DisordersHypertensiona 73 44 16 4Hypotension 9 2 2 0
Gastrointestinal DisordersDiarrhea 67 9 17 0Nausea 47 2 25 1Stomatitisb 41 5 8 0Vomiting 36 2 15 0Abdominal painc 31 2 11 1Constipation 29 0.4 15 1Oral paind 25 1 2 0Dry mouth 17 0.4 8 0Dyspepsia 13 0.4 4 0
General Disorders and Administration Site ConditionsFatiguee 67 11 35 4Edema peripheral 21 0.4 8 0
Musculoskeletal and Connective Tissue DisordersArthralgia/Myalgiaf 62 5 28 3
Metabolism and Nutrition DisordersWeight decreased 51 13 15 1Decreased appetite 54 7 18 1Dehydration 9 2 2 1
Nervous System DisordersHeadache 38 3 11 1Dysgeusia 18 0 3 0Dizziness 15 0.4 9 0
Renal and Urinary DisordersProteinuria 34 11 3 0
Skin and Subcutaneous Tissue DisordersPalmar-plantar erythrodysesthesia 32 3 1 0Rashg 21 0.4 3 0Alopecia 12 0 5 0Hyperkeratosis 7 0 2 0
Respiratory, Thoracic and Mediastinal DisordersDysphonia 31 1 5 0Cough 24 0 18 0Epistaxis 12 0 1 0
Psychiatric DisordersInsomnia 12 0 3 0
Infections and InfestationsDental and oral infectionsh 10 1 1 0Urinary tract infection 11 1 5 0
Cardiac Disorders
Electrocardiogram QT prolonged 9 2 2 0a Includes hypertension, hypertensive crisis, increased blood pressure diastolic, and increased
blood pressure b Includes aphthous stomatitis, stomatitis, glossitis, mouth ulceration, and mucosal inflammationc Includes abdominal discomfort, abdominal pain, abdominal pain lower, abdominal pain upper,
abdominal tenderness, epigastric discomfort, and gastrointestinal paind Includes oral pain, glossodynia, and oropharyngeal paine Includes asthenia, fatigue, and malaisef Includes musculoskeletal pain, back pain, pain in extremity, arthralgia, and myalgiag Includes rash macular, rash maculo-papular, rash generalized, and rash h Includes gingivitis, oral infection, parotitis, pericoronitis, periodontitis, sialoadenitis, tooth abscess,
and tooth infectionA clinically important adverse reaction occurring more frequently in LENVIMA-treated patients than patients receiving placebo, but with an incidence of less than 5% was pulmonary embolism (3%, including fatal reports vs 2%, respectively).
Table 3 Laboratory Abnormalities with a Difference of at Least ≥2% in Grade 3 - 4 Events and at a Higher Incidence in LENVIMA-Treated Patientsa
Laboratory Abnormality LENVIMA 24 mg N=258b
Placebo N=131b
Grades 3-4 (%)
Grades 3-4 (%)
ChemistryCreatinine increased 3 0Alanine aminotransferase (ALT) increased 4 0Aspartate aminotransferase (AST) increased 5 0Hypocalcemia 9 2Hypokalemia 6 1Lipase increased 4 1
HematologyPlatelet count decreased 2 0
a With at least 1 grade increase from baseline b Subject with at least 1 post baseline laboratory value
In addition the following laboratory abnormalities (all Grades) occurred in greater than 5% of LENVIMA-treated patients and at a rate that was two-fold or higher than in patients who received placebo: hypoalbuminemia, increased alkaline phosphatase, hypomagnesemia, hypoglycemia, hyperbilirubinemia, hypercalcemia, hypercholesterolemia, increased serum amylase, and hyperkalemia.7 DRUG INTERACTIONS7.1 Effect of Other Drugs on LenvatinibNo dose adjustment of LENVIMA is recommended when co-administered with CYP3A, P-glycoprotein (P-gp), and breast cancer resistance protein (BCRP) inhibitors and CYP3A and P-gp inducers.8 USE IN SPECIFIC POPULATIONS8.1 PregnancyRisk SummaryBased on its mechanism of action and data from animal reproduction studies, LENVIMA can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of lenvatinib during organogenesis at doses below the recommended human dose resulted in embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits. There are no available human data informing the drug-associated risk. Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown; however, the background risk in the U.S. general population of major birth defects is 2-4% and of miscarriage is 15-20% of clinically recognized pregnancies.DataAnimal DataIn an embryofetal development study, daily oral administration of lenvatinib mesylate at doses greater than or equal to 0.3 mg/kg [approximately 0.14 times the recommended human dose based on body surface area (BSA)] to pregnant rats during organogenesis resulted in dose-related decreases in mean fetal body weight, delayed fetal ossifications, and dose-related increases in fetal external (parietal edema and tail abnormalities), visceral, and skeletal anomalies. Greater than 80% postimplantation loss was observed at 1.0 mg/kg/day (approximately 0.5 times the recommended human dose based on BSA).Daily oral administration of lenvatinib mesylate to pregnant rabbits during organogenesis resulted in fetal external (short tail), visceral (retroesophageal subclavian artery), and skeletal anomalies at doses greater than or equal to 0.03 mg/kg (approximately 0.03 times the human dose of 24 mg based on body surface area). At the 0.03 mg/kg dose, increased post-implantation loss, including 1 fetal death, was also observed. Lenvatinib was abortifacient in rabbits, resulting in late abortions in approximately one-third of the rabbits treated at a dose level of 0.5 mg/kg/day (approximately 0.5 times the recommended clinical dose of 24 mg based on BSA).8.2 LactationRisk SummaryIt is not known whether LENVIMA is present in human milk. However, lenvatinib and its metabolites are excreted in rat milk at concentrations higher than in maternal plasma. Because of the potential for serious adverse reactions in nursing infants from LENVIMA, advise women to discontinue breastfeeding during treatment with LENVIMA.DataAnimal DataFollowing administration of radiolabeled lenvatinib to lactating Sprague Dawley rats, lenvatinib-related radioactivity was approximately 2 times higher (based on AUC) in milk compared to maternal plasma.8.3 Females and Males of Reproductive PotentialContraceptionBased on its mechanism of action, LENVIMA can cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy.InfertilityFemalesLENVIMA may result in reduced fertility in females of reproductive potential.MalesLENVIMA may result in damage to male reproductive tissues leading to reduced fertility of unknown duration.8.4 Pediatric UseThe safety and effectiveness of LENVIMA in pediatric patients have not been established.Juvenile Animal DataDaily oral administration of lenvatinib mesylate to juvenile rats for 8 weeks starting on postnatal day 21 (approximately equal to a human pediatric age of 2 years) resulted in growth retardation (decreased body weight gain, decreased food consumption, and decreases in the width and/or length of the femur and tibia) and secondary delays in physical development and reproductive organ immaturity at doses greater than or equal to 2 mg/kg (approximately 1.2 to 5 times the clinical exposure by AUC at the recommended human dose). Decreased length of the femur and tibia persisted following 4 weeks of recovery. In general, the toxicologic profile of lenvatinib was similar between juvenile and adult rats, though toxicities including broken teeth at all dose levels and mortality at the 10 mg/kg/day dose level (attributed to primary duodenal lesions) occurred at earlier treatment time-points in juvenile rats.8.5 Geriatric UseOf 261 patients who received LENVIMA in Study 1, 118 (45.2%) were greater than or equal to 65 years of age and 29 (11.1%) were greater than or equal to 75 years of age. No overall differences in safety or effectiveness were observed between these subjects and younger subjects.8.6 Renal ImpairmentNo dose adjustment is recommended in patients with mild or moderate renal impairment. In patients with severe renal impairment, the recommended dose is 14 mg taken once daily. Patients with end stage renal disease were not studied.8.7 Hepatic ImpairmentNo dose adjustment is recommended in patients with mild or moderate hepatic impairment. In patients with severe hepatic impairment, the recommended dose is 14 mg taken once daily.10 OVERDOSAGEThere is no specific antidote for overdose with LENVIMA. Due to the high plasma protein binding, lenvatinib is not expected to be dialyzable. Adverse reactions in patients receiving single doses of LENVIMA as high as 40 mg were similar to the adverse events reported in the clinical studies at the recommended dose.17 PATIENT COUNSELING INFORMATIONAdvise the patient to read the FDA-approved patient labeling (Patient Information).Hypertension:Advise patients to undergo regular blood pressure monitoring and to contact their health care provider if blood pressure is elevated.Cardiac Dysfunction:Advise patients that LENVIMA can cause cardiac dysfunction and to immediately contact their healthcare provider if they experience any clinical symptoms of cardiac dysfunction such as shortness of breath or swelling of ankles.Arterial Thrombotic EventsAdvise patients to seek immediate medical attention for new onset chest pain or acute neurologic symptoms consistent with myocardial infarction or stroke.Hepatotoxicity:Advise patients that they will need to undergo lab tests to monitor for liver function and to report any new symptoms indicating hepatic toxicity or failure.Proteinuria and Renal Failure/Impairment:Advise patients that they will need to undergo regular lab tests to monitor for kidney function and protein in the urine.Gastrointestinal perforation or fistula formation:Advise patients that LENVIMA can increase the risk of gastrointestinal perforation or fistula and to seek immediate medical attention for severe abdominal pain.Hemorrhagic Events:Advise patients that LENVIMA can increase the risk for bleeding and to contact their health care provider for bleeding or symptoms of severe bleeding.Embryofetal Toxicity:Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy. Lactation:Advise nursing women to discontinue breastfeeding during treatment with LENVIMA.
LENVIMA™ is a trademark of Eisai R&D Management Co., Ltd. and is licensed to Eisai Inc.© 2015 Eisai Inc. All rights reserved. Printed in USA/February 2015 LENV0176
S:14.375”
S:9.5”
T:14.625”
T:10”
Fold:7.3125”Safety:7.0625”
Fold:7.3125”Safety:7.0625”
78618ha_b.indd 4 2/23/15 4:50 PM
LENV-55860_M2_BriefSum_Islnd.indd2-19-2015 6:47 PM Vincent Jeffrey / Suke Yawata
Client CodeClient
LiveOverall TrimBleed
# of Colors
LENV0176 Eisai/LENVIMA
7.0625” x 9.5”7.3125” x 10”None
Black
Colors Black
FontsUnivers LT Std (67 Bold Condensed, 47 Light Con-densed, 47 Light Condensed Oblique)
Job info Fonts & ColorsImages
Saved at
None
from hsvjeffrey5552 by
Printed At
Eisai_K.ai (3.6%)
Notes Brief Summary - Island Spread
LENVIMA™ (lenvatinib) BRIEF SUMMARY – See package insert for full prescribing information.1 INDICATIONS AND USAGELENVIMA is indicated for the treatment of patients with locally recurrent or metastatic, progressive, radioactive iodine-refractory differentiated thyroid cancer (DTC).2 DOSAGE AND ADMINISTRATION2.1 Recommended DoseThe recommended daily dose of LENVIMA is 24 mg (two 10 mg capsules and one 4 mg capsule) orally taken once daily with or without food. Continue LENVIMA until disease progression or until unacceptable toxicity occurs.Take LENVIMA at the same time each day. If a dose is missed and cannot be taken within 12 hours, skip that dose and take the next dose at the usual time of administration.Severe Renal or Hepatic ImpairmentThe recommended dose of LENVIMA is 14 mg taken orally once daily in patients with severe renal impairment (creatinine clearance [CLcr] less than 30 mL/min calculated by the Cockroft-Gault equation) or severe hepatic impairment (Child-Pugh C).2.2 Dose ModificationsHypertension• Assess blood pressure prior to and periodically during treatment. Initiate or adjust medical management to
control blood pressure prior to and during treatment.• Withhold LENVIMA for Grade 3 hypertension that persists despite optimal antihypertensive therapy; resume
at a reduced dose (see Table 1) when hypertension is controlled at less than or equal to Grade 2.• Discontinue LENVIMA for life-threatening hypertension.Cardiac dysfunction or hemorrhage • Discontinue for a Grade 4 event.• Withhold LENVIMA for development of Grade 3 event until improved to Grade 0 or 1 or baseline.• Either resume at a reduced dose (see Table 1) or discontinue LENVIMA depending on the severity and
persistence of the adverse event.Arterial thrombotic event • Discontinue LENVIMA following an arterial thrombotic event.Renal failure and impairment or hepatotoxicity• Withhold LENVIMA for development of Grade 3 or 4 renal failure/impairment or hepatotoxicity until resolved
to Grade 0 to 1 or baseline.• Either resume at a reduced dose (see Table 1) or discontinue LENVIMA depending on the severity and
persistence of renal impairment or hepatotoxicity.• Discontinue LENVIMA for hepatic failure.Proteinuria• Withhold LENVIMA for ≥2 grams of proteinuria/24 hours.• Resume at a reduced dose (see Table 1) when proteinuria is <2 gm/24 hours.• Discontinue LENVIMA for nephrotic syndrome.Gastrointestinal perforation or fistula formation• Discontinue LENVIMA in patients who develop gastrointestinal perforation or life-threatening fistula.QT prolongation• Withhold LENVIMA for the development of Grade 3 or greater QT interval prolongation.• Resume LENVIMA at a reduced dose (see Table 1) when QT prolongation resolves to Grade 0 or 1 or baseline.Reversible posterior leukoencephalopathy syndrome (RPLS)• Withhold for RPLS until fully resolved.• Upon resolution, resume at a reduced dose or discontinue LENVIMA depending on the severity and
persistence of neurologic symptoms.Manage other adverse reactions according to the instructions in Table 1. Based on the absence of clinical experience, there are no recommendations on resumption of dosing in patients with Grade 4 clinical adverse reactions that resolve.
Table 1 Recommended Dose Modifications for Persistent and Intolerable Grade 2 or Grade 3 Adverse Reactions or Grade 4 Laboratory Abnormalitiesa
Adverse Reaction Modification Adjusted Doseb
First occurrence Interrupt until resolved to Grade 0-1 or baseline
20 mg (two 10 mg capsules) orally once daily
Second occurrencec Interrupt until resolved to Grade 0-1 or baseline
14 mg (one 10 mg capsule plus one 4 mg capsule)
orally once daily
Third occurrencec Interrupt until resolved to Grade 0-1 or baseline
10 mg (one 10 mg capsule) orally once daily
a Initiate medical management for nausea, vomiting, or diarrhea prior to interruption or dose reduction of LENVIMA
b Reduce dose in succession based on the previous dose level (24 mg, 20 mg, or 14 mg per day)c Refers to the same or a different adverse reaction that requires dose modification
4 CONTRAINDICATIONSNone.5 WARNINGS AND PRECAUTIONS5.1 HypertensionIn Study 1 hypertension was reported in 73% of LENVIMA-treated patients and 16% of patients in the placebo group. The median time to onset of new or worsening hypertension was 16 days for LENVIMA-treated patients. The incidence of Grade 3 hypertension was 44% as compared to 4% for placebo, and the incidence of Grade 4 hypertension was less than 1% in LENVIMA-treated patients and none in the placebo group.Control blood pressure prior to treatment with LENVIMA. Monitor blood pressure after 1 week, then every 2 weeks for the first 2 months, and then at least monthly thereafter during treatment with LENVIMA. Withhold LENVIMA for Grade 3 hypertension despite optimal antihypertensive therapy; resume at a reduced dose when hypertension is controlled at less than or equal to Grade 2. Discontinue LENVIMA for life-threatening hypertension.5.2 Cardiac DysfunctionIn Study 1, cardiac dysfunction, defined as decreased left or right ventricular function, cardiac failure, or pulmonary edema, was reported in 7% of LENVIMA-treated patients (2% Grade 3 or greater) and 2% (no Grade 3 or greater) of patients in the placebo group. The majority of these cases in LENVIMA-treated patients (14 of 17 cases) were based on findings of decreased ejection fraction as assessed by echocardiography. Six of 261 (2%) LENVIMA-treated patients in Study 1 had greater than 20% reduction in ejection fraction as measured by echocardiography compared to no patients who received placebo.Monitor patients for clinical symptoms or signs of cardiac decompensation. Withhold LENVIMA for development of Grade 3 cardiac dysfunction until improved to Grade 0 or 1 or baseline. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of cardiac dysfunction. Discontinue LENVIMA for Grade 4 cardiac dysfunction.5.3 Arterial Thromboembolic EventsIn Study 1, arterial thromboembolic events were reported in 5% of LENVIMA-treated patients and 2% of patients in the placebo group. The incidence of arterial thromboembolic events of Grade 3 or greater was 3% in LENVIMA-treated patients and 1% in the placebo group.Discontinue LENVIMA following an arterial thrombotic event. The safety of resuming LENVIMA after an arterial thromboembolic event has not been established and LENVIMA has not been studied in patients who have had an arterial thromboembolic event within the previous 6 months.5.4 HepatotoxicityIn Study 1, 4% of LENVIMA-treated patients experienced an increase in alanine aminotransferase (ALT) and 5% experienced an increase in aspartate aminotransferase (AST) that was Grade 3 or greater. No patients in the placebo group experienced Grade 3 or greater increases in ALT or AST. Across clinical studies in which 1108 patients received LENVIMA, hepatic failure (including fatal events) was reported in 3 patients and acute hepatitis was reported in 1 patient.Monitor liver function before initiation of LENVIMA, then every 2 weeks for the first 2 months, and at least monthly thereafter during treatment. Withhold LENVIMA for the development of Grade 3 or greater liver impairment until
resolved to Grade 0 to 1 or baseline. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of hepatotoxicity. Discontinue LENVIMA for hepatic failure.5.5 ProteinuriaIn Study 1, proteinuria was reported in 34% of LENVIMA-treated patients and 3% of patients in the placebo group. The incidence of Grade 3 proteinuria in LENVIMA-treated patients was 11% compared to none in the placebo group.Monitor for proteinuria before initiation of, and periodically throughout treatment. If urine dipstick proteinuria greater than or equal to 2+ is detected, obtain a 24 hour urine protein. Withhold LENVIMA for ≥2 grams of proteinuria/24 hours and resume at a reduced dose when proteinuria is <2 gm/24 hours. Discontinue LENVIMA for nephrotic syndrome.5.6 Renal Failure and ImpairmentIn Study 1, events of renal impairment were reported in 14% of LENVIMA-treated patients compared to 2% of patients in the placebo group. The incidence of Grade 3 or greater renal failure or impairment was 3% in LENVIMA-treated patients and 1% in the placebo group. The primary risk factor for severe renal impairment in LENVIMA-treated patients was dehydration/hypovolemia due to diarrhea and vomiting.Withhold LENVIMA for development of Grade 3 or 4 renal failure/impairment until resolved to Grade 0 to 1 or baseline. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of renal impairment.5.7 Gastrointestinal Perforation and Fistula FormationIn Study 1, events of gastrointestinal perforation or fistula were reported in 2% of LENVIMA-treated patients and 0.8% of patients in the placebo group.Discontinue LENVIMA in patients who develop gastrointestinal perforation or life-threatening fistula.5.8 QT Interval ProlongationIn Study 1, QT/QTc interval prolongation was reported in 9% of LENVIMA-treated patients and 2% of patients in the placebo group. The incidence of QT interval prolongation of Grade 3 or greater was 2% in LENVIMA-treated patients compared to no reports in the placebo group. Monitor electrocardiograms in patients with congenital long QT syndrome, congestive heart failure, bradyarrhythmias, or those who are taking drugs known to prolong the QT interval, including Class Ia and III antiarrhythmics.Monitor and correct electrolyte abnormalities in all patients. Withhold LENVIMA for the development of Grade 3 or greater QT interval prolongation. Resume LENVIMA at a reduced dose when QT prolongation resolves to Grade 0 or 1 or baseline.5.9 HypocalcemiaIn Study 1, 9% of LENVIMA-treated patients experienced Grade 3 or greater hypocalcemia compared to 2% in the placebo group. In most cases hypocalcemia responded to replacement and dose interruption/dose reduction.Monitor blood calcium levels at least monthly and replace calcium as necessary during LENVIMA treatment. Interrupt and adjust LENVIMA dosing as necessary depending on severity, presence of ECG changes, and persistence of hypocalcemia.5.10 Reversible Posterior Leukoencephalopathy SyndromeAcross clinical studies in which 1108 patients received LENVIMA, there were 3 reported events of reversible posterior leukoencephalopathy syndrome (RPLS). Confirm the diagnosis of RPLS with MRI. Withhold for RPLS until fully resolved. Upon resolution, resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of neurologic symptoms.5.11 Hemorrhagic EventsIn Study 1, hemorrhagic events occurred in 35% of LENVIMA-treated patients and in 18% of the placebo group. However, the incidence of Grade 3-5 hemorrhage was similar between arms at 2% and 3%, respectively. The most frequently reported hemorrhagic event was epistaxis (11% Grade 1 and 1% Grade 2). Discontinuation due to hemorrhagic events occurred in 1% of LENVIMA-treated patients.Across clinical studies in which 1108 patients received LENVIMA, Grade 3 or greater hemorrhage was reported in 2% of patients. In Study 1, there was 1 case of fatal intracranial hemorrhage among 16 patients who received lenvatinib and had CNS metastases at baseline.Withhold LENVIMA for the development of Grade 3 hemorrhage until resolved to Grade 0 to 1. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of hemorrhage. Discontinue LENVIMA in patients who experience Grade 4 hemorrhage.5.12 Impairment of Thyroid Stimulating Hormone SuppressionLENVIMA impairs exogenous thyroid suppression. In Study 1, 88% of all patients had a baseline thyroid stimulating hormone (TSH) level less than or equal to 0.5 mU/L. In those patients with a normal TSH at baseline, elevation of TSH level above 0.5 mU/L was observed post baseline in 57% of LENVIMA-treated patients as compared with 14% of patients receiving placebo.Monitor TSH levels monthly and adjust thyroid replacement medication as needed in patients with DTC.5.13 Embryofetal ToxicityBased on its mechanism of action and data from animal reproduction studies, LENVIMA can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of lenvatinib during organogenesis at doses below the recommended human dose resulted in embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy.6 ADVERSE REACTIONSThe following adverse reactions are discussed elsewhere in the label. Please see the Warnings and Precautions sections in the full prescribing information.• Hypertension• Cardiac Dysfunction• Arterial Thromboembolic Events• Hepatotoxicity• Proteinuria• Renal Failure and Impairment• Gastrointestinal Perforation and Fistula Formation• QT Interval Prolongation• Hypocalcemia• Reversible Posterior Leukoencephalopathy Syndrome• Hemorrhagic Events• Impairment of Thyroid Stimulating Hormone Suppression6.1 Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Safety data obtained in 1108 patients with advanced solid tumors who received LENVIMA as a single agent across multiple clinical studies was used to further characterize risks of serious adverse drug reactions. The median age was 60 years (range 21-89 years). The dose range was 0.2 mg to 32 mg. The median duration of exposure in the entire population was 5.5 months.The safety data described below are derived from Study 1 which randomized (2:1) patients with radioactive iodine-refractory differentiated thyroid cancer (RAI-refractory DTC) to LENVIMA (n=261) or placebo (n=131). The median treatment duration was 16.1 months for LENVIMA and 3.9 months for placebo. Among 261 patients who received LENVIMA in Study 1, median age was 64 years, 52% were women, 80% were White, 18% were Asian, and 2% were Black; 4% identified themselves as having Hispanic or Latino ethnicity.In Study 1, the most common adverse reactions observed in LENVIMA-treated patients (greater than or equal to 30%) were, in order of decreasing frequency, hypertension, fatigue, diarrhea, arthralgia/myalgia, decreased appetite, weight decreased, nausea, stomatitis, headache, vomiting, proteinuria, palmar-plantar erythrodysesthesia (PPE) syndrome, abdominal pain, and dysphonia. The most common serious adverse reactions (at least 2%) were pneumonia (4%), hypertension (3%), and dehydration (3%).Adverse reactions led to dose reductions in 68% of patients receiving LENVIMA and 5% of patients receiving placebo; 18% of patients discontinued LENVIMA and 5% discontinued placebo for adverse reactions. The most common adverse reactions (at least 10%) resulting in dose reductions of LENVIMA were hypertension (13%), proteinuria (11%), decreased appetite (10%), and diarrhea (10%); the most common adverse reactions (at least 1%) resulting in discontinuation of LENVIMA were hypertension (1%) and asthenia (1%).Table 2 presents the percentage of patients in Study 1 experiencing adverse reactions at a higher rate in LENVIMA-treated patients than patients receiving placebo in the double-blind phase of the DTC study.
Table 2 Adverse Reactions Occurring in Patients with a Between-Group Difference of Greater than or Equal to 5% All Grades or Greater than or Equal to 2% Grades 3 and 4
Adverse Reaction
LENVIMA 24 mg N=261
Placebo N=131
All Grades (%)
Grades 3-4 (%)
All Grades (%)
Grades 3-4 (%)
Vascular DisordersHypertensiona 73 44 16 4Hypotension 9 2 2 0
Gastrointestinal DisordersDiarrhea 67 9 17 0Nausea 47 2 25 1Stomatitisb 41 5 8 0Vomiting 36 2 15 0Abdominal painc 31 2 11 1Constipation 29 0.4 15 1Oral paind 25 1 2 0Dry mouth 17 0.4 8 0Dyspepsia 13 0.4 4 0
General Disorders and Administration Site ConditionsFatiguee 67 11 35 4Edema peripheral 21 0.4 8 0
Musculoskeletal and Connective Tissue DisordersArthralgia/Myalgiaf 62 5 28 3
Metabolism and Nutrition DisordersWeight decreased 51 13 15 1Decreased appetite 54 7 18 1Dehydration 9 2 2 1
Nervous System DisordersHeadache 38 3 11 1Dysgeusia 18 0 3 0Dizziness 15 0.4 9 0
Renal and Urinary DisordersProteinuria 34 11 3 0
Skin and Subcutaneous Tissue DisordersPalmar-plantar erythrodysesthesia 32 3 1 0Rashg 21 0.4 3 0Alopecia 12 0 5 0Hyperkeratosis 7 0 2 0
Respiratory, Thoracic and Mediastinal DisordersDysphonia 31 1 5 0Cough 24 0 18 0Epistaxis 12 0 1 0
Psychiatric DisordersInsomnia 12 0 3 0
Infections and InfestationsDental and oral infectionsh 10 1 1 0Urinary tract infection 11 1 5 0
Cardiac Disorders
Electrocardiogram QT prolonged 9 2 2 0a Includes hypertension, hypertensive crisis, increased blood pressure diastolic, and increased
blood pressure b Includes aphthous stomatitis, stomatitis, glossitis, mouth ulceration, and mucosal inflammationc Includes abdominal discomfort, abdominal pain, abdominal pain lower, abdominal pain upper,
abdominal tenderness, epigastric discomfort, and gastrointestinal paind Includes oral pain, glossodynia, and oropharyngeal paine Includes asthenia, fatigue, and malaisef Includes musculoskeletal pain, back pain, pain in extremity, arthralgia, and myalgiag Includes rash macular, rash maculo-papular, rash generalized, and rash h Includes gingivitis, oral infection, parotitis, pericoronitis, periodontitis, sialoadenitis, tooth abscess,
and tooth infectionA clinically important adverse reaction occurring more frequently in LENVIMA-treated patients than patients receiving placebo, but with an incidence of less than 5% was pulmonary embolism (3%, including fatal reports vs 2%, respectively).
Table 3 Laboratory Abnormalities with a Difference of at Least ≥2% in Grade 3 - 4 Events and at a Higher Incidence in LENVIMA-Treated Patientsa
Laboratory Abnormality LENVIMA 24 mg N=258b
Placebo N=131b
Grades 3-4 (%)
Grades 3-4 (%)
ChemistryCreatinine increased 3 0Alanine aminotransferase (ALT) increased 4 0Aspartate aminotransferase (AST) increased 5 0Hypocalcemia 9 2Hypokalemia 6 1Lipase increased 4 1
HematologyPlatelet count decreased 2 0
a With at least 1 grade increase from baseline b Subject with at least 1 post baseline laboratory value
In addition the following laboratory abnormalities (all Grades) occurred in greater than 5% of LENVIMA-treated patients and at a rate that was two-fold or higher than in patients who received placebo: hypoalbuminemia, increased alkaline phosphatase, hypomagnesemia, hypoglycemia, hyperbilirubinemia, hypercalcemia, hypercholesterolemia, increased serum amylase, and hyperkalemia.7 DRUG INTERACTIONS7.1 Effect of Other Drugs on LenvatinibNo dose adjustment of LENVIMA is recommended when co-administered with CYP3A, P-glycoprotein (P-gp), and breast cancer resistance protein (BCRP) inhibitors and CYP3A and P-gp inducers.8 USE IN SPECIFIC POPULATIONS8.1 PregnancyRisk SummaryBased on its mechanism of action and data from animal reproduction studies, LENVIMA can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of lenvatinib during organogenesis at doses below the recommended human dose resulted in embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits. There are no available human data informing the drug-associated risk. Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown; however, the background risk in the U.S. general population of major birth defects is 2-4% and of miscarriage is 15-20% of clinically recognized pregnancies.DataAnimal DataIn an embryofetal development study, daily oral administration of lenvatinib mesylate at doses greater than or equal to 0.3 mg/kg [approximately 0.14 times the recommended human dose based on body surface area (BSA)] to pregnant rats during organogenesis resulted in dose-related decreases in mean fetal body weight, delayed fetal ossifications, and dose-related increases in fetal external (parietal edema and tail abnormalities), visceral, and skeletal anomalies. Greater than 80% postimplantation loss was observed at 1.0 mg/kg/day (approximately 0.5 times the recommended human dose based on BSA).Daily oral administration of lenvatinib mesylate to pregnant rabbits during organogenesis resulted in fetal external (short tail), visceral (retroesophageal subclavian artery), and skeletal anomalies at doses greater than or equal to 0.03 mg/kg (approximately 0.03 times the human dose of 24 mg based on body surface area). At the 0.03 mg/kg dose, increased post-implantation loss, including 1 fetal death, was also observed. Lenvatinib was abortifacient in rabbits, resulting in late abortions in approximately one-third of the rabbits treated at a dose level of 0.5 mg/kg/day (approximately 0.5 times the recommended clinical dose of 24 mg based on BSA).8.2 LactationRisk SummaryIt is not known whether LENVIMA is present in human milk. However, lenvatinib and its metabolites are excreted in rat milk at concentrations higher than in maternal plasma. Because of the potential for serious adverse reactions in nursing infants from LENVIMA, advise women to discontinue breastfeeding during treatment with LENVIMA.DataAnimal DataFollowing administration of radiolabeled lenvatinib to lactating Sprague Dawley rats, lenvatinib-related radioactivity was approximately 2 times higher (based on AUC) in milk compared to maternal plasma.8.3 Females and Males of Reproductive PotentialContraceptionBased on its mechanism of action, LENVIMA can cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy.InfertilityFemalesLENVIMA may result in reduced fertility in females of reproductive potential.MalesLENVIMA may result in damage to male reproductive tissues leading to reduced fertility of unknown duration.8.4 Pediatric UseThe safety and effectiveness of LENVIMA in pediatric patients have not been established.Juvenile Animal DataDaily oral administration of lenvatinib mesylate to juvenile rats for 8 weeks starting on postnatal day 21 (approximately equal to a human pediatric age of 2 years) resulted in growth retardation (decreased body weight gain, decreased food consumption, and decreases in the width and/or length of the femur and tibia) and secondary delays in physical development and reproductive organ immaturity at doses greater than or equal to 2 mg/kg (approximately 1.2 to 5 times the clinical exposure by AUC at the recommended human dose). Decreased length of the femur and tibia persisted following 4 weeks of recovery. In general, the toxicologic profile of lenvatinib was similar between juvenile and adult rats, though toxicities including broken teeth at all dose levels and mortality at the 10 mg/kg/day dose level (attributed to primary duodenal lesions) occurred at earlier treatment time-points in juvenile rats.8.5 Geriatric UseOf 261 patients who received LENVIMA in Study 1, 118 (45.2%) were greater than or equal to 65 years of age and 29 (11.1%) were greater than or equal to 75 years of age. No overall differences in safety or effectiveness were observed between these subjects and younger subjects.8.6 Renal ImpairmentNo dose adjustment is recommended in patients with mild or moderate renal impairment. In patients with severe renal impairment, the recommended dose is 14 mg taken once daily. Patients with end stage renal disease were not studied.8.7 Hepatic ImpairmentNo dose adjustment is recommended in patients with mild or moderate hepatic impairment. In patients with severe hepatic impairment, the recommended dose is 14 mg taken once daily.10 OVERDOSAGEThere is no specific antidote for overdose with LENVIMA. Due to the high plasma protein binding, lenvatinib is not expected to be dialyzable. Adverse reactions in patients receiving single doses of LENVIMA as high as 40 mg were similar to the adverse events reported in the clinical studies at the recommended dose.17 PATIENT COUNSELING INFORMATIONAdvise the patient to read the FDA-approved patient labeling (Patient Information).Hypertension:Advise patients to undergo regular blood pressure monitoring and to contact their health care provider if blood pressure is elevated.Cardiac Dysfunction:Advise patients that LENVIMA can cause cardiac dysfunction and to immediately contact their healthcare provider if they experience any clinical symptoms of cardiac dysfunction such as shortness of breath or swelling of ankles.Arterial Thrombotic EventsAdvise patients to seek immediate medical attention for new onset chest pain or acute neurologic symptoms consistent with myocardial infarction or stroke.Hepatotoxicity:Advise patients that they will need to undergo lab tests to monitor for liver function and to report any new symptoms indicating hepatic toxicity or failure.Proteinuria and Renal Failure/Impairment:Advise patients that they will need to undergo regular lab tests to monitor for kidney function and protein in the urine.Gastrointestinal perforation or fistula formation:Advise patients that LENVIMA can increase the risk of gastrointestinal perforation or fistula and to seek immediate medical attention for severe abdominal pain.Hemorrhagic Events:Advise patients that LENVIMA can increase the risk for bleeding and to contact their health care provider for bleeding or symptoms of severe bleeding.Embryofetal Toxicity:Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy. Lactation:Advise nursing women to discontinue breastfeeding during treatment with LENVIMA.
LENVIMA™ is a trademark of Eisai R&D Management Co., Ltd. and is licensed to Eisai Inc.© 2015 Eisai Inc. All rights reserved. Printed in USA/February 2015 LENV0176
S:14.375”
S:9.5”
T:14.625”
T:10”
Fold:7.3125”Safety:7.0625”
Fold:7.3125”Safety:7.0625”
78618ha_b.indd 3 2/23/15 4:50 PM

ASCOPost.com | MARCH 25, 2015 PAGE 53
Announcements
cially incentivize high-quality, coordinat-ed care. Participating practices will also receive monthly care management pay-ments for each Medicare fee-for-service beneficiary during an episode to support oncology practice transformation, in-cluding the provision of comprehensive, coordinated patient care.
“With the Oncology Care Model,
CMS has the opportunity to achieve three goals in the care of this medically complex population who are facing a cancer diag-nosis: better care, smarter spending, and healthier people,” added Dr. Conway. “As a practicing physician and son of a Medi-care beneficiary who died of cancer, I know the importance of well-coordinated care focused on the patient’s needs.”
Application DetailsPhysician group practices and solo
practitioners who provide chemotherapy for cancer and are currently enrolled in Medicare may apply to participate. Other payers, including commercial insurers, state programs, and Medicaid managed-care plans, are also encouraged to apply.
To be considered, interested payers
must submit a letter of intent through the Oncology Care Model inbox at [email protected] by March 19, 2015. Interested practices must submit letters of intent by April 23, 2015. Practices and payers who submit a timely letter of intent will be sent an authenticated web link and password with which to sub-mit an electronic application. Applications must be submitted by June 18, 2015. n
Disclosure: Dr. Conway reported no potential conflicts of interest.
ASCO Expresses Concern Over New Oncology Care Model
While commending the Cen-ters for Medicare & Medic-
aid Services (CMS) for seeking new approaches to physician payment, ASCO expressed concerns over the model’s limited scope. “We are disappointed [CMS has] chosen to pursue only one model—and one that continues to rely on a broken fee-for-service system,” said ASCO Chief Medical Officer Richard L. Schilsky, MD, FACP, FASCO.
In comments submitted to CMS on a draft version of its mod-el, ASCO supported testing the Oncology Care Model as well as other payment reform models to determine new approaches to pay-ment for oncology care. Moreover, ASCO urged the Center to test models that include more funda-mental reform, which moves away from the fee-for-service system.
“ASCO looks forward to work-ing with both public and private payers to explore new payment strategies that better reflect mod-ern oncology practice and sup-port high-value, patient-centered care,” said Dr. Schilsky.
ASCO has developed a com-prehensive proposal that, it be-lieves, better matches payments to the work performed by can-cer care providers. In May 2014, ASCO released its Consolidated Payments for Oncology: Payment Reform to Support Patient-Centered Care for Cancer, outlining a new approach to physician payment for cancer care services under Medicare, and shared it with CMS and private insurance companies. The Society is also working on pi-loting its proposal in different U.S. oncology settings. n
LENV-55860_M2_BriefSum_Islnd.indd2-19-2015 6:47 PM Vincent Jeffrey / Suke Yawata
Client CodeClient
LiveOverall TrimBleed
# of Colors
LENV0176 Eisai/LENVIMA
7.0625” x 9.5”7.3125” x 10”None
Black
Colors Black
FontsUnivers LT Std (67 Bold Condensed, 47 Light Con-densed, 47 Light Condensed Oblique)
Job info Fonts & ColorsImages
Saved at
None
from hsvjeffrey5552 by
Printed At
Eisai_K.ai (3.6%)
Notes Brief Summary - Island Spread
LENVIMA™ (lenvatinib) BRIEF SUMMARY – See package insert for full prescribing information.1 INDICATIONS AND USAGELENVIMA is indicated for the treatment of patients with locally recurrent or metastatic, progressive, radioactive iodine-refractory differentiated thyroid cancer (DTC).2 DOSAGE AND ADMINISTRATION2.1 Recommended DoseThe recommended daily dose of LENVIMA is 24 mg (two 10 mg capsules and one 4 mg capsule) orally taken once daily with or without food. Continue LENVIMA until disease progression or until unacceptable toxicity occurs.Take LENVIMA at the same time each day. If a dose is missed and cannot be taken within 12 hours, skip that dose and take the next dose at the usual time of administration.Severe Renal or Hepatic ImpairmentThe recommended dose of LENVIMA is 14 mg taken orally once daily in patients with severe renal impairment (creatinine clearance [CLcr] less than 30 mL/min calculated by the Cockroft-Gault equation) or severe hepatic impairment (Child-Pugh C).2.2 Dose ModificationsHypertension• Assess blood pressure prior to and periodically during treatment. Initiate or adjust medical management to
control blood pressure prior to and during treatment.• Withhold LENVIMA for Grade 3 hypertension that persists despite optimal antihypertensive therapy; resume
at a reduced dose (see Table 1) when hypertension is controlled at less than or equal to Grade 2.• Discontinue LENVIMA for life-threatening hypertension.Cardiac dysfunction or hemorrhage • Discontinue for a Grade 4 event.• Withhold LENVIMA for development of Grade 3 event until improved to Grade 0 or 1 or baseline.• Either resume at a reduced dose (see Table 1) or discontinue LENVIMA depending on the severity and
persistence of the adverse event.Arterial thrombotic event • Discontinue LENVIMA following an arterial thrombotic event.Renal failure and impairment or hepatotoxicity• Withhold LENVIMA for development of Grade 3 or 4 renal failure/impairment or hepatotoxicity until resolved
to Grade 0 to 1 or baseline.• Either resume at a reduced dose (see Table 1) or discontinue LENVIMA depending on the severity and
persistence of renal impairment or hepatotoxicity.• Discontinue LENVIMA for hepatic failure.Proteinuria• Withhold LENVIMA for ≥2 grams of proteinuria/24 hours.• Resume at a reduced dose (see Table 1) when proteinuria is <2 gm/24 hours.• Discontinue LENVIMA for nephrotic syndrome.Gastrointestinal perforation or fistula formation• Discontinue LENVIMA in patients who develop gastrointestinal perforation or life-threatening fistula.QT prolongation• Withhold LENVIMA for the development of Grade 3 or greater QT interval prolongation.• Resume LENVIMA at a reduced dose (see Table 1) when QT prolongation resolves to Grade 0 or 1 or baseline.Reversible posterior leukoencephalopathy syndrome (RPLS)• Withhold for RPLS until fully resolved.• Upon resolution, resume at a reduced dose or discontinue LENVIMA depending on the severity and
persistence of neurologic symptoms.Manage other adverse reactions according to the instructions in Table 1. Based on the absence of clinical experience, there are no recommendations on resumption of dosing in patients with Grade 4 clinical adverse reactions that resolve.
Table 1 Recommended Dose Modifications for Persistent and Intolerable Grade 2 or Grade 3 Adverse Reactions or Grade 4 Laboratory Abnormalitiesa
Adverse Reaction Modification Adjusted Doseb
First occurrence Interrupt until resolved to Grade 0-1 or baseline
20 mg (two 10 mg capsules) orally once daily
Second occurrencec Interrupt until resolved to Grade 0-1 or baseline
14 mg (one 10 mg capsule plus one 4 mg capsule)
orally once daily
Third occurrencec Interrupt until resolved to Grade 0-1 or baseline
10 mg (one 10 mg capsule) orally once daily
a Initiate medical management for nausea, vomiting, or diarrhea prior to interruption or dose reduction of LENVIMA
b Reduce dose in succession based on the previous dose level (24 mg, 20 mg, or 14 mg per day)c Refers to the same or a different adverse reaction that requires dose modification
4 CONTRAINDICATIONSNone.5 WARNINGS AND PRECAUTIONS5.1 HypertensionIn Study 1 hypertension was reported in 73% of LENVIMA-treated patients and 16% of patients in the placebo group. The median time to onset of new or worsening hypertension was 16 days for LENVIMA-treated patients. The incidence of Grade 3 hypertension was 44% as compared to 4% for placebo, and the incidence of Grade 4 hypertension was less than 1% in LENVIMA-treated patients and none in the placebo group.Control blood pressure prior to treatment with LENVIMA. Monitor blood pressure after 1 week, then every 2 weeks for the first 2 months, and then at least monthly thereafter during treatment with LENVIMA. Withhold LENVIMA for Grade 3 hypertension despite optimal antihypertensive therapy; resume at a reduced dose when hypertension is controlled at less than or equal to Grade 2. Discontinue LENVIMA for life-threatening hypertension.5.2 Cardiac DysfunctionIn Study 1, cardiac dysfunction, defined as decreased left or right ventricular function, cardiac failure, or pulmonary edema, was reported in 7% of LENVIMA-treated patients (2% Grade 3 or greater) and 2% (no Grade 3 or greater) of patients in the placebo group. The majority of these cases in LENVIMA-treated patients (14 of 17 cases) were based on findings of decreased ejection fraction as assessed by echocardiography. Six of 261 (2%) LENVIMA-treated patients in Study 1 had greater than 20% reduction in ejection fraction as measured by echocardiography compared to no patients who received placebo.Monitor patients for clinical symptoms or signs of cardiac decompensation. Withhold LENVIMA for development of Grade 3 cardiac dysfunction until improved to Grade 0 or 1 or baseline. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of cardiac dysfunction. Discontinue LENVIMA for Grade 4 cardiac dysfunction.5.3 Arterial Thromboembolic EventsIn Study 1, arterial thromboembolic events were reported in 5% of LENVIMA-treated patients and 2% of patients in the placebo group. The incidence of arterial thromboembolic events of Grade 3 or greater was 3% in LENVIMA-treated patients and 1% in the placebo group.Discontinue LENVIMA following an arterial thrombotic event. The safety of resuming LENVIMA after an arterial thromboembolic event has not been established and LENVIMA has not been studied in patients who have had an arterial thromboembolic event within the previous 6 months.5.4 HepatotoxicityIn Study 1, 4% of LENVIMA-treated patients experienced an increase in alanine aminotransferase (ALT) and 5% experienced an increase in aspartate aminotransferase (AST) that was Grade 3 or greater. No patients in the placebo group experienced Grade 3 or greater increases in ALT or AST. Across clinical studies in which 1108 patients received LENVIMA, hepatic failure (including fatal events) was reported in 3 patients and acute hepatitis was reported in 1 patient.Monitor liver function before initiation of LENVIMA, then every 2 weeks for the first 2 months, and at least monthly thereafter during treatment. Withhold LENVIMA for the development of Grade 3 or greater liver impairment until
resolved to Grade 0 to 1 or baseline. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of hepatotoxicity. Discontinue LENVIMA for hepatic failure.5.5 ProteinuriaIn Study 1, proteinuria was reported in 34% of LENVIMA-treated patients and 3% of patients in the placebo group. The incidence of Grade 3 proteinuria in LENVIMA-treated patients was 11% compared to none in the placebo group.Monitor for proteinuria before initiation of, and periodically throughout treatment. If urine dipstick proteinuria greater than or equal to 2+ is detected, obtain a 24 hour urine protein. Withhold LENVIMA for ≥2 grams of proteinuria/24 hours and resume at a reduced dose when proteinuria is <2 gm/24 hours. Discontinue LENVIMA for nephrotic syndrome.5.6 Renal Failure and ImpairmentIn Study 1, events of renal impairment were reported in 14% of LENVIMA-treated patients compared to 2% of patients in the placebo group. The incidence of Grade 3 or greater renal failure or impairment was 3% in LENVIMA-treated patients and 1% in the placebo group. The primary risk factor for severe renal impairment in LENVIMA-treated patients was dehydration/hypovolemia due to diarrhea and vomiting.Withhold LENVIMA for development of Grade 3 or 4 renal failure/impairment until resolved to Grade 0 to 1 or baseline. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of renal impairment.5.7 Gastrointestinal Perforation and Fistula FormationIn Study 1, events of gastrointestinal perforation or fistula were reported in 2% of LENVIMA-treated patients and 0.8% of patients in the placebo group.Discontinue LENVIMA in patients who develop gastrointestinal perforation or life-threatening fistula.5.8 QT Interval ProlongationIn Study 1, QT/QTc interval prolongation was reported in 9% of LENVIMA-treated patients and 2% of patients in the placebo group. The incidence of QT interval prolongation of Grade 3 or greater was 2% in LENVIMA-treated patients compared to no reports in the placebo group. Monitor electrocardiograms in patients with congenital long QT syndrome, congestive heart failure, bradyarrhythmias, or those who are taking drugs known to prolong the QT interval, including Class Ia and III antiarrhythmics.Monitor and correct electrolyte abnormalities in all patients. Withhold LENVIMA for the development of Grade 3 or greater QT interval prolongation. Resume LENVIMA at a reduced dose when QT prolongation resolves to Grade 0 or 1 or baseline.5.9 HypocalcemiaIn Study 1, 9% of LENVIMA-treated patients experienced Grade 3 or greater hypocalcemia compared to 2% in the placebo group. In most cases hypocalcemia responded to replacement and dose interruption/dose reduction.Monitor blood calcium levels at least monthly and replace calcium as necessary during LENVIMA treatment. Interrupt and adjust LENVIMA dosing as necessary depending on severity, presence of ECG changes, and persistence of hypocalcemia.5.10 Reversible Posterior Leukoencephalopathy SyndromeAcross clinical studies in which 1108 patients received LENVIMA, there were 3 reported events of reversible posterior leukoencephalopathy syndrome (RPLS). Confirm the diagnosis of RPLS with MRI. Withhold for RPLS until fully resolved. Upon resolution, resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of neurologic symptoms.5.11 Hemorrhagic EventsIn Study 1, hemorrhagic events occurred in 35% of LENVIMA-treated patients and in 18% of the placebo group. However, the incidence of Grade 3-5 hemorrhage was similar between arms at 2% and 3%, respectively. The most frequently reported hemorrhagic event was epistaxis (11% Grade 1 and 1% Grade 2). Discontinuation due to hemorrhagic events occurred in 1% of LENVIMA-treated patients.Across clinical studies in which 1108 patients received LENVIMA, Grade 3 or greater hemorrhage was reported in 2% of patients. In Study 1, there was 1 case of fatal intracranial hemorrhage among 16 patients who received lenvatinib and had CNS metastases at baseline.Withhold LENVIMA for the development of Grade 3 hemorrhage until resolved to Grade 0 to 1. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of hemorrhage. Discontinue LENVIMA in patients who experience Grade 4 hemorrhage.5.12 Impairment of Thyroid Stimulating Hormone SuppressionLENVIMA impairs exogenous thyroid suppression. In Study 1, 88% of all patients had a baseline thyroid stimulating hormone (TSH) level less than or equal to 0.5 mU/L. In those patients with a normal TSH at baseline, elevation of TSH level above 0.5 mU/L was observed post baseline in 57% of LENVIMA-treated patients as compared with 14% of patients receiving placebo.Monitor TSH levels monthly and adjust thyroid replacement medication as needed in patients with DTC.5.13 Embryofetal ToxicityBased on its mechanism of action and data from animal reproduction studies, LENVIMA can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of lenvatinib during organogenesis at doses below the recommended human dose resulted in embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy.6 ADVERSE REACTIONSThe following adverse reactions are discussed elsewhere in the label. Please see the Warnings and Precautions sections in the full prescribing information.• Hypertension• Cardiac Dysfunction• Arterial Thromboembolic Events• Hepatotoxicity• Proteinuria• Renal Failure and Impairment• Gastrointestinal Perforation and Fistula Formation• QT Interval Prolongation• Hypocalcemia• Reversible Posterior Leukoencephalopathy Syndrome• Hemorrhagic Events• Impairment of Thyroid Stimulating Hormone Suppression6.1 Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Safety data obtained in 1108 patients with advanced solid tumors who received LENVIMA as a single agent across multiple clinical studies was used to further characterize risks of serious adverse drug reactions. The median age was 60 years (range 21-89 years). The dose range was 0.2 mg to 32 mg. The median duration of exposure in the entire population was 5.5 months.The safety data described below are derived from Study 1 which randomized (2:1) patients with radioactive iodine-refractory differentiated thyroid cancer (RAI-refractory DTC) to LENVIMA (n=261) or placebo (n=131). The median treatment duration was 16.1 months for LENVIMA and 3.9 months for placebo. Among 261 patients who received LENVIMA in Study 1, median age was 64 years, 52% were women, 80% were White, 18% were Asian, and 2% were Black; 4% identified themselves as having Hispanic or Latino ethnicity.In Study 1, the most common adverse reactions observed in LENVIMA-treated patients (greater than or equal to 30%) were, in order of decreasing frequency, hypertension, fatigue, diarrhea, arthralgia/myalgia, decreased appetite, weight decreased, nausea, stomatitis, headache, vomiting, proteinuria, palmar-plantar erythrodysesthesia (PPE) syndrome, abdominal pain, and dysphonia. The most common serious adverse reactions (at least 2%) were pneumonia (4%), hypertension (3%), and dehydration (3%).Adverse reactions led to dose reductions in 68% of patients receiving LENVIMA and 5% of patients receiving placebo; 18% of patients discontinued LENVIMA and 5% discontinued placebo for adverse reactions. The most common adverse reactions (at least 10%) resulting in dose reductions of LENVIMA were hypertension (13%), proteinuria (11%), decreased appetite (10%), and diarrhea (10%); the most common adverse reactions (at least 1%) resulting in discontinuation of LENVIMA were hypertension (1%) and asthenia (1%).Table 2 presents the percentage of patients in Study 1 experiencing adverse reactions at a higher rate in LENVIMA-treated patients than patients receiving placebo in the double-blind phase of the DTC study.
Table 2 Adverse Reactions Occurring in Patients with a Between-Group Difference of Greater than or Equal to 5% All Grades or Greater than or Equal to 2% Grades 3 and 4
Adverse Reaction
LENVIMA 24 mg N=261
Placebo N=131
All Grades (%)
Grades 3-4 (%)
All Grades (%)
Grades 3-4 (%)
Vascular DisordersHypertensiona 73 44 16 4Hypotension 9 2 2 0
Gastrointestinal DisordersDiarrhea 67 9 17 0Nausea 47 2 25 1Stomatitisb 41 5 8 0Vomiting 36 2 15 0Abdominal painc 31 2 11 1Constipation 29 0.4 15 1Oral paind 25 1 2 0Dry mouth 17 0.4 8 0Dyspepsia 13 0.4 4 0
General Disorders and Administration Site ConditionsFatiguee 67 11 35 4Edema peripheral 21 0.4 8 0
Musculoskeletal and Connective Tissue DisordersArthralgia/Myalgiaf 62 5 28 3
Metabolism and Nutrition DisordersWeight decreased 51 13 15 1Decreased appetite 54 7 18 1Dehydration 9 2 2 1
Nervous System DisordersHeadache 38 3 11 1Dysgeusia 18 0 3 0Dizziness 15 0.4 9 0
Renal and Urinary DisordersProteinuria 34 11 3 0
Skin and Subcutaneous Tissue DisordersPalmar-plantar erythrodysesthesia 32 3 1 0Rashg 21 0.4 3 0Alopecia 12 0 5 0Hyperkeratosis 7 0 2 0
Respiratory, Thoracic and Mediastinal DisordersDysphonia 31 1 5 0Cough 24 0 18 0Epistaxis 12 0 1 0
Psychiatric DisordersInsomnia 12 0 3 0
Infections and InfestationsDental and oral infectionsh 10 1 1 0Urinary tract infection 11 1 5 0
Cardiac Disorders
Electrocardiogram QT prolonged 9 2 2 0a Includes hypertension, hypertensive crisis, increased blood pressure diastolic, and increased
blood pressure b Includes aphthous stomatitis, stomatitis, glossitis, mouth ulceration, and mucosal inflammationc Includes abdominal discomfort, abdominal pain, abdominal pain lower, abdominal pain upper,
abdominal tenderness, epigastric discomfort, and gastrointestinal paind Includes oral pain, glossodynia, and oropharyngeal paine Includes asthenia, fatigue, and malaisef Includes musculoskeletal pain, back pain, pain in extremity, arthralgia, and myalgiag Includes rash macular, rash maculo-papular, rash generalized, and rash h Includes gingivitis, oral infection, parotitis, pericoronitis, periodontitis, sialoadenitis, tooth abscess,
and tooth infectionA clinically important adverse reaction occurring more frequently in LENVIMA-treated patients than patients receiving placebo, but with an incidence of less than 5% was pulmonary embolism (3%, including fatal reports vs 2%, respectively).
Table 3 Laboratory Abnormalities with a Difference of at Least ≥2% in Grade 3 - 4 Events and at a Higher Incidence in LENVIMA-Treated Patientsa
Laboratory Abnormality LENVIMA 24 mg N=258b
Placebo N=131b
Grades 3-4 (%)
Grades 3-4 (%)
ChemistryCreatinine increased 3 0Alanine aminotransferase (ALT) increased 4 0Aspartate aminotransferase (AST) increased 5 0Hypocalcemia 9 2Hypokalemia 6 1Lipase increased 4 1
HematologyPlatelet count decreased 2 0
a With at least 1 grade increase from baseline b Subject with at least 1 post baseline laboratory value
In addition the following laboratory abnormalities (all Grades) occurred in greater than 5% of LENVIMA-treated patients and at a rate that was two-fold or higher than in patients who received placebo: hypoalbuminemia, increased alkaline phosphatase, hypomagnesemia, hypoglycemia, hyperbilirubinemia, hypercalcemia, hypercholesterolemia, increased serum amylase, and hyperkalemia.7 DRUG INTERACTIONS7.1 Effect of Other Drugs on LenvatinibNo dose adjustment of LENVIMA is recommended when co-administered with CYP3A, P-glycoprotein (P-gp), and breast cancer resistance protein (BCRP) inhibitors and CYP3A and P-gp inducers.8 USE IN SPECIFIC POPULATIONS8.1 PregnancyRisk SummaryBased on its mechanism of action and data from animal reproduction studies, LENVIMA can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of lenvatinib during organogenesis at doses below the recommended human dose resulted in embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits. There are no available human data informing the drug-associated risk. Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown; however, the background risk in the U.S. general population of major birth defects is 2-4% and of miscarriage is 15-20% of clinically recognized pregnancies.DataAnimal DataIn an embryofetal development study, daily oral administration of lenvatinib mesylate at doses greater than or equal to 0.3 mg/kg [approximately 0.14 times the recommended human dose based on body surface area (BSA)] to pregnant rats during organogenesis resulted in dose-related decreases in mean fetal body weight, delayed fetal ossifications, and dose-related increases in fetal external (parietal edema and tail abnormalities), visceral, and skeletal anomalies. Greater than 80% postimplantation loss was observed at 1.0 mg/kg/day (approximately 0.5 times the recommended human dose based on BSA).Daily oral administration of lenvatinib mesylate to pregnant rabbits during organogenesis resulted in fetal external (short tail), visceral (retroesophageal subclavian artery), and skeletal anomalies at doses greater than or equal to 0.03 mg/kg (approximately 0.03 times the human dose of 24 mg based on body surface area). At the 0.03 mg/kg dose, increased post-implantation loss, including 1 fetal death, was also observed. Lenvatinib was abortifacient in rabbits, resulting in late abortions in approximately one-third of the rabbits treated at a dose level of 0.5 mg/kg/day (approximately 0.5 times the recommended clinical dose of 24 mg based on BSA).8.2 LactationRisk SummaryIt is not known whether LENVIMA is present in human milk. However, lenvatinib and its metabolites are excreted in rat milk at concentrations higher than in maternal plasma. Because of the potential for serious adverse reactions in nursing infants from LENVIMA, advise women to discontinue breastfeeding during treatment with LENVIMA.DataAnimal DataFollowing administration of radiolabeled lenvatinib to lactating Sprague Dawley rats, lenvatinib-related radioactivity was approximately 2 times higher (based on AUC) in milk compared to maternal plasma.8.3 Females and Males of Reproductive PotentialContraceptionBased on its mechanism of action, LENVIMA can cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy.InfertilityFemalesLENVIMA may result in reduced fertility in females of reproductive potential.MalesLENVIMA may result in damage to male reproductive tissues leading to reduced fertility of unknown duration.8.4 Pediatric UseThe safety and effectiveness of LENVIMA in pediatric patients have not been established.Juvenile Animal DataDaily oral administration of lenvatinib mesylate to juvenile rats for 8 weeks starting on postnatal day 21 (approximately equal to a human pediatric age of 2 years) resulted in growth retardation (decreased body weight gain, decreased food consumption, and decreases in the width and/or length of the femur and tibia) and secondary delays in physical development and reproductive organ immaturity at doses greater than or equal to 2 mg/kg (approximately 1.2 to 5 times the clinical exposure by AUC at the recommended human dose). Decreased length of the femur and tibia persisted following 4 weeks of recovery. In general, the toxicologic profile of lenvatinib was similar between juvenile and adult rats, though toxicities including broken teeth at all dose levels and mortality at the 10 mg/kg/day dose level (attributed to primary duodenal lesions) occurred at earlier treatment time-points in juvenile rats.8.5 Geriatric UseOf 261 patients who received LENVIMA in Study 1, 118 (45.2%) were greater than or equal to 65 years of age and 29 (11.1%) were greater than or equal to 75 years of age. No overall differences in safety or effectiveness were observed between these subjects and younger subjects.8.6 Renal ImpairmentNo dose adjustment is recommended in patients with mild or moderate renal impairment. In patients with severe renal impairment, the recommended dose is 14 mg taken once daily. Patients with end stage renal disease were not studied.8.7 Hepatic ImpairmentNo dose adjustment is recommended in patients with mild or moderate hepatic impairment. In patients with severe hepatic impairment, the recommended dose is 14 mg taken once daily.10 OVERDOSAGEThere is no specific antidote for overdose with LENVIMA. Due to the high plasma protein binding, lenvatinib is not expected to be dialyzable. Adverse reactions in patients receiving single doses of LENVIMA as high as 40 mg were similar to the adverse events reported in the clinical studies at the recommended dose.17 PATIENT COUNSELING INFORMATIONAdvise the patient to read the FDA-approved patient labeling (Patient Information).Hypertension:Advise patients to undergo regular blood pressure monitoring and to contact their health care provider if blood pressure is elevated.Cardiac Dysfunction:Advise patients that LENVIMA can cause cardiac dysfunction and to immediately contact their healthcare provider if they experience any clinical symptoms of cardiac dysfunction such as shortness of breath or swelling of ankles.Arterial Thrombotic EventsAdvise patients to seek immediate medical attention for new onset chest pain or acute neurologic symptoms consistent with myocardial infarction or stroke.Hepatotoxicity:Advise patients that they will need to undergo lab tests to monitor for liver function and to report any new symptoms indicating hepatic toxicity or failure.Proteinuria and Renal Failure/Impairment:Advise patients that they will need to undergo regular lab tests to monitor for kidney function and protein in the urine.Gastrointestinal perforation or fistula formation:Advise patients that LENVIMA can increase the risk of gastrointestinal perforation or fistula and to seek immediate medical attention for severe abdominal pain.Hemorrhagic Events:Advise patients that LENVIMA can increase the risk for bleeding and to contact their health care provider for bleeding or symptoms of severe bleeding.Embryofetal Toxicity:Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy. Lactation:Advise nursing women to discontinue breastfeeding during treatment with LENVIMA.
LENVIMA™ is a trademark of Eisai R&D Management Co., Ltd. and is licensed to Eisai Inc.© 2015 Eisai Inc. All rights reserved. Printed in USA/February 2015 LENV0176
S:14.375”
S:9.5”
T:14.625”
T:10”
Fold:7.3125”Safety:7.0625”
Fold:7.3125”Safety:7.0625”
78618ha_b.indd 4 2/23/15 4:50 PM
LENV-55860_M2_BriefSum_Islnd.indd2-19-2015 6:47 PM Vincent Jeffrey / Suke Yawata
Client CodeClient
LiveOverall TrimBleed
# of Colors
LENV0176 Eisai/LENVIMA
7.0625” x 9.5”7.3125” x 10”None
Black
Colors Black
FontsUnivers LT Std (67 Bold Condensed, 47 Light Con-densed, 47 Light Condensed Oblique)
Job info Fonts & ColorsImages
Saved at
None
from hsvjeffrey5552 by
Printed At
Eisai_K.ai (3.6%)
Notes Brief Summary - Island Spread
LENVIMA™ (lenvatinib) BRIEF SUMMARY – See package insert for full prescribing information.1 INDICATIONS AND USAGELENVIMA is indicated for the treatment of patients with locally recurrent or metastatic, progressive, radioactive iodine-refractory differentiated thyroid cancer (DTC).2 DOSAGE AND ADMINISTRATION2.1 Recommended DoseThe recommended daily dose of LENVIMA is 24 mg (two 10 mg capsules and one 4 mg capsule) orally taken once daily with or without food. Continue LENVIMA until disease progression or until unacceptable toxicity occurs.Take LENVIMA at the same time each day. If a dose is missed and cannot be taken within 12 hours, skip that dose and take the next dose at the usual time of administration.Severe Renal or Hepatic ImpairmentThe recommended dose of LENVIMA is 14 mg taken orally once daily in patients with severe renal impairment (creatinine clearance [CLcr] less than 30 mL/min calculated by the Cockroft-Gault equation) or severe hepatic impairment (Child-Pugh C).2.2 Dose ModificationsHypertension• Assess blood pressure prior to and periodically during treatment. Initiate or adjust medical management to
control blood pressure prior to and during treatment.• Withhold LENVIMA for Grade 3 hypertension that persists despite optimal antihypertensive therapy; resume
at a reduced dose (see Table 1) when hypertension is controlled at less than or equal to Grade 2.• Discontinue LENVIMA for life-threatening hypertension.Cardiac dysfunction or hemorrhage • Discontinue for a Grade 4 event.• Withhold LENVIMA for development of Grade 3 event until improved to Grade 0 or 1 or baseline.• Either resume at a reduced dose (see Table 1) or discontinue LENVIMA depending on the severity and
persistence of the adverse event.Arterial thrombotic event • Discontinue LENVIMA following an arterial thrombotic event.Renal failure and impairment or hepatotoxicity• Withhold LENVIMA for development of Grade 3 or 4 renal failure/impairment or hepatotoxicity until resolved
to Grade 0 to 1 or baseline.• Either resume at a reduced dose (see Table 1) or discontinue LENVIMA depending on the severity and
persistence of renal impairment or hepatotoxicity.• Discontinue LENVIMA for hepatic failure.Proteinuria• Withhold LENVIMA for ≥2 grams of proteinuria/24 hours.• Resume at a reduced dose (see Table 1) when proteinuria is <2 gm/24 hours.• Discontinue LENVIMA for nephrotic syndrome.Gastrointestinal perforation or fistula formation• Discontinue LENVIMA in patients who develop gastrointestinal perforation or life-threatening fistula.QT prolongation• Withhold LENVIMA for the development of Grade 3 or greater QT interval prolongation.• Resume LENVIMA at a reduced dose (see Table 1) when QT prolongation resolves to Grade 0 or 1 or baseline.Reversible posterior leukoencephalopathy syndrome (RPLS)• Withhold for RPLS until fully resolved.• Upon resolution, resume at a reduced dose or discontinue LENVIMA depending on the severity and
persistence of neurologic symptoms.Manage other adverse reactions according to the instructions in Table 1. Based on the absence of clinical experience, there are no recommendations on resumption of dosing in patients with Grade 4 clinical adverse reactions that resolve.
Table 1 Recommended Dose Modifications for Persistent and Intolerable Grade 2 or Grade 3 Adverse Reactions or Grade 4 Laboratory Abnormalitiesa
Adverse Reaction Modification Adjusted Doseb
First occurrence Interrupt until resolved to Grade 0-1 or baseline
20 mg (two 10 mg capsules) orally once daily
Second occurrencec Interrupt until resolved to Grade 0-1 or baseline
14 mg (one 10 mg capsule plus one 4 mg capsule)
orally once daily
Third occurrencec Interrupt until resolved to Grade 0-1 or baseline
10 mg (one 10 mg capsule) orally once daily
a Initiate medical management for nausea, vomiting, or diarrhea prior to interruption or dose reduction of LENVIMA
b Reduce dose in succession based on the previous dose level (24 mg, 20 mg, or 14 mg per day)c Refers to the same or a different adverse reaction that requires dose modification
4 CONTRAINDICATIONSNone.5 WARNINGS AND PRECAUTIONS5.1 HypertensionIn Study 1 hypertension was reported in 73% of LENVIMA-treated patients and 16% of patients in the placebo group. The median time to onset of new or worsening hypertension was 16 days for LENVIMA-treated patients. The incidence of Grade 3 hypertension was 44% as compared to 4% for placebo, and the incidence of Grade 4 hypertension was less than 1% in LENVIMA-treated patients and none in the placebo group.Control blood pressure prior to treatment with LENVIMA. Monitor blood pressure after 1 week, then every 2 weeks for the first 2 months, and then at least monthly thereafter during treatment with LENVIMA. Withhold LENVIMA for Grade 3 hypertension despite optimal antihypertensive therapy; resume at a reduced dose when hypertension is controlled at less than or equal to Grade 2. Discontinue LENVIMA for life-threatening hypertension.5.2 Cardiac DysfunctionIn Study 1, cardiac dysfunction, defined as decreased left or right ventricular function, cardiac failure, or pulmonary edema, was reported in 7% of LENVIMA-treated patients (2% Grade 3 or greater) and 2% (no Grade 3 or greater) of patients in the placebo group. The majority of these cases in LENVIMA-treated patients (14 of 17 cases) were based on findings of decreased ejection fraction as assessed by echocardiography. Six of 261 (2%) LENVIMA-treated patients in Study 1 had greater than 20% reduction in ejection fraction as measured by echocardiography compared to no patients who received placebo.Monitor patients for clinical symptoms or signs of cardiac decompensation. Withhold LENVIMA for development of Grade 3 cardiac dysfunction until improved to Grade 0 or 1 or baseline. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of cardiac dysfunction. Discontinue LENVIMA for Grade 4 cardiac dysfunction.5.3 Arterial Thromboembolic EventsIn Study 1, arterial thromboembolic events were reported in 5% of LENVIMA-treated patients and 2% of patients in the placebo group. The incidence of arterial thromboembolic events of Grade 3 or greater was 3% in LENVIMA-treated patients and 1% in the placebo group.Discontinue LENVIMA following an arterial thrombotic event. The safety of resuming LENVIMA after an arterial thromboembolic event has not been established and LENVIMA has not been studied in patients who have had an arterial thromboembolic event within the previous 6 months.5.4 HepatotoxicityIn Study 1, 4% of LENVIMA-treated patients experienced an increase in alanine aminotransferase (ALT) and 5% experienced an increase in aspartate aminotransferase (AST) that was Grade 3 or greater. No patients in the placebo group experienced Grade 3 or greater increases in ALT or AST. Across clinical studies in which 1108 patients received LENVIMA, hepatic failure (including fatal events) was reported in 3 patients and acute hepatitis was reported in 1 patient.Monitor liver function before initiation of LENVIMA, then every 2 weeks for the first 2 months, and at least monthly thereafter during treatment. Withhold LENVIMA for the development of Grade 3 or greater liver impairment until
resolved to Grade 0 to 1 or baseline. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of hepatotoxicity. Discontinue LENVIMA for hepatic failure.5.5 ProteinuriaIn Study 1, proteinuria was reported in 34% of LENVIMA-treated patients and 3% of patients in the placebo group. The incidence of Grade 3 proteinuria in LENVIMA-treated patients was 11% compared to none in the placebo group.Monitor for proteinuria before initiation of, and periodically throughout treatment. If urine dipstick proteinuria greater than or equal to 2+ is detected, obtain a 24 hour urine protein. Withhold LENVIMA for ≥2 grams of proteinuria/24 hours and resume at a reduced dose when proteinuria is <2 gm/24 hours. Discontinue LENVIMA for nephrotic syndrome.5.6 Renal Failure and ImpairmentIn Study 1, events of renal impairment were reported in 14% of LENVIMA-treated patients compared to 2% of patients in the placebo group. The incidence of Grade 3 or greater renal failure or impairment was 3% in LENVIMA-treated patients and 1% in the placebo group. The primary risk factor for severe renal impairment in LENVIMA-treated patients was dehydration/hypovolemia due to diarrhea and vomiting.Withhold LENVIMA for development of Grade 3 or 4 renal failure/impairment until resolved to Grade 0 to 1 or baseline. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of renal impairment.5.7 Gastrointestinal Perforation and Fistula FormationIn Study 1, events of gastrointestinal perforation or fistula were reported in 2% of LENVIMA-treated patients and 0.8% of patients in the placebo group.Discontinue LENVIMA in patients who develop gastrointestinal perforation or life-threatening fistula.5.8 QT Interval ProlongationIn Study 1, QT/QTc interval prolongation was reported in 9% of LENVIMA-treated patients and 2% of patients in the placebo group. The incidence of QT interval prolongation of Grade 3 or greater was 2% in LENVIMA-treated patients compared to no reports in the placebo group. Monitor electrocardiograms in patients with congenital long QT syndrome, congestive heart failure, bradyarrhythmias, or those who are taking drugs known to prolong the QT interval, including Class Ia and III antiarrhythmics.Monitor and correct electrolyte abnormalities in all patients. Withhold LENVIMA for the development of Grade 3 or greater QT interval prolongation. Resume LENVIMA at a reduced dose when QT prolongation resolves to Grade 0 or 1 or baseline.5.9 HypocalcemiaIn Study 1, 9% of LENVIMA-treated patients experienced Grade 3 or greater hypocalcemia compared to 2% in the placebo group. In most cases hypocalcemia responded to replacement and dose interruption/dose reduction.Monitor blood calcium levels at least monthly and replace calcium as necessary during LENVIMA treatment. Interrupt and adjust LENVIMA dosing as necessary depending on severity, presence of ECG changes, and persistence of hypocalcemia.5.10 Reversible Posterior Leukoencephalopathy SyndromeAcross clinical studies in which 1108 patients received LENVIMA, there were 3 reported events of reversible posterior leukoencephalopathy syndrome (RPLS). Confirm the diagnosis of RPLS with MRI. Withhold for RPLS until fully resolved. Upon resolution, resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of neurologic symptoms.5.11 Hemorrhagic EventsIn Study 1, hemorrhagic events occurred in 35% of LENVIMA-treated patients and in 18% of the placebo group. However, the incidence of Grade 3-5 hemorrhage was similar between arms at 2% and 3%, respectively. The most frequently reported hemorrhagic event was epistaxis (11% Grade 1 and 1% Grade 2). Discontinuation due to hemorrhagic events occurred in 1% of LENVIMA-treated patients.Across clinical studies in which 1108 patients received LENVIMA, Grade 3 or greater hemorrhage was reported in 2% of patients. In Study 1, there was 1 case of fatal intracranial hemorrhage among 16 patients who received lenvatinib and had CNS metastases at baseline.Withhold LENVIMA for the development of Grade 3 hemorrhage until resolved to Grade 0 to 1. Either resume at a reduced dose or discontinue LENVIMA depending on the severity and persistence of hemorrhage. Discontinue LENVIMA in patients who experience Grade 4 hemorrhage.5.12 Impairment of Thyroid Stimulating Hormone SuppressionLENVIMA impairs exogenous thyroid suppression. In Study 1, 88% of all patients had a baseline thyroid stimulating hormone (TSH) level less than or equal to 0.5 mU/L. In those patients with a normal TSH at baseline, elevation of TSH level above 0.5 mU/L was observed post baseline in 57% of LENVIMA-treated patients as compared with 14% of patients receiving placebo.Monitor TSH levels monthly and adjust thyroid replacement medication as needed in patients with DTC.5.13 Embryofetal ToxicityBased on its mechanism of action and data from animal reproduction studies, LENVIMA can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of lenvatinib during organogenesis at doses below the recommended human dose resulted in embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy.6 ADVERSE REACTIONSThe following adverse reactions are discussed elsewhere in the label. Please see the Warnings and Precautions sections in the full prescribing information.• Hypertension• Cardiac Dysfunction• Arterial Thromboembolic Events• Hepatotoxicity• Proteinuria• Renal Failure and Impairment• Gastrointestinal Perforation and Fistula Formation• QT Interval Prolongation• Hypocalcemia• Reversible Posterior Leukoencephalopathy Syndrome• Hemorrhagic Events• Impairment of Thyroid Stimulating Hormone Suppression6.1 Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Safety data obtained in 1108 patients with advanced solid tumors who received LENVIMA as a single agent across multiple clinical studies was used to further characterize risks of serious adverse drug reactions. The median age was 60 years (range 21-89 years). The dose range was 0.2 mg to 32 mg. The median duration of exposure in the entire population was 5.5 months.The safety data described below are derived from Study 1 which randomized (2:1) patients with radioactive iodine-refractory differentiated thyroid cancer (RAI-refractory DTC) to LENVIMA (n=261) or placebo (n=131). The median treatment duration was 16.1 months for LENVIMA and 3.9 months for placebo. Among 261 patients who received LENVIMA in Study 1, median age was 64 years, 52% were women, 80% were White, 18% were Asian, and 2% were Black; 4% identified themselves as having Hispanic or Latino ethnicity.In Study 1, the most common adverse reactions observed in LENVIMA-treated patients (greater than or equal to 30%) were, in order of decreasing frequency, hypertension, fatigue, diarrhea, arthralgia/myalgia, decreased appetite, weight decreased, nausea, stomatitis, headache, vomiting, proteinuria, palmar-plantar erythrodysesthesia (PPE) syndrome, abdominal pain, and dysphonia. The most common serious adverse reactions (at least 2%) were pneumonia (4%), hypertension (3%), and dehydration (3%).Adverse reactions led to dose reductions in 68% of patients receiving LENVIMA and 5% of patients receiving placebo; 18% of patients discontinued LENVIMA and 5% discontinued placebo for adverse reactions. The most common adverse reactions (at least 10%) resulting in dose reductions of LENVIMA were hypertension (13%), proteinuria (11%), decreased appetite (10%), and diarrhea (10%); the most common adverse reactions (at least 1%) resulting in discontinuation of LENVIMA were hypertension (1%) and asthenia (1%).Table 2 presents the percentage of patients in Study 1 experiencing adverse reactions at a higher rate in LENVIMA-treated patients than patients receiving placebo in the double-blind phase of the DTC study.
Table 2 Adverse Reactions Occurring in Patients with a Between-Group Difference of Greater than or Equal to 5% All Grades or Greater than or Equal to 2% Grades 3 and 4
Adverse Reaction
LENVIMA 24 mg N=261
Placebo N=131
All Grades (%)
Grades 3-4 (%)
All Grades (%)
Grades 3-4 (%)
Vascular DisordersHypertensiona 73 44 16 4Hypotension 9 2 2 0
Gastrointestinal DisordersDiarrhea 67 9 17 0Nausea 47 2 25 1Stomatitisb 41 5 8 0Vomiting 36 2 15 0Abdominal painc 31 2 11 1Constipation 29 0.4 15 1Oral paind 25 1 2 0Dry mouth 17 0.4 8 0Dyspepsia 13 0.4 4 0
General Disorders and Administration Site ConditionsFatiguee 67 11 35 4Edema peripheral 21 0.4 8 0
Musculoskeletal and Connective Tissue DisordersArthralgia/Myalgiaf 62 5 28 3
Metabolism and Nutrition DisordersWeight decreased 51 13 15 1Decreased appetite 54 7 18 1Dehydration 9 2 2 1
Nervous System DisordersHeadache 38 3 11 1Dysgeusia 18 0 3 0Dizziness 15 0.4 9 0
Renal and Urinary DisordersProteinuria 34 11 3 0
Skin and Subcutaneous Tissue DisordersPalmar-plantar erythrodysesthesia 32 3 1 0Rashg 21 0.4 3 0Alopecia 12 0 5 0Hyperkeratosis 7 0 2 0
Respiratory, Thoracic and Mediastinal DisordersDysphonia 31 1 5 0Cough 24 0 18 0Epistaxis 12 0 1 0
Psychiatric DisordersInsomnia 12 0 3 0
Infections and InfestationsDental and oral infectionsh 10 1 1 0Urinary tract infection 11 1 5 0
Cardiac Disorders
Electrocardiogram QT prolonged 9 2 2 0a Includes hypertension, hypertensive crisis, increased blood pressure diastolic, and increased
blood pressure b Includes aphthous stomatitis, stomatitis, glossitis, mouth ulceration, and mucosal inflammationc Includes abdominal discomfort, abdominal pain, abdominal pain lower, abdominal pain upper,
abdominal tenderness, epigastric discomfort, and gastrointestinal paind Includes oral pain, glossodynia, and oropharyngeal paine Includes asthenia, fatigue, and malaisef Includes musculoskeletal pain, back pain, pain in extremity, arthralgia, and myalgiag Includes rash macular, rash maculo-papular, rash generalized, and rash h Includes gingivitis, oral infection, parotitis, pericoronitis, periodontitis, sialoadenitis, tooth abscess,
and tooth infectionA clinically important adverse reaction occurring more frequently in LENVIMA-treated patients than patients receiving placebo, but with an incidence of less than 5% was pulmonary embolism (3%, including fatal reports vs 2%, respectively).
Table 3 Laboratory Abnormalities with a Difference of at Least ≥2% in Grade 3 - 4 Events and at a Higher Incidence in LENVIMA-Treated Patientsa
Laboratory Abnormality LENVIMA 24 mg N=258b
Placebo N=131b
Grades 3-4 (%)
Grades 3-4 (%)
ChemistryCreatinine increased 3 0Alanine aminotransferase (ALT) increased 4 0Aspartate aminotransferase (AST) increased 5 0Hypocalcemia 9 2Hypokalemia 6 1Lipase increased 4 1
HematologyPlatelet count decreased 2 0
a With at least 1 grade increase from baseline b Subject with at least 1 post baseline laboratory value
In addition the following laboratory abnormalities (all Grades) occurred in greater than 5% of LENVIMA-treated patients and at a rate that was two-fold or higher than in patients who received placebo: hypoalbuminemia, increased alkaline phosphatase, hypomagnesemia, hypoglycemia, hyperbilirubinemia, hypercalcemia, hypercholesterolemia, increased serum amylase, and hyperkalemia.7 DRUG INTERACTIONS7.1 Effect of Other Drugs on LenvatinibNo dose adjustment of LENVIMA is recommended when co-administered with CYP3A, P-glycoprotein (P-gp), and breast cancer resistance protein (BCRP) inhibitors and CYP3A and P-gp inducers.8 USE IN SPECIFIC POPULATIONS8.1 PregnancyRisk SummaryBased on its mechanism of action and data from animal reproduction studies, LENVIMA can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of lenvatinib during organogenesis at doses below the recommended human dose resulted in embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits. There are no available human data informing the drug-associated risk. Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown; however, the background risk in the U.S. general population of major birth defects is 2-4% and of miscarriage is 15-20% of clinically recognized pregnancies.DataAnimal DataIn an embryofetal development study, daily oral administration of lenvatinib mesylate at doses greater than or equal to 0.3 mg/kg [approximately 0.14 times the recommended human dose based on body surface area (BSA)] to pregnant rats during organogenesis resulted in dose-related decreases in mean fetal body weight, delayed fetal ossifications, and dose-related increases in fetal external (parietal edema and tail abnormalities), visceral, and skeletal anomalies. Greater than 80% postimplantation loss was observed at 1.0 mg/kg/day (approximately 0.5 times the recommended human dose based on BSA).Daily oral administration of lenvatinib mesylate to pregnant rabbits during organogenesis resulted in fetal external (short tail), visceral (retroesophageal subclavian artery), and skeletal anomalies at doses greater than or equal to 0.03 mg/kg (approximately 0.03 times the human dose of 24 mg based on body surface area). At the 0.03 mg/kg dose, increased post-implantation loss, including 1 fetal death, was also observed. Lenvatinib was abortifacient in rabbits, resulting in late abortions in approximately one-third of the rabbits treated at a dose level of 0.5 mg/kg/day (approximately 0.5 times the recommended clinical dose of 24 mg based on BSA).8.2 LactationRisk SummaryIt is not known whether LENVIMA is present in human milk. However, lenvatinib and its metabolites are excreted in rat milk at concentrations higher than in maternal plasma. Because of the potential for serious adverse reactions in nursing infants from LENVIMA, advise women to discontinue breastfeeding during treatment with LENVIMA.DataAnimal DataFollowing administration of radiolabeled lenvatinib to lactating Sprague Dawley rats, lenvatinib-related radioactivity was approximately 2 times higher (based on AUC) in milk compared to maternal plasma.8.3 Females and Males of Reproductive PotentialContraceptionBased on its mechanism of action, LENVIMA can cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy.InfertilityFemalesLENVIMA may result in reduced fertility in females of reproductive potential.MalesLENVIMA may result in damage to male reproductive tissues leading to reduced fertility of unknown duration.8.4 Pediatric UseThe safety and effectiveness of LENVIMA in pediatric patients have not been established.Juvenile Animal DataDaily oral administration of lenvatinib mesylate to juvenile rats for 8 weeks starting on postnatal day 21 (approximately equal to a human pediatric age of 2 years) resulted in growth retardation (decreased body weight gain, decreased food consumption, and decreases in the width and/or length of the femur and tibia) and secondary delays in physical development and reproductive organ immaturity at doses greater than or equal to 2 mg/kg (approximately 1.2 to 5 times the clinical exposure by AUC at the recommended human dose). Decreased length of the femur and tibia persisted following 4 weeks of recovery. In general, the toxicologic profile of lenvatinib was similar between juvenile and adult rats, though toxicities including broken teeth at all dose levels and mortality at the 10 mg/kg/day dose level (attributed to primary duodenal lesions) occurred at earlier treatment time-points in juvenile rats.8.5 Geriatric UseOf 261 patients who received LENVIMA in Study 1, 118 (45.2%) were greater than or equal to 65 years of age and 29 (11.1%) were greater than or equal to 75 years of age. No overall differences in safety or effectiveness were observed between these subjects and younger subjects.8.6 Renal ImpairmentNo dose adjustment is recommended in patients with mild or moderate renal impairment. In patients with severe renal impairment, the recommended dose is 14 mg taken once daily. Patients with end stage renal disease were not studied.8.7 Hepatic ImpairmentNo dose adjustment is recommended in patients with mild or moderate hepatic impairment. In patients with severe hepatic impairment, the recommended dose is 14 mg taken once daily.10 OVERDOSAGEThere is no specific antidote for overdose with LENVIMA. Due to the high plasma protein binding, lenvatinib is not expected to be dialyzable. Adverse reactions in patients receiving single doses of LENVIMA as high as 40 mg were similar to the adverse events reported in the clinical studies at the recommended dose.17 PATIENT COUNSELING INFORMATIONAdvise the patient to read the FDA-approved patient labeling (Patient Information).Hypertension:Advise patients to undergo regular blood pressure monitoring and to contact their health care provider if blood pressure is elevated.Cardiac Dysfunction:Advise patients that LENVIMA can cause cardiac dysfunction and to immediately contact their healthcare provider if they experience any clinical symptoms of cardiac dysfunction such as shortness of breath or swelling of ankles.Arterial Thrombotic EventsAdvise patients to seek immediate medical attention for new onset chest pain or acute neurologic symptoms consistent with myocardial infarction or stroke.Hepatotoxicity:Advise patients that they will need to undergo lab tests to monitor for liver function and to report any new symptoms indicating hepatic toxicity or failure.Proteinuria and Renal Failure/Impairment:Advise patients that they will need to undergo regular lab tests to monitor for kidney function and protein in the urine.Gastrointestinal perforation or fistula formation:Advise patients that LENVIMA can increase the risk of gastrointestinal perforation or fistula and to seek immediate medical attention for severe abdominal pain.Hemorrhagic Events:Advise patients that LENVIMA can increase the risk for bleeding and to contact their health care provider for bleeding or symptoms of severe bleeding.Embryofetal Toxicity:Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for at least 2 weeks following completion of therapy. Lactation:Advise nursing women to discontinue breastfeeding during treatment with LENVIMA.
LENVIMA™ is a trademark of Eisai R&D Management Co., Ltd. and is licensed to Eisai Inc.© 2015 Eisai Inc. All rights reserved. Printed in USA/February 2015 LENV0176
S:14.375”
S:9.5”T:14.625”
T:10”
Fold:7.3125”Safety:7.0625”
Fold:7.3125”Safety:7.0625”
78618ha_b.indd 3 2/23/15 4:50 PM

PAGE 54 The ASCO Post | MARCH 25, 2015
Avoiding Burnout and Maintaining Well-Being While Caring for Seriously Ill PatientsA Conversation With Sandra Sanchez-Reilly, MD, AGSF, FAAHPMBy Jo Cavallo
A variety of studies, including one published this past year in the Jour-
nal of Clinical Oncology,1 have showed that clinicians who care for seriously ill patients are at high risk for diminished personal well-being, including high rates of burnout; moral distress, defined as the inability to act in a manner consis-tent with one’s personal and professional values; and compassion fatigue, in which physicians experience diminished emo-tional energy to care for patients.
One key approach to mitigate the dis-tress and grief oncologists and palliative care specialists often experience when car-ing for terminally ill patients is to engage in self-care, according to Sandra Sanchez-Reilly, MD, AGSF, FAAHPM, Profes-sor, Palliative Medicine Section Chief, and Director of Palliative Medicine at the University of Texas Health Science Cen-ter at San Antonio; and Interprofessional Fellowship Program Director at the South Texas Veterans Health Care System. She is also the lead author of the article “Caring for Oneself to Care for Others: Physicians and Their Self-Care,” which appeared in The Journal of Supportive Oncology.2
“Many times, our patients are criti-cally or terminally ill and in very difficult situations, and we are right there with them,” said Dr. Sanchez-Reilly. “We are always in crisis mode, but to effectively care for our difficult patients, we need to take care of ourselves first.”
The ASCO Post talked with Dr. San-chez-Reilly about the factors that place oncologists at risk for diminished personal and professional well-being as well as the self-care tools and strategies oncologists can use to improve their overall well-being.
Self-Care Rounds and RetreatsPlease talk about how oncologists can
promote and maintain well-being while
caring for patients with life-limiting cancer.Taking care of patients with advanced
cancer is very difficult and all-consum-ing, often leading oncologists and pallia-tive care specialists to experience distress and grief in response to their patients’ suf-fering. Unless recognized and dealt with, these emotions can compromise clini-cians’ well-being and lead to burnout, moral distress, and compassion fatigue.
So, the first thing is to recognize that we actually need to take care of our-selves, which we often don’t do well, and to understand that we are human beings, and it is natural to experience feelings of sadness, frustration, and an-ger when we lose a patient.
Here at the University of Texas Health Science Center, we encourage physicians to use self-care strategies to maintain personal well-being through-out their professional lives. We also hold self-care rounds twice a month to give the medical staff an opportunity to talk about how their professional lives are af-fecting their personal lives.
During these sessions, the staff may present case stories and explain how a specific patient situation is impacting
them. For example, younger profes-sionals tend to have a difficult time tak-ing care of their younger patients with advanced cancer, because they identify with those patients. Or, a clinician may have lost a loved one to a particular can-cer and finds that taking care of a pa-tient with a similar diagnosis is difficult.
Other issues, such as problems with time management, are discussed as well. Making patient decisions takes a lot of time, not only to see and treat the pa-tient, but to document those decisions and meet the requirements from the dif-ferent hospitals or health-care systems that may be involved in that patient’s care. There may also be issues surround-ing coordinating care within the various medical teams in the hospital as well as dealing with family members. Some-
times, there are disagreements between oncologists and family members or the palliative care team members and on-cologists, and these disagreements can be cumbersome to deal with.
In addition to our twice-monthly ses-sions, once a year, we have a self-care re-treat held somewhere outside the hospital to get away from our work environment and give participants a chance to focus on themselves. During the retreat, we discuss how to take care of ourselves so we have more energy and dedication to bring to the care of our patients.
The Wellness WheelWhat are some self-care tools oncolo-
gists can employ to mitigate the stresses in their professional lives?
Personal self-care refers to activities that are performed independently by individuals or a team to promote and maintain well-being throughout their professional lives. Some of those activi-ties may include maintaining a healthy lifestyle through regular exercise, getting enough sleep, eating a healthy diet, and taking vacations; practicing mindfulness meditation; spending time with family
and friends; and pursuing spiritual devel-opment if that is personally significant.
One instrument clinicians may find helpful is the Wellness Wheel (www.vanderbilt.edu/recreationandwell-nesscenter/wellness/wellness-wheel). It includes seven areas of wellness to inte-grate into one’s life: physical, spiritual, so-cial, emotional, intellectual, environmen-tal, and occupational, depending on one’s specific needs.
Burnout, Moral Distress, and Compassion Fatigue
What are some consequences of expe-riencing burnout, moral distress, and com-passion fatigue?
The symptoms and consequences are similar for all three. The one that has been most closely studied is burnout, which
refers to a feeling of emotional exhaustion and may be associated with both high job turnover and poorer health. It is also linked to patient-related outcomes, such as medical errors by physicians and lower patient satisfaction, as well as unprofes-sional conduct and less altruistic values.
Less is known about moral distress and compassion fatigue, both of which can lead to burnout, but physicians experiencing moral distress may have increased irritability, decreased job satisfaction, insomnia, headaches, high blood pressure, and depression.
Compassion fatigue may manifest in symptoms similar to post-traumatic stress disorder, including hyperarousal and reliving events, such as clinical en-counters with suffering patients or fam-ily members.
Are all oncologists at risk for develop-ing burnout, moral distress, and compas-sion fatigue?
It is not only oncologists who are susceptible to these problems. Every health-care provider is at risk for devel-oping these syndromes. In oncology, there is a more than 30% chance pro-viders will experience these symptoms sometime during their professional
Education in Oncology
Professional Development
continued on page 57
Many times, our patients are critically or terminally ill and in very difficult situations, and we are right there with them. But to effectively care for them, we need to
take care of ourselves first. —Sandra Sanchez-Reilly, MD, AGSF, FAAHPM
Sandra Sanchez-Reilly, MD, AGSF, FAAHPM
GUEST EDITOR
Education in Oncology focuses on faculty development, med-
ical education curricula, fellow-ship training, and communication skills. The column is guest edited by Leora Horn, MD, MSc, Asso-ciate Professor of Medicine, Assis-tant Director of the Educator De-velopment Program, and Clinical Director of the Thoracic Oncology Program at Vanderbilt University School of Medicine, Nashville.
Leora Horn, MD, MSc
LYNPARZA is a trademark of the AstraZeneca group of companies.
©2015 AstraZeneca. All Rights Reserved. 3076602 Last updated 01/15
Please see next page for Important Safety Information.
The fi rst approved PARP inhibitorLYNPARZA is indicated as monotherapy in patients with deleterious or suspected
deleterious germline BRCA mutated (as detected by an FDA-approved test) advanced
ovarian cancer who have been treated with three or more prior lines
of chemotherapy.
The indication is approved under accelerated approval based on objective response rate
and duration of response. Continued approval for this indication may be contingent upon
verifi cation and description of clinical benefi t in confi rmatory trials.
• In clinical studies the most common adverse reactions (Grades 1-4) in ≥20% of patients
included anemia, nausea, fatigue (including asthenia), vomiting, diarrhea, dysgeusia,
dyspepsia, headache, decreased appetite, nasopharyngitis/pharyngitis/URI, cough, arthralgia/
musculoskeletal pain, myalgia, back pain, dermatitis/rash, and abdominal pain/discomfort
You are encouraged to report negative side effects of prescription drugs
to the FDA. Visit www.FDA.gov/medwatch or call 1-800-FDA-1088.
FOR MORE INFORMATION VISIT www.LYNPARZA.com
NOW APPROVED

LYNPARZA is a trademark of the AstraZeneca group of companies.
©2015 AstraZeneca. All Rights Reserved. 3076602 Last updated 01/15
Please see next page for Important Safety Information.
The fi rst approved PARP inhibitorLYNPARZA is indicated as monotherapy in patients with deleterious or suspected
deleterious germline BRCA mutated (as detected by an FDA-approved test) advanced
ovarian cancer who have been treated with three or more prior lines
of chemotherapy.
The indication is approved under accelerated approval based on objective response rate
and duration of response. Continued approval for this indication may be contingent upon
verifi cation and description of clinical benefi t in confi rmatory trials.
• In clinical studies the most common adverse reactions (Grades 1-4) in ≥20% of patients
included anemia, nausea, fatigue (including asthenia), vomiting, diarrhea, dysgeusia,
dyspepsia, headache, decreased appetite, nasopharyngitis/pharyngitis/URI, cough, arthralgia/
musculoskeletal pain, myalgia, back pain, dermatitis/rash, and abdominal pain/discomfort
You are encouraged to report negative side effects of prescription drugs
to the FDA. Visit www.FDA.gov/medwatch or call 1-800-FDA-1088.
FOR MORE INFORMATION VISIT www.LYNPARZA.com
NOW APPROVED

IMPORTANT SAFETY INFORMATION
There are no contraindications for LYNPARZA.
Myelodysplastic syndrome/Acute Myeloid Leukemia (MDS/AML) have been
confi rmed in 2% of patients enrolled in both a single arm monotherapy trial (6 out
of 298) and a randomized placebo controlled trial (3 out of 136). Overall, MDS/AML
were reported in <1% of patients (22 out of 2,618) treated with LYNPARZA.
The majority of MDS/AML cases were fatal (17 out of 22) and the duration of therapy
in patients who developed secondary MDS/AML varied from <6 months to >2 years.
Monitor complete blood count testing at baseline and monthly thereafter. For
prolonged hematological toxicities, interrupt LYNPARZA and monitor blood counts
weekly until recovery.
Pneumonitis, including fatal cases, occurred in <1% of patients treated with
LYNPARZA.
In clinical studies the most common adverse reactions (Grades 1-4) in ≥20% of patients
included anemia, nausea, fatigue (including asthenia), vomiting, diarrhea, dysgeusia,
dyspepsia, headache, decreased appetite, nasopharyngitis/pharyngitis/URI, cough,
arthralgia/musculoskeletal pain, myalgia, back pain, dermatitis/rash, and abdominal
pain/discomfort.
Coadministration of drugs which are potent inhibitors or inducers of CYP3A could
increase or decrease exposure to LYNPARZA and should be avoided. Clinical studies
of LYNPARZA in combination with other myelosuppressive anticancer agents,
including DNA damaging agents, indicate a potentiation and prolongation
of myelosuppressive toxicity.
LYNPARZA is Pregnancy Category D.
LYNPARZA is not recommended for use in patients with hepatic impairment (serum
bilirubin greater than 1.5 times upper limit of normal), moderate renal impairment or
severe renal impairment, since safety and effi cacy have not been established.
Please see adjacent Brief Summary of the full Prescribing Information.
LYNPARZA is a trademark of the AstraZeneca group of companies.
©2015 AstraZeneca. All Rights Reserved. 3076602 Last updated 01/15
LYNPARZA™ (olaparib) capsules, for oral use
Brief Summary of Prescribing Information. For complete prescribing information consult official package insert.
INDICATIONS AND USAGETreatment of gBRCA-mutated advanced ovarian cancerLynparza is indicated as monotherapy in patients with deleterious or suspected deleterious germline BRCA mutated (as detected by an FDA-approved test) advanced ovarian cancer who have been treated with three or more prior lines of chemotherapy.The indication is approved under accelerated approval based on objective response rate and duration of response [see Clinical Studies (14) in the full Prescribing Information]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
DOSAGE AND ADMINISTRATIONPatient SelectionSelect patients for the treatment of advanced ovarian cancer with Lynparza based on the presence of deleterious or suspected deleterious germline BRCA-mutations [see Indications and Usage (1) and Clinical Studies (14) in the full Prescribing Information]. Information on FDA-approved test for the detection of BRCA-mutations is available at http://www.fda.gov/companiondiagnostics.Recommended DosingThe recommended dose of Lynparza is 400 mg (eight 50 mg capsules) taken twice daily, for a total daily dose of 800 mg. Continue treatment until disease progression or unacceptable toxicity.If a patient misses a dose of Lynparza, instruct patients to take their next dose at its scheduled time.Swallow capsule whole. Do not chew, dissolve, or open capsule. Do not take capsules which appear deformed or show evidence of leakage [see How Supplied/Storage and Handling (16.2) in the full Prescribing Information].Dose Adjustments for Adverse ReactionsTo manage adverse reactions, consider dose interruption of treatment or dose reduction.The recommended dose reduction is to 200 mg (four 50 mg capsules) taken twice daily, for a total daily dose of 400 mg.If a further final dose reduction is required, then reduce to 100 mg (two 50 mg capsules) taken twice daily, for a total daily dose of 200 mg.Dose Modifications for Use with CYP3A InhibitorsAvoid concomitant use of strong and moderate CYP3A inhibitors and consider alternative agents with less CYP3A inhibition. If the inhibitor cannot be avoided, reduce the Lynparza dose to 150 mg (three 50 mg capsules) taken twice daily for a strong CYP3A inhibitor or 200 mg (four 50 mg capsules) taken twice daily for a moderate CYP3A inhibitor [see Drug Interactions (7.2) in the full Prescribing Information].
CONTRAINDICATIONSNone
WARNINGS AND PRECAUTIONSMyelodysplastic syndrome/Acute Myeloid LeukemiaMyelodysplastic syndrome/Acute Myeloid Leukemia (MDS/AML) have been confirmed in 6 out of 298 (2%) patients enrolled in a single arm trial of Lynparza monotherapy, in patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAm) advanced cancers. In a randomized placebo controlled trial, MDS/AML occurred in 3 out of 136 (2%) patients with advanced ovarian cancer treated with Lynparza. Overall, MDS/AML were reported in 22 of 2,618 (<1%) patients treated with Lynparza. The majority of MDS/AML cases (17 of 22 cases) were fatal, and the duration of therapy with Lynparza in patients who developed secondary MDS/cancer-therapy related AML varied from <6 months to >2 years. All patients had previous chemotherapy with platinum agents and/or other DNA damaging agents.Monitor complete blood count testing at baseline and monthly thereafter. Do not start Lynparza until patients have recovered from hematological toxicity caused by previous chemotherapy (�CTCAE Grade 1). For prolonged hematological toxicities, interrupt Lynparza and monitor blood counts weekly until recovery. If the levels have not recovered to CTCAE Grade 1 or less after 4 weeks, refer the patient to a hematologist for further investigations, including bone marrow analysis and blood sample for cytogenetics. If MDS/AML is confirmed, discontinue Lynparza.PneumonitisPneumonitis, including fatal cases, occurred in <1% of patients treated with Lynparza. If patients present with new or worsening respiratory symptoms such as dyspnea, fever, cough, wheezing, or a radiological abnormality occurs, interrupt treatment with Lynparza and initiate prompt investigation. If pneumonitis is confirmed, discontinue Lynparza.Embryo-Fetal ToxicityLynparza can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. Olaparib was teratogenic and caused embryo-fetal toxicity in rats at exposures below those in patients receiving the recommended human dose of 400 mg twice daily. If the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus [see Use in Specific Populations (8.1) in the full Prescribing Information].Advise females of reproductive potential to avoid becoming pregnant while taking Lynparza. If contraceptive methods are being considered, use effective contraception during treatment and for at least one month after receiving the last dose of Lynparza [see Use in Specific Populations (8.6) in the full Prescribing Information].
ADVERSE REACTIONSThe following adverse reactions are discussed elsewhere in the labeling:• Myelodysplastic syndrome/Acute Myeloid Leukemia [see Warnings and Precautions
(5.1) in the full Prescribing Information]• Pneumonitis [see Warnings and Precautions (5.2) in the full Prescribing Information]
Clinical Trial ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Lynparza 400 mg twice daily as monotherapy, has been studied in 300 patients with gBRCA-mutated advanced ovarian cancer, and 223 of these patients had received 3 or more prior lines of chemotherapy.In the 223 patients with gBRCA-mutated ovarian cancer who received 3 or more prior lines of chemotherapy (including 137 patients in Study 1 with measureable disease) [see Clinical Studies (14) in the full Prescribing Information] adverse reactions led to dose interruption in 40% of patients, dose reduction in 4%, and discontinuation in 7%. There were 8 (4%) patients with adverse reactions leading to death, two were attributed to acute leukemia, and one each was attributed to COPD, cerebrovascular accident, intestinal perforation, pulmonary embolism, sepsis, and suture rupture. Table 1 presents the frequency of adverse reactions reported in �20% of 223 patients (in 6 studies) with gBRCA-mutated advanced ovarian cancer who had received 3 or more prior lines of chemotherapy who were treated with Lynparza 400 mg twice daily. The median exposure to Lynparza in these patients was 158 days.
Table 1 Adverse Reactions Reported in �20% of Patients with gBRCA-Mutated Advanced Ovarian Cancer Receiving Lynparza
3 or more lines of prior chemotherapy
Adverse Reaction Grades 1-4 Grades 3-4 N=223 N=223 % % Blood and Lymphatic disorders
Anemia 34 18
Gastrointestinal disorders
Abdominal pain/discomfort 43 8
Decreased appetite 22 1
Nausea 64 3
Vomiting 43 4
Diarrhea 31 1
Dyspepsia 25 0
General disorders
Fatigue/asthenia 66 8
Infections and infestations
Nasopharyngitis/URI 26 0
Musculoskeletal and Connective Tissue disorders
Arthralgia/musculoskeletal pain 21 0
Myalgia 22 0
Table 2 presents the frequency of abnormal laboratory findings in the 223 patients with gBRCA-mutated advanced ovarian cancer who had received three or more prior lines of chemotherapy receiving Lynparza 400 mg twice daily.
Table 2 Laboratory Abnormalities Reported in Patients with gBRCA-Mutated Advanced Ovarian Cancer Receiving Lynparza
Laboratory Parameter* 3 or more lines of prior chemotherapy
Grades 1-4 Grades 3-4 N=223 N=223 % %
Decrease in hemoglobin (anemia) 90 15
Decrease in absolute neutrophil count 25 7 (neutropenia)
Decrease in platelets (thrombocytopenia) 30 3
Decrease in lymphocytes (lymphopenia) 56 17
Mean corpuscular volume elevation 57 -
Increase in creatinine* 30 2* Patients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
The following adverse reactions and laboratory abnormalities have been identified in �10 to <20% of the 223 patients receiving Lynparza and not included in the table: cough, constipation, dysgeusia, peripheral edema, back pain, dizziness, headache, urinary tract infection, dyspnea, and rash. The following adverse reactions and laboratory abnormalities have been identified in �1 to <10% of the 223 patients receiving Lynparza and not included in the table: leukopenia, stomatitis, peripheral neuropathy, pyrexia, hypomagnesemia, hyperglycemia, anxiety, depression, insomnia, dysuria, urinary incontinence, vulvovaginal disorder, dry skin/ eczema, pruritis, hypertension, venous thrombosis (including pulmonary embolism), and hot flush.Table 3 presents adverse reactions reported in �20% of patients from a randomized trial of Lynparza 400 mg twice daily as maintenance monotherapy compared to placebo in patients with platinum sensitive, relapsed, high-grade serous ovarian cancer following treatment with 2 or more platinum-containing regimens. Table 4 presents the laboratory abnormalities in patients from this randomized trial. Of the 96 patients with gBRCA- mutation, 53 received Lynparza, and 43 received placebo. The median duration on treatment with Lynparza was 11.1 months for patients with a gBRCA mutation compared to 4.4 months for patients with gBRCA mutation on placebo.Adverse reactions led to dose interruptions in 26% of those receiving Lynparza and 7% of those receiving placebo; dose reductions in 15% of Lynparza and 5% of placebo patients; and discontinuation in 9% of Lynparza and 0% in placebo patients. One (2%) patient on Lynparza died as a result of an adverse reaction.

ASCOPost.com | MARCH 25, 2015 PAGE 57
Education in Oncology
lives. Physicians in emergency medi-cine have more than a 50% chance of experiencing burnout.
Finding Middle GroundTo avoid these problems, would it be
helpful for oncologists to maintain some
emotional distance from their patients?I would say no. Physicians need to
feel empathy for their patients, and if they are emotionally distant from their patients, they won’t. Physicians have to find middle-ground territory, which is difficult, but that is where employing the elements in the Wellness Wheel can be helpful. It is important to main-
tain a balance in one’s physical, emo-tional, professional, social, and spiri-tual life to reduce the chances of stress and burnout.
Strategies for Self-Care NeededCurrent evidence shows that medical
students, residents, and fellows receive in-adequate self-care training. What can on-
cology professionals do to initiate personal and professional self-care?
The Accreditation Council for Graduate Medical Education (AC-GME) has incorporated some areas of self-reflection into its hematology and medical oncology fellowship program requirements, but the program does
Avoiding Burnoutcontinued from page 54
continued on page 58
IMPORTANT SAFETY INFORMATION
There are no contraindications for LYNPARZA.
Myelodysplastic syndrome/Acute Myeloid Leukemia (MDS/AML) have been
confi rmed in 2% of patients enrolled in both a single arm monotherapy trial (6 out
of 298) and a randomized placebo controlled trial (3 out of 136). Overall, MDS/AML
were reported in <1% of patients (22 out of 2,618) treated with LYNPARZA.
The majority of MDS/AML cases were fatal (17 out of 22) and the duration of therapy
in patients who developed secondary MDS/AML varied from <6 months to >2 years.
Monitor complete blood count testing at baseline and monthly thereafter. For
prolonged hematological toxicities, interrupt LYNPARZA and monitor blood counts
weekly until recovery.
Pneumonitis, including fatal cases, occurred in <1% of patients treated with
LYNPARZA.
In clinical studies the most common adverse reactions (Grades 1-4) in ≥20% of patients
included anemia, nausea, fatigue (including asthenia), vomiting, diarrhea, dysgeusia,
dyspepsia, headache, decreased appetite, nasopharyngitis/pharyngitis/URI, cough,
arthralgia/musculoskeletal pain, myalgia, back pain, dermatitis/rash, and abdominal
pain/discomfort.
Coadministration of drugs which are potent inhibitors or inducers of CYP3A could
increase or decrease exposure to LYNPARZA and should be avoided. Clinical studies
of LYNPARZA in combination with other myelosuppressive anticancer agents,
including DNA damaging agents, indicate a potentiation and prolongation
of myelosuppressive toxicity.
LYNPARZA is Pregnancy Category D.
LYNPARZA is not recommended for use in patients with hepatic impairment (serum
bilirubin greater than 1.5 times upper limit of normal), moderate renal impairment or
severe renal impairment, since safety and effi cacy have not been established.
Please see adjacent Brief Summary of the full Prescribing Information.
LYNPARZA is a trademark of the AstraZeneca group of companies.
©2015 AstraZeneca. All Rights Reserved. 3076602 Last updated 01/15
LYNPARZA™ (olaparib) capsules, for oral use
Brief Summary of Prescribing Information. For complete prescribing information consult official package insert.
INDICATIONS AND USAGETreatment of gBRCA-mutated advanced ovarian cancerLynparza is indicated as monotherapy in patients with deleterious or suspected deleterious germline BRCA mutated (as detected by an FDA-approved test) advanced ovarian cancer who have been treated with three or more prior lines of chemotherapy.The indication is approved under accelerated approval based on objective response rate and duration of response [see Clinical Studies (14) in the full Prescribing Information]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
DOSAGE AND ADMINISTRATIONPatient SelectionSelect patients for the treatment of advanced ovarian cancer with Lynparza based on the presence of deleterious or suspected deleterious germline BRCA-mutations [see Indications and Usage (1) and Clinical Studies (14) in the full Prescribing Information]. Information on FDA-approved test for the detection of BRCA-mutations is available at http://www.fda.gov/companiondiagnostics.Recommended DosingThe recommended dose of Lynparza is 400 mg (eight 50 mg capsules) taken twice daily, for a total daily dose of 800 mg. Continue treatment until disease progression or unacceptable toxicity.If a patient misses a dose of Lynparza, instruct patients to take their next dose at its scheduled time.Swallow capsule whole. Do not chew, dissolve, or open capsule. Do not take capsules which appear deformed or show evidence of leakage [see How Supplied/Storage and Handling (16.2) in the full Prescribing Information].Dose Adjustments for Adverse ReactionsTo manage adverse reactions, consider dose interruption of treatment or dose reduction.The recommended dose reduction is to 200 mg (four 50 mg capsules) taken twice daily, for a total daily dose of 400 mg.If a further final dose reduction is required, then reduce to 100 mg (two 50 mg capsules) taken twice daily, for a total daily dose of 200 mg.Dose Modifications for Use with CYP3A InhibitorsAvoid concomitant use of strong and moderate CYP3A inhibitors and consider alternative agents with less CYP3A inhibition. If the inhibitor cannot be avoided, reduce the Lynparza dose to 150 mg (three 50 mg capsules) taken twice daily for a strong CYP3A inhibitor or 200 mg (four 50 mg capsules) taken twice daily for a moderate CYP3A inhibitor [see Drug Interactions (7.2) in the full Prescribing Information].
CONTRAINDICATIONSNone
WARNINGS AND PRECAUTIONSMyelodysplastic syndrome/Acute Myeloid LeukemiaMyelodysplastic syndrome/Acute Myeloid Leukemia (MDS/AML) have been confirmed in 6 out of 298 (2%) patients enrolled in a single arm trial of Lynparza monotherapy, in patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAm) advanced cancers. In a randomized placebo controlled trial, MDS/AML occurred in 3 out of 136 (2%) patients with advanced ovarian cancer treated with Lynparza. Overall, MDS/AML were reported in 22 of 2,618 (<1%) patients treated with Lynparza. The majority of MDS/AML cases (17 of 22 cases) were fatal, and the duration of therapy with Lynparza in patients who developed secondary MDS/cancer-therapy related AML varied from <6 months to >2 years. All patients had previous chemotherapy with platinum agents and/or other DNA damaging agents.Monitor complete blood count testing at baseline and monthly thereafter. Do not start Lynparza until patients have recovered from hematological toxicity caused by previous chemotherapy (�CTCAE Grade 1). For prolonged hematological toxicities, interrupt Lynparza and monitor blood counts weekly until recovery. If the levels have not recovered to CTCAE Grade 1 or less after 4 weeks, refer the patient to a hematologist for further investigations, including bone marrow analysis and blood sample for cytogenetics. If MDS/AML is confirmed, discontinue Lynparza.PneumonitisPneumonitis, including fatal cases, occurred in <1% of patients treated with Lynparza. If patients present with new or worsening respiratory symptoms such as dyspnea, fever, cough, wheezing, or a radiological abnormality occurs, interrupt treatment with Lynparza and initiate prompt investigation. If pneumonitis is confirmed, discontinue Lynparza.Embryo-Fetal ToxicityLynparza can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. Olaparib was teratogenic and caused embryo-fetal toxicity in rats at exposures below those in patients receiving the recommended human dose of 400 mg twice daily. If the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus [see Use in Specific Populations (8.1) in the full Prescribing Information].Advise females of reproductive potential to avoid becoming pregnant while taking Lynparza. If contraceptive methods are being considered, use effective contraception during treatment and for at least one month after receiving the last dose of Lynparza [see Use in Specific Populations (8.6) in the full Prescribing Information].
ADVERSE REACTIONSThe following adverse reactions are discussed elsewhere in the labeling:• Myelodysplastic syndrome/Acute Myeloid Leukemia [see Warnings and Precautions
(5.1) in the full Prescribing Information]• Pneumonitis [see Warnings and Precautions (5.2) in the full Prescribing Information]
Clinical Trial ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.Lynparza 400 mg twice daily as monotherapy, has been studied in 300 patients with gBRCA-mutated advanced ovarian cancer, and 223 of these patients had received 3 or more prior lines of chemotherapy.In the 223 patients with gBRCA-mutated ovarian cancer who received 3 or more prior lines of chemotherapy (including 137 patients in Study 1 with measureable disease) [see Clinical Studies (14) in the full Prescribing Information] adverse reactions led to dose interruption in 40% of patients, dose reduction in 4%, and discontinuation in 7%. There were 8 (4%) patients with adverse reactions leading to death, two were attributed to acute leukemia, and one each was attributed to COPD, cerebrovascular accident, intestinal perforation, pulmonary embolism, sepsis, and suture rupture. Table 1 presents the frequency of adverse reactions reported in �20% of 223 patients (in 6 studies) with gBRCA-mutated advanced ovarian cancer who had received 3 or more prior lines of chemotherapy who were treated with Lynparza 400 mg twice daily. The median exposure to Lynparza in these patients was 158 days.
Table 1 Adverse Reactions Reported in �20% of Patients with gBRCA-Mutated Advanced Ovarian Cancer Receiving Lynparza
3 or more lines of prior chemotherapy
Adverse Reaction Grades 1-4 Grades 3-4 N=223 N=223 % % Blood and Lymphatic disorders
Anemia 34 18
Gastrointestinal disorders
Abdominal pain/discomfort 43 8
Decreased appetite 22 1
Nausea 64 3
Vomiting 43 4
Diarrhea 31 1
Dyspepsia 25 0
General disorders
Fatigue/asthenia 66 8
Infections and infestations
Nasopharyngitis/URI 26 0
Musculoskeletal and Connective Tissue disorders
Arthralgia/musculoskeletal pain 21 0
Myalgia 22 0
Table 2 presents the frequency of abnormal laboratory findings in the 223 patients with gBRCA-mutated advanced ovarian cancer who had received three or more prior lines of chemotherapy receiving Lynparza 400 mg twice daily.
Table 2 Laboratory Abnormalities Reported in Patients with gBRCA-Mutated Advanced Ovarian Cancer Receiving Lynparza
Laboratory Parameter* 3 or more lines of prior chemotherapy
Grades 1-4 Grades 3-4 N=223 N=223 % %
Decrease in hemoglobin (anemia) 90 15
Decrease in absolute neutrophil count 25 7 (neutropenia)
Decrease in platelets (thrombocytopenia) 30 3
Decrease in lymphocytes (lymphopenia) 56 17
Mean corpuscular volume elevation 57 -
Increase in creatinine* 30 2* Patients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
The following adverse reactions and laboratory abnormalities have been identified in �10 to <20% of the 223 patients receiving Lynparza and not included in the table: cough, constipation, dysgeusia, peripheral edema, back pain, dizziness, headache, urinary tract infection, dyspnea, and rash. The following adverse reactions and laboratory abnormalities have been identified in �1 to <10% of the 223 patients receiving Lynparza and not included in the table: leukopenia, stomatitis, peripheral neuropathy, pyrexia, hypomagnesemia, hyperglycemia, anxiety, depression, insomnia, dysuria, urinary incontinence, vulvovaginal disorder, dry skin/ eczema, pruritis, hypertension, venous thrombosis (including pulmonary embolism), and hot flush.Table 3 presents adverse reactions reported in �20% of patients from a randomized trial of Lynparza 400 mg twice daily as maintenance monotherapy compared to placebo in patients with platinum sensitive, relapsed, high-grade serous ovarian cancer following treatment with 2 or more platinum-containing regimens. Table 4 presents the laboratory abnormalities in patients from this randomized trial. Of the 96 patients with gBRCA- mutation, 53 received Lynparza, and 43 received placebo. The median duration on treatment with Lynparza was 11.1 months for patients with a gBRCA mutation compared to 4.4 months for patients with gBRCA mutation on placebo.Adverse reactions led to dose interruptions in 26% of those receiving Lynparza and 7% of those receiving placebo; dose reductions in 15% of Lynparza and 5% of placebo patients; and discontinuation in 9% of Lynparza and 0% in placebo patients. One (2%) patient on Lynparza died as a result of an adverse reaction.

PAGE 58 The ASCO Post | MARCH 25, 2015
Education in Oncology
not specify strategies to accomplish individual or team-based professional self-care.
Some examples of strategies for improving individual professional self-care include regularly appraising all aspects of work life; developing a
network of peers and mentors; seeking organizational engagement opportuni-ties; improving communication and management skills; increasing self-awareness in setting limits; prioritiz-ing personal relationships with family members and close friends; and pursu-ing reflective writing.
Strategies for improving team mem-
bers’ well-being include enhancing their skills to empathize with others; formalizing structures, policies, and procedures to guide team meetings; and sharing personal and professional sources of meaning and incorporating them into daily life. n
Disclosure: Dr. Sanchez-Reilly reported no potential conflicts of interest.
References1. Shanafelt TD, Gradishar WJ, Kosty
M, et al: Burnout and career satisfac-tion among US oncologists. J Clin Oncol 32:678-686, 2014.
2. Sanchez-Reilly S, Morrison LJ, Carey E, et al: Caring for oneself to care for oth-ers: Physicians and their self-care. J Support Oncol 11:75-81, 2013.
Avoiding Burnoutcontinued from page 57
Don’t Miss These Important
Reports in This Issue of
The ASCO Post
Hyman Muss, MD, on Caring for Older Patients with Cancer see page 1
Sandy Srinivas, MD, on Antiangiogenesis in Advanced Bladder Cancer see page 6
Jeff M. Michalski, MD, on High-Dose Radiotherapy in Localized Prostate Cancer see page 5
Visit The ASCO Post online at ASCOPost.com
LYNPARZATM (olaparib) capsules 2
Table 3 Adverse Reactions Reported in �20% of Patients with gBRCA-Mutated Ovarian Cancer in the Randomized Trial Adverse Reactions Lynparza Placebo N=53 N=43 Grades 1-4 Grades 3-4 Grades 1-4 Grades 3-4 % % % % Blood and Lymphatic disorders Anemia 25 4 7 2 Gastrointestinal disorders Abdominal pain/discomfort 47 0 58 2 Decreased appetite 25 0 14 0 Nausea 75 2 37 0 Vomiting 32 4 9 0 Diarrhea 28 4 21 2 Dyspepsia 25 0 14 0 Dysgeusia 21 0 9 0 General disorders Fatigue (including asthenia, lethargy) 68 6 53 2 Infections and infestations Nasopharyngitis/Pharyngitis/URI 43 0 16 0 Musculoskeletal and Connective tissue disorders Arthralgia/Musculoskeletal pain 32 4 21 0 Myalgia 25 2 12 0 Back pain 25 6 21 0 Nervous system disorder Headache 25 0 19 2 Respiratory, Thoracic, Mediastinal disorders Cough 21 0 14 0 Skin and Subcutaneous Tissue Dermatitis/Rash 25 0 14 0
Table 4 Laboratory Abnormalities in Patients with gBRCA-Mutated Ovarian Cancer in the Randomized Trial Laboratory parameter* Lynparza Placebo N=53 N=43 Grades 1-4 Grades 3-4 Grades 1-4 Grades 3-4 % % % % Decrease in hemoglobin 85 8 58 2 Decrease in absolute neutrophil count 32 8 23 0 Decrease in platelets 26 6 19 0 Mean corpuscular volume elevation 85 - 44 - Increase in creatinine* 26 0 5 0 * Patients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
DRUG INTERACTIONSOlaparib is primarily metabolized by CYP3A.Anticancer AgentsClinical studies of Lynparza in combination with other myelosuppressive anticancer agents, including DNA damaging agents, indicate a potentiation and prolongation of myelosuppressive toxicity.Drugs that may Increase Olaparib Plasma ConcentrationsIn patients (N=57), co-administration of itraconazole, a strong CYP3A inhibitor, increased AUC of olaparib by 2.7-fold. A moderate CYP3A inhibitor, fluconazole, is predicted to increase the AUC of olaparib by 2-fold.Avoid concomitant use of strong CYP3A inhibitors (e.g., itraconazole, telithromycin, clarithromycin, ketoconazole, voriconazole, nefazodone, posaconazole, ritinovir, lopinavir/ ritinovir, indinavir, saquinavir, nelfinavir, boceprevir, telaprevir) and moderate CYP3A inhibitors (e.g., amprenavir, aprepitant, atazanavir, ciprofloxacin, crizotinib, darunavir/ritonavir, diltiazem, erythromycin, fluconazole, fosamprenavir, imatinib, verapamil). If the strong or moderate CYP3A inhibitors must be co-administered, reduce the dose of Lynparza [see Dosage and Administration (2.4) in the full Prescribing Information].Avoid grapefruit and Seville oranges during Lynparza treatment [see Dosage and Adminis-tration (2.4) and Clinical Pharmacology (12.3) in the full Prescribing Information].Drugs that may Decrease Olaparib Plasma ConcentrationsIn patients (N=22), co-administration of rifampicin, a strong CYP3A inducer, decreased AUC of olaparib by 87%. A moderate CYP3A inducer, efavirenz, is predicted to decrease the AUC of olaparib by 50-60%.Avoid concomitant use of strong CYP3A inducers (e.g., phenytoin, rifampicin, carbamazepine, St. John’s Wort) and moderate CYP3A4 inducers (e.g., bosentan, efavirenz, etravirine, modafinil, nafcillin). If a moderate CYP3A inducer cannot be avoided, be aware of a potential for decreased efficacy of Lynparza [see Clinical Pharmacology (12.3) in the full Prescribing Information].USE IN SPECIFIC POPULATIONSPregnancyPregnancy Category D [see Warnings and Precautions (5.3) in the full Prescribing Information] Risk summaryLynparza can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. Olaparib was teratogenic and caused embryo-fetal toxicity in rats at exposures below those in patients receiving the recommended human dose of 400 mg twice daily. If this drug is used during pregnancy, or if a patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus and the potential risk for loss of the pregnancy.Animal DataIn a fertility and early embryonic development study in female rats, olaparib was administered orally for 14 days before mating through to day 6 of pregnancy, which
resulted in increased post-implantation loss at a dose level of 15 mg/kg/day (with maternal systemic exposures approximately 11% of the human exposure (AUC0-24h) at the recommended dose).In an embryo-fetal development study, pregnant rats received oral doses of 0.05 and 0.5 mg/kg/day olaparib during the period of organogenesis. A dose of 0.5 mg/kg/day (with maternal systemic exposures approximately 0.3% of human exposure (AUC0-24h) at the recommended dose) caused embryo-fetal toxicities including increased post-implantation loss and major malformations of the eyes (anophthalmia, microphthalmia), vertebrae/ribs (extra rib or ossification center; fused or absent neural arches, ribs, and sternebrae), skull (fused exoccipital) and diaphragm (hernia). Additional abnormalities or variants included incomplete or absent ossification (vertebrae/sternebrae, ribs, limbs) and other findings in the vertebrae/sternebrae, pelvic girdle, lung, thymus, liver, ureter and umbilical artery. Some findings noted above in the eyes, ribs and ureter were observed at a dose of 0.05 mg/kg/day olaparib at lower incidence.Nursing MothersIt is not known whether olaparib is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from olaparib, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.Pediatric UseThe safety and efficacy of Lynparza has not been established in pediatric patients.Geriatric UseIn clinical studies of Lynparza enrolling 735 patients with advanced solid tumors [the majority (69%) of whom had ovarian cancer] who received Lynparza 400 mg twice daily as monotherapy, 148 (20%) of patients were aged �65 years. The safety profile was similar irrespective of age with the exception of AEs of CTCAE �3 which were reported more frequently in patients aged �65 years (53.4%) than those <65 years (43.4%). No individual adverse event or System Organ Class accounted for this observed difference.Females of Reproductive PotentialLynparza can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1) in the full Prescribing Information]. Advise female patients of reproductive potential to avoid pregnancy while taking Lynparza. If contraceptive methods are being considered, use highly effective contraception during treatment with Lynparza and for at least one month following the last dose of Lynparza. Instruct patients to contact their health-care provider if they become pregnant, or if pregnancy is suspected, while taking Lynparza.Hepatic ImpairmentThe effect of hepatic impairment on exposure to Lynparza has not been studied. Patients with bilirubin >1.5 X ULN and AST/ALT �2.5 X ULN (�5 X ULN in the presence of liver metastases) were excluded from Lynparza clinical trials. There are no data in patients with baseline hepatic impairment (serum bilirubin >1.5 X ULN) [see Clinical Pharmacology (12.3) in the full Prescribing Information].Renal ImpairmentBased on preliminary data, a 1.5 fold increase in mean exposure (AUC) was observed in patients with mild renal impairment (CLcr = 50-80 mL/min) compared to patients with normal renal function (CLcr >80 mL/min). No dose adjustment to the starting dose is required in patients with CLcr of 50 to 80 mL/min, but patients should be monitored closely for toxicity. There are no data in patients with moderate or severe renal impairment (CLcr <50 mL/min) or patients on dialysis [see Clinical Pharmacology (12.3) in the full Prescribing Information].OVERDOSAGEThere is no specific treatment in the event of Lynparza overdose, and symptoms of overdose are not established. In the event of an overdose, physicians should follow general supportive measures and should treat symptomatically.17 PATIENT COUNSELING INFORMATIONSEE FDA-APPROVED PATIENT LABELING (MEDICATION GUIDE)• Dosing Instructions: Inform patients on how to take Lynparza [see Dosage and
Administration (2.1) in the full Prescribing Information]. Lynparza should be taken twice daily. Instruct patients that if they miss a dose of Lynparza, not to take an extra dose to make up for the one that they missed. They should take their next normal dose at the usual time. Each capsule should be swallowed whole. Do not chew, dissolve, or open the capsule. Patient should not take Lynparza with grapefruit or Seville oranges.
• MDS/AML: Advise patients to contact their healthcare provider if they experience weakness, feeling tired, fever, weight loss, frequent infections, bruising, bleeding easily, breathlessness, blood in urine or stool, and/or laboratory findings of low blood cell counts, or a need for blood transfusions. This may be a sign of hematological toxicity or a more serious uncommon bone marrow problem called ‘myelodysplastic syndrome’ (MDS) or ‘acute myeloid leukemia’ (AML) which have been reported in patients treated with Lynparza [see Warnings and Precautions (5.1) in the full Prescribing Information].
• Pneumonitis: Advise patients to contact their healthcare provider if they experience any new or worsening respiratory symptoms including shortness of breath, fever, cough, or wheezing [see Warnings and Precautions (5.2) in the full Prescribing Information].
• Pregnancy and Females of Reproductive Potential: Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Use in Specific Populations (8.1) in the full Prescribing Information]. Advise females of reproductive potential to use effective contraception during treatment with Lynparza and for at least one month after receiving the last dose of Lynparza [see Warnings and Precautions (5.3) and Use in Specific Populations (8.1, 8.6) in the full Prescribing Information].
• Nursing Mothers: Advise patients not to breastfeed while taking Lynparza [see Use in Special Populations (8.3) in the full Prescribing Information].
• Nausea/vomiting: Advise patients that mild or moderate nausea and/or vomiting is very common in patients receiving Lynparza and that they should contact their healthcare provider who will advise on available antiemetic treatment options.
Distributed by:AstraZeneca Pharmaceuticals LP, Wilmington, DE 198503079901 12/14 Issued: 12/2014

ASCOPost.com | MARCH 25, 2015 PAGE 59
Expert’s Corner
Five Questions Can Guide the Treatment of Relapsed MyelomaBy Caroline Helwick
Joseph Mikhael, MD, MEd, my-eloma expert at the Mayo Clinic
in Arizona, Scottsdale, and Associate Dean of the Mayo School of Gradu-ate Medical Education, considers five questions when selecting treatment for patients with multiple myeloma who relapse.
“With prolonged survival, which approaches 10 years in standard-risk patients, most patients will undergo many lines of therapy, and this will require a long-term approach that bal-ances disease control with the elimi-nation of clones,” Dr. Mikhael said in an interview with The ASCO Post.
“With many options available to the clinician, there is no simple or ideal se-quence of treatments that has been es-tablished for treating relapsed disease. Balancing efficacy, toxicity, and cost will become ever more challenging with our increased options,” he noted.
It is possible to do this, he main-tained, by asking five critical questions that form a practical algorithm. These questions will help clinicians differen-tiate this highly heterogeneous patient population. “We surely cannot treat all patients the same way,” he commented.
1. Does this patient really need to be treated now?
With slow, indolent relapse, cer-tain patients—especially those with low-risk features—may be monitored closely (usually monthly), without in-tervention. “Identifying the ‘indolent’ vs the ‘aggressive’ relapse is crucial,” he emphasized.
With indolent relapse, it is reason-able to introduce a sequential approach using one or two agents at a time, usually a proteasome inhibitor or an immuno-modulatory drug plus dexamethasone, eventually using all the active agents. This “control” approach is more likely to be useful in standard-risk patients. A
more aggressive relapse requires a more aggressive treatment regimen, often us-ing several agents in combination.
2. Should this patient be retreated with a previously used drug?
Retreatment with the same drugs that were used upfront is often neces-sary, and this may or may not be effec-tive. Several factors can be predictive of efficacy in the relapsed setting: depth of the initial response (the deeper the re-sponse, the more likely that re-exposure will produce a response); duration of first response (at least 6 months pre-dicts for a future response); and good tolerability of the agent. Bortezomib was approved for retreatment in 2014, he noted.
3. Have the ‘Big 5’ been used?Five “novel” agents remain the cen-
ter of myeloma therapy—thalidomide (Thalomid), lenalidomide (Revlimid), pomalidomide (Pomalyst), bortezomib (Velcade), and carfilzomib (Kyprolis). Although carfilzomib and pomalido-mide are newer-generation agents, the others remain a critical part of treat-ment and have not been replaced, Dr. Mikhael indicated.
For the immunomodulatory drug class, thalidomide is best used late in the disease course, in patients who cannot tolerate myelosuppression, and in combination (especially with bortezomib/dexamethasone or with cyclophosphamide/bortezomib/dexa-methasone). Lenalidomide has shown a survival advantage in large phase III trials and is successfully combined with bortezomib and carfilzomib. Pomalido-mide is highly active in combination with dexamethasone and has been ef-fectively combined with bortezomib and carfilzomib.
For the proteasome inhibitors, bort-ezomib can be effectively used alone or in combination with immunomodula-tory drugs (including pomalidomide) and cyclophosphamide. Carfilzomib can be widely used, even in bortezo-mib-refractory patients, alone and in combination with essentially all other myeloma agents.
Dr. Mikhael commented on the role of the two newest agents, carfilzomib and pomalidomide. “With very simi-lar indications for relapsed myeloma, deciding between these two agents is
a common clinical problem,” he noted. “It should be reassuring to clinicians that each myeloma patient is likely ulti-mately to see both agents, and the opti-mal sequence is not yet known.”
Several factors can help clinicians choose between these agents (both of which are effective in high-risk disease). Factors favoring carfilzomib include the presence of renal insufficiency and pre-existing neuropathy, whereas pomalid-omide is favored when convenience of administration is important and in pa-tients with poorly controlled heart fail-ure or hypertension.
“Class ‘switching’ is not necessarily supported by evidence,” he pointed out. This means that bortezomib-refractory patients may respond to carfilzomib, and lenalidomide-refractory patients may respond to pomalidomide.
Evidence is emerging that these two
drugs are effective when combined, and therefore this doublet should be con-sidered in aggressive relapse. The addi-tion of the second agent when patients do not respond or progress on the other can also be considered.
Elaborating on combinations of agents, Dr. Mikhael added that even when patients become resistant to a certain agent, they may become sensitive to it again, due to clonal evolution or because of something that occurs when the drug is combined with another. He noted that about 30% of patients who are resistant to
bortezomib and dexamethasone respond to the combination of bortezomib/ lenalidmide/dexamethasone (VRD).
4. Have the ‘add-on’ agents been used?
Although the five key agents dis-cussed here are considered the back-bone of myeloma treatment, several others have activity that can enhance their efficacy: corticosteroids (dexa-methasone weekly [20–40 mg], alter-nate-day prednisone (25–100 mg), alkylating agents (cyclophosphamide, melphalan), and liposomal doxorubi-cin. These agents can also be considered without a novel agent on board (ie, cy-clophosphamide/prednisone).
5. Has an individualized, risk-stratified approach been considered?
“A patient’s risk status may indeed in-fluence the selection of relapsed therapy. Standard-risk patients may only require single-agent approaches. Intermediate-risk patients will likely benefit from bortezomib-containing regimens. And high-risk patients require more intense combination regimens to effectively control their disease,” he indicated.
Elaborating on individual risk, Dr. Mikhael noted that relapse occurs sooner and more aggressively in high-risk pa-tients and that close monitoring and rap-id initiation of treatment in these patients
Hematology
Joseph Mikhael, MD, MEd
continued on page 62
A patient’s risk status may influence the selection of relapsed therapy. Standard-risk patients may only require
single-agent approaches, and high-risk patients require more intense combination regimens to
effectively control their disease. —Joseph Mikhael, MD, MEd
Fig. 1: Factors in Selecting Therapy for Relapsed Myeloma. Courtesy of Joseph Mikael, MD, MEd.
LYNPARZATM (olaparib) capsules 2
Table 3 Adverse Reactions Reported in �20% of Patients with gBRCA-Mutated Ovarian Cancer in the Randomized Trial Adverse Reactions Lynparza Placebo N=53 N=43 Grades 1-4 Grades 3-4 Grades 1-4 Grades 3-4 % % % % Blood and Lymphatic disorders Anemia 25 4 7 2 Gastrointestinal disorders Abdominal pain/discomfort 47 0 58 2 Decreased appetite 25 0 14 0 Nausea 75 2 37 0 Vomiting 32 4 9 0 Diarrhea 28 4 21 2 Dyspepsia 25 0 14 0 Dysgeusia 21 0 9 0 General disorders Fatigue (including asthenia, lethargy) 68 6 53 2 Infections and infestations Nasopharyngitis/Pharyngitis/URI 43 0 16 0 Musculoskeletal and Connective tissue disorders Arthralgia/Musculoskeletal pain 32 4 21 0 Myalgia 25 2 12 0 Back pain 25 6 21 0 Nervous system disorder Headache 25 0 19 2 Respiratory, Thoracic, Mediastinal disorders Cough 21 0 14 0 Skin and Subcutaneous Tissue Dermatitis/Rash 25 0 14 0
Table 4 Laboratory Abnormalities in Patients with gBRCA-Mutated Ovarian Cancer in the Randomized Trial Laboratory parameter* Lynparza Placebo N=53 N=43 Grades 1-4 Grades 3-4 Grades 1-4 Grades 3-4 % % % % Decrease in hemoglobin 85 8 58 2 Decrease in absolute neutrophil count 32 8 23 0 Decrease in platelets 26 6 19 0 Mean corpuscular volume elevation 85 - 44 - Increase in creatinine* 26 0 5 0 * Patients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
DRUG INTERACTIONSOlaparib is primarily metabolized by CYP3A.Anticancer AgentsClinical studies of Lynparza in combination with other myelosuppressive anticancer agents, including DNA damaging agents, indicate a potentiation and prolongation of myelosuppressive toxicity.Drugs that may Increase Olaparib Plasma ConcentrationsIn patients (N=57), co-administration of itraconazole, a strong CYP3A inhibitor, increased AUC of olaparib by 2.7-fold. A moderate CYP3A inhibitor, fluconazole, is predicted to increase the AUC of olaparib by 2-fold.Avoid concomitant use of strong CYP3A inhibitors (e.g., itraconazole, telithromycin, clarithromycin, ketoconazole, voriconazole, nefazodone, posaconazole, ritinovir, lopinavir/ ritinovir, indinavir, saquinavir, nelfinavir, boceprevir, telaprevir) and moderate CYP3A inhibitors (e.g., amprenavir, aprepitant, atazanavir, ciprofloxacin, crizotinib, darunavir/ritonavir, diltiazem, erythromycin, fluconazole, fosamprenavir, imatinib, verapamil). If the strong or moderate CYP3A inhibitors must be co-administered, reduce the dose of Lynparza [see Dosage and Administration (2.4) in the full Prescribing Information].Avoid grapefruit and Seville oranges during Lynparza treatment [see Dosage and Adminis-tration (2.4) and Clinical Pharmacology (12.3) in the full Prescribing Information].Drugs that may Decrease Olaparib Plasma ConcentrationsIn patients (N=22), co-administration of rifampicin, a strong CYP3A inducer, decreased AUC of olaparib by 87%. A moderate CYP3A inducer, efavirenz, is predicted to decrease the AUC of olaparib by 50-60%.Avoid concomitant use of strong CYP3A inducers (e.g., phenytoin, rifampicin, carbamazepine, St. John’s Wort) and moderate CYP3A4 inducers (e.g., bosentan, efavirenz, etravirine, modafinil, nafcillin). If a moderate CYP3A inducer cannot be avoided, be aware of a potential for decreased efficacy of Lynparza [see Clinical Pharmacology (12.3) in the full Prescribing Information].USE IN SPECIFIC POPULATIONSPregnancyPregnancy Category D [see Warnings and Precautions (5.3) in the full Prescribing Information] Risk summaryLynparza can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. Olaparib was teratogenic and caused embryo-fetal toxicity in rats at exposures below those in patients receiving the recommended human dose of 400 mg twice daily. If this drug is used during pregnancy, or if a patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus and the potential risk for loss of the pregnancy.Animal DataIn a fertility and early embryonic development study in female rats, olaparib was administered orally for 14 days before mating through to day 6 of pregnancy, which
resulted in increased post-implantation loss at a dose level of 15 mg/kg/day (with maternal systemic exposures approximately 11% of the human exposure (AUC0-24h) at the recommended dose).In an embryo-fetal development study, pregnant rats received oral doses of 0.05 and 0.5 mg/kg/day olaparib during the period of organogenesis. A dose of 0.5 mg/kg/day (with maternal systemic exposures approximately 0.3% of human exposure (AUC0-24h) at the recommended dose) caused embryo-fetal toxicities including increased post-implantation loss and major malformations of the eyes (anophthalmia, microphthalmia), vertebrae/ribs (extra rib or ossification center; fused or absent neural arches, ribs, and sternebrae), skull (fused exoccipital) and diaphragm (hernia). Additional abnormalities or variants included incomplete or absent ossification (vertebrae/sternebrae, ribs, limbs) and other findings in the vertebrae/sternebrae, pelvic girdle, lung, thymus, liver, ureter and umbilical artery. Some findings noted above in the eyes, ribs and ureter were observed at a dose of 0.05 mg/kg/day olaparib at lower incidence.Nursing MothersIt is not known whether olaparib is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from olaparib, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.Pediatric UseThe safety and efficacy of Lynparza has not been established in pediatric patients.Geriatric UseIn clinical studies of Lynparza enrolling 735 patients with advanced solid tumors [the majority (69%) of whom had ovarian cancer] who received Lynparza 400 mg twice daily as monotherapy, 148 (20%) of patients were aged �65 years. The safety profile was similar irrespective of age with the exception of AEs of CTCAE �3 which were reported more frequently in patients aged �65 years (53.4%) than those <65 years (43.4%). No individual adverse event or System Organ Class accounted for this observed difference.Females of Reproductive PotentialLynparza can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1) in the full Prescribing Information]. Advise female patients of reproductive potential to avoid pregnancy while taking Lynparza. If contraceptive methods are being considered, use highly effective contraception during treatment with Lynparza and for at least one month following the last dose of Lynparza. Instruct patients to contact their health-care provider if they become pregnant, or if pregnancy is suspected, while taking Lynparza.Hepatic ImpairmentThe effect of hepatic impairment on exposure to Lynparza has not been studied. Patients with bilirubin >1.5 X ULN and AST/ALT �2.5 X ULN (�5 X ULN in the presence of liver metastases) were excluded from Lynparza clinical trials. There are no data in patients with baseline hepatic impairment (serum bilirubin >1.5 X ULN) [see Clinical Pharmacology (12.3) in the full Prescribing Information].Renal ImpairmentBased on preliminary data, a 1.5 fold increase in mean exposure (AUC) was observed in patients with mild renal impairment (CLcr = 50-80 mL/min) compared to patients with normal renal function (CLcr >80 mL/min). No dose adjustment to the starting dose is required in patients with CLcr of 50 to 80 mL/min, but patients should be monitored closely for toxicity. There are no data in patients with moderate or severe renal impairment (CLcr <50 mL/min) or patients on dialysis [see Clinical Pharmacology (12.3) in the full Prescribing Information].OVERDOSAGEThere is no specific treatment in the event of Lynparza overdose, and symptoms of overdose are not established. In the event of an overdose, physicians should follow general supportive measures and should treat symptomatically.17 PATIENT COUNSELING INFORMATIONSEE FDA-APPROVED PATIENT LABELING (MEDICATION GUIDE)• Dosing Instructions: Inform patients on how to take Lynparza [see Dosage and
Administration (2.1) in the full Prescribing Information]. Lynparza should be taken twice daily. Instruct patients that if they miss a dose of Lynparza, not to take an extra dose to make up for the one that they missed. They should take their next normal dose at the usual time. Each capsule should be swallowed whole. Do not chew, dissolve, or open the capsule. Patient should not take Lynparza with grapefruit or Seville oranges.
• MDS/AML: Advise patients to contact their healthcare provider if they experience weakness, feeling tired, fever, weight loss, frequent infections, bruising, bleeding easily, breathlessness, blood in urine or stool, and/or laboratory findings of low blood cell counts, or a need for blood transfusions. This may be a sign of hematological toxicity or a more serious uncommon bone marrow problem called ‘myelodysplastic syndrome’ (MDS) or ‘acute myeloid leukemia’ (AML) which have been reported in patients treated with Lynparza [see Warnings and Precautions (5.1) in the full Prescribing Information].
• Pneumonitis: Advise patients to contact their healthcare provider if they experience any new or worsening respiratory symptoms including shortness of breath, fever, cough, or wheezing [see Warnings and Precautions (5.2) in the full Prescribing Information].
• Pregnancy and Females of Reproductive Potential: Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Use in Specific Populations (8.1) in the full Prescribing Information]. Advise females of reproductive potential to use effective contraception during treatment with Lynparza and for at least one month after receiving the last dose of Lynparza [see Warnings and Precautions (5.3) and Use in Specific Populations (8.1, 8.6) in the full Prescribing Information].
• Nursing Mothers: Advise patients not to breastfeed while taking Lynparza [see Use in Special Populations (8.3) in the full Prescribing Information].
• Nausea/vomiting: Advise patients that mild or moderate nausea and/or vomiting is very common in patients receiving Lynparza and that they should contact their healthcare provider who will advise on available antiemetic treatment options.
Distributed by:AstraZeneca Pharmaceuticals LP, Wilmington, DE 198503079901 12/14 Issued: 12/2014

Reference: 1. Melcher A, Parato, K, Rooney CM, Bell JC. Mol Ther. 2011;19:1008-1016.
©2014 Amgen Inc. All rights reserved. 10/14 USA-678-100791
Cancer cell
LyseOncolytic Immunotherapy is an innovative area of research that uses a modified virus designed to induce tumor cell lysis and expose tumor-derived antigens.
Amgen is researching ways to help T cells target cancer.
Activated dendritic cells can teach T cells to help find and fight cancer cells with a matching antigen profile.1
LearnDendritic cell
Explore what’s new at:OncolyticImmunotherapy.com
TVUS14CDNY6107_D_OI_JA_CompD_Spread_King_r8.indd All Pages 10/15/14 9:37 AM

Reference: 1. Melcher A, Parato, K, Rooney CM, Bell JC. Mol Ther. 2011;19:1008-1016.
©2014 Amgen Inc. All rights reserved. 10/14 USA-678-100791
Cancer cell
LyseOncolytic Immunotherapy is an innovative area of research that uses a modified virus designed to induce tumor cell lysis and expose tumor-derived antigens.
Amgen is researching ways to help T cells target cancer.
Activated dendritic cells can teach T cells to help find and fight cancer cells with a matching antigen profile.1
LearnDendritic cell
Explore what’s new at:OncolyticImmunotherapy.com
TVUS14CDNY6107_D_OI_JA_CompD_Spread_King_r8.indd All Pages 10/15/14 9:37 AM

PAGE 62 The ASCO Post | MARCH 25, 2015
Expert’s Corner
are critical. Risk factors may change since the time of first diagnosis, so patients should be reevaluated upon relapse.
In addition to risk tier, several other patient factors should be considered be-cause they may also influence the choice of treatment, especially age, performance status, renal insufficiency, bone marrow reserve (previous myelosuppression), preexisting neuropathy, and other co-morbidities, such as cardiac disease and diabetes (see Fig. 1 on page 59).
Other ApproachesAutologous stem cell transplanta-
tion remains the standard of care for
frontline therapy in eligible patients, although some may defer transplanta-tion until relapse. However, a second autologous stem cell transplantation is also feasible as salvage later in the dis-ease course assuming three criteria are met: (1) the patient responded to the first autologous stem cell transplanta-tion, (2) the patient tolerated the first autologous stem cell transplantation, and (3) the patient did not progress for at least 2 years after autologous stem cell transplantation. When these con-ditions are met, patients can expect a progression-free survival that is 50% to 70% that achieved with the first autolo-gous stem cell transplantation, he said.
Alternative approaches include al-
logeneic bone marrow transplant and high-dose chemotherapy. Allogeneic transplant remains controversial, based on the availability of novel agents and high treatment-related morbidity and mortality.
“My approach is that only patients meeting four requirements will be can-didates for allogeneic transplant,” he said. They must be “young,” have ge-notypically high-risk disease, and have phenotypically high-risk disease (ie, relapsing quickly), and the transplant should not be used as a “Hail Mary” af-ter all other strategies have failed.
High-dose chemotherapy (such as DT-PACE [dexamethasone, tha-lidomide, cisplatin, doxorubicin, cy-
clophosphamide, etoposide]) may produce responses in about 50% of pa-tients, although they tend to be short-lived. “This approach is likely best used as a bridge to a more definitive therapy or clinical trial,” he maintained.
Dr. Mikhael hailed the emergence of new agents, especially those with new mechanisms of action. Monoclonal an-tibodies, histone deacetylase inhibitors, immune therapies, and agents directed toward new targets will expand the landscape of treatment and will likely further improve survival in myeloma, he predicted. n
Disclosure: Dr. Mikhael has received institutional research funding from Onyx, Celgene, Sanofi, and AbbVie.
Relapsed Myelomacontinued from page 59
Fishing huts and gas pump on a lower harbor dock in Marquette, Michigan.
Photo by Steve Gualdoni, PA-C, UP Health System - Marquette Hematology Oncology, Marquette, Michigan.
Send your high-resolution photo and caption to [email protected]. All photos will be considered for publication in a future issue of The ASCO Post.
FDA Launches Drug Shortages Mobile App
Today, the U.S. Food and Drug Ad-ministration (FDA) launched
the agency’s first mobile application (app) specifically designed to speed pub-lic access to valuable information about drug shortages.
The app identifies current drug short-ages, resolved shortages, and discontinua-tions of drug products.
Drugs in short supply can delay or deny needed care for patients. Drug short-ages may also lead health care profession-als to rely on alternative drug products, which may be less effective or associated with higher risks than the drug in shortage.
“The FDA understands that health-care professionals and pharmacists need real-time information about drug short-ages to make treatment decisions,” said Valerie Jensen, RPh, Associate Director
of the Drug Shortage Staff in the FDA’s Center for Drug Evaluation and Research. “The new mobile app is an innovative tool that will offer easier and faster access to important drug shortage information.”
App users can search or browse by a
drug’s generic name or active ingredient, or browse by therapeutic category. The app can also be used to report a suspected drug shortage or supply issue to the FDA.
The agency developed the drug shortages app to improve access to in-formation about drug shortages, as part of the FDA’s efforts outlined in the “Stra-tegic Plan for Preventing and Mitigating Drug Shortages”.
The app is available for free download via iTunes (for Apple devices) and the Google Play store (for Android devices) by searching “FDA Drug Shortages.” n
The FDA understands that health-care professionals and pharmacists need real-time information about drug shortages to make treatment decisions.
—Valerie Jensen, RPh
AnnouncementsFishing Huts and Gas Pump, Marquette, Michigan
Visit The ASCO Post website at ASCOPost.com
JOB#: 40680 CLIENT: Genentech
DESC: Journal Ad 3-pg FILE NAM
E: GNH_HER_Q40680_JA_D03.indd DATE: 11-21-2014 6:50 PM
ROUND: 3PG: CordobaR/MerinoR
AD: R Vetrano-Pyke x4077 PM
: B Fu x4416 AE: K McGinty x3950
CW: L Gibbons
Last Saved: 11-21-2014 6:50 PMTRIM
: 15.5” x 10.5” BLEED: 17.5” x 11.5”
SAFETY: 14.75” x 9.875” PROD: M Haight x4245
INK Spec: 4C PRINT SCALE: 82.94%
FONTS: Myriad Pro (Bold, Regular, Light), Helvetica Neue LT Std (45 Light)
IMAGES: 40680_JA_1_fn.tif (CMYK; 300 ppi; 100%
), 40680_glow_fn.psd (CMYK; 300 ppi; 100%
), Perjeta_US_MBC_NEO_4C.eps (69.5%
)INKS:
Cyan, Magenta,
Yellow, Black
DOC PATH: Macintosh HD:Users:cordobar:De...sk_prep:GNH_HER_Q40680_JA_D03.inddNOTES: None
GNH_HER_Q40680_JA_D03.indd Galley: 1
S&H
PharmaGraphics
Disk
DATE
SIGNOFF
PGQC
TCAD
CDCW
AE/ASED
PROD
IndicationPERJETA® (pertuzumab) is a HER2/neu receptor antagonist indicated in combination with Herceptin® (trastuzumab) and docetaxel for the treatment of patients with HER2-positive metastatic breast cancer who have not received prior anti-HER2 therapy or chemotherapy for metastatic disease.
Boxed WARNINGS: Cardiomyopathy and Embryo-Fetal ToxicityPERJETA administration can result in subclinical and clinical cardiac failure. Evaluate left ventricular function in all patients prior to and during treatment with PERJETA. Discontinue PERJETA treatment for a confi rmed clinically signifi cant decrease in left ventricular function.
Exposure to PERJETA can result in embryo-fetal death and birth defects. Studies in animals have resulted in oligohydramnios, delayed renal development, and death. Advise patients of these risks and the need for eff ective contraception.
Please see Brief Summary of PERJETA full Prescribing Information including Boxed WARNINGS for additional Important Safety Information on the following pages.
STRENGTHEN HER DEFENSE
Treatment guidelines recommend PERJETA-based therapy as the preferred first-line option• NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) recommend pertuzumab (PERJETA) + trastuzumab (Herceptin) +
docetaxel as a (category 1) preferred option for the fi rst-line treatment of patients with HER2+ MBC1
• ASCO® Clinical Oncology Practice Guidelines recommend pertuzumab + trastuzumab + docetaxel as fi rst-line therapy for advanced HER2+ breast cancer2
NCCN®=National Comprehensive Cancer Network®; HER2=human epidermal growth factor receptor 2 ; ASCO®=American Society of Clinical Oncology®.
C M Y KCosmos Communications 1
1ej
29452a 11.25.14 133
Q1 Q2
S:6.875”
S:9.875”
T:7.75”
T:10.5”
B:8.75”
B:11.5”

ASCOPost.com | MARCH 25, 2015 PAGE 63
Announcements
Seattle Children’s Names Jeff Sperring, MD, New Chief Executive Officer
Seattle Children’s Hospital an-nounced that the Board of Trustees
has named Jeff Sperring, MD, Chief Executive Officer, effective early in May. Dr. Sperring, who currently serves as President and Chief Executive Officer of Riley Hospital for Children at Indi-ana University Health, will continue to implement Seattle Children’s 2012-2016 Strategic Plan, with the goal of carrying out the hospital’s mission to prevent, treat, and eliminate pediatric disease.
Dr. Sperring has worked at Riley Hos-pital for Children since 2002. As Chief Executive Officer of the hospital, he pro-vided pediatric strategic and policy over-
sight within the 18-hospital Indiana Uni-versity Health system, and he developed the framework to transform Riley from a children’s hospital to a statewide chil-dren’s health system. He also managed the completion of the 750,000 square foot, $500 million Simon Family Tower, and implemented lean-based process improve-ment programs. Prior to taking the role of President and Chief Executive Officer, he served as Riley’s Chief Medical Officer, Associate Chief Medical Officer, and Di-rector of Pediatric Hospital Medicine.
Passion and Dedication“We are thrilled that Dr. Sperring is as-
suming the role of CEO,”said Judy Holder, Chair, Board of Trustees. “He brings tre-mendous experience leading a highly ranked, complex healthcare organization, and he exudes a clear passion and dedica-tion to our mission at Seattle Children’s.”
“I am honored to have the opportunity to lead one of the best children’s hospitals in the country,” said Dr. Sperring. “Seattle Children’s already has a tremendous track record, and I look forward to carrying the torch to help bolster its ability to develop cures for pediatric disease and provide the highest level of care to every child in need,” Dr. Sperring said.
Dr. Sperring obtained his Bachelor of Science in Biology from Emory Uni-versity and his Doctor of Medicine from Vanderbilt University School of Medi-cine. He was a pediatric resident at the Naval Medical Center San Diego. After
residency, he served as a United States Navy Medical Corps officer in Twenty-nine Palms, California, for 3 years.
Former CEO to RetireDr. Sperring will replace current CEO
Thomas N. Hansen, MD, who will re-
tire in May after 10 years of service. Dr. Hansen is credited with growing Seattle Children’s Research Institute, opening a major outpatient and ambulatory sur-gery center in Bellevue, and overseeing the completion of the Building Hope expansion at the hospital. After stepping
down as CEO, Dr. Hansen plans to re-main an investigator at Seattle Children’s Research Institute.
Dr. Sperring will oversee Seattle Chil-dren’s Hospital, Research Institute, Hos-pital, and Research Foundation and Guild Association. n
Jeff Sperring, MD
JOB#: 40680 CLIENT: Genentech
DESC: Journal Ad 3-pg FILE NAM
E: GNH_HER_Q40680_JA_D03.indd DATE: 11-21-2014 6:50 PM
ROUND: 3PG: CordobaR/MerinoR
AD: R Vetrano-Pyke x4077 PM
: B Fu x4416 AE: K McGinty x3950
CW: L Gibbons
Last Saved: 11-21-2014 6:50 PMTRIM
: 15.5” x 10.5” BLEED: 17.5” x 11.5”
SAFETY: 14.75” x 9.875” PROD: M Haight x4245
INK Spec: 4C PRINT SCALE: 82.94%
FONTS: Myriad Pro (Bold, Regular, Light), Helvetica Neue LT Std (45 Light)
IMAGES: 40680_JA_1_fn.tif (CMYK; 300 ppi; 100%
), 40680_glow_fn.psd (CMYK; 300 ppi; 100%
), Perjeta_US_MBC_NEO_4C.eps (69.5%
)INKS:
Cyan, Magenta,
Yellow, Black
DOC PATH: Macintosh HD:Users:cordobar:De...sk_prep:GNH_HER_Q40680_JA_D03.inddNOTES: None
GNH_HER_Q40680_JA_D03.indd Galley: 1
S&H
PharmaGraphics
Disk
DATE
SIGNOFF
PGQC
TCAD
CDCW
AE/ASED
PROD
IndicationPERJETA® (pertuzumab) is a HER2/neu receptor antagonist indicated in combination with Herceptin® (trastuzumab) and docetaxel for the treatment of patients with HER2-positive metastatic breast cancer who have not received prior anti-HER2 therapy or chemotherapy for metastatic disease.
Boxed WARNINGS: Cardiomyopathy and Embryo-Fetal ToxicityPERJETA administration can result in subclinical and clinical cardiac failure. Evaluate left ventricular function in all patients prior to and during treatment with PERJETA. Discontinue PERJETA treatment for a confi rmed clinically signifi cant decrease in left ventricular function.
Exposure to PERJETA can result in embryo-fetal death and birth defects. Studies in animals have resulted in oligohydramnios, delayed renal development, and death. Advise patients of these risks and the need for eff ective contraception.
Please see Brief Summary of PERJETA full Prescribing Information including Boxed WARNINGS for additional Important Safety Information on the following pages.
STRENGTHEN HER DEFENSE
Treatment guidelines recommend PERJETA-based therapy as the preferred first-line option• NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) recommend pertuzumab (PERJETA) + trastuzumab (Herceptin) +
docetaxel as a (category 1) preferred option for the fi rst-line treatment of patients with HER2+ MBC1
• ASCO® Clinical Oncology Practice Guidelines recommend pertuzumab + trastuzumab + docetaxel as fi rst-line therapy for advanced HER2+ breast cancer2
NCCN®=National Comprehensive Cancer Network®; HER2=human epidermal growth factor receptor 2 ; ASCO®=American Society of Clinical Oncology®.
C M Y KCosmos Communications 1
1ej
29452a 11.25.14 133
Q1 Q2
S:6.875”
S:9.875”
T:7.75”
T:10.5”
B:8.75”
B:11.5”

PAGE 64 The ASCO Post | MARCH 25, 2015
Announcements
New Partnership Launches to Empower People Diagnosed With Lymphoma
The Leukemia & Lymphoma Society (LLS), together with the Lympho-
ma Research Foundation (LRF), Can-cerCare, the Association of Community Cancer Centers (ACCC), and Genentech announced the launch of a new partner-ship, the Alliance for Resource Collabora-
tion in Hematology (ARCH). ARCH was developed by these five leading organiza-tions to connect people with lymphoma, their caregivers, and loved ones to educa-tional resources and support.
“Many people diagnosed with lym-phoma, particularly in community can-
cer programs, are unaware of all the re-sources available to them,” said Louis J. DeGennaro, PhD, LLS’s President and Chief Executive Officer. “With this new initiative, our goal is to empower people with lymphoma to be active partners in their care, and ensure they don’t feel
isolated in their journey.”By visiting www.LymphomaResources
.com, people can connect to the partner organizations to learn about the different types of lymphoma, explore treatment op-tions, find information about specialists, and search financial support options. The
JOB#: 40680 CLIENT: Genentech
DESC: Journal Ad 3-pg FILE NAM
E: GNH_HER_Q40680_JA_D03.indd DATE: 11-21-2014 6:50 PM
ROUND: 3PG: CordobaR/MerinoR
AD: R Vetrano-Pyke x4077 PM
: B Fu x4416 AE: K McGinty x3950
CW: L Gibbons
Last Saved: 11-21-2014 6:50 PMTRIM
: 15.5” x 10.5” BLEED: 17.5” x 11.5”
SAFETY: 14.75” x 9.875” PROD: M Haight x4245
INK Spec: 4C PRINT SCALE: 82.94%
FONTS: Myriad Pro (Regular, Bold, Condensed, Condensed Italic, Bold Condensed)
IMAGES: 40680_JA_2_fn.tif (CMYK; 300 ppi; 100%
), Genentech_AMOTRG_4C.eps (96.6%
), Perjeta_US_MBC_NEO_4C.eps (65%
), 40680_KMchart_v5.eps (100%
), 40680_40062_oschartREV_v3.eps (100%)
INKS: Cyan,
Magenta, Yellow,
BlackDOC PATH: Macintosh HD:Users:cordobar:De...sk_prep:GNH_HER_Q40680_JA_D03.inddNOTES: None
GNH_HER_Q40680_JA_D03.indd Galley: 2
S&H
PharmaGraphics
Disk
DATE
SIGNOFF
PGQC
TCAD
CDCW
AE/ASED
PROD
PERJETA + Herceptin (trastuzumab) + docetaxel
Significantly extend progression-free survival (PFS) in first-line HER2+ metastatic breast cancerCombining PERJETA with Herceptin + docetaxel added 6 months median PFS3
Overall survival (OS) dataPERJETA demonstrated an OS improvement when combined with Herceptin + docetaxel at the final analysis4
6.1-month improvement in median PFS by independent review (primary endpoint)3
15.7-month improvement in median OS in the final analysis (secondary endpoint)4
HR=hazard ratio; CI=confidence interval.Median PFS was reached at the first interim analysis.3 Results of the phase III, randomized, double-blind, placebo-controlled CLEOPATRA trial in patients (N=808) with HER2+ locally recurrent, unresectable, or metastatic breast cancer previously untreated with a biologic or chemotherapy for metastatic disease. Patients received PERJETA + Herceptin + docetaxel or placebo + Herceptin + docetaxel every 3 weeks until progression or unacceptable toxicity. Primary endpoint: PFS, assessed by independent review.3
At the second interim analysis (30 months median follow-up, 1 year after the first interim analysis), the HR and P value for OS crossed the predefined efficacy stopping boundary (HR ≤0.739, P≤0.0138). OS improvement with PERJETA + trastuzumab + docetaxel was statistically significant at the second interim analysis (HR=0.66, P=0.0008).3 The final analysis was performed when 221 patient deaths occurred in the placebo-treated group and 168 in the PERJETA-treated group. The statistically significant OS benefit in favor of the PERJETA-treated group was maintained (HR=0.68, P=0.0002).4
• At the first interim analysis, PFS events occurred in 191 (47.5%) patients treated with PERJETA + Herceptin + docetaxel and 242 (59.6%) patients treated with Herceptin + docetaxel3
Select Important Safety Information: Discontinue/Interrupt/Withhold Withhold PERJETA and Herceptin and repeat left ventricular ejection fraction assessment within 3 weeks in patients with significant decrease in LVEF. Discontinue PERJETA and Herceptin if LVEF has not improved or has declined further. If a significant infusion reaction occurs, slow or interrupt the infusion and administer appropriate medical therapies. Consider permanent discontinuation in patients with severe infusion reactions. PERJETA treatment should be withheld or discontinued if Herceptin treatment is withheld or discontinued. Advise nursing mothers receiving PERJETA to discontinue treatment, taking into account the importance of the drug to the mother.
• At the final analysis, the OS benefit was maintained (HR=0.68, 95% CI: 0.56-0.84; P=0.0002) and the median was reached in the PERJETA + Herceptin + docetaxel arm (56.5 months vs 40.8 months in the Herceptin + docetaxel arm)4
• The most common adverse reactions (>30%) seen with the PERJETA-based regimen were diarrhea, alopecia, neutropenia, nausea, fatigue, rash, and peripheral neuropathy3
• Observe patients closely for 60 minutes after the first infusion and for 30 minutes after subsequent infusions of PERJETA. If a significant infusion reaction occurs, slow or interrupt the infusion and administer appropriate medical therapies. Monitor patients carefully until complete resolution of signs and symptoms. Consider permanent discontinuation in patients with severe infusion reactions
Hypersensitivity Reactions/Anaphylaxis• In Study 1, the overall frequency of hypersensitivity/anaphylaxis reactions was
10.8% in the PERJETA-treated group and 9.1% in the placebo-treated group. The incidence of Grades 3-4 hypersensitivity/anaphylaxis reactions was 2.0% in the PERJETA-treated group and 2.5% in the placebo-treated group according to NCI- CTCAE (version 3). Overall, 4 patients in PERJETA-treated group and 2 patients in the placebo-treated group experienced anaphylaxis
• Patients should be observed closely for hypersensitivity reactions. Severe hypersensitivity, including anaphylaxis, has been observed in clinical trials with treatment of PERJETA. Medications to treat such reactions, as well as emergency equipment, should be available for immediate use. PERJETA is contraindicated in patients with known hypersensitivity to pertuzumab or to any of its excipients
HER2 Testing• Detection of HER2 protein overexpression is necessary for selection of patients
appropriate for PERJETA therapy because these are the only patients studied and for whom benefit has been shown
• Patients were required to have evidence of HER2 overexpression, defined as 3+ IHC or FISH amplification ratio ≥2.0 in the clinical studies. Only limited data were available for patients whose breast cancer was positive by FISH but did not demonstrate protein overexpression by IHC
• Assessment of HER2 status should be performed by laboratories with demonstrated proficiency in the specific technology being utilized
Most Common Adverse Reactions• In MBC, the most common adverse reactions (>30%) seen with PERJETA in combination
with Herceptin and docetaxel were diarrhea, alopecia, neutropenia, nausea, fatigue, rash, and peripheral neuropathy. The most common NCI-CTCAE (version 3) Grade 3-4 adverse reactions (>2%) were neutropenia, febrile neutropenia, leukopenia, diarrhea, peripheral neuropathy, anemia, asthenia, and fatigue
You may report side effects to the FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. You may also report side effects to Genentech at 1-888-835-2555.
For more information about PERJETA, contact your local representative or visit www.PERJETA.com/hcp.
Please see Brief Summary of PERJETA full Prescribing Information including Boxed WARNINGS for additional Important Safety Information on the following pages.References: 1. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Breast Cancer V.3.2014. © National Comprehensive Cancer Network, Inc. 2014. All rights reserved. Accessed May 12, 2014. To view the most recent and complete version of the guideline, go online to NCCN.org. NATIONAL COMPREHENSIVE CANCER NETWORK®, NCCN®, NCCN GUIDELINES®, and all other NCCN Content are trademarks owned by the National Comprehensive Cancer Network, Inc. 2. Giordano SH, Temin S, Kirshner JJ, et al. Systemic therapy for patients with advanced human epidermal growth factor receptor 2-positive breast cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2014;32(19):2078-2099. 3. PERJETA Prescribing Information. Genentech, Inc. September 2013. 4. Data on file. Genentech, Inc. 5. Swain SM, Kim S-B, Cortes J, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013;14:461-471. 6. Baselga J, Cortés J, Kim S-B, et al; CLEOPATRA Study Group. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109-119.
Important Safety InformationBoxed WARNINGS: Cardiomyopathy and Embryo-Fetal Toxicity• PERJETA administration can result in subclinical and clinical cardiac
failure. Evaluate left ventricular function in all patients prior to and during treatment with PERJETA. Discontinue PERJETA treatment for a confirmed clinically significant decrease in left ventricular function
• Exposure to PERJETA can result in embryo-fetal death and birth defects. Studies in animals have resulted in oligohydramnios, delayed renal development, and death. Advise patients of these risks and the need for effective contraception—Verify pregnancy status prior to the initiation of PERJETA. Advise patients of the
risks of embryo-fetal death and birth defects and the need for contraception during and after treatment. Advise patients to contact their healthcare provider immediately if they suspect they may be pregnant
—If PERJETA is used during pregnancy or if a patient becomes pregnant while being treated with PERJETA, immediately report exposure to the Genentech Adverse Event Line at 1-888-835-2555. Encourage women who may be exposed during pregnancy to enroll in the MotHER Pregnancy Registry by contacting 1-800-690-6720
—Monitor patients who become pregnant during PERJETA therapy for oligohydramnios. If oligohydramnios occurs, perform fetal testing that is appropriate for gestational age and consistent with community standards of care. The efficacy of intravenous hydration in the management of oligohydramnios due to PERJETA exposure is not known
Additional Important Safety InformationPERJETA is contraindicated in patients with known hypersensitivity to pertuzumab or to any of its excipients.
Left Ventricular Dysfunction (LVD)• In Study 1, for patients with MBC, PERJETA in combination with Herceptin and
docetaxel was not associated with increases in the incidence of symptomatic left ventricular systolic dysfunction (LVSD) or decreases in left ventricular ejection fraction (LVEF) compared with placebo in combination with Herceptin and docetaxel
• Left ventricular dysfunction occurred in 4.4% of patients in the PERJETA-treated group and in 8.3% of patients in the placebo-treated group
• Symptomatic LVSD (congestive heart failure) occurred in 1.0% of patients in the PERJETA-treated group and in 1.8% of patients in the placebo-treated group
• Patients who have received prior anthracyclines or prior radiotherapy to the chest area may be at higher risk of decreased LVEF
• Assess LVEF prior to initiation of PERJETA and at regular intervals (eg, every 3 months in the metastatic setting) during treatment to ensure that LVEF is within your institution’s normal limits
• If LVEF is <45%, or is 45% to 49% with a 10% or greater absolute decrease below the pretreatment value, withhold PERJETA and Herceptin and repeat LVEF assessment within approximately 3 weeks. Discontinue PERJETA and Herceptin if LVEF has not improved or has declined further, unless benefits for the individual patient outweigh the risks
Infusion-Associated Reactions• PERJETA has been associated with infusion reactions• In Study 1, for patients with MBC, on the first day, when only PERJETA was
administered, the overall frequency of infusion reactions was 13.0% in the PERJETA-treated group and 9.8% in the placebo-treated group, with the majority being mild to moderate. The most common infusion reactions (≥1.0%) were pyrexia, chills, fatigue, headache, asthenia, hypersensitivity, and vomiting
• During the second cycle, when all drugs were administered on the same day, the most common infusion reactions in the PERJETA-treated group (≥1.0%) were fatigue, dysgeusia, hypersensitivity, myalgia, and vomiting
© 2014 Genentech USA, Inc. All rights reserved. PER/100114/0010 Printed in USA. 11/14
• In the second interim analysis, PERJETA improved both PFS and OS when combined with Herceptin + docetaxel in patients, including the visceral metastasis subgroup5,6 — There was an inability to show an OS benefit with PERJETA in patients with nonvisceral metastases
(n=178; HR=1.42 [95% CI: 0.71-2.84])3
• In the second interim analysis, there was a statistically significant improvement in OS (secondary endpoint)3 — Median not yet reached in the PERJETA-containing arm vs 37.6 months with Herceptin + docetaxel
(HR=0.66; 95% CI: 0.52-0.84; P=0.0008)
100
90
80
70
60
50
40
30
20
10
0
0 5 10 15 20 25 30 35 40
HR=0.6295% CI: 0.51-0.75
P<0.0001
402406
P+H+DPl+H+D
345311
267209
13993
8342
3217
107
00
00
Placebo + Herceptin + docetaxel
12.4MONTHS
MONTHS
PERJETA + Herceptin + docetaxel
18.5MONTHS
Patients at risk
PFS (
%) median line
MONTHS
Patients at risk
402406
P + H + D PI + H + D
371350
318289
268230
0
226179
10491
2823
10
Placebo + Herceptin + docetaxelPERJETA + Herceptin + docetaxel
56.5MONTHS
HR=0.68 95% CI: 0.56-0.84
P=0.0002
40.8MONTHS
median line
30
20
10
020 30 40 50 60 70
100
90
80
70
60
50
40
10
OS (%
)
00
80
C M Y KCosmos Communications 1
1ej
29452a 11.25.14 133
Q1 Q2
G:1.0”
S:9.875”
T:15.5”
T:10.5”
B:17.5”
B:11.5”
S:6.875” S:6.875”

ASCOPost.com | MARCH 25, 2015 PAGE 65
Announcements
ARCH website also helps in finding patient communities, peer support programs, and counseling services. The partner organiza-tions provide these resources in a variety of formats, including in-person programs, online materials, and telephone support.
It is estimated that only 15% of U.S. cancer patients are treated at the nation’s major academic cancer centers; the vast
majority receive care at cancer programs in or near their home community, where re-sources may be more limited. By bringing together ACCC’s network of community cancer programs with the quality resources provided by LLS, LRF and CancerCare, ARCH aims to have an immediate impact on access to information for lymphoma pa-tients where the need is greatest. n
With this new initiative, our goal is to empower people with lymphoma to be active
partners in their care, and ensure they don’t feel isolated in their journey.
—Louis J. DeGennaro, PhD
JOB#: 40680 CLIENT: Genentech
DESC: Journal Ad 3-pg FILE NAM
E: GNH_HER_Q40680_JA_D03.indd DATE: 11-21-2014 6:50 PM
ROUND: 3PG: CordobaR/MerinoR
AD: R Vetrano-Pyke x4077 PM
: B Fu x4416 AE: K McGinty x3950
CW: L Gibbons
Last Saved: 11-21-2014 6:50 PMTRIM
: 15.5” x 10.5” BLEED: 17.5” x 11.5”
SAFETY: 14.75” x 9.875” PROD: M Haight x4245
INK Spec: 4C PRINT SCALE: 82.94%
FONTS: Myriad Pro (Regular, Bold, Condensed, Condensed Italic, Bold Condensed)
IMAGES: 40680_JA_2_fn.tif (CMYK; 300 ppi; 100%
), Genentech_AMOTRG_4C.eps (96.6%
), Perjeta_US_MBC_NEO_4C.eps (65%
), 40680_KMchart_v5.eps (100%
), 40680_40062_oschartREV_v3.eps (100%)
INKS: Cyan,
Magenta, Yellow,
BlackDOC PATH: Macintosh HD:Users:cordobar:De...sk_prep:GNH_HER_Q40680_JA_D03.inddNOTES: None
GNH_HER_Q40680_JA_D03.indd Galley: 2
S&H
PharmaGraphics
Disk
DATE
SIGNOFF
PGQC
TCAD
CDCW
AE/ASED
PROD
PERJETA + Herceptin (trastuzumab) + docetaxel
Significantly extend progression-free survival (PFS) in first-line HER2+ metastatic breast cancerCombining PERJETA with Herceptin + docetaxel added 6 months median PFS3
Overall survival (OS) dataPERJETA demonstrated an OS improvement when combined with Herceptin + docetaxel at the final analysis4
6.1-month improvement in median PFS by independent review (primary endpoint)3
15.7-month improvement in median OS in the final analysis (secondary endpoint)4
HR=hazard ratio; CI=confidence interval.Median PFS was reached at the first interim analysis.3 Results of the phase III, randomized, double-blind, placebo-controlled CLEOPATRA trial in patients (N=808) with HER2+ locally recurrent, unresectable, or metastatic breast cancer previously untreated with a biologic or chemotherapy for metastatic disease. Patients received PERJETA + Herceptin + docetaxel or placebo + Herceptin + docetaxel every 3 weeks until progression or unacceptable toxicity. Primary endpoint: PFS, assessed by independent review.3
At the second interim analysis (30 months median follow-up, 1 year after the first interim analysis), the HR and P value for OS crossed the predefined efficacy stopping boundary (HR ≤0.739, P≤0.0138). OS improvement with PERJETA + trastuzumab + docetaxel was statistically significant at the second interim analysis (HR=0.66, P=0.0008).3 The final analysis was performed when 221 patient deaths occurred in the placebo-treated group and 168 in the PERJETA-treated group. The statistically significant OS benefit in favor of the PERJETA-treated group was maintained (HR=0.68, P=0.0002).4
• At the first interim analysis, PFS events occurred in 191 (47.5%) patients treated with PERJETA + Herceptin + docetaxel and 242 (59.6%) patients treated with Herceptin + docetaxel3
Select Important Safety Information: Discontinue/Interrupt/Withhold Withhold PERJETA and Herceptin and repeat left ventricular ejection fraction assessment within 3 weeks in patients with significant decrease in LVEF. Discontinue PERJETA and Herceptin if LVEF has not improved or has declined further. If a significant infusion reaction occurs, slow or interrupt the infusion and administer appropriate medical therapies. Consider permanent discontinuation in patients with severe infusion reactions. PERJETA treatment should be withheld or discontinued if Herceptin treatment is withheld or discontinued. Advise nursing mothers receiving PERJETA to discontinue treatment, taking into account the importance of the drug to the mother.
• At the final analysis, the OS benefit was maintained (HR=0.68, 95% CI: 0.56-0.84; P=0.0002) and the median was reached in the PERJETA + Herceptin + docetaxel arm (56.5 months vs 40.8 months in the Herceptin + docetaxel arm)4
• The most common adverse reactions (>30%) seen with the PERJETA-based regimen were diarrhea, alopecia, neutropenia, nausea, fatigue, rash, and peripheral neuropathy3
• Observe patients closely for 60 minutes after the first infusion and for 30 minutes after subsequent infusions of PERJETA. If a significant infusion reaction occurs, slow or interrupt the infusion and administer appropriate medical therapies. Monitor patients carefully until complete resolution of signs and symptoms. Consider permanent discontinuation in patients with severe infusion reactions
Hypersensitivity Reactions/Anaphylaxis• In Study 1, the overall frequency of hypersensitivity/anaphylaxis reactions was
10.8% in the PERJETA-treated group and 9.1% in the placebo-treated group. The incidence of Grades 3-4 hypersensitivity/anaphylaxis reactions was 2.0% in the PERJETA-treated group and 2.5% in the placebo-treated group according to NCI- CTCAE (version 3). Overall, 4 patients in PERJETA-treated group and 2 patients in the placebo-treated group experienced anaphylaxis
• Patients should be observed closely for hypersensitivity reactions. Severe hypersensitivity, including anaphylaxis, has been observed in clinical trials with treatment of PERJETA. Medications to treat such reactions, as well as emergency equipment, should be available for immediate use. PERJETA is contraindicated in patients with known hypersensitivity to pertuzumab or to any of its excipients
HER2 Testing• Detection of HER2 protein overexpression is necessary for selection of patients
appropriate for PERJETA therapy because these are the only patients studied and for whom benefit has been shown
• Patients were required to have evidence of HER2 overexpression, defined as 3+ IHC or FISH amplification ratio ≥2.0 in the clinical studies. Only limited data were available for patients whose breast cancer was positive by FISH but did not demonstrate protein overexpression by IHC
• Assessment of HER2 status should be performed by laboratories with demonstrated proficiency in the specific technology being utilized
Most Common Adverse Reactions• In MBC, the most common adverse reactions (>30%) seen with PERJETA in combination
with Herceptin and docetaxel were diarrhea, alopecia, neutropenia, nausea, fatigue, rash, and peripheral neuropathy. The most common NCI-CTCAE (version 3) Grade 3-4 adverse reactions (>2%) were neutropenia, febrile neutropenia, leukopenia, diarrhea, peripheral neuropathy, anemia, asthenia, and fatigue
You may report side effects to the FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. You may also report side effects to Genentech at 1-888-835-2555.
For more information about PERJETA, contact your local representative or visit www.PERJETA.com/hcp.
Please see Brief Summary of PERJETA full Prescribing Information including Boxed WARNINGS for additional Important Safety Information on the following pages.References: 1. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Breast Cancer V.3.2014. © National Comprehensive Cancer Network, Inc. 2014. All rights reserved. Accessed May 12, 2014. To view the most recent and complete version of the guideline, go online to NCCN.org. NATIONAL COMPREHENSIVE CANCER NETWORK®, NCCN®, NCCN GUIDELINES®, and all other NCCN Content are trademarks owned by the National Comprehensive Cancer Network, Inc. 2. Giordano SH, Temin S, Kirshner JJ, et al. Systemic therapy for patients with advanced human epidermal growth factor receptor 2-positive breast cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2014;32(19):2078-2099. 3. PERJETA Prescribing Information. Genentech, Inc. September 2013. 4. Data on file. Genentech, Inc. 5. Swain SM, Kim S-B, Cortes J, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013;14:461-471. 6. Baselga J, Cortés J, Kim S-B, et al; CLEOPATRA Study Group. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109-119.
Important Safety InformationBoxed WARNINGS: Cardiomyopathy and Embryo-Fetal Toxicity• PERJETA administration can result in subclinical and clinical cardiac
failure. Evaluate left ventricular function in all patients prior to and during treatment with PERJETA. Discontinue PERJETA treatment for a confirmed clinically significant decrease in left ventricular function
• Exposure to PERJETA can result in embryo-fetal death and birth defects. Studies in animals have resulted in oligohydramnios, delayed renal development, and death. Advise patients of these risks and the need for effective contraception—Verify pregnancy status prior to the initiation of PERJETA. Advise patients of the
risks of embryo-fetal death and birth defects and the need for contraception during and after treatment. Advise patients to contact their healthcare provider immediately if they suspect they may be pregnant
—If PERJETA is used during pregnancy or if a patient becomes pregnant while being treated with PERJETA, immediately report exposure to the Genentech Adverse Event Line at 1-888-835-2555. Encourage women who may be exposed during pregnancy to enroll in the MotHER Pregnancy Registry by contacting 1-800-690-6720
—Monitor patients who become pregnant during PERJETA therapy for oligohydramnios. If oligohydramnios occurs, perform fetal testing that is appropriate for gestational age and consistent with community standards of care. The efficacy of intravenous hydration in the management of oligohydramnios due to PERJETA exposure is not known
Additional Important Safety InformationPERJETA is contraindicated in patients with known hypersensitivity to pertuzumab or to any of its excipients.
Left Ventricular Dysfunction (LVD)• In Study 1, for patients with MBC, PERJETA in combination with Herceptin and
docetaxel was not associated with increases in the incidence of symptomatic left ventricular systolic dysfunction (LVSD) or decreases in left ventricular ejection fraction (LVEF) compared with placebo in combination with Herceptin and docetaxel
• Left ventricular dysfunction occurred in 4.4% of patients in the PERJETA-treated group and in 8.3% of patients in the placebo-treated group
• Symptomatic LVSD (congestive heart failure) occurred in 1.0% of patients in the PERJETA-treated group and in 1.8% of patients in the placebo-treated group
• Patients who have received prior anthracyclines or prior radiotherapy to the chest area may be at higher risk of decreased LVEF
• Assess LVEF prior to initiation of PERJETA and at regular intervals (eg, every 3 months in the metastatic setting) during treatment to ensure that LVEF is within your institution’s normal limits
• If LVEF is <45%, or is 45% to 49% with a 10% or greater absolute decrease below the pretreatment value, withhold PERJETA and Herceptin and repeat LVEF assessment within approximately 3 weeks. Discontinue PERJETA and Herceptin if LVEF has not improved or has declined further, unless benefits for the individual patient outweigh the risks
Infusion-Associated Reactions• PERJETA has been associated with infusion reactions• In Study 1, for patients with MBC, on the first day, when only PERJETA was
administered, the overall frequency of infusion reactions was 13.0% in the PERJETA-treated group and 9.8% in the placebo-treated group, with the majority being mild to moderate. The most common infusion reactions (≥1.0%) were pyrexia, chills, fatigue, headache, asthenia, hypersensitivity, and vomiting
• During the second cycle, when all drugs were administered on the same day, the most common infusion reactions in the PERJETA-treated group (≥1.0%) were fatigue, dysgeusia, hypersensitivity, myalgia, and vomiting
© 2014 Genentech USA, Inc. All rights reserved. PER/100114/0010 Printed in USA. 11/14
• In the second interim analysis, PERJETA improved both PFS and OS when combined with Herceptin + docetaxel in patients, including the visceral metastasis subgroup5,6 — There was an inability to show an OS benefit with PERJETA in patients with nonvisceral metastases
(n=178; HR=1.42 [95% CI: 0.71-2.84])3
• In the second interim analysis, there was a statistically significant improvement in OS (secondary endpoint)3 — Median not yet reached in the PERJETA-containing arm vs 37.6 months with Herceptin + docetaxel
(HR=0.66; 95% CI: 0.52-0.84; P=0.0008)
100
90
80
70
60
50
40
30
20
10
0
0 5 10 15 20 25 30 35 40
HR=0.6295% CI: 0.51-0.75
P<0.0001
402406
P+H+DPl+H+D
345311
267209
13993
8342
3217
107
00
00
Placebo + Herceptin + docetaxel
12.4MONTHS
MONTHS
PERJETA + Herceptin + docetaxel
18.5MONTHS
Patients at risk
PFS (
%) median line
MONTHS
Patients at risk
402406
P + H + D PI + H + D
371350
318289
268230
0
226179
10491
2823
10
Placebo + Herceptin + docetaxelPERJETA + Herceptin + docetaxel
56.5MONTHS
HR=0.68 95% CI: 0.56-0.84
P=0.0002
40.8MONTHS
median line
30
20
10
020 30 40 50 60 70
100
90
80
70
60
50
40
10
OS (%
)
00
80
C M Y KCosmos Communications 1
1ej
29452a 11.25.14 133
Q1 Q2
G:1.0”
S:9.875”
T:15.5”
T:10.5”
B:17.5”
B:11.5”
S:6.875” S:6.875”

PAGE 66 The ASCO Post | MARCH 25, 2015
FDA Update
Dinutuximab Combination Approved for Pediatric High-Risk Neuroblastoma
The U.S. Food and Drug Adminis-tration (FDA) has approved dinu-
tuximab (Unituxin), a monoclonal anti-body targeting glycolipid GD2, as part of first-line therapy for pediatric patients with high-risk neuroblastoma. A chime-ric monoclonal antibody that binds to
the surface of neuroblastoma cells, dinu-tuximab is being approved for use as part of a multimodality regimen, including surgery, chemotherapy, and radiation therapy for patients who have achieved at least a partial response to prior first-line multiagent, multimodality therapy.
“Dinutuximab marks the first approval for a therapy aimed specifically for the treatment of patients with high-risk neu-roblastoma,” said Richard Pazdur, MD, Director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Dinu-
tuximab fulfills a critical need by providing a treatment option that prolongs survival in children with high-risk neuroblastoma.”
The FDA granted dinutuximab prior-ity review and orphan product designa-tion. With this approval, the FDA also issued a rare pediatric disease priority
PERJETA® (pertuzumab)INJECTION, FOR INTRAVENOUS USEINITIAL U.S. APPROVAL: 2012
WARNING: CARDIOMYOPATHY and EMBRYO-FETAL TOXICITY
Cardiomyopathy PERJETA administration can result in subclinical and clinical cardiac failure. Evaluate left ventricular function in all patients prior to and during treatment with PERJETA. Discontinue PERJETA treatment for a confirmed clinically significant decrease in left ventricular function. (2.2, 5.2, 6.1)Embryo-Fetal Toxicity Exposure to PERJETA can result in embryo-fetal death and birth defects. Studies in animals have resulted in oligohydramnios, delayed renal development, and death. Advise patients of these risks and the need for effective contraception. (5.1, 8.1, 8.6)
1 INDICATIONS AND USAGE 1.1 Metastatic Breast Cancer (MBC)PERJETA is indicated for use in combination with trastuzumab and docetaxel for the treatment of patients with HER2-positive metastatic breast cancer who have not received prior anti-HER2 therapy or chemotherapy for metastatic disease.
1.2 Neoadjuvant Treatment of Breast CancerPERJETA is indicated for use in combination with trastuzumab and docetaxel for the neoadjuvant treatment of patients with HER2-positive, locally advanced, inflammatory, or early stage breast cancer (either greater than 2 cm in diameter or node positive) as part of a complete treatment regimen for early breast cancer. This indication is based on demonstration of an improvement in pathological complete response rate. No data are available demonstrating improvement in event-free survival or overall survival [see Clinical Studies (14.2) and Dosage and Administration (2.1)]. Limitations of Use: • The safety of PERJETA as part of a doxorubicin-containing
regimen has not been established. • The safety of PERJETA administered for greater than
6 cycles for early breast cancer has not been established.
4 CONTRAINDICATIONS PERJETA is contraindicated in patients with known hypersensitivity to pertuzumab or to any of its excipients.
5 WARNINGS AND PRECAUTIONS5.1 Embryo-Fetal Toxicity PERJETA can cause fetal harm when administered to a pregnant woman. Treatment of pregnant cynomolgus monkeys with pertuzumab resulted in oligohydramnios, delayed fetal kidney development, and embryo-fetal death. If PERJETA is administered during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to a fetus [see Use in Specific Populations (8.1)].Verify pregnancy status prior to the initiation of PERJETA. Advise patients of the risks of embryo-fetal death and birth defects and the need for contraception during and after treatment. Advise patients to contact their healthcare provider immediately if they suspect they may be pregnant. If PERJETA is administered during pregnancy or if a patient becomes pregnant while receiving PERJETA, immediately report exposure to the Genentech Adverse Event Line at 1-888-835-2555. Encourage women who may be exposed during pregnancy to enroll in the MotHER Pregnancy Registry by contacting 1-800-690-6720 [see Patient Counseling Information (17)].Monitor patients who become pregnant during PERJETA therapy for oligohydramnios. If oligohydramnios occurs, perform fetal testing that is appropriate for gestational age and consistent with community standards of care. The efficacy of intravenous hydration in the management of oligohydramnios due to PERJETA exposure is not known.
5.2 Left Ventricular Dysfunction Decreases in LVEF have been reported with drugs that block HER2 activity, including PERJETA. In Study 1, for patients with MBC, PERJETA in combination with trastuzumab and docetaxel was not associated with increases in the incidence of symptomatic left ventricular systolic dysfunction (LVSD) or decreases in LVEF compared with placebo in combination with trastuzumab and docetaxel [see Clinical Studies (14.1)]. Left ventricular dysfunction occurred in 4.4% of patients in the PERJETA-treated group and 8.3% of patients in the placebo-treated group. Symptomatic left ventricular systolic dysfunction (congestive heart failure) occurred in 1.0% of patients in the PERJETA-treated group and 1.8% of patients in the placebo-treated group [see Adverse Reactions (6.1)]. Patients who have received prior anthracyclines or prior radiotherapy to the chest area may be at higher risk of decreased LVEF.
In patients receiving neoadjuvant treatment in Study 2, the incidence of LVSD was higher in the PERJETA-treated groups compared to the trastuzumab- and docetaxel-treated group. An increased incidence of LVEF declines was observed in patients treated with PERJETA in combination with trastuzumab and docetaxel. In the overall treatment period, LVEF decline > 10% and a drop to less than 50% occurred in 1.9% of patients treated with neoadjuvant trastuzumab and docetaxel as compared to 8.4% of patients treated with neoadjuvant PERJETA in combination with trastuzumab and docetaxel. Symptomatic LVSD occurred in 0.9% of patients treated with neoadjuvant PERJETA in combination with trastuzumab and no patients in the other 3 arms. LVEF recovered to ≥ 50% in all patients.In patients receiving neoadjuvant PERJETA in Study 3, in the overall treatment period, LVEF decline > 10% and a drop to less than 50% occurred in 6.9% of patients treated with PERJETA plus trastuzumab and FEC followed by PERJETA plus trastuzumab and docetaxel, 16.0% of patients treated with PERJETA plus trastuzumab and docetaxel following FEC, and 10.5% of patients treated with PERJETA in combination with TCH. Symptomatic LVSD occurred in 4.0% of patients treated with PERJETA plus trastuzumab and docetaxel following FEC, 1.3% of patients treated with PERJETA in combination with TCH, and none of the patients treated with PERJETA plus trastuzumab and FEC followed by PERJETA plus trastuzumab and docetaxel. LVEF recovered to ≥ 50% in all but one patient.PERJETA has not been studied in patients with a pretreatment LVEF value of ≤ 50%, a prior history of CHF, decreases in LVEF to < 50% during prior trastuzumab therapy, or conditions that could impair left ventricular function such as uncontrolled hypertension, recent myocardial infarction, serious cardiac arrhythmia requiring treatment or a cumulative prior anthracycline exposure to > 360 mg/m2 of doxorubicin or its equivalent.Assess LVEF prior to initiation of PERJETA and at regular intervals (e.g., every three months in the metastatic setting and every six weeks in the neoadjuvant setting) during treatment to ensure that LVEF is within the institution’s normal limits. If LVEF is < 45%, or is 45% to 49% with a 10% or greater absolute decrease below the pretreatment value, withhold PERJETA and trastuzumab and repeat LVEF assessment within approximately 3 weeks. Discontinue PERJETA and trastuzumab if the LVEF has not improved or has declined further, unless the benefits for the individual patient outweigh the risks [see Dosage and Administration (2.2)].
5.3 Infusion-Related ReactionsPERJETA has been associated with infusion reactions [see Adverse Reactions (6.1)]. An infusion reaction was defined in Study 1 as any event described as hypersensitivity, anaphylactic reaction, acute infusion reaction, or cytokine release syndrome occurring during an infusion or on the same day as the infusion. The initial dose of PERJETA was given the day before trastuzumab and docetaxel to allow for the examination of PERJETA-associated reactions. On the first day, when only PERJETA was administered, the overall frequency of infusion reactions was 13.0% in the PERJETA-treated group and 9.8% in the placebo-treated group. Less than 1% were Grade 3 or 4. The most common infusion reactions (≥ 1.0%) were pyrexia, chills, fatigue, headache, asthenia, hypersensitivity, and vomiting.During the second cycle when all drugs were administered on the same day, the most common infusion reactions in the PERJETA-treated group (≥ 1.0%) were fatigue, dysgeusia, hypersensitivity, myalgia, and vomiting.In Study 2 and Study 3, PERJETA was administered on the same day as the other study treatment drugs. Infusion reactions were consistent with those observed in Study 1, with a majority of reactions being National Cancer Institute - Common Terminology Criteria for Adverse Events (NCI - CTCAE v3.0) Grade 1 – 2.Observe patients closely for 60 minutes after the first infusion and for 30 minutes after subsequent infusions of PERJETA. If a significant infusion-related reaction occurs, slow or interrupt the infusion, and administer appropriate medical therapies. Monitor patients carefully until complete resolution of signs and symptoms. Consider permanent discontinuation in patients with severe infusion reactions [see Dosage and Administration (2.2)].
5.4 Hypersensitivity Reactions/AnaphylaxisIn Study 1, the overall frequency of hypersensitivity/anaphylaxis reactions was 10.8% in the PERJETA-treated group and 9.1% in the placebo-treated group. The incidence of Grade 3 – 4 hypersensitivity/anaphylaxis reactions was 2.0% in the PERJETA-treated group and 2.5% in the placebo-treated group according to NCI - CTCAE v3.0. Overall, 4 patients in PERJETA-treated group and 2 patients in the placebo-treated group experienced anaphylaxis.
In Study 2 and Study 3, hypersensitivity/anaphylaxis events were consistent with those observed in Study 1. In Study 2, two patients in the PERJETA- and docetaxel-treated group experienced anaphylaxis. In Study 3, the overall frequency of hypersensitivity/anaphylaxis was highest in the PERJETA plus TCH treated group (13.2%), of which 2.6% were NCI-CTCAE (version 3) Grade 3 – 4.Patients should be observed closely for hypersensitivity reactions. Severe hypersensitivity, including anaphylaxis, has been observed in clinical trials with treatment of PERJETA [see Clinical Trials Experience (6.1)]. Medications to treat such reactions, as well as emergency equipment, should be available for immediate use. PERJETA is contraindicated in patients with known hypersensitivity to pertuzumab or to any of its excipients [see Contraindications (4)].
5.5 HER2 Testing Detection of HER2 protein overexpression is necessary for selection of patients appropriate for PERJETA therapy because these are the only patients studied and for whom benefit has been shown [see Indications and Usage (1) and Clinical Studies (14)]. Patients with breast cancer were required to have evidence of HER2 overexpression defined as 3+ IHC or FISH amplification ratio ≥ 2.0 in the clinical studies. Only limited data were available for patients whose breast cancer was positive by FISH, but did not demonstrate protein overexpression by IHC.Assessment of HER2 status should be performed by laboratories using FDA-approved tests with demonstrated proficiency in the specific technology being utilized. Improper assay performance, including use of sub-optimally fixed tissue, failure to utilize specified reagents, deviation from specific assay instructions, and failure to include appropriate controls for assay validation, can lead to unreliable results.
6 ADVERSE REACTIONS The following adverse reactions are discussed in greater detail in other sections of the label:• Embryo-Fetal Toxicity [see Warnings and Precautions
(5.1)] • Left Ventricular Dysfunction [see Warnings and
Precautions (5.2)]• Infusion-Related Reactions [see Warnings and
Precautions (5.3)]• Hypersensitivity Reactions/Anaphylaxis [see Warnings
and Precautions (5.4)]
6.1 Clinical Trials Experience Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.Metastatic Breast Cancer (MBC)The adverse reactions described in Table 1 were identified in 804 patients with HER2-positive metastatic breast cancer treated in Study 1. Patients were randomized to receive either PERJETA in combination with trastuzumab and docetaxel or placebo in combination with trastuzumab and docetaxel. The median duration of study treatment was 18.1 months for patients in the PERJETA-treated group and 11.8 months for patients in the placebo-treated group. No dose adjustment was permitted for PERJETA or trastuzumab. The rates of adverse events resulting in permanent discontinuation of all study therapy were 6.1% for patients in the PERJETA-treated group and 5.3% for patients in the placebo-treated group. Adverse events led to discontinuation of docetaxel alone in 23.6% of patients in the PERJETA-treated group and 23.2% of patients in the placebo-treated group. Table 1 reports the adverse reactions that occurred in at least 10% of patients in the PERJETA-treated group. The safety profile of PERJETA remained unchanged with an additional year of follow-up (median total follow-up of 30 months) in Study 1. The most common adverse reactions (> 30%) seen with PERJETA in combination with trastuzumab and docetaxel were diarrhea, alopecia, neutropenia, nausea, fatigue, rash, and peripheral neuropathy. The most common NCI - CTCAE v3.0 Grade 3 – 4 adverse reactions (> 2%) were neutropenia, febrile neutropenia, leukopenia, diarrhea, peripheral neuropathy, anemia, asthenia, and fatigue. An increased incidence of febrile neutropenia was observed for Asian patients in both treatment arms compared with patients of other races and from other geographic regions. Among Asian patients, the incidence of febrile neutropenia was higher in the pertuzumab-treated group (26%) compared with the placebo-treated group (12%).
Table 1 Summary of Adverse Reactions Occurring in ≥ 10% of Patients on the PERJETA Treatment Arm in Study 1
General disorders and administration site conditions Fatigue 37.6 2.2 36.8 3.3Asthenia 26.0 2.5 30.2 1.5Edema peripheral 23.1 0.5 30.0 0.8Mucosal inflammation 27.8 1.5 19.9 1.0Pyrexia 18.7 1.2 17.9 0.5Skin and subcutaneous tissue disorders Alopecia 60.9 0.0 60.5 0.3Rash 33.7 0.7 24.2 0.8Nail disorder 22.9 1.2 22.9 0.3Pruritus 14.0 0.0 10.1 0.0Dry skin 10.6 0.0 4.3 0.0Gastrointestinal disorders Diarrhea 66.8 7.9 46.3 5.0Nausea 42.3 1.2 41.6 0.5Vomiting 24.1 1.5 23.9 1.5Constipation 15.0 0.0 24.9 1.0Stomatitis 18.9 0.5 15.4 0.3Blood and lymphatic system disorders Neutropenia 52.8 48.9 49.6 45.8Anemia 23.1 2.5 18.9 3.5Leukopenia 18.2 12.3 20.4 14.6Febrile neutropenia* 13.8 13.0 7.6 7.3Nervous system disorders Neuropathy peripheral 32.4 3.2 33.8 2.0Headache 20.9 1.2 16.9 0.5Dysgeusia 18.4 0.0 15.6 0.0Dizziness 12.5 0.5 12.1 0.0Musculoskeletal and connective tissue disorders Myalgia 22.9 1.0 23.9 0.8Arthralgia 15.5 0.2 16.1 0.8Infections and infestationsUpper respiratory tract infectionNasopharyngitis 11.8 0.0 12.8 0.3Respiratory, thoracic, and mediastinal disorders Dyspnea 14.0 1.0 15.6 2.0Metabolism and nutrition disorders Decreased appetite 29.2 1.7 26.4 1.5Eye disorders Lacrimation increased 14.0 0.0 13.9 0.0Psychiatric disorders Insomnia 13.3 0.0 13.4 0.0
The following clinically relevant adverse reactions were reported in < 10% of patients in the PERJETA-treated group in Study 1:Skin and subcutaneous tissue disorders: Paronychia (7.1% in the PERJETA-treated group vs. 3.5% in the placebo-treated group)Respiratory, thoracic and mediastinal disorders: Pleural effusion (5.2% in the PERJETA-treated group vs. 5.8% in the placebo-treated group)Cardiac disorders: Left ventricular dysfunction (4.4% in the PERJETA-treated group vs. 8.3% in the placebo-treated group) including symptomatic left ventricular systolic dysfunction (CHF) (1.0% in the PERJETA-treated group vs. 1.8% in the placebo-treated group)Immune system disorders: Hypersensitivity (10.1% in the PERJETA-treated group vs. 8.6% in placebo-treated group)Adverse Reactions Reported in Patients Receiving PERJETA and Trastuzumab after Discontinuation of DocetaxelIn Study 1, adverse reactions were reported less frequently after discontinuation of docetaxel treatment. All adverse reactions in the PERJETA and trastuzumab treatment group occurred in < 10% of patients with the exception of diarrhea (19.1%), upper respiratory tract infection (12.8%), rash (11.7%), headache (11.4%), and fatigue (11.1%).Neoadjuvant Treatment of Breast Cancer (Study 2)In Study 2, the most common adverse reactions seen with PERJETA in combination with trastuzumab and docetaxel administered for 4 cycles were similar to those seen in the PERJETA-treated group in Study 1. The most common adverse reactions (> 30%) were alopecia, neutropenia, diarrhea, and nausea. The most common NCI – CTCAE v3.0 Grade 3 – 4 adverse reactions (> 2%) were neutropenia, febrile neutropenia, leukopenia, and diarrhea. In this group, one patient permanently discontinued neoadjuvant treatment due to an adverse event. Table 2 reports the adverse reactions that occurred in patients who received neoadjuvant treatment with PERJETA for breast cancer in Study 2.
Table 2 Summary of Adverse Reactions Occurring in ≥ 10% in the Neoadjuvant Setting for Patients Receiving PERJETA in Study 2
The following adverse reactions were reported in < 10% of patients receiving neoadjuvant treatment and occurred more frequently in PERJETA-treated groups in Study 2: (Ptz=pertuzumab; T=trastuzumab; D=docetaxel)Blood and lymphatic system disorders: Anemia (6.5% in the T+D arm, 2.8% in the Ptz+T+D arm, 4.6% in the Ptz+T arm and 8.5% in the Ptz+D arm), Febrile neutropenia (6.5% in the T+D arm, 8.4% in the Ptz+T+D arm, 0.0% in the Ptz+T arm and 7.4% in the Ptz+D arm)Immune system disorders: Hypersensitivity (1.9% in the T+D arm, 5.6% in the Ptz+T+D arm, 5.6% in the Ptz+T arm and 5.3% in the Ptz+D arm)Nervous system disorders: Dizziness (3.7% in the T+D arm, 2.8% in the Ptz+T+D arm, 5.6% in the Ptz+T arm and 3.2% in the Ptz+D arm)Infections and infestations: Upper respiratory tract infection (2.8% in the T+D arm, 4.7% in the Ptz+T+D arm, 1.9% in the Ptz+T arm and 7.4% in the Ptz+D arm)Respiratory, thoracic and mediastinal disorders: Dyspnea (3.7% in the T+D arm, 4.7% in the Ptz+T+D arm, 2.8% in the Ptz+T arm and 2.1% in the Ptz+D arm)Cardiac disorders: Left ventricular dysfunction (0.9% in the T+D arm, 2.8% in the Ptz+T+D arm, 0.0% in the Ptz+T arm, and 1.1% in the Ptz+D arm) including symptomatic left ventricular dysfunction (CHF) (0.9% in the Ptz+T arm and 0.0% in the T+D arm, Ptz+T+D arm, and Ptz+D arm)Eye disorders: Lacrimation increased (1.9% in the T+D arm, 3.7% in the Ptz+T+D arm, 0.9% in the Ptz+T arm, and 4.3% in the Ptz+D arm)Neoadjuvant Treatment of Breast Cancer (Study 3)In Study 3, when PERJETA was administered in combination with trastuzumab and docetaxel for 3 cycles following 3 cycles of FEC, the most common adverse reactions (> 30%) were diarrhea, nausea, alopecia, neutropenia, vomiting, and fatigue. The most common NCI-CTCAE (version 3) Grade 3 – 4 adverse reactions (> 2%) were neutropenia, leukopenia, febrile neutropenia, diarrhea, left ventricular dysfunction, anemia, dyspnea, nausea, and vomiting. Similarly, when PERJETA was administered in combination with docetaxel, carboplatin, and trastuzumab (TCH) for 6 cycles, the most common adverse reactions (> 30%) were diarrhea, alopecia, neutropenia, nausea, fatigue, vomiting, anemia, and thrombocytopenia. The most common NCI-CTCAE (version 3) Grade 3 – 4 adverse reactions (> 2%) were neutropenia, febrile neutropenia, anemia, leukopenia, diarrhea, thrombocytopenia, vomiting, fatigue, ALT increased, hypokalemia, and hypersensitivity. The rates of adverse events resulting in permanent discontinuation of any component of neoadjuvant treatment were 6.7% for patients receiving PERJETA in combination with trastuzumab and docetaxel following FEC and 7.9% for
patients receiving PERJETA in combination with TCH. Table 3 reports the adverse reactions that occurred in patients who received neoadjuvant treatment with PERJETA for breast cancer in Study 3.
Table 3 Summary of Adverse Reactions Occurring in ≥ 10% of Patients Receiving Neoadjuvant Treatment with PERJETA in Study 3
FEC=5-fluorouracil, epirubicin, cyclophosphamide, TCH=docetaxel, carboplatin, trastuzumabThe following selected adverse reactions were reported in < 10% of patients receiving neoadjuvant treatment in Study 3: (Ptz=pertuzumab; T=trastuzumab; D=docetaxel; FEC=fluorouracil, epirubicin, and cyclophosphamide; TCH=docetaxel, carboplatin, and trastuzumab)Skin and subcutaneous tissue disorders: Nail disorder (9.7% in the Ptz+T+FEC/Ptz+T+D arm, 6.7% in the FEC/Ptz+T+D arm, and 9.2% in the Ptz+TCH arm), Paronychia (0% in the Ptz+T+FEC/Ptz+T+D and 1.3% in both the FEC/Ptz+T+D and Ptz+TCH arms), Pruritis (2.8% in the Ptz+T+FEC/Ptz+T+D arm, 4.0% in the FEC/Ptz+T+D arm, and 3.9% in the Ptz+TCH arm)Infections and infestations: Upper respiratory tract infection (8.3% in the Ptz+T+FEC/Ptz+T+D arm, 4.0% in the FEC/Ptz+T+D arm, and 2.6% in the Ptz+TCH arm), Nasopharyngitis (6.9% in the Ptz+T+FEC/Ptz+T+D arm, 6.7% in the FEC/Ptz+T+D arm, and 7.9% in the Ptz+TCH arm)
Body System/ Adverse Reactions
Trastuzumab + docetaxel
n=107
PERJETA+ trastuzumab + docetaxel
n=107
PERJETA + trastuzumab
n=108
PERJETA + docetaxel
n=108Frequency rate
%Frequency rate
%Frequency rate
%Frequency rate
%All
Grades %
Grades 3–4 %
All Grades
%
Grades 3–4 %
All Grades
%
Grades 3–4 %
All Grades
%
Grades 3–4 %
General disorders and administration site conditionsFatigue 27.1 0.0 26.2 0.9 12.0 0.0 25.5 1.1Asthenia 17.8 0.0 20.6 1.9 2.8 0.0 16.0 2.1Edema peripheral 10.3 0.0 2.8 0.0 0.9 0.0 5.3 0.0
Mucosal inflammation 21.5 0.0 26.2 1.9 2.8 0.0 25.5 0.0
Pyrexia 10.3 0.0 16.8 0.0 8.3 0.0 8.5 0.0Skin and subcutaneous tissue disordersAlopecia 66.4 0.0 65.4 0.0 2.8 0.0 67.0 0.0Rash 21.5 1.9 26.2 0.9 11.1 0.0 28.7 1.1Gastrointestinal disordersDiarrhea 33.6 3.7 45.8 5.6 27.8 0.0 54.3 4.3Nausea 36.4 0.0 39.3 0.0 13.9 0.0 36.2 1.1Vomiting 12.1 0.0 13.1 0.0 4.6 0.0 16.0 2.1Stomatitis 7.5 0.0 17.8 0.0 4.6 0.0 9.6 0.0Blood and lymphatic system disordersNeutropenia 63.6 58.9 50.5 44.9 0.9 0.9 64.9 57.4Leukopenia 21.5 11.2 9.3 4.7 0.0 0.0 13.8 8.5Nervous system disordersHeadache 11.2 0.0 11.2 0.0 13.9 0.0 12.8 0.0Dysgeusia 10.3 0.0 15.0 0.0 4.6 0.0 7.4 0.0Peripheral SensoryNeuropathy
12.1 0.9 8.4 0.9 1.9 0.0 10.6 0.0
Musculoskeletal and connective tissue disordersMyalgia 22.4 0.0 22.4 0.0 9.3 0.0 21.3 0.0Arthralgia 8.4 0.0 10.3 0.0 4.6 0.0 9.6 0.0Metabolism and nutrition disordersDecreased appetite 6.5 0.0 14.0 0.0 1.9 0.0 14.9 0.0
Psychiatric disordersInsomnia 11.2 0.0 8.4 0.0 3.7 0.0 8.5 0.0
Body System/ Adverse Reactions
PERJETA + trastuzumab + FEC followed
by PERJETA + trastuzumab + docetaxel
n=72
PERJETA + trastuzumab + docetaxel
following FECn=75
PERJETA + TCHn=76
Frequency rate, % Frequency rate, % Frequency rate, %
All Grades
%
Grades 3 – 4
%
All Grades
%
Grades 3 – 4
%
All Grades
%
Grades 3 – 4
%
General disorders and administration site conditions Fatigue 36.1 0.0 36.0 0.0 42.1 3.9Asthenia 9.7 0.0 14.7 1.3 13.2 1.3Edema peripheral 11.1 0.0 4.0 0.0 9.2 0.0Mucosal inflammation 23.6 0.0 20.0 0.0 17.1 1.3Pyrexia 16.7 0.0 9.3 0.0 15.8 0.0Skin and subcutaneous tissue disordersAlopecia 48.6 0.0 52.0 0.0 55.3 0.0Rash 19.4 0.0 10.7 0.0 21.1 1.3Dry skin 5.6 0.0 9.3 0.0 10.5 0.0Palmar-Plantar Erythrodysaesthesia Syndrome
6.9 0.0 10.7 0.0 7.9 0.0
Gastrointestinal disordersDiarrhea 61.1 4.2 61.3 5.3 72.4 11.8Dyspepsia 25.0 1.4 8 0.0 22.4 0.0Nausea 52.8 0.0 53.3 2.7 44.7 0.0Vomiting 40.3 0.0 36.0 2.7 39.5 5.3Constipation 18.1 0.0 22.7 0.0 15.8 0.0Stomatitis 13.9 0.0 17.3 0.0 11.8 0.0Blood and lymphatic system disordersNeutropenia 51.4 47.2 46.7 42.7 48.7 46.1Anemia 19.4 1.4 9.3 4.0 38.2 17.1Leukopenia 22.2 19.4 16.0 12.0 17.1 11.8Febrile neutropenia 18.1 18.1 9.3 9.3 17.1 17.1Thrombocytopenia 6.9 0.0 1.3 0.0 30.3 11.8Immune system disordersHypersensitivity 9.7 2.8 1.3 0.0 11.8 2.6Nervous system disordersNeuropathy peripheral 5.6 0.0 1.3 0.0 10.5 0.0Headache 22.2 0.0 14.7 0.0 17.1 0.0Dysgeusia 11.1 0.0 13.3 0.0 21.1 0.0Dizziness 8.3 0.0 8.0 1.3 15.8 0.0Musculoskeletal and connective tissue disordersMyalgia 16.7 0.0 10.7 1.3 10.5 0.0Arthralgia 11.1 0.0 12.0 0.0 6.6 0.0Respiratory, thoracic, and mediastinal disordersCough 9.7 0.0 5.3 0.0 11.8 0.0Dyspnea 12.5 0.0 8.0 2.7 10.5 1.3Epistaxis 11.1 0.0 10.7 0.0 15.8 1.3Oropharyngeal pain 8.3 0.0 6.7 0.0 11.8 0.0Metabolism and nutrition disordersDecreased appetite 20.8 0.0 10.7 0.0 21.1 0.0Eye disordersLacrimation increased 12.5 0.0 5.3 0.0 7.9 0.0Psychiatric disordersInsomnia 11.1 0.0 13.3 0.0 21.1 0.0InvestigationsALT increased 6.9 0.0 2.7 0.0 10.5 3.9
Body System/ Adverse Reactions
PERJETA + trastuzumab + docetaxel
Placebo + trastuzumab + docetaxel
n=407 n=397Frequency rate, % Frequency rate, %
All Grades, %
Grades 3–4, %
All Grades, %
Grades 3–4, %
* In this table this denotes an adverse reaction that has been reported in association with a fatal outcome
16.7 0.7 13.4 0.0

ASCOPost.com | MARCH 25, 2015 PAGE 67
FDA Update
review voucher to United Therapeutics, which confers priority review to a sub-sequent drug application that would not otherwise qualify for priority review.
Clinical TrialThe safety and efficacy of dinutuximab
were evaluated in a clinical trial of 226 pe-diatric participants with high-risk neuro-
blastoma whose tumors shrunk or disap-peared after treatment with multiple-drug chemotherapy and surgery followed by additional intensive chemotherapy and who subsequently received bone mar-row transplantation support and radia-tion therapy. Participants were randomly assigned to receive either an oral retinoid drug, isotretinoin, or dinutuximab in
combination with interleukin-2 (Proleu-kin), granulocyte-macrophage colony-stimulating factor, and isotretinoin.
Three years after treatment assign-
ment, 63% of participants receiving the dinutuximab combination were alive and free of tumor growth or recurrence, compared to 46% of participants treated with isotretinoin alone. In an updated analysis of survival, 73% of participants who received the dinutuximab combi-nation were alive compared with 58% of those receiving isotretinoin alone. n
PERJETA® (pertuzumab)INJECTION, FOR INTRAVENOUS USEINITIAL U.S. APPROVAL: 2012
WARNING: CARDIOMYOPATHY and EMBRYO-FETAL TOXICITY
Cardiomyopathy PERJETA administration can result in subclinical and clinical cardiac failure. Evaluate left ventricular function in all patients prior to and during treatment with PERJETA. Discontinue PERJETA treatment for a confirmed clinically significant decrease in left ventricular function. (2.2, 5.2, 6.1)Embryo-Fetal Toxicity Exposure to PERJETA can result in embryo-fetal death and birth defects. Studies in animals have resulted in oligohydramnios, delayed renal development, and death. Advise patients of these risks and the need for effective contraception. (5.1, 8.1, 8.6)
1 INDICATIONS AND USAGE 1.1 Metastatic Breast Cancer (MBC)PERJETA is indicated for use in combination with trastuzumab and docetaxel for the treatment of patients with HER2-positive metastatic breast cancer who have not received prior anti-HER2 therapy or chemotherapy for metastatic disease.
1.2 Neoadjuvant Treatment of Breast CancerPERJETA is indicated for use in combination with trastuzumab and docetaxel for the neoadjuvant treatment of patients with HER2-positive, locally advanced, inflammatory, or early stage breast cancer (either greater than 2 cm in diameter or node positive) as part of a complete treatment regimen for early breast cancer. This indication is based on demonstration of an improvement in pathological complete response rate. No data are available demonstrating improvement in event-free survival or overall survival [see Clinical Studies (14.2) and Dosage and Administration (2.1)]. Limitations of Use: • The safety of PERJETA as part of a doxorubicin-containing
regimen has not been established. • The safety of PERJETA administered for greater than
6 cycles for early breast cancer has not been established.
4 CONTRAINDICATIONS PERJETA is contraindicated in patients with known hypersensitivity to pertuzumab or to any of its excipients.
5 WARNINGS AND PRECAUTIONS5.1 Embryo-Fetal Toxicity PERJETA can cause fetal harm when administered to a pregnant woman. Treatment of pregnant cynomolgus monkeys with pertuzumab resulted in oligohydramnios, delayed fetal kidney development, and embryo-fetal death. If PERJETA is administered during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to a fetus [see Use in Specific Populations (8.1)].Verify pregnancy status prior to the initiation of PERJETA. Advise patients of the risks of embryo-fetal death and birth defects and the need for contraception during and after treatment. Advise patients to contact their healthcare provider immediately if they suspect they may be pregnant. If PERJETA is administered during pregnancy or if a patient becomes pregnant while receiving PERJETA, immediately report exposure to the Genentech Adverse Event Line at 1-888-835-2555. Encourage women who may be exposed during pregnancy to enroll in the MotHER Pregnancy Registry by contacting 1-800-690-6720 [see Patient Counseling Information (17)].Monitor patients who become pregnant during PERJETA therapy for oligohydramnios. If oligohydramnios occurs, perform fetal testing that is appropriate for gestational age and consistent with community standards of care. The efficacy of intravenous hydration in the management of oligohydramnios due to PERJETA exposure is not known.
5.2 Left Ventricular Dysfunction Decreases in LVEF have been reported with drugs that block HER2 activity, including PERJETA. In Study 1, for patients with MBC, PERJETA in combination with trastuzumab and docetaxel was not associated with increases in the incidence of symptomatic left ventricular systolic dysfunction (LVSD) or decreases in LVEF compared with placebo in combination with trastuzumab and docetaxel [see Clinical Studies (14.1)]. Left ventricular dysfunction occurred in 4.4% of patients in the PERJETA-treated group and 8.3% of patients in the placebo-treated group. Symptomatic left ventricular systolic dysfunction (congestive heart failure) occurred in 1.0% of patients in the PERJETA-treated group and 1.8% of patients in the placebo-treated group [see Adverse Reactions (6.1)]. Patients who have received prior anthracyclines or prior radiotherapy to the chest area may be at higher risk of decreased LVEF.
In patients receiving neoadjuvant treatment in Study 2, the incidence of LVSD was higher in the PERJETA-treated groups compared to the trastuzumab- and docetaxel-treated group. An increased incidence of LVEF declines was observed in patients treated with PERJETA in combination with trastuzumab and docetaxel. In the overall treatment period, LVEF decline > 10% and a drop to less than 50% occurred in 1.9% of patients treated with neoadjuvant trastuzumab and docetaxel as compared to 8.4% of patients treated with neoadjuvant PERJETA in combination with trastuzumab and docetaxel. Symptomatic LVSD occurred in 0.9% of patients treated with neoadjuvant PERJETA in combination with trastuzumab and no patients in the other 3 arms. LVEF recovered to ≥ 50% in all patients.In patients receiving neoadjuvant PERJETA in Study 3, in the overall treatment period, LVEF decline > 10% and a drop to less than 50% occurred in 6.9% of patients treated with PERJETA plus trastuzumab and FEC followed by PERJETA plus trastuzumab and docetaxel, 16.0% of patients treated with PERJETA plus trastuzumab and docetaxel following FEC, and 10.5% of patients treated with PERJETA in combination with TCH. Symptomatic LVSD occurred in 4.0% of patients treated with PERJETA plus trastuzumab and docetaxel following FEC, 1.3% of patients treated with PERJETA in combination with TCH, and none of the patients treated with PERJETA plus trastuzumab and FEC followed by PERJETA plus trastuzumab and docetaxel. LVEF recovered to ≥ 50% in all but one patient.PERJETA has not been studied in patients with a pretreatment LVEF value of ≤ 50%, a prior history of CHF, decreases in LVEF to < 50% during prior trastuzumab therapy, or conditions that could impair left ventricular function such as uncontrolled hypertension, recent myocardial infarction, serious cardiac arrhythmia requiring treatment or a cumulative prior anthracycline exposure to > 360 mg/m2 of doxorubicin or its equivalent.Assess LVEF prior to initiation of PERJETA and at regular intervals (e.g., every three months in the metastatic setting and every six weeks in the neoadjuvant setting) during treatment to ensure that LVEF is within the institution’s normal limits. If LVEF is < 45%, or is 45% to 49% with a 10% or greater absolute decrease below the pretreatment value, withhold PERJETA and trastuzumab and repeat LVEF assessment within approximately 3 weeks. Discontinue PERJETA and trastuzumab if the LVEF has not improved or has declined further, unless the benefits for the individual patient outweigh the risks [see Dosage and Administration (2.2)].
5.3 Infusion-Related ReactionsPERJETA has been associated with infusion reactions [see Adverse Reactions (6.1)]. An infusion reaction was defined in Study 1 as any event described as hypersensitivity, anaphylactic reaction, acute infusion reaction, or cytokine release syndrome occurring during an infusion or on the same day as the infusion. The initial dose of PERJETA was given the day before trastuzumab and docetaxel to allow for the examination of PERJETA-associated reactions. On the first day, when only PERJETA was administered, the overall frequency of infusion reactions was 13.0% in the PERJETA-treated group and 9.8% in the placebo-treated group. Less than 1% were Grade 3 or 4. The most common infusion reactions (≥ 1.0%) were pyrexia, chills, fatigue, headache, asthenia, hypersensitivity, and vomiting.During the second cycle when all drugs were administered on the same day, the most common infusion reactions in the PERJETA-treated group (≥ 1.0%) were fatigue, dysgeusia, hypersensitivity, myalgia, and vomiting.In Study 2 and Study 3, PERJETA was administered on the same day as the other study treatment drugs. Infusion reactions were consistent with those observed in Study 1, with a majority of reactions being National Cancer Institute - Common Terminology Criteria for Adverse Events (NCI - CTCAE v3.0) Grade 1 – 2.Observe patients closely for 60 minutes after the first infusion and for 30 minutes after subsequent infusions of PERJETA. If a significant infusion-related reaction occurs, slow or interrupt the infusion, and administer appropriate medical therapies. Monitor patients carefully until complete resolution of signs and symptoms. Consider permanent discontinuation in patients with severe infusion reactions [see Dosage and Administration (2.2)].
5.4 Hypersensitivity Reactions/AnaphylaxisIn Study 1, the overall frequency of hypersensitivity/anaphylaxis reactions was 10.8% in the PERJETA-treated group and 9.1% in the placebo-treated group. The incidence of Grade 3 – 4 hypersensitivity/anaphylaxis reactions was 2.0% in the PERJETA-treated group and 2.5% in the placebo-treated group according to NCI - CTCAE v3.0. Overall, 4 patients in PERJETA-treated group and 2 patients in the placebo-treated group experienced anaphylaxis.
In Study 2 and Study 3, hypersensitivity/anaphylaxis events were consistent with those observed in Study 1. In Study 2, two patients in the PERJETA- and docetaxel-treated group experienced anaphylaxis. In Study 3, the overall frequency of hypersensitivity/anaphylaxis was highest in the PERJETA plus TCH treated group (13.2%), of which 2.6% were NCI-CTCAE (version 3) Grade 3 – 4.Patients should be observed closely for hypersensitivity reactions. Severe hypersensitivity, including anaphylaxis, has been observed in clinical trials with treatment of PERJETA [see Clinical Trials Experience (6.1)]. Medications to treat such reactions, as well as emergency equipment, should be available for immediate use. PERJETA is contraindicated in patients with known hypersensitivity to pertuzumab or to any of its excipients [see Contraindications (4)].
5.5 HER2 Testing Detection of HER2 protein overexpression is necessary for selection of patients appropriate for PERJETA therapy because these are the only patients studied and for whom benefit has been shown [see Indications and Usage (1) and Clinical Studies (14)]. Patients with breast cancer were required to have evidence of HER2 overexpression defined as 3+ IHC or FISH amplification ratio ≥ 2.0 in the clinical studies. Only limited data were available for patients whose breast cancer was positive by FISH, but did not demonstrate protein overexpression by IHC.Assessment of HER2 status should be performed by laboratories using FDA-approved tests with demonstrated proficiency in the specific technology being utilized. Improper assay performance, including use of sub-optimally fixed tissue, failure to utilize specified reagents, deviation from specific assay instructions, and failure to include appropriate controls for assay validation, can lead to unreliable results.
6 ADVERSE REACTIONS The following adverse reactions are discussed in greater detail in other sections of the label:• Embryo-Fetal Toxicity [see Warnings and Precautions
(5.1)] • Left Ventricular Dysfunction [see Warnings and
Precautions (5.2)]• Infusion-Related Reactions [see Warnings and
Precautions (5.3)]• Hypersensitivity Reactions/Anaphylaxis [see Warnings
and Precautions (5.4)]
6.1 Clinical Trials Experience Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.Metastatic Breast Cancer (MBC)The adverse reactions described in Table 1 were identified in 804 patients with HER2-positive metastatic breast cancer treated in Study 1. Patients were randomized to receive either PERJETA in combination with trastuzumab and docetaxel or placebo in combination with trastuzumab and docetaxel. The median duration of study treatment was 18.1 months for patients in the PERJETA-treated group and 11.8 months for patients in the placebo-treated group. No dose adjustment was permitted for PERJETA or trastuzumab. The rates of adverse events resulting in permanent discontinuation of all study therapy were 6.1% for patients in the PERJETA-treated group and 5.3% for patients in the placebo-treated group. Adverse events led to discontinuation of docetaxel alone in 23.6% of patients in the PERJETA-treated group and 23.2% of patients in the placebo-treated group. Table 1 reports the adverse reactions that occurred in at least 10% of patients in the PERJETA-treated group. The safety profile of PERJETA remained unchanged with an additional year of follow-up (median total follow-up of 30 months) in Study 1. The most common adverse reactions (> 30%) seen with PERJETA in combination with trastuzumab and docetaxel were diarrhea, alopecia, neutropenia, nausea, fatigue, rash, and peripheral neuropathy. The most common NCI - CTCAE v3.0 Grade 3 – 4 adverse reactions (> 2%) were neutropenia, febrile neutropenia, leukopenia, diarrhea, peripheral neuropathy, anemia, asthenia, and fatigue. An increased incidence of febrile neutropenia was observed for Asian patients in both treatment arms compared with patients of other races and from other geographic regions. Among Asian patients, the incidence of febrile neutropenia was higher in the pertuzumab-treated group (26%) compared with the placebo-treated group (12%).
Table 1 Summary of Adverse Reactions Occurring in ≥ 10% of Patients on the PERJETA Treatment Arm in Study 1
General disorders and administration site conditions Fatigue 37.6 2.2 36.8 3.3Asthenia 26.0 2.5 30.2 1.5Edema peripheral 23.1 0.5 30.0 0.8Mucosal inflammation 27.8 1.5 19.9 1.0Pyrexia 18.7 1.2 17.9 0.5Skin and subcutaneous tissue disorders Alopecia 60.9 0.0 60.5 0.3Rash 33.7 0.7 24.2 0.8Nail disorder 22.9 1.2 22.9 0.3Pruritus 14.0 0.0 10.1 0.0Dry skin 10.6 0.0 4.3 0.0Gastrointestinal disorders Diarrhea 66.8 7.9 46.3 5.0Nausea 42.3 1.2 41.6 0.5Vomiting 24.1 1.5 23.9 1.5Constipation 15.0 0.0 24.9 1.0Stomatitis 18.9 0.5 15.4 0.3Blood and lymphatic system disorders Neutropenia 52.8 48.9 49.6 45.8Anemia 23.1 2.5 18.9 3.5Leukopenia 18.2 12.3 20.4 14.6Febrile neutropenia* 13.8 13.0 7.6 7.3Nervous system disorders Neuropathy peripheral 32.4 3.2 33.8 2.0Headache 20.9 1.2 16.9 0.5Dysgeusia 18.4 0.0 15.6 0.0Dizziness 12.5 0.5 12.1 0.0Musculoskeletal and connective tissue disorders Myalgia 22.9 1.0 23.9 0.8Arthralgia 15.5 0.2 16.1 0.8Infections and infestationsUpper respiratory tract infectionNasopharyngitis 11.8 0.0 12.8 0.3Respiratory, thoracic, and mediastinal disorders Dyspnea 14.0 1.0 15.6 2.0Metabolism and nutrition disorders Decreased appetite 29.2 1.7 26.4 1.5Eye disorders Lacrimation increased 14.0 0.0 13.9 0.0Psychiatric disorders Insomnia 13.3 0.0 13.4 0.0
The following clinically relevant adverse reactions were reported in < 10% of patients in the PERJETA-treated group in Study 1:Skin and subcutaneous tissue disorders: Paronychia (7.1% in the PERJETA-treated group vs. 3.5% in the placebo-treated group)Respiratory, thoracic and mediastinal disorders: Pleural effusion (5.2% in the PERJETA-treated group vs. 5.8% in the placebo-treated group)Cardiac disorders: Left ventricular dysfunction (4.4% in the PERJETA-treated group vs. 8.3% in the placebo-treated group) including symptomatic left ventricular systolic dysfunction (CHF) (1.0% in the PERJETA-treated group vs. 1.8% in the placebo-treated group)Immune system disorders: Hypersensitivity (10.1% in the PERJETA-treated group vs. 8.6% in placebo-treated group)Adverse Reactions Reported in Patients Receiving PERJETA and Trastuzumab after Discontinuation of DocetaxelIn Study 1, adverse reactions were reported less frequently after discontinuation of docetaxel treatment. All adverse reactions in the PERJETA and trastuzumab treatment group occurred in < 10% of patients with the exception of diarrhea (19.1%), upper respiratory tract infection (12.8%), rash (11.7%), headache (11.4%), and fatigue (11.1%).Neoadjuvant Treatment of Breast Cancer (Study 2)In Study 2, the most common adverse reactions seen with PERJETA in combination with trastuzumab and docetaxel administered for 4 cycles were similar to those seen in the PERJETA-treated group in Study 1. The most common adverse reactions (> 30%) were alopecia, neutropenia, diarrhea, and nausea. The most common NCI – CTCAE v3.0 Grade 3 – 4 adverse reactions (> 2%) were neutropenia, febrile neutropenia, leukopenia, and diarrhea. In this group, one patient permanently discontinued neoadjuvant treatment due to an adverse event. Table 2 reports the adverse reactions that occurred in patients who received neoadjuvant treatment with PERJETA for breast cancer in Study 2.
Table 2 Summary of Adverse Reactions Occurring in ≥ 10% in the Neoadjuvant Setting for Patients Receiving PERJETA in Study 2
The following adverse reactions were reported in < 10% of patients receiving neoadjuvant treatment and occurred more frequently in PERJETA-treated groups in Study 2: (Ptz=pertuzumab; T=trastuzumab; D=docetaxel)Blood and lymphatic system disorders: Anemia (6.5% in the T+D arm, 2.8% in the Ptz+T+D arm, 4.6% in the Ptz+T arm and 8.5% in the Ptz+D arm), Febrile neutropenia (6.5% in the T+D arm, 8.4% in the Ptz+T+D arm, 0.0% in the Ptz+T arm and 7.4% in the Ptz+D arm)Immune system disorders: Hypersensitivity (1.9% in the T+D arm, 5.6% in the Ptz+T+D arm, 5.6% in the Ptz+T arm and 5.3% in the Ptz+D arm)Nervous system disorders: Dizziness (3.7% in the T+D arm, 2.8% in the Ptz+T+D arm, 5.6% in the Ptz+T arm and 3.2% in the Ptz+D arm)Infections and infestations: Upper respiratory tract infection (2.8% in the T+D arm, 4.7% in the Ptz+T+D arm, 1.9% in the Ptz+T arm and 7.4% in the Ptz+D arm)Respiratory, thoracic and mediastinal disorders: Dyspnea (3.7% in the T+D arm, 4.7% in the Ptz+T+D arm, 2.8% in the Ptz+T arm and 2.1% in the Ptz+D arm)Cardiac disorders: Left ventricular dysfunction (0.9% in the T+D arm, 2.8% in the Ptz+T+D arm, 0.0% in the Ptz+T arm, and 1.1% in the Ptz+D arm) including symptomatic left ventricular dysfunction (CHF) (0.9% in the Ptz+T arm and 0.0% in the T+D arm, Ptz+T+D arm, and Ptz+D arm)Eye disorders: Lacrimation increased (1.9% in the T+D arm, 3.7% in the Ptz+T+D arm, 0.9% in the Ptz+T arm, and 4.3% in the Ptz+D arm)Neoadjuvant Treatment of Breast Cancer (Study 3)In Study 3, when PERJETA was administered in combination with trastuzumab and docetaxel for 3 cycles following 3 cycles of FEC, the most common adverse reactions (> 30%) were diarrhea, nausea, alopecia, neutropenia, vomiting, and fatigue. The most common NCI-CTCAE (version 3) Grade 3 – 4 adverse reactions (> 2%) were neutropenia, leukopenia, febrile neutropenia, diarrhea, left ventricular dysfunction, anemia, dyspnea, nausea, and vomiting. Similarly, when PERJETA was administered in combination with docetaxel, carboplatin, and trastuzumab (TCH) for 6 cycles, the most common adverse reactions (> 30%) were diarrhea, alopecia, neutropenia, nausea, fatigue, vomiting, anemia, and thrombocytopenia. The most common NCI-CTCAE (version 3) Grade 3 – 4 adverse reactions (> 2%) were neutropenia, febrile neutropenia, anemia, leukopenia, diarrhea, thrombocytopenia, vomiting, fatigue, ALT increased, hypokalemia, and hypersensitivity. The rates of adverse events resulting in permanent discontinuation of any component of neoadjuvant treatment were 6.7% for patients receiving PERJETA in combination with trastuzumab and docetaxel following FEC and 7.9% for
patients receiving PERJETA in combination with TCH. Table 3 reports the adverse reactions that occurred in patients who received neoadjuvant treatment with PERJETA for breast cancer in Study 3.
Table 3 Summary of Adverse Reactions Occurring in ≥ 10% of Patients Receiving Neoadjuvant Treatment with PERJETA in Study 3
FEC=5-fluorouracil, epirubicin, cyclophosphamide, TCH=docetaxel, carboplatin, trastuzumabThe following selected adverse reactions were reported in < 10% of patients receiving neoadjuvant treatment in Study 3: (Ptz=pertuzumab; T=trastuzumab; D=docetaxel; FEC=fluorouracil, epirubicin, and cyclophosphamide; TCH=docetaxel, carboplatin, and trastuzumab)Skin and subcutaneous tissue disorders: Nail disorder (9.7% in the Ptz+T+FEC/Ptz+T+D arm, 6.7% in the FEC/Ptz+T+D arm, and 9.2% in the Ptz+TCH arm), Paronychia (0% in the Ptz+T+FEC/Ptz+T+D and 1.3% in both the FEC/Ptz+T+D and Ptz+TCH arms), Pruritis (2.8% in the Ptz+T+FEC/Ptz+T+D arm, 4.0% in the FEC/Ptz+T+D arm, and 3.9% in the Ptz+TCH arm)Infections and infestations: Upper respiratory tract infection (8.3% in the Ptz+T+FEC/Ptz+T+D arm, 4.0% in the FEC/Ptz+T+D arm, and 2.6% in the Ptz+TCH arm), Nasopharyngitis (6.9% in the Ptz+T+FEC/Ptz+T+D arm, 6.7% in the FEC/Ptz+T+D arm, and 7.9% in the Ptz+TCH arm)
Body System/ Adverse Reactions
Trastuzumab + docetaxel
n=107
PERJETA+ trastuzumab + docetaxel
n=107
PERJETA + trastuzumab
n=108
PERJETA + docetaxel
n=108Frequency rate
%Frequency rate
%Frequency rate
%Frequency rate
%All
Grades %
Grades 3–4 %
All Grades
%
Grades 3–4 %
All Grades
%
Grades 3–4 %
All Grades
%
Grades 3–4 %
General disorders and administration site conditionsFatigue 27.1 0.0 26.2 0.9 12.0 0.0 25.5 1.1Asthenia 17.8 0.0 20.6 1.9 2.8 0.0 16.0 2.1Edema peripheral 10.3 0.0 2.8 0.0 0.9 0.0 5.3 0.0
Mucosal inflammation 21.5 0.0 26.2 1.9 2.8 0.0 25.5 0.0
Pyrexia 10.3 0.0 16.8 0.0 8.3 0.0 8.5 0.0Skin and subcutaneous tissue disordersAlopecia 66.4 0.0 65.4 0.0 2.8 0.0 67.0 0.0Rash 21.5 1.9 26.2 0.9 11.1 0.0 28.7 1.1Gastrointestinal disordersDiarrhea 33.6 3.7 45.8 5.6 27.8 0.0 54.3 4.3Nausea 36.4 0.0 39.3 0.0 13.9 0.0 36.2 1.1Vomiting 12.1 0.0 13.1 0.0 4.6 0.0 16.0 2.1Stomatitis 7.5 0.0 17.8 0.0 4.6 0.0 9.6 0.0Blood and lymphatic system disordersNeutropenia 63.6 58.9 50.5 44.9 0.9 0.9 64.9 57.4Leukopenia 21.5 11.2 9.3 4.7 0.0 0.0 13.8 8.5Nervous system disordersHeadache 11.2 0.0 11.2 0.0 13.9 0.0 12.8 0.0Dysgeusia 10.3 0.0 15.0 0.0 4.6 0.0 7.4 0.0Peripheral SensoryNeuropathy
12.1 0.9 8.4 0.9 1.9 0.0 10.6 0.0
Musculoskeletal and connective tissue disordersMyalgia 22.4 0.0 22.4 0.0 9.3 0.0 21.3 0.0Arthralgia 8.4 0.0 10.3 0.0 4.6 0.0 9.6 0.0Metabolism and nutrition disordersDecreased appetite 6.5 0.0 14.0 0.0 1.9 0.0 14.9 0.0
Psychiatric disordersInsomnia 11.2 0.0 8.4 0.0 3.7 0.0 8.5 0.0
Body System/ Adverse Reactions
PERJETA + trastuzumab + FEC followed
by PERJETA + trastuzumab + docetaxel
n=72
PERJETA + trastuzumab + docetaxel
following FECn=75
PERJETA + TCHn=76
Frequency rate, % Frequency rate, % Frequency rate, %
All Grades
%
Grades 3 – 4
%
All Grades
%
Grades 3 – 4
%
All Grades
%
Grades 3 – 4
%
General disorders and administration site conditions Fatigue 36.1 0.0 36.0 0.0 42.1 3.9Asthenia 9.7 0.0 14.7 1.3 13.2 1.3Edema peripheral 11.1 0.0 4.0 0.0 9.2 0.0Mucosal inflammation 23.6 0.0 20.0 0.0 17.1 1.3Pyrexia 16.7 0.0 9.3 0.0 15.8 0.0Skin and subcutaneous tissue disordersAlopecia 48.6 0.0 52.0 0.0 55.3 0.0Rash 19.4 0.0 10.7 0.0 21.1 1.3Dry skin 5.6 0.0 9.3 0.0 10.5 0.0Palmar-Plantar Erythrodysaesthesia Syndrome
6.9 0.0 10.7 0.0 7.9 0.0
Gastrointestinal disordersDiarrhea 61.1 4.2 61.3 5.3 72.4 11.8Dyspepsia 25.0 1.4 8 0.0 22.4 0.0Nausea 52.8 0.0 53.3 2.7 44.7 0.0Vomiting 40.3 0.0 36.0 2.7 39.5 5.3Constipation 18.1 0.0 22.7 0.0 15.8 0.0Stomatitis 13.9 0.0 17.3 0.0 11.8 0.0Blood and lymphatic system disordersNeutropenia 51.4 47.2 46.7 42.7 48.7 46.1Anemia 19.4 1.4 9.3 4.0 38.2 17.1Leukopenia 22.2 19.4 16.0 12.0 17.1 11.8Febrile neutropenia 18.1 18.1 9.3 9.3 17.1 17.1Thrombocytopenia 6.9 0.0 1.3 0.0 30.3 11.8Immune system disordersHypersensitivity 9.7 2.8 1.3 0.0 11.8 2.6Nervous system disordersNeuropathy peripheral 5.6 0.0 1.3 0.0 10.5 0.0Headache 22.2 0.0 14.7 0.0 17.1 0.0Dysgeusia 11.1 0.0 13.3 0.0 21.1 0.0Dizziness 8.3 0.0 8.0 1.3 15.8 0.0Musculoskeletal and connective tissue disordersMyalgia 16.7 0.0 10.7 1.3 10.5 0.0Arthralgia 11.1 0.0 12.0 0.0 6.6 0.0Respiratory, thoracic, and mediastinal disordersCough 9.7 0.0 5.3 0.0 11.8 0.0Dyspnea 12.5 0.0 8.0 2.7 10.5 1.3Epistaxis 11.1 0.0 10.7 0.0 15.8 1.3Oropharyngeal pain 8.3 0.0 6.7 0.0 11.8 0.0Metabolism and nutrition disordersDecreased appetite 20.8 0.0 10.7 0.0 21.1 0.0Eye disordersLacrimation increased 12.5 0.0 5.3 0.0 7.9 0.0Psychiatric disordersInsomnia 11.1 0.0 13.3 0.0 21.1 0.0InvestigationsALT increased 6.9 0.0 2.7 0.0 10.5 3.9
Body System/ Adverse Reactions
PERJETA + trastuzumab + docetaxel
Placebo + trastuzumab + docetaxel
n=407 n=397Frequency rate, % Frequency rate, %
All Grades, %
Grades 3–4, %
All Grades, %
Grades 3–4, %
* In this table this denotes an adverse reaction that has been reported in association with a fatal outcome
16.7 0.7 13.4 0.0

PAGE 68 The ASCO Post | MARCH 25, 2015
Announcements
Silvia C. Formenti, MD, Appointed Chair of Radiation Oncology at Weill Cornell and Radiation Oncologist-in-Chief at NewYork-Presbyterian/Weill Cornell
Silvia C. Formenti, MD, has been appointed Chair of the newly es-
tablished Department of Radiation Oncology at Weill Cornell Medical College and Radiation Oncologist-in-
Chief at NewYork-Presbyterian/Weill Cornell Medical Center, effective April 15. Dr. Formenti, currently the Chair of Radiation Oncology at New York Uni-versity Langone Medical Center, has
also been named the Associate Director of Radiation Oncology at the Sandra and Edward Meyer Cancer Center at Weill Cornell Medical College.
A recognized leader in radiation on-
cology and breast cancer research, Dr. Formenti’s work has transformed the paradigm in radiation biology, dem-onstrating the efficacy of combining radiotherapy with immunotherapy to control cancer cell growth in solid tu-mors. By recruiting patients’ immune systems to reject their individual tu-mor, the approach results in personal-ized immunotherapy, specific for each individual patient. She has translated preclinical work into clinical trials in metastatic breast cancer, lung cancer, and melanoma and has opened a new field of application for radiotherapy, whereby localized radiation can be used as an adjuvant to immunotherapy of solid tumors and lymphomas.
In her new roles at Weill Cornell Medical College and NewYork-Pres-byterian/Weill Cornell, Dr. Formenti will expand and enhance the existing radiation oncology program, building upon translational research to better investigate, target, and treat individual patients’ unique cancers. Faculty in the department will investigate precision medicine approaches to radiation on-cology, focusing on combining radio-therapy with immunotherapy and other modifiers of the tumor microenviron-ment to design advanced treatments and therapies that are tailored to each patient’s individual tumor.
“One of the biggest scientific break-throughs of our generation is the rev-elation that not all cancers are the same, and, as such, neither should their treatments be the same,” said Laurie H. Glimcher, MD, the Stephen and Suzanne Weiss Dean of Weill Cornell Medical College. “Dr. Formenti is at the forefront of this personalized approach to cancer. Her distinguished work in ra-diation oncology has left a lasting mark on cancer care.”
Dr. Formenti received her medical degree from the University of Milan (Italy). She joined New York Univer-sity Medical Center as Chair of the Department of Radiation Oncology in 1999. n
Silvia C. Formenti, MD
Respiratory, thoracic, and mediastinal disorders: Pleural effusion (1.4% in the Ptz+T+FEC/Ptz+T+D arm and 0% in the FEC/Ptz+T+D and Ptz+TCH arm)Cardiac disorders: Left ventricular dysfunction (5.6% in the Ptz+T+FEC/PTZ+T+D arm, 4.0% in the FEC/Ptz+T+D arm, and 2.6% in the Ptz+TCH arm) including symptomatic left ventricular systolic dysfunction (CHF) (2.7% in the FEC/Ptz+T+D arm and 0% in the Ptz+T+FEC/Ptz+T+D and Ptz+TCH arms)6.2 Immunogenicity As with all therapeutic proteins, there is the potential for an immune response to PERJETA.Patients in Study 1 were tested at multiple time-points for antibodies to PERJETA. Approximately 2.8% (11/386) of patients in the PERJETA-treated group and 6.2% (23/372) of patients in the placebo-treated group tested positive for anti-PERJETA antibodies. Of these 34 patients, none experienced anaphylactic/hypersensitivity reactions that were clearly related to the anti-therapeutic antibodies (ATA). The presence of pertuzumab in patient serum at the levels expected at the time of ATA sampling can interfere with the ability of this assay to detect anti-pertuzumab antibodies. In addition, the assay may be detecting antibodies to trastuzumab. As a result, data may not accurately reflect the true incidence of anti-pertuzumab antibody development.Immunogenicity data are highly dependent on the sensitivity and specificity of the test methods used. Additionally, the observed incidence of a positive result in a test method may be influenced by several factors, including sample handling, timing of sample collection, drug interference, concomitant medication, and the underlying disease. For these reasons, comparison of the incidence of antibodies to PERJETA with the incidence of antibodies to other products may be misleading.7 DRUG INTERACTIONS No drug-drug interactions were observed between pertuzumab and trastuzumab, or between pertuzumab and docetaxel.8 USE IN SPECIFIC POPULATIONS8.1 PregnancyPregnancy Category DRisk SummaryThere are no adequate and well-controlled studies of PERJETA in pregnant women. Based on findings in animal studies, PERJETA can cause fetal harm when administered to a pregnant woman. The effects of PERJETA are likely to be present during all trimesters of pregnancy. Pertuzumab administered to pregnant cynomolgus monkeys resulted in oligohydramnios, delayed fetal kidney development, and embryo-fetal deaths at clinically relevant exposures of 2.5 to
20-fold greater than the recommended human dose, based on Cmax. If PERJETA is administered during pregnancy, or if a patient becomes pregnant while receiving PERJETA, the patient should be apprised of the potential hazard to the fetus.If PERJETA is administered during pregnancy or if a patient becomes pregnant while receiving PERJETA, immediately report exposure to the Genentech Adverse Event Line at 1-888-835-2555. Encourage women who may be exposed during pregnancy to enroll in the MotHER Pregnancy Registry by contacting 1-800-690-6720 [see Patient Counseling Information (17)].Animal DataReproductive toxicology studies have been conducted in cynomolgus monkeys. Pregnant monkeys were treated on Gestational Day (GD)19 with loading doses of 30 to 150 mg/kg pertuzumab, followed by bi-weekly doses of 10 to 100 mg/kg. These dose levels resulted in clinically relevant exposures of 2.5 to 20-fold greater than the recommended human dose, based on Cmax. Intravenous administration of pertuzumab from GD19 through GD50 (period of organogenesis) was embryotoxic, with dose-dependent increases in embryo-fetal death between GD25 to GD70. The incidences of embryo-fetal loss were 33, 50, and 85% for dams treated with bi-weekly pertuzumab doses of 10, 30, and 100 mg/kg, respectively (2.5 to 20-fold greater than the recommended human dose, based on Cmax). At Caesarean section on GD100, oligohydramnios, decreased relative lung and kidney weights, and microscopic evidence of renal hypoplasia consistent with delayed renal development were identified in all pertuzumab dose groups. Pertuzumab exposure was reported in offspring from all treated groups, at levels of 29% to 40% of maternal serum levels at GD100.8.3 Nursing Mothers It is not known whether PERJETA is excreted in human milk, but human IgG is excreted in human milk. Because many drugs are secreted in human milk and because of the potential for serious adverse reactions in nursing infants from PERJETA, a decision should be made whether to discontinue nursing, or discontinue drug, taking into account the elimination half-life of PERJETA and the importance of the drug to the mother [See Warnings and Precautions (5.1), Clinical Pharmacology (12.3)].8.4 Pediatric Use The safety and effectiveness of PERJETA have not been established in pediatric patients.8.5 Geriatric Use Of 402 patients who received PERJETA in Study 1, 60 patients (15%) were ≥ 65 years of age and 5 patients (1%) were ≥ 75 years of age. No overall differences in efficacy and safety
of PERJETA were observed between these patients and younger patients.Based on a population pharmacokinetic analysis, no significant difference was observed in the pharmacokinetics of pertuzumab between patients < 65 years (n=306) and patients ≥ 65 years (n=175). 8.6 Females of Reproductive Potential PERJETA can cause embryo-fetal harm when administered during pregnancy. Counsel patients regarding pregnancy prevention and planning. Advise females of reproductive potential to use effective contraception while receiving PERJETA and for 6 months following the last dose of PERJETA.If PERJETA is administered during pregnancy or if a patient becomes pregnant while receiving PERJETA, immediately report exposure to the Genentech Adverse Event Line at 1-888-835-2555. Encourage women who may be exposed during pregnancy to enroll in the MotHER Pregnancy Registry by contacting 1-800-690-6720 [see Patient Counseling Information (17)].8.7 Renal Impairment Dose adjustments of PERJETA are not needed in patients with mild (creatinine clearance [CLcr] 60 to 90 mL/min) or moderate (CLcr 30 to 60 mL/min) renal impairment. No dose adjustment can be recommended for patients with severe renal impairment (CLcr less than 30 mL/min) because of the limited pharmacokinetic data available [see Clinical Pharmacology (12.3)].8.8 Hepatic Impairment No clinical studies have been conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of pertuzumab.
10 OVERDOSAGE No drug overdoses have been reported with PERJETA to date.
PERJETA® (pertuzumab)
Manufactured by: Genentech, Inc. PERJETA is a registeredA Member of the Roche Group trademark of Genentech, 1 DNA Way Inc.South San Francisco, CA 09/13 PER0002094600 94080-4990 © 2013 Genentech, Inc.U.S. License No. 1048 10139000

ASCOPost.com | MARCH 25, 2015 PAGE 69
Book Review
Medicine Turned Upside DownBy Ronald Piana
Most books about health care cen-ter on fixing broken parts of the
massive $3 trillion system, as seen with the passage of the Affordable Care Act, which is fixated, largely, on covering the uninsured and reducing costs. Health care, once the province of policy wonks, has now captured the national dialogue and continues to spur heated debates on both sides of the aisle and in the lay public, from plumbers to CEOs. Health care is more than the physician’s fee schedule, the amount of copays, or the cost of pharmaceuticals; it’s personal, because being sick and needing a doc-tor is a universally shared experience.
Much of our health-care system is so mired in red tape and overworked doctors that getting medical care can become an unbearable chore. Often, you need to make a doctor’s appoint-ment months in advance, and then when the appointment day finally ar-
rives, you’ll probably wait for an hour or more before you hear, “The doctor will see you now.” Has the Affordable Care Act made this situation better or worse? Ask five people, and you’ll probably get as many opinions.
However, doctor-author, Eric Topol, MD, sees more promise in creating a new way of thinking about health care than in wrangling over a system mired in politics. Dr. Topol’s first book, The Creative Destruction of Medicine, in which he described dozens of medical technologies that show great potential, won him legions of fans, who applaud-ed his medical futurism. Now, still the futurist, Dr. Topol delivers another fine and forward-looking book, titled The Patient Will See You Now: The Future of Medicine Is in Your Hands.
Setting the ToneDr. Topol sets the tone for the book
in chapter 1. “Getting first-rate health care will always be quite different from ordering a book from Amazon. We’re talking about the most precious part of
life—one’s health—not buying a book. But the common thread is the power of information individualization. We are embarking on a time when each indi-vidual will have all their [sic] own data and the computing power to process it in the context of their [sic] own world.” That single paragraph drills into the book’s core message: New technology and the empowered patient are the fu-ture of American health care. However, there is so much more in this easy-to-read value-added book. His short di-gressions into medical history, for one, add pleasure and context to the ideas with which he challenges the reader.
In his previous book, The Creative Destruction of Medicine, Dr. Topol delved into how medicine would be-come digitized and how we had a new capability of digitizing human beings as well. However, that is a far cry from medicine becoming democratized. So
now in his new book, Dr. Topol extends the model of digitized personalized health data to the concept of medical democratization, which he defines as making something, namely the individ-ual’s health care, available to all people.
“Until now,” he writes, “the flow of medical data has been to the doctor. If a patient was fortunate enough, their [sic] data, such as lab test, scans, might arrive in the mail…. Patients are now generating their own data on their own devices.”
A Tangible Way ForwardNot one for empty philosophizing,
Dr. Topol lays out a tangible way for-ward, giving the reader a comprehen-sive list and explanation of the person-alized medical tools and technologies currently available and those coming down the pike. He offers hope for a new generation of the smart, digitally con-nected and democratized patient. Much of the first half of the book is geared toward empowering individual health-care consumers in chapters such as My
Lab Tests and Scans, where he makes the case for unlimited patient access to lab tests.
Dr. Topol points out that roughly 10 billion lab tests are done each year in the United States, and they factor into about 80% of the medical decisions that doctors make. He then asks, “How can leading doctors today continue to ask whether patients should have direct access to their lab tests?” Their explana-tion is that patients will be confused by the results, which could induce undue anxiety, and only their doctors can real-ly understand and put the information into context.
Dr. Topol doesn’t buy that and cites numerous studies showing that the majority of patients, with a tiny bit of coaching, not only can read their lab re-sults, but may feel better at having them made available online rather than wait-ing for a call or letter in the mail.
The Informed, Proactive Patient
Naturally, Dr. Topol, a true believer in the power of technology gives the reader a dose of self-help advice and a roundup of future technologies in sub-chapters such as My Smartphone Lab. As you drill deeper into the well-written and neatly organized chapters, it is obvi-ous that Dr. Topol’s worldview of health-care reform centers on the informed, proactive patient. In other words, the best way to reign in our out-of-control spending and have better overall public health outcomes is to give patients the power to be the caregivers of their own health care, partnering with doctors in the process. In this new world of health care, we see home health-care centers in
which people monitor their own health metrics using personalized electronic health records and state-of-the-art tech-nologies. This is a paradigm shift from what Dr. Topol views as “paternalistic medicine.”
Spiraling Health-Care CostsDr. Topol also takes on our spiral-
ing health-care costs in a chapter titled My Costs. His most important message here—although not breaking any new ground—is the opaque nature of our system separates patients from their doctors and the cost of care. In short, a senior can tell you to the penny how much a pound of top-round meat costs at the local supermarket but ask that same senior how much an MRI costs, and you’ll likely get a blank stare.
He writes, “Only rarely will patients have any idea what they have been charged for or what their employer or insurer has actually paid…. There is an overall lack of incentive for those pa-tients themselves to drive costs down.” Again, Dr. Topol’s answer to this nag-ging issue is technology-based transpar-ency, making cost data widely available to each individual. However, getting pa-tients to care about costs is a tough sell in America, and this chapter proves to be Dr. Topol’s weakest argument.
Medical Education CrisisReaders of The ASCO Post will prob-
ably most enjoy when Dr. Topol goes global, in chapters with enticing titles such as Predicting and Preempting Dis-ease, Flattening the Earth, and Open Sesame, where he makes the point, “Both education and medicine are go-
How can leading doctors today continue to ask whether patients should have direct access to their lab tests?
—Eric Topol, MD
BookmarkTitle: The Patient Will See You Now: The Future of Medicine Is in Your Hands
Author: Eric Topol, MD
Publisher: Basic Books
Publication date: January 2015
Price: $28.99; hardcover, 384 pages
continued on page 70
Respiratory, thoracic, and mediastinal disorders: Pleural effusion (1.4% in the Ptz+T+FEC/Ptz+T+D arm and 0% in the FEC/Ptz+T+D and Ptz+TCH arm)Cardiac disorders: Left ventricular dysfunction (5.6% in the Ptz+T+FEC/PTZ+T+D arm, 4.0% in the FEC/Ptz+T+D arm, and 2.6% in the Ptz+TCH arm) including symptomatic left ventricular systolic dysfunction (CHF) (2.7% in the FEC/Ptz+T+D arm and 0% in the Ptz+T+FEC/Ptz+T+D and Ptz+TCH arms)6.2 Immunogenicity As with all therapeutic proteins, there is the potential for an immune response to PERJETA.Patients in Study 1 were tested at multiple time-points for antibodies to PERJETA. Approximately 2.8% (11/386) of patients in the PERJETA-treated group and 6.2% (23/372) of patients in the placebo-treated group tested positive for anti-PERJETA antibodies. Of these 34 patients, none experienced anaphylactic/hypersensitivity reactions that were clearly related to the anti-therapeutic antibodies (ATA). The presence of pertuzumab in patient serum at the levels expected at the time of ATA sampling can interfere with the ability of this assay to detect anti-pertuzumab antibodies. In addition, the assay may be detecting antibodies to trastuzumab. As a result, data may not accurately reflect the true incidence of anti-pertuzumab antibody development.Immunogenicity data are highly dependent on the sensitivity and specificity of the test methods used. Additionally, the observed incidence of a positive result in a test method may be influenced by several factors, including sample handling, timing of sample collection, drug interference, concomitant medication, and the underlying disease. For these reasons, comparison of the incidence of antibodies to PERJETA with the incidence of antibodies to other products may be misleading.7 DRUG INTERACTIONS No drug-drug interactions were observed between pertuzumab and trastuzumab, or between pertuzumab and docetaxel.8 USE IN SPECIFIC POPULATIONS8.1 PregnancyPregnancy Category DRisk SummaryThere are no adequate and well-controlled studies of PERJETA in pregnant women. Based on findings in animal studies, PERJETA can cause fetal harm when administered to a pregnant woman. The effects of PERJETA are likely to be present during all trimesters of pregnancy. Pertuzumab administered to pregnant cynomolgus monkeys resulted in oligohydramnios, delayed fetal kidney development, and embryo-fetal deaths at clinically relevant exposures of 2.5 to
20-fold greater than the recommended human dose, based on Cmax. If PERJETA is administered during pregnancy, or if a patient becomes pregnant while receiving PERJETA, the patient should be apprised of the potential hazard to the fetus.If PERJETA is administered during pregnancy or if a patient becomes pregnant while receiving PERJETA, immediately report exposure to the Genentech Adverse Event Line at 1-888-835-2555. Encourage women who may be exposed during pregnancy to enroll in the MotHER Pregnancy Registry by contacting 1-800-690-6720 [see Patient Counseling Information (17)].Animal DataReproductive toxicology studies have been conducted in cynomolgus monkeys. Pregnant monkeys were treated on Gestational Day (GD)19 with loading doses of 30 to 150 mg/kg pertuzumab, followed by bi-weekly doses of 10 to 100 mg/kg. These dose levels resulted in clinically relevant exposures of 2.5 to 20-fold greater than the recommended human dose, based on Cmax. Intravenous administration of pertuzumab from GD19 through GD50 (period of organogenesis) was embryotoxic, with dose-dependent increases in embryo-fetal death between GD25 to GD70. The incidences of embryo-fetal loss were 33, 50, and 85% for dams treated with bi-weekly pertuzumab doses of 10, 30, and 100 mg/kg, respectively (2.5 to 20-fold greater than the recommended human dose, based on Cmax). At Caesarean section on GD100, oligohydramnios, decreased relative lung and kidney weights, and microscopic evidence of renal hypoplasia consistent with delayed renal development were identified in all pertuzumab dose groups. Pertuzumab exposure was reported in offspring from all treated groups, at levels of 29% to 40% of maternal serum levels at GD100.8.3 Nursing Mothers It is not known whether PERJETA is excreted in human milk, but human IgG is excreted in human milk. Because many drugs are secreted in human milk and because of the potential for serious adverse reactions in nursing infants from PERJETA, a decision should be made whether to discontinue nursing, or discontinue drug, taking into account the elimination half-life of PERJETA and the importance of the drug to the mother [See Warnings and Precautions (5.1), Clinical Pharmacology (12.3)].8.4 Pediatric Use The safety and effectiveness of PERJETA have not been established in pediatric patients.8.5 Geriatric Use Of 402 patients who received PERJETA in Study 1, 60 patients (15%) were ≥ 65 years of age and 5 patients (1%) were ≥ 75 years of age. No overall differences in efficacy and safety
of PERJETA were observed between these patients and younger patients.Based on a population pharmacokinetic analysis, no significant difference was observed in the pharmacokinetics of pertuzumab between patients < 65 years (n=306) and patients ≥ 65 years (n=175). 8.6 Females of Reproductive Potential PERJETA can cause embryo-fetal harm when administered during pregnancy. Counsel patients regarding pregnancy prevention and planning. Advise females of reproductive potential to use effective contraception while receiving PERJETA and for 6 months following the last dose of PERJETA.If PERJETA is administered during pregnancy or if a patient becomes pregnant while receiving PERJETA, immediately report exposure to the Genentech Adverse Event Line at 1-888-835-2555. Encourage women who may be exposed during pregnancy to enroll in the MotHER Pregnancy Registry by contacting 1-800-690-6720 [see Patient Counseling Information (17)].8.7 Renal Impairment Dose adjustments of PERJETA are not needed in patients with mild (creatinine clearance [CLcr] 60 to 90 mL/min) or moderate (CLcr 30 to 60 mL/min) renal impairment. No dose adjustment can be recommended for patients with severe renal impairment (CLcr less than 30 mL/min) because of the limited pharmacokinetic data available [see Clinical Pharmacology (12.3)].8.8 Hepatic Impairment No clinical studies have been conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of pertuzumab.
10 OVERDOSAGE No drug overdoses have been reported with PERJETA to date.
PERJETA® (pertuzumab)
Manufactured by: Genentech, Inc. PERJETA is a registeredA Member of the Roche Group trademark of Genentech, 1 DNA Way Inc.South San Francisco, CA 09/13 PER0002094600 94080-4990 © 2013 Genentech, Inc.U.S. License No. 1048 10139000

PAGE 70 The ASCO Post | MARCH 25, 2015
Book Review
Wrestling With the Challenges of Breast CancerBy Ronald Piana
Over the past decade or so, the on-cology community has increased
its understanding and recognition about issues and challenges faced by our grow-ing survivorship community. A universal challenge among survivors is the sudden recognition of mortality that comes with a diagnosis of cancer. From that moment on, a cancer patient moves into survivor-ship and views life through a lens that looks at the possibility of recurrence. Many cancer patients describe survivor-ship as a subconscious anticipation, wait-ing for the next shoe to drop.
Different disease sites also present with cancer-specific challenges. Breast cancer survivors wrestle with such is-sues as body image and sexual identity as they negotiate their way through life’s ups and downs, which are poignantly captured in the new book by Geralyn Lucas, Then Came Life: Living With Courage, Spirit, and Gratitude After Breast Cancer.
Ms. Lucas was only 27 years old when she was diagnosed with aggres-sive breast cancer. “Because of my age and the type of cancer, the prognosis wasn’t great: They expected me to have a recurrence within 2 years, and my fu-ture recurrence would more than likely be, as they said, treatable, not curable,” writes Ms. Lucas.
An Important RealityNow at age 45, she seemingly has a
lot to celebrate, first and foremost be-ing a happily married, 16-year breast cancer survivor with a great career as
a television producer and two terrific kids. However, early on in the book, she poignantly summed up her private struggle: “For 16 years, I have been walking that tightrope with cancer on
one side and life on the other.” This metaphor speaks to an impor-
tant reality that not just breast cancer pa-tients but also all cancer patients share. Ms. Lucas voices the identity quandary
ing through economic crisis, and neither have sustainable archetypes.”
To address this medical education cri-sis, Dr. Topol coined the term “MOOM,” for massive open online medicine. In a subchapter titled The Cancer MOOM, he writes, “Several cancer MOOMs are taking hold…. The American Society of Clinical Oncology is supporting such initiatives, and its CEO, Allen S. Lichter, MD, FASCO, said, ‘There’s a treasure trove of information inside case histories if we simply bring them together.’ They are, in a project called CancerLinQ.”
Once again, Dr. Topol has delivered a highly informative, futuristic book that is not only a pleasure to read but offers real-life and actionable advice on how we can improve our health-care system by empowering patients to take control of their own bodies. Moreover, Dr. Topol has no ax to grind; he’s simply a doctor who wants the best for his patients. This is a must-read for all doctors and those thinking of joining the profession. n
Medicine Turned Upside Downcontinued from page 69
www.bostonbiomedical.com
Malignancies, like normal adult tissue, have been shown to contain a subset
of cells that have the capacity to both self-renew and produce more differentiated progeny. Pre-clinical research suggests that these “cancer stem cells”, while a minority of the total cancer cell population, are part of a hierarchical structure within the tumor mass that retains the highest malignant potential.1,2 Cancer stem cells are also highly resistant to conventional chemotherapeutic drugs.3
Cancer stem cells may originate from either normal stem cells as a result of mutation or from daughter cells which are a progeny of stem cells that have acquired the ability to self-renew as a result of genetic and/or epigenetic changes. Cells that reproduce themselves are more likely
to live long enough to accumulate the mutations that lead to cancer.4 It has also been demonstrated that non-stem cancer cells can acquire stemness properties to become cancer stem cells.5,6
Our understanding of the role of cancer stem cells in the natural history of cancer is evolving. Cancer stem cells may not only lead to the development of the primary tumor, but migrate to distant sites and cause metastasis.7 Cancer stem cells not eradicated during chemotherapy may also lead to regrowth or recurrence.
Cancer stem cells have been positively identified and isolated from a variety of human cancers, including hematological malignancies as well as solid tumors.8 Most chemotherapeutics target actively
proliferating cells, resulting in bulk tumor shrinkage; however these agents are not as effective at killing relatively slowly proliferating cancer stem cells. Moreover, it has been shown that conventional chemotherapy and radiation therapy can induce stemness properties in non-stem cancer cells.9,10
Cancer stem cells at primary and metastatic sites can be activated by signals from the tumor microenvironment.11 Targeting these signaling pathways may disrupt aberrant signaling in cancer stem cells while reducing the toxicity to normal tissues associated with chemotherapy.12
1. Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522-526. 2. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730-737. 3. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275-285. 4. Al-Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev. 2004;14(1):43-47. 5. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15(9):1010-1012. 6. Gupta PB, Fillmore CM, Jiang G, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146(4):633-644. 7. Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355(12):1253-1261. 8. Korkaya H, Wicha MS. Selective targeting of cancer stem cells: a new concept in cancer therapeutics. BioDrugs. 2007;21(5):299-310. 9. Hu X, Ghisolfi L, Keates AC, et al. Induction of cancer cell stemness by chemotherapy. Cell Cycle. 2012;11(14):2691-2698. 10. Ghisolfi L, Keates AC, Hu X, Lee DK, Li CJ. Ionizing radiation induces stemness in cancer cells. PLoS One. 2012;7(8):e43628. 11. Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea – a paradigm shift. Cancer Res. 2006;66(4):1883-1890. 12. Boman BM, Huang E. Human colon cancer stem cells: a new paradigm in gastrointestinal oncology. J Clin Oncol. 2008;26(17):2828-2838.
Boston Biomedical’s mission is to develop the next generation of cancer therapeutics by creating drugs designed to target cancer stem cell pathways.
Understanding Cancer Stem CellsCancer Stem Cells Signal Pathways Regrowth Metastasis
EDU-NPS-0009 12/2014 ©2014 Boston Biomedical
ADVERTORIAL
06111A_R3_BBI_Advertorial_Island.indd 1 12/30/14 3:01 PM

ASCOPost.com | MARCH 25, 2015 PAGE 71
Book Review
BookmarkTitle: Then Came Life: Living With Courage, Spirit, and Gratitude After Breast Cancer
Author: Geralyn Lucas
Publisher: Gotham Books
Publication date: October 2, 2014
Price: $19.89; hardcover, 240 pages
continued on page 72
that cancer patients face, separating themselves from the disease. And even after 16 years of survivorship, she still walked the tightrope. She further drove home this central issue of survivorship as she stared at herself in the mirror on the anniversary of her mastectomy.
Ms. Lucas writes, “My huge faded-red mastectomy scar seems just as fresh and
tender now as it was at first. All these years later, I walk funny, holding one side of my body back protectively so no one can brush against me. My scar is a souvenir from that trauma. It is the badge I wear every day.”
A Sequel of SortsThen Came Life is a sequel of sorts
to her first memoir published in 2005,
Why I Wore Lipstick to My Mastectomy, which chronicles her experience as a 27-year-old, journalism school grad, having just landed her dream job as a reporter for the show 20/20, when she was diagnosed with breast cancer. But Ms. Lucas refused to capitulate to cancer and fall into the 24/7 gloom of victimhood. She penned Why I Wore
Lipstick to My Mastectomy, because in her words, “I really thought I was going to die. All the films, all the books, were just so gloomy and traumatic. I wanted to show women that they are so much more than their breasts, that there is life at the other side of the pain.”
So, whereas her first memoir dealt with the shock of a breast cancer diag-nosis, with all its rollercoaster, life-alter-ing effects, Then Came Life has a slower, less volatile pace, dealing more with middle-aged angst, which is magnified by the looming shadow of recurrence that all survivors live with. Ms. Lucas mentions this on just about every page, but it never feels redundant; it simply reinforces the reality of survivorship.
Compelling Details Mixed With Diary Entries
The fact that the arc of these two “cancer” memoirs spans 16 years is im-portant to readers of The ASCO Post, speaking directly to the remarkable ad-vances made in oncology. There are also some frank and well-informed talks with her oncologists, which offer in-sight into the territory of “difficult con-versations” between doctor and patient.
www.bostonbiomedical.com
Malignancies, like normal adult tissue, have been shown to contain a subset
of cells that have the capacity to both self-renew and produce more differentiated progeny. Pre-clinical research suggests that these “cancer stem cells”, while a minority of the total cancer cell population, are part of a hierarchical structure within the tumor mass that retains the highest malignant potential.1,2 Cancer stem cells are also highly resistant to conventional chemotherapeutic drugs.3
Cancer stem cells may originate from either normal stem cells as a result of mutation or from daughter cells which are a progeny of stem cells that have acquired the ability to self-renew as a result of genetic and/or epigenetic changes. Cells that reproduce themselves are more likely
to live long enough to accumulate the mutations that lead to cancer.4 It has also been demonstrated that non-stem cancer cells can acquire stemness properties to become cancer stem cells.5,6
Our understanding of the role of cancer stem cells in the natural history of cancer is evolving. Cancer stem cells may not only lead to the development of the primary tumor, but migrate to distant sites and cause metastasis.7 Cancer stem cells not eradicated during chemotherapy may also lead to regrowth or recurrence.
Cancer stem cells have been positively identified and isolated from a variety of human cancers, including hematological malignancies as well as solid tumors.8 Most chemotherapeutics target actively
proliferating cells, resulting in bulk tumor shrinkage; however these agents are not as effective at killing relatively slowly proliferating cancer stem cells. Moreover, it has been shown that conventional chemotherapy and radiation therapy can induce stemness properties in non-stem cancer cells.9,10
Cancer stem cells at primary and metastatic sites can be activated by signals from the tumor microenvironment.11 Targeting these signaling pathways may disrupt aberrant signaling in cancer stem cells while reducing the toxicity to normal tissues associated with chemotherapy.12
1. Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522-526. 2. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730-737. 3. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275-285. 4. Al-Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev. 2004;14(1):43-47. 5. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15(9):1010-1012. 6. Gupta PB, Fillmore CM, Jiang G, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146(4):633-644. 7. Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355(12):1253-1261. 8. Korkaya H, Wicha MS. Selective targeting of cancer stem cells: a new concept in cancer therapeutics. BioDrugs. 2007;21(5):299-310. 9. Hu X, Ghisolfi L, Keates AC, et al. Induction of cancer cell stemness by chemotherapy. Cell Cycle. 2012;11(14):2691-2698. 10. Ghisolfi L, Keates AC, Hu X, Lee DK, Li CJ. Ionizing radiation induces stemness in cancer cells. PLoS One. 2012;7(8):e43628. 11. Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea – a paradigm shift. Cancer Res. 2006;66(4):1883-1890. 12. Boman BM, Huang E. Human colon cancer stem cells: a new paradigm in gastrointestinal oncology. J Clin Oncol. 2008;26(17):2828-2838.
Boston Biomedical’s mission is to develop the next generation of cancer therapeutics by creating drugs designed to target cancer stem cell pathways.
Understanding Cancer Stem CellsCancer Stem Cells Signal Pathways Regrowth Metastasis
EDU-NPS-0009 12/2014 ©2014 Boston Biomedical
ADVERTORIAL
06111A_R3_BBI_Advertorial_Island.indd 1 12/30/14 3:01 PM

PAGE 72 The ASCO Post | MARCH 25, 2015
Book Review
Challenges of Breast Cancercontinued from page 71
For 16 years, I have been walking that tightrope with cancer on one side and life on the other.
—Geralyn Lucas
Then there’s a sudden tragedy right after Ms. Lucas takes the reader through the emotional ups and downs of her 16th “cancerversary.” Her 39-year-old cousin, Hallie, is diagnosed with stage IV breast cancer. Ms. Lucas nails the shock of diagnosis with pithy, well-in-formed experience. And she is a good cousin, full of support, giving a detailed description, replete with her own diag-nosis flashbacks, of the mind-numbing cancer-diagnosis experience.
However, that’s where, at least for a while, the interesting content for the readers of The ASCO Post ends. This book, compelling as it is at times for Ms.
Lucas’ sheer energy, wit, and moxy, has a tendency to read too much like a per-sonal diary, filling the pages with things only she and her tight circle of friends would appreciate.
Powerful Final ChaptersSuch grievances aside, the book
closes its final chapters with Ms. Lucas serving as one of the caregiv-ers for her cousin Hallie, who after a brave fight, succumbs to breast can-cer. Ms. Lucas, to her credit, treats this section, the book’s most power-ful and best written, with the care and decency that dying cancer patients deserve. She writes, “For several days we watch over Hallie in the hospital,
until we see the last pulsing vein in her neck…. The finality of it is stun-ning. My aunt Lydia crawls into bed with her daughter and hugs her baby for the last time…. She doesn’t want to let her go.”
No one wants to let go of a loved one, but sometimes we must, as Ms. Lucas ably described in her some-times soapy, but otherwise sturdy read, especially for women in or near the breast cancer experience. n
Fred Hutchinson Health Economist Gary Lyman, MD, MPH, Editor of Recently Released Oncology Handbook
Oncologist and health economist Gary Lyman, MD, MPH, Co-
director of the Hutchinson Institute of Cancer Outcomes Research at Fred Hutchinson Cancer Research Center, is the Editor of the second edition of the Oxford American Handbook of On-cology, published February 25, 2015, by Oxford University Press.
The textbook is designed to be an es-sential reference for medical students,
residents, and clinical oncologists seeking a current resource on state-of-the-art can-cer care. The reference aims to help guide clinical decision-making, from treatment choices to best-practice guidelines.
“This represents an updated and expanded edition of the handbook, which continues in the popular bul-leted style that the Oxford handbooks have made popular across a range of medical disciplines,” said Dr. Lyman, a
member of the Public Health Sciences Division at Fred Hutchinson.
The 864-page textbook covers can-cer biology, genetics, prevention and screening, treatment complications, and supportive care. The handbook also provides background on mo-lecular cancer biology, etiology, and epidemiology.
More than 50 oncology professionals contributed to the reference guide. n
Gary Lyman, MD, MPH
Download The ASCO Post iPad App
FREE from iTunes today!

ASCOPost.com | MARCH 25, 2015 PAGE 73
Clinical Trials Resource Guide
Clinical Trials Actively Recruiting Patients With Esophageal Cancer Compiled by Liz Janetschek
OBSERVATIONAL
Study Type: ObservationalStudy Title: Interrogation of Wnt,
Notch and Hedgehog Activity in Che-mo-naive Tumors Collected During the Staging Ultrasound of Patients Diag-nosed With Esophageal Cancer
Study Sponsor and Collaborators: University of Miami
Purpose: To investigate the elucidation of signaling mechanisms, control of cellular processes and discovery of small molecules that selectively target Wnt, Shh, and Notch signaling pathways that are fundamental to cancer stem or progenitor cells (CSCs).
Ages Eligible: 18 years and olderGenders Eligible: BothAccepts Healthy Volunteers: NoPrimary Outcome Measures: Per-
centage of samples with identifiable Notch, Wnt and Hedghog pathways in the chemo naive esophageal tumor as well as in cancer stem cell populations (CSC) (time frame: 24 months)
Principal Investigator: Anthony Capobianco, MD, University of Mi-ami; 305-243-6308, tcapobianco@med .miami.edu.
For More Information: Visit Clini-calTrials.gov and refer to this study by its identifier: NCT02221245.
Study Type: ObservationalStudy Title: A Study of Patient-
reported Outcomes in Patients With Lung or Esophageal Cancer
Study Sponsor and Collabora-tors: University of Virginia; Alliance for Clinical Trials in Oncology; Patient Centered Outcome Research Institute
Purpose: To determine if patient re-ported outcomes (using the PROMIS survey) will vary according to the pres-ence of recurrent or metastatic lung or esophageal cancer.
Ages Eligible: 18 years and olderGenders Eligible: BothAccepts Healthy Volunteers: No
Primary Outcome Measures: Pa-tient Reported Outcomes Measurement Information System (PROMIS) Scores (time frame: up to 30 day preoperatively for patients undergoing lung/esophageal
cancer surgery through a maximum of 5 years after 1 survey is completed)
Principal Investigator: Benjamin Kozower, MD, University of Virginia. Contact: Sandra Burks, RN, 434-243-
0315, [email protected] More Information: Visit Clini-
calTrials.gov and refer to this study by its identifier: NCT02239328.
The information contained in this Clinical Trials Resource
Guide includes actively recruiting observational, interventional, phase I, phase II, and phase III clinical studies for patients with newly di-agnosed or recurrent esophageal cancer. All of the studies are listed on the National Institutes of Health website at ClinicalTrials.gov.
continued on page 74
SAFETY:7”
SAFETY:10”
TRIM:7.875”
TRIM:10.5”
BLEED:8.375”
BLEED:11.125”
Patients (N=286)• Advanced, recurrent, or metastatic ALK-positive NSCLC
Alectinib1
CrizotinibRan
dom
ize
1:1
THIS ADVERTISEMENT PREPARED BY AREA23 Sig Date Time Time Spent Spell Check
Mac Op
QC
Editing
Copywriting
Art Director
Art Buying
Account
Traffic
Production
DQC
Client: GenentechProduct: Alectinib Job#: 100173873Colors: 4C Sizes: Bleed: 8.375"” W x 11.125" H
Sm Trim: 7.875" W x 10.5" H Live: 7" W x 10" H Gutter: N/A
Publications:Journal of Thoracic Oncology:
Oncology Times:
The ASCO Post:
Journal of Clinical Oncology:
HEM/ONC Today:
The New England Journal of Medicine:
A Size
A Randomized, Phase III Study Comparing Alectinib With Crizotinib in Treatment-Naïve Anaplastic Lymphoma Kinase (ALK)-Positive Advanced Non-Small Cell Lung Cancer (NSCLC) Patients
Now enrolling for alectinib
NCT02075840BO28984
1. Product under investigation has not been approved for use outside of the clinical trial setting. This information is presented only for the purpose of providing an overview of the clinical trial and should not be construed as a recommendation for the use of any product for unapproved purposes.
2. For more information on trial inclusion and exclusion criteria, visit clinicaltrials.gov.
For more information, please call the Genentech Trial Information Support Line at 1-888-662-6728 (US only), visit clinicaltrials.gov, or e-mail [email protected].
Primary Endpoint: • Progression-free survival (PFS), investigator-assessed,
by RECIST 1.1
Key Inclusion Criteria2: • Advanced, recurrent, or metastatic ALK-positive NSCLC
• Life expectancy ≥12 weeks
• ECOG performance status of 0-2
• No prior systemic therapy for advanced, recurrent, or metastatic disease
• Measurable disease by RECIST 1.1
Secondary Endpoints: • Objective response rate, investigator-assessed,
using RECIST 1.1
• Time to CNS progression, IRC-assessed, using RECIST 1.1
• PFS, IRC-assessed, using RECIST 1.1
• Duration of response
• Overall survival
• Safety: incidence of adverse events
• AUC of alectinib
• Patient-reported outcomes
Key Exclusion Criteria2: • Prior malignancy in past 3 years
• Any ≥ grade 3 toxicity (NCI CTCAE 4.0) from a prior therapy
• Baseline QTc >470 ms or symptomatic bradycardia <45 beats per minute
• Concomitant strong cytochrome P4503A inhibitors/inducers or QT-prolonging medications
© 2014 Genentech USA, Inc. All rights reserved. BIO0002445101 Printed in USA.
100173873_GEN_Phase3_JRN_ad_FR.indd 1 8/5/14 11:48 AM

PAGE 74 The ASCO Post | MARCH 25, 2015
Clinical Trials Resource Guide
Study Type: ObservationalStudy Title: Prospective Collection
of Health Information and Biospeci-mens in Esophageal Cancer
Study Sponsor and Collabora-tors: Washington University School of Medicine
Purpose: To create an esophageal cancer biospecimen repository that will collect, annotate, store, and distribute human esophageal cancer biospecimens.
Ages Eligible: 18 years and olderGenders Eligible: BothAccepts Healthy Volunteers: NoPrimary Outcome Measures: Pro-
spective Database (time frame: 5 years)Principal Investigator: Varun Puri,
MD, Washington University School of Medicine. Contact: Jasmine A. Lewis, BSBA; 314-362-4611; [email protected].
For More Information: Visit Clini-calTrials.gov and refer to this study by its identifier: NTC01780961.
PHASE I
Study Type: Phase I/interventional/nonrandomized/single-group assignment
Study Title: Epigenetically-Modi-fied Autologous Tumor Cell Vaccines With ISCOMATRIX(TM) Adjuvant and Oral Celecoxib in Patients Under-going Resection of Lung and Esopha-geal Cancers, Thymic Neoplasms, Tho-racic Sarcomas, and Malignant Pleural Mesotheliomas
Study Sponsor and Collaborators: National Cancer Institute
Purpose: To assess the safety and ef-fectiveness of tumor cell vaccines given with ISCOMATRIX and celecoxib in the treatment of lung and esophagus cancers.
Ages Eligible: 18 to 75 yearsGenders Eligible: BothAccepts Healthy Volunteers: NoPrimary Outcome Measures: Tab-
ulation of patient toxicities and their grades (time frame: 30 days after last vaccine up to 25 months)
Principal Investigator: David S. Schrump, MD, National Cancer Insti-tute; 301-496-2128, [email protected].
For More Information: Visit Clini-calTrials.gov and refer to this study by its identifier: NCT01258868.
Study Type: Phase I/interventionalStudy Title: Phase I Dose Esca-
lation of Neoadjuvant Proton Beam Radiotherapy With Concurrent Che-motherapy in Locally Advanced Esoph-ageal Cancer
Study Sponsor and Collaborators: Abramson Cancer Center of the Uni-versity of Pennsylvania
Purpose: To investigate the safety and efficacy of treating patients with esophageal cancer with concurrent pre-operative proton therapy, along with carboplatin and paclitaxel.
Ages Eligible: 18 years and olderGenders Eligible: BothAccepts Healthy Volunteers: NoPrimary Outcome Measures: Num-
ber of adverse events (time frame: 3 years)Principal Investigator: John Plasta-
ras, MD, Abramson Cancer Center of the University of Pennsylvania; 855-216-0098, [email protected].
For More Information: Visit Clini-calTrials.gov and refer to this study by its identifier: NCT02213497.
Study Type: Phase I/interventionalStudy Title: Allogeneic Tumor Cell
Vaccine With Metronomic Oral Cyclo-phosphamide and Celecoxib as Adjuvant Therapy for Lung and Esophageal Cancers, Thymic Neoplasms, Thoracic Sarcomas, and Malignant Pleural Mesotheliomas
Study Sponsor and Collaborators: National Cancer Institute
Purpose: To evaluate the safety and effectiveness of tumor cell vaccines in combination with cyclophosphamide and celecoxib in patients with cancers involving the chest.
Ages Eligible: 18 years and olderGenders Eligible: BothAccepts Healthy Volunteers: NoPrimary Outcome Measures:
Tabulation of toxicity type and grade (time frame: 2 years)
Principal Investigator: David S. Schrump, MD, National Cancer Insti-tute; 301-496-2128, [email protected].
For More Information: Visit Clini-calTrials.gov and refer to this study by its identifier: NCT01143545.
PHASE II
Study Type: Phase II/interventional/single-group assignment
Study Title: A Phase 2 Study of Pacli-taxel With Cisplatin Versus Fluoropyrim-idine With a Platinum Agent for Neoad-juvant Therapy in Operable Esophageal Cancer Based on CHFR Methylation Status in Diagnostic Biopsies
Study Sponsor and Collaborators: Sidney Kimmel Comprehensive Can-cer Center
Purpose: To determine the rate of pathological complete response when the inclusion of paclitaxel in neoadju-
vant therapy is based on the presence or absence of CHFR methylation in diag-nostic biopsy specimens.
Ages Eligible: 18 to 75 yearsGenders Eligible: BothAccepts Healthy Volunteers: NoPrimary Outcome Measures: Pro-
spective Database (time frame: 5 years)Principal Investigator: Ronan Kel-
ly, MD, Johns Hopkins University; 443-287-0005, rkelly25@jhmi@edu.
For More Information: Visit Clini-calTrials.gov and refer to this study by its identifier: NCT02239328.
Study Type: Phase II/interventional/randomized
Study Title: Randomized Phase II Trial of PET Scan-Directed Combined Modality Therapy in Esophageal Cancer
Study Sponsor and Collaborators: Cancer and Leukemia Group B; Na-tional Cancer Institute
Purpose: To study PET scan im-aging in assessing response in patients with esophageal cancer receiving com-bination chemotherapy.
Ages Eligible: 18 years and olderGenders Eligible: BothAccepts Healthy Volunteers: NoPrimary Outcome Measures: pCR
rate of PET/CT non-responders within each induction treatment group
Principal Investigator: Karyn A. Goodman, MD, Memorial Sloan Ket-tering Cancer Center. Contact: David H. Ilson, MD, PhD, 212-639-8895.
For More Information: Visit Clini-calTrials.gov and refer to this study by its identifier: NCT01333033.
Study Type: Phase II/interventional/single-group assignment
Study Title: A Phase II, Multi-Cen-ter Study of Interventional Spray Cryo-therapy for Early-Stage Esophageal Cancer (ICE-CANCER)
Study Sponsor and Collaborators: University of Maryland
Purpose: To evaluate the safety and efficacy of endoscopic spray cryo-therapy using the CSA Medical, Inc. truFreeze System for patients with previously untreated early-stage cancer (T1a, N0, M0) that are ineligible or refuse conventional therapy, including surgery, chemotherapy, radiation thera-py, and endoscopic resection.
Ages Eligible: 18 years and olderGenders Eligible: BothAccepts Healthy Volunteers: NoPrimary Outcome Measures: De-
termining the response rate to therapy (time frame: 5 years)
Principal Investigator: Bruce D. Greenwald, MD, University of Mary-
land; 410-328-8731, [email protected].
For More Information: Visit Clini-calTrials.gov and refer to this study by its identifier: NCT01868139.
PHASE III
Study Type: Phase III/interven-tional/randomized
Study Title: A Phase III Trial Evalu-ating the Addition of Trastuzumab to Trimodality Treatment of Her2-Over-expressing Esophageal Adenocarcinoma
Study Sponsor and Collaborators: National Cancer Institute
Purpose: To study how well radia-tion therapy, paclitaxel, and carbopla-tin with or without trastuzumab work in treating patients with esophageal cancer.
Ages Eligible: 18 years and olderGenders Eligible: BothAccepts Healthy Volunteers: NoPrimary Outcome Measures: Dis-
ease-free survival (time frame: from the date of randomization to the date of first failure or last follow-up, assessed up to 8 years)
Principal Investigator: Howard Safran, MD, NRG Oncology; 401-793-2224, [email protected]
For More Information: Visit Clini-calTrials.gov and refer to this study by its identifier: NCT01196390.
Study Type: Phase III/interventional/randomized
Study Title: Phase III Randomized Trial of Proton Beam Therapy Versus Intensity-Modulated Radiation Therapy for the Treatment of Esophageal Cancer
Study Sponsor and Collaborators: MD Anderson Cancer Center
Purpose: To study how safe and effective proton-beam therapy may be in comparison to intensity modulated radiation therapy in combination with chemotherapy in patients with esopha-geal cancer.
Ages Eligible: 18 years and olderGenders Eligible: BothAccepts Healthy Volunteers: NoPrimary Outcome Measures: Pro-
gression-free survival (time frame: 6 weeks after radiation therapy)
Principal Investigator: Steven H. Lin, MD, PhD, MD Anderson Cancer Center; 713-563-2300.
For More Information: Visit Clini-calTrials.gov and refer to this study by its identifier: NCT01512589. n
Editor’s Note: The clinical trials presented here do not represent all the trials listed on ClinicalTrials.gov. For the complete list, go to ClinicalTrials.gov.
Esophageal Cancer Trialscontinued from page 73
TrimSize 10.5”x14”1506US15BR00117-02-01: Publication:
Please visit www.OPDIVO.com for more information.
Announcing an Additional Indication
OPDIVO® and the related logo are trademarks of Bristol-Myers Squibb Company. ©2015 Bristol-Myers Squibb Company. All rights reserved. Printed in USA. 1506US15BR00117-02-01 03/15
NOW THERE’S
Website Call to ActionWebsite Call to Action
14538003_0011702_Ancmnt_JrnlAd_10_5x14_v1_m.indd 1 3/10/15 9:28 AM

TrimSize 10.5”x14”1506US15BR00117-02-01: Publication:
Please visit www.OPDIVO.com for more information.
Announcing an Additional Indication
OPDIVO® and the related logo are trademarks of Bristol-Myers Squibb Company. ©2015 Bristol-Myers Squibb Company. All rights reserved. Printed in USA. 1506US15BR00117-02-01 03/15
NOW THERE’S
Website Call to ActionWebsite Call to Action
14538003_0011702_Ancmnt_JrnlAd_10_5x14_v1_m.indd 1 3/10/15 9:28 AM

PAGE 76 The ASCO Post | MARCH 25, 2015
Integrative Oncology
Herbal Supplements: Increasing ProblemsBy Jyothi Gubili, MS, K. Simon Yeung, PharmD, MBA, LAc, and Barrie R. Cassileth, MS, PhD
In Don Quixote, the 1605 Spanish liter-ary masterpiece by Miguel Cervantes,
“Balsam of Fierabras” is mentioned often as a therapeutic panacea. It calls for mix-ing rosemary, wine, oil, and salt. As the story goes, the knight relied heavily on this herbal preparation to relieve him of pain from the many beatings he was sub-jected to following his exploits.1
Since ancient times, herbs have been used as medicine in cultures around the world. Today, herbalists continue to pre-scribe herbal formulas to treat a range of ailments. The 1990s saw a major change in the way herbal medicines were con-sumed, as herbal remedies were marketed as supplements following the passage of the Dietary Supplement Health Educa-tion Act.
A Question of QualityThe major downside of the Dietary
Supplement Health Education Act is that dietary supplements are not subjected to the same safety and effectiveness re-quirements as drugs are, by the U.S. Food and Drug Administration (FDA). With limited oversight, the quality of available products was found to vary substantially across manufacturers. Some contain too
much or too little of the labeled ingre-dient. Others are manufactured under poorly controlled conditions. Although companies are required to follow good manufacturing practices, lack of strict regulation has resulted in the proliferation of unscrupulous practices, including false health claims.
Examples include reports on adulter-ated herbal products that have surfaced in
the past 2 years. A 2013 study of 37 saw palmetto–containing supplements found that 6% contained species that cannot le-gally be sold as dietary supplements in the United States. The identity of an addition-al 9% of supplements remained inconclu-sive.2 A 2013 Canadian investigation of 44 herbal products sold by 12 companies revealed that the majority were of poor quality, including product substitution, contamination, and use of fillers.3
A recent case of questionable supple-ments was exposed by the New York At-torney General, which led to the issuance of cease-and-desist letters to four major retailers in the New York area, preventing them from selling those products.4 How-ever, the validity of the DNA barcoding technique used in the investigations is be-ing debated.5
Steps to Ensure SafetyThe popular belief that herbs are safer
than synthetic pharmaceuticals continues unabated. According to the most recent National Health Survey conducted in 2012, the most commonly used comple-mentary approach was the use of natu-ral products.6 Sales of herbal products reached $6 billion in 2013.7
What steps can consumers take to pur-chase products with some certainty that what’s on the label is indeed inside the bottle? A good way to ensure the quality of supplements is to purchase products with labels indicating they have been tested by either the U.S. Pharmacopoeia (USP) Convention Dietary Supplement Verification Program or ConsumerLab.com. These are two major independent
groups that routinely evaluate products submitted by manufacturers to ensure that they meet the minimum quality stan-dards.
A USP-verified product means it con-tains the listed ingredients at the strength indicated and that it has been screened for harmful contaminants. ConsumerLab.com is a company that collects several brands of a product and independently tests, reviews, compares, and rates them. The company’s process also involves as-certaining that the product contains the listed ingredients and is not contaminated or adulterated. In addition to labels on dietary supplement packaging, Consum-erLab.com provides detailed information about its findings in its paid-subscription offerings. However, neither USP nor ConsumerLab.com evaluates the efficacy of the products or product-prescription drug interactions.
Stricter Regulation NeededIn 2010, the FDA established a system
for regulators to track complaints about dietary supplements and to prevent prod-ucts in question from being sold. Given the estimated 65,000 products currently available,3 such checks and balances will barely impact the status quo.
There is clearly an urgent need for stricter regulation of the dietary supple-ment industry. Had the rosemary used to concoct the Balsam of Fierabras been inauthentic, Don Quixote would not have been reinvigorated to continue his whimsical quests, depriving us of a great story. n
Disclosure: Ms. Gubili, Dr. Yeung, and Dr. Cassileth reported no potential conflicts of interest.
References1. López-Muñoz F, Alamo C, García-Gar-
cía P: Psychotropic drugs in the Cervantine texts. J R Soc Med 101:226-234, 2008.
2. Little DP, Jeanson ML: DNA barcode authentication of saw palmetto herbal dietary supplements. Sci Rep 3:3518, 2013.
3. Newmaster SG, Grguric M, Shan-mughanandhan D, et al: DNA barcoding detects contamination and substitution in North American herbal products. BMC Med 11:222, 2013.
4. Press Release: A.G. Schneiderman asks major retailers to halt sales of certain herbal supplements as DNA tests fail to detect plant materials listed on majority of products tested. Press Release. Available at http://www.ag.ny.gov/press-release/ag-schneiderman-asks-major-retailers-halt-sales-certain-herbal-sup-plements-dna-tests. Accessed February 18, 2015.
5. Sarma N: DNA testing of herbal supple-ments—Does it work or doesn’t it? Available at http://qualitymatters.usp.org/dna-testing-herbal-supplements-does-it-work-or-doesnt-it. Accessed February 18, 2015.
6. National Center for Complementary and Integrative Health: What complementary and integrative approaches do Americans use? Key findings from the 2012 National Health Interview Survey. Available at https://nccih.nih.gov/research/statistics/NHIS/2012/key-findings. Accessed February 18, 2015.
7. American Botanical Coun-cil: Herbal dietary supplement re-tail sales up 7.9% in 2013. Available at h t t p : / / c m s . h e r b a l g r a m . o r g /press/2014/2013_Herb_Market_Report.html?ts=1411057336&signature=bd00edd66448806455e36de3bc333928. Accessed Febru-ary 18, 2015.
GUEST EDITOR
Integrative Oncology is guest edited by Barrie R. Cassileth, MS, PhD, Chief of the Integra-
tive Medicine Service and Laurance S. Rockefeller Chair in Integrative Medicine at Memorial Sloan Kettering Cancer Center, New York.
The Integrative Medicine Service at Memo-rial Sloan Kettering Cancer Center developed and maintains a free website—About Herbs (www.mskcc.org/aboutherbs)—that provides objective
and unbiased information about herbs, vitamins, minerals, and other dietary supplements, and unproved anticancer treatments. Each of the close to 300 and growing number of entries offer health-care professional and patient ver-sions, and entries are regularly updated with the latest research findings.
In addition, the About Herbs app, Memorial Sloan Kettering Cancer Cen-ter’s very first mobile application, can be downloaded at http://itunes.apple.com/us/app/about-herbs/id554267162?mt=8. The app is compatible with iPad, iPhone, and iPod Touch devices.
Barrie R. Cassileth, MS, PhD
The popular belief that herbs are safer than synthetic pharmaceuticals continues unabated. Sales of herbal
products reached $6 billion in 2013.
—Jyothi Gubili, MS, K. Simon Yeung, PharmD, MBA, LAc, and Barrie R. Cassileth, MS, PhD

ASCOPost.com | MARCH 25, 2015 PAGE 77
Journal Spotlight
Study Finds Websites That Market Personalized Cancer Care Services Overemphasize BenefitsBy Jo Cavallo
A recent analysis of 55 Internet web-sites marketing a broad range of tests
and services that promise the ability to per-sonalize cancer treatment has found that the websites often overemphasize their purported benefits and downplay their limitations. In addition, the study results show that the majority of companies that market somatic tests online promote tests
that do not have evidence of clinical utili-ty. The study by Stacy W. Gray, MD, AM, of Dana-Farber Cancer Institute, and col-leagues, is published in the Journal of the National Cancer Institute.1
Study MethodologyThe researchers screened the top 30
websites on Google, Yahoo, and Bing, using 54 search items related to person-alized or genomic cancer care, as well as websites identified through a literature review and an abstraction of exhibitor information from a national oncology conference. A Delphi Panel categorized Personalized Cancer Medicine as stan-dard or nonstandard based on evidence of clinical utility. To capture websites that market cancer-related germline testing, genomic interpretative services, and providers who advertise personal-ized care, the researchers defined Per-sonalized Cancer Medicine as products or services that could be used to tailor, personalize, or individualize care based on genomic or tumor-derived data.
While the majority of websites were sponsored by commercial enti-ties (56%), others were sponsored by academic institutions (20%), private institutions (15%), and individual phy-sicians (2%). Thirty-one percent of the websites offered multiple Personalized Cancer Medicine services; 58% offered somatic analysis, and 20% offered germ-line analysis. In addition, 44% of the sites offered some form of personalized can-cer care, and 15% offered interpretive services. About half of the commercial websites included the cost of testing, which ranged from $99 to $13,000.
Study FindingsThe researchers’ analysis found that
the vast majority (87%) of all websites included benefit information, and just
27% included limitation information. Websites included more information on the benefits of personalized cancer medicine than the limitations of per-
sonalized cancer medicine (P < .001). Compared with noncommercial web-sites, commercial websites were more
Genomics/Genetics
continued on page 78
Stacy W. Gray, MD, AM

PAGE 78 The ASCO Post | MARCH 25, 2015
Mobile Patient-Centered App Tracks Breast Cancer Survivors’ Experiences
Patricia Ganz, MD, Director of Cancer Prevention and Control
Research at the Jonsson Cancer Center of UCLA, and collaborators Apple and Sage Bionetworks, recently announced the launch of “Share the Journey: Mind, Body and Wellness after Breast Cancer,” a patient-centered mobile applica-tion (app) that empowers women to be partners in the research process by tracking their symptoms and successes.
Share the Journey was developed by
UCLA’s Jonsson Comprehensive Can-cer Center, Penn Medicine, Dana-Far-ber Cancer Institute, and Sage Bionet-works, and it is available for download now at the iTunes App Store. The app is an interactive research study that aims to understand why some breast can-cer survivors recover faster than oth-ers, why their symptoms vary over time, and what can be done to improve symptoms.
App Collects and Tracks Symptoms
Share the Journey marries science and technology by using surveys and sensor data on the iPhone to collect and track fatigue, mood and cognitive changes, sleep disturbances, and reduc-tions in exercise.
Share the Journey is one of five new
apps being launched in conjunction with Apple’s ResearchKit, an open-source tool that serves as a streamlined hub for iOS apps that can help speed scientific progress toward cures by am-plifying the patient voice in shaping re-search directions and outcomes.
Share the Journey shifts the center of care, healing, and intervention into the hands of women who have survived breast cancer. Women who have under-gone surgery, radiation, or drug thera-
py to treat breast cancer often experi-ence symptoms that affect their quality of life and impede recovery. The app’s creators say that collecting women’s experiences after breast cancer treat-ment will create a trove of data based on well-validated surveys and measure-ments, which will be continuously im-proved upon based on the participants’ feedback.
One Step Closer to More Personalized Care
“We’re excited to use these new Re-searchKit tools to expand participant recruitment and quickly gather even more data through the simple use of an app. The data it will provide takes us one step closer to developing more personalized care,” said Dr. Ganz, who also is a Professor at the UCLA Field-
ing School of Public Health. “Access to more diverse patient-reported health data will help us learn more about
long-term aftereffects of cancer treat-ments, and provide us with a better un-derstanding of breast cancer patients’ experience.”
Share the Journey is open to women between the ages of 18 and 80 who live in the United States, whether or not they have had breast cancer. Those who have not had breast cancer will contribute important data to the app that will help researchers understand which symptoms may be related to cancer treatment, and which may be part of the normal aging process. The developers also are creating a Span-ish language version of the app, and planning to expand the study to other countries.
“One reason to build these apps and run these studies is to see whether we can turn anecdotes into signals, and by generating signals, find windows for in-tervention,” said Stephen Friend, MD, PhD, President of Sage Bionetworks, and a Principal Investigator for Share the Journey. “We’re most interested in dis-ease variations and the hourly, daily, or weekly ebb and flow of symptoms that are not being tracked and are completely missed by biannual visits to the doctor.”
The platform is based on the concept
that if individuals’ experiences were at the center of the research process, researchers working in virtual teams might be able to get efficient, inexpen-sive, and ubiquitous ways of gathering information using websites, tablets, or an app. This technology will allow Sage and other teams to include patients and other study participants as owners of their own data and equal partners.
“We need to better understand some of the long-term negative treat-ment effects, such as fatigue, that can be associated with the disease control benefits of cancer therapies. What are the biological mechanisms that under-pin those effects, and why some survi-vors are more vulnerable to those ef-fects than others,” Dr. Ganz said. “With Share the Journey, women can tell us when something’s wrong, and the app has the potential to capture valuable information on the patient experience. Our current cancer care system lacks the ability to predict or treat these chronic and enduring symptoms, but Share the Journey can set us on a path toward understanding why some peo-ple recover, and some do not.”
CollaboratorsIn addition to Dr. Ganz, Apple and
Sage were advised in development of Share the Journey by Ann Partridge, MD, MPH, and Judy Garber, MD, MPH, of Dana-Farber Cancer Insti-tute; Kathryn Schmitz, PhD, MPH, at the University of Pennsylvania Perel-man School of Medicine; and Susan Love, MD, MBA, at UCLA and the Dr. Susan Love Research Foundation. n
Disclaimer: Dr. Ganz reported no potential conflicts of interest. Dr. Friend is President of Sage Bionetworks, and a Principal Investigator for Share the Journey.
Survivorship
The app has the potential to capture valuable information on the patient experience. Share the Journey can set us on a path toward understanding why some people recover, and some do not.
—Patricia Ganz, MD
Stephen Friend, MD, PhD
likely to provide information on the benefits of personalized cancer medi-cine (100% vs 67%, P < .001).
In addition, the analysis showed that the majority of companies that market somatic tests online promote tests that do not have evidence of clinical utility. According to the study
abstract, six websites offered chemo-therapy sensitivity testing despite the fact that technology assessments from ASCO show insufficient evidence to recommend the tests.
Currently, claims and other infor-mation posted on websites are not subject to regulation by the U.S. Food and Drug Administration (FDA) or the Federal Trade Commission, al-
though the FDA has said it intends to begin regulating genomic testing more broadly.
“Given the lack of uniform regula-tion over Internet marketing, dispro-portionate claims of benefit, and pro-motion of nonstandard technologies, it is essential that clinicians and pa-tients critically evaluate online prod-ucts,” concluded the researchers. n
Disclosure: The study was supported by grants from the American Cancer Society and the National Institutes of Health. The study authors reported no conflicts of interest.
Reference1. Gray SW, Cronin A, Bair E, et al:
Marketing of personalized cancer care on the Web: An analysis of Internet websites. J Natl Cancer Inst 107(5):djv030, 2015.
Marketing Personalized Carecontinued from page 77
Visit The ASCO Post website at ASCOPost.com

ASCOPost.com | MARCH 25, 2015 PAGE 79
Through the Lens of Oncology History
THE ANTISEPTIC ERA: 1876–1900
Dr. Halsted’s Breast Amputation Specimens, Johns Hopkins Hospital, 1894
In 1894, William Halsted, MD, il-lustrated his paper on the en bloc
resection for breast cancer with pho-tographs of the complete amputation (A, B). These images reflect the extent of surgical intervention necessary to give the patient a chance of survival. Many surgeons adopted his technique and saved thousands of lives. As the
new century dawned, Dr. Halsted’s suc-cess had a more important effect on the public’s mentality by the creation of hope for a cure rather than despair and expectation of an agonizing death. Patients slowly began to seek treatment for breast cancer while it was still in its early stages.
By the 1920s, however, survival rates
had peaked and remained unchanged for the next 2 decades. In an attempt to improve cures, the super-radical mas-tectomy was developed by Drs. Jerome Urban, Mario Margottini, and Umberto Veronesi. This procedure required even more extensive removal of tissue but still did not provide a significant increase in breast cancer survival rates. A new stan-
dard of care was instituted, which re-quired cancer surgeons to consult with oncologists preoperatively.
Excerpted from Oncology: Tumors & Treatment, A Photographic History, The Antiseptic Era 1876-1900 by Stanley B. Burns, MD, FACS. Photographs cour-tesy of Stanley B. Burns, MD, and the Burns Archive. n
A Century of ProgressThe text and photographs on this page represent the establishment of oncology as a viable medical specialty during the late 1800s and showcase the early medical advances and treatments in cancer. The images and captions are excerpted from a four-volume series of books titled Oncology Tumors & Treatment: A Photographic History by Stanley B. Burns, MD, FACS. To view additional photos from this series, visit burnsarchive.com.
“These images reflect the extent of surgical intervention necessary to give the patient a
chance of survival. Many surgeons adopted his technique and saved thousands of lives.”

TrimSize: 10.75”x14.25”
THERE’S A NATURAL KILLER
INSIDE EVERYONEWITH THE POTENTIAL TO TAKE ON
MULTIPLE MYELOMA
14536702_0094406_DSE_KING_JournalAD_OSFA_v1_M.indd 1 2/6/15 2:23 PM TrimSize: 10.75”x14.25”
© 2014 Bristol-Myers Squibb Company. All rights reserved. ONCUS14UB00944-06-01 12/14
Despite recent advances, multiple myeloma remains a largely incurable disease, with fewer than half of patients surviving � ve years after diagnosis.1
Natural killer cells are part of the body’s � rst line of defense against cancer, but myeloma cells evade and suppress a patient’s natural immune response, making further medical advances challenging.2-11
We need a new approach.
Bristol-Myers Squibb is deeply committed to furthering the science behind Immuno-Oncology. Leading the way in Immuno-Oncology research, Bristol-Myers Squibb is investigating the potential of the SLAMF7, KIR, and CD137 pathways to activate the body’s own natural killer cells to target myeloma cells.
www.explorenaturalkillercells.com
REFERENCES: 1. SEER Stat Fact Sheets: Myeloma, 2004-2010. http://seer.cancer.gov/statfacts/html/mulmy.html. Accessed July 17, 2014; 2. Godfrey J and Benson DM Jr. The role of natural killer cells in immunity against multiple myeloma. Leuk Lymphoma. 2012;53:1666-1676; 3. Cheng M, Chen Y, Xiao W et al. NK cell-based immunotherapy for malignant diseases. Cell Molec Immunol. 2013;10:230-252; 4. Bernal M, Garrido P, Jiménez P et al. Changes in activatory and inhibitory natural killer (NK) receptors may induce progression to multiple myeloma : implications for tumor evasion of T and NK cells. Human Immunol. 2009;70:854-857; 5. Jinushi M, Vanneman M, Munshi NC et al. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci USA. 2008;105:1285-1290; 6. Carbone E, Neri P, Mesuraca M et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood. 2005;105:251-258; 7. von Lilienfeld-Toal M, Frank S, Leyendecker C et al. Reduced immune effector cell NKG2D expression and increased levels of soluble NKG2D ligands in multiple myeloma may not be causally linked. Cancer Immunol Immunother. 2010;59:829-839; 8. Cook G, Campbell JDM, Carr CE et al. Transforming growth factor beta from multiple myeloma cells inhibits proliferation and IL-2 responsiveness in T lymphocytes. J Leukoc Biol. 1999;66:981-988; 9. Yu J, Wei M, Becknell B et al. Pro- and anti-infl ammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity. 2006;24:575-590; 10. Nielsen H, Nielsen HJ, Tvede N et al. Immune dysfunction in multiple myeloma. Reduced natural killer cell activity and increased levels of soluble interleukin-2 receptors. APMIS. 1991;99:340-346; 11. Tinhofer I, Marschitz I, Henn T et al. Expression of functional interleukin-15 receptor and autocrine production of interleukin-15 as mechanisms of tumor propagation in multiple myeloma. Blood. 2000;95:610-618.
14536702_0094406_DSE_KING_JournalAD_OSFA_v1_M.indd 2 2/6/15 2:23 PM

TrimSize: 10.75”x14.25”
THERE’S A NATURAL KILLER
INSIDE EVERYONEWITH THE POTENTIAL TO TAKE ON
MULTIPLE MYELOMA
14536702_0094406_DSE_KING_JournalAD_OSFA_v1_M.indd 1 2/6/15 2:23 PM TrimSize: 10.75”x14.25”
© 2014 Bristol-Myers Squibb Company. All rights reserved. ONCUS14UB00944-06-01 12/14
Despite recent advances, multiple myeloma remains a largely incurable disease, with fewer than half of patients surviving � ve years after diagnosis.1
Natural killer cells are part of the body’s � rst line of defense against cancer, but myeloma cells evade and suppress a patient’s natural immune response, making further medical advances challenging.2-11
We need a new approach.
Bristol-Myers Squibb is deeply committed to furthering the science behind Immuno-Oncology. Leading the way in Immuno-Oncology research, Bristol-Myers Squibb is investigating the potential of the SLAMF7, KIR, and CD137 pathways to activate the body’s own natural killer cells to target myeloma cells.
www.explorenaturalkillercells.com
REFERENCES: 1. SEER Stat Fact Sheets: Myeloma, 2004-2010. http://seer.cancer.gov/statfacts/html/mulmy.html. Accessed July 17, 2014; 2. Godfrey J and Benson DM Jr. The role of natural killer cells in immunity against multiple myeloma. Leuk Lymphoma. 2012;53:1666-1676; 3. Cheng M, Chen Y, Xiao W et al. NK cell-based immunotherapy for malignant diseases. Cell Molec Immunol. 2013;10:230-252; 4. Bernal M, Garrido P, Jiménez P et al. Changes in activatory and inhibitory natural killer (NK) receptors may induce progression to multiple myeloma : implications for tumor evasion of T and NK cells. Human Immunol. 2009;70:854-857; 5. Jinushi M, Vanneman M, Munshi NC et al. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci USA. 2008;105:1285-1290; 6. Carbone E, Neri P, Mesuraca M et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood. 2005;105:251-258; 7. von Lilienfeld-Toal M, Frank S, Leyendecker C et al. Reduced immune effector cell NKG2D expression and increased levels of soluble NKG2D ligands in multiple myeloma may not be causally linked. Cancer Immunol Immunother. 2010;59:829-839; 8. Cook G, Campbell JDM, Carr CE et al. Transforming growth factor beta from multiple myeloma cells inhibits proliferation and IL-2 responsiveness in T lymphocytes. J Leukoc Biol. 1999;66:981-988; 9. Yu J, Wei M, Becknell B et al. Pro- and anti-infl ammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity. 2006;24:575-590; 10. Nielsen H, Nielsen HJ, Tvede N et al. Immune dysfunction in multiple myeloma. Reduced natural killer cell activity and increased levels of soluble interleukin-2 receptors. APMIS. 1991;99:340-346; 11. Tinhofer I, Marschitz I, Henn T et al. Expression of functional interleukin-15 receptor and autocrine production of interleukin-15 as mechanisms of tumor propagation in multiple myeloma. Blood. 2000;95:610-618.
14536702_0094406_DSE_KING_JournalAD_OSFA_v1_M.indd 2 2/6/15 2:23 PM

PAGE 82 The ASCO Post | MARCH 25, 2015
2015 Oncology Meetings 2015MarchPersonalised Cancer Medicine and Big Data Analysis – 7th International Conference of Contemporary OncologyMarch 25-27 • Poznań, Poland For more information: www.termedia.pl/Konferencje?Invitation&e=372&p=2787
Society of Surgical Oncology Annual MeetingMarch 25-28 • Houston, Texas For more information: www.surgonc.org/
EORTC-EANO-ESMO 2015March 27-28 • Istanbul, Turkey For more information: www.ecco-org.eu/Events/EORTC_EANO_ESMO-2015
Northern New England Clinical Oncology Society March 27-28 • Portsmouth, New HampshireFor more information: http://nnecos.org/spring
46th Annual Meeting on Women’s Cancer March 28-31 • Chicago, Illinois For more information: www.sgo.org
AprilAmerican Brachytherapy Society Annual MeetingApril 9-11 • Orlando, Florida For more information: http://www.americanbrachytherapy .org/meetings/annual2015/index.cfm
Melanoma and Cutaneous Malignancies MeetingApril 10-11 • New York, New York For more information: www.healio.com/meeting/hemonctodaymelanoma/home?promocode689-8045
Hematologic Malignancies: New Therapies and the Evolving Role of TransplantApril 10-11 • Chicago, Illinois For more information: www.mayo.edu/cme/hematology-and-oncology-2015r919
2015 Business of Oncology Summit and 2015 Annual Meeting & Spring SessionApril 10-12 • Orlando, Florida For more information: www.flasco.org
International Society of Geriatric Oncology (SIOG) USA Forum April 11 • Tampa, Florida For more information: www.siog.org
HPV-Induced Head and Neck Cancer: Screening, Detection and Less Invasive TherapiesApril 11 • Miami, Florida For more information: http://cme.baptisthealth.net/headneckcancer/pages/index.aspx
OSMO Spring 2015 Oncology ConferenceApril 11 • Portland, Oregon For more information: www.osmo.org/events/view/26
ASCO/C-KIN Joint Session at Cancer & the Kidney International Network’s First Annual Conference (C-KIN 2015)April 14-15 • Brussels, Belgium For more information: www.c-kin.org/conference2015/
ASCO Multidisciplinary Cancer Management Course (MCMC)April 15 • Vina Del Mar, Chile For more information: www.asco.org/international-programs/multidisciplinary-cancer-management-courses
ESMO European Lung Cancer ConferenceApril 15-18 • Geneva, Switzerland For more information: www.esmo.org/Conferences/ELCC-2015-Lung-Cancer
Multidisciplinary Spine Oncology SymposiumApril 17-18 • New York, New York For more information: www.mskcc.org/events/cme/multidisciplinary-spine-oncology-symposium/form
VAHO Spring 2015 Membership Conference April 17-18 • Hot Springs, VirginiaFor more information: www.accc-cancer.org/ossn_network/VA/VAHO-meetings.asp
American Association for Cancer Research Annual Meeting April 18-22 • Philadelphia, Pennsylvania For more information: www.aacr.org
ONS 40th Annual Congress April 23-26 • Orlando, Florida For more information: www.ons.org/conferences/congress-2015
3rd ESTRO Forum April 23-28 • Barcelona, Spain For more information: http://www.estro.org/congresses-meetings/items/3rd-estro-forum
Fourth Annual Cancer Pain ConferenceApril 24-26 • Scottsdale, Arizona For more information: www.cancerpainconference.org
MayInternational Society of Geriatric Oncology (SIOG): Cancer Care of the Older Adult Across the Cancer ContinuumMay 1 • New York, New York For more information: www.siog.org
The 28th Annual Meeting of the American Society of Pediatric Hematology/OncologyMay 6-9 • Phoenix, Arizona For more information: www.aspho.org/education/content/annualmeeting.html
WCIO 2015May 6-9 • New York, New York For more information: http://www.wcioevents.org
Breast Cancer ConferenceMay 7-9 • Brussels, Belgium For more information: www.esmo.org/Conferences/ IMPAKT-2015-Breast-Cancer
13th Annual Meeting of the Association for Cancer Immunotherapy (CIMT)May 11-13 • Mainz, Germany For more information: www.meeting.cimt.eu
54th Annual Conference of the Particle Therapy Co-Operative GroupMay 18-23 • San Diego, California For more information: http://ptcog54.org
American Association for Cancer Research: Advances in Brain Cancer Research May 27-30 • Washington, DC For more information: www.aacr.org
ASCO Annual MeetingMay 29-June 2 • Chicago, Illinois For more information: http://am.asco.org/
2015 ASCO State Affiliates’ ReceptionMay 31 • Chicago, Illinois For more information: www.asco.org/about-asco/state-affiliate-leadership-conference
JuneInternational Cancer Screening Network (ICSN) Triennial MeetingJune 2-4 • Rotterdam, The Netherlands For more information: www.scgcorp.com/ICSN2015/
Save the Date
13th International Conference on Malignant
Lymphoma (ICML)June 17–20, 2015
Lugano, Switzerland
The 13-ICML will bring 3,000 physicians to Lugano from all over the world: hematologists, clinical oncologists, radiation on-cologists, pediatricians, patholo-gists, and leading researchers involved in the study and treat-ment of lymphoid neoplasms.
For more information, visit www.lymphcon.ch/imcl/index.php

ASCOPost.com | MARCH 25, 2015 PAGE 83
2015 Oncology Meetings 20152015 Clinical Update: 21st Century Prevention of HPV-Associated CancerJune 5-7 • Baltimore, Maryland For more information: www.asccp .org/Education/2015-21st-Century-Prevention-of-HPV-Associated-Cancer
Society of Nuclear Medicine and Molecular Imaging Annual MeetingJune 6-10 • Baltimore, Maryland For more information: www.snm.org
13th International Conference on Malignant Lymphoma (ICML)June 17-20 • Lugano, Switzerland For more information: www.lymphcon.ch/imcl/index.php2
Anticancer Drug Action and Resistance: From Cancer Biology to the ClinicJune 20-23 • Florence, Italy For more information: www.ecco-org.eu/Events/EAS2015
CAP-ACP 2015 Annual MeetingJune 20-23 • Montreal, Canada For more information: www.cap-acp.org/annual_meeting .php
International Society on Thrombosis and Haemostasis Annual MeetingJune 20-25 • Toronto, Canada For more information: www.isth.org/page/2015Microsite/
MASCC/ISOO Annual Meeting on Supportive Care in CancerJune 25-27 • Copenhagen, Denmark For more information: http://www.kenes.com/mascc2015/
July7th World Congress on Gastrointestinal CancerJuly 1-4 • Barcelona, Spain For more information: http://worldgicancer.com/WCGI/WGIC2015/index.asp
14th Annual International Congress on the Future of Breast Cancer®July 16-18 • Huntington Beach, California For more information: www.gotoper.com
The 13th Annual Scientific Meeting of JSMOJuly 16-18 • Sapporo, Japan For more information: www.congre.co.jp/jsmo2015/en/index .html
NRG Oncology MeetingJuly 16-19 • Denver, Colorado For more information: www.gog.org/meetinginformation .html
APOS 12th Annual Conference and IPOS 17th World Congress of Psycho-OncologyJuly 28-August 1 • Washington, DC For more information: http://www.apos-society.org
16th Annual International Lung Cancer Congress®July 30-August 1 • Huntington Beach, California For more information: www.gotoper.com/conferences/ilc/meetings/16th-International-Lung-Cancer-Congress
Best of ASCO® BostonJuly 31-August 1 • Boston, Massachusetts For more information: http://boa.asco.org/
AugustBest of ASCO - San FranciscoAugust 7-8 • San Francisco, California For more information: http://boa.asco.org/
World Congress on Cancer and Prevention MethodsAugust 27-29 • Dubai, United Arab Emirates For more information: http://scientificfuture.com/oncology-2015/
ASCO Multidisciplinary Cancer Management Course (MCMC)August 28-29 • Sao Paulo, Brazil For more information: www.asco.org/international-programs/multidisciplinary-cancer-management-courses
Best of ASCO - Chicago August 28-29 • Chicago, Illinois For more information: http://boa.asco.org/
European Society for Medical Oncology Academy 2015August 28-30 • Oxford, United Kingdom For more information: www.esmo.org/Conferences/ ESMO-Academy-2015
SeptemberInternational Palliative Care WorkshopSeptember 3-5 • Fez, Morocco For more information: www.asco.org/international-programs/international-palliative-care-workshops
25th World Congress of the International Association of Surgeons, Gastroenterologists and OncologistsSeptember 4-6 • Fuzhou, China For more information: www.csw-iasgo2015.org
ILCA 2015 —The International Liver Cancer Association’s 9th Annual ConferenceSeptember 4-6 • Paris, France For more information: www.ilca2015.org
16th World Conference on Lung CancerSeptember 6-9 • Denver, Colorado For more information: http://wclc2015.iaslc.org
ISEH 44th Annual Scientific MeetingSeptember 17-19 • Kyoto, Japan For more information: www.iseh.org/?page=Meeting
4th Annual Conference on Immunotherapy in Pediatric Oncology (CIPO2015)September 25-26 • Seattle, Washington For more information: www.seattlechildrens.org/research/childhood-cancer/CIPO-2015/
2015 Breast Cancer SymposiumSeptember 25-27 • San Francisco, California For more information: http://breastcasym.org
European Cancer Congress (ECC 2015)September 25-29 • Vienna, Austria For more information: www.esmo.org/Conferences/European-Cancer-Congress-2015
October30th Annual Harvard “Critical Issues in Tumor Microenvironment: Angiogenesis, Metastasis and Immunology”October 5-8 • Boston, Massachusetts For more information: http://steele.mgh.harvard.edu/tumorcourse
Palliative Care in Oncology SymposiumOctober 9-10 • Boston, Massachusetts For more information: http://pallonc.org
NovemberSITC 30th Anniversary Annual MeetingNovember 4-8 • National Harbor, Maryland For more information: www.ncer.org/sitc-meetings/sitc2015
December57th Annual ASH Meeting & ExpositionDecember 5-8 • Orlando, Florida For more information: www.hematology.org/ Annual-Meeting/
San Antonio Breast Cancer SymposiumDecember 8-12 • San Antonio, Texas For more information: www.sabcs.org

PAGE 84 The ASCO Post | MARCH 25, 2015
Announcements
Harold Varmus, MD, Steps Down as NCI Director, Douglas Lowy, MD, Named Acting Director
Harold Varmus, MD, who has led the National Cancer Institute
(NCI) at the National Institutes of Health (NIH) for nearly 5 years, has announced that he will step down from his post, effective March 31, 2015.
“It has been our great fortune to have Dr. Varmus at the helm of the NCI,” said NIH Director Francis S. Collins, MD, PhD. “His breadth and depth of expertise in biomedical research are un-paralleled, and he’s been a tremendous colleague, invaluable to the agency.”
Douglas Lowy, MD, who currently serves as the Deputy Director, will become Acting Director for NCI, beginning April 1, 2015. Dr. Lowy is a longtime NCI intra-mural researcher. He received the National Medal of Technology and Innovation from President Obama in 2014 for his research that led to the development of the human papillomavirus (HPV) vaccine.
Dr. Varmus to Join Weill CornellDr. Harold Varmus will join Weill Cor-
nell Medical College’s faculty as the Lewis Thomas University Professor of Medi-cine, effective April 1. In conjunction with his appointment at Weill Cornell, Dr. Var-mus will team up with the New York Ge-nome Center as a Senior Associate Core Member to promote the use of cancer ge-nomics throughout the New York region. In his new position at Weill Cornell, Dr. Varmus will continue to conduct research on fundamental aspects of cancer, in col-laboration with investigators at the Sandra and Edward Meyer Cancer Center, led by Meyer Director Lewis C. Cantley.
“This is a remarkable time in cancer research, Dr. Varmus said. “Technolog-
ical advances have enabled scientists to conduct comprehensive genomic studies that are revealing detailed por-traits of cancer cells, sparking new op-portunities to develop next-generation
therapies, diagnostics, and prevention strategies. I’m excited to join Weill Cornell Medical College and the New York Genome Center as we strive to reduce the burden of cancer and en-
hance human health in New York and around the world.”
Dr. Varmus will also serve as a senior advisor to Laurie H. Glimcher, MD, the Stephen and Suzanne Weiss Dean of
Harold Varmus, MD
Douglas Lowy, MD
25695_clovco_fa2_rash_ASCOpst.indd 1 9/23/14 3:31 PM
We want to change the face of EGFR-targeted therapy
In the treatment of EGFR mutation–positive non–small cell lung cancer (NSCLC), rash and other skin toxicities are well-established side effects of EGFR tyrosine kinase inhibitors.3,4
90% of patients treated with approved EGFR inhibitors experience rash3,4 In some studies, rash and paronychia were among the most frequent causes of dose modification, combining to cause dose reductions in as many as 33% of patients.3,4
Rash and its symptoms can negatively affect both patient quality of life and patient compliance, while its psychosocial impact contributes to the assessment of severity.5,6 Beyond the clinical symptom burden, rash visibility can cause significant patient distress even when it is not severe.5
At Clovis Oncology, we’re committed to exploring new approaches in EGFR therapy to advance the fight against NSCLC.
REFERENCES: 1. Lynch TJ Jr et al. Epidermal growth factor receptor inhibitor–associated cutaneous toxicities: an evolving paradigm in clinical management. Oncologist. 2007;12(5):610-621. 2. Pérez-Soler R et al. HER1/EGFR inhibitor-associated rash: future directions for management and investigation outcomes from the HER1/EGFR Inhibitor Rash Management Forum. Oncologist. 2005;10(5):345-356. 3. Tarceva [package insert]. Northbrook, IL: OSI Pharmaceuticals LLC; 2014. 4. Gilotrif [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc; 2014. 5. White KJ et al. Psychosocial impact of cutaneous toxicities associated with epidermal growth factor receptor–inhibitor treatment. Clin J Oncol Nurse. 2011;15(1):88-96. 6. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE), Version 4.0. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_8.5x11.pdf. Published May 28, 2009. Updated June 14, 2010. Accessed August 26, 2014.
Clovis Oncology is leading the fight
Rash is caused by inhibition of wild-type epidermal growth factor receptor (EGFR) and can be debilitating1,2
Copyright © 2014 Clovis Oncology. DARO-101 8/14
25695_clovco_fa2_rash_ASCOpst.indd 2 9/23/14 3:31 PM

ASCOPost.com | MARCH 25, 2015 PAGE 85
Announcements
ASCO Commends Dr. Varmus for Outstanding Leadership
Following news of Dr. Varmus’ planned departure from the
National Cancer Institute (NCI), ASCO issued the following statement:
ASCO is enormously grateful for the service and leadership of Harold Varmus, MD, as Director of the National Cancer Institute [NCI]. During Dr. Varmus’ tenure, the cancer community made im-portant strides in modernizing can-cer research for the molecular era.
“Dr. Varmus established valu-able initiatives that will help trans-form cancer research for the 21st century and improve cancer care on a global level,” said ASCO Chief Medical Officer Richard L. Schilsky, MD, FACP, FASCO. “He accomplished these tasks dur-ing a period when the NCI faced unprecedented financial instability.”
After becoming NCI Director in June 2010, Dr. Varmus’ efforts included creating the Institute’s Center for Global Health; establish-ing an initiative for asking provoca-tive questions on complex issues in cancer research; and developing the cancer component of the Precision Medicine Initiative, which was an-nounced in January 2015.
Dr. Varmus also led the trans-formation of the cooperative group program into the National Clinical Trials Network, which is now un-dertaking highly innovative clinical trials that aim to bring the benefits of precision medicine to patients. In addition, the Society was proud to have Dr. Varmus share his insights on decades of progress against can-cer and the future of cancer research at ASCO’s 2011 Annual Meeting.
“As NCI Director, Dr. Varmus drew on his deep knowledge of cancer biology and the nation’s biomedical research enterprise to focus the efforts of the cancer community on identifying and tackling the most vexing prob-lems in cancer research and care,” said Dr. Schilsky. “ASCO is grate-ful for his service to the cancer community and the country.” n
Weill Cornell and provost for medical af-fairs for Cornell University, and will have an appointment in the Weill Cornell Graduate School of Medical Sciences.
Career HighlightsDr. Varmus has had a longstanding
association with NIH, dating back to 1968–1970 when, as a young Public
Health Service Officer, he studied bac-terial gene expression with Ira Pastan, MD, who is currently Chief of NCI’s Laboratory of Molecular Biology.
In 1989, Dr. Varmus was Corecipi-ent of the Nobel Prize in Physiology or Medicine for “discovery of the cellular origin of retroviral oncogenes.” From 1993–1999, he served as the Director
of NIH under President Bill Clinton. After leaving NIH and before returning to run NCI in 2010, Dr. Varmus served as President of Memorial Sloan Ketter-ing Cancer Center. Before President Barack Obama appointed Dr. Varmus to lead NCI, he named him Co-Chair of the President’s Council of Advisors on Science and Technology. n
25695_clovco_fa2_rash_ASCOpst.indd 1 9/23/14 3:31 PM
We want to change the face of EGFR-targeted therapy
In the treatment of EGFR mutation–positive non–small cell lung cancer (NSCLC), rash and other skin toxicities are well-established side effects of EGFR tyrosine kinase inhibitors.3,4
90% of patients treated with approved EGFR inhibitors experience rash3,4 In some studies, rash and paronychia were among the most frequent causes of dose modification, combining to cause dose reductions in as many as 33% of patients.3,4
Rash and its symptoms can negatively affect both patient quality of life and patient compliance, while its psychosocial impact contributes to the assessment of severity.5,6 Beyond the clinical symptom burden, rash visibility can cause significant patient distress even when it is not severe.5
At Clovis Oncology, we’re committed to exploring new approaches in EGFR therapy to advance the fight against NSCLC.
REFERENCES: 1. Lynch TJ Jr et al. Epidermal growth factor receptor inhibitor–associated cutaneous toxicities: an evolving paradigm in clinical management. Oncologist. 2007;12(5):610-621. 2. Pérez-Soler R et al. HER1/EGFR inhibitor-associated rash: future directions for management and investigation outcomes from the HER1/EGFR Inhibitor Rash Management Forum. Oncologist. 2005;10(5):345-356. 3. Tarceva [package insert]. Northbrook, IL: OSI Pharmaceuticals LLC; 2014. 4. Gilotrif [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc; 2014. 5. White KJ et al. Psychosocial impact of cutaneous toxicities associated with epidermal growth factor receptor–inhibitor treatment. Clin J Oncol Nurse. 2011;15(1):88-96. 6. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE), Version 4.0. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_8.5x11.pdf. Published May 28, 2009. Updated June 14, 2010. Accessed August 26, 2014.
Clovis Oncology is leading the fight
Rash is caused by inhibition of wild-type epidermal growth factor receptor (EGFR) and can be debilitating1,2
Copyright © 2014 Clovis Oncology. DARO-101 8/14
25695_clovco_fa2_rash_ASCOpst.indd 2 9/23/14 3:31 PM

PAGE 86 The ASCO Post | MARCH 25, 2015
Reflections
The Center of Who We Are By Eric M. Genden, MD
There’s nothing more terrifying than having a cancer in the head
and neck region—it’s the core and cen-ter of who we are—and to understand that it has to be removed or radiated or burned.
No one marches for head and neck cancer. Unlike the pink-clad legions of breast cancer survivors, head and neck cancer patients often dwell in the shad-ows, dreading the workplace and avoid-ing the favorite restaurant or the friend-ly gin rummy game that was so much a part of their lives.
My specialty, otolaryngology, is argu-ably different from every other type of surgery—particularly when it encom-passes oncology—because there is no place you can do surgery on the human body that more affects the center of who we are. We express ourselves by speech and articulation, by the movement of our eyes and facial expressions. When we socialize with family and friends, we eat, we get a cup of coffee or something to drink; we break bread. Unfortunate as it is, much of how we do in life is based on our physical presentation.
Head and neck cancer surgery al-most always impacts all these things.
Whether it’s a surgical procedure on the tongue, the upper jaw, or the skull base, the ability to speak, socialize, move the face—to self-express—is affected, which is a reason suicide and depres-sion rank high among head and neck cancer patients. Unlike breast or ab-dominal surgery, you can’t put a shirt on and make these scars or this deformity disappear. There’s no way to hide it.
Advances in CareDuring the past decade, minimally
invasive surgical approaches and meth-ods of reconstruction have become critically important in addressing these terrible challenges. Probably most sig-nificant has been the introduction of robotic surgery.
In the past, if someone had a can-cer in the back of the throat, to get back there and remove it safely we had to undertake a 15-hour surgery: cutting through the lip, the mandible (jaw), and the muscles of the floor of the mouth to open up and swing the jaw, much the way you’d open a book. The patient would be hospitalized for 15 days, with a 10% to 20% possibility of a permanent feeding tube or breath-ing tube. All this to resect a tumor no larger than a walnut.
About 7 years ago, we introduced an approach in which a surgical robot (Intuitive’s daVinci™ system) is inserted through the mouth, and a laser is used to resect and remove the tumor. Instead of 14 hours, it’s a 2.5-hour surgery; the patient is hospitalized for 1 or 2 nights and is eating and drinking the next day. Most remarkably, it obviates the need for a potentially deforming facial inci-sion. As a surgeon, when you don’t have to essentially decouple a patient’s jaw and face, you become a believer [in this technology] very, very quickly.
The first two institutions in the world to adapt this technology—which was originally designed for cardiac, ab-dominal, and urologic applications—
were Mount Sinai (where I serve as Chairman of the Department of Otolar-yngology) and the University of Penn-sylvania (Penn Medicine).
We can’t do robotic surgery in ev-ery case, but there just happens to be an epidemic of human papillomavirus (HPV)–induced cancers in people rang-ing in age from 35 to 70. In the next 5 years, these cancers will surpass cervical cancer in women in terms of prevalence.
Treating HPV-Induced CancersFor 100 years, patients who de-
veloped throat cancers have typically been smokers and drinkers, genetically predisposed to cancer and unable to re-pair the ravages of tobacco and alcohol. Such squamous cell carcinomas are ag-gressive and can afflict multiple areas, so the treatment needs to be equally aggressive—big surgeries with chemo-therapy and radiation therapy.
Most cancers in the head and neck are classified as squamous cell carcinoma, but they are more than one disease. Un-fortunately, most oncologists are treat-ing patients with HPV-induced cancer with the regimens designed for smokers and drinkers with aggressive squamous cell carcinoma. In many cases, the side effects of these treatments dramatically
impair a patient’s quality of life.HPV-induced squamous cell can-
cers are focal. The virus inoculates an area of the tonsil and creates a chronic infection that leads to a cancer, but, typically, the whole upper aerodigestive tract is not afflicted. These cancers are amenable to surgical removal with the robot. As a result, 20% of HPV-induced cancer patients will not require any ra-diation; 40% require radiotherapy—but in a reduced dose that significantly reduces acute toxicity and long-term chronic toxicity.
How does this play out in terms of quality of life? I’ve seen a well-known chef with HPV-induced throat cancer who was scheduled to undergo high-
dose radiation and chemotherapy. He was 47 years old, a guy whose entire life revolved around his ability to taste the finest changes in food. Without question, chemotherapy and radiation would have destroyed his career. It’s no different from removing the vocal cords of a singer or amputating a surgeon’s hand. Since his procedure, he’s been cancer-free for 4 years and has opened two more successful restaurants. (See “It Sounds Crazy, but Cancer Has Made Me a Better Chef,” in the December 1, 2013, issue of The ASCO Post.)
Medicine Is Who I AmUltimately, our patients become
mirrors on which our own lives and ca-reers are reflected. My father was a Navy fighter pilot who served three tours dur-ing the Vietnam War. Like the character in the movie The Great Santini, he was a perfectionist who insisted I follow in his footsteps. When I told him it just wasn’t in the cards, he said, “Well, if you have to go into medicine, for God’s sake, be a surgeon! On the flight deck, the only people we respect are the flight surgeons!”
And so I did. I like to think I inher-ited my father’s passion and focus.
Like flying Phantom jets, medicine is not a job but a way of life. It becomes who you are. Some people go to work and leave work and can put it all be-hind them; then there are others whose work, life, and family all intersect. There is no division. It’s not a thing you can face or misinterpret. It is the essence of who we are as doctors.
Coming Face to Face With the Unknowable
As I’ve grown older, I find myself staring more deeply into the mirror. When I was a young surgeon, the chal-lenge was to get patients to survive, to beat the cancer, and to get them recon-structed. My questions were, “Are you swallowing food? Are you able to eat without difficulty? Is there much pain?”
Now I’m more focused on the deep-er quality of life. Do they go out in pub-lic and feel good about themselves? Are they interacting with their family? Do they wake up every morning and say, “Boy, I’m glad I’m alive! Life is good for me and better than I had expected!”?
Fulfillment comes with putting people back into society. With head and neck cancer, you can cure a lot of
continued on page 88
Eric M. Genden, MD, is Chairman of the Department of Otolaryngology and Profes-sor of Otolaryngology-Head and Neck Sur-gery at Mount Sinai Hospital in New York.
No one marches for head and neck cancer. Unlike the pink-clad legions of breast cancer survivors, head and neck cancer patients often dwell in the shadows.
—Eric M. Genden, MD
The following essay by Eric M. Genden, MD, is adapted from The Big Casino: America’s Best Cancer Doctors Share Their Most Power-ful Stories, which was coedited by Stan Winokur, MD, and Vincent Coppola and published in May 2014. The book is available on Amazon.com and thebigcasino.org.
Take a bite out of G-CSF acquisition costsBased on wholesale acquisition cost (WAC) of all short-acting G-CSF products as of November 11, 2013. WAC represents published catalogue or list prices and may not represent actual transactional prices. Please contact your supplier for actual prices.
GRANIX® is an option in short-acting G-CSF therapy» A 71% reduction in duration of severe neutropenia vs placebo (1.1 days vs 3.8 days, p<0.0001)1
– Efficacy was evaluated in a multinational, multicenter, randomized, controlled, Phase III study of chemotherapy-naïve patients with high-risk breast cancer receiving doxorubicin (60 mg/m2 IV bolus)/docetaxel (75 mg/m2)1
» The safety of GRANIX was established in 3 Phase III trials, with 680 patients receiving chemotherapy for either breast cancer, lung cancer, or non-Hodgkin lymphoma (NHL)1
» Now offering a new presentation for self-administration
Indication» GRANIX is a leukocyte growth factor indicated for reduction in the duration of severe neutropenia in patients
with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a clinically significant incidence of febrile neutropenia.
Important Safety Information» Splenic rupture: Splenic rupture, including fatal cases, can occur following the administration of human granulocyte
colony-stimulating factors (hG-CSFs). Discontinue GRANIX and evaluate for an enlarged spleen or splenic rupture in patients who report upper abdominal or shoulder pain after receiving GRANIX.
» Acute respiratory distress syndrome (ARDS): ARDS can occur in patients receiving hG-CSFs. Evaluate patients who develop fever and lung infiltrates or respiratory distress after receiving GRANIX, for ARDS. Discontinue GRANIX in patients with ARDS.
» Allergic reactions: Serious allergic reactions, including anaphylaxis, can occur in patients receiving hG-CSFs. Reactions can occur on initial exposure. Permanently discontinue GRANIX in patients with serious allergic reactions. Do not administer GRANIX to patients with a history of serious allergic reactions to filgrastim or pegfilgrastim.
» Use in patients with sickle cell disease: Severe and sometimes fatal sickle cell crises can occur in patients with sickle cell disease receiving hG-CSFs. Consider the potential risks and benefits prior to the administration of GRANIX in patients with sickle cell disease. Discontinue GRANIX in patients undergoing a sickle cell crisis.
» Capillary leak syndrome (CLS): CLS can occur in patients receiving hG-CSFs and is characterized by hypotension, hypoalbuminemia, edema and hemoconcentration. Episodes vary in frequency, severity and may be life-threatening if treatment is delayed. Patients who develop symptoms of CLS should be closely monitored and receive standard symptomatic treatment, which may include a need for intensive care.
» Potential for tumor growth stimulatory effects on malignant cells: The granulocyte colony-stimulating factor (G-CSF) receptor, through which GRANIX acts, has been found on tumor cell lines. The possibility that GRANIX acts as a growth factor for any tumor type, including myeloid malignancies and myelodysplasia, diseases for which GRANIX is not approved, cannot be excluded.
» Most common treatment-emergent adverse reaction: The most common treatment-emergent adverse reaction that occurred in patients treated with GRANIX at the recommended dose with an incidence of at least 1% or greater and two times more frequent than in the placebo group was bone pain.
Please see brief summary of Full Prescribing Information on adjacent page.
For more information, visit GRANIXhcp.com.Reference: 1. GRANIX® (tbo-� lgrastim) Injection Prescribing Information. North Wales, PA: Teva Pharmaceuticals; 2014.
©2015 Cephalon, Inc., a wholly-owned subsidiary of Teva Pharmaceutical Industries Ltd. GRANIX is a registered trademark of Teva Pharmaceutical Industries Ltd. All rights reserved. GRX-40582 January 2015.
K Job Number: 21089Revision No: 0Date: 01/20/15
YMC724-41865 Page 1 DIGITAL

Take a bite out of G-CSF acquisition costsBased on wholesale acquisition cost (WAC) of all short-acting G-CSF products as of November 11, 2013. WAC represents published catalogue or list prices and may not represent actual transactional prices. Please contact your supplier for actual prices.
GRANIX® is an option in short-acting G-CSF therapy» A 71% reduction in duration of severe neutropenia vs placebo (1.1 days vs 3.8 days, p<0.0001)1
– Efficacy was evaluated in a multinational, multicenter, randomized, controlled, Phase III study of chemotherapy-naïve patients with high-risk breast cancer receiving doxorubicin (60 mg/m2 IV bolus)/docetaxel (75 mg/m2)1
» The safety of GRANIX was established in 3 Phase III trials, with 680 patients receiving chemotherapy for either breast cancer, lung cancer, or non-Hodgkin lymphoma (NHL)1
» Now offering a new presentation for self-administration
Indication» GRANIX is a leukocyte growth factor indicated for reduction in the duration of severe neutropenia in patients
with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a clinically significant incidence of febrile neutropenia.
Important Safety Information» Splenic rupture: Splenic rupture, including fatal cases, can occur following the administration of human granulocyte
colony-stimulating factors (hG-CSFs). Discontinue GRANIX and evaluate for an enlarged spleen or splenic rupture in patients who report upper abdominal or shoulder pain after receiving GRANIX.
» Acute respiratory distress syndrome (ARDS): ARDS can occur in patients receiving hG-CSFs. Evaluate patients who develop fever and lung infiltrates or respiratory distress after receiving GRANIX, for ARDS. Discontinue GRANIX in patients with ARDS.
» Allergic reactions: Serious allergic reactions, including anaphylaxis, can occur in patients receiving hG-CSFs. Reactions can occur on initial exposure. Permanently discontinue GRANIX in patients with serious allergic reactions. Do not administer GRANIX to patients with a history of serious allergic reactions to filgrastim or pegfilgrastim.
» Use in patients with sickle cell disease: Severe and sometimes fatal sickle cell crises can occur in patients with sickle cell disease receiving hG-CSFs. Consider the potential risks and benefits prior to the administration of GRANIX in patients with sickle cell disease. Discontinue GRANIX in patients undergoing a sickle cell crisis.
» Capillary leak syndrome (CLS): CLS can occur in patients receiving hG-CSFs and is characterized by hypotension, hypoalbuminemia, edema and hemoconcentration. Episodes vary in frequency, severity and may be life-threatening if treatment is delayed. Patients who develop symptoms of CLS should be closely monitored and receive standard symptomatic treatment, which may include a need for intensive care.
» Potential for tumor growth stimulatory effects on malignant cells: The granulocyte colony-stimulating factor (G-CSF) receptor, through which GRANIX acts, has been found on tumor cell lines. The possibility that GRANIX acts as a growth factor for any tumor type, including myeloid malignancies and myelodysplasia, diseases for which GRANIX is not approved, cannot be excluded.
» Most common treatment-emergent adverse reaction: The most common treatment-emergent adverse reaction that occurred in patients treated with GRANIX at the recommended dose with an incidence of at least 1% or greater and two times more frequent than in the placebo group was bone pain.
Please see brief summary of Full Prescribing Information on adjacent page.
For more information, visit GRANIXhcp.com.Reference: 1. GRANIX® (tbo-� lgrastim) Injection Prescribing Information. North Wales, PA: Teva Pharmaceuticals; 2014.
©2015 Cephalon, Inc., a wholly-owned subsidiary of Teva Pharmaceutical Industries Ltd. GRANIX is a registered trademark of Teva Pharmaceutical Industries Ltd. All rights reserved. GRX-40582 January 2015.
K Job Number: 21089Revision No: 0Date: 01/20/15
YMC724-41865 Page 1 DIGITAL

PAGE 88 The ASCO Post | MARCH 25, 2015
Reflections
people, but that doesn’t mean you’re putting them back in the workforce. At Mount Sinai, we’ve been able to do that. We have a strong belief that quality of life and the ability to function are as im-portant as removing the cancer.
Finally, in the mirror, we come face
to face with the unknowable: things that seemingly defy the hard and fast dictates of science and medicine and carry us to some higher plane, where courage and determination, and yes, love are all that matter.
Early in my career, an Indian woman with a horrible oral cancer came to see me. She’d had about six or seven opera-
tions. Now the tumor had come back in quite an extensive way. These were the days when we first started remov-ing pieces of the jaw and the tongue and rebuilding them by transplanting tissue. We didn’t have a good way to do it. It would leave people so badly deformed they couldn’t eat or drink.
She’d been to all the local places and
had been turned away. I said, “Look, your tumor is too advanced. You need to have palliative care and pain control because there’s not much we can do.”
She begged and begged me to help her. “Why don’t you come back next week, and we’ll align you with our on-cologists,” I finally said. “They’ll give you palliative chemotherapy.” When she came back, she brought her 7-year-old daughter with her. At one point, the pa-tient stepped away for a moment and the little girl essentially said to me, “You have to help her. You have to operate on her because if you don’t, nobody else will.”
It was the first time, in fact, the only time, this has ever happened to me. I told her there was probably a 5% chance she’d actually beat this cancer, but we’d give it a try. We took out her jaw and half her tongue and rebuilt the jaw and tongue using the bone and skin from her lower leg.
Now, it’s 15 years later, and I still get cards from her and her daughter. The little girl has graduated from college. And she still has her mom with her. I can see them in the mirror. n
Eric M. Genden, MDcontinued from page 86
Follow us on
@ASCOPost
The ASCO Post
BRIEF SUMMARY OF PRESCRIBING INFORMATION FORGRANIX® (tbo-fi lgrastim) injection, for subcutaneous useSEE PACKAGE INSERT FOR FULL PRESCRIBING INFORMATION1 INDICATIONS AND USAGEGRANIX is indicated to reduce the duration of severe neutropenia in patients with non-myeloid malignancies receiving myelosuppressive anti-cancer drugs associated with a clinically signifi cant incidence of febrile neutropenia.4 CONTRAINDICATIONSNone.5 WARNINGS AND PRECAUTIONS5.1 Splenic RuptureSplenic rupture, including fatal cases, can occur following administration of human gran-ulocyte colony-stimulating factors. In patients who report upper abdominal or shoulder pain after receiving GRANIX, discontinue GRANIX and evaluate for an enlarged spleen or splenic rupture.5.2 Acute Respiratory Distress Syndrome (ARDS)Acute respiratory distress syndrome (ARDS) can occur in patients receiving human gran-ulocyte colony-stimulating factors. Evaluate patients who develop fever and lung infi ltrates or respiratory distress after receiving GRANIX, for ARDS. Discontinue GRANIX in patients with ARDS.5.3 Allergic ReactionsSerious allergic reactions including anaphylaxis can occur in patients receiving human granulocyte colony-stimulating factors. Reactions can occur on initial exposure. The administration of antihistamines‚ steroids‚ bronchodilators‚ and/or epinephrine may reduce the severity of the reactions. Permanently discontinue GRANIX in patients with serious allergic reactions. Do not administer GRANIX to patients with a history of serious allergic reactions to fi lgrastim or pegfi lgrastim.5.4 Use in Patients with Sickle Cell DiseaseSevere and sometimes fatal sickle cell crises can occur in patients with sickle cell disease receiving human granulocyte colony-stimulating factors. Consider the potential risks and ben-efi ts prior to the administration of human granulocyte colony-stimulating factors in patients with sickle cell disease. Discontinue GRANIX in patients undergoing a sickle cell crisis.5.5 Capillary Leak SyndromeCapillary leak syndrome (CLS) can occur in patients receiving human granulocyte colony-stimulating factors and is characterized by hypotension, hypoalbuminemia, edema and hemoconcentration. Episodes vary in frequency, severity and may be life-threatening if treatment is delayed. Patients who develop symptoms of capillary leak syndrome should be closely monitored and receive standard symptomatic treatment, which may include a need for intensive care.5.6 Potential for Tumor Growth Stimulatory Effects on Malignant CellsThe granulocyte colony-stimulating factor (G-CSF) receptor through which GRANIX acts has been found on tumor cell lines. The possibility that GRANIX acts as a growth factor for any tumor type, including myeloid malignancies and myelodysplasia, diseases for which GRANIX is not approved, cannot be excluded.6 ADVERSE REACTIONSThe following potential serious adverse reactions are discussed in greater detail in other sections of the labeling:• Splenic Rupture [see Warnings and Precautions (5.1)]• Acute Respiratory Distress Syndrome [see Warnings and Precautions (5.2)]• Serious Allergic Reactions [see Warnings and Precautions (5.3)]• Use in Patients with Sickle Cell Disease [see Warnings and Precautions (5.4)]• Capillary Leak Syndrome [see Warnings and Precautions (5.5)]• Potential for Tumor Growth Stimulatory Effects on Malignant Cells [see Warnings and
Precautions (5.6)]The most common treatment-emergent adverse reaction that occurred at an incidence of at least 1% or greater in patients treated with GRANIX at the recommended dose and was numerically two times more frequent than in the placebo group was bone pain.6.1 Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not refl ect the rates observed in clinical practice.GRANIX clinical trials safety data are based upon the results of three randomized clinical trials in patients receiving myeloablative chemotherapy for breast cancer (N=348), lung cancer (N=240) and non-Hodgkin’s lymphoma (N=92). In the breast cancer study, 99% of patients were female, the median age was 50 years, and 86% of patients were Caucasian. In the lung cancer study, 80% of patients were male, the median age was 58 years, and 95% of patients were Caucasian. In the non-Hodgkin’s lymphoma study, 52% of patients were male, the median age was 55 years, and 88% of patients were Caucasian. In all three studies a placebo (Cycle 1 of the breast cancer study only) or a non-US-approved fi lgras-tim product were used as controls. Both GRANIX and the non-US-approved fi lgrastim product were administered at 5 mcg/kg subcutaneously once daily beginning one day after chemotherapy for at least fi ve days and continued to a maximum of 14 days or until an ANC of ≥10,000 x 106/L after nadir was reached.
Bone pain was the most frequent treatment-emergent adverse reaction that occurred in at least 1% or greater in patients treated with GRANIX at the recommended dose and was numerically two times more frequent than in the placebo group. The overall incidence of bone pain in Cycle 1 of treatment was 3.4% (3.4% GRANIX, 1.4% placebo, 7.5% non-US-approved fi lgrastim product).LeukocytosisIn clinical studies, leukocytosis (WBC counts > 100,000 x 106/L) was observed in less than 1% patients with non-myeloid malignancies receiving GRANIX. No complications attribut-able to leukocytosis were reported in clinical studies.Additional Adverse ReactionsOther adverse reactions known to occur following administration of human granulocyte colony-stimulating factors include myalgia, headache, vomiting, Sweet’s syndrome (acute febrile neutrophilic dermatosis), cutaneous vasculitis and thrombocytopenia.6.2 ImmunogenicityAs with all therapeutic proteins, there is a potential for immunogenicity. The incidence of antibody development in patients receiving GRANIX has not been adequately determined.7 DRUG INTERACTIONS No formal drug interaction studies between GRANIX and other drugs have been per-formed.Drugs which may potentiate the release of neutrophils‚ such as lithium‚ should be used with caution.Increased hematopoietic activity of the bone marrow in response to growth factor therapy has been associated with transient positive bone imaging changes. This should be consid-ered when interpreting bone-imaging results.8 USE IN SPECIFIC POPULATIONS 8.1 PregnancyPregnancy Category CRisk SummaryThere are no adequate and well-controlled studies of GRANIX in pregnant women. In animal reproduction studies, treatment of pregnant rabbits with tbo-fi lgrastim resulted in increased spontaneous abortion and fetal malformations at systemic exposures substan-tially higher than the human exposure. GRANIX should be used during pregnancy only if the potential benefi t justifi es the potential risk to the fetus.Animal DataIn an embryofetal developmental study, pregnant rabbits were administered subcutaneous doses of tbo-fi lgrastim during the period of organogenesis at 1, 10 and 100 mcg/kg/day. Increased abortions were evident in rabbits treated with tbo-fi lgrastim at 100 mcg/kg/day. This dose was maternally toxic as demonstrated by reduced body weight. Other embry-ofetal fi ndings at this dose level consisted of post-implantation loss‚ decrease in mean live litter size and fetal weight, and fetal malformations such as malformed hindlimbs and cleft palate. The dose of 100 mcg/kg/day corresponds to a systemic exposure (AUC) of approximately 50-90 times the exposures observed in patients treated with the clinical tbo-fi lgrastim dose of 5 mcg/kg/day.8.3 Nursing Mothers It is not known whether tbo-fi lgrastim is secreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when GRANIX is administered to a nursing woman. Other recombinant G-CSF products are poorly secreted in breast milk and G-CSF is not orally absorbed by neonates.8.4 Pediatric Use The safety and effectiveness of GRANIX in pediatric patients have not been established.8.5 Geriatric Use Among 677 cancer patients enrolled in clinical trials of GRANIX, a total of 111 patients were 65 years of age and older. No overall differences in safety or effectiveness were observed between patients age 65 and older and younger patients.8.6 Renal ImpairmentThe safety and effi cacy of GRANIX have not been studied in patients with moderate or severe renal impairment. No dose adjustment is recommended for patients with mild renal impairment.8.7 Hepatic ImpairmentThe safety and effi cacy of GRANIX have not been studied in patients with hepatic impair-ment.10 OVERDOSAGENo case of overdose has been reported.
©2014 Cephalon, Inc., a wholly-owned subsidiary of Teva Pharmaceutical Industries Ltd. All rights reserved.GRANIX is a registered trademark of Teva Pharmaceutical Industries Ltd.Manufactured by: Distributed by:Sicor Biotech UAB Teva Pharmaceuticals USA, Inc.Vilnius, Lithuania North Wales, PA 19454U.S. License No. 1803Product of IsraelGRX-40581 January 2015This brief summary is based on TBO-004 GRANIX full Prescribing Information.
KJob Number: 21089Revision No: 1Date: 01/26/15
724_41865 Page 2 DIGITAL V2

ASCOPost.com | MARCH 25, 2015 PAGE 89
Patient’s Corner
Having Cancer So Early in Life Gave Me PurposeReceiving a diagnosis of choroid plexus carcinoma when I was 24 forced me to follow my passion. The experience has changed my life.By Sheri Sobrato Brisson, MA, as told to Jo Cavallo
I had every classic brain tumor symp-tom in the book—severe headaches,
dizziness, morning nausea—which plagued me for 16 years, starting when I was 8. In college, if I allowed myself to sleep more than 4 hours a night, the morning headaches, which were cen-tered on the top of my head, were so se-vere, I chose to sleep less to try to avoid the pain. This left me so exhausted dur-ing the day that it was difficult to con-centrate on my schoolwork.
Persistent SymptomsMy parents continually tried to find
the source of the problem, and over the ensuing years, I was diagnosed with vari-ous ailments, including migraine head-aches, food allergies, histamine sensi-tivities, and even a sleep disorder. No matter how I adjusted my diet and life-style, the headaches persisted, although their patterns changed over the years, sometimes giving me a 6-month re-prieve and then coming back with such
a vengeance, they would last 3 weeks. I was so young when the symptoms
started that no one ever suspected I could have a brain tumor. Finally, when I was 24, I had a CT scan of my brain, which showed a malignancy in the fourth ventricle. The pathology report said the diagnosis was choroid plexus carcinoma. The cancer was so aggres-sive, my oncologist said I probably had only 6 months to live.
Although the news was, of course,
frightening, since I had had this cancer for most of my life, I didn’t believe it was now about to kill me. I was prescribed an aggressive course of treatment, which included surgery, then radiation therapy followed by combinations of chemotherapy, including vincristine, carboplatin, cisplatin, and cytarabine, the last delivered via a shunt inserted directly to my brain. Because the tu-mor was lodged on my brain stem, the surgeon could not remove it all with-
out jeopardizing my functioning, so I still have a remnant of the tumor left. Thankfully, the mass has remained sta-ble for the past 28 years.
I know how lucky I am to have staved off recurrent disease for nearly 3 decades. And although I have some hearing loss and peripheral neuropathy in my hands from the chemotherapy, there are currently no other late effects from my treatment.
Process of Self-ReflectionDespite the lingering physical and
emotional effects of having cancer at such a young age, I have to say, cancer has given me much more than it has taken away. Driven to succeed in everything I did, early in my career I worked on Wall Street and then was the number-two employee for a company that developed worksite child care. However, I was on a path that at the core was not who I am. So at 24, I was forced to go through a process of self-reflection not many peo-ple that age go through, and it gave me the opportunity to follow my passion of working with children and families cop-ing with serious illness. The experience has changed my life.
I went to graduate school for a de-gree in psychology and for over 15 years facilitated support groups for children with cancer and their families for such organizations as the American Can-cer Society, the National Brain Tumor
Foundation, and Packard Children’s Hospital in California.
The Meaning of CancerWorking with other cancer survivors
and listening to their stories about how their disease affects their lives has made me aware of how important it is for on-cologists to ask their patients not just about the symptoms they are experienc-ing, but about what having the disease has meant to them. The information gleaned from these conversations will not only help oncologists gain an un-derstanding of how best to support their patients emotionally, it may also impact the treatment plan they prescribe.
Having cancer has made me a more authentic person and deepened my relationships with family members and friends. And although it initially stripped away my identity, it allowed me to concentrate on my core values, and that is how I rebuilt my life.
I’m so grateful to have learned these lessons at such a young age. I know that this is the life I was meant to live. n
Sheri Sobrato Brisson, MA, is the Found-er and Manager of Resonance House Publishing and the coauthor with Rose Offner, MFA, of Digging Deep: A Journal for Young People Facing Health Chal-lenges (Resonance House, 2014). She lives in Atherton, California, with her husband and two children.
Despite the lingering physical and emotional effects of having cancer at such a young age, I have to say, cancer has given me much more than it has taken away.
—Sheri Sobrato Brisson, MA
Patient Guides Available Through ASCO University Bookstore• ASCO Answers: Managing the Cost of Cancer Care explains the various costs associated with cancer treatment, including
health-care coverage through the Affordable Care Act. It also provides a list of financial resources available to help offset expenses related to care and tips for organizing financial paperwork. Learn more at www.cancer.net/managingcostofcare.
• ASCO Answers: Survivorship helps patients transition into life after active treatment has finished. In addition to in-formation on the challenges survivors may face and the importance of follow-up care, it includes a blank treatment summary and survivorship care form that patients can fill out with the help of their health-care team. Learn more at www.cancer.net/survivorship.
Copies of these booklets can be purchased through the ASCO University Bookstore at www.cancer.net/estore. All booklets ship for free, and ASCO members receive a 20% discount. n
The ASCO Post Wants to Hear
From YouWe encourage readers to share
their opinions and thoughts on issues of interest to the
oncology community.
Write to The ASCO Post at [email protected]
BRIEF SUMMARY OF PRESCRIBING INFORMATION FORGRANIX® (tbo-fi lgrastim) injection, for subcutaneous useSEE PACKAGE INSERT FOR FULL PRESCRIBING INFORMATION1 INDICATIONS AND USAGEGRANIX is indicated to reduce the duration of severe neutropenia in patients with non-myeloid malignancies receiving myelosuppressive anti-cancer drugs associated with a clinically signifi cant incidence of febrile neutropenia.4 CONTRAINDICATIONSNone.5 WARNINGS AND PRECAUTIONS5.1 Splenic RuptureSplenic rupture, including fatal cases, can occur following administration of human gran-ulocyte colony-stimulating factors. In patients who report upper abdominal or shoulder pain after receiving GRANIX, discontinue GRANIX and evaluate for an enlarged spleen or splenic rupture.5.2 Acute Respiratory Distress Syndrome (ARDS)Acute respiratory distress syndrome (ARDS) can occur in patients receiving human gran-ulocyte colony-stimulating factors. Evaluate patients who develop fever and lung infi ltrates or respiratory distress after receiving GRANIX, for ARDS. Discontinue GRANIX in patients with ARDS.5.3 Allergic ReactionsSerious allergic reactions including anaphylaxis can occur in patients receiving human granulocyte colony-stimulating factors. Reactions can occur on initial exposure. The administration of antihistamines‚ steroids‚ bronchodilators‚ and/or epinephrine may reduce the severity of the reactions. Permanently discontinue GRANIX in patients with serious allergic reactions. Do not administer GRANIX to patients with a history of serious allergic reactions to fi lgrastim or pegfi lgrastim.5.4 Use in Patients with Sickle Cell DiseaseSevere and sometimes fatal sickle cell crises can occur in patients with sickle cell disease receiving human granulocyte colony-stimulating factors. Consider the potential risks and ben-efi ts prior to the administration of human granulocyte colony-stimulating factors in patients with sickle cell disease. Discontinue GRANIX in patients undergoing a sickle cell crisis.5.5 Capillary Leak SyndromeCapillary leak syndrome (CLS) can occur in patients receiving human granulocyte colony-stimulating factors and is characterized by hypotension, hypoalbuminemia, edema and hemoconcentration. Episodes vary in frequency, severity and may be life-threatening if treatment is delayed. Patients who develop symptoms of capillary leak syndrome should be closely monitored and receive standard symptomatic treatment, which may include a need for intensive care.5.6 Potential for Tumor Growth Stimulatory Effects on Malignant CellsThe granulocyte colony-stimulating factor (G-CSF) receptor through which GRANIX acts has been found on tumor cell lines. The possibility that GRANIX acts as a growth factor for any tumor type, including myeloid malignancies and myelodysplasia, diseases for which GRANIX is not approved, cannot be excluded.6 ADVERSE REACTIONSThe following potential serious adverse reactions are discussed in greater detail in other sections of the labeling:• Splenic Rupture [see Warnings and Precautions (5.1)]• Acute Respiratory Distress Syndrome [see Warnings and Precautions (5.2)]• Serious Allergic Reactions [see Warnings and Precautions (5.3)]• Use in Patients with Sickle Cell Disease [see Warnings and Precautions (5.4)]• Capillary Leak Syndrome [see Warnings and Precautions (5.5)]• Potential for Tumor Growth Stimulatory Effects on Malignant Cells [see Warnings and
Precautions (5.6)]The most common treatment-emergent adverse reaction that occurred at an incidence of at least 1% or greater in patients treated with GRANIX at the recommended dose and was numerically two times more frequent than in the placebo group was bone pain.6.1 Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not refl ect the rates observed in clinical practice.GRANIX clinical trials safety data are based upon the results of three randomized clinical trials in patients receiving myeloablative chemotherapy for breast cancer (N=348), lung cancer (N=240) and non-Hodgkin’s lymphoma (N=92). In the breast cancer study, 99% of patients were female, the median age was 50 years, and 86% of patients were Caucasian. In the lung cancer study, 80% of patients were male, the median age was 58 years, and 95% of patients were Caucasian. In the non-Hodgkin’s lymphoma study, 52% of patients were male, the median age was 55 years, and 88% of patients were Caucasian. In all three studies a placebo (Cycle 1 of the breast cancer study only) or a non-US-approved fi lgras-tim product were used as controls. Both GRANIX and the non-US-approved fi lgrastim product were administered at 5 mcg/kg subcutaneously once daily beginning one day after chemotherapy for at least fi ve days and continued to a maximum of 14 days or until an ANC of ≥10,000 x 106/L after nadir was reached.
Bone pain was the most frequent treatment-emergent adverse reaction that occurred in at least 1% or greater in patients treated with GRANIX at the recommended dose and was numerically two times more frequent than in the placebo group. The overall incidence of bone pain in Cycle 1 of treatment was 3.4% (3.4% GRANIX, 1.4% placebo, 7.5% non-US-approved fi lgrastim product).LeukocytosisIn clinical studies, leukocytosis (WBC counts > 100,000 x 106/L) was observed in less than 1% patients with non-myeloid malignancies receiving GRANIX. No complications attribut-able to leukocytosis were reported in clinical studies.Additional Adverse ReactionsOther adverse reactions known to occur following administration of human granulocyte colony-stimulating factors include myalgia, headache, vomiting, Sweet’s syndrome (acute febrile neutrophilic dermatosis), cutaneous vasculitis and thrombocytopenia.6.2 ImmunogenicityAs with all therapeutic proteins, there is a potential for immunogenicity. The incidence of antibody development in patients receiving GRANIX has not been adequately determined.7 DRUG INTERACTIONS No formal drug interaction studies between GRANIX and other drugs have been per-formed.Drugs which may potentiate the release of neutrophils‚ such as lithium‚ should be used with caution.Increased hematopoietic activity of the bone marrow in response to growth factor therapy has been associated with transient positive bone imaging changes. This should be consid-ered when interpreting bone-imaging results.8 USE IN SPECIFIC POPULATIONS 8.1 PregnancyPregnancy Category CRisk SummaryThere are no adequate and well-controlled studies of GRANIX in pregnant women. In animal reproduction studies, treatment of pregnant rabbits with tbo-fi lgrastim resulted in increased spontaneous abortion and fetal malformations at systemic exposures substan-tially higher than the human exposure. GRANIX should be used during pregnancy only if the potential benefi t justifi es the potential risk to the fetus.Animal DataIn an embryofetal developmental study, pregnant rabbits were administered subcutaneous doses of tbo-fi lgrastim during the period of organogenesis at 1, 10 and 100 mcg/kg/day. Increased abortions were evident in rabbits treated with tbo-fi lgrastim at 100 mcg/kg/day. This dose was maternally toxic as demonstrated by reduced body weight. Other embry-ofetal fi ndings at this dose level consisted of post-implantation loss‚ decrease in mean live litter size and fetal weight, and fetal malformations such as malformed hindlimbs and cleft palate. The dose of 100 mcg/kg/day corresponds to a systemic exposure (AUC) of approximately 50-90 times the exposures observed in patients treated with the clinical tbo-fi lgrastim dose of 5 mcg/kg/day.8.3 Nursing Mothers It is not known whether tbo-fi lgrastim is secreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when GRANIX is administered to a nursing woman. Other recombinant G-CSF products are poorly secreted in breast milk and G-CSF is not orally absorbed by neonates.8.4 Pediatric Use The safety and effectiveness of GRANIX in pediatric patients have not been established.8.5 Geriatric Use Among 677 cancer patients enrolled in clinical trials of GRANIX, a total of 111 patients were 65 years of age and older. No overall differences in safety or effectiveness were observed between patients age 65 and older and younger patients.8.6 Renal ImpairmentThe safety and effi cacy of GRANIX have not been studied in patients with moderate or severe renal impairment. No dose adjustment is recommended for patients with mild renal impairment.8.7 Hepatic ImpairmentThe safety and effi cacy of GRANIX have not been studied in patients with hepatic impair-ment.10 OVERDOSAGENo case of overdose has been reported.
©2014 Cephalon, Inc., a wholly-owned subsidiary of Teva Pharmaceutical Industries Ltd. All rights reserved.GRANIX is a registered trademark of Teva Pharmaceutical Industries Ltd.Manufactured by: Distributed by:Sicor Biotech UAB Teva Pharmaceuticals USA, Inc.Vilnius, Lithuania North Wales, PA 19454U.S. License No. 1803Product of IsraelGRX-40581 January 2015This brief summary is based on TBO-004 GRANIX full Prescribing Information.
KJob Number: 21089Revision No: 1Date: 01/26/15
724_41865 Page 2 DIGITAL V2

Prevent bone complications longer
IMPORTANT SAFETY INFORMATION
Hypocalcemia• Pre-existing hypocalcemia must be corrected prior to
initiating therapy with XGEVA®. XGEVA® can cause severe symptomatic hypocalcemia, and fatal cases have been reported. Monitor calcium levels and administer calcium, magnesium, and vitamin D as necessary. Monitor levels more frequently when XGEVA® is administered with other drugs that can also lower calcium levels. Advise patients to contact a healthcare professional for symptoms of hypocalcemia.
• An increased risk of hypocalcemia has been observed in clinical trials of patients with increasing renal dysfunction, most commonly with severe dysfunction (creatinine clearance less than 30 mL/minute and/or on dialysis), and with inadequate/no calcium supplementation. Monitor calcium levels and calcium and vitamin D intake.
Hypersensitivity• XGEVA® is contraindicated in patients with known
clinically signifi cant hypersensitivity to XGEVA®, including anaphylaxis that has been reported with use of XGEVA®. Reactions may include hypotension, dyspnea, upper airway edema, lip swelling, rash, pruritus, and urticaria. If an anaphylactic or other clinically signifi cant allergic reaction occurs, initiate appropriate therapy and discontinue XGEVA® therapy permanently.
Drug Products with Same Active Ingredient• Patients receiving XGEVA® should not take Prolia® (denosumab).
Osteonecrosis of the Jaw• Osteonecrosis of the jaw (ONJ) can occur in patients
receiving XGEVA®, manifesting as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration, or gingival erosion. Persistent pain or slow healing of the mouth or jaw after dental surgery may also be manifestations of ONJ. In clinical trials in patients with osseous metastasis, the incidence of ONJ was higher with longer duration of exposure.
• Perform an oral examination and appropriate preventive dentistry prior to the initiation of XGEVA® and periodically during XGEVA® therapy. Advise patients regarding oral hygiene practices. Avoid invasive dental procedures during treatment with XGEVA®.
• Patients who are suspected of having or who develop ONJ while on XGEVA® should receive care by a dentist or an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition.
Atypical Subtrochanteric and Diaphyseal Femoral Fracture• Atypical femoral fracture has been reported with XGEVA®.
These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar fl are and are transverse or short oblique in orientation without evidence of comminution.
Median Time to First Bone Complication1
XGEVAVAV ®A®A 120 mg Q4W (n = 2,862)
zoledronic acid 4 mg Q4W (n = 2,861)
HR* = 0.83 (95% CI: 0.76-0.90) P < 0.001†
1 YEAR 2 YEARS
19.519.5months
2277..77months
Learn more at XGEVA.com
Data from a prespecifi ed integrated analysis of three international, phase 3, double-blind, double-dummy, active-controlled trials comparing XGEVA® with zoledronic acid for the prevention of bone complications in patients with bone metastases from solid tumors or multiple myeloma.1
*Hazard ratio (HR) is defi ned as the increase or decrease in likelihood of an event of interest (in this case a bone complication) for one group relative to that in a comparator group. †P value for superiority.
In a prespecifi ed integrated analysis of 3 pivotal trials (N = 5,723),XGEVA® was proven to delay the median time to fi rst bone complication by
XGEVA® is a convenient 120 mg subcutaneous injection administered onceevery 4 weeks.2
XGEVA® is indicated for the prevention of skeletal-related events in patients with bone metastases from solid tumors. XGEVA® is not indicated for the prevention of skeletal-related events in patients with multiple myeloma.
• RANK Ligand (RANKL) is produced by bone cells in the skeleton and is a key mediator of bone resorption4
• RANKL production is increased at sites of bone metastases, and stimulates osteoclasts to destroy bone4
• XGEVA® acts precisely to bind RANKL and inhibits osteoclast formation, function, and survival2
• Pre-existing hypocalcemia must be corrected prior to initiating therapy with XGEVA®2
©2014 Amgen Inc. All rights reserved. 04/14 80218-R1-V1 www.XGEVA.com
• Atypical femoral fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g. prednisone) at the time of fracture. During XGEVA® treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Any patient who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patient presenting with an atypical femur fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of XGEVA® therapy should be considered, pending a risk/benefi t assessment, on an individual basis.
Embryo-Fetal Toxicity• XGEVA® can cause fetal harm when administered to a
pregnant woman. Based on fi ndings in animals, XGEVA® is expected to result in adverse reproductive effects.
• Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after the last dose of XGEVA®. Apprise the patient of the potential hazard to a fetus if XGEVA® is used during pregnancy or if the patient becomes pregnant while patients are exposed to XGEVA®.
Adverse Reactions• The most common adverse reactions in patients receiving
XGEVA® with bone metastasis from solid tumors were fatigue/asthenia, hypophosphatemia, and nausea. The most common serious adverse reaction was dyspnea. The most common adverse reactions resulting in discontinuation were osteonecrosis and hypocalcemia.
Please see brief summary of Prescribing Information on the following page.
REFERENCES: 1. Lipton A, Fizazi K, Stopeck AT, et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: a combined analysis of 3 pivotal, randomised, phase 3 trials. Eur J Cancer. 2012;48:3082-3092. 2. XGEVA® (denosumab) prescribing information, Amgen. 3. Brodowicz T, O’Byrne K, Manegold C. Bone matters in lung cancer. Ann Oncol. 2012;23:2215-2222. 4. Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655-1664.
months longer vs zoledronic acid1
months longer vs zoledronic acid 8.2
Bone complications, or skeletal-related events (SREs), are defi ned as radiation to bone, pathologic fracture, surgery to bone, and spinal cord compression.2,3
For patients with bone metastases from solid tumors
DOUS14CDNY3469_A_ONCAd_Tabloid_r11.indd 1 9/19/14 11:47 AM

Prevent bone complications longer
IMPORTANT SAFETY INFORMATION
Hypocalcemia• Pre-existing hypocalcemia must be corrected prior to
initiating therapy with XGEVA®. XGEVA® can cause severe symptomatic hypocalcemia, and fatal cases have been reported. Monitor calcium levels and administer calcium, magnesium, and vitamin D as necessary. Monitor levels more frequently when XGEVA® is administered with other drugs that can also lower calcium levels. Advise patients to contact a healthcare professional for symptoms of hypocalcemia.
• An increased risk of hypocalcemia has been observed in clinical trials of patients with increasing renal dysfunction, most commonly with severe dysfunction (creatinine clearance less than 30 mL/minute and/or on dialysis), and with inadequate/no calcium supplementation. Monitor calcium levels and calcium and vitamin D intake.
Hypersensitivity• XGEVA® is contraindicated in patients with known
clinically signifi cant hypersensitivity to XGEVA®, including anaphylaxis that has been reported with use of XGEVA®. Reactions may include hypotension, dyspnea, upper airway edema, lip swelling, rash, pruritus, and urticaria. If an anaphylactic or other clinically signifi cant allergic reaction occurs, initiate appropriate therapy and discontinue XGEVA® therapy permanently.
Drug Products with Same Active Ingredient• Patients receiving XGEVA® should not take Prolia® (denosumab).
Osteonecrosis of the Jaw• Osteonecrosis of the jaw (ONJ) can occur in patients
receiving XGEVA®, manifesting as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration, or gingival erosion. Persistent pain or slow healing of the mouth or jaw after dental surgery may also be manifestations of ONJ. In clinical trials in patients with osseous metastasis, the incidence of ONJ was higher with longer duration of exposure.
• Perform an oral examination and appropriate preventive dentistry prior to the initiation of XGEVA® and periodically during XGEVA® therapy. Advise patients regarding oral hygiene practices. Avoid invasive dental procedures during treatment with XGEVA®.
• Patients who are suspected of having or who develop ONJ while on XGEVA® should receive care by a dentist or an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition.
Atypical Subtrochanteric and Diaphyseal Femoral Fracture• Atypical femoral fracture has been reported with XGEVA®.
These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar fl are and are transverse or short oblique in orientation without evidence of comminution.
Median Time to First Bone Complication1
XGEVAVAV ®A®A 120 mg Q4W (n = 2,862)
zoledronic acid 4 mg Q4W (n = 2,861)
HR* = 0.83 (95% CI: 0.76-0.90) P < 0.001†
1 YEAR 2 YEARS
19.519.5months
2277..77months
Learn more at XGEVA.com
Data from a prespecifi ed integrated analysis of three international, phase 3, double-blind, double-dummy, active-controlled trials comparing XGEVA® with zoledronic acid for the prevention of bone complications in patients with bone metastases from solid tumors or multiple myeloma.1
*Hazard ratio (HR) is defi ned as the increase or decrease in likelihood of an event of interest (in this case a bone complication) for one group relative to that in a comparator group. †P value for superiority.
In a prespecifi ed integrated analysis of 3 pivotal trials (N = 5,723),XGEVA® was proven to delay the median time to fi rst bone complication by
XGEVA® is a convenient 120 mg subcutaneous injection administered onceevery 4 weeks.2
XGEVA® is indicated for the prevention of skeletal-related events in patients with bone metastases from solid tumors. XGEVA® is not indicated for the prevention of skeletal-related events in patients with multiple myeloma.
• RANK Ligand (RANKL) is produced by bone cells in the skeleton and is a key mediator of bone resorption4
• RANKL production is increased at sites of bone metastases, and stimulates osteoclasts to destroy bone4
• XGEVA® acts precisely to bind RANKL and inhibits osteoclast formation, function, and survival2
• Pre-existing hypocalcemia must be corrected prior to initiating therapy with XGEVA®2
©2014 Amgen Inc. All rights reserved. 04/14 80218-R1-V1 www.XGEVA.com
• Atypical femoral fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g. prednisone) at the time of fracture. During XGEVA® treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Any patient who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patient presenting with an atypical femur fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of XGEVA® therapy should be considered, pending a risk/benefi t assessment, on an individual basis.
Embryo-Fetal Toxicity• XGEVA® can cause fetal harm when administered to a
pregnant woman. Based on fi ndings in animals, XGEVA® is expected to result in adverse reproductive effects.
• Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after the last dose of XGEVA®. Apprise the patient of the potential hazard to a fetus if XGEVA® is used during pregnancy or if the patient becomes pregnant while patients are exposed to XGEVA®.
Adverse Reactions• The most common adverse reactions in patients receiving
XGEVA® with bone metastasis from solid tumors were fatigue/asthenia, hypophosphatemia, and nausea. The most common serious adverse reaction was dyspnea. The most common adverse reactions resulting in discontinuation were osteonecrosis and hypocalcemia.
Please see brief summary of Prescribing Information on the following page.
REFERENCES: 1. Lipton A, Fizazi K, Stopeck AT, et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: a combined analysis of 3 pivotal, randomised, phase 3 trials. Eur J Cancer. 2012;48:3082-3092. 2. XGEVA® (denosumab) prescribing information, Amgen. 3. Brodowicz T, O’Byrne K, Manegold C. Bone matters in lung cancer. Ann Oncol. 2012;23:2215-2222. 4. Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655-1664.
months longer vs zoledronic acid1
months longer vs zoledronic acid 8.2
Bone complications, or skeletal-related events (SREs), are defi ned as radiation to bone, pathologic fracture, surgery to bone, and spinal cord compression.2,3
For patients with bone metastases from solid tumors
DOUS14CDNY3469_A_ONCAd_Tabloid_r11.indd 1 9/19/14 11:47 AM

Brief Summary: Consult package insert for complete Prescribing Information
INDICATIONS AND USAGE:Bone Metastasis from Solid Tumors. Xgeva is indicated for the prevention of skeletal-related events in patients with bone metastases from solid tumors.Important Limitation of Use. Xgeva is not indicated for the prevention of skeletal-related events in patients with multiple myeloma.DOSAGE AND ADMINISTRATION:Recommended Dosage. The recommended dose of Xgeva is 120 mg administered as a subcutaneous injection every 4 weeks in the upper arm, upper thigh, or abdomen. Administer calcium and vitamin D as necessary to treat or prevent hypocalcemia.Preparation and Administration. Visually inspect Xgeva for particulate matter and discoloration prior to administration. Xgeva is a clear, colorless to pale yellow solution that may contain trace amounts of translucent to white proteinaceous particles. Do not use if the solution is discolored or cloudy or if the solution contains many particles or foreign particulate matter. Prior to administration, Xgeva may be removed from the refrigerator and brought to room temperature (up to 25°C/77°F) by standing in the original container. This generally takes 15 to 30 minutes. Do not warm Xgeva in any other way. Use a 27-gauge needle to withdraw and inject the entire contents of the vial. Do not re-enter the vial. Discard vial after single-use or entry.CONTRAINDICATIONS: Hypocalcemia. Pre-existing hypocalcemia must be corrected prior to initiating therapy with Xgeva.Hypersensitivity. Xgeva is contraindicated in patients with known clinically significant hypersensitivity to Xgeva.WARNINGS AND PRECAUTIONS: Drug Products with Same Active Ingredient. Xgeva includes the same active ingredient (denosumab) found in Prolia. Patients receiving Xgeva should not take Prolia.Hypersensitivity. Clinically significant hypersensitivity including anaphylaxis has been reported with use of Xgeva. Reactions may include hypotension, dyspnea, upper airway edema, lip swelling, rash, pruritus, and urticaria. If an anaphylactic or other clinically significant allergic reaction occurs, initiate appropriate therapy and discontinue Xgeva therapy permanently.Hypocalcemia. Xgeva can cause severe symptomatic hypocalcemia, and fatal cases have been reported. Correct pre-existing hypocalcemia prior to Xgeva treatment. Monitor calcium levels and administer calcium, magnesium, and vitamin D as necessary. Monitor levels more frequently when Xgeva is administered with other drugs that can also lower calcium levels. In the postmarketing setting, severe symptomatic hypocalcemia has been reported. Advise patients to contact a healthcare professional for symptoms of hypocalcemia. An increased risk of hypocalcemia has been observed in clinical trials of patients with increasing renal dysfunction, most commonly with severe dysfunction (creatinine clearance less than 30 mL/minute and/or on dialysis), and with inadequate/no calcium supplementation. Monitor calcium levels and calcium and vitamin D intake.Osteonecrosis of the Jaw. Osteonecrosis of the jaw (ONJ) can occur in patients receiving Xgeva, manifesting as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration, or gingival erosion. Persistent pain or slow healing of the mouth or jaw after dental surgery may also be manifestations of ONJ. In clinical trials, in patients with osseous metastasis, the incidence of ONJ was higher with longer duration of exposure (see Adverse Reactions). Seventy-nine percent of patients with ONJ had a history of tooth extraction, poor oral hygiene, or use of a dental appliance as a predisposing factor. Perform an oral examination and appropriate preventive dentistry prior to the initiation of Xgeva and periodically during Xgeva therapy. Advise patients regarding oral hygiene practices. Avoid invasive dental procedures during treatment with Xgeva. Patients who are suspected of having or who develop ONJ while on Xgeva should receive care by a dentist or an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition.Atypical Subtrochanteric and Diaphyseal Femoral Fracture. Atypical femoral fracture has been reported with Xgeva. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution. Atypical femoral fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g. prednisone) at the time of fracture. During Xgeva treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Any patient who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patient presenting with an atypical femur fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of Xgeva therapy should be considered, pending a risk/benefit assessment, on an individual basis.EMBRYO-FETAL TOXICITY: Xgeva can cause fetal harm when administered to a pregnant woman. Based on findings in animals, Xgeva is expected to result in adverse reproductive effects. In utero denosumab exposure in cynomolgus monkeys resulted in increased fetal loss, stillbirths, and postnatal mortality, along with evidence of absent peripheral lymph nodes, abnormal bone growth, and decreased neonatal growth (see Use in Specific Populations). Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after with the last dose of Xgeva. Apprise the patient of the potential hazard to a fetus if Xgeva is used during pregnancy or if the patient becomes pregnant while patients are exposed to Xgeva. Advise patients to contact their healthcare provider if they become pregnant or a pregnancy is suspected during this time.ADVERSE REACTIONS: The following adverse reactions are discussed below and elsewhere in the labeling:• Hypocalcemia• Osteonecrosis of the JawThe most common adverse reactions in patients receiving Xgeva (per-patient incidence greater than or equal to 25%) were fatigue/asthenia, hypophosphatemia, and nausea (see Table 1). The most common serious adverse reaction in patients receiving Xgeva was dyspnea. The most common adverse reactions resulting in discontinuation of Xgeva were osteonecrosis and hypocalcemia.Clinical Trials Experience. Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in practice. The safety of Xgeva was evaluated in three randomized, double-blind, double-dummy trials in which a total of 2841 patients with bone metastasis from prostate cancer, breast cancer, or other solid tumors, or lytic bony lesions from multiple myeloma received at least one dose of Xgeva. In Trials 1, 2, and 3, patients were randomized to receive either 120 mg of Xgeva every 4 weeks as a subcutaneous injection or 4 mg (dose adjusted for reduced renal function) of zoledronic acid every 4 weeks by intravenous (IV) infusion. Entry criteria included serum calcium (corrected) from 8 to 11.5 mg/dL (2 to 2.9 mmol/L) and creatinine clearance 30 mL/min or greater. Patients who had received IV bisphosphonates were excluded, as were patients with prior history of ONJ or osteomyelitis of the
jaw, an active dental or jaw condition requiring oral surgery, non-healed dental/oral surgery, or any planned invasive dental procedure. During the study, serum chemistries including calcium and phosphorus were monitored every 4 weeks. Calcium and vitamin D supplementation was recommended but not required. The median duration of exposure to Xgeva was 12 months (range: 0.1 – 41) and median duration on-study was 13 months (range: 0.1 – 41). Of patients who received Xgeva, 46% were female. Eighty-five percent were White, 5% Hispanic/Latino, 6% Asian, and 3% Black. The median age was 63 years (range: 18 – 93). Seventy-five percent of patients who received Xgeva received concomitant chemotherapy.Table 1. Per-patient Incidence of Selecteda Adverse Reactions of Any Severity (Trials 1, 2, and 3)
a Adverse reactions reported in at least 10% of patients receiving Xgeva in Trials 1, 2, and 3, and meeting one of the following criteria:
• At least 1% greater incidence in Xgeva-treated patients, or • Between-group difference (either direction) of less than 1% and more than
5% greater incidence in patients treated with zoledronic acid compared to placebo (US Prescribing Information for zoledronic acid)
b Laboratory-derived and below the central laboratory lower limit of normal [8.3 – 8.5 mg/dL (2.075 – 2.125 mmol/L) for calcium and 2.2 – 2.8 mg/dL (0.71 – 0.9 mmol/L) for phosphorus]
Severe Mineral/Electrolyte Abnormalities• Severe hypocalcemia (corrected serum calcium less than 7 mg/dL or less
than 1.75 mmol/L) occurred in 3.1% of patients treated with Xgeva and 1.3% of patients treated with zoledronic acid. Of patients who experienced severe hypocalcemia, 33% experienced 2 or more episodes of severe hypocalcemia and 16% experienced 3 or more episodes.
• Severe hypophosphatemia (serum phosphorus less than 2 mg/dL or less than 0.6 mmol/L) occurred in 15.4% of patients treated with Xgeva and 7.4% of patients treated with zoledronic acid.
Osteonecrosis of the JawIn the primary treatment phases of Trials 1, 2, and 3, ONJ was confirmed in 1.8% of patients in the Xgeva group (median exposure of 12.0 months; range 0.1 – 40.5) and 1.3% of patients in the zoledronic acid group. The trials in patients with breast (Trial 1) or prostate (Trial 3) cancer included an Xgeva open label extension treatment phase where patients were offered Xgeva 120 mg once every 4 weeks (median overall exposure of 14.9 months; range 0.1 – 67.2). The patient-year adjusted incidence of confirmed ONJ was 1.1% during the first year of treatment and 4.1% thereafter. The median time to ONJ was 20.6 months (range: 4 – 53).Atypical Subtrochanteric and Diaphyseal FractureAtypical femoral fracture has been reported with Xgeva.Postmarketing Experience. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.The following adverse reactions have been identified during post approval use of Xgeva:• Hypocalcemia: Severe symptomatic hypocalcemia, including fatal cases.• Hypersensitivity, including anaphylactic reactions.• Musculoskeletal pain, including severe musculoskeletal pain. Positive
rechallenge has been reported.Immunogenicity. As with all therapeutic proteins, there is potential for immunogenicity. Using an electrochemiluminescent bridging immunoassay, less than 1% (7/2758) of patients with osseous metastases treated with denosumab doses ranging from 30 – 180 mg every 4 weeks or every 12 weeks for up to 3 years tested positive for binding antibodies. No patient with positive binding antibodies tested positive for neutralizing antibodies as assessed using a chemiluminescent cell-based in vitro biological assay. There was no evidence of altered pharmacokinetic profile, toxicity profile, or clinical response associated with binding antibody development. The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of a positive antibody (including neutralizing antibody) test result may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of antibodies to denosumab with the incidence of antibodies to other products may be misleading.DRUG INTERACTIONS: No formal drug-drug interaction trials have been conducted with Xgeva. In clinical trials in patients with breast cancer metastatic to bone, Xgeva was administered in combination with standard anticancer treatment. Serum denosumab concentrations at 1 and 3 months and reductions in the bone turnover marker uNTx/Cr (urinary N-terminal telopeptide corrected for creatinine) at 3 months were similar in patients with and without prior intravenous bisphosphonate therapy. There was no evidence that various anticancer treatments affected denosumab systemic exposure and pharmacodynamic effect. Serum denosumab concentrations at 1 and 3 months were not altered by concomitant chemotherapy and/or hormone therapy. The median reduction in uNTx/Cr from baseline to month 3 was similar between patients receiving concomitant chemotherapy and/or hormone therapy.USE IN SPECIFIC POPULATIONS:Pregnancy: Category D. Risk Summary: Xgeva can cause fetal harm when administered to a pregnant woman based on findings in animals. In utero denosumab exposure in cynomolgus monkeys resulted in increased fetal loss, stillbirths, and postnatal mortality, along with evidence of absent lymph nodes, abnormal bone growth and decreased neonatal growth. There are no adequate and well-controlled studies with Xgeva in pregnant women. Women should be advised not to become pregnant when taking Xgeva. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women who become pregnant during Xgeva treatment are encouraged to enroll in Amgen’s Pregnancy Surveillance Program. Patients or their physicians should call 1-800-77-AMGEN (1-800-772-6436) to enroll.Clinical Considerations: The effects of Xgeva are likely to be greater during the second and third trimesters of pregnancy. Monoclonal antibodies are transported across the placenta in a linear fashion as pregnancy progresses, with the largest amount transferred during the third trimester. If the patient becomes pregnant during Xgeva therapy, consider the risks and benefits in continuing or discontinuing treatment with Xgeva.
Animal Data: The effects of denosumab on prenatal development have been studied in both cynomolgus monkeys and genetically engineered mice in which RANK ligand (RANKL) expression was turned off by gene removal (a “knockout mouse”). In cynomolgus monkeys dosed subcutaneously with denosumab throughout pregnancy at a pharmacologically active dose, there was increased fetal loss during gestation, stillbirths, and postnatal mortality. Other findings in offspring included absence of axillary, inguinal, mandibular, and mesenteric lymph nodes; abnormal bone growth, reduced bone strength, reduced hematopoiesis, dental dysplasia and tooth malalignment; and decreased neonatal growth. At birth out to one month of age, infants had measurable blood levels of denosumab (22-621% of maternal levels). Following a recovery period from birth out to 6 months of age, the effects on bone quality and strength returned to normal; there were no adverse effects on tooth eruption, though dental dysplasia was still apparent; axillary and inguinal lymph nodes remained absent, while mandibular and mesenteric lymph nodes were present, though small; and minimal to moderate mineralization in multiple tissues was seen in one recovery animal. There was no evidence of maternal harm prior to labor; adverse maternal effects occurred infrequently during labor. Maternal mammary gland development was normal. There was no fetal NOAEL (no observable adverse effect level) established for this study because only one dose of 50 mg/kg was evaluated. In RANKL knockout mice, absence of RANKL (the target of denosumab) also caused fetal lymph node agenesis and led to postnatal impairment of dentition and bone growth. Pregnant RANKL knockout mice showed altered maturation of the maternal mammary gland, leading to impaired lactation.Nursing Mothers. It is not known whether Xgeva is excreted into human milk. Measurable concentrations of denosumab were present in the maternal milk of cynomolgus monkeys up to 1 month after the last dose of denosumab (≤ 0.5% milk:serum ratio). Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Xgeva, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother. Maternal exposure to Xgeva during pregnancy may impair mammary gland development and lactation based on animal studies in pregnant mice lacking the RANK/RANKL signaling pathway that have shown altered maturation of the maternal mammary gland, leading to impaired lactation postpartum. However, in cynomolgus monkeys treated with denosumab throughout pregnancy, maternal mammary gland development was normal, with no impaired lactation. Mammary gland histopathology at 6 months of age was normal in female offspring exposed to denosumab in utero; however, development and lactation have not been fully evaluated.Pediatric Use. Xgeva is not recommended in pediatric patients. The safety and effectiveness of Xgeva in pediatric patients have not been established. Treatment with Xgeva may impair bone growth in children with open growth plates and may inhibit eruption of dentition. In neonatal rats, inhibition of RANKL (the target of Xgeva therapy) with a construct of osteoprotegerin bound to Fc (OPG-Fc) at doses ≤ 10 mg/kg was associated with inhibition of bone growth and tooth eruption. Adolescent primates treated with denosumab at doses 5 and 25 times (10 and 50 mg/kg dose) higher than the recommended human dose of 120 mg administered once every 4 weeks, based on body weight (mg/kg), had abnormal growth plates, considered to be consistent with the pharmacological activity of denosumab. Cynomolgus monkeys exposed in utero to denosumab exhibited bone abnormalities, reduced hematopoiesis, tooth malalignment, decreased neonatal growth, and an absence of axillary, inguinal, mandibular, and mesenteric lymph nodes. Some bone abnormalities recovered once exposure was ceased following birth; however, axillary and inguinal lymph nodes remained absent 6 months post-birth.Geriatric Use. Of patients who received Xgeva in Trials 1, 2, and 3, 1260 (44%) were 65 years of age or older. No overall differences in safety or efficacy were observed between these patients and younger patients.Renal Impairment. Two clinical trials were conducted in patients without cancer and with varying degrees of renal function. In one study, patients (N=55) with varying degrees of renal function (ranging from normal through end-stage renal disease requiring dialysis) received a single 60 mg subcutaneous dose of denosumab. In a second study, patients (N=32) with severe renal dysfunction (creatinine clearance less than 30 mL/minute and/or on dialysis) were given two 120 mg subcutaneous doses of denosumab. In both studies, greater risk of developing hypocalcemia was observed with increasing renal impairment, and with inadequate/no calcium supplementation. Hypocalcemia was mild to moderate in severity in 96% of patients. Monitor calcium levels and, calcium and vitamin D intake.Females and Males of Reproductive Potential. ContraceptionFemales: Counsel patients on pregnancy planning and prevention. Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after the last dose of Xgeva. Advise patients to contact their healthcare provider if they become pregnant, or a pregnancy is suspected, during treatment or within 5 months after the last dose of Xgeva. Males: The extent to which denosumab is present in seminal fluid is unknown. There is potential for fetal exposure to denosumab when a male treated with Xgeva has unprotected sexual intercourse with a pregnant partner. Advise males of this potential risk. OVERDOSAGE: There is no experience with overdosage of Xgeva. HOW SUPPLIED/STORAGE AND HANDLING: Xgeva is supplied in a single-use vial. Store Xgeva in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton. Do not freeze. Once removed from the refrigerator, Xgeva must not be exposed to temperatures above 25°C/77°F or direct light and must be used within 14 days. Discard Xgeva if not used within the 14 days. Do not use Xgeva after the expiry date printed on the label. Protect Xgeva from direct light and heat. Avoid vigorous shaking of Xgeva.PATIENT COUNSELING INFORMATION:Advise patients to contact a healthcare professional for any of the following:• Symptoms of a hypersensitivity reaction, including rash, urticaria, pruritus,
lip swelling, shortness of breath, hypotension and respiratory tract edema• Symptoms of hypocalcemia, including paresthesias or muscle stiffness,
twitching, spasms, or cramps• Symptoms of ONJ, including pain, numbness, swelling of or drainage from
the jaw, mouth, or teeth• Persistent pain or slow healing of the mouth or jaw after dental surgery• Symptoms of atypical femoral fracture, including new or unusual thigh, hip,
or groin pain• Pregnancy or nursingAdvise patients of the need for:• Avoiding therapy with Xgeva if a serious allergic reaction occurred with prior
Xgeva or Prolia therapy• Proper oral hygiene and routine dental care• Informing their dentist that they are receiving Xgeva• Avoiding invasive dental procedures during treatment with Xgeva• The use of highly effective contraception during and for at least 5 months
after treatment with Xgeva for females of reproductive potentialAdvise patients that denosumab is also marketed as Prolia®. Patients should inform their healthcare provider if they are taking Prolia.
Amgen Manufacturing Limited, a subsidiary of Amgen Inc.One Amgen Center DriveThousand Oaks, California 91320-1799©2010-2014 Amgen Inc. All rights reserved.Printed in USA.
Body SystemXgeva
n = 2841 %
Zoledronic Acid n = 2836
%GA STROINTESTINAL
Nausea Diarrhea
31 20
32 19
GE NERAL Fatigue/ Asthenia
45
46
IN VESTIGATIONS Hypocalcemiab
Hypophosphatemiab
18 32
9
20NE UROLOGICAL
Headache
13
14RE SPIRATORY
Dyspnea Cough
21 15
18 15
S:9.5”S
:13
”
DOUS14CDNY4736_XGEVA_Tabloid_BS_V10_8pt_r13.indd 1 8/11/14 11:56 AM
DOUS14CDNY3469_A_ONCAd_Tabloid_BS_r11.indd 1 9/19/14 11:45 AM

ASCOPost.com | MARCH 25, 2015 PAGE 93
In the News
Recognizing Physical Signs Associated With Impending Death Can Assist Clinicians, Patients, and Caregivers With Complex Decisions By Charlotte Bath
In a recently published study of pa-tients with advanced cancer whose
status was systematically documented twice a day, from the time of admission to a palliative care unit until death or discharge, investigators identified eight physical signs associated with death within 3 days. Taken together with five physical signs reported earlier, “these objective bedside signs may assist clini-cians, family members, and researchers in recognizing when the patient has en-tered the final days of life,” David Hui, MD, MSc, and colleagues reported in Cancer.1 That recognition, in turn, can inform physicians’ recommendations concerning continued treatment, as well as personal decisions by caregivers about maintaining a constant presence at the patient’s bedside and alerting others fam-ily members of the impending death.
The eight identified signs, includ-ing seven neurologic conditions and one bleeding complication, had 95% or higher specificity and likelihood ratios
from 6.7 to 16.7 (with likelihood ratios > 5 suggesting good and > 10 suggesting excellent diagnostic value) for predict-ing death within 3 days.
Dr. Hui told The ASCO Post that he had been contacted by several reporters and that the study has been the subject of several editorials and perspectives. Moreover, he expects other articles to evolve over time as the study is dis-cussed in the context of end-of-life care in general. Dr. Hui is Assistant Profes-sor, Department of Palliative Care and
Rehabilitation Medicine and Depart-ment of General Oncology at The Uni-versity of Texas MD Anderson Cancer Center, Houston.
Investigating the Process of Dying
The study reported in Cancer was a planned secondary analysis of the In-vestigating the Process of Dying study, a prospective, longitudinal, observational study that systematically documented an array of clinical signs every 12 hours among 357 consecutive patients with
advanced cancer admitted to acute pal-liative care units at two tertiary care cancer centers—MD Anderson and Barretos Cancer Hospital in Brazil. The average age of the patients was 58; 55% were female, 65% were of Hispanic ori-gin, and 28% had a diagnosis of gastro-intestinal disorder. Of the 357 patients, 203 (57%) died at the end of the admis-sion, the investigators reported.
The researchers compiled a list of 62 clinical signs, selecting 10 as target signs based on their prevalence in the literature. They had also previously identified five physical signs “that were highly diagnostic of impend-ing death.” Based on their findings, which were published in The Oncolo-gist2 (and subsequently reviewed in The ASCO Post3), these five signs are pulselessness of the radial artery, de-creased urine output, Cheyne-Stokes breathing, respiration with mandibu-lar movement, and death rattle.
Caregivers May Want to Know About a Patient’s Impending Death but May Be Afraid to Ask By Charlotte Bath
The likelihood of impending death of patients with advanced
cancer “is one of those questions that many people want to know about, but they are too afraid to ask,” David Hui, MD, MSc, said in an in-terview with The ASCO Post. Dr. Hui is lead author of a study, published in Cancer, on clinical signs of impend-ing death and Assistant Professor, Department of Palliative Care and Rehabilitation Medicine and Depart-ment of General Oncology at The University of Texas MD Anderson Cancer Center, Houston.
Foreseeing and ForetellingPhysicians can use the clinical
signs to estimate probability of death within 3 days, “foreseeing” what is likely to happen to the patient and “foretelling this to the family,” Dr. Hui stated. Although sometimes afraid to bring up the topic of death, “a major-ity of families do want to understand what is happening to the patient,” Dr. Hui said. “Of course, we will ask them, ‘Do you want to know more about
his prognosis?’ If they say, ‘I really do not want to hear anything about that,’ then we will be careful not to poten-tially traumatize them.”
In many cases, Dr. Hui noted, fam-ily members already realize the pa-tient is dying, but will say, “I just don’t know when it is going to happen and I need to plan ahead. I need to be able to decide whether I am going to stay tonight. If he might die over the next few days, I really want to spend the time here. I want to make sure that I get my family members in. When is a good time to get them?”
Individualized Decisions“Of course, each patient is unqiue.
But when the probability of impend-ing death is very high, then we can start to think about making decisions based on this diagnosis,” Dr. Hui stat-ed. “For example, should we continue the daily blood work if the patient has only 3 days to live and the blood work is not going to contribute further to the patient care? This is a very individ-ualized decision that the team needs
to make together with the family. The family will get a better sense of what to do and plan ahead,” Dr. Hui said.
“[Impending death] is not just a simple diagnosis but one that is made with great care and often in consulta-tion with other members of the team looking after the patient. If we all agree this is happening, then we will feel confident in talking to the family about this. We can say, ‘Based on our understanding, there is a very high chance that your loved one will die within the next 3 days.’ That can help families plan ahead during this very stressful time,” Dr. Hui added.
“The more data we have, the more confident that we get. The diagnosis of impending death is one where we don’t want to make a mistake,” he stressed.
“Having data to back up that con-fidence helps me a lot,” Dr. Hui said. “Before this study, I would say, ‘I think this is going to happen. There is a high chance, but I can’t give you a number.’ Now I know there is an 80% to 90% chance that the patient is go-
ing to die in the next 3 days … or even potentially in the next hour,” Dr. Hui clarified.
Caregivers Often Notice the Signs
“When we explain the process of dying to families, we may tell them that the patient’s breathing might change or that it might be hard to recognize the pulse. And then the families say, ‘You know, I noticed that already,’” Dr. Hui said. “They are the ones who are by the bedside a lot of the time, and some of these signs are not difficult to detect.” Dr. Hui said that if the reliabil-ity of the signs to predict impending death is confirmed by further studies, they could prove more useful to fam-ily caregivers as well as clinicians.
“Some of the signs may contrib-ute to our better understanding of the very end of life and can help sup-port decision-making. But we need to recognize that these signs are just one piece of the big puzzle when we do make decisions like that,” Dr. Hui concluded. n
End-of-Life Care
continued on page 94
What is important about this study is that this is the first time we have systematically examined patients for these signs from the very first day they enter our unit.
—David Hui, MD, MSc
Brief Summary: Consult package insert for complete Prescribing Information
INDICATIONS AND USAGE:Bone Metastasis from Solid Tumors. Xgeva is indicated for the prevention of skeletal-related events in patients with bone metastases from solid tumors.Important Limitation of Use. Xgeva is not indicated for the prevention of skeletal-related events in patients with multiple myeloma.DOSAGE AND ADMINISTRATION:Recommended Dosage. The recommended dose of Xgeva is 120 mg administered as a subcutaneous injection every 4 weeks in the upper arm, upper thigh, or abdomen. Administer calcium and vitamin D as necessary to treat or prevent hypocalcemia.Preparation and Administration. Visually inspect Xgeva for particulate matter and discoloration prior to administration. Xgeva is a clear, colorless to pale yellow solution that may contain trace amounts of translucent to white proteinaceous particles. Do not use if the solution is discolored or cloudy or if the solution contains many particles or foreign particulate matter. Prior to administration, Xgeva may be removed from the refrigerator and brought to room temperature (up to 25°C/77°F) by standing in the original container. This generally takes 15 to 30 minutes. Do not warm Xgeva in any other way. Use a 27-gauge needle to withdraw and inject the entire contents of the vial. Do not re-enter the vial. Discard vial after single-use or entry.CONTRAINDICATIONS: Hypocalcemia. Pre-existing hypocalcemia must be corrected prior to initiating therapy with Xgeva.Hypersensitivity. Xgeva is contraindicated in patients with known clinically significant hypersensitivity to Xgeva.WARNINGS AND PRECAUTIONS: Drug Products with Same Active Ingredient. Xgeva includes the same active ingredient (denosumab) found in Prolia. Patients receiving Xgeva should not take Prolia.Hypersensitivity. Clinically significant hypersensitivity including anaphylaxis has been reported with use of Xgeva. Reactions may include hypotension, dyspnea, upper airway edema, lip swelling, rash, pruritus, and urticaria. If an anaphylactic or other clinically significant allergic reaction occurs, initiate appropriate therapy and discontinue Xgeva therapy permanently.Hypocalcemia. Xgeva can cause severe symptomatic hypocalcemia, and fatal cases have been reported. Correct pre-existing hypocalcemia prior to Xgeva treatment. Monitor calcium levels and administer calcium, magnesium, and vitamin D as necessary. Monitor levels more frequently when Xgeva is administered with other drugs that can also lower calcium levels. In the postmarketing setting, severe symptomatic hypocalcemia has been reported. Advise patients to contact a healthcare professional for symptoms of hypocalcemia. An increased risk of hypocalcemia has been observed in clinical trials of patients with increasing renal dysfunction, most commonly with severe dysfunction (creatinine clearance less than 30 mL/minute and/or on dialysis), and with inadequate/no calcium supplementation. Monitor calcium levels and calcium and vitamin D intake.Osteonecrosis of the Jaw. Osteonecrosis of the jaw (ONJ) can occur in patients receiving Xgeva, manifesting as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration, or gingival erosion. Persistent pain or slow healing of the mouth or jaw after dental surgery may also be manifestations of ONJ. In clinical trials, in patients with osseous metastasis, the incidence of ONJ was higher with longer duration of exposure (see Adverse Reactions). Seventy-nine percent of patients with ONJ had a history of tooth extraction, poor oral hygiene, or use of a dental appliance as a predisposing factor. Perform an oral examination and appropriate preventive dentistry prior to the initiation of Xgeva and periodically during Xgeva therapy. Advise patients regarding oral hygiene practices. Avoid invasive dental procedures during treatment with Xgeva. Patients who are suspected of having or who develop ONJ while on Xgeva should receive care by a dentist or an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition.Atypical Subtrochanteric and Diaphyseal Femoral Fracture. Atypical femoral fracture has been reported with Xgeva. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution. Atypical femoral fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g. prednisone) at the time of fracture. During Xgeva treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Any patient who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patient presenting with an atypical femur fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of Xgeva therapy should be considered, pending a risk/benefit assessment, on an individual basis.EMBRYO-FETAL TOXICITY: Xgeva can cause fetal harm when administered to a pregnant woman. Based on findings in animals, Xgeva is expected to result in adverse reproductive effects. In utero denosumab exposure in cynomolgus monkeys resulted in increased fetal loss, stillbirths, and postnatal mortality, along with evidence of absent peripheral lymph nodes, abnormal bone growth, and decreased neonatal growth (see Use in Specific Populations). Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after with the last dose of Xgeva. Apprise the patient of the potential hazard to a fetus if Xgeva is used during pregnancy or if the patient becomes pregnant while patients are exposed to Xgeva. Advise patients to contact their healthcare provider if they become pregnant or a pregnancy is suspected during this time.ADVERSE REACTIONS: The following adverse reactions are discussed below and elsewhere in the labeling:• Hypocalcemia• Osteonecrosis of the JawThe most common adverse reactions in patients receiving Xgeva (per-patient incidence greater than or equal to 25%) were fatigue/asthenia, hypophosphatemia, and nausea (see Table 1). The most common serious adverse reaction in patients receiving Xgeva was dyspnea. The most common adverse reactions resulting in discontinuation of Xgeva were osteonecrosis and hypocalcemia.Clinical Trials Experience. Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in practice. The safety of Xgeva was evaluated in three randomized, double-blind, double-dummy trials in which a total of 2841 patients with bone metastasis from prostate cancer, breast cancer, or other solid tumors, or lytic bony lesions from multiple myeloma received at least one dose of Xgeva. In Trials 1, 2, and 3, patients were randomized to receive either 120 mg of Xgeva every 4 weeks as a subcutaneous injection or 4 mg (dose adjusted for reduced renal function) of zoledronic acid every 4 weeks by intravenous (IV) infusion. Entry criteria included serum calcium (corrected) from 8 to 11.5 mg/dL (2 to 2.9 mmol/L) and creatinine clearance 30 mL/min or greater. Patients who had received IV bisphosphonates were excluded, as were patients with prior history of ONJ or osteomyelitis of the
jaw, an active dental or jaw condition requiring oral surgery, non-healed dental/oral surgery, or any planned invasive dental procedure. During the study, serum chemistries including calcium and phosphorus were monitored every 4 weeks. Calcium and vitamin D supplementation was recommended but not required. The median duration of exposure to Xgeva was 12 months (range: 0.1 – 41) and median duration on-study was 13 months (range: 0.1 – 41). Of patients who received Xgeva, 46% were female. Eighty-five percent were White, 5% Hispanic/Latino, 6% Asian, and 3% Black. The median age was 63 years (range: 18 – 93). Seventy-five percent of patients who received Xgeva received concomitant chemotherapy.Table 1. Per-patient Incidence of Selecteda Adverse Reactions of Any Severity (Trials 1, 2, and 3)
a Adverse reactions reported in at least 10% of patients receiving Xgeva in Trials 1, 2, and 3, and meeting one of the following criteria:
• At least 1% greater incidence in Xgeva-treated patients, or • Between-group difference (either direction) of less than 1% and more than
5% greater incidence in patients treated with zoledronic acid compared to placebo (US Prescribing Information for zoledronic acid)
b Laboratory-derived and below the central laboratory lower limit of normal [8.3 – 8.5 mg/dL (2.075 – 2.125 mmol/L) for calcium and 2.2 – 2.8 mg/dL (0.71 – 0.9 mmol/L) for phosphorus]
Severe Mineral/Electrolyte Abnormalities• Severe hypocalcemia (corrected serum calcium less than 7 mg/dL or less
than 1.75 mmol/L) occurred in 3.1% of patients treated with Xgeva and 1.3% of patients treated with zoledronic acid. Of patients who experienced severe hypocalcemia, 33% experienced 2 or more episodes of severe hypocalcemia and 16% experienced 3 or more episodes.
• Severe hypophosphatemia (serum phosphorus less than 2 mg/dL or less than 0.6 mmol/L) occurred in 15.4% of patients treated with Xgeva and 7.4% of patients treated with zoledronic acid.
Osteonecrosis of the JawIn the primary treatment phases of Trials 1, 2, and 3, ONJ was confirmed in 1.8% of patients in the Xgeva group (median exposure of 12.0 months; range 0.1 – 40.5) and 1.3% of patients in the zoledronic acid group. The trials in patients with breast (Trial 1) or prostate (Trial 3) cancer included an Xgeva open label extension treatment phase where patients were offered Xgeva 120 mg once every 4 weeks (median overall exposure of 14.9 months; range 0.1 – 67.2). The patient-year adjusted incidence of confirmed ONJ was 1.1% during the first year of treatment and 4.1% thereafter. The median time to ONJ was 20.6 months (range: 4 – 53).Atypical Subtrochanteric and Diaphyseal FractureAtypical femoral fracture has been reported with Xgeva.Postmarketing Experience. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.The following adverse reactions have been identified during post approval use of Xgeva:• Hypocalcemia: Severe symptomatic hypocalcemia, including fatal cases.• Hypersensitivity, including anaphylactic reactions.• Musculoskeletal pain, including severe musculoskeletal pain. Positive
rechallenge has been reported.Immunogenicity. As with all therapeutic proteins, there is potential for immunogenicity. Using an electrochemiluminescent bridging immunoassay, less than 1% (7/2758) of patients with osseous metastases treated with denosumab doses ranging from 30 – 180 mg every 4 weeks or every 12 weeks for up to 3 years tested positive for binding antibodies. No patient with positive binding antibodies tested positive for neutralizing antibodies as assessed using a chemiluminescent cell-based in vitro biological assay. There was no evidence of altered pharmacokinetic profile, toxicity profile, or clinical response associated with binding antibody development. The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of a positive antibody (including neutralizing antibody) test result may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of antibodies to denosumab with the incidence of antibodies to other products may be misleading.DRUG INTERACTIONS: No formal drug-drug interaction trials have been conducted with Xgeva. In clinical trials in patients with breast cancer metastatic to bone, Xgeva was administered in combination with standard anticancer treatment. Serum denosumab concentrations at 1 and 3 months and reductions in the bone turnover marker uNTx/Cr (urinary N-terminal telopeptide corrected for creatinine) at 3 months were similar in patients with and without prior intravenous bisphosphonate therapy. There was no evidence that various anticancer treatments affected denosumab systemic exposure and pharmacodynamic effect. Serum denosumab concentrations at 1 and 3 months were not altered by concomitant chemotherapy and/or hormone therapy. The median reduction in uNTx/Cr from baseline to month 3 was similar between patients receiving concomitant chemotherapy and/or hormone therapy.USE IN SPECIFIC POPULATIONS:Pregnancy: Category D. Risk Summary: Xgeva can cause fetal harm when administered to a pregnant woman based on findings in animals. In utero denosumab exposure in cynomolgus monkeys resulted in increased fetal loss, stillbirths, and postnatal mortality, along with evidence of absent lymph nodes, abnormal bone growth and decreased neonatal growth. There are no adequate and well-controlled studies with Xgeva in pregnant women. Women should be advised not to become pregnant when taking Xgeva. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women who become pregnant during Xgeva treatment are encouraged to enroll in Amgen’s Pregnancy Surveillance Program. Patients or their physicians should call 1-800-77-AMGEN (1-800-772-6436) to enroll.Clinical Considerations: The effects of Xgeva are likely to be greater during the second and third trimesters of pregnancy. Monoclonal antibodies are transported across the placenta in a linear fashion as pregnancy progresses, with the largest amount transferred during the third trimester. If the patient becomes pregnant during Xgeva therapy, consider the risks and benefits in continuing or discontinuing treatment with Xgeva.
Animal Data: The effects of denosumab on prenatal development have been studied in both cynomolgus monkeys and genetically engineered mice in which RANK ligand (RANKL) expression was turned off by gene removal (a “knockout mouse”). In cynomolgus monkeys dosed subcutaneously with denosumab throughout pregnancy at a pharmacologically active dose, there was increased fetal loss during gestation, stillbirths, and postnatal mortality. Other findings in offspring included absence of axillary, inguinal, mandibular, and mesenteric lymph nodes; abnormal bone growth, reduced bone strength, reduced hematopoiesis, dental dysplasia and tooth malalignment; and decreased neonatal growth. At birth out to one month of age, infants had measurable blood levels of denosumab (22-621% of maternal levels). Following a recovery period from birth out to 6 months of age, the effects on bone quality and strength returned to normal; there were no adverse effects on tooth eruption, though dental dysplasia was still apparent; axillary and inguinal lymph nodes remained absent, while mandibular and mesenteric lymph nodes were present, though small; and minimal to moderate mineralization in multiple tissues was seen in one recovery animal. There was no evidence of maternal harm prior to labor; adverse maternal effects occurred infrequently during labor. Maternal mammary gland development was normal. There was no fetal NOAEL (no observable adverse effect level) established for this study because only one dose of 50 mg/kg was evaluated. In RANKL knockout mice, absence of RANKL (the target of denosumab) also caused fetal lymph node agenesis and led to postnatal impairment of dentition and bone growth. Pregnant RANKL knockout mice showed altered maturation of the maternal mammary gland, leading to impaired lactation.Nursing Mothers. It is not known whether Xgeva is excreted into human milk. Measurable concentrations of denosumab were present in the maternal milk of cynomolgus monkeys up to 1 month after the last dose of denosumab (≤ 0.5% milk:serum ratio). Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Xgeva, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother. Maternal exposure to Xgeva during pregnancy may impair mammary gland development and lactation based on animal studies in pregnant mice lacking the RANK/RANKL signaling pathway that have shown altered maturation of the maternal mammary gland, leading to impaired lactation postpartum. However, in cynomolgus monkeys treated with denosumab throughout pregnancy, maternal mammary gland development was normal, with no impaired lactation. Mammary gland histopathology at 6 months of age was normal in female offspring exposed to denosumab in utero; however, development and lactation have not been fully evaluated.Pediatric Use. Xgeva is not recommended in pediatric patients. The safety and effectiveness of Xgeva in pediatric patients have not been established. Treatment with Xgeva may impair bone growth in children with open growth plates and may inhibit eruption of dentition. In neonatal rats, inhibition of RANKL (the target of Xgeva therapy) with a construct of osteoprotegerin bound to Fc (OPG-Fc) at doses ≤ 10 mg/kg was associated with inhibition of bone growth and tooth eruption. Adolescent primates treated with denosumab at doses 5 and 25 times (10 and 50 mg/kg dose) higher than the recommended human dose of 120 mg administered once every 4 weeks, based on body weight (mg/kg), had abnormal growth plates, considered to be consistent with the pharmacological activity of denosumab. Cynomolgus monkeys exposed in utero to denosumab exhibited bone abnormalities, reduced hematopoiesis, tooth malalignment, decreased neonatal growth, and an absence of axillary, inguinal, mandibular, and mesenteric lymph nodes. Some bone abnormalities recovered once exposure was ceased following birth; however, axillary and inguinal lymph nodes remained absent 6 months post-birth.Geriatric Use. Of patients who received Xgeva in Trials 1, 2, and 3, 1260 (44%) were 65 years of age or older. No overall differences in safety or efficacy were observed between these patients and younger patients.Renal Impairment. Two clinical trials were conducted in patients without cancer and with varying degrees of renal function. In one study, patients (N=55) with varying degrees of renal function (ranging from normal through end-stage renal disease requiring dialysis) received a single 60 mg subcutaneous dose of denosumab. In a second study, patients (N=32) with severe renal dysfunction (creatinine clearance less than 30 mL/minute and/or on dialysis) were given two 120 mg subcutaneous doses of denosumab. In both studies, greater risk of developing hypocalcemia was observed with increasing renal impairment, and with inadequate/no calcium supplementation. Hypocalcemia was mild to moderate in severity in 96% of patients. Monitor calcium levels and, calcium and vitamin D intake.Females and Males of Reproductive Potential. ContraceptionFemales: Counsel patients on pregnancy planning and prevention. Advise females of reproductive potential to use highly effective contraception during therapy, and for at least 5 months after the last dose of Xgeva. Advise patients to contact their healthcare provider if they become pregnant, or a pregnancy is suspected, during treatment or within 5 months after the last dose of Xgeva. Males: The extent to which denosumab is present in seminal fluid is unknown. There is potential for fetal exposure to denosumab when a male treated with Xgeva has unprotected sexual intercourse with a pregnant partner. Advise males of this potential risk. OVERDOSAGE: There is no experience with overdosage of Xgeva. HOW SUPPLIED/STORAGE AND HANDLING: Xgeva is supplied in a single-use vial. Store Xgeva in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton. Do not freeze. Once removed from the refrigerator, Xgeva must not be exposed to temperatures above 25°C/77°F or direct light and must be used within 14 days. Discard Xgeva if not used within the 14 days. Do not use Xgeva after the expiry date printed on the label. Protect Xgeva from direct light and heat. Avoid vigorous shaking of Xgeva.PATIENT COUNSELING INFORMATION:Advise patients to contact a healthcare professional for any of the following:• Symptoms of a hypersensitivity reaction, including rash, urticaria, pruritus,
lip swelling, shortness of breath, hypotension and respiratory tract edema• Symptoms of hypocalcemia, including paresthesias or muscle stiffness,
twitching, spasms, or cramps• Symptoms of ONJ, including pain, numbness, swelling of or drainage from
the jaw, mouth, or teeth• Persistent pain or slow healing of the mouth or jaw after dental surgery• Symptoms of atypical femoral fracture, including new or unusual thigh, hip,
or groin pain• Pregnancy or nursingAdvise patients of the need for:• Avoiding therapy with Xgeva if a serious allergic reaction occurred with prior
Xgeva or Prolia therapy• Proper oral hygiene and routine dental care• Informing their dentist that they are receiving Xgeva• Avoiding invasive dental procedures during treatment with Xgeva• The use of highly effective contraception during and for at least 5 months
after treatment with Xgeva for females of reproductive potentialAdvise patients that denosumab is also marketed as Prolia®. Patients should inform their healthcare provider if they are taking Prolia.
Amgen Manufacturing Limited, a subsidiary of Amgen Inc.One Amgen Center DriveThousand Oaks, California 91320-1799©2010-2014 Amgen Inc. All rights reserved.Printed in USA.
Body SystemXgeva
n = 2841 %
Zoledronic Acid n = 2836
%GA STROINTESTINAL
Nausea Diarrhea
31 20
32 19
GE NERAL Fatigue/ Asthenia
45
46
IN VESTIGATIONS Hypocalcemiab
Hypophosphatemiab
18 32
9
20NE UROLOGICAL
Headache
13
14RE SPIRATORY
Dyspnea Cough
21 15
18 15
S:9.5”
S:1
3”
DOUS14CDNY4736_XGEVA_Tabloid_BS_V10_8pt_r13.indd 1 8/11/14 11:56 AM
DOUS14CDNY3469_A_ONCAd_Tabloid_BS_r11.indd 1 9/19/14 11:45 AM

PAGE 94 The ASCO Post | MARCH 25, 2015
In the News
Powerful PredictorsThe secondary analysis looked at
the remaining 52 physical signs. “The 10 signs that we looked at initially were the more commonly cited signs that people were already pretty sure about, and for this study, we were looking into the things that we are not as sure about. We found that 8 of these 52 signs were quite powerful in predicting death,” Dr. Hui said in an interview with The ASCO Post.
The researchers explained: To determine the diagnostic utility of each sign, we computed the sensitivity, specificity, positive [likelihood ratio], and negative [likelihood ratio] for death within 3 days. Positive [likelihood ra-tios] indicate how many times the sign of interest appeared in patients who died within 3 days in comparison with patients who did not die within 3 days. Positive [likelihood ratios] > 5 and > 10 suggest good and excellent discrimina-tory test performance, respectively. The last 3 days was chosen as the cutoff for impending death because our previous study showed the emergence of many of the signs of impending death during this period, and knowing a patient is within the last 3 days of life could affect many medical decisions such as hospital dis-charge, discontinuation of prescription medications, artificial nutrition, and use of life- support measures.
The eight physical signs identi-fied as highly diagnostic of impend-ing death “occurred in 5% to 78% of the patients within the last 3 days of life, had a late onset, and had a high specificity (> 95%) and a high positive [likelihood ratio] for death within 3 days,” the authors noted. Listed from
high to low likelihood ratios, the 7 neurologic signs included nonreac-tive pupils (likelihood ratio = 16.7), inability to close eyelids (likelihood ratio = 13.6), grunting of vocal cords (likelihood ratio = 11.8), decreased response to verbal stimuli (likelihood ratio = 8.3), drooping of the nasolabial fold (likelihood ratio = 8.3), hyperex-tension of the neck (likelihood ratio = 7.3), and decreased response to visual stimuli (likelihood ratio = 6.7). In ad-dition, upper gastrointestinal bleeding had a likelihood ratio of 10.3.
On multivariate analysis, two physi-cal signs (ie, decreased response to verbal stimuli and drooping of the nasolabial fold) were identified as in-dependently significant, according to the study report. “However, the other physical signs with high positive [likeli-hood ratios], when present, could still be helpful in the diagnosis of impending death. This is particularly true because patients often do not present with all the physical signs at the same time.”
It is important for clinicians to real-ize, Dr. Hui noted, that “if they don’t see the signs, it doesn’t mean that their pa-tient won’t die in the next 3 days. These signs don’t always occur in patients. So the flip side is not always true.”
Systematic Examination From Day 1
Physicians have been aware of some of the signs of impending death, such as decreased level of consciousness, respon-siveness, grunting of vocal chords, non-reactive pupils, Dr. Hui acknowledged, “but prior to this study, these signs had not really been examined systematically. What is important about this study is that this is the first time we have system-atically examined patients for a large and comprehensive array of these signs in the
largest population to date from the very first day they enter our unit. Twice a day, we monitored these signs, and we con-tinued that monitoring until they either died or were discharged.”
Previous studies that have looked at signs of impending death “tended to start monitoring when people are al-ready recognized as dying, and so there may be potential bias. That is why we started out from the very beginning to try to get an unselected and consecutive patient population to make sure that our findings are valid,” Dr. Hui added.
Signs Add Up to Higher Probability
In addition to the reported results, further analysis of the data showed that a “higher number of signs tends to be associated with a higher probability of death,” Dr. Hui said. “Just to put things in context, let’s say that when patients come into a palliative care unit, there is maybe a 30% to 40% chance that they would die over the next 3 days.” If a pa-tient, however, displays one of the eight signs, and that sign has a likelihood ratio of 10, “it would increase the probability to about 80% to 90% that they would die in the next 3 days. Then, if another sign is added, the probability of death would rise above 90%,” Dr. Hui added.
Additional signs could raise the probability to 95% or 98%. “That is how we generally can use these signs,” Dr. Hui said. More signs can be confirma-tory, but “even just one or two of these signs can be very informative.”
Next Generation of Studies Dr. Hui and his colleagues cautioned
that the results concerning the reliability of the eight signs to predict death within 3 days among patients with advanced cancer should be considered preliminary.
“At this time, I would say that they pro-vide good preliminary information, but, of course, I always want more confirma-tory data,” Dr. Hui stated. “I do believe that this finding is at least applicable to the study population that we had, which was a unique population in the palliative care unit, as not too many hospitals have palliative care units.”
Further studies “are actively in the planning phase right now,” Dr. Hui said. “We are not enrolling patients yet, but based on the findings from both studies, we are crafting the next generation of studies to further confirm our findings.”
Whether the next round of studies will involve Dr. Hui’s study partners in Brazil or others “depends on funding op-portunities,” he noted, “but we definitely want to expand into other populations, such as those in hospice care. A large proportion of people are under hospice care in the last days of life, and I think it is very important to see whether these signs hold outside of a hospital. General-ly we believe they are universal in nature, but what distinguishes science from art is that we really want to get the numbers right by documenting things carefully.” n
Disclosure: Dr. Hui reported no potential conflicts of interest.
References1. Hui D, dos Santos R, Chisholm G,
et al: Bedside clinical signs associated with impending death in patients with advanced cancer: Preliminary findings of a prospec-tive, longitudinal cohort study. Cancer 121:960-967, 2015.
2. Hui D, dos Santos R, Chisholm G, et al: Clinical signs of impending death in can-cer patients. Oncologist 19:681-687, 2014.
3. Piana R: Identifying impending death helps patients and caregivers. A conversa-tion with David Hui, MD, MSc. ASCO Post, September 1, 2014.
Signs of Impending Deathcontinued from page 93
Like us on Facebook
facebook.com/TheASCOPost
The ASCO Post

ASCOPost.com | MARCH 25, 2015 PAGE 95
In the Literature
Emerging Clinical Data on Cancer Management
PROSTATE CANCER
Selenium Supplements After Diagnosis of Nonmetastatic Prostate Cancer May Raise Prostate Cancer Mortality Risk
“Selenium supplementation of 140 or more μg/d after diagnosis of non-metastatic prostate cancer may increase risk of prostate cancer mortality,” ac-cording to a prospective study follow-ing 4,459 men initially diagnosed with nonmetastatic prostate cancer in the Health Professionals Follow-up Study from 1988 through 2010. “Caution is warranted regarding usage of such supplements among men with prostate cancer,” Stacey A. Kenfield, ScD, of the University of California, San Fran-cisco, and coauthors advised in the Jour-nal of the National Cancer Institute.
The Health Professionals Follow-up Study is a prospective cohort study of 51,529 U.S. male health professionals be-tween the age of 40 and 75 at the time of enrollment in 1986. Participants complet-ed a baseline questionnaire that included extensive data, such as a medical history and lifestyle factors (diet and supplement use), and detailed information on the use and dose of supplements every 2 years.
“Total selenium supplement intake was calculated as the sum from multivi-tamins and selenium supplements,” the investigators explained. The categories of total selenium supplement dosage follow: nonuser, 1 to 24 μg/d, 25 to 139 μg/d, and 140 or more μg/d. “We also calculat-ed total duration of use to assess whether the association between selenium supple-ment use and prostate cancer mortality differed by duration,” the authors noted.
The primary outcome in the current study was prostate cancer mortality. Sec-ondary outcomes were biochemical re-currence, overall mortality, and cardiovas-cular mortality. After study participants reported a prostate cancer diagnosis, the investigators obtained medical records “to confirm the diagnosis and record the clin-ical T stage, Gleason score, treatments, prostate-specific antigen (PSA) values at diagnosis, PSA levels after treatment (to identify events of biochemical tumor recurrence), and metastasis. Participants also completed biennial follow-up ques-tionnaires to update data on treatments, PSA levels, and clinical progression,” the researchers explained.
“We documented 965 deaths, 226 (23.4%) from prostate cancer and 267 (27.7%) from cardiovascular disease, dur-ing a median follow-up of 8.9 years. In the
biochemical recurrence analysis, we doc-umented 762 tumor recurrences during a median follow-up of 7.8 years. Crude rates per 1,000 person-years for prostate cancer death were 5.6 among selenium nonusers and 10.5 among men who consumed 140
or more μg/d,” the researchers reported. “In multivariable analyses, men who
consumed 1 to 24 μg/d, 25 to 139 μg/d, and 140 or more μg/d of supplemental selenium had a 1.18 (95% confidence in-terval [CI] = 0.73–1.91), 1.33 (95% CI
= 0.77–2.30), and 2.60-fold (95% CI = 1.44–4.70) greater risk of prostate cancer mortality compared with nonusers, re-spectively, Ptrend = .001. There were no sta-tistically significant associations between
continued on page 96
ASCO in Action
ASCO in Action covers the latest news on policy developments and ASCO’s advocacy positions on everything from Medicare reimbursement to federal funding for clinical cancer research to patient access to high-quality care. Find timely information on clinical affairs, government relations, and quality of care issues that affect cancer practice, research, and patient care.
ASCO in Action also includes a variety of advocacy tools for ASCO members, including policy priorities, policy statements, and ASCO’s ACT (Alert Congress Now) Network.
Check out ASCO in Action today!ascoaction.asco.org
Check out ASCO in Action on ASCO.org
Access all the latest policy and advocacy news in the Advocacy section of ASCO.org.

PAGE 96 The ASCO Post | MARCH 25, 2015
In the Literature
the use of selenium supplements and bio-chemical recurrence, cardiovascular dis-ease mortality, and overall mortality.
The authors noted that their study “is the first study to examine the relation of selenium supplements taken after diag-nosis with risk of prostate cancer mortal-ity.” Findings from previous studies of se-lenium supplementation by healthy men have included an inverse relationship between selenium levels and incident-advanced prostate cancer, no associa-tion for incident prostate cancer but an increased risk for high-grade prostate cancer, and an increased risk of prostate cancer mortality when selenium supple-ments were taken prior to diagnosis.
“These data underscore the poten-tially complex and variable role that lifestyle factors may play in the long etiologic course of some cancers, in particular that risk factors for incidence may be very different from those for mortality,” the investigators observed. “Associations between a given exposure and incident prostate cancer presumably reflect biologic effects of the exposure in the prostate gland, whereas associations with metastatic/fatal disease may reflect effects of the exposure in the prostate (if the tumor is still present) and/or effects on other organs or systems that influ-ence the likelihood of metastatic disease developing and spreading. Additional studies with long-term follow-up of life-style factors before and after cancer diag-nosis are warranted.”
Kenfield SA, et al: J Natl Cancer Inst 107:360, 2014 (print January 2015).
BREAST CANCER
Nearly Half of Women Taking Tamoxifen for Primary Prevention Discontinue Its Use Before 5 Years
After 4.5 years of taking tamoxifen for primary prevention of breast cancer, 46% of women discontinued its use, ac-cording to research conducted within the Sister Study, a prospective cohort of women who had a sister diagnosed with breast cancer but did not have breast cancer themselves. Eligible women were between the ages of 35 and 74 at enroll-ment. Researchers examined charac-teristics associated with initiating and discontinuing tamoxifen for primary prevention of breast cancer.
Tamoxifen has been approved by the U.S. Food and Drug Administration for primary prevention of breast cancer since 1998. “The U.S. Preventive Servic-
es Task Force (USPSTF) recently pub-lished a recommendation statement for risk reduction of primary breast cancer that encourages clinicians to offer to pre-scribe tamoxifen or raloxifene to reduce breast cancer risk,” Hazel B. Nichols, PhD, of the University of North Caro-lina Gillings School of Global Public Health, Chapel Hill, noted in the Journal of the National Cancer Institute.
“National estimates indicate that less than 1% of eligible women use tamoxi-fen for prevention. It is unknown why so few women use tamoxifen for breast cancer prevention, although vasomo-tor symptoms, increased risk of adverse health effects, and difficulties in estimat-ing or communicating risk-benefit pro-files can be deterrents.”
From 2003 to 2009, the Sister Study recruited 50,884 women in the United States and Puerto Rico. The current trial “identified 788 tamoxifen users and 3,131 nonusers matched on age and year of enrollment who had no history of contraindicating factors (stroke, tran-sient ischemic attack, cataract, endome-trial or uterine cancer),” the investigators stated. Study participants self-reported tamoxifen use, ages when they started and stopped taking tamoxifen, and total duration of its use at enrollment.
Women were categorized according to a risk-benefit index recommended by the USPSTF and the National Com-prehensive Cancer Network (NCCN). “Briefly, the risk-benefit index classi-fies women according to the level of evidence (none, moderate, strong) for tamoxifen benefits (breast cancer and fracture prevention) to exceed the risk of serious side effects (endometrial can-cer, stroke, pulmonary embolism, deep vein thrombosis, and cataract) using age, 5-year projected risk of invasive breast cancer, hysterectomy, and race,” the au-thors explained.
“Overall, 74% of classified tamoxifen users had a favorable (defined as mod-erate to strong evidence for benefits to exceed risks) risk-benefit profile, and 20% had no evidence that the benefits exceeded risks; the risk-benefit index could not be calculated for 6%,” the re-searchers reported. “Absence of expect-ed benefit was most pronounced for older women, African Americans, and those with an intact uterus,” the authors added.
“Discontinuation of tamoxifen be-fore the recommended 5 years (46%) was somewhat greater than the 24% to 36% nonadherence reported in preven-tion trials,” the authors acknowledged, but it “is closely aligned with a recent metaregression estimate of 47.2% 5-year
discontinuation of tamoxifen as adjuvant endocrine therapy.”
The median duration of tamoxifen use among those who discontinued treatment was 3 years. “Raloxifene use after tamoxifen was associated with early discontinuation, potentially because of the availability of an alternative pre-ventive therapy,” the researchers wrote. Women who reported a family history of breast cancer, BRCA1/2 testing, or posi-tive mutation status appeared less likely to complete 5 years of tamoxifen.
The authors concluded: “While the majority of women who used tamoxifen for primary prevention of breast cancer were likely to benefit, substantial discon-tinuation of tamoxifen before 5 years and use by women at risk of serious side effects may attenuate benefits for breast cancer prevention.”
Nichols HB, et al: J Natl Cancer Inst 107:354, 2014 (print January 2015).
CHRONIC PAIN
Key Evidence Gaps and Research Priorities Should Be Addressed So Physicians Can Identify Patients Most Likely to Benefit From Opioids
Key evidence gaps and research pri-orities must be addressed “so that physi-cians can recognize patients for whom opioids are most appropriate and use optimal regimens for these patients,” according to the National Institutes of Health (NIH) Pathways to Prevention Workshop final report on the role of opi-oids in the treatment of chronic pain.
The workshop panel called for federal and nonfederal agencies to sponsor re-search “to identify which types of pain,
specific diseases, and patients are most likely to benefit and incur harm from opioids,” as well as the development of multidisciplinary pain interventions. “In the absence of definitive evidence, clini-cians and health-care systems should fol-low current guidelines by professional societies about which patients and which types of pain should be treated with opioids and about how best to monitor patients and mitigate risk for harm,” the panel recommended.
Among the clinical issues considered by the panel were the challenge of appro-priate patient selection and how the type of pain could influence its management. “Data were presented on three distinct pain mechanisms: peripheral nocicep-tive (caused by tissue damage or inflam-mation), peripheral neuropathic (caused by damage or dysfunction of peripheral nerves), and centralized (characterized by a disturbance in the processing of pain by the brain and spinal cord). Persons with more peripheral nociceptive pain (such as acute pain due to injury, rheumatoid ar-thritis, or cancer pain) may respond better to opioid analgesics,” the panel stated.
The lead author of the report was David B. Reuben, MD, of the David Geffen School of Medicine at the Uni-versity of California, Los Angeles. The abridged report is published in Annals of Internal Medicine, and the full re-port at is available at the NIH website https://prevention.nih.gov/programs-events/pathways -to-prevention/ workshops/opioids-chronic-pain/workshop-resources#finalreport. n
Reuben DB, et al: Ann Intern Med. January 13, 2015 (early release online).
In the Literature is compiled and written for The ASCO Post by Charlotte Bath.
©David Sipress/The New Yorker Collection/www.cartoonbank.com
Emerging Clinical Datacontinued from page 95

ASCOPost.com | MARCH 25, 2015 PAGE 97
Perspective
Issues in Oncology
Abuse Control Project was started by the Department of Health and Human Services. This project allocates funds and resources to fight federal health-care fraud and abuse, with much of the funding coming from financial recover-ies obtained the prior year. The program reports annually to the public on the fi-nancial success of the recovery efforts.
Comprehensive ReviewWe comprehensively reviewed major
recently completed health-care fraud cases (2006–2011), attempting to overcome limitations of prior published reviews of health-care fraud. These limitations in-cluded a focus on cases initiated by qui tam relators or on specific sectors (pharmaceu-ticals and Medicaid, in particular).1-4
All cases were recovered under the fed-eral False Claims Act, the primary legisla-tion used to investigate health care fraud and abuse. Data sources included Lexis/Nexis News (terms: “health care fraud,” “False Claims Act,” and “qui tam”), Tax-payers Against Fraud, and Department of Justice websites (2006–2011). Cases with recoveries over $5 million were included. Cases were evaluated as involving qui tam relators (whistle-blowers) or not, as rela-tor cases historically account for 90% of all cases and 90% of all recoveries. 3
As illustrated in Table 1 (page 98), we found that between 2006 and 2011, 123 qui tam health-care fraud cases con-cluded, totaling $15.7 billion in recover-ies (84% of the total recoveries). Phar-maceutical manufacturers accounted for 31% of these cases and $11.3 billion (72%) in recoveries. Health systems and related organizations accounted for 30 cases (24% of these cases), totaling $2.4 billion in recoveries (16% of recoveries). Hospitals accounted for 20 cases (16% of these cases), totaling $716 million in recoveries (5% of recoveries).
Fifty-two cases not involving qui tam relators (whistle-blowers) also concluded during these years, totaling $3.7 billion in federal recoveries (16% of the total recoveries). Pharmaceuti-cal manufacturers accounted for 31%
of these cases, and $2.6 billion (71% of non-qui-tam-related recoveries) in recoveries. Health systems and related organizations accounted for 13 cases (25% of these cases), totaling $384 mil-lion in recoveries (10% of recoveries). Hospitals accounted for 10 cases (20% of the cases), totaling $347 million in recoveries (5% of recoveries).
Important ImplicationsThese findings provide the first com-
prehensive look at the huge financial fines paid in the course of providing health care in the current era. In particular, financial recoveries from completed health-care fraud investigations returned an estimat-ed $15 billion to $16 billion to the fed-eral government between 2006 and 2011,
while qui tam relators (whistle-blowers) received an estimated $2 billion to $3 bil-lion for their collaboration.
Most importantly, the space of health-care fraud is dominated by phar-maceutical manufacturers—much more than they had previously. Pharmaceuti-cal manufacturers now account for 30%
Health-Care Fraud and Abusecontinued from page 1
Dr. Lu is Assistant Professor, Department of Clinical Pharmacy and Outcome Sciences, University of South Carolina College of Phar-macy, Columbia. Dr. Chen and Dr. Quereshi are Assistant Professors, Health Services Policy and Management, Arnold School of Public Health, Columbia, South Carolina. Dr. Sartor is Medical Director, Tulane Cancer Center, New Orleans. Dr. Bennett is Frank P. and Josie M. Fletcher Professor of Pharmacy, University of South Carolina College of Pharmacy, Columbia.
continued on page 98
NCCN Clinical Practice Guidelines in Oncology
(NCCN Guidelines®) NCCN has published the 20th Annual Editions of the NCCN Guidelines® for Acute Myeloid Leukemia, Breast Cancer, Colon Cancer, Non-Small Cell Lung Cancer, Ovarian Cancer, Prostate Cancer, Rectal Cancer, and Small Cell Lung Cancer.
NCCN.org/guidelines
JNCCN-N-0204-0315
Visit NCCN.org
for 20th Annual Editions
2015
th 20th ANNUAL EDITIONS NOW AVAILABLE!
Clinica
l Pra
ctice
Gui
delin
es in
Onc
olog
y (N
CCN G
uide
lines
® )
Colon
Can
cer
Vers
ion
2.20
15
Continu
e
NCCN
.org
Versi
on 2.
2015
, 10/0
3/14 ©
Nati
onal
Compr
ehen
sive C
ance
r Netw
ork,
Inc. 2
014,
All righ
ts re
serve
d. Th
e NCCN G
uideli
nes®
and t
his ill
ustra
tion m
ay no
t be r
epro
duce
d in a
ny fo
rm w
ithou
t the e
xpre
ss w
ritten
perm
ission
of N
CCN® .
NCCN Guid
eline
s for
Pati
ents
® avail
able
at www.nc
cn.or
g/pati
ents
Clinica
l Practi
ce Guidelin
es in O
ncology (N
CCN Guidelin
es® )
Colon Cance
r
Version 2.2015
Continue
NCCN.org
Version 2.2015, 10/03/14 © National C
omprehensive Cancer N
etwork, Inc. 2
014, All rights r
eserved. The NCCN Guidelines® and this i
llustration may n
ot be reproduced in any form without th
e express writte
n permission of N
CCN® .
NCCN Guidelines for Patients
® available at www.nccn.org/patients
Clinical Practice Guidelines in Oncology (NCCN Guidelines®)
Colon Cancer
Version 2.2015
Continue
NCCN.org
Version 2.2015, 10/03/14 © National Comprehensive Cancer Network, Inc. 2014, All rights reserved. The NCCN Guidelines® and this illustration may not be reproduced in any form without the express written permission of NCCN®.
NCCN Guidelines for Patients® available at www.nccn.org/patients
Clinical Practice Guidelines in Oncology (NCCN Guidelines®)
Colon CancerVersion 2.2015
Continue
NCCN.org
Version 2.2015, 10/03/14 © National Comprehensive Cancer Network, Inc. 2014, All rights reserved. The NCCN Guidelines® and this illustration may not be reproduced in any form without the express written permission of NCCN®.
NCCN Guidelines for Patients® available at www.nccn.org/patients
NCCN 9th Annual Congress:
Hematologic Malignancies™
R E C O R D E D P R E S E N TAT I O N S
These activities are approved for AMA PRA Category 1 Credit(s)™. Nursing and pharmacy (ACPE) credits are also provided. Complete accreditation details are provided at the beginning of the individual activities.
Supported by educational grants from Foundation Medicine, Inc.; Gilead Sciences, Inc.; Jazz Pharmaceuticals; Pharmacyclics, Inc. and Janssen Biotech, Inc; and Takeda Oncology.
Online CME/CE!
Save the Date!New location!
NCCN 10th Annual Congress: Hematologic MalignanciesOctober 16 – 17, 2015San Francisco, CASan Francisco Marriott Marquis
If you were unable to attend the meeting in September, visit
education.nccn.org/hem2014
to see what you missed and earn credits online.
JNCCN-N-0204-0315
07_JCN/JAD Ad Template.indd 2 2/26/15 4:48 PM

PAGE 98 The ASCO Post | MARCH 25, 2015
Perspective
of all federal health-care fraud cases and 70% of recoveries vs 4% of cases and 39% of recoveries previously (prior to 2006), 3 while cases not initiated by qui tam relators are increasingly important.
Specific fraudulent activities involving the pharmaceutical industry in both time periods included overcharging Medic-aid and off-label marketing. However, it should be noted that concealing find-ings on drug safety is a new fraudulent activity (accompanied by large fines and settlements) that has not been reported previously—as outlined in the $3 billion settlement with GlaxoSmithKline.4
Health-care provider systems and hos-pitals are emerging areas for False Claims Act investigation, now accounting for 25% of False Claims Act cases and 15% of recovered funds. Our findings suggest that accountability initiatives recently de-veloped for banks and their executives5 should be extended to include all sectors of health care; executives of these corpora-tions must be held personally liable if the corporate culture of the health-care indus-try is to be improved and fraud decreased.
These findings have important impli-cations for health care in general, and the
discipline of oncology in particular. Oncol-ogy pharmaceuticals account for the ma-jority of high-priced pharmaceuticals, and, hence, are the target of many of the federal investigations related to the False Claims Act. It is incumbent on oncologists to pay close attention to the medical necessity or safety of pharmaceuticals prescribed, and where kickbacks may be occurring. As on-cologists, we can play an important role in the health-care system—particularly when oncology pharmaceuticals are involved. n
Disclosure: This work was funded partly
by the National Cancer Institute, the American Cancer Society, the South Carolina SmartState Program, and an unrestricted grant from Doris Levkoff Meddin to the South Carolina College of Pharmacy Center for Medication Safety and Efficacy. Drs. Lu, Chen, Qureshi, Sartor, and Bennett reported no potential conflicts of interest.
References1. Institute of Medicine: Best care at
lower cost. Washington, DC, National Acad-emies Press, 2012. Available at www.iom.edu. Accessed February 9, 2015.
2. Almashat S, Wolfe S: Pharmaceutical in-dustry criminal and civil penalties. Available at www.citizen.org. Accessed February 9, 2015.
3. Kesselheim AS, Studdert DM: Whis-tleblower-initiated enforcement actions against health care fraud and abuse in the United States, 1996 to 2005. Ann Intern Med 149:342-349, 2008.
4. Department of Justice, Office of Pub-lic Affairs: GlaxoSmithKline to plead guilty and pay $3 billion to resolve fraud allegations and failure to report safety data. Available at www.justice.gov. Accessed February 9, 2015.
Health-Care Fraud and Abusecontinued from page 97
It is incumbent on oncologists to pay close attention to concerns such as off-label marketing and marketing of pharmaceuticals where safety considerations are not
appropriately disseminated, and where kickbacks may be occurring.
— Z. Kevin Lu, PhD, Brian Chen, JD, PhD, Zaina Qureshi, PhD, MPH, Oliver Sartor, MD, and Charles Bennett, MD, PhD, MPP (left to right)
Table 1: Frequency of Enforcement Actions by Industry Type and Qui Tam Relator Status
Qui Tam Relator Cases Non–Qui Tam Relator Cases
Industry Number of Total Recoveries Mean Recoveries Cases (%) (millions of $) (%) (millions of $)
Number of Total Recoveries Mean Recoveries Cases (%) (millions of $) (%) (millions of $)
Mean Difference a
Pharmaceutical manufacturer
38 (30.9%) 11,259 (71.6%) 296 16 (30.8% 2,636 (70.8%) 165 132
Health system 9 (7.3%) 1,385 (8.8%) 154 9 (17.3%) 304 (8.2%) 34 120
Other health-care related organization 21 (17.1%) 1,114 (7.1%) 53 4 (7.7%) 80 (2.1%) 20 33
Hospital 20 (16.3%) 716 (4.6%) 36 10 (19.2%) 200 (5.4%) 20 16
Medical equipment company
8 (6.5%) 294 (1.9%) 37 4 (7.7%) 347 (9.3%) 87 −50
Laboratory services provider
3 (2.4%) 363 (2.3%) 121 0 (0%) 0 (0%) 0 121
Pharmacy or pharmacy chain
5 (4.1%) 152 (1.0%) 30 3 (5.8%) 81 (2.2%) 27 3
Home health-care provider
3 (2.4%) 186 (1.2%) 62 2 (3.8%) 18 (0.5%) 9 53
Physician practice or individual physician group
10 (8.1%) 149 (0.9%) 15 0 (0%) 0 (0%) 0 15
Billing company 3 (2.4%) 71 (0.5%) 24 2 (3.8%) 33 (0.9%) 16 7
Long-term care facility 2 (1.6%) 22 (0.1%) 11 1 (1.9%) 20 (0.5%) 20 −9
Ambulance medical transportation company
1 (0.8%) 9 (0.1%) 9 1 (1.9%) 7 (0.2%) 7 2
Total 123 (100.0%) 15,720 (100.0%) 128 52 (100.0%) 3,725 (100.0%) 72 56
a Difference in mean recoveries for qui tam relator vs non–qui tam relator recoveries (millions of dollars).
ASCO’s New Journal of Global Oncology to Debut in 2015
Journal of Global Oncology fulfills a growing need for high-quality literature on the array of challenges that health care professionals in resource-constrained settings face in conducting research and caring for patients with cancer.• Open access, online-only journal.• Global, multidisciplinary editorial board.• Bimonthly. First issue to publish third quarter of 2015. • Published by ASCO, the most trusted source in oncology information.
CALLING ALL AUTHORS!Publish with ASCO Journals, the global leader in oncology research and practice. More information on submitting to JGO is coming soon. For inquiries, please email [email protected].
The Journal of Global Oncology is funded through the Conquer Cancer Foundation of the American Society of Clinical Oncology with the support of the Conquer Cancer Foundation Mission Endowment, the Doris Duke Charitable Foundation and Novartis Oncology.
David Kerr, MD, DSc Founding Editor-in-Chief, University of Oxford and Weill Cornell Medical College.

ASCO’s New Journal of Global Oncology to Debut in 2015
Journal of Global Oncology fulfills a growing need for high-quality literature on the array of challenges that health care professionals in resource-constrained settings face in conducting research and caring for patients with cancer.• Open access, online-only journal.• Global, multidisciplinary editorial board.• Bimonthly. First issue to publish third quarter of 2015. • Published by ASCO, the most trusted source in oncology information.
CALLING ALL AUTHORS!Publish with ASCO Journals, the global leader in oncology research and practice. More information on submitting to JGO is coming soon. For inquiries, please email [email protected].
The Journal of Global Oncology is funded through the Conquer Cancer Foundation of the American Society of Clinical Oncology with the support of the Conquer Cancer Foundation Mission Endowment, the Doris Duke Charitable Foundation and Novartis Oncology.
David Kerr, MD, DSc Founding Editor-in-Chief, University of Oxford and Weill Cornell Medical College.

Emerging research is directing focus on programmed death-ligand 1 (PD-L1) expressed on and around tumors
Tumor cell
Macrophage Dendritic cell
Cancer can evade immune destruction by upregulating the inhibitory ligand PD-L1 on tumor cells and tumor-in� ltrating immune cells, such as macrophages and dendritic cells.1,2 PD-L1 binds to B7.1 and PD-1 on cytotoxic T cells, disabling the anticancer immune response.1-3
Inactive T cell
PD-L1 expression
B7.1
PD-L1
PD-L1
PD-1
Exploring the PD-L1 pathway as a new direction in cancer immunotherapy research
References: 1. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1-10. 2. Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy––inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res. 2012;18:6580-6587. 3. Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207-212.
© 2014 Genentech USA, Inc. All rights reserved. BIO/092214/0027 Printed in USA.
Discover the PD-L1 pathway, a focus of investigation and cancer immunotherapy research by visiting
www.ResearchPDL1.com
78123ha_f.indd 1 10/29/14 10:50 PM





























