Embed Size (px)
Citation preview

Synthesis of OrganofluorineCompounds and AllenylboronicAcids - Applications IncludingFluorine-18 Labelling Denise N. Meyer
Denise N
. Meyer Syn
thesis of O
rganoflu
orine C
ompou
nds an
d Allenylboron
ic Acids - A
pplications In
cludin
g Fluorin
e-18 Labelling
Doctoral Thesis in Organic Chemistry at Stockholm University, Sweden 2021
Department of Organic Chemistry
ISBN 978-91-7911-490-9
Denise N. MeyerRaised in Lauterecken, South-WestGermany, Denise studied chemistryat Johannes Gutenberg UniversityMainz where she obtained herBachelor's and Master's degree. In2017, she moved to Stockholmwhere she pursued her doctoralstudies with Prof. Kálmán J. Szabó.


Synthesis of Organofluorine Compounds andAllenylboronic Acids - Applications IncludingFluorine-18 LabellingDenise N. Meyer
Academic dissertation for the Degree of Doctor of Philosophy in Organic Chemistry atStockholm University to be publicly defended on Friday 4 June 2021 at 10.00 in Magnélisalen,Kemiska övningslaboratoriet, Svante Arrhenius väg 16 B.
AbstractThis work is focused on two areas: the chemistry of organofluorine and organoboron compounds. In the first chapter, acopper-catalysed synthesis of tri- and tetrasubstituted allenylboronic acids is presented. Extension of the same methodleads to allenylboronic esters. The very reactive and moisture-sensitive allenylboronic acids are further applied to thereaction with aldehydes, ketones and imines to form homopropargyl alcohols and amines. In addition, an enantioselectivereaction catalysed by a BINOL organocatalyst was developed to form tertiary alcohols with adjacent quaternary carbonstereocenters.
The second chapter specialises in the functionalisation of 2,2-difluoro enol silyl ethers with electrophilic reagents undermild reaction conditions. This allowed for the formation of perfluoroethyl ketones, 2-aryl-2,2-difluoro ketones as well asthe novel -COCF2SCF3-moiety.
The third chapter continues with electrophilic fluorination. A synthesis of an electrophilic, 18F-labelled hypervalentiodine reagent from nucleophilic 18F-labelled tetrabutylammonium fluoride was devised. Then, the 18F-labelling of o-styrylbenzamides using the developed reagent afforded [18F]benzoxazepines in good radiochemical yields and molar activity.
The objective of the final chapter was the development of a method for the nucleophilic 18F-labelling of 2-halo-2,2-difluoro ketones derived from the corresponding 2,2-difluoro enol silyl ethers. The radiochemical utility of this method wasdemonstrated by fluorine-18 labelling of a potential neutrophil elastase ligand followed by successful preclinical studies.
Keywords: boron, allenylboronic acid, BINOL, homopropargyl alcohol, organocatalyst, organoboron, organofluorine,fluorine, fluorine-18, late-stage, labelling, labeling, nucleophilic, electrophilic, fluorination, positron emisiontomography, PET, radiotracer, hypervalent iodine, benziodoxole, benzoxazepines, diaryliodonium salt, metal-free,neutrophil elastase, NE, neutrophil elastase inhibitor, trifluoromethylation, trifluoromethylthiolation, difluoro enol silylethers.
Stockholm 2021http://urn.kb.se/resolve?urn=urn:nbn:se:su:diva-192156
ISBN 978-91-7911-490-9ISBN 978-91-7911-491-6
Department of Organic Chemistry
Stockholm University, 106 91 Stockholm


SYNTHESIS OF ORGANOFLUORINE COMPOUNDS ANDALLENYLBORONIC ACIDS - APPLICATIONS INCLUDINGFLUORINE-18 LABELLING
Denise N. Meyer


Synthesis of OrganofluorineCompounds and AllenylboronicAcids - Applications IncludingFluorine-18 Labelling
Denise N. Meyer

©Denise N. Meyer, Stockholm University 2021 ISBN print 978-91-7911-490-9ISBN PDF 978-91-7911-491-6 Front cover: A walk through the Palatinate forest by Marcus K. Meyer Back cover: Portrait by Tautvydas Kireilis Printed in Sweden by Universitetsservice US-AB, Stockholm 2021

For my brother.


By three methods we may learn wisdom:First, by reflection, which is noblest;second, by imitation, which is easiestand third by experience, which is the bitterest. - CONFUCIUS


i
Abstract
This work is focused on two areas: the chemistry of organofluorine and
organoboron compounds. In the first chapter, a copper-catalysed synthesis of tri-
and tetrasubstituted allenylboronic acids is presented. Extension of the same
method leads to allenylboronic esters. The very reactive and moisture-sensitive
allenylboronic acids are further applied to the reaction with aldehydes, ketones
and imines to form homopropargyl alcohols and amines. In addition, an
enantioselective reaction catalysed by a BINOL organocatalyst was developed to
form tertiary alcohols with adjacent quaternary carbon stereocenters.
The second chapter specialises in the functionalisation of 2,2-difluoro enol
silyl ethers with electrophilic reagents under mild reaction conditions. This
allowed for the formation of perfluoroethyl ketones, 2-aryl-2,2-difluoro ketones
as well as the novel -COCF2SCF3-moiety.
The third chapter continues with electrophilic fluorination. A synthesis of an
electrophilic, 18F-labelled hypervalent iodine reagent from nucleophilic 18F-labelled tetrabutylammonium fluoride was devised. Then, the 18F-labelling of
o-styryl benzamides using the developed reagent afforded [18F]benzoxazepines
in good radiochemical yields and molar activity.
The objective of the final chapter was the development of a method for the
nucleophilic 18F-labelling of 2-halo-2,2-difluoro ketones derived from the
corresponding 2,2-difluoro enol silyl ethers. The radiochemical utility of this
method was demonstrated by fluorine-18 labelling of a potential neutrophil
elastase ligand followed by successful preclinical studies.

ii
Populärvetenskaplig sammanfattning
Ju bättre vi förstår kroppen, hur den fungerar och hur den är strukturerad,
desto fler behandlingsalternativ och diagnostiska metoder finns. Som kemister
strävar vi efter att skapa och undersöka nya potentiellt farmakologiskt aktiva
molekyler. I många av de nya läkemedlen som är på väg till godkännande, har
grundämnet fluor en viktig funktion. Detta har att göra med fluors inneboende
egenskaper, såsom den största elektronegativiteten av alla grundämnen och
atomens storlek. Dessa egenskaper gör att tillsatsen av fluor till en molekyl kan
förändra till exempel lipofiliciteten, polariserbarheten, surhetsgraden och den
metaboliska stabiliteten. Den sistnämnda är ofta särskilt viktigt eftersom
mediciner ofta tas oralt och måste passera genom den sura miljön i magen innan
de når sitt mål.
Ett viktigt användningsområde för fluor är positronemissionstomografi
(PET). Här arbetar man med radioaktiva ämnen som till exempel fluor-18, som
har en kort halveringstid på bara 110 minuter. Om en molekyl är radioaktivt
märkt, till exempel med fluor-18, kan vissa biologiska processer följas i levande
organismer. Endast en minimal mängd av det radioaktiva ämnet krävs för detta,
och därför ger läkemedlet ingen verklig effekt. Metoden följer ”tracer”-principen.
Det vanligaste radiotracern, fluor-18-deoxyglukos, används till exempel för att
diagnostisera cancer.
I denna avhandling presenteras nya metoder inom organofluor- och
organoborkemi. Dessutom presenteras två nya metoder för radioaktiv märkning
av en elektrofil hypervalent jodförening och ett antal trifluormetylketoner. I
slutändan var det till och med möjligt att tillverka ett potentiellt
radiofarmaceutiskt läkemedel och använda det i prekliniska försök.

iii
Populärwissenschaftliche Zusammenfassung
Je besser wir den Körper verstehen, wie er funktioniert und wie er aufgebaut ist,
desto mehr Behandlungsmöglichkeiten und Diagnosemethoden stehen zur
Verfügung. Als Chemiker bemühen wir uns, neue potenziell pharmakologisch
aktive Moleküle zu entwickeln und zu untersuchen. In vielen der neuen
Medikamente, die auf dem Weg zur Zulassung sind, hat das Element Fluor eine
wichtige Funktion. Dies hat mit den inhärenten Eigenschaften von Fluor zu tun,
wie der höchsten Elektronegativität aller Elemente und der Größe des Atoms.
Diese Eigenschaften bedeuten, dass die Zugabe von Fluor zu einem Molekül
beispielsweise die Lipophilie, Polarisierbarkeit, Säurestärke und
Stoffwechselstabilität verändern können. Letzteres ist oft besonders wichtig, da
Medikamente oft oral eingenommen werden und die saure Umgebung des
Magens passieren müssen, bevor sie ihr Ziel erreichen.
Eine wichtiges Anwendungsgebiet für Fluor ist die Positronenemissions-
tomographie (PET). Hier arbeitet man mit radioaktiven Elementen wie Fluor-18,
welches eine recht kurze Halbwertszeit von nur 110 Minuten hat. Wenn ein
Molekül radioaktiv markiert ist, beispielsweise mit Fluor-18, können bestimmte
biologische Prozesse in lebenden Organismen verfolgt werden. Hierfür ist nur
eine minimale Menge der radioaktiven Substanz erforderlich, daher hat die
Markierungssubstanz keine pharmakologische Wirkung. Die Methode folgt dem
"Tracer"-Prinzip. Die häufigste Radiomarkierungssubstanz, Fluor-18-
desoxyglucose, wird beispielsweise zur Diagnose von Krebs eingesetzt.
Diese Dissertation präsentiert neue Methoden in der Organofluor- und
Organoborchemie. Darüber hinaus werden zwei neue Methoden zur
radioaktiven Markierung einer elektrophilen hypervalenten Iodverbindung und
einer Reihe von Trifluormethylketonen vorgestellt. Abschließend war es sogar
möglich, eine potenzielle Radiomarkierungssubstanz herzustellen und in
präklinischen Studien zu verwenden.

iv
List of Publications
This thesis is based on the following papers or manuscripts, referred to in the
text by Roman numerals I-IV.
I. Copper-catalyzed synthesis of allenylboronic acids. Access to
sterically encumbered homopropargylic alcohols and amines by
propargylboration
Jian Zhao,§ Sybrand J. T. Jonker,§ Denise N. Meyer, Göran Schulz, Duc C.
Tran, Lars Eriksson, Kálmán J. Szabó,*
Chem. Sci. 2018, 9, 3305-3312.
II. Trifluoromethylthiolation, Trifluoromethylation, and Arylation
Reactions of Difluoro Enol Silyl Ethers
Xingguo Jiang,§ Denise Meyer,§ Dominik Baran, Miguel A. Cortes
Gonzalez and Kálmán J. Szabo,*
J. Org. Chem. 2020, 85, 8311-8319. Featured article.
III. [18F]fluoro-benziodoxole: A no-carrier-added electrophilic
fluorinating reagent. Rapid, simple radiosynthesis, purification
and application for fluorine-18 labelling
Miguel A. Cortés González, Patrik Nordeman, Antonio Bermejo Gómez,
Denise N. Meyer, Gunnar Antoni, Magnus Schou and Kálmán J. Szabó,*
Chem. Commun. 2018, 54, 4286-4289.
IV. Base-catalysed 18F-labelling of trifluoromethyl ketones.
Application to the synthesis of a 18F-labelled neutrophil elastase
ligand
Denise N. Meyer,§ Miguel A. Cortés González,§ Xingguo Jiang,§ Linus
Johansson-Holm, Sergio Estrada, Patrik Nordeman, Gunnar Antoni
and Kálmán J. Szabó,*
Manuscript
§ Authors contributed equally to the publication.

v
Previous document is based on this work
This thesis builds partly on the author’s half-time report titled “Synthetic
Methodologies on Boron and Fluorine Incorporation” (defended on December
10, 2020). The literature review has been updated (Introduction and
Chapters 1-2). Of the papers included in this thesis, papers I-II were part of the
half-time report. By chapters, the contribution from the half-time report is as
follows:
Chapter 1: This chapter was included in the half-time report; for this thesis it has
been reviewed and updated, and around 20% of the text and references are new.
Chapter 2: This chapter was included in the half-time report; for this thesis it has
been reviewed and updated, and around 10% of the text and references are new.

vi
Contents
Abstract .................................................................................................................................................. i
Populärvetenskaplig sammanfattning ..................................................................................... ii
Populärwissenschaftliche Zusammenfassung.................................................................... iii
List of Publications ..........................................................................................................................iv
Previous document is based on this work ............................................................................. v
Contents ................................................................................................................................................vi
Abbreviations ..................................................................................................................................... ix
1. Introduction ...................................................................................................................... 1
1.1. Synthesis and application of fluorinating and fluoroalkylating reagents in organic synthesis ..................................................................................................................... 2
1.1.1. Nucleophilic and electrophilic fluorination methods ............................................... 2 1.1.2. Trifluoromethylation reagents and their synthetic application.......................... 5 1.1.3. Trifluoromethylthiolating reagents and their synthetic application ................ 6
1.2. 18F-labelling of small organic molecules .............................................................. 7 1.2.1. Positron emission tomography ........................................................................................... 7 1.2.2. Fluorine-18 containing radiotracers and application in PET ............................... 7 1.2.3. Methods for 18F-labelling ....................................................................................................... 8
1.3. Introduction to organoboron chemistry ........................................................... 10 1.3.1. Synthesis of allenylboron compounds .......................................................................... 10
1.4. Aims of this thesis ....................................................................................................... 12
2. Results and discussion .............................................................................................. 13
2.1. Allenylboronic accesstoapplicationandsynthesisacids:amines, demonstratedhomopropargylic alcohols and asymmetric
organocatalysis (Paper I) ....................................................................................................... 13 2.1.1. Variation of the reaction conditions for the synthesis of allenylboronic acid
15 2.1.2. Purification and isolation of allenylboronic acid 50a ........................................... 17 2.1.3. Scope of the synthesis of allenylboronic acids 50................................................... 17 2.1.4. Extension to other allenylboron derivatives ............................................................. 19 2.1.5. Racemic synthesis of propargyl alcohols and amines using allenylboronic
acids 20 2.1.6. Optimisation and substrate scope for the asymmetric organocatalysed
synthesis of homopropargylic alcohols........................................................................................... 22 2.1.7. Kinetic resolution experiment to form vicinal quaternary stereocenters .. 24

vii
2.1.8. Proposed stereoselection model and catalytic cycle for the asymmetric
synthesis of homopropargyl alcohol 57a ....................................................................................... 25 2.1.9. Conclusions and outlook ..................................................................................................... 26
2.2. Synthesis of 2,2-difluoro ketones from 2,2-difluoro enol silyl ethers and application to trifluoromethylthiolation, fluorohomologation, and arylation (Paper II) ....................................................................................................................................... 27
2.2.1. Electrophilic trifluoromethylthiolation of 2,2-difluoro enol silyl ethers ..... 29 2.2.2. Variation of reaction conditions for the trifluoromethylthiolation of 2,2-
difluoro enol silyl ethers......................................................................................................................... 29 2.2.3. Substrate scope for the trifluoromethylthiolation of 2,2-difluoro enol silyl
ethers 30 2.2.4. Electrophilic trifluoromethylation of 2,2-difluoro enol silyl ethers ............... 32 2.2.5. Optimisation of the reaction conditions for the trifluoromethylation of 66a.
33 2.2.6. Substrate scope for the iron(II) chloride-mediated trifluoromethylation of
66 35 2.2.7. Potassium fluoride-mediated arylation of 66 ........................................................... 36 2.2.8. Conclusion .................................................................................................................................. 37
2.3. Synthesis of a fluorine-18 labelled, electrophilic hypervalent iodine fluorinating reagent (Paper III) .......................................................................................... 38
2.3.1. From fluorine-19 to fluorine-18: Adaption of the synthesis of fluoro-
benziodoxole ................................................................................................................................................ 38 2.3.2. Application of [18F]5d to an electrophilic fluorocyclisation reaction ............ 42 2.3.3. Isolation and measurement of the molar activity of [18F]11b .......................... 43 2.3.4. Conclusions and outlook ..................................................................................................... 44
2.4. 18F-labelling of trifluoromethyl ketones including neutrophil elastase inhibitors (Paper IV) ................................................................................................................ 45
2.4.1. Development of the reaction forconditions
2,2,2-[18F]trifluoroacetophenones .................................................................................................... 47 2.4.2. Substrate theforscope labelling ofcircular
2,2,2-[18F]trifluoromethylketones ..................................................................................................... 48 2.4.3. The radio diagnostic utility of the circular labelling method ............................ 49
2.4.3.1. Synthesis of and studies with model compound 65i ......................................50 2.4.3.2. Fluorine-18 labelling of inhibitor 1a ....................................................................52 2.4.3.3. Isolation and autoradiography study of 1a ........................................................53
2.4.4. Conclusions and outlook ..................................................................................................... 56
Concluding remarks ...................................................................................................................... 57
Reprint permissions ...................................................................................................................... 58
Contribution list .............................................................................................................................. 59
Acknowledgements ....................................................................................................................... 60

viii
References ......................................................................................................................................... 63

ix
Abbreviations
Abbreviations are used in accordance with the standards of the subject.1
Additional or unconventional abbreviations are listed below.
18c6 1,4,7,10,13,16-Hexaoxacyclooctadecane ARDS Acute respiratory distress syndrome Bdan 2,3-Dihydro-1H-naphtho[1,8-de][1,3,2]diazaborinine bpy 2,2′-Bipyridine B2nep2 Bis(neopentyl glycolato)diboron B2pin2 Bis(pinacolato)diboron Bpin Pinacolatoboron B2pne2 Bis[(+)-pinanediolato]diboron (S)-Br2-BINOL (S)-3,3′-Dibromo-1,1′-bi-2-naphthol CuMes Mesitylcopper(I) DABCO 1,4-Diazabicyclo[2.2.2]octane dba (1E,4E)-1,5-Diphenylpenta-1,4-dien-3-one DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene DCM Dichloromethane DIPEA N,N-Diisopropylethylamine DMAP 4-(Dimethylamino)pyridine DMT 4,4′-Dimethoxytrityl d.r. Diastereomeric ratio EDC 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide equiv. Equivalent FDG Fluoro-deoxy-D-glucose F-DOPA Fluoro-L-3,4-dihydroxyphenylalanine HBcat Catecholborane K222 4,7,13,16,21,24-Hexaoxa-1,10-
diazabicyclo[8.8.8]hexacosane MeCN Acetonitrile MS Molecular sieves MTBD 7-Methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene NE Neutrophil elastase NET Neutrophil extracellular traps rt Room temperature TBAF Tetrabutylammonium fluoride TBAT Tetrabutylammonium difluorotriphenylsilicate TBD 1,5,7-Triazabicyclo[4.4.0]dec-5-ene T Temperature [°C] TFAA Trifluoroacetic anhydride TMEDA N,N,N′,N′-Tetramethylethylenediamine TREAT-HF Triethylamine trihydrofluoride Umemoto’s reagent
5-(Trifluoromethyl)dibenzothiophenium tetrafluoroborate
Xantphos 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene

x
Abbreviations of terms and units in radiochemistry
Due to the variety of nomenclature in radiochemistry, a definition of the scientific
terms was devised by an international working group in accordance with IUPAC
rules and SI units.2 A summary is given below.
Activity: Measures the radioactivity of a nuclide. The spontaneous decay of a
known amount of compound in a certain amount of time is quantified and
measured in the SI unit Becquerel [Bq = 1/s].
As: Specific activity is defined as the measured amount of activity per g of
compound [GBq/µg].
Am: Molar activity is defined as the measured amount of activity per mol of
compound [GBq/µmol].
For both units, the exact time of measurement must be reported.
RCY: Radiochemical yield is defined as the amount of activity of product
compared to starting material of the same radioisotope and is decay-corrected
with an identical reference time and given in [%]. It can refer to isolated and non-
isolated compounds and is comparable to what is understood as “yield” in
chemistry.
AY: Activity yield indicates the efficacy of a procedure. AY is defined as the amount of activity (in Bq) compared to the starting amount (end of bombardment) and is not decay-corrected [%].
RCP: Radiochemical purity is defined by the presence of product compared to
any other radioactive material [%].

1
1. Introduction
The elements boron and fluorine have a distinct distribution in nature.
Fluorine is the 13th most abundant element in the Earth’s crust and is mainly
found in water-insoluble minerals such as fluorospar (CaF2).3 In living
organisms, only a few naturally occurring organic compounds have been
discovered. Two of the reasons for their rarity are the high oxidation potential of
the fluoride ion (−3.06 V) and its high solvation energy (−117 kcal/mol) which
leads to a low nucleophilicity in aqueous media.4 Similarly, boron is present in
nature only in form of borate salts or as boric acid. However, it is essential for
the growth of vascular plants.5 Despite the scarcity of natural organofluorine and
organoboron compounds, they have an important role in life sciences.6
In general, organofluorines often occur as drug substances, while
organoboron compounds are important intermediates in the synthesis of
complex natural products and bioactive molecules.
Figure 1: Fluorine- and boron-containing bioactive molecules.
The introduction of fluorine into a molecule can enhance its biological activity
and modulate its pharmacological (metabolic stability, lipophilicity) as well as
physicochemical (pKa, conformation) properties.7 As a result, fluorine is
prominent in industry, but especially in drug design with more than 35% of new
drug candidates currently in phase ІІ-ІІІ clinical trials and more than 50% of

2
most-prescribed multibillion-dollar pharmaceuticals containing at least one
fluorine atom.8
Among fluorine-containing bioactive molecules, α-fluorinated ketone motifs
are often used as pharmacophores to inhibit serine proteases or related enzymes
(Figure 1).9 For example, the imbalance of a serine protease, neutrophil elastase
(NE) can cause a number of chronic diseases (such as chronic obstructive
pulmonary disease).10 For this reason there is a large demand to find inhibitors
of NE, where 1a was found to be a potential candidate.11 Also, perfluoroethyl
ketone 1b was found to be an inhibitor for phospholipase A2, which is involved
in several inflammatory disorders (such as arthritis or autoimmune diseases).6a
In contrast, a small number of trifluoromethylthiolated compounds are found
on the market, for example, the first-generation cephalosporin antibiotic
cefazaflur 1c.6e, 12
Nevertheless, there are organoboron-containing drugs such as bortezomib
1d, which is used for the treatment of relapsed and refractory multiple
myeloma.13
1.1. Synthesis and application of fluorinating and fluoroalkylating reagents in organic synthesis
With the increasing demand for new fluorinated pharmacophores, it is a
necessity that the chemical space of organofluorine chemistry is expanded
further. New reagents and methodologies are needed for further development of
late-stage fluorination methods. In the following chapter, complementary
electrophilic and nucleophilic reagents and their applications are introduced.
1.1.1. Nucleophilic and electrophilic fluorination methods The first discovered nucleophilic fluorination reaction to form aryl fluorides
from diazonium salts is the Balz-Schiemann reaction.14 Another example, even
performed on an industrial scale, is the halogen-exchange reaction (Halex
reaction). The first example features a substitution of chloride in 1-chloro-2,4-
dinitrobenzene with anhydrous KF under harsh conditions.15 Recently, the
application of transition-metal catalysis has led to milder reaction conditions
and a wider substrate scope.16
In addition, the fluorination of sp3-carbons has been widely explored.16a, b, f, 17
Among these, a special area of nucleophilic fluorination is the construction
of -CF3 from a -CF2X group. This reaction is particularly important for the late-
stage fluorination in fluorine-18 labelling.18 Sokolenko, Yagupolskii and
co-workers first reported a possible route for the introduction of fluorine
to -CF2Br, a carbon that already contained fluorine substituents.19 Here,
N-bromodifluoromethyl heterocycles were fluorinated to form

3
trifluoromethylated heterocycles under harsh conditions with TBAF or CsF. A
recent example for a copper-mediated fluorination of 2-bromo-2,2-
difluoromethyl arenes, ketones and acetamides was published by Szabó and
co-workers.20 Using this method, trifluoromethylacetamide 3 was obtained in
74% yield, in the presence of DBU (Scheme 1). These findings led to a second
study where the concept was adapted to fluorine-18 labelling. In the labelling
studies, [18F]TBAF [18F]4a was used as the fluorine source instead of CuF-
complexes.21
Scheme 1: Nucleophilic fluorination of acetamides 2.
In comparison to nucleophilic fluorinating reagents, such as alkali metals or
ammonium salts, electrophilic reagents are usually more complicated. The
electrophilic reagents used in the early days of organofluorine chemistry, such
as F2 gas, XeF2 etc. are very reactive and therefore unselective.22
In recent years, various N-F reagents have been developed and gave rise to
milder reaction conditions and diverse applications (Figure 2, 5a-c).16a, 23
Another type of electrophilic reagents are hypervalent iodine compounds
(Figure 2, 5d). These are air and moisture stable and react under mild conditions.
Therefore, these reagents have been broadly applied in selective organic
synthesis.23a, h, 24 Fluoro-benziodoxole 5d, which was first described by Prévost
and Legault,25 has been used as an electrophilic fluorinating reagent in many
synthetic applications.26 Its structure and reactivity were investigated27 as well
as a number of computational studies conducted to rationalised the mechanism
of the fluorination.28 In contrast to other electrophilic fluorinating reagents, 5d
can be synthesised in a single step using nucleophilic fluorine precursors (see
below section 2.3.1).

4
Figure 2: Electrophilic fluorinating reagents.
Reagent 5d was employed in a number of fluorocyclisation reactions. Stuart
and co-workers presented a fluorocyclisation of styrene derivatives with 5d and
5c.29 The authors observed an interesting regiodivergence depending on the
utilised fluorinating reagent. Carboxylic acid 6 reacted with 5d affording
γ-lactone 7a with a tertiary fluoride, whereas reaction with 5c instead formed
γ-lactone 7b containing a primary fluoride (Scheme 2).27a, 29
Scheme 2: Regiodivergent fluoro-lactonisation of carboxylic acids.
The Szabó group has gathered experience working with reagent 5d in several
studies.26, 30 For example, the intramolecular fluorocyclisation of alkenes
catalysed by zinc tetrafluoroborate was investigated (Scheme 3a).31 A successful
oxy-, carbo- and aminofluorination led to a diverse substrate scope. For example,
piperidine (9) was obtained in 82% yield. Later, Gulder and co-workers studied
the fluorocyclisation of o-styryl benzamides 10a, affording fluoro-
benzoxazepines 11a at room temperature within 5 minutes in 77% yield
(Scheme 3b).32 The substrate scope was further expanded by Gulder and co-
workers to the synthesis of indoles,33 fluoro-azabenzoxazepines and
azaindoles.34
Recently, several other fluorocyclisation reactions have been developed,27a, 35
and even elegant enantioselective routes for the fluorination with hypervalent
iodine reagents have been reported by several groups.24e, 36

5
Scheme 3: Fluorocyclisation reactions with fluoro-benziodoxole reagent 5d.
1.1.2. Trifluoromethylation reagents and their synthetic application The introduction of a -CF3 moiety can be achieved by either electrophilic,
nucleophilic, or radical pathways (Figure 3). Examples for nucleophilic
trifluoromethylating reagents are the Ruppert-Prakash reagent (TMSCF3)37 and
copper-CF3 complexes (Figure 3).38 In addition, Langlois and co-workers39
developed a reagent that initiates radical CF3 (NaSO2CF3). Examples for
electrophilic trifluoromethylating reagents are hypervalent iodine reagents
12a-c23h, 24c, 40 and Umemoto’s reagent 12d.41 These reagents also have the ability
to transfer the CF3 group in radical processes.
Figure 3: Recent and commonly used trifluoromethylating reagents.
Togni and co-workers reported 12a-b for the first time (Scheme 4).40a An
initial assessment through the reaction with β-ketoester 13 provided the
α-trifluoromethylated ketoester 14. A subsequent study by Gade and co-workers
presented an enantioselective version of this reaction.42 Reviews by the groups
of Togni43 and Postigo44 give a comprehensive summary of the development in
this area of research.

6
Scheme 4: Trifluoromethylation of β-ketoester.
1.1.3. Trifluoromethylthiolating reagents and their synthetic application
In the last decade, there has been growing interest in the development and
application of electrophilic trifluoromethylthiolating reagents.12b, 45 Compared to
the electron-withdrawing CF3 group, the SCF3 group is as electron-withdrawing,
but much more lipophilic. The Hansch-Leo lipophilicity parameter π is 1.44 for
the SCF3 group bound to an aromatic group, whereas for CF3 it is only 0.88.46 For
this reason, the groups of Munavalli,47 Shen,48 and several others49 have
dedicated remarkable effort towards the development of new, stable
SCF3-transfer reagents 15a-b (Figure 4). The relative electrophilicity of many
SCF3 transfer reagents was compared using DFT calculations by Xue, Cheng and
co-workers.23a, 50 Therein, N-trifluoromethylthio-dibenzene sulfonimide 15b had
a relatively high trifluoromethylthio cation-donating ability compared to other
O- or N-based reagents. Recently, Zhang and co-workers have published the first
cyclic, hypervalent iodine SCF3-transfer reagent 15c.51
Figure 4: Recent and commonly used trifluoromethylthiolating reagents.
Shen and co-workers developed a procedure for the direct
trifluoromethylthiolation of various electron-rich heteroarenes and activated
electron-deficient heteroarenes.48b Furthermore, they described a Lewis acid
catalysed dearomative trifluoromethylthiolation of 1,3-dimethyl-2-naphthol 16
(Scheme 5). Billard, Shen and others have extensively reviewed the application
of SCF3-transfer reagents using catalysis in synthetic applications.23g, 48c, d, 49e, 52

7
Scheme 5: Dearomative trifluoromethylthiolation of 1,3-dimethyl-2-naphthol.
1.2. 18F-labelling of small organic molecules
Bridging the gap between the development of new methodologies in synthetic
fluorine-19 chemistry and the adaptation of these methodologies to fluorine-18
chemistry requires tremendous efforts.53 Two main problems are the relatively
short half-time of the 18F-isotope (109.8 min)54 and the very small (nanomolar)
amounts of the 18F-sources available for radiosynthesis. Adaptation of mmol-
scale reactions developed for the natural fluorine isotope to nmol scale using on-
site produced 18F-sources is an important domain of translational science in the
development of imaging tracers for positron emission tomography.55
1.2.1. Positron emission tomography Positron emission tomography (PET) is a non-invasive, dynamic molecular
imaging technique utilising positron-emitting molecules as radiotracers for the
in vivo imaging of biological processes.54-56 PET can aid the early diagnosis of
diseases, monitor treatment as well as the development of new drug
candidates.55, 56b, 57 In comparison to other imaging techniques, such as MRI, CT,
ultrasound or X-ray, which give mostly structural and anatomical images, PET
allows for the visualisation of biological processes on cellular or even molecular
level.56c
1.2.2. Fluorine-18 containing radiotracers and application in PET The radionuclide fluorine-18 decays under emission of a positron, whereby
one proton is transformed into a neutron in the nucleus, forming the stable
oxygen-18 (Figure 5). This β+ decay has a specific energy (E) for each nuclide
and is quite small for fluorine-18, in comparison to other β+-decaying nuclides
(11C, 13N, 15O). The emitted positron (e+) scatters through the environment until
E is depleted. An annihilation with its antiparticle, the electron, leads to the
emission of two photons in opposite direction (180°) with the same energy
(Eγ = 511 keV). The photons can be detected via a coincidence measurement and
the origin of them extrapolated.54, 56a-d, 57c, 58

8
Figure 5: The PET principle: Decay of fluorine-18, annihilation process and coincidence measurement of the emitted photons.
The most commonly used radiopharmaceutical tracer is [18F]FDG (Figure 6a).
It enables imaging of the metabolism of glucose and thereby facilitates the
diagnosis of cancer and neurological diseases.55, 56b-d, 59 Another common
radiopharmaceutical tracer is [18F]F-L-DOPA. This PET tracer is used in
neurology for the imaging of the dopaminergic function (Figure 6b).56c, 59b
For the assessment of a new radiotracer, apart from synthesis criteria (see
below section 2.3.1), preclinical studies are essential. This includes in vitro
imaging studies (autoradiography) as well as in vivo studies, on animals and
humans, depending on the use of the tracer.60
Figure 6: 18F-labelled radiotracers.
1.2.3. Methods for 18F-labelling Nucleophilic [18F]fluoride can be conveniently produced with a cyclotron by
proton bombardment of [18O]H2O.54 However, in the presence of water the
fluoride ion is solvated and thereby a poor nucleophile (see introduction above).
For this reason, water and species solvating F- should be strictly avoided in 18F-labelling. The solution give crown ethers, such as K222, that are often added
to increase the nucleophilicity and solubility of [18F]fluoride in dry organic
solvents.16a After synthesis, [18F]KF/K222 is usually azeotropically dried with
MeCN. Then, [18F]KF/K222 can be employed to an array of reactions, for example
aliphatic (SN2) or aromatic (SNAr) substitutions, which are described in a number
of reviews.54-55, 56b, 57a, 61

9
Another example of a nucleophilic 18F-precursor is [18F]TBAF [18F]4a, which
has a good solubility in organic solvents.62 For example [18F]4a was applied in
the construction of a [18F]trifluoromethyl group from a 3-bromo-3,3-difluoro
imidazole 18 to form [18F]Celecoxib [18F]19 (Figure 7).38, 55, 63
Figure 7: Radiosynthesis of [18F]Celecoxib.
Another method to obtain the [18F]CF3 moiety is through transition metal
catalysis. For example, copper complexes have been utilised.64 Also, Gouverneur
and co-workers developed a method for the radiolabelling of Umemoto’s reagent
12d through a Halex reaction.65
Compared to the synthesis of nucleophilic 18F-sources, electrophilic reagents
are commonly derived from [18F]F2.54-56, 59c, d, 66 N-F reagents such as [18F]NFSI66g,
k or [18F]Selectfluor59c, d are examples for electrophilic 18F-reagents. However,
they inherit the low molar activity of their precursor [18F]F2. Significant efforts
have been made to find other routes to synthesise electrophilic 18F-reagents. For
example, a post-target synthesis of [18F]F2 from [18F]fluoride has been developed
by Solin and co-workers and extended in collaboration with Gouverneur and
co-workers to the synthesis of [18F]Selectfluor.59c, d, 67 The groups proved the
usefulness of their method by labelling enol silyl ether 20 with [18F]Selectfluor
(Scheme 5).
Scheme 6: Radiolabelling of enol silyl ether 20.
An elegant solution to circumvent low molar activities, but still perform an
electrophilic fluorination, is to employ a umpolung strategy. Ritter and
co-workers have developed a palladium-mediated 18F-fluorination of electron-
rich arenes.53a, 68
Another type of reagents known for their umpolung ability are the
hypervalent iodines. Li, Lu and co-workers developed an intramolecular

10
fluorocyclisation of unsaturated carbamates (Scheme 7).35b Starting from chloro-
benziodoxole 22, the electrophilic 18F-fluorinating reagent [18F]5d was produced
in situ from nucleophilic [18F]4a. After synthesis of [18F]5d, silver(I) triflate was
added and the oxazolidin-2-one [18F]24 formed through a proposed radical aryl
migration, cyclisation reaction.
Scheme 7: In situ synthesis and application of [18F]5d.
1.3. Introduction to organoboron chemistry
In synthetic chemistry, allyl- and allenylboron compounds are very valuable
building blocks.69 In comparison to the also well-known organozinc, lithium or
magnesium reagents, they do not undergo 1,3-metallotropic shifts.
1.3.1. Synthesis of allenylboron compounds The first synthesis of an allenylboronic acid has been reported by Yamamoto
and co-workers (Scheme 8a).70 Starting from a Grignard reagent and trimethyl
borate, the pyrophoric allenylboronic acid 27 was obtained. An alternative
procedure was presented by the group of Petasis.71 In this study sodium
periodate was used to convert allenyl-Bpin compounds to allenylboronic acids.
Scheme 8: Synthesis of a disubstituted allenylboronic acid.
Hayashi and co-workers published an asymmetric synthesis of axially chiral
allenylboronates (Scheme 9a).72 The palladium-catalysed synthesis used
catechol borane 29 as boron source to obtain the chiral allenylboronate 31. The
groups of Hoveyda and Engle have further explored the hydroboration route.73
Chiral, trisubstituted allenylboronates 34 were synthesised by a copper-
catalysed enantioselective 1,4-hydroboration of 1,3-enynes using HBpin 32 as
boron source (Scheme 9b).73b

11
Scheme 9: Enantioselective synthesis of allenylboronates.
Ito and co-workers studied a CuO-t-Bu-catalysed reaction of chiral
propargylic carbonates with B2pin2 (36), employing Xantphos as the ligand to
obtain allenyl boronates 37a (Scheme 10a).74 Szabó and co-workers have shown
that this reaction can be carried out with a Cu/Pd bicatalytic system without
using water sensitive CuO-t-Bu (Scheme 10b).75
Scheme 10: Examples of the synthesis of allenylboronic esters.
The first propargylboration with allenylboronates of aldehydes was reported
by Blais and co-workers.76 However, a mixture of homopropargyl and α-allenyl
alcohol was detected. Studies on the propargylboration of ketones were
conducted by Kobayashi and co-workers.77 Starting from an allenylboronate, the
desired product was obtained through a zinc-amide-catalysed reaction in good
regioselectivity. The synthesis and application of allenylboron species was
extensively reviewed by Pyne,78 Wang,79 Mao and Bose.80

12
1.4. Aims of this thesis
As mentioned in the introduction, fluorine-containing pharmacophores are of
great interest in drug design (see above section 1.1). Therefore, an extension of
the available methods to functionalise molecules with fluorine atoms is an
important field of research.
The first chapter aims to develop a method for the copper-catalysed synthesis
of sterically encumbered allenylboronic acids. Their application to ketones with
a chiral organocatalyst can lead to synthetically valuable tertiary homopropargyl
alcohols.
The second chapter aims for the application of electrophilic
trifluoromethylating, trifluoromethylthiolating and arylating reagents starting
from trifluoromethyl ketones.
The third and fourth part aim to bridge the gaps between the synthesis of
state-of-the-art fluorine-19 methods to fluorine-18 radiochemical applications.
As mentioned in the introduction, very few electrophilic reagents are easily
synthesised for 18F-labelling. The third chapter aims for the radiosynthesis of a
hypervalent iodine-based 18F-reagent. This reagent can be employed in
fluorocyclisation reactions for the synthesis of 18F-labelled heterocyclic
compounds.
In the fourth chapter, we aim to develop a 18F-labelling methodology of
trifluoromethyl ketones. The approach is based on a circular-defluorination-
halogenation-18F-labelling sequence. By this method, the COCF3 moiety can be
easily converted to the corresponding [18F]COCF3 analogues. As COCF3 is an
important pharmacophore in serine protease inhibitors, we hypothesised that
our method is suitable for the development of a new radioligand for these types
of enzymes.

13
2. Results and discussion
2.1. Allenylboronic acids: synthesis and application to access homopropargylic alcohols and amines, demonstrated asymmetric organocatalysis (Paper I)
Asymmetric allyl- and propargylboration of carbonyl compounds are very
useful reactions in stereoselective synthesis.81 The most widely used reaction
partners are aldehydes and ketones. Ketones are less reactive towards
allylboration with allylboronates than aldehydes. Schaus and co-workers found
the boronic ester isopropylboronate 38 superior in reactivity compared to the
pinacolyl ester (Scheme 11a).82 In addition, Szabó and co-workers developed an
asymmetric allylboration with a larger substrate scope, by using allylboronic
acids as substrates and 40a as organocatalyst (Scheme 11b).83 The
γ,γ-disubstituted allylboronic acid, geranylboronic acid 42, reacted smoothly
with p-Br-acetophenone (39b) to form tertiary homopropargyl alcohol 41b with
vicinal quarternary stereocenters in good yield and enantioselectivity.
Scheme 11: Examples of the catalytic asymmetric allylboration of ketones.
The environment of boron can influence the reactivity and stability of
organoboron compounds significantly. For example, allylboronates such as the
pinacol ester are bench-stable and have a relatively low reactivity towards
ketones and imines. On the other side, allylboronic acids such as 42 exhibit a high

14
reactivity towards these substrates. Compound 42 reacts readily with ketones
and imines but is also less stable and oxygen-sensitive than allylboronic esters.
Even more reactive are allyl boroxines, the anhydrides of allylboronic acids.84
Boroxines can be obtained by azeotropic removal of water or application of
molecular sieves in the reaction mixtures.13
Allenylboron reagents have received less attention than allylboron reagents.
Nevertheless, allenylboron compounds have great synthetic potential for many
racemic and asymmetric propargylboration applications. The group of
Yamamoto explored the asymmetric propargylboration of benzaldehyde (44a)
with dialkyl tartrate as the chiral auxiliary (Scheme 12a).70 The homopropargyl
alcohol 45a was obtained with a moderate ee of 63%. Fandrick, Senanayake and
co-workers proposed the first example of an asymmetric propargylboration of
ketones.85 Using propargylboronates with a chiral copper catalyst, propargylic
alcohols formed with high enantioselectivities.
In addition, Schaus and co-workers developed a catalytic, asymmetric
propargylboration of ketone 39a with allenylboronate 46 (Scheme 12b).86 The
groups of Roush and Chen offered a complementary procedure utilizing a chiral
phosphoric acid to yield homopropargyl alcohol 45c with excellent
enantioselectivities (Scheme 12c).87
Scheme 12: Catalytic asymmetric porpargylborations using allenylboronates.
As mentioned in the introduction (section 1.3), allenylboron species are very
versatile building blocks. The reaction with ketones and imines, in the presence
of a chiral catalyst, may lead to tertiary and quaternary stereocenters in sterically
crowded homopropargylic alcohols and amines. Based on the above-mentioned

15
strategies we aimed to develop a route for the synthesis of tri- and
tetrasubstituted boronic acids and to access homopropargylic alcohols and
amines. We also aimed to design an asymmetric synthesis to form vicinal
stereocenters on tertiary homopropargyl alcohols and amines.
2.1.1. Variation of the reaction conditions for the synthesis of allenylboronic acid
Allenyl-boronic esters such as 37 are bench stable and reactive towards
aldehydes, but do not react easily with ketones and imines (Scheme 13).74-75, 88
This underlines the need for a method to synthesise sterically encumbered
allenylboronic acids.
Scheme 13: Allenylboronic esters are unreactive towards ketones and imines.
Inspired by the previous work on the synthesis of allenylboronic esters by Ito
and co-workers74, and by our group75 (see above section 1.3.1), we developed a
copper-catalysed synthesis from propargylic carbonates. Tetrahydroxydiboron
(49) was chosen as a boron source, motivated by the earlier work of the Szabó
group on the synthesis of allylboronic acids.89 The optimisation required three
major innovations: First, the active catalyst copper(I) methoxide (CuOMe) was
generated in situ from mesitylcopper(I) (CuMes, 10 mol%) and MeOH. Sadighi
and co-workers have reported the transmetallation of copper(I) alkoxides with
B2pin2 and characterised the formed copper-Bpin species.90 Secondly, the weakly
coordinating and easy-to-remove trimethyl phosphite (P(OMe)3, 20 mol%) was
employed as ligand. And third, for the in situ protection of the generated boronic
acid, ethylene glycol (3.0 equivalents) was added.
With the optimal conditions in hand, allenylboronic acid 50a was generated
at −10 °C after 24 hours in 76% 1H NMR yield (Table 1, entry 1). Deviation from
these optimal conditions, such as using CuCl and alkali metal salts KOMe, NaOMe
or LiOMe instead of mesitylcopper(I) led to decreased yields of 52%, 49%, and
65%, respectively (entries 2-4). A drastic drop in yield to 29% was found when
CuI was employed with LiOMe (entry 5). A change of the ligand from P(OMe)3 to
PPh3 was possible, but the yield declined to 46% (entry 6). Other ligands such as
tricyclohexylphosphine (PCy3) and triisopropyl phosphite (P(O-i-Pr)3) did only

16
lead to allene formation (entry 7), which is probably caused by a copper-
catalysed protodeborylation of 50a. Replacing ethylene glycol with
1,3-propanediol led to a diminished yield of 49% (entry 8). Without any ethylene
glycol, the yield decreased slightly to 66% (entry 9). The reaction was performed
without 3 Å molecular sieves and a similar yield of 75% compared to the
standard conditions was obtained (entry 10). However, molecular sieves were
used for all further reactions, as we found an improved reproducibility of the
yield with them. With an elevated reaction temperature of 0 °C, only 16% of the
desired allenylboronic acid 50a was found together with a large amount of
protodeborylated allene (entry 11). Replacing the solvent MeOH with THF or
toluene inhibited the formation of the product altogether (entry 12).
Table 1: Optimisation of the synthesis of allenylboronic acid 50a.a
It is important to note, that allenylboronic acid 50a is sensitive to oxygen and
silica gel. The crude reaction mixture was quenched with a 0.5 M HCl solution,
which released the free allenylboronic acid from any ethylene glycol or methyl
esters. The boronic acid 50a was then extracted with degassed toluene.

17
Allenylboronic acids could be stored in toluene solution (over 3 Å molecular
sieves at −18 °C under Ar) for several weeks without decomposition.
2.1.2. Purification and isolation of allenylboronic acid 50a If necessary, allenylboronic acid 50a can be further purified (Scheme 14).
Inspired by the methodology of Santos and co-workers91 for the purification of
organoboron species, we added diethanolamine to 50a in toluene. Within
30 minutes a boronic ester 51a precipitated (Scheme 14a). This moisture-stable
boronic ester 51a was, unfortunately, decomposing on silica gel. However,
simple filtration and wash with diethyl ether provided an efficient alternative for
the purification. Afterward, degassed toluene was added to 51a and the
diethanolamine was released after the addition of 0.5 M HCl. Extraction with
degassed toluene provided the purified allenylboronic acid 50a in a slightly
decreased yield of 73%.
Scheme 14: Purification of allenylboronic acid 50a via esterification and deprotection (a) and attempted transesterification of the allenyl pinacolate 37a (b).
Santos and co-workers reported a transesterification procedure using alkyl
pinacol boronic esters, which were converted to the corresponding
diethanolamine-protected species within 30 minutes at room temperature.91 In
the case of allenylboronic esters such as 37a, no transesterification was
observed, even when the reaction mixture was heated to 40 °C for 48 hours
(Scheme 14b).
2.1.3. Scope of the synthesis of allenylboronic acids 50 With the optimal reaction conditions in hand, the functional group influence
of the reaction was explored (Table 2). First, the substituent on the terminal
position of the alkyne was changed to a n-butyl chain (Table 2, entry 1).
18
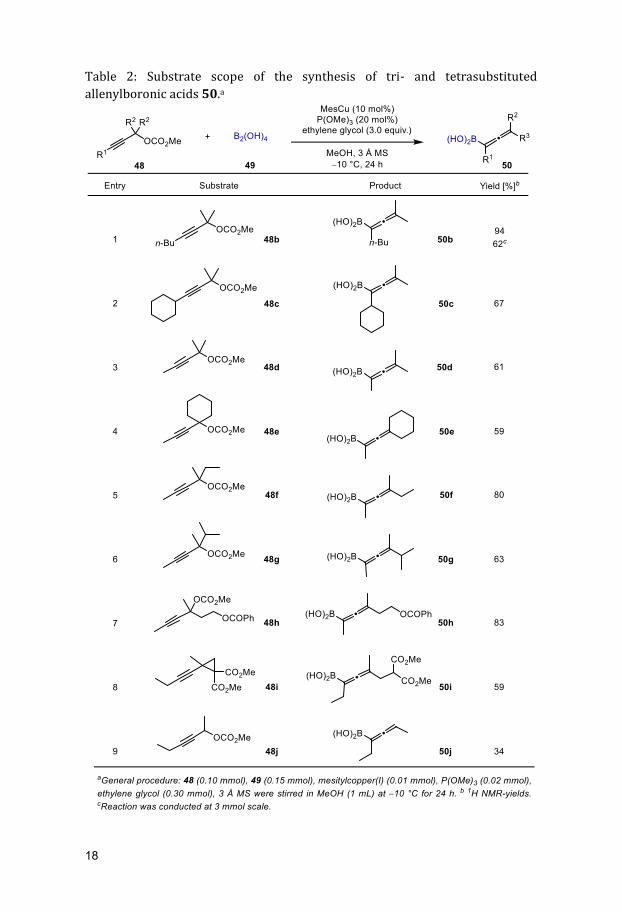
Table 2: Substrate scope of the synthesis of tri- and tetrasubstituted
allenylboronic acids 50.a

19
Allenylboronic acid 50b was obtained in a high 1H NMR yield of 94% at 0.1 mmol
scale. At a larger scale of 3 mmol, the yield decreased to 62%. Changing the
substituent to a cyclohexyl 48c or methyl group 48d provided the corresponding
allenylboronic acids 50c and 50d in 67% and 61% yield, respectively (entries
2-3).
Exchanging the two methyl substituents at the propargyl carbon to a
cyclohexyl ring 48e, provided allenylboronic acid 50e in a slightly decreased
yield of 59% (entry 4). When only one of the methyl substituents at the
propargylic carbon was substituted by either an ethyl 48f or an isopropyl moiety
48g, racemic allenylboronic acids 50f-g were obtained in 80% and 63% yield,
respectively (entries 5-6). Carbonates containing an aliphatic carboxylate group
48h were also well-tolerated and provided the desired product 50h in 83% yield
(entry 7). Inspired by a previous publication by the group,92 borylation of the
propargyl cyclopropane 48i opened the cyclopropane ring to afford
allenylboronic acid 50i in 59% (entry 8). Even the synthesis of the unstable
trisubstituted allenylboronic acid 50j was possible in a moderate yield of 34%
(entry 9).
2.1.4. Extension to other allenylboron derivatives We were very pleased to see, that the above-described method could be
extended to other diboron sources such as B2nep2, B2pne2 and B2pin2 (Scheme
15). The neopentyl glycolate boronic ester was formed in 67% NMR yield, by
exchanging B2(OH)4 to B2nep2, and omitting ethylene glycol (Scheme 15a). The
formed ester 53 was moisture and oxygen stable.
However, 53 decomposed on silica gel and therefore only the 1H NMR yield is
given. The reaction of carbonate 48a with the commercially available chiral
diboron reagent 54 provided allenylboronic ester 55 in 52% yield. The isolated
boronate 55 could be stored in the refrigerator for several weeks (Scheme 15b).
The third borylation provides allenyl pinacolates and offers an alternative to the
earlier methods by Ito and co-workers74 and our group75, 92 (Scheme 15c).
The first example of a hydrolysis of pinacolyl arylboronic esters to
arylboronic acids was reported by Falck and co-workers.93 Inspired by this
procedure, Petasis and co-workers applied this hydrolysis to mono- and
disubstituted allenylboronic acids.71 An excess of sodium periodate was used to
deprotect the Bpin group which was hydrolysed and protonated under acidic
conditions. The deprotection of allenylboronate ester 37a to 50a through
oxidative hydrolysis was also investigated (Scheme 15c). Allenylboronic acid
50a was obtained in 63% overall yield by this procedure.

20
Scheme 15: Extension of the methodology by using other boron sources. a 1H NMR yield; b Isolated yield.
2.1.5. Racemic synthesis of propargyl alcohols and amines using allenylboronic acids
Next, the reactivity of the synthesised allenylboronic acids 50 was studied.
The allenylboronic acids 50 were synthesised following the general procedure
in Table 2 and used without further purification. To a solution of 1.5 equivalents
of p-Br-benzaldehyde (44b) in toluene in the presence of 3 Å MS, was added
allenylboronic acid 50b in toluene (Table 3, entry 1). The reaction was
completed within 10 minutes resulting in 87% yield of the homopropargyl
alcohol 56. Applying the same reaction conditions to the allenylboronic ester
37b did not provide any product 56. Also, 37b was unreactive towards ketones
and imines (entries 2-3). The negative results with 37b further highlight the very
high reactivity of unprotected allenylboronic acids which is a consequence of
their ability to form boroxines.84
Allenylboronic acid 50b reacted with p-Br-acetophenone 39b in the presence
of 3 Å MS affording the corresponding homopropargyl alcohol 57b in 72% yield
(entry 2). Aldimines 47a-c reacted smoothly with allenylboronic acids 50b and
50e in yields of 63−83% (entries 3-5). To our delight, 3,4-dihydroisoquinoline
(47d) reacted readily to afford 58d in an excellent yield of 96% (entry 6). Indole
(47e) underwent propargylation with allenylboronic acid 50a in C-2 position94
providing indoline 58e in 82% yield (entry 7).

21
Table 3: Synthesis of racemic propargyl alcohols and amines.a

22
2.1.6. Optimisation and substrate scope for the asymmetric organocatalysed synthesis of homopropargylic alcohols
As mentioned in the introduction, the catalytic asymmetric propargylation of
ketones has been studied by several groups (see Scheme 12). Using 15 mol% of
(S)-Br2-BINOL 40a, tetrasubstituted allenylboronic acid 50b underwent
stereoselective propargylation with p-Br-acetophenone (39b). The
homopropargyl alcohol 57b formed in the presence of 3 Å MS with 2 equivalents
of ethanol as an additive in toluene within 48 hours at room temperature. The
product 57b was obtained in a high yield of 95% and excellent enantioselectivity
of 94% ee. (Table 4, entry 1). With equimolar amounts of (S)-Br2-BINOL 40a and
without the use of ethanol, the yield and enantioselectivity decreased
significantly to 82% and 77% ee (entry 2). The selectivity dropped to 44% ee
when catalytic amounts of 40a (15 mol%) were used, without any ethanol (entry
3). These findings underline the importance of an aliphatic alcohol in the reaction
mixture, which has been highlighted by previous studies.83, 95 Using t-BuOH
instead of ethanol afforded a similarly high yield of 91%, but unfortunately, a low
enantioselectivity of only 55% ee (entry 4). The role of ethanol will be further
discussed in section 2.1.8. The substitution of (S)-Br2-BINOL 40a to
(R)-Br2-BINOL 40b led to a high yield of 93% and a comparable ee of 94% of the
other enantiomer of 57b.
Table 4: Variation of conditions for the asymmetric synthesis of homopropargyl alcohols 57b.a
The substrate scope for the asymmetric propargylboration was explored.
Starting from acetophenone, the desired homopropargyl alcohol 57a was

23
obtained in 75% yield and 97% ee (Table 5, entry 1). The p-CN and
p-OAc-substituted acetophenones (entries 2-3) reacted smoothly with
allenylboronic acid 50b in good yields of 67−90% and excellent
enantioselectivities of 91−96% ee.
Table 5: Substrate scope for the asymmetric propargylation of ketones 39.a
Unfortunately, all the above-mentioned tertiary alcohols were oils. In order to
determine the absolute configuration of the homopropargyl alcohols 57 by X-ray

24
diffraction, we made several attempts to obtain a crystalline compound. The
sulfone derivatives 57e-g were obtained in good yields of 62−64% and excellent
enantioselectivities 94−99% (entries 4-6). The esterification of 57f with
biphenyl acyl chloride 59a provided the crystalline product 60 in 71% yield.
Single X-ray diffraction revealed the stereocenter has R configuration, therefore,
all products were then assigned accordingly (Scheme 16).
Scheme 16: Esterification of homopropargylic alcohol 57e.
2.1.7. Kinetic resolution experiment to form vicinal quaternary stereocenters
A report by Schaus and co-workers86 described the kinetic resolution of
racemic, disubstituted allylboronic acids. Here, we wanted to explore, if the same
applies to a racemic allenylboronic acid 50g (Scheme 17). To obtain the sterically
encumbered tertiary homopropargyl alcohol 61 the reaction conditions had to
be re-optimised, most likely because of the increased steric bulk of the isopropyl
group. The desired product formed with 2 equivalents of ketone 39b,
1 equivalent of the chiral organocatalyst 40a and 3 Å MS at an elevated
temperature of 45 °C in 22 h. We have found that only one of the enantiomers of
50g underwent the reaction.
Scheme 17: Kinetic resolution of boronic acid 50g affording homopropargylic alcohol 61 with two vicinal quaternary stereocenters.
Consequently, we were able to obtain only one single diastereomer of 61 with
an isolated yield of 31% and an excellent enantioselectivity of 96% ee. It is
noteworthy that without the BINOL catalyst 40a no product was formed. The
reason could be an activating effect of BINOL on 50g.84

25
2.1.8. Proposed stereoselection model and catalytic cycle for the asymmetric synthesis of homopropargyl alcohol 57a
Based on the reaction of allenylboronic acid 50b, ketone 39a, and
organocatalyst 40a we propose a stereoselection model (Scheme 18). First,
BINOL 40a esterifies with the boronic acid 50b. The ketone 39a has two
enantiotopic faces and can approach the BINOL ester 62 with either face to
undergo a 6-membered transition state, which has been reported by several
groups.86-87, 88, 96 If 39a is in the Re-face, it will clash with one of the bromine
substituents of BINOL. This transition state would lead to the (S)-enantiomer of
homopropargyl alcohol 57i. However, the favoured approach is from the Si-face.
There are no steric clashes and the (R)-enantiomer of 57a is detected as the
major product.
Scheme 18: Proposed stereoselection model for the propargylation of acetophenone.
The proposed mechanism of the catalytic process is given in Scheme 19. The
asymmetric propargylboration starts with a fast mono- or diesterification with
ethanol to allenylboronic ester 63, which has been observed by 1H NMR. It is
important to note, that 63 is not able to react with the ketone directly. Therefore,
the racemic reaction is efficiently prevented. This is in accordance with the
earlier findings from the optimisation above (Table 4, entries 2-3), where the
omission of alcohol decreased the enantioselectivity.83 In the second step, 63
undergoes transesterification to the BINOL ester 62. According to DFT studies
by our84 and other groups,97 the activation barrier of the reaction between a
ketone and a BINOL-ester, such as 62, is much lower than for the same reaction
with boronic esters such as 63. A reason could be the increased Lewis acidity of
62 compared to 63. The transesterification itself is likely to be much faster
between the aliphatic ester 63 and 62 than it is for the bidentate pinacolate 37b

26
to diethanolamine ester 51a (vide supra Scheme 14). The intermediate 62 is
highly reactive towards the ketone electrophile, it undergoes propargylation
(see stereoselection model, Scheme 18). Then, ethanol releases the product 57a
and the BINOL catalyst 40a.
Scheme 19: Proposed mechanism for the asymmetric propargylation exemplified with ketone 39a and boronic acid 50b.
2.1.9. Conclusions and outlook This study provides the first copper-catalysed synthesis of tetrasubstituted
allenylboronic acids. The developed procedure can be extended to other boron
sources, supplying an alternative to previous protocols. The high reactivity of
allenylboronic acids was beneficial for their smooth reaction with electrophiles
such as ketones and imines. The asymmetric propargylboration of ketones was
successful and propargyl alcohols with vicinal quaternary stereocenters were
accessed. Recently, the Szabó group has published two further studies on the
application of allenylboronic acids.95a, 98

27
2.2. Synthesis of 2,2-difluoro ketones from 2,2-difluoro enol silyl ethers and application totrifluoromethylthiolation, fluorohomologation, and arylation (Paper II)
The relevance of fluorine in pharmaceutical industry has increased drastically
over the last decade, as mentioned above (section 1.1).3, 99 For example,
organofluorine compounds with α-fluorinated ketone motifs are important
pharmacophores, in particular in serine protease inhibitors (see above Figure 1,
section 1). Nucleophilic attack by the hydroxy group of the serine moiety leads
to the formation of a tetrahedral hemiketal in which the ligand covalently binds
to the active site of the serine protease. A possible way to synthesise α-
fluorinated ketones is through the use of 2,2-difluoro enol silyl ethers as starting
material.6c, 9a, 10a, 11a, 100
A common protocol for the synthesis of 2,2-difluoro enol silyl ethers was
published by the groups of Uneyama101 and Prakash, Hu and Olah (Scheme
20).102 Activation of 2,2,2-trifluoroacetophenone (65a) with magnesium leads to
an enolate intermediate which is then protected by a trimethylsilyl group. After
filtration and evaporation, the 2,2-difluoro enol silyl ether 66a can be used
without further purification. Protonation of 66a results in the corresponding
difluoromethyl ketone 67a.102
Scheme 20: Synthesis and protonation of 2,2-difluoro enol silyl ethers.
Further applications required activation of 66a by tertiary amines, Lewis or
Brønsted acids. An early example by Uneyama and co-workers describes the
copper-mediated cross-coupling of 66a with furan 68 (Scheme 21).103 The
procedure was further expanded to thiophenes and pyrroles.
Scheme 21: Oxidative cross-coupling of 2,2-difluoro enol silyl ethers.
Several studies on the arylation of 66a have been reported.104 Shreeve and
co-workers published a cross-coupling reaction of 66a with aryl bromides 70
catalysed by Pd(OAc)2 (Scheme 22a).105 Additionally, Cai, Lu and co-workers
presented a photocatalysed difluoromethylation of diazonium salts 72 (Scheme

28
22b).106 Recently, Peng, Wang and co-workers studied the ortho-
difluoroalkylation of aryliodanes with 66a (Scheme 22c).107 In this reaction, a
simultaneous rearrangement and reduction of the aryliodane took place leading
to difluoroalkylated o-iodo arenes 74. The reaction was initiated by in situ
generation of the electrophilic Ar-I(OTf)2 from Ar-I(OAc)2 73 and TMSOTf.
Scheme 22: Examples of the arylation of 2,2-difluoro enol silyl ethers.
It is interesting to mention that Cahard and co-workers performed the first
trifluoromethylation of a non-fluorinated enol silyl ether 21 with Umemoto’s
reagent 12d.108
Scheme 23: Trifluoromethylation of enol silyl ether using reagent 12b.
A consecutive study by Togni and co-workers utilised reagent 12b for the
trifluoromethylation of non-fluorinated cyclic enol silyl ether to form a
β-trifluoromethylated ketone 75 (Scheme 23).109

29
2.2.1. Electrophilic trifluoromethylthiolation of 2,2-difluoro enol silyl ethers
This project aims for the development of new versatile functionalisations of
2,2-difluoro enol silyl ethers 66 with a variety of electrophiles. Szabó and
co-workers have studied the application of the dibenzene sulfonimide-based
SCF3 transfer reagent 15b48b for the bifunctionalisation of diazocarbonyl
compounds 76 (Scheme 24).110 We hypothesised that an activated 2,2-difluoro
enol silyl ether should also react with a reactive electrophilic reagent such as
N-trifluoromethylthio-dibenzene sulfonimide 15b.48b
Scheme 24: Trifluoromethylthiolation of diazo ketones by Szabó and co-workers.
2.2.2. Variation of reaction conditions for thetrifluoromethylthiolation of 2,2-difluoro enol silyl ethers
Our idea was based on the reaction of 66a with the SCF3 transfer reagent 15b
in the presence of an additive. We began our investigation by screening different
additives. Substrate 66a was freshly prepared from 2,2,2-trifluoroacetophenone
(65a) with magnesium and TMSCl (Table 6).101-102 The optimal reaction
conditions were found to be equimolar amounts of each 66a, 15b and potassium
fluoride in MeCN. The desired -CF2SCF3 product 79a was obtained in 78% yield,
within 3 hours at room temperature (entry 1). Replacing KF with other additives
or changing the solvent gave lower yields. Thus, changing from KF to CsF, slightly
decreased the yield to 61% (entry 2). In both cases, a small quantity of the
protonated side-product 67a was formed, 3% and 6%, respectively (entries 1-2).
When TBAF was used as an additive instead, the yield dropped significantly to
15% (entry 3). Also, the formation of 67a increased to 45% as TBAF contains
three mol of crystal water. The group of Prakash has reported the protonation of
2,2-difluoro enol silyl ether 66a in high yields in the presence of water (vide
supra Scheme 20).102 In entry 4, using TBAT as an additive brought a moderate
yield of 41% and a minor formation of 67a. In aldol reactions of 66a, tertiary
amines are used as additives.111 However, in this case, 10 mol% of DABCO did

30
not lead to any product formation (entry 5). Instead, 70% of unreacted 66a could
be detected. A similar result was obtained with 10 mol% of FeCl2 (entry 6). Only
8% of 79a formed. Changing the solvent from MeCN to DCM or omitting the
additive was both detrimental to product formation (entries 7 and 8). Without
any additives, only traces of trifluoromethylthiolated 79a formed and 75% of
unreacted 66a were detected.
Table 6: Additive screening for the trifluoromethylthiolation of 2,2-difluoro enol silyl ether 66a.a
2.2.3. Substrate scope for the trifluoromethylthiolation of 2,2-difluoro enol silyl ethers
With the optimal reaction conditions in hand (Table 6, entry 1), the
robustness towards varying substrates was explored (Table 7). First, it is
important to note that fluorine substituents in small molecules often lead to
lower boiling points and therefore increased volatility compared to their non-
fluorinated analogues. This is the case for 79a which was detected in 78% yield
in the crude reaction mixture, through 19F NMR with addition of an internal
standard. However, after isolation product 79a was obtained in only 25% yield.
As the p-phenyl-substituted acetophenone 79b was less volatile than 79a, it

31
could be isolated in a high yield of 78%. The reaction proved to be scalable
without significant loss, yielding 71% at 1.0 mmol scale. The 2-naphthyl-
substituted ketone 79c gave a high yield of 84% within a prolonged reaction time
of 4 h. A slightly decreased yield of 67% and 62%, respectively, was found for
substrates with the electron-donating substituents p-methoxy 79d and
p-ethylthio 79e.
Table 7: Substrate scope in the potassium fluoride-mediated trifluoromethylthiolation of 66.a
The p-fluoro-substituted ketone 79f was isolated in 35% yield. Its volatility
resulted in a reduced isolated yield compared to the NMR yield of 57%. Both the
2-methyl alkenyl naphthyl 79g and 2-methyl alkenyl thienyl 79h products
formed in good yields of 70% and 60%. We were also able to
trifluoromethylthiolate a synthetic precursor of the NE inhibitor (see above
Figure 1, section 1) For this purpose, the reaction time was prolonged to 18 h,
product 79i was then isolated in 35% yield.

32
2.2.4. Electrophilic trifluoromethylation of 2,2-difluoro enol silyl ethers
Inspired by the results of the electrophilic trifluoromethylthiolation, we
aimed to extend the project to trifluoromethylations. Since the discovery of
reagents 12a-b, the interest in trifluoromethylation has grown considerably and
many research groups have contributed to the field.43-44 There are numerous
studies to understand the structure, properties, and reactivity profile of
hypervalent iodine reagents 12a-b. Among them, it was found that activation of
12a-b is possible with Lewis and Brønsted acids.43-44 Shen, Liu and co-workers
reported the first copper-catalysed trifluoromethylation of organoboron
compounds with 12b using arylboronic and vinylboronic acids.112 Shortly after,
Buchwald and co-workers described an iron(ІІ)-catalysed trifluoromethylation
of vinylborates (Scheme 25).113
Scheme 25: Trifluoromethylation of vinylborates with 12a.
Our group has also gathered considerable experience in the synthesis,
reactivity and application of hypervalent iodine compounds.28f, 30a, 114 For
example the cyanotrifluoromethylation of styrenes 82 (Scheme 26a).114b And, as
the second example the rhodium-catalysed difunctionalisation of diazo ketones
76 (Scheme 26b).30a This study used a variety of alcohols and diazo ketones and
even allowed for fluorination reactions to take place under similar conditions.

33
Scheme 26: Cyanotrifluoromethylation of styrenes (a) and oxytrifluoromethylation of diazo ketones (b).
2.2.5. Optimisation of the reaction conditions for the trifluoromethylation of 66a.
The trifluoromethylation reaction was first attempted under the optimal
conditions of the above-described trifluoromethylthiolation of 2,2-difluoro enol
silyl ethers. Thus, the reaction was performed with equimolar amounts of the
trifluoromethyl-benziodoxolone 12a,43 2,2-difluoro enol silyl ether 66a and KF
in MeCN for 2 hours at room temperature (Table 8). The desired product,
pentafluoroethyl acetophenone (86a), was obtained in 35% yield (entry 1). The
protonated 2,2-difluoroacetophenone (67a) was formed in 10% yield and 32%
of trifluoromethylating reagent 12a remained unreacted. We realised that the
mode of activation or the reactivity of reagents 12a and 15b were different.

34
Table 8: Additive screening for the trifluoromethylation of 2,2-difluoro enol silyl ether 66a.a
When CsF was used instead of KF, the desired product 86a was formed in
24% yield (Table 8, entry 2), while 36% yield of reagent 12b remained
unreacted. A slight increase to 42% yield was observed when 1.5 equivalents of
KF were added (entry 3), also the side-product formation and recovery of 12a
remained similar. Employing a tertiary amine such as DABCO or TMEDA gave
comparable yields of 43% and 36%, respectively (entries 4 and 5). As mentioned
above (see section 2.2), in many reactions of 2,2-difluoro enol silyl ethers,
transition metals are used as catalysts or Lewis acid-type additives. Therefore
we tested several metal complexes.103-104 The use of 10 mol% CuOAc allowed a
moderate product formation of 37% and led to a recovery of 28% of 12a, and no
protonated side-product (entry 6). A significant increase in yield to 64% was
observed when 10 mol% of FeCl3115 were added. Gratifyingly, no side-product
was found and the trifluoromethylating reagent was completely consumed

35
(entry 7). In comparison, using 10 mol% of FeCl277, 113, 116 gave a similar yield of
62%, also without any formation of protonated ketone 67a (entry 8).
Replacement of 12a with other electrophilic trifluoromethylating reagents such
as trifluoromethyl-benziodoxole 12b43 resulted in a decreased yield of 20%
(entry 9). And Umemoto’s reagent 12d (5-(trifluoromethyl)dibenzo-
thiophenium tetrafluoroborate)41 gave only 1% yield of the desired product
(entry 10). In conclusion, FeCl2 was chosen as the catalyst over FeCl3 as it proved
to give an overall cleaner reaction.
2.2.6. Substrate scope for the iron(II) chloride-mediated trifluoromethylation of 66
With the optimised reaction conditions in hand, the scope of the reaction was
explored (Table 9). In comparison to the trifluoromethylthiolated ketones 79,
the perfluoroethyl ketones 86 are even more volatile. The increased tendency of
small fluorinated molecules to be volatile compared to their hydrocarbon
analogues originates from the properties of the C-F bond. The low polarizability
of the C-F bond is the reason for its weak cohesive forces between organofluorine
molecules (London dispersion force).117
Table 9: Substrate scope of perfluoroethyl ketones 86.a
For compound 86a only the 19F NMR yield of 62% could be obtained.
Exchanging acetonitrile to other, more volatile solvents such as
dichloromethane, did not lead to any product formation. p-Phenylacetophenone
66b reacted smoothly and 86b was isolated in 85% yield. A significant drop in
yield was found for the 2-naphthyl ketone 86c, which can be partially explained
by a difficult purification. The electron-donating p-EtS-acetophenone reacted in

36
a high yield of 70%. The p-fluoroacetophenone 86f was volatile and gave a 19F
NMR yield of 65%. Lower yields of 20% and 33%, respectively, were found for
the thienyl substrate 86h and the NE inhibitor analogue 86i.6c
After studying both trifluoromethylation and trifluoromethylthiolation of
66a, one may wonder, if it is possible to perform the two reactions in sequence.
The group of Nakamura118 had already published a study involving the synthesis
of a fluoro-trifluoromethyl enol silyl ether. Using slightly different conditions for
the synthesis of 66j, we successfully obtained the trifluoromethylthiolated
ketone 87 in an excellent yield of 86%. Interestingly, this molecule 87 has three
different fluorinated groups attached to the same carbon atom (Scheme 27).
Scheme 27: First synthesis of an acetophenone with three fluorine functional groups in β-position.
2.2.7. Potassium fluoride-mediated arylation of 66 After the successful use of the hypervalent iodine reagent 12a for the
transformation of 66, we wanted to further expand the scope of the reaction to
other iodine(III) reagents such as diaryliodonium salts and alkynylation
reagents.119 Unfortunately, the alkynylation did not result in any product
formation. On the other side, the arylation was successful. Reaction of equimolar
amounts of diphenyliodonium triflate (88a), 2,2-difluoro enol silyl ether 66a and
KF as additive yielded product 71a in 61% in MeCN at room temperature within
18 hours (Table 10). Other additives, such as FeCl2 or DABCO did not lead to any
product formation.
The reaction of 66a with bis(4-bromophenyl)iodonium triflate (88b) instead
gave a high yield of 72% for 71b. A slightly decreased yield to 56% was found
when 3-naphthyl substituted enol silyl ether 68c reacted with iodonium salt 88a.
When 88b was used instead, the yield increased noticeably to 71%. A smaller
difference was found for the p-F-substituted ketones 71e and 71f, here the
phenyl and p-Br-phenyl iodonium salts lead to 50% and 55% yield, respectively.
The p-SEt-acetophenone 71g was obtained in 56% yield and the 2-methyl-
thienyl substrate 71h in a high yield of 69%.

37
Table 10: Scope of the arylation of 2,2-difluoro enol silyl ethers 66.
2.2.8. Conclusion In summary, we have developed three new methods for the electrophilic
functionalisation of 2,2-difluoro enol silyl ethers. First, the
trifluoromethylthiolation for the construction of a new functional group,
a -COCF2SCF3 moiety. The electron-withdrawing character of the fluorine atoms
in combination with the lipophilicity of the thiol provide an interesting new
motif, especially for bioactive compounds. Second, the study was further
extended to an iron-catalysed difluoro homologation method. There, the well-
known, electrophilic trifluoromethyl-benziodoxolone 12a was used to provide
an easy method to synthesise perfluoroethyl ketones. The third part of the study
offers a transition-metal free arylation of 2,2-difluoro enol silyl ethers from
commercially available diaryliodonium salts.

38
2.3. Synthesis of a fluorine-18 labelled, electrophilic hypervalent iodine fluorinating reagent (Paper III)
As mentioned in the introduction above (see section 1.1.1), hypervalent iodine
reagents, such as 5d can be synthesised from nucleophilic fluorine sources. This
is especially interesting for 18F-labelling where only the nucleophilic
[18F]fluoride can be produced in large amounts of activity with high molar
activities. This chapter aims for the development of a new method to obtain an
electrophilic 18F-fluorinating reagent and its application to a cyclisation reaction.
2.3.1. From fluorine-19 to fluorine-18: Adaption of the synthesis of fluoro-benziodoxole
The development of a new 18F-fluorinating reagent bears several difficulties.
First, there is a time constraint with the half-life of fluorine-18 (t1/2 =
109.8 min).54 The whole process from bombardment, synthesis of the reagent,
application, isolation and quality control should not take longer than two half-
lives of the radionuclide.57c Second, there is a fundamental scale difference
between experiments performed with non-radioactive fluorine-19 and
radioactive fluorine-18. The labelled compound will always be the lowest in
concentration compared to all other components in the reaction mixture
(nmol scale). Third, some reagents, reaction conditions and purification methods
are not accessible in a radiochemical laboratory.64f
Inspired by previous studies from the Szabó group on the application of the
electrophilic fluorinating reagent fluoro-benziodoxole 5d,30-31 we aimed to
develop a 18F-labelled analogue. In contrast to other electrophilic fluorinating
reagents such as Selectfluor® 5c or NFSI 5a, which are obtained from F2, 5d can
be synthesised from a variety of nucleophilic fluorine sources through a
umpolung reaction.25, 40, 43, 120 This advantage could lead to a higher molar activity
than for 18F-reagents derived from [18F]F2.54, 56b, d, 58
Stuart and co-workers have developed a versatile procedure for the synthesis
of 5d starting from either the trifluoroacetoxy-benziodoxole 89 or the tosyl-
benziodoxole 90 (Scheme 28).120c The fluorination of 89 with TREAT-HF and
TBAF (1.0 M in THF) 4b in DCM afforded the desired reagent 5d in 63% yield
within 4 hours at room temperature. The fluorination of 90 under the same
conditions yielded 5d in 46%.

39
Scheme 28: Fluorination of trifluoroacetoxy- 89 or tosylbenziodoxole 90 with TREAT-HF and TBAF.
An advantage of this approach is the versatility of the reaction. Several
precursors are possible. However, there are two sources of fluorine used and the
reaction time of 4 hours is too long. To adapt the procedure by Stuart and
co-workers120c to 18F-labelling conditions, the reaction time was reduced to
20 min, the temperature was elevated to 70 °C and only TBAF hydrate 4c was
used as fluorine source (Scheme 29). Gratifyingly, we were able to obtain 99% of
5d determined by 19F NMR yield. Application of 89 as precursor instead yielded
only 33% of 5d under the same reaction conditions.
Scheme 29: Fluorination of 90 with 4c.
To mimic 18F-labelling conditions such as the large excess of all other reaction
components compared to the fluorine-18 source, we decreased the amount of 4c
gradually (Table 11). Using 0.5 equivalents of 4c instead of 1.2 equivalents, did
not decrease the yield (Table 11, entry 1). When the amount of 4c was lowered
to 0.1 equivalents the yield dropped to 72% (Table 11, entry 2). Elevating the
temperature to 80 °C increased the yield to 93% whereas further heating to 90 °C
led to a drop in yield to 75% (Table 11, entries 3-4).

40
Table 11: Optimisation of reaction conditions for the fluorination of 90.a
Fluoro-benziodoxole 5d is known for its affinity to silicon, aluminium and
alkali metal ions.121 This usually leads to the decomposition of the reagent while
performing most purifying techniques such as chromatography. After
evaporation of DCM, Stuart and co-workers performed an extraction into
n-hexane at 40 °C.120c This is an efficient method for the purification of the n-
hexane soluble 5d, as neither 90, 4c nor tetrabutylammonium salts, which are
formed during the reaction, are soluble in n-hexane.
Table 12 gives an overview of the performed optimisation for the synthesis of
5d (entries 1-3) and [18F]5d (entries 4-10). After the addition of 0.1 equivalents
of 4c to 90 in DCM (1.4 mL), the reaction mixture was stirred for 20 minutes at
80 °C. The solvent was evaporated and then n-hexane was added and stirred at
room temperature for 1 minute (entry 1). The supernatant was transferred and
evaporated, but the desired product 5d was not obtained. Performing the
reaction under the same conditions, but the extraction with n-hexane at 40 °C
yielded 81% of 5d detected by 19F NMR (entry 2). Elevating the extraction
temperature further to 70 °C led to a drop in yield to 48% (entry 3).
It cannot be expected that the use of the optimised conditions from the
fluorine-19 procedure (Table 12, entry 2) give identical results under 18F-conditions, where only minute amounts of the 18F-source are used. Using
freshly prepared [18F]TBAF [18F]4a and 90 in dry DCM the reaction mixture was
stirred for 20 minutes at 70 °C (Table 12, entry 4). Then, the extraction was
performed at 70 °C giving [18F]5d with an RCY of 40 ± 7%. Prolongation of the
reaction time to 40 minutes increased the RCY to 47 ± 2% (entry 5).
Concentrating the reaction mixture, by using 250 L of DCM instead of 500 L,
decreased the RCY to 37 ± 1% (entry 6). Interestingly, when the reaction
temperature was lowered to room temperature, an acceptable RCY of 40 ± 3%
was obtained (entry 7).

41
Table 12: Optimisation for the synthesis and purification of 5d and [18F]5d.a
When the reaction time was shortened from 20 minutes to 5 minutes an RCY
of 41 ± 1% was detected (entry 8). It was also found that the extraction
temperature had an important effect on the RCY. When the extraction was
performed at 40 °C or room temperature instead of 70 °C, the RCY dropped to
23 ± 3% and 20 ± 2%, respectively (entries 9-10). The overall best conditions are
given in entry 8, featuring short reaction times and a mild reaction temperature.

42
2.3.2. Application of [18F]5d to an electrophilic fluorocyclisation reaction
In order to demonstrate the radiosynthetic utility of [18F]5d, we performed a
fluorocyclisation reaction with o-styryl amides. This fluorocyclisation reaction
using the natural fluorine isotope was first reported by Gulder and co-workers
(Scheme 30).122 When freshly synthesised 5d and o-styryl amide 10a were
reacted at 70 °C, fluoro-benzoxazepine 11a was obtained in 2 minutes in 39%
yield.
Scheme 30: Application of 5d in the fluorocyclisation of an o-styryl amide.
These reaction conditions were also applied using [18F]5d affording
[18F]benzoxazepine [18F]11a in good RCY of 80 ± 1% (Table 13, entry 1). When
[18F]TBAF [18F]4a was used instead of [18F]5d, as expected [18F]11a was not
formed (entry 2). This control experiment was also indirect evidence for the
formation of [18F]5d from 90 and [18F]TBAF. This is an important finding, as
[18F]5d and 5d cannot be detected by any chromatography methods because of
their instability towards silica.
Then, we studied the substituent effects on the fluorocyclisation reaction. In
the presence of a p-chloro substituent, [18F]11b was formed in an RCY of 12 ± 1%
within 7 minutes at 50 °C (entry 3). When the temperature was increased to
90 °C the RCY increased to 76 ± 2% (entry 4). In the presence of a p-chloro
substituent on the phenacyl moiety of the o-styryl benzamide 10c, a high RCY of
88 ± 6% was obtained (entry 5). However, using an electron-donating
substituent such as a methyl group in para-position of the o-styryl benzamide
10d, led to a decreased RCY of the fluorocyclisation product 11d of 46 ± 3%
(entry 6). A reason could be the lower thermal stability of 7d compared to 11b-c.
Thus, the reaction was conducted at 70 °C and an elevated RCY of 74 ± 3% was
obtained (entries 7).

43
Table 13: Electrophilic fluorocyclisation leading to [18F]fluoro-benzoxazepines [18F]11.a
2.3.3. Isolation and measurement of the molar activity of [18F]11b As mentioned above, [18F]5d is decomposing in the presence of silica.
Therefore detection and isolation under the measures of a radiochemical
laboratory such as radio-HPLC, radio-TLC and radio-semi-prep-HPLC could not
be performed.

44
However, the isolation of [18F]fluoro-benzoxazepine [18F]11b was possible
(Scheme 31). Starting with 904 MBq of [18F]5d and o-styryl benzamide 10b, the 18F-labelled product [18F]11b was isolated with a semi-prep-radio HPLC system
in 92.4 MBq. A molar activity Am of 396 GBq/mol was measured, indicating that
[18F]5d is an efficient electrophilic 18F-transfer reagent for the synthesis of PET
imaging tracers.
Scheme 31: Isolation of [18F]11b.
2.3.4. Conclusions and outlook
We have developed a new 18F-fluorinating reagent. [18F]5d can be prepared
and purified in 2 hours from [18F]TBAF. The radiosynthetic utility of [18F]5d was
demonstrated in a fluorocyclisation of o-styryl amides affording 18F-labelled
benzoxazepines. The molar activity of [18F]benzoxazepine labelled with [18F]5d
was several magnitudes higher than the molar activities obtained for other
electrophilic 18F-fluorinating reagents derived from [18F]F2. The group123 has
developed another Rh-catalysed 18F-labelling method based on the application
of [18F]5d. A number of fluorination reactions and automation of the
radiosynthesis of [18F]5d are ongoing projects.

45
2.4. 18F-labelling of trifluoromethyl ketones including neutrophil elastase inhibitors (Paper IV)
As mentioned above (sections 1 and 2.2) trifluoromethyl ketones play an
important role as pharmacophores in serine proteases. The proteolytic activity
of a serine protease, neutrophil elastase (NE), is usually regulated by a number
of proteins (α1-protease inhibitor and secretory leukocyte protease inhibitor).
However, in certain pathophysiological states, these proteins fail to regulate the
elastases activity. These changes can lead to abnormal cell degradation, which
occurs for example in the diseases of cystic fibrosis and chronic bronchitis.6b, 124
Another disorder is connected to the formation of neutrophil extracellular
traps (NETs) in the innate immune response of the body. NETs are web-like
structures composed of several fibres among others also neutrophil elastase. It
was found that acute respiratory distress syndrome (ARDS) and other
inflammatory conditions could be caused by the excessive formation of NETs.7d,
10b, 125 ARDS is one of the leading death causes for COVID-19 patients.125b-d
Therefore, research to find a specific tracer to identify neutrophil elastase or
NETs is of great importance.
Electrophilic trifluoromethyl ketones are promising candidates for such
tracers as they can form a tetrahedral hemiketal intermediate covalently bound
to the active site Ser195 hydroxyl (Figure 8). This hemiketal intermediate is an
analogue to the non-fluorinated hemiketal that forms when the hydrolysis of
peptide bonds takes place. Hemiketal formation is followed by the cleavage of
the C-N bond of peptides. However, in the case of the trifluoromethyl hemiketal,
the C-C bond at the -CF3 group cannot be cleaved and thereby prevents further
metabolisation of the molecule.6c, 9a, c, 10b, 11a, 100a, 126
Figure 8: Proposed binding of NE inhibitor to the enzyme.
Many pyridone-based trifluoromethyl ketones have been evaluated as NE
inhibitors. These may differ considerably in their pharmacological properties
such as the inhibition constant Ki and the lipophilicity, examples of NE inhibitors
with a low Ki are carbamates 1a,e and sulfonamides 1f-g (Figure 9).6c
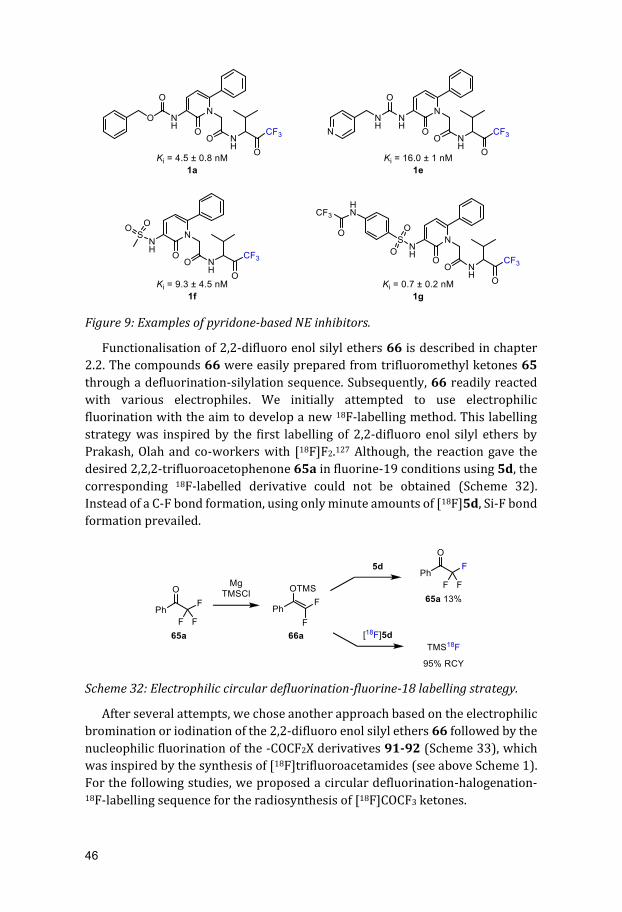
46
Figure 9: Examples of pyridone-based NE inhibitors.
Functionalisation of 2,2-difluoro enol silyl ethers 66 is described in chapter
2.2. The compounds 66 were easily prepared from trifluoromethyl ketones 65
through a defluorination-silylation sequence. Subsequently, 66 readily reacted
with various electrophiles. We initially attempted to use electrophilic
fluorination with the aim to develop a new 18F-labelling method. This labelling
strategy was inspired by the first labelling of 2,2-difluoro enol silyl ethers by
Prakash, Olah and co-workers with [18F]F2.127 Although, the reaction gave the
desired 2,2,2-trifluoroacetophenone 65a in fluorine-19 conditions using 5d, the
corresponding 18F-labelled derivative could not be obtained (Scheme 32).
Instead of a C-F bond formation, using only minute amounts of [18F]5d, Si-F bond
formation prevailed.
Scheme 32: Electrophilic circular defluorination-fluorine-18 labelling strategy.
After several attempts, we chose another approach based on the electrophilic
bromination or iodination of the 2,2-difluoro enol silyl ethers 66 followed by the
nucleophilic fluorination of the -COCF2X derivatives 91-92 (Scheme 33), which
was inspired by the synthesis of [18F]trifluoroacetamides (see above Scheme 1).
For the following studies, we proposed a circular defluorination-halogenation-18F-labelling sequence for the radiosynthesis of [18F]COCF3 ketones.

47
Scheme 33: Nucleophilic circular defluorination-18F-labelling strategy.
2.4.1. Development of the reaction conditions for 2,2,2-[18F]trifluoroacetophenones
For the optimisation of the 18F-labelling of [18F]COCF3 ketones, we chose
2-bromo-2,2-difluoroacetophenone 91a as a model compound. First, [18F]TBAF
4a was synthesised, dried and dissolved in dry DMF. Then, 150 L of the solution
was added to the precursor 91a in 150 L dry DMF and heated at 100 °C for
10 minutes (Table 14, entry 1).
Table 14: Optimisation for the synthesis of 2,2,2-[18F]trifluoroacetophenone [18F]65a.a

48
Encouragingly, the formation of [18F]65a was detected with an RCY of 5%.
Inspired by the conditions of a previous study by the Szabó group, we added N-
containing bases to help release the bromine.21 Addition of 60 mol DBU lead to
a considerable increase in RCY to 65 ± 9% (entry 2). Using TBD instead of DBU
increased the RCY further to a very good 92 ± 3% (entry 3), while lowering the
temperature to 75 °C gave a similar result of 90 ± 1% (entry 4). When MTBD was
used as an additive instead (also at 75 °C) the RCY dropped to 73 ± 13% (entry
5). Even milder temperatures of 50 °C and room temperature, with TBD as the
base, lead to RCYs of 84 ± 0% and 74 ± 2%, respectively (entries 6-7). We have
found that even [18F]KF/K222 could be used as 18F-source. In the presence of TBD,
[18F]65a was formed in an RCY of 90 ± 1% (entry 8).
2.4.2. Substrate scope for the circular labelling of2,2,2-[18F]trifluoromethylketones
With the optimised conditions found (Table 14, entry 4), we explored the
substrate scope of the 18F-labelling of 2,2,2-trifluoromethyl ketones. Starting
from 2-bromo-2,2-difluoromethyl-biphenyl ketone (91b) a very good RCY of
90 ± 1% was obtained for [18F]65b (Table 15). Similarly, 2-[18F]naphthyl ketone
[18F]65c was synthesised with an RCY of 84 ± 9%. Products with substituents in
the para-position such as p-[18F]methoxy [18F]65d or p-[18F]ethylthio [18F]65e
on the arene yielded 93 ± 3% and 20 ± 9%, respectively. A reason for the low RCY
of [18F]65e could be the instability of the ethylthio functional group under the
applied conditions. High RCYs of 90 ± 1% and 92 ± 3%, were obtained for
p-[18F]fluoro [18F]65f and 1-[18F]naphthyl [18F]65j substituted ketones. Good to
high RCYs of 71 ± 1% and 92 ± 1%, were obtained for p-[18F]methyl [18F]65k and
3,5-[18F]difluoro [18F]65l substituted ketones. The conjugated ketones
2-[18F]methyl alkenyl naphthyl [18F]65g and 2-[18F]methyl alkenyl thienyl
[18F]65h were obtained in RCYs of 43 ± 6% and 51 ± 1% RCY at standard
conditions. At an elevated temperature of 100 °C, the RCY decreased for [18F]65g
to 38 ± 1% but increased for [18F]65h to 67 ± 1%. 18F-labelling of [18F]aliphatic
ketone [18F]65l was successfully achieved in 51 ± 1% RCY and the RCY increased
to 64 ± 1% when using DIPEA instead of TBD.

49
Table 15: Substrate scope for the synthesis of 2,2,2-[18F]trifluoroacetophenones [18F]65. a
2.4.3. The radio diagnostic utility of the circular labelling method After the development of a circular 18F-labelling approach for various ketones
65, we were now ready to trial the concept on a potential NE inhibitor. The
synthesis of the desired molecule 1a begins with the synthesis of two fragments,
spanning over five steps each. The synthesis is described in the literature.6c, 11a, c,
128 However, several steps proceeded with very low yields, therefore the
syntheses had to be improved. The first fragment contains a pyridone 93 and the
second a valine moiety 94, which were combined in a condensation reaction to
form a molecule featuring a secondary alcohol 95 (Scheme 34). Then, the desired
ketone 1a was obtained via oxidation.129 In our preliminary studies, we choose

50
the valine fragment 65i as the model for the whole inhibitor, as it contains a
2,2,2-trifluoromethyl group already.
Scheme 34: Condensation reaction of the pyridone 93 and valine 94 fragments.
2.4.3.1. Synthesis of and studies with model compound 65i First, the N-terminus of DL-valine was protected with benzoyl chloride (59b)
(Scheme 35a). Then, oxazolone 98 was formed by the addition of acetic
anhydride.11c Both reactions gave yields above 89%. Following a literature
procedure, we obtained the desired trifluoromethyl ketone 65i in very low
yields.11c The reaction was optimised by addition of triethylamine as a base in
the first step and DMAP130 as an accelerator in the second step of the reaction
(Scheme 35b). With the employed changes 65i was isolated in 63% yield.
Scheme 35: Synthesis of a model compound of a NE inhibitor.
Next followed the defluorination, halogenation steps (Scheme 36). The
corresponding 2,2-difluoro enol silyl ether 66i formed from 65i within 4 hours
at 0 °C in high conversion. The crude product was filtered, dried, and employed
in the bromination reaction to give 2-bromo-2,2-difluoromethyl ketone 91i in
56% yield over two steps.

51
Scheme 36: Defluorination and bromination of the model compound 65i.
Subsequently, we studied the 18F-labelling of 91i (Table 16). Surprisingly, the
conditions optimised for 2,2,2-trifluoroacetophenone analogues [18F]65a-h and
[18F]65j-m did not give any radiolabelled product [18F]65i (Table 16, entry 1).
Using MTBD or DIPEA as a base, [18F]65i led to no product formation or only 6%
RCY, respectively (entries 2-3). Omitting the base or employing a lower reaction
temperature (rt) led to 0% RCY (entries 4-5). However, the application of
[18F]KF/K222 instead of [18F]TBAF as 18F-source gave respectable levels of RCY
for the formation of [18F]67i (entries 6-8). Both DIPEA and TBD proved to be
efficient (entries 6-7). However, the highest RCY of 55 ± 4% was obtained
without the use of any base (entry 8).
Table 16: Optimisation for the synthesis of labelled benzamide [18F]65i.a

52
2.4.3.2. Fluorine-18 labelling of inhibitor 1a After the successful labelling of model compound 65i, we attempted the
synthesis of tracer candidate 1a. During the synthesis of 2,2-difluoro enol silyl
ether 66n we encountered several difficulties. 66n did not form as easily as for
the smaller molecules. Therefore, the amount of Mg and TMSCl added were
doubled. Also, the reaction temperature was changed to 3 hours at 0 °C and with
an additional 1 hour at 25 °C. Besides, it is important to note, that this reaction is
more moisture sensitive than the syntheses of the above 2,2-difluoro enol silyl
ethers. Improvement of the reproducibility was found, when Mg was stirred
overnight in an Ar-filled glovebox to increase the surface area.131
Scheme 37: Synthesis of 2,2-difluoro enol silyl ether 66n.
The halogenation reaction was carried out immediately after the formation of
66n. 2-Bromo-2,2-difluoromethyl ketone 91n was isolated in a good yield of
59%, whereas the 2-iodo-2,2-difluoromethyl ketone 92n was obtained in a high
yield of 77% (Scheme 38).
Scheme 38: Iodination or bromination of 2,2-difluoro enol silyl ether 66n.
Then, the 18F-labelling of 91n and 92n was attempted. Using the reaction
conditions, which were optimal for the synthesis of 2,2,2-
[18F]trifluoroacetophenone ([18F]65a), [18F]1a formed only with an RCY of 4%
(Table 17, entry 1). Without any additive, at the temperatures of 75 °C, 50 °C and
100 °C also very low RCYs of 4%, 2% and 4%, respectively, were detected
(entries 2-4). Changing to the reaction conditions for the successful synthesis of
model compound [18F]65i, product [18F]1a was not formed. However, omission
of the additive led to an RCY of 5 ± 0% (entry 6). Using [18F]KF, but with crown
ether K18c6 instead of K222, [18F]1a was not detected (entry 7).

53
Table 17: Optimisation of the synthesis of labelled elastase inhibitor [18F]1a.a
Starting from the difluoroiodomethyl ketone 92n instead, with [18F]TBAF as 18F-source and with TBD as an additive, led to 0% RCY (entry 8). However, using
[18F]TBAF without any additive afforded [18F]1a with the acceptable RCY of
25 ± 1% (entry 9). The same conditions, but with [18F]KF/K222 yielded 15% RCY
for [18F]1a (entry 10). Accordingly, we chose the reaction of 92n with [18F]TBAF
(in DMF at 75 °C for 10 min, see entry 9) for the following isolation and molar
activity measurements.
2.4.3.3. Isolation and autoradiography study of 1a
Tracer candidate [18F]1a was isolated by a semi-preparative radio-HPLC in five
separate experiments, for which the average values of AY, RCY, Am and RCP are
given in Scheme 39. The isolated activity varied in a range from 30−128 MBq.
The AY was 0.8−3.4% and the RCY was 15 ± 12% (average time of the synthesis:

54
71 min). The calculated molar activity Am was 0.2−0.5 GBq/mol. This value is
rather low compared to the radiosynthesis of monofluorinated compounds.
However, for the construction of 18F-labelled trifluoromethyl groups, the low
molar activity is not unusual.64d, 132 Isotopic exchange between fluorine-19 (for
example in the starting material 92n) and fluorine-18 may lead to a lower Am.
Subsequently, we performed in vitro autoradiography studies with [18F]1a.
Scheme 39: Labelling and isolation of [18F]1a.
The binding of a radiopharmaceutical may occur in three forms: specific,
non-specific, and non-displaceable. First, specific binding, which means the
radioligand is bound to the active site of the enzyme or receptor. The second, an
unwanted trait, is non-specific binding, where the radiopharmaceutical binds
also to non-target receptors and other active sites of the enzyme. And third, there
is non-displaceable binding, where the radioligand binds indiscriminately to
lipids or membranes, which increases linearly with the increasing lipophilicity of
the radiopharmaceutical. The non-displaceable binding is a reason for many
radiopharmaceuticals to fail the preclinical trials.6c, 57b, 58
The first autoradiography experiment was performed on 20 µm sections of
tissue from a mouse with lung inflammation, and a control group. The samples
were prepared as follows. The first with only [18F]1d (Figure 10a,d). In the
second sample in addition to [18F]1a, the enzymes active sites blocked by the
commercially available inhibitor GW311616A 99 (Figure 10b,e)133 and in the
third sample, [18F]1a was applied together with the non-radioactive isotope 1a
(Figure 10c,f).

55
a)
b)
c)
d)
e)
f)
Figure 10: Autoradiography experiments with a 20 µm sections of a mouse lung with lung inflammation (a-c) and a control group (d-e), assay buffer PBS (phosphate-buffered saline, pH = 7.4); a,d) 0.022 nmol/mL of [18F]1a; b,e) 0.022 nmol/mL of [18F]1a and 2.3 nmol/mL of 99; c,f) 0.022 nmol/mL of [18F]1a and 12.0 nmol/mL of 1a.
Under the performed conditions we observed majorly non-displaceable
binding. Most likely [18F]1a was too lipophilic. It did not dissolve well in the
phosphate buffer solution, and therefore [18F]1a aggregated unselectively
throughout the whole sample.
An idea of how to circumvent this was the addition of bovine serum albumin
(BSA) to the phosphate buffer solution. We theorized that this would lead to an
increase in solubility, which would cause a better specificity.6c Indeed, the second
autoradiography, where 1% BSA was added to the phosphate buffer, gave very
encouraging results (Figure 11).6c Here, significant accumulation of [18F]1a was
only found in a).
a)
b)
c)
Figure 11: Autoradiography experiments with a 20 µm sections of mouse lung with lung inflammation, assay buffer PBS; with 1% bovine serum albumin (BSA); a) 0.02 nmol/mL of [18F]1a; b) 0.02 nmol/mL of [18F]1a and 2.3 nmol/mL of inhibitor 99; c) 0.02 nmol/mL of [18F]1a and 12.0 nmol/mL of 1a.
Conversely, samples b) and c), in which the active sites of the enzyme were
blocked with the non-radioactive ligands of 1a and 99, did not show significant
binding of [18F]1a. This indicates that there is selective binding with [18F]1a.

56
These results also suggest that a less lipophilic compound would be an excellent
tracer candidate for PET imaging of NE.
2.4.4. Conclusions and outlook In summary, we have developed a circular defluorination 18F-labelling
strategy. The method has been applied to various ketones and was further
extended to a model of an NE inhibitor ligand and even to the NE inhibitor ligand
itself. The molar activity of the fluorine-18 labelled COCF3 product was low, but
high enough to successfully perform the first preclinical studies. These
autoradiography studies showed selective binding to the enzyme, but the low
polarity of the molecule was an issue. Further studies will be conducted to find
another ligand with a lower lipophilicity (see above Figure 9).

57
Concluding remarks
In this work, an array of four organofunctionalisation methods is presented.
They range from fundamental research and method development to the
synthesis and application of a potential radiotracer. The two important areas
included therein are organoboron and organofluorine chemistry.
First, a versatile copper-catalysed synthesis of tri- and tetrasubstituted
allenylboronic acids and esters was devised. Application led to enantioenriched
tertiary homopropargyl alcohols. In addition, the allenylboronic acids have
already been utilised in two further studies by the Szabó group. The synthesised
allenylboronic acids are more reactive than the commonly used Bpin derivatives.
Accordingly, our new methodology provides a convenient approach for the
selective synthesis of complex organic molecules and natural products.
The second chapter was focused on the development of a versatile
electrophilic functionalisation of 2,2-difluoro enol silyl ethers through
trifluoromethylthiolation, trifluoromethylation and arylation reactions under
mild conditions. This lead to the novel -COCF2SCF3 functional group with
potentially interesting properties due to the high lipophilicity of the -SCF3
moiety, compared to other fluorine-containing functional groups.
The third chapter was able to adapt the fluorine-19 synthesis of a well-known
electrophilic, hypervalent fluorinating reagent to 18F-labelling conditions. The
[18F]fluoro-benziodoxole reagent was synthesised in high molar activity and its
first application to a fluorocyclisation of o-styryl benzamides was successful.
Recently, an additional application was published by the group. The presented
[18F]fluoro-benziodoxole is one of the very few easily accessible electrophilic 18F-transfer reagents in radiochemistry. Therefore this reagent will probably
find many new applications for electrophilic 18F-labelling of PET imaging tracers.
The last chapter uses the knowledge gained in chapters 2-3. Starting from
trifluoromethyl ketones, a defluorination lead to 2,2-difluoro enol silyl ethers,
which were further functionalised by halogenation. The obtained
2-halo-2,2-difluoroketones underwent nucleophilic 18F-labelling to form
[18F]trifluoromethyl ketones thereby closing the circle from -CF3 to -[18F]CF3. The
synthesis and 18F-labelling of a potential NE ligand was successful. Furthermore,
the first in vitro experiments provided evidence of the NE ligands aptitude for
specific binding. Subsequent studies will explore the derivatisation of the tracer
candidate to selectively modulate its pharmacological and physicochemical
properties.

58
Reprint permissions
Permission to reprint was obtained for each publication from the respective
publisher, referred to by Roman numerals I-III.
I. J. Zhao, S. J. T. Jonker, D. N. Meyer, G. Schulz, D. C. Tran, L. Eriksson,
K. J. Szabó,* Chem. Sci. 2018, 9, 3305-3312. Royal Society of Chemistry:
Open access article licensed a Creative Commons Attribution
3.0 Unported Licence.
II. X. Jiang, D. Meyer, D. Baran, M. A. Cortes Gonzalez, K. J. Szabo,*
J. Org. Chem. 2020, 85, 8311-8319. American Chemical Society: Open
access article published under a Creative Commons Attribution (CC-BY)
Licence.
III. M. A. Cortés González, P. Nordeman, A. Bermejo Gómez, D. N. Meyer,
G. Antoni, M. Schou, K. J. Szabó,* Chem. Commun. 2018, 54, 4286-4289.
Royal Society of Chemistry: Open access article licensed under a Creative
Commons Attribution 3.0 Unported Licence.

59
Contribution list
I. Participated in the synthesis of starting materials and small parts of
the substrate scope. Carried out derivatisation reactions to obtain a
crystal structure. Wrote part of the supporting information and
minor parts of the manuscript.
II. Participated in the synthesis of starting materials, in the substrate
scope and analysis. Developed the trifluoromethylation reaction
and scope. Wrote parts of the supporting information and
manuscript. Shared first authorship.
III. Participated in the synthesis of starting material, optimisation of the
synthesis and application of the reagent in fluorine-19 chemistry.
Wrote minor parts of the supporting information. IV. Participated in the synthesis and analysis of starting materials, in
the substrate scope and analysis. Developed parts of the starting
material syntheses and parts of the optimisation of the labelling
chemistry. Wrote parts of the supporting information and
manuscript. Shared first authorship. The publication of this
manuscript is dependent on the radiosynthesis and
autoradiography of a new tracer candidate, which is less lipophilic
than [18F]1a.

60
Acknowledgements
I would like to express my sincerest gratitude to:
My supervisor Prof. Kálmán J. Szabó for accepting me as a PhD student in his
research group and giving me the opportunity to learn under his excellent
guidance. Also for his optimism in discussions and his patience while solving
problems.
Prof. Belén Martín-Matute and Prof. Joseph Samec for taking the time to read
this report and their valuable feedback.
My sincerest gratitude goes towards all my collaborators in this thesis:
Dr. Xingguo Jiang, Dr. Miguel A. Cortés González, Dr. Sybrand S. T. Jonker, Linus
Johansson-Holm, Dominik Baran, Dr. Jian Zhao, Dr. Sergio Estrada, Dr. Patrik
Nordeman, Prof. Gunnar Antoni, Dr. Antonio Bermejo Gómez, Dr. Magnus Schou,
Göran Schulz and C. Duc Tran.
For the good working atmosphere, fruitful discussions, and pleasant time in
general, I would like to thank all past and present members of the Szabó group:
Antonio, Colin, Denis, Dina, Duc, Dominik, Dong, Erik, Göran, Jayarajan, Ken
(Jian), Kevin, Linus, Lujia, Maria, Marie, Martin H., Martin d. W., Marvin, Miguel,
Mihály, Monireh, Nadia, Soumitra, Tautvydas, Qiang, Weiming and Xingguo.
I would also like to thank my proofreaders, Marvin, Sybrand, Aleksandra, Paul
and Michael as well as all present group members for their valuable feedback.
Maria, thank you for helping me with the graphic.
Tautvydas thanks for the photoshoot to get a profile picture! Finally, a photo
in which my glasses weren't reflective!
With me from day 1 were my office-mates, and older (PhD-)brothers Marvin
and Sybrand. I am so very grateful to have had you by my side. So special thanks
to Marvin: A lab boss that was always offering a helping hand and co-worker that
amazingly remembers every name reaction and every speaker to ever enter the
department. And Sybrand, we started just two months apart and went through
most challenges together. Including sharing a computer (for a short time) and
the first successful and unsuccessful projects as PhD students! I am really
thankful to have made this journey with you!
Xingguo, it was an honour to work side-by-side with you on two projects. Your
dedication and work ethic are commendable. I will miss our conversations about
nothing and everything on the train. Thank you for teaching me the ropes in the
hot lab!

61
Special thanks to Miguel who introduced me to fluorine chemistry, working
in the group and the department when I joined his project as a master’s student.
Thank you for always helping me wherever you could, for keeping a clear head
and for your dedication and optimism to solve problems. I am very grateful to
have learned from you and being able to work with you on the latest fluorine-18
project.
Everyone working at the PET-center at Akademiska Sjukhuset Uppsala for the
very pleasant work atmosphere and everyone’s helpfulness. Thank you for
everything! Especially Mathias and Patrik for their support in running
fluorine-18 experiments and organising our work there. And Sergio for the
introduction to autoradiography!
Linus, my one and only master student! It was a pleasure to work with you. I
will for sure miss your baked goods, especially those snickers-inspired treats.
Good luck with all the upcoming challenges and projects. I am sure you will do
amazing!
Dominik and Denis, it was a pleasure to meet and supervise you in the lab.
Please come back to visit anytime! (but maybe when it is a little safer)
Dong, for introducing me to the wonderful world of allylboronic acids and
esters.
Marie, our new office mate, practically neighbour and new lab-responsible
person. I am really happy you decided to join the group! All the best for your
future adventures in boron chemistry!
Angela, thank you so much for letting Linus and I travel to Uppsala with you!
The car rides were a pleasure!
The TA staff: Carin, Gabriella, Isabella, Jenny, Jonas, Kristina, Louise, Martin,
Ola, Petra, Sigrid and Ulrika. Thank you for all your support, teamwork and effort
to keep the department running.
All the new friends I made in Sweden, for interesting lunch discussions, board-
game evenings, quizzes (Gabbi!), after-work laser tag, boat cruises, train trips,
evenings at Gröna Villa and all the other fun events: Aleksandra, Alex, Angela,
Arnar, Beichen, Catarina, Christoffer, Daniels, Davide, Denis, Dina, Dominik,
Dong, Elena, Finn, Gabbi, Göran, Jonas, Kevin, Kilian, Linus, Matic, Matteo, Marie,
Martin H., Martin d. W., Marvin, Melanie, Miguel, Paul, Peeter, Samuel, Stephanie,
Stefano, Sybrand, Tamás, Tautvydas, Thibault, Tony, Viola. Please forgive me, if I
missed someone!
And of course, I would like to thank everyone in the department of organic
chemistry, past and present, for the open and friendly work environment.
I would also like to acknowledge my former teachers at the Johannes
Gutenberg University Mainz. At the department of nuclear chemistry:
Prof. Dr. Tobias Reich who gave me the most interesting Bachelor project work.
Prof. Dr. Frank Rösch for his lectures and book teaching radiochemistry,
specifically PET. I would also like to thank my bachelor supervisors Dr. Nils

62
Stöbener and Dr. Pascal Schönberg for their support and for introducing me to
THE best department.
At the department of organic chemistry: Prof. Dr. Siegfried R. Waldvogel,
thank you for supporting me in going abroad! And Dr. Sebastian Resch for an
interesting project assignment. Also thanks to Prof. Dr. Heiner Detert for being
able to teach and discipline all the organic chemistry courses with just a bell.
Last but not least Dr. Martin Panthöfer for the advanced inorganic labs, who
taught us working under inert conditions, synthesising organometallic and
inorganic compounds and to clean glassware properly.
I like to pronounce my gratitude to all the scholarships that made it possible
to participate in conferences (if they were not postponed) from the
Apothekersocieteten, Gålöstiftelse, Gesellschaft Deutscher Chemiker, Helge Ax:son
Johnsson, Hilda Rietz, Längmanska and Knut och Alice Wallenbergs stiftelse.
Einen ganz herzlichen Dank an meine liebe Familie und meine Freunde. Ohne
eure fortwährende Unterstützung wäre ich nicht dort wo ich jetzt bin. Danke,
dass ihr immer für mich da seit und an mich glaubt. Ganz besonderen Dank an
meine Eltern Beate und Wolfgang, die mir jederzeit mit Rat und Tat zur Seite
stehen und mich auf dem Weg hierhin unterstützt haben.
Lieber Michael, danke dass du immer für mich da bist, mich immer aufbaust
und die richtigen Worte findest. Natürlich auch danke dafür, dass du mich dazu
überredet hast meinen Erasmus+ Aufenthalt in Stockholm zu verbringen!
Marcus, ich wünschte du wärst heute noch bei uns. Ich vermisse dich sehr und
habe gehofft, eines Tages auch die Möglichkeit zu haben deine Doktorarbeit lesen
zu können.

63
References
1. The ACS Style Guide, American Chemical Society, Oxford University Press, New York, 2006.
2. a) Coenen, H. H.; Gee, A. D.; Adam, M.; Antoni, G.; Cutler, C. S.; Fujibayashi, Y.; Jeong, J. M.; Mach, R. H.; Mindt, T. L.; Pike, V. W.; Windhorst, A. D., EJNMMI Radiopharm. Chem. 2019, 4, 1-5; b) Coenen, H. H.; Gee, A. D.; Adam, M.; Antoni, G.; Cutler, C. S.; Fujibayashi, Y.; Jeong, J. M.; Mach, R. H.; Mindt, T. L.; Pike, V. W.; Windhorst, A. D., Nucl. Med. Biol. 2017, 55, 5-11.
3. Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D., Chem. Rev. 2018, 118, 3887-3964.
4. Harper, D. B.; O'Hagan, D., Nat. Prod. Rep. 1994, 11, 123-133.
5. Groziak, M. P., Am. J. Ther. 2001, 8, 1075-2765.
6. a) Nikolaou, A.; Kokotou, M. G.; Vasilakaki, S.; Kokotos, G., Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2019, 1864, 941-956; b) Veale, C. A.; Bernstein, P. R.; Bohnert, C. M.; Brown, F. J.; Bryant, C.; Damewood, J. R.; Earley, R.; Feeney, S. W.; Edwards, P. D.; Gomes, B.; Hulsizer, J. M.; Kosmider, B. J.; Krell, R. D.; Moore, G.; Salcedo, T. W.; Shaw, A.; Silberstein, D. S.; Steelman, G. B.; Stein, M.; Strimpler, A.; Thomas, R. M.; Vacek, E. P.; Williams, J. C.; Wolanin, D. J.; Woolson, S., J. Med. Chem. 1997, 40, 3173-3181; c) Bernstein, P. R.; Andisik, D.; Bradley, P. K.; Bryant, C. B.; Ceccarelli, C.; Damewood, J. R.; Earley, R.; Edwards, P. D.; Feeney, S., J. Med. Chem. 1994, 37, 3313-3326; d) Cregge, R. J.; Durham, S. L.; Farr, R. A.; Gallion, S. L.; Hare, C. M.; Hoffman, R. V.; Janusz, M. J.; Kim, H.-O.; Koehl, J. R.; Mehdi, S.; Metz, W. A.; Peet, N. P.; Pelton, J. T.; Schreuder, H. A.; Sunder, S.; Tardif, C., J. Med. Chem. 1998, 41, 2461-2480; e) Counts, G. W.; Gregory, D.; Zeleznik, D.; Turck, M., Antimicrob. Agents Chemother. 1977, 11, 708-711.
7. a) Smart, B. E., J. Fluor. Chem. 2001, 109, 3-11; b) Böhm, H. J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M., Chembiochem. 2004, 5, 637-643; c) Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V., Chem. Soc. Rev. 2008, 37, 320-330; d) Johnson, B. M.; Shu, Y.-Z.; Zhuo, X.; Meanwell, N. A., J. Med. Chem. 2020, 63, 6315-6386.
8. a) Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H., Chem. Rev. 2016, 116, 422-518; b) Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D. M.; Santi, C.; Ruzziconi, R.; Soloshonok, V. A., Chem. Eur. J. 2019, 25, 11797-11819.

64
9. a) Gelb, M. H.; Svaren, J. P.; Abeles, R. H., Biochemistry 1985, 24, 1813-1817; b) Dreyer, G. B.; Metcalf, B. W., Tetrahedron Lett. 1988, 29, 6885-6888; c) Bégué, J.-P.; Bonnet-Delpon, D., Bioorganic and Medicinal Chemistry of Fluorine. Wiley: 2008; p. 223-278.
10. a) Crocetti, L.; Quinn, M. T.; Schepetkin, I. A.; Giovannoni, M. P., Exp. Opin. Ther. Pat. 2019, 29, 555-578; b) von Nussbaum, F.; Li, V. M., Bioorg. Med. Chem. Lett. 2015, 25, 4370-4381.
11. a) Damewood, J. R., Jr.; Edwards, P. D.; Feeney, S.; Gomes, B. C.; Steelman, G. B.; Tuthill, P. A.; Williams, J. C.; Warner, P.; Woolson, S. A.; Wolanin, D. J.; et al., J. Med. Chem. 1994, 37, 3303-3312; b) Bernstein, P. R.; Andisik, D.; Bradley, P. K.; Bryant, C. B.; Ceccarelli, C.; Damewood, J. R., Jr.; Earley, R.; Edwards, P. D.; Feeney, S.; Gomes, B. C.; et al., J. Med. Chem. 1994, 37, 3313-3326; c) Peet, N. P.; Burkhart, J. P.; Angelastro, M. R.; Giroux, E. L.; Mehdi, S.; Bey, P.; Kolb, M.; Neises, B.; Schirlin, D., J. Med. Chem. 1990, 33, 394-407.
12. a) Barata-Vallejo, S.; Bonesi, S.; Postigo, A., Org. Biomol. Chem. 2016, 14, 7150-7182; b) Xu, X.-H.; Matsuzaki, K.; Shibata, N., Chem. Rev. 2015, 115, 731-764.
13. Hall, D. G., Structure, Properties, and Preparation of Boronic Acid Derivatives. Overview of Their Reactions and Applications. In Boronic Acids. In Boronic Acids, Wiley-VCH: Weinheim, Germany, 2006; Vol. 2, p. 1-99.
14. Balz, G.; Schiemann, G., Ber. Dtsch. Chem. Ges. 1927, 60, 1186-1190.
15. Gottlieb, H. B., J. Am. Chem. Soc. 1936, 58, 532-533.
16. a) Liang, T.; Neumann, C. N.; Ritter, T., Angew. Chem. Int. Ed. 2013, 52, 8214-8164; b) Szpera, R.; Moseley, D. F. J.; Smith, L. B.; Sterling, A. J.; Gouverneur, V., Angew. Chem. Int. Ed. 2019, 58, 14824-14848; c) Sather, A. C.; Buchwald, S. L., Acc. Chem. Res. 2016, 49, 2146-2157; d) Grushin, V. V., Acc. Chem. Res. 2010, 43, 160-171; e) Watson, D. A.; Su, M.; Teverovskiy, G.; Zhang, Y.; García-Fortanet, J.; Kinzel, T.; Buchwald, S. L., 2009, 325, 1661-1664; f) Wilkinson, J. A., Chem. Rev. 1992, 92, 505-519.
17. Wu, J., Tetrahedron Lett. 2014, 55, 4289-4294.
18. a) Campbell, M. G.; Ritter, T., Org. Process Res. Dev. 2014, 18, 474-480; b) Neumann, C. N.; Ritter, T., Angew. Chem. Int. Ed. 2015, 54, 3216-3221.
19. Sokolenko, T. M.; Petko, K. I.; Yagupolskii, L. M., Chem. Heterocycl. Compd. 2009, 45, 430-435.
20. Bermejo Góme, A.; González, M. A. C.; Lübcke, M.; Johansson, M. J.; Schou, M.; Szabó, K. J., J. Fluor. Chem. 2017, 194, 51-57.
21. Gómez, A. B.; Cortés González, M. A.; Lübcke, M.; Johansson, M. J.; Halldin, C.; Szabó, K. J.; Schou, M., Chem. Comm. 2016, 52, 13963-13966.
22. Tius, M. A., Tetrahedron 1995, 51, 6605-6634.
23. a) Li, M.; Xue, X.-S.; Cheng, J.-P., Acc. Chem. Res. 2020, 53, 182-197; b) Barnette, W. E., J. Am. Chem. Soc. 1984, 106, 452-454; c) Umemoto, T.; Kawada,

65
K.; Tomita, K., Tetrahedron Lett. 1986, 27, 4465-4468; d) Umemoto, T.; Tomita, K., Tetrahedron Lett. 1986, 27, 3271-3274; e) Differding, E.; Ofner, H., Synlett 1991, 187-189; f) Banks, R. E.; Mohialdin-Khaffaf, S. N.; Lal, G. S.; Sharif, I.; Syvret, R. G., Chem. Comm. 1992, 595-596; g) Groult, H.; Leroux, F.; Tressaud, A., Modern Synthesis Processes and Reactivity of Fluorinated Compounds -Progress in Fluorine Science Elsevier: 2016; p. 1-765; h) Olofsson, B.; Marek, I.; Rappoport, Z., Patai's Chemistry of Functional Groups: The Chemistry of Hypervalent Halogen Compounds. John Wiley and Sons: Eds., 2019; p. 1-1094; i) Guyon, H.; Cahard, D., Selectfluor and Its Analogs Electrophilic Fluorination for Preparing Alkyl Fluorides. In Fluorination, Hu, J.; Umemoto, T., Eds. Springer: Singapore, 2018; p. 1-24.
24. a) Arnold, A. M.; Ulmer, A.; Gulder, T., Chemistry 2016, 22, 8728-8739; b) Kohlhepp, S. V.; Gulder, T., Chem. Soc. Rev. 2016, 45, 6270-6288; c) Yoshimura, A.; Zhdankin, V. V., Chem. Rev. 2016, 116, 3328-435; d) Li, Y.; Hari, D. P.; Vita, M. V.; Waser, J., Angew. Chem. Int. Ed. 2016, 55, 4436-4454; e) Han, Z. Z.; Zhang, C. P., Adv. Synth. Catal. 2020, 1-38; f) Murphy, G. K.; Gulder, T., Hypervalent Iodine Fluorination for Preparing Alkyl Fluorides (Stoichiometrically and Catalytically). In Fluorination, Hu, J.; Umemoto, T., Eds. Springer: Singapore, 2018; p. 1-32.
25. Legault, C. Y.; Prévost, J., Acta Cryst. 2012, E68, o1238.
26. Szabó, K. J., Fluorination, Trifluoromethylation, andTrifluoromethylthiolation of Alkenes, Cyclopropanes, and Diazo Compounds. In Organofluorine Chemistry, Szabó, K.; Selander, N., Eds. Wiley‐VCH: Weinheim, Germany, 2021; p. 201-223.
27. a) Minhas, H. K.; Riley, W.; Stuart, A. M.; Urbonaite, M., Org. Biomol. Chem. 2018, 16, 7170-7173; b) Sreenithya, A.; Sunoj, R. B., Dalton Trans. 2019, 48, 4086-4093; c) Sajith, P. K.; Suresh, C. H., Inorg. Chem. 2012, 51, 967-977; d) Pinto de Magalhães, H.; Togni, A.; Lüthi, H. P., J. Org. Chem. 2017, 82, 11799-11805; e) Ochiai, M.; Sueda, T.; Miyamoto, K.; Kiprof, P.; Zhdankin, V. V., Angew. Chem. Int. Ed. 2006, 45, 8203-8206.
28. a) Sun, T. Y.; Wang, X.; Geng, H.; Xie, Y.; Wu, Y. D.; Zhang, X.; Schaefer, H. F., 3rd, Chem. Commun. 2016, 52, 5371-8374; b) Zhou, B.; Yan, T.; Xue, X. S.; Cheng, J. P., Org. Lett. 2016, 18, 6128-6131; c) Zhou, B.; Xue, X.-s.; Cheng, J.-p., Tetrahedron Lett. 2017, 58, 1287-1291; d) Zhang, J. J.; Szabó, K. J.; Himo, F., ACS Catal. 2017, 7, 1093-1100; e) Yan, T.; Zhou, B.; Xue, X. S.; Cheng, J. P., J. Org. Chem. 2016, 81, 9006-9011; f) Mai, B. K.; Szabó, K. J.; Himo, F., ACS Catal. 2018, 8, 4483-4492; g) Mai, B. K.; Himo, F., Mechanisms of Metal-Catalyzed Electrophilic F/CF3/SCF3 Transfer Reactions from Quantum Chemical Calculations. In New Directions in the Modeling of Organometallic Reactions, Lledós, A.; Ujaque, G., Eds. Springer Nature: Cham, Switzerland, 2020; p. 39-56.
29. Geary, G. C.; Hope, E. G.; Stuart, A. M., Angew. Chem. Int. Ed. 2015, 54, 14911-14914.
30. a) Yuan, W.; Eriksson, L.; Szabó, K. J., Angew. Chem. Int. Ed. 2016, 55, 8410-8415; b) Ilchenko, N. O.; Szabó, K. J., J. Fluor. Chem. 2017, 203, 104-109; c)

66
Ilchenko, N. O.; Hedberg, M.; Szabó, K. J., Chem. Sci. 2017, 8, 1056-1061; d) Ilchenko, N. O.; Tasch, B. O.; Szabó, K. J., Angew. Chem. Int. Ed. 2014, 53, 12897-12901; e) Ilchenko, N. O.; Cortés, M. A.; Szabó, K. J., ACS Catal. 2015, 6, 447-450.
31. Yuan, W.; Szabó, K. J., Angew. Chem. Int. Ed. 2015, 54, 8533-8537.
32. Ulmer, A.; Brunner, C.; Arnold, A. M.; Pothig, A.; Gulder, T., Chem. Eur. J. 2016, 22, 3660-3664.
33. Andries-Ulmer, A.; Brunner, C.; Rehbein, J.; Gulder, T., J. Am. Chem. Soc. 2018, 140, 13034-13041.
34. Brunner, C.; Andries-Ulmer, A.; Kiefl, G. M.; Gulder, T., Eur. J. Org. Chem. 2018, 2615-2621.
35. a) Zhao, J.-F.; Duan, X.-H.; Yang, H.; Guo, L.-N., J. Org. Chem. 2015, 80, 11149-11155; b) Yang, B.; Chansaenpak, K.; Wu, H.; Zhu, L.; Wang, M.; Li, Z.; Lu, H., Chem. Commun. 2017, 53, 3497-3500; c) Yang, S.; Shi, S.; Chen, Y.; Ding, Z., J. Org. Chem. 2021, DOI: 10.1021/acs.joc.1c00159.
36. a) Kong, W.; Feige, P.; de Haro, T.; Nevado, C., Angew. Chem. Int. Ed. 2013, 52, 2469-2473; b) Suzuki, S.; Kamo, T.; Fukushi, K.; Hiramatsu, T.; Tokunaga, E.; Dohi, T.; Kita, Y.; Shibata, N., Chem. Sci. 2014, 5, 2754-2760; c) Wang, Q.; Lubcke, M.; Biosca, M.; Hedberg, M.; Eriksson, L.; Himo, F.; Szabó, K. J., J. Am. Chem. Soc. 2020, 142, 20048-20057; d) Mennie, K. M.; Banik, S. M.; Reichert, E. C.; Jacobsen, E. N., J. Am. Chem. Soc. 2018, 140, 4797-4802; e) Woerly, E. M.; Banik, S. M.; Jacobsen, E. N., J. Am. Chem. Soc. 2016, 138, 13858-13861.
37. a) Ruppert, I.; Schlich, K.; Volbach, W., Tetrahedron Lett. 1984, 25, 2195-2198; b) Prakash, G. K. S.; Krishnamurti, R.; Olah, G. A., J. Am. Chem. Soc. 1989, 111, 393-395.
38. Tomashenko, O. A.; Grushin, V. V., Chem. Rev. 2011, 111, 4475-4521.
39. Langlois, B. R.; Laurent, E.; Roidot, N., Tetrahedron Lett. 1991, 32, 7525-7528.
40. a) Eisenberger, P.; Gischig, S.; Togni, A., Chem. Eur. J. 2006, 12, 2579-2586; b) Kalim, J.; Duhail, T.; Le, T. N.; Vanthuyne, N.; Anselmi, E.; Togni, A.; Magnier, E., Chem. Sci. 2019, 10, 10516-10523.
41. Umemoto, T.; Ishihara, S., J. Am. Chem. Soc. 1993, 115, 2156-2164.
42. Deng, Q.-H.; Wadepohl, H.; Gade, L. H., J. Am. Chem. Soc. 2012, 134, 10769-10772.
43. Charpentier, J.; Früh, N.; Togni, A., Chem. Rev. 2015, 115, 650-682.
44. Barata-Vallejo, S.; Lantano, B.; Postigo, A., Chemistry 2014, 20, 16806-16829.
45. a) Emerging fluorinated motifs. Cahard, D.; Ma, J. A., Wiley-VCH: Weinheim, Germany, 2020; Vol. 1-2; b) Organofluorine Chemistry. Szabó, K.; Selander, N., Wiley‐VCH Weinheim, Germany, 2021.

67
46. a) Leo, A.; Hansch, C.; Elkins, D., Chem. Rev. 1971, 71, 525-616; b) Hansch, C.; Leo, A.; Taft, R. W., Chem. Rev. 1991, 91, 165-195; c) Hansch, C.; Leo, A.; Unger, S. H.; Kim, K. H.; Nikaitani, D.; Lien, E. J., J. Med. Chem. 1973, 16, 1207-1216.
47. Munavalli, S.; Rohrbaugh, D. K.; Rossman, D. I.; Berg, F. J.; Wagner, G. W.; Durst, H. D., Synth. Commun. 2000, 30, 2847-2854.
48. a) Kang, K.; Xu, C. F.; Shen, Q. L., Org. Chem. Front. 2014, 1, 294-297; b) Zhang, P.; Li, M.; Xue, X. S.; Xu, C.; Zhao, Q.; Liu, Y.; Wang, H.; Guo, Y.; Lu, L.; Shen, Q., J. Org. Chem. 2016, 81, 7486-7509; c) Liu, H.; Ge, H.; Shen, Q., Reagents for Direct Trifluoromethylthiolation. In Emerging fluorinated motifs, Cahard, D.; Ma, J. A., Eds. Wiley-VCH: Weinheim, Germany, 2020; Vol. 2, p. 307-341; d) Ge, H.; Liu, H.; Shen, Q., Introduction of Trifluoromethylthio Group into Organic Molecules. In Organofluorine Chemistry, Szabó, K.; Selander, N., Eds. Wiley‐VCH: Weinheim, Germany, 2021; p. 99-172.
49. a) Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N., J. Am. Chem. Soc. 2013, 135, 8782-8785; b) Alazet, S.; Zimmer, L.; Billard, T., Chem. Eur. J. 2014, 20, 8589-8593; c) Haas, A.; Möller, G., Chem. Ber. 1996, 129, 1383-1388; d) Zhang, J.; Yang, J.-D.; Zheng, H.; Xue, X.-S.; Mayr, H.; Cheng, J.-P., Angew. Chem. Int. Ed. 2018, 57, 12690-12695; e) Toulgoat, F.; Liger, F.; Billard, T., Chemistry of OCF3, SCF3, and SeCF3 Functional Groups. In Organofluorine Chemistry, Szabó, K.; Selander, N., Eds. Wiley‐VCH: Weinheim, Germany, 2021; p. 49-97.
50. Li, M.; Zheng, H. L.; Xue, X. S.; Cheng, J. P., Tetrahedron Lett. 2018, 59, 1278-1285.
51. Yang, X.-G.; Zheng, K.; Zhang, C., Org. Lett. 2020, 22, 2026-2031.
52. a) Yi, W.; Song, Z.; Liu, J.; Mumtaz, Y.; Zhang, W., Trifluoromethylthiolation of Aromatic and Heteroaromatic Compounds. In Emerging fluorinated motifs, Cahard, D.; Ma, J. A., Eds. Wiley-VCH: Weinheim, Germany, 2020; Vol. 2, p. 343-371; b) Liang, Y.; Cahard, D.; Shibata, N., Direct Trifluoromethylthiolation Toward C(sp3)–SCF3 Compounds. Cahard, D.; Ma, J. A., Eds. Wiley-VCH: Weinheim, Germany, 2020; Vol. 2, p. 403-447.
53. a) Campbell, M. G.; Mercier, J.; Genicot, C.; Gouverneur, V.; Hooker, J. M.; Ritter, T., Nat. Chem. 2017, 9, 1-3; b) Hoover, A. J.; Lazari, M.; Ren, H.; Narayanam, M. K.; Murphy, J. M.; van Dam, R. M.; Hooker, J. M.; Ritter, T., Organometallics 2016, 35, 1008-1014; c) Tredwell, M.; Gouverneur, V., Angew. Chem. Int. Ed. 2012, 51, 11426-11437.
54. Preshlock, S.; Tredwell, M.; Gouverneur, V., Chem. Rev. 2016, 116, 719-766.
55. Deng, X.; Rong, J.; Wang, L.; Vasdev, N.; Zhang, L.; Josephson, L.; Liang, S. H., Angew. Chem. Int. Ed. 2019, 58, 2580-2605.
56. a) Phelps, M. E., Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 9226-9233; b) Miller, P. W.; Long, N. J.; Vilar, R.; Gee, A. D., Angew. Chem. Int. Ed. 2008, 47, 8998-9033; c) Ametamey, S. M.; Honer, M.; Schubiger, P. A., Chem. Rev. 2008, 108, 1501-1516; d) Cole, E. L.; Stewart, M. N.; Littich, R.; Hoareau, R.; Scott, P. J.,

68
Curr. Top. Med. Chem. 2014, 14, 875-900; e) Chansaenpak, K.; Vabre, B.; Gabbai, F. P., Chem. Soc. Rev. 2016, 45, 954-971.
57. a) van der Born, D.; Pees, A.; Poot, A. J.; Orru, R. V. A.; Windhorst, A. D.; Vugts, D. J., Chem. Soc. Rev. 2017, 46, 4709-4773; b) Matthews, P. M.; Rabiner, E. A.; Passchier, J.; Gunn, R. N., Br. J. Clin. Pharmacol. 2012, 73, 175-186; c) Fowler, J. S.; Wolf, A. P., Acc. Chem. Res. 1997, 30, 181-188.
58. Lewis, J. S.; Windhorst, A. D.; Zeglis, B. M., Radiopharmaceutical Chemistry. Springer Nature: Cham, Switzerland, 2019; p. 1-651.
59. a) Namavari, M.; Bishop, A.; Satyamurthy, N.; Bida, G.; Barrio, J. R., Int. J. Rad. Appl. Instrum. A 1992, 43, 989-996; b) Collins, J.; Waldmann, C. M.; Drake, C.; Slavik, R.; Ha, N. S.; Sergeev, M.; Lazari, M.; Shen, B.; Chin, F. T.; Moore, M.; Sadeghi, S.; Phelps, M. E.; Murphy, J. M.; van Dam, R. M., Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 11309-11314; c) Teare, H.; Robins, E. G.; Kirjavainen, A.; Forsback, S.; Sandford, G.; Solin, O.; Luthra, S. K.; Gouverneur, V., Angew. Chem. Int. Ed. 2010, 49, 6821-6824; d) Stenhagen, I. S. R.; Kirjavainen, A. K.; Forsback, S. J.; Jørgensen, C. G.; Robins, E. G.; Luthra, S. K.; Solin, O.; Gouverneur, V., Chem. Comm. 2013, 49, 1386-1388.
60. Ido, T.; Irie, T.; Kasida, Y., J. Labelled Compd. Radiopharm. 1979, 16, 153-154.
61. a) Jacobson, O.; Kiesewetter, D. O.; Chen, X., Bioconjug. Chem. 2015, 26, 1-18; b) Rong, J.; Haider, A.; Liang, S., Precision Radiochemistry for Fluorine-18 Labeling of PET Tracers. In Organofluorine Chemistry, Szabó, K.; Selander, N., Eds. Wiley‐VCH: Weinheim, Germany, 2021; p. 397-425.
62. Wilkinson, J. A., Chem. Rev. 2002, 92, 505-519.
63. Prabhakaran, J.; Underwood, M. D.; Parsey, R. V.; Arango, V.; Majo, V. J.; Simpson, N. R.; Van Heertum, R.; Mann, J. J.; Kumar, J. S. D., Biorg. Med. Chem. 2007, 15, 1802-1807.
64. a) Huiban, M.; Tredwell, M.; Mizuta, S.; Wan, Z.; Zhang, X.; Collier, T. L.; Gouverneur, V.; Passchier, J., Nat. Chem. 2013, 5, 941-944; b) Huang, X.; Liu, W.; Ren, H.; Neelamegam, R.; Hooker, J. M.; Groves, J. T., J. Am. Chem. Soc. 2014, 136, 6842-6845; c) Rühl, T.; Rafique, W.; Lien, V. T.; Riss, P. J., Chem. Comm. 2014, 50, 6056-6059; d) Ivashkin, P.; Lemonnier, G.; Cousin, J.; Grégoire, V.; Labar, D.; Jubault, P.; Pannecoucke, X., Chem. Eur. J. 2014, 20, 9514-9518; e) van der Born, D.; Sewing, C.; Herscheid, J. D. M.; Windhorst, A. D.; Orru, R. V. A.; Vugts, D. J., Angew. Chem. Int. Ed. 2014, 53, 11046-11050; f) Brooks, A. F.; Topczewski, J. J.; Ichiishi, N.; Sanford, M. S.; Scott, P. J. H., Chem. Sci. 2014, 5, 4545-4553; g) Pees, A.; Windhorst, A. D.; Vosjan, M. J. W. D.; Tadino, V.; Vugts, D. J., Eur. J. Org. Chem. 2020, 2020, 1177-1185.
65. Verhoog, S.; Kee, C. W.; Wang, Y.; Khotavivattana, T.; Wilson, T. C.; Kersemans, V.; Smart, S.; Tredwell, M.; Davis, B. G.; Gouverneur, V., J. Am. Chem. Soc. 2018, 140, 1572-1575.
66. a) Adam, M. J.; Grierson, J. R.; Ruth, T. J.; Jivan, S., Int. J. Rad. Appl. Instrum. A 1986, 37, 877-882; b) Adam, M. J.; Ruth, T. J.; Grierson, J. R.; Abeysekera, B.; Pate, B. D., 1986, 27, 1462-1466; c) Chirakal, R.; Firnau, G.; Couse, J.; Garnett,

69
E. S., Int. J. Appl. Radiat. Isot. 1984, 35, 651-653; d) Coenen, H. H.; Franken, K.; Kling, P.; Stöcklin, G., Int. J. Rad. Appl. Instrum. A 1988, 39, 1243-1250; e) Jewett, D. M.; Potocki, J. F.; Ehrenkaufer, R. E., J. Fluor. Chem. 1984, 24, 477-484; f) Speranza, M.; Shiue, C.-Y.; Wolf, A. P.; Wilbur, D. S.; Angelini, G., Chem. Comm. 1984, 1448-1449; g) Buckingham, F.; Kirjavainen, A. K.; Forsback, S.; Krzyczmonik, A.; Keller, T.; Newington, I. M.; Glaser, M.; Luthra, S. K.; Solin, O.; Gouverneur, V., Angew. Chem. Int. Ed. 2015, 54, 13366-13369; h) Oberdorfer, F.; Hofmann, E.; Maier-Borst, W., J. Labelled Comp. Radiopharm. 1988, 25, 999-1005; i) Oberdorfer, F.; Hofmann, E.; Maier-Borst, W., Int. J. Rad. Appl. Instrum. A 1988, 39, 685-688; j) Satyamurthy, N.; Bida, G. T.; Phelps, M. E.; Barrio, J. R., Int. J. Rad. Appl. Instrum. A 1990, 41, 733-738; k) Teare, H.; Robins, E. G.; Arstad, E.; Luthra, S. K.; Gouverneur, V., Chem. Commun. 2007, 2330-2332; l) Rong, J.; Liang, S. H., Aliphatic [18F]Fluorination Chemistry for Positron Emission Tomography. In Fluorination, Hu, J.; Umemoto, T., Eds. Springer: Singapore, 2018; p. 1-14.
67. Bergman, J.; Solin, O., Nucl. Med. Biol. 1997, 24, 677-683.
68. Lee, E.; Kamlet, A. S.; Powers, D. C.; Neumann, C. N.; Boursalian, G. B.; Furuya, T.; Choi, D. C.; Hooker, J. M.; Ritter, T., Science 2011, 334, 639-642.
69. Diner, C.; Szabo, K. J., J. Am. Chem. Soc. 2017, 139, 2-14.
70. Haruta, R.; Ishiguro, M.; Ikeda, N.; Yamamoto, H., J. Am. Chem. Soc. 1982, 104, 7667-7669.
71. Liepouri, F.; Bernasconi, G.; Petasis, N. A., Org. Lett. 2015, 17, 1628-1631.
72. Matsumoto, Y.; Naito, M.; Uozumi, Y.; Hayashi, T., Chem. Comm. 1993, 1468-1469.
73. a) Gao, D. W.; Xiao, Y.; Liu, M.; Liu, Z.; Karunananda, M. K.; Chen, J. S.; Engle, K. M., ACS Catal. 2018, 8, 3650-3654; b) Huang, Y.; Del Pozo, J.; Torker, S.; Hoveyda, A. H., J. Am. Chem. Soc. 2018, 140, 2643-2655.
74. Ito, H.; Sasaki, Y.; Sawamura, M., J. Am. Chem. Soc. 2008, 130, 15774-15775.
75. Zhao, T. S.; Yang, Y.; Lessing, T.; Szabó, K. J., J. Am. Chem. Soc. 2014, 136, 7563-7566.
76. Blais, J.; L'Honore, A.; Soulie, J.; Cadiot, P., J. Organomet. Chem. 1974, 78, 323-337.
77. Yasuhiro Yamashita; Masaharu Ueno; Yokusu Kuriyama; Kobayashi, S., Adv. Synth. Catal. 2002, 344, 929-931.
78. Thaima, T.; Zamani, F.; Hyland, C. J. T.; Pyne, S. G., Synthesis 2017, 49, 1461-1480.
79. Chu, W.-D.; Zhang, Y.; Wang, J., Catal. Sci. Technol. 2017, 7, 4570-4579.
80. Mao, L.; Bose, S. K., Adv. Synth. Catal. 2020, 362, 4174-4188.
81. a) Diner, C.; Szabó, K. J., J. Am. Chem. Soc. 2017, 139, 2-14; b) Hall, D. G.; Lachance, H., Allylboration of Carbonyl Compounds. Wiley-VCH: Hoboken, NY, 2012; p. 1-574.

70
82. a) Lou, S.; Moquist, P. N.; Schaus, S. E., J. Am. Chem. Soc. 2006, 128, 12660-12661; b) Barnett, D. S.; Moquist, P. N.; Schaus, S. E., Org. Lett. 2009, 48, 8679-8682.
83. Alam, R.; Vollgraff, T.; Eriksson, L.; Szabo, K. J., J. Am. Chem. Soc. 2015, 137, 11262-11265.
84. Huang, G.; Diner, C.; Szabó, K. J.; Himo, F., Org. Lett. 2017, 19, 5904-5907.
85. Fandrick, K. R.; Fandrick, D. R.; Reeves, J. T.; Gao, J.; Ma, S.; Li, W.; Lee, H.; Grinberg, N.; Lu, B.; Senanayake, C. H., J. Am. Chem. Soc. 2011, 133, 10332-10335.
86. Barnett, D. S.; Schaus, S. E., Org. Lett. 2011, 13, 4020-4023.
87. a) Chen, M.; Roush, W. R., J. Am. Chem. Soc. 2012, 134, 10947-10952; b) Wang, M.; Khan, S.; Miliordos, E.; Chen, M., Org. Lett. 2018, 20, 3810-3814.
88. Sasaki, Y.; Sawamura, M.; Ito, H., Chem. Lett. 2011, 40, 1044-1046.
89. a) Raducan, M.; Alam, R.; Szabó, K. J., Angew. Chem. Int. Ed. 2012, 51, 13050-13053; b) Sebelius, S.; Olsson, V. J.; Szabó, K. J., J. Am. Chem. Soc. 2005, 127, 10478-10479; c) Olsson, V. J.; Sebelius, S.; Selander, N.; Szabó, K. J., J. Am. Chem. Soc. 2006, 128, 4588-4589.
90. Laitar, D. S.; Müller, P.; Sadighi, J. P., J. Am. Chem. Soc. 2005, 127, 17196-17197.
91. Sun, J.; Perfetti, M. T.; Santos, W. L., J. Org. Chem. 2011, 76, 3571-3575.
92. Zhao, J.; Szabó, K. J., Angew. Chem. Int. Ed. 2016, 55, 1502-1506.
93. Falck, J. R.; Bondlela, M.; Venkataraman, S. K.; Srinivas, D., J. Org. Chem. 2001, 66, 7148-7150.
94. Nowrouzi, F.; Batey, R. A., Angew. Chem. Int. Ed. 2013, 52, 892-895.
95. a) Jonker, S. J. T.; Diner, C.; Schulz, G.; Iwamoto, H.; Eriksson, L.; Szabó, K. J., Chem. Commun. 2018, 54, 12852-12855; b) Alam, R.; Diner, C.; Jonker, S.; Eriksson, L.; Szabo, K. J., Angew. Chem. Int. Ed. 2016, 55, 14417-14421.
96. Tsai, A. S.; Chen, M.; Roush, W. R., Org. Lett. 2013, 15, 1568-1571.
97. Paton, R. S.; Goodman, J. M.; Pellegrinet, S. C., Org. Lett. 2009, 11, 37-40.
98. Wang, D.; de Wit, M. J. M.; Szabó, K. J., J. Org. Chem. 2018, 83, 8786-8792.
99. Ni, C.; Hu, J., Chem. Soc. Rev. 2016, 45, 5441-5454.
100. a) Warner, P.; Green, R. C.; Gomes, B.; Strimpler, A. M., J. Med. Chem. 1994, 37, 3090-3099; b) Veale, C. A.; Bernstein, P. R.; Bryant, C.; Ceccarelli, C.; Damewood, J. R., Jr.; Earley, R.; Feeney, S. W.; Gomes, B.; Kosmider, B. J., J. Med. Chem. 1995, 38, 98-108; c) Metz, W. A.; Peet, N. P., Exp. Opin. Ther. Patents 1999, 9, 851-868.
101. Amii, H.; Kobayashi, T.; Hatamoto, Y.; Uneyama, K., Chem. Comm. 1999, 1323-1324.

71
102. Prakash, G. K. S.; Hu, J.; Olah, G. A., J. Fluor. Chem. 2001, 112, 355-360.
103. Uneyama, K.; Tanaka, H.; Kobayashi, S.; Shioyama, M.; Amii, H., Org. Lett. 2004, 6, 2733-2736.
104. a) Hu, X.-S.; Yu, J.-S.; Zhou, J., Chem. Comm. 2019, 55, 13638-13648; b) Decostanzi, M.; Campagne, J. M.; Leclerc, E., Org. Biomol. Chem. 2015, 13, 7351-7380.
105. Guo, Y.; Shreeve, J. M., Chem. Commun. 2007, 3583-3585.
106. Wu, Y.-B.; Lu, G.-P.; Zhou, B.-J.; Bu, M.-J.; Wan, L.; Cai, C., Chem. Comm. 2016, 52, 5965-5968.
107. Huang, X.; Zhang, Y.; Zhang, C.; Zhang, L.; Xu, Y.; Kong, L.; Wang, Z.-X.; Peng, B., Angew. Chem. Int. Ed. 2019, 58, 5956-5961.
108. Ma, J.-A.; Cahard, D., J. Org. Chem. 2003, 68, 8726-8729.
109. Kieltsch, I.; Eisenberger, P.; Stanek, K.; Togni, A., CHIMIA 2008, 62, 260-263.
110. a) Lübcke, M.; Bezhan, D.; Szabó, K. J., Chem. Sci. 2019, 10, 5990-5995; b) Lübcke, M.; Yuan, W.; Szabó, K. J., Org. Lett. 2017, 19, 4548-4551.
111. a) Liu, Y.-L.; Zhou, J., Chem. Comm. 2012, 48, 1919-1921; b) Liu, Y.-L.; Liao, F.-M.; Niu, Y.-F.; Zhao, X.-L.; Zhou, J., Org. Chem. Front. 2014, 1, 742-747.
112. Liu, T.; Shen, Q., Org. Lett. 2011, 13, 2342-2345.
113. Parsons, A. T.; Senecal, T. D.; Buchwald, S. L., Angew. Chem. Int. Ed. 2012, 51, 2947-2950.
114. a) Ilchenko, N. O.; Janson, P. G.; Szabó, K. J., Chem. Commun. 2013, 49, 6614-6616; b) Ilchenko, N. O.; Janson, P. G.; Szabó, K. J., J. Org. Chem. 2013, 78, 11087-11091.
115. Hao, Y.-J.; Hu, X.-S.; Yu, J.-S.; Zhou, F.; Zhou, Y.; Zhou, J., Tetrahedron 2018, 74, 7395-7398.
116. a) Yu, L.-Z.; Xu, Q.; Tang, X.-Y.; Shi, M., ACS Catal. 2016, 6, 526-531; b) Li, Z.-R.; Bao, X.-X.; Sun, J.; Shen, J.; Wu, D.-Q.; Liu, Y.-K.; Deng, Q.-H.; Liu, F., Org. Chem. Front. 2016, 3, 934-938.
117. Lemal, D. M., J. Org. Chem. 2004, 69, 1-11.
118. Nakamura, Y.; Ozeki, Y.; Uneyama, K., J. Fluor. Chem. 2008, 129, 274-279.
119. a) Dixon, L. I.; Carroll, M. A.; Gregson, T. J.; Ellames, G. J.; Harrington, R. W.; Clegg, W., Eur. J. Org. Chem. 2013, 2013, 2334-2345; b) Nicolai, S.; Piemontesi, C.; Waser, J., Angew. Chem. Int. Ed. 2011, 50, 4680-4683.
120. a) Eisenberger, P. K., I.; Koller, R.; Stanek, K.; Togni, A., Org. Synth. 2011, 88, 168-180; b) Matoušek, V.; Václavík, J.; Hájek, P.; Charpentier, J.; Blastik, Z. E.; Pietrasiak, E.; Budinská, A.; Togni, A.; Beier, P., Chem. Eur. J. 2016, 22, 417-424; c) Geary, G. C.; Hope, E. G.; Singh, K.; Stuart, A. M., RSC Adv. 2015, 5,

72
16501-16506; d) Matoušek, V.; Pietrasiak, E.; Schwenk, R.; Togni, A., J. Org. Chem. 2013, 78, 6763-6768.
121. Cortés González, M. A.; Nordeman, P.; Bermejo Gómez, A.; Meyer, D. N.; Antoni, G.; Schou, M.; Szabó, K. J., Chem. Commun. 2018, 54, 4286-4289.
122. Ulmer, A.; Brunner, C.; Arnold, A. M.; Pöthig, A.; Gulder, T., Chem. Eur. J. 2016, 22, 3660-3664.
123. Cortés González, M. A.; Jiang, X.; Nordeman, P.; Antoni, G.; Szabo, K. J., Chem. Commun. 2019, 55, 13358-13361.
124. Edwards, P. D.; Andisik, D. W.; Bryant, C. A.; Ewing, B.; Gomes, B.; Lewis, J. J.; Rakiewicz, D.; Steelman, G.; Strimpler, A.; Trainor, D. A.; Tuthill, P. A.; Mauger, R. C.; Veale, C. A.; Wildonger, R. A.; Williams, J. C.; Wolanin, D. J.; Zottola, M., J. Med. Chem. 1997, 40, 1876-1885.
125. a) Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y. J.; Long, A. T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; Kluge, S.; Panzer, U.; Mizuta, R.; Mannherz, H. G.; Kitamura, D.; Herrmann, M.; Napirei, M.; Fuchs, T. A., Science 2017, 358, 1202-1206; b) Mehta, P.; McAuley, D. F.; Brown, M.; Sanchez, E.; Tattersall, R. S.; Manson, J. J., The Lancet 2020, 395, 1033-1034; c) Barnes, B. J.; Adrover, J. M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J. M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J. S.; Loda, M.; Looney, M. R.; McAllister, F.; Rayes, R.; Renaud, S.; Rousseau, S.; Salvatore, S.; Schwartz, R. E.; Spicer, J. D.; Yost, C. C.; Weber, A.; Zuo, Y.; Egeblad, M., J. Exp. Med. 2020, 217, 1-7; d) Thierry, A.; Roch, B., Am. Chem. Soc. Div. Pet. Chem. Prepr. 2020, 2020040238; e) Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D. S.; Weinrauch, Y.; Zychlinsky, A., 2004, 303, 1532-1535.
126. a) Lucas, S. D.; Costa, E.; Guedes, R. C.; Moreira, R., 2013, 33, E73-E101; b) Skiles, J. W.; Jeng, A. Y., Exp. Opin. Ther. Patents 1999, 9, 869-895; c) Schirlin, D.; Tarnus, C.; Baltzer, S.; Rémy, J. M., Bioorg. Med. Chem. Lett. 1992, 2, 651-654.
127. Prakash, G. K. S.; Alauddin, M. M.; Hu, J.; Conti, P. S.; Olah, G. A., J. Labelled Compd. Radiopharm. 2003, 46, 1087-1092.
128. Badiola, E.; Fiser, B.; Gómez-Bengoa, E.; Mielgo, A.; Olaizola, I.; Urruzuno, I.; García, J. M.; Odriozola, J. M.; Razkin, J.; Oiarbide, M.; Palomo, C., J. Am. Chem. Soc. 2014, 136, 17869-17881.
129. a) Fearon, K.; Spaltenstein, A.; Hopkins, P. B.; Gelb, M. H., J. Med. Chem. 1987, 30, 1617-1622; b) Pfitzner, K. E.; Moffatt, J. G., J. Am. Chem. Soc. 1965, 87, 5661-5670.
130. Steglich, W.; Höfle, G., Angew. Chem. Int. Ed. 1969, 8, 981.
131. Baker, K. V.; Brown, J. M.; Hughes, N.; Skarnulis, A. J.; Sexton, A., J. Org. Chem. 1991, 56, 698-703.
132. a) Lien, V. T.; Riss, P. J., Biomed. Res. Int. 2014, 2014, 380124; b) Khotavivattana, T.; Verhoog, S.; Tredwell, M.; Pfeifer, L.; Calderwood, S.;

73
Wheelhouse, K.; Lee Collier, T.; Gouverneur, V., Angew. Chem. Int. Ed. 2015, 54, 9991-9995.
133. Macdonald, S. J. F.; Dowle, M. D.; Harrison, L. A.; Shah, P.; Johnson, M. R.; Inglis, G. G. A.; Clarke, G. D. E.; Smith, R. A.; Humphreys, D.; Molloy, C. R.; Amour, A.; Dixon, M.; Murkitt, G.; Godward, R. E.; Padfield, T.; Skarzynski, T.; Singh, O. M. P.; Kumar, K. A.; Fleetwood, G.; Hodgson, S. T.; Hardy, G. W.; Finch, H., Bioorg. Med. Chem. Lett. 2001, 11, 895-898.





























