Embed Size (px)
Citation preview

Structure
Article
Structural and Functional Analysisof HtrA1 and Its SubdomainsCharles Eigenbrot,1,* Mark Ultsch,1 Michael T. Lipari,2 Paul Moran,2 S. Jack Lin,2 Rajkumar Ganesan,2 Clifford Quan,2
Jeffrey Tom,2 Wendy Sandoval,3 Menno van Lookeren Campagne,4 and Daniel Kirchhofer2,*1Department of Structural Biology2Department of Early Discovery Biochemistry3Department of Protein Chemistry4Department of Immunology
Genentech, Inc., South San Francisco, CA 94080, USA
*Correspondence: [email protected] (D.K.), [email protected] (C.E.)DOI 10.1016/j.str.2012.03.021
SUMMARY
The homotrimeric human serine protease HtrA1 ishomologous to bacterial HtrA proteases regardingthe trypsin-like catalytic and PDZ domains butdiffers by the presence of an N-terminal domainwith IGFBP and Kazal homology. The crystal struc-tures and SAXS analysis presented herein reveal therare tandem of IGFBP- and Kazal-like modules, aprotease active site that adopts a competent confor-mation in the absence of substrate or inhibitor andamodel for the intact protein in solution. Highly sensi-tive enzymatic assays and binding studies demon-strate that the N-terminal tandem has no apparenteffect on protease activity, and in accordance withthe structure-based predictions, neither the IGFBP-nor Kazal-like module retains the function of theirprototype proteins. Our structures of the unligandedHtrA1 active site suggest two-state equilibriumand a ‘‘conformational selection’’ model, in whichsubstrate binds to the active conformer.
INTRODUCTION
The serine protease HtrA1 (PRSS11; Clan PA, family S1) belongs
to an evolutionarily conserved family of HtrA proteins (Clausen
et al., 2002, 2011). In humans, HtrA1, 3, and 4 share the same
domain architecture: an N-terminal IGFBP-like module and a
Kazal-like module, a protease domain with trypsin-like fold,
and a C-terminal PDZ domain. The identification of human
loss-of-function mutations, causing familial ischemic cerebral
small-vessel disease (Hara et al., 2009), suggested an important
role for HtrA1 in human physiology. The molecular mechanism
involves deficient TGF-b inhibition by HtrA1, resulting in
increased TGF-b signaling (Hara et al., 2009). Dysregulated
TGF-b signaling by aberrant HtrA1 expression may also
contribute to arthritic disease, perhaps in conjunction with
HtrA1-mediated degradation of various extracellular matrix
components (Chamberland et al., 2009; Grau et al., 2006;
Hadfield et al., 2008; Hu et al., 1998; Oka et al., 2004; Tocharus
et al., 2004; Tsuchiya et al., 2005) or indirectly via upregulation
of matrix metalloproteases (Grau et al., 2006). In addition, human
1040 Structure 20, 1040–1050, June 6, 2012 ª2012 Elsevier Ltd All ri
genetic studies identified a strong correlation between the
progression of age-related macular degeneration and a SNP in
the HtrA1 promoter region, which results in increasedHtrA1 tran-
script levels (Dewan et al., 2006; Yang et al., 2006).
The bacterial HtrA proteins DegP, DegS, and DegQ share
homology with HtrA1’s trypsin-like and PDZ domains, but they
lack the IGFBP and Kazal modules at their N-termini (Kim and
Kim, 2005; Meltzer et al., 2008). They form trimers, but DegP
can also form higher-order oligomers, including 24-mers (Krojer
et al., 2008). DegS is an allosteric enzyme with its PDZ domain
acting as a regulatory switch (Hasselblatt et al., 2007; Sohn
et al., 2007). The autoinhibited state was found to be relieved
by binding of small peptides to the PDZ domain or by domain
deletion (Sohn et al., 2007; Wilken et al., 2004). Enzyme regula-
tory function of the PDZ domain has been also reported for
DegP and the human HtrA1 and HtrA2 (Hadfield et al., 2008;
Martins et al., 2003;Meltzer et al., 2008;Murwantokoet al., 2004).
The IGFBP-like/Kazal-like tandem (referred to as the
N-domain) is rare and found only in HtrA1 and five other human
proteins (see The Family of N-Domain-Containing Proteins).
Kazal domains are tight binding inhibitors of serine proteases
(Lu et al., 2001) with three conserved disulfide bonds and are
exemplified by the pancreatic secretory trypsin inhibitor SPINK1
(Hecht et al., 1991), which was originally discovered by Kazal
et al. (1948). A loop between the second and third cysteines
binds to the protease active-site cleft and inserts its P1 residue
into the S1 specificity pocket (Hecht et al., 1991) in a fashion
similar to other standard-mechanism serine protease inhibitors,
such as the Kunitz domains (Arnoux et al., 1995; Kunitz and
Northrop, 1936). The IGFBP-like module of HtrA1 shares
sequence homology with the N-terminal lobe of the high affinity
IGF binding proteins IGFBP-1–IGFBP-6, which regulate IGF
signaling (Hwa et al., 1999). The N-terminal lobe of IGFBP
proteins is connected via a linker to the C-terminal lobe, and
both are usually required for high-affinity IGF binding (Bach
et al., 2005; Hwa et al., 1999; Sitar et al., 2006). A group of
proteins, including HtrA1,Mac-25 (Kato, 2000), the CCNproteins
Holbourn et al., 2008), and ESM-1 (Lassalle et al., 1996), all con-
tain segments with sequence homology to the N-terminal IGFBP
lobe of bona fide IGFBPs and have been categorized as low-
affinity IGF-binding proteins (Hwa et al., 1999; Kim et al., 1997).
Recent structural insight into the trimeric HtrA1 protease
domain suggested that catalytic activity is regulated by a
substrate-induced activation mechanism based on the finding
ghts reserved

A
B C D
Figure 1. HtrA1 Constructs and Their Enzy-
matic Activity
(A) Full-length HtrA1 (HtrA1), its catalytically in-
activated form HtrA1(S328A) and PDZ-deleted
form (HtrA_DPDZ), and the IGFBP/Kazal tandem
(N-domain) were expressed and purified from
insect cells. HtrA1 with N-domain deletion
(HtrA1_DN), the catalytic domain (HtrA1_Cat), and
its inactivated form HtrA1_Cat(S328A) were ex-
pressed and purified from Escherichia coli. All
catalytic domain-containing constructs eluted
as trimers by size-exclusion chromatography,
whereas the N-domain eluted as amonomer. Solid
and open diamonds indicate the positions of
disease-associated missense (A252T and V297M)
and nonsense (R302X and R370X) mutations,
respectively, as reported (Hara et al., 2009). See
also Table S1.
(B) Linear time-dependent cleavage of the fluo-
rescence-quenched peptide substrate H2-Opt
(Mca-IRRVSYSF(Dnp)KK) by increasing concen-
trations of HtrA1 (0.35–4 nM) versus 4 nM of
catalytically inactive HtrA1(S328A).
(C) Effect of N-domain deletion on H2-Opt
cleavage rates as a function of enzyme concen-
tration.
(D) Effect of N-domain deletion on time-dependent
enzyme inactivation by the active site-directed
irreversible inhibitor DFP. Calculated pseudo-first-
order rate constants k for inactivation of HtrA1 and HtrA_DN by DFP were 69.3 3 10�3 min�1 and 64.8 3 10�3 min�1, respectively. Error bars in (C) and (D)
represent the standard deviation of three independent measurements.
See also Table S1.
Structure
HtrA1 Structures
that the competent active-site conformation was only observed
when a peptidic substrate mimic was covalently bound to the
active site (Truebestein et al., 2011). Moreover, the study further
demonstrated that the PDZ domain does not regulate enzymatic
activity, raising the question of whether the N-domain could
serve as an activity modulator in lieu of the PDZ domain. There-
fore, we investigated the N-domain by structural and biochem-
ical approaches. The X-ray crystal structure of the N-domain at
2.0 A resolution showed an unprecedented arrangement of
two common folds. We report on the N-domain’s inability to
bind IGF-I or inhibit serine proteases, including HtrA1. Our
X-ray crystal structures of the apo-form of the HtrA1 protease
domain show a substantial degree of active-site conformational
plasticity. Contrary to a prior report, we see the enzymatically
competent active-site conformation in the absence of substrate,
which demonstrates that HtrA1 does not require substrate for its
activation. In addition, a low-resolution structure of full-length
HtrA1 was determined using small-angle X-ray scattering, which
allows approximate placement of the N-domain and PDZ
domains within the HtrA1 trimer.
RESULTS
Investigation of HtrA1 Activity Regulationby the N-DomainTo investigatewhether theHtrA1N-domainmay act as an activity
regulator in place of or in conjunction with the PDZ domain, we
established a sensitive enzymatic assay using the fluores-
cence-quenched peptide substrate Mca-IRRVSYSF(Dnp)KK
(H2-Opt), originally identified for the related HtrA2 (Martins
Structure 20, 10
et al., 2003), to measure catalytic activities of HtrA1 constructs
having N- and PDZ domain deletions (Figure 1A). H2-Opt was
an excellent substrate for HtrA1, allowing measurements at
pM HtrA1 concentrations (enzyme concentrations refer to the
HtrA1 monomer). Figure 1B shows the linear initial velocities of
H2-Opt cleavage with increasing enzyme concentrations (0.35–
4 nM). Replacing the catalytic serine with alanine (HtrA1(S328A))
completely abolished H2-Opt cleavage. Plots of the initial
velocities as a function of HtrA1 concentration showed a linear
relationship with a slope of 21.6 ± 0.2 nM product min�1 nM�1
enzyme (Figure 1C). The slope from HtrA1 lacking the N-domain
(HtrA1_DN) was identical (21.2 ± 0.4 nM product min�1 nM�1
enzyme), indicating that the N-domain neither enhanced nor
reduced HtrA1 enzymatic activity.
Diisopropyl fluorophosphate (DFP) is an irreversible active-site
inhibitor that forms a covalent bond with the catalytic serine and
occupies the S1 specificity pocket. It is an excellent probe to
investigate conformational states of active sites, because it
only binds to enzymatically competent active sites, as shown
for DegS (Sohn et al., 2007). Figure 1D shows that the pseudo-
first-order rate constants k for inactivation of HtrA1 and
HtrA1_DN by DFP were almost identical (69.3 3 10�3 min�1 vs.
64.8 3 10�3 min�1). In the absence of DFP, the enzyme activity
remained unchanged. The results suggested that the active
site readily adopts a competent conformation and that the
N-domain imparts neither activating nor inhibitory effects.
The recent study by Truebestein and colleagues (Truebestein
et al., 2011) used HtrA1 constructs lacking the N-domain to
assess the role of the PDZ domain for HtrA1 catalysis. Here,
we examined the role of the PDZ domain in the context of
40–1050, June 6, 2012 ª2012 Elsevier Ltd All rights reserved 1041
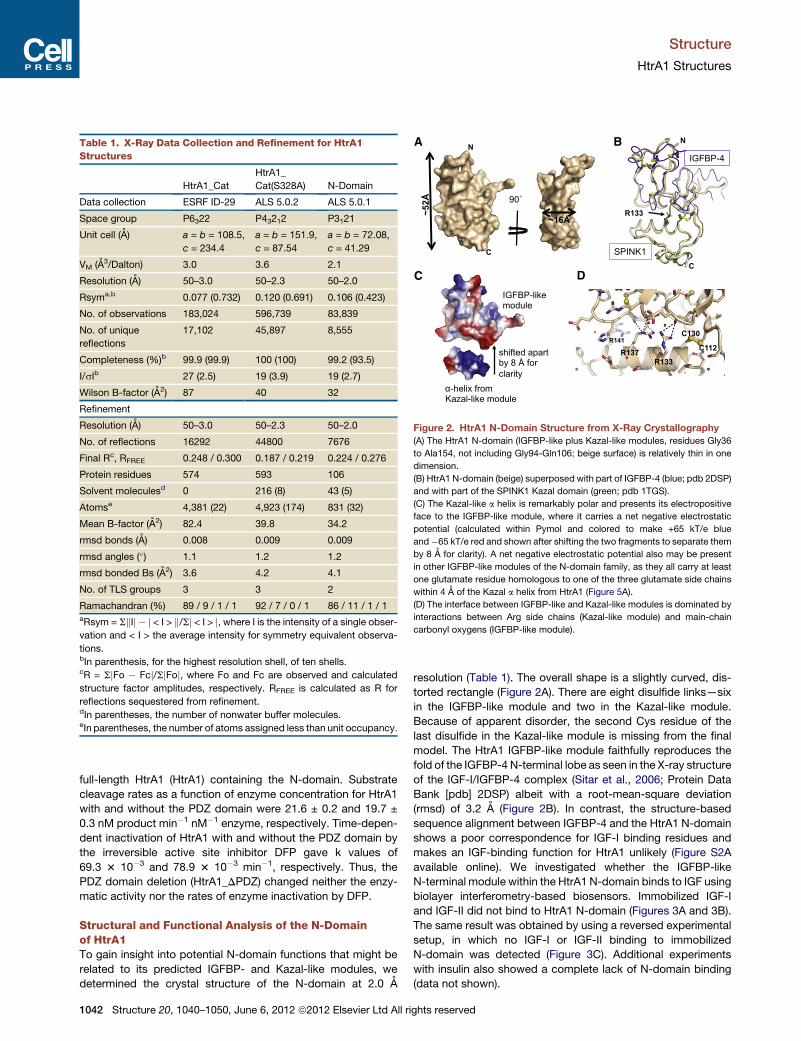
Table 1. X-Ray Data Collection and Refinement for HtrA1
Structures
HtrA1_Cat
HtrA1_
Cat(S328A) N-Domain
Data collection ESRF ID-29 ALS 5.0.2 ALS 5.0.1
Space group P6322 P43212 P3121
Unit cell (A) a = b = 108.5,
c = 234.4
a = b = 151.9,
c = 87.54
a = b = 72.08,
c = 41.29
VM (A3/Dalton) 3.0 3.6 2.1
Resolution (A) 50–3.0 50–2.3 50–2.0
Rsyma,b 0.077 (0.732) 0.120 (0.691) 0.106 (0.423)
No. of observations 183,024 596,739 83,839
No. of unique
reflections
17,102 45,897 8,555
Completeness (%)b 99.9 (99.9) 100 (100) 99.2 (93.5)
I/sIb 27 (2.5) 19 (3.9) 19 (2.7)
Wilson B-factor (A2) 87 40 32
Refinement
Resolution (A) 50–3.0 50–2.3 50–2.0
No. of reflections 16292 44800 7676
Final Rc, RFREE 0.248 / 0.300 0.187 / 0.219 0.224 / 0.276
Protein residues 574 593 106
Solvent moleculesd 0 216 (8) 43 (5)
Atomse 4,381 (22) 4,923 (174) 831 (32)
Mean B-factor (A2) 82.4 39.8 34.2
rmsd bonds (A) 0.008 0.009 0.009
rmsd angles (�) 1.1 1.2 1.2
rmsd bonded Bs (A2) 3.6 4.2 4.1
No. of TLS groups 3 3 2
Ramachandran (%) 89 / 9 / 1 / 1 92 / 7 / 0 / 1 86 / 11 / 1 / 1aRsym = SkIj � j < I > k/Sj < I > j, where I is the intensity of a single obser-
vation and < I > the average intensity for symmetry equivalent observa-
tions.bIn parenthesis, for the highest resolution shell, of ten shells.cR = SjFo � Fcj/SjFoj, where Fo and Fc are observed and calculated
structure factor amplitudes, respectively. RFREE is calculated as R for
reflections sequestered from refinement.dIn parentheses, the number of nonwater buffer molecules.eIn parentheses, the number of atoms assigned less than unit occupancy.
shifted apart
clarity
A B
DC
90˚
N
C
N
C
C130
C112
R133
R137
R141
IGFBP-4
SPINK1
IGFBP-like module
-helix from Kazal-like module
R133
Figure 2. HtrA1 N-Domain Structure from X-Ray Crystallography
(A) The HtrA1 N-domain (IGFBP-like plus Kazal-like modules, residues Gly36
to Ala154, not including Gly94-Gln106; beige surface) is relatively thin in one
dimension.
(B) HtrA1 N-domain (beige) superposed with part of IGFBP-4 (blue; pdb 2DSP)
and with part of the SPINK1 Kazal domain (green; pdb 1TGS).
(C) The Kazal-like a helix is remarkably polar and presents its electropositive
face to the IGFBP-like module, where it carries a net negative electrostatic
potential (calculated within Pymol and colored to make +65 kT/e blue
and�65 kT/e red and shown after shifting the two fragments to separate them
by 8 A for clarity). A net negative electrostatic potential also may be present
in other IGFBP-like modules of the N-domain family, as they all carry at least
one glutamate residue homologous to one of the three glutamate side chains
within 4 A of the Kazal a helix from HtrA1 (Figure 5A).
(D) The interface between IGFBP-like and Kazal-like modules is dominated by
interactions between Arg side chains (Kazal-like module) and main-chain
carbonyl oxygens (IGFBP-like module).
Structure
HtrA1 Structures
full-length HtrA1 (HtrA1) containing the N-domain. Substrate
cleavage rates as a function of enzyme concentration for HtrA1
with and without the PDZ domain were 21.6 ± 0.2 and 19.7 ±
0.3 nM product min�1 nM�1 enzyme, respectively. Time-depen-
dent inactivation of HtrA1 with and without the PDZ domain by
the irreversible active site inhibitor DFP gave k values of
69.3 3 10�3 and 78.9 3 10�3 min�1, respectively. Thus, the
PDZ domain deletion (HtrA1_DPDZ) changed neither the enzy-
matic activity nor the rates of enzyme inactivation by DFP.
Structural and Functional Analysis of the N-Domainof HtrA1To gain insight into potential N-domain functions that might be
related to its predicted IGFBP- and Kazal-like modules, we
determined the crystal structure of the N-domain at 2.0 A
1042 Structure 20, 1040–1050, June 6, 2012 ª2012 Elsevier Ltd All ri
resolution (Table 1). The overall shape is a slightly curved, dis-
torted rectangle (Figure 2A). There are eight disulfide links—six
in the IGFBP-like module and two in the Kazal-like module.
Because of apparent disorder, the second Cys residue of the
last disulfide in the Kazal-like module is missing from the final
model. The HtrA1 IGFBP-like module faithfully reproduces the
fold of the IGFBP-4 N-terminal lobe as seen in the X-ray structure
of the IGF-I/IGFBP-4 complex (Sitar et al., 2006; Protein Data
Bank [pdb] 2DSP) albeit with a root-mean-square deviation
(rmsd) of 3.2 A (Figure 2B). In contrast, the structure-based
sequence alignment between IGFBP-4 and the HtrA1 N-domain
shows a poor correspondence for IGF-I binding residues and
makes an IGF-binding function for HtrA1 unlikely (Figure S2A
available online). We investigated whether the IGFBP-like
N-terminal module within the HtrA1 N-domain binds to IGF using
biolayer interferometry-based biosensors. Immobilized IGF-I
and IGF-II did not bind to HtrA1 N-domain (Figures 3A and 3B).
The same result was obtained by using a reversed experimental
setup, in which no IGF-I or IGF-II binding to immobilized
N-domain was detected (Figure 3C). Additional experiments
with insulin also showed a complete lack of N-domain binding
(data not shown).
ghts reserved

A
B
C
D
E
Figure 3. N-Domain neither Binds to IGF
nor Inhibits Protease Activities
(A–C) Representative sensorgram with analyte
proteins binding to sensor surfaces immobilized
with biotinylated (A) IGF-I, (B) IGF-II, and (C) HtrA1
N-domain (N-domain). The concentrations of the
soluble analyte proteins used were 500 nM
N-domain, 200 nM IGFBP-1 and IGFBP-3, 500 nM
IGF-I and IGF-II, and rabbit polyclonal anti-N-
domain antibody.
(D) Inhibition of trypsin-like proteases by N-
domain (5 mM), aprotinin (5 mM), or a1-antitrypsin
(A1AT, 5 mM; 20 mM for HtrA1 and HtrA1_Cat).
Enzyme activity was determined by measuring
the rates of cleavage of H2-Opt (for HtrA1 and
HtrA1_Cat) or synthetic paranitroanilide sub-
strates. Activities are expressed as a percentage
of the uninhibited enzyme. Error bars represent
standard deviations of three or more independent
measurements.
(E) Effect of N-domain on b-casein digestion.
b-casein (50 mg/ml) digestion by full-length HtrA1
(FL, 15 nM; left panel) or HtrA1_Cat (15 nM; right
panel) in the presence of N-domain (N, 5 mM) or
DFP (DFP, 7.5 mM) was analyzed after 2 hr at
37�C by SDS-PAGE. Arrows indicate b-casein
cleavage products; arrowhead shows intact b-casein and asterisk shows N-domain. Lanes labeled ‘‘FL – DFP’’ and ‘‘Cat – DFP’’ are formal controls, where all
buffer components used for addition of DFP were added but without DFP.
See also Figure S1.
Structure
HtrA1 Structures
The HtrA1 Kazal-like a helix packs against the IGFBP-like
module, mediated by highly polar interactions. The Kazal-like
module’s helix is remarkably amphipathic, and the polar, electro-
positive side packs against the IGFBP-like module (Figure 2C).
Arginine side chains from residues 133, 137, and 141 are within
H-bonding distance to main-chain carbonyl oxygen atoms in
loops of the IGFBP-like module (Figure 2D). The disulfide link
between Cys112 and Cys130 acts as an additional tether for
the Kazal-like module’s helix.
The HtrA1 Kazal-like module differs from canonical Kazal
modules (e.g., SPINK1) by a longer a helix (one turn more) and
the presence of only two—instead of three—disulfide bonds.
The disulfide pattern of a canonical Kazal module is preserved
for the (Cys#2/Cys#4) and (Cys#3,Cys#6) cystines (Figures 4A
and 5A). The remaining canonical disulfide, (Cys#1/Cys#5), is
lacking in HtrA1 because the first Cys is missing and Cys #50sposition and role is supplanted by Arg133. The activin-binding
protein follistatin also includes noninhibitory Kazal-like modules,
and these include a noncanonical position for Cys#5, which
aligns with HtrA1’s Arg137. Thus, Arg133 and Arg137 serve
a role analogous to Cys residues in SPINK1 and follistatin,
respectively, by linking the Kazal-like a helix to a preceding poly-
peptide segment (Figures 2B, 4A, 4B, and 5A). In SPINK1 and fol-
listatin, the linkage is to the N-terminal part of the Kazal module.
In the N-domain, the Arg-mediated linkages are to the IGFBP-
like module. In this light, we note that other than the covalent
peptide link connecting them (Val111 – Cys112), all interactions
between the N-domain’s IGFBP- and Kazal-like modules are
polar in nature.
Despite sequence and structure differences, the HtrA1 Kazal
superposes with inhibitory Kazal modules (e.g., SPINK1 [pdb
1TGS]) with a rmsd less than 1 A when these features are
Structure 20, 10
ignored. Of 41 residues within our Kazal-like domain, 30 Ca
atoms superpose on SPINK1 with an rmsd of 0.8 A (Figure 2B).
However, the HtrA1 Kazal has two features, which are inconsis-
tent with the protease interactions seen for true inhibitors. The
nominal ‘‘P1’’ residue of HtrA1 (Ser114) is significantly out of
position, as can be seen by comparison to the Rhodniin complex
with thrombin (van de Locht et al., 1995; Figures 4C and S2C). In
addition, the adjacent IGFBP-like module would cause a major
steric clash with the target protease. The inhibitory activity of
the HtrA1 Kazal-like module was examined by treating a panel
of trypsin-like proteases with soluble N-domain at a concentra-
tion of 5 mM. The results showed that it did not interfere
with protease activities, whereas the serine protease inhibitors
a1-antitrypsin and aprotinin displayed inhibitory activities ac-
cording to their known specificities (Figure 3D). Likewise, HtrA1
and HtrA1_Cat were not inhibited by the N-domain but were
inhibited by a1-antitrypsin, as reported (Hara et al., 2009; Hou
et al., 2005; Hu et al., 1998). Additional experiments showed
that, whereas DFP completely inhibited b-casein digestion by
either HtrA1 or HtrA1_Cat, the N-domain had no apparent inhib-
itory activity (Figure 3E).
The Family of N-Domain-Containing ProteinsThe small family of IGFBP/Kazal tandem (= N-domain)-contain-
ing human proteins includes HtrA1, HtrA3, HtrA4, Mac25, Kazal
D1, and IGFBPL1 (Figure 5B). In each case the IGFBP/Kazal
tandem forms the N-terminal portion of the proteins, followed
by a catalytic domain (HtrA proteins) or an Ig-like domain. Other
proteins, such as members of the CCN family and follistatin,
contain one of the modules—IGFBP—or Kazal-like module—
but never both. Based on protein sequence alignment (Fig-
ure 5A), we expect good overall structural homology within the
40–1050, June 6, 2012 ª2012 Elsevier Ltd All rights reserved 1043

IGFBP-like EGF
A B
C
R137
C112 (Cys#2)
Cys#5
Cys#1 Cys#2
Cys#4
Cys#6
Cys#3
Thrombin catalytic triad
Asp
Ser His
Rhodniin P1 residue (His)
HtrA1 P1 residue (Ser114)
C119 (Cys#3)
C155 (Cys#6)
Ser114
Figure 4. N-Domain Structure Compared to Analogs
(A) The N-domain Kazal-like module uses Cys and Arg residues separated by
six amino acids to associate with the preceding IGFBP-like module. (Cys155 is
not observed because of disorder.)
(B) Follistatin (pdb 2ARP) uses two Cys residues separated by six amino acids
in an analogous fashion.
(C) Thrombin (gray) and Rhodniin’s Kazal1 (light blue) from the X-ray structure
of their complex (pdb 1TBR), showing the P1 residue from Rhodniin inserted in
the S1 pocket of thrombin, which blocks access of substrates to the active
site/catalytic triad. The N-domain (light brown), superposed on Rhodniin,
carries its ‘‘P1’’ residue on a loop, which tracks far from the S1 pocket,
consistent with a lack of inhibitory activity.
See also Figure S2.
Structure
HtrA1 Structures
‘‘N-domains.’’ The reported structure allows precise assignment
of the boundary between the two modules: there is no structural
correspondence between IGFBP-4 and the HtrA1 N-domain
after HtrA1 residue Cys110 and no structural correspondence
between inhibitory Kazal modules and the HtrA1 N-domain
before HtrA1 residue Val111. Therefore, the module boundary
in this family is identifiable by the Cys-X-Cys motif. The other
five N-domains share Kazal a helix sequences rich in Arg resi-
dues, mostly including ones homologous with Arg133 and
Arg137 and putative P1 residues that are either the same as in
HtrA1 (Ser) or other relatively uncommon P1 amino acids (e.g.,
Trp in HtrA3).
Enzymatically Competent and Incompetent Structuresof the Protease Domain Apo-FormWe report X-ray structures of the unliganded HtrA1 catalytic
domain with no added substrate mimic or inhibitor (apo protein;
1044 Structure 20, 1040–1050, June 6, 2012 ª2012 Elsevier Ltd All ri
Table 1). Our structures of the wild-type HtrA1 catalytic
domain (HtrA1_Cat, with protomers named chainA, chainB,
and chainD) and mutationally inactivated HtrA1 catalytic domain
(HtrA1_Cat(S328A), with protomers named chainX, chainY, and
chainZ) provide six crystallographically independent apo cata-
lytic domain conformations, and they differ in interesting ways
with respect to their apparent readiness to bind and process
substrate (summarized in Figure 6). A graphical summary of
crystal packing appears in Figure S3. For HtrA1_Cat, one proto-
mer (chainD) is relatively poorly ordered overall, and key residues
forming the oxyanion hole and S1 substrate specificity pocket
are not included in the final model. Protomers chainA and chainB
present two distinct conformations of the active-site region
(Figures 7A, 7B, and S4). For chainA, the side chain of the cata-
lytic triad residueHis220 is out of place relative to othermembers
of the triad (Ser328 and Asp250). Moreover, access to the S1
pocket is completely blocked by loop2, including Leu345, which
has a pronounced conformational difference relative to chainB
(see the next paragraph; Figures 7B and 7C). Residues 343–
348 do not adopt the b strand conformation observed for analo-
gous residues in active serine proteases and therefore cannot
form the usual main-chain H-bonds with substrate. This incom-
petent chainA conformation is essentially the same as seen for
apo HtrA1 structures (Truebestein et al., 2011) in pdb entries
3NUM and 3NWU.
For HtrA1_Cat chainB, the active-site residues are poised to
bind substrate and support amide bond cleavage. The catalytic
triad residues are properly positioned and Leu345 is now part of
an intact b strand and helps define the S1 pocket rather than
occlude it (Figures 7B, 7C, and S4). This chainB arrangement
is very similar to that observed in pdb entry 3NZI (Truebestein
et al., 2011), a structure of the HtrA1 protease with a substrate
surrogate covalently attached to Ser328. The rmsds for superpo-
sition of 190 Ca atom pairs and for 14 Ca pairs nearest the cata-
lytic site are 0.6 and 0.2 A, respectively (Figure 7D).
The three protomers in the HtrA1_Cat(S328A) structure
display the occluded S1 pocket seen for chainA and additional
features inconsistent with catalytic activity (see Supplemental
Results). As a measure of the low similarity of the loop2 confor-
mations of canonical and noncanonical forms of HtrA1 we
observe, the rmsd for superposition of Ca atoms of noncanonical
versions of residues 188–192 (chymotrypsinogen numbering
213–217) onto the canonical version is 1.723 ± 0.006 A. The anal-
ogous calculation for thrombin (pdb codes 1TBR and 1AFQ),
prostasin (pdb codes 3E0P and 3E1X), and HGFA (pdb codes
1YC0 and 1YBW) yields rmsd values of 0.819, 1.300, and
1.745 A, respectively.
Taken together, our apo HtrA1 protease structures reveal
large conformational differences in the active site region and
are easily classified as either incompetent or competent for
substrate binding. Because there are no large differences
between the number of H-bonds or of hydrophobic interactions
in the two conformations, and because of the disorder apparent
in chainD, we conclude that the two conformations are close in
free energy and therefore are both sampled frequently in
solution. We note that, whereas active sites of chainB and
chainD do not have intermolecular packing contacts likely to
have affected their conformations, chainA active sites have
contacts that are similar to those found for all three protomers
ghts reserved

Group 1
Group 2
IGFBP Kaz Catalytic PDZ
N L IGFBP family
HtrA1, HtrA3, HtrA4
Mac25, Kazal D1, IGFBPL1 Ig-like C2 type
N-domain Family
N-domain
IGFBP Kaz
IGFPB vWC
C
TSP-1 CT CCN family
EGF Kaz Follistatin repeat
B
A
Figure 5. N-Domain Family
(A) Sequence alignment of the six human proteins with the IGFBP-Kazal tandem. Included for reference are portions of IGFBP-4 N-lobe, Kazal (SPINK1), and
follistatin sequences, with canonical Kazal Cys residue linkages indicated with black lines. The pattern of disulfide links in HtrA1’s Kazal-like domain is unlike that
of SPINK1 and the follistatin Kazal-like domain for two reasons. First, the HtrA1 Kazal starts after the first canonical cysteine. Second, canonical Cys#50s role is
supplanted by Arg133, and follistatin’s noncanonical Cys#50s role is supplanted by Arg137. HtrA1-IGFBP-like residues within 4 A of the Kazal-like module (d)
include Glu residues, creating net negative charge.
(B) Domain arrangement of proteins with IGFBP- and Kazal-like modules (highlighted in green and blue, respectively). Not included here is ESM-1 (IGFBP-rP6),
which has an IGFPB-like domain followed by a nondefined region. Within the N-domain family, two groups of three proteins share identical domain architecture
(group 1: HtrA1, HtrA3, and HtrA4; group 2:Mac25, Kazal D1, and IGFBPL1). Group 2 differs from the group 1 in that the N-domain is followed by an Ig-like domain
(C2 type) instead of the catalytic/PDZ domains. Alternate names used for N-domain family members: HtrA1, IGFBP-rP5, and L56; Mac25, IGFBP-rP1, TAF, and
angiomodulin; IGFBPL1 and IGFBP-rP4. The six bona fide IGFPBs (IGFBP family) are composed of an N-terminal lobe (homologous to the IGFBP-like module of
the N-domain), a linker region (L), and a C-terminal lobe. The six CCN family proteins (cysteine-rich protein 61, connective tissue growth factor, and nephro-
blastoma overexpressed protein) have an N-terminal IGFBP-like domain, a von Willebrand factor type C repeat (vWC), a thrombospondin type-1 repeat (TSP-1),
and a cysteine-knot-containing module (CT) (except for CCN5).
Structure
HtrA1 Structures
of HtrA1_Cat(S328A) and the isomporphous pdb entry 3NWU.
However, we discount their influence in our analysis because
another recent HtrA1 catalytic domain structure, which lacks
such contacts (pdb 3NUM [Truebestein et al., 2011]),
has essentially the same incompetent conformation seen for
chainA.
Structure 20, 10
Low-Resolution Structure of Full-Length HtrA1by Small-Angle X-Ray ScatteringA low-resolution molecular envelope of intact Htra1 was
calculated using small-angle X-ray scattering (SAXS) analysis.
The central core was assigned to the protease trimer. Three
pronounced features protruding from the central core were
40–1050, June 6, 2012 ª2012 Elsevier Ltd All rights reserved 1045

distorted disordered/missing residues
distorted distorted + detergent distorted + disordered
competent active site in apo protein
Y325
L345
D250
H220
S328
Y325
L345
D250 H220
A328
Y325
L345
D250 H220
A328
Y325
L345
D250 H220
A328
! -OG
glycerol
Y325
L345
D250 H220
S328
D250
H220
S1
oxyanion hole
HtrA1_Cat chainA HtrA1_Cat chainB HtrA1_Cat chainD
HtrA1_Cat(S328A) chainX HtrA1_Cat(S328A) chainY HtrA1_Cat(S328A) chainZ
Figure 6. Six Crystallographically Unique
Catalytic Domain Protomers
Together, these apo structures demonstrate
significant conformational variability of the HtrA1
protease active-site region, including one version
that is poised for substrate binding (chainB). The
label ‘‘distorted’’ refers to the position of Leu345,
although His220 and Ser328 also adopt confor-
mations inconsistent with catalysis. ‘‘b-OG’’ is
b-octylglucoside.
See also Figure S3.
Structure
HtrA1 Structures
assigned to PDZ domains so that there are no significant
contacts between protease and PDZ domains (Figure 8). This
arrangement is reminiscent of the relationship between protease
and PDZ domains in DegP (pdb 1KY9) and is consistent with the
lack of ordered electron density seen for the PDZ domain of
an HtrA1 protease-PDZ construct (pdb 3NUM). Our best fit to
the experimental scattering curve was obtained when three
N-domains were placed within the central core with their short
dimension roughly parallel to the protease 3-fold axis and allow-
ing for covalent attachment to the protease domain N-termini. In
this position, there are no major contacts between N-domains
and they are adjacent to protease domains on the ‘‘back’’ side,
opposite the active sites. Experimental and calculated radii of
gyration were in good agreement (52.2 and 52.5 A, respectively).
Examination of Substrate SpecificityLike HtrA2 (Martins et al., 2003), HtrA1 cleaved H2-Opt at the
Val-Ser peptide bond, consistent with strong sequence conser-
vation among the specificity-determining residues between the
two proteases. A panel of H2-Opt peptides with variable P1 resi-
dues was tested in enzymatic assays with HtrA1 and HtrA1_Cat
and cleavage sites confirmed bymass spectrometry. The results
indicated a clear P1 preference for small- and medium-sized
hydrophobic residues (Val, Leu, Met, Ala, and Ile) but also for
the two polar amino acids Thr and Gln (Figure S1). None of
the substrates with charged P1 residues were cleaved. There
was no difference in substrate specificity between HtrA1 and
HtrA1_Cat, suggesting that the accessory domains did not influ-
ence substrate interactions. A similar P1 preference was found
for the HtrA1-cleavable macromolecular substrates b-casein,
biglycan, decorin, IGFBP-5, and osteoadherin (Table S1). Only
the P1 residues Glu (decorin) and Tyr (IGFBP-5) were not
observed in the panel of H2-Opt substrates. The P1 specificity
1046 Structure 20, 1040–1050, June 6, 2012 ª2012 Elsevier Ltd All rights reserved
profile is identical (except for Tyr from
IGFBP-5) to that reported recently by
Truebestein et al. (2011) and almost iden-
tical to that of HtrA2, which also has
strong preference for Val, Leu, Met, Ala,
Ile, Gln, and Ser (Martins et al., 2003).
DISCUSSION
The three human HtrA family members
HtrA1, 3, and 4 share an N-domain that
is notably absent in the well-described
bacterial homologs. Previous studies
recognized that the N-domain comprises two distinct modules
with sequences homologous to the N-terminal lobe of IGFBPs
and Kazal domains, respectively, giving rise to the notion
that the N-domain may recapitulate some functions associated
with these modules (Zumbrunn and Trueb, 1996). Furthermore,
a recent study indicated that, unlike bacterial HtrAs, the
HtrA1 catalytic activity may not be regulated by its PDZ
domain (Truebestein et al., 2011), raising the possibility of a regu-
latory role by the N-domain instead. The crystal structure
elucidated herein of the HtrA1 N-domain shows that it indeed
faithfully recapitulates the IGFBP- and Kazal-like folds, but
there are important structural deviations, which have functional
implications.
The Kazal-like module of HtrA1 is atypical in three important
aspects: it has only two instead of the typical three disulfide
bonds, the characteristic a helix is unusually long, and the
nominal P1 residue in the inhibitory loop is in a noncanonical
position. The missing disulfide bond per se does not preclude
protease inhibitory function, as shown by the two disulfide
bond-containing Kazal domain EPI1, which potently inhibits the
serine protease subtilisin A (Tian and Kamoun, 2005). Interest-
ingly, in HtrA1 the missing disulfide bond is replaced by H-bonds
from Arg133 of the Kazal-like module to main-chain atoms of
the IGFBP-like module. This H-bonding interaction and addi-
tional interactions from Arg137, also in the helix, strengthen the
connection between the two modules, thereby effectively re-
stricting access of the Kazal-like module to a target protease.
Indeed, in using relatively high concentrations of the N-domain,
we did not find inhibition of a diverse panel of serine proteases,
including HtrA1. The substitution of a disulfide bond, which is
present in both inhibitory and noninhibitory Kazal domains
(e.g., SPINK1 and follistatin), with H-bonding interactions in the
HtrA1 N-domain is probably also a feature of the other five

Y325
L345
D250
H220
S328
chainA distorted chainB competent
Y325
L345
D250
H220
S328
chainB with no substrate 3NZI with substrate
open S1
blocked S1
loopA
loop2
loop1
loopC
loopD
loopB
loop3
A
B
C
D
chainA
chainB
Figure 7. Competent and Incompetent
Conformations of the HtrA1 Protease
Domain from X-Ray Structures
(A) Apo protease domains have a loop2 confor-
mation that is inconsistent with substrate binding
(chainA [gray]) or that is poised to bind and
process substrate (chainB [orange]) Dotted
triangle and spheres represent Ca atoms of cata-
lytic triad residues His220, Asp250, and Ser328.
(B) Close-up view of active sites for chainA (gray)
and chainB (orange). The positions of His220 and
Leu345 in chainA are incorrect for substrate pro-
cessing. The large difference in the side chain of
Tyr325 is associated with an intermolecular
contact for chainA and is probably irrelevant.
Elements of the oxyanion hole for chainB are
illustrated as competent for catalytic activity, but
the resolution of our X-ray structure is too low to be
certain of this detail.
(C) The incompetence of chainA for substrate
binding arises from blockage of the S1 pocket
(green) by Leu345 (red).
(D) Active-site regions from the substrate-bound
structure (pdb 3NZI, light blue) and from chainB
(orange) show close correspondence of these
competent conformations. The large difference
between the two versions of Tyr325 is associated
with differences in their intermolecular contacts
and is probably irrelevant.
See also Figure S4.
Structure
HtrA1 Structures
human N-domain proteins. This prediction is based on the pres-
ence of a series of Arg residues in the Kazal a helix in all
N-domain proteins as well as the conserved pattern of Cys resi-
dues (Figure 5A). To the extent that these predicted interactions
preserve the relationship between IGFBP- and Kazal-like
modules, the other N-domain proteins would also lack inhibitory
activity.
We did not obtain any experimental evidence to suggest IGF-I,
IGF-II, or insulin binding activity for the N-domain. The IGFBP-
like module in HtrA1 is a reasonable facsimile of the homologous
domain from IGFBP-4 (Sitar et al., 2006) but only with respect to
the overall fold. The structural correspondence is quite imper-
fect, and the side chain character is very poorly analogous.
Despite initial evidence for interaction between another N-
domain family member, Mac25 (IGFBP-7) and IGF-I and IGF-II
(Oh et al., 1996), a sequence alignment of HtrA1 and Mac25
leads us to predict that no biologically relevant IGF-binding func-
tion pertains to Mac25 or other N-domain family members. This
notion is supported by a biosensor binding study that found no
appreciable Mac-25 interaction with IGF-I or IGF-II (Vorwerk
et al., 2002).
Another aspect of HtrA1 N-domain function is its potential
regulation of HtrA1 catalytic activity in the context of full-length
HtrA1. However, enzymatic assays with various HtrA1 con-
structs measuring activities and inhibitor incorporation rates
demonstrated that, like the PDZ domain, the N-domain is
dispensable for catalytic activity. Therefore, HtrA1 significantly
deviates from the bacterial HtrA paradigm in that neither of its
accessory domains, N-domain and PDZ, regulates catalytic
activity. This is further supported by the finding that the speci-
Structure 20, 10
ficity profile of a synthetic substrate panel was not influenced
by the accessory domains.
Our analysis of intact HtrA1 using SAXS yielded a saucer-like,
almost flat low-resolution molecular envelope that is consistent
with an HtrA1 trimer (Figures 8 and S5). In our best model ratio-
nalizing this envelope, the PDZ domains protrude from the
protease trimer and the N-domains lie flat against the side of
the protease trimer that is distal to the active sites, without
large contact areas between N-domains. If strong homotypic
interactions among N-domains exist in HtrA1, they might be
reasonably expected to be present in crystals of the N-domain.
The N-domains in our crystals are closely packed but are in an
arrangement that is inconsistent with integration into the
protease trimer (Figure S2B). Our molecular SAXS model does
not provide an intratrimer direct-contact mechanism by which
N-domains might influence protease activity, in accordance
with our biochemical results showing that the N-domain is
dispensable for activity. Additionally, the simple fact that the
N-terminal residues of the protease domains are found far from
the catalytic sites (Figure S3B) suggests N-domains are also
far from the catalytic sites in intact HtrA1. Our attempts to form
diffracting crystals of HtrA1_DPDZ or of intact HtrA1 were
unsuccessful.
Our finding of an apo catalytic domain with a competent
active-site conformation is contrary to the proposal made by
Truebestein et al. (2011) that HtrA1 is activated by substrate-
induced remodeling. Our experimental observations are
consistent with a low-energy barrier separating competent and
incompetent active-site conformers. First, the inactive con-
former would be unable to accept DFP inactivation because of
40–1050, June 6, 2012 ª2012 Elsevier Ltd All rights reserved 1047

PDZ
PDZ PDZ
CAT
CAT
CAT
N
N
N
90˚
active site face
loopA
PDZPDZ PDZ
N N
Figure 8. Molecular Envelope of Full-Length HtrA1 from SAXS and
Fit of Protein Domains
Two views rotated by 90�, semitransparent spheres represent the SAXS
envelope, and three HtrA1 molecules are shown schematically in blue, green,
and magenta. ‘‘CAT’’ is catalytic domain; ‘‘N’’ is N-domain.
See also Figure S5.
Structure
HtrA1 Structures
the occluded S1 pocket. However, 10 mM DFP inactivates 90%
of HtrA1 in amere 35min, which is only slightly longer than the 5–
10 min for inactivation of PDZ-peptide activated DegS by 10 mM
DFP (Sohn et al., 2007). Therefore, the active HtrA1 conformer
must be frequently available for DFP interaction. Second, the
mild detergent b-octylglucoside was observed to distort an
a-helical portion at the active site, including the catalytic
His220, suggesting a pronounced conformational lability of the
active site. Furthermore, we and others have observed apo
forms of trypsin-homology proteases with active-site regions in
incompetent active-site conformations, quite different from
those seen in complex with pseudosubstrate inhibitors (Carvalho
et al., 2002; Fujinaga and James, 1987; Gohara and Di Cera,
2011; Huntington, 2008; Johnson et al., 2005; Shia et al., 2005;
Spraggon et al., 2009). Five of the six crystallographically
unique HtrA1 protease domain structures fall into this category.
They share certain features with other apo forms of trypsin-
homology proteases, such as a distorted loop2 and blockade
of the S1 pocket. Therefore, the observed HtrA1 conformations
are best explained by an equilibrium between active (E) and
inactive states (E*), the latter of which may, for HtrA1, formally
include more than one conformation. Such an equilibrium is
a regulatory mechanism commonly observed in trypsin-like
serine proteases (Gohara and Di Cera, 2011). A comprehensive
survey of trypsin homology proteases found that E and E* are
associated with two distinct conformational states of loop2
(220 loop in chymotrypsinogen numbering; Gohara and Di
Cera, 2011), similar to those observed in apo forms of HtrA1
and the related DegS (Sohn et al., 2010). Thus, in following the
allosteric equilibrium scheme of trypsin-like serine proteases
(Gohara and Di Cera, 2011), we propose that the substrate (S)
simply samples the active HtrA1 conformer (HtrA1) out of an
equilibrium with inactive conformers (HtrA1*) to form the enzy-
me:substrate complex (HtrA1:S) for catalysis and product (P)
formation:
HtrA1�4HtrA14+S
HtrA1 : S/HtrA1+P:
In this sense, we view HtrA1 as being subject to ‘‘conforma-
tional selection’’ by substrate (Boehr et al., 2009) rather than
‘‘substrate-induced remodeling,’’ although there may be limits
1048 Structure 20, 1040–1050, June 6, 2012 ª2012 Elsevier Ltd All ri
to the substrate and enzyme concentrations over which such
a distinction is useful (Hammes et al., 2009).
Our results suggest that neither the PDZ nor the N-domain of
HtrA1 influence the equilibrium state, raising the possibility that
there exists a yet unknown allosteric regulator of HtrA1 catalytic
activity. The substantial intrinsic catalytic activity of HtrA1 points
toward the possibility that such a putative regulator may serve
as an allosteric switch by stabilizing the inactive rather than the
active state of HtrA1. The HtrA1* 4 HtrA1 equilibrium also
suggests potential therapeutic approaches aimed at HtrA1
neutralization, in that stabilization of HtrA1* may fully inhibit
enzyme activity by a competitive inhibition mechanism (Gohara
and Di Cera, 2011).
Human HtrA1 is linked to a variety of human diseases. In the
studies described herein, we have sought structural and
biochemical insights to its function so that its role in disease
may be better understood. We have determined a low-resolution
envelope for the structure of intact full-length HtrA1, to our
knowledge the first structure of any ‘‘N-domain,’’ and provided
clear evidence that its enzyme active site can exist in a conforma-
tion competent for substrate binding even in the absence of
substrate. Our functional studies have shown that the N-domain
neither regulates HtrA1 enzyme activity nor interacts with IGF or
proteases. As such, our current understanding of HtrA1 function
and regulation is still insufficient to understand its role in patho-
logical, as well as physiological, processes. Among the most
glaring, remaining unknowns about HtrA1 is the means by which
its protease activity is regulated, which would be helpful in
understanding its role in disease.
EXPERIMENTAL PROCEDURES
Experimental procedures, covering crystallographic analysis of the N-domain,
HtrA1_Cat and HtrA1_Cat(S328A), SAXS analysis of HtrA1, sources of
reagents, production of HtrA1 proteins, synthesis of fluorescence-quenched
peptide substrates, enzyme assays, and measurements using biolayer inter-
ferometry appear in the Supplemental Experimental Procedures.
ACCESSION NUMBERS
Coordinates and structure factors for crystal structures of the N-domain,
HtrA1_Cat, and HtrA1_Cat(S328S) have been deposited at the Protein Data
Bank with accession codes 3TJQ, 3TJN, and 3TJO, respectively, for imme-
diate release upon publication.
SUPPLEMENTAL INFORMATION
Supplemental Information includes one table, five figures, Supplemental
Results, and Supplemental Experimental Procedures and can be found with
this article online at doi:10.1016/j.str.2012.03.021.
ACKNOWLEDGMENTS
We thank J. Corn for assistance in building models of the N-domain for use
as molecular replacement probes, Y. Franke for baculovirus constructs,
K. Mortara for insect cell expression, A. Schoeffler for help with SAXS analysis,
P. Liu for mass spectra, and K. Stengel, J. Murray, and staff at MXPress for
collection of diffraction data. Portions of this research were conducted at
the Advanced Light Source operated on behalf of the U.S. Department of
Energy, Office of Basic Energy Sciences. At the time this work was conducted,
all authors were employees of Genentech, Inc.
ghts reserved

Structure
HtrA1 Structures
Received: December 2, 2011
Revised: March 15, 2012
Accepted: March 30, 2012
Published online: May 10, 2012
REFERENCES
Arnoux, B., Merigeau, K., Saludjian, P., Norris, F., Norris, K., Bjørn, S., Olsen,
O., Petersen, L., and Ducruix, A. (1995). The 1.6 A structure of Kunitz-type
domain from the alpha 3 chain of human type VI collagen. J. Mol. Biol. 246,
609–617.
Bach, L.A., Headey, S.J., and Norton, R.S. (2005). IGF-binding proteins—the
pieces are falling into place. Trends Endocrinol. Metab. 16, 228–234.
Boehr, D.D., Nussinov, R., andWright, P.E. (2009). The role of dynamic confor-
mational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796.
Carvalho, A.L., Sanz, L., Barettino, D., Romero, A., Calvete, J.J., and Romao,
M.J. (2002). Crystal structure of a prostate kallikrein isolated from stallion
seminal plasma: a homologue of human PSA. J. Mol. Biol. 322, 325–337.
Chamberland, A., Wang, E., Jones, A.R., Collins-Racie, L.A., LaVallie, E.R.,
Huang, Y., Liu, L., Morris, E.A., Flannery, C.R., and Yang, Z. (2009).
Identification of a novel HtrA1-susceptible cleavage site in human aggrecan:
evidence for the involvement of HtrA1 in aggrecan proteolysis in vivo.
J. Biol. Chem. 284, 27352–27359.
Clausen, T., Southan, C., and Ehrmann, M. (2002). The HtrA family of prote-
ases: implications for protein composition and cell fate. Mol. Cell 10, 443–455.
Clausen, T., Kaiser, M., Huber, R., and Ehrmann, M. (2011). HTRA proteases:
regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 12,
152–162.
Dewan, A., Liu, M., Hartman, S., Zhang, S.S., Liu, D.T., Zhao, C., Tam, P.O.,
Chan, W.M., Lam, D.S., Snyder, M., et al. (2006). HTRA1 promoter polymor-
phism in wet age-related macular degeneration. Science 314, 989–992.
Fujinaga, M., and James, M.N. (1987). Rat submaxillary gland serine protease,
tonin. Structure solution and refinement at 1.8 A resolution. J. Mol. Biol. 195,
373–396.
Gohara, D.W., and Di Cera, E. (2011). Allostery in trypsin-like proteases
suggests new therapeutic strategies. Trends Biotechnol. 29, 577–585.
Grau, S., Richards, P.J., Kerr, B., Hughes, C., Caterson, B., Williams, A.S.,
Junker, U., Jones, S.A., Clausen, T., and Ehrmann, M. (2006). The role of
human HtrA1 in arthritic disease. J. Biol. Chem. 281, 6124–6129.
Hadfield, K.D., Rock, C.F., Inkson, C.A., Dallas, S.L., Sudre, L., Wallis, G.A.,
Boot-Handford, R.P., and Canfield, A.E. (2008). HtrA1 inhibits mineral deposi-
tion by osteoblasts: requirement for the protease and PDZ domains. J. Biol.
Chem. 283, 5928–5938.
Hammes, G.G., Chang, Y.-C., and Oas, T.G. (2009). Conformational selection
or induced fit: a flux description of reaction mechanism. Proc. Natl. Acad. Sci.
USA 106, 13737–13741.
Hara, K., Shiga, A., Fukutake, T., Nozaki, H., Miyashita, A., Yokoseki, A.,
Kawata, H., Koyama, A., Arima, K., Takahashi, T., et al. (2009). Association
of HTRA1 mutations and familial ischemic cerebral small-vessel disease.
N. Engl. J. Med. 360, 1729–1739.
Hasselblatt, H., Kurzbauer, R., Wilken, C., Krojer, T., Sawa, J., Kurt, J., Kirk, R.,
Hasenbein, S., Ehrmann, M., and Clausen, T. (2007). Regulation of the sigmaE
stress response by DegS: how the PDZ domain keeps the protease inactive in
the resting state and allows integration of different OMP-derived stress signals
upon folding stress. Genes Dev. 21, 2659–2670.
Hecht, H.J., Szardenings, M., Collins, J., and Schomburg, D. (1991). Three-
dimensional structure of the complexes between bovine chymotrypsinogen
A and two recombinant variants of human pancreatic secretory trypsin inhib-
itor (Kazal-type). J. Mol. Biol. 220, 711–722.
Holbourn, K.P., Acharya, K.R., and Perbal, B. (2008). The CCN family of
proteins: structure-function relationships. Trends Biochem. Sci. 33, 461–473.
Hou, J., Clemmons, D.R., and Smeekens, S. (2005). Expression and character-
ization of a serine protease that preferentially cleaves insulin-like growth factor
binding protein-5. J. Cell. Biochem. 94, 470–484.
Structure 20, 10
Hu, S.I., Carozza, M., Klein, M., Nantermet, P., Luk, D., and Crowl, R.M. (1998).
Human HtrA, an evolutionarily conserved serine protease identified as a differ-
entially expressed gene product in osteoarthritic cartilage. J. Biol. Chem. 273,
34406–34412.
Huntington, J.A. (2008). How Na+ activates thrombin—a review of the func-
tional and structural data. Biol. Chem. 389, 1025–1035.
Hwa, V., Oh, Y., and Rosenfeld, R.G. (1999). The insulin-like growth factor-
binding protein (IGFBP) superfamily. Endocr. Rev. 20, 761–787.
Johnson, D.J., Adams, T.E., Li, W., and Huntington, J.A. (2005). Crystal
structure of wild-type human thrombin in the Na+-free state. Biochem. J.
392, 21–28.
Kato, M.V. (2000). A secreted tumor-suppressor, mac25, with activin-binding
activity. Mol. Med. 6, 126–135.
Kazal, L.A., Spicer, D.S., and Brahinsky, R.A. (1948). Isolation of a crystalline
trypsin inhibitor-anticoagulant protein from pancreas. J. Am. Chem. Soc. 70,
3034–3040.
Kim, D.Y., and Kim, K.K. (2005). Structure and function of HtrA family proteins,
the key players in protein quality control. J. Biochem. Mol. Biol. 38, 266–274.
Kim, H.S., Nagalla, S.R., Oh, Y., Wilson, E., Roberts, C.T., Jr., and Rosenfeld,
R.G. (1997). Identification of a family of low-affinity insulin-like growth factor
binding proteins (IGFBPs): characterization of connective tissue growth
factor as a member of the IGFBP superfamily. Proc. Natl. Acad. Sci. USA
94, 12981–12986.
Krojer, T., Sawa, J., Schafer, E., Saibil, H.R., Ehrmann, M., and Clausen, T.
(2008). Structural basis for the regulated protease and chaperone function of
DegP. Nature 453, 885–890.
Kunitz, M., and Northrop, J.H. (1936). Isolation from beef pancreas of
crystalline trypsinogen, trypsin, a trypsin inhibitor, and an inhibitor-trypsin
compound. J. Gen. Physiol. 19, 991–1007.
Lassalle, P., Molet, S., Janin, A., Heyden, J.V., Tavernier, J., Fiers, W., Devos,
R., and Tonnel, A.B. (1996). ESM-1 is a novel human endothelial cell-specific
molecule expressed in lung and regulated by cytokines. J. Biol. Chem. 271,
20458–20464.
Lu, S.M., Lu, W., Qasim, M.A., Anderson, S., Apostol, I., Ardelt, W., Bigler, T.,
Chiang, Y.W., Cook, J., James, M.N., et al. (2001). Predicting the reactivity of
proteins from their sequence alone: Kazal family of protein inhibitors of serine
proteinases. Proc. Natl. Acad. Sci. USA 98, 1410–1415.
Martins, L.M., Turk, B.E., Cowling, V., Borg, A., Jarrell, E.T., Cantley, L.C., and
Downward, J. (2003). Binding specificity and regulation of the serine protease
and PDZ domains of HtrA2/Omi. J. Biol. Chem. 278, 49417–49427.
Meltzer, M., Hasenbein, S., Hauske, P., Kucz, N., Merdanovic, M., Grau, S.,
Beil, A., Jones, D., Krojer, T., Clausen, T., et al. (2008). Allosteric activation
of HtrA protease DegP by stress signals during bacterial protein quality
control. Angew. Chem. Int. Ed. Engl. 47, 1332–1334.
Murwantoko, Y., Yano, M., Ueta, Y., Murasaki, A., Kanda, H., Oka, C., and
Kawaichi, M. (2004). Binding of proteins to the PDZ domain regulates proteo-
lytic activity of HtrA1 serine protease. Biochem. J. 381, 895–904.
Oh, Y., Nagalla, S.R., Yamanaka, Y., Kim, H.S., Wilson, E., and Rosenfeld, R.G.
(1996). Synthesis and characterization of insulin-like growth factor-binding
protein (IGFBP)-7. Recombinant human mac25 protein specifically binds
IGF-I and -II. J. Biol. Chem. 271, 30322–30325.
Oka, C., Tsujimoto, R., Kajikawa, M., Koshiba-Takeuchi, K., Ina, J., Yano, M.,
Tsuchiya, A., Ueta, Y., Soma, A., Kanda, H., et al. (2004). HtrA1 serine protease
inhibits signaling mediated by Tgfbeta family proteins. Development 131,
1041–1053.
Shia, S., Stamos, J., Kirchhofer, D., Fan, B., Wu, J., Corpuz, R.T., Santell, L.,
Lazarus, R.A., and Eigenbrot, C. (2005). Conformational lability in serine
protease active sites: structures of hepatocyte growth factor activator
(HGFA) alone and with the inhibitory domain from HGFA inhibitor-1B. J. Mol.
Biol. 346, 1335–1349.
Sitar, T., Popowicz, G.M., Siwanowicz, I., Huber, R., and Holak, T.A. (2006).
Structural basis for the inhibition of insulin-like growth factors by insulin-like
growth factor-binding proteins. Proc. Natl. Acad. Sci. USA 103, 13028–13033.
40–1050, June 6, 2012 ª2012 Elsevier Ltd All rights reserved 1049

Structure
HtrA1 Structures
Sohn, J., Grant, R.A., and Sauer, R.T. (2007). Allosteric activation of DegS,
a stress sensor PDZ protease. Cell 131, 572–583.
Sohn, J., Grant, R.A., and Sauer, R.T. (2010). Allostery is an intrinsic property of
the protease domain of DegS: implications for enzyme function and evolution.
J. Biol. Chem. 285, 34039–34047.
Spraggon, G., Hornsby, M., Shipway, A., Tully, D.C., Bursulaya, B., Danahay,
H., Harris, J.L., and Lesley, S.A. (2009). Active site conformational changes of
prostasin provide a newmechanism of protease regulation by divalent cations.
Protein Sci. 18, 1081–1094.
Tian, M., and Kamoun, S. (2005). A two disulfide bridge Kazal domain from
Phytophthora exhibits stable inhibitory activity against serine proteases of
the subtilisin family. BMC Biochem. 6, 15.
Tocharus, J., Tsuchiya, A., Kajikawa, M., Ueta, Y., Oka, C., and Kawaichi, M.
(2004). Developmentally regulated expression of mouse HtrA3 and its role as
an inhibitor of TGF-beta signaling. Dev. Growth Differ. 46, 257–274.
Truebestein, L., Tennstaedt, A., Monig, T., Krojer, T., Canellas, F., Kaiser, M.,
Clausen, T., and Ehrmann, M. (2011). Substrate-induced remodeling of the
active site regulates humanHTRA1 activity. Nat. Struct. Mol. Biol. 18, 386–388.
Tsuchiya, A., Yano, M., Tocharus, J., Kojima, H., Fukumoto, M., Kawaichi, M.,
and Oka, C. (2005). Expression of mouse HtrA1 serine protease in normal bone
1050 Structure 20, 1040–1050, June 6, 2012 ª2012 Elsevier Ltd All ri
and cartilage and its upregulation in joint cartilage damaged by experimental
arthritis. Bone 37, 323–336.
van de Locht, A., Lamba, D., Bauer, M., Huber, R., Friedrich, T., Kroger, B.,
Hoffken, W., and Bode, W. (1995). Two heads are better than one: crystal
structure of the insect derived double domain Kazal inhibitor rhodniin in
complex with thrombin. EMBO J. 14, 5149–5157.
Vorwerk, P., Hohmann, B., Oh, Y., Rosenfeld, R.G., and Shymko, R.M. (2002).
Binding properties of insulin-like growth factor binding protein-3 (IGFBP-3),
IGFBP-3 N- and C-terminal fragments, and structurally related proteins
mac25 and connective tissue growth factor measured using a biosensor.
Endocrinology 143, 1677–1685.
Wilken, C., Kitzing, K., Kurzbauer, R., Ehrmann, M., and Clausen, T. (2004).
Crystal structure of the DegS stress sensor: How a PDZ domain recognizes
misfolded protein and activates a protease. Cell 117, 483–494.
Yang, Z., Camp, N.J., Sun, H., Tong, Z., Gibbs, D., Cameron, D.J., Chen, H.,
Zhao, Y., Pearson, E., Li, X., et al. (2006). A variant of the HTRA1 gene
increases susceptibility to age-related macular degeneration. Science 314,
992–993.
Zumbrunn, J., and Trueb, B. (1996). Primary structure of a putative serine
protease specific for IGF-binding proteins. FEBS Lett. 398, 187–192.
ghts reserved





























