Embed Size (px)
Citation preview

Spectroscopic elucidation of energy transfer in hybridinorganic–biological organisms for solar-to-chemical productionNikolay Kornienkoa,1, Kelsey K. Sakimotoa,1, David M. Herlihya, Son C. Nguyena, A. Paul Alivisatosa,b,c,Charles. B. Harrisa, Adam Schwartzbergd, and Peidong Yanga,b,c,2
aDepartment of Chemistry, University of California, Berkeley, CA 94720; bDepartment of Materials Science and Engineering, University of California,Berkeley, CA 94720; cKavli Energy NanoSciences Institute, Berkeley, CA 94720; and dMolecular Foundry, Lawrence Berkeley National Laboratory, Berkeley,CA 94720
Edited by Thomas E. Mallouk, The Pennsylvania State University, University Park, PA, and approved August 30, 2016 (received for review June 28, 2016)
The rise of inorganic–biological hybrid organisms for solar-to-chemicalproduction has spurred mechanistic investigations into the dynamicsof the biotic–abiotic interface to drive the development of next-generation systems. The model system, Moorella thermoacetica–cadmium sulfide (CdS), combines an inorganic semiconductornanoparticle light harvester with an acetogenic bacterium to drivethe photosynthetic reduction of CO2 to acetic acid with high effi-ciency. In this work, we report insights into this unique electro-trophic behavior and propose a charge-transfer mechanism fromCdS to M. thermoacetica. Transient absorption (TA) spectroscopyrevealed that photoexcited electron transfer rates increase withincreasing hydrogenase (H2ase) enzyme activity. On the same timescale as the TA spectroscopy, time-resolved infrared (TRIR) spectros-copy showed spectral changes in the 1,700–1,900-cm−1 spectral re-gion. The quantum efficiency of this system for photosyntheticacetic acid generation also increased with increasing H2ase activityand shorter carrier lifetimes when averaged over the first 24 h ofphotosynthesis. However, within the initial 3 h of photosynthesis,the rate followed an opposite trend: The bacteria with the lowestH2ase activity photosynthesized acetic acid the fastest. These resultssuggest a two-pathway mechanism: a high quantum efficiencycharge-transfer pathway to H2ase generating H2 as a molecular in-termediate that dominates at long time scales (24 h), and a directenergy-transducing enzymatic pathway responsible for acetic acidproduction at short time scales (3 h). This work represents a prom-ising platform to utilize conventional spectroscopic methodology toextract insights from more complex biotic–abiotic hybrid systems.
energy conversion | spectroscopy | CO2 reduction | biohybrid systems |catalysis
The sluggish transduction of solar energy into chemical bondsthrough natural photosynthesis has strangled our efforts to
harvest the full bounty of the sun’s energy (1). We have temporarilysidestepped this limitation by tapping into large reserves of car-bonaceous energy to drive an exponential growth in manufacturing,agriculture, urbanization, and population. However, the growingscarcity of petrochemicals has called for a return to photosynthesis––rather a new form of photosynthesis capable of keeping pace withmodern society (2, 3). As a sign of progress, inorganic semicon-ductor light harvesters now routinely surpass the efficiency ofplants (4). In contrast, synthetic catalysts still struggle to replicatethe complex C–C bond formation of biology (5, 6). Significantstrides toward comprehensive solar-to-chemical production havebeen demonstrated through several inorganic–biological hybridsystems combining inorganic semiconductor light harvesters withmicrobial CO2 reduction (7–9). Recently, we have reported theself-photosensitized hybrid bacterium, Moorella thermoacetica–cadmium sulfide (M. thermoacetica-CdS), which photosynthesizesacetic acid from CO2 via bioprecipitated CdS nanoparticles (10).Although e− transfer from electrodes to bacteria has been
demonstrated across several genera, the mechanism remains in
contention (11). Spectroscopic investigations of bacterium-to-electrode anodic e− transfer in electrogenic microbial fuel cellshave implicated cytochrome-mediated mechanisms (12). How-ever, analogous studies of semiconductor-to-bacterium cathodice− transfer in electrotrophic organisms have remained sparse.Electron transfer first to membrane-bound or extracellularH2ase to generate molecular H2 as an intermediate followed byuptake into the native acetogenic Wood–Ljungdahl pathway(WLP) has been speculated or inferred (13–15). Still, detailedspectroscopic characterization has remained elusive due to thedifficulty of adapting previous techniques to solid electrodeplatforms. In contrast, our model system, M. thermoacetica-CdS,as a translucent colloidal suspension in which an optically ad-dressable CdS nanoparticle generates photoelectrons, elimi-nates the need for opaque electrodes, thereby enabling existingtransmittance-based spectroscopies in uncovering the molecularbasis of this charge-transfer mechanism (Fig. 1A). Here, we pre-sent transient absorption (TA) and time-resolved infrared (TRIR)spectroscopies correlated with biochemical activity to propose amodel of the dynamics of inorganic–biological charge and en-ergy transfer.Upon photoexcitation of CdS, cysteine (Cys) oxidation to cys-
tine (CySS) and H+ quenches the valence band h+, while theconduction band e− may transfer to membrane-bound proteins or
Significance
Solar-powered chemical production from CO2 promises to alle-viate petrochemical consumption. Hybrid systems of an in-organic semiconductor light harvester and a microbial catalystoffer a viable way forward. Whereas a number of such systemshave been described, the semiconductor-to-bacterium electrontransfer mechanism remains largely unknown, limiting rationalapproaches to improving their performance. In this work, welook at how a semiconductor nanoparticle-sensitized bacteriumtransforms CO2 and sunlight into acetic acid, a known precursorfor fuels, food, pharmaceuticals, and polymers. Using time-resolved spectroscopy and biochemical analysis, we concludethat multiple pathways facilitate electron and light energytransfer from semiconductor to bacterium. This foundationalstudy enables future investigation, understanding, and im-provement of complex biotic–abiotic hybrid systems.
Author contributions: N.K., K.K.S., A.P.A., C.B.H., A.S., and P.Y. designed research; N.K., K.K.S.,D.M.H., S.C.N., and A.P.A. performed research; N.K. and K.K.S. contributed new reagents/analytic tools; N.K. and K.K.S. analyzed data; and N.K. and K.K.S. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1N.K. and K.K.S contributed equally to this work.2To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1610554113/-/DCSupplemental.
11750–11755 | PNAS | October 18, 2016 | vol. 113 | no. 42 www.pnas.org/cgi/doi/10.1073/pnas.1610554113
Dow
nloa
ded
by g
uest
on
May
23,
202
0

soluble e− acceptors. Genomic mining, enzymology, and thermo-dynamic comparison of protein redox potentials have proposedseveral viable e− transfer pathways (Fig. 1B) (13, 14, 16, 17).Membrane-bound NiFe H2ases may play a significant role as H2
directly feeds into the WLP. Demonstrations of direct e− transferbetween metal chalcogenide nanoparticles and purified H2asesin vitro lend credibility to this pathway’s existence in complexwhole cells (18–20). Alternative pathways have implicated e−
transfer first to membrane-bound cytochromes, ferredoxin (Fd),flavoproteins (Fp), and menaquinones (MK) (13, 14). Althoughthese pathways generate reducing equivalents, implicitly, theymust also couple to the formation of a H+ gradient to facilitateATP synthesis by ATPase. The generation of this proton motiveforce, either through the transmembrane Ech complex or simply
through surface proximal Cys oxidation, may be crucial to elec-trotrophic behavior, as a related acetogen, Acetobacterium woodii,which instead uses a Na+ motive force, notably cannot engage inelectrotrophy (17, 21).
Results and DiscussionBiochemical Characterization. To investigate the possibility of H2asemediated e− transfer,M. thermoacetica-CdS was incubated from 0 to48 h on H2 (H2:CO2, 80:20), henceforth referred to as H2 incubated)or glucose (25 mM) to vary the expression and activity of H2ase.Activity was assayed photometrically by standard benzyl viologen(BV) reduction in the presence of H2 (Fig. 2A and Fig. S1) (22, 23).Consistent with previous characterizations of M. thermoacetica,
H2 oxidation–BV reduction activity increased under increasing H2
CB
VB
H2ase2H+
H2
CH2-THF-reductase
CH3-THFCH2-THF
ATPaseADP + Pi
ATPnH+
EchComplexH+
2H+
H2Fd2-
Fd
2Cys
CySS + 2H+CdS
e-
h+
MKH2↕
MK
Fd,Fp
Cyt. b560H+
A B
2CO2 + 8H+ + 8e-
CH3COOH + 2H2O
M. thermoacetica-CdS
TA
TRIR
Fig. 1. Schematic of M. thermoacetica-CdS photosynthetic charge transfer. (A) Visible light excitation of optically addressable CdS photosensitizing nano-particles enables photosynthetic acetic acid production from CO2, as well as characterization by TA and TRIR spectroscopy. (B) Potential e− pathways inM. thermoacetica-CdS.
012345678
Act
ivity
(×10
-15 a
tm-1 s
-1 c
ell-1
)
0 3 6 12 24 48Time H2 Incubation (hrs)
A
0.00
0.05
0.10
0.15
0.20
0.25
Rat
e (m
M C
H3C
OO
H h
r-1)
0 3 6 12 24 48Time H2 Incubation (hrs)
Initial 3 hrs ofPhotosynthesis
B
0.00
0.01
0.02
0.03
0.04
0.05
0.06
Rat
e (m
M C
H3C
OO
H h
r-1)
0 12 24 36Time H2 Incubation (hrs)
Initial 24 hrs ofPhotosynthesis
C
Fig. 2. Biochemical assays of M. thermoacetica-CdS. (A) H2ase activity with varying incubation time under H2. See Materials and Methods for details ofquantification. (B) CO2-to-acetic acid conversion rates averaged over the first 3 h of photosynthesis show a decreasing trend with increasing H2 incubationtime. For comparison, 24-h glucose grown cells had a measured rate of 0.47 ± 0.15 mM h−1. (C) CO2-to-acetic acid conversion rates averaged over the first 24 hof photosynthesis show an increasing trend with increasing H2 incubation time. Error bars represent the SD obtained from triplicate experiments.
Kornienko et al. PNAS | October 18, 2016 | vol. 113 | no. 42 | 11751
CHEM
ISTR
YBIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
Dow
nloa
ded
by g
uest
on
May
23,
202
0

incubation time, presumably through increased H2ase expression.Comparison of M. thermoacetica-CdS incubated for 24 h underglucose and H2 showed H2ase activity of 6.56 ± 0.92 × 10−15 (atm scell)−1 and 1.85 ± 8.39 × 10−17·(atm s cell)−1, respectively.To correlate enzyme activity with photosynthetic performance,
the same samples were subjected to simulated solar illumination(0.5% sun, AM1.5G) and analyzed for acetic acid production.During the initial 3 h of photosynthesis, the rate of CO2 reductionanticorrelated with H2ase activity, with the 0-h (previously grownon glucose only) incubated sample showing the highest activity(0.23 ± 0.02 mM h−1). The 24-h glucose incubated sample pro-duced acetic acid at 0.47 ± 0.15 mM h−1, one order of magnitudefaster than that of the 24-h H2 incubated sample (0.052 ±0.009 mM h−1). However, at longer illumination times, H2 in-cubated samples demonstrated the opposite trend. When averagedover 24 h of illumination, photosynthesis rates increased with in-creasing incubation time in H2 from 0.010 ± 0.001 mM h−1 (7.1 ±4.5% quantum yield of photons above CdS bandgap) at 0-h H2incubation to 0.047 ± 0.005 mM h−1 (32 ± 4% quantum yield) at36-h H2 incubation (Fig. 2C). We note that the rate of acetic acidgeneration is higher within the first 3 h than the 24-h average. The3-h rates are also not stoichiometric (e.g., 0-h H2 sample has anapparent quantum yield of 160 ± 10%), suggesting that despiteextensive washing and removal of residual glucose and H2, somereduced intermediate carries over from the preincubation period.The observed trend indicates that lower H2ase activity cells moreeffectively transfer CdS e− to terminally reduce these intermediates,perhaps through a later set of enzymes in the WLP. In contrast, the24-h averaged data agree in quantum yield with previous reportsshowing no such residual intermediates (10). These contrastingresults suggest two competing charge-transfer mechanisms: a non-H2ase mediated pathway dominant at short time scales (<3 h) anda H2ase mediated pathway dominant at long time scales (∼24 h).
TA Spectroscopy. To delve deeper into the molecular basis of thesetwo mechanisms, we turned to time-resolved spectroscopic tech-niques for a dynamic understanding of the activity trends. TAdecay kinetics followed the rate of photogenerated e− leaving CdSfor various preparations of M. thermoacetica-CdS (Fig. 3 and Fig.S2A). A transient bleach from 440 to 490 nm matched typicalspectra observed with CdS e− acceptor systems and was not ob-served in CdS-free M. thermoacetica. The spectrum of chemicallyprecipitated CdS alone decayed much slower than M. thermoace-tica-CdS, indicating that rapid quenching may result from aproximal e− acceptor. The H2 incubated M. thermoacetica-CdSdisplayed even faster decay kinetics than the glucose analog,correlating well with the higher H2ase activity. These observationspoint to likely either faster e− transfer to an acceptor site or moree− acceptors available in H2 incubated bacteria (20). Fitting each
of the TA data sets to a triexponential decay revealed three life-times: a fast component in the range of 2–10 ps (τ1), a longercomponent in the range of 20–80 ps (τ2), and an even longercomponent in the range of several hundred picoseconds (τ3) (TableS1). A multiexponential decay unsurprisingly indicates severalprocesses at play in the complex M. thermoacetica-CdS hybrids.Rapid picosecond decays were previously measured with colloidalCdS that featured molecular acceptors with fast e− transfer behavior(24–26). As hot e− relaxation in Cd-chalcogenide quantum dotsoccurs in the subpicosecond regime, this process does not likelycontribute to the TA kinetics in the measured time scale (27).However, previous studies of FeFe H2ase-CdS constructs reportedTA lifetimes in the range of 100 ns, significantly longer than thedata presented here (19, 20). The discrepancy may be attributed tothe presence of surface ligands (not present in M. thermoacetica-CdS) which present a charge-transfer barrier, differences in CdS-H2ase spatial proximity, solvent effects and reorganization energies,H-bonding networks, and the relatively impaired functionality ofpurified enzymes under in vitro vs. in vivo conditions (28–30). Amolecular carrier may also be at play, accepting charge and sub-sequently transferring it to H2ase, among other potential charge-transfer pathways.The differences in lifetimes between cell-free CdS, H2 incubated,
and glucose incubated cells suggests the importance of H2ase ex-pression in e− transfer kinetics. To correlate with the above H2aseactivity (Fig. 2A), the TA lifetimes of the H2 incubated time serieswere measured (Fig. 3 B and C). Both the fast τ1, τ2 and theirweighted average showed decreasing lifetimes with increasingH2ase activity, suggesting that the fast e− transfer kinetics was dueto an increase in H2ase e− acceptor sites or molecular carrierswhose appearance is correlated with H2ase expression. Inhibitionof the H2ase active site (H cluster) with CO did not significantlychange the TA kinetics, similar to previous works, in which e−
transfer initially proceeds through the FeS cluster chain rather thandirectly to the NiFe active site (Fig. S2B) (20, 31, 32).
TRIR Spectroscopy. Whereas kinetically efficient e− transfer to amembrane-bound H2ase may explain the increasing photosyn-thesis rates at long time scales (Fig. 2C), TRIR helped determinethe basis of the decreasing rates seen at short time scales (Fig. 2B).We observed changes in the 1,760–1,880-cm−1 spectral window,
roughly corresponding to the vibrational range of CO and CNdouble and triple bonds, among other IR active modes charac-teristic of amino acid residues (Fig. 4) (33–35). The peaks decayedon the same time scale as the TA signal (Figs. S3–S6), giving fur-ther evidence that the picosecond e− transfer resulted from amolecular, rather than purely physical, process. No significantchanges in the 1,900–2,100-cm−1 spectral window associated withthe catalytic cycle of the NiFe H2ase H cluster were observed,
τ 1 Life
time
(ps)
Time H2 Incubation (hrs)
τ2 L
ifetim
e (p
s)
B
Nor
mal
ized
Life
time
Time H2 Incubation (hrs)
C
0 10 20 30 40 50-0.2
0.0
0.2
0.4
0.6
0.8
1.0
1.2
Nor
mal
ized
Abs
.
Time (ps)
CdS only (cell free)Glucose Incubated H2 Incubated
A
0 3 6 12 24 480
5
10
15
20
01020304050607080
0 3 6 12 24 480.0
0.2
0.4
0.6
0.8
1.0
1.2
Fig. 3. TA spectroscopy of M. thermoacetica-CdS. (A) TA plots of CdS only (cell-free), 24-h glucose incubated, and 24-h H2 incubated. (B) Trends of expo-nential τ1 and τ2 lifetimes with increasing incubation time under H2. (C) Weighted averages of the normalized τ1 and τ2 lifetimes with increasing incubationtime under H2. Error bars represent the SE associated with the exponential fitting.
11752 | www.pnas.org/cgi/doi/10.1073/pnas.1610554113 Kornienko et al.
Dow
nloa
ded
by g
uest
on
May
23,
202
0

indicating that a significant quantity of photogenerated e− was nottransferred to the active site within this timeframe (18, 36, 37).Whereas the complexity of the whole-cell M. thermoacetica-CdSsystem renders unambiguous peak assignment beyond the scopeof these initial results, careful construction of controls yieldedvaluable insight into the nature of these vibrational changes.H2 and glucose incubated samples yielded different TRIR
spectral responses. Whereas several bleach features in the range of1,760–1,820 cm−1 on the picosecond time regime appeared for theH2 incubated sample (Fig. 4A and Fig. S3), in the same spectralwindow, glucose incubated samples showed long-lived peakgrowth (Fig. 4B). These features may indicate H2ase mediatedcharge transfer, with the differential response seen in glucosepotentially representing an alternate pathway. In the region of1,810–1,880 cm−1, signal bleaches of similar time scales appearedin both H2 and glucose incubated samples, implicating a similar e−
transfer pathway in both systems (Fig. 4 C and D). However,differences in their kinetic evolution point toward different utili-zation of this shared mechanism. Whereas the pair of bands at
1,823 and 1,827 cm−1 retained a roughly 1:1 ratio for the H2 in-cubated sample, in the glucose incubated sample, the 1,823-cm−1
feature did not grow in until 1–2 ps after the pump excitation, anddecayed back to zero faster than the 1,827-cm−1 feature. We con-clude that these spectral responses point toward different pre-dominant e− transfer pathways in H2 vs. glucose incubated samples.Further elucidation of the e− transfer pathways can be accomplishedthrough fluorescent labeling studies, 2D electron spectroscopy, andsynthetic biology (38–40).
Proposed e− Transfer Mechanism of M. thermoacetica-CdS. Takentogether, the biochemical, TAS, and TRIR data suggest twocompeting pathways for e− transfer within M. thermoacetica-CdS(Fig. 5). Initially, low H2ase expression likely favors e− injection toa membrane-bound e− acceptor (Fd, Fp, cytochrome, MK) thatgenerates a proton motive force for ATP generation, as well aspotentially directly reduces CH2–THF for the final stages of theWLP, using up any accumulated intermediates. However, slowercharge transfer leads to poorer quantum efficiency, likely due to e−–h+
1760 1770 1780 1790 1800 1810 1820Wavenumber (cm-1)
0
Rel
ativ
e In
tens
ity
0.25 ps0.5 ps1.75 ps4 ps8 ps15 ps50 ps100 ps
1760 1770 1780 1790Wavenumber (cm-1)
0
Rel
ativ
e In
tens
ity
0.25 ps0.75 ps1.25 ps1.75 ps2.75 ps
4 ps8 ps15.25 ps25 ps50 ps
A B
1810 1820 1830 1840 1850 1860 1870 1880
0
Rel
ativ
e In
tens
ity
Wavenumber (cm-1)
D1810 1820 1830 1840 1850 1860 1870 1880
0
Rel
ativ
e In
tens
ity
Wavenumber (cm-1)
C
0.25 ps0.5 ps1.75 ps4 ps8 ps15 ps50 ps100 ps
0.25 ps0.75 ps1.25 ps1.75 ps2.75 ps
4 ps8 ps15.25 ps25 ps50 ps
Fig. 4. TRIR spectra of M. thermoacetica-CdS. (A and C) TRIR of 24-h H2 incubated M. thermoacetica-CdS showing bleaching of several peaks in the region ofC, N, and O double and triple bonds. (B and D) TRIR of 24-h glucose incubated M. thermoacetica-CdS.
Kornienko et al. PNAS | October 18, 2016 | vol. 113 | no. 42 | 11753
CHEM
ISTR
YBIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
Dow
nloa
ded
by g
uest
on
May
23,
202
0

recombination losses. Additionally, this pathway cannot generatehigh energy reducing equivalents [H2, NAD(P)H, Fd] needed forthe first reductive steps of the full WLP. As H2ase expressionincreases, charge-transfer kinetics favors e− transfer to a membrane-bound H2ase to generate molecular H2. The possibility also existsof electron transfer to a molecular acceptor, which subsequentlytransfers an electron to H2ase. Whereas this H2ase mediatedpathway may display higher quantum efficiency, the higher pho-tosynthetic rates only kick in once a significant concentration ofextra(intra)cellular H2 accumulates. This results in lower photo-synthetic rates in the first 3 h with increasing H2ase activity (e−
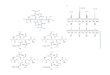
diverted from the non-H2ase pathway), but eventually higherrates after 24 h. This H2 then likely enters the normal WLP viaoxidation by the HydABC complex generating both ATP andreducing equivalents (17). Interestingly, the presence of CdSon M. thermoacetica may also induce novel, yet highly func-tional electron transfer pathways, different from what occursnaturally.
ConclusionsIn conclusion, the work here represents an initial experi-mental probe into the photosynthetic, electrotrophic behavior
of M. thermoacetica-CdS. With the careful construction of controlsand mild, biocompatible probing conditions, complex biologicalbehaviors may be studied through conventional spectroscopictechniques. Increasing H2ase activity correlated with more efficientlong-term photosynthetic rates of acetic acid generation. Evidenceprovided by TA and TRIR likewise supported the existence of acharge-transfer pathway that correlated with results from bio-chemical characterization. Our proposed two-pathway mechanismbears further investigation to probe more acutely the molecularand enzymatic basis of this biotic–abiotic charge transfer. Suchinsights will ultimately lead to a deeper understanding of theburgeoning complex nexus of inorganic materials and biologicalsystems, and provide a rational framework for the optimization anddesign of next-generation solar-to-chemical systems.
Materials and MethodsPreparation and Biochemical Characterization of M. thermoacetica-CdS.M. thermoacetica-CdS was prepared and assayed as previously describedwith modifications noted below (10). After full growth, M. thermoacetica-CdSwas centrifuged under N2 and resuspended in 20% of the original volume ofdefined photosynthesis media (DPM) with 0.1 wt % Cys·HCl and 2.5 atm H2:CO2 (80:20) or 25 mM glucose.
At various time points, aliquots of M. thermoacetica-CdS were sampledand stored at 4 °C under N2:CO2 (80:20).
BV reduction assays were conducted in a modified procedure as previouslydescribed (22). In short, 0.1 mL of M. thermoacetica-CdS was added to 5 mL of50 mM Pipes buffer (pH 7) with [BV dichloride] ranging from 0.5 to 8 mMunder a 2.5-atm H2:CO2 head space in a Hungate tube (Chemglass, Inc.). Forvarying [H2:CO2], volumes of H2:CO2 were syringe injected into a N2:CO2
headspace with [BV] = 4 mM. Kinetics of BV reduction was monitored at578 nm (Shimadzu UV3101PC UV-Vis-NIR Spectrophotometer with an in-tegrating sphere) for the initial 30 s. Rates were cell normalized by OD600
correlated with manual cell counting.H2ase activity was calculated assuming pseudo–first-order kinetics (constant
[BV] or [H2]). The activity is thus defined as mM BV formed (mM benzyl viol-ogen)-1·(atm H2)
-1 s−1 cell−1.For photosynthesis measurements, 1 mL of M. thermoacetica-CdS was di-
luted into 4 mL of DPMwith 0.1 wt % Cys·HCl. The suspension was illuminatedby a filtered 75-W xenon lamp (Newport Corp.; AM1.5G, 0.5% sun) withheating and stirring (55 °C, 150 rpm) under a 2.5-atm 80:20 N2:CO2 atmo-sphere. Time points were centrifuged to remove cells and nanoparticles andassayed by quantitative proton nuclear magnetic resonance spectroscopy.
TA Spectroscopy. Broadband TA spectra were obtained using an UltrafastSystems Helios TA system with a Coherent Libra amplified Ti:sapphire lasersystem and Coherent OPerA optical parametric amplifier (OPA) pump/probesource. Briefly, the samples were excited with ∼50-fs laser pulses generated bythe OPA at a repetition rate of 1 kHz. TA spectra were obtained by time-delaying a broadband supercontinuum probe pulse that is overlapped in timeand space with the femtosecond pump pulse. The supercontinuum is producedby focusing a small portion of the amplified laser fundamental into a sapphireplate. Multiwavelength TA spectra were recorded using dual spectrometers(signal and reference) equipped with fast Si array detectors. In all experiments,the fluence value was held constant at 0.6 μJ cm−2 to rule out effects fromexciton–exciton annihilation as a result of high-power excitation. TA datawere fit to a multiexponential decay.
TRIR Spectroscopy. TRIR spectroscopy was performed with a home-built setupdescribed elsewhere (41). A 400-nm pump pulse was used and the spectralregion of 1,700–2,800 cm−1 was probed. The sample was circulated in a N2
purged and sealed flow cell through 2 IR-transparent CaF2 windows spaced150 or 75 μm apart. To exclude the contributions of hot e− in the conductionband to the IR spectrum at early time scales as a broad positive absorbance,TRIR data were baseline-normalized by using a flat, featureless area of theTRIR spectrum as the baseline.
ACKNOWLEDGMENTS. This work was supported by the Office of Science,Office of Basic Energy Sciences, of the US Department of Energy (DOE),under Contract DE-AC02-05CH11231 (pchem). Solar-to-chemical produc-tion experiments were supported by the National Science Foundation un-der Grant DMR-1507914. TA measurements were performed in theMolecular Foundry. Work at the Molecular Foundry was supported bythe Office of Science, Office of Basic Energy Sciences, of the US DOE underContract DE-AC02-05CH11231.
CB
VB
CH2-THFreductase
CH3-THFCH2-THF
ATPaseADP + Pi
ATPnH+
2Cys
CySS + 2H+CdS
e-
h+
MKH2↕
MK
Fd,Fp
Cyt. b560H+
VVB
To Wood-LjungdahlPathway
e--h+
RecombinationLoss
Non-hydrogenase MediatedCharge Transfer
A
CB
VB
H2ase2H+
H2
EchComplexH+
2H+
H2Fd2-
Fd2Cys
CySS + 2H+CdS
e-
h+VVB
IntracellularH2 Pool
ExtracellularH2 Pool
4H+
2H2HydABC
Fd
Fd2-
NAD+
NADH
To Wood-LjungdahlPathway
Hydrogenase MediatedCharge Transfer
B
Fig. 5. Proposed dual pathway of charge and energy transfer in M. ther-moacetica-CdS. (A) The proposed non–H2ase-mediated pathway predominant inglucose incubated cells, transiently faster during the initial 3 h of photosyn-thesis. (B) Membrane-bound H2ase mediated pathway dominant in H2 in-cubated cells and photosynthetically faster at long time intervals.
11754 | www.pnas.org/cgi/doi/10.1073/pnas.1610554113 Kornienko et al.
Dow
nloa
ded
by g
uest
on
May
23,
202
0

1. Larkum AW (2010) Limitations and prospects of natural photosynthesis for bioenergyproduction. Curr Opin Biotechnol 21(3):271–276.
2. Ort DR, et al. (2015) Redesigning photosynthesis to sustainably meet global food andbioenergy demand. Proc Natl Acad Sci USA 112(28):8529–8536.
3. Faunce TA, et al. (2013) Energy and environment policy case for a global project onartificial photosynthesis. Energy Environ Sci 6(3):695–698.
4. Blankenship RE, et al. (2011) Comparing photosynthetic and photovoltaic efficienciesand recognizing the potential for improvement. Science 332(6031):805–809.
5. Kim D, Sakimoto KK, Hong D, Yang P (2015) Artificial photosynthesis for sustainablefuel and chemical production. Angew Chem Int Ed Engl 54(11):3259–3266.
6. Appel AM, et al. (2013) Frontiers, opportunities, and challenges in biochemical andchemical catalysis of CO2 fixation. Chem Rev 113(8):6621–6658.
7. Liu C, et al. (2015) Nanowire-bacteria hybrids for unassisted solar carbon dioxidefixation to value-added chemicals. Nano Lett 15(5):3634–3639.
8. Nichols EM, et al. (2015) Hybrid bioinorganic approach to solar-to-chemical conver-sion. Proc Natl Acad Sci USA 112(37):11461–11466.
9. Torella JP, et al. (2015) Efficient solar-to-fuels production from a hybrid microbial-water-splitting catalyst system. Proc Natl Acad Sci USA 112(8):2337–2342.
10. Sakimoto KK, Wong AB, Yang P (2016) Self-photosensitization of nonphotosyntheticbacteria for solar-to-chemical production. Science 351(6268):74–77.
11. RosenbaumM, Aulenta F, Villano M, Angenent LT (2011) Cathodes as electron donorsfor microbial metabolism: Which extracellular electron transfer mechanisms are in-volved? Bioresour Technol 102(1):324–333.
12. Kuzume A, et al. (2014) An in situ surface electrochemistry approach towards whole-cell studies: The structure and reactivity of a Geobacter sulfurreducens submonolayeron electrified metal/electrolyte interfaces. Phys Chem Chem Phys 16(40):22229–22236.
13. Kracke F, Vassilev I, Krömer JO (2015) Microbial electron transport and energy con-servation - the foundation for optimizing bioelectrochemical systems. Front Microbiol6:575.
14. Das A, Ljungdahl LG (2003) Electron-transport system in acetogens. Biochemistry andPhysiology of Anaerobic Bacteria (Springer, New York), pp 191–204.
15. Deutzmann JS, Sahin M, Spormann AM (2015) Extracellular enzymes facilitate elec-tron uptake in biocorrosion and bioelectrosynthesis. MBio 6(2):e00496–e00415.
16. Pierce E, et al. (2008) The complete genome sequence of Moorella thermoacetica(f. Clostridium thermoaceticum). Environ Microbiol 10(10):2550–2573.
17. Schuchmann K, Müller V (2014) Autotrophy at the thermodynamic limit of life: A modelfor energy conservation in acetogenic bacteria. Nat Rev Microbiol 12(12):809–821.
18. Greene BL, Joseph CA, Maroney MJ, Dyer RB (2012) Direct evidence of active-sitereduction and photodriven catalysis in sensitized hydrogenase assemblies. J Am ChemSoc 134(27):11108–11111.
19. Utterback JK, et al. (2015) Competition between electron transfer, trapping, andrecombination in CdS nanorod-hydrogenase complexes. Phys Chem Chem Phys 17(8):5538–5542.
20. Wilker MB, et al. (2014) Electron transfer kinetics in CdS nanorod-[FeFe]-hydrogenasecomplexes and implications for photochemical H2 generation. J Am Chem Soc 136(11):4316–4324.
21. Nevin KP, et al. (2011) Electrosynthesis of organic compounds from carbon dioxide iscatalyzed by a diversity of acetogenic microorganisms. Appl Environ Microbiol 77(9):2882–2886.
22. Drake HL (1982) Demonstration of hydrogenase in extracts of the homoacetate-fer-menting bacterium Clostridium thermoaceticum. J Bacteriol 150(2):702–709.
23. Daniel SL, Hsu T, Dean SI, Drake HL (1990) Characterization of the H2- and CO-
dependent chemolithotrophic potentials of the acetogens Clostridium thermoaceticum
and Acetogenium kivui. J Bacteriol 172(8):4464–4471.24. Tseng H-W, Wilker MB, Damrauer NH, Dukovic G (2013) Charge transfer dynamics
between photoexcited CdS nanorods and mononuclear Ru water-oxidation catalysts.
J Am Chem Soc 135(9):3383–3386.25. Nosaka Y, Miyama H, Terauchi M, Kobayashi T (1988) Photoinduced electron transfer
from colloidal cadmium sulfide to methylviologen: A picosecond transient absorption
study. J Phys Chem 92(2):255–256.26. Ben-Shahar Y, et al. (2016) Optimal metal domain size for photocatalysis with hybrid
semiconductor-metal nanorods. Nat Commun 7:10413.27. Kambhampati P (2011) Unraveling the structure and dynamics of excitons in semi-
conductor quantum dots. Acc Chem Res 44(1):1–13.28. King PW (2013) Designing interfaces of hydrogenase-nanomaterial hybrids for effi-
cient solar conversion. Biochim Biophys Acta 1827(8-9):949–957.29. Gray HB, Winkler JR (1996) Electron transfer in proteins. Annu Rev Biochem 65:
537–561.30. Gray HB, Winkler JR (2005) Long-range electron transfer. Proc Natl Acad Sci USA
102(10):3534–3539.31. Morra S, et al. (2011) Direct electrochemistry of an [FeFe]-hydrogenase on a TiO2
electrode. Chem Commun (Camb) 47(38):10566–10568.32. Flanagan LA, Parkin A (2016) Electrochemical insights into the mechanism of NiFe
membrane-bound hydrogenases. Biochem Soc Trans 44(1):315–328.33. De Lacey AL, Fernandez VM, Rousset M, Cammack R (2007) Activation and in-
activation of hydrogenase function and the catalytic cycle: spectroelectrochemical
studies. Chem Rev 107(10):4304–4330.34. Kong J, Yu S (2007) Fourier transform infrared spectroscopic analysis of protein sec-
ondary structures. Acta Biochim Biophys Sin (Shanghai) 39(8):549–559.35. Venyaminov SYu, Kalnin NN (1990) Quantitative IR spectrophotometry of peptide
compounds in water (H2O) solutions. I. Spectral parameters of amino acid residue
absorption bands. Biopolymers 30(13-14):1243–1257.36. Pandelia M-E, Ogata H, Currell LJ, Flores M, Lubitz W (2009) Probing intermediates in
the activation cycle of [NiFe] hydrogenase by infrared spectroscopy: The Ni-SIr state
and its light sensitivity. J Biol Inorg Chem 14(8):1227–1241.37. Pierik AJ, Roseboom W, Happe RP, Bagley KA, Albracht SP (1999) Carbon monoxide
and cyanide as intrinsic ligands to iron in the active site of [NiFe]-hydrogenases. NiFe
(CN)2CO, Biology’s way to activate H2. J Biol Chem 274(6):3331–3337.38. Flanagan ML, et al. (2016) Mutations to R. sphaeroides reaction center perturb energy
levels and vibronic coupling but not observed energy transfer rates. J Phys Chem A
120(9):1479–1487.39. Srikun D, Albers AE, Nam CI, Iavarone AT, Chang CJ (2010) Organelle-targetable
fluorescent probes for imaging hydrogen peroxide in living cells via SNAP-Tag protein
labeling. J Am Chem Soc 132(12):4455–4465.40. Bond-Watts BB, Bellerose RJ, Chang MC (2011) Enzyme mechanism as a kinetic control
element for designing synthetic biofuel pathways. Nat Chem Biol 7(4):222–227.41. Nguyen SC, et al. (2012) Chemistry of the triplet 14-electron complex Fe(CO)3 in so-
lution studied by ultrafast time-resolved IR spectroscopy. Organometallics 31(10):
3980–3984.
Kornienko et al. PNAS | October 18, 2016 | vol. 113 | no. 42 | 11755
CHEM
ISTR
YBIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
Dow
nloa
ded
by g
uest
on
May
23,
202
0