Embed Size (px)
Citation preview

Indian Journal of Chemistry
Vol. 52A, Aug-Sept 2013, pp. 973-991
Advances in Contemporary Research
Site-specific chemical modifications of proteins
Divya Agrawala,
* & Christian P R Hackenbergera, b,
*
aLeibniz-Institut für Molekulare Pharmakologie (FMP), Robert-Rössle-Str- 10, 13125, Berlin, Germany bHumboldt Universität zu Berlin, Institut für Organische und Bioorganische Chemie,
Institut für Chemie, Brook-Taylor-Str. 2, 12489 Berlin, Germany
Email: [email protected] (DA); [email protected] (CPRH)
Received 8 April 2013; accepted 10 May 2013
Since long, peptides and proteins have been recognized as targets for the development of synthetic methodologies.
There have been major advances in the development of methods for the site-specific modifications. The naturally occurring
functionalities in peptides and proteins are used to modify the less common amino acid targets. The most important benefit
of chemical bioconjugation techniques using naturally occurring amino acids is that they do not require additional
biochemical techniques to install unnatural functionalities for chemoselective reactions. However, unnatural functionalities
can well be incorporated into peptides and proteins followed by the establishment of bioorthogonal coupling methodologies.
The unnatural amino acids can be incorporated into proteins either in a site-directed or residue-specific fashion.
Bioorthogonal chemistry is an important tool for the development of synthetic methodologies and for further advances in
biological research. A variety of methods exist that tag cellular components with reporters not only for visualization but
also for isolation from biological samples. In this review, we present an overview of the efforts of both chemical and
biochemical approaches to functionalize peptides and proteins with the desired molecular components. We focus on the site-
directed methods to incorporate unnatural groups into biomolecules and the development of bioorthogonal transformations
involving unnatural functional groups.
Keywords: Protein modifications, Site-selective modifications, Protein functionalities, Bioorthogonal transformations,
Chemoselective reactions, Bioconjugation, Chemical bioconjugation, Copper catalysed reactions, Biomolecules,
Amino acids, Unnatural amino acids, Canonical amino acids, Azides, Alkynes, Ketones, Aldehydes
How can one probe diverse biological functions
of proteins as well as the impact of side-chain
functionalities in complex systems? Although
innumerable successful research projects have been
carried out, this is still a major challenge for chemical
biologists and chemists which they tend to address
by the introduction of various chemical modifications
in proteins1.
The site-selective introduction of chemical
functionalities into proteins has a particular focus.
A set of enzymes present in nature leads to
the introduction of bioorthogonal functional groups
and modifications of proteins in living cells2.
The enzymes such as biotin ligase (BirA), trans-
glutaminase (TGase) and lipoic acid ligase (LplA)
have been used to site-specifically modify proteins
with biophysical probes, short chain azido fatty acids
and fluorophores3. The formylglycine-generating
enzyme (FGE) consensus is exploited as a genetically
encoded aldehyde tag for site-specific protein
modification (Scheme 1)4. The fusion proteins of
O6-alkylguanine-DNA alkyltransferase (AGT) in
mammalian cells are specifically labeled with
fluorophores using guanine derivatives in a method
termed as ‘SNAP tag’5. The other tags used
for covalent labeling of proteins inside living cells
include SNAP/CLIP tag and Halotag6.
The techniques addressing naturally occurring
amino acids do not require additional biochemical
techniques to install unnatural functionalities
for chemoselective reactions7 and are important
in constructing the majority of man-made protein

INDIAN J CHEM, SEC A, AUG-SEPT 2013
974
conjugates. However, the presence of the large
number of electrophilic and nucleophilic sites
makes the selective functionalization using naturally
occurring functional groups complicated. In addition
to the functional groups present in the 22 canonical
amino acids, posttranslational modifications provide
proteins with additional chemical moieties to
carry out their natural functions. However, there still
remains the need to develop methodologies in order
to functionalize peptides and proteins both, in vitro
and in vivo. The attachment of non-natural
functionalities to specific locations on proteins
requires a set of chemoselective reactions. Metabolic
and genetic engineering methods have provided
an alternative strategy and allowed the incorporation
of abiotic functional groups like azides and alkynes
into proteins, nucleic acids, glycans and lipids8.
The functional group acting as a chemical reporter
is first installed on a target molecule to which
various probes can be delivered exogenously9.
Metabolic oligosaccharide engineering (MOE) is
another rapidly growing technique that helps to
deliver bioorthogonal chemistry for cell surface
modification10
.
Bioorthogonal chemistry addresses the challenge
of selectively combining two functional groups in
a chemically complex environment ranging from
aqueous solutions to living animals. The components
of bioorthogonal chemical reactions react rapidly
and selectively with each other under physiological
conditions in the presence of multitude
of functionality. Bioorthogonal protein modification
reactions target chemical reporters while
ignoring native protein functionalities (Fig. 1)11
.
The challenges include that the reactions must
proceed in water at physiological pH and temperature
providing good yield under reasonably fast reaction
rates at low reagent concentrations while remaining
inert to surrounding biological electrophiles and
nucleophiles. The components must be non-toxic
to cells and organisms. It is of importance if one
of the reactive groups is small, to minimally perturb
a biomolecule into which it has been introduced.
The bioorthogonal reactions of azides mark the first
successful story, azide being a small and biostable
functional group that can act either as a soft
electrophile or a 1,3-dipole. Alkynes are considered
bioorthogonal at physiological conditions due to their
high chemical inertness. Recently, Mootz et al.12
discovered an unexpected reaction between cysteine
proteases of the SUMO and ubiquitin family and
their propargylated substrate proteins forming a vinyl
sulfide linkage inhibiting the proteases. The reaction
was driven by the close proximity and alignment
of the two functional groups.
Incorporation of Unnatural Amino Acids
The replacement of a particular canonical amino
acid by genetic-code engineering (residue-specific incorporation) involves the use of an auxotrophic bacterial strain starved for the natural amino acid and supplemented with its analogue leading to the efficient incorporation of its analogues into bacterial proteins (Fig. 2)
13. The aminoacyl tRNA synthetases
(aaRSs) correctly charges tRNA with unnatural amino acids which are structurally and electronically similar to natural analogs
14. There have been various attempts
to enrich the variety of unnatural amino acids that can
Fig. 1—Bioorthogonal chemical reactions for the modification of peptides and proteins.
Fig. 2—Incorporation of unnatural amino acids using auxotrophic cell lines.

AGRAWAL & HACKENBERGER: SITE-SPECIFIC CHEMICAL MODIFICATIONS OF PROTEINS
975
be incorporated15
. Using this methodology, amino acid analogues have been incorporated not only for phase determination in X-ray crystallography and for spectroscopic studies, but also to study protein activity, folding
16, and stability. Non-proteinogenic
functional groups are introduced into proteins for further protein modification
15(d, f),17.
Genetic-code engineering providing one type of non-canonical amino acid (NCAA) per target
protein has also been used to change the biophysical properties of a certain protein, for example, fluorescence
18. The functional groups like alkynes,
alkenes and azides have been introduced into proteins using methionine (Met) surrogates homopropargylamine (Hpg), homoallylglycine (Hag),
allylcysteine and azidohomoalanine (Aha) (Fig. 3). The incorporated alkynes and azides have been further reacted with other molecules via Cu-catalyzed azide-alkyne cycloaddition (CuAAC) and Staudinger ligation
17f,19. Virus-like capsids have also been
functionalized with azides and alkynes by this
method20
. The selective identification of newly synthesized proteins in mammalian cells has been accomplished using this method in a process termed as bioorthogonal non-canonical amino acid tagging (BONCAT)
21.
The major disadvantage of genetic-code engineering is that it is restricted to the naturally occurring amino acids. The technique also leads to chemical modification at multiple sites. Although this is an important technology, there are other
circumstances where it is required to replace only a single residue while retaining access to all natural
amino acids. Schultz and coworkers22
developed the amber suppression method for introducing non-natural amino acids into specific positions of proteins. Significant advances have been made in the development of methods for the site-specific
modifications23
. Herein we focus on the methods to introduce unnatural functional groups by site-specific incorporation of NCAAs as well as a few other promising methods followed by the development of a series of bioorthogonal reactions involving these unnatural functional moieties.
Site-Directed Incorporation of Unnatural Amino
Acids Unnatural amino acids act as handles for
subsequent modification and help to tune protein
function. The use of the existing protein biosynthetic
machinery of the cell has allowed the development
of a number of in vitro methods to incorporate
NCAAs into proteins24
. Bertozzi and coworkers4
described a method for the site-specific introduction
of aldehyde groups into recombinant proteins using
the 6-amino-acid consensus sequence recognized by
the FGE. FGE oxidizes cysteine to formylglycine
(fGly). Coexpression of the tagged protein alongside
FGE directly produces the aldehyde-functionalized
protein. The aldehyde can then be modified by
using a variety of methods, such as condensations
with aminooxy or hydrazide probes4. However,
these methods suffer from major drawbacks and
thus, broader methods for the selective amino-
acylation of tRNAs were required. Nevertheless,
misaminoacylation of the tRNA with unnatural amino
acids using aaRSs as well as direct chemical acylation
of the tRNA are not practical25
. Consequently,
truncated tRNAs were enzymatically ligated to
chemically aminoacylated mono- and dinucleotides
with the development of semisynthetic methods26
.
The three degenerate stop codons, UAA, UAG, and
UGA, termed as nonsense codons do not encode
amino acids, but signal termination of polypeptide
synthesis by binding release factors27
. However, only
one stop codon is required for the termination of
protein synthesis thereby leaving two ‘blank’ codons
in the genetic code which can be used to uniquely
specify an unnatural amino acid. This approach
was used to report a general in vitro method for
the site-specific incorporation of a large number
of unnatural amino acids into proteins with
excellent translational conformity. The generation of
mutant β-lactamases containing p-nitrophenylalanine,
Fig. 3—Non-canonical amino acids as analogues of methionine
(Met).

INDIAN J CHEM, SEC A, AUG-SEPT 2013
976
p-fluorophenylalanine or homophenylalanine
substitutions at Phe 66 is the first demonstration of
this general approach for the site-specific
incorporation of unnatural amino acids into a protein
in vitro22
. Many unnatural amino acids have been
incorporated into proteins using this technique despite
low yields and considerable effort involved in
synthesizing the amino-acylated tRNA28
. Varieties of
proteins have been studied by this in vitro technique29
.
The microinjection of the engineered mRNA and a
chemically aminoacylated yeast tRNAPhe
-derived
amber suppressor has led to the extension of unnatural
amino acid mutagenesis to Xenopus oocytes30
.
A completely autonomous bacterium with a 21 amino
acid genetic code was generated. This bacterium
can biosynthesize a nonstandard amino acid from
basic carbon sources and incorporate this amino
acid into proteins in response to the amber nonsense
codon31
.
In vivo systems for site-specific mutagenesis
of unnatural amino acids involve the selection of
orthogonal tRNA and aaRSs recognizing the amber
stop codon and unnatural amino acid, respectively
(Fig. 4)32
. Subsequently, many other orthogonal
aaRS:tRNA pairs were reported33
. The adaptation of
this technique to yeast34
and mammalian cell culture35
has expanded the scope to in vivo functional
interrogation studies at multiple levels. By increasing
the production of the engineered tRNA and reducing
the rate of non-sense mediated mRNA decay,
Wang et al.36
have increased the efficiency of yeast
expression to E. coli-like levels (10–20 mg L−1
).
In vivo methods have been used for the
incorporation of a variety of unnatural amino acids
into proteins. Functional groups like azides11b,37
,
alkynes38
, alkenes39
, ketones40
, aniline41
, halides42
and
boronic acids43
have been successfully installed and
are further reacted. In an interesting example, Francis
et al.44
used bacteriophage MS2 to create a targeted,
multivalent photodynamic therapy vehicle for
Jurkat leukemia T cells. The unnatural amino acid,
p-aminophenylalanine (pAF) was introduced on the
external surface and the aniline groups of the side
chains were modified with N,N-dialkylphenylene
diamine derivatives through a highly chemoselective
oxidative coupling strategy (Scheme 2)45
. This
method was also used to attach porphyrins to pAF
side chains introduced using amber stop codon
suppression46
. The post-translational modifications
are efficiently incorporated by the in vivo system47
.
An orthogonal MjtRNATyr
CUA/MjTyrRS pair
was evolved for the selective incorporation of
p-carboxymethyl-L-phenylalanine (pCMF), a mimetic
of phosphotyrosine (pTyr) into proteins in response to
an amber codon in E. coli with high conformity and
efficiency. As pCMF is resistant to hydrolysis by
protein tyrosine phosphatases (PTPs), it provides a
useful tool for the generation of stable analogues of
selectively phosphorylated proteins and peptides48
.
The cross-linking groups49
and fluorophores50
are successfully installed using this approach. The
photocaged versions of cysteine, serine and tyrosine
residues have been genetically encoded to allow the
masking of important residues51
.
In a dual-labeling approach developed by Schultz
and coworkers52
, a cysteine residue and a NCAA,
p-acetyl-L-phenylalanine coded by an amber codon
Fig. 4—In vivo site-specific incorporation of unnatural amino
acids into proteins.

AGRAWAL & HACKENBERGER: SITE-SPECIFIC CHEMICAL MODIFICATIONS OF PROTEINS
977
were used for the site-selective dual labeling of
a protein. Chin and coworkers53
generated two
mutually orthogonal M. jannaschii TyrRS:tRNACUATyr
pairs capable of suppressing UAG and AGGA, thus
making possible multiple NCAA incorporation.
Liu and coworkers54
incorporated two chemically
distinct NCAAs into green fluorescent protein
(GFPuv) by combining two orthogonal pairs in a
single expression experiment. A UAG (amber) codon
was used in combination with a UAA (ochre) codon.
Later, they applied this double NCAA incorporation
method to genetically install two different bioorthogonal
functional groups into a protein allowing catalyst-free
and site-specific one-pot labeling of the protein with
a FRET (Förster resonance energy transfer) pair55
.
An overview of the attempts at the multiple
site-specific NCAA incorporation of two different
NCAAs by the reassignment of termination and
quadruplet codons has been presented by Hoesl and
Budisa56
. Furthermore, it is demonstrated that the
suppression-based incorporation of single NCAAs
can further be improved by the use of mutated
ribosomal proteins57
, elongation factors58
, and even
mutated ribosomal RNAs59
.
Bioorthogonal Reactions
The development of synthetic methodologies
and biological research rely largely on the expansion
of the field of bioorthogonal chemistry60
. A variety
of methods exist that tag biomolecules with reporters
enabling visualization and even isolation from
biological samples61
. However, as discussed
previously, bioorthogonal reactions easily allow
selective functionalization of peptides and
proteins in the presence of a pool of functionalities.
A variety of chemical reporters possessing the
requisite qualities include azides62
, terminal
alkynes63
and peptide sequences64
. The
bioorthogonal reactions help in the chemical
modification of biomolecules in vitro and enable
real time imaging of processes in cells and live
organisms. The noteworthy examples include
Staudinger ligation62a
and click chemistry (both
Cu-catalyzed65
and strain-promoted versions66
).
These bioorthogonal reactions help to probe a
biomolecule labeled with an azide using
complementary reagents in vitro, in cells and in live
organisms (Fig. 5)67
.
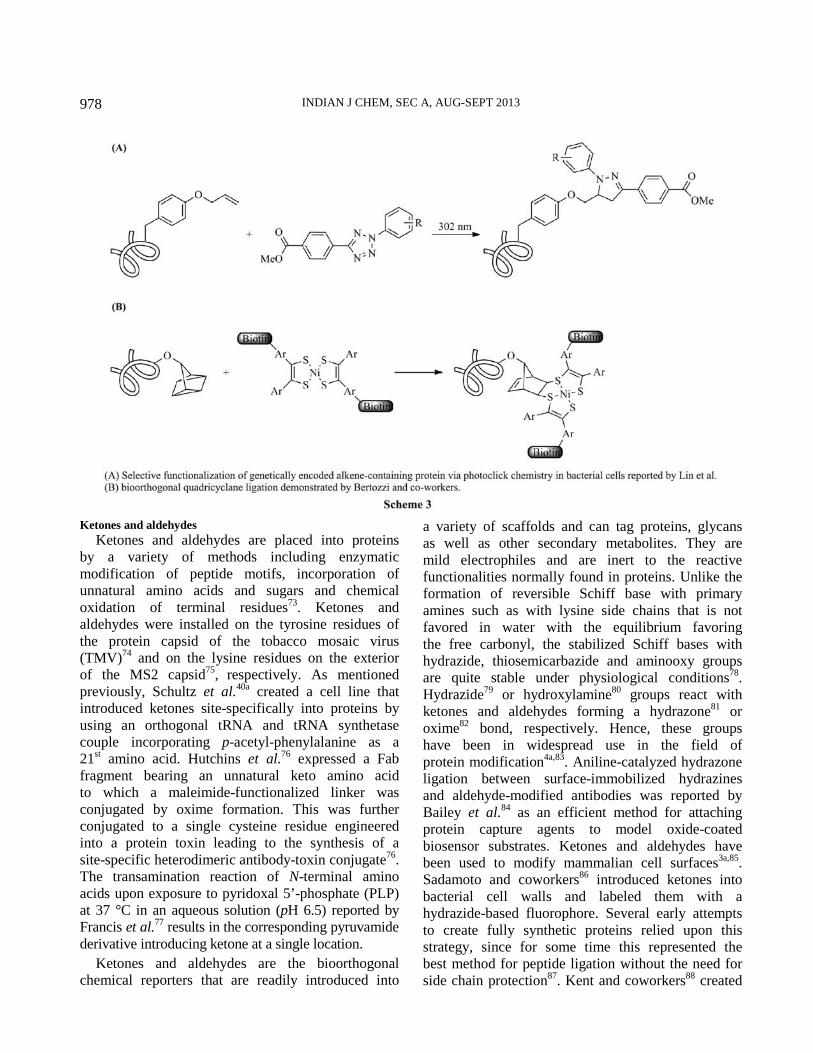
Recently, chemical transformations like 1,3-dipolar
cycloadditions (Scheme 3A)68
, Diels-Alder reactions69
,
metal-catalyzed couplings70
and nucleophilic
additions71
have also been exploited towards
bioorthogonal chemistry. For selective protein
labeling, Sletten and Bertozzi72
have demonstrated
quadricyclane ligation as a promising bioorthogonal
reaction (Scheme 3B). In the subsequent section we
present an overview of the existing chemical reporters
and bioorthogonal reactions.
Fig. 5—A phosphine-biotin (Phos-biotin) probe, FRET-based fluorogenic phosphine, phosphine-luciferin probe and cyclooctyne-biotin
(OCT-biotin) probe for detection of azides reported by Bertozzi and coworkers.
INDIAN J CHEM, SEC A, AUG-SEPT 2013
978
Ketones and aldehydes
Ketones and aldehydes are placed into proteins
by a variety of methods including enzymatic
modification of peptide motifs, incorporation of
unnatural amino acids and sugars and chemical
oxidation of terminal residues73
. Ketones and
aldehydes were installed on the tyrosine residues of
the protein capsid of the tobacco mosaic virus
(TMV)74
and on the lysine residues on the exterior
of the MS2 capsid75
,
respectively. As mentioned
previously, Schultz et al.40a
created a cell line that
introduced ketones site-specifically into proteins by
using an orthogonal tRNA and tRNA synthetase
couple incorporating p-acetyl-phenylalanine as a
21st amino acid. Hutchins et al.
76 expressed a Fab
fragment bearing an unnatural keto amino acid
to which a maleimide-functionalized linker was
conjugated by oxime formation. This was further
conjugated to a single cysteine residue engineered
into a protein toxin leading to the synthesis of a
site-specific heterodimeric antibody-toxin conjugate76
.
The transamination reaction of N-terminal amino
acids upon exposure to pyridoxal 5’-phosphate (PLP)
at 37 °C in an aqueous solution (pH 6.5) reported by
Francis et al.77
results in the corresponding pyruvamide
derivative introducing ketone at a single location.
Ketones and aldehydes are the bioorthogonal
chemical reporters that are readily introduced into
a variety of scaffolds and can tag proteins, glycans
as well as other secondary metabolites. They are
mild electrophiles and are inert to the reactive
functionalities normally found in proteins. Unlike the
formation of reversible Schiff base with primary
amines such as with lysine side chains that is not
favored in water with the equilibrium favoring
the free carbonyl, the stabilized Schiff bases with
hydrazide, thiosemicarbazide and aminooxy groups
are quite stable under physiological conditions78
.
Hydrazide79
or hydroxylamine80
groups react with
ketones and aldehydes forming a hydrazone81
or
oxime82
bond, respectively. Hence, these groups
have been in widespread use in the field of
protein modification4a,83
. Aniline-catalyzed hydrazone
ligation between surface-immobilized hydrazines
and aldehyde-modified antibodies was reported by
Bailey et al.84
as an efficient method for attaching
protein capture agents to model oxide-coated
biosensor substrates. Ketones and aldehydes have
been used to modify mammalian cell surfaces3a,85
.
Sadamoto and coworkers86
introduced ketones into
bacterial cell walls and labeled them with a
hydrazide-based fluorophore. Several early attempts
to create fully synthetic proteins relied upon this
strategy, since for some time this represented the
best method for peptide ligation without the need for
side chain protection87
. Kent and coworkers88
created

AGRAWAL & HACKENBERGER: SITE-SPECIFIC CHEMICAL MODIFICATIONS OF PROTEINS
979
a fully synthetic mimic of human erythropoietin,
a glycosylated protein hormone that stimulates the
production of erythroid cells. Native chemical ligation
(NCL) was used to create the full-length polypeptide
product with specifically placed ketones that were
used as attachment points for aminooxy terminated
polymers designed to mimic natural glycans.
Ketones and aldehydes (best considered as
‘biorestricted’ chemical reporters), although suitable
in the presence of cultured cells, have limitations in
living organisms. The optimum pH for condensation
reactions is 5−6 that cannot be achieved in most
tissues in vivo and are not truly bioorthogonal in more
complex physiological settings. However, ketones and
aldehydes serve as unique chemical reporters89
.
Paulson and coworkers90
introduced aldehydes
into cell-surface sialic acid residues by mild
periodate oxidation and then captured the modified
glycoproteins by reaction with aminooxybiotin
followed by streptavidin chromatography.
Azides
The aldehydes and ketones have limited
intracellular applicability. Azides, however, are
more versatile chemical reporters91
. They are totally
absent from biological systems except one naturally
occurring azido metabolite isolated from unialgal
cultures92
. The azide group being small, minimally
perturbs the modified substrate93
. Azides are
easily approachable synthetically. They are mild
electrophiles and do not react with hard nucleophiles
abundant in biological systems. Azides do not react
with water, are resistant to oxidation and stable at
physiological temperatures. Covalently bound azides
decompose at elevated temperatures and are
considered explosive classes of compounds. For
organic azides to be non-explosive, the rule is that
the number of nitrogen atoms must not exceed that of
carbon and that (NC + NO)/NN ≥ 3 (N = number of
atoms)94
. The azide anion is a widely used cytotoxin,
however, organic azides have no intrinsic toxicity.
Azides possess orthogonal reactivity to the
majority of biological functional groups. The large
energy content of azides is used for the development
of bioorthogonal reactions for selective labeling of
azide-functionalized biomolecules. Azides are largely
introduced by metabolic incorporation of unnatural
amino acids and monosaccharides17e,32a
. The diazo
transfer agents can also be used to introduce azide
group into both peptides and proteins. Triflyl azide
in the presence of divalent Cu ions can easily
convert solid-phase bound peptide amines into
azides95
. The water-soluble diazo transfer agents are
used to substitute the amines of both lysine residues
and the N-terminus by azide groups on proteins96
.
Pedersen and Johansson97
have nicely compiled
available methods for the synthesis of optically active
azide- and alkyne-functionalized α-amino acids.
Staudinger ligation
Staudinger98
reported the reaction of azides and
triphenylphosphines under mild conditions to produce
aza-ylide intermediates that can subsequently be
hydrolyzed in water or trapped by electrophiles to
produce an amine and the corresponding phosphine
oxide. Later, Bertozzi and Saxon62a
modified the
classic Staudinger reaction by introduction of an
intramolecular trap, a proximal ester into the phosphine,
now known as Staudinger ligation. The Staudinger
ligation can be used to covalently attach probes to
azide-bearing biomolecules99
. However, oxidation
of the phosphine may reduce the amount of probe
available in biological systems. The major limitation of
Staudinger ligation is relatively slow reaction kinetics
necessitating high concentrations of triarylphosphine100
.
All efforts to improve the reaction kinetics by
increasing the nucleophilicity of the phosphine reagents
lead to increased phosphine oxidation in air.
The Staudinger ligation has been used for
site-selective immobilization of azide-labeled proteins
on surfaces101
and to impart new functionality to
recombinant proteins102
. The selective in vivo covalent
modification of cell-surface glycans with chemical
probes was achieved in live mice by Staudinger
ligation103
. Lemieux et al.104
reported a fluorogenic
phosphine by replacing one of the aryl rings
with a coumarin dye104
. Phosphine oxidation during
Staudinger ligation relieves the quenching effect
caused by the lone pair of electrons on the phosphorus
atom producing a highly fluorescent biomolecule-
bound product (Scheme 4). However, the oxidation
of phosphine in air caused background fluorescence.
To overcome this problem, Bertozzi et al.105
incorporated a FRET quenching group at the ester
position. Recently, they reported bioluminogenic
phosphine reagent that releases luciferin during
Staudinger ligation. Luciferin readily enters cells
wherein heterologously expressed luciferase
catalyses its oxidation and the concomitant emission
of light106
. The Staudinger ligation was also used for
the site-specific PEGylation of azido-homoalanine
containing trombomodulin107
.

INDIAN J CHEM, SEC A, AUG-SEPT 2013
980
Hackenberger et al.108
demonstrated the Staudinger
reaction of unprotected azido-peptides with silylated
phosphinic acids and esters in solution as well as
on the solid support (Scheme 5A). The protocol
offers a straightforward acid-free entry to different
phosphonamidate peptide esters or acids under
mild conditions in high yield and purity108
.
They identified another Staudinger-type reaction
for the chemoselective transformations of azides
in peptides and proteins (Scheme 5B)109
.
This Staudinger-phosphite reaction (SPR) utilizes
phosphites and hence is very easy to perform.
The phosphites can be prepared by standard organic
synthesis protocols and are stable against oxidation
upon exposure to air. SPRs can be performed in
various solvents and buffers at room temperature,
conditions suitable for quantitative modification
reactions in proteins. They also showed that not

AGRAWAL & HACKENBERGER: SITE-SPECIFIC CHEMICAL MODIFICATIONS OF PROTEINS
981
only symmetrical but also unsymmetrical phosphites
can be used for the modification of proteins via
aqueous SPR (Scheme 5B)110
. Additionally, the first
example of Staudinger reaction of aryl-phosphonites
(Staudinger-phosphonite reaction) for the chemoselective
transformation of azido-containing peptides and
proteins in aqueous systems at room temperature in
high conversions was developed (Scheme 5A)111a
.
Since alkyl-phosphonites appeared to oxidize rapidly,
aryl-substituted analogues were used in which the
sp2-hybridized carbon at phosphorus accounts for
a higher stability upon air exposure. Also, the
phosphonites used were obtained as borane adducts
in a one-pot process to allow prolonged storage.
The phosphonite hydrolysis to a phosphinic acid ester
as observed by 31
P NMR was a limiting factor in the
Staudinger-phosphonite reaction. This limitation was
addressed by introducing OEG-(oligo ethylene glycol)
groups at phosphorus to further enhance the water
solubility and reduce the probability of nucleophilic
attack at the phosphorus. The Staudinger-phosphonite
reaction was then successfully applied to the
site-specific modification of the protein, calmodulin.
Along those lines, Hackenberger and coworkers
have also used alkyne-phosphonites in a modular
azide-azide coupling strategy by sequential CuAAC
and Staudinger reactions110b
.
Copper-catalyzed [3 + 2] azide-alkyne cycloaddition
Azides also act as 1,3-dipoles and undergo
electrocyclization reactions with dipolarophiles such
as activated alkynes112
to provide stable triazole
adducts, first described by Huisgen113
. Both the
reactants are stable under physiological conditions
and the reaction proceeds readily in water and
tolerates a wide range of functionalities. Despite the
exothermic nature of these reactions, without alkyne
activation the process requires elevated temperatures
or pressure incompatible with living systems114
and
produces a mixture of regioisomers. Sharpless and
coworkers65,115b
and Meldal and others115a
however,
demonstrated that the rate of cycloaddition can be
accelerated using catalytic amounts of Cu(I). The
cycloaddition proceeds with almost complete
regioselectivity (1,4-regioisomer) at physiological
temperatures in the presence of biological materials
providing 1,4-disubstituted triazoles62b
. Initially,
active Cu(I) was generated in situ from Cu(II)
salts, (Cu wire) and a reductant, ascorbic acid
or tris(2-carboxyethylphosphine) (TCEP). However,
Cu(I) salts in the presence of a ligand (commonly,
tris-triazole) have shown to increase the rate
of reaction quite significantly116
. The use of an
electrochemical cell to generate and protect
catalytically active-Cu-ligand species for CuAAC
bioconjugation and synthetic coupling reactions
has been reported by Finn and coworkers117
. The
Cu-accelerated azide-alkyne cycloaddition (CuAAC)
more appropriately termed as ‘click’ reaction118
has
found broad application as ligation tool in polymer
science119
and in combinatorial organic synthesis120
.
The azides incorporated within proteins121
,
fucosylated glycoproteins (Scheme 6)122
, nucleic
acids123
and virus particles124
are tagged using this
approach. The first examples of directly radiolabeled
([18
F]-glyco)proteins were reported by Davis and
coworkers125
. They used this ‘tag and modify’
approach to synthesize homogenous fluorinated
glyco-amino acids, peptides and proteins carrying
a fluorine label in the sugar.
Metalloproteases are a diverse class of enzymes
involved in many physiological and disease processes
and are regulated by post-translational mechanisms
that reduce the effectiveness of conventional
genomic and proteomic methods for their functional
characterization. Click chemistry is used for
the activity-based protein profiling (ABPP) of
metalloproteases126
. CuAAC finds excellent use
where high reaction yields are required at low
substrate concentrations. The major advantage of
click reaction is its faster rate compared to Staudinger
ligation, whereas the cellular toxicity of the
metal catalyst and copper-induced denaturation of
proteins are the biggest disadvantages17f,127
. Finn and
coworkers128
described two immobilized forms of
a Cu-binding ligand shown to accelerate triazole
formation under many different conditions using
different resin supports that are appropriate for
aqueous or organic solvents. CuAAC was used for
the site-specific PEGylation of human superoxide
dismutase-1 (SOD), a key enzyme in preventing

INDIAN J CHEM, SEC A, AUG-SEPT 2013
982
the formation of reactive oxygen species in cells129
.
There are also examples of CuAAC available
where the azide and alkyne functionalities switched
places130
. Bertozzi and coworkers130d
identified
potential fluorogenic azidofluoresceins. Later,
in order to evaluate them as biological imaging
reagents, Chinese hamster ovary (CHO) cells
were metabolically labeled with peracetylated
N-(4-pentynoyl)-mannosamine (Ac4ManNAl). These
cells convert Ac4ManNAl to the corresponding
alkynylsialic acid, which is then incorporated
into cell-surface glycoproteins. Control cells were
incubated with peracetylated N-acetylmannosamine
(Ac4ManNAc). The cells were then fixed
with paraformaldehyde and incubated with
azidofluorescein under CuAAC conditions.
After washing, the cells showed robust alkyne-
dependent labeling with azidofluorescein.
The low concentrations at which biomolecules
are manipulated bring challenges for any
ligation methodology. A generally applicable
CuAAC procedure solving many click bioconjugation
problems was described by Finn and coworkers131
.
They used the new CuAAC conditions to couple
a protein to the outer surface of the Qβ virus-like
particle. The major improvements involve
the addition of a water-soluble member of
the tris(triazolylmethyl)amine family, tris(3-hydroxy-
propyltriazolylmethyl) amine (THPTA) and
aminoguanidine. This allows ascorbate to be
used as reducing agent while eliminating problems
caused by copper ascorbate side-reactions.
The addition of at least five equivalents of THPTA
relative to Cu helps to quickly reduce reactive
oxygen species generated by the ascorbate-driven
reduction of dissolved O2 without affecting
the CuAAC reaction rate to a large extent.
Aminoguanidine intercepts byproducts of ascorbate
oxidation that can covalently modify or crosslink
proteins.
Strain-promoted [3 + 2] azide-alkyne cycloaddition
The use of ring strain to activate dipolarophile62c
for electrocyclization involves constraining the alkyne
within an eight-membered ring creating strain
that is released in the transition state upon [3+2]
cycloadditoon with an azide132
. The strain-promoted
cycloaddition of cyclooctynes with azides, commonly
referred to as Cu-free click chemistry, has been
used to label biomolecules both in vitro and on cell
surfaces without the need for a catalyst (Fig. 6)62c,133.
The major drawback, however, is the slow rate
of reaction. In order to improve reaction
kinetics cyclooctyne derivatives with added fluorines
were designed134
. Later, a series of cyclooctynes
were investigated in living systems through the
strain-promoted azide-alkyne cycloaddition (SPAAC)
(Fig. 6)135
. Three coumarin-cyclooctyne conjugates
were used to label proteins tagged with Aha in Rat-1
fibroblasts leading to the visualization of biomolecule
dynamics in living cells136
. BODIPY-cyclooctyne
(BDPY), a membrane-permeant fluorophore was used
to label intracellular proteins in live mammalian
cells137
.
Lemke and coworkers138
genetically encoded
strained alkynes into E. coli by use of an engineered
pyrrolysine amber suppressor tRNA/synthetase
pair from Methanosarcina mazei. As a result
cyclooctynyl lysine derivatives were incorporated
site-specifically in proteins allowing efficient labeling
with fluorogenic azide-bearing dyes by means of
a SPAAC under physiological conditions in vitro and
in vivo (Scheme 7A)138
. The difluorinated cyclooctyne
(DIFO) showed kinetics comparable to CuAAC
in biomolecule labeling experiments and has been
used for imaging azide-labeled biomolecules
within complex biological systems134,139
. Boons and
coworkers140
fused two aryl rings to the cyclooctyne
core to form dibenzocyclooctyne (DIBO) for
rate enhancement, and later Bertozzi and
coworkers141
added an amide bond to it forming
biarylazacyclooctynone (BARAC). Later, many
versions of DIBO and BARAC were reported142
.
Hinderlich and Hackenberger et al.143
tested the
potential of Ac3-4-azido-ManNAc as a tool for
glycan labeling in living animals. They injected
Ac3-4-azido-ManNAc into the hindbrain vesicle of
zebrafish larvae at 24 hpf (hours post fertilization).
AlexaFluor 488-conjugated DIBO which reacts
specifically with the azido group of modified sialic
acids was injected at 48 hpf and live embryos
were analyzed at 72 hpf. In embryos injected with
Ac3-4-azido-ManNAc, a distinct labeling of the
mid-brain and the hindbrain was detected. In addition,
a faint staining of the dorsal myosepta was observed,
which was absent in the control embryos.
The fluorescence staining of regions of the central
nervous system and the myosepta reflected high
incorporation of Ac3-4-azido-ManNAc into sialic
acids of heavily O-glycosylated proteins. The use of
dibenzocyclooctyn-ol in strain-promoted cyclization

AGRAWAL & HACKENBERGER: SITE-SPECIFIC CHEMICAL MODIFICATIONS OF PROTEINS
983
Fig. 6—Strain-promoted azide-alkyne cycloaddition; a series of cyclooctynes for copper-free click chemistry.

INDIAN J CHEM, SEC A, AUG-SEPT 2013
984
has been reported by Boons and coworkers140
(Scheme 7B). Ting et al.144
optimized two-step
enzymatic/chemical labeling scheme to tag and image
a variety of cellular proteins fused to a 13-amino acid
recognition sequence (LAP) in multiple mammalian
cell lines with diverse fluorophore conjugates to
aza-dibenzocyclooctyne (ADIBO). The theoretical
studies by Houk and coworkers145
enable to
predict and optimize cyclooctyne reactivity.
Studies also demonstrated that scaffolds like
monobenzocyclooctyne (MOBO) and bicyclononyne
(BCN) find an optimal balance between
strain enhancement and minimization of steric
hinderance146
. Difluorobenzocyclooctyne (DIFBO)
that was found to be almost 20 times more
reactive than MOBO, was unstable and prone
to oligomerization in concentrated solution147
.
Bertozzi and coworkers148
synthesized thiaOCT and
thiaDIFBO (the sulfur-containing analogues) in order
to assess the effects of an endocyclic sulfur atom
on cyclooctyne reactivity. Thiacycloheptynes also
form a promising class of reagents for bioorthogonal
Cu-free click chemistry. Banert and Plefka149
reported
that 3,3,6,6-tetramethylthiacycloheptyne (TMTH)
is more reactive than a DIBO-like cyclooctyne
in a 1,3-dipolar cycloaddition with nitrous oxide
(Scheme 7C). Bertozzi et al.148
tested the reactivity
of TMTH with azide-functionalized biomolecules.
Alkenes and iodides
Alkene acts a versatile chemical reporter/probe
capable of multiplexed applications due to
various possible organic transformations including
1,3-dipolar cycloaddition, Diels-Alder reaction
and olefin metathesis. Alkene-containing amino acids
incorporated site-specifically into proteins include
Hag19e
, O-allyl-tyrosine39
and dehydroalanine150
.
Several groups have investigated the potential
of strained bicyclic alkenes for bioconjugation151
.
Delft et al.152
described the reaction of
oxanorbornadienes with azides via a tandem
cycloaddition-retro-Diels-Alder (crDA) reaction and
used the technology for protein modification. Lin and
coworkers68a
developed photoinducible bioorthogonal
reactions known as ‘photoclick’ chemistry to modify
tetrazole-containing proteins in biological media by
their 1,3-dipolar cycloaddition with simple alkenes to
form pyrazolines. The reaction required light with a
wavelength of 302 nm to produce the nitrile-imine
dipole. In order to avoid photodamage caused
to exposed cells, they designed diaryltetrazoles that
could undergo ring-opening at 365 nm to generate
reactive nitrile imine dipoles153
. The utility of this
approach is demonstrated by the selective labeling
of an alkene-containing Z-domain protein in
E. coli with diaryl tetrazoles68e
. In addition to the mild
reaction conditions, the approach offers convenient
fluorescent monitoring due to the formation of
the fluorescent cycloadducts. An unusually
fast bioorthogonal reaction based on the inverse-
electron-demand hetero-Diels-Alder reaction between
dipyridyltetrazine and trans-cyclooctene (TCO)
was reported by Fox and coworkers69a
and higher
rate constant was reported in phosphate buffered
saline (PBS) at 37 oC (Scheme 8A)
154. Tetrazine-TCO
ligation was also utilized in a fluorogenic reaction
and 18
F-scintigraphic imaging155
. Hilderbrand and
coworkers69b
developed the reaction of norbornene-
conjugated antibody, SKBR3 and fluorescent
VT680-tethered tetrazine, both in serum and live
cells. Later, they reported the use of this reaction in
selectively imaging TCO-labeled cancer cells156
.
Prescher and coworkers157
used a new chemical
reporter–cyclopropene–to target biomolecules in vitro
and in live cells. The small size and selectivity make
cyclopropenes a versatile class of chemical reporters
to label biomolecules in living systems. A variety of
substituted cyclopropene scaffolds were synthesized
and were found to be stable in aqueous solution and
in the presence of biological nucleophiles. In order
to investigate whether cyclopropenes would be
useful for cellular labeling studies, they constructed
a methylcyclopropene–sialic acid conjugate
(9-Cp-NeuAc). These modified sialic acids were
metabolically introduced into cell surface glycans.
The presence of these cell surface cyclopropenes was
subsequently probed by reaction with a tetrazine–
biotin conjugate (Tz-Biotin) (Scheme 8B).
With the development of water-soluble olefin
metathesis catalysts, chemoselective modification
of alkenes by cross-metathesis is emerging as
an important bioorthogonal reaction158
. Davis and
coworkers19g,159
modified proteins containing allyl
sulfide groups through cross-metathesis (Scheme 9A).
Later,
allyl selenides were found to be the
most reactive substrates for olefin metathesis
(Scheme 9B)160
.
With the ability to genetically incorporate
cross-coupling partners on proteins, palladium
catalyzed cross-coupling has found applications
on biomolecules. Schultz and coworkers161
developed

AGRAWAL & HACKENBERGER: SITE-SPECIFIC CHEMICAL MODIFICATIONS OF PROTEINS
985

INDIAN J CHEM, SEC A, AUG-SEPT 2013
986
a method to site-specifically incorporate 4-iodo-
L-phenylalanine (iodoPhe) into proteins in response
to an amber TAG codon. Yokoyama and coworkers162
genetically engineered a Ras protein carrying an
iodophenylalanine that serves as an orthogonal
functionality for selective conjugation with a biotin-
tethered alkene via the Mizoruki-Heck reaction.
Later, a related site-specific biotinylation via the
Sonogashira reaction was reported70
. Davis et al.163
disclosed a convenient catalyst for Suzuki-Miyaura
cross-coupling on proteins under aqueous conditions
(Scheme 9C) and later demonstrated the use of small
molecule palladium scavenger, 3-mercaptopropionic
acid (3-MPrAc)164
.
Conclusions Till date a number of bioorthogonal strategies have
been reported, each with a particular strength and
utility, changing our ways of studying biomolecules
in vitro and in vivo. Among these, in vivo site-specific
incorporation of NCAAs proves to the most potential
method for ongoing development and widespread
application. However, the competition with naturally
occurring functional moieties leads to incomplete
substitution with the reporter-modified building
block. Also, some of the existing bioorthogonal
reactions proceed either at slow rates or require
high concentrations of reactants. There exists a need
for additional reactions that have faster reaction
rates and high overall yields. There is also a need
to develop additional orthogonal functional groups
as well as new chemical reporters to produce
multidimensional experiments. The methodology for
robust and efficient incorporation of unnatural amino
acids into proteins is evolving very rapidly and the
newer methods that take advantage of our present
knowledge are constantly underway.
Acknowledgement
The authors acknowledge financial support
from the German Science Foundation within the
Emmy-Noether program (HA4468/2–1), the SPP
1623, the Einstein Foundation Berlin (Leibniz-
Humboldt Professorship), the SFB 765, the
Boehringer-Ingelheim Foundation (Plus 3 award)
and the Fonds der chemischen Industrie (FCI).
References 1 (a) Hackenberger C P R & Schwarzer D, Angew Chem Int
Ed, 47 (2008) 10030; (b) Glazer A N, Annu Rev Biochem, 39
(1970) 101; (c) Hermanson G T, Bioconjugate Techniques,
2nd ed., (Academic Press, San Diego) 2008; (d) Joo C, Balci H,
Ishitsuka Y, Buranachai C & Ha T, Anuu Rev Biochem, 77
(2008) 51; (e) Evans M J & Cravatt B F, Chem Rev, 106
(2006) 3279; (f) Cecconi C, Shank E A, Bustamante C &
Marqusee S, Science, 309 (2005) 2057; (g) Muir T W,
Sondhi D & Cole P A, Proc Natl Acad Sci USA, 95 (1998)
6705; (h) Valiyaveetil F I, Sekedat M, MacKinnon R &
Muir T W, Proc Natl Acad Sci USA, 101 (2004) 17045;
(i) Cooper J A, Walker S B & Pollard T D, J Mus Res Cell
Motil, 4 (1983) 253.
2 Foley T L & Burkart M D, Curr Opin Chem Biol, 11 (2007)
12.
3 (a) Chen I, Howarth M, Lin W & Ting A Y, Nat Methods, 2
(2005) 99; (b) Lin C W & Ting A Y, J Am Chem Soc, 128
(2006) 4542; (c) Uttamapinant C, White K A, Baruah H,
Thompson S, Fernandez-Suarez M, Puthenveetil S &
Ting A Y, Proc Natl Acad Sci USA, 107 (2010) 10914.
4 (a) Carrico I S, Carlson B L & Bertozzi C R, Nat Chem Biol,
3 (2007) 321; (b) Rabuka D, Rush J S, deHart G W, Wu P &
Bertozzi C R, Nat Protoc, 7 (2012) 1052.
5 Keppler A, Pick H, Arrivoli C, Vogel H & Johnsson K, Proc
Natl Acad Sci USA, 101 (2004) 9955.
6 (a) Gautier A, Juillerat A, Heinis C, Correa I R, Kindermann
M, Beaufils F & Johnsson K, Chem Biol, 15 (2008) 128;
(b) Los G V, Encell L P, McDougall M G, Hartzell D D,
Karassina N, Zimprich C, Wood M G, Learish R, Ohana R F,
Urh M, Simpson D, Mendez J, Zimmerman K, Otto P,
Vidugiris G, Zhu J, Darzins A, Klaubert D H, Bulleit R F &
Wood K V, ACS Chem Biol, 3 (2008) 373.
7 Baslé E, Joubert N & Pucheault M, Chem Biol, 17 (2010) 213.
8 Johnson J A, Lu Y Y, van Deventer J A & Tirrell D A,
Curr Opin Chem Biol, 14 (2010) 774.
9 (a) Prescher J A & Bertozzi C R, Nat Chem Biol, 1 (2005)
13; (b) de Graaf A J, Kooijman M, Hennink W E &
Mastrobattista E, Bioconjugate Chem, 20 (2009) 1281;
(c) Lim R K V & Lin Q, Chem Commun, 46 (2010) 1589.
10 Dube D H & Bertozzi C R, Curr Opin Chem Biol, 7 (2003)
616.
11 (a) Sletten E M & Bertozzi C R, Angew Chem Int Ed, 48
(2009) 6974; (b) Deiters A, Cropp T A, Mukherji M,
Chin J W, Anderson J C & Schultz P G, J Am Chem Soc,
125 (2003) 11782.
12 Sommer S, Weikart N D, Linne U & Mootz H D, Bioorg Med
Chem, (2013) http://dx.doi.org/10.1016/j.bmc.2013.02.039.
13 (a) Munier R & Cohen G N, Biochim Biophys Acta, 21
(1956) 592; (b) Richmond M H, Bacterial Rev, 26 (1962)
398; (c) Cohen G N & Cowie D B, C R Hebd Seances Acad
Sci, 244 (1957) 680; (d) Cowie D B & Cohen G N, Biochim
Biophys Acta, 26 (1957) 252; (e) Hortin G & Boime I,
Methods Enzymol, 96 (1983) 777; (f) Brawerman G &
Ycas M, Arch Biochem Biophys, 68 (1957) 112;
(g) Richmond M H, Biochem J, 73 (1959) 261; (h) Cowie D B,
Cohen G N, Bolton E T & de Robichon-Szulmajster H,
Biochim Biophys Acta, 34 (1959) 39; (i) Rennert O M &
Anker H S, Biochemistry, 13 (1961) 471.
14 (a) Connor R E & Tirrell D A, Polym Rev, 47 (2007) 9;
(b) Link A J, Mock M L & Tirrell D A, Curr Opin
Biotechnol, 14 (2003) 603; (c) Budisa N, Engineering the
Genetic Code: Expanding the Amino Acid Repertoire for the
Design of Novel Proteins, (Wiley-VCH, Weinheim) 2006;
(d) Hendrickson T L, de Crecy-Lagard V & Schimmel P,

AGRAWAL & HACKENBERGER: SITE-SPECIFIC CHEMICAL MODIFICATIONS OF PROTEINS
987
Annu Rev Biochem, 73 (2004) 147; (e) Budisa N, Angew
Chem, 116 (2004) 6586; Angew Chem Int Ed, 43 (2004) 6426.
15 (a) Ibba M, Kast P & Hennecke H, Biochemistry, 33 (1994)
7107; (b) Ibba M & Hennecke H, FEBS Lett, 364 (1995)
272; (c) Sharma N, Furter R, Kast P & Tirrell D A, FEBS
Lett, 467 (2000) 37; (d) Kirshenbaum K, Carrico S I &
Tirrell A D, ChemBioChem, 3 (2002) 235; (e) Hamano-
Takaku F, Iwama T, Saito-Yano S, Takaku K, Monden Y,
Kitabatake M, Soll D & Nishimura S, J Biol Chem, 275
(2000) 40324; (f) Datta D, Wang P, Carrico I S, Mayo S L &
Tirrell D A, J Am Chem Soc, 124 (2002) 5652; (g) Kast P &
Hennecke H, J Mol Biol, 222 (1991) 99; (h) Budisa N,
Minks C, Medrano F J, Lutz J, Huber R & Moroder L,
Proc Natl Acad Sci USA, 95 (1998) 455.
16 (a) Renner C, Alefelder S, Bae J H, Budisa N, Huber R &
Moroder L, Angew Chem, 113 (2001) 949; Angew Chem
Int Ed, 40 (2001) 923; (b) Moroder L & Budisa N,
ChemPhysChem, 11 (2010) 1181; (c) Hoesl M G, Larregola M,
Cui H & Budisa N, J Pept Sci, 16 (2010) 589.
17 (a) Kothakota S, Mason T L, Tirrell D A & Fournier M J,
J Am Chem Soc, 117 (1995) 536; (b) Deming T J,
Fournier M J, Mason T L & Tirrell D A, J Macromol Sci
Pure Appl Chem, 34 (1997) 2143; (c) van Hest J C M &
Tirrell D A, FEBS Lett, 428 (1998) 68; (d) Kiick K L,
van Hest J C & Tirrell D A, Angew Chem, 112 (2000) 2232;
Angew Chem Int Ed, 39 (2000) 2148; (e) Kiick K L, Saxon
E, Tirrell D A & Bertozzi C R, Proc Natl Acad Sci USA,
99 (2002) 19; (f) Link A J & Tirrell D A, J Am Chem Soc,
125 (2003) 11164.
18 Lepthien S, Hoesl M G, Merkel L & Budisa N, Proc Natl
Acad Sci USA, 105 (2008) 16095.
19 (a) Beatty K E, Xie F, Wang Q & Tirrell D A, J Am Chem
Soc, 127 (2005) 14150; (b) Beatty K E, Liu J C, Xie F,
Dieterich D C, Schuman E M, Wang Q & Tirrell D A,
Angew Chem, 118 (2006) 7524; Angew Chem Int Ed, 45
(2006) 7364; (c) Link A J, Vink M K S, Agard N J,
Prescher J A, Bertozzi C R & Tirrell D A, Proc Natl Acad
Sci USA, 103 (2006) 10180; (d) Link A J, Vink M K S &
Tirrell D A, J Am Chem Soc, 126 (2004) 10598; (e) van Hest
J C M, Kiick K L & Tirrell D A, J Am Chem Soc, 122 (2000)
1282; (f) Schoffelen S, Lambermon M H L, van Eldijk M B
& van Hest J C M, Bioconjugate Chem, 19 (2008) 1127;
(g) Lin Y A, Chalker J M, Floyd N, Bernardes G J L &
Davis B G, J Am Chem Soc, 130 (2008) 9642.
20 Strable E, Prasuhn D E, Udit A K, Brown S, Link A J,
Ngo J T, Lander G, Quispe J, Potter C S, Carragher B,
Tirrell D A & Finn M G, Bioconjugate Chem, 19 (2008) 866.
21 Dieterich D C, Link A J, Graumann J, Tirrell D A &
Schuman E M, Proc Natl Acad Sci USA, 103 (2006) 9482.
22 Noren C J, Anthony-Cahill S J, Griffith M C & Schultz P G,
Science, 244 (1989) 182.
23 (a) Carrico I S, Chem Soc Rev, 37 (2008) 1423; (b) Chalker J M,
Bernardes G J L, Davis B G & Acc Chem Res, 44 (2011)
730.
24 (a) Crick F H C, Symp Soc Exp Biol, 12 (1958) 138;
(b) Chapeville F, Lipmann F, von Ehrenstein G, Weisblum B,
Ray W J & Benzer S, Proc Natl Acad Sci USA, 48 (1962)
1086; (c) Johnson A E, Woodward W R, Herbert E &
Menninger J R, Biochemistry, 15 (1976) 569.
25 (a) Schimmel P, Annu Rev Biochem, 56 (1987) 125; (b) Ibba M
& Soll D, Annu Rev Biochem, 69 (2000) 617.
26 (a) Heckler T G, Chang L H, Zama Y, Naka T, Chorghade M S
& Hecht S M, Biochemistry, 23 (1984) 1468; (b) Heckler T G,
Zama Y, Naka T & Hecht S M, J Biol Chem, 258 (1983)
4492; (c) Roesser J R, Chorghade M S & Hecht S M,
Biochemistry, 25 (1986) 6361; (d) Heckler T G, Roesser J R,
Xu C, Chang P I & Hecht S M, Biochemistry, 27 (1988)
7254; (e) Baldini G, Martoglio B, Schachenmann A &
Zugliani C, Brunner J, Biochemistry, 27(1988) 7951.
27 (a) Crick F H C, Barret L, Brenner S & Watts-Tobin R,
Nature, 192 (1961) 1227; (b) Brenner S, Stretton A O &
Kaplan S, Nature, 206 (1965) 994.
28 Ellman J, Mendel D, Anthony-Cahill S, Noren C J &
Schultz P G, Methods Enzymol, 202 (1991) 301.
29 (a) Ellman J A, Mendel D & Schultz P G, Science, 255 (1992)
197; (b) Koh J T, Cornish V W & Schultz P G, Biochemistry,
36 (1997) 11314; (c) Chapman E, Thorson J S & Schultz P G,
J Am Chem Soc, 119 (1997) 7151; (d) Ting A Y,
Shin I, Lucero C & Schultz P G, J Am Chem Soc, 120
(1998) 7135.
30 (a) Dougherty D A, Chem Rev, 108 (2008) 1642;
(b) Dougherty D A, Curr Opin Chem Biol, 4 (2000) 645.
31 Mehl R A, Anderson J C, Santoro S W, Wang L, Martin A B,
King D S, Horn D M & Schultz P G, J Am Chem Soc,
125 (2003) 935.
32 (a) Wang L & Schultz P G, Angew Chem, 117 (2005) 34;
Angew Chem Int Ed, 44 (2005) 34; (b) Xie J & Schultz P G,
Nat Rev Mol Cell Biol, 7 (2006) 775; (c) Wang L, Brock A,
Herberich B & Schultz P G, Science, 292 (2001) 498.
33 (a) Anderson J C & Schultz P G, Biochemistry, 42 (2003)
9598; (b) Santoro S W, Anderson J C, Lakshman V &
Schultz P G, Nucleic Acids Res, 31 (2003) 6700;
(c) Anderson J C, Wu N, Santoro S W, Lakshman V, King D S
& Schultz P G, Proc Natl Acad Sci USA, 101 (2004) 7566;
(d) Hughes R A & Ellington A D, Nucleic Acids Res, 38
(2010) 6813; (e) Liu C C & Schultz P G, Annu Rev Biochem,
79 (2010) 413; (f) Wang Q, Parrish A R & Wang L,
Chem Biol, 16 (2009) 323; (g) Wu X & Schultz P G,
J Am Chem Soc, 131 (2009) 12497.
34 (a) Chin J W, Cropp T A, Anderson J C, Mukherj M,
Zhang Z W & Schultz P G, Science, 301 (2003) 964;
(b) Young T S, Ahmad I, Brock A & Schultz P G,
Biochemistry, 48 (2009) 2643.
35 (a) Zhang Z, Alfonta L, Tian F, Bursulaya B, Uryu S, King D S
& Schultz P G, Proc Natl Acad Sci USA, 101 (2004) 8882;
(b) Ye S X, Köhrer C, Huber T, Kazmi M, Sachdev P,
Yan E C Y, Bhagat A, Raj Bhandary U L & Sakmar T P,
J Biol Chem, 283 (2008) 1525; (c) Liu W S, Brock A,
Chen S, Chen S B & Schultz P G, Nat Methods, 4 (2007)
239; (d) Köhrer C, Xie L, Kellerer S, Varshney U &
Raj Bhandary U L, Proc Natl Acad Sci USA, 98 (2001) 14310.
36 Wang Q & Wang L, J Am Chem Soc, 130 (2008) 6066.
37 Chin J W, Santoro S W, Martin A B, King D S, Wang L &
Schultz P G, J Am Chem Soc, 124 (2002) 9026.
38 Deiters A & Schultz P G, Bioorg Med Chem Lett, 15 (2005)
1521.
39 Zhang Z W, Wang L, Brock A & Schultz P G, Angew Chem,
114 (2002) 2964; Angew Chem Int Ed, 41 (2002) 2840.
40 (a) Zhang Z W, Smith B A C, Wang L, Brock A, Cho C &
Schultz P G, Biochemistry, 42 (2003) 6735; (b) Wang L,
Wang Z W, Brock A & Schultz P G, Proc Natl Acad Sci
USA,100 (2003) 56.

INDIAN J CHEM, SEC A, AUG-SEPT 2013
988
41 (a) Santoro S W, Wang L, Herberich B, King D S &
Schultz P G, Nat Biotechnol, 20 (2002) 1044; (b) Carrico Z M,
Romanini D W, Mehl R A & Francis M B, Chem Commun,
(2008) 1205; (c) Behrens C R, Hooker J M, Obermeyer A C,
Romanini D W, Katz E M & Francis M B, J Am Chem Soc,
133 (2011) 16398; (d) Tong G J, Hsiao S C, Carrico Z M &
Francis M B, J Am Chem Soc, 131 (2009) 11174.
42 Wang L, Xie J M, Deniz A A & Schultz P G, J Org Chem,
68 (2003) 174.
43 Brustad E, Bushey M L, Lee J W, Groff D, Liu W &
Schultz P G, Angew Chem, 120 (2008) 8344; Angew Chem
Int Ed, 47 (2008) 8220.
44 Stephanopoulos N, Tong G J, Hsiao S C & Francis M B,
ACS Nano, 4 (2010) 6014.
45 Hooker J M, Esser-Kahn A P & Francis M B, J Am Chem
Soc, 128 (2006) 15558.
46 Stephanopoulos N, Carrico Z M & Francis M B, Angew
Chem Int Ed, 48 (2009) 9498.
47 (a) Zhang Z W, Gildersleeve J, Yang Y Y, Xu R, Loo J A,
Uryu S, Wong C H & Schultz P G, Science, 303 (2004) 371;
(b) Liu C C & Schultz P G, Nat Biotechnol, 24 (2006) 1436;
(c) Guo J, Wang J, Lee J S & Schultz P G, Angew Chem Int
Ed, 47 (2008) 6399.
48 Xie J, Supekova L & Schultz P G, ACS Chem Biol, 2 (2007)
474.
49 (a) Chin J W, Martin A B, King D S, Wang L & Schultz P G, Proc Natl Acad Sci USA, 99 (2002) 11020; (b) Bose M, Groff D, Xie J M, Brustad E & Schultz P G, J Am Chem Soc, 128 (2006) 388.
50 (a) Summerer D, Chen S, Wu N, Deiters A, Chin J W &
Schultz P G, Proc Natl Acad Sci USA, 103 (2006) 9785;
(b) Wang J Y, Xie J M & Schultz P G, J Am Chem Soc,
128 (2006) 8738.
51 (a) Lemke E A, Summerer D, Geierstanger B H, Brittain S M
& Schultz P G, Nat Chem Biol, 3 (2007) 769; (b) Wu N,
Deiters A, Cropp T A, King D & Schultz P G, J Am Chem
Soc, 126 (2004) 14306; (c) Deiters A, Groff D, Ryu Y H,
Xie J M & Schultz P G, Angew Chem, 118 (2006) 2794;
Angew Chem Int Ed, 45 (2006) 2728.
52 Brustad E M, Lemke E A, Schultz P G & Deniz A A,
J Am Chem Soc, 130 (2008) 17664.
53 (a) Neumann H, Slusarczyk A L & Chin J W, J Am Chem
Soc, 132 (2010) 2142; (b) Neumann H, Wang K, Davis L,
Garcia-Alai M & Chin J W, Nature, 464 (2010) 441.
54 Wan W, Huang Y, Wang Z Y, Russell W K, Pai P J,
Russell D H & Liu W R, Angew Chem, 122 (2010) 3279;
Angew Chem Int Ed, 49 (2010) 3211.
55 Wu B, Wang Z, Huang Y & Liu W R, ChemBioChem, 13
(2012) 1405.
56 Hoesl M G & Budisa N, Angew Chem Int Ed, 50 (2011)
2896.
57 Huang Y, Russell W K, Wan W, Pai P J, Russell D H &
Liu W, Mol BioSyst, 6 (2010) 683.
58 Doi Y, Ohtsuki T, Shimizu Y, Ueda T & Sisido M,
J Am Chem Soc, 129 (2007) 14458.
59 Wang K H, Neumann H, Peak-Chew S Y & Chin J W,
Nat Biotechnol, 25 (2007) 770.
60 (a) Boyce M & Bertozzi C R, Nat Methods, 8 (2011) 638;
(b) Bertozzi C R, Acc Chem Res, 44 (2011) 651.
61 (a) Tsien R Y, Annu Rev Biochem, 67 (1998) 509;
(b) Lippincott-Schwartz J & Patterson G H, Science,
300 (2003) 87; (c) Zhang J, Campbell R E, Ting A Y &
Tsien R Y, Nat Rev Mol Cell Biol, 3 (2002) 906;
(d) Verkhusha V V & Lukyanov K A, Nat Biotechnol,
22 (2004) 289.
62 (a) Saxon E & Bertozzi C R, Science, 287 (2000) 2007;
(b) Kolb H C & Sharpless K B, Drug Discov Today,
8 (2003) 1128; (c) Agard N J, Prescher J A & Bertozzi C R,
J Am Chem Soc, 126 (2004) 15046.
63 Speers A E & Cravatt B F, Chem Biol, 11 (2004) 535.
64 Adams S A, Campbell R E, Gross L A, Martin B R,
Walkup G K, Yao Y, Llopis J & Tsien R Y, J Am Chem Soc,
124 (2002) 6063.
65 Rostovtsev V V, Green L G, Fokin V V & Sharpless K B,
Angew Chem, 114 (2002) 2708; Angew Chem Int Ed, 41
(2002) 2596.
66 Jewett J C & Bertozzi C R, Chem Soc Rev, 39 (2010) 1272.
67 Sletten E M & Bertozzi C R, Acc Chem Res, 44 (2011) 666.
68 (a) Song W, Wang Y, Qu J, Madden M M & Lin Q, Angew
Chem Int Ed, 47 (2008) 2832; (b) Sanders B C, Friscourt F,
Ledin P A, Mbua N E, Arumugam S, Guo J, Boltje T J,
Popik V V & Boons G-J, J Am Chem Soc, 133 (2011) 949;
(c) McKay C S, Moran J & Pezacki J P, Chem Commun, 46
(2010) 931; (d) Ning X, Temming R P, Dommerholt J,
Guo J, Ania D B, Debets M F, Wolfert M A, Boons G-J &
van Delft F L, Angew Chem, 122 (2010) 3129; Angew Chem
Int Ed, 49 (2010) 3065; (e) Song W, Wang Y, Qu J & Lin Q,
J Am Chem Soc, 130 (2008) 9654.
69 (a) Blackman M L, Royzen M & Fox J M, J Am Chem Soc, 130 (2008) 13518; (b) Devaraj N K, Weissleder R & Hilderbrand S A, Bioconjugate Chem, 19 (2008) 2297; (c) Pipkorn R, Waldeck W, Didinger B, Koch M, Mueller G, Wiessler M & Braun K, J Pept Sci, 15 (2009) 235; (d) Taylor M T, Blackman M L, Dmitrenko O & Fox J M, J Am Chem Soc, 133 (2011) 9646.
70 Kodama K, Fukuzawa S, Nakayama H, Sakamoto K, Kigawa T,
Yabuki T, Matsuda N, Shirouzu M, Takio K, Yokoyama S
& Tachibana K, ChemBioChem, 8 (2007) 232.
71 (a) Ren H, Xiao F, Zhan K, Kim Y-P, Xie H, Xia Z & Rao J,
Angew Chem Int Ed, 48 (2009) 9658; (b) Ou W, Uno T.
Chiu H-P, Grunewald J, Cellitti S E, Crossgrove T, Hao X,
Fan Q, Quinn L L, Patterson P, Okach L, Jones D H,
Lesley S A, Brock A & Geierstanger B H, Proc Natl Acad
Sci USA, 108 (2011) 10437.
72 Sletten E M & Bertozzi C R, J Am Chem Soc, 133 (2011)
17570.
73 Gaertner H F & Offord R E, Bioconjugate Chem, 7 (1996)
38.
74 Schlick T L, Ding Z, Kovacs E W & Francis M B, J Am
Chem Soc, 127 (2005) 3718.
75 Kovacs E W, Hooker J M, Romanini D W, Holder P G,
Barry K E & Francis M B, Bioconjugate Chem, 18 (2007)
1140.
76 Hutchins B M, Kazane S A, Staflin K, Forsyth J S,
Felding-Habermann B, Smider V V & Schultz P G,
Chem Biol, 18 (2011) 299.
77 (a) Scheck R A & Francis M B, ACS Chem Biol, 2 (2007) 247;
(b) Gilmore J M, Scheck R A, Esser-Kahn A P, Joshi N S
& Francis M B, Angew Chem, 118 (2006) 5433; Angew
Chem Int Ed, 45 (2006) 5307.
78 (a) Jencks W P, J Am Chem Soc, 81 (1959) 475; (b) Hang H C
& Bertozzi C R, Acc Chem Res, 34 (2001) 727.
79 Herman S, Loccufier J & Schacht E, Macromol Chem Phys,
95 (1994) 203.

AGRAWAL & HACKENBERGER: SITE-SPECIFIC CHEMICAL MODIFICATIONS OF PROTEINS
989
80 Joshi N S, Whitaker L R & Francis M B, J Am Chem Soc,
126 (2004) 15942.
81 Brocchini S, Godwin A, Balan S, Choi J W, Zloh M &
Shaunak S, Adv Drug Delivery Rev, 60 (2008) 3.
82 Tilley S D & Francis M B, J Am Chem Soc, 128 (2006) 1080.
83 (a) Cornish V W, Hahn K M & Schultz P G, J Am Chem Soc,
118 (1996) 8150; (b) Oshannessy D J, Dobersen M J &
Quarles R H, Immunol Lett, 8 (1984) 273.
84 Byeon J-Y, Limpoco F T & Bailey R C, Langmuir, 26 (2010)
15430.
85 (a) Luchansky S J, Goon S & Bertozzi C R, ChemBioChem,
5 (2004) 371; (b) Yarema K J, Mahal L K, Bruehl R E,
Rodriguez E C & Bertozzi C R, J Biol Chem, 273 (1998)
31168; (c) Lee J H, Baker T J, Mahal L K, Zabner J,
Bertozzi C R, Wiemer D F & Welsh M J, J Biol Chem, 274
(1999) 21878; (d) Mahal L K, Yarema K J & Bertozzi C R,
Science, 276 (1997) 1125.
86 Sadamoto R, Niikura K, Ueda T, Monde K, Fukuhara N &
Nishimura S-I, J Am Chem Soc, 126 (2004) 3755.
87 Gaertner H F, Bose K, Cotton R, Timms D, Camble R &
Offord R E, Bioconjugate Chem, 3 (1992) 262.
88 Kochendoerfer G G, Chen S Y, Mao F, Cressman S,
Traviglia S, Shao H, Hunter C L, Low D W, Cagle E N,
Carnevali M, Gueriguian V, Keogh P J, Porter H, Stratton S M,
Wiedeke M C, Wilken J, Tang J, Levy J J, Miranda L P,
Crnogorac M M, Kalbag S, Botti P, Schindler-Horvat J,
Savatski L, Adamson J W, Kung A, Kent S B & Bradburne J A,
Science, 299 (2003) 884.
89 Hang H C & Bertozzi C R, J Am Chem Soc, 123 (2001)
1242.
90 (a) Zeng Y, Ramya T N C, Dirksen A, Dawson P E &
Paulson J C, Nat Methods, 6 (2009) 207; (b) Dirksen A,
Hackeng T M & Dawson P E, Angew Chem, 118 (2006)
7743; Angew Chem Int Ed, 45 (2006) 7581.
91 (a) Debets M F, van der Doelen C W J, Rutjes F P J T &
van Delft F L, ChemBioChem, 11 (2010) 1168; (b) Bräse S,
Gil C, Knepper K & Zimmermann V, Angew Chem Int Ed,
44 (2005) 5188.
92 Griffin R J, Prog Med Chem, 31 (1994) 121.
93 (a) Hendricks S B & Pauling L, J Am Chem Soc, 47 (1925)
2904; (b) Sidgwick N V, Sutton L E & Thomas W, J Chem
Soc, (1933) 406; (c) Knaggs I E, Nature, 135 (1935) 268.
94 (a) Smith P A S, Open-Chain Nitrogen Compounds, Vol. 2,
(Benjamin, New York), 1966, 211; (b) Boyer J H,
Moriarty R, Darwent B de & Smith P A S, Chem Eng
News, 42 (1964) 6.
95 Rijkers D T S, van Vugt H H R, Jacobs H J F &
Liskamp R M J, Tetrahedron Lett, 43 (2002) 3657.
96 (a) Goddard-Borger E D & Stick R V, Org Lett, 9 (2007)
3797; (b) van Dongen S F M, Teeuwen R L M, Nallani M,
van Berkel S S, Cornelissen J J L M, Nolte R J M &
van Hest J C M, Bioconjugate Chem, 20 (2009) 20.
97 Johansson H & Pedersen D S, Eur J Org Chem, (2012) 4267.
98 (a) Staudinger H & Meyer J, Helv Chim Acta, 2 (1919) 635;
(b) Gololobov Y G & Kasukhin L F, Tetrahedron, 48 (1992)
1353.
99 Kohn M & Breinbauer R, Angew Chem Int Ed Engl, 43
(2004) 3106.
100 Lin F L, Hoyt H M, Van Halbeek H, Bergman R G &
Bertozzi C R, J Am Chem Soc, 127 (2005) 2686.
101 Kohn M, J Pept Sci, 15 (2009) 393.
102 Luchansky S J, Argade S, Hayes B K & Bertozzi C R,
Biochemistry, 43 (2004) 12358.
103 Prescher J A, Dube D H & Bertozzi C R, Nature, 430 (2004)
873.
104 Lemieux G A, de Graffenried C L & Bertozzi C R,
J Am Chem Soc, 125 (2003) 4708.
105 Hangauer M J & Bertozzi C R, Angew Chem, 120 (2008)
2428; Angew Chem Int Ed, 47 (2008) 2394.
106 Cohen A S, Dubikovskaya E ARush, J S & Bertozzi C R,
J Am Chem Soc, 132 (2010) 8563.
107 Cazalis C S, Haller C A, Sease-Cargo L & Chaikof E L,
Bioconjugate Chem, 15 (2004) 1005.
108 Wilkening I, del Signore G & Hackenberger C P R, Chem
Commun, 47 (2011) 349.
109 (a) Serwa R, Wilkening I, del Signore G, Mühlberg M,
Clauβnitzer I, Weise C, Gerrits M & Hackenberger C P R,
Angew Chem Int Ed, 48 (2009) 8234; (b) Serwa R, Majkut P,
Horstmann B, Swiecicki J-M, Gerrits M, Krause E &
Hackenberger C P R, Chem Sci, 1 (2010) 596; (c) Böhrsch V,
Mathew T, Zieringer M, Vallée M R J, Artner L M, Dernedde J,
Haag R & Hackenberger C P R, Org Biomol Chem, 10 (2012)
6211; (d) Wilkening I, del Signore G & Hackenberger C P R,
Chem Commun, (2008) 2932; (e) Jaradat D M M, Hamouda H
& Hackenberger C P R, Eur J Org Chem, 26 (2010) 5004;
f) Nischan N, Chakrabarti A, Serwa R A, Bovee-Geurts P H M,
Brock R & Hackenberger C P R, Angew Chem Int Ed, (2013)
DOI: 10.1002/anie.201303467.Böhrsch V, Serwa R, Majkut P,
Krause E & Hackenberger C P R, Chem Commun, 46 (2010)
3176.
110 (a) Vallée M R J, Majkut P, Wilkening I, Weise C,
Müller G & Hackenberger C P R, Org Lett, 13 (2011)
5440; (b) Vallée M R J, Artner L M, Dernedde J &
Hackenberger C P R, Angew Chem Int Ed, (2013) DOI:
10.1002/anie.201302462.
111 Huisgen R, 1,3-Dipolar Cycloaddition Chemistry, edited by
A Padwa, Wiley, New York) 1984.
112 Huisgen R, Angew Chem, 75 (1963) 604; Angew Chem Int
Ed, 2 (1963) 565.
113 Hartzel L W & Benson F R, J Am Chem Soc, 76 (1954) 667.
114 (a) Tornoe C W, Christensen C & Meldal M, J Org Chem, 67
(2002) 3057; (b) Himo F, Lovell T, Hilgraf R, Rostovtsev V V,
Noodleman L, Sharpless K B & Fokin V V, J Am Chem Soc,
127 (2005) 210.
115 (a) Rodionov V O, Presolski S I, Diaz D D, Fokin V V &
Finn M G, J Am Chem Soc, 129 (2007) 12705; (b) Chan T R,
Hilgraf R, Sharpless K B & Fokin V V, Org Lett, 6 (2004)
2853.
116 Hong V, Udit A K, Evans R A & Finn M G, ChemBioChem,
9 (2008) 1481.
117 (a) Kolb H C, Finn M G & Sharpless K B, Angew Chem, 113
(2001) 2056; Angew Chem Int Ed, 40 (2001) 2004;
(b) Hawker C J, Fokin V V, Finn M G & Sharpless K B,
Aust J Chem, 60 (2007) 381; (c) Presolski S I, Hong V P &
Finn M G, Curr Protoc Chem Biol, (2011) 153.
118 (a) Ladmiral V, Mantovani G, Clarkson G J, Cauet S, Irwin J L
& Haddleton D M, J Am Chem Soc, 128 (2006) 4823;
(b) Quémener D, Davis T P, Barner-Kowollik C & Stenzel M H,
Chem Commun, (2006) 5051; (c) Li H, Riva R, Jerome R &
Lecomte P, Macromolecules, 40 (2007) 824; (d) Lutz J-F,
Angew Chem, 119 (2007) 1036; Angew Chem Int Ed, 46
(2007) 1018.

INDIAN J CHEM, SEC A, AUG-SEPT 2013
990
119 (a) van der Peet P, Gannon C T, Walker I, Dinev Z, Angelin M,
Tam S, Ralton J E, McConville M J & Williams S J,
ChemBioChem, 7 (2006) 1384; (b) Goess B C, Hannoush R N,
Chan L K, Kirchhausen T & Shair M D, J Am Chem Soc, 128
(2006) 5391; (c) Xie J & Seto C T, Bioorg Med Chem, 15
(2007) 458.
120 a) Speers A E & Cravatt B F, ChemBioChem, 5 (2004) 41;
(b) Speers A E, Adam G C & Cravatt B F, J Am Chem Soc,
125 (2003) 4686.
121 See highlights on the topic in: Hackenberger C P R &
Hinderlich S, ChemBioChem, 8 (2007) 1763.
122 Seo T S, Bai X, Ruparel H, Li Z, Turro N J & Ju J, Proc Natl
Acad Sci USA, 101 (2004) 5488.
123 (a) Wang Q, Chan T R, Hilgraf R, Fokin V V, Sharpless K B
& Finn M G, J Am Chem Soc, 125 (2003) 3192;
(b) Prasuhn D E, Yeh R M, Obenaus A, Manchester M &
Finn M G, Chem Commun, (2007) 1269.
124 Boutureira O, D’Hooge F, Fernández-González M,
Bernardes G J L, Sánchez-Navarro M, Koeppe J R &
Davis B G, Chem Commun, 46 (2010) 8142.
125 Sieber S A, Niessen S, Hoover H S & Cravatt B F, Nat Chem
Biol, 2 (2006) 274.
126 a) Wolbers F, ter Braak P, Le Gac S, Luttge R, Andersson H,
Vermes I & van den Berg A, Electrophoresis, 27 (2006)
5073; (b) Stohs S J & Bagchi D, Free Radical Biol Med, 18
(1995) 321.
127 Presolski S I, Mamidyala S K, Manzenrieder F & Finn M G,
ACS Comb Sci, 14 (2012) 527.
128 Deiters A, Cropp T A, Summerer D, Mukherji M & Schultz
P G, Bioorg Med Chem Lett, 14 (2004) 5743.
129 (a) Dirks A J T, van Berkel S S, Hatzakis N S, Opsteen J A,
van Delft F L, Cornelissen J J L M, Rowan A E, van Hest J C M,
Rutjes F P J T & Nolte R J M, Chem Commun, 33 (2005)
4172; (b) Tzokova N, Fernyhouph C M, Topham P D,
Sandon N, Adams D J, Butler M F, Armes S P & Ryan A J,
Langmuir, 25 (2009) 2479; (c) Gupta S S, Kuzelka J, Singh P,
Lewis W G, Manchester M & Finn M G, Bioconjugate
Chem, 16 (2005) 1572; (d) Shieh P, Hangauer M J &
Bertozzi C R, J Am Chem Soc, 134 (2012) 17428;
(e) Artner L M, Merkel L, Bohlke N, Beceren-Braun F,
Weise C, Dernedde J, Budisa N & Hackenberger C P R,
Chem Commun, 48 (2012) 522.
130 Hong V, Presolski S I, Ma C & Finn M G, Angew Chem Int
Ed, 48 (2009) 9879.
131 Turner R, Jarrett A D, Goebel P & Mallon B J, J Am Chem
Soc, 95 (1972) 790.
132 (a) Wittig G & Krebs A, Chem Ber, 94 (1961) 3260;
(b) Lallana E, Riguera R & Fernandez-Megia E, Angew
Chem, 123 (2011) 8956; Angew Chem Int Ed, 50 (2011)
8794; (c) Debets M F, van Berkel S S, Dommerholt J,
Dirks A J, Rutjes F P J T & van Delft F L, Acc Chem Res,
44 (2011) 805.
133 Baskin J M, Prescher J A, Laughlin S T, Agard N J,
Chang P V, Miller I A, Lo A, Codelli J A & Bertozzi C R,
Proc Natl Acad Sci USA, 104 (2007) 16793.
134 (a) Codelli J A, Baskin J M, Agard N J & Bertozzi C R, J Am
Chem Soc, 130 (2008) 11486; (b) Agard N J, Baskin J M,
Prescher J A, Lo A & Bertozzi C R, ACS Chem Biol,
1 (2006) 644; (c) Sletten E M & Bertozzi C R, Org Lett,
10 (2008) 3097.
135 Beatty K E, Fisk J D, Smart B P, Lu Y Y, Szychowski J,
Hangauer M J, Baskin J M & Bertozzi C R, ChemBioChem,
11 (2010) 2092.
136 Beatty K E, Szychowski J, Fisk J D & Tirrell D A,
ChemBioChem, 12 (2011) 2137.
137 Plass T, Milles S, Koehler C, Schultz C & Lemke E A,
Angew Chem Int Ed, 50 (2011) 3878.
138 (a) Laughlin S T, Baskin J M, Amacher S L & Bertozzi C R,
Science, 320 (2008) 664; (b) Dehnert K W, Baskin J M,
Laughlin S T, Beahm B J, Naidu N N, Amacher S L &
Bertozzi C R, ChemBioChem, 13 (2012) 353.
139 Ning X H, Guo J, Wolfert M A & Boons G J, Angew Chem,
120 (2008) 2285; Angew Chem Int Ed, 47 (2008) 2253.
140 Jewett J C, Sletten E M & Bertozzi C R, J Am Chem Soc,
132 (2010) 3688.
141 (a) Debets M F, van Berkel S S, Schoffelen S, Rutjes F P J T,
van Hest J C M & van Delft F L, Chem Commun, 46 (2010)
97; (b) Kuzmin A, Poloukhtine A, Wolfert M A & Popik V V,
Bioconjugate Chem, 21 (2010) 2076; (c) Poloukhtine A A,
Mbua N E, Wolfert M A, Boons G-J & Popik V V, J Am
Chem Soc, 131 (2009) 15769; (d) Stockmann H, Neves A A,
Stairs S, Ireland-Zecchini H, Brindle K M & Leeper F J,
Chem Sci, 2 (2011) 932; (e) Mbua N E, Guo J, Wolfert M A,
Steet R & Boons G-J, ChemBioChem, 12 (2011) 1912;
(f) Jewett J C & Bertozzi C R, Org Lett, 13 (2011) 5937;
(g) Gordon C G, Mackey J L, Jewett J C, Sletten E M,
Houk K N & Bertozzi C R, J Am Chem Soc, 134 (2012)
9199.
142 Möller H, Böhrsch V, Bentrop J, Bender J, Hinderlich S &
Hackenberger C P R, Angew Chem Int Ed, 51 (2012) 5986.
143 Yao J Z, Uttamapinant C, Poloukhtine A, Baskin J M,
Codelli J A, Sletten E M, Bertozzi C R, Popik V V & Ting A Y,
J Am Chem Soc, 134 (2012) 3720.
144 (a) Ess D H, Jones G O & Houk K N, Org Lett, 10 (2008)
1633; (b) Schoenebeck F, Ess D H, Jones G O & Houk K N,
J Am Chem Soc, 131 (2009) 8121.
145 (a) Chenoweth K, Chenoweth D & Goddard W A, Org
Biomol Chem, 7 (2009) 5255; (b) Dommerholt J, Schmidt S,
Temming R, Hendriks L J A, Rutjes F P J T, van Hest J C M,
Lefeber D J, Friedl P & van Delft F L, Angew Chem, 122
(2010) 9612; Angew Chem Int Ed, 49 (2010) 9422.
146 Sletten E M, Nakamura H, Jewett J C & Bertozzi C R,
J Am Chem Soc, 132 (2010) 11799.
147 de Almeida G, Sletten E M, Nakamura H, Palaniappan K K
& Bertozzi C R, Angew Chem Int Ed, 51 (2012) 2443.
148 Banert K & Plefka O, Angew Chem, 123 (2011) 6295;
Angew Chem Int Ed, 50 (2011) 6171.
149 Bernardes G J L, Chalker J M, Errey J C & Davis B G,
J Am Chem Soc, 130 (2008) 5052.
150 (a) van Berkel S S, Dirks A J, Debets M F, van Delft F L,
Cornelissen J J L M, Nolte R J M & Rutjes F P J T,
ChemBioChem, 8 (2007) 1504; (b) Gutsmiedl K, Wirges C
T, Ehmke V & Carell T, Org Lett, 11 (2009) 2405.
151 van Berkel S S, Dirks A J, Meeuwissen S A, Pingen D L L,
Boerman O C, Laverman P, van Delft F L, Cornelissen J J L M
& Rutjes F P J T, ChemBioChem, 9 (2008) 1805.
152 Wang Y, Hu W J, Song W, Lim R K V & Lin Q, Org Lett,
10 (2008) 3725.
153 Rossin R, Verkerk P R, van den Bosch S M, Vulders R C M,
Verel I, Lub J & Robillard M S, Angew Chem Int Ed, 49
(2010) 3375.

AGRAWAL & HACKENBERGER: SITE-SPECIFIC CHEMICAL MODIFICATIONS OF PROTEINS
991
154 (a) Devaraj N K, Hilderbrand S, Upadhyay R, Mazitchek R
& Weissleder R, Angew Chem Int Ed, 49 (2010) 2869;
(b) Liu D S, Tangpeerachaikul A, Selvaraj R,
Taylor M T, Fox J M & Ting A Y, J Am Chem Soc, 133
(2011) 19769; (c) Li Z, Cai H, Kassink M, Blackman M L,
Brown R C D, Contia P S & Fox J M, Chem Commun,
46 (2010) 8043.
155 Devaraj N K, Upadhyay R, Haun J B, Hilderbrand S A &
Weissleder R, Angew Chem Int Ed, 48 (2009) 7013.
156 Patterson D M, Nazarova L A, Xie B, Kamber D N &
Prescher J A, J Am Chem Soc, 134 (2012) 18638.
157 (a) Hong S H & Grubbs R H, J Am Chem Soc, 128 (2006)
3508; (b) Jordan J P & Grubbs R H, Angew Chem, 119
(2007) 5244; Angew Chem Int Ed, 46 (2007) 5152;
(c) Binder J B, Blank J J & Raines R T, Org Lett, 9 (2007)
4885; (d) Gulajski L, Michrowska A, Naroznik J,
Kaczmarska Z, Rupnicki L & Grela K, ChemSusChem,
1 (2008) 103; (e) Burtscher D & Grela K, Angew Chem
Int Ed, 48 (2009) 442.
158 Chalker J M, Lin Y A, Boutureira O & Davis B G,
Chem Commun, (2009) 3714.
159 (a) Lin Y A, Chalker J M & Davis B G, J Am Chem Soc, 132
(2010) 16805; (b) Lin Y A & Davis B G, J Org Chem, 6
(2010) 1219; (c) Lin Y A, Chalker J M & Davis B G,
ChemBioChem, 10 (2009) 959.
160 Xie J, Wang L, Wu N, Brock A, Spraggon G & Schultz P G,
Nat Biotechnol, 22 (2004) 1297.
161 Kodama K, Fukuzawa S, Nakayama H, Kigawa T, Sakamoto
K, Yabuki T, Matsuda N, Shirouzu M, Takio K, Tachibana K
& Yokoyama S, ChemBioChem, 7 (2006) 134.
162 (a) Chalker J M, Wood C S C & Davis B G, J Am Chem Soc,
131 (2009) 16346; (b) Böhrsch V & Hackenberger C P R,
ChemCatChem, 2 (2010) 243.
163 Spicer C D & Davis B G, Chem Commun, 47 (2011) 1698.





























