Embed Size (px)
Citation preview

Simple and highly efficient BAC recombineering usinggalK selectionSoslashren Warming Nina Costantino1 Donald L Court1 Nancy A Jenkins and
Neal G Copeland
Mouse Cancer Genetics Program and 1Gene Regulation and Chromosome Biology LaboratoryNational Cancer Institute Frederick MD 21702-1201 USA
Received January 11 2005 Revised and Accepted February 4 2005
ABSTRACT
Recombineering allows DNA cloned in Escherichiacoli to be modified via lambda (l) Red-mediatedhomologous recombination obviating the need forrestriction enzymes and DNA ligases to modify DNAHere we describe the construction of three newrecombineering strains (SW102 SW105 and SW106)that allow bacterial artificial chromosomes (BACs) tobe modified using galK positivenegative selectionThis two-step selection procedure allows DNA to bemodified without introducing an unwanted selectablemarker at the modification site All three strains con-tain an otherwise complete galactose operon exceptfor a precise deletion of the galK gene and a defect-ive temperature-sensitive l prophage that makesrecombineering possible SW105 and SW106 cellsin addition carry L-arabinose-inducible Cre or Flpgenes respectively The galK function can be selec-ted both for and against This feature greatly reducesthe background seen in other negative-selectionschemes and galK selection is considerably moreefficient than other related selection methodspublished We also show how galK selection can beused to rapidly introduce point mutations deletionsand loxP sites into BAC DNA and thus facilitatefunctional studies of SNP andor disease-causingpoint mutations the identification of long-range regu-latory elements and the construction of conditionaltargeting vectors
INTRODUCTION
Bacterial artificial chromosomes (BACs) have become theDNA of choice for genomic sequencing due to their high
stability and large insert size (100ndash300 kb) (1) BACs arealso being used more and more for making transgenic micesince in many cases all of the important regulatory sequencesrequired for normal gene expression can be found on a singleBAC (23) Many laboratories also use BACs as the startingpoint for making gene-targeting constructs for manipulatingmouse genes using ES cell technology (knock-outs knock-insand conditional targeting using CreloxP) (45)
Recombineering (recombination-mediated genetic engi-neering) makes it possible to modify BAC DNA via homo-logous recombination [reviewed in (67)] Recombineering ismade possible through the use of three l Red-encoded genesexo bet and gam exo encodes a 50ndash30 exonuclease that pro-duces 30 overhangs from introduced double-stranded DNAtargeting cassettes (dsDNA) bet encodes a pairing proteinthat binds to the 30 overhangs and mediates its annealingand homologous recombination with complementary DNApresent on the BAC At the same time gam encodes an inhib-itor of the Escherichia coli RecBCD exonuclease and therebyprotects the linear DNA-targeting cassette from degradationby RecBCD l Red (or the corresponding RecE and RecTgenes of the prophage Rac) can be expressed from a multicopyplasmid using an inducible promoter (89) Alternatively thesegenes can be expressed from a stably integrated defectivel prophage where exo bet and gam are controlled by thestrong phage promoter pL under stringent control of thetemperature-sensitive repressor cI857 (1011) In the pro-phage system exo bet and gam are not expressed when thebacteria are kept at 32C By shifting the bacteria to 42C foras little as 15 min the genes are rapidly induced to very highlevels and homologous recombination is very efficient
Using recombineering one can easily subclone a genomicfragment from a BAC by gap repair either for use as atransgene directly or for subsequent manipulation to make agene-targeting construct The introduction of selectable mark-ers into a BAC is also very easy using recombineering How-ever a major limitation to the usefulness of BACs is the easeand efficiency with which one can make subtle and lsquoseamlessrsquo
To whom correspondence should be addressed at Mouse Cancer Genetics Program Center for Cancer Research National Cancer Institute West 7th Street at FortDetrick Bldg 539 PO Box B Frederick MD 21702-1201 USA Tel +1 301 846 1260 Fax +1 301 846 6666 Email Copelandncifcrfgov
ordf The Author 2005 Published by Oxford University Press All rights reserved
The online version of this article has been published under an open access model Users are entitled to use reproduce disseminate or display the open accessversion of this article for non-commercial purposes provided that the original authorship is properly and fully attributed the Journal and Oxford University Pressare attributed as the original place of publication with the correct citation details given if an article is subsequently reproduced or disseminated not in its entirety butonly in part or as a derivative work this must be clearly indicated For commercial re-use please contact journalspermissionsoupjournalsorg
Nucleic Acids Research 2005 Vol 33 No 4 e36doi101093nargni035
mutations like point mutations and clean deletions or introducein-frame fusions of cDNAs or epitope tags without leaving atthe same time a selectable marker or a loxPFrt site at themodification site A handful of methods for making such muta-tions all based on homologous recombination in Ecoli havebeen developed One method is RecA dependent and relies onthe use of a shuttle vector and two recombination steps integ-ration followed by the resolution of the co-integrate (212)This method requires considerable up-front effort using tradi-tional cloning with restriction enzymes and ligation A simplerand more widely used method is based on positivenegativeselection using eg a sacBndashneo fusion gene (8) neo (kana-mycin) resistance is used for positive selection while sucrosetoxicity resulting from sacB expression is used for negativeselection A major drawback of this selection system is thatspontaneous point mutations in the sacB portion of the sacB-neo fusion gene can occur without influencing the bacteriarsquosability to grow on kanamycin These sacB mutants signific-antly increase the background after negative selection Arelated method is based on counterselection using a recogni-tion site for a rare restriction enzyme like I-SceI (13) For thismethod to work efficiently the I-SceI restriction enzyme hasto be induced in trans and since there is no selection formaintaining the correct recognition sequence for this enzymethe background due to point mutation or deletion of the restric-tion site is high A method for BAC modification withoutselection has also been developed (14) Although relativelyefficient this method relies on a PCR-based screening of theresulting colonies to identify the desired clones Since there isno selection step the number of manipulations made possibleby this selection procedure is more limited
Here we report the development of a novel and highlyefficient galK-based positivenegative selection system forthe manipulation of BACs The Ecoli galactose operon con-sists of four genes galE galT galK and galM which arenecessary for growth and utilization of galactose as theonly carbon source The galK gene product galactokinasecatalyzes the first step in the galactose degradation pathwayphosphorylating galactose to galactose-1-phosphate Galacto-kinase also efficiently catalyzes the phosphorylation of agalactose analog 2-deoxy-galactose (DOG) The product ofthis reaction cannot be further metabolized leading to a toxicbuild-up of 2-deoxy-galactose-1-phosphate (15) Thus bothpositive and negative selection can be conferred by galKBecause galK is used for both selection steps backgroundfollowing negative selection is reduced and no colonyscreening is necessary The small size of the galK cassette(around 1200 bp plus homology arms) also makes it easier toamplify by PCR and to introduce into bacteria using electro-poration
MATERIALS AND METHODS
General recombineering and galK selection
Recombineering was performed as follows 500 ml of anovernight culture was diluted in 25 ml LuriandashBertani(LB) medium with or without chloramphenicol selection(125 mgml) in a 50 ml baffled conical flask and grown at32C in a shaking waterbath to an OD600 of 06 Then 10 mlwas transferred to another baffled 50 ml conical flask and
heat-shocked at 42C for exactly 15 min in a shakingwaterbath The remaining culture was left at 32C as theuninduced control After 15 min the two samples inducedand uninduced were briefly cooled in an icewaterbathslurry and then transferred to two 15 ml Falcon tubes (BDBiosciences) and pelleted using 5000 rpm (eppendorf cen-trifuge 5804R rotor A-4-44) at 0C for 5 min The supernatantwas poured off and the pellet was resuspended in 1 ml ice-coldddH2O by gently swirling the tubes in an icewaterbath slurrySubsequently 9 ml ice-cold ddH2O was added and the samplespelleted again This step was repeated once more After thesecond washing and centrifugation step the supernatant wasremoved and the pellet (50 ml each) was kept on ice untilelectroporated with PCR product gel-purified fragment ordouble-stranded oligo An aliquot of 25 ml was used foreach electroporation in a 01 cm cuvette (BioRad) at 25 mF175 kV and 200 V After electroporation the bacteria wererecovered in 1 ml LB (15 ml Falcon tube) for 1 h in a 32Cshaking waterbath For the counterselection step (see below)the bacteria were recovered in 10 ml LB in a 50 ml baffledconical flask and incubated for 45 h in a 32C shaking water-bath After the recovery period the bacteria were washedtwice in 1middot M9 salts as follows 1 ml culture was pelletedin an eppendorf tube at 13 200 rpm for 15 s and the super-natant was removed with a pipette The pellet was resuspendedin 1 ml of 1middot M9 salts and pelleted again This washing stepwas repeated once more After the second wash the supernat-ant was removed and the pellet was resuspended in 1 ml of 1middotM9 salts before plating serial dilutions (100 ml 100 ml each of110 1100 and 11000 dilutions) on minimal medium plates(see below) Washing in M9 salts is necessary to remove anyrich media from the bacteria prior to selection on minimalmedium The uninduced samples routinely had a higher degreeof lysisbacterial death after electroporation so the uninducedsamples were diluted in 025ndash075 ml 1middot M9 salts in the finalstep to make up for the difference Detailed protocolsfor recombineering can also be found at our website (httprecombineeringncifcrfgov)
Minimal media and indicator plates
Minimal media and the indicator plates were prepared usingstandard methods (16) We added supplements as indicated
Washing solution 1middot M9 mediumGal positive selection M63 + agar (15 gl Difco BDBiosciences) + D-galactose (02 Sigma) + D-biotin(1 mgl Sigma) + L-leucine (45 mgl Sigma) and ndashchloramphenicol (125 mgml Sigma)Gal counterselection M63 + agar + glycerol (02Fischer) + D-biotin (1 mgl) + L-leucine (45 mgl) +DOG (02 Ferro Pfanstiehl) and ndash chloramphenicol(125 mgml)Gal indicator plates MacConkey agar (Difco BDBiosciences) + D-galactose (1) and ndash chloramphenicol(125 mgml) Plates were prepared using manufacturerrsquosinstructions
Bacterial strains
The strains used in this paper are listed in Table 1 SW101was derived from DY380 by a homologous recombinationalexchange of the mutated gal operon leader sequence with a
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 2 OF 12
441 bp PCR product (Expand High-Fidelity PCR SystemRoche Applied Science) containing the wild-type gal operonleader sequence made using W3110 (17) bacteria as the tem-plate The primers used to amplify the wild-type leader were(Integrated DNA Technologies Inc) F 50-CGACGCATG-CAGGCATGAA-30 and R 50-AGTGGATCACGGTGTC-GATA-30 and the PCR conditions were 94C for 15 s 60Cfor 30 s and 72C for 30 s for 35 cycles The PCR product wasgel purified (GFX kit Amersham Biosciences) and eluted in50 ml ddH2O An aliquot of 25 ml was used in the recombin-eering experiment as described above and the bacteria wereplated on M63 minimal medium + galactose + leucine + biotinThe plates were incubated at 32C for 2ndash3 days A few of themany resulting colonies were streaked for single colonies onindicator plates and a single dark red Gal+ colony was used inthe next step SW102 was derived from SW101 in thefollowing way two homology arms flanking the galK openreading frame (ORF) were PCR amplified using SW101 bac-teria as template and the following primers (Integrated DNATechnologies Inc) recognition sites for restriction enzymesare underlined 50arm F 50-AAATAACTCGAGCAGCTGC-ACGCGCACTTT-30 50arm R 50-AAATAAGAATTCTTCT-TACACTCCGGATTCGC-30 30arm F 50-AAATAAGAAT-TCTGTAAACCATCACAAGGAGCAG-30 30arm R 50-AA-TAAAGCGGCCGCCAGCTGGTTAACGCCCTGA-30 PCRconditions were 94C for 15 s 60C for 30 s and 72C for30 s for 35 cycles The 50 homology arm (171 bp) was digestedwith XhoI and EcoRI and the 30 arm (335 bp) with EcoRI andNotI The digested PCR products were gel purified and triple-ligated into a vector digested with XhoI and NotI The targetingcassette was then released from the backbone using XhoI andNotI digestion followed by gel purification The cassette waseluted in 50 ml ddH2O and 25 ml was used for recombineeringas described above After 45 h of outgrowth in LB media andtwo washes in 1middot M9 salts serial dilutions of the bacteria wereplated on minimal plates containing glycerol as carbon sourceleucine and biotin plus DOG for selection against galK Theplates were incubated for 3 days at 32C A few of the manyresulting colonies were streaked for single whitecolorless(Gal) colonies on indicator plates followed by PCR analysis
and sequencing to check for correct deletion of the galK geneA single verified colony was expanded and a glycerol stockwas made and used for initiation of all subsequent experimentsSW103 and SW104 were derived from EL250 and EL350respectively using the same method as for the derivation ofSW101 SW105 and SW106 were derived from SW103 andSW104 respectively using the same method as for the deriva-tion of SW102 from SW101 SW105 expresses Flp under thecontrol of an arabinose-inducible promoter and SW106expresses Cre under the same promoter
Construction of pgalK
Two rounds of PCR were performed to construct a galK ORFdriven by the prokaryotic em7 promoter The first round wasperformed using galK ORF 1st F and galK ORF R primersusing W3110 bacteria (17) as template The resulting PCRproduct was then used as template for a second round ofPCR using galK ORF 2nd F and galK ORF R primers The pro-duct from the second round of PCR was gel purified anddigested with EcoRI and BamHI and cloned into EcoRI andBamHI digested pBluescript SK pgalK was verified bysequencing [the plasmid sequence is available fromour website (httprecombineeringncifcrfgov)]The primersequences were (restriction sites and the galK ATG are under-lined and the em7 promoter is in italics) galK ORF 1st F50-CCCAGGAGGCAGATCATGAGTCTGAAAGAAAAAA-CTGAAAGAAAAAACACAATCTCTGT-30 galK ORF2nd F AATAAAGAATTCCTGTTGACAATTAATCATCG-GCATAGTATATCGGCATAGTATAATACGACAAGGTGA-GGAACTAAACCCAGGAGGCAGATCATG galK ORF RAATAAAGGATCCTCAGCACTGTCCTGCTCCTT-30 PCRconditions for both rounds were 94C for 15 s 60C for30 s and 72C for 15 min for 30 cycles Primers werefrom Invitrogen
PCR amplification of the galK targeting cassette
Sequences of all galK primers (Invitrogen) used in the experi-ments described in this paper are given below em7-galK wasPCR amplified using 1 ng pgalK as template and the following
Table 1 Recombineering reagents used in this work
Genotype Phenotype Reference
StrainsW3110 Gal+ (17)DH10B mcrA D(mrr-hsdRMS-mcrBC) DlacX74 deoR
endA1 araD139 D(ara leu) 7697 rpsLrecA1 nupG f80dlacZDM15 galU galK
Gal (18)
DY380 DH10B [lc1857 (cro-bioA)ltgtTet] galK+ gal490 Gal (11)SW101 DY380 gal+ Gal+ This workSW102 SW101 DgalK Gal This workEL250 DY380 (cro-bioA)ltgtaraC-PBAD Flpe Gal (11)EL350 DY380 (cro-bioA)ltgtaraC-PBAD Cre Gal (11)SW103 EL250 gal+ Gal+ This workSW104 EL350 gal+ Gal+ This workSW105 SW103 DgalK Gal This workSW106 SW104 DgalK Gal This work
Selection cassettepgalK galK wild-type gene driven by the em 7 promoter This work
ltgt indicates the result of a homologous recombination event Gal+ the ability to use galactose as the sole carbon source Gal resistance to DOG and lack of the abilityto grow on galactose as the sole carbon source
PAGE 3 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
conditions 94C for 15 s 60C for 30 s and 72C for 1 min for30 cycles After completion of PCR 2 ml DpnI was added toeach 25 ml reaction and incubated for 2 h at 37C to removeany plasmid template The DpnI-digested reaction mix was runon a 1 agarose gel over night and the PCR product waspurified and eluted in 50 ml ddH2O An aliquot of 25 ml wasused for each experiment For making the Nras G12D substi-tution the following primers were used for the first step intro-ducing galK Homology to Nras sequence is in italics and thesequence recognizing em7-galK is underlined Nras galK F50-TTTTTGCTGGTGTGAAATGACTGAGTACAAACTGGTGG-TGGTTGGAGCAGCCTGTTGACAATTAATCATCCGCA-30Nras galK R 50-CAAAGTGGTTCTGGATTAGCTGGATCG-TCAAGGCGCTTTTCCCAACACCATCAGCACTGTCCTGC-TCCTT-30 For making 50 75 and 100 kb deletions in theRP23-341F12 BAC the following primers were used (homo-logy to BAC sequence is in italics and the sequence recog-nizing em7-galK is underlined) galK F 341F12 start50-ACTCCCACTGGAAGCTTTTTACAAAACATGTGTTGCT-GACATGTTGACAGCCTGTTGACAATTAATCATCGGCA-30 galK R 341F12 50 kb 50-ACCCAAACCAAACAACAT-CCAAACCAAAAACACAGACAAAACCAAATATGTCAGCAC-TGTCCTGCTCCTT-30 galK R 341F12 75 kb 50-ACACTAAGC-CAAACTCCTTGCCTGGGCTATTTCTCTTTGTTTTTCCAAATTCAGC-ACTGTCCTGCTCCTT-30 galK R 341F12 100 kb 50-TATGT-GTCTGTGTGTGTATGTACAGTTCTTTGTTTTTGTTTTTTTT-CTTTTCAGCACTGTCCTGCTCCTT-30 For insertion of aloxP511 site in the RP23-341F12 BAC the following primerswere used for the first step (introduction of galK) Homology toBAC sequence is in italics and the sequence recognizing em7-galK is underlined 95 kb loxP511 galK F 50-GGACA-GAGGCGTGACAGACGTGTGAGCTCCGTGGACAACTCTC-CCCGAAGCCTGTTGACAATTAATCATCGGCA-30 95 kbloxP511 galK R 50-GACTCTGAGCAGCAACGGCTGAGCCT-CACTTGAGAGGGTCCCTGAGTCACTCAGCACTGTCCTG-CTCCTT-30
Oligos for recombineering
Oligos used to replace the galK-targeting cassette wereobtained from Invitrogen dsDNA was used and the oligos(sense and antisense) annealed in vitro 10 mg of each oligo(sense and antisense) was mixed in an eppendorf tube in a totalvolume of 100 ml of 1middot PCR buffer (Expand High FidelityPCR kit Roche Applied Science) and boiled for 5 minallowed to cool to room temperature for 30 min ethanol pre-cipitated and resuspended in 100 ml ddH2O to a final concen-tration of 200 ngml double-stranded oligo An aliquot of 1 ml(200 ng) was used in the recombineering experiments Tointroduce the G12D (GltgtA) point mutation in the NrasBAC CITB 50J2 the following oligos were used for thesecond step (the introduced adenosinethymidine base pairis underlined the flanking sequences are homologous to theNras BAC sequence) G12D S 50-TTTTTGCTGGTGT-GAAATGACTGAGTACAAACTGGTGGTGGTTGGAGC-AGATGGTGTTGGGAAAAGCGCCTTGACGATCCAGC-TAATCCAGAACCACTTT-30 G12D AS 50-AAAGTGGT-TCTGGATTAGCTGGATCGTCAAGGCGCTTTTCCCAA-CACCATCTGCTCCAACCACCACCAGTTTGTACTCAG-TCATTTCACACCAGCAAAAA-30 To introduce a loxP511site in the RP23-341F12 BAC the following oligos were used
for the second step (loxP511 is underlined the flankingsequences are homologous to the BAC sequence aroundposition 95 kb) 95 kb loxP511 S 50-ACGTGTGAGCTC-CGTGGACAACTCTCCCCGAAGATAACTTCGTATAGT-ATACATTATACGAAGTTATGTGACTCAGGGACCCTC-TCAAGTGAGGCTCAGC-30 95 kb loxP511 AS 50-GCTG-AGCCTCACTTGAGAGGGTCCCTGAGTCACATAACTT-CGTATAATGTATACTATACGAAGTTATCTTCGGGGA-GAGTTGTCCACGGAGCTCACACGT-30
Verification of positive recombinants
In the G12D Nras experiment the selected Gal clones wereanalyzed by SpeI digestion of BAC miniprep DNA usingunmodified CITB 50J2 BAC DNA as a control Clones with-out rearrangements were analyzed by PCR using 1 ml BACminiprep DNA as the template The PCR products were gelpurified and sequenced using the same primers as were usedfor PCR Primers flanking the targeted mutation were Nrastest F 50-CACTCATCTGCAAGGAATGCT-30 Nras test R50-CCTCAGTAAGCACGAACTTGT-30 PCR conditionswere 94C for 15 s 60C for 30 s and 72C for 30 s for30 cycles Modifications of the RP23-341F12 BAC (50 75and 100 kb deletions and the introduction of a loxP511 site)were tested by SpeI restriction analysis of BAC miniprep DNAand compared with unmodified 341F12 BAC DNA In theloxP511 experiment clones 3 5 and 6 were further testedfor correct insertion of the loxP511 site by transforming1 ml of BAC miniprep DNA into electrocompetent andarabinose-induced EL350 cells (11) and plating on LB plateswith chloramphenicol Two colonies from each starting clonewere tested by SpeI digestion of BAC miniprep DNA for the95 kb Cre-mediated deletion Finally the Cre-recombinedclones were tested by PCR with one primer mapping to theend of the pBACe36 BAC backbone and the other mapping toa position 95 kb away on the wild-type BAC The primers(Invitrogen) used for this analysis were 95 kb loxP511 checkF 50-GCGGATGAATGGCAGAAATTC-30 95 kb LoxP511check R 50-TTTGCCAGACTGGTGCCTAA-30 PCR condi-tions were 94C for 15 s 60C for 30 s and 72C for 30 s for30 cycles The resulting PCR bands were gel purified andconfirmed by sequencing using the same primers as wereused for the PCR amplification The follow-up experimentfor testing the source of the observed BAC deletions wasdone as described above
BACs
Mouse BACs were obtained from Invitrogen The RP23-341F12 BAC (C57BL6 DNA) was chosen based on positionwithin the mouse Ebfaz gene using the UCSC genome browser(httpgenomeucscedu) The CITB 50J2 BAC (CJ7129SVDNA) was identified by screening a CITB mouse BAC library(Invitrogen) with an Nras genomic probe using standardhybridization methods The BAC clones were streaked forsingle colonies and characterized by PCR and restriction ana-lysis before proceeding to the recombineering experimentsTwo methods were used for DNA preparation For BAC mini-preps (1ndash15 mg) we used the following protocol 5 ml over-night LB culture (15 ml Falcon tube) was pelleted for 5 min at5000 rpm the supernatant removed and the pellet wasdissolved in 250 ml buffer P1 (miniprep kit Qiagen) and
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 4 OF 12
transferred to an eppendorf tube An aliquot of 250 ml P2buffer was added followed by mixing by inversion and incuba-tion for lt5 min at room temperature An aliquot of 250 ml N3buffer was added followed by mixing and incubation on ice for5 min The supernatant was cleared by two rounds of centri-fugation at 13 200 rpm for 5 min in a tabletop centrifugeEach time the supernatant was transferred to a new tube DNAwas precipitated by adding 750 ml isopropanol mixing andincubating on ice for 10 min and centrifugation for 10 min at13 200 rpm The pellet was washed once in 70 ethanol andthe dry pellet was dissolved in 50 ml TE An aliquot of 40 ml(1 mg) was used for restriction analysis and 1 ml was used astemplate for PCR analysis or transformation of electrocom-petent bacteria Large-scale preparations of BAC DNA(25ndash100 mg) were done using the Nucleobond BAC maxikit from Clontech (BD Biosciences) following the manufac-turerrsquos protocol
RESULTS
Generation of SW102 cells
To see whether we could develop a more efficient selectivesystem for BAC recombineering we looked for a single select-able marker that could be used for both positive and negativeselection We focused on Ecoli galK because both selectionsteps could be done using galK and its small size makes it easyto amplify by PCR We previously reported the developmentof a bacterial strain DY380 that is readily transformable withBAC DNA due to its DH10B origin (1) DY380 cells alsoharbor the defective l prophage required for recombineeringThe defective prophage in DY380 was transferred into DH10Bwith a P1 phage lysate obtained from DY363 cells [for detailssee (11)] DY363 cells carry a 1200 bp IS2 insertion elementgal490 in the mRNA leader sequence of the galactose operonpreventing gal gene transcription Since the galactose operonis directly proximal to the site of insertion of the defective
prophage the gal490 mutation was also transferred to DY380during P1 transduction Thus DY380 is phenotypically Gal
(galactose minus) and therefore unable to grow on galactoseminimal medium
Recombineering was used to correct this problem From thewild-type Ecoli strain W3110 (17) we PCR amplified a 441bp fragment from the wild-type gal promoter which spans theregion containing the IS2 insertion element in DY380 ThisPCR product was introduced into DY380 and Gal+ recombin-ants were selected for growth on galactose minimal medium(see Materials and Methods) We named this strain SW101(Table 1) This strain is identical to DY380 except that it lacksthe IS2 insertion element and has been made wild type for thegal operon We then made a precise deletion of the galK geneleaving all other genes in the galactose operon intact This wasachieved by PCR amplifying two homology arms of 171 and335 bp respectively flanking the galK ORF and cloning themtogether into a plasmid using three-way ligation Since theORF of galK overlaps that of galM we left behind the last33 bp of galK (Figure 1) to allow for proper translation ofgalM The targeting cassette consisting of a 512 bp linearfragment was electroporated into heat-induced and electro-competent SW101 cells and the recombinant clones selectedon minimal medium containing glycerol as the carbon sourceand DOG for selection against galK The genotype of theresulting strain SW102 (Table 1) was confirmed by PCRanalysis and sequencing of the modified region (Figure 1)This strain now harbors not only the defective l prophagebut also a functional gal operon except for the deletion ofgalK The l prophage is located between the galactose operonand the biotin operon and the bio operon in these cells isnonfunctional causing a biotin requirement DH10B andall derived strains including SW102 are also deficient inleucine metabolism (18) When using minimal medium onetherefore needs to add both biotin and leucine to the plates toallow SW102 growth (see Materials and Methods)
Figure 1 Sequence analysis of the galactose operon in strains DY380 (A) SW101 (B) and SW102 (C) In SW102 the ORF of galK was deleted leaving only 33 bp ofgalK behind to make sure that translation of galM is initiated properly EcoRI the restriction site used to clone the 50 and 30 homology arms flanking galK
PAGE 5 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
galK expression cassette
Having produced a bacterial strain SW102 which is galKdefective the next step was to make a galK expression cassettethat could be used to restore the bacteriarsquos ability to grow ongalactose by providing galK in trans This was achieved byPCR amplification of the wild-type galK ORF from W3110cells We then added a minimal bacterial promoter em7 usinga two-step PCR approach (see Materials and Methods) andcloned the expression cassette into pBluescript SK We callthis plasmid pgalK (Table 1) The constitutively active galKexpression cassette can easily be amplified by PCRwith homology arms added to the primers (see Materialsand Methods)
Making a single base pair substitution
The general scheme for making mutations in BACs using galKselection is depicted in Figure 2 To test the galK selectionsystem for BAC recombineering we decided to introduce apoint mutation into a BAC containing the murine Nras gene(CITB clone 50J2) The sequence of the glycine-coding codon12 is GGT By changing this codon to GAT we would obtainthe desired mutation G12D In order to introduce this muta-tion into the BAC we first amplified the galK expressioncassette by PCR using primers with 50 bp of homology toeither side of the second position of the GGT codon Followinghomologous recombination this targeting would introduce a1 bp deletion into codon 12 in addition to inserting the galKselection cassette Instead of deleting the basepair the galKcassette could have been inserted right next to the basepairinstead SW102 cells containing the 50J2 BAC were heat-induced and made electrocompetent and then electroporatedwith the galK cassette Gal+ recombinant colonies were selec-ted for growth on galactose minimal medium with chloram-phenicol to maintain the BAC Bacteria grow more slowly onminimal media than on rich media and we generally pickcolonies after 2ndash3 days For this first step we do not expectany background colonies on the non-induced control plates ifthe pgalK plasmid is properly eliminated (see Materials andMethods) To purify the Gal+ colonies we streaked a fewcolonies on MacConkey galactose indicator plates to obtainsingle bright pinkred Gal+ colonies One of these single-cloned colonies was picked to initiate a culture for thenext step counterselection We find that there is no need toanalyze the Gal+ colonies further before proceeding tocounterselection
A 100 bp dsDNA oligo was then prepared by annealing twocomplementary oligos having 49 and 50 bp homologyrespectively to either side of the desired mutation a singleAT bp An aliquot of 200 ng of this oligo was then electro-porated into heat-induced and electrocompetent SW102 Gal+
cells containing the galK modified 50J2 BAC After electro-poration the bacteria were allowed 45 h outgrowth in a 32Cshaking waterbath The bacteria were then washed in M9 saltsto remove any rich medium and plated on minimal mediumwith glycerol DOG and chloramphenicol The 45 h out-growth is necessary to obtain complete segregation of therecombinant BACs containing the mutation After 3 dayswe obtained colonies with a ratio of 10ndash1001 when comparingplates with heat-induced to non-induced bacteria We picked12 colonies from the heat-induced plates for BAC minipreps
followed by SpeI restriction analysis (Figure 3A) Ten out ofthe twelve clones had the same restriction pattern as theunmodified 50J2 BAC DNA suggesting that the desiredreplacement of the galK gene by the point mutation hadoccurred This was confirmed by PCR amplification followed
Figure 2 Overview of the galK selection scheme The result of the firsttargeting event is the insertion of constitutively active galK into a definedposition on the BAC by selection on minimal medium containing galactoseand chloramphenicol to select for the maintenance of the BAC The bacteria arenow phenotypically Gal+ Next the galK cassette is replaced by a dsDNA oligoa PCR product or a cloned dsDNA fragment carrying a desired mutation(indicated by a star) and flanked by the same homology arms used in thefirst selection step This is achieved by negative selection using minimalmedium containing 2-deoxy-galactose (DOG) with glycerol as the sole carbonsource The bacteria become phenotypically Gal H1 and H2 homology arms1 and 2 respectively cat chloramphenicol acetyl transferase gene ori2 BACorigin of replication galK Ecoli galactokinase gene driven by a minimalpromoter
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 6 OF 12
by sequencing of the modified region of the 10 BAC clones(Figure 3B) All 10 sequenced clones had the desired pointmutation The effective recombination efficiency in thisexperiment however was 812 (67) since 2 of the 10 clonesalso had an additional single base pair deletion (clones 9 and11) These deletions probably occurred during oligo synthesissince we did not purify the oligos beyond desalting (19)Because of the high efficiency in this system there was noneed to pre-screen the selected colonies prior to picking forminipreps
Large BAC deletions
A drawback of using BACs for the production of transgenicmice is the frequent presence of other genes on the BAC inaddition to the gene of interest This is especially a problem forBAC complementation used in positional cloning where aBAC is tested for its ability to rescue a loss-of-function mutantphenotype by making BAC transgenic mice If complementa-tion is achieved it is impossible to know which gene on theBAC is responsible for the rescue Therefore we decided to
see whether galK selection could be used to make largespecific and clean deletions in BACs so as to remove unwantedgenes (a process called BAC trimming) As a model systemwe chose a mouse BAC from the C57BL6-derived libraryRP23 RP23-341F12 since we could obtain the BAC sequencedirectly from the UCSC genomic web browser (see Materialsand Methods) We then used PCR to amplify the galK selec-tion cassette with primers containing homology to the BACThe forward primer contained 50 bp of homology to the very 50
end of the BAC insert and this primer was combined withthree different reverse primers having 50 bp of homology topositions 50 75 and 100 kb away from the forward primerrsquoshomology (Figure 4A) SW102 bacteria containing the 341F12BAC were heat-induced and made electrocompetent and thentransformed with the three different selection cassettes fol-lowed by the selection on minimal medium with galactoseFrom the many resulting Gal+ colonies we picked four fromeach experiment and then did a SpeI restriction analysis of theBAC miniprep DNAs from them (Figure 4B) All four colon-ies from each experiment had the desired deletion (50 75 and100 kb respectively) The galK selection cassette can then be
Figure 3 Introduction of a G12D mutation in the Nras gene (A) SpeI restriction analysis of BAC miniprep DNA First lane is the unmodified CITB-50J2 Nras BACLanes 1ndash12 show digestion patterns of 12 clones counterselected for the substitution of galK with an oligo containing the GA substitution for the second position ofcodon 12 of Nras Clones 7 and 10 had internal deletions indicating that DOG resistance was achieved by spontaneous deletion and not homologous recombinationThese two clones were not analyzed further (B) Sequence analysis of a PCR product spanning the modified region from clones 1ndash6 8ndash9 and 11ndash12 All clones had theintended substitution (highlighted) However clones 9 and 11 also had an internal basepair deletion indicated by a minus (highlighted) The Nras ATG and codon 12are indicated (shadow)
PAGE 7 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
removed with an oligo using galK negative selection to cleanlydelete the galK cassette from the BAC as described in theprevious experiment (data not shown)
Insertion of a mutant loxP511 site into a BAC
Encouraged by the efficiency of this selection system wedecided to see whether we could introduce a single 34 bploxP511 site cleanly into BAC DNA using galK positiveneg-ative selection It has been previously shown that loxP andloxP511 sites cannot recombine with each other therefore the
introduced loxP511 site can only recombine with the loxP511site present in the BAC vector backbone and not the wild-typesite (Figure 5A) (20) We used galK positive selection to inserta PCR-amplified galK cassette flanked by 50 bp homologyarms to a position 95 kb away from the mutant loxP511 site inthe BAC RP23-341F12 vector backbone (Figure 5A) We thenreplaced the galK cassette using DOG counterselection with adouble-stranded 100 bp oligo containing a 34 bp loxP511 siteflanked by two 33 bp homology arms In this experiment weobserved less than a 10-fold difference in the number of col-onies on the plates from heat-induced and non-induced bac-teria suggesting fewer recombinants This lower frequency ofrecombinants when compared with the Nras G12D substitu-tion experiment is likely explained by the shorter homologyarms used in this latter experiment as it has been shown thatthe efficiency of recombination increase four orders of mag-nitude when homology length is increased from 20 to 40 bp(10) We analyzed six BAC minipreps from potential recom-binants by digesting with SpeI and comparing the restrictionpatterns with that of wild-type RP23-341F12 BAC DNA(Figure 5B) Three of six (50) colonies had exactly thesame restriction pattern as the unmodified RP23-341F12BAC suggesting that DOG resistance had selected for thedesired homologous recombination products in these threecases whereas the other three clones apparently becameDOG resistant due to the selection of BACs that carry largedeletions spanning galK
The 100 bp oligo was designed so that the loxP511 sitewould be inserted in the same orientation as the loxP511site in the BAC vector backbone To confirm that the BACswith the wild-type restriction pattern (3 5 and 6) had theloxP511 site correctly inserted we electroporated thesethree BACs into electrocompetent and L-arabinose inducedEL350 cells which carry an L-arabinose-inducible Cregene and then plated the cells on LB plates containing chlor-amphenicol to select for the BACs Two colonies generatedfrom each recombinant loxP511 clone were then tested bySpeI digestion of BAC miniprep DNA and all had the expec-ted Cre-mediated 95 kb deletion (Figure 5C) Recombinationwas confirmed by PCR using a forward primer from the BACvector backbone and a reverse primer mapping to a positiondistal to the reverse homology arm used to insert the galKcassette These primers are 95 kb apart on wild-type RP23-341F12 DNA and only 378 bp apart on CreloxP511 recom-bined DNA PCR analysis of all six clones produced a band ofthe expected length whereas no product could be amplifiedfrom unmodified RP23-341F12 BAC DNA (Figure 5D)
Source of background deletions following DOG selection
In the Nras G12D substitution and loxP511 insertion experi-ments a background of DOG-resistant bacteria which lackedthe desired mutation and instead carried unwanted deletionsthat spanned the galK selection cassette were observed Therelative ratio of colonies containing BACs with deletions wasalso higher when short homology arms were used This wasexpected since homologous recombination is more efficientwith longer homology arms (10) that would increase the rel-ative frequency of correctly targeted BACs The source of thedeletions could be l Red-mediated since proteins capable ofmediating recombination are expressed when the Red genes
Figure 4 BAC trimming using galK selection (A) Illustration of the design ofthe deletion experiment Homology arm 1 (H1) was held constant and H2 wasseparated from H1 by either 50 75 or 100 kb (B) SpeI restriction analysis ofBAC miniprep DNA from 12 clones showing deletions of 50 75 and 100 kbrespectively after the insertion of the galK selection cassette The first lane isunmodified RP23-341F12 BAC DNA which was included as a control Alltested clones had the intended deletion
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 8 OF 12
are activated Alternatively the deletions could be induced bythe large amount of oligos (200 ng) used in these experimentsor they could simply represent spontaneous deletions thatoccur at low level during normal BAC replication and thatare found by DOG selection To distinguish between thesepossibilities we repeated the DOG selection step used to insertthe loxP511 oligo into BAC RP23-341F12 DNA using fourdifferently treated bacteria samples non-induced bacteriawithout the 100 bp loxP511 oligo (Figure 6A) non-inducedbacteria with the oligo (Figure 6B) induced bacteria without
the oligo (Figure 6C) and induced bacteria with the oligo(Figure 6D) The number of resistant colonies obtainedfrom the four experiments varied by lt10-fold (data notshown) Ten colonies from each electroporation were thenanalyzed by SpeI digestion of BAC miniprep DNA and com-pared with unmodified RP23-341F12 BAC DNA (Figure 6AndashD) Unwanted galK region deletions were observed in all 10colonies for both of the non-induced samples (Figure 6A andB) These deletions are therefore unlikely to be Red-mediatedsince these samples were not heat-induced We also observed
Figure 5 Insertion of a loxP511 site (A) The location of the wild-type and mutant loxP sites in the BAC backbone are indicated along with the extra mutant loxP511site that was introduced into the BAC genomic insert via galK counterselection The 95 kb region deleted by Cre-mediated recombination between the two loxP511sites is indicated and PCR primers used to confirm the deletion are shown as small arrows (B) SpeI restriction analysis of six miniprep clones selected forthe replacement of galK with a dsDNA oligo containing the mutant loxP511 site Clones 3 5 and 6 (circles) had the same restriction pattern as the unmodifiedBAC indicating that DOG resistance occurred due to the intended homologous recombination event Clones 1 2 and 4 had large deletions and were not analyzedfurther (C) SpeI restriction analysis of BAC miniprep DNA from clones 3 5 and 6 after transformation into Cre-induced EL350 cells Two clones from each parentalclone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all clones analyzed confirming the correctinsertion of loxP511 in clones 3 5 and 6 (D) PCR analysis of the six clones from (C) with one primer mapping to the BAC backbone and the other to a position distalto the inserted loxP511 site
PAGE 9 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
deletions in both the absence and presence of oligo indicatingthat the deletions are not oligo induced We therefore concludethat these are spontaneous deletions that occur at low levelduring normal BAC replication in the DH10B background
Non-deleted colonies with the parental SpeI fingerprintwere only observed in the Red-induced sample that received
oligo (Figure 6D clones 33 and 35) These clones were sub-sequently electroporated into EL350 Cre-expressing cells toconfirm they contained the introduced loxP511 site Two col-onies from each original clone were tested by SpeI digestion ofthe BAC miniprep DNA As shown in Figure 6E each colonyhas undergone Cre-mediated deletion confirming that these
Figure 6 Same experiment as in Figure 5 with modifications as indicated at the top of each panel (A) SpeI digest of 10 minipreps from a control experiment withoutheat-induction and without the loxP511 dsDNA oligo (B) SpeI digest of 10 minipreps from a control experiment without heat-induction but with the loxP511 dsDNAoligo (C) SpeI digest of 10 minipreps from a control experiment with heat-induction but without the loxP511 dsDNA oligo (D) SpeI digest of 10 minipreps from anexperiment with heat-induction and with the loxP511 dsDNA oligo (comparable with Figure 5B) Clones with the parental digestion pattern indicating DOGresistance due to homologous recombination (clones 33 and 35 circles) are only seen in (D) DOG resistance in all other clones likely occurred due to internaldeletions of the BACs (E) SpeI restriction analysis of BAC miniprep DNA from clones 33 and 35 after transformation into Cre-induced EL350 cells Two clonesfrom each parental clone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all the clones analyzedconfirming the correct insertion of loxP511 in clones 33 and 35 (compare with Figure 5C)
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 10 OF 12
colonies were correctly targeted and contain the introducedloxP511 site (2 out of 10 clones a 20 efficiency in thisexperiment)
Generation of SW105 and SW106 cells
The two DY380-derived bacterial strains EL250 and EL350contain the defective l prophage needed for recombineering inaddition to L-arabinose inducible Flp or Cre genes respect-ively (11) Both strains have proven to be very useful for BACmodification (11) and EL350 is now used routinely for mak-ing conditional targeting vectors for ES cell knock-out experi-ments (4) To further enhance the usefulness of these strainswe decided to transfer the galK selection system into themThis was done as previously described for SW102 The galKIS2 element present in both strains was replaced with the wild-type gal promoter and the galK gene in the gal operon deletedusing DOG selection The resulting strains SW105 (Flp) andSW106 (Cre) (Table 1) were then tested to confirm that theystill contained inducible Flp and Cre genes by transformingarabinose-induced and electrocompetent cells with plasmidscontaining a neo gene flanked by Frt or loxP sites PL451 andPL452 (4) respectively Using SW105 or SW106 it is nowpossible to introduce a point mutation or an informativerestriction site into a BAC retrieve a fragment from thisBAC containing the introduced mutation(s) into a plasmidbackbone using gap repair and turn the retrieved fragmentinto a conditional targeting vectormdashall using only one bac-terial strain and only a single initial BAC transformation
DISCUSSION
Here we describe a new recombineering-based Ecoli BACmodification system that makes use of galK positive selectionfor growth on galactose minimal medium and galK negativeselection (counterselection) for growth on DOG This modi-fication system has several advantages compared with theother related BAC modification systems First the galK selec-tion cassette is small (1231 bp + homology arms) comparedwith for example the sacBndashneo cassette (3 kb) making PCRamplification and transformation into bacteria easier Twohomology arms are easily added to the galK cassette by includ-ing these sequences in the 50 ends of the primers used foramplification (see Materials and Methods) Second sincegalK is used for both selection steps mutations occurringin galK during PCR amplification will be selected againstduring positive selection significantly reducing the risk ofDOG resistance from PCR mutations in galK during negativeselection Third even when very short (33 bp) homology armswere used the frequency of recombinants among the analyzedDOG-resistant clones was still 20ndash50 To our knowledgesuccessful BAC modification with such short homology armshas never been reported Finally since galK recombineering isso efficient little screening is required following selectionreducing the overall hands-on-time to a minimum galK selec-tion requires growth on minimal medium in both selectionsteps although perhaps not used routinely in most molecularbiology laboratories where BAC modification is needed theseplates are fairly simple to make and should not prevent anyonefrom using this method
For all such counterselection schemes there will bebackground since during negative selection any event leadingto the loss of the counterselectable marker will result in sur-vival In our system virtually all background appears to resultfrom deletions that span the inserted galK gene These dele-tions likely occur during BAC replication in the 45 h out-growth phase since we have shown that these deletions occurindependently of Red induction and in the absence or presenceof oligo It is well established that in recA defective bacteriaBACs are very stable compared with other large insert vectorssuch as yeast artificial chromosomes (YACs) However usingcounterselection rare spontaneous deletions are seen becauseof the strong selection force This spontaneous deletion back-ground is not a problem however due to the high frequency ofhomologous recombination obtained with the defective lprophage system
Increasing the length of homology arms used for recombin-eering will reduce the relative number of background deletionsobserved following negative selection since the percentage ofcolonies containing BACs that eliminated galK by homolog-ous recombination will be increased Oligos with longerhomology arms can be produced by annealing two oligoswith overlapping 30 ends (21) These overlapping oligos (50
single strand overhangs) are then filled in by DNA polymerasein vivo (or in vitro) and subsequently serve as the substrate forthe l Red proteins exo and bet Longer homology arms canalso be added by traditional cloning using restriction enzymesand DNA ligase (4)
A number of uses can be imagined for the strains describedhere In principle our strains can be used to make virtually anykind of BAC modification one can imagine including pointmutations or clean deletions or insertions of everything fromcDNA to loxP sites or small epitope tags BAC trimming thespecific deletion of BAC DNA flanking a gene of interest couldalso be used to remove genes from BACs prior to making BACtransgenic mice so that only the gene of interest on the BAC isanalyzed This should be of particular interest for BAC rescueexperiments in positional cloning Of course trimming andmodification can be combined since the galK selection cassettecan be recycled so that the same BAC can be modified severaltimes BAC modification could also be the starting point forconstructing a gene-targeting vector to allow for more soph-isticated gene targeting in mouse ES cells The desired muta-tion(s) could first be created in the BAC followed by retrievalinto a plasmid vector containing a negative-selection markerlike thymidine kinase using gap repair (4) Finally a neo mar-ker could be introduced to allow for positive selection in EScells Alternatively the retrieved fragment could be modifiedso that the end result is a conditional targeting vector StrainSW106 which in addition to galK selection can be induced toexpress Cre recombinase makes it possible to perform all ofthe steps needed to construct a conditional targeting vectorstarting with only a single BAC transformation step
Furthermore in experiments where BAC transgenic miceare used to analyze the effect of deleting long-range regulatorsof gene expression galK DOG counterselection can even beused in the absence of an oligo to generate a series of dele-tions around the galK insertion site and the effect on generegulation studied
The Flp or Cre ORFs contained in strains SW105 andSW106 respectively can also be replaced for any desired
PAGE 11 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
gene thus creating tight arabinose-inducible expression forany gene of interest This is done by first replacing the CreFlpe ORF with galK and select for Gal+ recombinants ThegalK cassette is then replaced by the ORF of the gene ofinterest by DOG selection for Gal recombinants
Finally the strains described here should be very useful forgenetic manipulation not only of BACs but also the Ecoligenome itself
All recombineering reagents discussed in this workare freely available upon request To obtain these materialsplease follow the directions listed on our website (httprecombineeringncifcrfgov) Detailed protocols for recom-bineering including galK selection can also be downloadeddirectly from our website
ACKNOWLEDGEMENTS
We thank Allen Kane and Carolyn Whistler from ScientificPublications Graphics amp Media NCI-Frederick for help withillustrations and figure preparation Funding for this research aswell as the Open Access publication charges was provided byDHHS NIH NCI
REFERENCES
1 ShizuyaH BirrenB KimUJ MancinoV SlepakTTachiiriY and SimonM (1992) Cloning and stable maintenance of300-kilobase-pair fragments of human DNA in Escherichia coli using anF-factor-based vector Proc Natl Acad Sci USA 89 8794ndash8797
2 YangXW ModelP and HeintzN (1997) Homologous recombinationbased modification in Escherichia coli and germline transmission intransgenic mice of a bacterial artificial chromosome Nat Biotechnol15 859ndash865
3 AntochMP SongEJ ChangAM VitaternaMH ZhaoYWilsbacherLD SangoramAM KingDP PintoLH andTakahashiJS (1997) Functional identification of the mouse circadianClock gene by transgenic BAC rescue Cell 89 655ndash667
4 LiuP JenkinsNA and CopelandNG (2003) A highly efficientrecombineering-based method for generating conditional knockoutmutations Genome Res 13 476ndash484
5 Cotta-de-AlmeidaV SchonhoffS ShibataT LeiterA andSnapperSB (2003) A new method for rapidly generating gene-targetingvectors by engineering BACs through homologous recombination inbacteria Genome Res 13 2190ndash2194
6 CopelandNG JenkinsNA and CourtDL (2001) Recombineeringa powerful new tool for mouse functional genomics Nature RevGenet 2 769ndash779
7 CourtDL SawitzkeJA and ThomasonLC (2002) Geneticengineering using homologous recombination Annu Rev Genet 36361ndash388
8 ZhangY BuchholzF MuyrersJP and StewartAF (1998) A newlogic for DNA engineering using recombination in Escherichia coliNature Genet 20 123ndash128
9 MuyrersJP ZhangY TestaG and StewartAF (1999) Rapidmodification of bacterial artificial chromosomes by ET-recombinationNucleic Acids Res 27 1555ndash1557
10 YuD EllisHM LeeEC JenkinsNA CopelandNG andCourtDL (2000) An efficient recombination system for chromosomeengineering in Escherichia coli Proc Natl Acad Sci USA 975978ndash5983
11 LeeEC YuD Martinez de VelascoJ TessarolloL SwingDACourtDL JenkinsNA and CopelandNG (2001) A highly efficientEscherichia coli-based chromosome engineering system adapted forrecombinogenic targeting and subcloning of BAC DNA Genomics73 56ndash65
12 GongS YangXW LiC and HeintzN (2002) Highly efficientmodification of bacterial artificial chromosomes (BACs) using novelshuttle vectors containing the R6Kgamma origin of replicationGenome Res 12 1992ndash1998
13 JamsaiD OrfordM NefedovM FucharoenS WilliamsonR andIoannouPA (2003) Targeted modification of a human beta-globin locusBAC clone using GET Recombination and an I-SceI counterselectioncassette Genomics 82 68ndash77
14 SwaminathanS EllisHM WatersLS YuD LeeECCourtDL and SharanSK (2001) Rapid engineering of bacterialartificial chromosomes using oligonucleotides Genesis 2914ndash21
15 AlperMD and AmesBN (1975) Positive selection of mutants withdeletions of the gal-chl region of the Salmonella chromosome as ascreening procedure for mutagens that cause deletions J Bacteriol121 259ndash266
16 AusubelFM BrentR KingstonRE MooreDD SeidmanJGSmithJA and StruhlK (1987) Current Protocols in Molecular BiologyGreene Publishing Associates and Wiley-Interscience John Wiley ampSons Hoboken NJ
17 BachmannBJ (1996) Derivations and genotypes of some mutantderivatives of Escherichia coli K-12 In NeidhardtFC CurtissRIIIIngrahamJL LinECC LowKB MagasanikB ReznikoffWSRileyM SchaechterM and UmbargerHE (eds) Escherichia coli andSalmonella Cellular and Molecular Biology ASM press WashingtonDC Vol 2 pp 2460ndash2488
18 GrantSG JesseeJ BloomFR and HanahanD (1990) Differentialplasmid rescue from transgenic mouse DNAs into Escherichia colimethylation-restriction mutants Proc Natl Acad Sci USA 874645ndash4649
19 OppenheimAB RattrayAJ BubunenkoM ThomasonLC andCourtDL (2004) In vivo recombineering of bacteriophage lambda byPCR fragments and single-strand oligonucleotides Virology 319185ndash189
20 HoessRH WierzbickiA and AbremskiK (1986) The role ofthe loxP spacer region in P1 site-specific recombination NucleicAcids Res 14 2287ndash2300
21 YuD SawitzkeJA EllisH and CourtDL (2003) Recombineeringwith overlapping single-stranded DNA oligonucleotides testing arecombination intermediate Proc Natl Acad Sci USA 1007207ndash7212
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 12 OF 12

mutations like point mutations and clean deletions or introducein-frame fusions of cDNAs or epitope tags without leaving atthe same time a selectable marker or a loxPFrt site at themodification site A handful of methods for making such muta-tions all based on homologous recombination in Ecoli havebeen developed One method is RecA dependent and relies onthe use of a shuttle vector and two recombination steps integ-ration followed by the resolution of the co-integrate (212)This method requires considerable up-front effort using tradi-tional cloning with restriction enzymes and ligation A simplerand more widely used method is based on positivenegativeselection using eg a sacBndashneo fusion gene (8) neo (kana-mycin) resistance is used for positive selection while sucrosetoxicity resulting from sacB expression is used for negativeselection A major drawback of this selection system is thatspontaneous point mutations in the sacB portion of the sacB-neo fusion gene can occur without influencing the bacteriarsquosability to grow on kanamycin These sacB mutants signific-antly increase the background after negative selection Arelated method is based on counterselection using a recogni-tion site for a rare restriction enzyme like I-SceI (13) For thismethod to work efficiently the I-SceI restriction enzyme hasto be induced in trans and since there is no selection formaintaining the correct recognition sequence for this enzymethe background due to point mutation or deletion of the restric-tion site is high A method for BAC modification withoutselection has also been developed (14) Although relativelyefficient this method relies on a PCR-based screening of theresulting colonies to identify the desired clones Since there isno selection step the number of manipulations made possibleby this selection procedure is more limited
Here we report the development of a novel and highlyefficient galK-based positivenegative selection system forthe manipulation of BACs The Ecoli galactose operon con-sists of four genes galE galT galK and galM which arenecessary for growth and utilization of galactose as theonly carbon source The galK gene product galactokinasecatalyzes the first step in the galactose degradation pathwayphosphorylating galactose to galactose-1-phosphate Galacto-kinase also efficiently catalyzes the phosphorylation of agalactose analog 2-deoxy-galactose (DOG) The product ofthis reaction cannot be further metabolized leading to a toxicbuild-up of 2-deoxy-galactose-1-phosphate (15) Thus bothpositive and negative selection can be conferred by galKBecause galK is used for both selection steps backgroundfollowing negative selection is reduced and no colonyscreening is necessary The small size of the galK cassette(around 1200 bp plus homology arms) also makes it easier toamplify by PCR and to introduce into bacteria using electro-poration
MATERIALS AND METHODS
General recombineering and galK selection
Recombineering was performed as follows 500 ml of anovernight culture was diluted in 25 ml LuriandashBertani(LB) medium with or without chloramphenicol selection(125 mgml) in a 50 ml baffled conical flask and grown at32C in a shaking waterbath to an OD600 of 06 Then 10 mlwas transferred to another baffled 50 ml conical flask and
heat-shocked at 42C for exactly 15 min in a shakingwaterbath The remaining culture was left at 32C as theuninduced control After 15 min the two samples inducedand uninduced were briefly cooled in an icewaterbathslurry and then transferred to two 15 ml Falcon tubes (BDBiosciences) and pelleted using 5000 rpm (eppendorf cen-trifuge 5804R rotor A-4-44) at 0C for 5 min The supernatantwas poured off and the pellet was resuspended in 1 ml ice-coldddH2O by gently swirling the tubes in an icewaterbath slurrySubsequently 9 ml ice-cold ddH2O was added and the samplespelleted again This step was repeated once more After thesecond washing and centrifugation step the supernatant wasremoved and the pellet (50 ml each) was kept on ice untilelectroporated with PCR product gel-purified fragment ordouble-stranded oligo An aliquot of 25 ml was used foreach electroporation in a 01 cm cuvette (BioRad) at 25 mF175 kV and 200 V After electroporation the bacteria wererecovered in 1 ml LB (15 ml Falcon tube) for 1 h in a 32Cshaking waterbath For the counterselection step (see below)the bacteria were recovered in 10 ml LB in a 50 ml baffledconical flask and incubated for 45 h in a 32C shaking water-bath After the recovery period the bacteria were washedtwice in 1middot M9 salts as follows 1 ml culture was pelletedin an eppendorf tube at 13 200 rpm for 15 s and the super-natant was removed with a pipette The pellet was resuspendedin 1 ml of 1middot M9 salts and pelleted again This washing stepwas repeated once more After the second wash the supernat-ant was removed and the pellet was resuspended in 1 ml of 1middotM9 salts before plating serial dilutions (100 ml 100 ml each of110 1100 and 11000 dilutions) on minimal medium plates(see below) Washing in M9 salts is necessary to remove anyrich media from the bacteria prior to selection on minimalmedium The uninduced samples routinely had a higher degreeof lysisbacterial death after electroporation so the uninducedsamples were diluted in 025ndash075 ml 1middot M9 salts in the finalstep to make up for the difference Detailed protocolsfor recombineering can also be found at our website (httprecombineeringncifcrfgov)
Minimal media and indicator plates
Minimal media and the indicator plates were prepared usingstandard methods (16) We added supplements as indicated
Washing solution 1middot M9 mediumGal positive selection M63 + agar (15 gl Difco BDBiosciences) + D-galactose (02 Sigma) + D-biotin(1 mgl Sigma) + L-leucine (45 mgl Sigma) and ndashchloramphenicol (125 mgml Sigma)Gal counterselection M63 + agar + glycerol (02Fischer) + D-biotin (1 mgl) + L-leucine (45 mgl) +DOG (02 Ferro Pfanstiehl) and ndash chloramphenicol(125 mgml)Gal indicator plates MacConkey agar (Difco BDBiosciences) + D-galactose (1) and ndash chloramphenicol(125 mgml) Plates were prepared using manufacturerrsquosinstructions
Bacterial strains
The strains used in this paper are listed in Table 1 SW101was derived from DY380 by a homologous recombinationalexchange of the mutated gal operon leader sequence with a
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 2 OF 12
441 bp PCR product (Expand High-Fidelity PCR SystemRoche Applied Science) containing the wild-type gal operonleader sequence made using W3110 (17) bacteria as the tem-plate The primers used to amplify the wild-type leader were(Integrated DNA Technologies Inc) F 50-CGACGCATG-CAGGCATGAA-30 and R 50-AGTGGATCACGGTGTC-GATA-30 and the PCR conditions were 94C for 15 s 60Cfor 30 s and 72C for 30 s for 35 cycles The PCR product wasgel purified (GFX kit Amersham Biosciences) and eluted in50 ml ddH2O An aliquot of 25 ml was used in the recombin-eering experiment as described above and the bacteria wereplated on M63 minimal medium + galactose + leucine + biotinThe plates were incubated at 32C for 2ndash3 days A few of themany resulting colonies were streaked for single colonies onindicator plates and a single dark red Gal+ colony was used inthe next step SW102 was derived from SW101 in thefollowing way two homology arms flanking the galK openreading frame (ORF) were PCR amplified using SW101 bac-teria as template and the following primers (Integrated DNATechnologies Inc) recognition sites for restriction enzymesare underlined 50arm F 50-AAATAACTCGAGCAGCTGC-ACGCGCACTTT-30 50arm R 50-AAATAAGAATTCTTCT-TACACTCCGGATTCGC-30 30arm F 50-AAATAAGAAT-TCTGTAAACCATCACAAGGAGCAG-30 30arm R 50-AA-TAAAGCGGCCGCCAGCTGGTTAACGCCCTGA-30 PCRconditions were 94C for 15 s 60C for 30 s and 72C for30 s for 35 cycles The 50 homology arm (171 bp) was digestedwith XhoI and EcoRI and the 30 arm (335 bp) with EcoRI andNotI The digested PCR products were gel purified and triple-ligated into a vector digested with XhoI and NotI The targetingcassette was then released from the backbone using XhoI andNotI digestion followed by gel purification The cassette waseluted in 50 ml ddH2O and 25 ml was used for recombineeringas described above After 45 h of outgrowth in LB media andtwo washes in 1middot M9 salts serial dilutions of the bacteria wereplated on minimal plates containing glycerol as carbon sourceleucine and biotin plus DOG for selection against galK Theplates were incubated for 3 days at 32C A few of the manyresulting colonies were streaked for single whitecolorless(Gal) colonies on indicator plates followed by PCR analysis
and sequencing to check for correct deletion of the galK geneA single verified colony was expanded and a glycerol stockwas made and used for initiation of all subsequent experimentsSW103 and SW104 were derived from EL250 and EL350respectively using the same method as for the derivation ofSW101 SW105 and SW106 were derived from SW103 andSW104 respectively using the same method as for the deriva-tion of SW102 from SW101 SW105 expresses Flp under thecontrol of an arabinose-inducible promoter and SW106expresses Cre under the same promoter
Construction of pgalK
Two rounds of PCR were performed to construct a galK ORFdriven by the prokaryotic em7 promoter The first round wasperformed using galK ORF 1st F and galK ORF R primersusing W3110 bacteria (17) as template The resulting PCRproduct was then used as template for a second round ofPCR using galK ORF 2nd F and galK ORF R primers The pro-duct from the second round of PCR was gel purified anddigested with EcoRI and BamHI and cloned into EcoRI andBamHI digested pBluescript SK pgalK was verified bysequencing [the plasmid sequence is available fromour website (httprecombineeringncifcrfgov)]The primersequences were (restriction sites and the galK ATG are under-lined and the em7 promoter is in italics) galK ORF 1st F50-CCCAGGAGGCAGATCATGAGTCTGAAAGAAAAAA-CTGAAAGAAAAAACACAATCTCTGT-30 galK ORF2nd F AATAAAGAATTCCTGTTGACAATTAATCATCG-GCATAGTATATCGGCATAGTATAATACGACAAGGTGA-GGAACTAAACCCAGGAGGCAGATCATG galK ORF RAATAAAGGATCCTCAGCACTGTCCTGCTCCTT-30 PCRconditions for both rounds were 94C for 15 s 60C for30 s and 72C for 15 min for 30 cycles Primers werefrom Invitrogen
PCR amplification of the galK targeting cassette
Sequences of all galK primers (Invitrogen) used in the experi-ments described in this paper are given below em7-galK wasPCR amplified using 1 ng pgalK as template and the following
Table 1 Recombineering reagents used in this work
Genotype Phenotype Reference
StrainsW3110 Gal+ (17)DH10B mcrA D(mrr-hsdRMS-mcrBC) DlacX74 deoR
endA1 araD139 D(ara leu) 7697 rpsLrecA1 nupG f80dlacZDM15 galU galK
Gal (18)
DY380 DH10B [lc1857 (cro-bioA)ltgtTet] galK+ gal490 Gal (11)SW101 DY380 gal+ Gal+ This workSW102 SW101 DgalK Gal This workEL250 DY380 (cro-bioA)ltgtaraC-PBAD Flpe Gal (11)EL350 DY380 (cro-bioA)ltgtaraC-PBAD Cre Gal (11)SW103 EL250 gal+ Gal+ This workSW104 EL350 gal+ Gal+ This workSW105 SW103 DgalK Gal This workSW106 SW104 DgalK Gal This work
Selection cassettepgalK galK wild-type gene driven by the em 7 promoter This work
ltgt indicates the result of a homologous recombination event Gal+ the ability to use galactose as the sole carbon source Gal resistance to DOG and lack of the abilityto grow on galactose as the sole carbon source
PAGE 3 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
conditions 94C for 15 s 60C for 30 s and 72C for 1 min for30 cycles After completion of PCR 2 ml DpnI was added toeach 25 ml reaction and incubated for 2 h at 37C to removeany plasmid template The DpnI-digested reaction mix was runon a 1 agarose gel over night and the PCR product waspurified and eluted in 50 ml ddH2O An aliquot of 25 ml wasused for each experiment For making the Nras G12D substi-tution the following primers were used for the first step intro-ducing galK Homology to Nras sequence is in italics and thesequence recognizing em7-galK is underlined Nras galK F50-TTTTTGCTGGTGTGAAATGACTGAGTACAAACTGGTGG-TGGTTGGAGCAGCCTGTTGACAATTAATCATCCGCA-30Nras galK R 50-CAAAGTGGTTCTGGATTAGCTGGATCG-TCAAGGCGCTTTTCCCAACACCATCAGCACTGTCCTGC-TCCTT-30 For making 50 75 and 100 kb deletions in theRP23-341F12 BAC the following primers were used (homo-logy to BAC sequence is in italics and the sequence recog-nizing em7-galK is underlined) galK F 341F12 start50-ACTCCCACTGGAAGCTTTTTACAAAACATGTGTTGCT-GACATGTTGACAGCCTGTTGACAATTAATCATCGGCA-30 galK R 341F12 50 kb 50-ACCCAAACCAAACAACAT-CCAAACCAAAAACACAGACAAAACCAAATATGTCAGCAC-TGTCCTGCTCCTT-30 galK R 341F12 75 kb 50-ACACTAAGC-CAAACTCCTTGCCTGGGCTATTTCTCTTTGTTTTTCCAAATTCAGC-ACTGTCCTGCTCCTT-30 galK R 341F12 100 kb 50-TATGT-GTCTGTGTGTGTATGTACAGTTCTTTGTTTTTGTTTTTTTT-CTTTTCAGCACTGTCCTGCTCCTT-30 For insertion of aloxP511 site in the RP23-341F12 BAC the following primerswere used for the first step (introduction of galK) Homology toBAC sequence is in italics and the sequence recognizing em7-galK is underlined 95 kb loxP511 galK F 50-GGACA-GAGGCGTGACAGACGTGTGAGCTCCGTGGACAACTCTC-CCCGAAGCCTGTTGACAATTAATCATCGGCA-30 95 kbloxP511 galK R 50-GACTCTGAGCAGCAACGGCTGAGCCT-CACTTGAGAGGGTCCCTGAGTCACTCAGCACTGTCCTG-CTCCTT-30
Oligos for recombineering
Oligos used to replace the galK-targeting cassette wereobtained from Invitrogen dsDNA was used and the oligos(sense and antisense) annealed in vitro 10 mg of each oligo(sense and antisense) was mixed in an eppendorf tube in a totalvolume of 100 ml of 1middot PCR buffer (Expand High FidelityPCR kit Roche Applied Science) and boiled for 5 minallowed to cool to room temperature for 30 min ethanol pre-cipitated and resuspended in 100 ml ddH2O to a final concen-tration of 200 ngml double-stranded oligo An aliquot of 1 ml(200 ng) was used in the recombineering experiments Tointroduce the G12D (GltgtA) point mutation in the NrasBAC CITB 50J2 the following oligos were used for thesecond step (the introduced adenosinethymidine base pairis underlined the flanking sequences are homologous to theNras BAC sequence) G12D S 50-TTTTTGCTGGTGT-GAAATGACTGAGTACAAACTGGTGGTGGTTGGAGC-AGATGGTGTTGGGAAAAGCGCCTTGACGATCCAGC-TAATCCAGAACCACTTT-30 G12D AS 50-AAAGTGGT-TCTGGATTAGCTGGATCGTCAAGGCGCTTTTCCCAA-CACCATCTGCTCCAACCACCACCAGTTTGTACTCAG-TCATTTCACACCAGCAAAAA-30 To introduce a loxP511site in the RP23-341F12 BAC the following oligos were used
for the second step (loxP511 is underlined the flankingsequences are homologous to the BAC sequence aroundposition 95 kb) 95 kb loxP511 S 50-ACGTGTGAGCTC-CGTGGACAACTCTCCCCGAAGATAACTTCGTATAGT-ATACATTATACGAAGTTATGTGACTCAGGGACCCTC-TCAAGTGAGGCTCAGC-30 95 kb loxP511 AS 50-GCTG-AGCCTCACTTGAGAGGGTCCCTGAGTCACATAACTT-CGTATAATGTATACTATACGAAGTTATCTTCGGGGA-GAGTTGTCCACGGAGCTCACACGT-30
Verification of positive recombinants
In the G12D Nras experiment the selected Gal clones wereanalyzed by SpeI digestion of BAC miniprep DNA usingunmodified CITB 50J2 BAC DNA as a control Clones with-out rearrangements were analyzed by PCR using 1 ml BACminiprep DNA as the template The PCR products were gelpurified and sequenced using the same primers as were usedfor PCR Primers flanking the targeted mutation were Nrastest F 50-CACTCATCTGCAAGGAATGCT-30 Nras test R50-CCTCAGTAAGCACGAACTTGT-30 PCR conditionswere 94C for 15 s 60C for 30 s and 72C for 30 s for30 cycles Modifications of the RP23-341F12 BAC (50 75and 100 kb deletions and the introduction of a loxP511 site)were tested by SpeI restriction analysis of BAC miniprep DNAand compared with unmodified 341F12 BAC DNA In theloxP511 experiment clones 3 5 and 6 were further testedfor correct insertion of the loxP511 site by transforming1 ml of BAC miniprep DNA into electrocompetent andarabinose-induced EL350 cells (11) and plating on LB plateswith chloramphenicol Two colonies from each starting clonewere tested by SpeI digestion of BAC miniprep DNA for the95 kb Cre-mediated deletion Finally the Cre-recombinedclones were tested by PCR with one primer mapping to theend of the pBACe36 BAC backbone and the other mapping toa position 95 kb away on the wild-type BAC The primers(Invitrogen) used for this analysis were 95 kb loxP511 checkF 50-GCGGATGAATGGCAGAAATTC-30 95 kb LoxP511check R 50-TTTGCCAGACTGGTGCCTAA-30 PCR condi-tions were 94C for 15 s 60C for 30 s and 72C for 30 s for30 cycles The resulting PCR bands were gel purified andconfirmed by sequencing using the same primers as wereused for the PCR amplification The follow-up experimentfor testing the source of the observed BAC deletions wasdone as described above
BACs
Mouse BACs were obtained from Invitrogen The RP23-341F12 BAC (C57BL6 DNA) was chosen based on positionwithin the mouse Ebfaz gene using the UCSC genome browser(httpgenomeucscedu) The CITB 50J2 BAC (CJ7129SVDNA) was identified by screening a CITB mouse BAC library(Invitrogen) with an Nras genomic probe using standardhybridization methods The BAC clones were streaked forsingle colonies and characterized by PCR and restriction ana-lysis before proceeding to the recombineering experimentsTwo methods were used for DNA preparation For BAC mini-preps (1ndash15 mg) we used the following protocol 5 ml over-night LB culture (15 ml Falcon tube) was pelleted for 5 min at5000 rpm the supernatant removed and the pellet wasdissolved in 250 ml buffer P1 (miniprep kit Qiagen) and
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 4 OF 12
transferred to an eppendorf tube An aliquot of 250 ml P2buffer was added followed by mixing by inversion and incuba-tion for lt5 min at room temperature An aliquot of 250 ml N3buffer was added followed by mixing and incubation on ice for5 min The supernatant was cleared by two rounds of centri-fugation at 13 200 rpm for 5 min in a tabletop centrifugeEach time the supernatant was transferred to a new tube DNAwas precipitated by adding 750 ml isopropanol mixing andincubating on ice for 10 min and centrifugation for 10 min at13 200 rpm The pellet was washed once in 70 ethanol andthe dry pellet was dissolved in 50 ml TE An aliquot of 40 ml(1 mg) was used for restriction analysis and 1 ml was used astemplate for PCR analysis or transformation of electrocom-petent bacteria Large-scale preparations of BAC DNA(25ndash100 mg) were done using the Nucleobond BAC maxikit from Clontech (BD Biosciences) following the manufac-turerrsquos protocol
RESULTS
Generation of SW102 cells
To see whether we could develop a more efficient selectivesystem for BAC recombineering we looked for a single select-able marker that could be used for both positive and negativeselection We focused on Ecoli galK because both selectionsteps could be done using galK and its small size makes it easyto amplify by PCR We previously reported the developmentof a bacterial strain DY380 that is readily transformable withBAC DNA due to its DH10B origin (1) DY380 cells alsoharbor the defective l prophage required for recombineeringThe defective prophage in DY380 was transferred into DH10Bwith a P1 phage lysate obtained from DY363 cells [for detailssee (11)] DY363 cells carry a 1200 bp IS2 insertion elementgal490 in the mRNA leader sequence of the galactose operonpreventing gal gene transcription Since the galactose operonis directly proximal to the site of insertion of the defective
prophage the gal490 mutation was also transferred to DY380during P1 transduction Thus DY380 is phenotypically Gal
(galactose minus) and therefore unable to grow on galactoseminimal medium
Recombineering was used to correct this problem From thewild-type Ecoli strain W3110 (17) we PCR amplified a 441bp fragment from the wild-type gal promoter which spans theregion containing the IS2 insertion element in DY380 ThisPCR product was introduced into DY380 and Gal+ recombin-ants were selected for growth on galactose minimal medium(see Materials and Methods) We named this strain SW101(Table 1) This strain is identical to DY380 except that it lacksthe IS2 insertion element and has been made wild type for thegal operon We then made a precise deletion of the galK geneleaving all other genes in the galactose operon intact This wasachieved by PCR amplifying two homology arms of 171 and335 bp respectively flanking the galK ORF and cloning themtogether into a plasmid using three-way ligation Since theORF of galK overlaps that of galM we left behind the last33 bp of galK (Figure 1) to allow for proper translation ofgalM The targeting cassette consisting of a 512 bp linearfragment was electroporated into heat-induced and electro-competent SW101 cells and the recombinant clones selectedon minimal medium containing glycerol as the carbon sourceand DOG for selection against galK The genotype of theresulting strain SW102 (Table 1) was confirmed by PCRanalysis and sequencing of the modified region (Figure 1)This strain now harbors not only the defective l prophagebut also a functional gal operon except for the deletion ofgalK The l prophage is located between the galactose operonand the biotin operon and the bio operon in these cells isnonfunctional causing a biotin requirement DH10B andall derived strains including SW102 are also deficient inleucine metabolism (18) When using minimal medium onetherefore needs to add both biotin and leucine to the plates toallow SW102 growth (see Materials and Methods)
Figure 1 Sequence analysis of the galactose operon in strains DY380 (A) SW101 (B) and SW102 (C) In SW102 the ORF of galK was deleted leaving only 33 bp ofgalK behind to make sure that translation of galM is initiated properly EcoRI the restriction site used to clone the 50 and 30 homology arms flanking galK
PAGE 5 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
galK expression cassette
Having produced a bacterial strain SW102 which is galKdefective the next step was to make a galK expression cassettethat could be used to restore the bacteriarsquos ability to grow ongalactose by providing galK in trans This was achieved byPCR amplification of the wild-type galK ORF from W3110cells We then added a minimal bacterial promoter em7 usinga two-step PCR approach (see Materials and Methods) andcloned the expression cassette into pBluescript SK We callthis plasmid pgalK (Table 1) The constitutively active galKexpression cassette can easily be amplified by PCRwith homology arms added to the primers (see Materialsand Methods)
Making a single base pair substitution
The general scheme for making mutations in BACs using galKselection is depicted in Figure 2 To test the galK selectionsystem for BAC recombineering we decided to introduce apoint mutation into a BAC containing the murine Nras gene(CITB clone 50J2) The sequence of the glycine-coding codon12 is GGT By changing this codon to GAT we would obtainthe desired mutation G12D In order to introduce this muta-tion into the BAC we first amplified the galK expressioncassette by PCR using primers with 50 bp of homology toeither side of the second position of the GGT codon Followinghomologous recombination this targeting would introduce a1 bp deletion into codon 12 in addition to inserting the galKselection cassette Instead of deleting the basepair the galKcassette could have been inserted right next to the basepairinstead SW102 cells containing the 50J2 BAC were heat-induced and made electrocompetent and then electroporatedwith the galK cassette Gal+ recombinant colonies were selec-ted for growth on galactose minimal medium with chloram-phenicol to maintain the BAC Bacteria grow more slowly onminimal media than on rich media and we generally pickcolonies after 2ndash3 days For this first step we do not expectany background colonies on the non-induced control plates ifthe pgalK plasmid is properly eliminated (see Materials andMethods) To purify the Gal+ colonies we streaked a fewcolonies on MacConkey galactose indicator plates to obtainsingle bright pinkred Gal+ colonies One of these single-cloned colonies was picked to initiate a culture for thenext step counterselection We find that there is no need toanalyze the Gal+ colonies further before proceeding tocounterselection
A 100 bp dsDNA oligo was then prepared by annealing twocomplementary oligos having 49 and 50 bp homologyrespectively to either side of the desired mutation a singleAT bp An aliquot of 200 ng of this oligo was then electro-porated into heat-induced and electrocompetent SW102 Gal+
cells containing the galK modified 50J2 BAC After electro-poration the bacteria were allowed 45 h outgrowth in a 32Cshaking waterbath The bacteria were then washed in M9 saltsto remove any rich medium and plated on minimal mediumwith glycerol DOG and chloramphenicol The 45 h out-growth is necessary to obtain complete segregation of therecombinant BACs containing the mutation After 3 dayswe obtained colonies with a ratio of 10ndash1001 when comparingplates with heat-induced to non-induced bacteria We picked12 colonies from the heat-induced plates for BAC minipreps
followed by SpeI restriction analysis (Figure 3A) Ten out ofthe twelve clones had the same restriction pattern as theunmodified 50J2 BAC DNA suggesting that the desiredreplacement of the galK gene by the point mutation hadoccurred This was confirmed by PCR amplification followed
Figure 2 Overview of the galK selection scheme The result of the firsttargeting event is the insertion of constitutively active galK into a definedposition on the BAC by selection on minimal medium containing galactoseand chloramphenicol to select for the maintenance of the BAC The bacteria arenow phenotypically Gal+ Next the galK cassette is replaced by a dsDNA oligoa PCR product or a cloned dsDNA fragment carrying a desired mutation(indicated by a star) and flanked by the same homology arms used in thefirst selection step This is achieved by negative selection using minimalmedium containing 2-deoxy-galactose (DOG) with glycerol as the sole carbonsource The bacteria become phenotypically Gal H1 and H2 homology arms1 and 2 respectively cat chloramphenicol acetyl transferase gene ori2 BACorigin of replication galK Ecoli galactokinase gene driven by a minimalpromoter
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 6 OF 12
by sequencing of the modified region of the 10 BAC clones(Figure 3B) All 10 sequenced clones had the desired pointmutation The effective recombination efficiency in thisexperiment however was 812 (67) since 2 of the 10 clonesalso had an additional single base pair deletion (clones 9 and11) These deletions probably occurred during oligo synthesissince we did not purify the oligos beyond desalting (19)Because of the high efficiency in this system there was noneed to pre-screen the selected colonies prior to picking forminipreps
Large BAC deletions
A drawback of using BACs for the production of transgenicmice is the frequent presence of other genes on the BAC inaddition to the gene of interest This is especially a problem forBAC complementation used in positional cloning where aBAC is tested for its ability to rescue a loss-of-function mutantphenotype by making BAC transgenic mice If complementa-tion is achieved it is impossible to know which gene on theBAC is responsible for the rescue Therefore we decided to
see whether galK selection could be used to make largespecific and clean deletions in BACs so as to remove unwantedgenes (a process called BAC trimming) As a model systemwe chose a mouse BAC from the C57BL6-derived libraryRP23 RP23-341F12 since we could obtain the BAC sequencedirectly from the UCSC genomic web browser (see Materialsand Methods) We then used PCR to amplify the galK selec-tion cassette with primers containing homology to the BACThe forward primer contained 50 bp of homology to the very 50
end of the BAC insert and this primer was combined withthree different reverse primers having 50 bp of homology topositions 50 75 and 100 kb away from the forward primerrsquoshomology (Figure 4A) SW102 bacteria containing the 341F12BAC were heat-induced and made electrocompetent and thentransformed with the three different selection cassettes fol-lowed by the selection on minimal medium with galactoseFrom the many resulting Gal+ colonies we picked four fromeach experiment and then did a SpeI restriction analysis of theBAC miniprep DNAs from them (Figure 4B) All four colon-ies from each experiment had the desired deletion (50 75 and100 kb respectively) The galK selection cassette can then be
Figure 3 Introduction of a G12D mutation in the Nras gene (A) SpeI restriction analysis of BAC miniprep DNA First lane is the unmodified CITB-50J2 Nras BACLanes 1ndash12 show digestion patterns of 12 clones counterselected for the substitution of galK with an oligo containing the GA substitution for the second position ofcodon 12 of Nras Clones 7 and 10 had internal deletions indicating that DOG resistance was achieved by spontaneous deletion and not homologous recombinationThese two clones were not analyzed further (B) Sequence analysis of a PCR product spanning the modified region from clones 1ndash6 8ndash9 and 11ndash12 All clones had theintended substitution (highlighted) However clones 9 and 11 also had an internal basepair deletion indicated by a minus (highlighted) The Nras ATG and codon 12are indicated (shadow)
PAGE 7 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
removed with an oligo using galK negative selection to cleanlydelete the galK cassette from the BAC as described in theprevious experiment (data not shown)
Insertion of a mutant loxP511 site into a BAC
Encouraged by the efficiency of this selection system wedecided to see whether we could introduce a single 34 bploxP511 site cleanly into BAC DNA using galK positiveneg-ative selection It has been previously shown that loxP andloxP511 sites cannot recombine with each other therefore the
introduced loxP511 site can only recombine with the loxP511site present in the BAC vector backbone and not the wild-typesite (Figure 5A) (20) We used galK positive selection to inserta PCR-amplified galK cassette flanked by 50 bp homologyarms to a position 95 kb away from the mutant loxP511 site inthe BAC RP23-341F12 vector backbone (Figure 5A) We thenreplaced the galK cassette using DOG counterselection with adouble-stranded 100 bp oligo containing a 34 bp loxP511 siteflanked by two 33 bp homology arms In this experiment weobserved less than a 10-fold difference in the number of col-onies on the plates from heat-induced and non-induced bac-teria suggesting fewer recombinants This lower frequency ofrecombinants when compared with the Nras G12D substitu-tion experiment is likely explained by the shorter homologyarms used in this latter experiment as it has been shown thatthe efficiency of recombination increase four orders of mag-nitude when homology length is increased from 20 to 40 bp(10) We analyzed six BAC minipreps from potential recom-binants by digesting with SpeI and comparing the restrictionpatterns with that of wild-type RP23-341F12 BAC DNA(Figure 5B) Three of six (50) colonies had exactly thesame restriction pattern as the unmodified RP23-341F12BAC suggesting that DOG resistance had selected for thedesired homologous recombination products in these threecases whereas the other three clones apparently becameDOG resistant due to the selection of BACs that carry largedeletions spanning galK
The 100 bp oligo was designed so that the loxP511 sitewould be inserted in the same orientation as the loxP511site in the BAC vector backbone To confirm that the BACswith the wild-type restriction pattern (3 5 and 6) had theloxP511 site correctly inserted we electroporated thesethree BACs into electrocompetent and L-arabinose inducedEL350 cells which carry an L-arabinose-inducible Cregene and then plated the cells on LB plates containing chlor-amphenicol to select for the BACs Two colonies generatedfrom each recombinant loxP511 clone were then tested bySpeI digestion of BAC miniprep DNA and all had the expec-ted Cre-mediated 95 kb deletion (Figure 5C) Recombinationwas confirmed by PCR using a forward primer from the BACvector backbone and a reverse primer mapping to a positiondistal to the reverse homology arm used to insert the galKcassette These primers are 95 kb apart on wild-type RP23-341F12 DNA and only 378 bp apart on CreloxP511 recom-bined DNA PCR analysis of all six clones produced a band ofthe expected length whereas no product could be amplifiedfrom unmodified RP23-341F12 BAC DNA (Figure 5D)
Source of background deletions following DOG selection
In the Nras G12D substitution and loxP511 insertion experi-ments a background of DOG-resistant bacteria which lackedthe desired mutation and instead carried unwanted deletionsthat spanned the galK selection cassette were observed Therelative ratio of colonies containing BACs with deletions wasalso higher when short homology arms were used This wasexpected since homologous recombination is more efficientwith longer homology arms (10) that would increase the rel-ative frequency of correctly targeted BACs The source of thedeletions could be l Red-mediated since proteins capable ofmediating recombination are expressed when the Red genes
Figure 4 BAC trimming using galK selection (A) Illustration of the design ofthe deletion experiment Homology arm 1 (H1) was held constant and H2 wasseparated from H1 by either 50 75 or 100 kb (B) SpeI restriction analysis ofBAC miniprep DNA from 12 clones showing deletions of 50 75 and 100 kbrespectively after the insertion of the galK selection cassette The first lane isunmodified RP23-341F12 BAC DNA which was included as a control Alltested clones had the intended deletion
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 8 OF 12
are activated Alternatively the deletions could be induced bythe large amount of oligos (200 ng) used in these experimentsor they could simply represent spontaneous deletions thatoccur at low level during normal BAC replication and thatare found by DOG selection To distinguish between thesepossibilities we repeated the DOG selection step used to insertthe loxP511 oligo into BAC RP23-341F12 DNA using fourdifferently treated bacteria samples non-induced bacteriawithout the 100 bp loxP511 oligo (Figure 6A) non-inducedbacteria with the oligo (Figure 6B) induced bacteria without
the oligo (Figure 6C) and induced bacteria with the oligo(Figure 6D) The number of resistant colonies obtainedfrom the four experiments varied by lt10-fold (data notshown) Ten colonies from each electroporation were thenanalyzed by SpeI digestion of BAC miniprep DNA and com-pared with unmodified RP23-341F12 BAC DNA (Figure 6AndashD) Unwanted galK region deletions were observed in all 10colonies for both of the non-induced samples (Figure 6A andB) These deletions are therefore unlikely to be Red-mediatedsince these samples were not heat-induced We also observed
Figure 5 Insertion of a loxP511 site (A) The location of the wild-type and mutant loxP sites in the BAC backbone are indicated along with the extra mutant loxP511site that was introduced into the BAC genomic insert via galK counterselection The 95 kb region deleted by Cre-mediated recombination between the two loxP511sites is indicated and PCR primers used to confirm the deletion are shown as small arrows (B) SpeI restriction analysis of six miniprep clones selected forthe replacement of galK with a dsDNA oligo containing the mutant loxP511 site Clones 3 5 and 6 (circles) had the same restriction pattern as the unmodifiedBAC indicating that DOG resistance occurred due to the intended homologous recombination event Clones 1 2 and 4 had large deletions and were not analyzedfurther (C) SpeI restriction analysis of BAC miniprep DNA from clones 3 5 and 6 after transformation into Cre-induced EL350 cells Two clones from each parentalclone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all clones analyzed confirming the correctinsertion of loxP511 in clones 3 5 and 6 (D) PCR analysis of the six clones from (C) with one primer mapping to the BAC backbone and the other to a position distalto the inserted loxP511 site
PAGE 9 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
deletions in both the absence and presence of oligo indicatingthat the deletions are not oligo induced We therefore concludethat these are spontaneous deletions that occur at low levelduring normal BAC replication in the DH10B background
Non-deleted colonies with the parental SpeI fingerprintwere only observed in the Red-induced sample that received
oligo (Figure 6D clones 33 and 35) These clones were sub-sequently electroporated into EL350 Cre-expressing cells toconfirm they contained the introduced loxP511 site Two col-onies from each original clone were tested by SpeI digestion ofthe BAC miniprep DNA As shown in Figure 6E each colonyhas undergone Cre-mediated deletion confirming that these
Figure 6 Same experiment as in Figure 5 with modifications as indicated at the top of each panel (A) SpeI digest of 10 minipreps from a control experiment withoutheat-induction and without the loxP511 dsDNA oligo (B) SpeI digest of 10 minipreps from a control experiment without heat-induction but with the loxP511 dsDNAoligo (C) SpeI digest of 10 minipreps from a control experiment with heat-induction but without the loxP511 dsDNA oligo (D) SpeI digest of 10 minipreps from anexperiment with heat-induction and with the loxP511 dsDNA oligo (comparable with Figure 5B) Clones with the parental digestion pattern indicating DOGresistance due to homologous recombination (clones 33 and 35 circles) are only seen in (D) DOG resistance in all other clones likely occurred due to internaldeletions of the BACs (E) SpeI restriction analysis of BAC miniprep DNA from clones 33 and 35 after transformation into Cre-induced EL350 cells Two clonesfrom each parental clone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all the clones analyzedconfirming the correct insertion of loxP511 in clones 33 and 35 (compare with Figure 5C)
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 10 OF 12
colonies were correctly targeted and contain the introducedloxP511 site (2 out of 10 clones a 20 efficiency in thisexperiment)
Generation of SW105 and SW106 cells
The two DY380-derived bacterial strains EL250 and EL350contain the defective l prophage needed for recombineering inaddition to L-arabinose inducible Flp or Cre genes respect-ively (11) Both strains have proven to be very useful for BACmodification (11) and EL350 is now used routinely for mak-ing conditional targeting vectors for ES cell knock-out experi-ments (4) To further enhance the usefulness of these strainswe decided to transfer the galK selection system into themThis was done as previously described for SW102 The galKIS2 element present in both strains was replaced with the wild-type gal promoter and the galK gene in the gal operon deletedusing DOG selection The resulting strains SW105 (Flp) andSW106 (Cre) (Table 1) were then tested to confirm that theystill contained inducible Flp and Cre genes by transformingarabinose-induced and electrocompetent cells with plasmidscontaining a neo gene flanked by Frt or loxP sites PL451 andPL452 (4) respectively Using SW105 or SW106 it is nowpossible to introduce a point mutation or an informativerestriction site into a BAC retrieve a fragment from thisBAC containing the introduced mutation(s) into a plasmidbackbone using gap repair and turn the retrieved fragmentinto a conditional targeting vectormdashall using only one bac-terial strain and only a single initial BAC transformation
DISCUSSION
Here we describe a new recombineering-based Ecoli BACmodification system that makes use of galK positive selectionfor growth on galactose minimal medium and galK negativeselection (counterselection) for growth on DOG This modi-fication system has several advantages compared with theother related BAC modification systems First the galK selec-tion cassette is small (1231 bp + homology arms) comparedwith for example the sacBndashneo cassette (3 kb) making PCRamplification and transformation into bacteria easier Twohomology arms are easily added to the galK cassette by includ-ing these sequences in the 50 ends of the primers used foramplification (see Materials and Methods) Second sincegalK is used for both selection steps mutations occurringin galK during PCR amplification will be selected againstduring positive selection significantly reducing the risk ofDOG resistance from PCR mutations in galK during negativeselection Third even when very short (33 bp) homology armswere used the frequency of recombinants among the analyzedDOG-resistant clones was still 20ndash50 To our knowledgesuccessful BAC modification with such short homology armshas never been reported Finally since galK recombineering isso efficient little screening is required following selectionreducing the overall hands-on-time to a minimum galK selec-tion requires growth on minimal medium in both selectionsteps although perhaps not used routinely in most molecularbiology laboratories where BAC modification is needed theseplates are fairly simple to make and should not prevent anyonefrom using this method
For all such counterselection schemes there will bebackground since during negative selection any event leadingto the loss of the counterselectable marker will result in sur-vival In our system virtually all background appears to resultfrom deletions that span the inserted galK gene These dele-tions likely occur during BAC replication in the 45 h out-growth phase since we have shown that these deletions occurindependently of Red induction and in the absence or presenceof oligo It is well established that in recA defective bacteriaBACs are very stable compared with other large insert vectorssuch as yeast artificial chromosomes (YACs) However usingcounterselection rare spontaneous deletions are seen becauseof the strong selection force This spontaneous deletion back-ground is not a problem however due to the high frequency ofhomologous recombination obtained with the defective lprophage system
Increasing the length of homology arms used for recombin-eering will reduce the relative number of background deletionsobserved following negative selection since the percentage ofcolonies containing BACs that eliminated galK by homolog-ous recombination will be increased Oligos with longerhomology arms can be produced by annealing two oligoswith overlapping 30 ends (21) These overlapping oligos (50
single strand overhangs) are then filled in by DNA polymerasein vivo (or in vitro) and subsequently serve as the substrate forthe l Red proteins exo and bet Longer homology arms canalso be added by traditional cloning using restriction enzymesand DNA ligase (4)
A number of uses can be imagined for the strains describedhere In principle our strains can be used to make virtually anykind of BAC modification one can imagine including pointmutations or clean deletions or insertions of everything fromcDNA to loxP sites or small epitope tags BAC trimming thespecific deletion of BAC DNA flanking a gene of interest couldalso be used to remove genes from BACs prior to making BACtransgenic mice so that only the gene of interest on the BAC isanalyzed This should be of particular interest for BAC rescueexperiments in positional cloning Of course trimming andmodification can be combined since the galK selection cassettecan be recycled so that the same BAC can be modified severaltimes BAC modification could also be the starting point forconstructing a gene-targeting vector to allow for more soph-isticated gene targeting in mouse ES cells The desired muta-tion(s) could first be created in the BAC followed by retrievalinto a plasmid vector containing a negative-selection markerlike thymidine kinase using gap repair (4) Finally a neo mar-ker could be introduced to allow for positive selection in EScells Alternatively the retrieved fragment could be modifiedso that the end result is a conditional targeting vector StrainSW106 which in addition to galK selection can be induced toexpress Cre recombinase makes it possible to perform all ofthe steps needed to construct a conditional targeting vectorstarting with only a single BAC transformation step
Furthermore in experiments where BAC transgenic miceare used to analyze the effect of deleting long-range regulatorsof gene expression galK DOG counterselection can even beused in the absence of an oligo to generate a series of dele-tions around the galK insertion site and the effect on generegulation studied
The Flp or Cre ORFs contained in strains SW105 andSW106 respectively can also be replaced for any desired
PAGE 11 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
gene thus creating tight arabinose-inducible expression forany gene of interest This is done by first replacing the CreFlpe ORF with galK and select for Gal+ recombinants ThegalK cassette is then replaced by the ORF of the gene ofinterest by DOG selection for Gal recombinants
Finally the strains described here should be very useful forgenetic manipulation not only of BACs but also the Ecoligenome itself
All recombineering reagents discussed in this workare freely available upon request To obtain these materialsplease follow the directions listed on our website (httprecombineeringncifcrfgov) Detailed protocols for recom-bineering including galK selection can also be downloadeddirectly from our website
ACKNOWLEDGEMENTS
We thank Allen Kane and Carolyn Whistler from ScientificPublications Graphics amp Media NCI-Frederick for help withillustrations and figure preparation Funding for this research aswell as the Open Access publication charges was provided byDHHS NIH NCI
REFERENCES
1 ShizuyaH BirrenB KimUJ MancinoV SlepakTTachiiriY and SimonM (1992) Cloning and stable maintenance of300-kilobase-pair fragments of human DNA in Escherichia coli using anF-factor-based vector Proc Natl Acad Sci USA 89 8794ndash8797
2 YangXW ModelP and HeintzN (1997) Homologous recombinationbased modification in Escherichia coli and germline transmission intransgenic mice of a bacterial artificial chromosome Nat Biotechnol15 859ndash865
3 AntochMP SongEJ ChangAM VitaternaMH ZhaoYWilsbacherLD SangoramAM KingDP PintoLH andTakahashiJS (1997) Functional identification of the mouse circadianClock gene by transgenic BAC rescue Cell 89 655ndash667
4 LiuP JenkinsNA and CopelandNG (2003) A highly efficientrecombineering-based method for generating conditional knockoutmutations Genome Res 13 476ndash484
5 Cotta-de-AlmeidaV SchonhoffS ShibataT LeiterA andSnapperSB (2003) A new method for rapidly generating gene-targetingvectors by engineering BACs through homologous recombination inbacteria Genome Res 13 2190ndash2194
6 CopelandNG JenkinsNA and CourtDL (2001) Recombineeringa powerful new tool for mouse functional genomics Nature RevGenet 2 769ndash779
7 CourtDL SawitzkeJA and ThomasonLC (2002) Geneticengineering using homologous recombination Annu Rev Genet 36361ndash388
8 ZhangY BuchholzF MuyrersJP and StewartAF (1998) A newlogic for DNA engineering using recombination in Escherichia coliNature Genet 20 123ndash128
9 MuyrersJP ZhangY TestaG and StewartAF (1999) Rapidmodification of bacterial artificial chromosomes by ET-recombinationNucleic Acids Res 27 1555ndash1557
10 YuD EllisHM LeeEC JenkinsNA CopelandNG andCourtDL (2000) An efficient recombination system for chromosomeengineering in Escherichia coli Proc Natl Acad Sci USA 975978ndash5983
11 LeeEC YuD Martinez de VelascoJ TessarolloL SwingDACourtDL JenkinsNA and CopelandNG (2001) A highly efficientEscherichia coli-based chromosome engineering system adapted forrecombinogenic targeting and subcloning of BAC DNA Genomics73 56ndash65
12 GongS YangXW LiC and HeintzN (2002) Highly efficientmodification of bacterial artificial chromosomes (BACs) using novelshuttle vectors containing the R6Kgamma origin of replicationGenome Res 12 1992ndash1998
13 JamsaiD OrfordM NefedovM FucharoenS WilliamsonR andIoannouPA (2003) Targeted modification of a human beta-globin locusBAC clone using GET Recombination and an I-SceI counterselectioncassette Genomics 82 68ndash77
14 SwaminathanS EllisHM WatersLS YuD LeeECCourtDL and SharanSK (2001) Rapid engineering of bacterialartificial chromosomes using oligonucleotides Genesis 2914ndash21
15 AlperMD and AmesBN (1975) Positive selection of mutants withdeletions of the gal-chl region of the Salmonella chromosome as ascreening procedure for mutagens that cause deletions J Bacteriol121 259ndash266
16 AusubelFM BrentR KingstonRE MooreDD SeidmanJGSmithJA and StruhlK (1987) Current Protocols in Molecular BiologyGreene Publishing Associates and Wiley-Interscience John Wiley ampSons Hoboken NJ
17 BachmannBJ (1996) Derivations and genotypes of some mutantderivatives of Escherichia coli K-12 In NeidhardtFC CurtissRIIIIngrahamJL LinECC LowKB MagasanikB ReznikoffWSRileyM SchaechterM and UmbargerHE (eds) Escherichia coli andSalmonella Cellular and Molecular Biology ASM press WashingtonDC Vol 2 pp 2460ndash2488
18 GrantSG JesseeJ BloomFR and HanahanD (1990) Differentialplasmid rescue from transgenic mouse DNAs into Escherichia colimethylation-restriction mutants Proc Natl Acad Sci USA 874645ndash4649
19 OppenheimAB RattrayAJ BubunenkoM ThomasonLC andCourtDL (2004) In vivo recombineering of bacteriophage lambda byPCR fragments and single-strand oligonucleotides Virology 319185ndash189
20 HoessRH WierzbickiA and AbremskiK (1986) The role ofthe loxP spacer region in P1 site-specific recombination NucleicAcids Res 14 2287ndash2300
21 YuD SawitzkeJA EllisH and CourtDL (2003) Recombineeringwith overlapping single-stranded DNA oligonucleotides testing arecombination intermediate Proc Natl Acad Sci USA 1007207ndash7212
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 12 OF 12
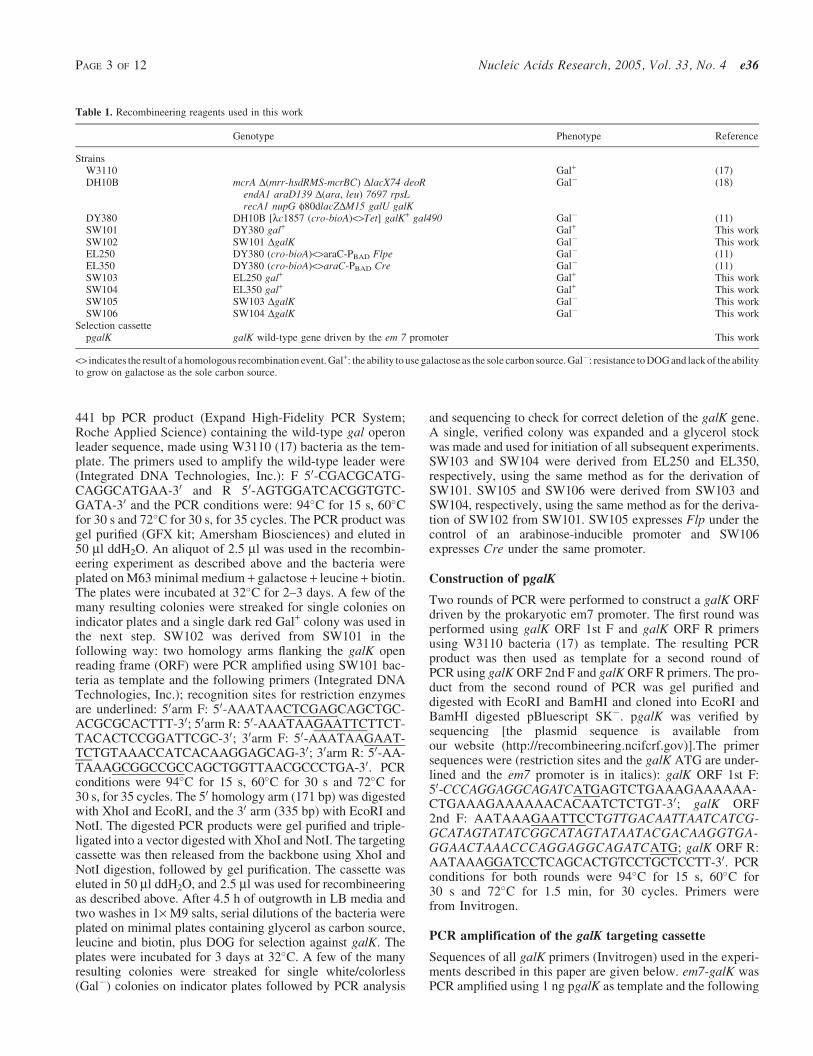
441 bp PCR product (Expand High-Fidelity PCR SystemRoche Applied Science) containing the wild-type gal operonleader sequence made using W3110 (17) bacteria as the tem-plate The primers used to amplify the wild-type leader were(Integrated DNA Technologies Inc) F 50-CGACGCATG-CAGGCATGAA-30 and R 50-AGTGGATCACGGTGTC-GATA-30 and the PCR conditions were 94C for 15 s 60Cfor 30 s and 72C for 30 s for 35 cycles The PCR product wasgel purified (GFX kit Amersham Biosciences) and eluted in50 ml ddH2O An aliquot of 25 ml was used in the recombin-eering experiment as described above and the bacteria wereplated on M63 minimal medium + galactose + leucine + biotinThe plates were incubated at 32C for 2ndash3 days A few of themany resulting colonies were streaked for single colonies onindicator plates and a single dark red Gal+ colony was used inthe next step SW102 was derived from SW101 in thefollowing way two homology arms flanking the galK openreading frame (ORF) were PCR amplified using SW101 bac-teria as template and the following primers (Integrated DNATechnologies Inc) recognition sites for restriction enzymesare underlined 50arm F 50-AAATAACTCGAGCAGCTGC-ACGCGCACTTT-30 50arm R 50-AAATAAGAATTCTTCT-TACACTCCGGATTCGC-30 30arm F 50-AAATAAGAAT-TCTGTAAACCATCACAAGGAGCAG-30 30arm R 50-AA-TAAAGCGGCCGCCAGCTGGTTAACGCCCTGA-30 PCRconditions were 94C for 15 s 60C for 30 s and 72C for30 s for 35 cycles The 50 homology arm (171 bp) was digestedwith XhoI and EcoRI and the 30 arm (335 bp) with EcoRI andNotI The digested PCR products were gel purified and triple-ligated into a vector digested with XhoI and NotI The targetingcassette was then released from the backbone using XhoI andNotI digestion followed by gel purification The cassette waseluted in 50 ml ddH2O and 25 ml was used for recombineeringas described above After 45 h of outgrowth in LB media andtwo washes in 1middot M9 salts serial dilutions of the bacteria wereplated on minimal plates containing glycerol as carbon sourceleucine and biotin plus DOG for selection against galK Theplates were incubated for 3 days at 32C A few of the manyresulting colonies were streaked for single whitecolorless(Gal) colonies on indicator plates followed by PCR analysis
and sequencing to check for correct deletion of the galK geneA single verified colony was expanded and a glycerol stockwas made and used for initiation of all subsequent experimentsSW103 and SW104 were derived from EL250 and EL350respectively using the same method as for the derivation ofSW101 SW105 and SW106 were derived from SW103 andSW104 respectively using the same method as for the deriva-tion of SW102 from SW101 SW105 expresses Flp under thecontrol of an arabinose-inducible promoter and SW106expresses Cre under the same promoter
Construction of pgalK
Two rounds of PCR were performed to construct a galK ORFdriven by the prokaryotic em7 promoter The first round wasperformed using galK ORF 1st F and galK ORF R primersusing W3110 bacteria (17) as template The resulting PCRproduct was then used as template for a second round ofPCR using galK ORF 2nd F and galK ORF R primers The pro-duct from the second round of PCR was gel purified anddigested with EcoRI and BamHI and cloned into EcoRI andBamHI digested pBluescript SK pgalK was verified bysequencing [the plasmid sequence is available fromour website (httprecombineeringncifcrfgov)]The primersequences were (restriction sites and the galK ATG are under-lined and the em7 promoter is in italics) galK ORF 1st F50-CCCAGGAGGCAGATCATGAGTCTGAAAGAAAAAA-CTGAAAGAAAAAACACAATCTCTGT-30 galK ORF2nd F AATAAAGAATTCCTGTTGACAATTAATCATCG-GCATAGTATATCGGCATAGTATAATACGACAAGGTGA-GGAACTAAACCCAGGAGGCAGATCATG galK ORF RAATAAAGGATCCTCAGCACTGTCCTGCTCCTT-30 PCRconditions for both rounds were 94C for 15 s 60C for30 s and 72C for 15 min for 30 cycles Primers werefrom Invitrogen
PCR amplification of the galK targeting cassette
Sequences of all galK primers (Invitrogen) used in the experi-ments described in this paper are given below em7-galK wasPCR amplified using 1 ng pgalK as template and the following
Table 1 Recombineering reagents used in this work
Genotype Phenotype Reference
StrainsW3110 Gal+ (17)DH10B mcrA D(mrr-hsdRMS-mcrBC) DlacX74 deoR
endA1 araD139 D(ara leu) 7697 rpsLrecA1 nupG f80dlacZDM15 galU galK
Gal (18)
DY380 DH10B [lc1857 (cro-bioA)ltgtTet] galK+ gal490 Gal (11)SW101 DY380 gal+ Gal+ This workSW102 SW101 DgalK Gal This workEL250 DY380 (cro-bioA)ltgtaraC-PBAD Flpe Gal (11)EL350 DY380 (cro-bioA)ltgtaraC-PBAD Cre Gal (11)SW103 EL250 gal+ Gal+ This workSW104 EL350 gal+ Gal+ This workSW105 SW103 DgalK Gal This workSW106 SW104 DgalK Gal This work
Selection cassettepgalK galK wild-type gene driven by the em 7 promoter This work
ltgt indicates the result of a homologous recombination event Gal+ the ability to use galactose as the sole carbon source Gal resistance to DOG and lack of the abilityto grow on galactose as the sole carbon source
PAGE 3 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
conditions 94C for 15 s 60C for 30 s and 72C for 1 min for30 cycles After completion of PCR 2 ml DpnI was added toeach 25 ml reaction and incubated for 2 h at 37C to removeany plasmid template The DpnI-digested reaction mix was runon a 1 agarose gel over night and the PCR product waspurified and eluted in 50 ml ddH2O An aliquot of 25 ml wasused for each experiment For making the Nras G12D substi-tution the following primers were used for the first step intro-ducing galK Homology to Nras sequence is in italics and thesequence recognizing em7-galK is underlined Nras galK F50-TTTTTGCTGGTGTGAAATGACTGAGTACAAACTGGTGG-TGGTTGGAGCAGCCTGTTGACAATTAATCATCCGCA-30Nras galK R 50-CAAAGTGGTTCTGGATTAGCTGGATCG-TCAAGGCGCTTTTCCCAACACCATCAGCACTGTCCTGC-TCCTT-30 For making 50 75 and 100 kb deletions in theRP23-341F12 BAC the following primers were used (homo-logy to BAC sequence is in italics and the sequence recog-nizing em7-galK is underlined) galK F 341F12 start50-ACTCCCACTGGAAGCTTTTTACAAAACATGTGTTGCT-GACATGTTGACAGCCTGTTGACAATTAATCATCGGCA-30 galK R 341F12 50 kb 50-ACCCAAACCAAACAACAT-CCAAACCAAAAACACAGACAAAACCAAATATGTCAGCAC-TGTCCTGCTCCTT-30 galK R 341F12 75 kb 50-ACACTAAGC-CAAACTCCTTGCCTGGGCTATTTCTCTTTGTTTTTCCAAATTCAGC-ACTGTCCTGCTCCTT-30 galK R 341F12 100 kb 50-TATGT-GTCTGTGTGTGTATGTACAGTTCTTTGTTTTTGTTTTTTTT-CTTTTCAGCACTGTCCTGCTCCTT-30 For insertion of aloxP511 site in the RP23-341F12 BAC the following primerswere used for the first step (introduction of galK) Homology toBAC sequence is in italics and the sequence recognizing em7-galK is underlined 95 kb loxP511 galK F 50-GGACA-GAGGCGTGACAGACGTGTGAGCTCCGTGGACAACTCTC-CCCGAAGCCTGTTGACAATTAATCATCGGCA-30 95 kbloxP511 galK R 50-GACTCTGAGCAGCAACGGCTGAGCCT-CACTTGAGAGGGTCCCTGAGTCACTCAGCACTGTCCTG-CTCCTT-30
Oligos for recombineering
Oligos used to replace the galK-targeting cassette wereobtained from Invitrogen dsDNA was used and the oligos(sense and antisense) annealed in vitro 10 mg of each oligo(sense and antisense) was mixed in an eppendorf tube in a totalvolume of 100 ml of 1middot PCR buffer (Expand High FidelityPCR kit Roche Applied Science) and boiled for 5 minallowed to cool to room temperature for 30 min ethanol pre-cipitated and resuspended in 100 ml ddH2O to a final concen-tration of 200 ngml double-stranded oligo An aliquot of 1 ml(200 ng) was used in the recombineering experiments Tointroduce the G12D (GltgtA) point mutation in the NrasBAC CITB 50J2 the following oligos were used for thesecond step (the introduced adenosinethymidine base pairis underlined the flanking sequences are homologous to theNras BAC sequence) G12D S 50-TTTTTGCTGGTGT-GAAATGACTGAGTACAAACTGGTGGTGGTTGGAGC-AGATGGTGTTGGGAAAAGCGCCTTGACGATCCAGC-TAATCCAGAACCACTTT-30 G12D AS 50-AAAGTGGT-TCTGGATTAGCTGGATCGTCAAGGCGCTTTTCCCAA-CACCATCTGCTCCAACCACCACCAGTTTGTACTCAG-TCATTTCACACCAGCAAAAA-30 To introduce a loxP511site in the RP23-341F12 BAC the following oligos were used
for the second step (loxP511 is underlined the flankingsequences are homologous to the BAC sequence aroundposition 95 kb) 95 kb loxP511 S 50-ACGTGTGAGCTC-CGTGGACAACTCTCCCCGAAGATAACTTCGTATAGT-ATACATTATACGAAGTTATGTGACTCAGGGACCCTC-TCAAGTGAGGCTCAGC-30 95 kb loxP511 AS 50-GCTG-AGCCTCACTTGAGAGGGTCCCTGAGTCACATAACTT-CGTATAATGTATACTATACGAAGTTATCTTCGGGGA-GAGTTGTCCACGGAGCTCACACGT-30
Verification of positive recombinants
In the G12D Nras experiment the selected Gal clones wereanalyzed by SpeI digestion of BAC miniprep DNA usingunmodified CITB 50J2 BAC DNA as a control Clones with-out rearrangements were analyzed by PCR using 1 ml BACminiprep DNA as the template The PCR products were gelpurified and sequenced using the same primers as were usedfor PCR Primers flanking the targeted mutation were Nrastest F 50-CACTCATCTGCAAGGAATGCT-30 Nras test R50-CCTCAGTAAGCACGAACTTGT-30 PCR conditionswere 94C for 15 s 60C for 30 s and 72C for 30 s for30 cycles Modifications of the RP23-341F12 BAC (50 75and 100 kb deletions and the introduction of a loxP511 site)were tested by SpeI restriction analysis of BAC miniprep DNAand compared with unmodified 341F12 BAC DNA In theloxP511 experiment clones 3 5 and 6 were further testedfor correct insertion of the loxP511 site by transforming1 ml of BAC miniprep DNA into electrocompetent andarabinose-induced EL350 cells (11) and plating on LB plateswith chloramphenicol Two colonies from each starting clonewere tested by SpeI digestion of BAC miniprep DNA for the95 kb Cre-mediated deletion Finally the Cre-recombinedclones were tested by PCR with one primer mapping to theend of the pBACe36 BAC backbone and the other mapping toa position 95 kb away on the wild-type BAC The primers(Invitrogen) used for this analysis were 95 kb loxP511 checkF 50-GCGGATGAATGGCAGAAATTC-30 95 kb LoxP511check R 50-TTTGCCAGACTGGTGCCTAA-30 PCR condi-tions were 94C for 15 s 60C for 30 s and 72C for 30 s for30 cycles The resulting PCR bands were gel purified andconfirmed by sequencing using the same primers as wereused for the PCR amplification The follow-up experimentfor testing the source of the observed BAC deletions wasdone as described above
BACs
Mouse BACs were obtained from Invitrogen The RP23-341F12 BAC (C57BL6 DNA) was chosen based on positionwithin the mouse Ebfaz gene using the UCSC genome browser(httpgenomeucscedu) The CITB 50J2 BAC (CJ7129SVDNA) was identified by screening a CITB mouse BAC library(Invitrogen) with an Nras genomic probe using standardhybridization methods The BAC clones were streaked forsingle colonies and characterized by PCR and restriction ana-lysis before proceeding to the recombineering experimentsTwo methods were used for DNA preparation For BAC mini-preps (1ndash15 mg) we used the following protocol 5 ml over-night LB culture (15 ml Falcon tube) was pelleted for 5 min at5000 rpm the supernatant removed and the pellet wasdissolved in 250 ml buffer P1 (miniprep kit Qiagen) and
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 4 OF 12
transferred to an eppendorf tube An aliquot of 250 ml P2buffer was added followed by mixing by inversion and incuba-tion for lt5 min at room temperature An aliquot of 250 ml N3buffer was added followed by mixing and incubation on ice for5 min The supernatant was cleared by two rounds of centri-fugation at 13 200 rpm for 5 min in a tabletop centrifugeEach time the supernatant was transferred to a new tube DNAwas precipitated by adding 750 ml isopropanol mixing andincubating on ice for 10 min and centrifugation for 10 min at13 200 rpm The pellet was washed once in 70 ethanol andthe dry pellet was dissolved in 50 ml TE An aliquot of 40 ml(1 mg) was used for restriction analysis and 1 ml was used astemplate for PCR analysis or transformation of electrocom-petent bacteria Large-scale preparations of BAC DNA(25ndash100 mg) were done using the Nucleobond BAC maxikit from Clontech (BD Biosciences) following the manufac-turerrsquos protocol
RESULTS
Generation of SW102 cells
To see whether we could develop a more efficient selectivesystem for BAC recombineering we looked for a single select-able marker that could be used for both positive and negativeselection We focused on Ecoli galK because both selectionsteps could be done using galK and its small size makes it easyto amplify by PCR We previously reported the developmentof a bacterial strain DY380 that is readily transformable withBAC DNA due to its DH10B origin (1) DY380 cells alsoharbor the defective l prophage required for recombineeringThe defective prophage in DY380 was transferred into DH10Bwith a P1 phage lysate obtained from DY363 cells [for detailssee (11)] DY363 cells carry a 1200 bp IS2 insertion elementgal490 in the mRNA leader sequence of the galactose operonpreventing gal gene transcription Since the galactose operonis directly proximal to the site of insertion of the defective
prophage the gal490 mutation was also transferred to DY380during P1 transduction Thus DY380 is phenotypically Gal
(galactose minus) and therefore unable to grow on galactoseminimal medium
Recombineering was used to correct this problem From thewild-type Ecoli strain W3110 (17) we PCR amplified a 441bp fragment from the wild-type gal promoter which spans theregion containing the IS2 insertion element in DY380 ThisPCR product was introduced into DY380 and Gal+ recombin-ants were selected for growth on galactose minimal medium(see Materials and Methods) We named this strain SW101(Table 1) This strain is identical to DY380 except that it lacksthe IS2 insertion element and has been made wild type for thegal operon We then made a precise deletion of the galK geneleaving all other genes in the galactose operon intact This wasachieved by PCR amplifying two homology arms of 171 and335 bp respectively flanking the galK ORF and cloning themtogether into a plasmid using three-way ligation Since theORF of galK overlaps that of galM we left behind the last33 bp of galK (Figure 1) to allow for proper translation ofgalM The targeting cassette consisting of a 512 bp linearfragment was electroporated into heat-induced and electro-competent SW101 cells and the recombinant clones selectedon minimal medium containing glycerol as the carbon sourceand DOG for selection against galK The genotype of theresulting strain SW102 (Table 1) was confirmed by PCRanalysis and sequencing of the modified region (Figure 1)This strain now harbors not only the defective l prophagebut also a functional gal operon except for the deletion ofgalK The l prophage is located between the galactose operonand the biotin operon and the bio operon in these cells isnonfunctional causing a biotin requirement DH10B andall derived strains including SW102 are also deficient inleucine metabolism (18) When using minimal medium onetherefore needs to add both biotin and leucine to the plates toallow SW102 growth (see Materials and Methods)
Figure 1 Sequence analysis of the galactose operon in strains DY380 (A) SW101 (B) and SW102 (C) In SW102 the ORF of galK was deleted leaving only 33 bp ofgalK behind to make sure that translation of galM is initiated properly EcoRI the restriction site used to clone the 50 and 30 homology arms flanking galK
PAGE 5 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
galK expression cassette
Having produced a bacterial strain SW102 which is galKdefective the next step was to make a galK expression cassettethat could be used to restore the bacteriarsquos ability to grow ongalactose by providing galK in trans This was achieved byPCR amplification of the wild-type galK ORF from W3110cells We then added a minimal bacterial promoter em7 usinga two-step PCR approach (see Materials and Methods) andcloned the expression cassette into pBluescript SK We callthis plasmid pgalK (Table 1) The constitutively active galKexpression cassette can easily be amplified by PCRwith homology arms added to the primers (see Materialsand Methods)
Making a single base pair substitution
The general scheme for making mutations in BACs using galKselection is depicted in Figure 2 To test the galK selectionsystem for BAC recombineering we decided to introduce apoint mutation into a BAC containing the murine Nras gene(CITB clone 50J2) The sequence of the glycine-coding codon12 is GGT By changing this codon to GAT we would obtainthe desired mutation G12D In order to introduce this muta-tion into the BAC we first amplified the galK expressioncassette by PCR using primers with 50 bp of homology toeither side of the second position of the GGT codon Followinghomologous recombination this targeting would introduce a1 bp deletion into codon 12 in addition to inserting the galKselection cassette Instead of deleting the basepair the galKcassette could have been inserted right next to the basepairinstead SW102 cells containing the 50J2 BAC were heat-induced and made electrocompetent and then electroporatedwith the galK cassette Gal+ recombinant colonies were selec-ted for growth on galactose minimal medium with chloram-phenicol to maintain the BAC Bacteria grow more slowly onminimal media than on rich media and we generally pickcolonies after 2ndash3 days For this first step we do not expectany background colonies on the non-induced control plates ifthe pgalK plasmid is properly eliminated (see Materials andMethods) To purify the Gal+ colonies we streaked a fewcolonies on MacConkey galactose indicator plates to obtainsingle bright pinkred Gal+ colonies One of these single-cloned colonies was picked to initiate a culture for thenext step counterselection We find that there is no need toanalyze the Gal+ colonies further before proceeding tocounterselection
A 100 bp dsDNA oligo was then prepared by annealing twocomplementary oligos having 49 and 50 bp homologyrespectively to either side of the desired mutation a singleAT bp An aliquot of 200 ng of this oligo was then electro-porated into heat-induced and electrocompetent SW102 Gal+
cells containing the galK modified 50J2 BAC After electro-poration the bacteria were allowed 45 h outgrowth in a 32Cshaking waterbath The bacteria were then washed in M9 saltsto remove any rich medium and plated on minimal mediumwith glycerol DOG and chloramphenicol The 45 h out-growth is necessary to obtain complete segregation of therecombinant BACs containing the mutation After 3 dayswe obtained colonies with a ratio of 10ndash1001 when comparingplates with heat-induced to non-induced bacteria We picked12 colonies from the heat-induced plates for BAC minipreps
followed by SpeI restriction analysis (Figure 3A) Ten out ofthe twelve clones had the same restriction pattern as theunmodified 50J2 BAC DNA suggesting that the desiredreplacement of the galK gene by the point mutation hadoccurred This was confirmed by PCR amplification followed
Figure 2 Overview of the galK selection scheme The result of the firsttargeting event is the insertion of constitutively active galK into a definedposition on the BAC by selection on minimal medium containing galactoseand chloramphenicol to select for the maintenance of the BAC The bacteria arenow phenotypically Gal+ Next the galK cassette is replaced by a dsDNA oligoa PCR product or a cloned dsDNA fragment carrying a desired mutation(indicated by a star) and flanked by the same homology arms used in thefirst selection step This is achieved by negative selection using minimalmedium containing 2-deoxy-galactose (DOG) with glycerol as the sole carbonsource The bacteria become phenotypically Gal H1 and H2 homology arms1 and 2 respectively cat chloramphenicol acetyl transferase gene ori2 BACorigin of replication galK Ecoli galactokinase gene driven by a minimalpromoter
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 6 OF 12
by sequencing of the modified region of the 10 BAC clones(Figure 3B) All 10 sequenced clones had the desired pointmutation The effective recombination efficiency in thisexperiment however was 812 (67) since 2 of the 10 clonesalso had an additional single base pair deletion (clones 9 and11) These deletions probably occurred during oligo synthesissince we did not purify the oligos beyond desalting (19)Because of the high efficiency in this system there was noneed to pre-screen the selected colonies prior to picking forminipreps
Large BAC deletions
A drawback of using BACs for the production of transgenicmice is the frequent presence of other genes on the BAC inaddition to the gene of interest This is especially a problem forBAC complementation used in positional cloning where aBAC is tested for its ability to rescue a loss-of-function mutantphenotype by making BAC transgenic mice If complementa-tion is achieved it is impossible to know which gene on theBAC is responsible for the rescue Therefore we decided to
see whether galK selection could be used to make largespecific and clean deletions in BACs so as to remove unwantedgenes (a process called BAC trimming) As a model systemwe chose a mouse BAC from the C57BL6-derived libraryRP23 RP23-341F12 since we could obtain the BAC sequencedirectly from the UCSC genomic web browser (see Materialsand Methods) We then used PCR to amplify the galK selec-tion cassette with primers containing homology to the BACThe forward primer contained 50 bp of homology to the very 50
end of the BAC insert and this primer was combined withthree different reverse primers having 50 bp of homology topositions 50 75 and 100 kb away from the forward primerrsquoshomology (Figure 4A) SW102 bacteria containing the 341F12BAC were heat-induced and made electrocompetent and thentransformed with the three different selection cassettes fol-lowed by the selection on minimal medium with galactoseFrom the many resulting Gal+ colonies we picked four fromeach experiment and then did a SpeI restriction analysis of theBAC miniprep DNAs from them (Figure 4B) All four colon-ies from each experiment had the desired deletion (50 75 and100 kb respectively) The galK selection cassette can then be
Figure 3 Introduction of a G12D mutation in the Nras gene (A) SpeI restriction analysis of BAC miniprep DNA First lane is the unmodified CITB-50J2 Nras BACLanes 1ndash12 show digestion patterns of 12 clones counterselected for the substitution of galK with an oligo containing the GA substitution for the second position ofcodon 12 of Nras Clones 7 and 10 had internal deletions indicating that DOG resistance was achieved by spontaneous deletion and not homologous recombinationThese two clones were not analyzed further (B) Sequence analysis of a PCR product spanning the modified region from clones 1ndash6 8ndash9 and 11ndash12 All clones had theintended substitution (highlighted) However clones 9 and 11 also had an internal basepair deletion indicated by a minus (highlighted) The Nras ATG and codon 12are indicated (shadow)
PAGE 7 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
removed with an oligo using galK negative selection to cleanlydelete the galK cassette from the BAC as described in theprevious experiment (data not shown)
Insertion of a mutant loxP511 site into a BAC
Encouraged by the efficiency of this selection system wedecided to see whether we could introduce a single 34 bploxP511 site cleanly into BAC DNA using galK positiveneg-ative selection It has been previously shown that loxP andloxP511 sites cannot recombine with each other therefore the
introduced loxP511 site can only recombine with the loxP511site present in the BAC vector backbone and not the wild-typesite (Figure 5A) (20) We used galK positive selection to inserta PCR-amplified galK cassette flanked by 50 bp homologyarms to a position 95 kb away from the mutant loxP511 site inthe BAC RP23-341F12 vector backbone (Figure 5A) We thenreplaced the galK cassette using DOG counterselection with adouble-stranded 100 bp oligo containing a 34 bp loxP511 siteflanked by two 33 bp homology arms In this experiment weobserved less than a 10-fold difference in the number of col-onies on the plates from heat-induced and non-induced bac-teria suggesting fewer recombinants This lower frequency ofrecombinants when compared with the Nras G12D substitu-tion experiment is likely explained by the shorter homologyarms used in this latter experiment as it has been shown thatthe efficiency of recombination increase four orders of mag-nitude when homology length is increased from 20 to 40 bp(10) We analyzed six BAC minipreps from potential recom-binants by digesting with SpeI and comparing the restrictionpatterns with that of wild-type RP23-341F12 BAC DNA(Figure 5B) Three of six (50) colonies had exactly thesame restriction pattern as the unmodified RP23-341F12BAC suggesting that DOG resistance had selected for thedesired homologous recombination products in these threecases whereas the other three clones apparently becameDOG resistant due to the selection of BACs that carry largedeletions spanning galK
The 100 bp oligo was designed so that the loxP511 sitewould be inserted in the same orientation as the loxP511site in the BAC vector backbone To confirm that the BACswith the wild-type restriction pattern (3 5 and 6) had theloxP511 site correctly inserted we electroporated thesethree BACs into electrocompetent and L-arabinose inducedEL350 cells which carry an L-arabinose-inducible Cregene and then plated the cells on LB plates containing chlor-amphenicol to select for the BACs Two colonies generatedfrom each recombinant loxP511 clone were then tested bySpeI digestion of BAC miniprep DNA and all had the expec-ted Cre-mediated 95 kb deletion (Figure 5C) Recombinationwas confirmed by PCR using a forward primer from the BACvector backbone and a reverse primer mapping to a positiondistal to the reverse homology arm used to insert the galKcassette These primers are 95 kb apart on wild-type RP23-341F12 DNA and only 378 bp apart on CreloxP511 recom-bined DNA PCR analysis of all six clones produced a band ofthe expected length whereas no product could be amplifiedfrom unmodified RP23-341F12 BAC DNA (Figure 5D)
Source of background deletions following DOG selection
In the Nras G12D substitution and loxP511 insertion experi-ments a background of DOG-resistant bacteria which lackedthe desired mutation and instead carried unwanted deletionsthat spanned the galK selection cassette were observed Therelative ratio of colonies containing BACs with deletions wasalso higher when short homology arms were used This wasexpected since homologous recombination is more efficientwith longer homology arms (10) that would increase the rel-ative frequency of correctly targeted BACs The source of thedeletions could be l Red-mediated since proteins capable ofmediating recombination are expressed when the Red genes
Figure 4 BAC trimming using galK selection (A) Illustration of the design ofthe deletion experiment Homology arm 1 (H1) was held constant and H2 wasseparated from H1 by either 50 75 or 100 kb (B) SpeI restriction analysis ofBAC miniprep DNA from 12 clones showing deletions of 50 75 and 100 kbrespectively after the insertion of the galK selection cassette The first lane isunmodified RP23-341F12 BAC DNA which was included as a control Alltested clones had the intended deletion
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 8 OF 12
are activated Alternatively the deletions could be induced bythe large amount of oligos (200 ng) used in these experimentsor they could simply represent spontaneous deletions thatoccur at low level during normal BAC replication and thatare found by DOG selection To distinguish between thesepossibilities we repeated the DOG selection step used to insertthe loxP511 oligo into BAC RP23-341F12 DNA using fourdifferently treated bacteria samples non-induced bacteriawithout the 100 bp loxP511 oligo (Figure 6A) non-inducedbacteria with the oligo (Figure 6B) induced bacteria without
the oligo (Figure 6C) and induced bacteria with the oligo(Figure 6D) The number of resistant colonies obtainedfrom the four experiments varied by lt10-fold (data notshown) Ten colonies from each electroporation were thenanalyzed by SpeI digestion of BAC miniprep DNA and com-pared with unmodified RP23-341F12 BAC DNA (Figure 6AndashD) Unwanted galK region deletions were observed in all 10colonies for both of the non-induced samples (Figure 6A andB) These deletions are therefore unlikely to be Red-mediatedsince these samples were not heat-induced We also observed
Figure 5 Insertion of a loxP511 site (A) The location of the wild-type and mutant loxP sites in the BAC backbone are indicated along with the extra mutant loxP511site that was introduced into the BAC genomic insert via galK counterselection The 95 kb region deleted by Cre-mediated recombination between the two loxP511sites is indicated and PCR primers used to confirm the deletion are shown as small arrows (B) SpeI restriction analysis of six miniprep clones selected forthe replacement of galK with a dsDNA oligo containing the mutant loxP511 site Clones 3 5 and 6 (circles) had the same restriction pattern as the unmodifiedBAC indicating that DOG resistance occurred due to the intended homologous recombination event Clones 1 2 and 4 had large deletions and were not analyzedfurther (C) SpeI restriction analysis of BAC miniprep DNA from clones 3 5 and 6 after transformation into Cre-induced EL350 cells Two clones from each parentalclone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all clones analyzed confirming the correctinsertion of loxP511 in clones 3 5 and 6 (D) PCR analysis of the six clones from (C) with one primer mapping to the BAC backbone and the other to a position distalto the inserted loxP511 site
PAGE 9 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
deletions in both the absence and presence of oligo indicatingthat the deletions are not oligo induced We therefore concludethat these are spontaneous deletions that occur at low levelduring normal BAC replication in the DH10B background
Non-deleted colonies with the parental SpeI fingerprintwere only observed in the Red-induced sample that received
oligo (Figure 6D clones 33 and 35) These clones were sub-sequently electroporated into EL350 Cre-expressing cells toconfirm they contained the introduced loxP511 site Two col-onies from each original clone were tested by SpeI digestion ofthe BAC miniprep DNA As shown in Figure 6E each colonyhas undergone Cre-mediated deletion confirming that these
Figure 6 Same experiment as in Figure 5 with modifications as indicated at the top of each panel (A) SpeI digest of 10 minipreps from a control experiment withoutheat-induction and without the loxP511 dsDNA oligo (B) SpeI digest of 10 minipreps from a control experiment without heat-induction but with the loxP511 dsDNAoligo (C) SpeI digest of 10 minipreps from a control experiment with heat-induction but without the loxP511 dsDNA oligo (D) SpeI digest of 10 minipreps from anexperiment with heat-induction and with the loxP511 dsDNA oligo (comparable with Figure 5B) Clones with the parental digestion pattern indicating DOGresistance due to homologous recombination (clones 33 and 35 circles) are only seen in (D) DOG resistance in all other clones likely occurred due to internaldeletions of the BACs (E) SpeI restriction analysis of BAC miniprep DNA from clones 33 and 35 after transformation into Cre-induced EL350 cells Two clonesfrom each parental clone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all the clones analyzedconfirming the correct insertion of loxP511 in clones 33 and 35 (compare with Figure 5C)
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 10 OF 12
colonies were correctly targeted and contain the introducedloxP511 site (2 out of 10 clones a 20 efficiency in thisexperiment)
Generation of SW105 and SW106 cells
The two DY380-derived bacterial strains EL250 and EL350contain the defective l prophage needed for recombineering inaddition to L-arabinose inducible Flp or Cre genes respect-ively (11) Both strains have proven to be very useful for BACmodification (11) and EL350 is now used routinely for mak-ing conditional targeting vectors for ES cell knock-out experi-ments (4) To further enhance the usefulness of these strainswe decided to transfer the galK selection system into themThis was done as previously described for SW102 The galKIS2 element present in both strains was replaced with the wild-type gal promoter and the galK gene in the gal operon deletedusing DOG selection The resulting strains SW105 (Flp) andSW106 (Cre) (Table 1) were then tested to confirm that theystill contained inducible Flp and Cre genes by transformingarabinose-induced and electrocompetent cells with plasmidscontaining a neo gene flanked by Frt or loxP sites PL451 andPL452 (4) respectively Using SW105 or SW106 it is nowpossible to introduce a point mutation or an informativerestriction site into a BAC retrieve a fragment from thisBAC containing the introduced mutation(s) into a plasmidbackbone using gap repair and turn the retrieved fragmentinto a conditional targeting vectormdashall using only one bac-terial strain and only a single initial BAC transformation
DISCUSSION
Here we describe a new recombineering-based Ecoli BACmodification system that makes use of galK positive selectionfor growth on galactose minimal medium and galK negativeselection (counterselection) for growth on DOG This modi-fication system has several advantages compared with theother related BAC modification systems First the galK selec-tion cassette is small (1231 bp + homology arms) comparedwith for example the sacBndashneo cassette (3 kb) making PCRamplification and transformation into bacteria easier Twohomology arms are easily added to the galK cassette by includ-ing these sequences in the 50 ends of the primers used foramplification (see Materials and Methods) Second sincegalK is used for both selection steps mutations occurringin galK during PCR amplification will be selected againstduring positive selection significantly reducing the risk ofDOG resistance from PCR mutations in galK during negativeselection Third even when very short (33 bp) homology armswere used the frequency of recombinants among the analyzedDOG-resistant clones was still 20ndash50 To our knowledgesuccessful BAC modification with such short homology armshas never been reported Finally since galK recombineering isso efficient little screening is required following selectionreducing the overall hands-on-time to a minimum galK selec-tion requires growth on minimal medium in both selectionsteps although perhaps not used routinely in most molecularbiology laboratories where BAC modification is needed theseplates are fairly simple to make and should not prevent anyonefrom using this method
For all such counterselection schemes there will bebackground since during negative selection any event leadingto the loss of the counterselectable marker will result in sur-vival In our system virtually all background appears to resultfrom deletions that span the inserted galK gene These dele-tions likely occur during BAC replication in the 45 h out-growth phase since we have shown that these deletions occurindependently of Red induction and in the absence or presenceof oligo It is well established that in recA defective bacteriaBACs are very stable compared with other large insert vectorssuch as yeast artificial chromosomes (YACs) However usingcounterselection rare spontaneous deletions are seen becauseof the strong selection force This spontaneous deletion back-ground is not a problem however due to the high frequency ofhomologous recombination obtained with the defective lprophage system
Increasing the length of homology arms used for recombin-eering will reduce the relative number of background deletionsobserved following negative selection since the percentage ofcolonies containing BACs that eliminated galK by homolog-ous recombination will be increased Oligos with longerhomology arms can be produced by annealing two oligoswith overlapping 30 ends (21) These overlapping oligos (50
single strand overhangs) are then filled in by DNA polymerasein vivo (or in vitro) and subsequently serve as the substrate forthe l Red proteins exo and bet Longer homology arms canalso be added by traditional cloning using restriction enzymesand DNA ligase (4)
A number of uses can be imagined for the strains describedhere In principle our strains can be used to make virtually anykind of BAC modification one can imagine including pointmutations or clean deletions or insertions of everything fromcDNA to loxP sites or small epitope tags BAC trimming thespecific deletion of BAC DNA flanking a gene of interest couldalso be used to remove genes from BACs prior to making BACtransgenic mice so that only the gene of interest on the BAC isanalyzed This should be of particular interest for BAC rescueexperiments in positional cloning Of course trimming andmodification can be combined since the galK selection cassettecan be recycled so that the same BAC can be modified severaltimes BAC modification could also be the starting point forconstructing a gene-targeting vector to allow for more soph-isticated gene targeting in mouse ES cells The desired muta-tion(s) could first be created in the BAC followed by retrievalinto a plasmid vector containing a negative-selection markerlike thymidine kinase using gap repair (4) Finally a neo mar-ker could be introduced to allow for positive selection in EScells Alternatively the retrieved fragment could be modifiedso that the end result is a conditional targeting vector StrainSW106 which in addition to galK selection can be induced toexpress Cre recombinase makes it possible to perform all ofthe steps needed to construct a conditional targeting vectorstarting with only a single BAC transformation step
Furthermore in experiments where BAC transgenic miceare used to analyze the effect of deleting long-range regulatorsof gene expression galK DOG counterselection can even beused in the absence of an oligo to generate a series of dele-tions around the galK insertion site and the effect on generegulation studied
The Flp or Cre ORFs contained in strains SW105 andSW106 respectively can also be replaced for any desired
PAGE 11 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
gene thus creating tight arabinose-inducible expression forany gene of interest This is done by first replacing the CreFlpe ORF with galK and select for Gal+ recombinants ThegalK cassette is then replaced by the ORF of the gene ofinterest by DOG selection for Gal recombinants
Finally the strains described here should be very useful forgenetic manipulation not only of BACs but also the Ecoligenome itself
All recombineering reagents discussed in this workare freely available upon request To obtain these materialsplease follow the directions listed on our website (httprecombineeringncifcrfgov) Detailed protocols for recom-bineering including galK selection can also be downloadeddirectly from our website
ACKNOWLEDGEMENTS
We thank Allen Kane and Carolyn Whistler from ScientificPublications Graphics amp Media NCI-Frederick for help withillustrations and figure preparation Funding for this research aswell as the Open Access publication charges was provided byDHHS NIH NCI
REFERENCES
1 ShizuyaH BirrenB KimUJ MancinoV SlepakTTachiiriY and SimonM (1992) Cloning and stable maintenance of300-kilobase-pair fragments of human DNA in Escherichia coli using anF-factor-based vector Proc Natl Acad Sci USA 89 8794ndash8797
2 YangXW ModelP and HeintzN (1997) Homologous recombinationbased modification in Escherichia coli and germline transmission intransgenic mice of a bacterial artificial chromosome Nat Biotechnol15 859ndash865
3 AntochMP SongEJ ChangAM VitaternaMH ZhaoYWilsbacherLD SangoramAM KingDP PintoLH andTakahashiJS (1997) Functional identification of the mouse circadianClock gene by transgenic BAC rescue Cell 89 655ndash667
4 LiuP JenkinsNA and CopelandNG (2003) A highly efficientrecombineering-based method for generating conditional knockoutmutations Genome Res 13 476ndash484
5 Cotta-de-AlmeidaV SchonhoffS ShibataT LeiterA andSnapperSB (2003) A new method for rapidly generating gene-targetingvectors by engineering BACs through homologous recombination inbacteria Genome Res 13 2190ndash2194
6 CopelandNG JenkinsNA and CourtDL (2001) Recombineeringa powerful new tool for mouse functional genomics Nature RevGenet 2 769ndash779
7 CourtDL SawitzkeJA and ThomasonLC (2002) Geneticengineering using homologous recombination Annu Rev Genet 36361ndash388
8 ZhangY BuchholzF MuyrersJP and StewartAF (1998) A newlogic for DNA engineering using recombination in Escherichia coliNature Genet 20 123ndash128
9 MuyrersJP ZhangY TestaG and StewartAF (1999) Rapidmodification of bacterial artificial chromosomes by ET-recombinationNucleic Acids Res 27 1555ndash1557
10 YuD EllisHM LeeEC JenkinsNA CopelandNG andCourtDL (2000) An efficient recombination system for chromosomeengineering in Escherichia coli Proc Natl Acad Sci USA 975978ndash5983
11 LeeEC YuD Martinez de VelascoJ TessarolloL SwingDACourtDL JenkinsNA and CopelandNG (2001) A highly efficientEscherichia coli-based chromosome engineering system adapted forrecombinogenic targeting and subcloning of BAC DNA Genomics73 56ndash65
12 GongS YangXW LiC and HeintzN (2002) Highly efficientmodification of bacterial artificial chromosomes (BACs) using novelshuttle vectors containing the R6Kgamma origin of replicationGenome Res 12 1992ndash1998
13 JamsaiD OrfordM NefedovM FucharoenS WilliamsonR andIoannouPA (2003) Targeted modification of a human beta-globin locusBAC clone using GET Recombination and an I-SceI counterselectioncassette Genomics 82 68ndash77
14 SwaminathanS EllisHM WatersLS YuD LeeECCourtDL and SharanSK (2001) Rapid engineering of bacterialartificial chromosomes using oligonucleotides Genesis 2914ndash21
15 AlperMD and AmesBN (1975) Positive selection of mutants withdeletions of the gal-chl region of the Salmonella chromosome as ascreening procedure for mutagens that cause deletions J Bacteriol121 259ndash266
16 AusubelFM BrentR KingstonRE MooreDD SeidmanJGSmithJA and StruhlK (1987) Current Protocols in Molecular BiologyGreene Publishing Associates and Wiley-Interscience John Wiley ampSons Hoboken NJ
17 BachmannBJ (1996) Derivations and genotypes of some mutantderivatives of Escherichia coli K-12 In NeidhardtFC CurtissRIIIIngrahamJL LinECC LowKB MagasanikB ReznikoffWSRileyM SchaechterM and UmbargerHE (eds) Escherichia coli andSalmonella Cellular and Molecular Biology ASM press WashingtonDC Vol 2 pp 2460ndash2488
18 GrantSG JesseeJ BloomFR and HanahanD (1990) Differentialplasmid rescue from transgenic mouse DNAs into Escherichia colimethylation-restriction mutants Proc Natl Acad Sci USA 874645ndash4649
19 OppenheimAB RattrayAJ BubunenkoM ThomasonLC andCourtDL (2004) In vivo recombineering of bacteriophage lambda byPCR fragments and single-strand oligonucleotides Virology 319185ndash189
20 HoessRH WierzbickiA and AbremskiK (1986) The role ofthe loxP spacer region in P1 site-specific recombination NucleicAcids Res 14 2287ndash2300
21 YuD SawitzkeJA EllisH and CourtDL (2003) Recombineeringwith overlapping single-stranded DNA oligonucleotides testing arecombination intermediate Proc Natl Acad Sci USA 1007207ndash7212
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 12 OF 12

conditions 94C for 15 s 60C for 30 s and 72C for 1 min for30 cycles After completion of PCR 2 ml DpnI was added toeach 25 ml reaction and incubated for 2 h at 37C to removeany plasmid template The DpnI-digested reaction mix was runon a 1 agarose gel over night and the PCR product waspurified and eluted in 50 ml ddH2O An aliquot of 25 ml wasused for each experiment For making the Nras G12D substi-tution the following primers were used for the first step intro-ducing galK Homology to Nras sequence is in italics and thesequence recognizing em7-galK is underlined Nras galK F50-TTTTTGCTGGTGTGAAATGACTGAGTACAAACTGGTGG-TGGTTGGAGCAGCCTGTTGACAATTAATCATCCGCA-30Nras galK R 50-CAAAGTGGTTCTGGATTAGCTGGATCG-TCAAGGCGCTTTTCCCAACACCATCAGCACTGTCCTGC-TCCTT-30 For making 50 75 and 100 kb deletions in theRP23-341F12 BAC the following primers were used (homo-logy to BAC sequence is in italics and the sequence recog-nizing em7-galK is underlined) galK F 341F12 start50-ACTCCCACTGGAAGCTTTTTACAAAACATGTGTTGCT-GACATGTTGACAGCCTGTTGACAATTAATCATCGGCA-30 galK R 341F12 50 kb 50-ACCCAAACCAAACAACAT-CCAAACCAAAAACACAGACAAAACCAAATATGTCAGCAC-TGTCCTGCTCCTT-30 galK R 341F12 75 kb 50-ACACTAAGC-CAAACTCCTTGCCTGGGCTATTTCTCTTTGTTTTTCCAAATTCAGC-ACTGTCCTGCTCCTT-30 galK R 341F12 100 kb 50-TATGT-GTCTGTGTGTGTATGTACAGTTCTTTGTTTTTGTTTTTTTT-CTTTTCAGCACTGTCCTGCTCCTT-30 For insertion of aloxP511 site in the RP23-341F12 BAC the following primerswere used for the first step (introduction of galK) Homology toBAC sequence is in italics and the sequence recognizing em7-galK is underlined 95 kb loxP511 galK F 50-GGACA-GAGGCGTGACAGACGTGTGAGCTCCGTGGACAACTCTC-CCCGAAGCCTGTTGACAATTAATCATCGGCA-30 95 kbloxP511 galK R 50-GACTCTGAGCAGCAACGGCTGAGCCT-CACTTGAGAGGGTCCCTGAGTCACTCAGCACTGTCCTG-CTCCTT-30
Oligos for recombineering
Oligos used to replace the galK-targeting cassette wereobtained from Invitrogen dsDNA was used and the oligos(sense and antisense) annealed in vitro 10 mg of each oligo(sense and antisense) was mixed in an eppendorf tube in a totalvolume of 100 ml of 1middot PCR buffer (Expand High FidelityPCR kit Roche Applied Science) and boiled for 5 minallowed to cool to room temperature for 30 min ethanol pre-cipitated and resuspended in 100 ml ddH2O to a final concen-tration of 200 ngml double-stranded oligo An aliquot of 1 ml(200 ng) was used in the recombineering experiments Tointroduce the G12D (GltgtA) point mutation in the NrasBAC CITB 50J2 the following oligos were used for thesecond step (the introduced adenosinethymidine base pairis underlined the flanking sequences are homologous to theNras BAC sequence) G12D S 50-TTTTTGCTGGTGT-GAAATGACTGAGTACAAACTGGTGGTGGTTGGAGC-AGATGGTGTTGGGAAAAGCGCCTTGACGATCCAGC-TAATCCAGAACCACTTT-30 G12D AS 50-AAAGTGGT-TCTGGATTAGCTGGATCGTCAAGGCGCTTTTCCCAA-CACCATCTGCTCCAACCACCACCAGTTTGTACTCAG-TCATTTCACACCAGCAAAAA-30 To introduce a loxP511site in the RP23-341F12 BAC the following oligos were used
for the second step (loxP511 is underlined the flankingsequences are homologous to the BAC sequence aroundposition 95 kb) 95 kb loxP511 S 50-ACGTGTGAGCTC-CGTGGACAACTCTCCCCGAAGATAACTTCGTATAGT-ATACATTATACGAAGTTATGTGACTCAGGGACCCTC-TCAAGTGAGGCTCAGC-30 95 kb loxP511 AS 50-GCTG-AGCCTCACTTGAGAGGGTCCCTGAGTCACATAACTT-CGTATAATGTATACTATACGAAGTTATCTTCGGGGA-GAGTTGTCCACGGAGCTCACACGT-30
Verification of positive recombinants
In the G12D Nras experiment the selected Gal clones wereanalyzed by SpeI digestion of BAC miniprep DNA usingunmodified CITB 50J2 BAC DNA as a control Clones with-out rearrangements were analyzed by PCR using 1 ml BACminiprep DNA as the template The PCR products were gelpurified and sequenced using the same primers as were usedfor PCR Primers flanking the targeted mutation were Nrastest F 50-CACTCATCTGCAAGGAATGCT-30 Nras test R50-CCTCAGTAAGCACGAACTTGT-30 PCR conditionswere 94C for 15 s 60C for 30 s and 72C for 30 s for30 cycles Modifications of the RP23-341F12 BAC (50 75and 100 kb deletions and the introduction of a loxP511 site)were tested by SpeI restriction analysis of BAC miniprep DNAand compared with unmodified 341F12 BAC DNA In theloxP511 experiment clones 3 5 and 6 were further testedfor correct insertion of the loxP511 site by transforming1 ml of BAC miniprep DNA into electrocompetent andarabinose-induced EL350 cells (11) and plating on LB plateswith chloramphenicol Two colonies from each starting clonewere tested by SpeI digestion of BAC miniprep DNA for the95 kb Cre-mediated deletion Finally the Cre-recombinedclones were tested by PCR with one primer mapping to theend of the pBACe36 BAC backbone and the other mapping toa position 95 kb away on the wild-type BAC The primers(Invitrogen) used for this analysis were 95 kb loxP511 checkF 50-GCGGATGAATGGCAGAAATTC-30 95 kb LoxP511check R 50-TTTGCCAGACTGGTGCCTAA-30 PCR condi-tions were 94C for 15 s 60C for 30 s and 72C for 30 s for30 cycles The resulting PCR bands were gel purified andconfirmed by sequencing using the same primers as wereused for the PCR amplification The follow-up experimentfor testing the source of the observed BAC deletions wasdone as described above
BACs
Mouse BACs were obtained from Invitrogen The RP23-341F12 BAC (C57BL6 DNA) was chosen based on positionwithin the mouse Ebfaz gene using the UCSC genome browser(httpgenomeucscedu) The CITB 50J2 BAC (CJ7129SVDNA) was identified by screening a CITB mouse BAC library(Invitrogen) with an Nras genomic probe using standardhybridization methods The BAC clones were streaked forsingle colonies and characterized by PCR and restriction ana-lysis before proceeding to the recombineering experimentsTwo methods were used for DNA preparation For BAC mini-preps (1ndash15 mg) we used the following protocol 5 ml over-night LB culture (15 ml Falcon tube) was pelleted for 5 min at5000 rpm the supernatant removed and the pellet wasdissolved in 250 ml buffer P1 (miniprep kit Qiagen) and
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 4 OF 12
transferred to an eppendorf tube An aliquot of 250 ml P2buffer was added followed by mixing by inversion and incuba-tion for lt5 min at room temperature An aliquot of 250 ml N3buffer was added followed by mixing and incubation on ice for5 min The supernatant was cleared by two rounds of centri-fugation at 13 200 rpm for 5 min in a tabletop centrifugeEach time the supernatant was transferred to a new tube DNAwas precipitated by adding 750 ml isopropanol mixing andincubating on ice for 10 min and centrifugation for 10 min at13 200 rpm The pellet was washed once in 70 ethanol andthe dry pellet was dissolved in 50 ml TE An aliquot of 40 ml(1 mg) was used for restriction analysis and 1 ml was used astemplate for PCR analysis or transformation of electrocom-petent bacteria Large-scale preparations of BAC DNA(25ndash100 mg) were done using the Nucleobond BAC maxikit from Clontech (BD Biosciences) following the manufac-turerrsquos protocol
RESULTS
Generation of SW102 cells
To see whether we could develop a more efficient selectivesystem for BAC recombineering we looked for a single select-able marker that could be used for both positive and negativeselection We focused on Ecoli galK because both selectionsteps could be done using galK and its small size makes it easyto amplify by PCR We previously reported the developmentof a bacterial strain DY380 that is readily transformable withBAC DNA due to its DH10B origin (1) DY380 cells alsoharbor the defective l prophage required for recombineeringThe defective prophage in DY380 was transferred into DH10Bwith a P1 phage lysate obtained from DY363 cells [for detailssee (11)] DY363 cells carry a 1200 bp IS2 insertion elementgal490 in the mRNA leader sequence of the galactose operonpreventing gal gene transcription Since the galactose operonis directly proximal to the site of insertion of the defective
prophage the gal490 mutation was also transferred to DY380during P1 transduction Thus DY380 is phenotypically Gal
(galactose minus) and therefore unable to grow on galactoseminimal medium
Recombineering was used to correct this problem From thewild-type Ecoli strain W3110 (17) we PCR amplified a 441bp fragment from the wild-type gal promoter which spans theregion containing the IS2 insertion element in DY380 ThisPCR product was introduced into DY380 and Gal+ recombin-ants were selected for growth on galactose minimal medium(see Materials and Methods) We named this strain SW101(Table 1) This strain is identical to DY380 except that it lacksthe IS2 insertion element and has been made wild type for thegal operon We then made a precise deletion of the galK geneleaving all other genes in the galactose operon intact This wasachieved by PCR amplifying two homology arms of 171 and335 bp respectively flanking the galK ORF and cloning themtogether into a plasmid using three-way ligation Since theORF of galK overlaps that of galM we left behind the last33 bp of galK (Figure 1) to allow for proper translation ofgalM The targeting cassette consisting of a 512 bp linearfragment was electroporated into heat-induced and electro-competent SW101 cells and the recombinant clones selectedon minimal medium containing glycerol as the carbon sourceand DOG for selection against galK The genotype of theresulting strain SW102 (Table 1) was confirmed by PCRanalysis and sequencing of the modified region (Figure 1)This strain now harbors not only the defective l prophagebut also a functional gal operon except for the deletion ofgalK The l prophage is located between the galactose operonand the biotin operon and the bio operon in these cells isnonfunctional causing a biotin requirement DH10B andall derived strains including SW102 are also deficient inleucine metabolism (18) When using minimal medium onetherefore needs to add both biotin and leucine to the plates toallow SW102 growth (see Materials and Methods)
Figure 1 Sequence analysis of the galactose operon in strains DY380 (A) SW101 (B) and SW102 (C) In SW102 the ORF of galK was deleted leaving only 33 bp ofgalK behind to make sure that translation of galM is initiated properly EcoRI the restriction site used to clone the 50 and 30 homology arms flanking galK
PAGE 5 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
galK expression cassette
Having produced a bacterial strain SW102 which is galKdefective the next step was to make a galK expression cassettethat could be used to restore the bacteriarsquos ability to grow ongalactose by providing galK in trans This was achieved byPCR amplification of the wild-type galK ORF from W3110cells We then added a minimal bacterial promoter em7 usinga two-step PCR approach (see Materials and Methods) andcloned the expression cassette into pBluescript SK We callthis plasmid pgalK (Table 1) The constitutively active galKexpression cassette can easily be amplified by PCRwith homology arms added to the primers (see Materialsand Methods)
Making a single base pair substitution
The general scheme for making mutations in BACs using galKselection is depicted in Figure 2 To test the galK selectionsystem for BAC recombineering we decided to introduce apoint mutation into a BAC containing the murine Nras gene(CITB clone 50J2) The sequence of the glycine-coding codon12 is GGT By changing this codon to GAT we would obtainthe desired mutation G12D In order to introduce this muta-tion into the BAC we first amplified the galK expressioncassette by PCR using primers with 50 bp of homology toeither side of the second position of the GGT codon Followinghomologous recombination this targeting would introduce a1 bp deletion into codon 12 in addition to inserting the galKselection cassette Instead of deleting the basepair the galKcassette could have been inserted right next to the basepairinstead SW102 cells containing the 50J2 BAC were heat-induced and made electrocompetent and then electroporatedwith the galK cassette Gal+ recombinant colonies were selec-ted for growth on galactose minimal medium with chloram-phenicol to maintain the BAC Bacteria grow more slowly onminimal media than on rich media and we generally pickcolonies after 2ndash3 days For this first step we do not expectany background colonies on the non-induced control plates ifthe pgalK plasmid is properly eliminated (see Materials andMethods) To purify the Gal+ colonies we streaked a fewcolonies on MacConkey galactose indicator plates to obtainsingle bright pinkred Gal+ colonies One of these single-cloned colonies was picked to initiate a culture for thenext step counterselection We find that there is no need toanalyze the Gal+ colonies further before proceeding tocounterselection
A 100 bp dsDNA oligo was then prepared by annealing twocomplementary oligos having 49 and 50 bp homologyrespectively to either side of the desired mutation a singleAT bp An aliquot of 200 ng of this oligo was then electro-porated into heat-induced and electrocompetent SW102 Gal+
cells containing the galK modified 50J2 BAC After electro-poration the bacteria were allowed 45 h outgrowth in a 32Cshaking waterbath The bacteria were then washed in M9 saltsto remove any rich medium and plated on minimal mediumwith glycerol DOG and chloramphenicol The 45 h out-growth is necessary to obtain complete segregation of therecombinant BACs containing the mutation After 3 dayswe obtained colonies with a ratio of 10ndash1001 when comparingplates with heat-induced to non-induced bacteria We picked12 colonies from the heat-induced plates for BAC minipreps
followed by SpeI restriction analysis (Figure 3A) Ten out ofthe twelve clones had the same restriction pattern as theunmodified 50J2 BAC DNA suggesting that the desiredreplacement of the galK gene by the point mutation hadoccurred This was confirmed by PCR amplification followed
Figure 2 Overview of the galK selection scheme The result of the firsttargeting event is the insertion of constitutively active galK into a definedposition on the BAC by selection on minimal medium containing galactoseand chloramphenicol to select for the maintenance of the BAC The bacteria arenow phenotypically Gal+ Next the galK cassette is replaced by a dsDNA oligoa PCR product or a cloned dsDNA fragment carrying a desired mutation(indicated by a star) and flanked by the same homology arms used in thefirst selection step This is achieved by negative selection using minimalmedium containing 2-deoxy-galactose (DOG) with glycerol as the sole carbonsource The bacteria become phenotypically Gal H1 and H2 homology arms1 and 2 respectively cat chloramphenicol acetyl transferase gene ori2 BACorigin of replication galK Ecoli galactokinase gene driven by a minimalpromoter
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 6 OF 12
by sequencing of the modified region of the 10 BAC clones(Figure 3B) All 10 sequenced clones had the desired pointmutation The effective recombination efficiency in thisexperiment however was 812 (67) since 2 of the 10 clonesalso had an additional single base pair deletion (clones 9 and11) These deletions probably occurred during oligo synthesissince we did not purify the oligos beyond desalting (19)Because of the high efficiency in this system there was noneed to pre-screen the selected colonies prior to picking forminipreps
Large BAC deletions
A drawback of using BACs for the production of transgenicmice is the frequent presence of other genes on the BAC inaddition to the gene of interest This is especially a problem forBAC complementation used in positional cloning where aBAC is tested for its ability to rescue a loss-of-function mutantphenotype by making BAC transgenic mice If complementa-tion is achieved it is impossible to know which gene on theBAC is responsible for the rescue Therefore we decided to
see whether galK selection could be used to make largespecific and clean deletions in BACs so as to remove unwantedgenes (a process called BAC trimming) As a model systemwe chose a mouse BAC from the C57BL6-derived libraryRP23 RP23-341F12 since we could obtain the BAC sequencedirectly from the UCSC genomic web browser (see Materialsand Methods) We then used PCR to amplify the galK selec-tion cassette with primers containing homology to the BACThe forward primer contained 50 bp of homology to the very 50
end of the BAC insert and this primer was combined withthree different reverse primers having 50 bp of homology topositions 50 75 and 100 kb away from the forward primerrsquoshomology (Figure 4A) SW102 bacteria containing the 341F12BAC were heat-induced and made electrocompetent and thentransformed with the three different selection cassettes fol-lowed by the selection on minimal medium with galactoseFrom the many resulting Gal+ colonies we picked four fromeach experiment and then did a SpeI restriction analysis of theBAC miniprep DNAs from them (Figure 4B) All four colon-ies from each experiment had the desired deletion (50 75 and100 kb respectively) The galK selection cassette can then be
Figure 3 Introduction of a G12D mutation in the Nras gene (A) SpeI restriction analysis of BAC miniprep DNA First lane is the unmodified CITB-50J2 Nras BACLanes 1ndash12 show digestion patterns of 12 clones counterselected for the substitution of galK with an oligo containing the GA substitution for the second position ofcodon 12 of Nras Clones 7 and 10 had internal deletions indicating that DOG resistance was achieved by spontaneous deletion and not homologous recombinationThese two clones were not analyzed further (B) Sequence analysis of a PCR product spanning the modified region from clones 1ndash6 8ndash9 and 11ndash12 All clones had theintended substitution (highlighted) However clones 9 and 11 also had an internal basepair deletion indicated by a minus (highlighted) The Nras ATG and codon 12are indicated (shadow)
PAGE 7 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
removed with an oligo using galK negative selection to cleanlydelete the galK cassette from the BAC as described in theprevious experiment (data not shown)
Insertion of a mutant loxP511 site into a BAC
Encouraged by the efficiency of this selection system wedecided to see whether we could introduce a single 34 bploxP511 site cleanly into BAC DNA using galK positiveneg-ative selection It has been previously shown that loxP andloxP511 sites cannot recombine with each other therefore the
introduced loxP511 site can only recombine with the loxP511site present in the BAC vector backbone and not the wild-typesite (Figure 5A) (20) We used galK positive selection to inserta PCR-amplified galK cassette flanked by 50 bp homologyarms to a position 95 kb away from the mutant loxP511 site inthe BAC RP23-341F12 vector backbone (Figure 5A) We thenreplaced the galK cassette using DOG counterselection with adouble-stranded 100 bp oligo containing a 34 bp loxP511 siteflanked by two 33 bp homology arms In this experiment weobserved less than a 10-fold difference in the number of col-onies on the plates from heat-induced and non-induced bac-teria suggesting fewer recombinants This lower frequency ofrecombinants when compared with the Nras G12D substitu-tion experiment is likely explained by the shorter homologyarms used in this latter experiment as it has been shown thatthe efficiency of recombination increase four orders of mag-nitude when homology length is increased from 20 to 40 bp(10) We analyzed six BAC minipreps from potential recom-binants by digesting with SpeI and comparing the restrictionpatterns with that of wild-type RP23-341F12 BAC DNA(Figure 5B) Three of six (50) colonies had exactly thesame restriction pattern as the unmodified RP23-341F12BAC suggesting that DOG resistance had selected for thedesired homologous recombination products in these threecases whereas the other three clones apparently becameDOG resistant due to the selection of BACs that carry largedeletions spanning galK
The 100 bp oligo was designed so that the loxP511 sitewould be inserted in the same orientation as the loxP511site in the BAC vector backbone To confirm that the BACswith the wild-type restriction pattern (3 5 and 6) had theloxP511 site correctly inserted we electroporated thesethree BACs into electrocompetent and L-arabinose inducedEL350 cells which carry an L-arabinose-inducible Cregene and then plated the cells on LB plates containing chlor-amphenicol to select for the BACs Two colonies generatedfrom each recombinant loxP511 clone were then tested bySpeI digestion of BAC miniprep DNA and all had the expec-ted Cre-mediated 95 kb deletion (Figure 5C) Recombinationwas confirmed by PCR using a forward primer from the BACvector backbone and a reverse primer mapping to a positiondistal to the reverse homology arm used to insert the galKcassette These primers are 95 kb apart on wild-type RP23-341F12 DNA and only 378 bp apart on CreloxP511 recom-bined DNA PCR analysis of all six clones produced a band ofthe expected length whereas no product could be amplifiedfrom unmodified RP23-341F12 BAC DNA (Figure 5D)
Source of background deletions following DOG selection
In the Nras G12D substitution and loxP511 insertion experi-ments a background of DOG-resistant bacteria which lackedthe desired mutation and instead carried unwanted deletionsthat spanned the galK selection cassette were observed Therelative ratio of colonies containing BACs with deletions wasalso higher when short homology arms were used This wasexpected since homologous recombination is more efficientwith longer homology arms (10) that would increase the rel-ative frequency of correctly targeted BACs The source of thedeletions could be l Red-mediated since proteins capable ofmediating recombination are expressed when the Red genes
Figure 4 BAC trimming using galK selection (A) Illustration of the design ofthe deletion experiment Homology arm 1 (H1) was held constant and H2 wasseparated from H1 by either 50 75 or 100 kb (B) SpeI restriction analysis ofBAC miniprep DNA from 12 clones showing deletions of 50 75 and 100 kbrespectively after the insertion of the galK selection cassette The first lane isunmodified RP23-341F12 BAC DNA which was included as a control Alltested clones had the intended deletion
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 8 OF 12
are activated Alternatively the deletions could be induced bythe large amount of oligos (200 ng) used in these experimentsor they could simply represent spontaneous deletions thatoccur at low level during normal BAC replication and thatare found by DOG selection To distinguish between thesepossibilities we repeated the DOG selection step used to insertthe loxP511 oligo into BAC RP23-341F12 DNA using fourdifferently treated bacteria samples non-induced bacteriawithout the 100 bp loxP511 oligo (Figure 6A) non-inducedbacteria with the oligo (Figure 6B) induced bacteria without
the oligo (Figure 6C) and induced bacteria with the oligo(Figure 6D) The number of resistant colonies obtainedfrom the four experiments varied by lt10-fold (data notshown) Ten colonies from each electroporation were thenanalyzed by SpeI digestion of BAC miniprep DNA and com-pared with unmodified RP23-341F12 BAC DNA (Figure 6AndashD) Unwanted galK region deletions were observed in all 10colonies for both of the non-induced samples (Figure 6A andB) These deletions are therefore unlikely to be Red-mediatedsince these samples were not heat-induced We also observed
Figure 5 Insertion of a loxP511 site (A) The location of the wild-type and mutant loxP sites in the BAC backbone are indicated along with the extra mutant loxP511site that was introduced into the BAC genomic insert via galK counterselection The 95 kb region deleted by Cre-mediated recombination between the two loxP511sites is indicated and PCR primers used to confirm the deletion are shown as small arrows (B) SpeI restriction analysis of six miniprep clones selected forthe replacement of galK with a dsDNA oligo containing the mutant loxP511 site Clones 3 5 and 6 (circles) had the same restriction pattern as the unmodifiedBAC indicating that DOG resistance occurred due to the intended homologous recombination event Clones 1 2 and 4 had large deletions and were not analyzedfurther (C) SpeI restriction analysis of BAC miniprep DNA from clones 3 5 and 6 after transformation into Cre-induced EL350 cells Two clones from each parentalclone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all clones analyzed confirming the correctinsertion of loxP511 in clones 3 5 and 6 (D) PCR analysis of the six clones from (C) with one primer mapping to the BAC backbone and the other to a position distalto the inserted loxP511 site
PAGE 9 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
deletions in both the absence and presence of oligo indicatingthat the deletions are not oligo induced We therefore concludethat these are spontaneous deletions that occur at low levelduring normal BAC replication in the DH10B background
Non-deleted colonies with the parental SpeI fingerprintwere only observed in the Red-induced sample that received
oligo (Figure 6D clones 33 and 35) These clones were sub-sequently electroporated into EL350 Cre-expressing cells toconfirm they contained the introduced loxP511 site Two col-onies from each original clone were tested by SpeI digestion ofthe BAC miniprep DNA As shown in Figure 6E each colonyhas undergone Cre-mediated deletion confirming that these
Figure 6 Same experiment as in Figure 5 with modifications as indicated at the top of each panel (A) SpeI digest of 10 minipreps from a control experiment withoutheat-induction and without the loxP511 dsDNA oligo (B) SpeI digest of 10 minipreps from a control experiment without heat-induction but with the loxP511 dsDNAoligo (C) SpeI digest of 10 minipreps from a control experiment with heat-induction but without the loxP511 dsDNA oligo (D) SpeI digest of 10 minipreps from anexperiment with heat-induction and with the loxP511 dsDNA oligo (comparable with Figure 5B) Clones with the parental digestion pattern indicating DOGresistance due to homologous recombination (clones 33 and 35 circles) are only seen in (D) DOG resistance in all other clones likely occurred due to internaldeletions of the BACs (E) SpeI restriction analysis of BAC miniprep DNA from clones 33 and 35 after transformation into Cre-induced EL350 cells Two clonesfrom each parental clone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all the clones analyzedconfirming the correct insertion of loxP511 in clones 33 and 35 (compare with Figure 5C)
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 10 OF 12
colonies were correctly targeted and contain the introducedloxP511 site (2 out of 10 clones a 20 efficiency in thisexperiment)
Generation of SW105 and SW106 cells
The two DY380-derived bacterial strains EL250 and EL350contain the defective l prophage needed for recombineering inaddition to L-arabinose inducible Flp or Cre genes respect-ively (11) Both strains have proven to be very useful for BACmodification (11) and EL350 is now used routinely for mak-ing conditional targeting vectors for ES cell knock-out experi-ments (4) To further enhance the usefulness of these strainswe decided to transfer the galK selection system into themThis was done as previously described for SW102 The galKIS2 element present in both strains was replaced with the wild-type gal promoter and the galK gene in the gal operon deletedusing DOG selection The resulting strains SW105 (Flp) andSW106 (Cre) (Table 1) were then tested to confirm that theystill contained inducible Flp and Cre genes by transformingarabinose-induced and electrocompetent cells with plasmidscontaining a neo gene flanked by Frt or loxP sites PL451 andPL452 (4) respectively Using SW105 or SW106 it is nowpossible to introduce a point mutation or an informativerestriction site into a BAC retrieve a fragment from thisBAC containing the introduced mutation(s) into a plasmidbackbone using gap repair and turn the retrieved fragmentinto a conditional targeting vectormdashall using only one bac-terial strain and only a single initial BAC transformation
DISCUSSION
Here we describe a new recombineering-based Ecoli BACmodification system that makes use of galK positive selectionfor growth on galactose minimal medium and galK negativeselection (counterselection) for growth on DOG This modi-fication system has several advantages compared with theother related BAC modification systems First the galK selec-tion cassette is small (1231 bp + homology arms) comparedwith for example the sacBndashneo cassette (3 kb) making PCRamplification and transformation into bacteria easier Twohomology arms are easily added to the galK cassette by includ-ing these sequences in the 50 ends of the primers used foramplification (see Materials and Methods) Second sincegalK is used for both selection steps mutations occurringin galK during PCR amplification will be selected againstduring positive selection significantly reducing the risk ofDOG resistance from PCR mutations in galK during negativeselection Third even when very short (33 bp) homology armswere used the frequency of recombinants among the analyzedDOG-resistant clones was still 20ndash50 To our knowledgesuccessful BAC modification with such short homology armshas never been reported Finally since galK recombineering isso efficient little screening is required following selectionreducing the overall hands-on-time to a minimum galK selec-tion requires growth on minimal medium in both selectionsteps although perhaps not used routinely in most molecularbiology laboratories where BAC modification is needed theseplates are fairly simple to make and should not prevent anyonefrom using this method
For all such counterselection schemes there will bebackground since during negative selection any event leadingto the loss of the counterselectable marker will result in sur-vival In our system virtually all background appears to resultfrom deletions that span the inserted galK gene These dele-tions likely occur during BAC replication in the 45 h out-growth phase since we have shown that these deletions occurindependently of Red induction and in the absence or presenceof oligo It is well established that in recA defective bacteriaBACs are very stable compared with other large insert vectorssuch as yeast artificial chromosomes (YACs) However usingcounterselection rare spontaneous deletions are seen becauseof the strong selection force This spontaneous deletion back-ground is not a problem however due to the high frequency ofhomologous recombination obtained with the defective lprophage system
Increasing the length of homology arms used for recombin-eering will reduce the relative number of background deletionsobserved following negative selection since the percentage ofcolonies containing BACs that eliminated galK by homolog-ous recombination will be increased Oligos with longerhomology arms can be produced by annealing two oligoswith overlapping 30 ends (21) These overlapping oligos (50
single strand overhangs) are then filled in by DNA polymerasein vivo (or in vitro) and subsequently serve as the substrate forthe l Red proteins exo and bet Longer homology arms canalso be added by traditional cloning using restriction enzymesand DNA ligase (4)
A number of uses can be imagined for the strains describedhere In principle our strains can be used to make virtually anykind of BAC modification one can imagine including pointmutations or clean deletions or insertions of everything fromcDNA to loxP sites or small epitope tags BAC trimming thespecific deletion of BAC DNA flanking a gene of interest couldalso be used to remove genes from BACs prior to making BACtransgenic mice so that only the gene of interest on the BAC isanalyzed This should be of particular interest for BAC rescueexperiments in positional cloning Of course trimming andmodification can be combined since the galK selection cassettecan be recycled so that the same BAC can be modified severaltimes BAC modification could also be the starting point forconstructing a gene-targeting vector to allow for more soph-isticated gene targeting in mouse ES cells The desired muta-tion(s) could first be created in the BAC followed by retrievalinto a plasmid vector containing a negative-selection markerlike thymidine kinase using gap repair (4) Finally a neo mar-ker could be introduced to allow for positive selection in EScells Alternatively the retrieved fragment could be modifiedso that the end result is a conditional targeting vector StrainSW106 which in addition to galK selection can be induced toexpress Cre recombinase makes it possible to perform all ofthe steps needed to construct a conditional targeting vectorstarting with only a single BAC transformation step
Furthermore in experiments where BAC transgenic miceare used to analyze the effect of deleting long-range regulatorsof gene expression galK DOG counterselection can even beused in the absence of an oligo to generate a series of dele-tions around the galK insertion site and the effect on generegulation studied
The Flp or Cre ORFs contained in strains SW105 andSW106 respectively can also be replaced for any desired
PAGE 11 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
gene thus creating tight arabinose-inducible expression forany gene of interest This is done by first replacing the CreFlpe ORF with galK and select for Gal+ recombinants ThegalK cassette is then replaced by the ORF of the gene ofinterest by DOG selection for Gal recombinants
Finally the strains described here should be very useful forgenetic manipulation not only of BACs but also the Ecoligenome itself
All recombineering reagents discussed in this workare freely available upon request To obtain these materialsplease follow the directions listed on our website (httprecombineeringncifcrfgov) Detailed protocols for recom-bineering including galK selection can also be downloadeddirectly from our website
ACKNOWLEDGEMENTS
We thank Allen Kane and Carolyn Whistler from ScientificPublications Graphics amp Media NCI-Frederick for help withillustrations and figure preparation Funding for this research aswell as the Open Access publication charges was provided byDHHS NIH NCI
REFERENCES
1 ShizuyaH BirrenB KimUJ MancinoV SlepakTTachiiriY and SimonM (1992) Cloning and stable maintenance of300-kilobase-pair fragments of human DNA in Escherichia coli using anF-factor-based vector Proc Natl Acad Sci USA 89 8794ndash8797
2 YangXW ModelP and HeintzN (1997) Homologous recombinationbased modification in Escherichia coli and germline transmission intransgenic mice of a bacterial artificial chromosome Nat Biotechnol15 859ndash865
3 AntochMP SongEJ ChangAM VitaternaMH ZhaoYWilsbacherLD SangoramAM KingDP PintoLH andTakahashiJS (1997) Functional identification of the mouse circadianClock gene by transgenic BAC rescue Cell 89 655ndash667
4 LiuP JenkinsNA and CopelandNG (2003) A highly efficientrecombineering-based method for generating conditional knockoutmutations Genome Res 13 476ndash484
5 Cotta-de-AlmeidaV SchonhoffS ShibataT LeiterA andSnapperSB (2003) A new method for rapidly generating gene-targetingvectors by engineering BACs through homologous recombination inbacteria Genome Res 13 2190ndash2194
6 CopelandNG JenkinsNA and CourtDL (2001) Recombineeringa powerful new tool for mouse functional genomics Nature RevGenet 2 769ndash779
7 CourtDL SawitzkeJA and ThomasonLC (2002) Geneticengineering using homologous recombination Annu Rev Genet 36361ndash388
8 ZhangY BuchholzF MuyrersJP and StewartAF (1998) A newlogic for DNA engineering using recombination in Escherichia coliNature Genet 20 123ndash128
9 MuyrersJP ZhangY TestaG and StewartAF (1999) Rapidmodification of bacterial artificial chromosomes by ET-recombinationNucleic Acids Res 27 1555ndash1557
10 YuD EllisHM LeeEC JenkinsNA CopelandNG andCourtDL (2000) An efficient recombination system for chromosomeengineering in Escherichia coli Proc Natl Acad Sci USA 975978ndash5983
11 LeeEC YuD Martinez de VelascoJ TessarolloL SwingDACourtDL JenkinsNA and CopelandNG (2001) A highly efficientEscherichia coli-based chromosome engineering system adapted forrecombinogenic targeting and subcloning of BAC DNA Genomics73 56ndash65
12 GongS YangXW LiC and HeintzN (2002) Highly efficientmodification of bacterial artificial chromosomes (BACs) using novelshuttle vectors containing the R6Kgamma origin of replicationGenome Res 12 1992ndash1998
13 JamsaiD OrfordM NefedovM FucharoenS WilliamsonR andIoannouPA (2003) Targeted modification of a human beta-globin locusBAC clone using GET Recombination and an I-SceI counterselectioncassette Genomics 82 68ndash77
14 SwaminathanS EllisHM WatersLS YuD LeeECCourtDL and SharanSK (2001) Rapid engineering of bacterialartificial chromosomes using oligonucleotides Genesis 2914ndash21
15 AlperMD and AmesBN (1975) Positive selection of mutants withdeletions of the gal-chl region of the Salmonella chromosome as ascreening procedure for mutagens that cause deletions J Bacteriol121 259ndash266
16 AusubelFM BrentR KingstonRE MooreDD SeidmanJGSmithJA and StruhlK (1987) Current Protocols in Molecular BiologyGreene Publishing Associates and Wiley-Interscience John Wiley ampSons Hoboken NJ
17 BachmannBJ (1996) Derivations and genotypes of some mutantderivatives of Escherichia coli K-12 In NeidhardtFC CurtissRIIIIngrahamJL LinECC LowKB MagasanikB ReznikoffWSRileyM SchaechterM and UmbargerHE (eds) Escherichia coli andSalmonella Cellular and Molecular Biology ASM press WashingtonDC Vol 2 pp 2460ndash2488
18 GrantSG JesseeJ BloomFR and HanahanD (1990) Differentialplasmid rescue from transgenic mouse DNAs into Escherichia colimethylation-restriction mutants Proc Natl Acad Sci USA 874645ndash4649
19 OppenheimAB RattrayAJ BubunenkoM ThomasonLC andCourtDL (2004) In vivo recombineering of bacteriophage lambda byPCR fragments and single-strand oligonucleotides Virology 319185ndash189
20 HoessRH WierzbickiA and AbremskiK (1986) The role ofthe loxP spacer region in P1 site-specific recombination NucleicAcids Res 14 2287ndash2300
21 YuD SawitzkeJA EllisH and CourtDL (2003) Recombineeringwith overlapping single-stranded DNA oligonucleotides testing arecombination intermediate Proc Natl Acad Sci USA 1007207ndash7212
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 12 OF 12

transferred to an eppendorf tube An aliquot of 250 ml P2buffer was added followed by mixing by inversion and incuba-tion for lt5 min at room temperature An aliquot of 250 ml N3buffer was added followed by mixing and incubation on ice for5 min The supernatant was cleared by two rounds of centri-fugation at 13 200 rpm for 5 min in a tabletop centrifugeEach time the supernatant was transferred to a new tube DNAwas precipitated by adding 750 ml isopropanol mixing andincubating on ice for 10 min and centrifugation for 10 min at13 200 rpm The pellet was washed once in 70 ethanol andthe dry pellet was dissolved in 50 ml TE An aliquot of 40 ml(1 mg) was used for restriction analysis and 1 ml was used astemplate for PCR analysis or transformation of electrocom-petent bacteria Large-scale preparations of BAC DNA(25ndash100 mg) were done using the Nucleobond BAC maxikit from Clontech (BD Biosciences) following the manufac-turerrsquos protocol
RESULTS
Generation of SW102 cells
To see whether we could develop a more efficient selectivesystem for BAC recombineering we looked for a single select-able marker that could be used for both positive and negativeselection We focused on Ecoli galK because both selectionsteps could be done using galK and its small size makes it easyto amplify by PCR We previously reported the developmentof a bacterial strain DY380 that is readily transformable withBAC DNA due to its DH10B origin (1) DY380 cells alsoharbor the defective l prophage required for recombineeringThe defective prophage in DY380 was transferred into DH10Bwith a P1 phage lysate obtained from DY363 cells [for detailssee (11)] DY363 cells carry a 1200 bp IS2 insertion elementgal490 in the mRNA leader sequence of the galactose operonpreventing gal gene transcription Since the galactose operonis directly proximal to the site of insertion of the defective
prophage the gal490 mutation was also transferred to DY380during P1 transduction Thus DY380 is phenotypically Gal
(galactose minus) and therefore unable to grow on galactoseminimal medium
Recombineering was used to correct this problem From thewild-type Ecoli strain W3110 (17) we PCR amplified a 441bp fragment from the wild-type gal promoter which spans theregion containing the IS2 insertion element in DY380 ThisPCR product was introduced into DY380 and Gal+ recombin-ants were selected for growth on galactose minimal medium(see Materials and Methods) We named this strain SW101(Table 1) This strain is identical to DY380 except that it lacksthe IS2 insertion element and has been made wild type for thegal operon We then made a precise deletion of the galK geneleaving all other genes in the galactose operon intact This wasachieved by PCR amplifying two homology arms of 171 and335 bp respectively flanking the galK ORF and cloning themtogether into a plasmid using three-way ligation Since theORF of galK overlaps that of galM we left behind the last33 bp of galK (Figure 1) to allow for proper translation ofgalM The targeting cassette consisting of a 512 bp linearfragment was electroporated into heat-induced and electro-competent SW101 cells and the recombinant clones selectedon minimal medium containing glycerol as the carbon sourceand DOG for selection against galK The genotype of theresulting strain SW102 (Table 1) was confirmed by PCRanalysis and sequencing of the modified region (Figure 1)This strain now harbors not only the defective l prophagebut also a functional gal operon except for the deletion ofgalK The l prophage is located between the galactose operonand the biotin operon and the bio operon in these cells isnonfunctional causing a biotin requirement DH10B andall derived strains including SW102 are also deficient inleucine metabolism (18) When using minimal medium onetherefore needs to add both biotin and leucine to the plates toallow SW102 growth (see Materials and Methods)
Figure 1 Sequence analysis of the galactose operon in strains DY380 (A) SW101 (B) and SW102 (C) In SW102 the ORF of galK was deleted leaving only 33 bp ofgalK behind to make sure that translation of galM is initiated properly EcoRI the restriction site used to clone the 50 and 30 homology arms flanking galK
PAGE 5 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
galK expression cassette
Having produced a bacterial strain SW102 which is galKdefective the next step was to make a galK expression cassettethat could be used to restore the bacteriarsquos ability to grow ongalactose by providing galK in trans This was achieved byPCR amplification of the wild-type galK ORF from W3110cells We then added a minimal bacterial promoter em7 usinga two-step PCR approach (see Materials and Methods) andcloned the expression cassette into pBluescript SK We callthis plasmid pgalK (Table 1) The constitutively active galKexpression cassette can easily be amplified by PCRwith homology arms added to the primers (see Materialsand Methods)
Making a single base pair substitution
The general scheme for making mutations in BACs using galKselection is depicted in Figure 2 To test the galK selectionsystem for BAC recombineering we decided to introduce apoint mutation into a BAC containing the murine Nras gene(CITB clone 50J2) The sequence of the glycine-coding codon12 is GGT By changing this codon to GAT we would obtainthe desired mutation G12D In order to introduce this muta-tion into the BAC we first amplified the galK expressioncassette by PCR using primers with 50 bp of homology toeither side of the second position of the GGT codon Followinghomologous recombination this targeting would introduce a1 bp deletion into codon 12 in addition to inserting the galKselection cassette Instead of deleting the basepair the galKcassette could have been inserted right next to the basepairinstead SW102 cells containing the 50J2 BAC were heat-induced and made electrocompetent and then electroporatedwith the galK cassette Gal+ recombinant colonies were selec-ted for growth on galactose minimal medium with chloram-phenicol to maintain the BAC Bacteria grow more slowly onminimal media than on rich media and we generally pickcolonies after 2ndash3 days For this first step we do not expectany background colonies on the non-induced control plates ifthe pgalK plasmid is properly eliminated (see Materials andMethods) To purify the Gal+ colonies we streaked a fewcolonies on MacConkey galactose indicator plates to obtainsingle bright pinkred Gal+ colonies One of these single-cloned colonies was picked to initiate a culture for thenext step counterselection We find that there is no need toanalyze the Gal+ colonies further before proceeding tocounterselection
A 100 bp dsDNA oligo was then prepared by annealing twocomplementary oligos having 49 and 50 bp homologyrespectively to either side of the desired mutation a singleAT bp An aliquot of 200 ng of this oligo was then electro-porated into heat-induced and electrocompetent SW102 Gal+
cells containing the galK modified 50J2 BAC After electro-poration the bacteria were allowed 45 h outgrowth in a 32Cshaking waterbath The bacteria were then washed in M9 saltsto remove any rich medium and plated on minimal mediumwith glycerol DOG and chloramphenicol The 45 h out-growth is necessary to obtain complete segregation of therecombinant BACs containing the mutation After 3 dayswe obtained colonies with a ratio of 10ndash1001 when comparingplates with heat-induced to non-induced bacteria We picked12 colonies from the heat-induced plates for BAC minipreps
followed by SpeI restriction analysis (Figure 3A) Ten out ofthe twelve clones had the same restriction pattern as theunmodified 50J2 BAC DNA suggesting that the desiredreplacement of the galK gene by the point mutation hadoccurred This was confirmed by PCR amplification followed
Figure 2 Overview of the galK selection scheme The result of the firsttargeting event is the insertion of constitutively active galK into a definedposition on the BAC by selection on minimal medium containing galactoseand chloramphenicol to select for the maintenance of the BAC The bacteria arenow phenotypically Gal+ Next the galK cassette is replaced by a dsDNA oligoa PCR product or a cloned dsDNA fragment carrying a desired mutation(indicated by a star) and flanked by the same homology arms used in thefirst selection step This is achieved by negative selection using minimalmedium containing 2-deoxy-galactose (DOG) with glycerol as the sole carbonsource The bacteria become phenotypically Gal H1 and H2 homology arms1 and 2 respectively cat chloramphenicol acetyl transferase gene ori2 BACorigin of replication galK Ecoli galactokinase gene driven by a minimalpromoter
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 6 OF 12
by sequencing of the modified region of the 10 BAC clones(Figure 3B) All 10 sequenced clones had the desired pointmutation The effective recombination efficiency in thisexperiment however was 812 (67) since 2 of the 10 clonesalso had an additional single base pair deletion (clones 9 and11) These deletions probably occurred during oligo synthesissince we did not purify the oligos beyond desalting (19)Because of the high efficiency in this system there was noneed to pre-screen the selected colonies prior to picking forminipreps
Large BAC deletions
A drawback of using BACs for the production of transgenicmice is the frequent presence of other genes on the BAC inaddition to the gene of interest This is especially a problem forBAC complementation used in positional cloning where aBAC is tested for its ability to rescue a loss-of-function mutantphenotype by making BAC transgenic mice If complementa-tion is achieved it is impossible to know which gene on theBAC is responsible for the rescue Therefore we decided to
see whether galK selection could be used to make largespecific and clean deletions in BACs so as to remove unwantedgenes (a process called BAC trimming) As a model systemwe chose a mouse BAC from the C57BL6-derived libraryRP23 RP23-341F12 since we could obtain the BAC sequencedirectly from the UCSC genomic web browser (see Materialsand Methods) We then used PCR to amplify the galK selec-tion cassette with primers containing homology to the BACThe forward primer contained 50 bp of homology to the very 50
end of the BAC insert and this primer was combined withthree different reverse primers having 50 bp of homology topositions 50 75 and 100 kb away from the forward primerrsquoshomology (Figure 4A) SW102 bacteria containing the 341F12BAC were heat-induced and made electrocompetent and thentransformed with the three different selection cassettes fol-lowed by the selection on minimal medium with galactoseFrom the many resulting Gal+ colonies we picked four fromeach experiment and then did a SpeI restriction analysis of theBAC miniprep DNAs from them (Figure 4B) All four colon-ies from each experiment had the desired deletion (50 75 and100 kb respectively) The galK selection cassette can then be
Figure 3 Introduction of a G12D mutation in the Nras gene (A) SpeI restriction analysis of BAC miniprep DNA First lane is the unmodified CITB-50J2 Nras BACLanes 1ndash12 show digestion patterns of 12 clones counterselected for the substitution of galK with an oligo containing the GA substitution for the second position ofcodon 12 of Nras Clones 7 and 10 had internal deletions indicating that DOG resistance was achieved by spontaneous deletion and not homologous recombinationThese two clones were not analyzed further (B) Sequence analysis of a PCR product spanning the modified region from clones 1ndash6 8ndash9 and 11ndash12 All clones had theintended substitution (highlighted) However clones 9 and 11 also had an internal basepair deletion indicated by a minus (highlighted) The Nras ATG and codon 12are indicated (shadow)
PAGE 7 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
removed with an oligo using galK negative selection to cleanlydelete the galK cassette from the BAC as described in theprevious experiment (data not shown)
Insertion of a mutant loxP511 site into a BAC
Encouraged by the efficiency of this selection system wedecided to see whether we could introduce a single 34 bploxP511 site cleanly into BAC DNA using galK positiveneg-ative selection It has been previously shown that loxP andloxP511 sites cannot recombine with each other therefore the
introduced loxP511 site can only recombine with the loxP511site present in the BAC vector backbone and not the wild-typesite (Figure 5A) (20) We used galK positive selection to inserta PCR-amplified galK cassette flanked by 50 bp homologyarms to a position 95 kb away from the mutant loxP511 site inthe BAC RP23-341F12 vector backbone (Figure 5A) We thenreplaced the galK cassette using DOG counterselection with adouble-stranded 100 bp oligo containing a 34 bp loxP511 siteflanked by two 33 bp homology arms In this experiment weobserved less than a 10-fold difference in the number of col-onies on the plates from heat-induced and non-induced bac-teria suggesting fewer recombinants This lower frequency ofrecombinants when compared with the Nras G12D substitu-tion experiment is likely explained by the shorter homologyarms used in this latter experiment as it has been shown thatthe efficiency of recombination increase four orders of mag-nitude when homology length is increased from 20 to 40 bp(10) We analyzed six BAC minipreps from potential recom-binants by digesting with SpeI and comparing the restrictionpatterns with that of wild-type RP23-341F12 BAC DNA(Figure 5B) Three of six (50) colonies had exactly thesame restriction pattern as the unmodified RP23-341F12BAC suggesting that DOG resistance had selected for thedesired homologous recombination products in these threecases whereas the other three clones apparently becameDOG resistant due to the selection of BACs that carry largedeletions spanning galK
The 100 bp oligo was designed so that the loxP511 sitewould be inserted in the same orientation as the loxP511site in the BAC vector backbone To confirm that the BACswith the wild-type restriction pattern (3 5 and 6) had theloxP511 site correctly inserted we electroporated thesethree BACs into electrocompetent and L-arabinose inducedEL350 cells which carry an L-arabinose-inducible Cregene and then plated the cells on LB plates containing chlor-amphenicol to select for the BACs Two colonies generatedfrom each recombinant loxP511 clone were then tested bySpeI digestion of BAC miniprep DNA and all had the expec-ted Cre-mediated 95 kb deletion (Figure 5C) Recombinationwas confirmed by PCR using a forward primer from the BACvector backbone and a reverse primer mapping to a positiondistal to the reverse homology arm used to insert the galKcassette These primers are 95 kb apart on wild-type RP23-341F12 DNA and only 378 bp apart on CreloxP511 recom-bined DNA PCR analysis of all six clones produced a band ofthe expected length whereas no product could be amplifiedfrom unmodified RP23-341F12 BAC DNA (Figure 5D)
Source of background deletions following DOG selection
In the Nras G12D substitution and loxP511 insertion experi-ments a background of DOG-resistant bacteria which lackedthe desired mutation and instead carried unwanted deletionsthat spanned the galK selection cassette were observed Therelative ratio of colonies containing BACs with deletions wasalso higher when short homology arms were used This wasexpected since homologous recombination is more efficientwith longer homology arms (10) that would increase the rel-ative frequency of correctly targeted BACs The source of thedeletions could be l Red-mediated since proteins capable ofmediating recombination are expressed when the Red genes
Figure 4 BAC trimming using galK selection (A) Illustration of the design ofthe deletion experiment Homology arm 1 (H1) was held constant and H2 wasseparated from H1 by either 50 75 or 100 kb (B) SpeI restriction analysis ofBAC miniprep DNA from 12 clones showing deletions of 50 75 and 100 kbrespectively after the insertion of the galK selection cassette The first lane isunmodified RP23-341F12 BAC DNA which was included as a control Alltested clones had the intended deletion
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 8 OF 12
are activated Alternatively the deletions could be induced bythe large amount of oligos (200 ng) used in these experimentsor they could simply represent spontaneous deletions thatoccur at low level during normal BAC replication and thatare found by DOG selection To distinguish between thesepossibilities we repeated the DOG selection step used to insertthe loxP511 oligo into BAC RP23-341F12 DNA using fourdifferently treated bacteria samples non-induced bacteriawithout the 100 bp loxP511 oligo (Figure 6A) non-inducedbacteria with the oligo (Figure 6B) induced bacteria without
the oligo (Figure 6C) and induced bacteria with the oligo(Figure 6D) The number of resistant colonies obtainedfrom the four experiments varied by lt10-fold (data notshown) Ten colonies from each electroporation were thenanalyzed by SpeI digestion of BAC miniprep DNA and com-pared with unmodified RP23-341F12 BAC DNA (Figure 6AndashD) Unwanted galK region deletions were observed in all 10colonies for both of the non-induced samples (Figure 6A andB) These deletions are therefore unlikely to be Red-mediatedsince these samples were not heat-induced We also observed
Figure 5 Insertion of a loxP511 site (A) The location of the wild-type and mutant loxP sites in the BAC backbone are indicated along with the extra mutant loxP511site that was introduced into the BAC genomic insert via galK counterselection The 95 kb region deleted by Cre-mediated recombination between the two loxP511sites is indicated and PCR primers used to confirm the deletion are shown as small arrows (B) SpeI restriction analysis of six miniprep clones selected forthe replacement of galK with a dsDNA oligo containing the mutant loxP511 site Clones 3 5 and 6 (circles) had the same restriction pattern as the unmodifiedBAC indicating that DOG resistance occurred due to the intended homologous recombination event Clones 1 2 and 4 had large deletions and were not analyzedfurther (C) SpeI restriction analysis of BAC miniprep DNA from clones 3 5 and 6 after transformation into Cre-induced EL350 cells Two clones from each parentalclone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all clones analyzed confirming the correctinsertion of loxP511 in clones 3 5 and 6 (D) PCR analysis of the six clones from (C) with one primer mapping to the BAC backbone and the other to a position distalto the inserted loxP511 site
PAGE 9 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
deletions in both the absence and presence of oligo indicatingthat the deletions are not oligo induced We therefore concludethat these are spontaneous deletions that occur at low levelduring normal BAC replication in the DH10B background
Non-deleted colonies with the parental SpeI fingerprintwere only observed in the Red-induced sample that received
oligo (Figure 6D clones 33 and 35) These clones were sub-sequently electroporated into EL350 Cre-expressing cells toconfirm they contained the introduced loxP511 site Two col-onies from each original clone were tested by SpeI digestion ofthe BAC miniprep DNA As shown in Figure 6E each colonyhas undergone Cre-mediated deletion confirming that these
Figure 6 Same experiment as in Figure 5 with modifications as indicated at the top of each panel (A) SpeI digest of 10 minipreps from a control experiment withoutheat-induction and without the loxP511 dsDNA oligo (B) SpeI digest of 10 minipreps from a control experiment without heat-induction but with the loxP511 dsDNAoligo (C) SpeI digest of 10 minipreps from a control experiment with heat-induction but without the loxP511 dsDNA oligo (D) SpeI digest of 10 minipreps from anexperiment with heat-induction and with the loxP511 dsDNA oligo (comparable with Figure 5B) Clones with the parental digestion pattern indicating DOGresistance due to homologous recombination (clones 33 and 35 circles) are only seen in (D) DOG resistance in all other clones likely occurred due to internaldeletions of the BACs (E) SpeI restriction analysis of BAC miniprep DNA from clones 33 and 35 after transformation into Cre-induced EL350 cells Two clonesfrom each parental clone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all the clones analyzedconfirming the correct insertion of loxP511 in clones 33 and 35 (compare with Figure 5C)
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 10 OF 12
colonies were correctly targeted and contain the introducedloxP511 site (2 out of 10 clones a 20 efficiency in thisexperiment)
Generation of SW105 and SW106 cells
The two DY380-derived bacterial strains EL250 and EL350contain the defective l prophage needed for recombineering inaddition to L-arabinose inducible Flp or Cre genes respect-ively (11) Both strains have proven to be very useful for BACmodification (11) and EL350 is now used routinely for mak-ing conditional targeting vectors for ES cell knock-out experi-ments (4) To further enhance the usefulness of these strainswe decided to transfer the galK selection system into themThis was done as previously described for SW102 The galKIS2 element present in both strains was replaced with the wild-type gal promoter and the galK gene in the gal operon deletedusing DOG selection The resulting strains SW105 (Flp) andSW106 (Cre) (Table 1) were then tested to confirm that theystill contained inducible Flp and Cre genes by transformingarabinose-induced and electrocompetent cells with plasmidscontaining a neo gene flanked by Frt or loxP sites PL451 andPL452 (4) respectively Using SW105 or SW106 it is nowpossible to introduce a point mutation or an informativerestriction site into a BAC retrieve a fragment from thisBAC containing the introduced mutation(s) into a plasmidbackbone using gap repair and turn the retrieved fragmentinto a conditional targeting vectormdashall using only one bac-terial strain and only a single initial BAC transformation
DISCUSSION
Here we describe a new recombineering-based Ecoli BACmodification system that makes use of galK positive selectionfor growth on galactose minimal medium and galK negativeselection (counterselection) for growth on DOG This modi-fication system has several advantages compared with theother related BAC modification systems First the galK selec-tion cassette is small (1231 bp + homology arms) comparedwith for example the sacBndashneo cassette (3 kb) making PCRamplification and transformation into bacteria easier Twohomology arms are easily added to the galK cassette by includ-ing these sequences in the 50 ends of the primers used foramplification (see Materials and Methods) Second sincegalK is used for both selection steps mutations occurringin galK during PCR amplification will be selected againstduring positive selection significantly reducing the risk ofDOG resistance from PCR mutations in galK during negativeselection Third even when very short (33 bp) homology armswere used the frequency of recombinants among the analyzedDOG-resistant clones was still 20ndash50 To our knowledgesuccessful BAC modification with such short homology armshas never been reported Finally since galK recombineering isso efficient little screening is required following selectionreducing the overall hands-on-time to a minimum galK selec-tion requires growth on minimal medium in both selectionsteps although perhaps not used routinely in most molecularbiology laboratories where BAC modification is needed theseplates are fairly simple to make and should not prevent anyonefrom using this method
For all such counterselection schemes there will bebackground since during negative selection any event leadingto the loss of the counterselectable marker will result in sur-vival In our system virtually all background appears to resultfrom deletions that span the inserted galK gene These dele-tions likely occur during BAC replication in the 45 h out-growth phase since we have shown that these deletions occurindependently of Red induction and in the absence or presenceof oligo It is well established that in recA defective bacteriaBACs are very stable compared with other large insert vectorssuch as yeast artificial chromosomes (YACs) However usingcounterselection rare spontaneous deletions are seen becauseof the strong selection force This spontaneous deletion back-ground is not a problem however due to the high frequency ofhomologous recombination obtained with the defective lprophage system
Increasing the length of homology arms used for recombin-eering will reduce the relative number of background deletionsobserved following negative selection since the percentage ofcolonies containing BACs that eliminated galK by homolog-ous recombination will be increased Oligos with longerhomology arms can be produced by annealing two oligoswith overlapping 30 ends (21) These overlapping oligos (50
single strand overhangs) are then filled in by DNA polymerasein vivo (or in vitro) and subsequently serve as the substrate forthe l Red proteins exo and bet Longer homology arms canalso be added by traditional cloning using restriction enzymesand DNA ligase (4)
A number of uses can be imagined for the strains describedhere In principle our strains can be used to make virtually anykind of BAC modification one can imagine including pointmutations or clean deletions or insertions of everything fromcDNA to loxP sites or small epitope tags BAC trimming thespecific deletion of BAC DNA flanking a gene of interest couldalso be used to remove genes from BACs prior to making BACtransgenic mice so that only the gene of interest on the BAC isanalyzed This should be of particular interest for BAC rescueexperiments in positional cloning Of course trimming andmodification can be combined since the galK selection cassettecan be recycled so that the same BAC can be modified severaltimes BAC modification could also be the starting point forconstructing a gene-targeting vector to allow for more soph-isticated gene targeting in mouse ES cells The desired muta-tion(s) could first be created in the BAC followed by retrievalinto a plasmid vector containing a negative-selection markerlike thymidine kinase using gap repair (4) Finally a neo mar-ker could be introduced to allow for positive selection in EScells Alternatively the retrieved fragment could be modifiedso that the end result is a conditional targeting vector StrainSW106 which in addition to galK selection can be induced toexpress Cre recombinase makes it possible to perform all ofthe steps needed to construct a conditional targeting vectorstarting with only a single BAC transformation step
Furthermore in experiments where BAC transgenic miceare used to analyze the effect of deleting long-range regulatorsof gene expression galK DOG counterselection can even beused in the absence of an oligo to generate a series of dele-tions around the galK insertion site and the effect on generegulation studied
The Flp or Cre ORFs contained in strains SW105 andSW106 respectively can also be replaced for any desired
PAGE 11 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
gene thus creating tight arabinose-inducible expression forany gene of interest This is done by first replacing the CreFlpe ORF with galK and select for Gal+ recombinants ThegalK cassette is then replaced by the ORF of the gene ofinterest by DOG selection for Gal recombinants
Finally the strains described here should be very useful forgenetic manipulation not only of BACs but also the Ecoligenome itself
All recombineering reagents discussed in this workare freely available upon request To obtain these materialsplease follow the directions listed on our website (httprecombineeringncifcrfgov) Detailed protocols for recom-bineering including galK selection can also be downloadeddirectly from our website
ACKNOWLEDGEMENTS
We thank Allen Kane and Carolyn Whistler from ScientificPublications Graphics amp Media NCI-Frederick for help withillustrations and figure preparation Funding for this research aswell as the Open Access publication charges was provided byDHHS NIH NCI
REFERENCES
1 ShizuyaH BirrenB KimUJ MancinoV SlepakTTachiiriY and SimonM (1992) Cloning and stable maintenance of300-kilobase-pair fragments of human DNA in Escherichia coli using anF-factor-based vector Proc Natl Acad Sci USA 89 8794ndash8797
2 YangXW ModelP and HeintzN (1997) Homologous recombinationbased modification in Escherichia coli and germline transmission intransgenic mice of a bacterial artificial chromosome Nat Biotechnol15 859ndash865
3 AntochMP SongEJ ChangAM VitaternaMH ZhaoYWilsbacherLD SangoramAM KingDP PintoLH andTakahashiJS (1997) Functional identification of the mouse circadianClock gene by transgenic BAC rescue Cell 89 655ndash667
4 LiuP JenkinsNA and CopelandNG (2003) A highly efficientrecombineering-based method for generating conditional knockoutmutations Genome Res 13 476ndash484
5 Cotta-de-AlmeidaV SchonhoffS ShibataT LeiterA andSnapperSB (2003) A new method for rapidly generating gene-targetingvectors by engineering BACs through homologous recombination inbacteria Genome Res 13 2190ndash2194
6 CopelandNG JenkinsNA and CourtDL (2001) Recombineeringa powerful new tool for mouse functional genomics Nature RevGenet 2 769ndash779
7 CourtDL SawitzkeJA and ThomasonLC (2002) Geneticengineering using homologous recombination Annu Rev Genet 36361ndash388
8 ZhangY BuchholzF MuyrersJP and StewartAF (1998) A newlogic for DNA engineering using recombination in Escherichia coliNature Genet 20 123ndash128
9 MuyrersJP ZhangY TestaG and StewartAF (1999) Rapidmodification of bacterial artificial chromosomes by ET-recombinationNucleic Acids Res 27 1555ndash1557
10 YuD EllisHM LeeEC JenkinsNA CopelandNG andCourtDL (2000) An efficient recombination system for chromosomeengineering in Escherichia coli Proc Natl Acad Sci USA 975978ndash5983
11 LeeEC YuD Martinez de VelascoJ TessarolloL SwingDACourtDL JenkinsNA and CopelandNG (2001) A highly efficientEscherichia coli-based chromosome engineering system adapted forrecombinogenic targeting and subcloning of BAC DNA Genomics73 56ndash65
12 GongS YangXW LiC and HeintzN (2002) Highly efficientmodification of bacterial artificial chromosomes (BACs) using novelshuttle vectors containing the R6Kgamma origin of replicationGenome Res 12 1992ndash1998
13 JamsaiD OrfordM NefedovM FucharoenS WilliamsonR andIoannouPA (2003) Targeted modification of a human beta-globin locusBAC clone using GET Recombination and an I-SceI counterselectioncassette Genomics 82 68ndash77
14 SwaminathanS EllisHM WatersLS YuD LeeECCourtDL and SharanSK (2001) Rapid engineering of bacterialartificial chromosomes using oligonucleotides Genesis 2914ndash21
15 AlperMD and AmesBN (1975) Positive selection of mutants withdeletions of the gal-chl region of the Salmonella chromosome as ascreening procedure for mutagens that cause deletions J Bacteriol121 259ndash266
16 AusubelFM BrentR KingstonRE MooreDD SeidmanJGSmithJA and StruhlK (1987) Current Protocols in Molecular BiologyGreene Publishing Associates and Wiley-Interscience John Wiley ampSons Hoboken NJ
17 BachmannBJ (1996) Derivations and genotypes of some mutantderivatives of Escherichia coli K-12 In NeidhardtFC CurtissRIIIIngrahamJL LinECC LowKB MagasanikB ReznikoffWSRileyM SchaechterM and UmbargerHE (eds) Escherichia coli andSalmonella Cellular and Molecular Biology ASM press WashingtonDC Vol 2 pp 2460ndash2488
18 GrantSG JesseeJ BloomFR and HanahanD (1990) Differentialplasmid rescue from transgenic mouse DNAs into Escherichia colimethylation-restriction mutants Proc Natl Acad Sci USA 874645ndash4649
19 OppenheimAB RattrayAJ BubunenkoM ThomasonLC andCourtDL (2004) In vivo recombineering of bacteriophage lambda byPCR fragments and single-strand oligonucleotides Virology 319185ndash189
20 HoessRH WierzbickiA and AbremskiK (1986) The role ofthe loxP spacer region in P1 site-specific recombination NucleicAcids Res 14 2287ndash2300
21 YuD SawitzkeJA EllisH and CourtDL (2003) Recombineeringwith overlapping single-stranded DNA oligonucleotides testing arecombination intermediate Proc Natl Acad Sci USA 1007207ndash7212
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 12 OF 12

galK expression cassette
Having produced a bacterial strain SW102 which is galKdefective the next step was to make a galK expression cassettethat could be used to restore the bacteriarsquos ability to grow ongalactose by providing galK in trans This was achieved byPCR amplification of the wild-type galK ORF from W3110cells We then added a minimal bacterial promoter em7 usinga two-step PCR approach (see Materials and Methods) andcloned the expression cassette into pBluescript SK We callthis plasmid pgalK (Table 1) The constitutively active galKexpression cassette can easily be amplified by PCRwith homology arms added to the primers (see Materialsand Methods)
Making a single base pair substitution
The general scheme for making mutations in BACs using galKselection is depicted in Figure 2 To test the galK selectionsystem for BAC recombineering we decided to introduce apoint mutation into a BAC containing the murine Nras gene(CITB clone 50J2) The sequence of the glycine-coding codon12 is GGT By changing this codon to GAT we would obtainthe desired mutation G12D In order to introduce this muta-tion into the BAC we first amplified the galK expressioncassette by PCR using primers with 50 bp of homology toeither side of the second position of the GGT codon Followinghomologous recombination this targeting would introduce a1 bp deletion into codon 12 in addition to inserting the galKselection cassette Instead of deleting the basepair the galKcassette could have been inserted right next to the basepairinstead SW102 cells containing the 50J2 BAC were heat-induced and made electrocompetent and then electroporatedwith the galK cassette Gal+ recombinant colonies were selec-ted for growth on galactose minimal medium with chloram-phenicol to maintain the BAC Bacteria grow more slowly onminimal media than on rich media and we generally pickcolonies after 2ndash3 days For this first step we do not expectany background colonies on the non-induced control plates ifthe pgalK plasmid is properly eliminated (see Materials andMethods) To purify the Gal+ colonies we streaked a fewcolonies on MacConkey galactose indicator plates to obtainsingle bright pinkred Gal+ colonies One of these single-cloned colonies was picked to initiate a culture for thenext step counterselection We find that there is no need toanalyze the Gal+ colonies further before proceeding tocounterselection
A 100 bp dsDNA oligo was then prepared by annealing twocomplementary oligos having 49 and 50 bp homologyrespectively to either side of the desired mutation a singleAT bp An aliquot of 200 ng of this oligo was then electro-porated into heat-induced and electrocompetent SW102 Gal+
cells containing the galK modified 50J2 BAC After electro-poration the bacteria were allowed 45 h outgrowth in a 32Cshaking waterbath The bacteria were then washed in M9 saltsto remove any rich medium and plated on minimal mediumwith glycerol DOG and chloramphenicol The 45 h out-growth is necessary to obtain complete segregation of therecombinant BACs containing the mutation After 3 dayswe obtained colonies with a ratio of 10ndash1001 when comparingplates with heat-induced to non-induced bacteria We picked12 colonies from the heat-induced plates for BAC minipreps
followed by SpeI restriction analysis (Figure 3A) Ten out ofthe twelve clones had the same restriction pattern as theunmodified 50J2 BAC DNA suggesting that the desiredreplacement of the galK gene by the point mutation hadoccurred This was confirmed by PCR amplification followed
Figure 2 Overview of the galK selection scheme The result of the firsttargeting event is the insertion of constitutively active galK into a definedposition on the BAC by selection on minimal medium containing galactoseand chloramphenicol to select for the maintenance of the BAC The bacteria arenow phenotypically Gal+ Next the galK cassette is replaced by a dsDNA oligoa PCR product or a cloned dsDNA fragment carrying a desired mutation(indicated by a star) and flanked by the same homology arms used in thefirst selection step This is achieved by negative selection using minimalmedium containing 2-deoxy-galactose (DOG) with glycerol as the sole carbonsource The bacteria become phenotypically Gal H1 and H2 homology arms1 and 2 respectively cat chloramphenicol acetyl transferase gene ori2 BACorigin of replication galK Ecoli galactokinase gene driven by a minimalpromoter
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 6 OF 12
by sequencing of the modified region of the 10 BAC clones(Figure 3B) All 10 sequenced clones had the desired pointmutation The effective recombination efficiency in thisexperiment however was 812 (67) since 2 of the 10 clonesalso had an additional single base pair deletion (clones 9 and11) These deletions probably occurred during oligo synthesissince we did not purify the oligos beyond desalting (19)Because of the high efficiency in this system there was noneed to pre-screen the selected colonies prior to picking forminipreps
Large BAC deletions
A drawback of using BACs for the production of transgenicmice is the frequent presence of other genes on the BAC inaddition to the gene of interest This is especially a problem forBAC complementation used in positional cloning where aBAC is tested for its ability to rescue a loss-of-function mutantphenotype by making BAC transgenic mice If complementa-tion is achieved it is impossible to know which gene on theBAC is responsible for the rescue Therefore we decided to
see whether galK selection could be used to make largespecific and clean deletions in BACs so as to remove unwantedgenes (a process called BAC trimming) As a model systemwe chose a mouse BAC from the C57BL6-derived libraryRP23 RP23-341F12 since we could obtain the BAC sequencedirectly from the UCSC genomic web browser (see Materialsand Methods) We then used PCR to amplify the galK selec-tion cassette with primers containing homology to the BACThe forward primer contained 50 bp of homology to the very 50
end of the BAC insert and this primer was combined withthree different reverse primers having 50 bp of homology topositions 50 75 and 100 kb away from the forward primerrsquoshomology (Figure 4A) SW102 bacteria containing the 341F12BAC were heat-induced and made electrocompetent and thentransformed with the three different selection cassettes fol-lowed by the selection on minimal medium with galactoseFrom the many resulting Gal+ colonies we picked four fromeach experiment and then did a SpeI restriction analysis of theBAC miniprep DNAs from them (Figure 4B) All four colon-ies from each experiment had the desired deletion (50 75 and100 kb respectively) The galK selection cassette can then be
Figure 3 Introduction of a G12D mutation in the Nras gene (A) SpeI restriction analysis of BAC miniprep DNA First lane is the unmodified CITB-50J2 Nras BACLanes 1ndash12 show digestion patterns of 12 clones counterselected for the substitution of galK with an oligo containing the GA substitution for the second position ofcodon 12 of Nras Clones 7 and 10 had internal deletions indicating that DOG resistance was achieved by spontaneous deletion and not homologous recombinationThese two clones were not analyzed further (B) Sequence analysis of a PCR product spanning the modified region from clones 1ndash6 8ndash9 and 11ndash12 All clones had theintended substitution (highlighted) However clones 9 and 11 also had an internal basepair deletion indicated by a minus (highlighted) The Nras ATG and codon 12are indicated (shadow)
PAGE 7 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
removed with an oligo using galK negative selection to cleanlydelete the galK cassette from the BAC as described in theprevious experiment (data not shown)
Insertion of a mutant loxP511 site into a BAC
Encouraged by the efficiency of this selection system wedecided to see whether we could introduce a single 34 bploxP511 site cleanly into BAC DNA using galK positiveneg-ative selection It has been previously shown that loxP andloxP511 sites cannot recombine with each other therefore the
introduced loxP511 site can only recombine with the loxP511site present in the BAC vector backbone and not the wild-typesite (Figure 5A) (20) We used galK positive selection to inserta PCR-amplified galK cassette flanked by 50 bp homologyarms to a position 95 kb away from the mutant loxP511 site inthe BAC RP23-341F12 vector backbone (Figure 5A) We thenreplaced the galK cassette using DOG counterselection with adouble-stranded 100 bp oligo containing a 34 bp loxP511 siteflanked by two 33 bp homology arms In this experiment weobserved less than a 10-fold difference in the number of col-onies on the plates from heat-induced and non-induced bac-teria suggesting fewer recombinants This lower frequency ofrecombinants when compared with the Nras G12D substitu-tion experiment is likely explained by the shorter homologyarms used in this latter experiment as it has been shown thatthe efficiency of recombination increase four orders of mag-nitude when homology length is increased from 20 to 40 bp(10) We analyzed six BAC minipreps from potential recom-binants by digesting with SpeI and comparing the restrictionpatterns with that of wild-type RP23-341F12 BAC DNA(Figure 5B) Three of six (50) colonies had exactly thesame restriction pattern as the unmodified RP23-341F12BAC suggesting that DOG resistance had selected for thedesired homologous recombination products in these threecases whereas the other three clones apparently becameDOG resistant due to the selection of BACs that carry largedeletions spanning galK
The 100 bp oligo was designed so that the loxP511 sitewould be inserted in the same orientation as the loxP511site in the BAC vector backbone To confirm that the BACswith the wild-type restriction pattern (3 5 and 6) had theloxP511 site correctly inserted we electroporated thesethree BACs into electrocompetent and L-arabinose inducedEL350 cells which carry an L-arabinose-inducible Cregene and then plated the cells on LB plates containing chlor-amphenicol to select for the BACs Two colonies generatedfrom each recombinant loxP511 clone were then tested bySpeI digestion of BAC miniprep DNA and all had the expec-ted Cre-mediated 95 kb deletion (Figure 5C) Recombinationwas confirmed by PCR using a forward primer from the BACvector backbone and a reverse primer mapping to a positiondistal to the reverse homology arm used to insert the galKcassette These primers are 95 kb apart on wild-type RP23-341F12 DNA and only 378 bp apart on CreloxP511 recom-bined DNA PCR analysis of all six clones produced a band ofthe expected length whereas no product could be amplifiedfrom unmodified RP23-341F12 BAC DNA (Figure 5D)
Source of background deletions following DOG selection
In the Nras G12D substitution and loxP511 insertion experi-ments a background of DOG-resistant bacteria which lackedthe desired mutation and instead carried unwanted deletionsthat spanned the galK selection cassette were observed Therelative ratio of colonies containing BACs with deletions wasalso higher when short homology arms were used This wasexpected since homologous recombination is more efficientwith longer homology arms (10) that would increase the rel-ative frequency of correctly targeted BACs The source of thedeletions could be l Red-mediated since proteins capable ofmediating recombination are expressed when the Red genes
Figure 4 BAC trimming using galK selection (A) Illustration of the design ofthe deletion experiment Homology arm 1 (H1) was held constant and H2 wasseparated from H1 by either 50 75 or 100 kb (B) SpeI restriction analysis ofBAC miniprep DNA from 12 clones showing deletions of 50 75 and 100 kbrespectively after the insertion of the galK selection cassette The first lane isunmodified RP23-341F12 BAC DNA which was included as a control Alltested clones had the intended deletion
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 8 OF 12
are activated Alternatively the deletions could be induced bythe large amount of oligos (200 ng) used in these experimentsor they could simply represent spontaneous deletions thatoccur at low level during normal BAC replication and thatare found by DOG selection To distinguish between thesepossibilities we repeated the DOG selection step used to insertthe loxP511 oligo into BAC RP23-341F12 DNA using fourdifferently treated bacteria samples non-induced bacteriawithout the 100 bp loxP511 oligo (Figure 6A) non-inducedbacteria with the oligo (Figure 6B) induced bacteria without
the oligo (Figure 6C) and induced bacteria with the oligo(Figure 6D) The number of resistant colonies obtainedfrom the four experiments varied by lt10-fold (data notshown) Ten colonies from each electroporation were thenanalyzed by SpeI digestion of BAC miniprep DNA and com-pared with unmodified RP23-341F12 BAC DNA (Figure 6AndashD) Unwanted galK region deletions were observed in all 10colonies for both of the non-induced samples (Figure 6A andB) These deletions are therefore unlikely to be Red-mediatedsince these samples were not heat-induced We also observed
Figure 5 Insertion of a loxP511 site (A) The location of the wild-type and mutant loxP sites in the BAC backbone are indicated along with the extra mutant loxP511site that was introduced into the BAC genomic insert via galK counterselection The 95 kb region deleted by Cre-mediated recombination between the two loxP511sites is indicated and PCR primers used to confirm the deletion are shown as small arrows (B) SpeI restriction analysis of six miniprep clones selected forthe replacement of galK with a dsDNA oligo containing the mutant loxP511 site Clones 3 5 and 6 (circles) had the same restriction pattern as the unmodifiedBAC indicating that DOG resistance occurred due to the intended homologous recombination event Clones 1 2 and 4 had large deletions and were not analyzedfurther (C) SpeI restriction analysis of BAC miniprep DNA from clones 3 5 and 6 after transformation into Cre-induced EL350 cells Two clones from each parentalclone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all clones analyzed confirming the correctinsertion of loxP511 in clones 3 5 and 6 (D) PCR analysis of the six clones from (C) with one primer mapping to the BAC backbone and the other to a position distalto the inserted loxP511 site
PAGE 9 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
deletions in both the absence and presence of oligo indicatingthat the deletions are not oligo induced We therefore concludethat these are spontaneous deletions that occur at low levelduring normal BAC replication in the DH10B background
Non-deleted colonies with the parental SpeI fingerprintwere only observed in the Red-induced sample that received
oligo (Figure 6D clones 33 and 35) These clones were sub-sequently electroporated into EL350 Cre-expressing cells toconfirm they contained the introduced loxP511 site Two col-onies from each original clone were tested by SpeI digestion ofthe BAC miniprep DNA As shown in Figure 6E each colonyhas undergone Cre-mediated deletion confirming that these
Figure 6 Same experiment as in Figure 5 with modifications as indicated at the top of each panel (A) SpeI digest of 10 minipreps from a control experiment withoutheat-induction and without the loxP511 dsDNA oligo (B) SpeI digest of 10 minipreps from a control experiment without heat-induction but with the loxP511 dsDNAoligo (C) SpeI digest of 10 minipreps from a control experiment with heat-induction but without the loxP511 dsDNA oligo (D) SpeI digest of 10 minipreps from anexperiment with heat-induction and with the loxP511 dsDNA oligo (comparable with Figure 5B) Clones with the parental digestion pattern indicating DOGresistance due to homologous recombination (clones 33 and 35 circles) are only seen in (D) DOG resistance in all other clones likely occurred due to internaldeletions of the BACs (E) SpeI restriction analysis of BAC miniprep DNA from clones 33 and 35 after transformation into Cre-induced EL350 cells Two clonesfrom each parental clone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all the clones analyzedconfirming the correct insertion of loxP511 in clones 33 and 35 (compare with Figure 5C)
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 10 OF 12
colonies were correctly targeted and contain the introducedloxP511 site (2 out of 10 clones a 20 efficiency in thisexperiment)
Generation of SW105 and SW106 cells
The two DY380-derived bacterial strains EL250 and EL350contain the defective l prophage needed for recombineering inaddition to L-arabinose inducible Flp or Cre genes respect-ively (11) Both strains have proven to be very useful for BACmodification (11) and EL350 is now used routinely for mak-ing conditional targeting vectors for ES cell knock-out experi-ments (4) To further enhance the usefulness of these strainswe decided to transfer the galK selection system into themThis was done as previously described for SW102 The galKIS2 element present in both strains was replaced with the wild-type gal promoter and the galK gene in the gal operon deletedusing DOG selection The resulting strains SW105 (Flp) andSW106 (Cre) (Table 1) were then tested to confirm that theystill contained inducible Flp and Cre genes by transformingarabinose-induced and electrocompetent cells with plasmidscontaining a neo gene flanked by Frt or loxP sites PL451 andPL452 (4) respectively Using SW105 or SW106 it is nowpossible to introduce a point mutation or an informativerestriction site into a BAC retrieve a fragment from thisBAC containing the introduced mutation(s) into a plasmidbackbone using gap repair and turn the retrieved fragmentinto a conditional targeting vectormdashall using only one bac-terial strain and only a single initial BAC transformation
DISCUSSION
Here we describe a new recombineering-based Ecoli BACmodification system that makes use of galK positive selectionfor growth on galactose minimal medium and galK negativeselection (counterselection) for growth on DOG This modi-fication system has several advantages compared with theother related BAC modification systems First the galK selec-tion cassette is small (1231 bp + homology arms) comparedwith for example the sacBndashneo cassette (3 kb) making PCRamplification and transformation into bacteria easier Twohomology arms are easily added to the galK cassette by includ-ing these sequences in the 50 ends of the primers used foramplification (see Materials and Methods) Second sincegalK is used for both selection steps mutations occurringin galK during PCR amplification will be selected againstduring positive selection significantly reducing the risk ofDOG resistance from PCR mutations in galK during negativeselection Third even when very short (33 bp) homology armswere used the frequency of recombinants among the analyzedDOG-resistant clones was still 20ndash50 To our knowledgesuccessful BAC modification with such short homology armshas never been reported Finally since galK recombineering isso efficient little screening is required following selectionreducing the overall hands-on-time to a minimum galK selec-tion requires growth on minimal medium in both selectionsteps although perhaps not used routinely in most molecularbiology laboratories where BAC modification is needed theseplates are fairly simple to make and should not prevent anyonefrom using this method
For all such counterselection schemes there will bebackground since during negative selection any event leadingto the loss of the counterselectable marker will result in sur-vival In our system virtually all background appears to resultfrom deletions that span the inserted galK gene These dele-tions likely occur during BAC replication in the 45 h out-growth phase since we have shown that these deletions occurindependently of Red induction and in the absence or presenceof oligo It is well established that in recA defective bacteriaBACs are very stable compared with other large insert vectorssuch as yeast artificial chromosomes (YACs) However usingcounterselection rare spontaneous deletions are seen becauseof the strong selection force This spontaneous deletion back-ground is not a problem however due to the high frequency ofhomologous recombination obtained with the defective lprophage system
Increasing the length of homology arms used for recombin-eering will reduce the relative number of background deletionsobserved following negative selection since the percentage ofcolonies containing BACs that eliminated galK by homolog-ous recombination will be increased Oligos with longerhomology arms can be produced by annealing two oligoswith overlapping 30 ends (21) These overlapping oligos (50
single strand overhangs) are then filled in by DNA polymerasein vivo (or in vitro) and subsequently serve as the substrate forthe l Red proteins exo and bet Longer homology arms canalso be added by traditional cloning using restriction enzymesand DNA ligase (4)
A number of uses can be imagined for the strains describedhere In principle our strains can be used to make virtually anykind of BAC modification one can imagine including pointmutations or clean deletions or insertions of everything fromcDNA to loxP sites or small epitope tags BAC trimming thespecific deletion of BAC DNA flanking a gene of interest couldalso be used to remove genes from BACs prior to making BACtransgenic mice so that only the gene of interest on the BAC isanalyzed This should be of particular interest for BAC rescueexperiments in positional cloning Of course trimming andmodification can be combined since the galK selection cassettecan be recycled so that the same BAC can be modified severaltimes BAC modification could also be the starting point forconstructing a gene-targeting vector to allow for more soph-isticated gene targeting in mouse ES cells The desired muta-tion(s) could first be created in the BAC followed by retrievalinto a plasmid vector containing a negative-selection markerlike thymidine kinase using gap repair (4) Finally a neo mar-ker could be introduced to allow for positive selection in EScells Alternatively the retrieved fragment could be modifiedso that the end result is a conditional targeting vector StrainSW106 which in addition to galK selection can be induced toexpress Cre recombinase makes it possible to perform all ofthe steps needed to construct a conditional targeting vectorstarting with only a single BAC transformation step
Furthermore in experiments where BAC transgenic miceare used to analyze the effect of deleting long-range regulatorsof gene expression galK DOG counterselection can even beused in the absence of an oligo to generate a series of dele-tions around the galK insertion site and the effect on generegulation studied
The Flp or Cre ORFs contained in strains SW105 andSW106 respectively can also be replaced for any desired
PAGE 11 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
gene thus creating tight arabinose-inducible expression forany gene of interest This is done by first replacing the CreFlpe ORF with galK and select for Gal+ recombinants ThegalK cassette is then replaced by the ORF of the gene ofinterest by DOG selection for Gal recombinants
Finally the strains described here should be very useful forgenetic manipulation not only of BACs but also the Ecoligenome itself
All recombineering reagents discussed in this workare freely available upon request To obtain these materialsplease follow the directions listed on our website (httprecombineeringncifcrfgov) Detailed protocols for recom-bineering including galK selection can also be downloadeddirectly from our website
ACKNOWLEDGEMENTS
We thank Allen Kane and Carolyn Whistler from ScientificPublications Graphics amp Media NCI-Frederick for help withillustrations and figure preparation Funding for this research aswell as the Open Access publication charges was provided byDHHS NIH NCI
REFERENCES
1 ShizuyaH BirrenB KimUJ MancinoV SlepakTTachiiriY and SimonM (1992) Cloning and stable maintenance of300-kilobase-pair fragments of human DNA in Escherichia coli using anF-factor-based vector Proc Natl Acad Sci USA 89 8794ndash8797
2 YangXW ModelP and HeintzN (1997) Homologous recombinationbased modification in Escherichia coli and germline transmission intransgenic mice of a bacterial artificial chromosome Nat Biotechnol15 859ndash865
3 AntochMP SongEJ ChangAM VitaternaMH ZhaoYWilsbacherLD SangoramAM KingDP PintoLH andTakahashiJS (1997) Functional identification of the mouse circadianClock gene by transgenic BAC rescue Cell 89 655ndash667
4 LiuP JenkinsNA and CopelandNG (2003) A highly efficientrecombineering-based method for generating conditional knockoutmutations Genome Res 13 476ndash484
5 Cotta-de-AlmeidaV SchonhoffS ShibataT LeiterA andSnapperSB (2003) A new method for rapidly generating gene-targetingvectors by engineering BACs through homologous recombination inbacteria Genome Res 13 2190ndash2194
6 CopelandNG JenkinsNA and CourtDL (2001) Recombineeringa powerful new tool for mouse functional genomics Nature RevGenet 2 769ndash779
7 CourtDL SawitzkeJA and ThomasonLC (2002) Geneticengineering using homologous recombination Annu Rev Genet 36361ndash388
8 ZhangY BuchholzF MuyrersJP and StewartAF (1998) A newlogic for DNA engineering using recombination in Escherichia coliNature Genet 20 123ndash128
9 MuyrersJP ZhangY TestaG and StewartAF (1999) Rapidmodification of bacterial artificial chromosomes by ET-recombinationNucleic Acids Res 27 1555ndash1557
10 YuD EllisHM LeeEC JenkinsNA CopelandNG andCourtDL (2000) An efficient recombination system for chromosomeengineering in Escherichia coli Proc Natl Acad Sci USA 975978ndash5983
11 LeeEC YuD Martinez de VelascoJ TessarolloL SwingDACourtDL JenkinsNA and CopelandNG (2001) A highly efficientEscherichia coli-based chromosome engineering system adapted forrecombinogenic targeting and subcloning of BAC DNA Genomics73 56ndash65
12 GongS YangXW LiC and HeintzN (2002) Highly efficientmodification of bacterial artificial chromosomes (BACs) using novelshuttle vectors containing the R6Kgamma origin of replicationGenome Res 12 1992ndash1998
13 JamsaiD OrfordM NefedovM FucharoenS WilliamsonR andIoannouPA (2003) Targeted modification of a human beta-globin locusBAC clone using GET Recombination and an I-SceI counterselectioncassette Genomics 82 68ndash77
14 SwaminathanS EllisHM WatersLS YuD LeeECCourtDL and SharanSK (2001) Rapid engineering of bacterialartificial chromosomes using oligonucleotides Genesis 2914ndash21
15 AlperMD and AmesBN (1975) Positive selection of mutants withdeletions of the gal-chl region of the Salmonella chromosome as ascreening procedure for mutagens that cause deletions J Bacteriol121 259ndash266
16 AusubelFM BrentR KingstonRE MooreDD SeidmanJGSmithJA and StruhlK (1987) Current Protocols in Molecular BiologyGreene Publishing Associates and Wiley-Interscience John Wiley ampSons Hoboken NJ
17 BachmannBJ (1996) Derivations and genotypes of some mutantderivatives of Escherichia coli K-12 In NeidhardtFC CurtissRIIIIngrahamJL LinECC LowKB MagasanikB ReznikoffWSRileyM SchaechterM and UmbargerHE (eds) Escherichia coli andSalmonella Cellular and Molecular Biology ASM press WashingtonDC Vol 2 pp 2460ndash2488
18 GrantSG JesseeJ BloomFR and HanahanD (1990) Differentialplasmid rescue from transgenic mouse DNAs into Escherichia colimethylation-restriction mutants Proc Natl Acad Sci USA 874645ndash4649
19 OppenheimAB RattrayAJ BubunenkoM ThomasonLC andCourtDL (2004) In vivo recombineering of bacteriophage lambda byPCR fragments and single-strand oligonucleotides Virology 319185ndash189
20 HoessRH WierzbickiA and AbremskiK (1986) The role ofthe loxP spacer region in P1 site-specific recombination NucleicAcids Res 14 2287ndash2300
21 YuD SawitzkeJA EllisH and CourtDL (2003) Recombineeringwith overlapping single-stranded DNA oligonucleotides testing arecombination intermediate Proc Natl Acad Sci USA 1007207ndash7212
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 12 OF 12

by sequencing of the modified region of the 10 BAC clones(Figure 3B) All 10 sequenced clones had the desired pointmutation The effective recombination efficiency in thisexperiment however was 812 (67) since 2 of the 10 clonesalso had an additional single base pair deletion (clones 9 and11) These deletions probably occurred during oligo synthesissince we did not purify the oligos beyond desalting (19)Because of the high efficiency in this system there was noneed to pre-screen the selected colonies prior to picking forminipreps
Large BAC deletions
A drawback of using BACs for the production of transgenicmice is the frequent presence of other genes on the BAC inaddition to the gene of interest This is especially a problem forBAC complementation used in positional cloning where aBAC is tested for its ability to rescue a loss-of-function mutantphenotype by making BAC transgenic mice If complementa-tion is achieved it is impossible to know which gene on theBAC is responsible for the rescue Therefore we decided to
see whether galK selection could be used to make largespecific and clean deletions in BACs so as to remove unwantedgenes (a process called BAC trimming) As a model systemwe chose a mouse BAC from the C57BL6-derived libraryRP23 RP23-341F12 since we could obtain the BAC sequencedirectly from the UCSC genomic web browser (see Materialsand Methods) We then used PCR to amplify the galK selec-tion cassette with primers containing homology to the BACThe forward primer contained 50 bp of homology to the very 50
end of the BAC insert and this primer was combined withthree different reverse primers having 50 bp of homology topositions 50 75 and 100 kb away from the forward primerrsquoshomology (Figure 4A) SW102 bacteria containing the 341F12BAC were heat-induced and made electrocompetent and thentransformed with the three different selection cassettes fol-lowed by the selection on minimal medium with galactoseFrom the many resulting Gal+ colonies we picked four fromeach experiment and then did a SpeI restriction analysis of theBAC miniprep DNAs from them (Figure 4B) All four colon-ies from each experiment had the desired deletion (50 75 and100 kb respectively) The galK selection cassette can then be
Figure 3 Introduction of a G12D mutation in the Nras gene (A) SpeI restriction analysis of BAC miniprep DNA First lane is the unmodified CITB-50J2 Nras BACLanes 1ndash12 show digestion patterns of 12 clones counterselected for the substitution of galK with an oligo containing the GA substitution for the second position ofcodon 12 of Nras Clones 7 and 10 had internal deletions indicating that DOG resistance was achieved by spontaneous deletion and not homologous recombinationThese two clones were not analyzed further (B) Sequence analysis of a PCR product spanning the modified region from clones 1ndash6 8ndash9 and 11ndash12 All clones had theintended substitution (highlighted) However clones 9 and 11 also had an internal basepair deletion indicated by a minus (highlighted) The Nras ATG and codon 12are indicated (shadow)
PAGE 7 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
removed with an oligo using galK negative selection to cleanlydelete the galK cassette from the BAC as described in theprevious experiment (data not shown)
Insertion of a mutant loxP511 site into a BAC
Encouraged by the efficiency of this selection system wedecided to see whether we could introduce a single 34 bploxP511 site cleanly into BAC DNA using galK positiveneg-ative selection It has been previously shown that loxP andloxP511 sites cannot recombine with each other therefore the
introduced loxP511 site can only recombine with the loxP511site present in the BAC vector backbone and not the wild-typesite (Figure 5A) (20) We used galK positive selection to inserta PCR-amplified galK cassette flanked by 50 bp homologyarms to a position 95 kb away from the mutant loxP511 site inthe BAC RP23-341F12 vector backbone (Figure 5A) We thenreplaced the galK cassette using DOG counterselection with adouble-stranded 100 bp oligo containing a 34 bp loxP511 siteflanked by two 33 bp homology arms In this experiment weobserved less than a 10-fold difference in the number of col-onies on the plates from heat-induced and non-induced bac-teria suggesting fewer recombinants This lower frequency ofrecombinants when compared with the Nras G12D substitu-tion experiment is likely explained by the shorter homologyarms used in this latter experiment as it has been shown thatthe efficiency of recombination increase four orders of mag-nitude when homology length is increased from 20 to 40 bp(10) We analyzed six BAC minipreps from potential recom-binants by digesting with SpeI and comparing the restrictionpatterns with that of wild-type RP23-341F12 BAC DNA(Figure 5B) Three of six (50) colonies had exactly thesame restriction pattern as the unmodified RP23-341F12BAC suggesting that DOG resistance had selected for thedesired homologous recombination products in these threecases whereas the other three clones apparently becameDOG resistant due to the selection of BACs that carry largedeletions spanning galK
The 100 bp oligo was designed so that the loxP511 sitewould be inserted in the same orientation as the loxP511site in the BAC vector backbone To confirm that the BACswith the wild-type restriction pattern (3 5 and 6) had theloxP511 site correctly inserted we electroporated thesethree BACs into electrocompetent and L-arabinose inducedEL350 cells which carry an L-arabinose-inducible Cregene and then plated the cells on LB plates containing chlor-amphenicol to select for the BACs Two colonies generatedfrom each recombinant loxP511 clone were then tested bySpeI digestion of BAC miniprep DNA and all had the expec-ted Cre-mediated 95 kb deletion (Figure 5C) Recombinationwas confirmed by PCR using a forward primer from the BACvector backbone and a reverse primer mapping to a positiondistal to the reverse homology arm used to insert the galKcassette These primers are 95 kb apart on wild-type RP23-341F12 DNA and only 378 bp apart on CreloxP511 recom-bined DNA PCR analysis of all six clones produced a band ofthe expected length whereas no product could be amplifiedfrom unmodified RP23-341F12 BAC DNA (Figure 5D)
Source of background deletions following DOG selection
In the Nras G12D substitution and loxP511 insertion experi-ments a background of DOG-resistant bacteria which lackedthe desired mutation and instead carried unwanted deletionsthat spanned the galK selection cassette were observed Therelative ratio of colonies containing BACs with deletions wasalso higher when short homology arms were used This wasexpected since homologous recombination is more efficientwith longer homology arms (10) that would increase the rel-ative frequency of correctly targeted BACs The source of thedeletions could be l Red-mediated since proteins capable ofmediating recombination are expressed when the Red genes
Figure 4 BAC trimming using galK selection (A) Illustration of the design ofthe deletion experiment Homology arm 1 (H1) was held constant and H2 wasseparated from H1 by either 50 75 or 100 kb (B) SpeI restriction analysis ofBAC miniprep DNA from 12 clones showing deletions of 50 75 and 100 kbrespectively after the insertion of the galK selection cassette The first lane isunmodified RP23-341F12 BAC DNA which was included as a control Alltested clones had the intended deletion
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 8 OF 12
are activated Alternatively the deletions could be induced bythe large amount of oligos (200 ng) used in these experimentsor they could simply represent spontaneous deletions thatoccur at low level during normal BAC replication and thatare found by DOG selection To distinguish between thesepossibilities we repeated the DOG selection step used to insertthe loxP511 oligo into BAC RP23-341F12 DNA using fourdifferently treated bacteria samples non-induced bacteriawithout the 100 bp loxP511 oligo (Figure 6A) non-inducedbacteria with the oligo (Figure 6B) induced bacteria without
the oligo (Figure 6C) and induced bacteria with the oligo(Figure 6D) The number of resistant colonies obtainedfrom the four experiments varied by lt10-fold (data notshown) Ten colonies from each electroporation were thenanalyzed by SpeI digestion of BAC miniprep DNA and com-pared with unmodified RP23-341F12 BAC DNA (Figure 6AndashD) Unwanted galK region deletions were observed in all 10colonies for both of the non-induced samples (Figure 6A andB) These deletions are therefore unlikely to be Red-mediatedsince these samples were not heat-induced We also observed
Figure 5 Insertion of a loxP511 site (A) The location of the wild-type and mutant loxP sites in the BAC backbone are indicated along with the extra mutant loxP511site that was introduced into the BAC genomic insert via galK counterselection The 95 kb region deleted by Cre-mediated recombination between the two loxP511sites is indicated and PCR primers used to confirm the deletion are shown as small arrows (B) SpeI restriction analysis of six miniprep clones selected forthe replacement of galK with a dsDNA oligo containing the mutant loxP511 site Clones 3 5 and 6 (circles) had the same restriction pattern as the unmodifiedBAC indicating that DOG resistance occurred due to the intended homologous recombination event Clones 1 2 and 4 had large deletions and were not analyzedfurther (C) SpeI restriction analysis of BAC miniprep DNA from clones 3 5 and 6 after transformation into Cre-induced EL350 cells Two clones from each parentalclone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all clones analyzed confirming the correctinsertion of loxP511 in clones 3 5 and 6 (D) PCR analysis of the six clones from (C) with one primer mapping to the BAC backbone and the other to a position distalto the inserted loxP511 site
PAGE 9 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
deletions in both the absence and presence of oligo indicatingthat the deletions are not oligo induced We therefore concludethat these are spontaneous deletions that occur at low levelduring normal BAC replication in the DH10B background
Non-deleted colonies with the parental SpeI fingerprintwere only observed in the Red-induced sample that received
oligo (Figure 6D clones 33 and 35) These clones were sub-sequently electroporated into EL350 Cre-expressing cells toconfirm they contained the introduced loxP511 site Two col-onies from each original clone were tested by SpeI digestion ofthe BAC miniprep DNA As shown in Figure 6E each colonyhas undergone Cre-mediated deletion confirming that these
Figure 6 Same experiment as in Figure 5 with modifications as indicated at the top of each panel (A) SpeI digest of 10 minipreps from a control experiment withoutheat-induction and without the loxP511 dsDNA oligo (B) SpeI digest of 10 minipreps from a control experiment without heat-induction but with the loxP511 dsDNAoligo (C) SpeI digest of 10 minipreps from a control experiment with heat-induction but without the loxP511 dsDNA oligo (D) SpeI digest of 10 minipreps from anexperiment with heat-induction and with the loxP511 dsDNA oligo (comparable with Figure 5B) Clones with the parental digestion pattern indicating DOGresistance due to homologous recombination (clones 33 and 35 circles) are only seen in (D) DOG resistance in all other clones likely occurred due to internaldeletions of the BACs (E) SpeI restriction analysis of BAC miniprep DNA from clones 33 and 35 after transformation into Cre-induced EL350 cells Two clonesfrom each parental clone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all the clones analyzedconfirming the correct insertion of loxP511 in clones 33 and 35 (compare with Figure 5C)
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 10 OF 12
colonies were correctly targeted and contain the introducedloxP511 site (2 out of 10 clones a 20 efficiency in thisexperiment)
Generation of SW105 and SW106 cells
The two DY380-derived bacterial strains EL250 and EL350contain the defective l prophage needed for recombineering inaddition to L-arabinose inducible Flp or Cre genes respect-ively (11) Both strains have proven to be very useful for BACmodification (11) and EL350 is now used routinely for mak-ing conditional targeting vectors for ES cell knock-out experi-ments (4) To further enhance the usefulness of these strainswe decided to transfer the galK selection system into themThis was done as previously described for SW102 The galKIS2 element present in both strains was replaced with the wild-type gal promoter and the galK gene in the gal operon deletedusing DOG selection The resulting strains SW105 (Flp) andSW106 (Cre) (Table 1) were then tested to confirm that theystill contained inducible Flp and Cre genes by transformingarabinose-induced and electrocompetent cells with plasmidscontaining a neo gene flanked by Frt or loxP sites PL451 andPL452 (4) respectively Using SW105 or SW106 it is nowpossible to introduce a point mutation or an informativerestriction site into a BAC retrieve a fragment from thisBAC containing the introduced mutation(s) into a plasmidbackbone using gap repair and turn the retrieved fragmentinto a conditional targeting vectormdashall using only one bac-terial strain and only a single initial BAC transformation
DISCUSSION
Here we describe a new recombineering-based Ecoli BACmodification system that makes use of galK positive selectionfor growth on galactose minimal medium and galK negativeselection (counterselection) for growth on DOG This modi-fication system has several advantages compared with theother related BAC modification systems First the galK selec-tion cassette is small (1231 bp + homology arms) comparedwith for example the sacBndashneo cassette (3 kb) making PCRamplification and transformation into bacteria easier Twohomology arms are easily added to the galK cassette by includ-ing these sequences in the 50 ends of the primers used foramplification (see Materials and Methods) Second sincegalK is used for both selection steps mutations occurringin galK during PCR amplification will be selected againstduring positive selection significantly reducing the risk ofDOG resistance from PCR mutations in galK during negativeselection Third even when very short (33 bp) homology armswere used the frequency of recombinants among the analyzedDOG-resistant clones was still 20ndash50 To our knowledgesuccessful BAC modification with such short homology armshas never been reported Finally since galK recombineering isso efficient little screening is required following selectionreducing the overall hands-on-time to a minimum galK selec-tion requires growth on minimal medium in both selectionsteps although perhaps not used routinely in most molecularbiology laboratories where BAC modification is needed theseplates are fairly simple to make and should not prevent anyonefrom using this method
For all such counterselection schemes there will bebackground since during negative selection any event leadingto the loss of the counterselectable marker will result in sur-vival In our system virtually all background appears to resultfrom deletions that span the inserted galK gene These dele-tions likely occur during BAC replication in the 45 h out-growth phase since we have shown that these deletions occurindependently of Red induction and in the absence or presenceof oligo It is well established that in recA defective bacteriaBACs are very stable compared with other large insert vectorssuch as yeast artificial chromosomes (YACs) However usingcounterselection rare spontaneous deletions are seen becauseof the strong selection force This spontaneous deletion back-ground is not a problem however due to the high frequency ofhomologous recombination obtained with the defective lprophage system
Increasing the length of homology arms used for recombin-eering will reduce the relative number of background deletionsobserved following negative selection since the percentage ofcolonies containing BACs that eliminated galK by homolog-ous recombination will be increased Oligos with longerhomology arms can be produced by annealing two oligoswith overlapping 30 ends (21) These overlapping oligos (50
single strand overhangs) are then filled in by DNA polymerasein vivo (or in vitro) and subsequently serve as the substrate forthe l Red proteins exo and bet Longer homology arms canalso be added by traditional cloning using restriction enzymesand DNA ligase (4)
A number of uses can be imagined for the strains describedhere In principle our strains can be used to make virtually anykind of BAC modification one can imagine including pointmutations or clean deletions or insertions of everything fromcDNA to loxP sites or small epitope tags BAC trimming thespecific deletion of BAC DNA flanking a gene of interest couldalso be used to remove genes from BACs prior to making BACtransgenic mice so that only the gene of interest on the BAC isanalyzed This should be of particular interest for BAC rescueexperiments in positional cloning Of course trimming andmodification can be combined since the galK selection cassettecan be recycled so that the same BAC can be modified severaltimes BAC modification could also be the starting point forconstructing a gene-targeting vector to allow for more soph-isticated gene targeting in mouse ES cells The desired muta-tion(s) could first be created in the BAC followed by retrievalinto a plasmid vector containing a negative-selection markerlike thymidine kinase using gap repair (4) Finally a neo mar-ker could be introduced to allow for positive selection in EScells Alternatively the retrieved fragment could be modifiedso that the end result is a conditional targeting vector StrainSW106 which in addition to galK selection can be induced toexpress Cre recombinase makes it possible to perform all ofthe steps needed to construct a conditional targeting vectorstarting with only a single BAC transformation step
Furthermore in experiments where BAC transgenic miceare used to analyze the effect of deleting long-range regulatorsof gene expression galK DOG counterselection can even beused in the absence of an oligo to generate a series of dele-tions around the galK insertion site and the effect on generegulation studied
The Flp or Cre ORFs contained in strains SW105 andSW106 respectively can also be replaced for any desired
PAGE 11 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
gene thus creating tight arabinose-inducible expression forany gene of interest This is done by first replacing the CreFlpe ORF with galK and select for Gal+ recombinants ThegalK cassette is then replaced by the ORF of the gene ofinterest by DOG selection for Gal recombinants
Finally the strains described here should be very useful forgenetic manipulation not only of BACs but also the Ecoligenome itself
All recombineering reagents discussed in this workare freely available upon request To obtain these materialsplease follow the directions listed on our website (httprecombineeringncifcrfgov) Detailed protocols for recom-bineering including galK selection can also be downloadeddirectly from our website
ACKNOWLEDGEMENTS
We thank Allen Kane and Carolyn Whistler from ScientificPublications Graphics amp Media NCI-Frederick for help withillustrations and figure preparation Funding for this research aswell as the Open Access publication charges was provided byDHHS NIH NCI
REFERENCES
1 ShizuyaH BirrenB KimUJ MancinoV SlepakTTachiiriY and SimonM (1992) Cloning and stable maintenance of300-kilobase-pair fragments of human DNA in Escherichia coli using anF-factor-based vector Proc Natl Acad Sci USA 89 8794ndash8797
2 YangXW ModelP and HeintzN (1997) Homologous recombinationbased modification in Escherichia coli and germline transmission intransgenic mice of a bacterial artificial chromosome Nat Biotechnol15 859ndash865
3 AntochMP SongEJ ChangAM VitaternaMH ZhaoYWilsbacherLD SangoramAM KingDP PintoLH andTakahashiJS (1997) Functional identification of the mouse circadianClock gene by transgenic BAC rescue Cell 89 655ndash667
4 LiuP JenkinsNA and CopelandNG (2003) A highly efficientrecombineering-based method for generating conditional knockoutmutations Genome Res 13 476ndash484
5 Cotta-de-AlmeidaV SchonhoffS ShibataT LeiterA andSnapperSB (2003) A new method for rapidly generating gene-targetingvectors by engineering BACs through homologous recombination inbacteria Genome Res 13 2190ndash2194
6 CopelandNG JenkinsNA and CourtDL (2001) Recombineeringa powerful new tool for mouse functional genomics Nature RevGenet 2 769ndash779
7 CourtDL SawitzkeJA and ThomasonLC (2002) Geneticengineering using homologous recombination Annu Rev Genet 36361ndash388
8 ZhangY BuchholzF MuyrersJP and StewartAF (1998) A newlogic for DNA engineering using recombination in Escherichia coliNature Genet 20 123ndash128
9 MuyrersJP ZhangY TestaG and StewartAF (1999) Rapidmodification of bacterial artificial chromosomes by ET-recombinationNucleic Acids Res 27 1555ndash1557
10 YuD EllisHM LeeEC JenkinsNA CopelandNG andCourtDL (2000) An efficient recombination system for chromosomeengineering in Escherichia coli Proc Natl Acad Sci USA 975978ndash5983
11 LeeEC YuD Martinez de VelascoJ TessarolloL SwingDACourtDL JenkinsNA and CopelandNG (2001) A highly efficientEscherichia coli-based chromosome engineering system adapted forrecombinogenic targeting and subcloning of BAC DNA Genomics73 56ndash65
12 GongS YangXW LiC and HeintzN (2002) Highly efficientmodification of bacterial artificial chromosomes (BACs) using novelshuttle vectors containing the R6Kgamma origin of replicationGenome Res 12 1992ndash1998
13 JamsaiD OrfordM NefedovM FucharoenS WilliamsonR andIoannouPA (2003) Targeted modification of a human beta-globin locusBAC clone using GET Recombination and an I-SceI counterselectioncassette Genomics 82 68ndash77
14 SwaminathanS EllisHM WatersLS YuD LeeECCourtDL and SharanSK (2001) Rapid engineering of bacterialartificial chromosomes using oligonucleotides Genesis 2914ndash21
15 AlperMD and AmesBN (1975) Positive selection of mutants withdeletions of the gal-chl region of the Salmonella chromosome as ascreening procedure for mutagens that cause deletions J Bacteriol121 259ndash266
16 AusubelFM BrentR KingstonRE MooreDD SeidmanJGSmithJA and StruhlK (1987) Current Protocols in Molecular BiologyGreene Publishing Associates and Wiley-Interscience John Wiley ampSons Hoboken NJ
17 BachmannBJ (1996) Derivations and genotypes of some mutantderivatives of Escherichia coli K-12 In NeidhardtFC CurtissRIIIIngrahamJL LinECC LowKB MagasanikB ReznikoffWSRileyM SchaechterM and UmbargerHE (eds) Escherichia coli andSalmonella Cellular and Molecular Biology ASM press WashingtonDC Vol 2 pp 2460ndash2488
18 GrantSG JesseeJ BloomFR and HanahanD (1990) Differentialplasmid rescue from transgenic mouse DNAs into Escherichia colimethylation-restriction mutants Proc Natl Acad Sci USA 874645ndash4649
19 OppenheimAB RattrayAJ BubunenkoM ThomasonLC andCourtDL (2004) In vivo recombineering of bacteriophage lambda byPCR fragments and single-strand oligonucleotides Virology 319185ndash189
20 HoessRH WierzbickiA and AbremskiK (1986) The role ofthe loxP spacer region in P1 site-specific recombination NucleicAcids Res 14 2287ndash2300
21 YuD SawitzkeJA EllisH and CourtDL (2003) Recombineeringwith overlapping single-stranded DNA oligonucleotides testing arecombination intermediate Proc Natl Acad Sci USA 1007207ndash7212
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 12 OF 12

removed with an oligo using galK negative selection to cleanlydelete the galK cassette from the BAC as described in theprevious experiment (data not shown)
Insertion of a mutant loxP511 site into a BAC
Encouraged by the efficiency of this selection system wedecided to see whether we could introduce a single 34 bploxP511 site cleanly into BAC DNA using galK positiveneg-ative selection It has been previously shown that loxP andloxP511 sites cannot recombine with each other therefore the
introduced loxP511 site can only recombine with the loxP511site present in the BAC vector backbone and not the wild-typesite (Figure 5A) (20) We used galK positive selection to inserta PCR-amplified galK cassette flanked by 50 bp homologyarms to a position 95 kb away from the mutant loxP511 site inthe BAC RP23-341F12 vector backbone (Figure 5A) We thenreplaced the galK cassette using DOG counterselection with adouble-stranded 100 bp oligo containing a 34 bp loxP511 siteflanked by two 33 bp homology arms In this experiment weobserved less than a 10-fold difference in the number of col-onies on the plates from heat-induced and non-induced bac-teria suggesting fewer recombinants This lower frequency ofrecombinants when compared with the Nras G12D substitu-tion experiment is likely explained by the shorter homologyarms used in this latter experiment as it has been shown thatthe efficiency of recombination increase four orders of mag-nitude when homology length is increased from 20 to 40 bp(10) We analyzed six BAC minipreps from potential recom-binants by digesting with SpeI and comparing the restrictionpatterns with that of wild-type RP23-341F12 BAC DNA(Figure 5B) Three of six (50) colonies had exactly thesame restriction pattern as the unmodified RP23-341F12BAC suggesting that DOG resistance had selected for thedesired homologous recombination products in these threecases whereas the other three clones apparently becameDOG resistant due to the selection of BACs that carry largedeletions spanning galK
The 100 bp oligo was designed so that the loxP511 sitewould be inserted in the same orientation as the loxP511site in the BAC vector backbone To confirm that the BACswith the wild-type restriction pattern (3 5 and 6) had theloxP511 site correctly inserted we electroporated thesethree BACs into electrocompetent and L-arabinose inducedEL350 cells which carry an L-arabinose-inducible Cregene and then plated the cells on LB plates containing chlor-amphenicol to select for the BACs Two colonies generatedfrom each recombinant loxP511 clone were then tested bySpeI digestion of BAC miniprep DNA and all had the expec-ted Cre-mediated 95 kb deletion (Figure 5C) Recombinationwas confirmed by PCR using a forward primer from the BACvector backbone and a reverse primer mapping to a positiondistal to the reverse homology arm used to insert the galKcassette These primers are 95 kb apart on wild-type RP23-341F12 DNA and only 378 bp apart on CreloxP511 recom-bined DNA PCR analysis of all six clones produced a band ofthe expected length whereas no product could be amplifiedfrom unmodified RP23-341F12 BAC DNA (Figure 5D)
Source of background deletions following DOG selection
In the Nras G12D substitution and loxP511 insertion experi-ments a background of DOG-resistant bacteria which lackedthe desired mutation and instead carried unwanted deletionsthat spanned the galK selection cassette were observed Therelative ratio of colonies containing BACs with deletions wasalso higher when short homology arms were used This wasexpected since homologous recombination is more efficientwith longer homology arms (10) that would increase the rel-ative frequency of correctly targeted BACs The source of thedeletions could be l Red-mediated since proteins capable ofmediating recombination are expressed when the Red genes
Figure 4 BAC trimming using galK selection (A) Illustration of the design ofthe deletion experiment Homology arm 1 (H1) was held constant and H2 wasseparated from H1 by either 50 75 or 100 kb (B) SpeI restriction analysis ofBAC miniprep DNA from 12 clones showing deletions of 50 75 and 100 kbrespectively after the insertion of the galK selection cassette The first lane isunmodified RP23-341F12 BAC DNA which was included as a control Alltested clones had the intended deletion
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 8 OF 12
are activated Alternatively the deletions could be induced bythe large amount of oligos (200 ng) used in these experimentsor they could simply represent spontaneous deletions thatoccur at low level during normal BAC replication and thatare found by DOG selection To distinguish between thesepossibilities we repeated the DOG selection step used to insertthe loxP511 oligo into BAC RP23-341F12 DNA using fourdifferently treated bacteria samples non-induced bacteriawithout the 100 bp loxP511 oligo (Figure 6A) non-inducedbacteria with the oligo (Figure 6B) induced bacteria without
the oligo (Figure 6C) and induced bacteria with the oligo(Figure 6D) The number of resistant colonies obtainedfrom the four experiments varied by lt10-fold (data notshown) Ten colonies from each electroporation were thenanalyzed by SpeI digestion of BAC miniprep DNA and com-pared with unmodified RP23-341F12 BAC DNA (Figure 6AndashD) Unwanted galK region deletions were observed in all 10colonies for both of the non-induced samples (Figure 6A andB) These deletions are therefore unlikely to be Red-mediatedsince these samples were not heat-induced We also observed
Figure 5 Insertion of a loxP511 site (A) The location of the wild-type and mutant loxP sites in the BAC backbone are indicated along with the extra mutant loxP511site that was introduced into the BAC genomic insert via galK counterselection The 95 kb region deleted by Cre-mediated recombination between the two loxP511sites is indicated and PCR primers used to confirm the deletion are shown as small arrows (B) SpeI restriction analysis of six miniprep clones selected forthe replacement of galK with a dsDNA oligo containing the mutant loxP511 site Clones 3 5 and 6 (circles) had the same restriction pattern as the unmodifiedBAC indicating that DOG resistance occurred due to the intended homologous recombination event Clones 1 2 and 4 had large deletions and were not analyzedfurther (C) SpeI restriction analysis of BAC miniprep DNA from clones 3 5 and 6 after transformation into Cre-induced EL350 cells Two clones from each parentalclone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all clones analyzed confirming the correctinsertion of loxP511 in clones 3 5 and 6 (D) PCR analysis of the six clones from (C) with one primer mapping to the BAC backbone and the other to a position distalto the inserted loxP511 site
PAGE 9 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
deletions in both the absence and presence of oligo indicatingthat the deletions are not oligo induced We therefore concludethat these are spontaneous deletions that occur at low levelduring normal BAC replication in the DH10B background
Non-deleted colonies with the parental SpeI fingerprintwere only observed in the Red-induced sample that received
oligo (Figure 6D clones 33 and 35) These clones were sub-sequently electroporated into EL350 Cre-expressing cells toconfirm they contained the introduced loxP511 site Two col-onies from each original clone were tested by SpeI digestion ofthe BAC miniprep DNA As shown in Figure 6E each colonyhas undergone Cre-mediated deletion confirming that these
Figure 6 Same experiment as in Figure 5 with modifications as indicated at the top of each panel (A) SpeI digest of 10 minipreps from a control experiment withoutheat-induction and without the loxP511 dsDNA oligo (B) SpeI digest of 10 minipreps from a control experiment without heat-induction but with the loxP511 dsDNAoligo (C) SpeI digest of 10 minipreps from a control experiment with heat-induction but without the loxP511 dsDNA oligo (D) SpeI digest of 10 minipreps from anexperiment with heat-induction and with the loxP511 dsDNA oligo (comparable with Figure 5B) Clones with the parental digestion pattern indicating DOGresistance due to homologous recombination (clones 33 and 35 circles) are only seen in (D) DOG resistance in all other clones likely occurred due to internaldeletions of the BACs (E) SpeI restriction analysis of BAC miniprep DNA from clones 33 and 35 after transformation into Cre-induced EL350 cells Two clonesfrom each parental clone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all the clones analyzedconfirming the correct insertion of loxP511 in clones 33 and 35 (compare with Figure 5C)
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 10 OF 12
colonies were correctly targeted and contain the introducedloxP511 site (2 out of 10 clones a 20 efficiency in thisexperiment)
Generation of SW105 and SW106 cells
The two DY380-derived bacterial strains EL250 and EL350contain the defective l prophage needed for recombineering inaddition to L-arabinose inducible Flp or Cre genes respect-ively (11) Both strains have proven to be very useful for BACmodification (11) and EL350 is now used routinely for mak-ing conditional targeting vectors for ES cell knock-out experi-ments (4) To further enhance the usefulness of these strainswe decided to transfer the galK selection system into themThis was done as previously described for SW102 The galKIS2 element present in both strains was replaced with the wild-type gal promoter and the galK gene in the gal operon deletedusing DOG selection The resulting strains SW105 (Flp) andSW106 (Cre) (Table 1) were then tested to confirm that theystill contained inducible Flp and Cre genes by transformingarabinose-induced and electrocompetent cells with plasmidscontaining a neo gene flanked by Frt or loxP sites PL451 andPL452 (4) respectively Using SW105 or SW106 it is nowpossible to introduce a point mutation or an informativerestriction site into a BAC retrieve a fragment from thisBAC containing the introduced mutation(s) into a plasmidbackbone using gap repair and turn the retrieved fragmentinto a conditional targeting vectormdashall using only one bac-terial strain and only a single initial BAC transformation
DISCUSSION
Here we describe a new recombineering-based Ecoli BACmodification system that makes use of galK positive selectionfor growth on galactose minimal medium and galK negativeselection (counterselection) for growth on DOG This modi-fication system has several advantages compared with theother related BAC modification systems First the galK selec-tion cassette is small (1231 bp + homology arms) comparedwith for example the sacBndashneo cassette (3 kb) making PCRamplification and transformation into bacteria easier Twohomology arms are easily added to the galK cassette by includ-ing these sequences in the 50 ends of the primers used foramplification (see Materials and Methods) Second sincegalK is used for both selection steps mutations occurringin galK during PCR amplification will be selected againstduring positive selection significantly reducing the risk ofDOG resistance from PCR mutations in galK during negativeselection Third even when very short (33 bp) homology armswere used the frequency of recombinants among the analyzedDOG-resistant clones was still 20ndash50 To our knowledgesuccessful BAC modification with such short homology armshas never been reported Finally since galK recombineering isso efficient little screening is required following selectionreducing the overall hands-on-time to a minimum galK selec-tion requires growth on minimal medium in both selectionsteps although perhaps not used routinely in most molecularbiology laboratories where BAC modification is needed theseplates are fairly simple to make and should not prevent anyonefrom using this method
For all such counterselection schemes there will bebackground since during negative selection any event leadingto the loss of the counterselectable marker will result in sur-vival In our system virtually all background appears to resultfrom deletions that span the inserted galK gene These dele-tions likely occur during BAC replication in the 45 h out-growth phase since we have shown that these deletions occurindependently of Red induction and in the absence or presenceof oligo It is well established that in recA defective bacteriaBACs are very stable compared with other large insert vectorssuch as yeast artificial chromosomes (YACs) However usingcounterselection rare spontaneous deletions are seen becauseof the strong selection force This spontaneous deletion back-ground is not a problem however due to the high frequency ofhomologous recombination obtained with the defective lprophage system
Increasing the length of homology arms used for recombin-eering will reduce the relative number of background deletionsobserved following negative selection since the percentage ofcolonies containing BACs that eliminated galK by homolog-ous recombination will be increased Oligos with longerhomology arms can be produced by annealing two oligoswith overlapping 30 ends (21) These overlapping oligos (50
single strand overhangs) are then filled in by DNA polymerasein vivo (or in vitro) and subsequently serve as the substrate forthe l Red proteins exo and bet Longer homology arms canalso be added by traditional cloning using restriction enzymesand DNA ligase (4)
A number of uses can be imagined for the strains describedhere In principle our strains can be used to make virtually anykind of BAC modification one can imagine including pointmutations or clean deletions or insertions of everything fromcDNA to loxP sites or small epitope tags BAC trimming thespecific deletion of BAC DNA flanking a gene of interest couldalso be used to remove genes from BACs prior to making BACtransgenic mice so that only the gene of interest on the BAC isanalyzed This should be of particular interest for BAC rescueexperiments in positional cloning Of course trimming andmodification can be combined since the galK selection cassettecan be recycled so that the same BAC can be modified severaltimes BAC modification could also be the starting point forconstructing a gene-targeting vector to allow for more soph-isticated gene targeting in mouse ES cells The desired muta-tion(s) could first be created in the BAC followed by retrievalinto a plasmid vector containing a negative-selection markerlike thymidine kinase using gap repair (4) Finally a neo mar-ker could be introduced to allow for positive selection in EScells Alternatively the retrieved fragment could be modifiedso that the end result is a conditional targeting vector StrainSW106 which in addition to galK selection can be induced toexpress Cre recombinase makes it possible to perform all ofthe steps needed to construct a conditional targeting vectorstarting with only a single BAC transformation step
Furthermore in experiments where BAC transgenic miceare used to analyze the effect of deleting long-range regulatorsof gene expression galK DOG counterselection can even beused in the absence of an oligo to generate a series of dele-tions around the galK insertion site and the effect on generegulation studied
The Flp or Cre ORFs contained in strains SW105 andSW106 respectively can also be replaced for any desired
PAGE 11 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
gene thus creating tight arabinose-inducible expression forany gene of interest This is done by first replacing the CreFlpe ORF with galK and select for Gal+ recombinants ThegalK cassette is then replaced by the ORF of the gene ofinterest by DOG selection for Gal recombinants
Finally the strains described here should be very useful forgenetic manipulation not only of BACs but also the Ecoligenome itself
All recombineering reagents discussed in this workare freely available upon request To obtain these materialsplease follow the directions listed on our website (httprecombineeringncifcrfgov) Detailed protocols for recom-bineering including galK selection can also be downloadeddirectly from our website
ACKNOWLEDGEMENTS
We thank Allen Kane and Carolyn Whistler from ScientificPublications Graphics amp Media NCI-Frederick for help withillustrations and figure preparation Funding for this research aswell as the Open Access publication charges was provided byDHHS NIH NCI
REFERENCES
1 ShizuyaH BirrenB KimUJ MancinoV SlepakTTachiiriY and SimonM (1992) Cloning and stable maintenance of300-kilobase-pair fragments of human DNA in Escherichia coli using anF-factor-based vector Proc Natl Acad Sci USA 89 8794ndash8797
2 YangXW ModelP and HeintzN (1997) Homologous recombinationbased modification in Escherichia coli and germline transmission intransgenic mice of a bacterial artificial chromosome Nat Biotechnol15 859ndash865
3 AntochMP SongEJ ChangAM VitaternaMH ZhaoYWilsbacherLD SangoramAM KingDP PintoLH andTakahashiJS (1997) Functional identification of the mouse circadianClock gene by transgenic BAC rescue Cell 89 655ndash667
4 LiuP JenkinsNA and CopelandNG (2003) A highly efficientrecombineering-based method for generating conditional knockoutmutations Genome Res 13 476ndash484
5 Cotta-de-AlmeidaV SchonhoffS ShibataT LeiterA andSnapperSB (2003) A new method for rapidly generating gene-targetingvectors by engineering BACs through homologous recombination inbacteria Genome Res 13 2190ndash2194
6 CopelandNG JenkinsNA and CourtDL (2001) Recombineeringa powerful new tool for mouse functional genomics Nature RevGenet 2 769ndash779
7 CourtDL SawitzkeJA and ThomasonLC (2002) Geneticengineering using homologous recombination Annu Rev Genet 36361ndash388
8 ZhangY BuchholzF MuyrersJP and StewartAF (1998) A newlogic for DNA engineering using recombination in Escherichia coliNature Genet 20 123ndash128
9 MuyrersJP ZhangY TestaG and StewartAF (1999) Rapidmodification of bacterial artificial chromosomes by ET-recombinationNucleic Acids Res 27 1555ndash1557
10 YuD EllisHM LeeEC JenkinsNA CopelandNG andCourtDL (2000) An efficient recombination system for chromosomeengineering in Escherichia coli Proc Natl Acad Sci USA 975978ndash5983
11 LeeEC YuD Martinez de VelascoJ TessarolloL SwingDACourtDL JenkinsNA and CopelandNG (2001) A highly efficientEscherichia coli-based chromosome engineering system adapted forrecombinogenic targeting and subcloning of BAC DNA Genomics73 56ndash65
12 GongS YangXW LiC and HeintzN (2002) Highly efficientmodification of bacterial artificial chromosomes (BACs) using novelshuttle vectors containing the R6Kgamma origin of replicationGenome Res 12 1992ndash1998
13 JamsaiD OrfordM NefedovM FucharoenS WilliamsonR andIoannouPA (2003) Targeted modification of a human beta-globin locusBAC clone using GET Recombination and an I-SceI counterselectioncassette Genomics 82 68ndash77
14 SwaminathanS EllisHM WatersLS YuD LeeECCourtDL and SharanSK (2001) Rapid engineering of bacterialartificial chromosomes using oligonucleotides Genesis 2914ndash21
15 AlperMD and AmesBN (1975) Positive selection of mutants withdeletions of the gal-chl region of the Salmonella chromosome as ascreening procedure for mutagens that cause deletions J Bacteriol121 259ndash266
16 AusubelFM BrentR KingstonRE MooreDD SeidmanJGSmithJA and StruhlK (1987) Current Protocols in Molecular BiologyGreene Publishing Associates and Wiley-Interscience John Wiley ampSons Hoboken NJ
17 BachmannBJ (1996) Derivations and genotypes of some mutantderivatives of Escherichia coli K-12 In NeidhardtFC CurtissRIIIIngrahamJL LinECC LowKB MagasanikB ReznikoffWSRileyM SchaechterM and UmbargerHE (eds) Escherichia coli andSalmonella Cellular and Molecular Biology ASM press WashingtonDC Vol 2 pp 2460ndash2488
18 GrantSG JesseeJ BloomFR and HanahanD (1990) Differentialplasmid rescue from transgenic mouse DNAs into Escherichia colimethylation-restriction mutants Proc Natl Acad Sci USA 874645ndash4649
19 OppenheimAB RattrayAJ BubunenkoM ThomasonLC andCourtDL (2004) In vivo recombineering of bacteriophage lambda byPCR fragments and single-strand oligonucleotides Virology 319185ndash189
20 HoessRH WierzbickiA and AbremskiK (1986) The role ofthe loxP spacer region in P1 site-specific recombination NucleicAcids Res 14 2287ndash2300
21 YuD SawitzkeJA EllisH and CourtDL (2003) Recombineeringwith overlapping single-stranded DNA oligonucleotides testing arecombination intermediate Proc Natl Acad Sci USA 1007207ndash7212
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 12 OF 12
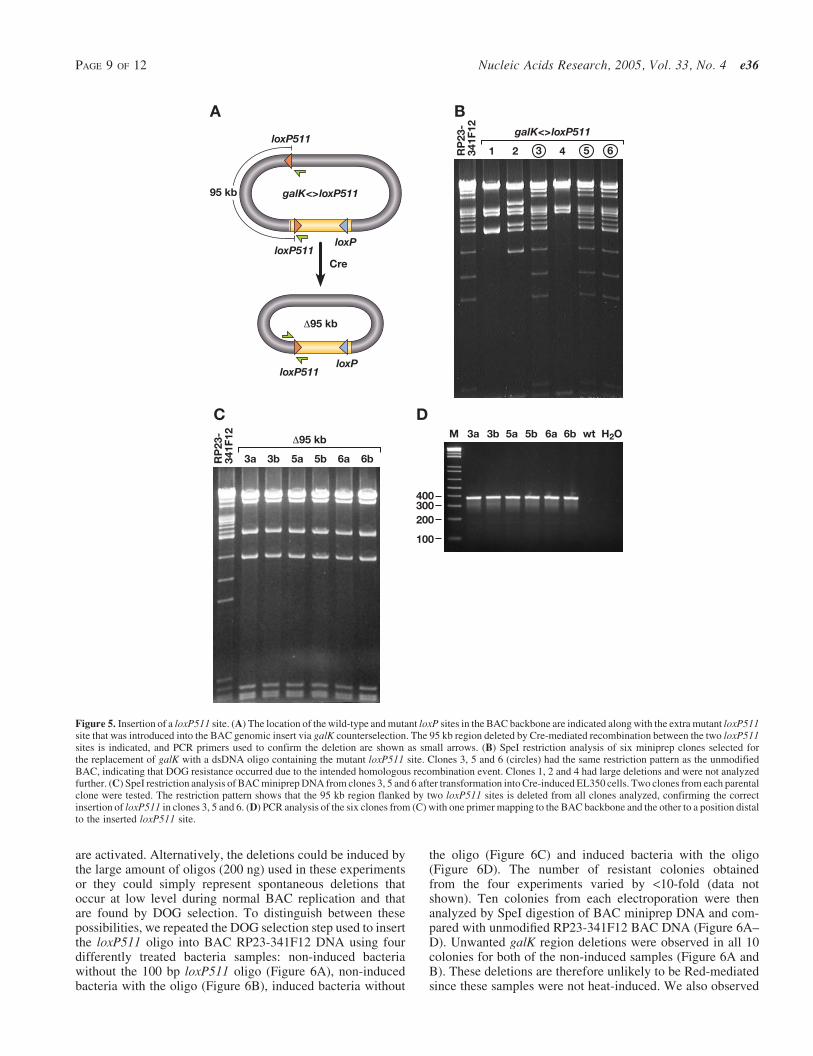
are activated Alternatively the deletions could be induced bythe large amount of oligos (200 ng) used in these experimentsor they could simply represent spontaneous deletions thatoccur at low level during normal BAC replication and thatare found by DOG selection To distinguish between thesepossibilities we repeated the DOG selection step used to insertthe loxP511 oligo into BAC RP23-341F12 DNA using fourdifferently treated bacteria samples non-induced bacteriawithout the 100 bp loxP511 oligo (Figure 6A) non-inducedbacteria with the oligo (Figure 6B) induced bacteria without
the oligo (Figure 6C) and induced bacteria with the oligo(Figure 6D) The number of resistant colonies obtainedfrom the four experiments varied by lt10-fold (data notshown) Ten colonies from each electroporation were thenanalyzed by SpeI digestion of BAC miniprep DNA and com-pared with unmodified RP23-341F12 BAC DNA (Figure 6AndashD) Unwanted galK region deletions were observed in all 10colonies for both of the non-induced samples (Figure 6A andB) These deletions are therefore unlikely to be Red-mediatedsince these samples were not heat-induced We also observed
Figure 5 Insertion of a loxP511 site (A) The location of the wild-type and mutant loxP sites in the BAC backbone are indicated along with the extra mutant loxP511site that was introduced into the BAC genomic insert via galK counterselection The 95 kb region deleted by Cre-mediated recombination between the two loxP511sites is indicated and PCR primers used to confirm the deletion are shown as small arrows (B) SpeI restriction analysis of six miniprep clones selected forthe replacement of galK with a dsDNA oligo containing the mutant loxP511 site Clones 3 5 and 6 (circles) had the same restriction pattern as the unmodifiedBAC indicating that DOG resistance occurred due to the intended homologous recombination event Clones 1 2 and 4 had large deletions and were not analyzedfurther (C) SpeI restriction analysis of BAC miniprep DNA from clones 3 5 and 6 after transformation into Cre-induced EL350 cells Two clones from each parentalclone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all clones analyzed confirming the correctinsertion of loxP511 in clones 3 5 and 6 (D) PCR analysis of the six clones from (C) with one primer mapping to the BAC backbone and the other to a position distalto the inserted loxP511 site
PAGE 9 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
deletions in both the absence and presence of oligo indicatingthat the deletions are not oligo induced We therefore concludethat these are spontaneous deletions that occur at low levelduring normal BAC replication in the DH10B background
Non-deleted colonies with the parental SpeI fingerprintwere only observed in the Red-induced sample that received
oligo (Figure 6D clones 33 and 35) These clones were sub-sequently electroporated into EL350 Cre-expressing cells toconfirm they contained the introduced loxP511 site Two col-onies from each original clone were tested by SpeI digestion ofthe BAC miniprep DNA As shown in Figure 6E each colonyhas undergone Cre-mediated deletion confirming that these
Figure 6 Same experiment as in Figure 5 with modifications as indicated at the top of each panel (A) SpeI digest of 10 minipreps from a control experiment withoutheat-induction and without the loxP511 dsDNA oligo (B) SpeI digest of 10 minipreps from a control experiment without heat-induction but with the loxP511 dsDNAoligo (C) SpeI digest of 10 minipreps from a control experiment with heat-induction but without the loxP511 dsDNA oligo (D) SpeI digest of 10 minipreps from anexperiment with heat-induction and with the loxP511 dsDNA oligo (comparable with Figure 5B) Clones with the parental digestion pattern indicating DOGresistance due to homologous recombination (clones 33 and 35 circles) are only seen in (D) DOG resistance in all other clones likely occurred due to internaldeletions of the BACs (E) SpeI restriction analysis of BAC miniprep DNA from clones 33 and 35 after transformation into Cre-induced EL350 cells Two clonesfrom each parental clone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all the clones analyzedconfirming the correct insertion of loxP511 in clones 33 and 35 (compare with Figure 5C)
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 10 OF 12
colonies were correctly targeted and contain the introducedloxP511 site (2 out of 10 clones a 20 efficiency in thisexperiment)
Generation of SW105 and SW106 cells
The two DY380-derived bacterial strains EL250 and EL350contain the defective l prophage needed for recombineering inaddition to L-arabinose inducible Flp or Cre genes respect-ively (11) Both strains have proven to be very useful for BACmodification (11) and EL350 is now used routinely for mak-ing conditional targeting vectors for ES cell knock-out experi-ments (4) To further enhance the usefulness of these strainswe decided to transfer the galK selection system into themThis was done as previously described for SW102 The galKIS2 element present in both strains was replaced with the wild-type gal promoter and the galK gene in the gal operon deletedusing DOG selection The resulting strains SW105 (Flp) andSW106 (Cre) (Table 1) were then tested to confirm that theystill contained inducible Flp and Cre genes by transformingarabinose-induced and electrocompetent cells with plasmidscontaining a neo gene flanked by Frt or loxP sites PL451 andPL452 (4) respectively Using SW105 or SW106 it is nowpossible to introduce a point mutation or an informativerestriction site into a BAC retrieve a fragment from thisBAC containing the introduced mutation(s) into a plasmidbackbone using gap repair and turn the retrieved fragmentinto a conditional targeting vectormdashall using only one bac-terial strain and only a single initial BAC transformation
DISCUSSION
Here we describe a new recombineering-based Ecoli BACmodification system that makes use of galK positive selectionfor growth on galactose minimal medium and galK negativeselection (counterselection) for growth on DOG This modi-fication system has several advantages compared with theother related BAC modification systems First the galK selec-tion cassette is small (1231 bp + homology arms) comparedwith for example the sacBndashneo cassette (3 kb) making PCRamplification and transformation into bacteria easier Twohomology arms are easily added to the galK cassette by includ-ing these sequences in the 50 ends of the primers used foramplification (see Materials and Methods) Second sincegalK is used for both selection steps mutations occurringin galK during PCR amplification will be selected againstduring positive selection significantly reducing the risk ofDOG resistance from PCR mutations in galK during negativeselection Third even when very short (33 bp) homology armswere used the frequency of recombinants among the analyzedDOG-resistant clones was still 20ndash50 To our knowledgesuccessful BAC modification with such short homology armshas never been reported Finally since galK recombineering isso efficient little screening is required following selectionreducing the overall hands-on-time to a minimum galK selec-tion requires growth on minimal medium in both selectionsteps although perhaps not used routinely in most molecularbiology laboratories where BAC modification is needed theseplates are fairly simple to make and should not prevent anyonefrom using this method
For all such counterselection schemes there will bebackground since during negative selection any event leadingto the loss of the counterselectable marker will result in sur-vival In our system virtually all background appears to resultfrom deletions that span the inserted galK gene These dele-tions likely occur during BAC replication in the 45 h out-growth phase since we have shown that these deletions occurindependently of Red induction and in the absence or presenceof oligo It is well established that in recA defective bacteriaBACs are very stable compared with other large insert vectorssuch as yeast artificial chromosomes (YACs) However usingcounterselection rare spontaneous deletions are seen becauseof the strong selection force This spontaneous deletion back-ground is not a problem however due to the high frequency ofhomologous recombination obtained with the defective lprophage system
Increasing the length of homology arms used for recombin-eering will reduce the relative number of background deletionsobserved following negative selection since the percentage ofcolonies containing BACs that eliminated galK by homolog-ous recombination will be increased Oligos with longerhomology arms can be produced by annealing two oligoswith overlapping 30 ends (21) These overlapping oligos (50
single strand overhangs) are then filled in by DNA polymerasein vivo (or in vitro) and subsequently serve as the substrate forthe l Red proteins exo and bet Longer homology arms canalso be added by traditional cloning using restriction enzymesand DNA ligase (4)
A number of uses can be imagined for the strains describedhere In principle our strains can be used to make virtually anykind of BAC modification one can imagine including pointmutations or clean deletions or insertions of everything fromcDNA to loxP sites or small epitope tags BAC trimming thespecific deletion of BAC DNA flanking a gene of interest couldalso be used to remove genes from BACs prior to making BACtransgenic mice so that only the gene of interest on the BAC isanalyzed This should be of particular interest for BAC rescueexperiments in positional cloning Of course trimming andmodification can be combined since the galK selection cassettecan be recycled so that the same BAC can be modified severaltimes BAC modification could also be the starting point forconstructing a gene-targeting vector to allow for more soph-isticated gene targeting in mouse ES cells The desired muta-tion(s) could first be created in the BAC followed by retrievalinto a plasmid vector containing a negative-selection markerlike thymidine kinase using gap repair (4) Finally a neo mar-ker could be introduced to allow for positive selection in EScells Alternatively the retrieved fragment could be modifiedso that the end result is a conditional targeting vector StrainSW106 which in addition to galK selection can be induced toexpress Cre recombinase makes it possible to perform all ofthe steps needed to construct a conditional targeting vectorstarting with only a single BAC transformation step
Furthermore in experiments where BAC transgenic miceare used to analyze the effect of deleting long-range regulatorsof gene expression galK DOG counterselection can even beused in the absence of an oligo to generate a series of dele-tions around the galK insertion site and the effect on generegulation studied
The Flp or Cre ORFs contained in strains SW105 andSW106 respectively can also be replaced for any desired
PAGE 11 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
gene thus creating tight arabinose-inducible expression forany gene of interest This is done by first replacing the CreFlpe ORF with galK and select for Gal+ recombinants ThegalK cassette is then replaced by the ORF of the gene ofinterest by DOG selection for Gal recombinants
Finally the strains described here should be very useful forgenetic manipulation not only of BACs but also the Ecoligenome itself
All recombineering reagents discussed in this workare freely available upon request To obtain these materialsplease follow the directions listed on our website (httprecombineeringncifcrfgov) Detailed protocols for recom-bineering including galK selection can also be downloadeddirectly from our website
ACKNOWLEDGEMENTS
We thank Allen Kane and Carolyn Whistler from ScientificPublications Graphics amp Media NCI-Frederick for help withillustrations and figure preparation Funding for this research aswell as the Open Access publication charges was provided byDHHS NIH NCI
REFERENCES
1 ShizuyaH BirrenB KimUJ MancinoV SlepakTTachiiriY and SimonM (1992) Cloning and stable maintenance of300-kilobase-pair fragments of human DNA in Escherichia coli using anF-factor-based vector Proc Natl Acad Sci USA 89 8794ndash8797
2 YangXW ModelP and HeintzN (1997) Homologous recombinationbased modification in Escherichia coli and germline transmission intransgenic mice of a bacterial artificial chromosome Nat Biotechnol15 859ndash865
3 AntochMP SongEJ ChangAM VitaternaMH ZhaoYWilsbacherLD SangoramAM KingDP PintoLH andTakahashiJS (1997) Functional identification of the mouse circadianClock gene by transgenic BAC rescue Cell 89 655ndash667
4 LiuP JenkinsNA and CopelandNG (2003) A highly efficientrecombineering-based method for generating conditional knockoutmutations Genome Res 13 476ndash484
5 Cotta-de-AlmeidaV SchonhoffS ShibataT LeiterA andSnapperSB (2003) A new method for rapidly generating gene-targetingvectors by engineering BACs through homologous recombination inbacteria Genome Res 13 2190ndash2194
6 CopelandNG JenkinsNA and CourtDL (2001) Recombineeringa powerful new tool for mouse functional genomics Nature RevGenet 2 769ndash779
7 CourtDL SawitzkeJA and ThomasonLC (2002) Geneticengineering using homologous recombination Annu Rev Genet 36361ndash388
8 ZhangY BuchholzF MuyrersJP and StewartAF (1998) A newlogic for DNA engineering using recombination in Escherichia coliNature Genet 20 123ndash128
9 MuyrersJP ZhangY TestaG and StewartAF (1999) Rapidmodification of bacterial artificial chromosomes by ET-recombinationNucleic Acids Res 27 1555ndash1557
10 YuD EllisHM LeeEC JenkinsNA CopelandNG andCourtDL (2000) An efficient recombination system for chromosomeengineering in Escherichia coli Proc Natl Acad Sci USA 975978ndash5983
11 LeeEC YuD Martinez de VelascoJ TessarolloL SwingDACourtDL JenkinsNA and CopelandNG (2001) A highly efficientEscherichia coli-based chromosome engineering system adapted forrecombinogenic targeting and subcloning of BAC DNA Genomics73 56ndash65
12 GongS YangXW LiC and HeintzN (2002) Highly efficientmodification of bacterial artificial chromosomes (BACs) using novelshuttle vectors containing the R6Kgamma origin of replicationGenome Res 12 1992ndash1998
13 JamsaiD OrfordM NefedovM FucharoenS WilliamsonR andIoannouPA (2003) Targeted modification of a human beta-globin locusBAC clone using GET Recombination and an I-SceI counterselectioncassette Genomics 82 68ndash77
14 SwaminathanS EllisHM WatersLS YuD LeeECCourtDL and SharanSK (2001) Rapid engineering of bacterialartificial chromosomes using oligonucleotides Genesis 2914ndash21
15 AlperMD and AmesBN (1975) Positive selection of mutants withdeletions of the gal-chl region of the Salmonella chromosome as ascreening procedure for mutagens that cause deletions J Bacteriol121 259ndash266
16 AusubelFM BrentR KingstonRE MooreDD SeidmanJGSmithJA and StruhlK (1987) Current Protocols in Molecular BiologyGreene Publishing Associates and Wiley-Interscience John Wiley ampSons Hoboken NJ
17 BachmannBJ (1996) Derivations and genotypes of some mutantderivatives of Escherichia coli K-12 In NeidhardtFC CurtissRIIIIngrahamJL LinECC LowKB MagasanikB ReznikoffWSRileyM SchaechterM and UmbargerHE (eds) Escherichia coli andSalmonella Cellular and Molecular Biology ASM press WashingtonDC Vol 2 pp 2460ndash2488
18 GrantSG JesseeJ BloomFR and HanahanD (1990) Differentialplasmid rescue from transgenic mouse DNAs into Escherichia colimethylation-restriction mutants Proc Natl Acad Sci USA 874645ndash4649
19 OppenheimAB RattrayAJ BubunenkoM ThomasonLC andCourtDL (2004) In vivo recombineering of bacteriophage lambda byPCR fragments and single-strand oligonucleotides Virology 319185ndash189
20 HoessRH WierzbickiA and AbremskiK (1986) The role ofthe loxP spacer region in P1 site-specific recombination NucleicAcids Res 14 2287ndash2300
21 YuD SawitzkeJA EllisH and CourtDL (2003) Recombineeringwith overlapping single-stranded DNA oligonucleotides testing arecombination intermediate Proc Natl Acad Sci USA 1007207ndash7212
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 12 OF 12

deletions in both the absence and presence of oligo indicatingthat the deletions are not oligo induced We therefore concludethat these are spontaneous deletions that occur at low levelduring normal BAC replication in the DH10B background
Non-deleted colonies with the parental SpeI fingerprintwere only observed in the Red-induced sample that received
oligo (Figure 6D clones 33 and 35) These clones were sub-sequently electroporated into EL350 Cre-expressing cells toconfirm they contained the introduced loxP511 site Two col-onies from each original clone were tested by SpeI digestion ofthe BAC miniprep DNA As shown in Figure 6E each colonyhas undergone Cre-mediated deletion confirming that these
Figure 6 Same experiment as in Figure 5 with modifications as indicated at the top of each panel (A) SpeI digest of 10 minipreps from a control experiment withoutheat-induction and without the loxP511 dsDNA oligo (B) SpeI digest of 10 minipreps from a control experiment without heat-induction but with the loxP511 dsDNAoligo (C) SpeI digest of 10 minipreps from a control experiment with heat-induction but without the loxP511 dsDNA oligo (D) SpeI digest of 10 minipreps from anexperiment with heat-induction and with the loxP511 dsDNA oligo (comparable with Figure 5B) Clones with the parental digestion pattern indicating DOGresistance due to homologous recombination (clones 33 and 35 circles) are only seen in (D) DOG resistance in all other clones likely occurred due to internaldeletions of the BACs (E) SpeI restriction analysis of BAC miniprep DNA from clones 33 and 35 after transformation into Cre-induced EL350 cells Two clonesfrom each parental clone were tested The restriction pattern shows that the 95 kb region flanked by two loxP511 sites is deleted from all the clones analyzedconfirming the correct insertion of loxP511 in clones 33 and 35 (compare with Figure 5C)
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 10 OF 12
colonies were correctly targeted and contain the introducedloxP511 site (2 out of 10 clones a 20 efficiency in thisexperiment)
Generation of SW105 and SW106 cells
The two DY380-derived bacterial strains EL250 and EL350contain the defective l prophage needed for recombineering inaddition to L-arabinose inducible Flp or Cre genes respect-ively (11) Both strains have proven to be very useful for BACmodification (11) and EL350 is now used routinely for mak-ing conditional targeting vectors for ES cell knock-out experi-ments (4) To further enhance the usefulness of these strainswe decided to transfer the galK selection system into themThis was done as previously described for SW102 The galKIS2 element present in both strains was replaced with the wild-type gal promoter and the galK gene in the gal operon deletedusing DOG selection The resulting strains SW105 (Flp) andSW106 (Cre) (Table 1) were then tested to confirm that theystill contained inducible Flp and Cre genes by transformingarabinose-induced and electrocompetent cells with plasmidscontaining a neo gene flanked by Frt or loxP sites PL451 andPL452 (4) respectively Using SW105 or SW106 it is nowpossible to introduce a point mutation or an informativerestriction site into a BAC retrieve a fragment from thisBAC containing the introduced mutation(s) into a plasmidbackbone using gap repair and turn the retrieved fragmentinto a conditional targeting vectormdashall using only one bac-terial strain and only a single initial BAC transformation
DISCUSSION
Here we describe a new recombineering-based Ecoli BACmodification system that makes use of galK positive selectionfor growth on galactose minimal medium and galK negativeselection (counterselection) for growth on DOG This modi-fication system has several advantages compared with theother related BAC modification systems First the galK selec-tion cassette is small (1231 bp + homology arms) comparedwith for example the sacBndashneo cassette (3 kb) making PCRamplification and transformation into bacteria easier Twohomology arms are easily added to the galK cassette by includ-ing these sequences in the 50 ends of the primers used foramplification (see Materials and Methods) Second sincegalK is used for both selection steps mutations occurringin galK during PCR amplification will be selected againstduring positive selection significantly reducing the risk ofDOG resistance from PCR mutations in galK during negativeselection Third even when very short (33 bp) homology armswere used the frequency of recombinants among the analyzedDOG-resistant clones was still 20ndash50 To our knowledgesuccessful BAC modification with such short homology armshas never been reported Finally since galK recombineering isso efficient little screening is required following selectionreducing the overall hands-on-time to a minimum galK selec-tion requires growth on minimal medium in both selectionsteps although perhaps not used routinely in most molecularbiology laboratories where BAC modification is needed theseplates are fairly simple to make and should not prevent anyonefrom using this method
For all such counterselection schemes there will bebackground since during negative selection any event leadingto the loss of the counterselectable marker will result in sur-vival In our system virtually all background appears to resultfrom deletions that span the inserted galK gene These dele-tions likely occur during BAC replication in the 45 h out-growth phase since we have shown that these deletions occurindependently of Red induction and in the absence or presenceof oligo It is well established that in recA defective bacteriaBACs are very stable compared with other large insert vectorssuch as yeast artificial chromosomes (YACs) However usingcounterselection rare spontaneous deletions are seen becauseof the strong selection force This spontaneous deletion back-ground is not a problem however due to the high frequency ofhomologous recombination obtained with the defective lprophage system
Increasing the length of homology arms used for recombin-eering will reduce the relative number of background deletionsobserved following negative selection since the percentage ofcolonies containing BACs that eliminated galK by homolog-ous recombination will be increased Oligos with longerhomology arms can be produced by annealing two oligoswith overlapping 30 ends (21) These overlapping oligos (50
single strand overhangs) are then filled in by DNA polymerasein vivo (or in vitro) and subsequently serve as the substrate forthe l Red proteins exo and bet Longer homology arms canalso be added by traditional cloning using restriction enzymesand DNA ligase (4)
A number of uses can be imagined for the strains describedhere In principle our strains can be used to make virtually anykind of BAC modification one can imagine including pointmutations or clean deletions or insertions of everything fromcDNA to loxP sites or small epitope tags BAC trimming thespecific deletion of BAC DNA flanking a gene of interest couldalso be used to remove genes from BACs prior to making BACtransgenic mice so that only the gene of interest on the BAC isanalyzed This should be of particular interest for BAC rescueexperiments in positional cloning Of course trimming andmodification can be combined since the galK selection cassettecan be recycled so that the same BAC can be modified severaltimes BAC modification could also be the starting point forconstructing a gene-targeting vector to allow for more soph-isticated gene targeting in mouse ES cells The desired muta-tion(s) could first be created in the BAC followed by retrievalinto a plasmid vector containing a negative-selection markerlike thymidine kinase using gap repair (4) Finally a neo mar-ker could be introduced to allow for positive selection in EScells Alternatively the retrieved fragment could be modifiedso that the end result is a conditional targeting vector StrainSW106 which in addition to galK selection can be induced toexpress Cre recombinase makes it possible to perform all ofthe steps needed to construct a conditional targeting vectorstarting with only a single BAC transformation step
Furthermore in experiments where BAC transgenic miceare used to analyze the effect of deleting long-range regulatorsof gene expression galK DOG counterselection can even beused in the absence of an oligo to generate a series of dele-tions around the galK insertion site and the effect on generegulation studied
The Flp or Cre ORFs contained in strains SW105 andSW106 respectively can also be replaced for any desired
PAGE 11 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
gene thus creating tight arabinose-inducible expression forany gene of interest This is done by first replacing the CreFlpe ORF with galK and select for Gal+ recombinants ThegalK cassette is then replaced by the ORF of the gene ofinterest by DOG selection for Gal recombinants
Finally the strains described here should be very useful forgenetic manipulation not only of BACs but also the Ecoligenome itself
All recombineering reagents discussed in this workare freely available upon request To obtain these materialsplease follow the directions listed on our website (httprecombineeringncifcrfgov) Detailed protocols for recom-bineering including galK selection can also be downloadeddirectly from our website
ACKNOWLEDGEMENTS
We thank Allen Kane and Carolyn Whistler from ScientificPublications Graphics amp Media NCI-Frederick for help withillustrations and figure preparation Funding for this research aswell as the Open Access publication charges was provided byDHHS NIH NCI
REFERENCES
1 ShizuyaH BirrenB KimUJ MancinoV SlepakTTachiiriY and SimonM (1992) Cloning and stable maintenance of300-kilobase-pair fragments of human DNA in Escherichia coli using anF-factor-based vector Proc Natl Acad Sci USA 89 8794ndash8797
2 YangXW ModelP and HeintzN (1997) Homologous recombinationbased modification in Escherichia coli and germline transmission intransgenic mice of a bacterial artificial chromosome Nat Biotechnol15 859ndash865
3 AntochMP SongEJ ChangAM VitaternaMH ZhaoYWilsbacherLD SangoramAM KingDP PintoLH andTakahashiJS (1997) Functional identification of the mouse circadianClock gene by transgenic BAC rescue Cell 89 655ndash667
4 LiuP JenkinsNA and CopelandNG (2003) A highly efficientrecombineering-based method for generating conditional knockoutmutations Genome Res 13 476ndash484
5 Cotta-de-AlmeidaV SchonhoffS ShibataT LeiterA andSnapperSB (2003) A new method for rapidly generating gene-targetingvectors by engineering BACs through homologous recombination inbacteria Genome Res 13 2190ndash2194
6 CopelandNG JenkinsNA and CourtDL (2001) Recombineeringa powerful new tool for mouse functional genomics Nature RevGenet 2 769ndash779
7 CourtDL SawitzkeJA and ThomasonLC (2002) Geneticengineering using homologous recombination Annu Rev Genet 36361ndash388
8 ZhangY BuchholzF MuyrersJP and StewartAF (1998) A newlogic for DNA engineering using recombination in Escherichia coliNature Genet 20 123ndash128
9 MuyrersJP ZhangY TestaG and StewartAF (1999) Rapidmodification of bacterial artificial chromosomes by ET-recombinationNucleic Acids Res 27 1555ndash1557
10 YuD EllisHM LeeEC JenkinsNA CopelandNG andCourtDL (2000) An efficient recombination system for chromosomeengineering in Escherichia coli Proc Natl Acad Sci USA 975978ndash5983
11 LeeEC YuD Martinez de VelascoJ TessarolloL SwingDACourtDL JenkinsNA and CopelandNG (2001) A highly efficientEscherichia coli-based chromosome engineering system adapted forrecombinogenic targeting and subcloning of BAC DNA Genomics73 56ndash65
12 GongS YangXW LiC and HeintzN (2002) Highly efficientmodification of bacterial artificial chromosomes (BACs) using novelshuttle vectors containing the R6Kgamma origin of replicationGenome Res 12 1992ndash1998
13 JamsaiD OrfordM NefedovM FucharoenS WilliamsonR andIoannouPA (2003) Targeted modification of a human beta-globin locusBAC clone using GET Recombination and an I-SceI counterselectioncassette Genomics 82 68ndash77
14 SwaminathanS EllisHM WatersLS YuD LeeECCourtDL and SharanSK (2001) Rapid engineering of bacterialartificial chromosomes using oligonucleotides Genesis 2914ndash21
15 AlperMD and AmesBN (1975) Positive selection of mutants withdeletions of the gal-chl region of the Salmonella chromosome as ascreening procedure for mutagens that cause deletions J Bacteriol121 259ndash266
16 AusubelFM BrentR KingstonRE MooreDD SeidmanJGSmithJA and StruhlK (1987) Current Protocols in Molecular BiologyGreene Publishing Associates and Wiley-Interscience John Wiley ampSons Hoboken NJ
17 BachmannBJ (1996) Derivations and genotypes of some mutantderivatives of Escherichia coli K-12 In NeidhardtFC CurtissRIIIIngrahamJL LinECC LowKB MagasanikB ReznikoffWSRileyM SchaechterM and UmbargerHE (eds) Escherichia coli andSalmonella Cellular and Molecular Biology ASM press WashingtonDC Vol 2 pp 2460ndash2488
18 GrantSG JesseeJ BloomFR and HanahanD (1990) Differentialplasmid rescue from transgenic mouse DNAs into Escherichia colimethylation-restriction mutants Proc Natl Acad Sci USA 874645ndash4649
19 OppenheimAB RattrayAJ BubunenkoM ThomasonLC andCourtDL (2004) In vivo recombineering of bacteriophage lambda byPCR fragments and single-strand oligonucleotides Virology 319185ndash189
20 HoessRH WierzbickiA and AbremskiK (1986) The role ofthe loxP spacer region in P1 site-specific recombination NucleicAcids Res 14 2287ndash2300
21 YuD SawitzkeJA EllisH and CourtDL (2003) Recombineeringwith overlapping single-stranded DNA oligonucleotides testing arecombination intermediate Proc Natl Acad Sci USA 1007207ndash7212
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 12 OF 12

colonies were correctly targeted and contain the introducedloxP511 site (2 out of 10 clones a 20 efficiency in thisexperiment)
Generation of SW105 and SW106 cells
The two DY380-derived bacterial strains EL250 and EL350contain the defective l prophage needed for recombineering inaddition to L-arabinose inducible Flp or Cre genes respect-ively (11) Both strains have proven to be very useful for BACmodification (11) and EL350 is now used routinely for mak-ing conditional targeting vectors for ES cell knock-out experi-ments (4) To further enhance the usefulness of these strainswe decided to transfer the galK selection system into themThis was done as previously described for SW102 The galKIS2 element present in both strains was replaced with the wild-type gal promoter and the galK gene in the gal operon deletedusing DOG selection The resulting strains SW105 (Flp) andSW106 (Cre) (Table 1) were then tested to confirm that theystill contained inducible Flp and Cre genes by transformingarabinose-induced and electrocompetent cells with plasmidscontaining a neo gene flanked by Frt or loxP sites PL451 andPL452 (4) respectively Using SW105 or SW106 it is nowpossible to introduce a point mutation or an informativerestriction site into a BAC retrieve a fragment from thisBAC containing the introduced mutation(s) into a plasmidbackbone using gap repair and turn the retrieved fragmentinto a conditional targeting vectormdashall using only one bac-terial strain and only a single initial BAC transformation
DISCUSSION
Here we describe a new recombineering-based Ecoli BACmodification system that makes use of galK positive selectionfor growth on galactose minimal medium and galK negativeselection (counterselection) for growth on DOG This modi-fication system has several advantages compared with theother related BAC modification systems First the galK selec-tion cassette is small (1231 bp + homology arms) comparedwith for example the sacBndashneo cassette (3 kb) making PCRamplification and transformation into bacteria easier Twohomology arms are easily added to the galK cassette by includ-ing these sequences in the 50 ends of the primers used foramplification (see Materials and Methods) Second sincegalK is used for both selection steps mutations occurringin galK during PCR amplification will be selected againstduring positive selection significantly reducing the risk ofDOG resistance from PCR mutations in galK during negativeselection Third even when very short (33 bp) homology armswere used the frequency of recombinants among the analyzedDOG-resistant clones was still 20ndash50 To our knowledgesuccessful BAC modification with such short homology armshas never been reported Finally since galK recombineering isso efficient little screening is required following selectionreducing the overall hands-on-time to a minimum galK selec-tion requires growth on minimal medium in both selectionsteps although perhaps not used routinely in most molecularbiology laboratories where BAC modification is needed theseplates are fairly simple to make and should not prevent anyonefrom using this method
For all such counterselection schemes there will bebackground since during negative selection any event leadingto the loss of the counterselectable marker will result in sur-vival In our system virtually all background appears to resultfrom deletions that span the inserted galK gene These dele-tions likely occur during BAC replication in the 45 h out-growth phase since we have shown that these deletions occurindependently of Red induction and in the absence or presenceof oligo It is well established that in recA defective bacteriaBACs are very stable compared with other large insert vectorssuch as yeast artificial chromosomes (YACs) However usingcounterselection rare spontaneous deletions are seen becauseof the strong selection force This spontaneous deletion back-ground is not a problem however due to the high frequency ofhomologous recombination obtained with the defective lprophage system
Increasing the length of homology arms used for recombin-eering will reduce the relative number of background deletionsobserved following negative selection since the percentage ofcolonies containing BACs that eliminated galK by homolog-ous recombination will be increased Oligos with longerhomology arms can be produced by annealing two oligoswith overlapping 30 ends (21) These overlapping oligos (50
single strand overhangs) are then filled in by DNA polymerasein vivo (or in vitro) and subsequently serve as the substrate forthe l Red proteins exo and bet Longer homology arms canalso be added by traditional cloning using restriction enzymesand DNA ligase (4)
A number of uses can be imagined for the strains describedhere In principle our strains can be used to make virtually anykind of BAC modification one can imagine including pointmutations or clean deletions or insertions of everything fromcDNA to loxP sites or small epitope tags BAC trimming thespecific deletion of BAC DNA flanking a gene of interest couldalso be used to remove genes from BACs prior to making BACtransgenic mice so that only the gene of interest on the BAC isanalyzed This should be of particular interest for BAC rescueexperiments in positional cloning Of course trimming andmodification can be combined since the galK selection cassettecan be recycled so that the same BAC can be modified severaltimes BAC modification could also be the starting point forconstructing a gene-targeting vector to allow for more soph-isticated gene targeting in mouse ES cells The desired muta-tion(s) could first be created in the BAC followed by retrievalinto a plasmid vector containing a negative-selection markerlike thymidine kinase using gap repair (4) Finally a neo mar-ker could be introduced to allow for positive selection in EScells Alternatively the retrieved fragment could be modifiedso that the end result is a conditional targeting vector StrainSW106 which in addition to galK selection can be induced toexpress Cre recombinase makes it possible to perform all ofthe steps needed to construct a conditional targeting vectorstarting with only a single BAC transformation step
Furthermore in experiments where BAC transgenic miceare used to analyze the effect of deleting long-range regulatorsof gene expression galK DOG counterselection can even beused in the absence of an oligo to generate a series of dele-tions around the galK insertion site and the effect on generegulation studied
The Flp or Cre ORFs contained in strains SW105 andSW106 respectively can also be replaced for any desired
PAGE 11 OF 12 Nucleic Acids Research 2005 Vol 33 No 4 e36
gene thus creating tight arabinose-inducible expression forany gene of interest This is done by first replacing the CreFlpe ORF with galK and select for Gal+ recombinants ThegalK cassette is then replaced by the ORF of the gene ofinterest by DOG selection for Gal recombinants
Finally the strains described here should be very useful forgenetic manipulation not only of BACs but also the Ecoligenome itself
All recombineering reagents discussed in this workare freely available upon request To obtain these materialsplease follow the directions listed on our website (httprecombineeringncifcrfgov) Detailed protocols for recom-bineering including galK selection can also be downloadeddirectly from our website
ACKNOWLEDGEMENTS
We thank Allen Kane and Carolyn Whistler from ScientificPublications Graphics amp Media NCI-Frederick for help withillustrations and figure preparation Funding for this research aswell as the Open Access publication charges was provided byDHHS NIH NCI
REFERENCES
1 ShizuyaH BirrenB KimUJ MancinoV SlepakTTachiiriY and SimonM (1992) Cloning and stable maintenance of300-kilobase-pair fragments of human DNA in Escherichia coli using anF-factor-based vector Proc Natl Acad Sci USA 89 8794ndash8797
2 YangXW ModelP and HeintzN (1997) Homologous recombinationbased modification in Escherichia coli and germline transmission intransgenic mice of a bacterial artificial chromosome Nat Biotechnol15 859ndash865
3 AntochMP SongEJ ChangAM VitaternaMH ZhaoYWilsbacherLD SangoramAM KingDP PintoLH andTakahashiJS (1997) Functional identification of the mouse circadianClock gene by transgenic BAC rescue Cell 89 655ndash667
4 LiuP JenkinsNA and CopelandNG (2003) A highly efficientrecombineering-based method for generating conditional knockoutmutations Genome Res 13 476ndash484
5 Cotta-de-AlmeidaV SchonhoffS ShibataT LeiterA andSnapperSB (2003) A new method for rapidly generating gene-targetingvectors by engineering BACs through homologous recombination inbacteria Genome Res 13 2190ndash2194
6 CopelandNG JenkinsNA and CourtDL (2001) Recombineeringa powerful new tool for mouse functional genomics Nature RevGenet 2 769ndash779
7 CourtDL SawitzkeJA and ThomasonLC (2002) Geneticengineering using homologous recombination Annu Rev Genet 36361ndash388
8 ZhangY BuchholzF MuyrersJP and StewartAF (1998) A newlogic for DNA engineering using recombination in Escherichia coliNature Genet 20 123ndash128
9 MuyrersJP ZhangY TestaG and StewartAF (1999) Rapidmodification of bacterial artificial chromosomes by ET-recombinationNucleic Acids Res 27 1555ndash1557
10 YuD EllisHM LeeEC JenkinsNA CopelandNG andCourtDL (2000) An efficient recombination system for chromosomeengineering in Escherichia coli Proc Natl Acad Sci USA 975978ndash5983
11 LeeEC YuD Martinez de VelascoJ TessarolloL SwingDACourtDL JenkinsNA and CopelandNG (2001) A highly efficientEscherichia coli-based chromosome engineering system adapted forrecombinogenic targeting and subcloning of BAC DNA Genomics73 56ndash65
12 GongS YangXW LiC and HeintzN (2002) Highly efficientmodification of bacterial artificial chromosomes (BACs) using novelshuttle vectors containing the R6Kgamma origin of replicationGenome Res 12 1992ndash1998
13 JamsaiD OrfordM NefedovM FucharoenS WilliamsonR andIoannouPA (2003) Targeted modification of a human beta-globin locusBAC clone using GET Recombination and an I-SceI counterselectioncassette Genomics 82 68ndash77
14 SwaminathanS EllisHM WatersLS YuD LeeECCourtDL and SharanSK (2001) Rapid engineering of bacterialartificial chromosomes using oligonucleotides Genesis 2914ndash21
15 AlperMD and AmesBN (1975) Positive selection of mutants withdeletions of the gal-chl region of the Salmonella chromosome as ascreening procedure for mutagens that cause deletions J Bacteriol121 259ndash266
16 AusubelFM BrentR KingstonRE MooreDD SeidmanJGSmithJA and StruhlK (1987) Current Protocols in Molecular BiologyGreene Publishing Associates and Wiley-Interscience John Wiley ampSons Hoboken NJ
17 BachmannBJ (1996) Derivations and genotypes of some mutantderivatives of Escherichia coli K-12 In NeidhardtFC CurtissRIIIIngrahamJL LinECC LowKB MagasanikB ReznikoffWSRileyM SchaechterM and UmbargerHE (eds) Escherichia coli andSalmonella Cellular and Molecular Biology ASM press WashingtonDC Vol 2 pp 2460ndash2488
18 GrantSG JesseeJ BloomFR and HanahanD (1990) Differentialplasmid rescue from transgenic mouse DNAs into Escherichia colimethylation-restriction mutants Proc Natl Acad Sci USA 874645ndash4649
19 OppenheimAB RattrayAJ BubunenkoM ThomasonLC andCourtDL (2004) In vivo recombineering of bacteriophage lambda byPCR fragments and single-strand oligonucleotides Virology 319185ndash189
20 HoessRH WierzbickiA and AbremskiK (1986) The role ofthe loxP spacer region in P1 site-specific recombination NucleicAcids Res 14 2287ndash2300
21 YuD SawitzkeJA EllisH and CourtDL (2003) Recombineeringwith overlapping single-stranded DNA oligonucleotides testing arecombination intermediate Proc Natl Acad Sci USA 1007207ndash7212
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 12 OF 12

gene thus creating tight arabinose-inducible expression forany gene of interest This is done by first replacing the CreFlpe ORF with galK and select for Gal+ recombinants ThegalK cassette is then replaced by the ORF of the gene ofinterest by DOG selection for Gal recombinants
Finally the strains described here should be very useful forgenetic manipulation not only of BACs but also the Ecoligenome itself
All recombineering reagents discussed in this workare freely available upon request To obtain these materialsplease follow the directions listed on our website (httprecombineeringncifcrfgov) Detailed protocols for recom-bineering including galK selection can also be downloadeddirectly from our website
ACKNOWLEDGEMENTS
We thank Allen Kane and Carolyn Whistler from ScientificPublications Graphics amp Media NCI-Frederick for help withillustrations and figure preparation Funding for this research aswell as the Open Access publication charges was provided byDHHS NIH NCI
REFERENCES
1 ShizuyaH BirrenB KimUJ MancinoV SlepakTTachiiriY and SimonM (1992) Cloning and stable maintenance of300-kilobase-pair fragments of human DNA in Escherichia coli using anF-factor-based vector Proc Natl Acad Sci USA 89 8794ndash8797
2 YangXW ModelP and HeintzN (1997) Homologous recombinationbased modification in Escherichia coli and germline transmission intransgenic mice of a bacterial artificial chromosome Nat Biotechnol15 859ndash865
3 AntochMP SongEJ ChangAM VitaternaMH ZhaoYWilsbacherLD SangoramAM KingDP PintoLH andTakahashiJS (1997) Functional identification of the mouse circadianClock gene by transgenic BAC rescue Cell 89 655ndash667
4 LiuP JenkinsNA and CopelandNG (2003) A highly efficientrecombineering-based method for generating conditional knockoutmutations Genome Res 13 476ndash484
5 Cotta-de-AlmeidaV SchonhoffS ShibataT LeiterA andSnapperSB (2003) A new method for rapidly generating gene-targetingvectors by engineering BACs through homologous recombination inbacteria Genome Res 13 2190ndash2194
6 CopelandNG JenkinsNA and CourtDL (2001) Recombineeringa powerful new tool for mouse functional genomics Nature RevGenet 2 769ndash779
7 CourtDL SawitzkeJA and ThomasonLC (2002) Geneticengineering using homologous recombination Annu Rev Genet 36361ndash388
8 ZhangY BuchholzF MuyrersJP and StewartAF (1998) A newlogic for DNA engineering using recombination in Escherichia coliNature Genet 20 123ndash128
9 MuyrersJP ZhangY TestaG and StewartAF (1999) Rapidmodification of bacterial artificial chromosomes by ET-recombinationNucleic Acids Res 27 1555ndash1557
10 YuD EllisHM LeeEC JenkinsNA CopelandNG andCourtDL (2000) An efficient recombination system for chromosomeengineering in Escherichia coli Proc Natl Acad Sci USA 975978ndash5983
11 LeeEC YuD Martinez de VelascoJ TessarolloL SwingDACourtDL JenkinsNA and CopelandNG (2001) A highly efficientEscherichia coli-based chromosome engineering system adapted forrecombinogenic targeting and subcloning of BAC DNA Genomics73 56ndash65
12 GongS YangXW LiC and HeintzN (2002) Highly efficientmodification of bacterial artificial chromosomes (BACs) using novelshuttle vectors containing the R6Kgamma origin of replicationGenome Res 12 1992ndash1998
13 JamsaiD OrfordM NefedovM FucharoenS WilliamsonR andIoannouPA (2003) Targeted modification of a human beta-globin locusBAC clone using GET Recombination and an I-SceI counterselectioncassette Genomics 82 68ndash77
14 SwaminathanS EllisHM WatersLS YuD LeeECCourtDL and SharanSK (2001) Rapid engineering of bacterialartificial chromosomes using oligonucleotides Genesis 2914ndash21
15 AlperMD and AmesBN (1975) Positive selection of mutants withdeletions of the gal-chl region of the Salmonella chromosome as ascreening procedure for mutagens that cause deletions J Bacteriol121 259ndash266
16 AusubelFM BrentR KingstonRE MooreDD SeidmanJGSmithJA and StruhlK (1987) Current Protocols in Molecular BiologyGreene Publishing Associates and Wiley-Interscience John Wiley ampSons Hoboken NJ
17 BachmannBJ (1996) Derivations and genotypes of some mutantderivatives of Escherichia coli K-12 In NeidhardtFC CurtissRIIIIngrahamJL LinECC LowKB MagasanikB ReznikoffWSRileyM SchaechterM and UmbargerHE (eds) Escherichia coli andSalmonella Cellular and Molecular Biology ASM press WashingtonDC Vol 2 pp 2460ndash2488
18 GrantSG JesseeJ BloomFR and HanahanD (1990) Differentialplasmid rescue from transgenic mouse DNAs into Escherichia colimethylation-restriction mutants Proc Natl Acad Sci USA 874645ndash4649
19 OppenheimAB RattrayAJ BubunenkoM ThomasonLC andCourtDL (2004) In vivo recombineering of bacteriophage lambda byPCR fragments and single-strand oligonucleotides Virology 319185ndash189
20 HoessRH WierzbickiA and AbremskiK (1986) The role ofthe loxP spacer region in P1 site-specific recombination NucleicAcids Res 14 2287ndash2300
21 YuD SawitzkeJA EllisH and CourtDL (2003) Recombineeringwith overlapping single-stranded DNA oligonucleotides testing arecombination intermediate Proc Natl Acad Sci USA 1007207ndash7212
e36 Nucleic Acids Research 2005 Vol 33 No 4 PAGE 12 OF 12

![py galk ml npy galk ml nsl slslsl - Meditation for Beginners · PDF filepy galk ml npy galk ml nsl slslsl Page 3 of 97 gh j[xs h4 ¬okHkfod g, fd eu dh fdlh Hh f∂;k ls] tks eu ds](https://img.dokumen.tips/doc/110x75/5a7a73567f8b9a2d788b768e/py-galk-ml-npy-galk-ml-nsl-slslsl-meditation-for-beginners-galk-ml-npy-galk-ml.jpg)



























