Embed Size (px)
Citation preview

Secondary Structure Prediction
Protein Analysis Workshop 2008
Bioinformatics groupInstitute of BiotechnologyUniversity of helsinki
Hung Ta

Overview
Hierarchy of protein structure. Introduction to structure prediction:
• Different approaches.• Prediction of 1D strings of structural elements.
Server/soft review:• COILS, MPEx, …• The PredictProtein metaserver.
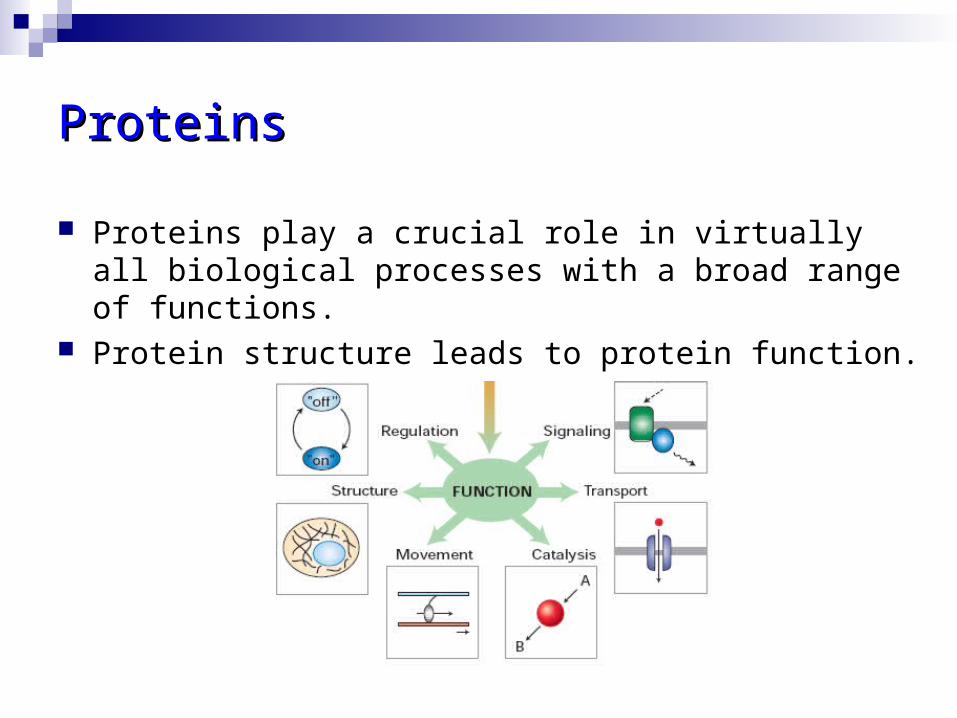
ProteinsProteins
Proteins play a crucial role in virtually all biological processes with a broad range of functions.
Protein structure leads to protein function.

Hierachy of Protein Structure Hierachy of Protein Structure

Primary Structure: a Primary Structure: a Linear Arrangement Linear Arrangement of Amino Acidsof Amino Acids
An amino acid has several structural components: a central carbon atom (C), an amino group (NH2), a carboxyl group (COOH), a hydrogen atom (H), a side chain (R). There are 20 amino acids
The peptide bond is formed as the cacboxyl group of an aa bind to the amino group of the adjacent aa.
The primary structure of a protein is simply the linear arrangement, or sequence, of the amino acid residues that compose it

Secondary Structure: Secondary Structure: Core Elements of Core Elements of Protein ArchitectureProtein Architecture
resulted from the folding of localized parts of a
polypeptide chain.
α-helix
β-sheet
Coils, turns,
} major internal supportive elements, 60 percent of the polypeptide chain

αα-Helix-Helix
Hydrogen-bonded
3.6 residues per turn
Axial dipole moment
Side chains point outward
Average length is 10 amino acids
(3 turns).
Typically, rich of Analine,
Glutamine, Leucine, Methione;
and poor of Proline, Glycine,
Tyrosine and Serine.

ββ-Sheet-Sheet
parallel anti-parallel
Formed due to hydrogen bonds
between β-strands which are short
polypeptide segments (5-8
residues).
Adjacent β-strands run in the
same directions -> parallel sheet.
Adjacent β-strands run in the
oposite directions -> anti-parallel
sheet.Ribbon diagram

Turns, loops, coils…Turns, loops, coils…
A turn, composed of 3-4 residues, forms
sharp bends that redirect the polypeptide
backbone back toward the interior.
A loop is similar with turns but can form
longer bends
Turns and loops help large proteins fold into
compact structures.
A random coil is a class of conformations
that indicate an absence of regular
secondary structure. Turn

Tertiary Structure: Overall Folding of Tertiary Structure: Overall Folding of Polypeptide Chain.Polypeptide Chain.
stabilized by hydrophobic interactions between the nonpolar side chains,
hydrogen bonds between polar side chains, and peptide bonds

Quaternary Structure: Arrangement of Quaternary Structure: Arrangement of Multiple Folded Protein Molecules.Multiple Folded Protein Molecules.
HemoglobinDNA polymerase

Structure PredictionStructure Prediction
GPSRYIVDL… ?
High importance in medicine (for example,
in drug design) and biotechnology (for
example, in the design of novel enzymes)

Structure PredictionStructure Prediction
Why: experimental methods, X-ray crystallography or NMR
spectroscopy, are very time-consuming and relatively expensive.
Challenges:
Extremely large number of possible structures.
the physical basis of protein structural stability is not fully
understood.
In this lecture, discuss about the protein secondary strutures
prediction.

Secondary Structure PredictionSecondary Structure Prediction
Primary: MSEGEDDFPRKRTPWCFDDEHMC
Secondary: CCHHHHHHCCCCEEEEEECCCCC
Why: the first level of structural organization.
The tasks:
• H: α-helix
• E: β- strand
• T: turn
• C: coil
aa
?

Secondary Structure PredictionSecondary Structure Prediction
Single residue statistical analysis (Chou-Fasman -1974): For each amino acid type, assign its ‘propensity’ to be in a helix, β-
sheet, or coil.
Based on 15 proteins of known conformation, 2473 total amino
acids.
Limited accuracy: ~55-60% on average.
Eg: Chou-Fasman (1974), not used any more

Secondary Structure PredictionSecondary Structure Prediction
Segment-based statistics: Look for correlations (within 11-21 aa windows).
Many algorithms have been tried.
Most performant: Neural Networks:
Input: a number of protein sequences with their known secondary
structure.
Output: a trained network that predicts secondary structure elements for
given query sequences.
Accuracy < 70%.

POPULAR SERVERS
FOR DEALING WITH
SECONDARY STRUCTURES
• Coiled-coils• Transmembrane helices• Secondary structure • Metaservers

Prediction of coiled-coilsPrediction of coiled-coils
Coiled-coils are generally solvent exposed multi-stranded helix structures:
Helix periodicity and solvent exposure imposespecial pattern of heptad repeat:
… abcdefg … hydrophobic residues hydrophilic residues
two-stranded
(From Wikipedia Leucine zipper article)
Helical diagram of2 interacting helices:

Compares a sequence to a database of known, parallel two-stranded coiled-coils, and derives a similarity score.
By comparing this score to the distribution of scores in globular and coiled-coil proteins, the program then calculates the probability that the sequence will adopt a coiled-coil conformation.
Options:• scoring matrices,• window size (score may vary),• weighting options.
The COILS server at EMBnetThe COILS server at EMBnet

The program works well for parallel two-stranded structures that are solvent-exposed but runs progressively into problems with the addition of more helices, their antiparallel orientation and their decreasing length.
The program fails entirely on buried structures.
COILS LimitationsCOILS Limitations

COILS DemoCOILS Demo
Let us submit the sequence
to the COILS server at EMBnet:
http://www.ch.embnet.org/software/COILS_form.html
>1jch_AVAAPVAFGFPALSTPGAGGLAVSISAGALSAAIADIMAALKGPFKFGLWGVALYGVLPSQIAKDDPNMMSKIVTSLPADDITESPVSSLPLDKATVNVNVRVVDDVKDERQNISVVSGVPMSVPVVDAKPTERPGVFTASIPGAPVLNISVNNSTPAVQTLSPGVTNNTDKDVRPAFGTQGGNTRDAVIRFPKDSGHNAVYVSVSDVLSPDQVKQRQDEENRRQQEWDATHPVEAAERNYERARAELNQANEDVARNQERQAKAVQVYNSRKSELDAANKTLADAIAEIKQFNRFAHDPMAGGHRMWQMAGLKAQRAQTDVNNKQAAFDAAAKEKSDADAALSSAMESRKKKEDKKRSAENNLNDEKNKPRKGFKDYGHDYHPAPKTENIKGLGDLKPGIPKTPKQNGGGKRKRWTGDKGRKIYEWDSQHGELEGYRASDGQHLGSFDPKTGNQLKGPDPKRNIKKYL


Transmembrane regions: Usually contain residues with hydrophobic side
chains (surface must be hydrophobic). Usually ~20 residues long, can be up to 30 if
not perpendicular through membrane.
Methods: Hydropathy plots (historical, better methods now available)
Threading (TMpred, MEMSAT), Hidden Markov Model (TMHMM), Neural Network (PHDhtm).
Transmembrane Region PredictionTransmembrane Region Prediction

Hydropathy Plots (Kyte-Doolittle)
The hydropathy index of an amino acid is a number
representing the hydrophobic or hydrophilic properties of
its side-chain
compute an average hydropathy value for each position
in the query sequence,
window length of 19 usually chosen for membrane-
spanning region prediction.

>sp|P06010|RCEM_RHOVI Reaction center protein M chain (Photosynthetic reaction center M subunit) - Rhodopseudomonas viridis. ADYQTIYTQIQARGPHITVSGEWGDNDRVGKPFYSYWLGKIGDAQIGPIYLGASGIAAFAFGSTAILIILFNMAAEVHFDPLQFFRQFFWLGLYPPKAQYGMGIPPLHDGGWWLMAGLFMTLSLGSWWIRVYSRARALGLGTHIAWNFAAAIFFVLCIGCIHPTLVGSWSEGVPFGIWPHIDWLTAFSIRYGNFYYCPWHGFSIGFAYGCGLLFAAHGATILAVARFGGDREIEQITDRGTAVERAALFWRWTIGFNATIESVHRWGWFFSLMVMVSASVGILLTGTFVDNWYLWCVKHG AAPDYPAYLPATPDPASLPGAPK
Hydropathy Plot ServersHydropathy Plot Servers
Let us submit the sequence
to
Membrane Explorer (also as standalone MPEx), Grease (http://fasta.bioch.virginia.edu/fasta/grease.htm)

Hydropathy PlotHydropathy Plot
The larger the number is, the more hydrophobic the amino acid

Scans a candidate sequence for matches to a sequence scoring matrix, obtained by aligning the sequences of all transmembrane alpha-helical regions that are known from structures.
These sequences are collected in a database called TMBase.
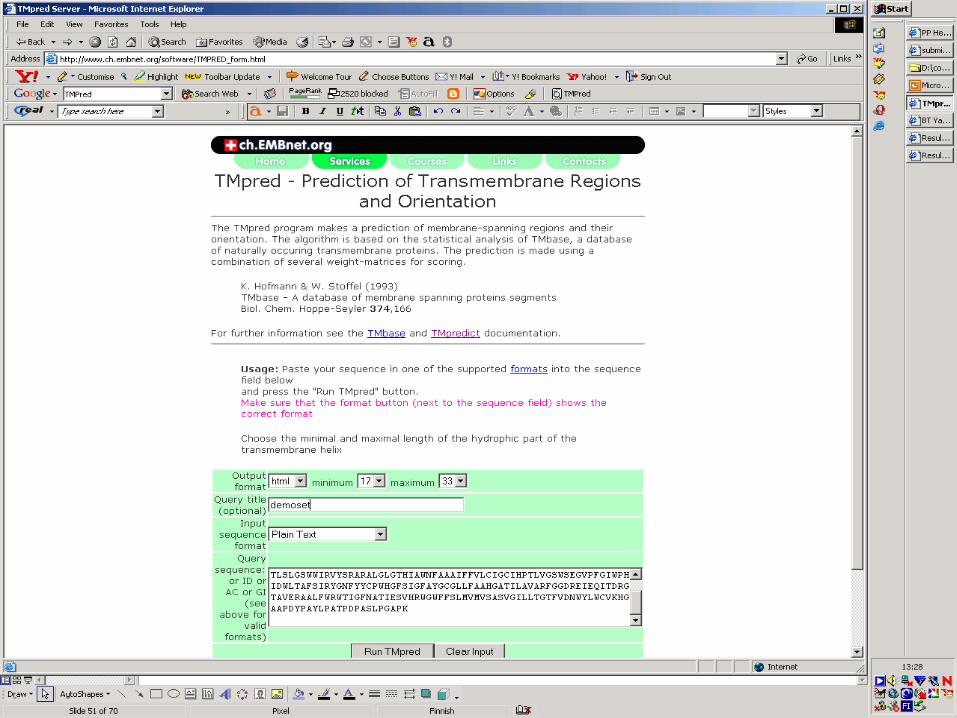
TM PredTM Pred
Method summary:
Remark: Authors do not suggest this method for genomic sequences. Automatic methods recommended, eg, TMHMM, PHDhtm.
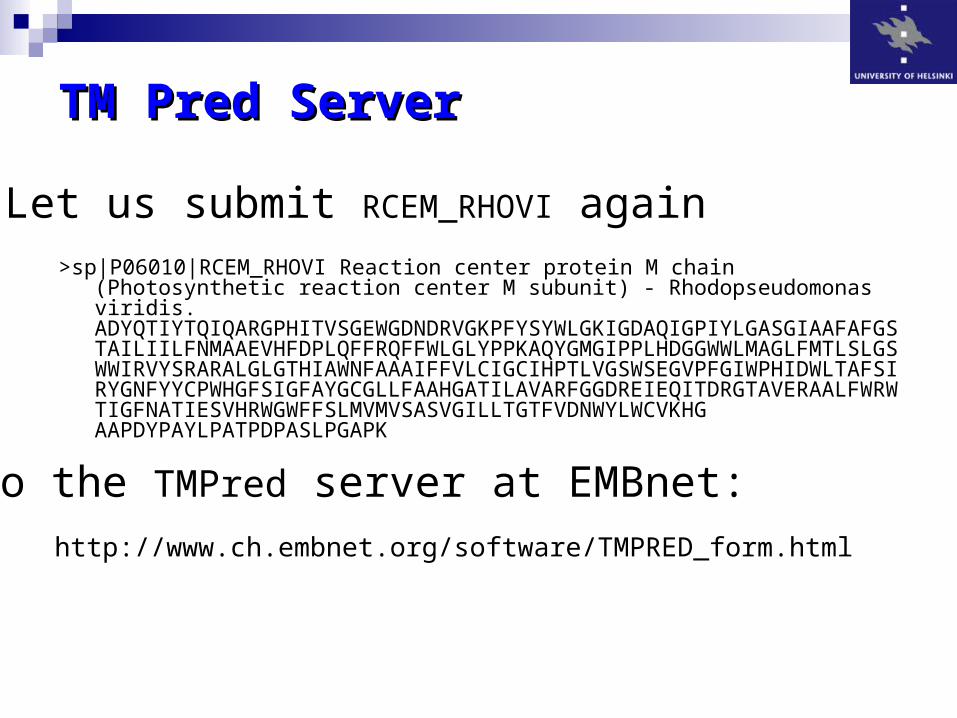
TM Pred ServerTM Pred Server
>sp|P06010|RCEM_RHOVI Reaction center protein M chain (Photosynthetic reaction center M subunit) - Rhodopseudomonas viridis. ADYQTIYTQIQARGPHITVSGEWGDNDRVGKPFYSYWLGKIGDAQIGPIYLGASGIAAFAFGSTAILIILFNMAAEVHFDPLQFFRQFFWLGLYPPKAQYGMGIPPLHDGGWWLMAGLFMTLSLGSWWIRVYSRARALGLGTHIAWNFAAAIFFVLCIGCIHPTLVGSWSEGVPFGIWPHIDWLTAFSIRYGNFYYCPWHGFSIGFAYGCGLLFAAHGATILAVARFGGDREIEQITDRGTAVERAALFWRWTIGFNATIESVHRWGWFFSLMVMVSASVGILLTGTFVDNWYLWCVKHG AAPDYPAYLPATPDPASLPGAPK
Let us submit RCEM_RHOVI again
to the TMPred server at EMBnet:
http://www.ch.embnet.org/software/TMPRED_form.html


allows you to obtain many informations based on your sequence including structure predictions, motif or domain search… The predictions are based on several methods.
PredictProtein: http://predictprotein.org
Meta-ServersMeta-Servers
A server which

For sequence analysis, structure and function prediction. When you submit
any protein sequence PredictProtein retrieves similar sequences in the
database and predicts aspects of protein structure and function
SEG: finds low complexity regions.
ProSite: database of functional motifs, ie, biologically relevant short patterns
ProDom: a comprehensive set of protein domain families automatically generated
from the SWISS-PROT and TrEMBL sequence databases.
PROFsec (PHDsec): secondary structure,
PROFacc (PHDacc): solvent accessibility,
PHDhtm: transmembrane helices.
Sequence database is scanned for similar sequences (Blast, Psi-Blast).
Multiple sequence alignment profiles are generated by weighted dynamic
programming (MaxHom).
The PredictProtein meta-server

PredictProtein Demo
Let´s submit again
to http://predictprotein.org/
>uniprot|P00772|ELA1_PIG Elastase-1 precursor MLRLLVVASLVLYGHSTQDFPETNARVVGGTEAQRNSWPSQISLQYRSGSSWAHTCGGTLIRQNWVMTAAHCVDRELTFRVVVGEHNLNQNDGTEQYVGVQKIVVHPYWNTDDVAAGYDIALLRLAQSVTLNSYVQLGVLPRAGTILANNSPCYITGWGLTRTNGQLAQTLQQAYLPTVDYAICSSSSYWGSTVKNSMVCAGGDGVRSGCQGDSGGPLHCLVNGQYAVHGVTSFVSRLGCNVTRKPTVFTRVSAYISWINNVIASN
For a list of mirror sites: http://predictprotein.org/newwebsite/doc/mirrors.html



Results

Low-complexity regions
Marked by ’X’

Secondary structure prediction results

Documentation:• COILS: http://www.ch.embnet.org/software/coils/COILS_doc.html
• TMPred: http://www.ch.embnet.org/software/tmbase/TMBASE_doc.html
• MPEx: http://blanco.biomol.uci.edu/mpex/MPEXdoc.html
Articles: B. Rost: Evolution teaches neural networks. In Scientific applications of neural nets. Ed.
J.W.Clark, T.Lindenau, M.L. Ristig, 207-223 (1999).
D.T Jones: Protein Secondary Structure Prediction Based on Position-specific Scoring Matrices. J.Mol.Biol. 292, 195-202 (1999).
B. Rost: Prediction in 1D: Secondary Structure, Membrane Helices, and Accessibility. In Structural Bioinformatics (reference below).
Books: P.E. Bourne, H. Weissig: Structural Bioinformatics. Wiley-Liss, 2003.
A. Tramontano: Protein Structure Prediction. Wiley-VCH, 2006.
References




![Matts Roos arXiv:1208.3662v1 [astro-ph.CO] 17 Aug …richard/ASTRO620/DM_History.pdfMatts Roos Department of Physics, FI-00014 University of Helsinki, Finland E-mail: matts.roos@helsinki.fi](https://img.dokumen.tips/doc/110x75/5ecdf8b208634901be1f43e1/matts-roos-arxiv12083662v1-astro-phco-17-aug-richardastro620dm-matts-roos.jpg)

















![Basics of colour & lightness 9.10.2014 / Tarja Peromaa PsyL Tarja Peromaa [tarja.peromaa@helsinki.fi] University of Helsinki Department of Behavioral Sciences](https://img.dokumen.tips/doc/110x75/56649d0e5503460f949e3b17/basics-of-colour-lightness-9102014-tarja-peromaa-psyl-tarja-peromaa-tarjaperomaahelsinkifi.jpg)






