Embed Size (px)
Citation preview

Mah and Lee Biomarker Research 2014, 2:5http://www.biomarkerres.org/content/2/1/5
REVIEW Open Access
DNA methylation: potential biomarker inHepatocellular CarcinomaWay-Champ Mah1,2,3 and Caroline GL Lee1,2,3,4*
Abstract
Hepatocellular Carcinoma (HCC) is one of the most common cancers in the world and it is often associated withpoor prognosis. Liver transplantation and resection are two currently available curative therapies. However, mostpatients cannot be treated with such therapies due to late diagnosis. This underscores the urgent need to identifypotential markers that ensure early diagnosis of HCC. As more evidences are suggesting that epigenetic changescontribute hepatocarcinogenesis, DNA methylation was poised as one promising biomarker. Indeed, genome wideprofiling reveals that aberrant methylation is frequent event in HCC. Many studies showed that differentiallymethylated genes and CpG island methylator phenotype (CIMP) status in HCC were associated with clinicopathologicaldata. Some commonly studied hypermethylated genes include p16, SOCS1, GSTP1 and CDH1. In addition,studies have also revealed that methylation markers could be detected in patient blood samples and associatedwith poor prognosis of the disease. Undeniably, increasing number of methylation markers are being discoveredthrough high throughput genome wide data in recent years. Proper and systematic validation of thesecandidate markers in prospective cohort is required so that their actual prognostication and surveillance valuecould be accurately determined. It is hope that in near future, methylation marker could be translate into clinicaluse, where patients at risk could be diagnosed early and that the progression of disease could be more correctlyassessed.
Keywords: Epigenetics, Methylation, Biomarker, CIMP, Hepatocellular carcinoma, Prognosis, Diagnosis
IntroductionHepatocellular Carcinoma (HCC) is one of the most fre-quent cancers in the world and annually, about 600,000patients died of liver cancer [1]. This disease is often as-sociated with poor prognosis because patients are eitherdiagnosed at very late stage or experienced recurrenceafter resection [2]. In fact, more than half of HCC pa-tients died within 12 months post diagnosis, and lessthan 6% of them have an average survival rate of 5 years[3]. Liver transplantation and resection are the only twocurative therapies available; however, in order to qualifyfor such therapies, patients need to be diagnosed earlywith HCC [4]. Presently, serum alpha-fetoprotein (AFP)concentration and hepatic ultrasonography are used in
* Correspondence: [email protected] of Biochemistry, Yong Loo Lin School of Medicine, NationalUniversity of Singapore, Singapore 117597, Singapore2Division of Medical Sciences, Humphrey Oei Institute of Cancer Research,National Cancer Centre Singapore, Level 6, Lab 5, 11 Hospital Drive,Singapore 169610, SingaporeFull list of author information is available at the end of the article
© 2014 Mah and Lee; licensee BioMed CentraCommons Attribution License (http://creativecreproduction in any medium, provided the orDedication waiver (http://creativecommons.orunless otherwise stated.
HCC surveillance program, where high risk patients arescreened for HCC in every six months [5]. As for actualdiagnosis, invasive biopsy and expensive imaging toolssuch as ultrasonography, spiral computed tomography(CT) and magnetic resonance imaging (MRI) are used[4]. AFP measurement is merely used as adjunct diag-nostic tool because of its variability in specificity andsensitivity [5,6]. Equally important to note is that apartfrom AFP level and tumor staging classification such asthe Barcelona Clinic Liver Cancer (BCLC) staging sys-tem, there is no good prognostic marker that can classifypatients and predict survival outcome [7-9]. The largenumber of HCC associated deaths clearly reflects theshortcomings of current diagnostic and prognostic tools.This underscores the importance of discovering noveland effective biomarkers that can improve overall cli-nical management of HCC.With the advance of genomic technologies, plethora of
molecular data is now available for translational research.Gene expression signatures and microRNA profiles are
l Ltd. This is an Open Access article distributed under the terms of the Creativeommons.org/licenses/by/2.0), which permits unrestricted use, distribution, andiginal work is properly credited. The Creative Commons Public Domaing/publicdomain/zero/1.0/) applies to the data made available in this article,

Mah and Lee Biomarker Research 2014, 2:5 Page 2 of 13http://www.biomarkerres.org/content/2/1/5
just some examples of molecular data that were activelyexplored as potential biomarkers for HCC [10-15]. In thisreview, we will focus specifically on DNA methylation,another potential biomarker that was shown to be impli-cated in HCC.
ReviewAberrant DNA methylation in HCCStudies have identified a few somatic mutations in HCC,for instance, mutations in TP53 [16,17], CTNNB1 [18,19],and AXIN1 [20,21]. However, frequencies of these muta-tions are inconsistent and rare, some occur only in certainsubtypes of tumor [22]. The lack of common genetic mar-ker associated with HCC cases strongly suggests that epi-genetic alterations such as aberrant DNA methylationcould be the alternative factor contributing towards livercarcinogenesis. DNA methylation occurs when a methylgroup is attached to the 5th carbon of cytosine nucleotideand this process is catalyzed by DNA methyltransferases(DNMTs), in which S-adenosyl-methionine (SAM) acts asa methyl donor (Figure 1) [23]. Deregulation of DNAmethylation was shown to be associated with many can-cers, as was first proposed by Andrew Feinberg and BertVogelstein in 1983 [24]. The two most common forms ofaberrant methylation are global hypomethylation and site-specific hypermethylation. In HCC, such deregulations arefrequently observed as well. Global hypomethylation inliver cancer affects the structural-nuclear function by pro-moting chromosomal and genomic instability, while re-gional hypermethylation is often associated with silencingof tumor suppressor genes [25]. Studies have revealed thatetiological factors like Hepatitis virus infection may leadto aberrant DNA methylation in cancerous tissues [26,27].DNA methyltransferases such as DNMT1, DNMT3A andDNMT3B were also shown to be up-regulated in livercancer [28,29]. Whether increased expression of DNMTsassociated with aberrant methylation of genes is still amatter of controversy as the exact mechanism has yet tobe elucidated [29,30]. Subsequent sections will summarize
Figure 1 Structures of deoxycytidine and 5-methyl-deoxycytidine. DNcarbon of cytosine, where DNMT serves as enzyme and SAM acts as the m
respective studies on aberrant DNA methylation in hepa-tocarcinogenesis and the full list of studies can be foundin Additional file 1: Table S1.
Genome wide studies on methylation profile of HCCPresent technologies allow researchers to profile methyla-tion in a genome wide manner. Two commonly usedmethods in HCC methylation profiling include hybri-dization of bisulfite converted DNA on beadarray [31,32]and enrichment of methylated DNA either by enzymaticdigestion [33,34] or antibody pull-down [35], followed bypromoter array profiling. Even though high throughputsequencing is becoming more available, presently, nostudy has yet to use this approach to map the methylomeof HCC. Table 1 briefly summarizes the strengths andlimitations of each profiling method. A few pivotal ge-nome wide methylation studies using these approachesare highlighted below.In 2008, Gao et al. adopted methylated CpG island amp-
lification microarray (MCAM) method to identify 719genes that were differentially methylated between tumorsand adjacent non-tumors [36]. They used pyrosequencingto validate their observations found by MCAM. Genessuch as RASSF1A, CDKN2A and CCNA1 were success-fully validated to be highly methylated in cancer tissuecompared to adjacent non-tumor and normal liver tissues.In subsequent year, Lu et al. used differential methylationhybridization (DMH) method to locate 38 hypomethylatedand 27 hypermethylated regions. Using Methylation spe-cific PCR (MSP) method, they validated the methylationstatus of KLK10 and OXGR1 in tumors, and found thathypermethylation of KLK10 was associated with HepatitisC virus (HCV) infection and cirrhosis [37]. Around thesame time, studies by Deng et al. and Stefanska et al. useda slightly different method called methylated DNA immu-noprecipitation microarray (MeDIP-chip) to locate aber-rant methylation in HCC. Deng et al. used MassArray®method to validate hypermethylation of DUSP4, NPR1and CYP24A1 in HCC, and correlate methylation status
A methylation occurs when a methyl group is attached to the 5th
ethyl group donor.

Table 1 Different methods in genome-wide methylation profiling
Platform Features Number ofregionsanalysedper sample
Methylationinformationon sitespecific CpGloci
Methylationinformationon non-CpGloci
Advantages Disadvantages
Microarraybased
Methylated CpGIsland Amplificationand Microarray(MCAM-chip)
Enzyme-based techniquesthat rely on restrictionenzymes (SmaI and XmaI)followed by profiling onpromoter array
~25,000 humanpromoters (depends onarray density)
No No Do not require bisulfiteconversion, good coverage onregion with low CpG density.
Require substantial quantities ofinput genomic DNA, low samplethroughput, do not reportmethylation status at singlenucleotide level, bias mayoccur due to genomic distributionof CpG loci, limited to mostlypromoter regions.
Differential MethylationHybridization andMicroarray (DMH-chip)
Enzyme-based techniquesthat rely on restrictionenzymes (MseI and BstUI)followed by profiling onpromoter array
Methylated DNAImmunoprecipitationand Microarray(MeDIP-chip)
Immunoprecipitation ofmethylated DNA with amonoclonal antibodyfollowed by profiling onpromoter array
Beadarraybased
GoldenGate Bisulfite convertion of DNAfollowed by microbeadbased microarray
~1,500 CpG sites Yes No Require minimum inputgenomic DNA, highsample throughput,provide methylationstatus at CpG loci,fairly accurate andreproducible.
Bisulfite treatment may not becomplete, bisulfite treatmentcaused DNA degradation, limitedto mostly promoter regions.
Infinium 27K ~27,000 CpG sites
Infinium 450K ~450,000 CpG sites
Highthroughputsequencing
Bisulfite sequencing Bisulphite conversion ofDNA followed by captureand high throughputsequencing
Whole genome Yes Yes High resolution mapping ofmethylation status at singlenucleotide level, no crosshybridization bias.
Bisulfite treatment may not becomplete, bisulfite treatmentcaused DNA degradation, lowsample throughput, expensive,complex bioinformatic analysis.
Mah
andLee
Biomarker
Research2014,2:5
Page3of
13http://w
ww.biom
arkerres.org/content/2/1/5

Mah and Lee Biomarker Research 2014, 2:5 Page 4 of 13http://www.biomarkerres.org/content/2/1/5
of these genes with recurrence free survival [38]. Stefanskaet al., on the other hand, delineated the profile of pro-moter hypomethylation in HCC and validated AKR1B10,CENPH, MMP9, MMP12, PAGE4, S100A5, MMP2 andNUPR1 to be hypomethylated in liver cancer using pyro-sequencing [39]. Earlier genome wide studies utilised pro-moter microarray to map out differentially methylatedregions. As a result, such arrays could not provide in-formation on site specific CpG dinucleotides that wereaberrantly methylated. Additional validation steps such aspyrosequencing and MassArray® were required before onecould locate the exact deregulated CpG sites. Nonetheless,as technology of methylation profiling matures over recentyears, many studies could now report genome wide me-thylation status of HCC at single-nucleotide resolution(Table 2). These studies mainly used the beadarray tech-nology developed by Illumina®. As shown in Table 2, themost recent studies by Song et al., Zhang et al. and Shenet al. reported the mapping of more than 485000 CpGsites, the highest throughput so far, in HCC.Genome wide methylation profiling provides wealth of
information for downstream analysis. Using these highthroughput data, researchers could efficiently separate tu-mors from adjacent non-tumors [39-47], cirrhotic liverfrom HCC [48,49], and cluster the tumors according totheir risk factors such as viral infection [38,43,46] and al-cohol consumption [42,43]. Novel tumor suppressor genesthat were silenced by methylation could also be uncoveredthrough genome wide studies, as shown by studies in[37,50-53]. As reports on genome wide methylation profileof HCC patients using serum DNA begin to emerge[41,42], it is hoped that these high throughput data couldaccelerate the process of biomarker discovery.
Methylation as prognostic markerFrom 2003 to 2013, many studies have published on theprognostic values of DNA methylation in HCC. Thesestudies are summarized in Additional file 1: Table S1. Dueto space constraints, within this review, only a few exam-ples will be highlighted. One of the early studies was re-ported by Yang et al. Their group profiled methylationstatus of 9 genes, namely GSTP1, SOCS1, CDH1, APC,p15, p16, p14, p73 and RAR-β in 51 HCC samples usingmethylation-specific polymerase chain reaction (MSP)[54]. Among these genes, methylation of SOCS1, APCand p15 were shown to be more frequently observed inHCV-positive HCC patients compared to HBV/HCV-negative HCC. Another group from Korea, Lee et al.examined the methylation level of CpG loci in 14 genes insixty HCC paired samples and found that methylation ofGSTP1 and CDH1 were associated with poorer overallsurvival [55]. Similarly, Yu et al. used MSP to identifymethylation level of 24 genes in 28 HCC samples froma Chinese population. They successfully showed that
methylation of AR, DBCCR1, IRF7, OCT6, p73, and p16were associated with late stage HCC [56]. These threestudies laid a strong basis for subsequent methylation ana-lysis. Many studies have since then attempted to associateclinical parameters with DNA methylation, particularly ongenes that were validated in these three studies. Subse-quent paragraphs will outline 4 of these genes, namely,p16, SOCS1, GSTP1 and CDH1 (Table 3).
Commonly studied methylation marker genesp16 (CDKN2A) is one of the most reported genes thatwas shown to be hypermethylated and associated with clin-ical parameters in HCC. It is a tumor suppressor gene thatplays a role in cell cycle regulation [57]. It was methylatedin many other cancers as well [58]. Beside earlier study byYu et al. [56], Shim et al. [59] and Su et al. [60] also re-ported that methylation level of p16 was associated withadvanced stage of HCC. They showed that methylation ofp16 gene increased from cirrhotic tissue to HCC. Studieshave also shown that hepatitis virus positive HCC sampleshave higher p16 methylation compared to HCC with noviral infection [61-64]. Zhu et al. even further showed thatHBx gene, a protein coded by HBV, was associated withmethylation of p16 in HBV positive HCC samples [65].Clearly, environment factors such as viral infection couldpossibly disturb the epigenetic profile of the liver and con-tribute towards carcinogenesis. In addition, vascular inva-sion [61] and tumor differentiation [66] were also shownto be associated with p16 methylation. As vascular inva-siveness and tumor differentiation were both strong predic-tors of survival in HCC [67-69], it is not surprising thathypermethylation of p16 in HCC patients was shown tohave worse disease free survival as well [70].Another frequently studied prognostic marker is SOCS1
methylation. SOCS1 gene was shown to be negativeregulator of JAK/STAT pathway and its suppression byhypermethylation promotes cell growth [71]. SOCS1 me-thylation was correlated with progression of HCC [72],age [73,74] and tumor size [73,75]. Moreover, as men-tioned in earlier paragraph, SOCS1 methylation wasshown by Yang et al. to be associated with HCV infectedHCC [54]. Concurring their study, Nishida et al. [76] andKo et al. [73] also revealed that SOCS1 methylation wasmore prevalent in HCV infected HCC compared to non-infected HCC. Interestingly, Chu et al. [75] and Okochiet al. [77] did not find this association to be significant intheir studies; instead, they found liver cirrhosis in HCC tobe closely linked to hypermethylation of SOCS1. As HCVinfection may lead to liver cirrhosis [78], more studies arerequired to ascertain this pathological link between HCVinfection, SOCS1 methylation and HCC progression.GSTP1 belongs to Glutathione S-transferases family,
where it plays a role in protecting cells against damage in-duced by carcinogens, and modulating signal transduction

Table 2 Genome wide methylation profiling in HCC
Promoter microarray
Discovery method HCC patients (n) Validation method Validated genes Publication Year
MCAM-chip 10 Pyrosequencing RASSF1A, p16, TBX4, MMP14, GNA14, SLC16A5, CCNA1 Gao et al. [36] 2008
16 Pyrosequencing KLHL35, PAX5, PENK, SPDYA, LINE-1 Shitani et al. [40] 2012
DMH-chip 21 MSP KLK10, OXGR1 Lu et al. [37] 2008
MeDIP-chip 6 MassArray CYP24A1, DLX1, ZNF141, RASGRF2, ZNF382, TUBB6, NPR1,RRAD, RUNX3, LOX, JAKMIP1, SFRP4, DUSP4, PARQ8, CYP7B1
Deng et al. [38] 2010
11 Pyrosequencing AKR1B10, CENPH, MMP9, MMP12, PAGE4, S100A5, MMP2, NUPR1 Stefanska et al. [39] 2011
Beadarray
Discovery method HCC patients (n) Validation method Validated genes Publication Year
GoldenGate 20 Methylight assay APC Archer et al. [49] 2010
38 Pyrosequencing RASSF1, GSTP1, APC, GABRA5, LINE-1 Hernandez-Vargas et al. [43] 2010
45 Bisulfite sequencing ERG, HOXA9 Hou et al. [47] 2013
Infinium 27K 3 COBRA and bisulfite sequencing WNK2, EMILIN2, TLX3, TM6SF1, TRIM58, HIST1H4F, GRASP Tao et al. [44] 2011
62 NIL NIL Yang et al. [118] 2011
13 NIL NIL Ammerpohl et al. [48] 2012
62 Pyrosequencing CDKL2, STEAP4, HIST1H3G, CDKN2A, ZNF154 Shen et al. [42] 2012
63 Pyrosequencing PER3 Neumann et al. [50] 2012
71 Pyrosequencing NEFH, SMPD3 Revill et al. [53] 2013
Infinium 450K 66 NIL NIL Shen et al. [46] 2013
27 Pyrosequencing GSTP1, RASSF1, BMP4, DLGAP1, GPR35 Song et al. [45] 2013
6 Bisulfite sequencing DBX2, THY1 Zhang et.al. [41] 2013
COBRA, Combined bisulfite restriction analysis; MSP, Methylation specific PCR.
Mah
andLee
Biomarker
Research2014,2:5
Page5of
13http://w
ww.biom
arkerres.org/content/2/1/5

Table 3 Commonly studied methylation markers in HCC
Gene HCC patients (n) Clinicopathological correlation Validation method Publication Year
p16 28 Tumor stage MSP Yu et al. [56] 2003
18 Tumor stage MSP Shim et al. [59] 2003
20 Tumor differentiation MSP Qin et al. [66] 2004
50 Age, gender, virus infection (HBV/HCV) MSP Li et al. [62] 2004
44 HBV infection MSP Jicai et al. [63] 2006
60 Age, tumor stage, vascular invasion, virus infection (HBV/HCV) MSP Katoh et al. [61] 2006
58 Tumor stage MSP Su et al. [60] 2007
23 HBV infection (HBx) MSP Zhu et al. [65] 2007
265 Disease free survival MSP Ko et al. [70] 2008
118 Gender Methylscreen Wang et al. [119] 2012
SOCS1 50 Liver cirrhosis MSP Okochi et al. [77] 2003
51 HCV infection MSP Yang et al. [54] 2003
284 Age, tumor size, virus infection (HBV/HCV) MSP Ko et al. [73] 2008
77 HCV infection COBRA Nishida et al. [76] 2008
46 Liver cirrhosis, tumor size MSP Chu et al. [75] 2010
46 Tumor stage MethyLight Um et al. [72] 2011
29 Chemotherapy treatment MSP Saelee et al. [120] 2012
116 Age and gender Methylscreen Zhang et al. [74] 2013
GSTP1 60 Overall survival MSP Lee et al. [55] 2003
83 Alcohol consumption, gender MSP Zhang et al. [82] 2005
60 Gender, viral infection (HBV/HCV) MSP Katoh et al. [61] 2006
58 HBV infection, tumor stage MSP Su et al. [60] 2007
77 HCV infection COBRA Nishida et al. [76] 2008
166 HBV infection Pyrosequencing Lambert et al. [81] 2011
CDH1 60 Overall survival MSP Lee et al. [55] 2003
32 Vascular invasion, recurrence MSP Ghee [91] 2005
COBRA, Combined Bisulfite Restriction Analysis; HBV, Hepatitis B virus; HCV, Hepatitis C virus; HBx, Hepatitis B virus X protein; MSP, Methylation-specific PCR.
Mah and Lee Biomarker Research 2014, 2:5 Page 6 of 13http://www.biomarkerres.org/content/2/1/5
pathways that control cell proliferation and cell death[79]. Promoter methylation of GSTP1 was first reportedin prostatic carcinoma back in 1994 [80]. Since then,many groups reported such observation in other cancers,including HCC. Analogous to earlier mentioned twogenes, GSTP1 was also found to be highly methylated inHCC infected with either HBV or HCV compared to non-infected HCC [60,61,76,81]. Interestingly, methylation ofGSTP1 was significantly associated with gender [61,82]and alcohol intake [82]. Also, study by Lee et al. managedto show that patients with high GSTP1 methylation levelhave worse overall survival outcome [55]. Although manystudies examined the association of GSTP1 methylationwith clinicopathological characteristics, only a few foundassociations suggesting that GSTP1 methylation alonemay not be sufficient to serve as good single prognosticpredictor for HCC.CDH1 is another well-known tumor suppressor gene
that was found methylated in many cancers [83-88]. Itwas frequently methylated in HCC as well. Despite many
studies showed that methylation of CDH1 was higher inHCC than adjacent non-tumors, it often was not signifi-cantly associated with clinical parameters [54,61,76,89,90].Only Lee et al. reported that methylation of CDH1 waslinked with worse overall survival [55], and Ghee et al.found that it was associated with vascular invasion and re-currence [91]. These reports suggest that probably CDH1alone may not have the power to be an independentprognostic factor. Notably, the concurrent methylation ofCDH1, GSTP1 and a few other genes were found to besignificantly associated with levels of AFP, recurrence freesurvival (RFS) and tumor numbers (Table 4). This con-cordant methylation of a group of genes associated withspecific tumor characteristics is known as CIMP or CpGisland methylator phenotype [92]. Presently, many studieshave attempted to elucidate CIMP in various cancers,including G-CIMP for gliomas [93], B-CIMP for breastcancer [94], and C-CIMP for colorectal cancer [95]. InHCC, concurrent methylation of various genes has beenassociated with various clinical phenotypes (Table 4) and
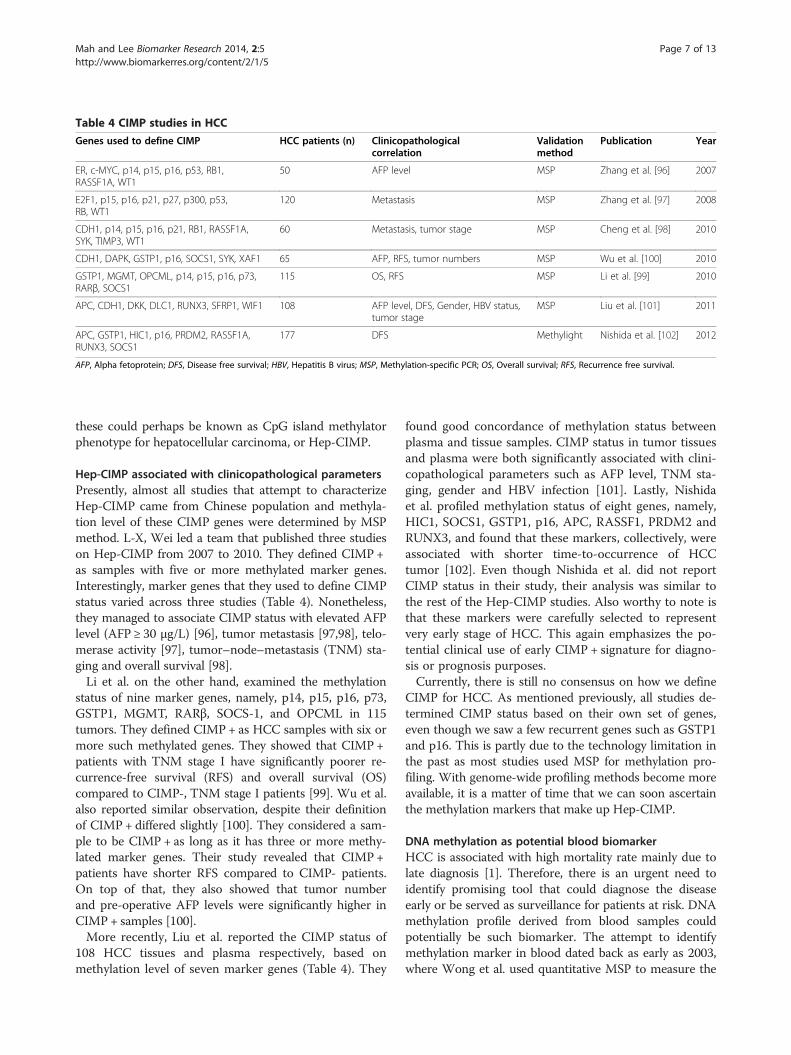
Table 4 CIMP studies in HCC
Genes used to define CIMP HCC patients (n) Clinicopathologicalcorrelation
Validationmethod
Publication Year
ER, c-MYC, p14, p15, p16, p53, RB1,RASSF1A, WT1
50 AFP level MSP Zhang et al. [96] 2007
E2F1, p15, p16, p21, p27, p300, p53,RB, WT1
120 Metastasis MSP Zhang et al. [97] 2008
CDH1, p14, p15, p16, p21, RB1, RASSF1A,SYK, TIMP3, WT1
60 Metastasis, tumor stage MSP Cheng et al. [98] 2010
CDH1, DAPK, GSTP1, p16, SOCS1, SYK, XAF1 65 AFP, RFS, tumor numbers MSP Wu et al. [100] 2010
GSTP1, MGMT, OPCML, p14, p15, p16, p73,RARβ, SOCS1
115 OS, RFS MSP Li et al. [99] 2010
APC, CDH1, DKK, DLC1, RUNX3, SFRP1, WIF1 108 AFP level, DFS, Gender, HBV status,tumor stage
MSP Liu et al. [101] 2011
APC, GSTP1, HIC1, p16, PRDM2, RASSF1A,RUNX3, SOCS1
177 DFS Methylight Nishida et al. [102] 2012
AFP, Alpha fetoprotein; DFS, Disease free survival; HBV, Hepatitis B virus; MSP, Methylation-specific PCR; OS, Overall survival; RFS, Recurrence free survival.
Mah and Lee Biomarker Research 2014, 2:5 Page 7 of 13http://www.biomarkerres.org/content/2/1/5
these could perhaps be known as CpG island methylatorphenotype for hepatocellular carcinoma, or Hep-CIMP.
Hep-CIMP associated with clinicopathological parametersPresently, almost all studies that attempt to characterizeHep-CIMP came from Chinese population and methyla-tion level of these CIMP genes were determined by MSPmethod. L-X, Wei led a team that published three studieson Hep-CIMP from 2007 to 2010. They defined CIMP +as samples with five or more methylated marker genes.Interestingly, marker genes that they used to define CIMPstatus varied across three studies (Table 4). Nonetheless,they managed to associate CIMP status with elevated AFPlevel (AFP ≥ 30 μg/L) [96], tumor metastasis [97,98], telo-merase activity [97], tumor–node–metastasis (TNM) sta-ging and overall survival [98].Li et al. on the other hand, examined the methylation
status of nine marker genes, namely, p14, p15, p16, p73,GSTP1, MGMT, RARβ, SOCS-1, and OPCML in 115tumors. They defined CIMP + as HCC samples with six ormore such methylated genes. They showed that CIMP +patients with TNM stage I have significantly poorer re-currence-free survival (RFS) and overall survival (OS)compared to CIMP-, TNM stage I patients [99]. Wu et al.also reported similar observation, despite their definitionof CIMP + differed slightly [100]. They considered a sam-ple to be CIMP + as long as it has three or more methy-lated marker genes. Their study revealed that CIMP+patients have shorter RFS compared to CIMP- patients.On top of that, they also showed that tumor numberand pre-operative AFP levels were significantly higher inCIMP + samples [100].More recently, Liu et al. reported the CIMP status of
108 HCC tissues and plasma respectively, based onmethylation level of seven marker genes (Table 4). They
found good concordance of methylation status betweenplasma and tissue samples. CIMP status in tumor tissuesand plasma were both significantly associated with clini-copathological parameters such as AFP level, TNM sta-ging, gender and HBV infection [101]. Lastly, Nishidaet al. profiled methylation status of eight genes, namely,HIC1, SOCS1, GSTP1, p16, APC, RASSF1, PRDM2 andRUNX3, and found that these markers, collectively, wereassociated with shorter time-to-occurrence of HCCtumor [102]. Even though Nishida et al. did not reportCIMP status in their study, their analysis was similar tothe rest of the Hep-CIMP studies. Also worthy to note isthat these markers were carefully selected to representvery early stage of HCC. This again emphasizes the po-tential clinical use of early CIMP + signature for diagno-sis or prognosis purposes.Currently, there is still no consensus on how we define
CIMP for HCC. As mentioned previously, all studies de-termined CIMP status based on their own set of genes,even though we saw a few recurrent genes such as GSTP1and p16. This is partly due to the technology limitation inthe past as most studies used MSP for methylation pro-filing. With genome-wide profiling methods become moreavailable, it is a matter of time that we can soon ascertainthe methylation markers that make up Hep-CIMP.
DNA methylation as potential blood biomarkerHCC is associated with high mortality rate mainly due tolate diagnosis [1]. Therefore, there is an urgent need toidentify promising tool that could diagnose the diseaseearly or be served as surveillance for patients at risk. DNAmethylation profile derived from blood samples couldpotentially be such biomarker. The attempt to identifymethylation marker in blood dated back as early as 2003,where Wong et al. used quantitative MSP to measure the
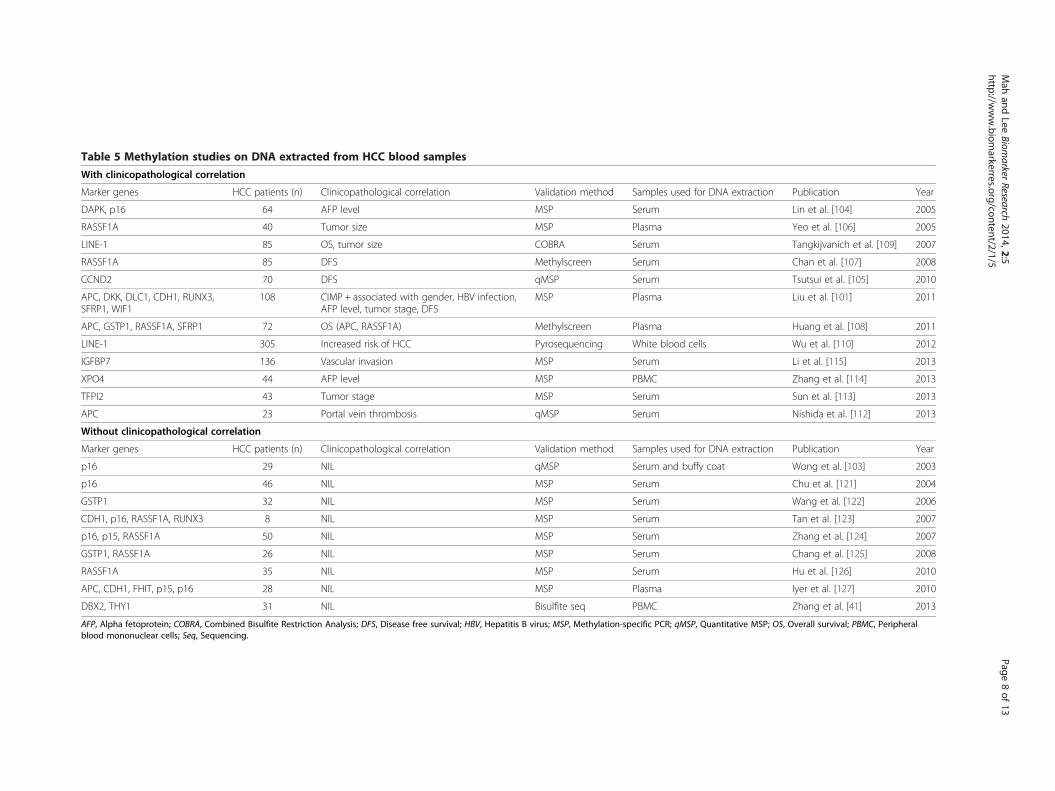
Table 5 Methylation studies on DNA extracted from HCC blood samples
With clinicopathological correlation
Marker genes HCC patients (n) Clinicopathological correlation Validation method Samples used for DNA extraction Publication Year
DAPK, p16 64 AFP level MSP Serum Lin et al. [104] 2005
RASSF1A 40 Tumor size MSP Plasma Yeo et al. [106] 2005
LINE-1 85 OS, tumor size COBRA Serum Tangkijvanich et al. [109] 2007
RASSF1A 85 DFS Methylscreen Serum Chan et al. [107] 2008
CCND2 70 DFS qMSP Serum Tsutsui et al. [105] 2010
APC, DKK, DLC1, CDH1, RUNX3,SFRP1, WIF1
108 CIMP + associated with gender, HBV infection,AFP level, tumor stage, DFS
MSP Plasma Liu et al. [101] 2011
APC, GSTP1, RASSF1A, SFRP1 72 OS (APC, RASSF1A) Methylscreen Plasma Huang et al. [108] 2011
LINE-1 305 Increased risk of HCC Pyrosequencing White blood cells Wu et al. [110] 2012
IGFBP7 136 Vascular invasion MSP Serum Li et al. [115] 2013
XPO4 44 AFP level MSP PBMC Zhang et al. [114] 2013
TFPI2 43 Tumor stage MSP Serum Sun et al. [113] 2013
APC 23 Portal vein thrombosis qMSP Serum Nishida et al. [112] 2013
Without clinicopathological correlation
Marker genes HCC patients (n) Clinicopathological correlation Validation method Samples used for DNA extraction Publication Year
p16 29 NIL qMSP Serum and buffy coat Wong et al. [103] 2003
p16 46 NIL MSP Serum Chu et al. [121] 2004
GSTP1 32 NIL MSP Serum Wang et al. [122] 2006
CDH1, p16, RASSF1A, RUNX3 8 NIL MSP Serum Tan et al. [123] 2007
p16, p15, RASSF1A 50 NIL MSP Serum Zhang et al. [124] 2007
GSTP1, RASSF1A 26 NIL MSP Serum Chang et al. [125] 2008
RASSF1A 35 NIL MSP Serum Hu et al. [126] 2010
APC, CDH1, FHIT, p15, p16 28 NIL MSP Plasma Iyer et al. [127] 2010
DBX2, THY1 31 NIL Bisulfite seq PBMC Zhang et al. [41] 2013
AFP, Alpha fetoprotein; COBRA, Combined Bisulfite Restriction Analysis; DFS, Disease free survival; HBV, Hepatitis B virus; MSP, Methylation-specific PCR; qMSP, Quantitative MSP; OS, Overall survival; PBMC, Peripheralblood mononuclear cells; Seq, Sequencing.
Mah
andLee
Biomarker
Research2014,2:5
Page8of
13http://w
ww.biom
arkerres.org/content/2/1/5

Mah and Lee Biomarker Research 2014, 2:5 Page 9 of 13http://www.biomarkerres.org/content/2/1/5
methylation status of p16 in 29 HCC patients [103]. How-ever, they did not perform any clinical association withtheir data. In fact, many studies successfully measure theaberrant methylation level of a marker gene in blood butdid not associate it with clinicopathological parameters.These studies can be found in Table 5.In this section, we will only highlight reports with sig-
nificant clinical association. As shown in Table 5, Linet al. showed that among 64 patients, about 77% of themhave p16 methylation and 41% of them have DAPKmethylation. Both markers were associated with AFPlevels but no other parameters [104]. Tsutsui et al. foundthat the serum of 39 out of 70 patients were positive formethylated CCND2 gene. Patients of this group hadshorter disease free survival [105].Yeo et al. used MSP and found that 17 out of 40 pa-
tients’ plasma (42.5%) had RASSF1A hypermethylationand their methylation status was associated with tumorsize [106]. Chan et al. used another method, methylation-sensitive restriction enzyme-mediated real-time PCR sys-tem, to detect RASSF1A methylation status in 85 HCCsera. They found that 93% of them have hypermethylationand their methylated status was associated with shorterdisease free survival and time-to-occurrence for HCC[107]. Using similar detection method, Huang et al. alsoshowed that RASSF1A gene in 72 patients’ blood washypermethylated compared to normal controls and thatits methylation level was associated with poorer overallsurvival [108].Tangkijvanich et al. used Combined Bisulfite Restriction
Analysis (COBRA) method to measure the hypomethyla-tion of LINE-1 in 85 patients’ sera. They reported that hy-pomethylation of LINE-1 was associated with HBVinfection, larger tumor size and more advance diseasestage [109]. Their study was further validated by Wu et al.,where they used pyrosequencing to determine the methy-lation level of LINE-1 in 305 patients’ white blood cellDNA [110]. They used logistic regression model to showthat hypomethylation of LINE-1 increased overall risk ofdeveloping HCC. Recently, Gao et al. also reported thatLINE-1 was hypomethylated in 71 HCC tissues and wasassociated with poorer prognosis [111]. These are fewstudies that showed hypomethylation instead of hyperme-thylation as potential prognostic biomarker.Following the availability of genome-wide methylation
profile, we also saw a sudden surge of methylation studiesbased on patients’ sera. Within year 2013, four studies re-ported prognostic value of four different hypermethylatedgenes. Briefly, Nishida et al. performed quantitative MSPand showed that APC was more methylated in 23 HCCsera compared to healthy volunteers. They also showedthat patients with higher APC methylation were associ-ated with portal vein thrombosis [112]. Sun et al. detectedTFPI2 to be more methylated in 43 HCC sera and its level
was associated with TNM stage [113]. Zhang et al. on theother hand found XPO4 to be frequently methylated in 44patients’ peripheral blood mononuclear cells. Their dataindicated that higher XPO4 methylation was associatedwith higher AFP level [114]. Lastly, Li et al. discoveredthat in HBV-associated HCC, IGFBP7 was more methy-lated compared to chronic hepatitis B patients and normalcontrols. Also, its methylation status was associated withvascular invasion in HCC [115].Clearly, methylation of marker gene in HCC blood
DNA has potential prognostic value as shown by its as-sociation with clinicopathological data. However, mostcurrent studies drawn conclusion from a retrospectivecohort. In order to translate these markers into actualclinical use, proper prospective studies and validationmethod are required.
ConclusionsMany genome wide methylation studies have confirmedthat HCC has distinct methylation profile (Table 2), andsome even showed that it is associated with different etio-logical factors such as HBV infection and alcohol con-sumption. Undeniably, the availability of these genomewide data has allowed the discovery of many novel geneswith aberrant methylation, especially in recent years. Asshown in Additional file 1: Table S1, apart from the com-monly studied genes mentioned in this review, there isplethora of genes that were differentially methylated andassociated with clinicopathological data. Future studiesneed to focus on collating current available data, shortlis-ting potential methylation markers by conducting propervalidation method [116,117] and defining well-character-ized CIMP status of HCC. It is hoped that emergingmethylation markers can be used as diagnostic or prog-nostic marker for HCC in near future.
Additional file
Additional file 1: Table S1. List of methylation studies on HCC fromyear 2003–2013.
AbbreviationsAFP: Alpha fetoprotein; CIMP: CpG island methylator phenotype;COBRA: Combined bisulfite restriction analysis; DFS: Disease free survival;DMH-chip: Differential methylation hybridization on microarray; DNMT: DNAmethyltransferase; HBV: Hepatitis B virus; HBx: Hepatitis B virus X protein;HCC: Hepatocellular Carcinoma; HCV: Hepatitis C virus; MCAM: MethylatedCpG island amplification microarray; MeDIP-Chip: Methylated DNAimmunoprecipitation microarray; MSP: Methylation-specific PCR; OS: Overallsurvival; PBMC: Peripheral blood mononuclear cells; PCR: Polymerase chainreaction; QMSP: Quantitative MSP; RFS: Recurrence free survival.
Competing interestsAll authors declare that they have no conflicts of interests.
Authors’ contributionsCGL and W-C researched data and wrote the manuscript. Both authors readand approved the final manuscript.

Mah and Lee Biomarker Research 2014, 2:5 Page 10 of 13http://www.biomarkerres.org/content/2/1/5
AcknowledgementsThis work is supported by grants from the National Medical Research Council(NMRC) (NMRC/1131/2007 and NMRC/1238/2009), the BioMedical ResearchCouncil (BMRC) (BMRC06/1/21/19/449) of Singapore and the SingaporeMillennium Foundation (SMF) as well as block fundings from the NationalCancer Centre, SINGAPORE and the DUKE-NUS Graduate Medical School toA/Prof Caroline Lee.
Author details1Department of Biochemistry, Yong Loo Lin School of Medicine, NationalUniversity of Singapore, Singapore 117597, Singapore. 2Division of MedicalSciences, Humphrey Oei Institute of Cancer Research, National Cancer CentreSingapore, Level 6, Lab 5, 11 Hospital Drive, Singapore 169610, Singapore.3NUS Graduate School for Integrative Sciences and Engineering, NationalUniversity of Singapore, Singapore 117456, Singapore. 4Duke-NUS GraduateMedical School, Singapore 169857, Singapore.
Received: 2 February 2014 Accepted: 7 March 2014Published: 17 March 2014
References1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D: Global cancer
statistics. CA Cancer J Clin 2011, 61(2):69–90.2. Villanueva A, Minguez B, Forner A, Reig M, Llovet JM: Hepatocellular
carcinoma: novel molecular approaches for diagnosis, prognosis, andtherapy. Annu Rev Med 2010, 61:317–328.
3. Hoofnagle JH: Hepatocellular carcinoma: summary andrecommendations. Gastroenterology 2004, 127(5 Suppl 1):S319–S323.
4. Yang JD, Roberts LR: Hepatocellular carcinoma: a global view. Nat RevGastroenterol Hepatol 2010, 7(8):448–458.
5. Poon D, Anderson BO, Chen LT, Tanaka K, Lau WY, van Cutsem E, Singh H,Chow WC, Ooi LL, Chow P, Khin MW, Koo WH: Management ofhepatocellular carcinoma in Asia: consensus statement from the AsianOncology Summit 2009. Lancet Oncol 2009, 10(11):1111–1118.
6. Colli A, Fraquelli M, Casazza G, Massironi S, Colucci A, Conte D, Duca P:Accuracy of ultrasonography, spiral CT, magnetic resonance, andalpha-fetoprotein in diagnosing hepatocellular carcinoma: a systematicreview: CME. Am J Gastroenterol 2006, 101(3):513–523.
7. Terentiev AA, Moldogazieva NT: Alpha-fetoprotein: a renaissance.Tumour Biol 2013, 34(4):2075–2091.
8. Song P, Tobe RG, Inagaki Y, Kokudo N, Hasegawa K, Sugawara Y, Tang W:The management of hepatocellular carcinoma around the world:a comparison of guidelines from 2001 to 2011. Liver Int 2012,32(7):1053–1063.
9. Llovet JM, Bru C, Bruix J: Prognosis of hepatocellular carcinoma: the BCLCstaging classification. Semin Liver Dis 1999, 19(3):329–338.
10. Borel F, Konstantinova P, Jansen PLM: Diagnostic and therapeuticpotential of miRNA signatures in patients with hepatocellular carcinoma.J Hepatol 2012, 56(6):1371–1383.
11. Budhu A, Jia HL, Forgues M, Liu CG, Goldstein D, Lam A, Zanetti KA, Ye QH,Qin LX, Croce CM, Tang ZY, Xin WW: Identification of metastasis-relatedmicroRNAs in hepatocellular carcinoma. Hepatology 2008, 47(3):897–907.
12. Hoshida Y, Nijman SMB, Kobayashi M, Chan JA, Brunet JP, Chiang DY,Villanueva A, Newell P, Ikeda K, Hashimoto M, Watanabe G, Gabriel S,Friedman SL, Kumada H, Llovet JM, Golub TR: Integrative transcriptomeanalysis reveals common molecular subclasses of human hepatocellularcarcinoma. Cancer Res 2009, 69(18):7385–7392.
13. Lee JS, Thorgeirsson SS: Genome-scale profiling of gene expression inhepatocellular carcinoma: classification, survival prediction, andidentification of therapeutic targets. Gastroenterology 2004,127(SUPPL):S51–S55.
14. Lemmer ER, Friedman SL, Llovet JM: Molecular diagnosis of chronic liverdisease and hepatocellular carcinoma: the potential of gene expressionprofiling. Semin Liver Dis 2006, 26(4):373–384.
15. Villanueva A, Hoshida Y, Battiston C, Tovar V, Sia D, Alsinet C, Cornella H,Liberzon A, Kobayashi M, Kumada H, Thung SN, Bruix J, Newell P, April C,Fan J, Roayaie S, Mazzaferro V, Schwartz ME, Llovet JM: Combining clinical,pathology, and gene expression data to predict recurrence ofhepatocellular carcinoma. Gastroenterology 2011, 140(5):1501–1512. e1502.
16. Minouchi K, Kaneko S, Kobayashi K: Mutation of p53 gene in regenerativenodules in cirrhotic liver. J Hepatol 2002, 37(2):231–239.
17. Bressac B, Kew M, Wands J, Ozturk M: Selective G to T mutations of p53gene in hepatocellular carcinoma from southern Africa. Nature 1991,350(6317):429–431.
18. Ishizaki Y, Ikeda S, Fujimori M, Shimizu Y, Kurihara T, Itamoto T, Kikuchi A,Okajima M, Asahara T: Immunohistochemical analysis and mutationalanalyses of beta-catenin, Axin family and APC genes in hepatocellularcarcinomas. Int J Oncol 2004, 24(5):1077–1083.
19. Edamoto Y, Hara A, Biernat W, Terracciano L, Cathomas G, Riehle HM,Matsuda M, Fujii H, Scoazec JY, Ohgaki H: Alterations of RB1, p53 and Wntpathways in hepatocellular carcinomas associated with hepatitis C,hepatitis B and alcoholic liver cirrhosis. Int J Cancer 2003,106(3):334–341.
20. Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T,Ishiguro H, Fujita M, Tokino T, Sasaki Y, Imaoka S, Murata M, Shimano T,Yamaoka Y, Nakamura Y: AXIN1 mutations in hepatocellular carcinomas,and growth suppression in cancer cells by virus-mediated transfer ofAXIN1. Nat Genet 2000, 24(3):245–250.
21. Taniguchi K, Roberts LR, Aderca IN, Dong X, Qian C, Murphy LM, NagorneyDM, Burgart LJ, Roche PC, Smith DI, Ross JA, Liu W: Mutational spectrumof beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas andhepatoblastomas. Oncogene 2002, 21(31):4863–4871.
22. Han ZG: Functional genomic studies: insights into the pathogenesis ofliver cancer. Annu Rev Genom Hum G 2012, 13:171–205.
23. Pradhan S, Bacolla A, Wells RD, Roberts RJ: Recombinant human DNA(cytosine-5) methyltransferase. I. Expression, purification, andcomparison of de novo and maintenance methylation. J Biol Chem 1999,274(46):33002–33010.
24. Feinberg AP, Vogelstein B: Hypomethylation distinguishes genes of somehuman cancers from their normal counterparts. Nature 1983,301(5895):89–92.
25. Tischoff I, Tannapfel A: DNA methylation in hepatocellular carcinoma.World J Gastroentero 2008, 14(11):1741–1748.
26. Park IY, Sohn BH, Yu E, Suh DJ, Chung Y, Lee J, Surzycki SJ, Lee YI: Aberrantepigenetic modifications in hepatocarcinogenesis induced by hepatitis Bvirus X protein. Gastroenterology 2007, 132(4):1476–1494.
27. Lim JS, Park SH, Jang KL: Hepatitis C virus core protein overcomesstress-induced premature senescence by down-regulating p16expression via DNA methylation. Cancer Lett 2012, 321(2):154–161.
28. Arora P, Kim EO, Jung JK, Jang KL: Hepatitis C virus core protein downregulatesE-cadherin expression via activation of DNA methyltransferase 1 and 3b.Cancer Lett 2008, 261(2):244–252.
29. Nagai M, Nakamura A, Makino R, Mitamura K: Expression of DNA (5-cytosin)-methyltransferases (DNMTs) in hepatocellular carcinomas. Hepatol Res 2003,26(3):186–191.
30. Park HJ, Yu E, Shim YH: DNA methyltransferase expression and DNAhypermethylation in human hepatocellular carcinoma. Cancer Lett 2006,233(2):271–278.
31. Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, Gunderson KL:Genome-wide DNA methylation profiling using Infinium(R) assay.Epigenomics 2009, 1(1):177–200.
32. Touleimat N, Tost J: Complete pipeline for Infinium((R)) HumanMethylation 450 K BeadChip data processing using subset quantilenormalization for accurate DNA methylation estimation. Epigenomics2012, 4(3):325–341.
33. Estécio MRH, Yan PS, Ibrahim AEK, Tellez CS, Shen L, Huang THM, Issa JPJ:High-throughput methylation profiling by MCA coupled to CpG islandmicroarray. Genome Res 2007, 17(10):1529–1536.
34. Huang TH, Perry MR, Laux DE: Methylation profiling of CpG islands inhuman breast cancer cells. Hum Mol Genet 1999, 8(3):459–470.
35. Mohn F, Weber M, Schubeler D, Roloff TC: Methylated DNAimmunoprecipitation (MeDIP). Methods Mol Biol 2009, 507:55–64.
36. Gao W, Kondo Y, Shen L, Shimizu Y, Sano T, Yamao K, Natsume A, Goto Y,Ito M, Murakami H, Osada H, Zhang J, Issa JPJ, Sekido Y: Variable DNAmethylation patterns associated with progression of disease inhepatocellular carcinomas. Carcinogenesis 2008, 29(10):1901–1910.
37. Lu CY, Hsieh SY, Lu YJ, Wu CS, Chen LC, Lo SJ, Wu CT, Chou MY, HuangTHM, Chang YS: Aberrant DNA methylation profile and frequentmethylation of KLK10 and OXGR1 genes in hepatocellular carcinoma.Genes Chromosomes Cancer 2009, 48(12):1057–1068.
38. Deng YB, Nagae G, Midorikawa Y, Yagi K, Tsutsumi S, Yamamoto S,Hasegawa K, Kokudo N, Aburatani H, Kaneda A: Identification of genes

Mah and Lee Biomarker Research 2014, 2:5 Page 11 of 13http://www.biomarkerres.org/content/2/1/5
preferentially methylated in hepatitis C virus-related hepatocellularcarcinoma. Cancer Sci 2010, 101(6):1501–1510.
39. Stefanska B, Huang J, Bhattacharyya B, Suderman M, Hallett M, Han ZG,Szyf M: Definition of the landscape of promoter DNA hypomethylation inliver cancer. Cancer Res 2011, 71(17):5891–5903.
40. Shitani M, Sasaki S, Akutsu N, Takagi H, Suzuki H, Nojima M, Yamamoto H,Tokino T, Hirata K, Imai K, Toyota M, Shinomura Y: Genome-wide analysisof DNA methylation identifies novel cancer-related genes inhepatocellular carcinoma. Tumor Biol 2012, 33(5):1307–1317.
41. Zhang P, Wen X, Gu F, Deng X, Li J, Dong J, Jiao J, Tian Y: Methylationprofiling of serum DNA from hepatocellular carcinoma patients using anInfinium Human Methylation 450 BeadChip. Hepatol Int 2013,7(3):893–900.
42. Shen J, Wang S, Zhang YJ, Kappil M, Wu HC, Kibriya MG, Wang Q, Jasmine F,Ahsan H, Lee PH, Yu MW, Chen CJ, Santella RM: Genome-wide DNAmethylation profiles in hepatocellular carcinoma. Hepatology 2012,55(6):1799–1808.
43. Hernandez-Vargas H, Lambert MP, le Calvez-Kelm F, Gouysse G, McKay-Chopin S, Tavtigian SV, Scoazec JY, Herceg Z: Hepatocellular carcinomadisplays distinct DNA methylation signatures with potential as clinicalpredictors. PLoS One 2010, 5(3):e9749.
44. Tao R, Li J, Xin J, Wu J, Guo J, Zhang L, Jiang L, Zhang W, Yang Z, Li L:Methylation profile of single hepatocytes derived from hepatitis Bvirus-related hepatocellular carcinoma. PLoS One 2011, 6(5):e19862.
45. Song MA, Tiirikainen M, Kwee S, Okimoto G, Yu H, Wong LL: Elucidatingthe landscape of aberrant DNA methylation in hepatocellular carcinoma.PLoS One 2013, 8(2):e55761.
46. Shen J, Wang S, Zhang YJ, Wu HC, Kibriya MG, Jasmine F, Ahsan H,Wu DPH, Siegel AB, Remotti H, Santella RM: Exploring genome-wideDNA methylation profiles altered in hepatocellular carcinoma usingInfinium Human Methylation 450 BeadChips. Epigenetics 2013,8(1):34–43.
47. Hou X, Peng JX, Hao XY, Cai JP, Liang LJ, Zhai JM, Zhang KS, Lai JM, Yin XY:DNA methylation profiling identifies EYA4 gene as a prognosticmolecular marker in hepatocellular carcinoma. Ann Surg Oncol 2013.in press.
48. Ammerpohl O, Pratschke J, Schafmayer C, Haake A, Faber W, von Kampen O,Brosch M, Sipos B, von Schönfels W, Balschun K, Röcken C, Arlt A, Schniewind B,Grauholm J, Kalthoff H, Neuhaus P, Stickel F, Schreiber S, Becker T, Siebert R,Hampe J: Distinct DNA methylation patterns in cirrhotic liver andhepatocellular carcinoma. Int J Cancer 2012, 130(6):1319–1328.
49. Archer KJ, Mas VR, Maluf DG, Fisher RA: High-throughput assessment ofCpG site methylation for distinguishing between HCV-cirrhosis andHCV-associated hepatocellular carcinoma. Mol Genet Genomics 2010,283(4):341–349.
50. Neumann O, Kesselmeier M, Geffers R, Pellegrino R, Radlwimmer B,Hoffmann K, Ehemann V, Schemmer P, Schirmacher P, Bermejo JL,Longerich T: Methylome analysis and integrative profiling of humanHCCs identify novel protumorigenic factors. Hepatology 2012,56(5):1817–1827.
51. Matsumura S, Imoto I, Kozaki KI, Matsui T, Muramatsu T, Furuta M, Tanaka S,Sakamoto M, Arii S, Inazawa J: Integrative array-based approach identifiesMZB1 as a frequently methylated putative tumor suppressor inhepatocellular carcinoma. Clin Cancer Res 2012, 18(13):3541–3551.
52. Okamura Y, Nomoto S, Hayashi M, Hishida M, Nishikawa Y, Yamada S, Fujii T,Sugimoto H, Takeda S, Kodera Y, Nakao A: Identification of the bleomycinhydrolase gene as a methylated tumor suppressor gene inhepatocellular carcinoma using a novel triple-combination array method.Cancer Lett 2011, 312(2):150–157.
53. Revill K, Wang T, Lachenmayer A, Kojima K, Harrington A, Li J, Hoshida Y,Llovet JM, Powers S: Genome-wide methylation analysis and epigeneticunmasking identify tumor suppressor genes in hepatocellular carcinoma.Gastroenterology 2013, 145(6):1424–1435.
54. Yang B, Guo M, Herman JG, Clark DP: Aberrant promoter methylationprofiles of tumor suppressor genes in hepatocellular carcinoma.Am J Pathol 2003, 163(3):1101–1107.
55. Lee S, Lee HJ, Kim JH, Lee HS, Jang JJ, Kang GH: Aberrant CpG islandhypermethylation along multistep hepatocarcinogenesis. Am J Pathol2003, 163(4):1371–1378.
56. Yu J, Zhang HY, Ma ZZ, Lu W, Wang YF, Zhu J: Methylation profiling oftwenty four genes and the concordant methylation behaviours of
nineteen genes that may contribute to hepatocellular carcinogenesis.Cell Res 2003, 13(5):319–333.
57. Liggett WH, Sidransky D: Role of the p16 tumor suppressor gene incancer. J Clin Oncol 1998, 16(3):1197–1206.
58. Esteller M, Corn PG, Baylin SB, Herman JG: A gene hypermethylationprofile of human cancer. Cancer Res 2001, 61(8):3225–3229.
59. Shim YH, Yoon GS, Choi HJ, Chung YH, Yu E: p16 Hypermethylation in theearly stage of hepatitis B virus-associated hepatocarcinogenesis.Cancer Lett 2003, 190(2):213–219.
60. Su PF, Lee TC, Lin PJ, Lee PH, Jeng YM, Chen CH, Liang JD, Chiou LL,Huang GT, Lee HS: Differential DNA methylation associated withhepatitis B virus infection in hepatocellular carcinoma. Int J Cancer 2007,121(6):1257–1264.
61. Katoh H, Shibata T, Kokubu A, Ojima H, Fukayama M, Kanai Y, Hirohashi S:Epigenetic instability and chromosomal instability in hepatocellularcarcinoma. Am J Pathol 2006, 168(4):1375–1384.
62. Li X, Hei AM, Sun L, Hasegawa K, Torzilli G, Minagawa M, Takayama T,Makuuchi M: p16INK4A hypermethylation is associated with hepatitisvirus infection, age, and gender in hepatocellular carcinoma. Clin CancerRes 2004, 10(22):7484–7489.
63. Jicai Z, Zongtao Y, Jun L, Haiping L, Jianmin W, Lihua H: Persistentinfection of hepatitis B virus is involved in high rate of p16 methylationin hepatocellular carcinoma. Mol Carcinog 2006, 45(7):530–536.
64. Feng Q, Stern JE, Hawes SE, Lu H, Jiang M, Kiviat NB: DNA methylationchanges in normal liver tissues and hepatocellular carcinoma withdifferent viral infection. Exp Mol Pathol 2010, 88(2):287–292.
65. Zhu R, Li BZ, Li H, Ling YQ, Hu XQ, Zhai WR, Zhu HG: Association ofp16INK4A hypermethylation with hepatitis B virus X protein expressionin the early stage of HBV-associated hepatocarcinogenesis. Pathol Int2007, 57(6):328–336.
66. Qin Y, Liu JY, Li B, Sun ZL, Sun ZF: Association of low p16INK4a andp15INK4b mRNAs expression with their CpG islands methylation withhuman hepatocellular carcinogenesis. World J Gastroenterol 2004,10(9):1276–1280.
67. Kikuchi LO, Paranagua-Vezozzo DC, Chagas AL, Mello ES, Alves VA,Farias AQ, Pietrobon R, Carrilho FJ: Nodules less than 20 mm and vascularinvasion are predictors of survival in small hepatocellular carcinoma.J Clin Gastroenterol 2009, 43(2):191–195.
68. Tamura S, Kato T, Berho M, Misiakos EP, O’Brien C, Reddy KR, Nery JR,Burke GW, Schiff ER, Miller J, Tzakis AG: Impact of histological grade ofhepatocellular carcinoma on the outcome of liver transplantation.Arch Surg 2001, 136(1):25–30.
69. Jonas S, Bechstein WO, Steinmuller T, Herrmann M, Radke C, Berg T,Settmacher U, Neuhaus P: Vascular invasion and histopathologic gradingdetermine outcome after liver transplantation for hepatocellularcarcinoma in cirrhosis. Hepatology 2001, 33(5):1080–1086.
70. Ko E, Kim Y, Kim SJ, Joh JW, Song S, Park CK, Park J, Kim DH: Promoterhypermethylation of the p16 gene is associated with poor prognosis inrecurrent early-stage hepatocellular carcinoma. Cancer EpidemiolBiomarkers Prev 2008, 17(9):2260–2267.
71. Yoshikawa H, Matsubara K, Qian GS, Jackson P, Groopman JD, Manning JE,Harris CC, Herman JG: SOCS-1, a negative regulator of the JAK/STATpathway, is silenced by methylation in human hepatocellular carcinomaand shows growth-suppression activity. Nat Genet 2001, 28(1):29–35.
72. Um TH, Kim H, Oh BK, Kim MS, Kim KS, Jung G, Park YN: Aberrant CpGisland hypermethylation in dysplastic nodules and early HCC of hepatitisB virus-related human multistep hepatocarcinogenesis. J Hepatol 2011,54(5):939–947.
73. Ko E, Kim SJ, Joh JW, Park CK, Park J, Kim DH: CpG island hypermethylationof SOCS-1 gene is inversely associated with HBV infection inhepatocellular carcinoma. Cancer Lett 2008, 271(2):240–250.
74. Zhang X, Wang J, Cheng J, Ding S, Li M, Sun S, Zhang L, Liu S, Chen X,Zhuang H, Lu F: An integrated analysis of SOCS1 down-regulation in HBVinfection-related hepatocellular carcinoma. J Viral Hepat 2013. in press.
75. Chu PY, Yeh CM, Hsu NC, Chang YS, Chang JG, Yeh KT: Epigeneticalteration of the SOCS1 gene in hepatocellular carcinoma. Swiss MedWkly 2010, 140.
76. Nishida N, Nagasaka T, Nishimura T, Ikai I, Boland CR, Goel A: Aberrantmethylation of multiple tumor suppressor genes in aging liver,chronic hepatitis, and hepatocellular carcinoma. Hepatology 2008,47(3):908–918.

Mah and Lee Biomarker Research 2014, 2:5 Page 12 of 13http://www.biomarkerres.org/content/2/1/5
77. Okochi O, Hibi K, Sakai M, Inoue S, Takeda S, Kaneko T, Nakao A:Methylation-mediated silencing of SOCS-1 gene in hepatocellularcarcinoma derived from cirrhosis. Clin Cancer Res 2003, 9(14):5295–5298.
78. Freeman AJ, Dore GJ, Law MG, Thorpe M, von Overbeck J, Lloyd AR,Marinos G, Kaldor JM: Estimating progression to cirrhosis in chronichepatitis C virus infection. Hepatology 2001, 34(4 Pt 1):809–816.
79. Laborde E: Glutathione transferases as mediators of signaling pathwaysinvolved in cell proliferation and cell death. Cell Death Differ 2010,17(9):1373–1380.
80. Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, Bova GS, Hsieh WS,Isaacs WB, Nelson WG: Cytidine methylation of regulatory sequences nearthe pi-class glutathione S-transferase gene accompanies human prostaticcarcinogenesis. Proc Natl Acad Sci USA 1994, 91(24):11733–11737.
81. Lambert MP, Paliwal A, Vaissière T, Chemin I, Zoulim F, Tommasino M,Hainaut P, Sylla B, Scoazec JY, Tost J, Herceg Z: Aberrant DNA methylationdistinguishes hepatocellular carcinoma associated with HBV and HCVinfection and alcohol intake. J Hepatol 2011, 54(4):705–715.
82. Zhang YJ, Chen Y, Ahsan H, Lunn RM, Chen SY, Lee PH, Chen CJ, Santella RM:Silencing of glutathione S-transferase P1 by promoter hypermethylationand its relationship to environmental chemical carcinogens inhepatocellular carcinoma. Cancer Lett 2005, 221(2):135–143.
83. Chang HW, Chow V, Lam KY, Wei WI, WingYuen AP: Loss of E-cadherinexpression resulting from promoter hypermethylation in oral tonguecarcinoma and its prognostic significance. Cancer 2002, 94(2):386–392.
84. Kanazawa T, Watanabe T, Kazama S, Tada T, Koketsu S, Nagawa H: Poorlydifferentiated adenocarcinoma and mucinous carcinoma of the colonand rectum show higher rates of loss of heterozygosity and loss ofE-cadherin expression due to methylation of promoter region.Int J Cancer 2002, 102(3):225–229.
85. Graziano F, Arduini F, Ruzzo A, Bearzi I, Humar B, More H, Silva R, Muretto P,Guilford P, Testa E, Mari D, Magnani M, Cascinu S: Prognostic analysis ofE-cadherin gene promoter hypermethylation in patients with surgicallyresected, node-positive, diffuse gastric cancer. Clin Cancer Res 2004,10(8):2784–2789.
86. Takeno S, Noguchi T, Fumoto S, Kimura Y, Shibata T, Kawahara K:E-cadherin expression in patients with esophageal squamos cellcarcinoma: Promoter hypermethylation, Snail overexpression, andclinicopathologic implications. Am J Clin Pathol 2004, 122(1):78–84.
87. Shimamoto T, Ohyashiki JH, Ohyashiki K: Methylation of p15INK4b andE-cadherin genes is independently correlated with poor prognosis inacute myeloid leukemia. Leuk Res 2005, 29(6):653–659.
88. Caldeira JRF, Prando EC, Quevedo FC, Moraes Neto FA, Rainho CA, Rogatto SR:CDH1 promoter hypermethylation and E-cadherin protein expression ininfiltrating breast cancer. BMC Cancer 2006, 6:48.
89. Lim SO, Gu JM, Kim MS, Kim HS, Park YN, Park CK, Cho JW, Park YM, Jung G:Epigenetic changes induced by reactive oxygen species inhepatocellular carcinoma: methylation of the E-cadherin promoter.Gastroenterology 2008, 135(6):2128–2140.
90. Oh BK, Kim H, Park HJ, Shim YH, Choi J, Park C, Park YN: DNAmethyltransferase expression and DNA methylation in humanhepatocellular carcinoma and their clinicopathological correlation.Int J Mol Med 2007, 20(1):65–73.
91. Ghee YK, Byung CY, Kwang CK, Jae WC, Won SP, Cheol KP: Promotermethylation of E-cadherin in hepatocellular carcinomas and dysplasticnodules. J Korean Med Sci 2005, 20(2):242–247.
92. Hughes LA, Melotte V, de Schrijver J, de Maat M, Smit VT, Bovee JV, French PJ,van den Brandt PA, Schouten LJ, de Meyer T, van Criekinge W, Ahuja N,Herman JG, Weijenberg MP, van Engeland M: The CpG island methylatorphenotype: what’s in a name? Cancer Res 2013, 73(19):5858–5868.
93. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K,Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG,Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK,van den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J,Herman JG, Baylin SB, Laird PW, Aldape K: Identification of a CpGisland methylator phenotype that defines a distinct subgroup ofglioma. Cancer Cell 2010, 17(5):510–522.
94. Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R,Seshan V, Mo Q, Heguy A, Baylin SB, Ahuja N, Viale A, Massague J,Norton L, Vahdat LT, Moynahan ME, Chan TA: Breast cancermethylomes establish an epigenomic foundation for metastasis.Sci Transl Med 2011, 3(75):75ra25.
95. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP: CpG islandmethylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999,96(15):8681–8686.
96. Zhang C, Li Z, Cheng Y, Jia F, Li R, Wu M, Li K, Wei L: CpG islandmethylator phenotype association with elevated serum α-fetoproteinlevel in hepatocellular carcinoma. Clin Cancer Res 2007, 13(3):944–952.
97. Zhang C, Guo X, Jiang G, Zhang L, Yang Y, Shen F, Wu M, Wei L:CpG island methylator phenotype association with upregulatedtelomerase activity in hepatocellular carcinoma. Int J Cancer 2008,123(5):998–1004.
98. Cheng Y, Zhang C, Zhao J, Wang C, Xu Y, Han Z, Jiang G, Guo X, Li R,Bu X, Wu M, Wei L: Correlation of CpG island methylator phenotypewith poor prognosis in hepatocellular carcinoma. Exp Mol Pathol2010, 88(1):112–117.
99. Li B, Liu W, Wang L, Li M, Wang J, Huang L, Huang P, Yuan Y: CpG islandmethylator phenotype associated with tumor recurrence in tumor-node-metastasis stage I hepatocellular carcinoma. Ann Surg Oncol 2010,17(7):1917–1926.
100. Wu LM, Zhang F, Zhou L, Yang Z, Xie HY, Zheng SS: Predictive valueof CpG island methylator phenotype for tumor recurrence inhepatitis B virus-associated hepatocellular carcinoma following livertransplantation. BMC Cancer 2010, 10:399.
101. Liu JB, Zhang YX, Zhou SH, Shi MX, Cai J, Liu Y, Chen KP, Qiang FL: CpGIsland methylator phenotype in plasma is associated with hepatocellularcarcinoma prognosis. World J Gastroenterol 2011, 17(42):4718–4724.
102. Nishida N, Kudo M, Nagasaka T, Ikai I, Goel A: Characteristic patterns ofaltered DNA methylation predict emergence of human hepatocellularcarcinoma. Hepatology 2012, 56(3):994–1003.
103. Wong IHN, Zhang J, Lai PBS, Lau WY, Lo YMD: Quantitative analysis oftumor-derived methylated p16INK4a sequences in plasma, serum, andblood cells of hepatocellular carcinoma patients. Clin Cancer Res 2003,9(3):1047–1052.
104. Lin Q, Chen LB, Tang YM, Wang J: Promoter hypermethylation of p16gene and DAPK gene in sera from hepatocellular carcinoma (HCC)patients. Chin J Cancer Res 2005, 17(4):250–254.
105. Tsutsui M, Iizuka N, Moribe T, Miura T, Kimura N, Tamatsukuri S, Ishitsuka H,Fujita Y, Hamamoto Y, Tsunedomi R, Iida M, Tokuhisa Y, Sakamoto K,Tamesa T, Sakaida I, Oka M: Methylated cyclin D2 gene circulating in theblood as a prognosis predictor of hepatocellular carcinoma. Clin ChimActa 2010, 411(7–8):516–520.
106. Yeo W, Wong N, Wong WL, Lai PBS, Zhong S, Johnson PJ: High frequencyof promoter hypermethylation of RASSF1A in tumor and plasma ofpatients with hepatocellular carcinoma. Liver Int 2005, 25(2):266–272.
107. Chan KCA, Lai PBS, Mok TSK, Chan HLY, Ding C, Yeung SW, Lo YMD:Quantitative analysis of circulating methylated DNA as a biomarker forhepatocellular carcinoma. Clin Chem 2008, 54(9):1528–1536.
108. Huang ZH, Hu Y, Hua D, Wu YY, Song MX, Cheng ZH: Quantitative analysisof multiple methylated genes in plasma for the diagnosis and prognosisof hepatocellular carcinoma. Exp Mol Pathol 2011, 91(3):702–707.
109. Tangkijvanich P, Hourpai N, Rattanatanyong P, Wisedopas N, Mahachai V,Mutirangura A: Serum LINE-1 hypomethylation as a potential prognosticmarker for hepatocellular carcinoma. Clin Chim Acta 2007,379(1–2):127–133.
110. Wu HC, Wang Q, Yang HI, Tsai W, Chen CJ, Santella RM: Global dnamethylation levels in white blood cells as a biomarker for hepatocellularcarcinoma risk: a nested case–control study. Carcinogenesis 2012,33(7):1340–1345.
111. Gao XD, Qu JH, Chang XJ, Lu YY, Bai WL, Wang H, Xu ZX, An LJ,Wang CP, Zeng Z, Yang YP: Hypomethylation of long interspersednuclear element-1 promoter is associated with poor outcomes forcurative resected hepatocellular carcinoma. Liver Int 2013. in press.
112. Nishida N, Arizumi T, Takita M, Nagai T, Kitai S, Yada N, Hagiwara S, Inoue T,Minami Y, Ueshima K, Sakurai T, Ida H, Kudo M: Quantification of tumorDNA in serum and vascular invasion of human hepatocellular carcinoma.Oncology 2013, 84(SUPPL.1):82–87.
113. Sun FK, Fan YC, Zhao J, Zhang F, Gao S, Zhao ZH, Sun Q, Wang K:Detection of TFPI2 methylation in the serum of hepatocellularcarcinoma patients. Dig Dis Sci 2013, 58(4):1010–1015.
114. Zhang F, Fan YC, Mu NN, Zhao J, Sun FK, Zhao ZH, Gao S, Wang K:Exportin 4 gene expression and DNA promoter methylation status inchronic hepatitis B virus infection. J Viral Hepat 2013. in press.

Mah and Lee Biomarker Research 2014, 2:5 Page 13 of 13http://www.biomarkerres.org/content/2/1/5
115. Li F, Fan YC, Gao S, Sun FK, Yang Y, Wang K: Methylation of serum insulin-likegrowth factor-binding protein 7 promoter in hepatitis B virus-associatedhepatocellular carcinoma. Genes Chromosomes Cancer 2014, 53(1):90–97.
116. McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM:REporting recommendations for tumor MARKer prognostic studies(REMARK). Nat Clin Prac Urol 2005, 2(8):416–422.
117. Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig LM,Lijmer JG, Moher D, Rennie D, de Vet HC: Toward complete and accuratereporting of studies of diagnostic accuracy. The STARD initiative.Am J Clin Pathol 2003, 119(1):18–22.
118. Yang JD, Seol SY, Leem SH, Kim YH, Sun Z, Lee JS, Thorgeirsson SS, Chu IS,Roberts LR, Kang KJ: Genes associated with recurrence of hepatocellularcarcinoma: Integrated analysis by gene expression and methylationprofiling. J Korean Med Sci 2011, 26(11):1428–1438.
119. Wang Y, Cheng J, Xu C, Liu S, Jiang S, Xu Q, Chen X, Zhuang H, Lu F:Quantitative methylation analysis reveals gender and age differences inp16INK4a hypermethylation in hepatitis B virus-related hepatocellularcarcinoma. Liver Int 2012, 32(3):420–428.
120. Saelee P, Chuensumran U, Wongkham S, Chariyalertsak S, Tiwawech D,Petmitr S: Hypermethylation of suppressor of cytokine signaling 1 inhepatocellular carcinoma patients. Asian Pac J Cancer Prev 2012,13(7):3489–3493.
121. Chu HJ, Heo J, Seo SB, Kim GH, Kang DH, Song GA, Cho M, Yang US:Detection of aberrant p16INK4A methylation in sera of patients withliver cirrhosis and hepatocellular carcinoma. J Korean Med Sci 2004,19(1):83–86.
122. Wang J, Qin Y, Li B, Sun Z, Yang B: Detection of aberrant promotermethylation of GSTP1 in the tumor and serum of Chinese humanprimary hepatocellular carcinoma patients. Clin Biochem 2006,39(4):344–348.
123. Tan SH, Ida H, Lau QC, Goh BC, Chieng WS, Loh M, Ito Y: Detection ofpromoter hypermethylation in serum samples of cancer patients bymethylation-specific polymerase chain reaction for tumour suppressorgenes including RUNX3. Oncol Rep 2007, 18(5):1225–1230.
124. Zhang YJ, Wu HC, Shen J, Ahsan H, Wei YT, Yang HI, Wang LY, Chen SY,Chen CJ, Santella RM: Predicting hepatocellular carcinoma by detectionof aberrant promoter methylation in serum DNA. Clin Cancer Res 2007,13(8):2378–2384.
125. Chang H, Yi B, Li L, Zhang HY, Sun F, Dong SQ, Cao Y: Methylation oftumor associated genes in tissue and plasma samples from liver diseasepatients. Exp Mol Pathol 2008, 85(2):96–100.
126. Hu L, Chen G, Yu H, Qiu X: Clinicopathological significance of RASSF1Areduced expression and hypermethylation in hepatocellular carcinoma.Hepatol Int 2010, 4(1):423–432.
127. Iyer P, Zekri AR, Hung CW, Schiefelbein E, Ismail K, Hablas A, Seifeldin IA,Soliman AS: Concordance of DNA methylation pattern in plasma andtumor DNA of Egyptian hepatocellular carcinoma patients. Exp MolPathol 2010, 88(1):107–111.
doi:10.1186/2050-7771-2-5Cite this article as: Mah and Lee: DNA methylation: potential biomarkerin Hepatocellular Carcinoma. Biomarker Research 2014 2:5.
Submit your next manuscript to BioMed Centraland take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit





























