Embed Size (px)
Citation preview

Proteomics and metabolomics in biological and medical applications Liudmila Shiryaeva
Akademisk avhandling
som med vederbörligt tillstånd av Rektor vid Umeå universitet för avläggande av filosofie doktorsexamen framläggs till offentligt försvar i sal KB3A9, KBC, Onsdagen den 25/5 2011, kl. 10:00.
Avhandlingen kommer att försvaras på engelska.
Fakultetsopponent: Professor Peter Roepstorff, Institut för Biokemi och Molekylärbiologi, Syddansk Universitet, Odense, Denmark.
Department of Chemistry, Umeå University 901 87 Umeå Umeå 2011 Sweden

Organization Document type Date of publication Umeå University Doctoral thesis 28 April 2011 Department of Chemistry
Author Liudmila Shiryaeva
Title Proteomics and metabolomics in biological and medical applications. Abstract Biological processes in living organisms consist of a vast number of different molecular networks and interactions, which are complex and often hidden from our understanding. This work is focused onrecovery of such details for two quite distant examples: acclimation to extreme freezing tolerance in Siberian spruce (Picea obovata) and detection of proteins associated with prostate cancer. The first biological system in the study, upon P. obovata, is interesting by this species ability to adapt and sustain extremely low temperatures, such as -60⁰C or below. Despite decades of investigations, the essential features and mechanisms of the amazing ability of this species still remains unclear. To enhance knowledge about extreme freezing tolerance, the metabolome and proteome of P. obovata’s needles were collected during the tree’s acclimation period, ranging from mid August to January, and have been analyzed. The second system within this study is the plasma proteome analysis of high risk prostate cancer(PCa) patients, with and without bone metastases. PCa is one of the most common cancers among Swedish men, which can abruptly develop into an aggressive, lethal disease. The diagnostic tools,including PSA-tests, are insufficient in predicting the disease’s aggressiveness and novel prognostic markers are urgently required. Both biological systems have been analyzed following similar steps: by two-dimensional difference gel electrophoresis (2D-DIGE) techniques, followed by protein identification using mass spectrometry (MS) analysis and multivariate methods. Data processing has been utilized for searching for proteins that serve as unique indicators for characterizing the status of the systems. In addition, the gas chromatography-mass spectrometry (GC-MS) study of the metabolic content of P.obovata’s needles, from the extended observation period, has been performed. The studies of both systems, combined with thorough statistical analysis of experimental outcomes, have resulted in novel insights and features for both P. obovata and prostate cancer. In particular, it has been shown that dehydrins, Hsp70s, AAA+ ATPases, lipocalin and severalproteins involved in cellular metabolism etc., can be uniquely associated with acclimation to extremefreezing in conifers. Metabolomic analysis of P. obovata needles has revealed systematic metabolic changes in carbohydrate and lipid metabolism. Substantial increase of raffinose, accumulation of desaturated fatty acids, sugar acids, sugar alcohols, amino acids and polyamines that may act ascompatible solutes or cryoprotectants have all been observed during the acclimation process. Relevant proteins for prostate cancer progression and aggressiveness have been identified in the plasma proteome study, for patients with and without bone metastasis. Proteins associated with lipid transport, coagulation, inflammation and immune response have been found among them. Conclusions Proteomic and metabolomic approaches, which include qualitative and quantitative analysis of proteins and metabolites, are widely used for studies of complex biological systems and for screening in medical applications. These approaches are an effective way to retrieve information and acquire a firmer understanding of biological processes and to highlight potential diagnostic and prognostic markers. In this work it has been demonstrated, through the use of a combination of ‘omics’ techniques and uni- and multivariate analyses, that one can attain erudition about theacclimation in boreal conifers to extreme freezing tolerance and how prognostic and diagnostic markers, for high risk prostate cancer, can be detected. Keywords: 2D-DIGE, biomarkers, cold-acclimation, conifer, freezing tolerance, GC-MS, metabolomics, multiple hypothesis test, multivariate analysis, OPLS-DA, Picea obovata, plasma, prostate cancer, proteomics, ProteoMiner, Siberian spruce
Language ISBN: 978-91-7459-204-7 Number of pagesEnglish 44 + 4 papers

Proteomics and metabolomics in biological and medical applications Liudmila Shiryaeva
Department of Chemistry, Umeå University 901 87 Umeå Umeå 2011 Sweden

Copyright © Liudmila Shiryaeva ISBN: 978-91-7459-204-7 Front cover: Spruce at forestline on mount Akkanålke, Arvidsjaur, Northern Sweden, by Anna Shevtsova Printed by VMC, KBC, Umeå University Umeå, Sweden 2011

Organization Document type Date of publication Umeå University Doctoral thesis 28 April 2011 Department of Chemistry
Author Liudmila Shiryaeva
Title Proteomik- och metabolomikstudier i biologiska och medicinska applikationer. Abstract Biologiska processer i levande organismer, är uppbyggda av stora nätverk av olika molekyler sominteragerar med varandra, vilka är komplexa och svåra att förstå. Denna avhandling fokuserar på attförstå sådana molekylära interaktioner kring två inbördes väldigt olika processer: anpassning avsibirisk gran till extrem kyla och uttrycksmönster av proteiner i plasma hos patienter medprostatacancer. Det första biologiska systemet i studien är intressant genom dess förmåga att anpassa sig tillextremt låga temperaturer som -60⁰C och ännu kallare. Trots årtionden av undersökningar så ärmekanismerna bakom denna häpnadsväckande förmåga hos träd fortfarande oklara. För att ökakunskaperna om extrem frystolerans så analyserades metabolomet och proteomet i barr insamladeunder trädens anpasningssperiod från mitten på augusti till januari. Det andra exemplet i studien är analys av plasmaproteomet hos patienter med aggressivprostatacancer, med och utan benmetastaser. Prostatacancer är en av de vanligaste cancerformernahos svenska män, vilken ibland utvecklas till en aggressiv dödlig sjukdom. De diagnostiska verktygsom används idag (inklusive “PSA-testet”) är otillräckliga för bedömning av sjukdomens aggressivitetoch det behövs därför nya prognostiska markörer. De båda biologiska systemen analyserades på följande sätt: 2D-DIGE-tekniken följd av MS-analysför proteinidentifiering och multivariata metoder för dataanalys och identifiering av proteinerassocierade till systemens unika särdrag. Dessutom utfördes en GC-MS-analys av barrensmetaboliter under den aktuella observationsperiden. Studierna av båda systemen resulterade i tidigare okända insikter. Mer specifikt så kundedehydriner, Hsp70s, AAA+ ATPases, lipocalin och flera andra proteiner associeras till barrträdsanpassning till extrem kyla. Metabolitanalysen av barren visade på förändringar i kolhydrat och fett-ömnesomsättningen under anpassnings processen, med kraftig ökning av sockersorterna raffinose(en oligosaccharid), sockersyror, socker alkoholer och dessutom ökade andelen av omättadefettsyror, aminosyror och polyaminer, vilkat a alla gör att den siberisk gran klara den extrema kylan. Proteiner relevanta för utvecklandet av prostatacancer detekterades i plasmaproteomet hospatienter med aggressive prostatacancer. Bland dessa kan nämnas proteiner viktiga förlipidtransport, koagulering, inflammation och immunrespons. Sammanfattning Tillämpningar av proteomik och metabolomik, vilka inkluderar kvalitativ ochkvantitativ analys av proteiner och metaboliter, används ofta för studier av komplexa biologiskasystem och för identifiering av medicinska biomarkörer. Detta tillvägagångssätt är effektivt då manvill erhålla ny kunskap om biologiska processer eller identifiera nya möjliga prognos markörer. Dettaavhandlingsarbete visar hur kombinationen av ‘omics’-tekniker och multivariat dataanalys kan ge nyinsikt om hur barrträd acklimatiserar sig till extrem kyla samt användas för att identifiera möjligadiagnostiska/prognostiska markörer för aggressiv prostatacancer. Keywords: 2D-DIGE, biomarkers, cold-acclimation, conifer, freezing tolerance, GC-MS,metabolomics, multiple hypothesis test, multivariate analysis, OPLS-DA, Picea obovata, plasma,prostate cancer, proteomics, ProteoMiner, Siberian spruce
Language ISBN: 978-91-7459-204-7 Number of pagesEnglish 44 + 4 papers

Copyright © Liudmila Shiryaeva ISBN: 978-91-7459-204-7 Front cover: Spruce at forestline on mount Akkanålke, Arvidsjaur, Northern Sweden, by Anna Shevtsova Printed by VMC, KBC, Umeå University Umeå, Sweden 2011

dedicated to my Mother….
Посвящаю моей Маме…….


1
Table of Contents
Table of Contents 1 List of Papers 2 Abbreviations 3 INTRODUCTION 5
‘Omics’ philosophy 5 Proteomics 6 Metabolomics 7
Biological Application 8 Siberian spruce (Picea obovata) 8 Molecular aspects of freezing stress 9
Medical Application 12 Prostate Cancer 12 Prostate cancer diagnosis and prognosis 13 Biomarkers for prostate cancer 13
AIMS OF THIS STUDY 16 MATERIALS AND METHODS 17
Samples and sample preparation 17 Plant material 18 Plasma samples 19
Proteomics workflow 20 2D-DIGE 20 Image Analysis 21 Protein identification by MS 21
Metabolomics workflow 22 GC-MS 22
Data processing 23 Analysis of GC-MS data 23 Statistical analysis 25
RESULTS AND DISCUSSION OF PAPERS 27 PAPER I 27 PAPER II 29 PAPER III 31 PAPER IV 32
CONCLUSIONS 34 References 37 Acknowledgments 44

LIST OF PAPERS
2
List of Papers
This thesis is based on the following papers, referred to in the text by their roman numerals in bold font:
I. Kjellsen TD, Shiryaeva L, Schröder WP, Strimbeck GR (2010).
Proteomics of extreme freezing tolerance in Siberian Spruce (Picea
obovata). J Proteomics, Mar 10; 73(5):965-75.
II. Shiryaeva L, Antti H, Kjellsen TD, Strimbeck GR, Schröder WP.
Metabolomic analysis of extreme freezing tolerance in Siberian
Spruce (Picea obovata). Manuscript.
III. Shiryaeva L, Antti H, Schröder WP, Strimbeck GR, Shiriaev AS.
Pair-wise multicomparison and OPLS analyses of cold-acclimation
phases in Siberian spruce. (2011) J. Metabolomics in press.
IV. Shiryaeva L, Antti H, Kieselbach T, Schröder WP, Stattin P,
Wikström P. Two-dimensional difference gel electrophoresis to
reveal proteins associated with high risk prostate cancer.
Manuscript
Paper I was reprinted with kind permission from Elsevier; Paper III was reprinted with kind permission from Springer.

ABBREVIATIONS
3
Abbreviations
2D-GE Two-dimensional gel electrophoresis
ACN Acetonitrile
ANOVA Analysis of variance
BPH benign prostatic hyperplasia
CV Cross validation
CyeDye Cyanine dye
DA Discriminant analysis
DHN Dehydrin
DIGE Difference gel electrophoresis
DNA Deoxyribonucleic acid
FWER Family-wise error rate
GC Gas chromatography
GS Gleason score
HABP2 Hyaluronic acid binding protein 2
IEF Isoelectric focusing
LC Liquid chromatography
LDH Lactate dehydrogenase
m/z Mass to charge ratio
M0 High risk prostate cancer
M1 High risk prostate cancer with bone metastases
MALDI Matrix assisted laser desorption/ionization
mRNA Messenger ribonucleic acid
MS Mass spectrometry
MVA Multivariate data analysis
N2 Liquid nitrogen
NMR Nuclear magnetic resonance

ABBREVIATIONS
4
OPLS Orthogonal projections to latent structures
PAGE Polyacrylamide gel electrophoresis
PCA Principal component analysis
PCa Prostate cancer
pH Potential of hydrogen
PLS Partial least squares
PSA Prostate specific antigen
PTM Post-translational modifications
RI Retention index
ROS Reactive oxygen species
RuBisCO Ribulose-1,5-bisphosphate carboxylase oxygenase
SDS Sodium dodecyl sulphate
SELDI Surface enhanced laser desorption/ionization
SMC Smooth muscle cells
TCA Trichloroacetic acid
TFA Trifluoric acid
TMS Trimethylsilyl
TOF Time of flight
UVA Univariate analysis

INTRODUCTION
5
INTRODUCTION
This chapter outlines the concept of ‘omics’ approaches, focusing on proteomics and metabolomics, as well as their application in plant and medical sciences. It gives a brief overview of the Siberian spruce (P. obovata) and the mechanisms of freezing tolerance within plants. Furthermore, it explains why plasma protein markers are in strong demand for the early diagnosis of high risk prostate cancer.
‘Omics’ philosophy
To understand the whole, one must study the whole… Henrik Kascer
The word ‘omics’, is derived from the Greek suffix, ‘ome’ (meaning all, every, whole, or complete) and refers to the scientific areas in molecular biology, which aim for a global view on biological systems in order to understand life and its organization as holistic existence. In the mid 1990’s, rapid evolution and growing knowledge concerning genomes (the complete genetic sequence of an organism) led to a quick development of the full range of ‘omics’ approaches (genomics, transcriptomics, proteomics, metabolomics and more recently, degradomics, lipidomics, interactomics, bacteriomics, eukaryomics, glycomics etc.). These methodologies are high-throughput screening and biological sample classification technologies, which allow one to get answers to such of the following questions:
− What is happening inside the cell/organ/tissue?
− What is a key player in the biological process being studied?
− Why do these biomolecules behave so differently?
− What new can one learn from this knowledge?
The ‘omics’ approaches are often applied to investigate samples collected from different populations or upon different physiological states (e.g. disease vs healthy, treated vs untreated, temperature stressed vs unstressed, etc.), aiming to find molecules that would differentiate between these classes of samples. Once the panel of differentially changed markers (genes, transcripts, proteins or metabolites) is characterized, they can be used for future sample differentiation and classification or as molecules indicating the processes that are involved in the scientific inquiry. The main power of ‘omics’ approaches lies in the generation and testing of new hypotheses. In this thesis, several new hypotheses have been put together and analyzed: two

INTRODUCTION
6
‘omics’ approaches, i.e. proteomics and metabolomics, have been used to investigate molecular processes involved in freezing tolerance development in boreal conifer P. obovata, when fully acclimated. Furthermore, this approach has been utilized to demonstrate how plasma proteomics can be applied to discover new biomarkers for high risk prostate cancer.
Proteomics
The term ‘proteome’ refers to the PROTEin complement that is expressed by the genOME (Wasinger et al. 1995). While the genome is relatively static and remains identical over the lifespan of a living organism, the proteome is highly dynamic, constantly changing and responding to internal and external stimuli. The proteome represents not only the expression part of the genome, but also the variation of several active forms and post-translational modifications (PTMs) of proteins for a single gene. This makes proteome studies increasingly more complex than expected, on the basis of the number of genes in the genome.
Based on the central dogma, that the sequential information is transferred from deoxyribonucleic acid (DNA) by transcription via messenger ribonucleic acid (mRNA) and then by translation to proteins (Crick 1970), it could be expected that proteome expression (the quantitative and qualitative measurement of the global protein levels) would remain consistent with mRNA expression. At the same time, the evaluation of the correlation between mRNA levels and corresponding protein levels in cells indicates a coherence, mostly for prokaryotes (Scherl et al. 2005) and a very poor correlation for eukaryotes, as was shown in several studies (Gygi et al. 1999). There is still little known of these processes and since genomics cannot answer all the questions, proteomics is a great complementary tool to convey knowledge to the life sciences.
Proteomics involves the systematic qualitative and quantitative study of sets of proteins and protein variations on a large scale, as a result of biological processes and perturbations within a cell or tissue. “Proteomics includes not only the identification and quantification of proteins, but also the determination of their localization, modifications, interactions, activities, and, ultimately, their function” - Stan Fields in Science, 2001 (Fields 2001).
Proteome analysis began with techniques based on two-dimensional gel electrophoresis (2D-GE), which was introduced by Klose and O’Farrell in 1975 (Klose 1975, O'Farrell 1975). This was expanded upon by application of gel-free techniques, such as liquid chromatography-mass spectrometry (LC-

INTRODUCTION
7
MS). Both gel-based and gel-free methods have relative merits and weaknesses and the choice of the preferable strategy is varying. While gel-based proteomics is time-consuming and difficult to automate (having limited dynamic range of protein detection as well as a restricted ability to separate membrane proteins), it nevertheless is an effective analytical tool for the global overview and quantitative comparison of thousands of proteins simultaneously within their complex biological systems. To achieve an in-depth proteomic analysis, pre-fractionation and depletion/enrichment procedures have been developed. Advances in instrumentation (application of narrow range immobilized pH gradient gels, which increase resolution of analysis; fluorescence imaging; mass spectrometry), methodologies (protein microarrays, MS-based methods, chromatography) and bioinformatics have expanded the application of proteomics research.
In the studies presented in this thesis (paper I and IV), the gel-based proteomics strategy has been used specifically for the study of these biological systems – the combination of two-dimensional difference gel electrophoresis (2D-DIGE) coupled with mass spectrometry (MS). Both proteomic studies have been successfully performed and resulted in an updated panel of the proteins crucial for the aforementioned studied processes. The principles of the selected gel-based proteomic approach that was utilized in this study are described in more details in the Materials and Methods chapter.
Metabolomics
Metabolomics was coined relatively recently, in 1999 (Fiehn et al. 2000, Nicholson et al. 1999). Similar to how proteomics is aimed at the global qualitative and quantitative characterization of all the proteins within the proteome, metabolomics is directed at the global, high-throughput measurements and characterization of the hundreds of metabolites (e.g. amino acids, fatty acids, mono- and di-carbohydrates etc.) within the metabolome. Metabolome refers to the set of endogenous and exogenous small-molecules, i.e. metabolites, that can be found in a living cell or organism (Oliver et al. 1998). Metabolites reflect the events that occur at different levels of the organism and can be regarded as the end-point of the chain beginning with gene expression in response to internal or external stimuli, through to mRNA and protein interactions. Thus, metabolites are believed to be able to describe the current state of an organism in a more comprehensive way than that of mRNA transcripts or proteins. However, for a complete understanding of the mechanisms involved in the processes

INTRODUCTION
8
taking place in a living organism, all levels of the information pathways should be considered.
Metabolomic analysis requires a set of analytical tools that can explore the overall ‘signatures’ of the chemically diverse cellular metabolites and characterize each metabolic variation. This can be achieved by a combination of several approaches: proper and optimized isolation/extraction procedures, separation techniques (e.g. GC, LC) coupled with MS identification and/or spectroscopic techniques without prior-separation (MS, NMR). Suitable software is required for rapid and correct spectral-chromatographic data processing.
In this metabolomic study of dynamic metabolic alterations during the acclimation process in Siberian spruce (paper II), GC-MS technique was used. This is one of the most powerful and sensitive high throughput methods, which can detect and identify the largest possible number of metabolite profiles. The principles of this technique are described in detail within the Materials and Methods chapter.
The amount of information and complexity of the data produced by metabolomics and proteomics gives rise to many new challenges. To recover the metabolite interconnection through metabolic reactions and their implication into metabolic pathways, the joined interrelation of several disciplines - computational science, statistics and bioinformatics - are required. The development of suitable tools for data analysis, interpretation and visualization, in order to reach a holistic view of living processes, is needed. Paper III is dedicated to this point. It proposes a discussion of new questions, where not only in the description of significantly changed metabolites are of main interest, but rather their coherent dynamics for particular intervals of time. The detailed strategy of data analysis is ascribed further in the Materials and Methods chapter.
Biological Application
Siberian spruce (Picea obovata)
Siberian spruce (P. obovata) is one of the most dominating species of dark coniferous forests in Russia (Siberia) and Canada. Within the territory of Middle Siberia (Krasnoyarsk, Evenkia, Khakasia, Tuva) it occupies more than 10 million hectares, which constitutes about 10.5% of the forest-covered territory (Rysin and Savel’eva 2002). Growing in the wide geographic range

INTRODUCTION
9
and in the areas where the summers are very hot, reaching over +35 ºC and mid-winter temperatures can drop below -60 °C and may remain below -40˚C for two to three months (Bonan and Shugart 1989). P. obovata has found its own unique mechanisms to survive such extreme conditions and to keep its needles alive.
Previously it has been shown by Strimbeck (2008), that P. obovata is already acclimated by the end of October and that this ability is sustained throughout the winter. When it is fully acclimated, its needles can survive immersion in liquid nitrogen at -196˚C, if at first they are slowly cooled to an intermediate temperature, typically -20 to -30˚C. This amazing ability to withstand the freezing stress makes this species attractive for the investigation of acclimation to extreme frost hardiness in living tissues. Although several proteomic and metabolomic studies gave valuable insight in the field of freezing stress in the Arabidopsis thaliana (Amme et al. 2006, Kaplan et al. 2004), this model organism does not achieve the same level of freezing tolerance as boreal conifers can, once fully acclimated. The physical properties of intracellular membranes and fluids at the point of full frost hardiness are not fully understood.
Despite the limited sequence information available for P. obovata, it is a suitable species for exploration of mechanisms of extreme freezing tolerance. Increase in published genome sequences from other conifers and boreal plants, coupled with continuous updating of protein and metabolite databases, as well as MS development and advances in the field of 2D-DIGE techniques, have all made it possible to obtain detailed proteomics and metabolomics data from non-model organisms like P. obovata (paper I, III).
Siberian spruce is one of the traditional subjects of forest genetic and breeding research. This species has a great economic and ecological significance and is part of intensive breeding programs. For this reason, the study of the molecular processes occurring in this tree during cold-acclimation may yield valuable knowledge on plant adaptation and evolution leading to the acquirement of freezing tolerance.
Molecular aspects of freezing stress
Boreal conifers, naturally growing in the regions where winter can arrive early, must be well prepared in advance for the first frost event and for avoidance of the ice-formation in the extracellular matrix and within the cell. This preparation/acclimation is undertaken by the triggering of complex cellular machinery. It includes:

INTRODUCTION
10
(1) changes in membrane fluidity which can be acquired by fatty acid desaturation and changes in lipid composition (Oquist 1982, Quinn 1985, Steponkus et al. 1990); 2) alterations in carbohydrate metabolism with accumulation of sucrose and oligosaccharides (Frankow-Lindberg 2001, Hansen and Beck 1994);
(3) synthesis and accumulation of proteins with probable cryoprotective functions (Close 1997, Shinozaki and Yamaguchi-Shinozaki 2000); (4) accumulation of lower molecular weight compatible solutes, such as proline and glycine betaine (Li et al. 2004);
(5) increases in the ability to withstand oxidative stress (Tao et al. 1998).
Freezing temperatures cause cellular water deficiency, which is driven by crystallization of unbound water in the intercellular space. As a result, cellular water moves down its potential gradient, crossing the plasma membrane into the cell wall and the intercellular space. In turn, living cells dehydrate, crenate and possibly die, if uncontrolled by the cellular machinery. Various models have been proposed to understand how cellular processes act to prevent injury to membranes and proteins, for example by promoting intracellular vitrification (i.e. glass transition) or by preventing membrane phase changes.
Nevertheless, there is no commonly accepted biochemical model that quantitatively and qualitatively describes the mechanism of acquired freezing tolerance on a molecular level. Furthermore, there are no known clear triggers or sensors of this process that are uniquely associated with response to a decrease in temperature. This study is an attempt to contribute to this fundamental question in nature.

INTRODUCTION
‘Frost talk’. Forestline on mount Akkanålke, Arvidsjaur, Northern Sweden, by Anna Shevtsova
11
INTRODUCTION
Medical Application
Prostate Cancer
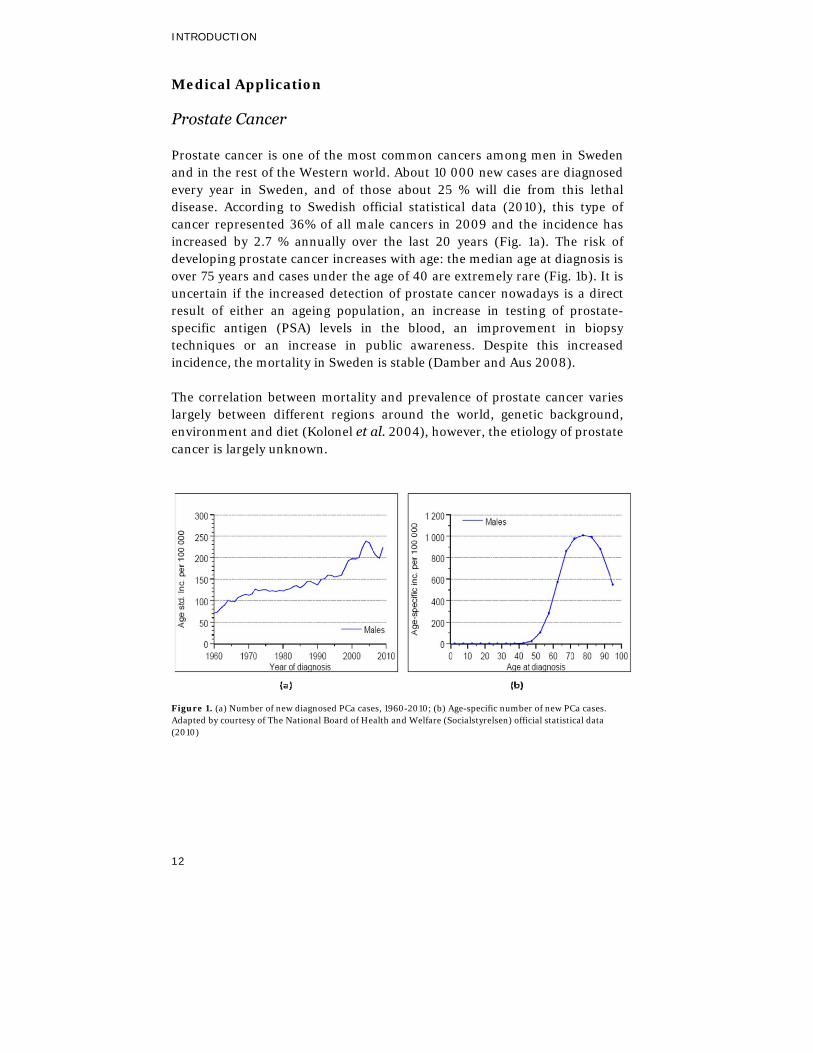
Prostate cancer is one of the most common cancers among men in Sweden and in the rest of the Western world. About 10 000 new cases are diagnosed every year in Sweden, and of those about 25 % will die from this lethal disease. According to Swedish official statistical data (2010), this type of cancer represented 36% of all male cancers in 2009 and the incidence has increased by 2.7 % annually over the last 20 years (Fig. 1a). The risk of developing prostate cancer increases with age: the median age at diagnosis is over 75 years and cases under the age of 40 are extremely rare (Fig. 1b). It is uncertain if the increased detection of prostate cancer nowadays is a direct result of either an ageing population, an increase in testing of prostate-specific antigen (PSA) levels in the blood, an improvement in biopsy techniques or an increase in public awareness. Despite this increased incidence, the mortality in Sweden is stable (Damber and Aus 2008).
The correlation between mortality and prevalence of prostate cancer varies largely between different regions around the world, genetic background, environment and diet (Kolonel et al. 2004), however, the etiology of prostate cancer is largely unknown.
Figure 1. (a) Number of new diagnosed PCa cases, 1960-2010; (b) Age-specific number of new PCa cases. Adapted by courtesy of The National Board of Health and Welfare (Socialstyrelsen) official statistical data (2010)
12

INTRODUCTION
13
Prostate cancer diagnosis and prognosis
At present, prostate cancer diagnostics is based upon testing for serum levels of prostate specific antigen (PSA), digital rectal examination and sampling of transrectal ultrasound guided biopsies. PSA testing remains the standard method for detection of prostate cancer, despite the fact that it has low specificity and limited prognostic value. It is considered normal if the level of PSA is in the range between 0 and 3.0 ng/ml, while values above 10.0 ng/ml indicate a substantial risk for having prostate cancer. Of major concern is that a majority of men with moderately elevated levels of PSA (3-10 ng/ml) do not have PCa, but are instead diagnosed with inflammation, prostatitis or benign prostatic hyperplasia (BPH) (Schröder 2009). Another issue is that there exists populations of men, with prostate cancer, who all actually have PSA values lower than 3.0 ng/ml (Thompson et al. 2004).
Furthermore, a large fraction of men diagnosed with prostate cancer have indolent tumors, which are unlikely to progress for many years. Prognosis for a patient is predicted by evaluating tumor grade (on the Gleason score scale) and clinical stage (T1-4) in combination with the PSA value (Fiorentino et al. 2010). Patients with high tumor grade (GS 8-10) and/or non-localized tumors (T3-4) have a high risk of developing metastases, primarily in the bone, leading to fatal disease. Low-risk tumors (T1, GS <6) are asymptomatic and remain confined in the gland for many years until they can be detected. The majority of newly diagnosed cases however, have intermediate-risk tumors (T2, GS 6-7) and the ability to predict tumor behavior in these cases is very limited. Thus, patients with benign and low-risk tumors can undergo a huge amount of unnecessary biopsies and over-treatment, while other patients get therapy too little too late. When the cancer has spread outside the prostate organ and metastasized, only palliative therapeutic options are available.
New markers to improve diagnostics and prognostics of prostate cancer are therefore crucial.
Biomarkers for prostate cancer
The prostate is a walnut-sized and shaped male gland, which surrounds the urethra just below the urinary bladder. A healthy male prostate has a high degree of cellular organization and is composed of ducts lined with basal epithelial cells, secretory luminal epithelial cells and neuroendocrine cells, surrounded by a fibromuscular stroma (Schalken and van Leenders 2003). Fibromuscular stroma is composed of different types of cells: smooth muscle

INTRODUCTION
14
cells (SMC), fibroblasts, mast cells, and macrophages, all embedded in a collagenous matrix together with nerves, lymphatic and blood vessels. Prostate tumors (malignant as well as benign) consist of abnormal cells that are different from the healthy prostate not only in the cell composition, but also in the expression of growth factors, angiogenetic factors and proteolytic enzymes etc. (Josson et al. 2010). Malignant tumors are often multifocal and heterogeneous with respect to gene and protein expression/utilization. In addition, the degree of aggressiveness results in the probability of finding biomarkers in tissue and blood, with prognostic information being highest in patients with this kind of tumor.
The ‘omics’ approaches aforementioned above, are widely used in cancer research. These approaches compare samples of diverse origins (for instance, from patients with different degrees of tumor aggressiveness) and allow discrimination and characterization of markers that can be used to differentiate clinically significant tumors. As a result of rapid ‘omics’-technologies, a few potential molecular markers for prostate cancer have been detected (Leman and Getzenberg 2009, Makarov et al. 2009). Their application for early detection of disease, prognostication, prediction of treatment response, or as a measure of response to treatment is currently under testing and validation phases.
An optimal tumor marker should be tumor-specific and easily assessable by non-invasive tests in order to precisely determine if there is a need for further biopsy analysis. Blood is one of the most suitable biofluids available for biomarker discovery. It reflects diverse physiological and pathological states and can contain various cellular elements, circulating tumor cells, secreted proteins/peptides, metabolites and cell-free DNAse/RNAse. Since the prostate tumor volume is minute, expected tumor-specific or metastatic-specific markers will be present at very low concentrations in the blood. Therefore, detection of biomarkers in plasma, especially for early stages of the disease, is extremely challenging. Since the research presented in this thesis was focused on the discovery of PCa relevant biomarkers using proteomics, the discussion of metabolomics application in this field was omitted. Successive application of metabolomics, in the search for PCa markers within human blood and tissue, was however, recently published by (Sreekumar et al. 2009, Thysell et al. 2010).
Plasma proteins have a high dynamic range, which makes plasma proteomic analysis particularly challenging. Identification of low abundant proteins within plasma is extremely difficult, since in the analysis highly abundant proteins can mask them. The difficulties caused by the complexity and broad protein abundance range in plasma are gradually being overcome with

INTRODUCTION
15
proper experimental design, novel experimental approaches and data analysis. Several comparative proteomic analyses of plasma or serum have revealed potential biomarkers for prostate cancer (Byrne et al. 2008, Lam et al. 2005, Qin et al. 2005). Several in-depth approaches, including plasma depletion (Chromy et al. 2004, Moritz et al. 2005) and extensive fractionation, using the ProteoMiner normalization technology (Righetti et al. 2006), have increased sensitivity of plasma proteome analysis (Pernemalm et al. 2008, Sihlbom et al. 2008). In this study, a combination of plasma fractionation with ProteoMiner technology and 2D-DIGE has been utilized, with the aim of finding new potential biomarkers in blood plasma for high risk prostate cancer patients.

AIMS OF THIS STUDY
16
AIMS OF THIS STUDY
The overall aims of this study were (i) to investigate the molecular changes of cold-acclimation effects in Siberian spruce (Picea obovata), and (ii) to identify potential plasma biomarkers for high risk prostate cancer.
The specific aims to accomplish this were:
I. To use proteomic and metabolomic approaches for finding specific proteins and metabolites associated with cold-acclimation and extreme freezing tolerance ability in P. obovata.
II. To explore different methods to detect the presence of distinctive phases in seasonal-responsive changes in metabolic patterns of P. obovata during cold acclimation.
III. To investigate proteomic differences between plasma samples corresponding to patients with high risk prostate cancer and patients with benign diseases.

MATERIALS AND METHODS
17
MATERIALS AND METHODS
This thesis is based on gel-based proteomic and GC-MS-based metabolomic analysis of two different types of biological samples. In papers I, II and III, the needles from Siberian spruce (Picea obovata) collected in time-course, were studied. In paper IV, plasma samples from patients diagnosed with high-risk prostate cancer were subjected for proteome analysis. The following subsections will briefly describe the main steps in proteomic and metabolomic analysis: (1) samples and sample preparation; (2) the key techniques in proteomics - 2D-DIGE coupled with MS-identification; and metabolomics - GC-ToF/MS; and (3) the subsequent data processing and statistical analyses used in this study.
Samples and sample preparation
Sample preparation is one of the most crucial steps in obtaining high-quality data for proteomic and metabolomic analysis. The major problem in extensive cell/tissue extraction of proteins or metabolites lies in the high dynamic range of expression/abundance and diversity of these molecules with respect to their chemical and physical properties (molecular weight, isoelectric point, solubility, polarity, volatility, etc.). One-step sample preparation procedures would be highly desirable with respect to simplicity, reproducibility and minimization of artifacts, but it is hardly achievable.
Proteins or metabolites can be extracted after cell disruption processes, achieved mainly by freezing, grinding, homogenization, detergent lyses, and other procedures. Samples should be kept cold during cell breakage to inhibit cellular metabolism and to prevent possible degradation and modifications during the sample preparation step. Interfering substances, e.g. proteolytic enzymes, DNAses/RNAses, salts, plant phenols and many others, should be inactivated by appropriate inhibitors and/or removed, keeping in mind that each additional sample preparation step can result in the possible loss of analyzed molecules.
Pre-fractionation steps are especially advantageous for proteome analysis in order to reduce complexity of the sample and to enrich the proteins of interest before sample extraction/solubilization. The buffer optimization and compatibility are crucial for both proteomic and metabolomic analysis. In this study (paper I and IV) a urea/thiourea buffer, compatible with 2D-DIGE, was used for protein solubilization and in paper III - an optimized methanol : chloroform buffer was used for metabolite extraction.

MATERIALS AND METHODS
18
Labeling of proteins with cyanine dyes (CyDyeTM DIGE Fluors) prior to electrophoretic protein separation, is also preferable. It allows obtaining highly accurate qualitative and quantitative proteomics, mainly because of (1) minimization of gel-to-gel variation, caused by distortion of 2D-patterns (i.e. it is possible to separate up to three samples on the same 2D-DIGE gel), and (2) the use of internal standard (i.e. the pooled mixture of all samples labeled with one specific CyDye, usually Cy2) facilitating image analysis.
After extraction of metabolites, additional derivatization procedures should be carried out before analysis with GC-MS, in order to reduce polarity of the metabolites and facilitate their chromatographic separation. Metabolites containing functional groups with active hydrogens (-OH, -SH, -NH) are inactivated, for example by a silylation reaction. In addition, keto (oxo-) groups are oximated in order to prevent enolization reactions, which can lead to generation of multiple products.
Plant material
Three Siberian spruce trees, growing in The Ringve Botanical Garden in Trondheim, were selected for repeated needle sampling every two to four weeks, from August 2006 to April 2007. It has been shown by Strimbeck et al. (2007) that these trees develop extreme freezing tolerance and the main interest in this study was to analyze protein and metabolite changes in needle cells during its cold acclimation.
Protein extraction, solubilization and labeling
Analysis of protein expression (paper I) in samples representing non-acclimated (5 September), partially acclimated (8 October) and fully acclimated (5 November) stages were chosen on the basis of freezing tolerance data from the same individual trees, sampled from August 2006 to April 2007 (Strimbeck et al. 2008). Total protein content was extracted from 80 mg ground needle tissue. Conifer tissues are very rich in polysaccharides and phenols, which can interact with proteins and as a result, give rise to streaky 2D – patterns and a poor quality of resolved protein spots. Thus, combined TCA/acetone and methanol washes and phenol extraction procedures were performed as described by Wang et al. (2006). The resulting pellets, from a total of four extractions of each sample, were dissolved in urea/thiourea buffer, compatible with minimal labeling from CyDye™ Fluors (Cy2, Cy3, and Cy5) prior to 2D-DIGE.

MATERIALS AND METHODS
19
Metabolite extraction and derivatization
Samples were also collected during broader time-course acclimation, representing nine different time points, every two to four weeks, from August 2006 to January 2007 (paper II and III). These were analyzed by a metabolomic approach. The metabolites were extracted from ~10 mg of P. obovata needle, ground to powder in N2 with normalized volumes of extraction mixture, consisting of chloroform : methanol : H2O (1:3:1) and a series of stable isotope compounds as described in (Gullberg et al. 2004). Samples were evaporated to dryness and then derivatized by methoxyamination and afterwards by trimethylsilylation, prior to GC-ToF/MS.
Plasma samples
Plasma proteome analysis is challenging because of the huge disproportion in protein abundance with the dynamic range of over ten orders of magnitude (Anderson and Anderson 2002). This prevents the simultaneous analysis of high- and low-abundant proteins (probably the most interesting plasma proteins, in terms of marker selection). Albumin alone represents ~60% of the total protein content and the nine most abundant human serum proteins (albumin, immunoglobulin G, haptoglobin, transferrin, transthyretin, a1-antitrypsin, a1-acidic glycoprotein, hemopexin, and a2-macroglobulin) constitute about 90% of the entire serum proteome (Pieper et al. 2003). In order to increase the probability of finding specific tumor markers in blood for aggressive prostate cancer, extensive ProteoMiner fractionation of plasma, in combination with DIGE, was carried out in this study.
Plasma fractionation using the ProteoMiner normalization technology (BioRad) (Thulasiraman et al. 2005) is based on the interaction of protein samples with a highly diversible library of hexameric peptide ligands bound to the surface of the beads. Each bead has millions of copies of a single peptide, and each bead has a potentially different peptide ligand. Since the binding capacity is limited by bead capacity, the high-abundant proteins quickly saturate all available ligands, while the low-abundant proteins are concentrated on their specific ligands. Thereby, the dynamic range of protein concentration is decreasing and low-abundant proteins are enriched, meaning quantitative information is preserved.
Several studies have shown (Pernemalm et al. 2008, Sihlbom et al. 2008) that ProteoMiner technology can have increased sensitivity of plasma

MATERIALS AND METHODS
20
analysis, by surface enhanced laser desorption and ionization time of flight mass spectrometry (SELDI-TOF/MS) and 2D-DIGE in comparison with other fractionation techniques. On the other hand, comparison of immuno-depletion strategies with the ProteoMiner technique by Bandow (2010) did not reveal substantial differences in protein patterns. The critical point against application of beads was published by Keidel et al. (2010), where it was claimed that the ProteoMiner bead interaction with protein mixtures was caused more likely by general hydrophobic binding mechanism similar to Sepabeads. Hence, the diversity in beads surface ligands plays only a negligible role.
In this study, plasma proteome patterns that corresponded to high risk prostate cancer patients with (M1, N=7) and without (M0, N=14) established bone metastasis, as well as patients with benign prostate disease (B, N=15) were obtained by 2D - DIGE. Selected samples were first ProteoMiner enriched and eluted using a series of buffers according to the manufacturer´s protocol and performed by scientists at BioRad (Hercules, CA) : fraction (1) - 1M NaCl, 20 mM, HEPES, pH 7.5; fraction (2) - 200 mM glycin, pH 2.4; fraction (3) - 60 % ethylene glycol; fraction (4) - 33.3 % isopropanol, 16.7 % acetonitrile (ACN), 0.1 % trifluoric acid (TFA). All four fractions corresponding to one person were subsequently cleaned-up and pooled before protein quantification. A pool of all samples corresponding to an internal standard sample was labelled with Cy2, and samples from different patients were labeled by either Cy3 or Cy5. Protein content of plasma samples was separated in the first dimension in the 3 – 11 pH range and in the second dimension on 10-15% gradient sodium dodecyl sulfate – polyacrylamide gel electrophoresis (SDS-PAGE).
Proteomics workflow
2D-DIGE
2D electrophoresis, introduced by O'Farrell (1975), is a widely used gel-based proteomic technique applied to analysis of complex protein mixtures, originated from cells, tissues or biofluids. The electrophoretic protein separation occurs in two dimensions, according to different protein properties. In the first-dimension, isoelectric focusing (IEF), proteins are separated in the gel matrix according to their isoelectric point (pI) and in the second-dimension, using SDS-PAGE, proteins are separated according to their mass. The innovation of immobilized pH gradients by Görg et al. (1988), (1995) made a great improvement in reproducibility of gels between labs and gave rise to gel-based proteomics in general. The further developed

MATERIALS AND METHODS
21
2D-DIGE method by Ünlü et al. (1997), facilitated quantitative and qualitative analysis of differences in protein patterns between groups of samples. In DIGE, proteins are labeled/stained with fluorescent dyes, like the cyanine dyes (CyDyes) used in our study, prior to 2D separation. Having an NHS-ester reactive group, cyanine dyes are covalently bound to an amino group of a single lysine residue, per protein molecule, via an amide linkage. The ratio between dye to protein was designed to ensure the staining of one dye per protein; thus, it is so called, "minimal labelling". Protein mixtures (up to three samples), labeled with CyDyes (Cy2, Cy3, Cy5) can be separated in one gel. Due to the different excitation wavelengths of each CyDye it is possible to acquire up to three images from one gel.
Image Analysis
After 2D-DIGE, the gel images with differentially labeled and co-resolved proteins need to be obtained on a multi-wavelength scanner and analysed. In general, image analysis is very time consuming and encompasses multiple steps. Several software packages for DIGE image analysis are available nowadays and the REDFIN software (Ludesi AB, Lund, Sweden) was used in this study. The images were matched using all-to-all spot matching and manually edited where needed. Quantification was achieved by measuring the cumulative spot intensity with subsequent background subtraction and a two-step normalization procedure. Within the first step, normalization was done according to a calculated normalization factor across all three images obtained from each gel. In the second step, spot volumes were normalized by the normalization factors that were calculated for all internal standards. The package also included several standard statistical tools for preliminary grouping of the matched spots based on pre-processing their relative intensities including Student t-test, Mann-Whitney test and Analysis of Variance (ANOVA).
Protein identification by MS
Mass spectrometry (MS) is the key analytical technique in proteomics. It provides information about protein identification by measuring mass to charge ratio (m/z) of ions generated in the gas phase. Therefore, protein digests (peptides) should be turned into the gas phase, than ionized and afterwards analyzed for mass to charge ratios. A mass spectrometer typically consists of three integral parts: (i) an ion source for peptide ionization; (ii) a mass analyzer where ions are separated according to their m/z ratio; (iii) a

MATERIALS AND METHODS
22
detector, which registers the amount of separated ions. All integral parts are connected to a computer to enable control, registration and evaluation of obtained data.
In the present study, identification of proteins from 2D gel spots was performed by two different MS-techniques. In paper I, protein identification was done by MALDI-TOF/TOF analysis at the W.M. Keck Foundation Biotechnology Resource Laboratory, Yale University, USA. Protein identification, in paper IV, was performed by LC-MS/MS at the Umeå Protein Analysis (UPA) Facility, Umeå University, Sweden.
Metabolomics workflow
GC-MS Several analytical methods can be used for analysis of the chemically diverse cellular metabolites and characterization of the metabolic variations. Cellular metabolites are usually analyzed using LC-MS or GC-MS, despite the fact that nuclear magnetic resonance spectroscopy (NMR) offers cost-effective and quick sample analysis. LC-MS has a number of advantages over GC-MS, since more polar compounds are covered with this technique and there is usually no need for derivatization. LC-MS chromatograms are however more difficult to process and deconvolute than is the case with GC-MS. Furthermore, nowadays the identification of compounds is increasingly easier for GC-MS, thanks to many commercial and public GC-MS libraries establishing. GC-MS has also a higher sensitivity, as compared to nuclear magnetic resonance spectroscopy (NMR) (Lisec et al. 2006), which makes it one of the most commonly used analytical techniques in metabolomics.
In GC-MS, compounds in the gas phase are separated on the column. They are carried through a column with an inert gas (such as helium) and column effluent flows into the mass spectrometer, where separated analytes are ionized and fragmented. The mass to charge ratios (m/z) of obtained fragments, together with their relative abundances, are measured. Libraries of known mass spectra are used for metabolite identification.
GC-TOF/MS analysis in papers II and III was undertaken according to a standardized protocol (Gullberg et al. 2004). Metabolites were annotated by comparing their mass spectra to spectral databases containing standard compounds and maintained by the Umeå Plant Science Center1 in Umeå, Sweden, as well as the Max Planck Institute2 in Golm, Germany. 1 http://www.upsc.se/Technology/Metabolomics-Facility/metabolomics-facility.html 2 http://csbdb.mpimp-golm.mpg.de/csbdb/gmd/gmd.html

MATERIALS AND METHODS
23
Data processing
‘Omics’ approaches generate large and multivariate data sets and the appropriate data pre-processing and hypothesis testing are required to construct new and valuable insights into the ‘overall picture’ of cellular complexity.
Analysis of GC-MS data
Several approaches have been successfully utilized for qualitative data pre-processing and quantitative analysis of information contained in data (Boccard et al. 2010). Data pre-processing encompasses normalization procedures, alignment and deconvolution of peaks across samples, multiple peak recalculations and metabolite annotation. Most of these steps can be done with the help of commercial or in-house developed softwares and with a minimal need in supervision. However, there are no perfect deconvolution algorithms and the quality of spectrum largely depends on the level of compound in the spectrum (i.e. low level – bad spectrum). Thus, the task to computerize and automate the processes of metabolite annotation is rather difficult and dependent on a subjective factor and the substantial involvement of a scientist.
Annotation of metabolites can bring further challenges, due to several other factors, including improper design of experiment, instrumental drift and poor quality of measurements or insufficient number of independent measurements. Even though the origins of inconsistency of metabolite identification are often difficult to detect, some of these problems can be resolved by appropriate modification of experimental conditions (e.g. different running modes), which were used for GC-MS analysis in papers II and III. They are briefly discussed below.
In the ideal situation, the relative metabolite profiles obtained by different running conditions should be the same. However, in practice, because of different reasons, e.g. broad dynamic range of metabolites present in the studied samples, profiles might substantially differ. Among the many reasons, two are apparent: (1) relative metabolite profiles are different, if a metabolite is highly abundant, then saturation of measurements (chromatographic peaks areas) can occur; (2) relative metabolite profiles can be different, if the measurement of a low abundant metabolite (for one of the running conditions) is below the threshold.

MATERIALS AND METHODS
To avoid these cases, GC-MS chromatograms in the presented study were recorded in two different running modes - splitless and split (i.e. metabolic extracts were injected in reduced proportion 1:20). Such measurements for both modes are depicted for raffinose and phytol in Fig.2 (A, B). As it is shown, relative levels of raffinose measured by both modes are almost the same during August - October, while they substantially differ (more than 2.5X) for observations obtained between 05/11 and 02/01. This observation indicates that the splitless data for raffinose are likely to be saturated and are deficient, despite the narrow ranges of confidence intervals voting for consistent and accurately computed measurements.
(A)
(B)
Figure 2. Time-course dynamics of relative raffinose (A) and phytol (B) levels in needles of P. obovata during cold-acclimation: the solid black line are the measurements obtained by GC-MS analysis performed in splitless mode and the dashed blue line - in split (1/20) mode.
The saturated peaks for raffinose, at several time points, were already observed during chromatogram evaluation and the metabolite annotation tasks. However, analysis of relative metabolite profiles, depicted in Fig.2, suggests a basis for developing a procedure for automatic evaluation and data filtering. Such analysis might provide comprehensive information about
24

MATERIALS AND METHODS
25
consistency of data, for both split and splitless modes, for the whole observation interval, i.e for which period of observations the results are similar and at what time they started to deviate etc.
Similarities in the relative metabolite profiles obtained in different modes are difficult to detect and quantify. Overlapping of measurements, as it appears for raffinose in the beginning of the observed time interval (i.e. between the dates 15/08 and 23/10), see Fig. 2 (A) — allows fusing of these independent data sets. This improves the measurements quality, via reducing the variance. However, there are many situations where fusing different measurements is a challenge. One such case, is depicted at Fig. 2 (B). The confidence intervals for most of the points of the two data sets do not overlap, meaning that fusing of data following the classical settings cannot be done. Meanwhile, it is clear that measurements obtained, in split and splitless mode, are rather similar. They possibly differ, merely by appropriate scaling. The formalization of this similarity and automatic detection of a common pattern is quite difficult.
These observations can be used for optimization and re-design of experiment or additional data pre-processing (i.e. with the aim to deduce what measurements are ‘as correct as possible’) for further statistical analysis and hypothesis testing.
Statistical analysis
The common statistical approach, used in metabolomics data analysis, is based on multivariate analysis (MVA), including methods such as principal component analysis (PCA) (Wold et al. 1987), partial least squares discriminant analysis (PLS-DA) and its extension - orthogonal projections to latent structures-discriminant analysis (OPLS-DA) (Trygg and Wold 2002).
These statistical methods are often applied for clustering and visualization of essential trends and changes among large subsets of metabolites. To a certain extent, these methods are insensitive to typical perturbations (e.g. random noise) and expected to draw similar conclusions. This robustness in analysis could be a limitation in the testing for delicate properties of subsets or even individual metabolites.
For example, the insensitivity of a statistical method to saturation in levels of raffinose, shown in Fig. 2 (A), potentially implies that this method is rather approximate and that:

MATERIALS AND METHODS
26
• it cannot provide comprehensive and detailed information on a pattern that this metabolite followed, in course of cold acclimation; • it cannot point out metabolites that shared a similar behavior; • it can be limited in analysis and reconstruction of biochemical networks and timing of critical events; and so on.
As a result, the conclusions drawn by MVA, regarding biochemical properties of individual metabolites, might be vague or inconsistent. Hence, if such dynamics are of interest and multivariate methods are primarily applied, they should be complemented by testing relevant hypotheses for individual metabolites of particular interest with univariate analysis (UVA). This can be done using a variety of available algorithms used for testing simple hypotheses, such as Student t-test, Mann-Whitney U-test, Kolmogorov-Smirnov test, and others. The UVA can be applied for testing several alternative hypotheses at once, which are important, for instance, in characterizing a pattern of time-course changes of metabolite and searching its homogeneity intervals. Again, various algorithms and software packages are available for these purposes, where the classical one-way Analysis of Variance (ANOVA) and non-parametric Kruskal-Wallis test are the immediate and well-known procedures to mention. This study went above and beyond this classical framework and used advanced statistical tools – the so-called step-up and step-down procedures (Holm 1979, Lehmann and Romano 2005) – for controlling the level of family-wise error rate (FWER) in pair-wise multiple comparisons. The motivation to employ these advanced tools arose from the analysis of acclimation period, performed in paper II, by the multivariate method (OPLS-DA) and the concluded segmentation of this period into four phases for majority of putative metabolites. The pair-wise multiple comparisons (Holm 1979, Lehmann and Romano 2005) were used for reconstructing homogeneity intervals for all metabolites that were found, to validate and complement the OPLS-DA classification. The results of this investigation were collected and are represented in paper III. In addition to analysis of metabolite patterns, it was proposed to re-use the pair-wise multiple comparisons to recover a set of metabolites that displayed a similar behavior over a time course.

RESULTS AND DISCUSSION OF PAPERS
27
RESULTS AND DISCUSSION OF PAPERS
PAPER I
Objective: To study proteome dynamics during acclimation to extreme freezing tolerance in Siberian spruce (Picea obovata).
Results: Two-dimensional difference gel electrophoresis (2D-DIGE) was used to analyze protein expression profiles of extreme freezing tolerant conifers, during September to November. Proteins associated with the acclimation process were clustered into five distinct expression profiles, using multivariate analysis (MVA). Forty three protein spots were identified by matrix-assisted laser desorption/ionization tandem time-of-flight mass spectrometry (MALDI-TOF/TOF). Twelve of these were previously found to be associated with various biotic and abiotic stress responses in other plants. Dehydrins, Hsp70s, AAA+ ATPases, lipocalin, cyclophilins, glycine-rich protein (GNP) and several reactive, oxygen intermediate scavenging proteins showed elevated levels from September to November.
Comments and discussion: The first 2D-proteome map of the needle tissue of P. obovata was presented in this paper. It provides a reference map for further studies on the freezing tolerance in boreal conifers. Several of the proteins, including their functions associated with acclimation to freezing tolerance in the needle tissue of P. obovata, are briefly discussed below.
Dehydrins (DHNs) were the most interesting finding and seem to be uniquely associated with the biochemical changes related to freezing tolerance. With a very low abundance found in the September samples, DHNs were strongly up-accumulated during October and November. Their biochemical functions are still unclear. Most of the studies suggest that DHNs prevent ice crystal growth (Wisniewski et al. 1999), stabilize membranes and macromolecules (Close 1996), and may interact with sugars to promote low-temperature cytoplasmic vitrification (Buitink and Leprince 2004).
The heat-shock class protein, HSP70, is a molecular chaperone involved in protein importation, translocation, proteolysis, aggregation prevention and refolding of non-native proteins under normal and stress conditions (Frydman 2001, Hartl 1996). Accumulation of HSP70, found in four protein spots in course of acclimation, indicates that this protein is involved in

RESULTS AND DISCUSSION OF PAPERS
28
maintaining proper protein biogenesis and may contribute to freezing tolerance by stabilizing proteins during frost induced protein denaturation.
Several proteins involved in cellular metabolism, such as photosynthesis, carbohydrate and lipid metabolism, protein and secondary metabolite synthesis, were also found in the present study. These include down-regulation of malate dehydrogenase, glyceraldehyde-3-phosphate dehydrogenase, glutamine synthetase, peptide methionine sulfoxide reductase, RuBisCO (Ribulose-1,5-bisphosphate carboxylase oxygenase) activase and the up-reagulation of nucleoside-diphosphate-sugar epimerases, proteasome subunit alpha type-2-B, PsbP, and dihydroflavonol 4-reductase.
The spot, identified as a lipocalin-like protein in this study, showed a 3.87 fold increase during the acclimation period. It was shown by Charron et al. (2005) that a temperature induced lipocalin from arabidopsis (atTIL) is associated with the plasma membrane and that lipocalins and lipocalin-like-proteins are up-regulated during cold acclimation (Charron et al. 2005). A later study reported that atTIL is involved in the plants ability to cope with oxidative stress (Charron et al. 2008). The biochemical function of lipocalin in plants is still unknown and therefore this protein is a strong candidate for further studies.
Many of the proteins found in the present paper belong to large protein families, typically found to fulfill a wide range of functions in the cells. Given the substantial structural and functional knowledge published to date, it is believed that one can now begin to observe the structure of the complex cellular machinery at work during acclimation and extreme freeze tolerance in P. obovata.

RESULTS AND DISCUSSION OF PAPERS
29
PAPER II
Objective: To study the metabolic alterations occurring during acclimation to extreme freezing tolerance in Siberian spruce (P. obovata) and to define when and how this acclimation takes place.
Results: Gas chromatography coupled with mass spectrometry (GC-MS) was used for metabolic profiling of needle contents of P. obovata to reveal metabolic alterations during cold acclimation in response to natural temperature fluctuations during August to January. Significant differences in metabolite profiles occurred in several distinct phases representing the next consecutive time intervals: I - pre-acclimation (around mid-August); II - early acclimation (from the beginning of September to middle of October); III - late acclimation (around the last decade of October); and IV - full acclimation (from the beginning of November to January). The comparison of pre-acclimation (phase I) with full acclimation (phase IV) P. obovata samples revealed almost three hundred putative metabolites, where 223 of these were found accumulated and 53 depleted respectively. Systematic metabolic changes in carbohydrate and lipid metabolism were observed, including changes associated with increased raffinose family oligosaccharide (RFO) synthesis and accumulation, desaturation of fatty acids in membrane lipids, and accumulation of sugar acids, sugar alcohols, and protein and non-protein amino acids and polyamines that may act as compatible solutes or cryoprotectants during acclimation process.
Comments and discussion: This metabolomic study contained some serious difficulties for GC-MS analysis, since concentrations of many of the metabolites varied over a wide range, during the observation period. To find an appropriate balance, GC-MS analysis of samples was done in two modes: split (1/20) and splitless. This improved the quality of the GC-MS data, but increased the amount and complexity of metabolite annotation and further statistical analysis. Combination of univariate and multivariate analyses for data processing proposed in this paper allowed successful handling for both split and splitless data. The orthogonal projections to latent structures-discriminant analysis (OPLS-DA) grouped samples, collected in different times, according to their profiles within the four acclimation phases.
The study has brought about new understanding and a deep insight into the mechanism of acclimation to extreme freezing tolerance in P. obovata. The

RESULTS AND DISCUSSION OF PAPERS
30
important features of the nature of metabolite alterations and characteristics of the acclimation process found in the work are briefly discussed below.
Carbohydrates Increases in sugar concentrations (e.g. sucrose increased by 24x) are the most consistent change associated with low-temperature (LT) acclimation in plants (Sakai and Larcher 1987). In the overall transition from pre-acclimation, to full acclimation in P. obovata, raffinose increased to over 100x its initial level, the single largest increase in any metabolite detected in this study. Raffinose was shown closely associated with LT in conifers and proposed to have important cryoprotectant functions (Fischer and Höll 1991, Strimbeck et.al 2008). The overall increases in melibiose (8x) and galactinol (11x) were likely due to their important roles in RFO metabolism.
Trehalose was found to increase by about 1.9x and was shown by Müller to be associated with desiccation tolerance in plants and animals (Müller et al. 1995). Various studies have suggested that trehalose has special protective properties involved in either direct interaction with proteins or membranes (Crowe et al. 1984, Crowe et al. 1996) or by promoting intracellular vitrification (Koster et al. 2000).
Myo-inositol decreased ≈0.8x, while pinitol and ononitol, both methylated forms of myo-inositol, increased 2x and decreased ≈0.3x respectively, during acclimation. These findings indicate that pinitol has some cryoprotectant function in P. obovata, while the decreases in myo-inositol and ononitol may be a result of their conversion to pinitol.
Amino acids Significant increases in tryptophan (21x) and ornithine (3.3x) were observed rather continuously during acclimation. It was shown by (Kim and Glerum 1995) that tryptophan was the only one out of 19 amino acids that consistently accumulated during acclimation in both Picea glauca and Pinus resinosa (Kim and Glerum 1995). Ornithine is also known to be an intermediate in polyamine synthesis, and various studies indicate that some polyamines may have a role in stress tolerance, either as protectants (Groppa and Benavides 2008) or as signaling molecules (Alcázar et al.). Hence, combining presented findings with these earlier studies suggest that tryptophan and ornithine may have some cryoprotectant function in P. obovata.
Fatty acids and lipid components Like sugar accumulation, fatty acid desaturation and changes in lipid composition are broadly linked to acclimation in both chilling and freezing temperatures. Across the entire acclimation period and in individual stages,

RESULTS AND DISCUSSION OF PAPERS
31
significant changes in seven fatty acids and several compounds (that are common components of lipid head groups) were observed. These findings provided new insight into the mechanism of freezing tolerance development at the metabolic level and highlighted the cryoprotectant role of previously known and new unknown compounds involved in acclimation to extreme freezing tolerance in P. obovata.
PAPER III
Objective: To evaluate the quality and the predictive power of conclusions given by multivariate analysis method (OPLS-DA), a commonly used in metabolomic studies.
Results: The OPLS-DA applied to the GC-MS data of Siberian spruce needles illustrated the division of the observation period from mid-August to January into four consecutive intervals (acclimation phases). This conclusion was approached and tested by univariate analyses for each individual metabolite found in the study. The complimentary analysis of dynamics of individual metabolites rejected the OPLS-DA conclusion in a strong sense: a majority of the metabolites did not follow this four phase change. However, the complimentary analysis confirmed that many metabolites did experience changes in one or several time points, predicted by OPLS-DA.
Comments and discussion: The main focus of this paper was to show that there are clear discrepancies in the conclusions made using multivariate and univarite methods in testing important hypotheses on features and transitions in metabolic modifications. The main efforts of this study were in the proper choice of statistical analysis, the organization of accurate data processing and the proper evaluation of findings to avoid trivial and/or inconsistent statements. Both methods are well known and successful in solving different tasks, so both can provide affirmative answers. With the exception of an analysis of the acclimation phases for each individual metabolite or the whole data set, both the univarite and multivariate methods can be used for searching metabolites with similar dynamics. However, for the hypothesis investigated in the paper, both methods came to two quite different conclusions, meaning that none of them was reliable and that the truth is still hidden within the data.
In this work, apparent weakness of a multivariate method was shown, since it strongly voted for a particular separation of the observation period, which could not be supported by an alternative approach. This investigation

RESULTS AND DISCUSSION OF PAPERS
32
motivates for a developing of new methods for processing and hypothesis testing that would incorporate multivariate and univariate techniques for reliable evaluation of experimental data.
PAPER IV
Objective: To examine plasma samples, corresponding to patients with high risk prostate cancer (PCa) and patients with benign diseases, by a proteomic approach, in order to reveal proteins that could serve as potential biomarkers for high risk prostate cancer.
Results: Plasma proteome analysis of high risk prostate cancer patients with (M1) and without (M0) established bone metastasis, as well as patients with benign prostate disease (B), was performed by 2D-DIGE analysis. This was undertaken after the low-abundant plasma proteins were enriched using the ProteoMiner enrichment technique. About fifty protein identities were detected as differentially expressed, compared between the defined study groups, using both univarite and multivariate analysis in combination. Protein identities that showed a consistently higher or lower plasma level in patients with high risk PCa (despite metastasis status) compared to benign disease (B) were considered as PCa associated proteins. Protein identities with higher or lower plasma levels in metastasis positive patients, compared to other PCa and control groups, were considered as metastasis-associated proteins and these were of specific interest. Twenty-five putative proteins were revealed as metastasis-associated markers with increased plasma levels in M1 patients, including apolipoprotein A1 (APOA1), hyaluronic acid binding protein 2 (HABP2), and inter-alpha-trypsin inhibitor heavy chain H4 (ITIH4). Certain PCa associated proteins were consistently observed to be down-regulated in M1 patients compared to M0 patients and therefore were of notable mention. Among these PCa and metastasis associated proteins are coagulation factor XIII, complement component 4B, plasminogen, and clusterin.
Comments and discussion: Combination of 2D-DIGE with ProteoMiner enrichment technique only allowed for the detection of proteins present in moderate to high abundances in plasma. The identified proteins, associated with high risk prostate cancer and bone metastasis, were mostly common plasma proteins involved in key functions, such as lipid transport, coagulation, inflammation and immune response.

RESULTS AND DISCUSSION OF PAPERS
33
Increased plasma levels of apolipoprotein A1 in patients with established bone metastases recorded in this study are consistent with the recent findings by Fan et al. (2011). It has been illustrated that increased apolipoprotein A1 levels were observed in patients with non-organ confined tumors compared to organ confined prostate tumors. Apolipoprotein A1 is involved in efflux of cholesterol to the liver (Zannis et al. 2004). Thus, higher levels of apolipoprotein in plasma could be indicative of a high cholesterol metabolism in prostate cancer bone metastases (Thysell et al, 2010).
Coagulation factors and fibrinogen were found to be deregulated in patients with high risk prostate cancer. This might be related to an extensive coagulation that often occurs in cancer patients, sometimes with resulting venous thrombosis (Sampson and Kakkar 2002). High levels of hyaluronic acid binding protein 2 (HABP2) and inter-α-trypsin inhibitor heavy chain H4 (ITIH4) were seen in patients with established metastases. Both are known to bind hyaluronic acid, a glycosaminoglycan of extracellular matrix, that has been associated with aggressive behavior of adenocarcinomas (Sironen et al. 2011). HABP2 activates factor VII during the coagulation process and is also termed factor VII activating protease (Romisch 2002). In addition, coagulation factors and regulating proteases could have direct tumor stimulating effects and their plasma levels may not only reflect coagulation but also the biology of the underlying cancer.
The consistently decreased levels of clusterin in patients with high risk tumors verify the previous results observed, of decreased levels in patients with an increasing tumor grade (Byrne et al. 2009). Clusterin is a multifunctional protein which has been implicated in a number of biological processes, such as having both anti- and pro-apoptotic effects (Rizzi and Bettuzzi 2010). Its relevance for prostate cancer progression, however, merits further investigation.
Combination of a ProteoMiner enrichment technique coupled with 2D-DIGE revealed that there were mainly plasma proteins present in moderate concentrations. This is indicative that tumor-derived proteins may still be out of reach. To reach a goal in plasma biomarker research screening, improved fractionation methods and techniques are greatly required.

CONCLUSIONS
34
CONCLUSIONS
This thesis presents the discussion of experimental studies and biochemical analyses for two applications: acquired freezing tolerance in Siberian spruce (P. obovata) and detection of plasma proteins associated with high risk prostate cancer. Both biological systems, being conceptually different, have been investigated using similar experimental techniques and the results have been analyzed by comparative statistical tools. The observed outcomes are briefly reiterated and emphasized below.
Study of extreme freezing tolerance in Siberian spruce
In order to characterize P. obovata’s protein and metabolite alterations and explore their dynamics during acclimation to extreme freezing tolerance, a combination of proteomic (2D-DIGE combined with MALDI-TOF/TOF) and metabolomic (GC-TOF/MS) studies have been performed. The large amount of data generated by these approaches has been carefully analyzed by utilizing appropriate choices of both univariate pair-wise comparison procedures and multivariate projection methods.
The proteomic analysis of needle tissues of P. obovata, during cold acclimation, has revealed differences in protein profiles associated with cold acclimation. These findings highlight the importance of several proteins, which are strongly believed to be involved in acclimation and freezing tolerance development in boreal conifers. The list of proteins associated with the acclimation processes, includes dehydrins (DHNs), heat-shock proteins, proteins related to reactive oxygen species (ROS) regulation, proteins related to cellular metabolism and photosynthesis light reaction.
The proteomic study of needle tissues has been followed by the analysis of metabolite alterations during the acclimation process. At least 275 putative metabolites have been found responsive to cold acclimation. Among other findings, it is worthwhile to emphasize the increases in sugar concentrations: for instance, in the overall transition, from pre-acclimation to the full acclimation state, in P. obovata. Raffinose, known for its cryoprotectant function, was observed to increase by over 100x its initial level, showing the single largest increase in any metabolite detected in this study.
Notable changes in other sugars that are not known to have cryoprotectant functions, but are related to cell wall synthesis or degradation, have been detected. Rhamnose (increased ≈2x) is best known as a component of cell wall polysaccharides, while cellobiose (increased ≈5x) and gentobiose

CONCLUSIONS
35
(decreased ≈0.4x) occur primarily as hydrolysis products of cellulose and hemicelluloses, respectively. Despite its relatively high overall increase, it is unclear what function cellobiose (or cellulase activity) might have in acclimating spruce needles. Myo-inositol decreased ≈0.8x, while pinitol and ononitol, both methylated forms of myo-inositol, have increased ≈2x and decreased ≈0.3x respectively, during acclimation. Other groups of metabolites, such as amino acids, polyamines, fatty acids and lipids have been found strongly associated with the acclimation process in P. obovata.
As a separate contribution in the presented thesis, it is worth mentioning several arguments that could be used in regard to the search and analysis of the phases involved in the development of freezing tolerance in P. obovata done with the help of the MVA. It turned out that this particular question is an example of the conceptual problem present in many metabolomics investigations: To what extent a conclusion, drawn from multivariate analysis and reformulated as a new hypothesis for each individual metabolite, will be valid for the majority of metabolites.
In the study, MVA resulted in separation of the observation period from mid August to January into four phases, in between which, the metabolome of P. obovata needles experienced substantial changes. Thus, a natural question appeared: Can this four phase pattern be observed in the dynamics of individual metabolites?
Comprehensive analysis and discussion led to an unexpected answer: it is likely, that for most of the metabolites, the pattern was not followed. A rather weak correlation existed between the MVA conclusion and the majority of metabolites. It is hoped that such results will stimulate the development of new statistical analysis tools that can provide comparative interpretations both for whole data sets and for a substantial number of individual metabolites.
Overall, the presented proteomic and metabolomic analyses are in-line and coherent with the available results on freezing tolerance. It is known that freeze dehydration causes cell wall shrinkage by forcing together the internal cell membranes and macromolecules. The structure of the membrane in the living cell is a result of the dielectric constant of water (Tanford 1978). To maintain cell integrity, during acclimation and freezing, molecules other than water could maintain the dielectric constant. It was suggested that sugars and dehydrins might carry out this function (Crowe et al. 1997) and in fact, both sugars and dehydrins have been found in this study.

CONCLUSIONS
36
Biomarker detection for aggressive prostate cancer
Plasma proteome analysis of patients with high risk PCa tumours and patients with benign prostate hyperplasia, the second biological system in the study, have revealed several proteins that could potentially serve as candidates for biomarkers. Namely, a high level of hyaluronic acid binding protein (HABP2) has been observed in patients with established metastases. HABP2 activates factor VII during the coagulation process and is also termed factor VII activating protease (Romisch 2002). It has been recently shown that hyaluronic acid content in the micromilieu of prostate tumors is associated with poor prognosis of prostate cancer patients (Josefsson et. al, 2011), and furthermore low molecular fragments of hyaluronic acid are known to activate HABP2, resulting in vascular leakage (Mambetsariev et al. 2011). If tumor levels of hyaluronic acid are mirrored in plasma and/or related to the HABP2 plasma levels, this could imply a possible biomarker that could be used for screening for high risk PCa. However, this has not been validated as of yet and further research is required.
In addition, coagulation factors (with deregulated levels in the patients with high risk prostate cancer) and regulating proteases could have direct tumor stimulating effects. Their plasma levels may not only reflect coagulation, but also the biology of the underlying cancer. The consistently decreased levels of clusterin in patients with high risk tumors concurs with previously recorded results, that showed a decrease in clusterin levels was observed in patients with an increasing tumor grade (Byrne et al. 2009). Clusterin is a multifunctional protein, which has been implicated in a number of biological processes, such as having both anti- and pro-apoptotic effects (Rizzi and Bettuzzi 2010). Its relevance for prostate cancer progression however, merits further investigation.
Protein markers associated with high risk prostate cancer progression, have the potential to improve prognosis of prostate cancer progression. Since this study has been based on a limited number of plasma samples, their value as biomarkers should be further validated. The findings presented in this study may also provide information regarding the mechanisms behind disease progression and thus, possible therapeutic targets, if their functional roles could be revealed in the future.

REFERENCES
37
References
Alcázar, R., Altabella, T., Marco, F., Bortolotti, C., Reymond, M.,
Koncz, C., Carrasco, P. and Tiburcio, A.F. Polyamines: molecules with regulatory functions in plant abiotic stress tolerance. Planta, 231, 1237-1249.
Amme, S., Matros, A., Schlesier, B. and Mock, H.P. (2006) Proteome analysis of cold stress response in Arabidopsis thaliana using DIGE-technology. Journal of experimental botany, 57, 1537.
Anderson, N.L. and Anderson, N.G. (2002) The human plasma proteome - History, character, and diagnostic prospects. Molecular & Cellular Proteomics, 1, 845-867.
Bandow, J.E. (2010) Comparison of protein enrichment strategies for proteome analysis of plasma. Proteomics, 10, 1416-1425.
Boccard, J., Veuthey, J. and Rudaz, S. (2010) Knowledge discovery in metabolomics: An overview of MS data handling. Journal of separation science, 33, 290-304.
Bonan, G.B. and Shugart, H.H. (1989) Environmental factors and ecological processes in boreal forests. Annual Review of Ecology and Systematics, 20, 1-28.
Buitink, J. and Leprince, O. (2004) Glass formation in plant anhydrobiotes: survival in the dry state. Cryobiology, 48, 215-228.
Byrne, J.C., Downes, M.R., O'Donoghue, N., O'Keane, C., O'Neill, A., Fan, Y., Fitzpatrick, J.M., Dunn, M. and Watson, R.W. (2009) 2D-DIGE as a strategy to identify serum markers for the progression of prostate cancer. J Proteome Res, 8, 942-957.
Byrne, J.C., Downes, M.R., O’Donoghue, N., O’Keane, C., O’Neill, A., Fan, Y., Fitzpatrick, J.M., Dunn, M.J. and Watson, R.W.G. (2008) 2D-DIGE as a strategy to identify serum markers for the progression of prostate cancer. Journal of Proteome Research, 8, 942-957.
Charron, J.B.F., Ouellet, F., Houde, M. and Sarhan, F. (2008) The plant Apolipoprotein D ortholog protects Arabidopsis against oxidative stress. BMC Plant Biology, 8, 86.
Chromy, B.A., Gonzales, A.D., Perkins, J., Choi, M.W., Corzett, M.H., Chang, B.C., Corzett, C.H. and McCutchen-Maloney, S.L. (2004) Proteomic analysis of human serum by two-dimensional differential gel electrophoresis after depletion of high-abundant proteins. Journal of Proteome Research, 3, 1120-1127.
Close, T.J. (1996) Dehydrins: Emergence of a biochemical role of a family of plant dehydration proteins. Physiologia Plantarum, 97, 795-803.
Close, T.J. (1997) Dehydrins: A commonality in the response of plants to dehydration and low temperature. Physiologia Plantarum, 100, 291-296.
Crick, F. (1970) Central dogma of molecular biology. Nature, 227, 561-563.

REFERENCES
38
Crowe, J.H., Crowe, L.M. and Chapman, D. (1984) Preservation of membranes in anhydrobiotic organisms: the role of trehalose. Science, 223, 701.
Crowe, J.H., Oliver, A.E., Hoekstra, F.A. and Crowe, L.M. (1997) Stabilization of dry membranes by mixtures of hydroxyethyl starch and glucose: The role of vitrification. Cryobiology, 35, 20-30.
Crowe, L.M., Reid, D.S. and Crowe, J.H. (1996) Is trehalose special for preserving dry biomaterials? Biophysical Journal, 71, 2087-2093.
Damber, J.E. and Aus, G. (2008) Prostate cancer. Lancet, 371, 1710-1721.
Fan, Y., Murphy, T.B., Byrne, J.C., Brennan, L., Fitzpatrick, J.M. and Watson, R.W.G. (2011) Applying Random Forests To Identify Biomarker Panels in Serum 2D-DIGE Data for the Detection and Staging of Prostate Cancer. Journal of Proteome Research, 10, 1361-1373.
Fiehn, O., Kopka, J., Dörmann, P., Altmann, T., Trethewey, R.N. and Willmitzer, L. (2000) Metabolite profiling for plant functional genomics. Nature Biotechnology, 18, 1157-1161.
Fields, S. (2001) Proteomics in genomeland. Science, 291, 1221. Fiorentino, M., Capizzi, E. and Loda, M. (2010) Blood and Tissue
Biomarkers in Prostate Cancer: State of the Art. Urologic Clinics of North America, 37, 131-+.
Fischer, C. and Höll, W. (1991) Food reserves of Scots pine (Pinus sylvestris L.). Trees-Structure and Function, 5, 187-195.
Frankow-Lindberg, B.E. (2001) Adaptation to winter stress in nine white clover populations: Changes in non-structural carbohydrates during exposure to simulated winter conditions and 'spring' regrowth potential. Annals of Botany, 88, 745-751.
Frenette Charron, J.B., Ouellet, F., Pelletier, M., Danyluk, J., Chauve, C. and Sarhan, F. (2005) Identification, expression, and evolutionary analyses of plant lipocalins. Plant Physiology, 139, 2017.
Frydman, J. (2001) Folding of newly translated proteins in vivo: the role of molecular chaperones. Annual review of biochemistry, 70, 603-647.
Görg, A., Boguth, G.Ü.N., Obermaier, C., Posch, A. and Weiss, W. (1995) Two dimensional polyacrylamide gel electrophoresis with immobilized pH gradients in the first dimensin (IPG Dalt): The state of the art and the controversy of vertical versus horizontal systems. Electrophoresis, 16, 1079-1086.
Görg, A., Postel, W. and Günther, S. (1988) The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis, 9, 531.
Groppa, M. and Benavides, M. (2008) Polyamines and abiotic stress: recent advances. Amino Acids, 34, 35-45.
Gullberg, J., Jonsson, P., Nordstrom, A., Sjostrom, M. and Moritz, T. (2004) Design of experiments: an efficient strategy to identify

REFERENCES
39
factors influencing extraction and derivatization of Arabidopsis thaliana samples in metabolomic studies with gas chromatography/mass spectrometry. Anal Biochem, 331, 283-295.
Gygi, S.P., Rochon, Y., Franza, B.R. and Aebersold, R. (1999) Correlation between protein and mRNA abundance in yeast. Molecular and cellular biology, 19, 1720.
Hansen, J. and Beck, E. (1994) Seasonal-Changes in the Utilization and Turnover of Assimilation Products in 8-Year-Old Scots Pine (Pinus-Sylvestris L) Trees. Trees-Structure and Function, 8, 172-182.
Hartl, F.U. (1996) Molecular chaperones in cellular protein folding. Holm, S. (1979) A Simple Sequentially Rejective Multiple Test Procedure.
Scandinavian Journal of Statistics, 6, 65-70. Josefsson A, Adamo H, Hammarstedt P, Granfors T, Stattin P,
Egevad L, Engstrm Laurent A, Wikstrm P, Bergh A. Prostate cancer increases hyaluronan in surrounding non-malignant stroma and this response is associated with tumor growth and an unfavorable outcome. Am. J. Pathol., under revision.
Josson, S., Matsuoka, Y., Chung, L.W.K., Zhau, H.E. and Wang, R. Tumor-stroma co-evolution in prostate cancer progression and metastasis. Seminars in Cell & Developmental Biology, 21, 26-32.
Kaplan, F., Kopka, J., Haskell, D.W., Zhao, W., Schiller, K.C., Gatzke, N., Sung, D.Y. and Guy, C.L. (2004) Exploring the temperature-stress metabolome of Arabidopsis. Plant Physiol, 136, 4159-4168.
Keidel, E.M., Ribitsch, D. and Lottspeich, F. (2010) Equalizer technology - Equal rights for disparate beads. Proteomics, 10, 2089-2098.
Kim, Y. and Glerum, C. (1995) Seasonal free amino acid fluctuations in red pine and white spruce needles. Canadian journal of forest research, 25, 697-703.
Klose, J. (1975) Protein mapping by combined isoelectric focusing and electrophoresis of mouse tissues. Human Genetics, 26, 231-243.
Kolonel, L.N., Altshuler, D. and Henderson, B.E. (2004) The multiethnic cohort study: exploring genes, lifestyle and cancer risk. Nature Reviews Cancer, 4, 519-527.
Koster, K.L., Lei, Y.P., Anderson, M., Martin, S. and Bryant, G. (2000) Effects of vitrified and nonvitrified sugars on phosphatidylcholine fluid-to-gel phase transitions. Biophysical Journal, 78, 1932-1946.
Lam, Y.W., Mobley, J.A., Evans, J.E., Carmody, J.F. and Ho, S.M. (2005) Mass profiling-directed isolation and identification of a stage-specific serologic protein biomarker of advanced prostate cancer. Proteomics, 5, 2927–2938.
Lehmann, E. and Romano, J. (2005) Testing statistical hypotheses: Springer Verlag.

REFERENCES
40
Leman, E.S. and Getzenberg, R.H. (2009) Biomarkers for prostate cancer. Journal of cellular biochemistry, 108, 3-9.
Li, C.Y., Junttila, O. and Palva, E.T. (2004) Environmental regulation and physiological basis of freezing tolerance in woody plants. Acta Physiologiae Plantarum, 26, 213-222.
Lisec, J., Schauer, N., Kopka, J., Willmitzer, L. and Fernie, A.R. (2006) Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat Protoc, 1, 387-396.
Makarov, D.V., Loeb, S., Getzenberg, R.H. and Partin, A.W. (2009) Biomarkers for prostate cancer. Annual review of medicine, 60, 139-151.
Mambetsariev, N., Mirzapoiazova, T., Mambetsariev, B., Sammani, S., Lennon, F.E., Garcia, J.G. and Singleton, P.A. Hyaluronic Acid binding protein 2 is a novel regulator of vascular integrity. Arterioscler Thromb Vasc Biol, 30, 483-490.
Moritz, R.L., Clippingdale, A.B., Kapp, E.A., Eddes, J.S., Ji, H., Gilbert, S., Connolly, L.M. and Simpson, R.J. (2005) Application of 2 D free flow electrophoresis/RP HPLC for proteomic analysis of human plasma depleted of multi high abundance proteins. Proteomics, 5, 3402-3413.
Müller, J., Boller, T. and Wiemken, A. (1995) Trehalose and trehalase in plants: recent developments. Plant science, 112, 1-9.
Nicholson, J.K., Lindon, J.C. and Holmes, E. (1999) Metabonomics': understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica, 29, 1181-1189.
O'Farrell, P.H. (1975) High resolution two-dimensional electrophoresis of proteins. Journal of biological chemistry, 250, 4007.
Oliver, S.G., Winson, M.K., Kell, D.B. and Baganz, F. (1998) Systematic functional analysis of the yeast genome. TRENDS in Biotechnology, 16, 373-378.
Öquist, G. (1982) Seasonally Induced Changes in Acyl Lipids and Fatty Acids of Chloroplast Thylakoids of Pinus silvestris: A correlation between the level of unsaturation of monogalactosyldiglyceride and the rate of electron transport. Plant Physiol, 69, 869-875.
Pernemalm, M., Orre, L.M., Lengqvist, J., Wikström, P., Lewensohn, R. and Lehtio, J. (2008) Evaluation of three principally different intact protein prefractionation methods for plasma biomarker discovery. Journal of Proteome Research, 7, 2712-2722.
Pieper, R., Gatlin, C.L., Makusky, A.J., Russo, P.S., Schatz, C.R., Miller, S.S., Su, Q., McGrath, A.M., Estock, M.A., Parmar, P.P., Zhao, M., Huang, S.-T., Zhou, J., Wang, F., Esquer-Blasco, R., Anderson, N.L., Taylor, J. and Steiner, S. (2003) The human serum proteome: Display of nearly 3700 chromatographically separated protein spots on two-dimensional

REFERENCES
41
electrophoresis gels and identification of 325 distinct proteins. Proteomics, 3, 1345-1364.
Qin, S., Ferdinand, A.S., Richie, J.P., O'Leary, M.P., Mok, S.C. and Liu, B.C.S. (2005) Chromatofocusing fractionation and two dimensional difference gel electrophoresis for low abundance serum proteins. Proteomics, 5, 3183-3192.
Quinn, P.J. (1985) A lipid-phase separation model of low-temperature damage to biological membranes. Cryobiology, 22, 128-146.
Righetti, P.G., Boschetti, E., Lomas, L. and Citterio, A. (2006) Protein Equalizer™ Technology: The quest for a “democratic proteome”. Proteomics, 6, 3980-3992.
Rizzi, F. and Bettuzzi, S. (2010) The clusterin paradigm in prostate and breast carcinogenesis. Endocr Relat Cancer, 17, R1-17.
Romisch, J. (2002) Factor VII activating protease (FSAP): a novel protease in hemostasis. Biol Chem, 383, 1119-1124.
Rysin, L.P. and Savel eva, L.I. (2002) Elovye lesa Rossii: Nauka. Sahni, A., Simpson-Haidaris, P.J., Sahni, S.K., Vaday, G.G. and
Francis, C.W. (2008) Fibrinogen synthesized by cancer cells augments the proliferative effect of fibroblast growth factor-2 (FGF-2). J Thromb Haemost, 6, 176-183.
Sakai, A. and Larcher, W. (1987) Frost survival of plants. Responses and adaptation to freezing stress: Springer-Verlag.
Sampson, M.T. and Kakkar, A.K. (2002) Coagulation proteases and human cancer. Biochem Soc Trans, 30, 201-207.
Schalken, J.A. and van Leenders, G. (2003) Cellular and molecular biology of the prostate: stem cell biology. Urology, 62, 11-20.
Scherl, A., Francois, P., Bento, M., Deshusses, J.M., Charbonnier, Y., Converset, V., Huyghe, A., Walter, N., Hoogland, C. and Appel, R.D. (2005) Correlation of proteomic and transcriptomic profiles of Staphylococcus aureus during the post-exponential phase of growth. Journal of microbiological methods, 60, 247-257.
Schröder, F.H. (2009) PSA screening--a review of recent studies. Eur J Cancer, 45 Suppl 1, 402-404.
Shinozaki, K. and Yamaguchi-Shinozaki, K. (2000) Molecular responses to dehydration and low temperature: differences and cross-talk between two stress signaling pathways. Current Opinion in Plant Biology, 3, 217-223.
Sihlbom, C., Kanmert, I., Bahr, H. and Davidsson, P. (2008) Evaluation of the combination of bead technology with SELDI-TOF-MS and 2-D DIGE for detection of plasma proteins. J Proteome Res, 7, 4191-4198.
Sironen, R.K., Tammi, M., Tammi, R., Auvinen, P.K., Anttila, M. and Kosma, V.M. Hyaluronan in human malignancies. Exp Cell Res, 317, 383-391.
Sreekumar, A., Poisson, L.M., Rajendiran, T.M., Khan, A.P., Cao, Q., Yu, J., Laxman, B., Mehra, R., Lonigro, R.J. and Li, Y.

REFERENCES
42
(2009) Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature, 457, 910.
Steponkus, P.L., Lynch, D.V. and Uemura, M. (1990) The Influence of Cold-Acclimation on the Lipid-Composition and Cryobehavior of the Plasma-Membrane of Isolated Rye Protoplasts. Philosophical Transactions of the Royal Society of London Series B-Biological Sciences, 326, 571-583.
Strimbeck, G.R., Kjellsen, T.D., Schaberg, P.G. and Murakami, P.F. (2007) Cold in the common garden: comparative low-temperature tolerance of boreal and temperate conifer foliage. Trees-Structure and Function, 21, 557-567.
Strimbeck, G.R., Kjellsen, T.D., Schaberg, P.G. and Murakami, P.F. (2008) Dynamics of low-temperature acclimation in temperate and boreal conifer foliage in a mild winter climate. Tree Physiol, 28, 1365-1374.
Tanford, C. (1978) The hydrophobic effect and the organization of living matter. Science, 200, 1012-1018.
Tao, D.L., Oquist, G. and Wingsle, G. (1998) Active oxygen scavengers during cold acclimation of Scots pine seedlings in relation to freezing tolerance. Cryobiology, 37, 38-45.
Thompson, I.M., Pauler, D.K., Goodman, P.J., Tangen, C.M., Lucia, M.S., Parnes, H.L., Minasian, L.M., Ford, L.G., Lippman, S.M. and Crawford, E.D. (2004) Prevalence of prostate cancer among men with a prostate-specific antigen level 4.0 ng per milliliter. New England Journal of Medicine, 350, 2239-2246.
Thulasiraman, V., Lin, S., Gheorghiu, L., Lathrop, J., Lomas, L., Hammond, D. and Boschetti, E. (2005) Reduction of the concentration difference of proteins in biological liquids using a library of combinatorial ligands. Electrophoresis, 26, 3561-3571.
Thysell, E., Surowiec, I., Hornberg, E., Crnalic, S., Widmark, A., Johansson, A.I., Stattin, P., Bergh, A., Moritz, T., Antti, H. and Wikstro, P. (2010) Metabolomic Characterization of Human Prostate Cancer Bone Metastases Reveals Increased Levels of Cholesterol. PLoS One, 5, -.
Trygg, J. and Wold, S. (2002) Orthogonal projections to latent structures (O-PLS). Journal of Chemometrics, 16, 119-128.
Ünlü, M., Morgan, M.E. and Minden, J.S. (1997) Difference gel electrophoresis. A single gel method for detecting changes in protein extracts. Electrophoresis, 18, 2071-2077.
Wang, W., Vignani, R., Scali, M. and Cresti, M. (2006) A universal and rapid protocol for protein extraction from recalcitrant plant tissues for proteomic analysis. Electrophoresis, 27, 2782-2786.
Wasinger, V.C., Cordwell, S.J., Cerpa Poljak, A., Yan, J.X., Gooley, A.A., Wilkins, M.R., Duncan, M.W., Harris, R., Williams, K.L. and Humphery Smith, I. (1995) Progress with gene product

REFERENCES
43
mapping of the Mollicutes: Mycoplasma genitalium. Electrophoresis, 16, 1090-1094.
Wisniewski, M., Webb, R., Balsamo, R., Close, T.J., Yu, X.M. and Griffith, M. (1999) Purification, immunolocalization, cryoprotective, and antifreeze activity of PCA60: a dehydrin from peach (Prunus persica). Physiologia Plantarum, 105, 600-608.
Wold, S., Esbensen, K. and Geladi, P. (1987) Principal Component Analysis. Chemometrics and Intelligent Laboratory Systems, 2, 37-52.
Zannis, V.I., Chroni, A., Kypreos, K.E., Kan, H.Y., Cesar, T.B., Zanni, E.E. and Kardassis, D. (2004) Probing the pathways of chylomicron and HDL metabolism using adenovirus-mediated gene transfer. Curr Opin Lipidol, 15, 151-166.

ACKNOWLEDGMENTS
44
Acknowledgments This work would not have been possible without the help of many people, who have patiently shared their expertise, time, and effort with me throughout my Ph.D years.
First of all, I would like to acknowledge Prof. Peter M. Larsen and Prof. Stephen Fey who introduced me to the world of proteomics ten years ago and gave me the unique chance to assist in their research projects at the Centre for Proteome Analysis (SDU, Odense, Denmark). I would also like to thank Prof. Erik Lundgren from the Department of Molecular Biology at Umea University, for sharing his deep and broad knowledge in melanoma, interferons and molecular biology in general. Special appreciation goes also to my supervisor Prof. Wolfgang Schröder, who constantly encouraged and guided me to take independent steps in my research and academic career. I thank my co-supervisor Assoc. Prof. Pernilla Wikström from the Department of Medical Biosciences & Pathology at Umea University, for providing the opportunity to join her research team and contribute to challenging biomarker discovery in prostate cancer research.
My rather spontaneous and exciting team work with Dr. Trygve K. and Prof. Richard Strimbeck from the Norwegian University of Science and Technology, Norway, turned out to be critical for the success of my studies. I deeply appreciate our truly open collaboration and the discussions we have kept going over many years. The competence and help of many active researchers from chemometric and metabolomic groups were essential in my metabolomics investigations. Iza, Krister, Henrik, Thomas, and IngaBritt, who shared with me their broad knowledge in the topic, helped me with organization of experiments and brought me to understanding of math principles they have developed for statistical data analysis – I thank you all. Finally, to my colleagues and friends at KBC-center, UPSC, SLU, Chemistry Dept, Mol.Biol Dept, Pathology Dept, former and present WS, CF, JM and PW group members, journal-club members of EL group, members of cancer-‘omics’ meetings – thank you all for creating a nice environment, for scientific discussions, for friendly chats and for your every day support.
Adriana, Barbara, Irene, Iza – WE ARE THE BEST! ☺ Thanks for your friendship, sense of reality, laugh, glamour,
dinners, Stockholm and fruitful discussions of my projects in coffe-room and ‘garden’.
Антон и Мама, без Вас бы я не справилась… Благодарю Вас за Вашу поддержку, заботу и любовь. Мои дорогие и самые-самые любимые детки Любочка, Артёмка и Алёшенька... Благодарю Вас за Вашу любовь и за то счастье которые Вы мне принесли. Люблю Вас Всех….
Людмила

REFERENCES
45

REFERENCES
46





























