Embed Size (px)
Citation preview

C H A
P T E R15
Sample Depletion,Fractionation, and Enrichment
for Biomarker DiscoveryHaleem J. Issaq, Timothy D. Veenstra
Laboratory of Proteomics and Analytical Technologies, Advanced Technology Program,Frederick National Laboratory for Cancer Research, Frederick, MD, USA
Ph
O U T L I N E
Introduction 23
7Depletion 238
Fractionation Procedures for Proteinsand Metabolites 238
Affinity Chromatography 241
roteomic and Metabolomic Approaches to Biomarker Discoveryttp://dx.doi.org/10.1016/B978-0-12-394446-7.00015-7 237
Isoelectric Focusing 24
1Size Exclusion Chromatography 242
Conclusions 243
References 243
INTRODUCTION
The strategy for the discovery of protein ormetabolite biomarkers aims for the identificationof all the proteins and/ormetabolites in a sample.Obviously, complete proteome and metabolomecoverage is presently impossible due to a numberof factors, including their dynamic nature. Thecomplexity of the proteome and metabolomerequires the development of efficient analyticalmethods that involve depletion, fractionation,
concentration, and separation procedures in thehope of detecting the largest possible number ofcompounds. Many attempts have been made inthe last 20 years by analytical chemists to developmethods that possess sufficient resolution to sepa-rate the components of the proteome and metab-olome as well as enough sensitivity to detectcompounds present at the lowest abundances.Because the physicochemical nature of the metab-olome and proteome differ, they require differentfractionation and separation procedures. The
Copyright � 2013 Elsevier Inc. All rights reserved.

15. SAMPLE DEPLETION, FRACTIONATION, AND ENRICHMENT FOR BIOMARKER DISCOVERY238
proteome ismade up of tens of thousands of poly-peptides/proteins that possess the same aminoacid building blocks; however, they vary in sizeand cover a wide range of physical and chemicalproperties (e.g., size, acidity, basicity, hydropho-bicity, hydrophilicity, etc.). Unlike the proteome,the metabolome contains compounds that arestructurallyunrelatedbut alsopossess awide rangeof different chemical and physical properties.Metabolites such as carbohydrates, salts, lipids,steroid hormones, hydrocarbons, and so on oftenrequire different procedures for enrichment, extrac-tion, fractionation, separation, and detection.
Sample fractionation, a procedure for simpli-fying complex mixtures, is an important step inthe search for biomarkers in clinical samples.Fractionationof a sample is different fromaliquot-ing a sample. Aliquoting is a simple procedure inwhich the sample (blood, urine) is divided intosmaller portions (aliquots); fractionation is thedivision of the sample’s “contents” (peptides,proteins, metabolites) into several “groups” (frac-tions) based on their chemical and physical prop-erties, generally using chromatography orelectrophoresis. Extraction, unlike depletion, isa purification step with the aim of selecting andenriching a single or group of compounds ofinterest from a mixture, such as extraction ofestrogen metabolites from urine or serum. Thepurpose of extraction, depletion, and fraction-ation in proteomic and metabolomic analysis isto simplify the complexity of the mixture andallow the detection of low-abundance proteinsand metabolites, resulting in an increase in thenumber of compounds that can be identified.
DEPLETION
Depletion is an essential step in the analysisof the proteome and metabolome. It is a processfor the removal of proteins and other compoundsfrom the sample that can interfere with theanalytical procedure and affect the accuracyof the results or the ability to detect
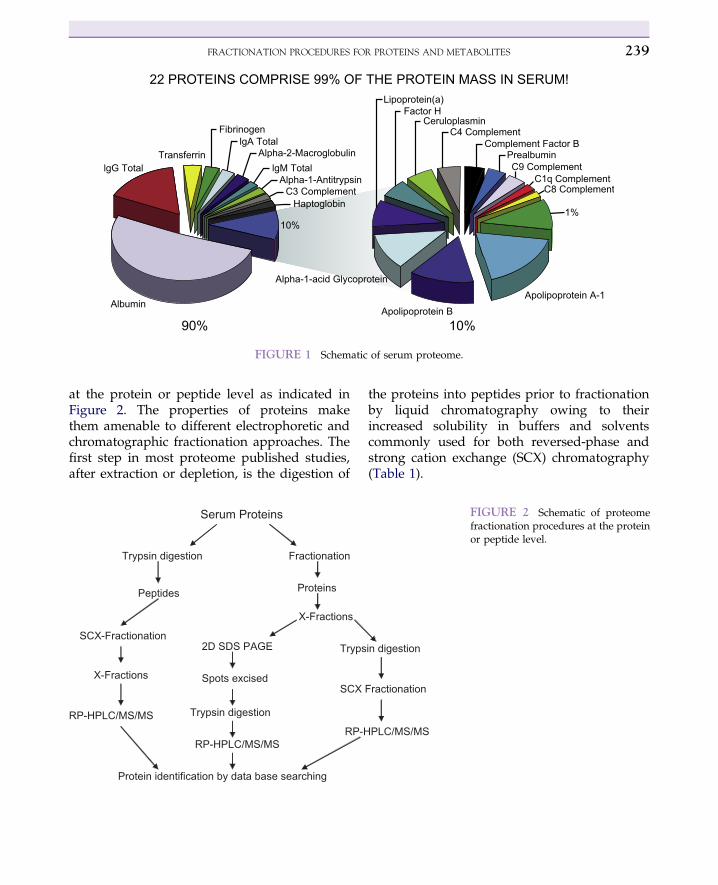
lower-abundance species. Examples of depletionsteps include the removal of human serumalbumin from blood (serum and plasma) andsalts and lipids from urine. The benefits of deple-tion include: (a) the detection of low-abundanceproteins and low-level metabolites that aremasked by the presence of the depletedcompounds, (b) the disruption of the binding ofmetabolites to proteins, and (c) minimization ofionization suppression when using mass spec-trometry (MS) as a detection device. The proteincontent of blood (serum, plasma) is dominatedby high-abundance proteins (albumin, trans-ferrin, a-1 acid glycoprotein, fibrinogen, cerulo-plasmin, a2-macroglobulin, a1-antitrypsin,apolipoprotein, plasminogen, haptoglobin, andprealbumin) that constitute 99% of the proteincontent of blood (Figure 1). Therefore, only 1%of the entire blood proteome is made up ofproteins that are considered to be in low abun-dance and of particular interest in the search forpotential biomarkers. Therefore, depletion of thehigh-abundance proteins is necessary for thedetection of the low-abundance proteins in blood.The most popular methods for depleting albuminand other high-abundance proteins from bloodinclude immunoaffinity columns or membranefilters.1
Analysis of the metabolome of biologicalsamples such as urine and serum requires theprecipitation of proteins followed by the analysisof the supernatant. Precipitation of proteins canbe achieved by heat or the addition of an acidor organic solvent such as acetonitrile, ethanol,or methanol. Removal of salts, lipids, and so onfrom the sample can be accomplished by columnchromatography, solid phase extraction, orliquideliquid extraction.
FRACTIONATION PROCEDURESFOR PROTEINS AND METABOLITES
Fractionation of protein mixtures prior toseparation and MS analysis can be carried out
22 PROTEINS COMPRISE 99% OF THE PROTEIN MASS IN SERUM!
Fibrinogen
Lipoprotein(a)Factor H
CeruloplasminC4 Complement
Complement Factor BPrealbuminC9 Complement
C1q ComplementC8 Complement
Apolipoprotein A-1
1%
TransferrinlgG Total
lgA TotalAlpha-2-Macroglobulin
lgM TotalAlpha-1-Antitrypsin
C3 ComplementHaptoglobin
10%
Alpha-1-acid Glycoprotein
AlbuminApolipoprotein B
90% 10%
FIGURE 1 Schematic of serum proteome.
FRACTIONATION PROCEDURES FOR PROTEINS AND METABOLITES 239
at the protein or peptide level as indicated inFigure 2. The properties of proteins makethem amenable to different electrophoretic andchromatographic fractionation approaches. Thefirst step in most proteome published studies,after extraction or depletion, is the digestion of
Serum Proteins
Trypsin digestion
SCX-Fractionation
Peptides
X-Fractions
RP-HPLC/MS/MS
Fractionation
X-Fractions
Tryps2D SDS PAGE
RP-H
Protein identification by data base searching
Spots excised
Trypsin digestion
RP-HPLC/MS/MS
SCX
Proteins
the proteins into peptides prior to fractionationby liquid chromatography owing to theirincreased solubility in buffers and solventscommonly used for both reversed-phase andstrong cation exchange (SCX) chromatography(Table 1).
in digestion
PLC/MS/MS
Fractionation
FIGURE 2 Schematic of proteomefractionation procedures at the proteinor peptide level.

TABLE 1 Proteome analysis: intact proteins or proteindigests (peptides)?
Protein mixtures arecomplex
Peptide mixtures are morecomplex
Proteins are less soluble Peptides are more soluble
Proteins are harder tohandle
Peptides are easier to workwith
Loss of one proteinis loss of information
Loss of one peptide is notloss of a protein orinformation
Protein analysis by MSrequires sophisticatedinstruments
Peptide analysis is donewith simpler MSinstrumentation that is moreaccessible
15. SAMPLE DEPLETION, FRACTIONATION, AND ENRICHMENT FOR BIOMARKER DISCOVERY240
Protein and metabolite mixtures are fraction-ated based on their physical and chemical prop-erties by a variety of chromatographic andelectrophoretic methods (Table 2).
TABLE 2 Various proteomic and metabolomicfractionation procedures based onchemical/physical properties
Fractionation Method Property
Ultracentrifugation Density
Size exclusionchromatography (SEX)
Stoke’s radius
Isoelectric focusing (IEF) Isoelectric point
Hydrophobic interactionchromatography (HIC)
Hydrophobicity
Hydrophilic interactionchromatography (HILIC)
Polarity
Reversed-phasechromatography (RP)
Hydrophobicity
Ion exchangechromatography (IEX)
Charge
Affinity chromatography Specific biomolecularinteraction
Gel electrophoresis (GE) Stoke’s radius
Fractionation is achieved by manipulation of(a) the mobile phase properties such as slope(time) of the gradient, organic modifier concen-tration, buffer pH, salt concentration, and/orampholyte pH range; (b) type of column:reversed-phase (e.g., C-18, C-8), ion exchange,size exclusion, normal phase (e.g., silica), oraffinity (use of antibody, lectin, aptamers, metal,DNA, etc.); and (c) gel properties. Therefore,experiments are usually designed to separateproteins and metabolites based on their size,hydrophobicity, polarity, charge, isoelectricpoint, or affinity.
Chromatography, especially HPLC in itsdifferent modes of separation, has been usedextensively for the fractionation of complexmixtures of proteins, peptides, and metabolites.An advantage of HPLC is that it allowsthe selective fractionation of a mixture bymanipulation of the mobile and/or stationaryphases. The principle of chromatographic frac-tionation is based on the interaction of thecompounds of interest with the solid support(stationary phase) and the mobile phase. Theinteraction may be adsorption on silica surfaces,partitioning on reversed phase materials, or ionexchange based on effective charge of theproteins, peptides, and metabolites. Fraction-ation is achieved by using mobile phase gradi-ents, whereby the compounds that areintroduced into the head of the column aredifferentially eluted by changing the organicmodifier concentration with time (RP andHILIC chromatography), the salt content withtime (hydrophobic interaction chromatographyand ion exchange [IEX]), or by mobile phasepH gradient (IEX chromatography). The frac-tionation of peptides has been mostly con-ducted using SCX, a procedure that waspopularized by John Yates and his coworkers2
when they introduced multidimensionalprotein identification technology (MudPIT).The acidified peptide mixture is loaded ontoa biphasic SCX/RP column. Discrete peptidefractions are displaced from the SCX column

ISOELECTRIC FOCUSING 241
directly and are trapped at the beginning RPsection of the biphasic column for separation.This iterative process is repeated multiple timesusing increasing salt gradients. An advantage ofthis separation technology is that the entiresystem is coupled directly online with themass spectrometer enabling a large number ofpeptides to be directly identified in a high-throughput manner. Other chromatographicmodes have been used for peptides, proteins,and metabolites fractionation, as given inTable 2.
Although most fractionation techniques havefocused on the development of methods to frac-tionate the entire proteome samples, methodshave also been developed to interrogate a specificsubset of compounds. For example, Regnier andcoworkers 3 used lectin affinity chromatographyto select glycosylated peptides, significantlyreducing the complexity of the peptide mixtureprior to MS analysis. Dr. Xia Xu et al.4 havedeveloped liquideliquid extraction and MSmethods for analyzing 15 different estrogenmetabolites in biofluid samples such as serumand urine.
AFFINITY CHROMATOGRAPHY
Affinity chromatography is primarily used tocapture a specific protein or a class of proteinswithin complex mixtures. The principle ofaffinity chromatography is based on the abilityof a biologically active molecule to bind specifi-cally and reversibly to a complementary mole-cule, which is often bound to a solid support.These ligand molecules may include antibodies,metals, lectins, biotin, aptamers, and other mole-cules. The binding sites of the immobilizedsubstances must be sterically accessible aftertheir coupling to the solid support and shouldnot be deformed by immobilization. In the caseof specific proteins, an affinant is attached tothe active surface of the column packing materialor column surface. The sample is injected onto
the column and the protein(s) of interest iscaptured by the affinant. Proteins that do notpossess a complementary binding site for thebound ligand will either pass directly throughthe column or be eluted by a low-stringencywashing step. The bound proteins are recoveredby washing the column with a competitivesubstrate or a solution that disrupts the interac-tion between the protein and the affinant (e.g.,denaturants). Although an antibody is directedto a specific protein, many other affinitymethods have been developed to capture a classof proteins. These methods include immobilizedmetal affinity columns containing nickel tocapture histidine-containing peptides,5,6 galliumto isolate phosphopeptides,7 or titanium di-oxides for phosphopeptides.8 In addition,affinity methods have been developed to selectpeptides containing specific types of residuessuch as cysteine, tryptophan, or methionine.9,10
There are a variety of different lectins that havebeen used to selectively separate glycoproteinsbased on the composition of the carbohydrateside-chain.11 A method was also developedthat utilizes phosphoprotein isotope affinitytags, combining stable isotope and biotinlabeling for proteome-wide affinity separationand quantitation of phosphoproteins.12
ISOELECTRIC FOCUSING
The principle of isoelectric focusing (IEF) issimple to understand and perform using eithergel or liquid phase medium. The protein sampleis mixed with the desired pH range carrierampholyte mixture, or other carrier buffer, ina focusing cell or spotted on an SDS-PAGE plate.If an electric potential is applied, the proteinsmigrate to a position in the established pHgradient equivalent to their respective isoelectricpoint (pI). If a protein diffuses away from thispH region, its net charge will change and theresulting electrophoretic forces will influence itsmigration back to its pI point. The net result is

15. SAMPLE DEPLETION, FRACTIONATION, AND ENRICHMENT FOR BIOMARKER DISCOVERY242
the “focusing” of proteins into narrow bands attheir pI values. Liquid-phase IEF allows the frac-tionation of a complexmixture of proteins accord-ing to their pIs in a nongel medium. The fractionscan be collected and further analyzed, if needed,using electrophoresis or chromatography. Thedisadvantages of liquid-phase IEF are that highconcentrations of “neutral” proteins (e.g., whenfocusedat their pI) oftenprecipitate fromsolution.Additionally, the ampholytesused to establish thepH gradient may interfere with subsequent elec-trospray ionization MS analysis. Also, highlyhydrophobic proteinsmay be lost in sample prep-aration or during focusing when the proteinsreach their isoelectric point.13 There are severalIEF devices that can be used to fractionatea complex mixture of proteins at the preparativelevel: (a) Rotofor� cell apparatus, Figure 314; (b)Multicompartment Electrolyzer�with isoelectricmembranes15; (c) recycling IEF (RIEF), pioneeredby Bier et al.16; and (d) free-flow (FF) IEF,17 inwhich the sample medium is recycled througha cooling chamber; in (FF)IEF, samples are contin-uously injected into a carrier ampholyte solution.Complex protein solutions can be easily fraction-ated into a large number of fractions that can becollected and subsequently fractionated using an
FIGURE 3 Rotofor is a liquid-phase isoelectric focusingdevice for the fractionation of a proteome into 20 fractions.
orthogonal chromatographic or electrophoretictechnique.18,19 For comprehensive fractionationand separation, a combination of chromato-graphic and electrophoretic procedures areemployed,20e22 for example, fractionation by gelelectrophoresis followed by digestion of theprotein bands into peptides, which are subse-quently separated using RP chromatography. Itis also possible to fractionate the protein mixtureusing liquid-phase IEF. In this mode, intactproteins separated by IEX are excised from thegel and digested into peptides. These peptidemixtures are then separated by RP-HPLC.Although the combinations of fractionations thatcan be used are almost unlimited, some combina-tions are not particularly efficient, and too manymanipulations can lead to sample loss.
SIZE EXCLUSIONCHROMATOGRAPHY
Size exclusion chromatography (SEC) andelectrophoresis separate proteins according totheir size. Although SEC is an old technique, ithas not been used extensively for proteome frac-tionation due to its comparatively low resolu-tion. In a recent proteomic study by Kosanamet al.,20 SEC was used for the fractionation ofproteins in bronchoalveolar lavage fluid fromlung transplant patients with and withoutchronic graft dysfunction. The proteomes ofbronchoalveolar lavage fluids collected fromfour asymptomatic post-transplant patients andthree patients with symptoms of chronic graftdysfunction were each fractionated into six frac-tions by SEC. The fractions were desalted,concentrated, and analyzed by reversed-phaseHPLC/MS/MS. The study resulted in the identi-fication of 531 proteins. A total of 30 and 39proteins detected exclusively in chronic graftdysfunction and nonchronic graft dysfunction,respectively, were identified as potential candi-dates for the verification phase of this biomarkerdiscovery project. It is not unusual to combine

REFERENCES 243
different extraction procedures in the same studyto achieve the largest number of metabolites/proteins; for example, the pancreatic ascitic fluidproteome was fractionated using a multidimen-sional chromatographic approach that includedsize exclusion, ion exchange, and lectin-affinitychromatography.21 The study resulted in theidentification of 816 proteins, of which 20 over-expressed proteins were identified as putativebiomarker candidates. Also, fractionation ofprotein mixtures may be carried out at theprotein or peptide level using multiple fraction-ation procedures, as mentioned earlier. Metabo-lites are extracted using different solvents thatare compatible with the properties of the metab-olites; different extraction solvents would resultin different metabolic profiles that may affectthe interpretation of the data.22
CONCLUSIONS
The search for protein or metabolite diseasebiomarkers requires the detection and identifi-cation of the largest possible number of thesecompounds in the proteome and metabolomeof clinical samples. To achieve this mission,analytical chemists have devised different chro-matographic and electrophoretic methods forthe depletion, fractionation, and concentrationprior to molecular identification. Depletionand fractionation are essential approaches thatare used to simplify the complexity of the pro-teome and the metabolome and to increase theefficiency of the mass spectrometer. It is notunusual to use a combination of chromato-graphic techniques to achieve comprehensiveproteomic analysis and detection of low-abundance proteins. Although MS and otherinstrumental technologies are often given themost credit for driving proteomics and metabo-lomics discovery, it is a certainty that neither ofthese fields would exist without the invention ofsome of the fractionation techniques discussedin this chapter.
References1. Millioni R, Tolin S, Puricelli L, et al. High abundance
protein depletion vs low abundance protein enrich-ment: Comparison of methods to reduce plasma pro-teome complexity. PloS ONE 2011;6(5):e19603.
2. Washburn MP, Wolters D, Yates JR. Large-scale anal-ysis of the yeast proteome by multidimensional proteinidentification technology. Nat Biotechnol 2001;19(3):242e7.
3. Geng M, Ji J, Regnier F. Signature-peptide approach todetecting proteins in complex mixtures. J Chromatogr A2000;870:295e313.
4. Xu X, Roman JM, Issaq HJ, et al. Quantitative measure-ment of endogenous estrogens and estrogen metabolitesin human serum by liquid chromatography-tandemmass spectrometry. Anal Chem 2007;79(20):7813e21.
5. Nakagawa Y, Yip T, Belew M, Porath J. Anal Biochem1981;75:168e72.
6. Ji J, Chakraborty A, Geng M, Zhang X, et al. Strategyfor qualitative and quantitative analysis in proteomicsbased on signature peptides. J Chromatogr B 2000;745:197e210.
7. Posewitz MC, Cooper J. Immobilized gallium(III)affinity chromatography of phosphopeptides. AnalChem 1999;71:2883e92.
8. Yu LR, Zhu Z, Chan KC, Issaq HJ, Dimitrov DS,Veenstra TD. Improved titanium dioxide enrichment ofphosphopeptides from HeLa cells and high confidentphosphopeptide identification by cross-validation ofMS/MS and MS/MS/MS spectra. J Proteome Res 2007;6(11):4150e62.
9. Barnard G, Bayer E, Wilchek M, Amir-Zaltsman Y.bioluminescence assay using avidin-biotin technology.Meth Enzymol 1986;133:284e8.
10. Coligan JE. In: Coligan JE,DunnB, PloeghH, SpeicherD,Wingfield P, editors. Current Protocols in Protein Science.New York: John Wiley and Sons; 1999. p. A.1A.
11. Schachter H. The “yellow brick road” to branchedcomplex N-glycans. Glycobiology 1991;1:453e61.
12. Goshe MB, Conrads TP, Panisko EA, et al. Phospho-protein isotope-coded affinity tag approach for isolatingand quantitating phosphopeptides in proteome-wideanalyses. Anal Chem 2001;73:2578e86.
13. Harry JL, Wilkins MR, Herbert BR. Proteomics:capacity versus utility. Electrophoresis 2000;21:1071e81.
14. Kachman MK, Wang H, Schwartz DR, Cho KR, et al.A 2-D liquid separations/mass mapping method forinterlysate comparison of ovarian cancers. Anal Chem2002;74:1779e91.
15. Herbert B, Righetti PG. A turning point in proteomeanalysis: sample prefractionation via multicompart-ment electrolyzers with isoelectric membranes. Electro-phoresis 2000;21:3639e48.

15. SAMPLE DEPLETION, FRACTIONATION, AND ENRICHMENT FOR BIOMARKER DISCOVERY244
16. Bier M. Recycling isoelectric focusing and iso-tachophoresis. Electrophoresis 1998;19:1057e63.
17. Hanning K. New aspects in preparative and analyticalcontinuous free-flow cell electrophoresis. Electrophoresis1982;3:235e43.
18. Wall DB, Kachman MT, Gong S, et al. Isoelectricfocusing nonporous RP HPLC: a two-dimensionalliquid-phase separation method for mapping ofcellular proteins with identification using MALDI-TOFmass spectrometry. Anal Chem 2000;72:1099e111.
19. Hoffmann P, Ji H, Moritz PL. Continuous free-flowelectrophoresis separation of cytosolic proteins fromthe human colon carcinoma cell line LIM 1215: a non
two-dimensional gel electrophoresis-based proteomeanalysis strategy. Proteomics 2001;1:807e18.
20. Kosanam H, Sato M, Batruch I, et al. Differential pro-teomic analysis of bronchoalveolar lavage fluid fromlung transplant patients with and without chronic graftdysfunction. Clin Biochem 2012;45(3):223e30.
21. Kosanam H, Makawita S, Judd B, et al. Mining themalignant ascites proteome for pancreatic cancerbiomarkers. Proteomics 2011;11(23):4551e8.
22. Duportet X, Mereschi RBA, Carneiro S, et al. The bio-logical interpretation of metabolomics data can bemisled by the extraction method used. Metabolomics2012;8:410e21.


![Metabolomic studies of human gastric cancer: Review · 2017-04-25 · biomarker discovery[39,56,57] and natural product drug discovery[18]. However, none of them have focused on a](https://img.dokumen.tips/doc/110x75/5ecff38006783c7a596b3a06/metabolomic-studies-of-human-gastric-cancer-review-2017-04-25-biomarker-discovery395657.jpg)


























