Embed Size (px)
Citation preview

Protein Structure and Function
Andrew HowardFourth LectureIntroductory Biochemistry
31 January 2008

IIT Biochemistry: 31 Jan 2008 Slide 2 of 60
Protein Structure Helps us Understand Protein Function If we do know what a protein does, its structure will tell us how it does it.
If we don’t know what a protein does, its structure might give us what we need to know to figure out its function.

IIT Biochemistry: 31 Jan 2008 Slide 3 of 60
Let’s finish up peptides, though! We need to talk about Ramachandran angles, peptide bonds, and oligopeptide and polypeptide chemistry before we go on to techniques for determining structures.

IIT Biochemistry: 31 Jan 2008 Slide 4 of 60
Ramachandran angles
G.N. Ramachandran

IIT Biochemistry: 31 Jan 2008 Slide 5 of 60
Preferred Values of and
Steric hindrance makes some values unlikely
Specific values are characteristic of particular types of secondary structure
Most structures with forbidden values of and turn out to be errors

IIT Biochemistry: 31 Jan 2008 Slide 6 of 60
How far from 180º can vary? Remember what we said about the partial double bond character of the C-N main-chain bond
That imposes planarity In practice it rarely varies by more than a few degrees from 180º.
IIT Biochemistry: 31 Jan 2008 Slide 7 of 60
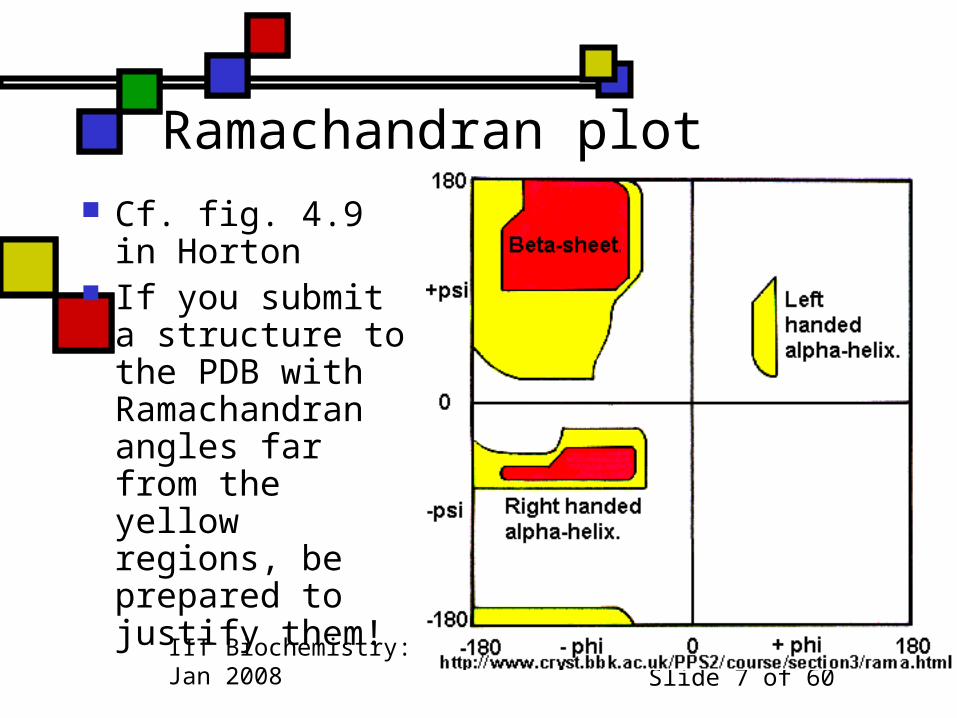
Ramachandran plot Cf. fig. 4.9 in Horton
If you submit a structure to the PDB with Ramachandran angles far from the yellow regions, be prepared to justify them!

IIT Biochemistry: 31 Jan 2008 Slide 8 of 60
How are oligo- and polypeptides synthesized? Formation of the peptide linkages occurs in the ribosome under careful enzymatic control
Polymerization is endergonic and requires energy in the form of GTP (like ATP, only with guanosine):
GTP + n-length-peptide + amino acid GDP + Pi + (n+1)-length peptide

IIT Biochemistry: 31 Jan 2008 Slide 9 of 60
What happens at the ends? Usually there’s a free amino end and a free carboxyl end:
H3N+-CHR-CO-(peptide)n-NH-COO-
Cyclic peptides do occur Cyclization doesn’t happen at the ribosome: it involves a separate, enzymatic step.

IIT Biochemistry: 31 Jan 2008 Slide 10 of 60
Reactivity in peptides & proteins
Main-chain acid-base reactivity unavailable except on the ends
Side-chain reactivity available but with slightly modified pKas.
Terminal main-chain pKavalues modified too
Environment of protein side chain is often hydrophobic, unlike free amino acid side chain’s neighborhood

IIT Biochemistry: 31 Jan 2008 Slide 11 of 60
Discussion questionWhat’s the net charge in ELVISat pH 7?
(a) 0 (b) +1 (c) -1 (d) +2 (e) -2
IIT Biochemistry: 31 Jan 2008 Slide 12 of 60
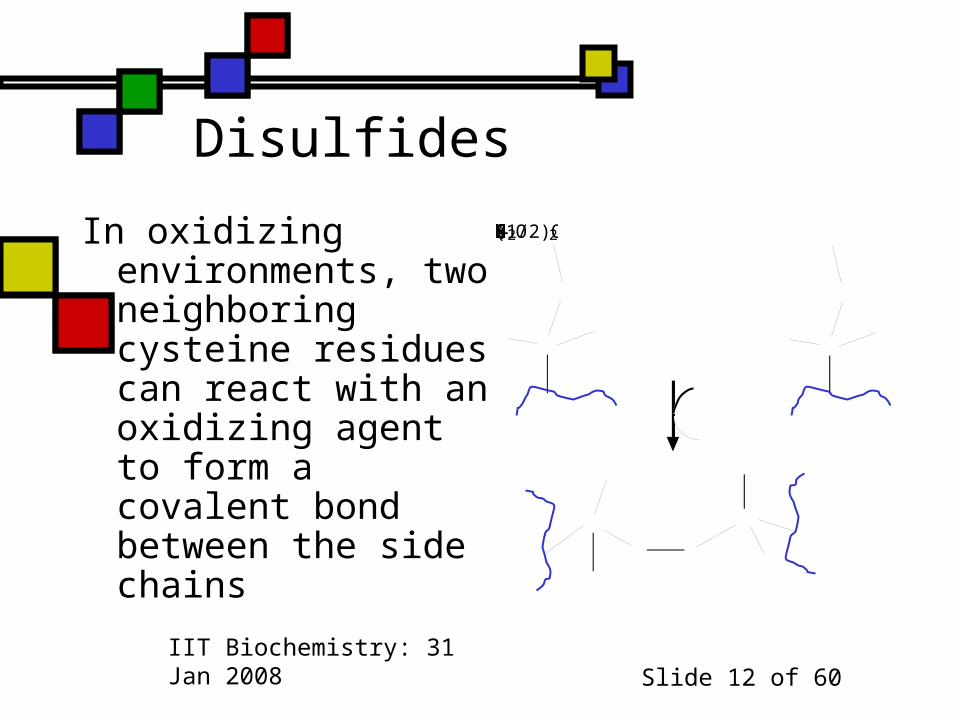
Disulfides
In oxidizing environments, two neighboring cysteine residues can react with an oxidizing agent to form a covalent bond between the side chains
CHHSHCHHSH+(1/2)O2SSHCHHCHH2O

IIT Biochemistry: 31 Jan 2008 Slide 13 of 60
What could this do? Can bring portions of a protein that are distant in amino acid sequence into close proximity with one another
This can influence protein stability

IIT Biochemistry: 31 Jan 2008 Slide 14 of 60
… and now, onward to protein structure methods and results We’ll look at several techniques, and then focus on what we know on the basis of the application of those techniques.

IIT Biochemistry: 31 Jan 2008 Slide 15 of 60
Plans for Today
Methods of Determining Protein Structure Crystallography
NMR CryoEM Specialty techniques
Levels of Protein Structure
Hydrogen Bonds Secondary structure in globular proteins
Tertiary Structure
Domains

IIT Biochemistry: 31 Jan 2008 Slide 16 of 60
Warning: Specialty Content! I determine protein structures (and develop methods for determining protein structures) as my own research focus
So it’s hard for me to avoid putting a lot of emphasis on this material
But today I’m allowed to do that, because it’s the stated topic of the day.

IIT Biochemistry: 31 Jan 2008 Slide 17 of 60
How do we determine structure? We can distinguish between methods that require little prior knowledge (crystallography, NMR, ?CryoEM?)and methods that answer specific questions (XAFS, fiber, …)
This distinction isn’t entirely clear-cut

IIT Biochemistry: 31 Jan 2008 Slide 18 of 60
Crystallography: overview Crystals are translationally ordered 3-D arrays of molecules
Conventional solids are usually crystals
Proteins have to be coerced into crystallizing
… but once they’re crystals, they behave like other crystals, mostly

IIT Biochemistry: 31 Jan 2008 Slide 19 of 60
How are protein crystals unusual? Aqueous interactions required for crystal integrity: they disintegrate if dried
Bigger unit cells (~10nm, not 1nm) Small # of unit cells and static disorder means they don’t scatter terribly well
So using them to determine 3D structures is feasible but difficult

IIT Biochemistry: 31 Jan 2008 Slide 20 of 60
Crystal structures: Fourier transforms of diffraction results Position of spots tells you how big the unit cell is
Intensity tells you what the contents are We’re using electromagnetic radiation, which behaves like a wave, exp(2ik•x)
Therefore intensity Ihkl = C*|Fhkl|2
Fhkl is a complex coefficient in the Fourier transform of the electron density in the unit cell:(r) = (1/V) hkl Fhkl exp(-2ih•r)

IIT Biochemistry: 31 Jan 2008 Slide 21 of 60
The phase problem Note that we said Ihkl = C*|Fhkl|2
That means we can figure out|Fhkl| = (1/C)√Ihkl
But we can’t figure out the direction of F:Fhkl = ahkl + ibhkl = |Fhkl|exp(ihkl)
This direction angle is called a phase angle
Because we can’t get it from Ihkl, we have a problem: it’s the phase problem!
F
ab

IIT Biochemistry: 31 Jan 2008 Slide 22 of 60
What can we learn? Electron density map + sequence we can determine the positions of all the non-H atoms in the protein—maybe!
Best resolution possible: Dmin = / 2 Often the crystal doesn’t diffract that well, so Dmin is larger—1.5Å, 2.5Å, worse
Dmin ~ 2.5Å tells us where backbone and most side-chain atoms are
Dmin ~ 1.2Å: all protein atoms, most solvent, some disordered atoms

IIT Biochemistry: 31 Jan 2008 Slide 23 of 60
What does this look like? Takes some experience to interpret
Automated fitting programs work pretty well with Dmin < 2.1Å
ATP binding to a protein of unknown function: S.H.Kim

IIT Biochemistry: 31 Jan 2008 Slide 24 of 60
How’s the field changing?
1990: all structures done by professionals
Now: many biochemists and molecular biologists are launching their own structure projects as part of broader functional studies
Fearless prediction: by 2020, crystallographers will be either technicians or methods developers

IIT Biochemistry: 31 Jan 2008 Slide 25 of 60
Macromolecular NMR NMR is a mature field Depends on resonant interaction between EM fields and unpaired nucleons (1H, 15N, 31S)
Raw data yield interatomic distances Conventional spectra of proteins are too muddy to interpret
Multi-dimensional (2-4D) techniques:initial resonances coupled with additional ones

IIT Biochemistry: 31 Jan 2008 Slide 26 of 60
Typical protein 2-D spectrum
Challenge: identify whichH-H distance is responsible for a particular peak
Enormous amount of hypothesis testing required
Prof. Mark Searle,University of Nottingham

IIT Biochemistry: 31 Jan 2008 Slide 27 of 60
Results
Often there’s a family of structures that satisfy the NMR data equally well
Can be portrayed as a series of threads tied down at unambiguous assignments
They portray the protein’s structure in solution

IIT Biochemistry: 31 Jan 2008 Slide 28 of 60
Comparing NMR to X-ray NMR family of structures often reflects real
conformational heterogeneity Nonetheless, it’s hard to visualize what’s
happening at the active site at any instant Hydrogens sometimes well-located;
they’re often the least defined atoms in an X-ray structure
The NMR structure is obtained in solution! Hard to make NMR work if MW > 25 kDa

IIT Biochemistry: 31 Jan 2008 Slide 29 of 60
What does it mean when NMR and X-ray structures differ?
Lattice forces may have tied down or moved surface amino acids in X-ray structure
NMR may have errors in it X-ray may have errors in it (measurable)
X-ray structure often closer to true atomic resolution
X-ray structure has built-in reliability checks

IIT Biochemistry: 31 Jan 2008 Slide 30 of 60
Cryoelectron microscopy
Like X-ray crystallography,EM damages the samples
Samples analyzed < 100Ksurvive better
2-D arrays of molecules Spatial averaging to improve resolution
Discerning details ~ 4Å resolution
Can be used with crystallography

IIT Biochemistry: 31 Jan 2008 Slide 31 of 60
Circular dichroism
Proteins in solution can rotate polarized light
Amount of rotation varies with
Effect depends on interaction with secondary structure elements, esp.
Presence of characteristic patterns in presence of other stuff enables estimate of helical content

IIT Biochemistry: 31 Jan 2008 Slide 32 of 60
Poll question: discuss! Which protein would yield a more interpretable CD spectrum? (a) myoglobin (b) Fab fragment of immunoglobulin G
(c) both would be fully interpretable
(d) CD wouldn’t tell us anything about either protein

IIT Biochemistry: 31 Jan 2008 Slide 33 of 60
Ultraviolet spectroscopy
Tyr, trp absorb and fluoresce:abs ~ 280-274 nm; f = 348 (trp), 303nm (tyr)
Reliable enough to use for estimating protein concentration via Beer’s law
UV absorption peaks for cofactors in various states are well-understood
More relevant for identification of moieties than for structure determination
Quenching of fluorescence sometimes provides structural information





























