Embed Size (px)
Citation preview

of September 17, 2018.This information is current as Not IL-17
Associated with IL-23-Dependent IL-22, butEnteritidis in the Absence of IL-12 Is
serovarSalmonella entericawith Attenuated Protective Immunity to Systemic Infection
Gottfried AlberChristoph Holscher, Uwe Müller, Robert A. Kastelein and Ellen Witte, Kerstin Wolk, Robert Sabat, Yoichiro Iwakura,Knauer, Reinhard K. Straubinger, Alissa A. Chackerian, Silke M. Schulz, Gabriele Köhler, Nicole Schütze, Jens
http://www.jimmunol.org/content/181/11/7891doi: 10.4049/jimmunol.181.11.7891
2008; 181:7891-7901; ;J Immunol
Referenceshttp://www.jimmunol.org/content/181/11/7891.full#ref-list-1
, 28 of which you can access for free at: cites 81 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2008 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Protective Immunity to Systemic Infection with AttenuatedSalmonella enterica serovar Enteritidis in the Absence of IL-12Is Associated with IL-23-Dependent IL-22, but Not IL-171
Silke M. Schulz,* Gabriele Kohler,† Nicole Schutze,* Jens Knauer,* Reinhard K. Straubinger,*Alissa A. Chackerian,‡ Ellen Witte,§ Kerstin Wolk,§ Robert Sabat,§ Yoichiro Iwakura,¶
Christoph Holscher,� Uwe Muller,* Robert A. Kastelein,‡ and Gottfried Alber2*
IL-12 is essential for protective T cell-mediated immunity against Salmonella infection. To characterize the role of the relatedcytokine IL-23, wild-type (WT) C57BL/6 and p19�/� mice were infected systemically with an attenuated strain of Salmonellaenterica serovar Enteritidis (S. Enteritidis). IL-23-deficient mice controlled infection with S. Enteritidis similarly as WT mice.Similar IFN-� production as compared with WT mice, but defective IL-17A and IL-22 production was found in the absence ofIL-23. Nevertheless, although IL-23 is required for T cell-dependent cytokine responses, IL-23 is dispensable for protection againstS. Enteritidis when IL-12 is present. To analyze the role of IL-23 in the absence of IL-12, low doses of S. Enteritidis wereadministered to p35�/� mice (lacking IL-12), p35/19�/� mice (lacking IL-12 and IL-23), p35/40�/� mice (lacking IL-12, IL-23, andhomodimeric IL-12p40), or p35/IL-17A�/� mice (lacking IL-12 and IL-17A). We found survival of p35�/� and p35/IL-17A�/�
mice, whereas p35/19�/� and p35/40�/� mice died within 3–6 wk and developed liver necrosis. This indicates that IL-23, but nothomodimeric IL-12p40, is required for protection, which, surprisingly, is independent of IL-17A. Moreover, protection wasassociated with IL-22, but not IL-17F or IL-21 expression or with neutrophil recruitment. Finally, anti-IL-22 treatment of S.Enteritidis-infected p35�/� mice resulted in liver necrosis, indicating a central role of IL-22 in hepatocyte protection duringsalmonellosis. In conclusion, IL-23-dependent IL-22, but not IL-17 production is associated with protection against systemicinfection with S. Enteritidis in the absence of IL-12. The Journal of Immunology, 2008, 181: 7891–7901.
I t is very well documented that IL-12-dependent Th1 immu-nity is essential for control of Salmonella infection in miceand humans (1, 2). It remains to be studied whether Salmo-
nella infection is able to activate IL-23-dependent Th17 responsesassociated with IL-17 and IL-22 production and whether this con-tributes to protective immunity. Th17 cells play a dominant role inhost defense against bacterial infection at the mucosal body bar-riers (reviewed in Ref. 3). However, the significance of these cellsduring systemic infection is currently unknown. In particular, it isof interest whether IL-17A and IL-22 produced by Th17 cells havedistinct or common functions.
Bacteriae of the genus Salmonella are Gram-negative Enter-obacteriaceae that can infect humans and animals. The symp-toms induced by Salmonella can range from acute self-limitinggastroenteritis (termed nontyphoidal salmonellosis) to systemicenteric fever (termed typhoid-like disease) (4, 5). Experimental
systemic infection of mice with different serovars of Salmonellaenterica mimics human typhoid-like disease. We established amodel of systemic infection in mice with an attenuated strain ofSalmonella enterica serovar Enteritidis (S. Enteritidis)3 (6). Thepathogenicity of this auxotrophic strain of S. Enteritidis formice depends on the size of inoculum used. It is sublethal forwild-type (WT) mice when given at inocula lower than 107
CFU. In addition, this strain of S. Enteritidis can be controlledeven by Th1-compromised mice (e.g., IL-12-deficient mice)when given at low doses (up to 2.5 � 103 CFU/mouse). Thus,using an attenuated strain of S. Enteritidis in defined doseranges allowed us to establish a long-term infection model foreach of both immunocompetent and immunodeficient mice toanalyze protective adaptive immune mechanisms. After i.p. ap-plication of a high sublethal infective dose of this attenuated S.Enteritidis strain (i.e., 2.5 � 106 CFU for WT C57BL/6 mice)and of a low sublethal infective dose (i.e., 2.5 � 103 CFU forIL-12-deficient C57BL/6 mice), either mouse strain survivesand does not develop a typhoid-like disease. These two differentexperimental models are suitable for studies comparing immu-noregulatory mechanisms in the presence and particularly in theabsence of IL-12, as is the case in human patients with IL-12/IL-23 defects leading to recurrent salmonellosis (7).
*Institute of Immunology, College of Veterinary Medicine, University of Leipzig,Leipzig, Germany; †Gerhard Domagk Institute of Pathology, University of Munster,Munster, Germany; ‡Discovery Research, Schering-Plough Biopharma, Palo Alto,CA 94304; §Interdisciplinary Group of Molecular Immunopathology, Dermatology/Medical Immunology, University Hospital Charite, Berlin, Germany; ¶University ofTokyo, Tokyo, Japan; and �Research Center Borstel, Borstel, Germany
Received for publication July 18, 2008. Accepted for publication September 22, 2008.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by Research Grant Al 371/3-3 from the Deutsche For-schungsgemeinschaft (to G.A.) and Bundesministerium fur Bildung, Wissenschaft,Forschung und Technologie Grant EXC306 “Inflammation at Interfaces” (to C.H.).2 Address correspondence and reprint requests to Dr. Gottfried Alber, Institute ofImmunology, College of Veterinary Medicine, University of Leipzig, An den Tier-kliniken 11, 04103 Leipzig, Germany. E-mail address: [email protected]
3 Abbreviations used in this paper: S. Enteritidis, Salmonella enterica serovar Enter-itidis; dpi, days postinfection; hk, heat killed; HPRT, hypoxanthine phosphoribosyl-transferase; NACE, naphthol AS-D-chloracetate; PMN, polymorphonuclear cell; WT,wild type.
Copyright © 2008 by The American Association of Immunologists, Inc. 0022-1767/08/$2.00
The Journal of Immunology
www.jimmunol.org
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

IL-12 produced by Salmonella-infected dendritic cells and mac-rophages is the key cytokine for Th1 cell development. Th1-de-rived IFN-� is responsible for activation of macrophages with in-ducible NO synthase-dependent production of NO (1, 8, 9). In theabsence of IL-12, IFN-� production is reduced and bacterial con-trol is impaired (2). IL-12 is a heterodimeric cytokine composed ofthe two subunits p40 and p35. It shares the p40 subunit with therecently discovered IL-23 (10). The p40 subunit of IL-12 can alsoform homodimers (p40)2 that were shown to act as antagonists forIL-12 at the IL-12R (11) or as agonists (12, 13).
IL-23 was found to have functions different from IL-12. It actson the CD4� Th17 cell lineage (14–16) to induce proliferation andmaintenance of these cells. Very recently, we were able to dem-onstrate that Salmonella infection, in addition to Th1 induction,activates Th17 cells and is associated with IL-17A production byclassical Th17 cells, �� T cells, and other CD4� lymphocytes (17).IL-17A produced by Th17 cells is a proinflammatory cytokine thatinduces the expression of IL-6, CXCL8, G-CSF, and TNF (18–20), and influences the activation and migration of polymorpho-nuclear cells (PMN) by induction of CXC chemokines (21, 22).The receptor for IL-17A is ubiquitously expressed in different tis-sues (23), leading to pleiotropic effects of the cytokine (reviewedin Refs. 24–26). IL-17-mediated recruitment of PMN is requiredfor protective immunity after infection with several pathogens(27–30). In addition, the IL-23/IL-17A pathway has been found tomediate pathophysiological processes and the development of or-gan-dependent autoimmunity, such as experimental autoimmuneencephalomyelitis, collagen-induced arthritis, and inflammatorybowel disease (31–34). IL-17F is another member of the IL-17family and is most closely related to IL-17A. Its expression wasfound to be similarly regulated as IL-17A in activated Th17 cells(32, 35–37). Little is known yet about the biological function ofIL-17F, but it was shown to share functions of IL-17A to a weakerextent (e.g., induction of various cytokines and pulmonary neutro-phil recruitment) (38, 39).
Only recently, Th17 cells were found to coexpress IL-17Aand IL-22 in response to IL-23 (40, 41). IL-22 is a novel IL-10family member, originally termed IL-10-related T cell-derivedinducible factor (42, 43). Neither resting nor activated immunecells express the functional IL-22R, and IL-22 does not haveany effects on these cells (44, 45). In contrast, tissue cells of theskin, digestive, and respiratory system tracts are putative targetsof this cytokine. IL-22 can induce the production of antimicro-bial molecules, such as �-defensins and S100 proteins (45) andacute-phase proteins (e.g., serum amyloid A and LPS-bindingprotein) (46, 47). Furthermore, IL-22 expression has beenlinked to proinflammatory processes in psoriasis, rheumatoidarthritis, and inflammatory bowel disease (45, 47–52). In con-trast, IL-22 can protect hepatocytes from apoptosis after ConA-induced hepatitis (53–55). Therefore, IL-22 represents anovel type of immune mediator that, although produced by Tcells, regulates tissue protection and homeostasis, and enhancesthe innate immunity of tissue cells (56).
The goal of this study was to define the role of IL-23 in immu-nity to systemic infection with an attenuated strain of S. Enteritidis.It was of interest to: 1) distinguish between the presence and ab-sence of IL-12-dependent Th1 responses, and 2) characterize therole of IL-17A and IL-22 production stimulated by IL-23. Our dataprovide evidence for an essential role of a protective IL-23/IL-22axis in a Th1-compromised state that may be relevant for patientswith an inherited defect in the IL-12/IL-12R/IFN-� pathway suf-fering from recurrent infections (reviewed in Ref. 7).
Materials and MethodsAnimals
C57BL/6 WT, IL-23p19�/� (p19�/�) (31), IL-12p35�/� (p35�/�), p35/40�/� (57, 58), and p35/19�/� (59) mice were bred and kept at the animalfacility of the Max Planck Institute of evolutionary Anthropology. Thep35/IL-17A�/� mice were obtained by crossing p35�/� and IL-17A�/�
mice (60). Only females at an initial age of 8–12 wk were used for ex-periments. Mice were housed under specific pathogen-free conditions. Allexperiments were conducted according to the German animal protectionlaw (approved by the Regierungsprasidium Leipzig) and the safety guide-lines for S1 organisms.
Bacteria and infection model
The attenuated strain of S. Enteritidis (ade�/his�; SALMOVAC SE) (6,61–63) was provided by J. Selbitz (Impfstoffwerke Dessau-Tornau GmbH,Rosslau, Germany). The strain has been characterized by us before in amurine infection model (6) and in a murine vaccination model (63). In-fection of mice with high inocula leads to typhoid-like disease associatedwith mortality depending on the infective dose. WT and p19�/� mice werei.p. infected with the highest sublethal infection dose for WT mice, whichwas determined to be 2.5 � 106 CFU. For infection of p35�/�, p35/40�/�,p35/19�/�, and p35/IL-17�/� mice, the highest sublethal inoculum forp35�/� mice (2.5 � 103 CFU) was used.
Bacteria were grown in Luria-Bertani medium for 5 h to the log phase,and aliquots with defined CFU of S. Enteritidis were suspended in FCS/10% DMSO and stored at �70°C. For infection, aliquots were thawed,washed twice, and diluted in PBS. The indicated inocula were administeredin a volume of 500 �l. For ex vivo stimulation of splenocytes, heat-killed(hk) bacteria of the same batch were used (at 60°C for 60 min in a waterbath).
In vivo neutralization of IL-22
The p35�/� mice were i.p. injected with 50 �g of purified goat anti-mouse-IL-22 IgG 1 day before infection (0 days postinfection (dpi)). Mice of thecontrol group received 50 �g of normal goat IgG instead (both Abs fromR&D Systems). At 7 dpi, all mice were sacrificed and the indicated anal-yses were performed.
Survival and bacterial counts in organs
Infected mice were monitored daily for survival until 80 dpi. For experi-ments with WT and p19�/� mice, seven mice per group were sacrificed byCO2 asphyxiation 20 and 80 days after infection. For experiments withp35�/�, p35/40�/�, p35/19�/�, and p35/IL-17�/� mice, five to seven miceper group were sacrificed 20 days after infection. For experiments for invivo neutralization of IL-22 in p35�/� mice, four mice per group weresacrificed 7 days after infection. Blood was collected by cardiac puncture,and spleen and liver were removed under sterile conditions. Organs wereweighed, and pieces of them were homogenized and diluted 1/10 in PBS(w/v). Log10 serial dilutions were plated onto xylose-lysine-desoxycholateagar (Sifin). After 24 h of incubation at 37°C, the number of CFU wasdetermined and corrected for the whole organ weight.
Isolation and ex vivo restimulation of splenocytes
Single-cell suspensions of the removed spleens of individual mice werecleared from erythrocytes by treatment with Gey�s solution and washedand resuspended in ISCOVE�s medium (PAA Laboratories) supplementedwith 10% heat-inactivated FCS, 100 U/ml penicillin, and 100 �g/ml strep-tomycin. The cells of each group were pooled by taking equal cell numbersfrom individual mice, and adjusted to a concentration of 107 cells/ml. Avolume of 500 �l of this splenocyte suspension was dispensed into eachwell of a 24-well plate. After 12 h of incubation at 37°C under a humidifiedatmosphere containing 5% CO2, the cells were restimulated for 48 h byaddition of either 500 �l of Con A (final concentration 5 �g/ml) for poly-clonal T cell stimulation, 108 hk CFU S. Enteritidis per ml for Ag-specificstimulation, or medium for negative control. Cell-free supernatants wereharvested and stored at �20°C for cytokine and NO measurement.
ELISA for cytokine determination and colorimetric assay fordetection of NO
Mouse IFN-�, IL-17A, and IL-22 were quantified using Duo-Set ELISA(R&D Systems), according to recommended standard protocols. IL-12p40was measured using the mAb 5C3 (5 �g/ml) as capture Ab and biotinylatedpolyclonal goat anti-mouse IL-12p40-purified IgG (1:1000; both Ab pro-vided by M. Gately, Hoffmann-LaRoche, Nutley, NJ) as detection Ab,
7892 IL-23-DEPENDENT IL-22 IS PROTECTIVE AGAINST Salmonella INFECTION
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

followed by addition of streptavidin-peroxidase (Southern BiotechnologyAssociates). Tetramethylbenzidine substrate was used for the colorimetricdevelopment (Kirkegaard & Perry Laboratories). Mouse rIL-12 was usedas standard (provided by M. Gately, Hoffmann-LaRoche, Nutley, NJ). ODmeasurement of ELISA was performed with an ELISA reader (Spectramax340 PC; Molecular Devices). NO was measured in cell culture supernatantswith Griess reagent, as described (64). The cytokine and NO concentra-tions were estimated from the standard curves with SoftmaxPro (MolecularDevices).
Real-time RT-PCR
Murine tissue samples, snap frozen in Invisorb lysing solution (Invitek),were homogenized during thawing by means of Ultraturrax tissue homog-enizer (Jahnke and Kunkel) and then treated with 4 mg/ml proteinase K for1 h (BD Clontech). Isolation of total cellular RNA was done by use ofInvisorb RNA kit II (Invitek). mRNA was reverse transcribed and analyzedin triplicate assays by TaqMan PCR using the ABI Prism 7700 SequenceDetection System (Applied Biosystems), as described previously (44, 65).The appropriate assays, including double-fluorescent probes in combina-tion with assay for the murine housekeeping gene hypoxanthine phospho-ribosyltransferase (HPRT), were developed by ourselves (IL-22, IFN-�,IL-10, and IL-22BP) or purchased from Applied Biosystems. Expressionlevels were calculated relative to the data for HPRT obtained with theevery matching assay.
FACS analysis of splenocyte cell subsets
Single-cell suspensions of the spleens were prepared, as described above.A total of 105 cells per staining was pretreated with anti-CD16/CD32 Fcblock (clone 2.4G2; BD Biocsiences) and subsequently stained with theindicated Abs and their isotype controls coupled to PE, FITC, or allophy-cocyanin. Cells were washed twice with FACS buffer (3% FCS, 0.1%NaN3 in PBS) and fixed with PBS/4% formaldehyde (v/v). Fluorescencewas detected using flow cytometry (FACSCalibur flow cytometer; BDBiocsiences). Analyses were performed with the software CellQuestPro(BD Immunocytometry Systems).
Histology
Parts of livers from sacrificed mice were fixed in 4% buffered formalin,embedded in paraffin, processed routinely for light microscopy, andstained with H&E. Additional naphtol-AS-D-chloracetate esterase(NACE) staining was performed for the detection of PMN. PMN appearred in the stainings. For the analysis displayed in Fig. 7B (livers ofanti-IL-22- and goat IgG-treated p35�/� mice), sections were preparedin a serial fashion. For that, four consecutive levels of two distant partsof each liver (eight levels/organ in total) were used. Livers and spleenswere analyzed and assorted into groups in a single-blind trial by thepathologist. All studies were done with an Olympus BX51 microscope.
Statistical analyses
For comparison of two independent groups, the Mann-Whitney rank sumtest was used. For statistical analyses of differences between more than twoindependent groups, a Kruskal-Wallis statistic followed by Dunn’s posttestwas performed. Differences were considered to be significant at p � 0.05.
ResultsIn the presence of IL-12, IL-23 is dispensable for survival andbacterial control following infection of mice with S. Enteritidis
IL-12 has been shown to play a major role for protective T cell-mediated immunity against intracellular pathogens such as S. En-teritidis (6, 8). As shown earlier, IL-12-deficient C57BL/6 p35�/�
mice were highly susceptible to S. Enteritidis infection with a doseof �104 CFU leading to death within 4–5 wk (data not shown).Several studies demonstrated a major role of IL-23 for protectiveimmunity after mucosal infection (3). To investigate whetherIL-23 plays an essential role in immunity against intracellularpathogens such as S. Enteritidis during systemic infection, WT andp19�/� mice on a C57BL/6 background were infected with in-creasing doses of S. Enteritidis and monitored for at least 80 days.IL-23-deficient mice survived the largest sublethal inocula (up to2.5 � 106 CFU S. Enteritidis) exactly like WT mice (100% sur-vival; data not shown). This was associated with the same effi-ciency in bacterial control compared with WT mice at a low (2.5 �
103 CFU; data not shown) and the highest possible inoculum(2.5 � 106 CFU). In fact, there were no statistically significantdifferences in bacterial burden in spleen and liver at 20 and 80 dpi,and mice of both groups strongly reduced bacteria in these organsuntil 80 dpi (Fig. 1). Higher inocula of 2.5 � 107 CFU caused100% mortality within 1 wk even in WT mice (data not shown).These data demonstrate that IL-23 is not required for protectiveimmunity to S. Enteritidis when IL-12 is present.
Splenocytes from IL-23-deficient mice show normal IFN-�, butabrogated Ag-specific IL-17A and IL-22 production at 20 dpi
A T cell-mediated immune response with the differentiation ofIFN-�-producing Th1 cells is essential for control of salmonellosis(1, 8). IFN-� secretion causes NO production by activated mac-rophages, which leads to the killing of the pathogen (9). BecauseIL-23-deficient mice survived infection with S. Enteritidis exactlylike WT mice, we were interested whether the secretion of IFN-�and NO or other molecules secreted by T cells with a potential rolein protective immunity is influenced when IL-23 is missing. There-fore, we restimulated splenocytes from naive and from S. Enteri-tidis-infected WT and p19�/� mice ex vivo with either hk S. En-teritidis or Con A for 48 h at 20 and 80 dpi, and analyzed thesupernatants. We found that splenocytes from S. Enteritidis-in-fected WT mice responded strongly with IFN-� production uponAg-specific and polyclonal restimulation at 20 dpi as comparedwith cells of naive mice. Interestingly, splenocytes from p19�/�
mice showed almost identical IFN-� responses as WT splenocytesat 20 dpi (Fig. 2A). Splenocytes of infected p19�/� mice alsoproduced similar amounts of IL-12p40 and antibacterial NO asWT splenocytes (data not shown). These data indicate that IL-23is dispensable for regulation of IFN-� during the initial phase ofadaptive immunity against infection with S. Enteritidis. However,at a late time point during infection, when most of the bacteria arealready cleared from the organs (80 dpi, Fig. 1), IFN-� productionis �2-fold lower in splenocytes of p19�/� mice as compared withWT splenocytes. Ag-specific NO is not produced in both groups at80 dpi (data not shown).
IL-23 has been shown to activate Th17 cells (37, 66) for pro-duction of IL-17A (67). Therefore, we also determined the Ag-specific and polyclonal production of IL-17A (Fig. 2B). Spleno-cytes of infected WT mice produced significant amounts ofAg-specific and polyclonal IL-17A. We have shown earlier that S.Enteritidis induces a Th17 response in C57BL/6 mice with one-third of IL-17A-producing cells being CD4 positive (17). In con-trast to WT splenocytes, splenocytes from infected p19�/� micehad a complete lack of Ag-specific IL-17A production and a partial
FIGURE 1. No differences in bacterial clearance of WT and p19–/– micein spleen and liver. Mice were i.p. infected with 2.5 � 106 CFU S. Enter-itidis and sacrificed at day 20 or 80 after infection, and organ burden wasdetermined. Each symbol represents an individual mouse. Shown is onerepresentative of two independent experiments (seven mice/group). Thedifference found in spleen of p19�/� mice infected for 80 days did notreach statistical significance.
7893The Journal of Immunology
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

reduction of polyclonally induced IL-17A production. Recently,Th17 cells were found to coexpress IL-17A and IL-22 in mice(40). Therefore, we investigated IL-22 production by splenocytesof p19�/� mice upon ex vivo stimulation. We found induction ofAg-specific IL-22 production in response to infection in WT aswell as in p19�/� mice at 20 dpi. However, there is a �2-foldreduction of IL-22 production in splenocytes from p19�/� mice ascompared with WT splenocytes. At the same time, IL-22 produc-tion of p19�/� splenocytes upon polyclonal T cell stimulation washardly increased as compared with naive splenocytes. At 80 dpi,the Ag-specific IL-22 production of p19�/� splenocytes was backto baseline level, whereas WT splenocytes still responded to stim-ulation (Fig. 2C). Therefore, we find reduced, but not abrogatedAg-specific IL-22 production in the spleen when IL-23 is absent.These data indicate that besides Th1 cells, also Th17 cells developupon S. Enteritidis infection. The Th1-mediated production of cy-tokines is not affected by the absence of IL-23 at 20 dpi, but ap-pears to be reduced at 80 dpi. Interestingly, during the infectionwith S. Enteritidis, IL-17A production was strictly and IL-22 pro-duction only partially IL-23 dependent.
We also measured IL-22 in the sera and found significantly re-duced levels close to the detection limit (0.012 ng/ml) in the ab-sence of IL-23 at 20 dpi (Fig. 2D). At 80 dpi, the IL-22 levels ofWT mice decreased compared with day 20 and reached the detec-tion limit. Concentrations of IL-17A and IFN-� were below de-tection limit in the sera (data not shown). Together, these in vivodata support the evidence of reduced Th17 responses in the ab-sence of IL-23 following infection with S. Enteritidis.
In the absence of IL-12, IL-23, but not (p40)2, providesprotection against S. Enteritidis infection that is independentof IL-17A
Absence of IL-12-dependent Th1 responses has been found in pa-tients with an inherited defect in the IL-12/IL-12R/IFN-� axis andwas associated with recurrent nontuberculous mycobacterial orSalmonella infections (7). Thus, it was of interest to us to study thefunction of IL-23 in the absence of IL-12-dependent Th1 re-sponses. Therefore, in contrast to the studies described above andin Figs. 1 and 2, the following studies were undertaken to analyzethe role of IL-23 and/or (p40)2 in the absence of IL-12, i.e., in aTh1-compromised situation. In addition to IL-12, the homodimer(p40)2 can be protective in immunity to mycobacterial infection
(12, 13). We reported previously that BALB/c and 129Sv/Ev micelacking all p40-containing members of the IL-12 cytokine familyshow a higher susceptibility to infection with S. Enteritidis thanmice only deficient in IL-12 (6). In accordance with these previousresults, more recent experiments corroborate these findings inC57BL/6 mice. Mice lacking IL-12 (p35�/� mice) completely sur-vived inocula of 2.5 � 102 CFU (data not shown), and 94% of allp35�/� mice (33 of 35 mice) survived infection with 2.5 � 103
CFU until the end of the experiment (80 dpi), whereas all p35/40�/� mice (lacking all p40-containing members of the IL-12 cy-tokine family) died after infection with either of those low inocula(Fig. 3A). After infection with a 10-fold higher dose of 2.5 � 104
CFU, p35�/� mice died significantly later than p35/40�/� mice(data not shown). With even higher inocula, the difference in sur-vival of mice lacking only IL-12 or all p40-dependent cytokinesdisappeared (data not shown). These data indicate that in the ab-sence of IL-12, a p40-dependent cytokine is essential for survivalof C57BL/6 mice after low-dose infection with S. Enteritidis. Theyleave open though whether IL-23 and/or (p40)2 account for thestriking phenotypic difference, especially at an infective dose of2.5 � 103 CFU. To analyze a potential protective role of IL-23and/or (p40)2 in the absence of IL-12, p35�/� mice, p35/40�/�
mice, and p35/19�/� mice (producing only free p40, but neitherIL-12 nor IL-23), all on a C57BL/6 background, were infectedwith 2.5 � 103 CFU S. Enteritidis. This inoculum is sublethal forp35�/� mice and 1000-fold lower than the inocula used for WTand p19�/� mice. Strikingly, both p35/19�/� mice and p35/40�/�
mice died within 4–6 wk after infection with S. Enteritidis (Fig.3A). This demonstrates that IL-23 is required for protective im-munity to S. Enteritidis when IL-12 is absent, as has been shownearlier for mycobacterial and Toxoplasma infection (68, 69).Moreover, these data unambiguously exclude a protective role ofmonomeric and homodimeric IL-12p40 during Salmonellainfection.
Because p19�/� mice showed significantly reduced IL-17Aproduction (see Fig. 2B), we suspected IL-17A to be responsi-ble for protection of low-dose infected mice in the absence ofIL-12. Therefore, we generated p35/IL-17A�/� mice and in-cluded them in the experiment. Surprisingly, these mice com-pletely survived the low-dose infection (Fig. 3A). This indicates
FIGURE 2. Normal Ag-specificIFN-� production (A), but abrogated IL-17A (B) and IL-22 (C) production andreduced IL-22 serum levels (D) in the ab-sence of IL-23. Mice were infected i.p.with 2.5 � 106 CFU S. Enteritidis andsacrificed at day 20 or 80 after infection.Splenocytes of each group were pooled,plated (5 � 106 cells/well), and stimu-lated with either hk S. Enteritidis or ConA. Culture supernatants were tested forIFN-� (A), IL-17A (B), and IL-22 (C) byELISA. Medium levels of supernatantsof naive mice were below detection level.Bars represent the mean � SEM (n � 3).D, IL-22 concentrations in the serum ofcardiac blood were determined. Shown isone representative of two independentexperiments (five to seven mice/group).Detection limits lie at 0.016 ng/ml for theIFN-� and IL-17A ELISAs, and at 0.012ng/ml for the IL-22 ELISA.
7894 IL-23-DEPENDENT IL-22 IS PROTECTIVE AGAINST Salmonella INFECTION
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
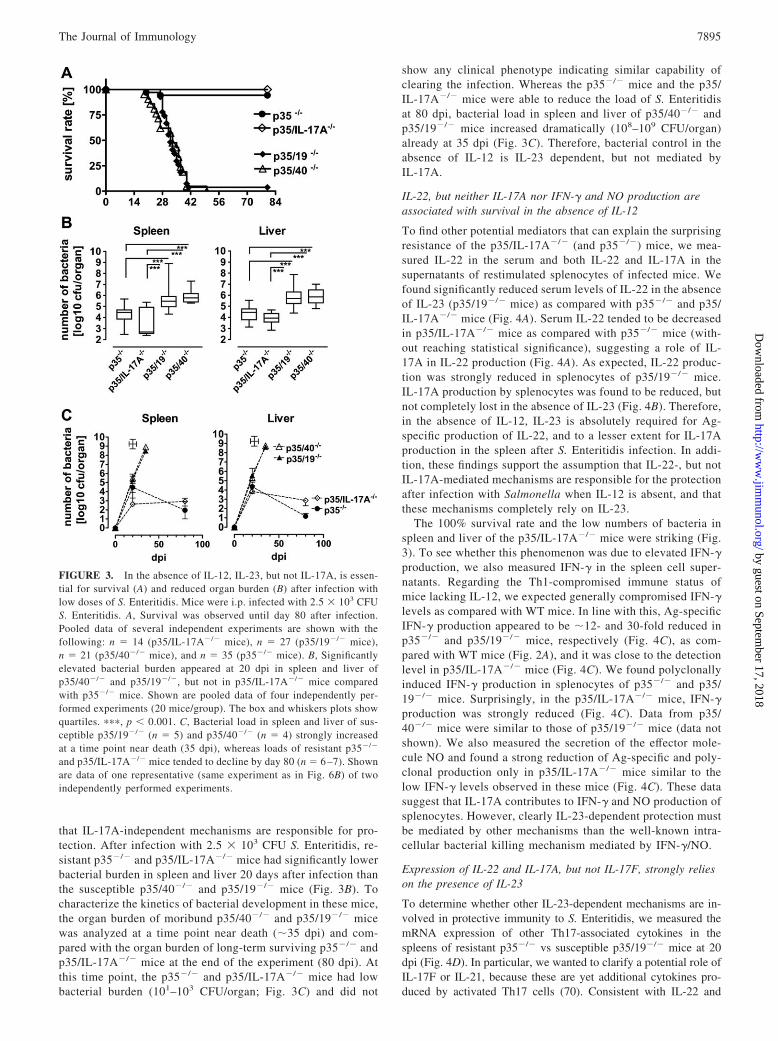
that IL-17A-independent mechanisms are responsible for pro-tection. After infection with 2.5 � 103 CFU S. Enteritidis, re-sistant p35�/� and p35/IL-17A�/� mice had significantly lowerbacterial burden in spleen and liver 20 days after infection thanthe susceptible p35/40�/� and p35/19�/� mice (Fig. 3B). Tocharacterize the kinetics of bacterial development in these mice,the organ burden of moribund p35/40�/� and p35/19�/� micewas analyzed at a time point near death (�35 dpi) and com-pared with the organ burden of long-term surviving p35�/� andp35/IL-17A�/� mice at the end of the experiment (80 dpi). Atthis time point, the p35�/� and p35/IL-17A�/� mice had lowbacterial burden (101–103 CFU/organ; Fig. 3C) and did not
show any clinical phenotype indicating similar capability ofclearing the infection. Whereas the p35�/� mice and the p35/IL-17A�/� mice were able to reduce the load of S. Enteritidisat 80 dpi, bacterial load in spleen and liver of p35/40�/� andp35/19�/� mice increased dramatically (108–109 CFU/organ)already at 35 dpi (Fig. 3C). Therefore, bacterial control in theabsence of IL-12 is IL-23 dependent, but not mediated byIL-17A.
IL-22, but neither IL-17A nor IFN-� and NO production areassociated with survival in the absence of IL-12
To find other potential mediators that can explain the surprisingresistance of the p35/IL-17A�/� (and p35�/�) mice, we mea-sured IL-22 in the serum and both IL-22 and IL-17A in thesupernatants of restimulated splenocytes of infected mice. Wefound significantly reduced serum levels of IL-22 in the absenceof IL-23 (p35/19�/� mice) as compared with p35�/� and p35/IL-17A�/� mice (Fig. 4A). Serum IL-22 tended to be decreasedin p35/IL-17A�/� mice as compared with p35�/� mice (with-out reaching statistical significance), suggesting a role of IL-17A in IL-22 production (Fig. 4A). As expected, IL-22 produc-tion was strongly reduced in splenocytes of p35/19�/� mice.IL-17A production by splenocytes was found to be reduced, butnot completely lost in the absence of IL-23 (Fig. 4B). Therefore,in the absence of IL-12, IL-23 is absolutely required for Ag-specific production of IL-22, and to a lesser extent for IL-17Aproduction in the spleen after S. Enteritidis infection. In addi-tion, these findings support the assumption that IL-22-, but notIL-17A-mediated mechanisms are responsible for the protectionafter infection with Salmonella when IL-12 is absent, and thatthese mechanisms completely rely on IL-23.
The 100% survival rate and the low numbers of bacteria inspleen and liver of the p35/IL-17A�/� mice were striking (Fig.3). To see whether this phenomenon was due to elevated IFN-�production, we also measured IFN-� in the spleen cell super-natants. Regarding the Th1-compromised immune status ofmice lacking IL-12, we expected generally compromised IFN-�levels as compared with WT mice. In line with this, Ag-specificIFN-� production appeared to be �12- and 30-fold reduced inp35�/� and p35/19�/� mice, respectively (Fig. 4C), as com-pared with WT mice (Fig. 2A), and it was close to the detectionlevel in p35/IL-17A�/� mice (Fig. 4C). We found polyclonallyinduced IFN-� production in splenocytes of p35�/� and p35/19�/� mice. Surprisingly, in the p35/IL-17A�/� mice, IFN-�production was strongly reduced (Fig. 4C). Data from p35/40�/� mice were similar to those of p35/19�/� mice (data notshown). We also measured the secretion of the effector mole-cule NO and found a strong reduction of Ag-specific and poly-clonal production only in p35/IL-17A�/� mice similar to thelow IFN-� levels observed in these mice (Fig. 4C). These datasuggest that IL-17A contributes to IFN-� and NO production ofsplenocytes. However, clearly IL-23-dependent protection mustbe mediated by other mechanisms than the well-known intra-cellular bacterial killing mechanism mediated by IFN-�/NO.
Expression of IL-22 and IL-17A, but not IL-17F, strongly relieson the presence of IL-23
To determine whether other IL-23-dependent mechanisms are in-volved in protective immunity to S. Enteritidis, we measured themRNA expression of other Th17-associated cytokines in thespleens of resistant p35�/� vs susceptible p35/19�/� mice at 20dpi (Fig. 4D). In particular, we wanted to clarify a potential role ofIL-17F or IL-21, because these are yet additional cytokines pro-duced by activated Th17 cells (70). Consistent with IL-22 and
FIGURE 3. In the absence of IL-12, IL-23, but not IL-17A, is essen-tial for survival (A) and reduced organ burden (B) after infection withlow doses of S. Enteritidis. Mice were i.p. infected with 2.5 � 103 CFUS. Enteritidis. A, Survival was observed until day 80 after infection.Pooled data of several independent experiments are shown with thefollowing: n � 14 (p35/IL-17A�/� mice), n � 27 (p35/19�/� mice),n � 21 (p35/40�/� mice), and n � 35 (p35�/� mice). B, Significantlyelevated bacterial burden appeared at 20 dpi in spleen and liver ofp35/40�/� and p35/19�/�, but not in p35/IL-17A�/� mice comparedwith p35�/� mice. Shown are pooled data of four independently per-formed experiments (20 mice/group). The box and whiskers plots showquartiles. ���, p � 0.001. C, Bacterial load in spleen and liver of sus-ceptible p35/19�/� (n � 5) and p35/40�/� (n � 4) strongly increasedat a time point near death (35 dpi), whereas loads of resistant p35�/�
and p35/IL-17A�/� mice tended to decline by day 80 (n � 6 –7). Shownare data of one representative (same experiment as in Fig. 6B) of twoindependently performed experiments.
7895The Journal of Immunology
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

IL-17A protein production (see Fig. 4B), we found significantlyreduced mRNA expression of IL-22 ( p � 0.002) and IL-17A ( p �0.03) when IL-23 was absent. In contrast, IL-17F as well as IL-21expression was independent of IL-23 and did not differ betweenprotected p35�/� and susceptible p35/19�/� mice. Therefore, IL-17F and IL-21 do not appear to be potential mediators of IL-23-dependent protection. In accordance with IFN-� protein produc-tion (Fig. 4C), there was also no difference in the expression ofIFN-� mRNA between infected p35�/� and p35/19�/� mice at 20dpi. Furthermore, no difference was found for mRNA productionof IL-27, IL-1ß, IL-6, TNF-�, or IL-10. These data strengthenthe predominant association of IL-23-dependent IL-22 withprotection.
Reduced CD11b�Gr1� cells in the absence of IL-17A, but notin the absence of IL-23
Neutrophilic granulocytes have been shown to have a protectivefunction during salmonellosis (71). IL-17A can contribute torecruitment of PMN to infected tissues (72). We analyzed thenumber of PMN in the spleens of p35�/�, p35/IL-17A�/�, and
p35/19�/� mice by FACS and found significantly reduced num-bers and percentages in the spleens of p35/IL-17A�/� mice. Incontrast, the percentages (Fig. 5A) and numbers (Fig. 5B) ofPMN in p35/19�/� mice were comparable to those of p35�/�
mice. Therefore, this indicates that IL-23-dependent protectionis independent of neutrophils. Moreover, PMN recruitment insystemic salmonellosis is not due to regulation of granulopoi-esis by the IL-23/IL-17 axis, as it has been shown for the lungsof mice infected with Mycoplasma pneumoniae (29). The anal-ysis of PMN recruitment to the spleen was confirmed by neu-trophil-specific staining (NACE) of liver sections derived fromresistant p35�/� and p35/IL-17A�/� vs susceptible p35/19�/�
and p35/40�/� mice (Fig. 5C). In livers of infected p35�/�,p35/19�/�, and p35/40�/� mice were similar frequencies ofPMN, but fewer PMN were detectable in livers of infected p35/IL-17A�/� mice, indicating again IL-17A-, but not IL-23-de-pendent neutrophil recruitment in the absence of IL-12. Thesedata also point to a role of IL-23-independent IL-17A in PMNrecruitment (Figs. 4B and 5).
FIGURE 4. Reduced expression of IL-22 and IL-17A, but not IL-17F, in the absence of IL-23. Mice were infected i.p. with 2.5 � 103 CFU S. Enteritidis andsacrificed at day 20 after infection. A, Significantly reduced serum concentrations of IL-22 in p35/19�/� mice. Shown are pooled data of two independentexperiments. Each symbol represents an individual mouse. �, p � 0.05; ���, p � 0.001. B and C, Splenocytes of each group were pooled, plated (5 � 106
cells/well), and stimulated with either hk S. Enteritidis or Con A. Serum and culture supernatants were tested for IL-22, IL-17A, and IFN-� by ELISA. NO wasdetermined using the Griess reagent. Shown is one representative of two independent experiments (five mice/group). D, mRNA expression of indicated moleculesin the spleens of infected p35�/� and p35/19�/� mice. Shown is the x-fold up-regulation compared with the HPRT expression. Shown are data from oneexperiment. Each symbol represents an individual mouse (n � 5–6).
7896 IL-23-DEPENDENT IL-22 IS PROTECTIVE AGAINST Salmonella INFECTION
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

In the absence of IL-23/IL-22, liver necrosis, fibrin thrombi,and aberrant granulomas develop
Histopathological analysis of the liver of infected resistant p35�/�
and p35/IL-17A�/� mice and susceptible p35/19�/� and p35/40�/� mice was undertaken to characterize the inflammatory re-sponse after infection with S. Enteritidis. Resistant p35�/� andp35/IL-17A�/� mice showed granuloma formation, whereas sus-ceptible p35/19�/� and p35/40�/� mice developed a more diffuseinflammation (Fig. 6A). Susceptible p35/19�/� and p35/40�/�
mice, but not resistant p35�/� and p35/IL-17A�/� mice, alsoshowed the occurrence of fibrin thrombi (Fig. 6A, bottom row) andnecrotic tissue areas (Fig. 6A, middle row) at 20 dpi. Interestingly,necrotic tissue damage of p35/19�/� and p35/40�/� mice wasrestricted to liver and did not occur in spleen, which is another siteof Salmonella infection (data not shown). This is consistent withthe expression of IL-22R1 in liver, but not spleen (45). Therefore,the absence of IL-23-dependent IL-22 is associated with the de-velopment of liver necrosis. Development of liver necrosis became
very striking at a time point near death (35 dpi). Broad areas withconfluent single cell necrosis of the liver cells had developed inp35/19�/� and p35/40�/� mice (Fig. 6B). In contrast, we foundonly few well-defined granulomas and no necrotic tissue in thelivers of resistant p35�/� and p35/IL-17A�/� mice at the end ofthe experiment at 80 dpi (Fig. 6B), indicating that IL-17A is notinvolved in protection from liver damage.
To examine the role of IL-22 in the development of liver ne-crosis during Salmonella infection more directly, we treated
FIGURE 5. Significantly reduced neutrophils in spleen and liver in theabsence of IL-17A, but not IL-23. Mice were i.p. infected with 2.5 � 103
CFU S. Enteritidis, sacrificed at day 20 after infection, and prepared forFACS staining. Percentages (A) and absolute cell numbers (B) of PMN(Gr1�CD11b�) in the spleen. Shown are pooled data of two independentlyperformed experiments (five mice/group). Each symbol represents an in-dividual mouse. �, p � 0.05; ��, p � 0.01; ���, p � 0.001. C, Represen-tative NACE-stained sections of the liver 20 days after S. Enteritidis in-fection show that livers from p35/IL-17A�/� mice contain fewer NACE�
neutrophils than livers from p35�/�, p35/19�/�, and p35/40�/� mice uponinfection with S. Enteritidis.
FIGURE 6. Hepatocyte necrosis and fibrin thrombi in p35/19–/– andp35/40–/– mice, but not in p35�/� or p35/IL-17A�/� mice. Mice were i.p.infected with 2.5 � 103 CFU S. Enteritidis and sacrificed at different timepoints, and livers were removed and prepared for histological H&E stain-ing. A, At 20 dpi in livers of susceptible p35/19�/� and p35/40�/� mice,necrotic tissue areas (middle row) and fibrin thrombi (lower row) occurredand granuloma formation was more diffuse compared with resistant p35�/�
and p35/IL-17A�/� mice, which did not show necrosis (�60 objective). B,At 80 dpi, resistant mice showed only few, but well-confined granulomaswithout necrosis. Susceptible p35/19�/� and p35/40�/� mice were sacri-ficed at a time point near death (35 dpi; same mice as shown in Fig. 3C).They had developed areas with confluent single cell necrosis of the livercells with a more diffuse infiltrate of inflammatory cells. Shown are rep-resentative stainings of two independently performed experiments (fivemice/group) (�60 objective).
7897The Journal of Immunology
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

p35�/� mice with neutralizing anti-IL-22 or normal goat IgG 1day before S. Enteritidis infection. At 7 dpi, mice were sacrificed,bacterial organ burden was determined, and serial sections of thelivers were prepared and analyzed in a single-blind fashion. Bac-terial organ burden at 7 dpi was quite low (Fig. 7A) and did not yetsignificantly differ between mice treated with anti-IL-22 or controlIgG. However, already at this early time point, all mice were cor-rectly grouped by histopathological examination in a single-blindapproach. Anti-IL-22-treated mice developed single cell necrosisin the liver. In contrast, smaller and fewer inflammatory foci with-out necrosis were found in the livers of mice treated with normalgoat IgG (Fig. 7B). These data point to a role of IL-22 in theprotection of hepatocytes in the Salmonella-infected liver.
DiscussionIn the present study, we show that the absence of IL-23 does notcompromise protective immunity to systemic infection with even ahigh sublethal dose of S. Enteritidis inducing a typhoid-like dis-ease. IL-23-deficient mice mounted normal IFN-� responses, and,therefore, it can be concluded that the IL-12/IFN-� pathway is stillintact in the absence of IL-23. IL-23-dependent Th17-associatedresponses (e.g., production of IL-17A and IL-22) are dispensablein the presence of an effective Th1 response. In contrast, in theabsence of IL-12-dependent Th1 responses, protection against low,yet sublethal doses of S. Enteritidis is associated with IL-23-de-pendent IL-22, but not IL-17A production, enabling protectionfrom liver damage. Neither residual production of IL-12-indepen-dent IFN-�/ (see Fig. 4C) nor IL-17A-dependent neutrophilrecruitment (Fig. 5) is associated with protection.
In other models of intracellular infection (e.g., Toxoplasma gon-dii, Mycobacterium tuberculosis, Mycobacterium bovis bacillus
Calmette-Guerin), IL-23-dependent protection was also found inthe absence of IL-12 (59, 68, 69). Interestingly, IL-23-dependentgeneration of IL-17 responses was found, but did not contributephenotypically to the course of the infection. IL-17A was experi-mentally excluded to have a functional role in immunity to infec-tion with M. bovis bacillus Calmette-Guerin by neutralizing IL-17A in WT, p35�/� mice, and p40�/� mice (59). Previously, inthe murine S. Enteritidis and M. tuberculosis infection models,IL-12p40/IL-23-dependent IFN-� was shown in the absence ofIL-12 (6, 68). With our present study, IL-23-dependent IL-22comes into the focus for systemic Salmonella infection both forliver cell survival and for pathogen defense. However, in murinetuberculosis and toxoplasmosis, IL-22 remains to be studied. In-terestingly, very recently in human tuberculosis patients, IL-17-and IL-22-producing CD4� cells and bronchoalveolar lavagecontaining IL-22, but not IL-17, were found (73). In contrast for ex-tracellularly multiplying pathogens (e.g., Klebsiella, Citrobacter), in-duction of Th17 responses was shown (74, 75). Extracellular multi-plication of a pathogen may be a prerequisite for being efficientlyrejected by the elements of Th17 responses. At the moment it seems,however, too early to draw conclusions as to a link between extra-cellularly and intracellularly replicating pathogens and the role ofTh17 responses for protection against extracellular stages.
Recently, it was shown that Th17 cells producing IL-17A arerequired for protective immunity after mucosal infection of ma-caques and streptomycin-pretreated mice with S. Typhimurium(76). Evidence was obtained that in the absence of IL-17RA ex-pression, mucosal barrier function of the gut is compromised andbacterial dissemination increased early on after oral infection. Incontrast, IL-17A in systemic Salmonella infection is dispensable inthe absence of IL-12. However, IL-22 becomes important in thissituation because it is required for protection against hepatocytenecrosis during salmonellosis.
Recently, it was demonstrated that IL-22 did not influence thefunction of immune cells, but can induce antimicrobial moleculessuch as �-defensins and S100 proteins in epithelial cells of bodybarriers and enhance the innate immunity of tissues cells (45). Anessential role of IL-23-dependent IL-22 during innate immunityhas been found for protection against infection of mice withCitrobacter rodentium, a model of human infection with attachingand effacing (A/C) pathogens (74). In this study, IL-22-inducedproduction of Reg family members acting as antimicrobial proteinsappeared to contribute to protection. Thus, various mechanisms(e.g., stability of mucosal barrier function, induction of antimicro-bial proteins) induced by IL-22 may be involved in innate protec-tion against intestinal Gram-negative bacteria. This notion isfurther strengthened and extended by data from a pulmonaryGram-negative infection with Klebsiella pneumoniae (77). In thisstudy, it was shown that although both IL-22 and IL-17A are in-volved in mucosal host defense, the role of IL-22 is more impor-tant than IL-17A. In particular, IL-22 enhanced the clonogenicityof human bronchial epithelial cells and recovery of resistance fol-lowing experimental creation of a wound. In addition, IL-22 in-duced the expression of chemokines and other host defense genessuch as lipocalin-2. Together, this points to a major function ofIL-22 in innate immunity by mucosal epithelial cells (78). It re-mains to be shown which of these mechanisms are initiated byIL-22 in our study. Preliminary data suggest that production ofacute-phase proteins, such as LPS-binding protein and serum amy-loid A, is not the essential protective mechanism initiated by IL-22after i.p. infection of mice with S. Enteritidis (data not shown).
Salmonella is able to invade both the liver and spleen followingsystemic or mucosal infection (as reviewed in Ref. 8). Already 7
FIGURE 7. Hepatocyte necrosis in anti-IL-22-treated p35–/– mice. Thep35�/� mice were treated once with anti-IL-22 Ab or goat IgG Ab asisotype control before infection (four mice/group). A, At 7 dpi, numbers ofbacteria of anti-IL-22-treated as well as goat IgG-treated mice were lowand did not significantly differ yet. B, Anti-IL-22-treated mice showedbigger and more numerous inflammatory infiltrates with hepatocyte necro-sis as compared with livers from goat IgG-treated mice, which displayed nonecrotic cells in the liver. Shown are representative sections (�40objective).
7898 IL-23-DEPENDENT IL-22 IS PROTECTIVE AGAINST Salmonella INFECTION
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

days after i.p. infection, we found necrosis of liver cells after neu-tralizing IL-22. This is a striking result in light of the low bacterialloads at this early time point. Necrosis of the liver was accompa-nied at 20 dpi by the occurrence of fibrin thrombi in the absence ofIL-22 (p35/19�/� and p35/40�/� mice; Fig. 6). All of this was notthe case in spleen, indicating an organ-specific effect of IL-22.Liver necrosis may be causally involved in mortality of p35/19�/�
and p35/40�/� mice unable to produce IL-22 after low-dose in-fection with S. Enteritidis. Survival of Salmonella-infected p35/IL-17A�/� mice indicates that IL-17A is not involved in protec-tion of hepatocytes. This is consistent with a recent study of ConA-induced hepatitis in which also IL-22, but not IL-17, was shownto protect hepatocytes from the effects of the immune-mediatedinflammation (55). Interestingly, C. rodentium infection was alsofound to be associated with hepatic embolic microabscesses in theabsence of IL-22 (75). IL-22 was shown to protect hepatocytesfrom apoptosis involving STAT3 activation (54). The IL-22R canactivate anti-inflammatory STAT3 activation indistinguishablefrom IL-10 (79).
Besides IL-23-dependent IL-17A and IL-22, IL-17F and IL-21could have also been likely candidates to mediate the observedprotection of p35/IL-17A�/� mice because they can also be ex-pressed by activated Th17 cells (32, 35, 70). No differences ofmRNA expression for IL-17F and IL-21 could, however, be foundbetween resistant (p35�/�) and susceptible (p35/19�/�) mice,pointing once more to a unique role of IL-22 for protection.
We find low levels of IL-23-independent IL-17A in the absenceof IL-12, but not in the presence of IL-12 (see Fig. 4 vs Fig. 2B).In contrast, IL-22 production was only partially IL-23 dependent inthe presence of IL-12 and completely IL-23 dependent in the ab-sence of IL-12 (see Fig. 2C vs Fig. 4B). It is known that IL-17Acan be produced independently of IL-23 (80). Therefore, it was tobe expected that neutrophil recruitment can occur independent ofIL-23 (see Fig. 5). Presently, it is unclear what the exact molecularbasis is for the regulatory effect of IL-12 in IL-23-dependent andIL-23-independent IL-17 and IL-22 production. From our data itappears that IL-12 directly or indirectly suppresses IL-23-indepen-dent IL-17A production. The opposite holds true for IL-22 pro-duction. To fully answer this question, further studies have tobe done.
Our findings from this experimental murine systemic Salmo-nella infection model may be relevant to the immune mechanismsthat are active during human typhoid disease. We propose that theIL-12/IFN-� axis is highly effective in control of high Salmonellainocula provoking otherwise liver damage. With a functional IL-12/IFN-� pathway, the IL-23/IL-17/IL-22 axis is even dispensable.If, however, the IL-12/IFN-� pathway is defective (as is the casefor p35�/� mice), only low infective doses are tolerated, and thisrequires the IL-23/IL-22 cascade. In contrast to IL-22 production,IL-17A is functionally dispensable for protective immunity to S.Enteritidis infection in a compromised Th1 status, although IL-17A is also generated by the IL-23/IL-22 cascade and is associatedwith more effective PMN recruitment.
It is intriguing to note that in a Th1-compromised situation asseen in resistant p35/IL-17A�/� mice, the role of the IFN-�/NOpathway appears to become dispensable (see Fig. 4C). The under-lying mechanism mediating the reduction of IFN-�/NO levels dur-ing IL-17A deficiency is presently unclear. However, this phenom-enon is interesting in light of the observation that Salmonellainfections are more frequent in human patients with an IL-12/IL-23 defect than with an IFN-� defect (81). IFN-�-independentactivities of IL-23 such as IL-22 induction could be protective im-mune mechanisms operative in these patients, as shown in this studyin the murine Salmonella infection model of IL-23 deficiency.
AcknowledgmentsWe thank Petra Meier for preparing the histological sections; Sabine Siege-mund and Juliane Richter for help with the dissection of animals and sub-sequent analysis; Eva Marquardt, Norman Kirchoff, and Rowina Voigt-lander for help with the animal husbandry; and Tanja Sonntag forgenotyping.
DisclosuresThe authors have no financial conflict of interest.
References1. Jouanguy, E., R. Doffinger, S. Dupuis, A. Pallier, F. Altare, and J. L. Casanova.
1999. IL-12 and IFN-� in host defense against mycobacteria and salmonella inmice and men. Curr. Opin. Immunol. 11: 346–351.
2. Mastroeni, P., J. A. Harrison, J. H. Robinson, S. Clare, S. Khan, D. J. Maskell,G. Dougan, and C. E. Hormaeche. 1998. Interleukin-12 is required for control ofthe growth of attenuated aromatic-compound-dependent salmonellae in BALB/cmice: role of � interferon and macrophage activation. Infect. Immun. 66:4767–4776.
3. Aujla, S. J., P. J. Dubin, and J. K. Kolls. 2007. Th17 cells and mucosal hostdefense. Semin. Immunol. 19: 377–382.
4. Hornick, R. B., S. E. Greisman, T. E. Woodward, H. L. DuPont, A. T. Dawkins,and M. J. Snyder. 1970. Typhoid fever: pathogenesis and immunologic control.2. N. Engl. J. Med. 283: 739–746.
5. Rabsch, W., H. Tschape, and A. J. Baumler. 2001. Non-typhoidal salmonellosis:emerging problems. Microbes Infect. 3: 237–247.
6. Lehmann, J., S. Bellmann, C. Werner, R. Schroder, N. Schutze, and G. Alber.2001. IL-12p40-dependent agonistic effects on the development of protectiveinnate and adaptive immunity against Salmonella enteritidis. J. Immunol. 167:5304–5315.
7. Filipe-Santos, O., J. Bustamante, A. Chapgier, G. Vogt, L. de Beaucoudrey,J. Feinberg, E. Jouanguy, S. Boisson-Dupuis, C. Fieschi, C. Picard, andJ. L. Casanova. 2006. Inborn errors of IL-12/23- and IFN-�-mediated immunity:molecular, cellular, and clinical features. Semin. Immunol. 18: 347–361.
8. Mastroeni, P. 2002. Immunity to systemic Salmonella infections. Curr. Mol.Med. 2: 393–406.
9. Vazquez-Torres, A., J. Jones-Carson, P. Mastroeni, H. Ischiropoulos, andF. C. Fang. 2000. Antimicrobial actions of the NADPH phagocyte oxidase andinducible nitric oxide synthase in experimental salmonellosis. I. Effects on mi-crobial killing by activated peritoneal macrophages in vitro. J. Exp. Med. 192:227–236.
10. Oppmann, B., R. Lesley, B. Blom, J. C. Timans, Y. Xu, B. Hunte, F. Vega,N. Yu, J. Wang, K. Singh, et al. 2000. Novel p19 protein engages IL-12p40 toform a cytokine, IL-23, with biological activities similar as well as distinct fromIL-12. Immunity 13: 715–725.
11. Mattner, F., S. Fischer, S. Guckes, S. Jin, H. Kaulen, E. Schmitt, E. Rude, andT. Germann. 1993. The interleukin-12 subunit p40 specifically inhibits effects ofthe interleukin-12 heterodimer. Eur. J. Immunol. 23: 2202–2208.
12. Holscher, C., R. A. Atkinson, B. Arendse, N. Brown, E. Myburgh, G. Alber, andF. Brombacher. 2001. A protective and agonistic function of IL-12p40 in myco-bacterial infection. J. Immunol. 167: 6957–6966.
13. Khader, S. A., S. Partida-Sanchez, G. Bell, D. M. Jelley-Gibbs, S. Swain,J. E. Pearl, N. Ghilardi, F. J. Desauvage, F. E. Lund, and A. M. Cooper. 2006.Interleukin 12p40 is required for dendritic cell migration and T cell priming afterMycobacterium tuberculosis infection. J. Exp. Med. 203: 1805–1815.
14. Cua, D. J., and R. A. Kastelein. 2006. TGF-�, a “double agent” in the immunepathology war. Nat. Immunol. 7: 557–559.
15. Dong, C. 2006. Diversification of T-helper-cell lineages: finding the family rootof IL-17-producing cells. Nat. Rev. Immunol. 6: 329–333.
16. Weaver, C. T., L. E. Harrington, P. R. Mangan, M. Gavrieli, and K. M. Murphy.2006. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity24: 677–688.
17. Schulz, S. M., G. Kohler, C. Holscher, Y. Iwakura, and G. Alber. 2008. IL-17Ais produced by Th17, �� T cells and other CD4� lymphocytes during infectionwith Salmonella enterica serovar Enteritidis and has a mild effect in bacterialclearance. Int. Immunol. 20: 1129–1138.
18. Fossiez, F., O. Djossou, P. Chomarat, L. Flores-Romo, S. Ait-Yahia, C. Maat,J. J. Pin, P. Garrone, E. Garcia, S. Saeland, et al. 1996. T cell interleukin-17induces stromal cells to produce proinflammatory and hematopoietic cytokines.J. Exp. Med. 183: 2593–2603.
19. Jovanovic, D. V., J. A. Di Battista, J. Martel-Pelletier, F. C. Jolicoeur, Y. He,M. Zhang, F. Mineau, and J. P. Pelletier. 1998. IL-17 stimulates the productionand expression of proinflammatory cytokines, IL-� and TNF-�, by human mac-rophages. J. Immunol. 160: 3513–3521.
20. Yao, Z., S. L. Painter, W. C. Fanslow, D. Ulrich, B. M. Macduff, M. K. Spriggs,and R. J. Armitage. 1995. Human IL-17: a novel cytokine derived from T cells.J. Immunol. 155: 5483–5486.
21. Laan, M., Z. H. Cui, H. Hoshino, J. Lotvall, M. Sjostrand, D. C. Gruenert,B. E. Skoogh, and A. Linden. 1999. Neutrophil recruitment by human IL-17 viaC-X-C chemokine release in the airways. J. Immunol. 162: 2347–2352.
22. Ye, P., F. H. Rodriguez, S. Kanaly, K. L. Stocking, J. Schurr,P. Schwarzenberger, P. Oliver, W. Huang, P. Zhang, J. E. Shellito, et al. 2001.Requirement of interleukin 17 receptor signaling for lung CXC chemokine and
7899The Journal of Immunology
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

granulocyte colony-stimulating factor expression, neutrophil recruitment, andhost defense. J. Exp. Med. 194: 519–527.
23. Yao, Z., W. C. Fanslow, M. F. Seldin, A. M. Rousseau, S. L. Painter,M. R. Comeau, J. I. Cohen, and M. K. Spriggs. 1995. Herpesvirus Saimiri en-codes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity3: 811–821.
24. Aggarwal, S., and A. L. Gurney. 2002. IL-17: prototype member of an emergingcytokine family. J. Leukocyte Biol. 71: 1–8.
25. Alber, G., and T. Kamradt. 2007. Regulation of protective and pathogenic Th17responses. Curr. Immunol. Rev. 3: 3–16.
26. Ouyang, W., J. K. Kolls, and Y. Zheng. 2008. The biological functions of Thelper 17 cell effector cytokines in inflammation. Immunity 28: 454–467.
27. Huang, W., L. Na, P. L. Fidel, and P. Schwarzenberger. 2004. Requirement ofinterleukin-17A for systemic anti-Candida albicans host defense in mice. J. In-fect. Dis. 190: 624–631.
28. Shibata, K., H. Yamada, H. Hara, K. Kishihara, and Y. Yoshikai. 2007. ResidentV�1� �� T cells control early infiltration of neutrophils after Escherichia coliinfection via IL-17 production. J. Immunol. 178: 4466–4472.
29. Wu, Q., R. J. Martin, J. G. Rino, R. Breed, R. M. Torres, and H. W. Chu. 2007.IL-23-dependent IL-17 production is essential in neutrophil recruitment and ac-tivity in mouse lung defense against respiratory Mycoplasma pneumoniae infec-tion. Microbes Infect. 9: 78–86.
30. Kelly, M. N., J. K. Kolls, K. Happel, J. D. Schwartzman, P. Schwarzenberger,C. Combe, M. Moretto, and I. A. Khan. 2005. Interleukin-17/Interleukin-17 re-ceptor-mediated signaling is important for generation of an optimal polymorpho-nuclear response against Toxoplasma gondii infection. Infect. Immun. 73:617–621.
31. Cua, D. J., J. Sherlock, Y. Chen, C. A. Murphy, B. Joyce, B. Seymour, L. Lucian,W. To, S. Kwan, T. Churakova, et al. 2003. Interleukin-23 rather than interleu-kin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature421: 744–748.
32. Langrish, C. L., Y. Chen, W. M. Blumenschein, J. Mattson, B. Basham,J. D. Sedgwick, T. McClanahan, R. A. Kastelein, and D. J. Cua. 2005. IL-23drives a pathogenic T cell population that induces autoimmune inflammation.J. Exp. Med. 201: 233–240.
33. Murphy, C. A., C. L. Langrish, Y. Chen, W. Blumenschein, T. McClanahan,R. A. Kastelein, J. D. Sedgwick, and D. J. Cua. 2003. Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation.J. Exp. Med. 198: 1951–1957.
34. Yen, D., J. Cheung, H. Scheerens, F. Poulet, T. McClanahan, B. McKenzie,M. A. Kleinschek, A. Owyang, J. Mattson, W. Blumenschein, et al. 2006. IL-23is essential for T cell-mediated colitis and promotes inflammation via IL-17 andIL-6. J. Clin. Invest. 116: 1310–1316.
35. Kolls, J. K., and A. Linden. 2004. Interleukin-17 family members and inflam-mation. Immunity 21: 467–476.
36. Harrington, L. E., R. D. Hatton, P. R. Mangan, H. Turner, T. L. Murphy,K. M. Murphy, and C. T. Weaver. 2005. Interleukin 17-producing CD4� effectorT cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat.Immunol. 6: 1123–1132.
37. Harrington, L. E., P. R. Mangan, and C. T. Weaver. 2006. Expanding the effectorCD4 T-cell repertoire: the Th17 lineage. Curr. Opin. Immunol. 18: 349–356.
38. Hurst, S. D., T. Muchamuel, D. M. Gorman, J. M. Gilbert, T. Clifford, S. Kwan,S. Menon, B. Seymour, C. Jackson, T. T. Kung, et al. 2002. New IL-17 familymembers promote Th1 or Th2 responses in the lung: in vivo function of the novelcytokine IL-25. J. Immunol. 169: 443–453.
39. Oda, N., P. B. Canelos, D. M. Essayan, B. A. Plunkett, A. C. Myers, andS. K. Huang. 2005. Interleukin-17F induces pulmonary neutrophilia and amplifiesantigen-induced allergic response. Am. J. Respir. Crit. Care Med. 171: 12–18.
40. Liang, S. C., X. Y. Tan, D. P. Luxenberg, R. Karim, K. Dunussi-Joannopoulos,M. Collins, and L. A. Fouser. 2006. Interleukin (IL)-22 and IL-17 are coex-pressed by Th17 cells and cooperatively enhance expression of antimicrobialpeptides. J. Exp. Med. 203: 2271–2279.
41. Zheng, Y., D. M. Danilenko, P. Valdez, I. Kasman, J. Eastham-Anderson, J. Wu,and W. Ouyang. 2007. Interleukin-22, a TH17 cytokine, mediates IL-23-induceddermal inflammation and acanthosis. Nature 445: 648–651.
42. Dumoutier, L., E. Van Roost, D. Colau, and J. C. Renauld. 2000. Human inter-leukin-10-related T cell-derived inducible factor: molecular cloning and func-tional characterization as an hepatocyte-stimulating factor. Proc. Natl. Acad. Sci.USA 97: 10144–10149.
43. Dumoutier, L., J. Louahed, and J. C. Renauld. 2000. Cloning and characterizationof IL-10-related T cell-derived inducible factor (IL-TIF), a novel cytokine struc-turally related to IL-10 and inducible by IL-9. J. Immunol. 164: 1814–1819.
44. Wolk, K., S. Kunz, K. Asadullah, and R. Sabat. 2002. Cutting edge: immune cellsas sources and targets of the IL-10 family members? J. Immunol. 168:5397–5402.
45. Wolk, K., S. Kunz, E. Witte, M. Friedrich, K. Asadullah, and R. Sabat. 2004.IL-22 increases the innate immunity of tissues. Immunity 21: 241–254.
46. Dumoutier, L., E. Van Roost, D. Colau, and J. C. Renauld. 2000. Human inter-leukin-10-related T cell-derived inducible factor: molecular cloning and func-tional characterization as an hepatocyte-stimulating factor. Proc. Natl. Acad. Sci.USA 97: 10144–10149.
47. Wolk, K., E. Witte, U. Hoffmann, W. D. Doecke, S. Endesfelder, K. Asadullah,W. Sterry, H. D. Volk, B. M. Wittig, and R. Sabat. 2007. IL-22 induces lipopo-lysaccharide-binding protein in hepatocytes: a potential systemic role of IL-22 inCrohn’s disease. J. Immunol. 178: 5973–5981.
48. Andoh, A., Z. Zhang, O. Inatomi, S. Fujino, Y. Deguchi, Y. Araki, T. Tsujikawa,K. Kitoh, S. Kim-Mitsuyama, A. Takayanagi, et al. 2005. Interleukin-22, a mem-
ber of the IL-10 subfamily, induces inflammatory responses in colonic subepi-thelial myofibroblasts. Gastroenterology 129: 969–984.
49. Boniface, K., J. C. Lecron, F. X. Bernard, G. Dagregorio, G. Guillet, F. Nau, andF. Morel. 2005. Keratinocytes as targets for interleukin-10-related cytokines: aputative role in the pathogenesis of psoriasis. Eur. Cytokine Network 16:309–319.
50. Brand, S., F. Beigel, T. Olszak, K. Zitzmann, S. T. Eichhorst, J. M. Otte,H. Diepolder, A. Marquardt, W. Jagla, A. Popp, et al. 2006. IL-22 is increased inactive Crohn’s disease and promotes proinflammatory gene expression and in-testinal epithelial cell migration. Am. J. Physiol. 290: G827–G838.
51. Ikeuchi, H., T. Kuroiwa, N. Hiramatsu, Y. Kaneko, K. Hiromura, K. Ueki, andY. Nojima. 2005. Expression of interleukin-22 in rheumatoid arthritis: potentialrole as a proinflammatory cytokine. Arthritis Rheum. 52: 1037–1046.
52. Wolk, K., E. Witte, E. Wallace, W. D. Docke, S. Kunz, K. Asadullah, H. D. Volk,W. Sterry, and R. Sabat. 2006. IL-22 regulates the expression of genes respon-sible for antimicrobial defense, cellular differentiation, and mobility in keratin-ocytes: a potential role in psoriasis. Eur. J. Immunol. 36: 1309–1323.
53. Pan, H., F. Hong, S. Radaeva, and B. Gao. 2004. Hydrodynamic gene delivery ofinterleukin-22 protects the mouse liver from concanavalin A-, carbon tetrachlo-ride-, and Fas ligand-induced injury via activation of STAT3. Cell. Mol. Immu-nol. 1: 43–49.
54. Radaeva, S., R. Sun, H. N. Pan, F. Hong, and B. Gao. 2004. Interleukin 22(IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is asurvival factor for hepatocytes via STAT3 activation. Hepatology 39: 1332–1342.
55. Zenewicz, L. A., G. D. Yancopoulos, D. M. Valenzuela, A. J. Murphy,M. Karow, and R. A. Flavell. 2007. Interleukin-22 but not interleukin-17 providesprotection to hepatocytes during acute liver inflammation. Immunity 27:647–659.
56. Wolk, K., and R. Sabat. 2006. Interleukin-22: a novel T- and NK-cell derivedcytokine that regulates the biology of tissue cells. Cytokine Growth Factor Rev.17: 367–380.
57. Magram, J., S. E. Connaughton, R. R. Warrier, D. M. Carvajal, C. Y. Wu,J. Ferrante, C. Stewart, U. Sarmiento, D. A. Faherty, and M. K. Gately. 1996.IL-12-deficient mice are defective in IFN� production and type 1 cytokine re-sponses. Immunity 4: 471–481.
58. Mattner, F., J. Magram, J. Ferrante, P. Launois, K. Di Padova, R. Behin,M. K. Gately, J. A. Louis, and G. Alber. 1996. Genetically resistant mice lackinginterleukin-12 are susceptible to infection with Leishmania major and mount apolarized Th2 cell response. Eur. J. Immunol. 26: 1553–1559.
59. Chackerian, A. A., S. J. Chen, S. J. Brodie, J. D. Mattson, T. K. McClanahan,R. A. Kastelein, and E. P. Bowman. 2006. Neutralization or absence of the in-terleukin-23 pathway does not compromise immunity to mycobacterial infection.Infect. Immun. 74: 6092–6099.
60. Nakae, S., Y. Komiyama, A. Nambu, K. Sudo, M. Iwase, I. Homma,K. Sekikawa, M. Asano, and Y. Iwakura. 2002. Antigen-specific T cell sensiti-zation is impaired in IL-17-deficient mice, causing suppression of allergic cellularand humoral responses. Immunity 17: 375–387.
61. Springer, S., J. Lehmann, T. Lindner, J. Thielebein, G. Alber, and H. J. Selbitz.2000. A new live Salmonella enteritidis vaccine for chickens: experimental ev-idence of its safety and efficacy. Berl. Munch. Tierarztl. Wochenschr. 113:246–252.
62. Martin, G., U. Methner, G. Steinbach, and H. Meyer. 1996. Immunization withpotential Salmonella enteritidis mutants. 2. Investigations on the attenuation andimmunogenicity for mice and young hens. Berl. Munch. Tierarztl. Wochenschr.109: 369–374.
63. Lehmann, J., S. Springer, C. E. Werner, T. Lindner, S. Bellmann,R. K. Straubinger, H. J. Selbitz, and G. Alber. 2006. Immunity induced with aSalmonella enterica serovar Enteritidis live vaccine is regulated by Th1-cell-dependent cellular and humoral effector mechanisms in susceptible BALB/cmice. Vaccine 24: 4779–4793.
64. Boudard, F., N. Vallot, C. Cabaner, and M. Bastide. 1994. Chemiluminescenceand nitrite determinations by the MALU macrophage cell line. J. Immunol. Meth-ods 174: 259–268.
65. Kunz, S., K. Wolk, E. Witte, K. Witte, W. D. Doecke, H. D. Volk, W. Sterry,K. Asadullah, and R. Sabat. 2006. Interleukin (IL)-19, IL-20 and IL-24 are pro-duced by and act on keratinocytes and are distinct from classical ILs. Exp. Der-matol. 15: 991–1004.
66. Park, H., Z. Li, X. O. Yang, S. H. Chang, R. Nurieva, Y. H. Wang, Y. Wang,L. Hood, Z. Zhu, Q. Tian, and C. Dong. 2005. A distinct lineage of CD4 T cellsregulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6:1133–1141.
67. Aggarwal, S., N. Ghilardi, M. H. Xie, F. J. de Sauvage, and A. L. Gurney. 2003.Interleukin-23 promotes a distinct CD4 T cell activation state characterized by theproduction of interleukin-17. J. Biol. Chem. 278: 1910–1914.
68. Khader, S. A., J. E. Pearl, K. Sakamoto, L. Gilmartin, G. K. Bell,D. M. Jelley-Gibbs, N. Ghilardi, F. deSauvage, and A. M. Cooper. 2005. IL-23compensates for the absence of IL-12p70 and is essential for the IL-17 responseduring tuberculosis but is dispensable for protection and antigen-specific IFN-�responses if IL-12p70 is available. J. Immunol. 175: 788–795.
69. Lieberman, L. A., F. Cardillo, A. M. Owyang, D. M. Rennick, D. J. Cua,R. A. Kastelein, and C. A. Hunter. 2004. IL-23 provides a limited mechanism ofresistance to acute toxoplasmosis in the absence of IL-12. J. Immunol. 173:1887–1893.
70. Chen, Z., and J. J. O’Shea. 2008. Th17 cells: a new fate for differentiating helperT cells. Immunol. Res. 41: 87–102.
7900 IL-23-DEPENDENT IL-22 IS PROTECTIVE AGAINST Salmonella INFECTION
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

71. Johansson, C., M. Ingman, and W. M. Jo. 2006. Elevated neutrophil, macrophageand dendritic cell numbers characterize immune cell populations in mice chron-ically infected with Salmonella. Microb. Pathog. 41: 49–58.
72. Schwarzenberger, P., V. La Russa, A. Miller, P. Ye, W. Huang, A. Zieske,S. Nelson, G. J. Bagby, D. Stoltz, R. L. Mynatt, et al. 1998. IL-17 stimulatesgranulopoiesis in mice: use of an alternate, novel gene therapy-derived methodfor in vivo evaluation of cytokines. J. Immunol. 161: 6383–6389.
73. Scriba, T. J., B. Kalsdorf, D. A. Abrahams, F. Isaacs, J. Hofmeister, G. Black,H. Y. Hassan, R. J. Wilkinson, G. Walzl, S. J. Gelderbloem, et al. 2008. Distinct,specific IL-17- and IL-22-producing CD4� T cell subsets contribute to the humananti-mycobacterial immune response. J. Immunol. 180: 1962–1970.
74. Aujla, S. J., Y. R. Chan, M. Zheng, M. Fei, D. J. Askew, D. A. Pociask,T. A. Reinhart, F. McAllister, J. Edeal, K. Gaus, et al. 2008. IL-22 mediatesmucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 14:275–281.
75. Zheng, Y., P. A. Valdez, D. M. Danilenko, Y. Hu, S. M. Sa, Q. Gong,A. R. Abbas, Z. Modrusan, N. Ghilardi, F. J. De Sauvage, and W. Ouyang. 2008.Interleukin-22 mediates early host defense against attaching and effacing bacte-rial pathogens. Nat. Med. 14: 282–289.
76. Raffatellu, M., R. L. Santos, D. E. Verhoeven, M. D. George, R. P. Wilson,S. E. Winter, I. Godinez, S. Sankaran, T. A. Paixao, M. A. Gordon, et al. 2008.
Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency pro-motes Salmonella dissemination from the gut. Nat. Med. 14: 421–428.
77. Zheng, Y., P. A. Valdez, D. M. Danilenko, Y. Hu, S. M. Sa, Q. Gong,A. R. Abbas, Z. Modrusan, N. Ghilardi, F. J. De Sauvage, and W. Ouyang. 2008.Interleukin-22 mediates early host defense against attaching and effacing bacte-rial pathogens. Nat. Med. 14: 282–289.
78. Zheng, Y., P. A. Valdez, D. M. Danilenko, Y. Hu, S. M. Sa, Q. Gong,A. R. Abbas, Z. Modrusan, N. Ghilardi, F. J. De Sauvage, et al. 2008. Interleu-kin-22 mediates early host defense against attaching and effacing bacterial patho-gens. Nat. Med. 14: 282–289.
79. El Kasmi, K. C., J. Holst, M. Coffre, L. Mielke, A. de Pauw, N. Lhocine,A. M. Smith, R. Rutschman, D. Kaushal, Y. Shen, et al. 2006. General nature ofthe STAT3-activated anti-inflammatory response. J. Immunol. 177: 7880–7888.
80. Mangan, P. R., L. E. Harrington, D. B. O’Quinn, W. S. Helms, D. C. Bullard,C. O. Elson, R. D. Hatton, S. M. Wahl, T. R. Schoeb, and C. T. Weaver. 2006.Transforming growth factor-� induces development of the TH17 lineage. Nature441: 231–234.
81. MacLennan, C., C. Fieschi, D. A. Lammas, C. Picard, S. E. Dorman, O. Sanal,J. M. MacLennan, S. M. Holland, T. H. Ottenhoff, J. L. Casanova, andD. S. Kumararatne. 2004. Interleukin (IL)-12 and IL-23 are key cytokines forimmunity against Salmonella in humans. J. Infect. Dis. 190: 1755–1757.
7901The Journal of Immunology
by guest on September 17, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from





























