Embed Size (px)
Citation preview

PROGNOSIS FOR SEIZURE CONTROL AND REMISSION IN CHILDREN WITH MYELOMENINGOCELE
Michael J . Noetzel Jeffrey N. Blake
Seizures in children with myelomen- ingocele have been recognized as a relatively common occurrence (Hosking 1974, Varfis et al. 1977, Blaauw 1978). Recent studies have examined the relative contribution of various factors, including hydrocephalus, that may play a part in the origin of a seizure disorder in these children (Bartoshesky et al. 1985, Stellman et al. 1986, Chadduck and Adametz 1988, Faillace and Canady 1990, Hack et al. 1990). There is little in- formation, however, about the outcome of seizures in patients with myelo- meningocele. In particular, no data are available on the prognosis for these children with seizures after discontinu- ation of anti-epileptic drugs. We therefore designed a prospective study in which all children with myelomeningocele were evaluated to determine (a) the incidence of seizures in this population, (b) the prognosis for seizure control and (c) which factors correlate with continued seizure remission after anticonvulsant therapy is discontinued.
Patients and method One hundred and forty children with myelomeningocele followed in the Birth Defects Center at St. Louis Children’s Hospital during the years 1962 to 1989 were the subjects of this combined
retrospective and prospective study. All were considered to have primary neural- tube defects (Noetzel and Volpe 1987). Hydrocephalus requiring shunting of cerebrospinal fluid (CSF) was diagnosed in 109 patients (76.4 per cent), either by ventriculography before 1976 or by CT scan subsequently. The criteria used to determine the need for shunting included clinical evidence of increased intracranial pressure in combination with either serial radiological studies revealing progressive ventricular enlargement or an initial study demonstrating marked ventricular enlarge- ment, compressed sulci or site of CSF obstruction. Over 70 per cent of the children with hydrocephalus had their first ventricular shunt inserted before one month of age, and nearly 96 per cent had it inserted before the age of six months.
In 1980 the medical records of all 126 children with myelomeningocele then receiving medical care through the Birth Defects Center were examined and the patients were enrolled in the prospective phase of the study. 120 of the children had been treated during the newborn period at St. Louis Children’s Hospital and then followed through the Birth Defects Center, so the complete medical history of each of these patients was available for review. The remaining six patients began to receive their care
m- m 3
OI Q‘
h 3 h B
803

b $
L;
9 .- 3
c .-
-0 9
804
through the center at two to six months of age (four children) and at three and six years (one child each). Subsequently 21 children with myelomeningocele born during the years 1980 to 1982 were enrolled in the study as their care was assumed by physicians in the Birth Defects Center: all 21 were treated as newborn infants at Children’s Hospital.
Information extracted from the medical records included race, sex, prenatal and perinatal history, family history of non- febrile seizures, developmental history and school performance, level of myelomeningocele, presence or absence of seizures, age at onset of seizures, EEG results, type of shunt procedure, number of shunt revisions and number of shunt infections. Shunt infections were diag- nosed by growth of bacterial organisms cultured from CSF, blood, peritoneal fluid or catheter tips, depending on the type of shunt.
All children enrolled in the study had at least one CT scan performed during the prospective phase of the study (1980 to 1989). Central nervous system (CNS) malformations involving the cerebral cortex, including porencephaly, agenesis of the corpus callosum and disorders of neuronal migration such as lissencephaly and pachygyria were documented from these scans.
Of the 147 patients enrolled in the study, seven were lost to follow-up within the first 24 months and were excluded from analysis. The remaining 140 children have been studied prospectively for up to 10 years (minimum five years, mean 8 .1 years), during which time serial neuro- logical evaluations and developmental testing were performed. Information from CT scans obtained during the prospective phase of the study was updated. Seizure frequency in patients with previously documented convulsions was monitored and the onset of seizures in previously unaffected children was recorded. Seizure activity had been observed and documented either by a Birth Defect Center or emergency-room physician or a family member. One patient, initially considered to have seizures, later demonstrated decerebrate posturing secondary to the slit ventricle syndrome, with resultant transient
cerebral herniation. Ninety-five (67.8 per cent) of the 140
children were evaluated by detailed psychometric testing by a group of specially trained examiners. Each was examined individually and testing was performed when the children attended as outpatients at the Birth Defects Center. The type of evaluation depended on the age of the child (Noetzel et al. 1985). The Peabody Picture Vocabulary Test (1965) was used for children two years of age and older. An appropriate general intelligence test was also administered: Cattell Infant Intelligence Scale (1 960), Stanford-Binet Intelligence Scale (1960), or Wechsler Intelligence Scales for Children-Revised (1972). Academic skills of children five years and older were evaluated by the Jastek Wide Range Achievement Test (1965). The Vineland Social Maturity Scale (1965) was used to assess the self- help skills and social and language development of children younger than five years. Generally evaluations were performed every six months for the first two years of life and yearly thereafter until age five, but some children were tested only at ages 18 to 24 months and five years. No child was given the diagnosis of mental retardation (IQ < 70) in the absence of a complete psychometric evaluation. Previous studies from our institution have demonstrated that this protocol for psychometric testing does have predictive value for later intelligence (Fishman and Palkes 1974). In addition, 43 children (31 per cent) who did not receive formal psychometric testing were judged to be of average intelligence because of their academic performance and progress in regular school up to at least the fourth grade. Some in this group were diagnosed by their special school district as having mild learning disabilities. Finally, two children less than five years of age were not formally evaluated by psychometric testing: even though they were achieving intellectual m.ilestones at a fairly normal rate, no conclusions were drawn as to the presence or absence of mental retardation.
The statistical significance of the differences in each subgroup was analysed using the Student t test. All p values are two-tailed.

Results Of the 147 children enrolled in the prospective phase of the study, 140 were available with a mean follow-up of 8.1 years. On the basis of information obtained at the time of entry to the study, there were no differences between the seven children lost to follow-up and the study population. In addition, a retro- spective review of 25 former patients whose care through the Birth Defects Center was discontinued before 1980 revealed no statistically significant difference in patient characteristics compared with the children enrolled in the study. Thus, while our investigation does not represent a true population sample, we feel that the patient group is very representative of children with myelo- meningocele referred to a comprehensive care center.
Seizures were documented in 24 (17.1 per cent) of the 140 children, 19 of whom had had two or more seizures before the initiation of anti-epileptic medication. The other five patients' treatment had been started after a single but prolonged seizure (minimum 15 minutes) and in four of these cases further seizures occurred while on medication. Five children had their first convulsion between birth and six months of age, another six between seven and 11 years, but the most common ages at seizure onset were two to five years (13 patients).
Of the 109 patients with myelomenin- gocele and shunted hydrocephalus, seizures were documented in 18 (16.5 per cent) (Table I). Shunt infections and shunt revisions per patient occurred with greater frequency among children with seizures. However, if one includes only those infections occurring before the first seizure, the frequency of infection was not statistically higher than that for children who did not have seizures (Table I). The number of shunt revisions before the first seizure, calculated per patient or per patient-year, was statistically in- creased for children with seizures. Age at the time of shunt insertion, location of shunt catheter and family history of seizures were not predictive of seizures. Of the 31 children whose myelomeningo- cele was not associated with hydro- cephalus, six (19.5 per cent) also
TABLE I Myelomeningocele and hydrocephalus (N = 109)
Seizures Absent Present
N 91 (83.5%) 18 (16.5%) Years of follow-up 16.3 ( + 5 ) 17.1 (+5) M:F ratio 42:49 8: 10 Shunt infection? 12.1% 16.6% Shunt revision/Ptt 1.12 1 * 89* Average age at shunt insertion (mths) 3 .2 2 . 5 Level of lesion
Thoracic 14 (15.4%) 4 (22.2%) Thoracolumbar 13 (14-3'70) 3 (16.7%) Lumbar 32 (35.2%) 6 (33.3%) Lumbosacral 28 (30.7%) 4 (22.2%) Sacral 4 (4.4%) 1 (5.6%)
*p < 0.05. tDetermined using only shunt complications preceding initial seizure.
TABLE11 ,
Myelomeningocele without hydrocephalus (N = 31)
Seizures Absent Present
N 25 (81.6%) 6 (19.4%) Years of follow-up 15.8 (+6) 17.2 (+5) M:F ratio 13:12 2:4 Level of lesion
Thoracic 1 (4%) 0 Thoracolumbar 3 (12%) 1 (16.7%) Lumbar 8 (32%) 2 (33.3%) Lumbosacral 10 (40%) 2 (33.3%)
Sacral 3 (12%) 1 (16.7%)
TABLE I11 Variables associated with onset of seizures (N=140)
Seizures Absent Present
N 116 24 Mental retardation 14 (12%) 9 (37.5%)* Malformation 1 (0.9%) 5 (21%)** Family history of seizures 5 (4.3%) 1 (4.2%)
0 00 3
s m m
b B
*p<0*02; **p<0.05. 805
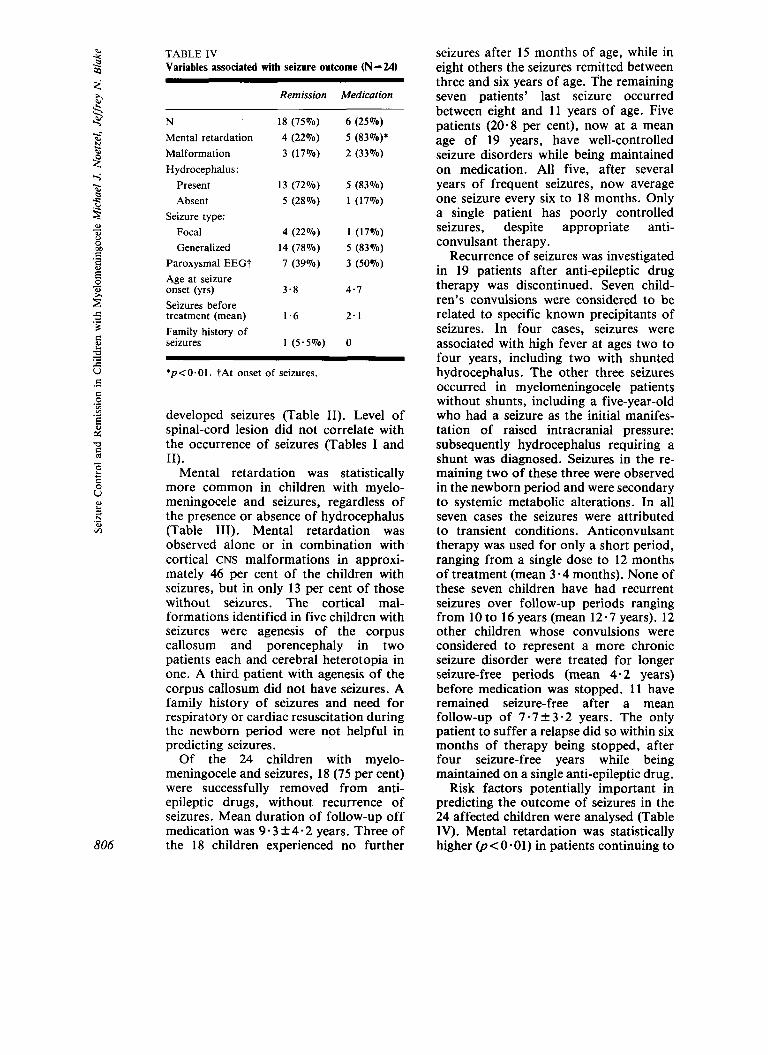
TABLE 1V Variables associated with seizure outcome (N = 24)
s 3 .-
.- E
806
Remission Medication
N Mental retardation Malformation Hydrocephalus:
Present Absent
Seizure type: Focal Generalized
Paroxysmal EEGt Age at seizure onset (yrs) Seizures before treatment (mean) Family history of seizures
18 (75%) 4 (22%) 3 (17%)
13 (72%) 5 ( 2 8 % )
4 (22%) 14 (78%) 7 (39%)
3 - 8
1.6
1 ( 5 . 5 % )
6 ( 2 5 % ) 5 (83%)* 2 (33%)
5 (83%) 1 (17%)
1 (17%) 5 (83%) 3 (50%)
4.7
2.1
0
*p<O.Ol. tAt onset of seizures.
developed seizures (Table 11). Level of spinal-cord lesion did not correlate with the occurrence of seizures (Tables I and 11).
Mental retardation was statistically more common in children with myelo- meningocele and seizures, regardless of the presence or absence of hydrocephalus (Table 111). Mental retardation was observed alone or in combination with cortical CNS malformations in approxi- mately 46 per cent of the children with seizures, but in only 13 per cent of those without seizures. The cortical mal- formations identified in five children with seizures were agenesis of the corpus callosum and porencephaly in two patients each and cerebral heterotopia in one. A third patient with agenesis of the corpus callosum did not have seizures. A family history of seizures and need for respiratory or cardiac resuscitation during the newborn period were not helpful in predicting seizures.
Of the 24 children with myelo- meningocele and seizures, 18 (75 per cent) were successfully removed from anti- epileptic drugs, without recurrence of seizures. Mean duration of follow-up off medication was 9 - 3 f 4 - 2 years. Three of the 18 children experienced no further
seizures after 15 months of age, while in eight others the seizures remitted between three and six years of age. The remaining seven patients’ last seizure occurred between eight and 1 1 years of age. Five patients (20-8 per cent), now at a mean age of 19 years, have well-controlled seizure disorders while being maintained on medication. All five, after several years of frequent seizures, now average one seizure every six to 18 months. Only a single patient has poorly controlled seizures, despite appropriate anti- convulsant therapy.
Recurrence of seizures was investigated in 19 patients after anti-epileptic drug therapy was discontinued. Seven child- ren’s convulsions were considered to be related to specific known precipitants of seizures. In four cases, seizures were associated with high fever at ages two to four years, including two with shunted hydrocephalus. The other three seizures occurred in myelomeningocele patients without shunts, including a five-year-old who had a seizure as the initial manifes- tation of raised intracranial pressure: subsequently hydrocephalus requiring a shunt was diagnosed. Seizures in the re- maining two of these three were observed in the newborn period and were secondary to systemic metabolic alterations. In all seven cases the seizures were attributed to transient conditions. Anticonvulsant therapy was used for only a short period, ranging from a single dose to 12 months of treatment (mean 3.4 months). None of these seven children have had recurrent seizures over follow-up periods ranging from 10 to 16 years (mean 12-7 years). 12 other children whose convulsions were considered to represent a more chronic seizure disorder were treated for longer seizure-free periods (mean 4 . 2 years) before medication was stopped. 1 1 have remained seizure-free after a mean follow-up of 7 - 7 f 3 . 2 years. The only patient to suffer a relapse did so within six months of therapy being stopped, after four seizure-free years while being maintained on a single anti-epileptic drug.
Risk factors potentially important in predicting the outcome of seizures in the 24 affected children were analysed (Table IV). Mental retardation was statistically higher @<0-01) in patients continuing to

require medicine for their seizures compared with those whose seizures remitted and for whom treatment was discontinued. Five of the six children requiring continued anti-epileptic treat- ment have an IQ <70. In contrast, all but four of the 18 children whose anti- convulsant medication was successfully discontinued were of at least low-normal intelligence. No other factor was useful in predicting whether a child’s seizures would be effectively controlled with treatment and whether medication subsequently could be discontinued without recurrence of seizures.
It is interesting that most children, including three of the five with CNS malformations, were reported to have generalized seizures, although the common EEG finding was of focal paroxysmal features, suggesting that a focal onset of the seizure may not have been recognized. The total number of shunt revisions and/or infections in patients with hydrocephalus was also not predictive of seizure control. In fact all five patients who had a shunt infection were among the group of children who have remained seizure-free off medication.
Discussion The present study was performed to assess the outcome of seizures in patients with myelomeningocele, one of the most common and clinically significant con- genital malformations of the central nervous system (Noetzel 1989). Previous investigations have suggested that the incidence of seizures in this population varies according to whether or not they also have hydrocephalus. For patients with hydrocephalus, the reported in- cidence of seizures ranges from 14.7 to 29 per cent (Hosking 1974, Varfis et al. 1977, Blaauw 1978, Bartoshesky et al. 1985, Stellman et al. 1986, Hack et al. 1990), whereas a much lower incidence-roughly 2 to 8 per cent-has been reported for children with myelomeningocele but without shunts (Bartoshesky et al. 1985, Chadduck and Adametz 1988). The increased incidence of seizures in patients with myelomeningocele and hydro- cephalus may result from (a) minor cortical injury at the time of shunt insertion (Hosking 1974, Varfis et al.
1977), (b) shunt complications such as 2 infection or malfunction causing at least transient raised intracranial pressure z (Hosking 1974, Blaauw 1978, Barto- m shesky et al. 1985), or (c) the pathology underlying or associated with the develop-
d ment of hydrocephalus (Bartoshesky et al. 1985). P
In our patients, seizures (either single or B recurrent) were not commonly associated 2 with shunt malfunction, thus confirming 2 the results of other investigations 3 (Stellman et al. 1986, Hack et al. 1990). 3 The number of shunt revisions, but not shunt infections, was statistically higher 3
9 in our patients who developed seizures (see Table 1). These findings contrast with those of Bartoshesky et al. (1985), who
patients with seizures were five times 3 B
m m
m - m 2
Ql .s
- - determined that myelomeningocele i!
2 more likely to have had a shunt infection, whereas isolated shunt revisions were not increased in their patients. This dis- crepancy in results may reflect differences in the ages of patients at the time of infection, virulence of organisms invading the CNS, or other factors contributing to the severity of shunt infection. Such differences would alter the risk of cerebral damage and hence the later development of seizures. Other investigators have suggested that both shunt infection and number of shunt revisions significantly increased the likelihood of a child with myelomeningocele developing seizures, but it is not clear whether shunt complications occurring after the first seizure were included in their analysis of data (Chadduck and Adametz 1988). Finally, some studies have failed to show any association between number of shunt revisions and occurrence of seizures (Blaauw 1978, Stellman et al. 1986).
In contrast to the findings of other studies, we observed an equivalent incidence of seizures in our patients without hydrocephalus compared with those requiring shunts. These two groups were combined to determine whether factors other than hydrocephalus and shunt complications were helpful in predicting the occurrence of convulsions. Our analysis suggests that mental retardation and/or CNS malformation may correlate with seizures in myelo- meningocele patients. This is not 807

C .-
U 9
2 2 .- 0 vl
808
surprising, since a high frequency of both gross and microscopic cortical mal- formations has been found in autopsy studies of patients with myelomeningocele (Gilbert et al. 1986). These abnormalities may contribute to the development of mental retardation, and also serve as epileptic foci. Other investigators have also observed an increased incidence of seizures in myelomeningocele patients with developmental delay and/or brain malformation (Bartoshesky et al. 1985, Stellman et al. 1986).
From our results and those of other investigators, it would appear that many factors may be involved in the develop- ment of seizures in patients with myelomeningocele. A better under- standing of the genesis of seizures in these children might be gained by more detailed studies examining, for example, severity of shunt infection, duration and degree of raised intracranial pressure accompanying shunt malfunction, and number of shunt revisions or replacements in which there is manipulation of the ventricular catheter. We are now using MRI scanning of patients with myelomeningocele to deter- mine the occurrence of migrational disorders. We hope to define better the apparent association between these abnormalities and developmental out- come and the occurrence of seizures.
Previous studies have suggested that seizures in many patients with myelo- meningocele respond well to usual anti- epileptic drug treatment (Varfis et al. 1977, Bartoshesky et al. 1985), although less encouraging results have been observed (Blaauw 1978). Our findings indicate that the prognosis is excellent, not only for seizure control but also for subsequent remission, for children with myelomeningocele. The single most useful predictor of good outcome was normal intellectual development (see Table IV).
Seven children had only short courses of anti-epileptic drug therapy since their convulsions were attributed to known precipitates of seizures. These patients have remained seizure-free off treatment for between 10 and 16 years. This suggests that not every initial seizure in a child with myelomeningocele (with or without hy- drocephalus) requires long-term anti- epileptic drug treatment, and that in some
cases no treatment at all may be necessary. A similar conclusion appears to have been reached by Varfis et al. (1977), who indicated that two of their four patients with new onset of seizures did not require treatment and neither had a recurrence over roughly four years.
Our study also provides data strongly suggesting that many children with myelomeningocele and chronic seizures subsequently will do well. 1 1 patients have been removed from medication without recurrent seizures over a mean follow-up period of 7.7 years. Based on previous reports, it can be estimated that approxi- mately 85 to 98 per cent of seizure relapses should have occurred during this length of follow-up (Holowach et al. 1972, Emerson et al. 1981, Matricardi et al. 1989). We conclude, therefore, that myelomeningocele patients with well- controlled seizures have an excellent chance of remaining seizure-free after withdrawal of medication.
Most recent studies on discontinuing treatment of children with epilepsy have evaluated patients who were seizure-free on medication for two or four years (Emerson et al. 198 1 , Holowach Thurston et al. 1982, Matricardi et al. 1989). These studies have concluded that while anti- epileptic drugs can be safely withdrawn for most children, there are specific factors associated with an increased risk of seizure relapse. Patients with mental retardation and those with known organic causes for their convulsions were more likely to suffer a recurrence, as were children who had had epilepsy for a long period before control was established (Holowach et al. 1972, Emerson et al. 1981, Matricardi et al. 1989). It was not possible in our study to determine factors statistically predictive of seizure recur- rence after discontinuation of anti- epileptic drugs, since only a single child had a relapse. From our results, however, no single variable should be viewed as an absolute deterrent to attempting dis- continuation after a long seizure-free interval. For example three of our patients who met this criterion were mentally retarded, but each successfully had their anticonvulsants stopped. Fur- thermore, in our study the risk of seizure recurrence was statistically no different

for patients SeiZUre-free on medication for Investigator Development Award (1 K08NS01010) two to five years (10 per cent) than for from the National Institute of Neurological and
Communicative Disorders and Stroke (to M. J.N.); those controlled for Seven or more years (0 The authors are grateful to Drs. Philip R. Dodge per cent). Therefore it seems reasonable to and Jean Holowach Thurston for their helpful consider stopping treatment when a child comments in reviewing the manuscript, and they
thank Ms. Debby O’Leary for typing the with myelomeningocele has been seizure- manuscript. free for two to four years, which is in keeping with recommendations for the Authors’ Appointments majority of children with chronic seizure *Michael J. Noetzel, M.D., Edward Mallinckrodt disorders (Gordon 1982, Holowach Department of Pediatrics and Department of
Neurology; Thurston et ale 1982, Matricardi et ale Jeffrey N. Blake, Edward Mallinckrodt Department of Pediatrics; Washington University School of Medicine, 400 S.
1989). Accepted for publication 9th April 1991. Kingshighway, St. Louis, MO 63110.
Acknowledgements This study was supported in part by a Clinical *Correspondence to first author.
SUMMARY A combined retrospective and prospective study was designed to determine the incidence of seizures in 140 children with myelomeningocele, as well as the potential for seizure control and remission. The incidence of seizures in 109 patients with myelomeningocele and hydrocephalus was 16.5 per cent, and 19-4 per cent in a further 31 patients without hydrocephalus. Mental retardation, often in combination with cerebral malformations, was significantly more common in children with seizures, regardless of presence or absence of hydrocephalus. Of the 24 patients with convulsions, three- quarters had anti-epileptic medication discontinued, without recurrence of seizures. An additional five children’s seizures are well controlled with medication. Mental retardation was the only significant predictor of long-term outcome. These results indicate that children with myelomeningocele have an excellent prognosis for seizure control and subsequent remission off medication.
Pronostic du contrble des crises comitiales et des rkmissions chez les enfants porteurs d’un myPlomkningoc&le Une etude combinke retrospective et prospective a CtC Ctablie pour determiner Y’incidence des crises comitiales chez 140 enfants porteurs d’un myelomhingoctle, ainsi que les possibilitts de contrde des crises et de remission. L’incidence des crises chez 109 patients avec myelomCningocele porteurs d’hydrockphalie a ttC de 16,5 pour cent auquel il faut ajouter 19,4 pour cent dans un groupe complementaire de 31 patients sans hydrockphalie. Le retard mental souvent associe aux malformations cerebrales ttait significativement plus habitue1 chez les enfants avec crises comitiales, qu’il y ait ou non hydroctphalie. Parmi les 24 patients presentant des convulsions, trois quarts d’entre eux avaient eu une medication anti-epileptique suspendue sans rkcidive des crises. Cinq autres enfants avaient un bon contrale des crises par medication. Le retard mental Ctait le seul caractere significatif du devenir a long terme. Ces rksultats indiquent que chez I’enfant porteur d’un myClomCningocCle le pronostic de contrBle des crises et de remission ultkrieure permettant l’arrst de la medication est excellent.
ZUSAMMENFASSUNG Prognose fur An fallskontrolle und Remission bei Kindern rnit Myelomeningocele Es wurde eine sowohl retrospektive, als auch prospektive Studie entwickelt, um die Anfallshaufigkeit bei 140 Kindern mit Myelomeningocele, sowie die Prognose fur Anfallskontrolle und Remission zu bestimmen. Bei 109 Patienten rnit Myelomeningocele und Hydrozephalus betrug die Anfallshaufigkeit 16.5 Prozent und bei weiteren 3 1 Patienten ohne Hydrozephalus 19.4 Prozent. Geistige Retardierung, oft im Zusammenhang rnit HirnmiRbildungen, war bei Kindern mit Anfallen signifikant hoher, unabhangig davon, ob ein Hydrozephslus vorlag oder nicht. Bei drei Viertel der 24 Kindern mit Anfdlen war die antikonvulsive Therapie abgesetzt worden, ohne daR wieder Anfallen auftraten. Weitere funf Kinder hatten eine gute Anfallskontrolle unter Medikation. Geistige Retardierung war der einzige prognostische Parameter fur die weitere Entwicklung. Diese Ergebnisse zeigen, daR die Prognose fur Anfallskontrolle und anschlienende Remission ohne Medikation bei Kindern rnit Myelomeningocele sehr gut ist.
RESUMEN Pronostico respecto a1 control y a la remisidn de las convulsiones en nifios con mielomeningocele Se disefl6 un estudio combinado retrospectivo y prospectivo para determinar la incidencia de convulsiones en 140 niflos con mielomeingocele. asi como la posibilidad potencial del control y remisi6n de las convulsiones. La incidencia de convulsiones en 109 pacientes con mielomeningocele e hidrocefalia fue del 16,5 por ciento, y del 19,4 por ciento en otros 31 pacientes sin hidrocefalia.
RESUME
2
z m m
m rn
809

B P Q
C E
u 9 a
e .-
El retraso mental, a menudo en combinacion con malformaciones cerebrales, era significativamente mas frecuente en niiios con convulsiones, tanto si habia como si n o hidrocefalia. De 10s 24 pacientes con convulsiones, las tres cuartas partes seguian una medicacion antiepiltptica discontinuada sin recurrencia de las convulsiones. Otros cinco niiios tenian convulsiones bien controladas con medicacion. El retraso mental fue el unico predictor significativo de un curso a largo plazo. Estos resultados indican que en niilos con mielomeningocele el pronostico del control y subsiguiente remision de las convulsiones es excelente.
References Bartoshesky, L. E., Haller, J., Scott, R. M.,
Wojick, C. (1985) ‘Seizures in children with meningomyelocele.’ American Journal of Diseases of Children, 139, 400-402.
Blaauw, G. (1978) ‘Hydrocephalus and epilepsy.’ Zeitschrifr fur Kinderchirurgie, 25, 341-345.
Chadduck, W., Adametz, J. (1988) ‘Incidence of seizures in patients with myelomeningocele: a multifactorial analysis.’ Surgical Neurology, 30,
Emerson, R., D’Souza, B. J., Vining, E. P., Holden, K. R., Mellits, E. D., Freeman, J. M. (1981) ‘Stopping medication in, children with epilepsy: predictors of outcome. New England Journal of Medicine, 304, 1125-1129.
Faillace, W. J., Canady, A. I. (1990) ‘Cerebrospinal fluid shunt malfunction signaled by new or recurrent seizures.’ Child’s Nervous System, 6, 37-40.
Fishman, M. A., Palkes, H. S. (1974) ‘The validity of psychometric testing in children with congenital malformations of the central nervous system.’ Developmental Medicine and Child Neurology, 16, 180-185.
Gilbert, J. N., Jones, K. L., Rorke, L. B., Chernoff, G. F., James, H. E. (1986) ‘Central nervous system anomalies associated with meningo- myelocele, hydrocephalus and the Arnold-Chiari malformation.’ Neurosurgery, 18, 559-563.
Gordon, N. (1982) ‘Duration of treatment for childhood epilepsy.’ Developmental Medicine and Child Neurology, 24, 84-88.
Hack, C. H., Enrile, B. G., Donat, J. F., Kosnik, E. (1990) ‘Seizures in relation to shunt dysfunction in children with meningomyelocele.’ Journal of Pediatrics, 116, 57-60.
281-285.
Holowach, J., Thurston, D. L. O’Leary, J. (1972) ‘Prognosis of childhood epilepsy: follow-up study of 148 cases in which therapy had been suspended after prolonged anticonvulsant control.’ New England Journal of Medicine, 286, 169-1 74.
Holowach Thurston, J. , Thurston, D. L., Hixon, B. B., Keller, A. J. (1982) ‘Prognosis in childhood epilepsy: additional follow-up of 148 children 15 to 23 years after withdrawal of anticonvulsant therapy.’ New England Journal of Medicine, 306,
Hosking, G. P. (1974) ‘Fits in hydrocephalic children.’ Archives of Disease in Childhood, 49,
Matricardi, M., Brinciotti, M., Benedetti, P. (1989) ‘Outcome after discontinuation of antieuileptic
831-836.
633-635.
drug therapy in children with epilepsy.’ E$/epsiu, 30. 582-589.
Noeizel, M. J. (1989) ‘Myelomeningocele: current concepts of management.’ Clinics in Peri- natology, 16, 311-329.
- Volpe, J. J. (1987) ‘Neural tube defects.’ In Davidoff, R. A. (Ed.) Handbook of the Spinal Cord. New York: Marcel Dekker.
- Marsh, J. L. Palkes, H., Gado, M. (1985) ‘Hydrocephalus and mental retardation in craniosynostosis.’ Journal of Pediatrics, 107, 885-892.
Stellman, G. R., Bannister, C. M., Hillier, V. (1986) ‘The incidence of seizure disorder in children with acquired and congenital hydrocephalus.’ Zeitschrut fur Kinderchirurgie, 41 (Suppl. l), 38-41.
Varfis, G., Berney, J., Beaumanoir, A. (1977) ‘Electroclinical follow-up of shunted hydro- cephalic children.’ Child’s Brain, 3, 129-139.
810





























