Embed Size (px)
Citation preview

Primitive Neuroectodermal Tumor Cell Lines: Chromosomal Analysis of Five Cases
Venkateswara R. Potluri, Fred Gilbert, Christiane Helsen, and Lawrence Helson
ABSTRACT: A cytogenetic analysis of primitive neuroectodermal tumor (PNET) cell lines was under- taken. PNET are presumed to be embryologically related to. but clinically and histologically distinct from, other tumors of neuroectodermal origin, including neuroblastoma and retino- blastoma. No single chromosome abnormality was found in all five of the tumors studied. In three of the five cases, however, additional lq material [either as extra chromosome #1 or i(lq)] was found in all cells, and in two of the five, monosomy 13 was noted in all cells; the possible significance of these findings is discussed.
INTRODUCTION
Embryonic neuroectoderm gives rise to a number of cell types in the central and peripheral nervous systems, as well as to certain non-neural tissues (including pig- ment epithelium and portions of the cranium) in the intact organism [1]. The cel- lular derivatives of neuroectoderm may become transformed and among the more common of such tumors arising in children are neuroblastoma and retinoblastoma [2]. A much rarer entity, also presumed to originate from nouroectoderm, is the so- called primitive neuroectodermal tumor (PNET) [3].
PNET can be distinguished from neuroblastoma on clinical and histologic grounds [3, 4]. The latter usually arise within the adrenal medulla or in the auto- nomic ganglia (of the abdominal, mediastinal, or cervical regions), whereas, the former have been reported in peripheral sites (e.g., dorsum of the foot, knee, thigh, testis, ovary, kidney) without evident involvement of the adrenals or autonomic ganglia. In neuroblastoma, the tumor will generally contain ganglion cells in vary- ing stages of maturation (some of which may include neurosecretory granules) and neurofibrils; PNET, on the other hand, have been found to contain primitive neural tubular structures, sheets of small, darkly staining cells with limited cytoplasm (in which neurosecretory granules may be scant or absent), and neuroglial elements and rare ganglion cells, all set in a prominent mesenchymal stroma. In the PNET reported to date, the average age at diagnosis was approximately 7 years (although cases have been reported from birth to 24 years); in neuroblastoma, the median age at diagnosis is less than 2 years (with greater than 70% of cases diagnosed before age 5) [5, 6].
From the Division of Medical Genetics, Mount Sinai School of Medicine (V. R. P., F. G.}; and Memorial- Sloan Kettering Cancer Center (C. H., L. H.), New York, NY.
Address requests for reprints to Dr. Fred Gilbert, Medical Genetics/Annenberg 17-76, Mount Sinai School of Medicine, Fifth Avenue and 100th Street, New York, NY 10029.
Received July 8, 1985; accepted January 6, 1986.
75
© 1987 Elsevier Science Publishing Co., Inc. Cancer Genet Cytogenet 24:75-86(1987) 52 Vanderbilt Ave., New York, NY 10017 0165-4608/87/$03.50
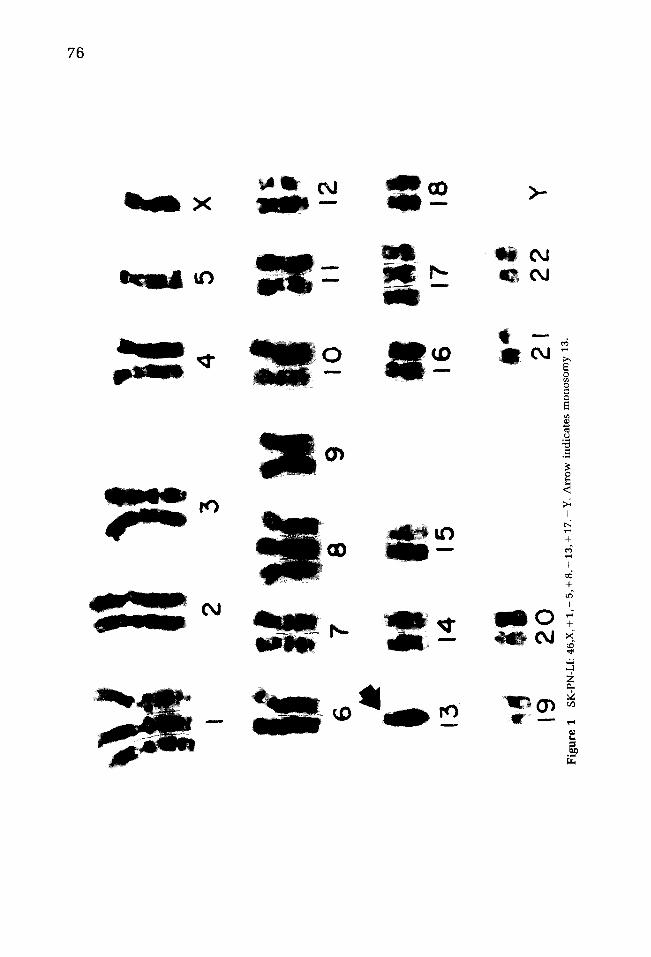
76
X CO m
ur~ m
r~ m
W ~
q~
r~
C~J
0
0~
QD u~j m
o C~
I
Jr L~ I
/ C~
6 7 8 9 I0 II 12
W iim iH '3 14 15 16 17 18
19 20 21 22 Y
J M
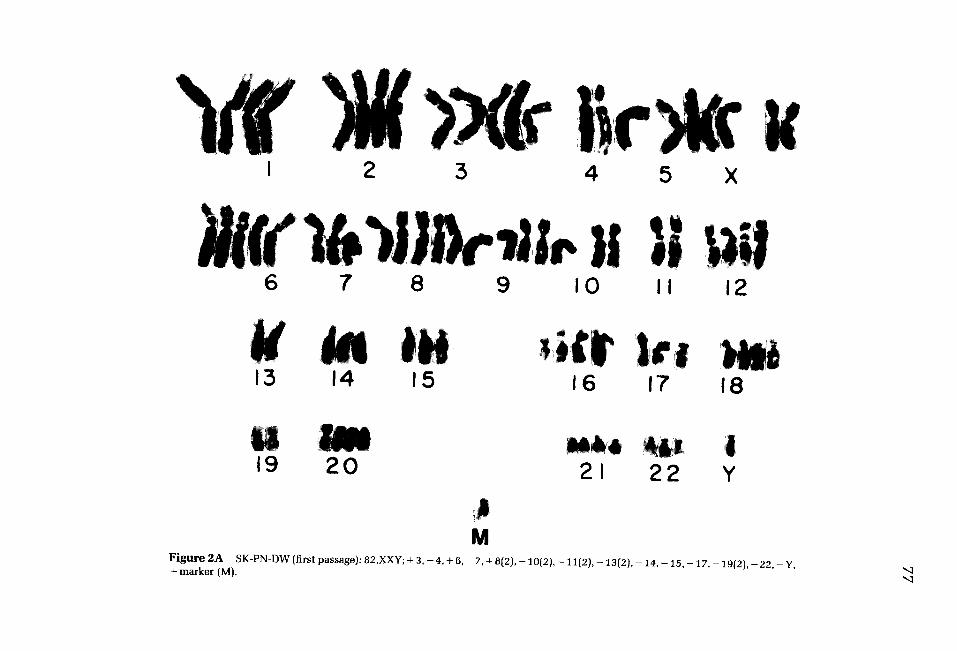
Figure 2 A S K - P N - D W ( f i r s t p a s s a g e ) : 8 2 , X X Y ; + 3 , - 4 , + 6 , - 7 , + 8 ( 2 ) , - 1 0 ( 2 ) , - 1 1 ( 2 ) , - 1 3 { 2 ) , - 1 4 , - 1 5 , - 1 7 , - 1 9 ( 2 ) , - 2 2 , - Y , + m a r k e r ( M ) . " ~

78
~ X
if)
/..®
0
I
J
A l t - - - J
i
i o-
i
~" ~ 0 ~
M
I I r~
O.C o

Chromosomal Analysis of PNET 79
q
Figure 2C SK-PN-DW (metaphase for DM). Arrows indicate double minutes.
In a recent review of karyotypes from neuroblastoma, a particular chromosome abnormality [rearrangements resulting in the loss of material from the short arm of chromosome #1 (lp)] was identified in some 70% of the collected cases [7]. In reports of karyotypes from a more primitive tumor of neuroectodermal origin, the so-called peripheral neuroepithelioma,~a particular marker rearrangement, a recip- rocal translocation between chromosomes #11 and #22, has been noted [8]. We now present a cytogenetic analysis of an additional five examples of primitive neu- roectodermal tumors.
MATERIALS AND METHODS
Tumor samples were transported from the operating room in sterile saline or me- dium (RPM1 1640), minced with scissors and scalpel, and allowed to grow in RPM1 1640 medium containing 20% fetal calf serum.
SK-PN-LI is a permanent cell line derived from a tumor in the area of the right scapula, removed September 1979. SK-PN-DW, also a permanent cell line, was es-

8 0 V.R. Potluri et el.
tablished from a presacral mass, excised January 1978. SK-PN-AG is a permanent line established from a chest wall mass, removed in September 1983. SK-PN-WA, a fourth permanent cell line, was established from a thoracic wall tumor, excised in August 1979. Patient BN was initially operated in 1980, at which time a mass ad- jacent to the left ovary, pathologically identified as a PNET, was removed; at reop- eration in 1985, a recurrence at the primary site (designated SK-PN-BW) was re- moved.
Chromosome preparations were made from recent reconstitutions of the perma- nent cell lines from cases LI, DW, AG, and WA, frozen within months of their establishment (the exception for case DW is described in ~SULTS), and from over- night cultures of the recurrent tumor from case BN. The methods of preparation were as previously described [9, 10]. The chromosomes were banded using a mod- ified Seabright technique [11] and karyotyped [12].
RESULTS
No single chromosomal abnormality was found in every cell in all five of the PNET analyzed. As illustrated in Figures 1-5 and Table 1, three of the five tumors con- tained additional lq material [as extra chromosomes #1, extra del(lp) markers, or iso(lq)], and two of the five contained monosomy 13. Each of these abnormalities found in greater than 20% of cells in individual tumors included trisomy 17, iso(17q), extra chromosomes #3, #8, and #14, missing #5, #9, #10, #11, and #18, and structural abnormalities of chromosomes #5, #6, #7, and #12.
In one case (SK-PN-DW), karyotypes were prepared from a reconstituted frozen ampule of the first passage of the tumor (Fig. 2A) and from the permanent line established from the tumor (Fig. 2B). Individual cells of the original tumor had chromosome numbers between 47 and 96, with a mode of 74. There were two cop- ies of chromosome #13 in each hyperdiploid cell, two fewer than the number ex- pected for a tetraploid cell. No consistent deletions or translocations could be iden- tified. Approximately 0-5 double minutes (DM) were noted per cell. In the permanent cell line, after at least 2 years of continuous culture, the chromosome number per cell was reduced to between 37 and 43 per cell, with a mode of 40 to 41. Monosomy 13 was found in 100% of cells; iso(17q) and monosomy 9, 10, 11, and 18 were found in greater than 20% of cells analyzed. In a large proportion of the cells of the permanent line, the DM per cell were too numerous to count (Fig. 2C).
DISCUSSION
Our analysis of karyotypes from primary tumor cells and tumor cell lines from five PNET cases, suggests that one may be able to distinguish between PNET and the embryologically-related neuroblastoma on cytogenetic grounds. None of the five PNET studied contained the marker chromosome abnormality identified in more than 66% of neuroblastomas, namely, deletions of lp (in the absence of trisomy lq) [7].
Cytogenetic analysis also makes it possible to separate the more primitive tumors of neuroectodermal origin that develop in peripheral sites, into two groups: those with a balanced t(11;22) for which we suggest the term "peripheral neuroepithe- lioma" should be reserved [8], and the remainder, with different karyotypic changes (the significance of which is discussed below), which we would designate as PNET. (Whether or not this classification will have additional clinical relevance--e.g., in terms of prognosis, patterns of drug responsiveness, etc.--will become clear as both patient groups are followed over time.)
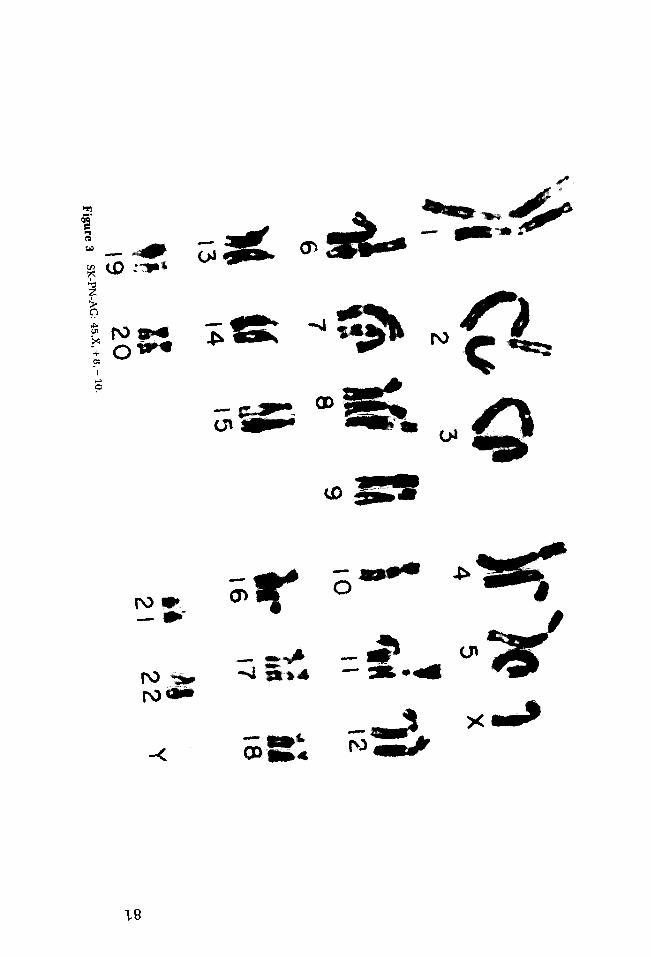
r ~ j
~b
c~
Q~
QU
f ~
~ . I D ~
O~
B
m ~
w ~ ° . I I
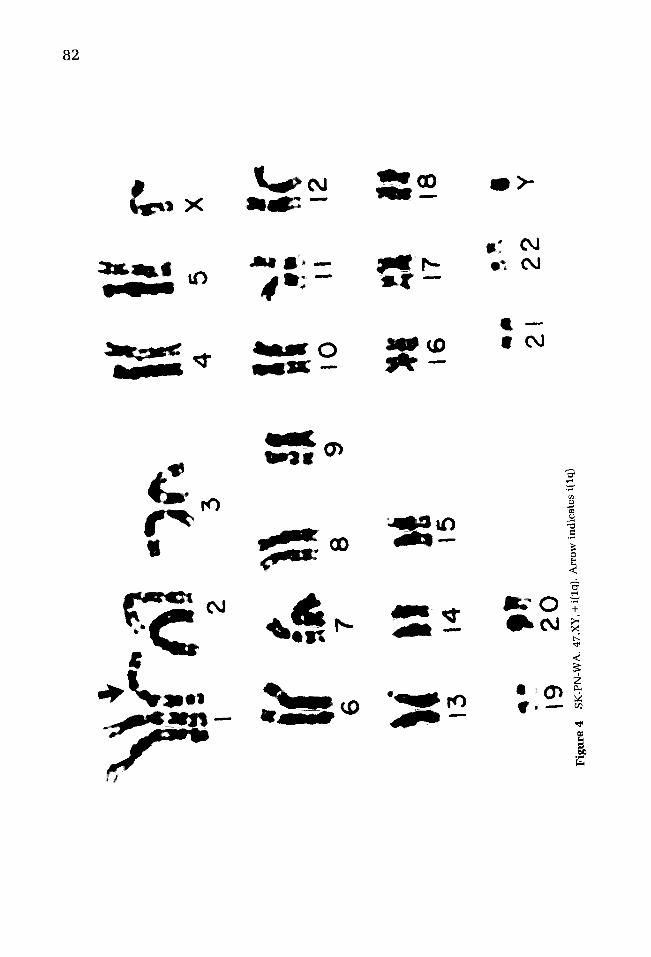
82
~M~'LI X I I 1 ~ - " S l W - -
4
amJO NS[--
a~ 0~ t~ oJ
11 1~'-" QO
qr.--- ~
<

83
~ X
L~r)
t
~ C )
a')
~jk~D O0
(,D
d i d LC)
d l B r ? )
• >-
o , ( ~ l t l ( ~ l
c~J
4bo (~j
e ~
& --F
0
+
v ~
+
. o
÷
z
CJ~
ol,,~

84 v . R . Potluri et al.
Table 1 Chromosome abnormal i t ies in pr imi t ive neuroectodermal tumors
Chromosome abnormalities
PNET Age/Sex Rx d Mode (Range) A (100% cells) B (>20% cells)
SK-PN-LI 3/M CT 46 (45-47) + 1, - 13 SK-PN-DW a 17/M CT 74 (47-96) - 13
SK-PN-DW b 46, 41 (37-43) - 1 3
SK-PN-AG 13/F CT 48 (8 cells) + 8, - 10 65 {1 cell)
SK-PN-WA 19/M No 47 + iso(lq) SK-PN-BN 24C/F CT/RT 47 + lq
+ 5, + 8, i(5q) -9 , -10 , -11, -17,
- 19, DMs - 9, - 10, - 11, i(17q),
-18, DM
t(7;6), 12q+, +14, + marker
aOriginal tumor. bPermanent cell line. COriginal tumor discovered at age 19; recurrence, reoperation at age 24. ~Treatment: CT, chemotherapy; RT, radiotherapy.
Two of the chromosome abnormal i t ies ident if ied in the PNET, extra l q mater ial and DM, however, have been repor ted in neuroblastomas, as well as in many other cancers in humans [7, 13-15]. The lack of specif ici ty of these anomalies for single tumor types suggests that such changes are not required for tumorigenesis . Instead, they are l ikely to p lay a secondary role in tumor development , p resumably by con- ferring a selective growth advantage on the cells in which they are carried [16].
Consistent With this hypothes is was a previous report of neuroblas toma cell karyotypes obtained ser ial ly from the same pat ient [17]. When compared with the original pr imary tumor, karyotypes from mul t ip le metastases sampled 11 months later demonst ra ted a reduct ion in modal chromosome number per cell and t r isomy for a lq marker chromosome.
Case SK-PN-DW is l ikely to represent an in vitro example of the same phenom- enon: When compared with the ini t ial tumor sample, the cont inuous ly growing cell l ine had a sharper and lower modal chromosome number per cell and also con- ta ined more DM per cell.
In all instances s tud ied to date, DM have been shown to represent sites of gene amplif icat ion, containing mul t ip le copies of one or a small number of genes [15]. In most (if not all) pa thologica l ly confirmed neuroblas tomas in which gene amplif ica- t ion has been demonstra ted, a par t icular oncogene, N-myc (mapped to 2p23-24 [18]) has been inc luded in the amplif icat ion unit [19, 20]. In SK-PN-DW, however, an- other member of the myc gene family (c-myc (mapped to 8q24) [21]) is present in mul t ip le copies (H. Rovigatti, personal communicat ion) .
The finding of monosomy 13 in two of the five PNET cases is interest ing in light of the associat ion between abnormal i t ies of chromosome #13 and another neuroec- todermal tumor, ret inoblastoma. Ret inoblastoma has been repor ted in certain pa- t ients who carry dele t ions of 13q14 in all somatic t issues [221. A number of labo- ratories also have repor ted the loss of 13q mater ial (either as delet ions of 13q or monosomy 13) in karyotypes from ret inoblas toma cells from ind iv idua ls whose const i tu t ional karyotypes were normal [23-25]. The actual fraction of ret inoblasto- mas containing monosomy 13 or del(13q) is low: fewer than 20% of all publ i shed cases. However, the combinat ion of cytogenetic data and studies of restr ict ion frag-

Chromosomal Analysis of PNET 8 5
ment length polymorphisms of DNA sequences mapped along 13q has led to the conclusion that the homozygous loss of genetic formation from 13q14 is necessary for tumorigenesis in all cases of retinoblastoma [26].
Monosomy 13 and structural abnormalit ies of chromosome 13q also have been identified in isolated examples of many tumor types [14, 27]. This finding in PNET, is intriguing, however, because it raises the possibility that this segment contains one or more genes that play a role in the differentiation of more than one neuroec- todermal derivative: genes whose loss may affect the proliferative capacity, and contribute to the transformation, of cells other than retinoblasts.
Supported by USPHS Grant CA 36122 (FG).
REFERENCES
1. Weston JA (1970): The migration and differentiation of the neural crest cells. In: Advances in Morphogenesis, Vol. 8, TJ King, ed. Publisher, NY, pp. 41-114.
2. Bolande RP (1974): The neurocristopathies. A unifying concept disease arising in neural crest maldevelopment. Hum Pathol 5:409--429.
3. Seemayer TA, Telmo WL, Bolande RP, Wiglesworth FW (1975): Peripheral neuroectoder- mal tumors. Persp Pediat Pathol 2:151-172.
4. Becker LE, Hinton D (1983): Primitive neuroectodermal tumors of the central nervous system. Hum Pathol 14:538-550.
5. Ashwal S, Hinshaw DB, Bedros A. (1984): CNS Primitive neuroectodermal tumors of childhood. Med Pediat Oncol 12:180-188.
6. Kinnear Wilson LM, Draper GJ (1974): Neuroblastoma: Its natural history and prognosis: A study of 487 Cases. Br Med J 3:3Ol-307.
7. Gilbert F, Feder M, Balaban G, Brangman D, Lurie DK, Podolsky R, Rinaldt V, Vinikoor N, Weisband J (1984): Human neuroblastomas and abnormalities of chromosomes 1 and 17. Cancer Res 44:5444-5449.
8. Whang-Peng J, Triche TJ, Knutsen T, Miser J, Douglass E, Israel MA (1984): Chromosome translocation in peripheral neuroepithelioma. N Engl J Med 311:584-585.
9. Gilbert F, Balaban G, Moorhead P, Bianchi D, and Schlesinger H (1982): Abnormalities of chromosome lp in human neuroblastoma tumors and cell lines. Cancer Genet Cytogenet 7:33-42.
10. Potluri VR, Gilbert F (1985): A cytogenetic study of embryonal rhabdomyosarcoma. Cancer Genet Cytogenet 14:169-173.
11. Seabright M (1971): A rapid banding technique for human chromosomes. Lancet ii: 971- 972.
12. International System for Human Cytogenetic Nomenclature 1978. Birth Defects: Original Article Series. XIV:313-404.
13. Rowley JD (1977): Mapping of human chromosomal regions related to neoplasia: Evidence from chromosomes 1 and 17. Proc Natl Acad Sci USA 74:5729-5733.
14. Sandberg AA (1980): The Chromosomes in Human Cancer and Leukemia. Elsevier North- Holland, NY.
15. Cowell JK (1982): Double minutes and homogenously staining regions: Gene amplification in mammalian cells. Ann Rev Genet 16:21-59.
16. Gilbert F (1983) Chromosomes, genes and cancer: A classification of chromosome changes in cancer. J Natl Cancer Inst 71:1107-1114.
17. Feder M, Gilbert F (1983): Clonal evolution in a human neuroblastoma. J Natl Cancer Inst 70:1051-1056.
18. Schwab M, Varmus HE, Bishop JM, Grzeschik K-H, Naylor SL, Sakaguchi AY, Brodeur G, Trent J (1984): Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature fLond) 308:288-291.

86 v . R . Potluri et al.
19. Kohl NE, Kanda N, Schreck R, Bruns G, Latt SA, Gilbert F, Aft F (1983): Transposition and amplification of oncogene related sequences in human neuroblastomas. Cell 35:359- 367.
20. Schwab M, Alitalo K, Klemphauer KH, Varmus HE, Bishop JM, Gilbert F, Brodeur G, Goldstein M, Trent J (1983): Amplified DNA domain with limited homology to the rnyc cellular oncogene is shared by human neuroblastoma cell lines and a human neuroblas- toma tumor. Nature (Lond) 305:245-248.
21. Neel BG, Jhanwar SC, Chaganti RSK, Hayward WS (1982): Two human c-onc genes are located on the long arm of chromosome 8. Proc Natl Acad Sci USA 79:7842-7846.
22. Yunis JJ, Ramsay N (1978). Retinoblastoma and subband deletion of chromosome 13. Am J Dis Child 132:161-163.
23. Balaban G, Gilbert F, Nichols W, Meadows A, Shields J (1982): Abnormalities of chromo- some 13 in retinoblastomas from individuals with normal constitutional karyotypes. Can- cer Genet Cytogenet 6:213-221.
24. Benedict WF, Banerjee A, Mark C, Murphree AL {1983): Nonrandom chromosomal changes in untreated retinoblastomas. Cancer Genet Cytogenet 10:311-333.
25. Workman ML, Soukup SW (1984): Chromosome features of two retinoblastomas. Cancer Genet Cytogenet 12:365-370.
26. Cavenee WK, Dryja TP, Phillips RA, Benedict WF, Godbout R, Gallie BL, Murphree AL, Strong LC, White RL (1983): Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature (Lond) 305:779-784.
27. Mitelman F (1983): Catalogue of Chromosome Aberrations in Cancer. Cytogenet Cell Genet 36:1-515.





























