Embed Size (px)
Citation preview

TRATAMIENTO FARMACOLÓGICO DEL
ESTATUS EPILÉPTICO
Lizeth K. Barrera ClavijoResidente de Primer año
Farmacología Clínica
Departamento de Farmacología Clínica y Terapéutica
Universidad de La Sabana


Contexto Epidemiológico
Incidencia en EEUU: 50-200 mil casos por año (más frecuente durante la infancia).
Tasas de prevalencia en América Latina: con estudios publicados entre 1974 y 1998 mostró una amplia variabilidad 5,8 por 1 000 habitantes, hasta un máximo de 57 por 1 000 habitantes
Aproximadamente 1-5% de los epilépticos y 16% de los niños epilépticos presentarán status epiléptico en algún momento de su vida.

Contexto Epidemiológico
• SE Convulsivo:– Mortalidad:
• Alta hospitalaria: 9-21%
• A 30 días: 19-27%
• A 90 días 19%
– Morbilidad: • Secuelas cognitivas o neurológicas severas: 11-16%
• Deterioro estado funcional: 23-26%
• SE No convulsivo:– Mortalidad:
• Alta hospitalaria: 23-61%
• A 90 días: 39%

Definición
• Guía: 5 min o más de actividad convulsivaclínica continua y/o electrográfica óactividad convulsiva recurrente sinrecuperación entre convulsiones.

Definición y ClasificaciónSE Convulsivo
• Convulsiones asociadas amovimientos rítmicos de lasextremidades
• Hallazgos característicos de GCSE:
– Movimientos tónico-clónicos generalizados de las extremidades
– Alteración estado de consciencia
– Puede haber déficit neurológico focal en periodo post ictal
SE No Convulsivo
• Actividad convulsiva detectada en EEGsin hallazgos clínicos de GCSE
• Espectro semiológico no convulsivo:– Síntomas (+): agitación, agresión,
automatismos, llanto, delirium, risa excesiva,náuseas, vómito, nistagmos, psicosis.
– Síntomas (-): anorexia, afasia, mutismo,amnesia, catatonia, coma, confusión.
• Tiene 2 fenotipos:– Paciente confundido que se presenta a
urgencias con pronóstico relativamente buenoó con síndrome epiléptico crónico.
– Paciente con estado mental gravementeafectado, con o sin movimientos motores sutiles(contracciones musculares rítmicas odesviación ocular

Causas
• Procesos agudos:
– Alteraciones metabólicas: electrolitos, hipoglicemia, falla renal
– Infecciones SNC
– ACV
– Trauma
– Medicamentos: toxicidad, retiro de opioides, BZD, barbitúricos, noadherencia a tratamiento AC
– Hipoxia, paro cardiaco, encefalopatía hipertensiva
• Procesos crónicos:
– Epilepsia
– Abuso de OH: Intoxicación por etanol o síndrome de abstinencia
– Tumores SNC

Fisiopatología
Desequilibrio en sistema GABAérgico
Aumento de descargas neurales endógenas mediadas por Ca y Na
Desequilibrio en mecanismos excitatorios sinápticos mediados por Glutamato y Acetilcolina

Fisiopatología
Disminución de péptidos inhibitorios o anticonvulsivos endógenos: galanina
Aumento de péptidos pro convulsivantes: Sustancia P, neuroquininas, taquininas




Diagnóstico

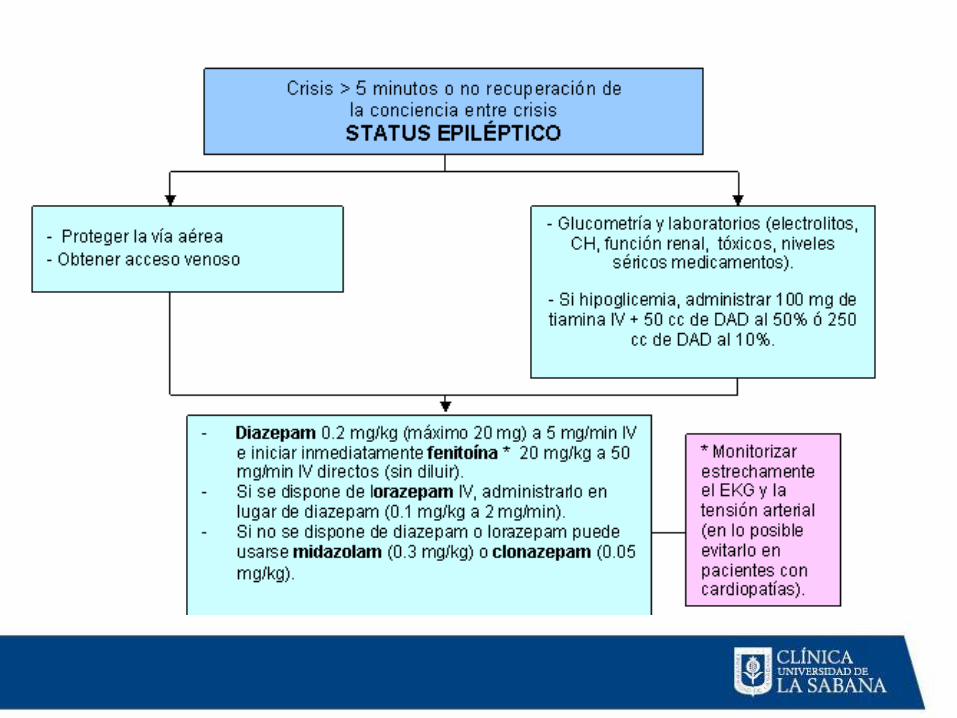
Tratamiento
Objetivo primario: detener actividad convulsiva
clínica y electrográfica
Control definitivo de SE debe ser establecido en los
primeros 60 minutos de su comienzo
Estrategias:
A: Valoración y manejo de vía aérea
B: Garantizar respiración
C: Circulación acceso IV
Tratamiento farmacológico
Detectar causa de SE

Tratamiento

Tratamiento

• Terapia Inicial Emergente
– Benzodiacepinas
– Vía de administración:• IV: Lorazepam
• IM: Midazolam
• Rectal: Diazepam
– Estudios controlados evaluaron uso de Lorazepam vsDiazepam, Fenobarbital, Fenitoina y Midazolam IM
– Midazolam IM: Igual de efectivo a Lorazepam
Tratamiento

• Terapia de control Urgente
– El tratamiento con BZD de control urgente después de la administración de BDZ decorta acción se requiere en todos los pacientes con SE, a menos que la causainmediata del SE se conozca y sea corregida.
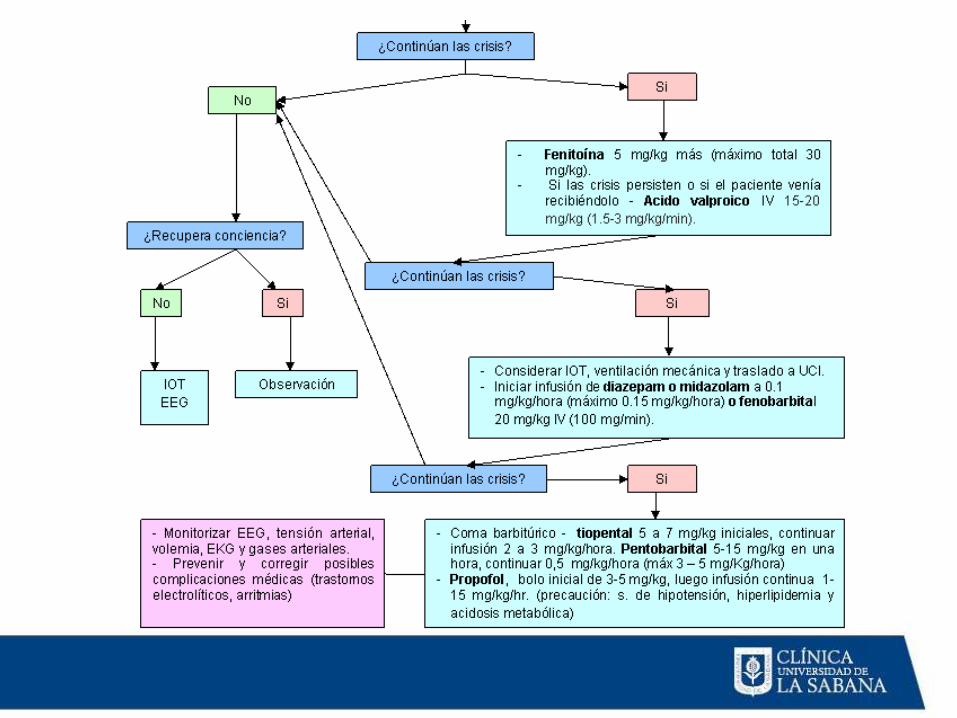
Tratamiento
Pacientes que respondieron aterapia inicial emergente y quetienen resolución del SE
Meta: lograr niveles terapéuticos de medicamentos anticonvulsivantes (MAC) y dar dosis de mantenimiento
Pacientes que NO respondieron aterapia inicial emergente
Meta: detener SE Fenitoína,valproato de Na, fenobarbital,infusión continua de midazolam
Pacientes con epilepsia conocida:Bolo de su MAC

Tratamiento

• Presentaciones incluidas en el POS:– Tabletas 1 mg
– Tabletas 2 mg
• Ampollas: 4 mg/ml
• Mecanismo de Acción:– BZD de corta acción
– Se une al sitio alostérico sobre los receptores GABA-A potencia actividad de GABA genera apertura de canales de Cl hiperpolarización
• Dosis: – 0.1 mg/kg IV, se puede repetir después de 5–10 min
• Categoría en Embarazo: DC15H10N2Cl2O2
Pka: 11.5
LORAZEPAM
(Ativan)

LORAZEPAM
(Ativan)
• Absorción:
– BD: 90%
– Comienzo de acción: 1-3 min (IV)
– Pico plasmático: VO: 2 horas; IM: < 3 horas
• Distribución:
– Unión a proteínas: 85-93%
– Vd: 1.3 L/Kg en adultos
• Metabolismo
– Conjugación con ácido glucorónido
• Excreción:
– Vida ½: 14 horas en adultos; IRC terminal: 18 horas
– Orina 88% (metabolitos inactivos); heces 7%.
FARMACOCINÉTICA:

• Interacciones mayores:
– Incremento acción sedante: Dexmedetomidina, BZD, etanol,fenobarbital, propofol
• Interacciones menores:– Omeprazol y Esomeprazol: aumenta niveles de Lorazepam por
disminución de su metabolismo (CYP2C19)
– Acetaminofén: Lorazepam disminuye niveles de acetaminofénporque aumenta su metabolismo (CYP2E1)
LORAZEPAM
(Ativan)

• Reacciones Adversas:
– Sedación
– Mareo
– Depresión respiratoria Propilenglicol
– Hipotensión
– Amnesia proceso de consolidación de memoria
– Desorientación
– Confusión
– Ataxia
– Astenia
LORAZEPAM
(Ativan)
GABA

MIDAZOLAM
(Dormicum)
C18H13CLFN3
Pka: 6.15
• Presentaciones incluidas en el POS:– Ampollas 1 mg/ml
– Ampollas 5 mg/5 ml
– Tabletas 7,5 mg
• Mecanismo de Acción:– BZD de corta acción
– Se une al sitio alostérico sobre los receptores GABA-A potencia actividad de GABA genera apertura de canales de Cl hiperpolarización
• Dosis: 0.2 mg/kg IM
• Categoría en Embarazo: C

• Absorción:
– BD: VO 40-50%; IM > 90%
– Comienzo de acción: 3-5 min (IV); 15-20 min (VO, IM)
– Pico plasmático: IM 30 min
• Distribución:
– Unión a proteínas: 97%
– Vd: 1.0 - 3.1 L/Kg en adultos
• Metabolismo
– Hepático: CYP3A4
– Metabolitos: 1-hidroximetilmidazolam
• Excreción:
– Vida ½: 2-6 horas
– Aclaramiento: 0.25-0.54 L/hr/kg
– Orina 90%; heces 2%.
MIDAZOLAM
(Dormicum)FARMACOCINÉTICA:

• Interacciones:
– Carbamazepina, Rifampicina, dexametasona, metilprednisolona:disminuye efecto de midazolam porque interactúa conmetabolismo de CYP3A4
– Claritromicina: aumenta efectos de midazolam por interacción demetabolismo de CYP3A4
– Incremento acción sedante: Dexmedetomidina, BZD, etanol,fenobarbital, propofol
MIDAZOLAM
(Dormicum)


• Reacciones Adversas:
– Bradipnea (23%)
– Apnea (15%)
– Dolor en sitio de aplicación (4-5%)
– Sedación
– Desorientación
– Confusión
GABA
MIDAZOLAM
(Dormicum)

DIAZEPAM
(Valium)• Presentaciones incluidas en el POS:
– Ampollas 5 mg/ml
– Tabletas 10 mg
– Cápsulas 5 y 10 mg
• Mecanismo de Acción:– BZD de acción prolongada
– Se une al sitio alostérico sobre los receptores GABA-A potencia actividad de GABA genera apertura de canales de Cl hiperpolarización
• Dosis: 0.15 mg/kg IV, máximo 10 mg/dosis, se puede
repetir después de 5 min.
• Categoría en Embarazo: D
C16H13CLN2O
PKb: 3.3
BD

• Absorción:
– BD: 90% Rectal
– Pico plasmático: VO: 30-90 min; Rectal: 5-90 min
• Distribución:
– Unión a proteínas: 98%
– Vd: 0.8-1 L/kg en adultos
• Metabolismo
– Hepático: CYP3A4, CYP2C19
– Metabolitos: N-desmetildiazepam, 3-hidroxdiazepam, oxazepam
• Excreción:
– Vida ½: 20-70 horas
– Aclaramiento: 20-30 ml/min
– Orina
FARMACOCINÉTICA:
DIAZEPAM
(Valium)

• Interacciones:
– Claritromicina: aumenta niveles de diazepamCYP3A4
– Incremento acción sedante: Dexmedetomidina, BZD, etanol,fenobarbital, propofol
– Carbamazepina: disminuye niveles de diazepamCYP3A4
DIAZEPAM
(Valium)

• Reacciones Adversas:
– Euforia (3%)
– Ataxia (3%)
– Incoordinación (3%)
– Somnolencia (1%)
– Diarrea (4%)
– Hipotensión
– Depresión respiratoria
DIAZEPAM
(Valium)

FENITOÍNA
(Epamin)
C15H12N2O2
Pka: 8.3
• Presentaciones incluidas en el POS:– Ampollas 50 mg/ml
– Tabletas 100 mg
– Suspensión 125/5ml
• Mecanismo de Acción:– Bloquea canales de Na en las neuronas de la corteza cerebral motora
efecto sobre las corrientes de calcio. Inhibe la liberación de NT’sde AcH y Glutamato.
• Dosis: 20 mg/kg IV, se puede dar adicional 5–10 mg/kg
– Mantenimiento 100 mg IV/VO cada 6-8 horas
– Rango terapéutico: 10-20 mcg/L (total) o 1-2 mcg/L(medicamento libre)
• Categoría en Embarazo: D

• Absorción:
– BD: varía según manufactura
– Comienzo de acción: 0.5 – 1 Hora (IV); 2-24 horas (VO con dosis de carga)
– Pico plasmático: 1.5 – 3 horas
• Distribución:
– Unión a proteínas: 95%
– Vd: 0.6-0.7 L/kg en adultos
• Metabolismo
– Hepático Citocromo P450 CYP2C9
– Inductor de CYP3A4
• Excreción:
– Vida ½: 10-15 Horas IV; 22 horas en adultos VO
– Orina
FARMACOCINÉTICA:
FENITOÍNA
(Epamin)

INTERACCIONES:
Es inductor de casi todas las isoenzimas microsomales del citocromo
hepático P450, en particular las CYP3A4, CYP2D6, CYP1A2, CYP2C9
y CYP2C19 acelera metabolismo de los fármacos
Amiodarona, atorvastatina, Clopidogrel CYP3A4
Carvedilol, Metoprolol CYP2D6
Acetaminofén CYP1A2
Losartán, Valsartán, ibuprofeno CYP2C9
Warfarina, Omeprazol CYP 2C19
RAM’S: Fatiga, ataxia, cefalea, ansiedad, vértigo, hipotensión, arritmias,
toxicidad local en sitio de infusión (extravasación?)
FENITOÍNA
(Epamin)
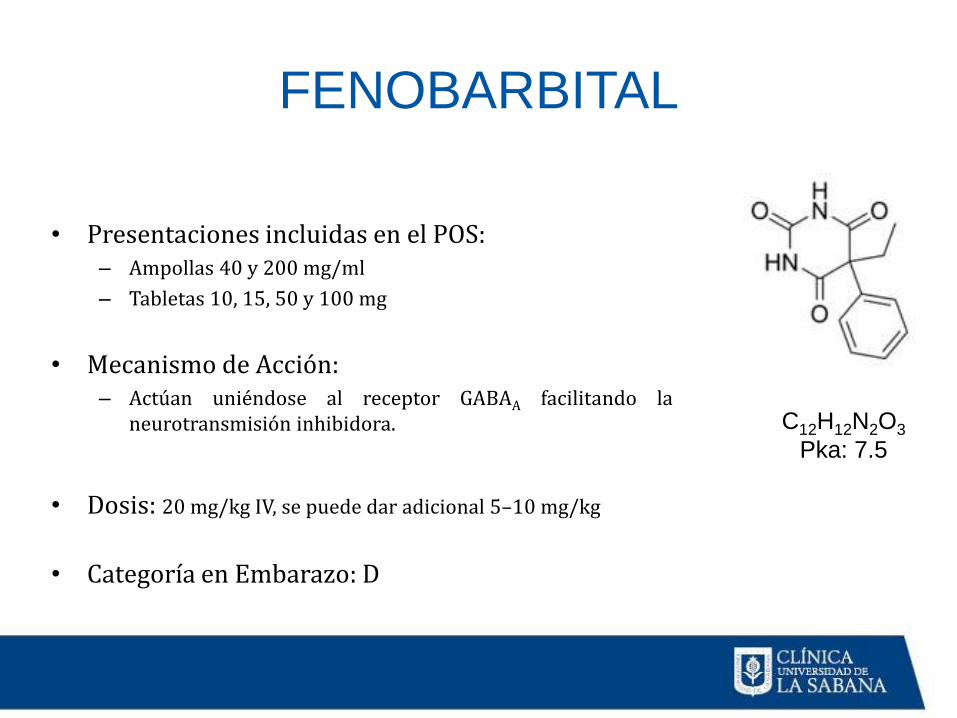
FENOBARBITAL
C12H12N2O3
Pka: 7.5
• Presentaciones incluidas en el POS:– Ampollas 40 y 200 mg/ml
– Tabletas 10, 15, 50 y 100 mg
• Mecanismo de Acción:– Actúan uniéndose al receptor GABAA facilitando la
neurotransmisión inhibidora.
• Dosis: 20 mg/kg IV, se puede dar adicional 5–10 mg/kg
• Categoría en Embarazo: D

• Absorción:
– BD: 70-90%
– Comienzo de acción: 5 min IV
– Pico plasmático: 8 horas VO
– Rango terapéutico por VO: 3-4 semanas 10-40 mcg/ml
• Distribución:
– Unión a proteínas: 20-45%
– Vd: 0.5 L/kg en adultos
• Metabolismo
– Hepático Citocromo P450 induce CYP1A2, CYP2C6, CYP2C19, CYP2C9/10, CYP3A4
• Excreción:
– Vida ½: 50-140 horas
– Orina
FARMACOCINÉTICA:
FENOBARBITAL

• Interacciones:
– Warfarina: CYP3A4 disminuye actividad anticoagulante
– Hidrocortosona-Dexametasona: aumenta metabolismo de corticoides por inducciónde CYP2D6, CYP3A4
– Doxiciclina: Fenobarbital disminuye la vida media, ya que induce CYP3A4
– BZD, etanol, antihistamínicos H1 efectos aditivos depresivos
• Reacciones Adversas:
– Depresión respiratoria IV
– Ataxia
– Mareo
– Disartria
– Cefalea
FENOBARBITAL

ÁCIDO VALPROICO
(Depakene)
• Presentaciones incluidas en el POS:– Tabletas 200 y 400 mg
– Jarabe 250 mg/5 ml
• Mecanismo de Acción:– Aumenta niveles de GABA en receptores postsinápticos
– Inhibe canales de Na y Ca.
• Dosis: : 20–40 mg/kg IV, puede darse adicional de 20 mg/kg
• Categoría en Embarazo: D
C8H16O2
Pka: 4.8

• Absorción:
– BD: 81-89%
– Pico plasmático: 2 horas VO
• Distribución:
– Unión a proteínas: 80-90%
– Vd: 0,15 a 0,29 L/kg
• Metabolismo
– Hepático: oxidación mitocondrial
– CYP2C9, CYP2A6, CYP2B6
• Excreción:
– Vida ½: 9-16 horas
– Orina 30-50%
FARMACOCINÉTICA:
ÁCIDO VALPROICO
(Depakene)

• Interacciones:
– Warfarina: CYP2C9 disminuye actividad anticoagulante
– Depresión SNC: Dexmedetomidina, BZD, etanol, fenobarbital
– Fenitoína: compiten por la unión a proteínastoxicidad por fenitoína
– Disminuye metabolismo de AINES inhibición de CYP2C9
– Disminuye metabolismo de ARA II inhibición de CYP2C9
• Reacciones Adversas:
– Náuseas (31%)
– Cefalea (< 31%)
– Trombocitopenia (26-30%) por supresión en médula ósea, efecto tóxico en precursores hematopoyéticos
– Alopecia (24%) ocurre porque el 85% de los pelos está en Fase Anágeno (índice mitótico alto y más sensible a noxas)
ÁCIDO VALPROICO
(Depakene)