Embed Size (px)
Citation preview

Das erste Mikroskop wurde bereits 1595 von Hansund Zacharias Hansen entwickelt. Jedoch werden
eher Antonie van Leeuwenhoek, der für die Fertigungseiner Mikroskope berühmt war, und Robert Hooke mitdieser bahnbrechenden Erfindung in Verbindung ge-bracht (Abbildung 2a). Hooke veröffentlichte 1665 seinrichtungsweisendes Werk Micrographia [10], welchesdetaillierte Zeichnungen von pflanzlichen und tieri-schen Präparaten enthielt. Hier fällt auch zum erstenMal der Begriff der Zelle, mit dem Hooke die poröseStruktur von Kork beschrieb (Abbildung 2b). DieseStrukturen sind selbstverständlich keine Zellen nachheutiger Definition, dennoch geht der Begriff der Zelledarauf zurück. Mit diesen einfachen Mikroskopen ge-lang es van Leeuwenhoek bereits im 17. Jahrhundert alserster Bakterien, Protozoen und Spermien zu beobach-ten und zu beschreiben. Durch permanente Verbesse-rung in der Fertigung der Mikroskope, technologischeWeiterentwicklungen wie die der Phasenkontrastmi-kroskopie, das Prinzip des � Differentialinterferenzkon-trastes und die Entdeckung geeigneter Färbelösungen,wurde es schließlich möglich, immer kleiner werdendeObjekte und Strukturen auch innerhalb von Zellen zuuntersuchen. Allerdings sind auch moderne Mikro-skope in ihrer Auflösung begrenzt.
Ernst Abbe und die AuflösungsgrenzeDer deutsche Physiker Ernst Abbe publizierte bereits1873 die Theorie der Auflösungsgrenze und zeigte mitden Formeln Auflösungx,y = λ / (2n × sin α) und Auflö-sungz = 2 λ / (n × sin α)2 die Grenzen konventionellerLicht- oder Fluoreszenzmikroskopie auf [1]. Hierbeisteht λ für die Wellenlänge des Lichts, n für die Brech-zahl des Mediums zwischen der Probe und dem Objek-tiv und a für den halben Öffnungswinkel des Objektivs.Ohne die Formeln im Einzelnen abzuleiten oder zu dis-kutieren, sollen sie nun angewendet werden, um dieAuflösungsgrenze zu berechnen. Geht man von kurz-welligem UV-nahem oder blauem Licht mit einer Wel-lenlänge von circa 380 nm beziehungsweise 400 nmaus und setzt diese in die Gleichungen ein, so wird klar,dass eine Auflösung von maximal 200 nm in der xy-Achse und 500 nm in der z-Achse möglich ist (Abbil-
Die Auflösung konventioneller Licht- undFluoreszenzmikroskope ist durch die Gesetzeder Physik limitiert. Genauer gesagt ist dasLicht hierbei der limitierende Faktor. Mit sogenannten Super-Resolution- Mikroskopie-techniken wie der Photoactivated Localiza-tion Microscopy (PALM) ist es auch mittelsFluoreszenzmikroskopie möglich, eine bisauf wenige Nanometer genaue Auflösung zuerzielen.
Photoactivated Localization Microscopy (PALM)
Mikroskopie jenseits derAuflösungsgrenzeRAPHAEL MANCK | REINHARD FISCHER
DOI:10.1002/ biuz.201210485
2 | Biol. Unserer Zeit | 4/2012 (42) © 2012 Wiley-VCH Verlag GmbH & Co. KGaA, WeinheimOnline-Ausgabe unter:wileyonlinelibrary.com
Die mit einem grünen Pfeil markierten Begriffewerden im Glossarauf Seite n erklärt.
A B B . 1 PALM-Aufnahme einer Escherichia coli-Zelle mit fluoreszent markier-tem Chemotaxis Rezeptor. Jeder Bildpunkt entspricht einem einzelnen Protein. Bild: http://newscenter.lbl.gov/feature-stories/2009/07/06/spontaneous-assembly/

S U P E R R E S O L U T I O N - M I K R O S K O P I E | I M FO KU S
dung 3a). Im Klartext bedeutet das, dass zwei Objekte,die näher als 200 nm in der xy-Achse und 500 nm in derz-Achse beieinander liegen, für den Betrachter als eineinziger verwaschener Bildpunkt erscheinen (Abbil-dung 3b). Dieses Phänomen ist generell unter dem Aus-druck point spread function (PSF) bekannt (Abbil-dung 3a). Eine solche Auflösung nahe der theoretischenGrenze war erst mit moderneren Mikroskopen mög-lich. Jedoch unterliegen selbst moderne Geräte diesenphysikalischen Beschränkungen. Da viele intrazelluläreKomponenten wie das aus Mikrotubuli, Aktin und In-termediärfilamenten bestehende Cytoskelett oder se-kretorische Vesikel erheblich kleiner sind oder zu dichtzusammen liegen, um sie mit konventioneller Mikro-skopie voneinander getrennt auflösen zu können (Ab-bildung 3c), sind Wissenschaftler schon lange bestrebt,die Auflösungsgrenze zu durchbrechen.
Eine Methode, um die Auflösungsgrenze im Ver-gleich zur Lichtmikroskopie zu verbessern, ist in derElektronenmikroskopie (EM) realisiert. Aus den Glei-chungen von Abbe wird klar, dass die beschränkendeGröße bei lichtoptischen Mikroskopen die Wellenlängedes eingesetzten Lichts ist. Der denkbar einfache Ansatzin der Elektronenmikroskopie ist es, die eingesetzteWellenlänge zu verringern, indem ein Elektronenstrahlanstelle eines Lichtstrahls verwendet wird. Trotz der un-glaublich hohen Auflösung, die mit Elektronenmikro-skopen erreicht wird, sind diese vor allem in der Zell-biologie nur begrenzt einsetzbar. In der Rasterelektro-nenmikroskopie werden die Präparate fixiert und mit
Gold bedampft, bevor sie unter Hochvakuum im Elek-tronenmikroskop betrachtet werden können. In derDurchlichtelektronenmikroskopie werden die Objekteebenfalls fixiert, entwässert und in ein Polymermaterialeingebettet. Dieses wird anschließend zu Dünnschnit-ten verarbeitet, die im Elektronenmikroskop analysiertwerden können. In beiden Anwendung werden alsonur abgetötete Objekte beobachtet und dynamischeProzesse können nur diskontinuierlich untersucht wer-
© 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.biuz.de 4/2012 (42) | Biol. Unserer Zeit | 3
Tubus
Objektiv
Öllampe Wasser-kolben
Proben-halter
Fokussier-schraube
Hooke Mikroskop(circa 1670)
Okulara) b)
a) Schematischer Aufbau eines von Robert Hooke verwendeten Mikro-skops. Bild: modifiziert nach http://micro.magnet.fsu.edu/primer/museum/hooke.htmlb) Originalzeichnung der porösen Struktur („Zellen“) von Korkpräparaten,angefertigt von Robert Hooke [10].
ABB. 2 D I E A N F Ä N G E D E R M I K ROS KO PI E
Pixel Pixel
PixelPixel
Point-SpreadFunction
CCDDisplay Gaussian
Fit
10 μm 1 μm 100 nm 10 nm 1 nm 1 ÅDiffraction limit
a)
b)
c)
a) Links: Lichtpunkt in der konventionellen Mikrosko-pie. Aufgrund der von E. Abbe postulierten Auflösungs-grenze hat ein Lichtpunkt in der konventionellen Mikro-skopie eine ellipsoide Ausdehnung von 250 nm in derxy-Achse und 500 nm in der z-Achse. Rechts: Eine Gaus-sian-Oberflächenanpassung des ellipsoiden Signals er-laubt die Lokalisierung dessen Ursprungs. Bild: modifi-ziert nach http://zeiss-campus.magnet.fsu.edu/articles/superresolution/palm/introduction.htmlb) Links: Sind zwei Lichtpunkte weiter als 250 nm in xy-Achse und 500 nm in z-Achse voneinander entfernt, sokönnen sie als zwei getrennte Signale detektiert wer-den. Rechts: Liegen die beiden Lichtpunkte näher zu-sammen, überlagern sich die Signale und können nurals ein Signal detektiert werden [11].c) Schematische Darstellung der Größen verschiedenerbiologischer Strukturen mit eingezeichneter Auflö-sungsgrenze. Von links nach rechts: eine eukaryotischeSäugetierzelle, eine Bakterienzelle, ein Mitochondrium,ein Influenzavirus, ein Ribosom, ein grün fluoreszentesProtein (GFP) und Thymin [modifiziert nach 12].
ABB. 3 D I E AU F L Ö S U N G S G R E N Z E – E I N E G R E N Z E D E R WA H R N E H M U N G

den. Zudem können die Proben bei der Präparation be-schädigt werden, was zu � Artefakten führen kann.
Eine weitere Methode zur Überwindung der Auflö-sungsgrenze ist die Rasterkraftmikroskopie (AtomicForce Microscopy oder kurz AFM). Bei der AFM wirdeine nanoskopisch kleine Messnadel – ein Cantilever –welche an der Spitze aus nur noch einem einzigenAtom besteht, in einem definierten Raster über das Prä-parat gezogen. So können Höhenunterschiede im Prä-parat visualisiert werden und eine Auflösung von bis zu5 nm erreicht werden. Durch die physische Art der Bild-aufnahme können zwar auch lebende Präparate ver-wendet werden, jedoch ist es nicht möglich, intrazellu-läre Vorgänge zu beobachten.
Somit ist die lichtoptische Mikroskopie immer nochdie wichtigste Methode, um intrazelluläre Vorgänge invivo mit einer Genauigkeit im Nanometerbereich zu vi-sualisieren. Für viele dieser Methoden war zunächsteine weitere Neuerung nötig, nämlich die Etablierungder Fluoreszenzmikroskopie.
Leuchtende Farbstoffe – Die Fluoreszenzmikroskopie
Bei der Fluoreszenzmikroskopie [8] spielen Fluoro-phore eine zentrale Rolle. Diese Atome oder Molekülewerden hierbei mit Licht einer spezifischen Wellen-länge angeregt, welches sie absorbieren können. Dasdaraufhin von den Fluorophoren emittierte energieär-mere, längerwellige Licht kann anschließend detektiertund schlussendlich auf die menschliche Retina abge-bildet werden. Fluorophore, die in der Fluoreszenzmi-kroskopie Anwendung finden, können vielfältiger Na-tur sein. Im einfachsten Fall wird die Autofluoreszenzvon Stoffen wie Chlorophyll in Pflanzen ausgenutzt,wobei diese Art von Fluoreszenz generell nicht ge-wünscht ist. Sehr viele Stoffe oder Organellen weisenAutofluoreszenz auf und erzeugen so starkes, unspezi-fisches � Hintergrundrauschen.
Um jedoch bestimmte Moleküle innerhalb von Zel-len zu visualisieren, werden diese im Normalfall durchzusätzliche Stoffe markiert. Durch zum Beispiel mitRhodamin fluoreszent markierte Liganden wie Phalloi-din, die spezifisch an intrazelluläre Komponenten bin-den, können gezielt Organellen, Strukturen oder Ein-zelmoleküle gefärbt werden. Ebenfalls gängige Praxisist die Immunfluoreszenz oder Immunohistochemie,bei der Zellbestandteile mit markierten Antikörpernsichtbar gemacht werden. Eine weitere Methode, umProteine in vivo fluoreszent zu markieren, ist sie schonauf DNA Level mit einem so genannten Tag zu verse-hen, das zusammen mit dem Protein exprimiert wirdund ein Fusionsprotein bildet (Abbildung 4a). DieseDNA Tags codieren beispielsweise für fluoreszente Pro-teine wie das grün fluoreszierende Protein (GFP) ausder Qualle Aequorea victoria [5] oder das rot fluores-zierende Protein (RFP) aus der Koralle Acropora mille-pora [13]. Ist die so veränderte DNA die einzige Kopieeines Gens in dem Organismus, so ist automatisch dasProtein, das durch dieses Gen codiert wird, markiertund kann mikroskopisch untersucht werden. DieseTechnologie hat in der Biologie zu einem echten Quan-tensprung der Analysemöglichkeiten geführt undwurde vor einiger Zeit mit dem Nobelpreis honoriert.
Inzwischen ist eine breite Palette fluoreszierenderProteine aus verschiedenen Organismen in unter-schiedlichen Farben kommerziell verfügbar, um auchmehrere Proteine simultan untersuchen und unter-scheiden zu können. Durch intensive Suche und Muta-genese-Experimente bekannter Proteine wurden auchbesondere Fluorophore entdeckt oder entwickelt wiezum Beispiel DsRed-E5. Bei dieser DsRed Variante dau-ert die Faltung und die autokatalytische Bildung desChromophors sehr lange, sodass erst nach einiger Zeitdie richtige Konformation erreicht wird, die eine rote� Emission erlaubt. Bevor es zu dieser Konformationkommt, bildet sich ein grün fluoreszentes Intermediat.Diese „Timer“-Eigenschaft ermöglicht es, das unge-
4 | Biol. Unserer Zeit | 4/2012 (42) www.biuz.de © 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Ar+
LASER
DM1
CS
OBJ
DM2
L1TL
F
CCDX405LASER
a) b)
(a) Schematischer Aufbau eines inversen Fluoreszenzmikroskops. Der Aktivierungslaser (X405) und Anregungslaser (Ar+) werden über einen � dichroitischen Spiegel (DM1) parallelisiert. Eine Linse (L1) fokussiert dieLaser und leitet sie auf einen zweiten dichroitischen Spiegel (DM2), der sienach oben durch das Objektiv (OBJ) auf das Präparat (CS) übermittelt. Dieemittierte Fluoreszenz wird im Objektiv gesammelt und durch denselbenStrahlengang zu DM2 zurückgeleitet, durch den auch Aktivierungs- undAnregungslaser gehen. Die emittierten Photonen sind in der Lage DM2 zupassieren, werden gefiltert (F) und mittels einer Linse (TL) fokussiert. DiePhotonen werden von einer EMCCD (Electron multiplying charge coupled devices)-Kamera gesammelt und können zu einem Bild verrechnet werden[9].(b) Beugungsbegrenztes Fluoreszenzbild eines mit GFP markierten Motor-proteins, welches selektiv nur an einer Subpopulation von Mikrotubulientlangläuft. Aufgrund einer Mutation in der ATP-Bindedomäne sind dieMotorproteine unbeweglich und bedecken so den Mikrotubulus. Auf-grund der PSF können keine einzelnen Moleküle voneinander unterschie-den werden. Stattdessen ist nur ein einzelner durchgehender Strang zu erkennen. [14]
ABB. 4 F LU O R E S Z E N Z M I K ROS KO PI E

S U P E R R E S O L U T I O N - M I K R O S K O P I E | I M FO KU S
fähre Alter eines Proteins zu be-stimmen und so Rückschlüsse aufProteinschicksale zu schließen. FürSuper Resolution Mikroskopietech-niken wie PALM, die in der Lagesind, die Auflösungsgrenze zu über-winden, sind vor allem die Fluoro-phore von Bedeutung, deren Fluo-reszenz sich zuverlässig kontrollie-ren lässt.
Fluorophore für die SuperResolution Mikroskopie
Oft hängt das Gelingen eines Expe-rimentes von der Wahl des richti-gen Fluorophors ab. Es existierenzur Zeit drei Hauptgruppen vonFluorophoren, die für die Super Re-solution Mikroskopie Verwendungfinden. Die erste Gruppe wird generell als photoaktivierbareFluorophore bezeichnet. Sie wa-ren die ersten Fluorophore, derenEmission kontrolliert beeinflusstwerden konnte. Das Besondere andiesen fluoreszenten Farbstoffenist, dass ihr � Absorptionsspektrum,also die Wellenlänge des Lichts, diebenötigt wird um eine Fluoreszenzhervorzurufen, verschoben werdenkann. Dies wird durch die Bestrah-lung mit violettem oder ultravioletten Licht erreicht.Dieser Effekt geht meistens mit einem bis zu 100fachenAnstieg der Fluoreszenz einher.
Die zweite Gruppe besteht aus photoschaltbarenFluorophoren, die sozusagen einen optischen „An“und „Aus“ Schalter besitzen. Bestrahlung mit einer be-stimmten Wellenlänge hat zur Folge, dass die Fluoreszenzverschwindet und das Fluorophor „abgeschaltet“ wird.Nach Anregung mit einer anderen bestimmten Wellen-länge lässt sich die Fluoreszenz wiederum „anschalten“.
Fluorophore der dritten Gruppe werden im Allge-meinen als photokonvertierbare Fluorophore be-zeichnet. Diese Farbstoffe sind in der Lage, nach Anre-gung mit einer bestimmten Wellenlänge ihre Emissi-onswellenlänge zu einer anderen zu verschieben. Siekönnen somit nach Anregung eine grüne Emission insrote verschieben.
Synthetische Fluorophore und Quantumpunkte
Synthetische Farbstoffe und anorganische Quantum-punkte sind Fluorophore, die ebenfalls Anwendung inder Super Resolution Mikroskopie finden. SynthetischeFluorophore, die die oben genannten Eigenschaftenaufweisen, sind Varianten von Rhodaminderivaten, Cya-
ninfarbstoffen und Alexa Fluor©-Farbstoffen. Diese or-ganischen Kohlenwasserstoffe gehören zu den Polyme-thin-Farbstoffen, die eine quartäre Ammoniumgruppeals Elektronenakzeptor und eine tertiäre Aminogruppeals Elektronendonator besitzen. Sie sind in der Lage,durch die besondere Anordnung der Elektronen (delo-kalisiertes π-Elektronensystem) Licht zu absorbieren.Quantumpunkte sind anorganische Nanokristalle, dieaus einem CdSe-Kern in einer ZnS-Hülle und hydrophi-ler Beschichtung bestehen (Abbildung 5c). Bei diesenNanokristallen bestimmt die Größe des Kerns das Emis-sionsprofil.
Diese Farbstoffe können entweder reversibel pho-togeschaltet oder irreversibel photoaktiviert werden.Sie zeichnen sich vor allem durch ihre Helligkeit, ihrePhotostabilität und gutem Kontrast gegenüber demHintergrund aus. Die Menge an emittierten Photonenpro Molekül entspricht zudem einem Vielfachem derMenge, die fluoreszente Proteine emittieren können.Da die Genauigkeit, mit der ein Molekül lokalisiert wer-den kann, von der Anzahl an detektierten Photonen ab-hängt, sind diese Fluorophore ideal, da eine großeMenge an Photonen detektiert werden kann. Das Pro-blem der synthetischen Fluorophore und Quantum-punkte liegt darin, sie an das gewünschte Ziel zu bin-
© 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.biuz.de 4/2012 (42) | Biol. Unserer Zeit | 5
SynthetischesFluorophor
(Cy5)
(a)
IgGAntikörper
(b)5 Nanometer
GelberQuantumpunkt
(c)
Fluoreszentes Protein(d)
CdSe
ZnSe
HydrophilicCoating
Cy5 ist ein synthetisches Fluorophor mit einer Emission im roten Bereich bei 670 nm (a). Es ist signifikant kleiner als beispielsweise ein Quantumpunkt (c) mit durchschnittlich 5 nm imDurchmesser oder die typische röhrenförmige Struktur (beta-Fass) eines fluoreszenten Pro-teins (d). Diese Marker sind im Vergleich zu einem primären oder sekundären IgG Antikörper(b) mit einer durchschnittlichen Größe von 12 bis 15 nm jedoch verhältnismäßig klein. Bild: modifiziert nach http://zeiss-campus.magnet.fsu.edu/articles/superresolution/palm/introduction.html.
ABB. 5 D U RC H S C H N I T T L I C H E G R Ö ß E N VO N F LU O RO PH O R E N U N D A N T I K Ö R PE R N

den. Es gibt zwar synthetische Fluorophore, die ohnezusätzliche Adaptoren spezifisch an ein bestimmtesZiel binden können, jedoch müssen die meisten von ih-nen und alle Quantumpunkte an primäre oder sekun-däre Antikörper konjugiert werden. Erst durch spezifi-sche Antikörper ist es möglich, die Farbstoffe gezielt anein bestimmtes Ziel zu binden. Da Antikörper nichtdurch die Zellmembran frei diffundieren können, müs-sen die Zellen dafür fixiert und permeabilisiert wer-den. Das Problem hierbei ist, dass Zellen den Fixie-rungsprozess mit zum Beispiel Paraformaldehyd nichtüberleben und es zu Artefakten durch Quervernetzun-gen der Zellkomponenten oder erhöhtem Hinter-grundrauschen durch Autofluoreszenz kommt.
Ein weiteres Problem ist, dass das Markieren mit An-tikörpern nicht sehr effizient ist und so nur ein Teil derin der Zelle eigentlich vorhandenen Moleküle beob-achtet werden kann. Problematisch ist auch der Anti-körper selbst. Die durchschnittliche Größe eines Anti-körpers beträgt circa 12 bis 15 nm (Abbildung 5). In derPraxis bedeutet das, dass das eigentliche Fluoreszenz-signal sich nicht direkt am Ziel befindet. Der Antikörperschafft somit einen Raum zwischen Ziel und Fluoro-phor in der Größe des Antikörpers in potenziell allendrei möglichen räumlichen Achsen. Der Einsatz von An-tikörpern zur Markierung zellulärer Komponenten istin der konventionellen Lichtmikroskopie weit verbrei-tet. Hier ist jedoch die Auflösung nicht hoch genug, sodass dieser Abstand zwischen Ziel und Fluorophornicht ins Gewicht fällt. Im Maßstab der Super Resolu-tion Mikroskopie sind 10 nm allerdings eine Distanz,die in den gewonnenen Daten Interpretationsspiel-raum offen lässt.
Fluoreszente ProteineUm lebende Zellen beobachten zu können, muss auffluoreszente Proteine zurückgegriffen werden, derenFluoreszenz ebenfalls kontrolliert werden kann. Im Ge-gensatz zu den synthetischen Fluorophoren oderQuantumpunkten, bei welchen nur photoschaltbareoder photoaktivierbare Fluorophore zu finden sind,können fluoreszente Proteine auch photokonvertier-bare Eigenschaften besitzen. Da fluoreszente Proteinewie alle Proteine von DNA codiert sind, müssen sienicht als Protein in die Zellen eingeschleust werden. Andas gewünschte Protein fusioniert sind sie kovalent ge-bunden, was zur Folge hat, dass auch alle so exprimier-ten Proteine in der Zelle markiert sind und beobachtetwerden können. Neben den photokontrollierbaren Ei-genschaften müssen sie auch genügend hohen Kon-trast gegenüber dem Hintergrund besitzen, was einesihrer größten Probleme darstellt, da sie je nach Fluoro-phor um ein Vielfaches weniger Photonen emittierenals synthetische Fluorophore oder Quantumpunkte.Des Weiteren sollen sie sich möglichst schnell und zu-verlässig in vivo in die richtige Konformation falten.
Die photokonvertierbaren Proteine sind potenzielldie Fluorophore, die am geeignetsten für die Super Re-solution-Mikroskopie sind. Jedoch sind die meistenProteine dieser Gruppe Dimere oder Tetramere und da-her für das Markieren von anderen Proteinen nicht im-mer unproblematisch. Hier besteht durch die tetramereAssoziation der Fusionsproteine die Gefahr, dass dasProtein in seiner Funktionalität extrem behindert odergar ganz blockiert wird. Es gibt jedoch bereits photo-konvertierbare Fluorophore, die durch Mutagenese soverändert wurden, dass sie nur noch eine monomereStruktur aufweisen und somit Probleme wie Mislokali-sationen oder Dysfunktion minimieren.
Richtungsweisend ist ein Hybridprotein, das so-wohl photoschaltbare als auch photokonvertierbare Ei-genschaften aufweist [2]. Das nach der griechischenRegenbogengöttin benannte IrisFP-Fluorophor wurdeursprünglich durch Zufallsmutagenese des EosFP ausder Steinkoralle Lobophyllia hemprichii entwickelt.Das ursprünglich tetramere EosFP besitzt photokon-vertierbare Eigenschaften, die es ermöglichen, einegrüne Fluoreszenz nach Anregung mit Licht einer Wel-lenlänge von 390 nm irreversibel ins rote zu verschie-ben. Aus diesem Grund wurde zuerst eine tetramereVersion von IrisFP verfügbar. Durch weiterführendeMutagenese Experimente konnten später eine dimereund schließlich eine monomere Variante entwickeltwerden.
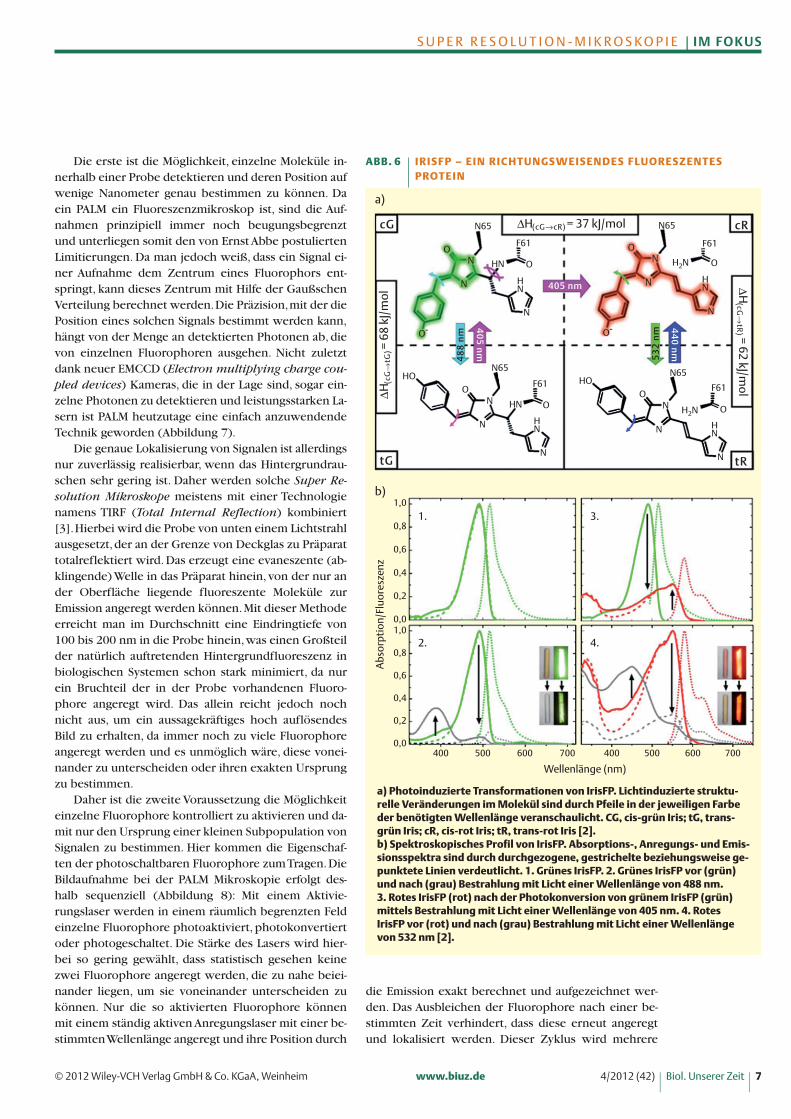
Nach Anregung mit einem schwachen Laser bei 405nm wechselt IrisFP reversibel von einem nicht-fluores-zenten zu einem grün-fluoreszenten Zustand. Wird dieIntensität des 405 nm Lasers verstärkt, so wird das grüneEmissionsspektrum irreversibel nach rot verschoben.Die rote Form von IrisFP kann wiederum photoreversi-bel in eine nicht-fluoreszente Form überführt werden,die wieder aktiviert werden kann (Abbildung 6). Diesezahlreichen kontrollierbaren Zustände machen IrisFPzu einem der potenziell leistungsfähigsten Fluorophoreder Super Resolution Mikroskopie.
Superauflösende lichtoptische MikroskopieZur Zeit existieren zwei Herangehensweisen, um mitlichtoptischen Geräten die durch die Wellenlänge desLichts abhängige Auflösungsgrenze zu durchbrechen.Die erste Variante ist ein System, das auf einer struktu-rierten Beleuchtung basiert, wie sie in der StructuredIllumination Microscopy (SIM) oder Stimulated Emis-sion Depletion (STED)-Mikroskopie realisiert ist [11].
Die zweite Variante nutzt die Eigenschaften photo-schaltbarer oder photoaktivierbarer Fluorophore, umsogar einzelne Moleküle innerhalb einer Probe zu de-tektieren. Ein solches System, das sich diese Technik zu-nutze macht um die Auflösungsgrenze zu durchbre-chen, bezeichnet man als Photoactivated LocalizationMicroscopy oder kurz PALM [4]. Es beruht prinzipiellauf zwei einfachen Voraussetzungen:
6 | Biol. Unserer Zeit | 4/2012 (42) www.biuz.de © 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
S U P E R R E S O L U T I O N - M I K R O S K O P I E | I M FO KU S
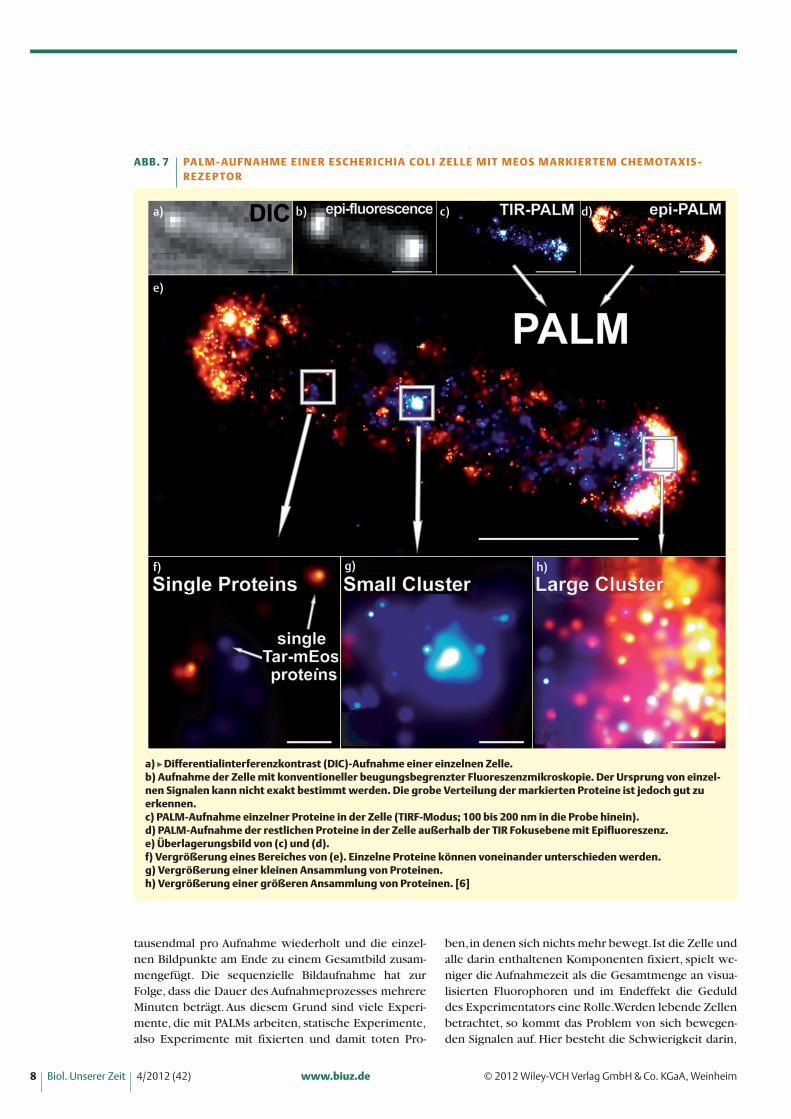
Die erste ist die Möglichkeit, einzelne Moleküle in-nerhalb einer Probe detektieren und deren Position aufwenige Nanometer genau bestimmen zu können. Daein PALM ein Fluoreszenzmikroskop ist, sind die Auf-nahmen prinzipiell immer noch beugungsbegrenztund unterliegen somit den von Ernst Abbe postuliertenLimitierungen. Da man jedoch weiß, dass ein Signal ei-ner Aufnahme dem Zentrum eines Fluorophors ent-springt, kann dieses Zentrum mit Hilfe der GaußschenVerteilung berechnet werden. Die Präzision, mit der diePosition eines solchen Signals bestimmt werden kann,hängt von der Menge an detektierten Photonen ab, dievon einzelnen Fluorophoren ausgehen. Nicht zuletztdank neuer EMCCD (Electron multiplying charge cou-pled devices) Kameras, die in der Lage sind, sogar ein-zelne Photonen zu detektieren und leistungsstarken La-sern ist PALM heutzutage eine einfach anzuwendendeTechnik geworden (Abbildung 7).
Die genaue Lokalisierung von Signalen ist allerdingsnur zuverlässig realisierbar, wenn das Hintergrundrau-schen sehr gering ist. Daher werden solche Super Re-solution Mikroskope meistens mit einer Technologienamens TIRF (Total Internal Reflection) kombiniert[3]. Hierbei wird die Probe von unten einem Lichtstrahlausgesetzt, der an der Grenze von Deckglas zu Präparattotalreflektiert wird. Das erzeugt eine evaneszente (ab-klingende) Welle in das Präparat hinein, von der nur ander Oberfläche liegende fluoreszente Moleküle zurEmission angeregt werden können. Mit dieser Methodeerreicht man im Durchschnitt eine Eindringtiefe von100 bis 200 nm in die Probe hinein, was einen Großteilder natürlich auftretenden Hintergrundfluoreszenz inbiologischen Systemen schon stark minimiert, da nurein Bruchteil der in der Probe vorhandenen Fluoro-phore angeregt wird. Das allein reicht jedoch nochnicht aus, um ein aussagekräftiges hoch auflösendesBild zu erhalten, da immer noch zu viele Fluorophoreangeregt werden und es unmöglich wäre, diese vonei-nander zu unterscheiden oder ihren exakten Ursprungzu bestimmen.
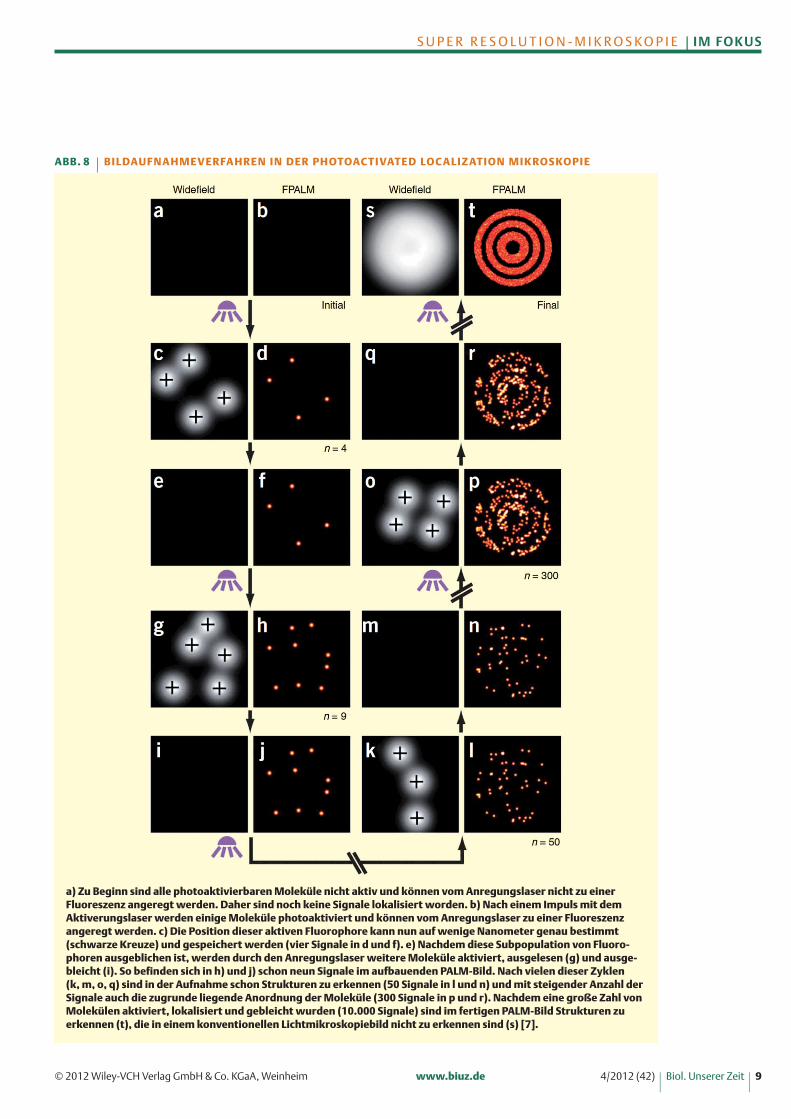
Daher ist die zweite Voraussetzung die Möglichkeiteinzelne Fluorophore kontrolliert zu aktivieren und da-mit nur den Ursprung einer kleinen Subpopulation vonSignalen zu bestimmen. Hier kommen die Eigenschaf-ten der photoschaltbaren Fluorophore zum Tragen. DieBildaufnahme bei der PALM Mikroskopie erfolgt des-halb sequenziell (Abbildung 8): Mit einem Aktivie-rungslaser werden in einem räumlich begrenzten Feldeinzelne Fluorophore photoaktiviert, photokonvertiertoder photogeschaltet. Die Stärke des Lasers wird hier-bei so gering gewählt, dass statistisch gesehen keinezwei Fluorophore angeregt werden, die zu nahe beiei-nander liegen, um sie voneinander unterscheiden zukönnen. Nur die so aktivierten Fluorophore könnenmit einem ständig aktiven Anregungslaser mit einer be-stimmten Wellenlänge angeregt und ihre Position durch
die Emission exakt berechnet und aufgezeichnet wer-den. Das Ausbleichen der Fluorophore nach einer be-stimmten Zeit verhindert, dass diese erneut angeregtund lokalisiert werden. Dieser Zyklus wird mehrere
© 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.biuz.de 4/2012 (42) | Biol. Unserer Zeit | 7
1,0
0,8
0,6
0,4
0,2
0,01,0
0,8
0,6
0,4
0,2
0,0400 500 600 700 400 500 600 700
1. 3.
2. 4.
Wellenlänge (nm)
Abs
orpt
ion/
Fluo
resz
enz
N65
F61HO HO
O
N
N
N N
N
N
HN
HN
HN
OO
O
N65
F61
F61 F61
N65 N65
H2N
H2N
OHN O
HN
N
HN
O
O-O-
N
NO
N
N
N
ΔH = 37 kJ/mol(cG→cR)
ΔH
= 62 kJ/mol
(cG→
tR)
ΔH
= 68
kJ/
mol
(cG
→tG
)
405 nm
48
8 n
m
53
2 n
m
40
5 n
m
44
0 n
m
cG
tG
cR
tR
a)
b)
a) Photoinduzierte Transformationen von IrisFP. Lichtinduzierte struktu-relle Veränderungen im Molekül sind durch Pfeile in der jeweiligen Farbeder benötigten Wellenlänge veranschaulicht. CG, cis-grün Iris; tG, trans-grün Iris; cR, cis-rot Iris; tR, trans-rot Iris [2].b) Spektroskopisches Profil von IrisFP. Absorptions-, Anregungs- und Emis-sionsspektra sind durch durchgezogene, gestrichelte beziehungsweise ge-punktete Linien verdeutlicht. 1. Grünes IrisFP. 2. Grünes IrisFP vor (grün)und nach (grau) Bestrahlung mit Licht einer Wellenlänge von 488 nm. 3. Rotes IrisFP (rot) nach der Photokonversion von grünem IrisFP (grün)mittels Bestrahlung mit Licht einer Wellenlänge von 405 nm. 4. RotesIrisFP vor (rot) und nach (grau) Bestrahlung mit Licht einer Wellenlängevon 532 nm [2].
ABB. 6 I R I S F P – E I N R I C H T U N G S W E I S E N D E S F LU O R E S Z E N T E S PROT E I N
tausendmal pro Aufnahme wiederholt und die einzel-nen Bildpunkte am Ende zu einem Gesamtbild zusam-mengefügt. Die sequenzielle Bildaufnahme hat zurFolge, dass die Dauer des Aufnahmeprozesses mehrereMinuten beträgt. Aus diesem Grund sind viele Experi-mente, die mit PALMs arbeiten, statische Experimente,also Experimente mit fixierten und damit toten Pro-
ben, in denen sich nichts mehr bewegt. Ist die Zelle undalle darin enthaltenen Komponenten fixiert, spielt we-niger die Aufnahmezeit als die Gesamtmenge an visua-lisierten Fluorophoren und im Endeffekt die Gedulddes Experimentators eine Rolle. Werden lebende Zellenbetrachtet, so kommt das Problem von sich bewegen-den Signalen auf. Hier besteht die Schwierigkeit darin,
8 | Biol. Unserer Zeit | 4/2012 (42) www.biuz.de © 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
a) b) c) d)
e)
f) g) h)
a) � Differentialinterferenzkontrast (DIC)-Aufnahme einer einzelnen Zelle. b) Aufnahme der Zelle mit konventioneller beugungsbegrenzter Fluoreszenzmikroskopie. Der Ursprung von einzel-nen Signalen kann nicht exakt bestimmt werden. Die grobe Verteilung der markierten Proteine ist jedoch gut zu erkennen. c) PALM-Aufnahme einzelner Proteine in der Zelle (TIRF-Modus; 100 bis 200 nm in die Probe hinein). d) PALM-Aufnahme der restlichen Proteine in der Zelle außerhalb der TIR Fokusebene mit Epifluoreszenz. e) Überlagerungsbild von (c) und (d). f) Vergrößerung eines Bereiches von (e). Einzelne Proteine können voneinander unterschieden werden.g) Vergrößerung einer kleinen Ansammlung von Proteinen.h) Vergrößerung einer größeren Ansammlung von Proteinen. [6]
ABB. 7 PA L M - AU F N A H M E E I N E R E S C H E R I C H I A CO L I Z E L L E M I T M EOS M A R K I E R T E M C H E M OTA X I S - R E Z E P TO R
S U P E R R E S O L U T I O N - M I K R O S K O P I E | I M FO KU S
© 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.biuz.de 4/2012 (42) | Biol. Unserer Zeit | 9
a) Zu Beginn sind alle photoaktivierbaren Moleküle nicht aktiv und können vom Anregungslaser nicht zu einerFluor eszenz angeregt werden. Daher sind noch keine Signale lokalisiert worden. b) Nach einem Impuls mit dem Aktiverungslaser werden einige Moleküle photoaktiviert und können vom Anregungslaser zu einer Fluoreszenz angeregt werden. c) Die Position dieser aktiven Fluorophore kann nun auf wenige Nanometer genau bestimmt(schwarze Kreuze) und gespeichert werden (vier Signale in d und f). e) Nachdem diese Subpopulation von Fluoro-phoren ausgeblichen ist, werden durch den Anregungslaser weitere Moleküle aktiviert, ausgelesen (g) und ausge-bleicht (i). So befinden sich in h) und j) schon neun Signale im aufbauenden PALM-Bild. Nach vielen dieser Zyklen (k, m, o, q) sind in der Aufnahme schon Strukturen zu erkennen (50 Signale in l und n) und mit steigender Anzahl derSignale auch die zugrunde liegende Anordnung der Moleküle (300 Signale in p und r). Nachdem eine große Zahl vonMolekülen aktiviert, lokalisiert und gebleicht wurden (10.000 Signale) sind im fertigen PALM-Bild Strukturen zu erkennen (t), die in einem konventionellen Lichtmikroskopiebild nicht zu erkennen sind (s) [7].
ABB. 8 B I L DAU F N A H M E V E R FA H R E N I N D E R PH OTOAC T I VAT E D LO C A L I Z AT I O N M I K ROS KO PI E

zwischen bereits aufgenommenen und neuen Signalenzu unterscheiden. Deshalb muss die Bildaufnahme hierschneller erfolgen als bei fixierten Zellen. Eine schnel-lere Bildaufnahme heißt in diesem Fall unter anderem,dass die Stärke des Lasers erhöht wird. Starke Laser-strahlung kann allerdings Zellen erheblich schädigenund so zu verfälschten Informationen führen. Dahersind lebende Objekte prinzipiell auch für PALM-Mikro-skopie geeignet, sie sind jedoch wesentlich schwierigerzu handhaben, da die Vitalität und Fitness der Zellen im-mer kritisch beurteilt werden muss.
Eine Kuh oder nur ein paar Bildpunkte? Die Dichte an Signalen macht den Unterschied
Eine Besonderheit von PALM ist, dass der Experimenta-tor bestimmen kann, wie viele Moleküle in der endgül-tigen Aufnahme dargestellt werden sollen. So ist es zumBeispiel möglich, nur Moleküle im fertigen Bild er-scheinen zu lassen, deren Position auf weniger als 10 nm genau bestimmt werden konnte.Allerdings musshierbei beachtet werden, dass man mit steigender Ge-nauigkeit dieser Grenze natürlich weniger Bildpunkteim endgültigen Bild erhält. Werden beispielsweise Pro-teine untersucht, die im Zellkern lokalisiert sind, undzieht nur Signale in Betracht, die mit höchster Genau-igkeit lokalisiert werden konnten, wird man zwarexakte Informationen über die Verteilung und Positionvon einzelnen Molekülen innerhalb des Zellkerns er-halten – der Zellkern als Ganzes wird jedoch nicht zuerkennen sein. Erst wenn unspezifischere Signale hin-zukommen, wird der Zellkern theoretisch sichtbar (Ab-bildung 9). Eine solche Dichte an Signalen ist jedochbei der Super Resolution Mikroskopie praktisch nichtrealisierbar. Zieht man die durchschnittliche Größe ei-nes Fluorophors in Betracht, so müsste die kompletteEbene nur aus photoaktivierten Fluorophoren beste-hen, was in Zellen nie der Fall sein kann. Dies ist jedochauch nicht nötig, da das Ziel eines PALM-Experimentesin der Regel ist, möglichst akkurate Informationen zurLokalisation von Molekülen zu erhalten.
Es war ein langer Weg bis zu diesem Stand der Tech-nik. Viele einzelne Aspekte und Voraussetzungen wiedie mathematischen und physikalischen Grundlagen,Kameratechnik und Fluorophore mussten erst er-forscht, entwickelt und etabliert werden, um PALM soanwendbar zu machen, wie es heute von Wissenschaft-lern tagtäglich getan wird. Aufgrund der langen Bildauf-nahmeprozedur sind Videos, die vor allem in der Zell-biologie von großer Bedeutung sind, nur begrenzt möglich. Verbesserte Auflösung und schnellere Bildse-quenzen könnten es sogar ermöglichen, Proteinkom-plexe wie DNA oder RNA-Polymerasen in Aktion zu be-obachten. Durch die ständige Weiterentwicklung derMikroskope und immer neue Fluorophore mit besse-ren Eigenschaften darf man gespannt sein, was die Zu-kunft hier noch bringen wird.
10 | Biol. Unserer Zeit | 4/2012 (42) www.biuz.de © 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
G LOSSA R
Artefakt: Veränderung einer Probe durch den Präparations-prozess oder das Auftreten falscher Signale durch Autofluo-reszenz in der Fluoreszenzmikroskopie.
Absorption: Aufnahme von Energie aus einfallender Strah-lung bestimmter Wellenlänge.
dichroitischer Spiegel: ein Spiegel, der nur einen Teil desLichtspektrums reflektiert und den Rest passieren lässt.
Differentialinterferenzkontrast: eine Methode der Lichtmi-kroskopie, bei der die Unterschiede in der optischen Weg-länge in der Probe vom Mikroskop in Helligkeitsunterschiedeumgewandelt und sichtbar gemacht werden.
Emission: Aussendung von Licht einer bestimmten Wellen-länge.
Hintergrundrauschen: Fluoreszenz, die von Fluorophorenoder autofluoreszenten Komponenten außerhalb der Fokus-ebene stammt.
a) b)
c) d)
In a) sind nur Signale dargestellt, die mit einer hohen Genauigkeit lokali-siert werden konnten. Durch die geringe Dichte an Signalen ist es nichtmöglich zu erkennen, um was es sich bei dem Bild handelt. Kommen mehrunspezifische Signale hinzu (b und c), wird der grobe Umriss einer Kuhsichtbar. Details der Kuh können jedoch erst erkannt werden, nachdemnoch mehr unspezifische Signale hinzugefügt werden (d). Ein Bild kannnur aus präzise lokalisierten Signalen bestehen, aber das Potential, Fein-heiten des eigentlichen Bildes aufzulösen korreliert direkt mit der Dichtean Signalen. Somit ist der Unterschied zwischen dem Bild einer Kuh und einer willkürlichen Ansammlung von Punkten die Menge und Dichte vonBildpunkten [12].
ABB. 9 PR Ä Z I S I O N , AU F L Ö S U N G U N D D I C H T E VO N S I G N A L E N I N PA L M - AU F N A H M E N

S U P E R R E S O L U T I O N - M I K R O S K O P I E | I M FO KU S
ZusammenfassungDie Auflösung konventioneller lichtoptischer Mikroskope istaufgrund physikalischer Gesetze durch die Wellenlänge deseingesetzten Lichts begrenzt. Die Größe vieler essentiellerintrazelluläre Komponenten wie das Cytoskelett oder sekre-torische Vesikel liegt jedoch weit unter dieser Auflösungs-grenze. Super Resolution Mikroskopie Techniken wie Photo-activated Localization Microscopy ermöglichen es dieseGrenze zu durchbrechen. Durch kontrolliertes Aktivieren ei-ner Subpopulation von photoaktivierbaren Fluorophoren ineiner Probe kann die Position eines jeden Moleküls auf we-nige Nanometer genau bestimmt werden.
SummarySuper Resolution MicroscopyResolution in conventional light microscopy is restricted bythe borders of physics. The limiting factor is the wavelengthof the used light. Nevertheless, many essential intracellularcomponents such as the cytoskeleton or secretory vesiclesare in size well below this diffraction limit. Super ResolutionMicroscopy techniques such as Photoactivated LocalizationMicroscopy make it possible to break through this diffrac-tion barrier. Controlled activation and sampling of a sub-population of photoactivatable fluorophores enables local-ization of probes in a nanometric scale.
Schlagworte PALM, Mikroskopie, Fluorophor, Super Resolution
Literatur[1] E. Abbe, Beiträge zur Theorie des Mikroskops und der mikroskopi-
schen Wahrnehmung, Archiv für Mikroskopische Anatomie 1873,9, 413–418.
[2] V. Adam et al., Structural characterization of IrisFP, an optical high-lighter undergoing multiple photo-induced transformations, PNAS2008, 105(47), 18343–18348.
[3] D. Axelrod, Cell-substrate contacts illuminated by total internal reflection fluorescence, J. Cell Biol. 1981, 89, 141–145.
[4] E. Betzig et al., Imaging intracellular fluorescent proteins at nano-meter resolution, Science 2006, 313(5793), 1642–1645.
[5] M. Chalfie et al., Green fluorescent protein as a marker for gene expression, Science 1994, 263, 802–805.
[6] D. Greenfield et al., Self-organization of the Escherichia coli chemo-taxis network imaged with super-resolution light microscopy, PLoSBiol. 2009, 7(6), e1000137.
[7] T. J. Gould, V. V. Verkhusha, S. T. Hess, Imaging biological structu-res with fluorescence photoactivation localization microscopy, Nature Prot. 2009, 4(3), 291–308.
[8] O. Heimstädt, Das Fluoreszenzmikroskop. Z. Wiss. Mikrosk. 28,1911, 330–337.
[9] S. T. Hess, T. P. K. Girirajan, M. D. Mason, Ultra-high resolution ima-ging by fluorescence photoactivation localization microscopy, Bio-phys. J. 2006, 91(11), 4258–4272.
[10] R. Hooke, Micrographia: or, Some physiological descriptions of mi-nute bodies made by magnifying glasses. London: J. Martyn and J. Allestry, 1665.
[11] B. Huang, H. Babcock, X. Zhuang, Breaking the diffraction barrier:super-resolution imaging of cells, Cell 2010, 143(7), 1047–1058.
[12] G. Patterson, M. Davidson, S. Manley, J. Lippincott-Schwartz, Superresolution imaging using single-molecule localization, Annu. Rev. Phys. Chem 2010, 61, 345–367.
[13] D. Veith, M. Veith, Biologie fluoreszierender Proteine: Ein Regen -bogen aus dem Ozean. Biol. Unserer Zeit 2005, 35, 394–404.
[14] N. Zekert, R. Fischer, The Aspergillus nidulans kinesin-3 UncA motormoves vesicles along a subpopulation of microtubules, Mol. Biol.Cell 2009, 20(2), 673–684.
Die AutorenRaphael Manck, geb. 1987 in Speyer, studierte Bio-logie am Karlsruher Institut für Technologie (KIT)und beschäftigte sich in seiner Bachelor Thesis mitdem polaren Wachstum von filamentösen Pilzen.Zur Zeit vertieft er diese Studien in seiner MasterThesis, die er wie seine Bachelor Thesis am Institutfür Angewandte Biowissenschaften bei ProfessorReinhard Fischer anfertigt. Bei diesen zellbiologi-schen Fragestellungen spielen mikroskopische Arbeiten mit Fluoreszenzmikroskopen eine wichtige Rolle.
Reinhard Fischer, geb. 1962 in Bad Berleburg, studierte Biologie an der Philipps-Universität inMarburg und promovierte 1990 unter der Anlei-tung von Prof. Dr. R.K. Thauer. 1991 Postdoc amgleichen Institut. 1992-1993 DFG-Stipendiat im Labor von Prof. Dr. W.E. Timberlake am Dept. ofGenetics, University of Georgia, Athens, USA.1994–2004 leitete er eine Forschungsgruppe ander Philipps Universität und am MPI für terrestri-sche Mikrobiologie. Er habilitierte sich 1998 undwar bis 2004 als Hochschuldozent tätig. 2004 er-hielt er eine C3-Professur für Angewandte Mikro-biologie an der Universität Karlsruhe und hat seit2007 hat eine W3-Professur für Mikrobiologie amKarlsruher Institut für Technologie (KIT) inne.
Korrespondenz: Raphael ManckKarlsruher Institut für Technologie (KIT)Institut für Angewandte BiowissenschaftenMikrobiologieHertzstr. 1676187 KarlsruheE-Mail: [email protected]
© 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.biuz.de 4/2012 (42) | Biol. Unserer Zeit | 11
Die Autoren sindMitglieder bei derVereinigung für Allgemeine und Angewandte Mikrobiologie(VAAM).www.vaam.de




























