Embed Size (px)
Citation preview

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
22Pharmacology of Hemostasis andThrombosis
April Wang Armstrong and David E. Golan
IntroductionCasePhysiology of Hemostasis
VasoconstrictionPrimary Hemostasis
Platelet AdhesionPlatelet Granule Release ReactionPlatelet Aggregation and Consolidation
Secondary Hemostasis: The Coagulation CascadeRegulation of Hemostasis
Pathogenesis of ThrombosisEndothelial InjuryAbnormal Blood FlowHypercoagulability
Pharmacologic Classes and AgentsAntiplatelet Agents
Cyclooxygenase InhibitorsPhosphodiesterase InhibitorsADP Receptor Pathway InhibitorsGPIIb–IIIa Antagonists
INTRODUCTION
Blood carries oxygen and nutrients to tissues and takes meta-bolic waste products away from tissues. Humans have devel-oped a well-regulated system of hemostasis to keep theblood fluid and clot-free in normal vessels and to form alocalized plug rapidly in injured vessels. Thrombosis de-scribes a pathologic state in which normal hemostatic pro-cesses are activated inappropriately. For example, a bloodclot (thrombus) may form as the result of a relatively minorvessel injury and occlude a section of the vascular tree. Thischapter presents the normal physiology of hemostasis, thepathophysiology of thrombosis, and the pharmacology ofdrugs that can be used to prevent or reverse a thromboticstate. Drugs introduced in this chapter are used to treat a
387
AnticoagulantsWarfarinUnfractionated and Low Molecular Weight
HeparinsSelective Factor Xa InhibitorsDirect Thrombin InhibitorsRecombinant Activated Protein C (r-APC)
Thrombolytic AgentsStreptokinaseRecombinant Tissue Plasminogen Activator (t-
PA)TenecteplaseReteplase
Inhibitors of Anticoagulation and FibrinolysisProtamineSerine-Protease InhibitorsLysine Analogues
Conclusion and Future DirectionsSuggested Readings
variety of cardiovascular diseases, such as deep vein throm-bosis and myocardial infarction.
Case
Mr. Soprano, a 55-year-old man with a history of hyperten-sion and cigarette smoking, is awakened in the middle ofthe night with substernal chest pressure, sweating, andshortness of breath. He calls 911 and is taken to the emer-gency room. An EKG shows deep T-wave inversions inleads V2 to V5. A cardiac biomarker panel shows a creatinekinase level of 400 U/L (normal, �200 U/L) with 10% MBfraction (the heart-specific isoform), suggesting myocardialinfarction. He is treated with IV nitroglycerin, aspirin, un-fractionated heparin, and eptifibatide, but his chest

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
388 III Principles of Cardiovascular Pharmacology
pain persists. He is taken to the cardiac catheterization labo-ratory, where he is found to have a 90% mid-LAD (leftanterior descending artery) thrombus with sluggish distalflow. He undergoes successful angioplasty and stent place-ment. At the time of stent placement, an intravenous load-ing dose of clopidogrel is administered. The heparin isstopped, the eptifibatide is continued for 18 more hours,and he is transferred to the telemetry ward. Six hours later,Mr. Soprano is noted to have an expanding hematoma (anarea of localized hemorrhage) in his right thigh below thearterial access site. The eptifibatide is stopped and pressureis applied to the access site, and the hematoma ceases toexpand. He is discharged 2 days later with prescriptions forclopidogrel and aspirin, which are administered to preventsubacute thrombosis of the stent.
QUESTIONS
■ 1. How did a blood clot arise in Mr. Soprano’s coronaryartery?
■ 2. If low molecular weight heparin had been usedinstead of unfractionated heparin, how would themonitoring of the patient’s coagulation status duringthe procedure have been affected?
■ 3. What accounts for the efficacy of eptifibatide (aplatelet GPIIb–IIIa antagonist) in inhibiting plateletaggregation?
■ 4. When the expanding hematoma was observed,could any measure other than stopping theeptifibatide have been used to reverse the effect ofthis agent?
■ 5. How do aspirin, heparin, clopidogrel, and epti-fibatide act in the attempt to treat Mr. Soprano’sblood clot and to prevent recurrent thrombusformation?
PHYSIOLOGY OF HEMOSTASIS
An injured blood vessel must induce the formation of a bloodclot to prevent blood loss and to allow healing. Clot forma-tion must also remain localized to prevent widespread clot-ting within intact vessels. The formation of a localized clotat the site of vessel injury is accomplished in four temporallyoverlapping stages (Fig. 22-1). First, localized vasocon-striction occurs as a response to a reflex neurogenic mecha-nism and to the secretion of endothelium-derived vasocon-strictors such as endothelin. Immediately followingvasoconstriction, primary hemostasis occurs. During thisstage, platelets are activated and adhere to the exposed sube-ndothelial matrix. Platelet activation involves both a changein shape of the platelet and the release of secretory granulecontents from the platelet. The secreted granule substancesrecruit other platelets, causing more platelets to adhere tothe subendothelial matrix and to aggregate with one anotherat the site of vascular injury. Primary hemostasis ultimatelyresults in the formation of a primary hemostatic plug.
The goal of the final two stages of hemostasis is to forma stable, permanent plug. During secondary hemostasis,also known as the coagulation cascade, the activated endo-thelium and other nearby cells (see below) express a mem-brane-bound procoagulant factor called tissue factor, whichcomplexes with coagulation factor VII to initiate the coagu-lation cascade. The end result of this cascade is the activationof thrombin, a critical enzyme. Thrombin serves two pivotalfunctions in hemostasis: (1) it converts soluble fibrinogento an insoluble fibrin polymer that forms the matrix of theclot; and (2) it induces more platelet recruitment and activa-tion. Recent evidence indicates that fibrin clot formation(secondary hemostasis) overlaps temporally with plateletplug formation (primary hemostasis), and that each processreinforces the other. During the final stage, platelet aggrega-tion and fibrin polymerization lead to the formation of astable, permanent plug. In addition, antithrombotic mech-anisms restrict the permanent plug to the site of vessel in-jury, ensuring that the permanent plug does not inappro-priately extend to occlude the vascular tree.
VASOCONSTRICTION
Transient arteriolar vasoconstriction occurs immediatelyafter vascular injury. This vasoconstriction is mediated bya poorly understood reflex neurogenic mechanism. Localendothelial secretion of endothelin, a potent vasoconstric-tor, potentiates the reflex vasoconstriction. Because thevasoconstriction is transient, bleeding would continue if pri-mary hemostasis were not activated.
PRIMARY HEMOSTASIS
The goal of primary hemostasis is to form a platelet plugthat rapidly stabilizes vascular injury. Platelets play a pivotalrole in primary hemostasis. Platelets are cell fragments thatarise by budding from megakaryocytes in the bone marrow;these small, membrane-bound discs contain cytoplasm butlack nuclei. Glycoprotein receptors in the platelet plasmamembrane are the primary mediators by which platelets areactivated. Primary hemostasis involves the transformationof platelets into a hemostatic plug through three reactions:(1) adhesion; (2) the granule release reaction; and (3) aggre-gation and consolidation.
Platelet AdhesionIn the first reaction, platelets adhere to subendothelial colla-gen that is exposed after vascular injury (Fig. 22-2). Thisadhesion is mediated by von Willebrand factor (vWF), alarge multimeric protein that is secreted by both activatedplatelets and the injured endothelium. vWF binds both tosurface receptors (especially glycoprotein Ib [GPIb]) on theplatelet membrane and to the exposed collagen; this ‘‘bridg-ing’’ action mediates adhesion of platelets to the collagen.The GPIb:vWF:collagen interaction is critical for initiationof primary hemostasis, because it is the only known molecu-lar mechanism by which platelets can adhere to the injuredvessel wall.

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 389
E
Resting platelets Activated spreadplatelet
Activated contractedplatelet
2μm
Figure 22-1. Sequence of events in hemostasis. The hemostaticprocess can be divided conceptually into four stages—vasoconstric-tion, primary hemostasis, secondary hemostasis, and resolution—although recent evidence suggests that these stages are temporallyoverlapping and may be nearly simultaneous. A. Vascular injurycauses endothelial denudation. Endothelin, released by activated en-dothelium, and neurohumoral factor(s) induce transient vasocon-striction. B. Injury-induced exposure of the subendothelial matrix (1)provides a substrate for platelet adhesion and activation (2). In thegranule release reaction, activated platelets secrete thromboxane A2
(TxA2) and ADP (3). TxA2 and ADP released by activated plateletscause nearby platelets to become activated; these newly activatedplatelets undergo shape change (4) and are recruited to the site ofinjury (5). The aggregation of activated platelets at the site of injuryforms a primary hemostatic plug (6). C. Tissue factor expressed onactivated endothelial cells (1) and leukocyte microparticles (notshown), together with acidic phospholipids expressed on activatedplatelets and activated endothelial cells (2), initiate the steps of thecoagulation cascade, culminating in the activation of thrombin (3).Thrombin proteolytically activates fibrinogen to form fibrin, whichpolymerizes around the site of injury, resulting in the formation of adefinitive (secondary) hemostatic plug (4). D. Natural anticoagulantand thrombolytic factors limit the hemostatic process to the site ofvascular injury. These factors include tissue plasminogen activator(t-PA), which activates the fibrinolytic system (1); thrombomodulin,which activates inhibitors of the coagulation cascade (2); prosta-cyclin, which inhibits both platelet activation and vasoconstriction(3); and surface heparin-like molecules, which catalyze the inactiva-tion of coagulation factors (4). E. Scanning electron micrographs ofresting platelets (1), a platelet undergoing cell spreading shortly aftercell activation (2), and a fully activated platelet after actin filamentbundling and crosslinking and myosin contraction (3).
3. Platelet granule release
Fibrin
A
B
C
D
PGI2
t-PA
1. Tissue factor expression on activated endothelium
4. Fibrin polymerization
1. Release of t-PA (fibrinolysis)
2. Thrombomodulin (blocks coagulation cascade)
3. Release of prostacyclin (inhibits platelet aggregation and vasoconstriction)
4. Surface heparin-like molecules (blocks coagulation cascade)
3. Thrombin activation
2. Phospholipid complex expression
2. Platelet adhesion and activation
1. Subendothelial matrix exposure
4. Platelet shape changeTxA2
ADP5. Platelet recruitment
6. Platelet aggregation (hemostatic plug)
Endothelial cells
Site of vascular injury(denuded endothelium)
Reflexvasoconstriction
Basement membrane
Vascular smooth muscle
Endothelin releaseby activated endothelium
LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
390 III Principles of Cardiovascular Pharmacology
Endothelium
Platelet Fibrinogen
von Willebrandfactor
GPIb
GPIIb-IIIa
Collagen (subendothelium)
Collagen
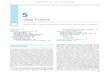
Figure 22-2. Platelet adhesion and aggregation. von Willebrandfactor mediates platelet adhesion to the subendothelium by bindingboth to the platelet membrane glycoprotein GPIb and to exposedsubendothelial collagen. During platelet aggregation, fibrinogencrosslinks platelets to one another by binding to GPIIb–IIIa receptorson platelet membranes.
Platelet Granule Release ReactionAdherent platelets undergo a process of activation (Fig. 22-3) during which the cells’ granule contents are released. Therelease reaction is initiated by agonist binding to cell surfacereceptors, which activates intracellular protein phosphoryla-tion cascades and ultimately causes release of granule con-tents. Specifically, stimulation by ADP, epinephrine, andcollagen leads to activation of platelet membrane phospholi-pase A2 (PLA2). PLA2 cleaves membrane phospholipids andliberates arachidonic acid, which is converted into a cyclicendoperoxide by platelet cyclooxygenase. Thromboxanesynthase subsequently converts the cyclic endoperoxide intothromboxane A2 (TxA2). TxA2, via a G protein-coupledreceptor, causes vasoconstriction at the site of vascular in-jury by inducing a decrease in cAMP levels within vascularsmooth muscle cells. TxA2 also stimulates the granule re-lease reaction within platelets, thereby propagating the cas-cade of platelet activation and vasoconstriction.
During the release reaction, large amounts of ADP, Ca2�,ATP, serotonin, vWF, and platelet factor 4 are actively se-
Resting platelet
Prothrombin
Thrombin
TXA2
ADP
Tissue factor
von Willebrandfactor
CollagenActivated endothelium
Endothelium
Collagen
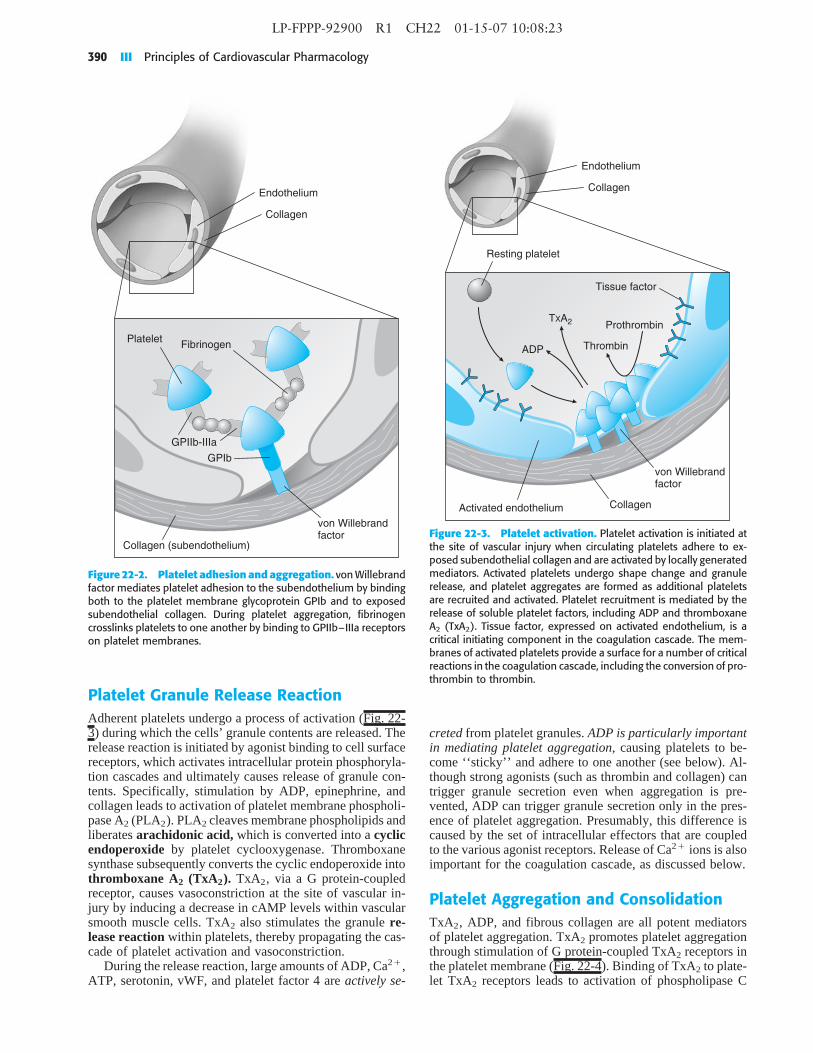
Figure 22-3. Platelet activation. Platelet activation is initiated atthe site of vascular injury when circulating platelets adhere to ex-posed subendothelial collagen and are activated by locally generatedmediators. Activated platelets undergo shape change and granulerelease, and platelet aggregates are formed as additional plateletsare recruited and activated. Platelet recruitment is mediated by therelease of soluble platelet factors, including ADP and thromboxaneA2 (TxA2). Tissue factor, expressed on activated endothelium, is acritical initiating component in the coagulation cascade. The mem-branes of activated platelets provide a surface for a number of criticalreactions in the coagulation cascade, including the conversion of pro-thrombin to thrombin.
creted from platelet granules. ADP is particularly importantin mediating platelet aggregation, causing platelets to be-come ‘‘sticky’’ and adhere to one another (see below). Al-though strong agonists (such as thrombin and collagen) cantrigger granule secretion even when aggregation is pre-vented, ADP can trigger granule secretion only in the pres-ence of platelet aggregation. Presumably, this difference iscaused by the set of intracellular effectors that are coupledto the various agonist receptors. Release of Ca2� ions is alsoimportant for the coagulation cascade, as discussed below.
Platelet Aggregation and ConsolidationTxA2, ADP, and fibrous collagen are all potent mediatorsof platelet aggregation. TxA2 promotes platelet aggregationthrough stimulation of G protein-coupled TxA2 receptors inthe platelet membrane (Fig. 22-4). Binding of TxA2 to plate-let TxA2 receptors leads to activation of phospholipase C

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 391
PKC
PKC(active)
PIP2
PLA2
IP3
DAG
βγ
GTP
αq
GDP
αq
GPIIb-IIIa
PLC
Ca2+
Ca2+
TXA2
TXA2-R
Arachidonic acid
CyclooxygenaseGeneration ofthromboxane A2by activatedplatelets
1
Activation ofthromboxane A2receptor
2
G protein-mediatedactivation ofphospholipase C
3
PLC hydrolyzes PIP2to yield IP3 and DAG
4
Increase in cytosoliccalcium concentration
5
Activation of proteinkinase C
6
Activation ofphospholipase A2
Fibrinogen
7
Activation ofGPIIb-IIIa
8
Binding of fibrinogento GPIIb-IIIa
9
Platelet aggregation10
Figure 22-4. Platelet activation by thromboxane A2. 1. Thromboxane A2 (TxA2) is generated from arachidonic acid in activated platelets;cyclooxygenase catalyzes the committed step in this process. 2. Secreted TxA2 binds to the cell surface TxA2 receptor (TxA2-R), a G protein-coupled receptor. 3. The G� isoform G�q activates phospholipase C (PLC). 4. PLC hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2)to yield inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). 5. IP3 raises the cytosolic Ca2� concentration by promoting vesicularrelease of Ca2� into the cytosol. 6. DAG activates protein kinase C (PKC). 7. PKC activates phospholipase A2 (PLA2). 8. Through a poorlyunderstood mechanism, activation of PLA2 leads to the activation of GPIIb–IIIa. 9. Activated GPIIb–IIIa binds to fibrinogen. 10. Fibrinogencrosslinks platelets by binding to GPIIb–IIIa receptors on other platelets. This crosslinking leads to platelet aggregation and formation of aprimary hemostatic plug.
(PLC), which hydrolyzes phosphatidylinositol 4,5-bisphos-phate (PI[4,5]P2) to yield inositol 1,4,5-trisphosphate (IP3)and diacylglycerol (DAG). IP3 raises the cytosolic Ca2�
concentration and DAG activates protein kinase C (PKC),which in turn promotes the activation of PLA2. Through apoorly understood mechanism, PLA2 activation induces theexpression of functional GPIIb–IIIa, the membrane integrinthat mediates platelet aggregation. ADP triggers platelet acti-vation by binding to G protein-coupled ADP receptors on theplatelet surface (Fig. 22-5). The two subtypes of G protein-coupled platelet ADP receptors are termed P2Y1 receptorsand P2Y(ADP) receptors. P2Y1, a Gq-coupled receptor,releases intracellular calcium stores through activation ofphospholipase C. P2Y(ADP), a Gi-coupled receptor, inhibitsadenylyl cyclase. The P2Y(ADP) receptor is the target of theantiplatelet agents ticlopidine and clopidogrel (see below).
Activation of ADP receptors mediates platelet shape changeand expression of functional GPIIb–IIIa. Fibrous collagenactivates platelets by binding directly to platelet glycoproteinVI (GPVI). Ligation of GPVI by collagen leads to phospholi-pase C activation and platelet activation, as described above.
Platelets aggregate with one another through a bridgingmolecule, fibrinogen, which has multiple binding sites forfunctional GPIIb–IIIa (Fig. 22-2). Just as the vWF:GPIbinteraction is important for platelet adhesion to exposed su-bendothelial collagen, the fibrinogen:GPIIb–IIIa interac-tion is critical for platelet aggregation. Platelet aggregationultimately leads to the formation of a reversible clot, or aprimary hemostatic plug.
Activation of the coagulation cascade proceeds nearlysimultaneously with the formation of the primary hemostaticplug, as described below. Activation of the coagulation cas-cade leads to the generation of fibrin, initially at the periph-

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
392 III Principles of Cardiovascular Pharmacology
βγ
GDP
αiGTP
αiβγ
βγ
GTP
αq
GDP
αq
GDP
αq
PKA
Adenylyl cyclase
P2Y(ADP) receptor
Thrombinreceptor
P2Y1 receptor
Thrombin
AMP
PLC
ADP ADP
1
Decreased PKA activityleads to platelet activation
3
Increased PLC activityleads to platelet activation
7
ATPcAMP
PDE2
4
5
6
Figure 22-5. Platelet activation by ADP and thrombin. Left panel: 1. Binding of ADP to the P2Y(ADP) receptor activates a Gi protein,which inhibits adenylyl cyclase. 2. Inhibition of adenylyl cyclase decreases the synthesis of cAMP, and hence decreases protein kinase A (PKA)activation (dashed arrow). cAMP is metabolized to AMP by phosphodiesterase (PDE). 3. PKA inhibits platelet activation through a series ofpoorly understood steps. Therefore, the decreased PKA activation that results from ADP binding to the P2Y(ADP) receptor causes plateletactivation. Right panel: 4. Thrombin proteolytically cleaves the extracellular domain of its receptor. This cleavage creates a new N-terminus,which binds to an activation site on the thrombin receptor to activate a Gq protein. 5. ADP also activates Gq by binding to the P2Y1 receptor.6. Gq activation (by either thrombin or ADP) activates phospholipase C (PLC). 7. PLC activity leads to platelet activation, as shown in Figure22-4. Note that ADP can activate platelets by binding to either the P2Y(ADP) receptor or the P2Y1 receptor, although recent evidence suggeststhat full platelet activation requires the participation of both receptors.
ery of the primary hemostatic plug. Platelet pseudopods at-tach to the fibrin strands at the periphery of the plug andcontract. Platelet contraction yields a compact, solid, irrever-sible clot, or a secondary hemostatic plug.
SECONDARY HEMOSTASIS: THECOAGULATION CASCADE
Secondary hemostasis is also termed the coagulation cas-cade. The goal of this cascade is to form a stable fibrinclot at the site of vascular injury. Details of the coagulationcascade are presented schematically in Figure 22-6. Severalgeneral principles should be noted.
First, the coagulation cascade is a sequence of enzymaticevents. Most plasma coagulation factors circulate as inactiveproenzymes, which are synthesized by the liver. These pro-enzymes are proteolytically cleaved, and thereby activated,by the activated factors that precede them in the cascade.
The activation reaction is catalytic and not stoichiometric.For example, one ‘‘unit’’ of activated factor X can poten-tially generate 40 ‘‘units’’ of thrombin. This robust amplifi-cation process rapidly generates large amounts of fibrin ata site of vascular injury.
Second, the major activation reactions in the cascadeoccur at sites where a phospholipid-based protein–proteincomplex has formed (Fig. 22-7). This complex is composedof a membrane surface (provided by activated platelets, acti-vated endothelial cells, and possibly activated leukocyte mi-croparticles [see below]), an enzyme (an activated coagula-tion factor), a substrate (the proenzyme form of thedownstream coagulation factor), and a cofactor. The pres-ence of negatively charged phospholipids, especially phos-phatidylserine, is critical for assembly of the complex. Phos-phatidylserine, which is normally sequestered in the innerleaflet of the plasma membrane, translocates to the outerleaflet of the membrane in response to agonist stimulationof platelets, endothelial cells, or leukocytes. Calcium is re-quired for the enzyme, substrate, and cofactor to adopt the

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 393
Common pathway
Thrombin (IIa)
IXaVIIaVIIIa
XaVa
Intrinsic pathway
XII
XI
VIII
IX
X
Xa
V
XIII
Prothrombin (II)
XIIIa
Fibrinogen Fibrin Fibrinpolymer
Crosslinkedfibrin polymer
Thrombin (IIa)
XIa
Kallikrein
Ca2+
Ca2+
Ca2+
Tissue injury
Tissuefactor
Prekallikrein
Thrombin (IIa)
Thrombin (IIa)
VIIa
Xa
XIIa
HMWK
Extrinsic pathway
Ca2+
VII
Ca2+
Ca2+
Figure 22-6. Coagulation cascade. The coagulation cascade isarbitrarily divided into the intrinsic pathway, the extrinsic pathway,and the common pathway. The intrinsic and extrinsic pathways con-verge at the level of factor X activation. The intrinsic pathway is largelyan in vitro pathway, while the extrinsic pathway accounts for themajority of in vivo coagulation. The extrinsic pathway is initiated atsites of vascular injury by the expression of tissue factor on severaldifferent cell types, including activated endothelial cells, activatedleukocytes (and leukocyte microparticles), subendothelial vascularsmooth muscle cells, and subendothelial fibroblasts. Note that Ca2�
is a cofactor in many of the steps, and that a number of the stepsoccur on phospholipid surfaces provided by activated platelets, acti-vated endothelial cells, and activated leukocytes (and leukocyte mi-croparticles). Activated coagulation factors are shown in blue andindicated with a lower case ‘‘a.’’ HMWK, high-molecular–weight ki-ninogen.
proper conformation for the proteolytic cleavage of a coagu-lation factor proenzyme to its activated form.
Third, the coagulation cascade has been divided tradition-ally into the intrinsic and extrinsic pathways (Fig. 22-6).This division is a result of in vitro testing and is essentiallyarbitrary. The intrinsic pathway is activated in vitro by factorXII (Hageman factor), while the extrinsic pathway is initi-ated in vivo by tissue factor, a lipoprotein expressed byactivated leukocytes (and microparticles derived from acti-vated leukocytes; see below), activated endothelial cells, su-bendothelial smooth muscle cells, and subendothelial fibro-
VIIIaVIIIa
VaVa
XIXa
Xa
Xa
IXa
Proteolytic cleavage(activation) of factor X
Proteolytic cleavage(activation) of prothrombin
Ca2+
Ca2+
Ca2+
Ca2+
Prothrombin (II)
Thrombin (IIa)
Figure 22-7. Coagulation factor activation on phospholipidsurfaces. Surface catalysis is critical for a number of the activationreactions in the coagulation cascade. Each activation reaction con-sists of an enzyme (e.g., factor IXa), a substrate (e.g., factor X), anda cofactor or reaction accelerator (e.g., factor VIIIa), all of which areassembled on the phospholipid surface of activated platelets, endo-thelial cells, and leukocytes. Ca2� allows the enzyme and substrateto adopt the proper conformation in each activation reaction. In theexample shown, factor VIIIa and Ca2� act as cofactors in the factorIXa-mediated cleavage of factor X to factor Xa. Factor Va and Ca2�
then act as cofactors in the factor Xa-mediated cleavage of prothrom-bin to thrombin.
blasts at the site of vascular injury. Although these twopathways converge at the activation of factor X, there alsoexists much interconnection between the two pathways. Be-cause factor VII (activated by the extrinsic pathway) canproteolytically activate factor IX (a key factor in the intrinsicpathway), the extrinsic pathway is regarded as the primarypathway for the initiation of coagulation in vivo.
Fourth, both the intrinsic and extrinsic coagulation path-ways lead to the activation of factor X. In an important reac-tion that requires factor V, activated factor X proteolyticallycleaves prothrombin (factor II) to thrombin (factor IIa) (Fig.22-8). Thrombin is a multifunctional enzyme that acts in thecoagulation cascade in four important ways: (1) it convertsthe soluble plasma protein fibrinogen into fibrin, which thenforms long insoluble polymer fibers; (2) it activates factorXIII, which crosslinks the fibrin polymers into a highly sta-ble meshwork or clot; (3) it amplifies the clotting cascadeby catalyzing the feedback activation of factors VIII and V;and (4) it strongly activates platelets, causing granule re-lease, platelet aggregation, and platelet-derived microparti-cle generation. In addition to its procoagulant properties,thrombin acts to modulate the coagulation response. Throm-bin binds to thrombin receptors on the intact vascular endo-thelial cells adjacent to the area of vascular injury, and stimu-lates these cells to release the platelet inhibitors prostacyclin(PGI2) and nitric oxide (NO), the profibrinolytic protein tis-sue-type plasminogen activator (t-PA), and the endogenoust-PA modulator plasminogen activator inhibitor 1 (PAI-1)(see below).
The thrombin receptor, a protease-activated G protein-coupled receptor, is expressed in the plasma membrane of

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
394 III Principles of Cardiovascular Pharmacology
Prothrombin (II)
V
Ca2+
PL
Va
XaVa
VIII
Activatedendothelialcells
Restingendothelialcells
Restingplatelets
Fibrinogen
Fibrin
Fibrin polymer Crosslinkedfibrin polymer
Activatedplatelets
XIII XIIIa
VIIIaVII VIIaXI XIa
Thrombin (IIa)
Figure 22-8. Central role of thrombin in the coagulation cas-cade. In the coagulation cascade, prothrombin is cleaved to thrombinby factor Xa; factor Va and Ca2� act as cofactors in this reaction,and the reaction takes place on an activated (phosphatidylserine-expressing) phospholipid surface (PL). Thrombin converts the solu-ble plasma protein fibrinogen to fibrin, which spontaneously poly-merizes. Thrombin also activates factor XIII, a transglutaminase thatcrosslinks the fibrin polymers into a highly stable meshwork or clot.Thrombin also activates co-factors V and VIII, as well as coagulationfactors VII and XI. In addition, thrombin activates both platelets andendothelial cells. Finally, thrombin stimulates the release of severalantithrombotic factors—including PGI2, NO, and t-PA—from resting(intact) endothelial cells near the site of vascular injury; these factorslimit primary and secondary hemostasis to the injured site (notshown).
platelets, vascular endothelial cells, smooth muscle cells, andfibroblasts. Activation of the thrombin receptor involves pro-teolytic cleavage of an extracellular domain of the receptorby thrombin. The new NH2-terminal-tethered ligand bindsintramolecularly to a discrete site within the receptor andinitiates intracellular signaling. Activation of the thrombinreceptor results in G protein-mediated activation of PLC(Fig. 22-5) and inhibition of adenylyl cyclase.
Finally, recent evidence from intravital (in vivo) micros-copy experiments suggests that leukocyte-derived micropar-ticles have an important role in coupling platelet plug forma-tion (primary hemostasis) to fibrin clot formation (secondaryhemostasis). A subpopulation of these microparticles, re-leased from monocytes that are activated in the context oftissue injury and inflammation, appears to express both tis-sue factor and PSGL-1, a protein that binds to the P-selectinadhesion receptor expressed on activated platelets. By re-cruiting tissue factor-bearing microparticles throughout thedeveloping platelet plug (primary hemostasis), thrombingeneration and fibrin clot formation (secondary hemostasis)could be greatly accelerated within the plug itself. Indeed, itappears that vessel wall tissue factor (expressed by activatedendothelial cells and subendothelial fibroblasts and smoothmuscle cells) and microparticle tissue factor are both impor-tant for formation of a stable clot.
REGULATION OF HEMOSTASIS
Hemostasis is exquisitely regulated for two major reasons.First, hemostasis must be restricted to the local site of vascu-lar injury. That is, activation of platelets and coagulationfactors in the plasma should occur only at the site of endothe-lial damage, tissue factor expression, and procoagulant phos-pholipid exposure. Second, the size of the primary and sec-ondary hemostatic plugs must be restricted so that thevascular lumen remains patent. After vascular injury, intactendothelium in the immediate vicinity of the injury becomes‘‘activated.’’ This activated endothelium presents a set ofprocoagulant factors that promote hemostasis at the site ofinjury, and anticoagulant factors that restrict propagation ofthe clot beyond the site of injury. The procoagulant factors,such as tissue factor and phosphatidylserine, tend to be mem-brane-bound and localized to the site of injury — thesefactors provide a surface on which the coagulation cascadecan proceed. In contrast, the anticoagulant factors are gener-ally secreted by the endothelium and are soluble in the blood.Thus, the activated endothelium maintains a balance of pro-coagulant and anticoagulant factors to limit hemostasis tothe site of vascular injury.
After vascular injury, the endothelium surrounding theinjured area participates in five separate mechanisms thatlimit the initiation and propagation of the hemostatic processto the immediate vicinity of the injury. These mechanismsinvolve prostacyclin (PGI2), antithrombin III, proteins C andS, tissue factor pathway inhibitor (TFPI), and tissue-typeplasminogen activator (t-PA).
Prostacyclin (PGI2) is an eicosanoid (i.e., a metaboliteof arachidonic acid) that is synthesized and secreted by theendothelium. By acting through Gs protein-coupled plateletsurface PGI2 receptors, this metabolite increases cAMP lev-els within platelets and thereby inhibits platelet aggregationand platelet granule release. PGI2 also has potent vasodila-tory effects; this mediator induces vascular smooth musclerelaxation by increasing cAMP levels within the vascularsmooth muscle cells. (Note that these mechanisms are physi-ologically antagonistic to those of TxA2, which inducesplatelet activation and vasoconstriction by decreasing intra-cellular cAMP levels.) Therefore, PGI2 both prevents plate-lets from adhering to the intact endothelium that surroundsthe site of vascular injury and maintains vascular patencyaround the site of injury.
Antithrombin III inactivates thrombin and other coagu-lation factors (IXa, Xa, XIa, and XIIa, where ‘‘a’’ denotesan ‘‘activated’’ factor) by forming a stoichiometric complexwith the coagulation factor (Fig. 22-9). These interactionsare enhanced by a heparin-like molecule that is expressedat the surface of intact endothelial cells, ensuring that thismechanism is operative at all locations in the vascular treeexcept where endothelium is denuded at the site of vascularinjury. (These endothelial cell surface proteoglycans are re-ferred to as ‘‘heparin-like’’ because they are the physiologicequivalent of the pharmacologic agent heparin, discussedbelow.) Heparin-like molecules on the endothelial cells bindto and activate antithrombin III, which is then primed tocomplex with (and thereby inactivate) the activated coagula-tion factors.

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 395
+ +
Thrombin
Xa
IXa
XIa
XIIa
Activecoagulation factors
Inactivecoagulation factors
A
B
+ATIII ATIII
Antithrombin III
Endogenous heparin-likemolecules orexogenous
unfractionated heparin
Heparin
Heparin
ATIII
ATIII
ATIII
Thrombin
Xa
IXaXIaXIIa
Xa
IXaXIaXIIa
ATIII
ATIII
Thrombin
ATIII
Heparin Heparin
Heparin
Heparin
Heparin
ATIII
Figure 22-9. Antithrombin III action. Antithrombin III (ATIII) inactivates thrombin and factors IXa, Xa, XIa, and XIIa by forming a stoichiomet-ric complex with these coagulation factors. These reactions are catalyzed physiologically by heparin-like molecules expressed on healthyendothelial cells; sites of vascular injury do not express heparin-like molecules because the endothelium is denuded or damaged. Pharmacolog-ically, these reactions are catalyzed by exogenously administered heparin. In more detail, the binding of heparin to ATIII induces a conforma-tional change in ATIII (A) that allows the ATIII to bind thrombin or coagulation factors IXa, Xa, XIa or XIIa. The stoichiometric complex betweenATIII and the coagulation factor is highly stable, allowing heparin to dissociate without breaking up the complex (B).
Protein C and protein S are vitamin K-dependent pro-teins that slow the coagulation cascade by inactivating coag-ulation factors Va and VIIIa. Protein C and protein S are partof a feedback control mechanism, in which excess thrombingeneration leads to activation of protein C which, in turn,helps to prevent the enlarging fibrin clot from occludingthe vascular lumen. Specifically, the endothelial cell surfaceprotein thrombomodulin is a receptor for both thrombinand protein C in the plasma. Thrombomodulin binds theseproteins in such a way that thrombomodulin-bound thrombincleaves protein C to activated protein C (also known as pro-tein Ca). In a reaction that requires the cofactor protein S,activated protein C then inhibits clotting by cleaving (andthereby inactivating) factors Va and VIIIa.
Tissue factor pathway inhibitor (TFPI), as its nameindicates, limits the action of tissue factor (TF). The coagula-tion cascade is initiated when factor VIIa complexes withTF at the site of vascular injury (Fig. 22-6). The resultingVIIa:TF complex catalyzes the activation of factors IX andX. After limited quantities of factors IXa and Xa are gener-ated, the VIIa:TF complex becomes feedback inhibited by
TFPI in a two-step reaction. First, TFPI binds to factor Xaand neutralizes its activity in a Ca2�-independent reaction.Subsequently, the TFPI:Xa complex interacts with the VIIa:TF complex via a second domain on TFPI, so that a quater-nary Xa:TFPI:VIIa:TF complex is formed. The molecular‘‘knots’’ of the TFPI molecule hold the quaternary complextightly together and thereby inactivate the VIIa:TF complex.In this manner, TFPI prevents excessive TF-mediated activa-tion of factors IX and X.
Plasmin exerts its anticoagulant effect by proteolyticallycleaving fibrin into fibrin degradation products. Becauseplasmin has powerful antithrombotic effects, the formationof plasmin has intrigued researchers for many years, and anumber of pharmacologic agents have been developed totarget the plasmin formation pathway (Fig. 22-10). Plasminis generated by the proteolytic cleavage of plasminogen, aplasma protein that is synthesized in the liver. The proteoly-tic cleavage is catalyzed by tissue-type plasminogen acti-vator (t-PA), which is synthesized and secreted by the endo-thelium. Plasmin activity is carefully modulated by threeregulatory mechanisms in order to restrict plasmin action to

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
396 III Principles of Cardiovascular Pharmacology
+
Tissue-type orurokinase-typeplasminogen
activator(inactive)
Tissue-type orurokinase-typeplasminogen
activator
Plasminogenactivator
inhibitor 1 or 2
Plasmin
Inactivatedplasmin
α2-antiplasmin α2-antiplasmin
Plasminogenactivator
inhibitor 1 or 2
Plasminogen
Crosslinkedfibrin polymer
Fibrin degradationproducts
Figure 22-10. The Fibrinolytic System. Plasmin is formed by theproteolytic cleavage of plasminogen by tissue-type or urokinase-typeplasminogen activator. Plasmin formation can be inhibited by plas-minogen activator inhibitor 1 or 2, which binds to and inactivatesplasminogen activators. In the fibrinolytic reaction, plasmin cleavescrosslinked fibrin polymers into fibrin degradation products. �2-Anti-plasmin, which circulates in the bloodstream, neutralizes freeplasmin in the circulation.
the site of clot formation. First, t-PA is most effective whenit is bound to a fibrin meshwork. Second, t-PA activity can beinhibited by plasminogen activator inhibitor (PAI). Whenlocal concentrations of thrombin and inflammatory cyto-kines (such as IL-1 and TNF-�) are high, endothelial cellsincrease the release of PAI, preventing t-PA from activatingplasmin. This ensures that a stable fibrin clot forms at thesite of vascular injury. Third, �2-antiplasmin is a plasmaprotein that neutralizes free plasmin in the circulation andthereby prevents random degradation of plasma fibrinogen.Plasma fibrinogen is important for platelet aggregation inprimary hemostasis (see above), and it is also the precursorfor the fibrin polymer that is required to form a stable clot.
PATHOGENESIS OF THROMBOSIS
Thrombosis is the pathologic extension of hemostasis. Inthrombosis, coagulation reactions are inappropriately regu-lated so that a clot uncontrollably enlarges and occludes thelumen of a blood vessel. The pathologic clot is now termeda thrombus. Three major factors predispose to thrombusformation—endothelial injury, abnormal blood flow, andhypercoagulability. These three factors influence one an-other, and are collectively known as Virchow’s triad (Fig.22-11).
Thrombosis
Endothelialinjury
HypercoagulabilityAbnormalblood flow
Figure 22-11. Virchow’s triad. Endothelial injury, abnormal bloodflow, and hypercoagulability are three factors that predispose tothrombus formation. These three factors are interrelated; endothelialinjury predisposes to abnormal blood flow and hypercoagulability,while abnormal blood flow can cause both endothelial injury andhypercoagulability.
ENDOTHELIAL INJURY
Endothelial injury is the most dominant influence on throm-bus formation in the heart and the arterial circulation. Thereare many possible causes of endothelial injury, includingchanges in shear stress associated with hypertension or tur-bulent flow, hyperlipidemia, elevated blood glucose in dia-betes mellitus, traumatic vascular injury, and some infec-tions. (Recall that Mr. Soprano developed coronary arterythrombosis, which was probably attributable to endothelialinjury secondary to hypertension and cigarette smoking.)
Endothelial injury predisposes the vascular lumen tothrombus formation through three mechanisms. First, plate-let activators, such as exposed subendothelial collagen, pro-mote platelet adhesion to the injured site. Second, exposureof tissue factor on injured endothelium initiates the coagula-tion cascade. Third, natural antithrombotics, such as t-PAand PGI2, become depleted at the site of vascular injurybecause these mechanisms rely on the functioning of an in-tact endothelial cell layer.
ABNORMAL BLOOD FLOW
Abnormal blood flow refers to a state of turbulence or stasisrather than laminar flow. Atherosclerotic plaques commonlypredispose to turbulent blood flow in the vicinity of theplaque. Bifurcations of blood vessels can also create areasof turbulent flow. Turbulent blood flow causes endothelialinjury, forms countercurrents, and creates local pockets ofstasis. Local stasis can also result from formation of an aneu-rysm (a focal out-pouching of a vessel or a cardiac chamber)and from myocardial infarction. In the latter condition, aregion of noncontractile (infarcted) myocardium serves as afavored site for stasis. Cardiac arrhythmias, such as atrialfibrillation, can also generate areas of local stasis. Stasis isthe major cause for the formation of venous thrombi.
Disruption of normal blood flow by turbulence or stasispromotes thrombosis by three major mechanisms. First, theabsence of laminar blood flow allows platelets to come intoclose proximity to the vessel wall. Second, stasis inhibitsthe flow of fresh blood into the vascular bed, so that activatedcoagulation factors in the region are not removed or diluted.

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 397
Third, abnormal blood flow promotes endothelial cell activa-tion, which leads to a prothrombotic state.
HYPERCOAGULABILITY
Hypercoagulability is generally less important than endothe-lial injury and abnormal blood flow in predisposing to throm-bosis, but this condition can be an important factor in somepatients. Hypercoagulability refers to an abnormally height-ened coagulation response to vascular injury, resulting fromeither: (1) primary (genetic) disorders; or (2) secondary(acquired) disorders (see Table 22-1). (Hypocoagulablestates, or hemorrhagic disorders, can also result from pri-mary or secondary causes; see Box 22-1 for an example.)
Among the genetic causes of hypercoagulability, the mostprevalent known mutation resides in the gene for coagulationfactor V. It is estimated that 6% of the Caucasian populationin the U.S. carries mutations in the factor V gene. The mostcommon mutation is the Leiden mutation, in which gluta-mine is substituted for arginine at position 506. This positionis important because it is part of a site in factor Va that ismarked for proteolytic cleavage by activated protein C. Themutant factor V Leiden protein is resistant to proteolyticcleavage by activated protein C. As a result of the Leidenmutation, factor Va is allowed to accumulate and thereby topromote coagulation.
A second common mutation (2% incidence) is the pro-thrombin G20210A mutation, in which adenine (A) is sub-stituted for guanine (G) in the 3′-untranslated region of the
TABLE 22-1 Major Causes of Hypercoagulability
CONDITION MECHANISM OF HYPERCOAGULABILITY
Primary (Genetic)Factor V Leiden mutation (factor Resistance to activated protein C → excess factor Va
V R506Q) (common)Hyperhomocysteinemia Endothelial damage due to accumulation of homocysteine
(common)Prothrombin G20210A mutation Increased prothrombin level and activity
(common)Antithrombin III deficiency (less Decreased inactivation of factors IIa, IXa, and Xa
common)Protein C or S deficiency (less Decreased proteolytic inactivation of factors VIIIa and Va
common)
Secondary (Acquired: Disease or Drug-Induced)Antiphospholipid syndrome Autoantibodies to negatively charged phospholipids → ↑ platelet adhesionHeparin-induced Antibodies to platelet factor 4 → platelet activation
thrombocytopeniaMalignancy Tumor cell induction of tissue factor expressionMyeloproliferative syndromes Elevated blood viscosity, altered plateletsNephrotic syndrome Loss of antithrombin III in urine, ↑ fibrinogen, ↑ platelet activationOral contraceptive use, estrogen ↑ Hepatic synthesis of coagulation factors and/or effects of estrogen on endothelium (effect
replacement therapy may be more prominent in patients with underlying primary hypercoagulability)Paroxysmal nocturnal Unknown, possibly ‘‘leaky’’ platelets
hemoglobinuriaPostpartum period Venous stasis, increased coagulation factors, tissue traumaSurgery/trauma Venous stasis, immobilization, tissue injury
prothrombin gene. This mutation leads to a 30% increase inplasma prothrombin levels. Both the factor V Leiden muta-tion and the prothrombin G20210A mutation are associatedwith a significantly increased risk of venous thrombosis anda modestly increased risk of arterial thrombosis. Other ge-netic disorders that predispose some individuals to thrombo-sis include mutations in the fibrinogen, protein C, proteinS, and antithrombin III genes. Although the latter disordersare relatively uncommon (less than 1% incidence), patientswith a genetic deficiency of protein C, protein S, or anti-thrombin III often present with spontaneous venous throm-bosis.
Hypercoagulability can sometimes be acquired (second-ary) rather than genetic. An example of acquired hypercoa-gulability is the heparin-induced thrombocytopenia syn-drome. In some patients, administration of the anticoagulantheparin stimulates the immune system to generate circulat-ing antibodies directed against a complex consisting of hepa-rin and platelet factor 4. Because platelet factor 4 is presenton platelet and endothelial cell surfaces, antibody bindingto the heparin:platelet factor 4 complex results in antibody-mediated removal of platelets from the circulation; that is,in thrombocytopenia. In some patients, however, antibodybinding also causes platelet activation, endothelial injury,and a prothrombotic state. Although both unfractionated andlow molecular weight heparin (see below) can cause throm-bocytopenia, it appears that low molecular weight heparinhas a lower incidence of thrombocytopenia than unfraction-ated heparin.

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
398 III Principles of Cardiovascular Pharmacology
BOX 22-1. Hemorrhagic Disorders: The Caseof Hemophilia A
When the vascular endothelium is injured, the hemostaticprocess ensures localized, stable clot formation withoutobstruction of the vascular lumen. Just as thrombosisconstitutes a pathologic variation on this otherwiseorchestrated physiologic process, disorders involvinginsufficient levels of functional platelets or coagulationfactors can lead to a hypocoagulable state characterizedclinically by episodes of uncontrolled hemorrhage.Hemorrhagic disorders result from a multitude of causes,including disorders of the vasculature, vitamin Kdeficiency, and disorders or deficiencies of platelets,coagulation factors, and von Willebrand factor.Hemophilia A serves as an example of a hemorrhagicdisorder in which hypocoagulability is the underlyingpathology.
Hemophilia A is the most common genetic disorder ofserious bleeding. The hallmark of the disorder is areduction in the amount or activity of coagulation factorVIII. The syndrome has an X-linked mode oftransmission, and the majority of patients are males orhomozygous females. Thirty percent of patients have nofamily history of hemophilia A and presumably representspontaneous mutations. The severity of the diseasedepends on the type of mutation in the factor VIII gene.Patients with 6% to 50% of normal factor VIII activitymanifest a mild form of the disease; those with 2% to 5%activity manifest moderate disease; patients with less than1% activity develop severe disease. All symptomaticpatients demonstrate easy bruisability and can developmassive hemorrhage after trauma or surgery. Spontaneoushemorrhage can occur in body areas that are normallysubjected to minor trauma, including joint spaces, wherespontaneous hemorrhage leads to the formation ofhemarthroses. Petechiae (microhemorrhages involvingcapillaries and small vessels, especially in mucocutaneousareas), which are usually an indication of plateletdisorders, are absent in patients with hemophilia.
Patients with hemophilia A are currently treated withinfusions of factor VIII that is either recombinant orderived from human plasma. Factor VIII infusion therapyis sometimes complicated in patients who developantibodies against factor VIII. HIV infection was a seriouscomplication of infusion therapy in patients who receivedfactor VIII products before the institution of routinescreening of blood for HIV infection (before the mid-1980s). Some sources suggest that the entire cohort ofhemophilioes who received factor VIII concentrates(factor VIII concentrated from the blood of manyindividuals) between 1981 and 1985 has been infectedwith HIV. With current blood screening practices and thedevelopment of recombinant factor VIII, the risk ofcontracting HIV through factor VIII infusions is nowvirtually zero.
PHARMACOLOGIC CLASSES ANDAGENTS
Drugs have been developed to prevent and/or reverse throm-bus formation. These drugs fall into three classes: antiplate-let agents, anticoagulants, and thrombolytic agents. Hemo-static agents, discussed at the end of the chapter, areoccasionally used to reverse the effects of anticoagulants orto inhibit endogenous fibrinolysis.
ANTIPLATELET AGENTS
As described above, formation of a localized platelet plugin response to endothelial injury is the initial step in arterialthrombosis. Therefore, inhibition of platelet function is auseful prophylactic and therapeutic strategy against myocar-dial infarction and stroke caused by thrombosis in coronaryand cerebral arteries, respectively. The classes of antiplateletagents in current clinical use include cyclooxygenase (COX)inhibitors, phosphodiesterase inhibitors, ADP receptor path-way inhibitors, and GPIIb–IIIa antagonists.
Cyclooxygenase InhibitorsAspirin inhibits the synthesis of prostaglandins, thereby in-hibiting the platelet release reaction and interfering with nor-mal platelet aggregation.
The biochemistry of prostaglandin synthesis in plateletsand endothelial cells provides a basis for understanding themechanism of action of aspirin as an antiplatelet agent. Fig-ure 22-12 depicts the prostaglandin synthesis pathway,which is discussed in more detail in Chapter 41, Pharmacol-ogy of Eicosanoids. Briefly, activation of both platelets andendothelial cells induces phospholipase A2 (PLA2) to cleavemembrane phospholipids and release arachidonic acid. Ara-chidonic acid is then transformed into a cyclic endoperoxide(also known as prostaglandin G2 or PGG2) by the enzymeCOX. In platelets, the cyclic endoperoxide is converted intothromboxane A2 (TxA2). Acting through cell surface TxA2
receptors, TxA2 causes localized vasoconstriction and is apotent inducer of platelet aggregation and the platelet releasereaction. In endothelial cells, the cyclic endoperoxide is con-verted into prostacyclin (PGI2). PGI2, in turn, causes local-ized vasodilation and inhibits platelet aggregation and theplatelet release reaction.
Aspirin acts by covalently acetylating a serine residuenear the active site of the COX enzyme, thereby inhibitingthe synthesis of the cyclic endoperoxide and the various me-tabolites of the cyclic endoperoxide. In the absence of TxA2,there is a marked decrease in platelet aggregation and theplatelet release reaction (Fig. 22-13A). Because platelets donot contain DNA or RNA, these cells cannot regenerate newCOX enzyme once aspirin has permanently inactivated allof the available COX enzyme. That is, the platelets becomeirreversibly ‘‘poisoned’’ for the lifetime of these cells (7–10days). Although aspirin also inhibits the COX enzyme inendothelial cells, its action is not permanent in endothelialcells because these cells are able to synthesize new COXmolecules. Thus, the endothelial cell production of prosta-

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 399
OOH
COOH
COOHO
COOH
S
HN
O
HN COOH
OH2N
COOH
OH
COOH
O
O
OOH
COOH
O
O
OH
COOH
O
COOH
OHOH
HH
O
OCOOH
OH OHHO
O
COOH
NSAIDs(aspirin, others)
Phospholipase A2
Cyclooxygenase
Peroxidase
Lipoxygenase
5-HPETE
Leukotriene A4
Leukotriene C4
Arachidonic acid
Membrane phospholipids
Prostaglandin E2
Otherprostaglandins
Prostaglandin H2
Prostaglandin G2
Thromboxane A2
Prostacyclin (PGI )2
Dehydrase
PGE2 synthase
PGI2 synthase Prostaglandin synthases
TxA2 synthase
Glutathione-S-transferase
Figure 22-12. Overview of Prostaglandin Synthesis. Membrane phospholipids are cleaved by phospholipase A2 to release free arachidonicacid. Arachidonic acid can be metabolized through either of two major pathways, the cyclooxygenase pathway or the lipoxygenase pathway.The cyclooxygenase pathway, which is inhibited by aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs), converts arachidonicacid into prostaglandins and thromboxanes. Platelets express TxA2 synthase and synthesize the pro-aggregatory mediator thromboxane A2;endothelial cells express PGI2 synthase and synthesize the anti-aggregatory mediator prostacyclin. The lipoxygenase pathway converts arachi-donic acid into leukotrienes, which are potent inflammatory mediators. (See Chapter 41, Pharmacology of Eicosanoids, for a detailed discussionof the lipoxygenase and cyclooxygenase pathways.) Aspirin inhibits cyclooxygenase by covalent acetylation of the enzyme near its active site.Because platelets lack the capability to synthesize new proteins, aspirin inhibits thromboxane synthesis for the life of the platelet.

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
400 III Principles of Cardiovascular Pharmacology
PKC
PKC(active)
PIP2
PLA2
IP3
DAG
βγ
GTP
αq
GDP
αq
GPIIb-IIIa
βγ
GDP
α i
α iβ
γ
βγ
GTP
αq
GDP
αq
GDP
αq
PKA
GTP
PLC
Ca2+
Ca2+
TXA2 (released by activated platelets)
NSAIDs(aspirin, others)
Abciximab
TXA2-R
Arachidonic acid
Cyclooxygenase
Fibrinogen
A
Clopidogrel,ticlopidine
Adenylyl cyclase
P2Y(ADP) receptor
Thrombinreceptor
P2Y1 receptor
Thrombin
AMP
PLC
ADP ADP
Platelet activation
Platelet activation
ATPcAMP
PDE
Dipyridamole
B
Figure 22-13. Mechanism of action of antiplatelet agents. A. NSAIDs and GPIIb-IIIa antagonists inhibit steps in thromboxane A2 (TxA2)-mediated platelet activation. Aspirin inhibits cyclooxygenase by covalent acetylation of the enzyme near its active site, leading to decreasedTxA2 production. The effect is profound because platelets lack the ability to synthesize new enzyme molecules. GPIIb–IIIa antagonists, suchas the monoclonal antibody abciximab and the small-molecule antagonists eptifibatide and tirofiban (not shown), inhibit platelet aggregationby preventing activation of GpIIb–IIIa (dashed line), leading to decreased platelet crosslinking by fibrinogen. B. Clopidogrel, ticlopidine, anddipyridamole inhibit steps in ADP-mediated platelet activation. Clopidogrel and ticlopidine are antagonists of the P2Y(ADP) receptor. Dipyrida-mole inhibits phosphodiesterase (PDE), thereby preventing the breakdown of cAMP and increasing cytoplasmic cAMP concentration.

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 401
cyclin is relatively unaffected by aspirin at pharmacologi-cally low doses (see below).
Aspirin is most often used as an antiplatelet agent to pre-vent arterial thrombosis leading to stroke, transient ischemicattack, and myocardial infarction. Because the action of aspi-rin on platelets is permanent, it is most effective as a selectiveantiplatelet agent when taken in low doses and/or at infre-quent intervals. For example, aspirin is often used as anantiplatelet agent at a dose of 81 mg once daily, while atypical anti-inflammatory dose of this agent could be 650mg three to four times daily. Taken at high doses, aspirincan inhibit prostacyclin production without increasing theeffectiveness of the drug as an antiplatelet agent. A moreextended discussion of the uses and toxicities of aspirin isfound in Chapter 41. Compared with aspirin, other nonsteroi-dal anti-inflammatory drugs (NSAIDs) are not as widelyused in the prevention of arterial thrombosis because theinhibitory action of these drugs on cyclooxygenase is notpermanent.
COX-1 is the predominant COX isoform in platelets, butendothelial cells appear to express both COX-1 and COX-2 under physiologic conditions. Because aspirin inhibitsCOX-1 and COX-2 nonselectively, this drug serves as aneffective antiplatelet agent. In contrast, the newer selectiveCOX-2 inhibitors cannot be used as antiplatelet agents be-cause they are poor inhibitors of COX-1. Furthermore, useof the selective COX-2 inhibitors appears to be associatedwith increased cardiovascular risk, most likely because theseagents inhibit endothelial production of PGI2 without inhib-iting platelet generation of TxA2. The adverse impact ofthe selective COX-2 inhibitors on cardiovascular risk hasresulted in the recent withdrawal of most of these agentsfrom the market (see Chapter 41).
Phosphodiesterase InhibitorsIn platelets, an increase in the concentration of intracellularcAMP leads to a decrease in platelet aggregability. PlateletcAMP levels are regulated physiologically by TxA2 andPGI2, among other mediators (see above). The mechanismby which increased intracellular cAMP concentration leadsto decreased platelet aggregability is not well understood.cAMP activates protein kinase A, which, through incom-pletely elucidated mechanisms, decreases availability of theintracellular Ca2� necessary for platelet aggregation (Fig.22-13B). Inhibitors of platelet phosphodiesterase decreaseplatelet aggregability by inhibiting cAMP degradation, whileactivators of platelet adenylyl cyclase decrease platelet ag-gregability by increasing cAMP synthesis. (There are cur-rently no direct adenylyl cyclase activators in clinical use.)
Dipyridamole is an inhibitor of platelet phosphodiester-ase that decreases platelet aggregability (Fig. 22-13B). Dipy-ridamole by itself has only weak antiplatelet effects, and istherefore usually administered in combination with warfarinor aspirin. The combination of dipyridamole and warfarincan be used to inhibit embolization from prosthetic heartvalves, while the combination of dipyridamole and aspirincan be used to reduce the likelihood of thrombosis in patientswith a thrombotic diathesis. Dipyridamole also has vasodila-tory properties. It may paradoxically induce angina in pa-tients with coronary artery disease by causing the coronary
steal phenomenon, which involves intense dilation of coro-nary arterioles (see Chapter 21, Pharmacology of VascularTone).
ADP Receptor Pathway InhibitorsBoth ticlopidine and clopidogrel are derivatives of thieno-pyridine. These agents, which irreversibly inhibit the ADP-dependent pathway of platelet activation, have antiplateleteffects in vitro and in vivo. Ticlopidine and clopidogrel arethought to act by covalently modifying and inactivating theplatelet P2Y(ADP) receptor (also called P2Y12), which isphysiologically coupled to the inhibition of adenylyl cyclase(Fig. 22-13B). Ticlopidine is a prodrug that requires conver-sion to active thiol metabolites in the liver. Maximal plateletinhibition is observed 8 to 11 days after initiating therapywith the drug; used in combination with aspirin, 4 to 7 daysare needed to achieve maximal platelet inhibition. Adminis-tration of a loading dose can produce a more rapid antiplate-let response. Ticlopidine is approved in the U.S. for twoindications: (1) secondary prevention of thrombotic strokesin patients intolerant of aspirin, and (2) in combination withaspirin, prevention of stent thrombosis for up to 30 days afterplacement of coronary artery stents. In general, ticlopidineis considered to be less safe than clopidogrel. The use ofticlopidine has occasionally been associated with neutro-penia, thrombocytopenia, and thrombotic thrombocytopenicpurpura (TTP); for this reason, blood counts must be moni-tored frequently when using ticlopidine.
Clopidogrel, a thienopyridine closely related to ticlopi-dine, has been used widely in combination with aspirin forimproved platelet inhibition during and after elective percu-taneous coronary intervention. Clopidogrel is a prodrug thatmust undergo oxidation by hepatic P450 3A4 to the activedrug form; it may therefore interact with statins and otherdrugs metabolized by this P450 enzyme. Clopidogrel is ap-proved for secondary prevention in patients with recent myo-cardial infarction, stroke, or peripheral vascular disease. Itis also approved for use in acute coronary syndromes thatare treated with either percutaneous coronary interventionor coronary artery bypass grafting. Like ticlopidine, clopido-grel requires a loading dose to achieve a maximal antiplateleteffect rapidly. For this reason, Mr. Soprano was given anintravenous loading dose of clopidogrel in the context of hismyocardial infarction. The adverse effect profile of clopido-grel is more favorable than that of ticlopidine: the gastroin-testinal side effects of clopidogrel are similar to those ofaspirin, and clopidogrel lacks the significant bone marrowtoxicity associated with ticlopidine.
GPIIb–IIIa AntagonistsAs noted above, platelet membrane GPIIb–IIIa receptors areimportant because they constitute the final common pathwayof platelet aggregation, serving to bind fibrinogen moleculesthat bridge platelets to one another. A variety of stimuli (e.g.,TxA2, ADP, epinephrine, collagen, and thrombin), actingthrough diverse signaling molecules, are capable of inducingthe expression of functional GPIIb–IIIa on the platelet sur-face. It could therefore be predicted that antagonists ofGPIIb–IIIa would prevent fibrinogen binding to theGPIIb–IIIa receptor and thus serve as powerful inhibitors of

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
402 III Principles of Cardiovascular Pharmacology
platelet aggregation. Eptifibatide, the GPIIb– IIIa receptorantagonist used in the opening case, is a highly efficaciousinhibitor of platelet aggregation. A synthetic peptide, eptifi-batide antagonizes the platelet GPIIb–IIIa receptor with highaffinity. This drug has been used to reduce ischemic eventsin patients undergoing percutaneous coronary interventionand to treat unstable angina and non-ST elevation myocar-dial infarction.
Abciximab is a chimeric mouse–human monoclonal an-tibody directed against the human GPIIb–IIIa receptor. Ex-periments in vitro have shown that occupation of 50% ofplatelet GPIIb–IIIa receptors by abciximab significantly re-duces platelet aggregation. The binding of abciximab toGPIIb–IIIa is essentially irreversible, with a dissociationhalf-time of 18 to 24 hours. In clinical trials, adding abcixi-mab to conventional antithrombotic therapy reduces bothlong-term and short-term ischemic events in patientsundergoing high-risk percutaneous coronary intervention.
Tirofiban is a nonpeptide tyrosine analogue that reversi-bly antagonizes fibrinogen binding to the platelet GPIIb–IIIareceptor. Both in vitro and in vivo studies have demonstratedthe ability of tirofiban to inhibit platelet aggregation. Tirofi-ban has been approved for use in patients with acute coro-nary syndromes.
Because of their mechanism of action as antiplateletagents, all of the GPIIb–IIIa antagonists can cause bleedingas an adverse effect. In the opening case, Mr. Soprano devel-oped a hematoma in his right thigh near the arterial accesssite at which eptifibatide was being infused. The expandinghematoma was caused by the excessive antiplatelet effect ofa very high local concentration of eptifibatide at the infusionsite. Importantly, the ability to reverse the effect ofGPIIb–IIIa receptor antagonists differs for the differentagents. Because abciximab is an irreversible inhibitor ofplatelet function, and all the abciximab previously infusedis already bound to platelets, infusion of fresh platelets afterthe drug has been stopped can reverse the antiplatelet effect.In contrast, because the two small-molecule antagonists (ep-tifibatide and tirofiban) bind the receptor reversibly and areinfused in great stoichiometric excess of receptor number,infusion of fresh platelets simply offers new sites to whichthe drug can bind, and it is not practical to deliver a sufficientnumber of platelets to overwhelm the vast excess of drugpresent. Therefore, one must stop the drug infusion and waitfor platelet function to return to normal as the drug is cleared.In the case of Mr. Soprano, no other measure could havebeen taken to reverse the effect of eptifibatide at the timehis hematoma was recognized.
ANTICOAGULANTS
As with antiplatelet agents, anticoagulants are used both toprevent and treat thrombotic disease. There are four classesof anticoagulants: warfarin, unfractionated and low molecu-lar weight heparins, selective factor Xa inhibitors, and directthrombin inhibitors. Anticoagulants target various factors inthe coagulation cascade, thereby interrupting the cascade andpreventing the formation of a stable fibrin meshwork (sec-ondary hemostatic plug). In this section, the four classes ofanticoagulants are discussed in order of selectivity, from theleast selective agents (warfarin and unfractionated heparin)
to the most selective agents (selective factor Xa inhibitorsand direct thrombin inhibitors). Recombinant activated pro-tein C also has anticoagulant activity, although its clinicalindication is severe sepsis. Because of the mechanisms ofaction of these drugs, bleeding is an adverse effect commonto all anticoagulants.
WarfarinIn the early 1900s, farmers in Canada and the North Dakotaplains adopted the practice of planting sweet clover insteadof corn for fodder. In the winter months of 1921 to 1922, afatal hemorrhagic disease was reported in cattle that hadforaged on the sweet clover. In almost every case, it wasfound that the affected cattle had foraged on sweet cloverthat had been spoiled by the curing process. After an inten-sive investigation, scientist K. P. Link reported that thespoiled clover contained the natural anticoagulant 3,3′-meth-ylene-bis-(4-hydroxycoumarin) or ‘‘dicumarol.’’ Dicumaroland warfarin (a potent synthetic congener) were introducedduring the 1940s as rodenticides and as oral anticoagulants.Because the oral anticoagulants act by affecting vitaminK-dependent reactions, it is important to understand howvitamin K functions.
Mechanism of Action of Vitamin K
Vitamin K (‘‘K’’ is derived from the German word ‘‘Koagu-lation’’) is required for the normal hepatic synthesis of fourcoagulation factors (II, VII, IX, and X), protein C, and pro-tein S. The coagulation factors, protein C, and protein S arebiologically inactive as unmodified polypeptides followingprotein synthesis on ribosomes. These proteins gain biologi-cal activity by post-translational carboxylation of their 9 to12 amino-terminal glutamic acid residues. The �-carboxyl-ated glutamate residues (but not the unmodified glutamateresidues) are capable of binding Ca2� ions. Ca2� bindinginduces a conformational change in these proteins that isrequired for efficient binding of the proteins to phospholipidsurfaces. The ability of the �-carboxylated molecules to bindCa2� increases the enzymatic activity of coagulation factorsIIa, VIIa, IXa, Xa, and protein Ca by approximately 1,000-fold. Thus, vitamin K-dependent carboxylation is crucial forthe enzymatic activity of the four coagulation factors andprotein C, and for the cofactor function of protein S.
The carboxylation reaction requires (1) a precursor formof the target protein with its 9 to 12 amino-terminal glutamicacid residues, (2) carbon dioxide, (3) molecular oxygen, and(4) reduced vitamin K. The carboxylation reaction is sche-matically presented in Figure 22-14. During this reaction,vitamin K is oxidized to the inactive 2,3-epoxide. An en-zyme, epoxide reductase, is then required to convert the inac-tive 2,3-epoxide into the active, reduced form of vitamin K.Thus, the regeneration of reduced vitamin K is essentialfor the sustained synthesis of biologically functional clottingfactors II, VII, IX, and X, all of which are critical componentsof the coagulation cascade.
Mechanism of Action of Warfarin
Warfarin acts on the carboxylation pathway, not by inhibit-ing the carboxylase directly, but by blocking the epoxide

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 403
Figure 22-14. Mechanism of action of warfarin.Vitamin K is a necessary cofactor in the post-transla-tional carboxylation of glutamate residues on factors II,VII, IX, and X. During the carboxylation reaction, vitaminK is oxidized to the inactive 2,3-epoxide. The enzymeepoxide reductase converts the inactive vitamin K 2,3-epoxide into the active, reduced form of vitamin K. Theregeneration of reduced vitamin K is essential for thesustained synthesis of biologically functional coagula-tion factors II, VII, IX, and X. Warfarin acts on the carbox-ylation pathway by inhibiting the epoxide reductasethat is required for the regeneration of reduced (active)vitamin K. Dicumarol is the natural anticoagulantformed in spoiled clover. Both warfarin and dicumarolare orally bioavailable, and are often termed ‘‘oral anti-coagulants.’’
O
OH
OO
OH
OO
OH
O O
R
OH
OH
R
O
O
O
HN
NH
O
COOH
COOH
HN
NH
O
COOH
Vitamin K-dependentcarboxylase
Epoxidereductase
WarfarinDicumarol
Oral anticoagulants
NADHNAD+
Vitamin K-reduced(active form)
Vitamin K 2,3-epoxide(inactive form)
Glutamate residue incoagulation factor
CO2
O2
γ-Carboxy glutamate residue incoagulation factor
reductase that mediates the regeneration of reduced vitaminK (Fig. 22-14). Because depletion of reduced vitamin K inthe liver prevents the �-carboxylation reaction that is re-quired for the synthesis of biologically active coagulationfactors, the onset of action of the oral anticoagulants parallelsthe half-life of these coagulation factors in the circulation.Of the four affected clotting factors (II, VII, IX, and X),factor VII has the shortest half-life (6 hours). Thus, the phar-macologic effect of a single dose of warfarin is not mani-fested for approximately 18 to 24 hours (i.e., for 3 to 4 factorVII half-lives). This delayed action is one pharmacologicproperty that distinguishes the warfarin class of anticoagu-lants from all the other classes of anticoagulants.
Evidence from studies of long-term rodenticide usesupports the hypothesis that the epoxide reductase is themolecular target of oral anticoagulant action. The use oforal anticoagulants as rodenticides has been a widespreadpractice in farming communities. In some areas of theUnited States, heavy rodenticide use has selected for apopulation of wild rodents that is resistant to 4-hydrox-ycoumarins. In vitro studies of tissues from these rodentshave demonstrated a mutation in the rodent epoxide re-ductase that renders the enzyme resistant to inhibition bythe anticoagulant. Similarly, a small population of patientsis genetically resistant to warfarin because of mutationsin their epoxide reductase gene. These patients require 10

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
404 III Principles of Cardiovascular Pharmacology
TABLE 22-2A Examples of Drugs That Diminish Warfarin’s Anticoagulant Effect
DRUG OR DRUG CLASS MECHANISM
Cholestyramine Inhibits warfarin absorption in the GI tractBarbiturates, carbamazepine, Accelerate warfarin metabolism by inducing hepatic
phenytoin, rifampin P450 enzymes (especially P450 2C9)Vitamin K (reduced) Bypasses warfarin’s inhibition of epoxide reductase
GI, gastrointestinal.
to 20 times the usual dose of warfarin to achieve thedesired anticoagulant effect.
Clinical Uses of Warfarin
Warfarin is often administered to complete a course of anti-coagulation that has been initiated with heparin (see below)and to prevent thrombosis in predisposed patients. Orallyadministered warfarin is nearly 100% bioavailable, and itslevels in the blood peak at 0.5 to 4 hours after administration.In the plasma, 99% of racemic warfarin is bound to plasmaprotein (albumin). Warfarin has a relatively long eliminationhalf-life (approximately 36 hours). The drug is hydroxylatedby the cytochrome P450 system in the liver to inactive me-tabolites that are subsequently eliminated in the urine.
Drug–drug interactions must be carefully considered inpatients taking warfarin. Because warfarin is highly albu-min-bound in the plasma, co-administration of warfarin withother albumin-bound drugs can increase the free (unbound)plasma concentrations of both drugs. In addition, becausewarfarin is metabolized by P450 enzymes in the liver, co-administration of warfarin with drugs that induce and/orcompete for P450 metabolism can affect the plasma concen-trations of both drugs. Table 22-2 lists some of the majorinteractions between warfarin and other drugs.
Among the adverse effects of warfarin, bleeding is themost serious and predictable toxicity. Withdrawal of thedrug may be recommended for patients who suffer fromrepeated bleeding episodes at otherwise therapeutic drug
TABLE 22-2B Examples of Drugs That Enhance Warfarin’s Anticoagulant Effect
DRUG OR DRUG CLASS MECHANISM
Chloral hydrate Displaces warfarin from plasma albuminAmiodarone, clopidogrel, Decrease warfarin metabolism by inhibiting hepatic
ethanol (intoxicating dose), P450 enzymes (especially P450 2C9)fluconazole, fluoxetine,metronidazole,sulfamethoxazole
Broad-spectrum antibiotics Eliminate gut bacteria and thereby reduce availabilityof vitamin K in the GI tract
Anabolic steroids (testosterone) Inhibit synthesis and increase degradation ofcoagulation factors
GI, gastrointestinal.
concentrations. For severe hemorrhage, patients shouldpromptly receive fresh frozen plasma, which contains bio-logically functional clotting factors II, VII, IX, and X. War-farin should never be administered to pregnant women be-cause it can cross the placenta and cause a hemorrhagicdisorder in the fetus. In addition, newborns exposed to warfa-rin in utero may have serious congenital defects character-ized by abnormal bone formation (certain bone matrix pro-teins are �-carboxylated). Rarely, warfarin causes skinnecrosis as a result of widespread thrombosis in the micro-vasculature. The fact that warfarin can cause thrombosis mayseem paradoxical. Recall that, in addition to inhibiting thesynthesis of biologically active coagulation factors II, VII,IX, and X, warfarin also prevents the synthesis of biologi-cally active proteins C and S, which are natural anticoagu-lants. In patients who are genetically deficient in protein Cor protein S (most commonly, patients who are heterozygousfor protein C deficiency), an imbalance between warfarin’seffects on coagulation factors and its effects on proteins Cand S may lead to microvascular thrombosis and skin ne-crosis.
Because warfarin has a narrow therapeutic index and par-ticipates in numerous drug–drug interactions, the pharmaco-dynamic (functional) effect of chronic warfarin therapy mustbe monitored regularly (on the order of every 2 to 4 weeks).Monitoring is most easily performed using the prothrombintime (PT), which is a simple test of the extrinsic and com-mon pathways of coagulation. In this test, the patient’splasma is added to a crude preparation of tissue factor (called

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 405
thromboplastin), and the time for formation of a fibrin clotis measured. Warfarin prolongs the PT mainly because itdecreases the amount of biologically functional factor VIIin the plasma. (Recall that factor VII is the vitamin K-depen-dent coagulation factor with the shortest half-life.) Measure-ment of the PT has been standardized worldwide, and isexpressed as the International Normalized Ratio (INR) ofthe prothrombin time in the patient sample to that in a controlsample, normalized for the international sensitivity index(ISI) of the laboratory’s thromboplastin preparation com-pared to the World Health Organization’s reference throm-boplastin preparation. The formula used to calculate the INRis as follows: INR � [PTpatient / PTcontrol]ISI.
Unfractionated and Low Molecular WeightHeparinsStructure of Heparin
Heparin is a sulfated mucopolysaccharide stored in the se-cretory granules of mast cells. It is a highly sulfated polymerof alternating uronic acid and D-glucosamine. Heparin mole-cules are highly negatively charged; indeed, endogenousheparin is the strongest organic acid in the human body.Commercial preparations of heparin are quite heterogene-ous, with molecular weights ranging from 1 to 30 kDa. Con-ventionally, commercially prepared heparins have been cate-gorized into unfractionated (standard) heparin and lowmolecular weight (LMW) heparin. Unfractionated hepa-rin, which is often prepared from bovine lung and porcineintestinal mucosa, ranges in molecular weight from 5 to 30kDa. LMW heparins are prepared from standard heparinby gel filtration chromatography; their molecular weightsrange from 1 to 5 kDa.
Mechanism of Action of Heparin
Heparin’s mechanism of action depends on the presence ofa specific plasma protease inhibitor, antithrombin III. Anti-thrombin III is actually a misnomer because, in additionto inactivating thrombin, antithrombin III inactivates otherserine proteases including factors IXa, Xa, XIa, and XIIa.Antithrombin III can be considered as a stoichiometric ‘‘sui-cide trap’’ for these serine proteases. When one of the pro-teases encounters an antithrombin III molecule, the serineresidue at the active site of the protease attacks a specificArg–Ser peptide bond in the reactive site of the antithrom-bin. The result of this nucleophilic attack is the formationof a covalent ester bond between the serine residue on theprotease and the arginine residue on the antithrombin III.This results in a stable 1:1 complex between the proteaseand antithrombin molecules, which prevents the proteasefrom further participation in the coagulation cascade.
In the absence of heparin, the binding reaction betweenthe proteases and antithrombin III proceeds slowly. Heparin,acting as a cofactor, accelerates the reaction by 1,000-fold.Heparin has two important physiologic functions: (1) itserves as a catalytic surface to which both antithrombin IIIand the serine proteases bind; and (2) it induces a conforma-tional change in antithrombin III that makes the reactive siteof this molecule more accessible to the attacking protease.The first step of the reaction involves the binding of thenegatively charged heparin to a lysine-rich region (a region
of positive charge) on antithrombin III. Thus, the interactionbetween heparin and antithrombin III is partly electrostatic.During the conjugation reaction between the protease andthe antithrombin, heparin may be released from antithrombinIII and become available to catalyze additional pro-tease–antithrombin III interactions (i.e., heparin is not con-sumed by the conjugation reaction [Fig. 22-9]). In practice,however, heparin’s high negative charge often causes this‘‘sticky’’ molecule to remain electrostatically bound to pro-tease, antithrombin or another nearby molecule in the vicin-ity of a thrombus.
Interestingly, heparins of different molecular weightshave divergent anticoagulant activities. These divergent ac-tivities derive from the differential requirements for heparinbinding exhibited by the inactivation of thrombin and factorXa by antithrombin III (Fig. 22-15). To catalyze most effi-ciently the inactivation of thrombin by antithrombin III, asingle molecule of heparin must bind simultaneously to boththrombin and antithrombin. This ‘‘scaffolding’’ function isrequired in addition to the heparin-induced conformationalchange in antithrombin III that renders the antithrombin sus-ceptible to conjugation with thrombin. In contrast, to cata-lyze the inactivation of factor Xa by antithrombin III, theheparin molecule must bind only to the antithrombin, be-cause the conformational change in antithrombin III inducedby heparin binding is sufficient by itself to render the anti-thrombin susceptible to conjugation with factor Xa. Thus,LMW heparins, which have an average molecular weight of3 to 4 kDa and contain fewer than 18 monosaccharide units,efficiently catalyze the inactivation of factor Xa by anti-thrombin III but less efficiently catalyze the inactivation ofthrombin by antithrombin III. In contrast, unfractionatedheparin, which has an average molecular weight of 20 kDaand contains more than 18 monosaccharide units, is of suffi-cient length to bind simultaneously to thrombin and anti-thrombin III, and therefore efficiently catalyzes the inactiva-tion of both thrombin and factor Xa by antithrombin III.Quantitatively, LMW heparin has a threefold higher ratio ofanti-Xa to anti-thrombin (anti-IIa) activity than does unfrac-tionated heparin. LMW heparin is therefore a more selectivetherapeutic agent than unfractionated heparin. Both LMWheparin and unfractionated heparin use a pentasaccharidestructure of high negative charge to bind antithrombin IIIand to induce the conformational change in antithrombinrequired for the conjugation reactions. This pentasaccharidehas recently been approved for use as a highly selectiveinhibitor of factor Xa (fondaparinux; see below).
Clinical Uses of Heparins
Heparins are used for both prophylaxis and treatment ofthromboembolic diseases. Both unfractionated and LMWheparins are used to prevent propagation of establishedthromboembolic disease such as deep vein thrombosis andpulmonary embolism. For prophylaxis against thrombosis,heparins are used at much lower doses than those indicatedfor the treatment of established thromboembolic disease. Be-cause the enzymatic coagulation cascade functions as an am-plification system (e.g., 1 unit of factor Xa generates 40 unitsof thrombin), the administration of relatively small amountsof circulating heparin at the first generation of factor Xa ishighly effective. Heparins are highly negatively charged, and

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
406 III Principles of Cardiovascular Pharmacology
Unfractionated heparin
Binds to antithrombin III (ATIII)and thrombin(inactivates thrombin)
(about 45 saccharide units,MW ~ 13,500)
Low molecular weight (LMW) heparins
Selective factor Xa inhibitors
Direct thrombin inhibitors
No effect on thrombin
No effect on Xa
(about 15 saccharide units,MW ~ 4,500)
Effect on ThrombinAnticoagulant class Effect on Factor Xa
Binds to antithrombin III (ATIII)via pentasaccharide(sufficient to inactivate Xa)
Binds to antithrombin III (ATIII)but not to thrombin(poorly inactivates thrombin)
Selectively inactivate thrombin
Binds to antithrombin III (ATIII)via pentasaccharide(sufficient to inactivate Xa)
Binds to antithrombin III (ATIII)via pentasaccharide(sufficient to inactivate Xa)
Fondaparinux
Lepirudin
Argatroban
ATIII ATIII
LMWH LMWH
Thrombin
Thrombin
ATIIIATIII
Heparin
Xa
Xa
ATIII
Xa
Heparin
Thrombin
Thrombin
Figure 22-15. Differential effects of unfractionated heparin and low molecular weight heparin on coagulation factor inactivation.Effect on thrombin: To catalyze the inactivation of thrombin, heparin must bind both to antithrombin III via a high-affinity pentasaccharideunit (5) and to thrombin via an additional 13 saccharide unit (here labeled ‘‘Heparin’’). Low molecular weight heparin (LMWH) does notcontain a sufficient number of saccharide units to bind thrombin, and therefore is a poor catalyst for thrombin inactivation. Effect on factorXa: Inactivation of factor Xa requires only the binding of antithrombin III to the high-affinity pentasaccharide unit (5). Both unfractionatedheparin and low molecular weight heparin (and fondaparinux; not shown) are therefore able to catalyze the inactivation of factor Xa.

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 407
neither unfractionated heparin nor LMW heparin can crossthe epithelial cell layer of the gastrointestinal tract. Hence,heparin must be administered parenterally, usually via intra-venous or subcutaneous routes.
Unfractionated heparin is often used in combinationwith antiplatelet agents in the treatment of acute coronarysyndromes. For example, Mr. Soprano was treated with theantiplatelet agents aspirin and eptifibatide and with unfrac-tionated heparin in an attempt to limit the extent of his myo-cardial infarction. Monitoring of unfractionated heparin ther-apy is important for maintaining the anticoagulant effectwithin the therapeutic range, because excessive heparin ad-ministration significantly increases the risk of bleeding.Monitoring is usually performed using the activated partialthromboplastin time (aPTT) assay. The aPTT is a simpletest of the intrinsic and common pathways of coagulation.The patient’s plasma is added to an excess of phospholipid,and fibrin forms at a normal rate only if the factors in theintrinsic and common pathways are present at normal levels.Increasing amounts of unfractionated heparin in the plasmaprolong the time required for the formation of a fibrin clot.
As is true of the other anticoagulants, the major adverseeffect of heparin is bleeding. Thus, it is critical to maintainthe anticoagulant effect of unfractionated heparin within thetherapeutic range in order to prevent the rare, devastatingadverse effect of intracranial hemorrhage. In addition, asmall fraction of patients taking heparin develop heparin-induced thrombocytopenia (HIT). In this syndrome, pa-tients develop antibodies to a hapten created when heparinmolecules bind to the platelet surface. In HIT type 1, theantibody-coated platelets are targeted for removal from thecirculation, and the platelet count decreases by 50% to 75%approximately 5 days into the course of heparin therapy.The thrombocytopenia in HIT type 1 is transient and rapidlyreversible upon heparin withdrawal. In HIT type 2, however,the heparin-induced antibodies not only target the plateletsfor destruction but also act as agonists to activate the plate-lets, leading to platelet aggregation, endothelial injury, andpotentially fatal thrombosis. There is a higher incidence ofHIT in patients receiving unfractionated heparin than inthose receiving LMW heparin.
The LMW heparins enoxaparin, dalteparin and tinza-parin are each fractionated heparins of low molecularweight. As discussed above, these agents are relatively selec-tive for anti-Xa compared to anti-IIa (anti-thrombin) activity.All LMW heparins are approved for use in the preventionand treatment of deep vein thrombosis. Additionally, enoxa-parin and dalteparin have been studied in the treatment ofacute myocardial infarction and as adjuncts to percutaneouscoronary intervention. LMW heparins have a higher thera-peutic index than unfractionated heparin, especially whenused for prophylaxis. For this reason, it is generally not nec-essary to monitor blood activity levels of LMW heparins.Accurate measurement of the anticoagulant effect of LMWheparins requires a specialized assay for anti-factor Xa activ-ity. Because LMW heparins are excreted via the kidneys,care should be taken to avoid excessive anticoagulation inpatients with renal insufficiency.
Selective Factor Xa InhibitorsFondaparinux is a synthetic pentasacharide molecule thatcontains the sequence of five essential carbohydrates neces-
sary for binding to antithrombin III and inducing the confor-mational change in antithrombin required for conjugation tofactor Xa (see above). This agent is therefore is a specificinhibitor of Xa, with negligible anti-IIa (anti-thrombin) ac-tivity. Fondaparinux is approved for prevention and treat-ment of deep vein thrombosis, and is available as a once-daily subcutaneous injection. It is excreted via the kidneysand should not be administered to patients with renal insuffi-ciency.
Direct Thrombin InhibitorsAs discussed above, thrombin plays a number of criticalroles in the hemostatic process (Fig. 22-8). Among othereffects, this clotting factor (1) proteolytically converts fibrin-ogen to fibrin, (2) activates factor XIII, which crosslinksfibrin polymers to form a stable clot, (3) activates platelets,and (4) induces endothelial release of PGI2, t-PA, and PAI-1. Thus, direct thrombin inhibitors would be expected tohave profound effects on coagulation. The currently ap-proved direct thrombin inhibitors include lepirudin, desiru-din, bivalirudin, and argatroban. These agents are specificinhibitors of thrombin, with negligible anti-factor Xa ac-tivity.
Lepirudin, a recombinant 65-amino acid polypeptide de-rived from the medicinal leech protein hirudin, is the proto-typical direct thrombin inhibitor. For years, surgeons haveused medicinal leeches to prevent thrombosis in the finevessels of reattached digits. Lepirudin binds with high affin-ity to two sites on the thrombin molecule — the enzymaticactive site and the ‘‘exosite,’’ a region of the thrombin pro-tein that orients substrate proteins. Lepirudin binding tothrombin prevents the thrombin-mediated activation of fi-brinogen and factor XIII. Lepirudin is a highly effectiveanticoagulant because it can inhibit both free and fibrin-bound thrombin in developing clots, and because lepirudinbinding to thrombin is essentially irreversible. It is approvedfor use in the treatment of heparin-induced thrombocyto-penia. Lepirudin has a short half-life, is available parenter-ally, and is renally excreted. It can be administered withrelative safety to patients with hepatic insufficiency. As withall direct thrombin inhibitors, bleeding is the major side ef-fect of lepirudin, and clotting times must be monitoredclosely. A small percentage of patients may develop anti-hirudin antibodies, limiting the long-term effectiveness ofthis agent as an anticoagulant. Another recombinant formu-lation of hirudin, desirudin, has been approved for prophy-laxis against deep vein thrombosis in patients undergoinghip replacement.
Bivalirudin is a synthetic 20-amino acid peptide that,like lepirudin and desirudin, binds to both the active site andexosite of thrombin and thereby inhibits thrombin activity.Thrombin slowly cleaves an arginine–proline bond in bivali-rudin, leading to reactivation of the thrombin. Bivalirudinis approved for anticoagulation in patients undergoing coro-nary angiography and angioplasty, and may have reducedrates of bleeding relative to heparin for this indication. Thedrug is excreted renally and has a short half-life (25 minutes).
Argatroban is a small-molecule inhibitor of thrombinthat is approved for the treatment of patients with heparin-induced thrombocytopenia. Unlike other direct thrombin in-hibitors, argatroban binds only to the active site of thrombin

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
408 III Principles of Cardiovascular Pharmacology
(i.e., it does not interact with the exosite). Also unlike otherdirect thrombin inhibitors, argatroban is excreted by biliarysecretion, and can therefore be administered with relativesafety to patients with renal insufficiency.
Recombinant Activated Protein C (r-APC)As described above, endogenously activated protein C(APC) exerts an anticoagulant effect by proteolyticallycleaving factors Va and VIIIa. APC also reduces the amountof circulating plasminogen activator inhibitor 1, thereby en-hancing fibrinolysis. Finally, APC reduces inflammation byinhibiting the release of tumor necrosis factor � (TNF-�) bymonocytes. Because enhanced coagulability and inflamma-tion are both hallmarks of septic shock, APC has been testedboth in animal models of this disorder and in humans. Re-combinant activated protein C (r-APC) has been foundto significantly reduce mortality in patients at high risk ofdeath from septic shock, and the U. S. Food and Drug Ad-ministration (FDA) has approved r-APC for the treatmentof patients with severe sepsis who demonstrate evidence ofacute organ dysfunction, shock, oliguria, acidosis, and hypo-xemia. r-APC is not indicated for the treatment of patientswith severe sepsis and a lower risk of death, however. Asis the case with other anticoagulants, r-APC increases therisk of bleeding. This agent is therefore contraindicated inpatients who have recently undergone a surgical procedureand in those with chronic liver failure, kidney failure, orthrombocytopenia.
THROMBOLYTIC AGENTS
Although warfarin, unfractionated and low molecular weightheparins, selective factor Xa inhibitors, and direct thrombininhibitors are effective in preventing the formation and prop-agation of thrombi, these agents are generally ineffectiveagainst pre-existing clots. Thrombolytic agents are used tolyse already-formed clots, and thereby to restore the patencyof an obstructed vessel before distal tissue necrosis occurs.Thrombolytic agents act by converting the inactive zymogenplasminogen to the active protease plasmin (Fig. 22-10). Asnoted above, plasmin is a relatively nonspecific protease thatdigests fibrin to fibrin degradation products. Unfortunately,thrombolytic therapy has the potential to dissolve not onlypathologic thrombi, but also physiologically appropriate fi-brin clots that have formed in response to vascular injury.Thus, the use of thrombolytic agents can lead to hemorrhageof varying severity.
StreptokinaseStreptokinase is a protein produced by �-hemolytic strepto-cocci as a component of that organism’s tissue-destroyingmachinery. The pharmacologic action of streptokinase in-volves two steps — complexation and cleavage. In the com-plexation reaction, streptokinase forms a stable, noncovalent1:1 complex with plasminogen. The complexation reactionproduces a conformational change in plasminogen that ex-poses this protein’s proteolytically active site. Streptokinase-complexed plasminogen, with its active site exposed andavailable, can then proteolytically cleave other plasminogen
molecules to plasmin. In fact, the thermodynamically stablestreptokinase:plasminogen complex is the most catalyticallyefficient plasminogen activator in vitro.
Although streptokinase exerts its most dramatic and po-tentially beneficial effects in fresh thrombi, its use has beenlimited by two factors. First, streptokinase is a foreign pro-tein that is capable of eliciting antigenic responses in humansupon repeated administration. Previous administration ofstreptokinase is a contraindication to its use, because of therisk of anaphylaxis. Second, the thrombolytic actions ofstreptokinase are relatively nonspecific and can result in sys-temic fibrinolysis. Currently, streptokinase is approved fortreatment of ST elevation myocardial infarction and for treat-ment of life-threatening pulmonary embolism.
Recombinant Tissue PlasminogenActivator (t-PA)An ideal thrombolytic agent would be nonantigenic andwould cause local fibrinolysis only at the site of a pathologicthrombus. Tissue plasminogen activator (t-PA) approxi-mates these goals. t-PA is a serine protease produced byhuman endothelial cells; therefore, t-PA is not antigenic.t-PA binds to newly formed (fresh) thrombi with high affin-ity, causing fibrinolysis at the site of a thrombus. Once boundto the fresh thrombus, t-PA undergoes a conformationalchange that renders it a potent activator of plasminogen. Incontrast, t-PA is a poor activator of plasminogen in the ab-sence of fibrin-binding.
Recombinant DNA technology has allowed the produc-tion of recombinant t-PA, generically referred to as al-teplase. Recombinant t-PA is effective at recanalizing oc-cluded coronary arteries, limiting cardiac dysfunction, andreducing mortality following an ST elevation myocardialinfarction. At pharmacologic doses, however, recombinantt-PA can generate a systemic lytic state and (as with otherthrombolytic agents) cause unwanted bleeding, including ce-rebral hemorrhage. Thus, its use is contraindicated in pa-tients who have had a recent hemorrhagic stroke. Like strep-tokinase, t-PA is approved for use in the treatment of patientswith ST elevation myocardial infarction or life-threateningpulmonary embolism. It is also approved for the treatmentof acute ischemic stroke.
TenecteplaseTenecteplase is a genetically engineered variant of t-PA.The molecular modifications in tenecteplase increase its fi-brin specificity relative to t-PA and make tenecteplase moreresistant to plasminogen activator inhibitor 1. Large trialshave shown that tenecteplase is identical in efficacy tot-PA, with similar (and possibly decreased) risk of bleeding.Additionally, tenecteplase has a longer half-life than t-PA.This pharmacokinetic property allows tenecteplase to be ad-ministered as a single weight-based bolus, thus simplifyingadministration.
ReteplaseSimilar to tenecteplase, reteplase is a genetically engineeredvariant of t-PA with longer half-life and increased specificity

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 409
for fibrin. Its efficacy and adverse effect profile are similarto those of streptokinase and t-PA. Because of its longerhalf-life, reteplase can be administered as a ‘‘double bolus’’(two boluses, 30 minutes apart).
INHIBITORS OF ANTICOAGULATION ANDFIBRINOLYSIS
ProtamineProtamine, a low molecular weight polycationic protein, isa chemical antagonist of heparin. This agent rapidly formsa stable complex with the negatively charged heparin mole-cule through multiple electrostatic interactions. Protamine isadministered intravenously to reverse the effects of heparinin situations of life-threatening hemorrhage or great heparinexcess (for example, at the conclusion of coronary arterybypass graft surgery). Protamine is most active against thelarge heparin molecules in unfractionated heparin and it canpartially reverse the anticoagulant effects of low molecularweight heparins, but it is inactive against fondaparinux.
Serine-Protease InhibitorsAprotinin, a naturally occurring polypeptide, is an inhibitorof the serine proteases plasmin, t-PA, and thrombin. By in-hibiting fibrinolysis, aprotinin promotes clot stabilization.Inhibition of thrombin may also promote platelet activity bypreventing platelet hyperstimulation. At higher doses, aprot-inin may also inhibit kallikrein and thereby (paradoxically)inhibit the coagulation cascade. Clinical trials have demon-strated decreased perioperative bleeding and erythrocytetransfusion requirement in patients treated with aprotininduring cardiac surgery. However, these positive findingshave been tempered by recent evidence suggesting that,compared to other antifibrinolytic agents, aprotinin may in-crease the risk of postoperative acute renal failure.
Lysine AnaloguesAminocaproic acid and tranexamic acid are analogues oflysine that bind to and inhibit plasminogen and plasmin.Like aprotinin, these agents are used to reduce perioperativebleeding during coronary artery bypass grafting. Unlikeaprotinin, these agents may not increase the risk of postoper-ative acute renal failure.
Conclusion and Future Directions
Hemostasis is a highly regulated process that maintains thefluidity of blood in normal vessels and initiates rapid forma-tion of a stable fibrin-based clot in response to vascular in-jury. Pathologic thrombosis results from endothelial injury,abnormal blood flow, and hypercoagulability. Antiplateletagents, anticoagulants, and thrombolytic agents target differ-ent stages of thrombosis and thrombolysis. Antiplateletagents interfere with platelet adhesion, the platelet releasereaction, and platelet aggregation; these agents can providepowerful prophylaxis against thrombosis in susceptible indi-
viduals. Anticoagulants primarily target plasma coagulationfactors and disrupt the coagulation cascade by inhibiting cru-cial intermediates. After a fibrin clot has been established,thrombolytic agents mediate dissolution of the clot by pro-moting the conversion of plasminogen to plasmin. Theseclasses of pharmacologic agents can be administered eitherindividually or in combination, to prevent or disrupt throm-bosis and to restore the patency of blood vessels occludedby thrombus.
Future development of new antiplatelet, anticoagulant,and thrombolytic agents will be forced to contend with twomajor constraints. First, for many clinical indications in thisfield, highly effective, orally bioavailable, and inexpensivetherapeutic agents are already available: these include theantiplatelet drug aspirin and the anticoagulant warfarin. Sec-ond, virtually every antithrombotic and thrombolytic agentis associated with the mechanism-based toxicity of bleeding,and this side effect is likely to plague new agents underdevelopment. Nonetheless, opportunities remain for the de-velopment of safer and more effective therapies. It is likelythat pharmacogenomic techniques (see Chapter 52, Pharma-cogenomics) will be capable of identifying individuals inthe population who carry an elevated genetic risk of throm-bosis, and such individuals may benefit from long-term anti-thrombotic treatment. Combinations of antiplatelet agents,low molecular weight heparins, orally bioavailable directthrombin inhibitors (such as the snake-venom–derived prod-rug ximelagatran, which is not approved for use in theU.S.), and new agents that target currently unexploited com-ponents of hemostasis (such as inhibitors of the factor VIIa/tissue factor pathway) could all be useful in these settings.At the other end of the spectrum, there remains a great needfor new agents that can achieve rapid, noninvasive, conven-ient, and selective lysis of acute thromboses associated withlife-threatening emergencies such as ST elevation myocar-dial infarction and stroke. Carefully designed clinical trialswill be critical to optimize the indications, dose, and durationof treatment for such drugs and drug combinations.
Suggested Readings
Baggish AL, Sabatine MS. Clopidogrel use in coronary artery dis-ease. Exp Rev Cardiovasc Ther 2006;4:7–15. (Reviews pharma-cology and expanding clinical applications for clopidogrel.)
Bates SM, Ginsberg JS. Treatment of deep-vein thrombosis. N EnglJ Med 2004;351:268–277. (Reviews treatment options for deepvein thrombosis.)
Bauer KA. New anticoagulants: anti IIa vs anti Xa—is one better?J Thromb Thrombolysis 2006;21:67–72. (Summarizes clinicaltrial data on selective factor Xa inhibitors and direct thrombininhibitors.)
Brass LF. The molecular basis for platelet activation. In: HoffmanR, Benz EJ, Shatill SJ, et al, eds. Hematology: basic principlesand practice. 4th ed. Philadelphia: Churchill Livingstone; 2004.(Detailed and mechanistic description of platelet activation.)
Di Nisio M, Middeldorp S, Buller HR. Direct thrombin inhibitors.N Engl J Med 2005;353:1028–1040. (Reviews mechanism ofaction and clinical indications for direct thrombin inhibitors.)
Franchini M, Veneri D, Salvagno GL, et al. Inherited thrombophi-lia. Crit Rev Clin Lab Sci 2006;43:249–290. (Reviews epidemiol-ogy, pathophysiology, and treatment of hypercoagulable states.)

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
410 III Principles of Cardiovascular Pharmacology
Furie B, Furie BC. Thrombus formation in vivo. J Clin Invest 2005;115:3355–3362. (Reviews molecular and cellular mechanismsof primary and secondary hemostasis in vivo.)
Grosser T, Fries S, FitzGerald GA. Biological basis for the cardio-vascular consequences of COX-2 inhibition: therapeutic chal-lenges and opportunities. J Clin Invest 2006;116:4–15. (Reviews
effects of COX-2 inhibition in cellular, animal, and humanstudies.)
Hirsh J, O’Donnell M, Weitz JI. New anticoagulants. Blood 2005;105:453–463. (Reviews anticoagulants in clinical development.)
Levy JH. Hemostatic agents. Transfusion 2004;44:58S–62S. (Re-views aprotinin, aminocaproic acid, and tranexamic acid.)

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 411
Dru
gSu
mm
ary
Tabl
eCh
apte
r22
Ph
arm
acol
ogy
ofH
emos
tasi
san
dTh
rom
bos
is
Seri
ous
and
Com
mon
Dru
gCl
inic
alA
pplic
atio
nsA
dver
seEf
fect
sCo
ntra
indi
cati
ons
Ther
apeu
tic
Cons
ider
atio
nsA
NTI
PLA
TELE
TA
GEN
TSCy
cloo
xyge
nase
Inhi
bito
rsM
echa
nism
—In
hibi
tpl
atel
etcy
cloo
xyge
nase
,th
ereb
ybl
ocki
ngth
rom
boxa
neA
2ge
nera
tion
and
inhi
bitin
gpl
atel
etre
leas
ere
actio
nan
dpl
atel
etag
greg
atio
n
Asp
irin
Prop
hyla
xis
agai
nst
tran
sien
tG
Ibl
eedi
ng,
acut
ere
nal
insu
ffic
ienc
y,N
SAID
-ind
uced
sens
itivi
tyIn
hibi
tsC
OX
-1an
dC
OX
-2no
nsel
ectiv
ely
isch
emic
atta
ck,
myo
card
ial
thro
mbo
cyto
peni
a,he
pati
tis,
angi
oede
ma,
reac
tions
Use
caut
ious
lyin
patie
nts
with
GI
lesi
ons,
infa
rctio
n,an
das
thm
a,R
eye’
ssy
ndro
me
Chi
ldre
nw
ithch
icke
npox
orfl
u-im
pair
edre
nal
func
tion,
hypo
thro
mbi
nem
ia,
thro
mbo
embo
licdi
sord
ers
Tin
nitu
s,dy
spep
sia,
occu
ltbl
eedi
ng,
like
synd
rom
esvi
tam
inK
defi
cien
cy,
thro
mbo
ticT
reat
men
tof
acut
eco
rona
rypr
olon
ged
blee
ding
time,
rash
G6P
Dde
fici
ency
thro
mbo
cyto
peni
cpu
rpur
a,or
hepa
ticim
pair
men
tsy
ndro
mes
Ble
edin
gdi
sord
ers
such
asC
o-ad
min
istr
atio
nw
itham
inog
lyco
side
s,Pr
even
tion
ofre
occl
usio
nin
hem
ophi
lia,
von
Will
ebra
nd’s
bum
etan
ide,
capr
eom
ycin
,ci
spla
tin,
coro
nary
reva
scul
ariz
atio
ndi
seas
e,or
imm
une
eryt
hrom
ycin
,et
hacr
ynic
acid
,fu
rose
mid
e,or
proc
edur
esan
dst
ent
thro
mbo
cyto
peni
ava
ncom
ycin
may
pote
ntia
teot
otox
icef
fect
sim
plan
tatio
nC
o-ad
min
istr
atio
nw
itham
mon
ium
chlo
ride
orA
rthr
itis,
juve
nile
arth
ritis
,ot
her
urin
eac
idif
iers
may
lead
toas
piri
nto
xici
tyrh
eum
atic
feve
rA
spir
inan
tago
nize
sur
icos
uric
effe
cts
ofM
ildpa
inor
feve
rph
enyl
buta
zone
,pr
oben
ecid
,an
dsu
lfin
pyra
zone
;av
oid
co-a
dmin
istr
atio
nw
ithth
ese
agen
ts
Phos
phod
iest
eras
eIn
hibi
tors
Mec
hani
sm—
Inhi
bit
plat
elet
cAM
Pde
grad
atio
nan
dth
ereb
yde
crea
sepl
atel
etag
greg
abili
ty
Dip
yrid
amol
ePr
ophy
laxi
sag
ains
tE
xace
rbat
ion
ofan
gina
(IV
rout
e),
rare
Hyp
erse
nsiti
vity
todi
pyri
dam
ole
Wea
kan
tipla
tele
tef
fect
thro
mbo
embo
licdi
sord
ers;
myo
card
ial
infa
rcti
on,
rare
vent
ricu
lar
Usu
ally
adm
inis
tere
din
com
bina
tion
with
war
fari
nA
ltern
ativ
eto
exer
cise
inar
rhyt
hmia
,ra
rebr
onch
ospa
smor
aspi
rin
thal
lium
myo
card
ial
perf
usio
nA
bnor
mal
EC
G,
hypo
tens
ion
(IV
rout
e),
Has
vaso
dila
tory
prop
ertie
s;m
aypa
rado
xica
llyim
agin
gab
dom
inal
disc
omfo
rt(o
ral
rout
e),
indu
cean
gina
byca
usin
gth
eco
rona
ryst
eal
dizz
ines
s,he
adac
heph
enom
enon
AD
PR
ecep
tor
Path
way
Inhi
bito
rsM
echa
nism
—C
oval
ently
mod
ify
plat
elet
AD
Pre
cept
or,
ther
eby
prev
entin
gre
cept
orsi
gnal
ing
and
irre
vers
ibly
inhi
bitin
gA
DP
-dep
ende
ntpl
atel
etac
tivat
ion
path
way
Tic
lopi
dine
Seco
ndar
ypr
even
tion
ofA
plas
tic
anem
ia,
neut
rope
nia,
thro
mbo
tic
Act
ive
blee
ding
diso
rder
Use
islim
ited
byas
soci
ated
mye
loto
xici
tyth
rom
botic
stro
kes
inpa
tient
sth
rom
bocy
tope
nic
purp
ura
Neu
trop
enia
,th
rom
bocy
tope
nia
Req
uire
sa
load
ing
dose
toac
hiev
eim
med
iate
anti-
into
lera
ntof
aspi
rin
Prur
itus,
rash
,dy
spep
sia,
abno
rmal
liver
Seve
reliv
erdy
sfun
ctio
npl
atel
etef
fect
Prev
entio
nof
sten
tth
rom
bosi
sfu
nctio
nte
sts,
dizz
ines
s(i
nco
mbi
natio
nw
ithas
piri
n)
Clo
pido
grel
Seco
ndar
ypr
even
tion
ofA
tria
lfi
bril
lati
on,
hear
tfa
ilur
e,er
ythe
ma
Act
ive
blee
ding
diso
rder
Mor
efa
vora
ble
adve
rse
effe
ctpr
ofile
than
athe
rosc
lero
ticev
ents
inm
ulti
form
e,G
Ihe
mor
rhag
e(i
ntic
lopi
dine
;si
gnif
ican
tlyle
ssm
yelo
toxi
city
than
patie
nts
with
rece
ntco
mbi
nati
onw
ith
aspi
rin)
,ve
ryra
retic
lopi
dine
myo
card
ial
infa
rctio
n,st
roke
,an
emia
orne
utro
peni
a,ra
rein
trac
rani
alR
equi
res
alo
adin
gdo
seto
achi
eve
imm
edia
tean
ti-or
peri
pher
alva
scul
ardi
seas
ehe
mor
rhag
e,ab
norm
alre
nal
func
tion
plat
elet
effe
ctA
cute
coro
nary
synd
rom
esC
hest
pain
,ed
ema,
hype
rten
sion
,pu
rpur
a,Pr
even
tion
ofst
ent
thro
mbo
sis
rare
abno
rmal
liver
func
tion
test
s,G
I(i
nco
mbi
natio
nw
ithas
piri
n)di
scom
fort
,ar
thra
lgia
,di
zzin
ess
(Con
tinue
d)

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
412 III Principles of Cardiovascular Pharmacology
Dru
gSu
mm
ary
Tabl
eCh
apte
r22
Ph
arm
acol
ogy
ofH
emos
tasi
san
dTh
rom
bos
is(C
ontin
ued
)
Seri
ous
and
Com
mon
Dru
gCl
inic
alA
pplic
atio
nsA
dver
seEf
fect
sCo
ntra
indi
cati
ons
Ther
apeu
tic
Cons
ider
atio
nsG
PIIb
–II
IaA
ntag
onis
tsM
echa
nism
—B
ind
topl
atel
etre
cept
orG
PII
b–II
Iaan
dth
ereb
ypr
even
tbi
ndin
gof
fibr
inog
enan
dot
her
adhe
sive
ligan
ds
Ept
ifib
atid
eA
cute
coro
nary
synd
rom
esM
ajor
blee
ding
,in
trac
ereb
ral
hem
orrh
age,
His
tory
ofbl
eedi
ngdi
athe
sis
orA
void
co-a
dmin
istr
atio
nw
itha
seco
ndG
PIIb
-III
aPe
rcut
aneo
usco
rona
ryth
rom
bocy
tope
nia
rece
ntab
norm
albl
eedi
ngan
tago
nist
inte
rven
tion
Hyp
oten
sion
,bl
eedi
ngC
onco
mita
ntad
min
istr
atio
nof
aM
inim
ize
use
ofar
teri
alan
dve
nous
punc
ture
s,se
cond
glyc
opro
tein
IIb–
IIIa
urin
ary
cath
eter
s,an
dna
sotr
ache
alan
dan
tago
nist
naso
gast
ric
tube
sR
ecen
tm
ajor
surg
ery
Ept
ifib
atid
eis
asy
nthe
ticpe
ptid
ede
liver
edvi
aR
ecen
tst
roke
orhi
stor
yof
pare
nter
alad
min
istr
atio
nhe
mor
rhag
icst
roke
Intr
acra
nial
hem
orrh
age,
mas
s,or
arte
riov
enou
sm
alfo
rmat
ion
Seve
reun
cont
rolle
dhy
pert
ensi
on
Abc
ixim
abA
djun
ctto
perc
utan
eous
Sam
eas
epti
fiba
tide
Sam
eas
eptif
ibat
ide
Sam
eth
erap
eutic
cons
ider
atio
nsas
epiti
fiba
tide,
coro
nary
inte
rven
tion
orex
cept
abci
xim
abis
ach
imer
icm
ouse
–hum
anat
here
ctom
yto
prev
ent
acut
em
onoc
lona
lan
tibod
yca
rdia
cis
chem
icco
mpl
icat
ions
Add
ing
abci
xim
abto
conv
entio
nal
antit
hrom
botic
Uns
tabl
ean
gina
not
resp
ondi
ngth
erap
yre
duce
sbo
thlo
ng-t
erm
and
shor
t-te
rmto
conv
entio
nal
ther
apy
inis
chem
icev
ents
inpa
tient
sun
derg
oing
high
-ris
kpa
tient
ssc
hedu
led
for
coro
nary
angi
opla
sty
perc
utan
eous
coro
nary
inte
rven
tion
Tir
ofib
anA
cute
coro
nary
synd
rom
esin
Sam
eas
epti
fiba
tide
;A
ddit
iona
lly,
Sam
eas
eptif
ibat
ide
Sam
eth
erap
eutic
cons
ider
atio
nsas
eptif
ibat
ide,
patie
ntun
derg
oing
angi
opla
sty
coro
nary
arte
rydi
ssec
tion
isra
rely
exce
pttir
ofib
anis
ano
npep
tide
tyro
sine
anal
ogue
orat
here
ctom
yor
man
aged
obse
rved
med
ical
ly
AN
TICO
AG
ULA
NTS
War
fari
nM
echa
nism
—In
hibi
the
patic
epox
ide
redu
ctas
eth
atca
taly
zes
the
rege
nera
tion
ofre
duce
dvi
tam
inK
,w
hich
isre
quir
edfo
rsy
nthe
sis
ofbi
olog
ical
lyac
tive
coag
ulat
ion
fact
ors
II,
VII
,IX
,an
dX
and
antic
oagu
lant
prot
eins
Can
dS
War
fari
nPr
ophy
laxi
san
dtr
eatm
ent
ofC
hole
ster
olem
boli
zati
onsy
ndro
me,
skin
Preg
nanc
yM
onito
ring
isre
quir
edby
usin
gth
epr
othr
ombi
npu
lmon
ary
embo
lism
and
othe
rti
ssue
necr
osis
,he
mor
rhag
e,H
emor
rhag
icte
nden
cyor
bloo
dtim
e(P
T),
expr
esse
das
the
inte
rnat
iona
lPr
ophy
laxi
san
dtr
eatm
ent
ofhe
pati
tis,
hype
rsen
siti
vity
reac
tion
dysc
rasi
ano
rmal
ized
ratio
(IN
R)
deep
vein
thro
mbo
sis
Ble
edin
gte
nden
cyas
soci
ated
with
Dru
g–dr
ugin
tera
ctio
nsm
ust
beca
refu
llyPr
ophy
laxi
san
dtr
eatm
ent
ofac
tive
ulce
ratio
nor
blee
ding
due
cons
ider
edw
ithw
arfa
rin
(ref
erto
Tab
le22
-2fo
rsy
stem
icem
bolis
maf
ter
tom
ucos
alle
sion
s,ex
ampl
esof
impo
rtan
tin
tera
ctio
ns);
Co-
myo
card
ial
infa
rctio
nce
rebr
ovas
cula
rhe
mor
rhag
e,ad
min
istr
atio
nof
war
fari
nw
ithot
her
albu
min
-Pr
ophy
laxi
san
dtr
eatm
ent
ofce
rebr
alor
aort
ican
eury
sm,
boun
ddr
ugs
can
incr
ease
the
free
(unb
ound
)sy
stem
icem
bolis
mas
soci
ated
peri
card
itis
and
peri
card
ial
plas
ma
conc
entr
atio
nsof
both
drug
s;C
o-w
ithat
rial
fibr
illat
ion,
effu
sion
,ba
cter
ial
endo
card
itis
adm
inis
trat
ion
ofdr
ugs
that
indu
cean
d/or
rheu
mat
iche
art
dise
ase
with
Rec
ent
eye,
brai
n,or
spin
alsu
rger
yco
mpe
tefo
rP4
50m
etab
olis
mca
naf
fect
the
hear
tva
lve
dam
age,
orSe
vere
unco
ntro
lled
hype
rten
sion
plas
ma
conc
entr
atio
nsof
both
drug
spr
osth
etic
mec
hani
cal
hear
tva
lve

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 413
Thr
eate
ned
abor
tion,
ecla
mps
ia,
War
fari
nsh
ould
neve
rbe
give
nto
preg
nant
pree
clam
psia
wom
enbe
caus
eit
can
caus
ea
hem
orrh
agic
Reg
iona
lor
lum
bar
bloc
kdi
sord
eran
d/or
cong
enita
lde
fect
sin
the
fetu
san
esth
esia
War
fari
nca
nca
use
skin
necr
osis
asa
resu
ltof
His
tory
ofw
arfa
rin-
indu
ced
wid
espr
ead
thro
mbo
sis
inth
em
icro
vasc
ulat
ure
necr
osis
For
seve
rehe
mor
rhag
edu
eto
war
fari
n,pa
tient
sU
nsup
ervi
sed
patie
nts
with
shou
ldpr
ompt
lyre
ceiv
efr
esh
froz
enpl
asm
aps
ycho
sis,
seni
lity,
alco
holis
m,
orla
ckof
coop
erat
ion,
and
espe
cial
lyth
ose
with
risk
offa
lling
Unf
ract
iona
ted
Hep
arin
and
Low
Mol
ecul
arW
eigh
tH
epar
ins
Mec
hani
sm—
Unf
ract
iona
ted
hepa
rin:
com
bine
sw
ithan
tithr
ombi
nII
Ian
din
hibi
tsse
cond
ary
hem
osta
sis
via
nons
elec
tive
inac
tivat
ion
ofth
rom
bin
(fac
tor
IIa)
,fa
ctor
Xa,
fact
orIX
a,fa
ctor
XIa
,an
dfa
ctor
XII
a.L
MW
hepa
rins
:co
mbi
new
ithan
tithr
ombi
nII
Ian
din
hibi
tse
cond
ary
hem
osta
sis
via
rela
tivel
y(3
-fol
d)se
lect
ive
inac
tivat
ion
offa
ctor
Xa.
Unf
ract
iona
ted
Prev
entio
nan
dtr
eatm
ent
of:
Hem
orrh
age,
hepa
rin-
indu
ced
Hep
arin
-ind
uced
thro
mbo
cyto
peni
aT
here
isa
high
erin
cide
nce
ofhe
pari
n-in
duce
dhe
pari
n—
Pulm
onar
yem
bolis
mth
rom
bocy
tope
nia,
hype
rsen
siti
vity
Act
ive
maj
orbl
eedi
ngth
rom
bocy
tope
nia
inpa
tient
sre
ceiv
ing
—D
eep
vein
thro
mbo
sis
reac
tion
sin
clud
ing
anap
hyla
ctoi
dB
leed
ing
tend
enci
essu
chas
unfr
actio
nate
dhe
pari
nth
anin
thos
ere
ceiv
ing
—C
ereb
ral
thro
mbo
sis
reac
tion
she
mop
hilia
,th
rom
bocy
tope
nia,
orL
MW
hepa
rin
—L
eft
vent
ricu
lar
thro
mbu
sO
verl
ypr
olon
ged
clot
ting
time,
muc
osal
hepa
ticdi
seas
ew
ithA
ntih
ista
min
es,
card
iac
glyc
osid
es,
nico
tine,
and
Prev
entio
nof
syst
emic
ulce
ratio
n,he
mat
oma
hypo
prot
hrom
bine
mia
tetr
acyc
lines
may
part
ially
coun
tera
ctem
bolis
mas
soci
ated
with
Susp
ecte
din
trac
rani
alhe
mor
rhag
ean
ticoa
gula
ntef
fect
myo
card
ial
infa
rctio
nO
pen
ulce
rativ
ew
ound
s,ex
tens
ive
Cep
halo
spor
ins,
peni
cilli
ns,
oral
antic
oagu
lant
s,U
nsta
ble
angi
nade
nuda
tion
ofsk
inan
dpl
atel
etin
hibi
tors
may
incr
ease
antic
oagu
lant
Ope
n-he
art
surg
ery
Con
ditio
nsth
atca
use
incr
ease
def
fect
Dis
sem
inat
edin
trav
ascu
lar
capi
llary
perm
eabi
lity
Dis
cour
age
conc
omita
ntus
eof
herb
ssu
chas
dong
coag
ulat
ion
Bac
teri
alen
doca
rditi
squ
ai,
garl
ic,
ging
er,
gink
go,
mot
herw
ort,
and
red
Mai
ntai
npa
tenc
yof
IVSe
vere
hype
rten
sion
clov
erdu
eto
incr
ease
dri
skof
blee
ding
cath
eter
s
LM
Whe
pari
ns:
Prev
entio
nan
dtr
eatm
ent
ofH
emor
rhag
e,th
rom
bocy
tope
nia,
abno
rmal
Act
ive
maj
orbl
eedi
ngA
dmin
iste
red
asw
eigh
t-ba
sed
subc
utan
eous
Eno
xapa
rin
deep
vein
thro
mbo
sis
(all
live
rfu
ncti
onte
sts,
anap
hyla
ctoi
dH
epar
in-i
nduc
edth
rom
bocy
tope
nia
inje
ctio
nD
alte
pari
nL
MW
hepa
rins
)re
acti
on,
spin
alhe
mat
oma
Hyp
erse
nsiti
vity
tohe
pari
nor
pork
Avo
idex
cess
ive
antic
oagu
latio
nin
patie
nts
with
Tin
zapa
rin
Tre
atm
ent
ofac
ute
coro
nary
Ede
ma,
diar
rhea
,na
usea
,he
mat
oma,
prod
ucts
rena
lin
suff
icie
ncy
synd
rom
esan
dad
junc
tto
norm
ocyt
ichy
poch
rom
ican
emia
,R
enal
insu
ffic
ienc
y(r
elat
ive
perc
utan
eous
coro
nary
conf
usio
n,pa
in,
dysp
nea,
feve
r,lo
cal
cont
rain
dica
tion)
inte
rven
tion
(eno
xapa
rin
and
irri
tatio
nda
ltepa
rin)
Sele
ctiv
eFa
ctor
XaIn
hibi
tors
Mec
hani
sm—
Com
bine
with
antit
hrom
bin
III
and
inhi
bits
seco
ndar
yhe
mos
tasi
svi
ahi
ghly
sele
ctiv
ein
activ
atio
nof
fact
orX
a
Fon
dapa
rinu
xPr
ophy
laxi
san
dtr
eatm
ent
ofH
emor
rhag
e,th
rom
bocy
tope
nia,
abno
rmal
Act
ive
maj
orbl
eedi
ngFo
ndap
arin
uxis
ape
ntas
acha
ride
com
pose
dof
the
deep
vein
thro
mbo
sis
live
rfu
ncti
onte
sts,
anap
hyla
ctoi
dSe
vere
rena
lim
pair
men
tes
sent
ial
five
carb
ohyd
rate
sne
cess
ary
for
bind
ing
Prop
hyla
xis
and
trea
tmen
tof
reac
tion
,sp
inal
hem
atom
aB
acte
rial
endo
card
itis
antit
hrom
bin
III;
Itis
asp
ecif
icin
dire
ctin
hibi
tor
pulm
onar
yem
bolis
mE
dem
a,di
arrh
ea,
naus
ea,
hem
atom
a,of
fact
orX
a,w
ithne
glig
ible
anti-
thro
mbi
n(a
nti-
norm
ocyt
ichy
poch
rom
ican
emia
,II
a)ac
tivity
conf
usio
n,pa
in,
dysp
nea,
feve
r,lo
cal
Avo
idex
cess
ive
antic
oagu
latio
nin
patie
nts
with
irri
tatio
nre
nal
insu
ffic
ienc
yFo
ndap
arin
uxus
eha
sno
tbe
enas
soci
ated
with
hepa
rin-
indu
ced
thro
mbo
cyto
peni
a
(Con
tinue
d)

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
414 III Principles of Cardiovascular Pharmacology
Dru
gSu
mm
ary
Tabl
eCh
apte
r22
Ph
arm
acol
ogy
ofH
emos
tasi
san
dTh
rom
bos
is(C
ontin
ued
)
Seri
ous
and
Com
mon
Dru
gCl
inic
alA
pplic
atio
nsA
dver
seEf
fect
sCo
ntra
indi
cati
ons
Ther
apeu
tic
Cons
ider
atio
nsD
irec
tTh
rom
bin
Inhi
bito
rsM
echa
nism
—B
ind
dire
ctly
toth
rom
bin
and
ther
eby
inhi
bit
seco
ndar
yhe
mos
tasi
s
Hir
udin
-rel
ated
Hep
arin
-ind
uced
Hea
rtfa
ilur
e,ga
stro
inte
stin
alhe
mor
rhag
e,A
ctiv
em
ajor
blee
ding
Rec
ombi
nant
poly
pept
ides
base
don
the
med
icin
alag
ents
:th
rom
bocy
tope
nia
(lep
irud
in)
blee
ding
,ab
norm
alli
ver
func
tion
test
s,Pr
egna
ncy
leec
hpr
otei
nhi
rudi
n;bi
ndto
both
activ
esi
tean
dL
epir
udin
Prop
hyla
xis
agai
nst
deep
vein
anap
hyla
xis,
hype
rten
sion
,hy
pote
nsio
n,Se
vere
unco
ntro
lled
hype
rten
sion
exos
iteof
thro
mbi
nD
esir
udin
thro
mbo
sis
(des
irud
in)
cere
bral
isch
emia
,in
trac
rani
alSe
vere
rena
lim
pair
men
tL
epir
udin
inhi
bits
both
free
and
fibr
in-b
ound
Biv
alir
udin
Ant
icoa
gula
tion
inpa
tient
she
mor
rhag
e,pe
riph
eral
nerv
epa
raly
sis,
thro
mbi
nun
derg
oing
coro
nary
faci
alne
rve
para
lysi
s,he
mat
uria
,re
nal
Aft
erbi
valir
udin
bind
sto
thro
mbi
n,th
eth
rom
bin
angi
ogra
phy
and
angi
opla
sty
fail
ure,
extr
insi
cal
lerg
icre
spir
ator
ysl
owly
clea
ves
anar
gini
ne–p
rolin
ebo
ndin
(biv
alir
udin
)di
seas
e,pn
eum
onia
,se
psis
biva
lirud
in,
lead
ing
tore
activ
atio
nof
thro
mbi
nC
utan
eous
hype
rsen
sitiv
ity,
anem
ia,
feve
rD
ose
adju
stm
ent
isre
quir
edin
patie
nts
with
rena
lin
suff
icie
ncy
beca
use
thes
eag
ents
are
excr
eted
via
the
kidn
eys
Arg
atro
ban
Cor
onar
yar
tery
thro
mbo
sis
Car
diac
arre
st,
cere
brov
ascu
lar
diso
rder
,A
ctiv
em
ajor
blee
ding
Bin
dsto
activ
esi
tebu
tno
tex
osite
ofth
rom
bin
Prop
hyla
xis
inpe
rcut
aneo
usve
ntri
cula
rta
chyc
ardi
a,se
psis
,Se
vere
liver
impa
irm
ent
Dos
ead
just
men
tis
requ
ired
inpa
tient
sw
ithliv
erco
rona
ryin
terv
entio
nhy
pote
nsio
ndi
seas
ebe
caus
ear
gatr
oban
isex
cret
edin
the
bile
Hep
arin
-ind
uced
thro
mbo
cyto
peni
a
Rec
ombi
nant
Act
ivat
edPr
otei
nC
(r-A
PC)
Mec
hani
sm—
Pro
teol
ytic
ally
inac
tivat
esfa
ctor
sV
aan
dV
IIIa
;m
ayal
soex
ert
anti-
infl
amm
ator
yef
fect
byin
hibi
ting
tum
orne
cros
isfa
ctor
prod
uctio
nan
dbl
ocki
ngle
ukoc
yte
adhe
sion
tose
lect
ins
Rec
ombi
nant
Seve
rese
psis
with
orga
nH
emor
rhag
eA
ctiv
ein
tern
albl
eedi
ngPr
olon
gsac
tivat
edpa
rtia
lth
rom
bopl
astin
time
acti
vate
dpr
otei
ndy
sfun
ctio
nan
dhi
ghri
skof
Intr
acra
nial
mas
s(a
PTT
)bu
tha
slit
tleef
fect
onpr
othr
ombi
ntim
eC
(r-A
PC
)de
ath
Hem
orrh
agic
stro
kew
ithin
3(P
T)
mon
ths
Rec
ent
intr
acra
nial
orin
tras
pina
lsu
rger
yor
seve
rehe
adtr
aum
aw
ithin
2m
onth
sPr
esen
ceof
anep
idur
alca
thet
erM
ajor
trau
ma
with
anin
crea
sed
risk
oflif
e-th
reat
enin
gbl
eedi
ng
THR
OM
BO
LYTI
CA
GEN
TSM
echa
nism
—P
rote
olyt
ical
lyac
tivat
epl
asm
inog
ento
form
plas
min
,w
hich
dige
sts
fibr
into
fibr
inde
grad
atio
npr
oduc
ts
Stre
ptok
inas
eST
elev
atio
nm
yoca
rdia
lC
ardi
acar
rhyt
hmia
,ch
oles
tero
lem
bolu
sA
ctiv
ein
tern
albl
eedi
ngor
know
nSt
rept
okin
ase
isa
fore
ign
bact
eria
lpr
otei
nth
atca
nin
farc
tion
synd
rom
e,m
ajor
blee
ding
,an
aphy
lact
oid
blee
ding
diat
hesi
sel
icit
antig
enic
resp
onse
sin
hum
ans
upon
Art
eria
lth
rom
bosi
sre
acti
on,
poly
neur
opat
hy,
non-
card
ioge
nic
Intr
acra
nial
orin
tras
pina
lsu
rger
yre
peat
edad
min
istr
atio
n;pr
ior
adm
inis
trat
ion
ofD
eep
vein
thro
mbo
sis
pulm
onar
yed
ema,
hypo
tens
ion
ortr
aum
aw
ithin
2m
onth
sst
rept
okin
ase
isa
cont
rain
dica
tion
tous
edu
eto
Pulm
onar
yem
bolis
mFe
ver,
shiv
erin
gC
ereb
rova
scul
arac
cide
ntw
ithin
2th
eri
skof
anap
hyla
xis
Intr
a-ar
teri
alor
intr
aven
ous
mon
ths
Thr
ombo
lytic
actio
nsof
stre
ptok
inas
ear
eca
thet
eroc
clus
ion
Intr
acra
nial
mas
sre
lativ
ely
nons
peci
fic
and
can
resu
ltin
syst
emic
Seve
reun
cont
rolle
dhy
pert
ensi
onfi
brin
olys
is

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Chapter 22 Pharmacology of Hemostasis and Thrombosis 415
Rec
ombi
nant
tiss
ueA
cute
myo
card
ial
infa
rctio
nC
ardi
acar
rhyt
hmia
,ch
oles
tero
lem
bolu
sSa
me
asst
rept
okin
ase
Bin
dsto
new
lyfo
rmed
(fre
sh)
thro
mbi
with
high
plas
min
ogen
Acu
tece
rebr
ovas
cula
rsy
ndro
me,
gast
roin
test
inal
hem
orrh
age,
affi
nity
,ca
usin
gfi
brin
olys
isat
the
site
ofa
acti
vato
r(t
-PA
)th
rom
bosi
sra
real
lerg
icre
acti
on,
intr
acra
nial
thro
mbu
s(A
ltep
lase
)Pu
lmon
ary
embo
lism
hem
orrh
age,
seps
isA
sw
ithot
her
thro
mbo
lytic
agen
ts,
t-PA
can
Cen
tral
veno
usca
thet
erge
nera
tea
syst
emic
lytic
stat
ean
dca
use
occl
usio
nun
wan
ted
blee
ding
Ten
ecte
plas
eA
cute
myo
card
ial
infa
rctio
nC
ardi
acar
rhyt
hmia
,ch
oles
tero
lem
bolu
sSa
me
asst
rept
okin
ase
Gen
etic
ally
engi
neer
edva
rian
tsof
t-PA
with
Ret
epla
sesy
ndro
me,
maj
orbl
eedi
ng,
alle
rgic
incr
ease
dsp
ecif
icity
for
fibr
inre
acti
on,
anap
hyla
xis,
cere
brov
ascu
lar
Lon
ger
half
-lif
eth
ant-
PA;
Ten
ecte
plas
eis
acci
dent
,in
trac
rani
alhe
mor
rhag
ead
min
iste
red
asa
sing
lew
eigh
t-ba
sed
bolu
s;re
tepl
ase
isad
min
iste
red
asa
doub
lebo
lus
INH
IBIT
OR
SO
FA
NTI
COA
GU
LATI
ON
AN
DFI
BR
INO
LYSI
SPr
otam
ine
Mec
hani
sm—
Inac
tivat
eshe
pari
nby
form
ing
ast
able
1:1
prot
amin
e:he
pari
nco
mpl
ex
Pro
tam
ine
Hep
arin
over
dose
Bra
dyar
rhyt
hmia
,hy
pote
nsio
n,H
yper
sens
itivi
tyto
prot
amin
ePr
otam
ine
can
also
part
ially
reve
rse
the
anap
hyla
ctoi
dre
acti
on,
circ
ulat
ory
antic
oagu
lant
effe
ctof
low
mol
ecul
arw
eigh
tco
llap
se,
capi
llar
yle
ak,
nonc
ardi
ogen
iche
pari
n,bu
tit
cann
otre
vers
eth
ean
ticoa
gula
ntpu
lmon
ary
edem
aef
fect
offo
ndap
arin
uxFl
ushi
ng,
naus
ea,
vom
iting
,dy
spep
sia
Seri
ne-P
rote
ase
Inhi
bito
rM
echa
nism
—In
hibi
tsse
rine
prot
ease
s,in
clud
ing
plas
min
,t-
PA
,an
dth
rom
bin
Apr
otin
inR
educ
epe
riop
erat
ive
blee
ding
Hea
rtfa
ilur
e,m
yoca
rdia
lin
farc
tion
,sh
ock,
Hyp
erse
nsiti
vity
toap
rotin
inA
thi
gher
dose
s,ap
rotin
inm
ayal
soin
hibi
tdu
ring
coro
nary
arte
ryby
pass
thro
mbo
tic
diso
rder
,an
aphy
laxi
sw
ith
kalli
krei
nan
dth
ereb
ypa
rado
xica
llyin
hibi
tth
egr
aft
surg
ery
reex
posu
re,
cere
bral
arte
ryoc
clus
ion,
coag
ulat
ion
casc
ade
rena
lfa
ilur
eA
prot
inin
may
incr
ease
the
risk
ofpo
stop
erat
ive
acut
ere
nal
failu
rere
lativ
eto
othe
ran
tifib
rino
lytic
agen
ts
Lysi
neA
nalo
gues
Mec
hani
sm—
Ana
logu
esof
lysi
neth
atbi
ndto
and
inhi
bit
plas
min
ogen
and
plas
min
Am
inoc
apro
icac
idD
isor
der
invo
lvin
gth
eB
rady
arrh
ythm
ia,
hypo
tens
ion,
thro
mbo
tic
Dis
sem
inat
edin
trav
ascu
lar
May
caus
ele
ssac
ute
rena
lfa
ilure
rela
tive
toT
rane
xam
icac
idfi
brin
olyt
icsy
stem
diso
rder
,dr
ug-i
nduc
edm
yopa
thy
coag
ulat
ion
apro
tinin
Hem
orrh
age
from
incr
ease
d(a
min
ocap
roic
acid
),ra
rere
nal
fail
ure
Hyp
erse
nsiti
vity
toam
inoc
apro
icfi
brin
olys
isac
id

LP-FPPP-92900 R1 CH22 01-15-07 10:08:23
Page 416 Blank