Embed Size (px)
Citation preview



Organo ufficiale della SocietàItaliana per le Malattie RespiratorieInfantili (SIMRI)
Volume 9, n. 35 - Settembre 2009Spedizione in A.P. - 45%art. 2 comma 20/blegge 662/96 - N. 1047 del 12/07/2002 - PisaReg.Trib. PI n. 12 del 3 giugno 2002
Direttore scientificoBaraldi Eugenio (Padova)
Codirettori scientificiRusconi Franca (Firenze)Santamaria Francesca (Napoli)
Segreteria scientificaCarraro Silvia (Padova)
Comitato editorialeBarbato Angelo (Padova)Bernardi Filippo (Bologna)Cutrera Renato (Roma)de Benedictis Fernando Maria (Ancona)Peroni Diego (Verona)Rusconi Franca (Firenze)Santamaria Francesca (Napoli)Tripodi Salvatore (Roma)
Gruppo Allergologiacoord. Marseglia Gianluigi (Pavia)
Gruppo Disturbi respiratori nel sonnocoord. Brunetti Luigia (Bari)
Gruppo Educazionecoord. La Grutta Stefania (Palermo)
Gruppo Endoscopia bronchiale edelle Urgenze respiratoriecoord. Midulla Fabio (Roma)
Gruppo Fisiopatologia respiratoriacoord.Verini Marcello (Chieti)
Gruppo Riabilitazione respiratoriacoord.Tancredi Giancarlo (Roma)
Gruppo Il polmone suppurativocoord. Canciani Mario (Udine)
Direttore responsabileBaraldi Eugenio (Padova)
© Copyright 2009 by Primula Multimedia
EditorePrimula Multimedia S.r.L.Via G. Ravizza, 22/b56121 Pisa - Loc. OspedalettoTel. 050 9656242; fax 050 3163810e-mail: [email protected]
RedazioneWalker Manuella
Realizzazione EditorialePrimula Multimedia S.r.L.
StampaLitografia VARO - San Giuliano Terme (PI)
Inserto speciale: Paediatric Hermes
Editoriale: Paediatric HERMES syllabus:
un’importante tappa per armonizzare la
Medicina Respiratoria Pediatrica in Europa5
Paediatric HERMES:
a European Syllabus in Paediatric
Respiratory Medicine7
Gappa M, Noël JL, Séverin T, Baraldi E, Bush A, Carlsen KH,de Jongste J, Eber E, Fauroux B, McKenzie S, Palange P,Pohunek P, Priftis K, Wildhaber J, Zivkovic Z, Zach M, Paton J
Al confine fra pneumologia e immuno-allergologiapediatrica
Editoriale21
La suscettibilità genetica alle
infezioni respiratorie22
F. Cardinale, F. Cristofori, I. Chinellato, F. Di Domenico,A. Cappiello, F. Carella, P. Piccarreta, M. F. Mastrototaro
Allergia alimentare e asma34
A. Martelli, A. Caddeo, S. El Oksha, E. Calcinai,L. Terracciano, A. Fiocchi
Rinite allergica ed asma in età pediatrica42
M. Tosca, G. Ciprandi
Aspergillosi broncopolmonare50
V. Raia, F. De Gregorio, P. Buonpensiero, A. Sepe
Asma e apoptosi55
F. M. de Benedictis, D. de Benedictis, C. Gabriele
Le malattie respiratorie ad eosinofili
nel bambino67
N. Fuiano
R U B R I C A : j o u r n a l c l u b
Corticosteroidi per via orale in bambini dietà pre-scolare con wheezing episodico virale:cosa resta di questa terapia?
79E. Opocher, M. Balzani, M. Zambolin, L. Bartolini, E. Baraldi
Congressi83
Articoli del prossimo numero85
INDICESUMMARY

Il sottoscritto, CHIEDE AL PRESIDENTE della Società Italiana per le Malattie Respiratorie Infantili di essereammesso quale socio ordinario. Pertanto, riporta i seguenti dati personali:
DATI PERSONALI
Cognome Nome
Luogo e data di nascita
Domicilio (via/piazza)
CAP Città Prov. Regione
Sede di lavoro Reparto
Indirizzo
Recapiti telefonici: Casa Studio Fax
Ospedale Cellulare e-mail
Laurea in Medicina e Chirurgia - Anno di laurea
Specializzazioni
Altri titoli
CATEGORIA
Universitario Ospedaliero Pediatra di libera scelta
QUALIFICA UNIVERSITARIA
Professore Ordinario Professore Associato Ricercatore Altro
QUALIFICA OSPEDALIERA
Dirigente di 2º Livello Dirigente di 1º Livello Altro
Con la presente autorizzo la Società Italiana per le Malattie Respiratorie Infantili al trattamento dei miei dati personali ai sensi del D.L. del 30giugno 2003 n. 196.
Data Firma del Richiedente
Soci presentatori (cognome e nome) Firma
1)
2)
Compilare in stampatello e spedire insieme con la copia dell’avvenuto versamento(quota sociale di euro 30,00. Specializzandi euro 10,00) a:
Biomedia srl - Segreteria Amministrativa SIP - Via Libero Temolo 4, 20126 Milanoc/c postale N. 67412643 intestato a: Società Italiana di Pediatria
È obbligatoria l’iscrizione anche alla SIP (quota sociale di euro 90,00), può essere fatto un unicoversamento indicando chiaramente nella causale per quali società affiliate viene effettuato il versamento.
Per informazioni: Biomedia srl - tel. 02/45498282 - fax 02/45498199 e-mail: [email protected]
Domanda di ammissione per nuovi Soci

Paediatric HERMES syllabus: un’importante tappa perarmonizzare la Medicina Respiratoria Pediatrica in Europa
Il progetto Paediatric HERMES (Harmonised Education inRespiratory Medicine for European Specialists) nasce nel 2007 comeTask Force dell’Assemblea Pediatrica della European RespiratorySociety (ERS) con lo scopo di aggiornare il precedente Syllabus del2002, sviluppare materiale educazionale e porre le basi per creare unCurriculum ed un esame Europeo per specialisti in MedicinaRespiratoria Pediatrica (PRM). Il nuovo Syllabus Europeo viene pub-blicato integralmente in questo numero della nostra rivista.La PRM è una giovane sottospecialità della Pediatria che ha ini-
ziato a svilupparsi negli Stati Uniti e in Australia e, più di recente, inalcune nazioni europee come Inghilterra, Olanda, Austria, Germaniae Svizzera. Grazie all’impegno del professor Max Zach, affiancato dalprofessor Attilio Boner, la pneumologia pediatrica è stata fra le primesottospecialità riconosciute in Europa dalla Union of EuropeanMedical Specialists (UEMS). In Italia la sottospecialità in PRM non èancora formalmente riconosciuta anche se numerosi centri diPneumologia Pediatrica hanno ottenuto l’accreditamento dall’ERS. LaSIMRI è attivamente impegnata con varie iniziative per la formazionedei giovani che si avvicinano alla pneumologia pediatrica ed è coin-volta nei programmi di accreditamento Europeo.Il nuovo Syllabus Europeo definisce le competenze che un tiroci-
nante in PRM dovrebbe acquisire per diventare uno specialista inquesta disciplina. Il Syllabus è stato sviluppato utilizzando un proces-so interattivo standardizzato chiamato “Delphi”, in stretta collabora-zione con esperti del settore di 20 nazioni europee. Il training in PRMinclude 21 moduli obbligatori e alcuni moduli opzionali. La flessibilitàdel Syllabus è uno degli elementi su cui si è basata l’iniziativa in mododa facilitarne l’applicabilità nelle diverse realtà delle nazioni europee.I prossimi traguardi del progetto HERMES Pediatrico, coordinato
dalla professoressa Monika Gappa, sono lo sviluppo di un Curriculumche descrive in dettaglio i metodi raccomandati per il training e imetodi di valutazione del tirocinante. A questo seguirà un program-ma di visite per l’accreditamento dei centri di training e lo sviluppo diuna rete europea di centri riconosciuti di Pneumologia pediatrica.
EditorialeView point
Pneumologia Pediatrica 2009; 33: 5-6 5

EditorialeView point
6
Infine verrà sviluppato un esame facoltativo per un diploma europeoche permetterà la libera circolazione degli specialisti in PRM.Le malattie respiratorie pediatriche sono fra le cause più impor-
tanti di morbilità e mortalità nei bambini. Il progetto HERMESPediatrico ha posto le basi per portare al meglio la qualità dellamedicina respiratoria pediatrica in Europa, un occasione che nonpossiamo lasciarci sfuggire!
Eugenio [email protected]
Giovanni [email protected]

7Pneumologia Pediatrica 2009; 35: 7-19
Dati tratti da:
Gappa M, Noël JL, Séverin T, Baraldi E, Bush A, Carlsen KH, deJongste J, Eber E, Fauroux B, McKenzie S, Palange P, Pohunek P,Priftis K, Wildhaber J, Zivkovic Z, Zach M, Paton J. Paediatric HER-MES: a European Syllabus in Paediatric Respiratory Medicine.Breathe 2009; 5 (3): 237-247.
InsertoInsert













Terra di confine fra Pneumologia e Immuno-allergologiapediatrica
Gli articoli proposti in questo numero di Pneumologia Pediatricaaffrontano gli aspetti immunologici e allergologici di alcune patologierespiratorie pediatriche.L’articolo del gruppo di Fabio Cardinale e collaboratori affronta
in modo puntuale il tema dei fattori genetici che condizionano lasuscettibilità dell’ospite alle infezioni, in particolare a quelle del trattorespiratorio, portando un utile aggiornamento su questo interessan-te argomento.Il gruppo milanese di Alessandro Fiocchi ci propone un tema
interessante e sempre attuale, quello del rapporto tra allergia ali-mentare e asma, da un lato considerando i sintomi asmatici comepossibile manifestazione di allergia alimentare, dall’altro valutando ilrapporto temporale tra allergia alimentare e asma nella cosiddettamarcia atopica.I contributi del gruppo genovese e del gruppo di Napoli sono
caratterizzati da un taglio prettamente clinico. Maria Angela Tosca eGiorgio Ciprandi descrivono i rapporti tra asma e rinite allergica sot-tolineando come queste due condizioni siano intimamente connes-se e dovrebbero essere considerate due aspetti della medesima enti-tà clinica.Valeria Raia e collaboratori affrontano invece il tema dell’a-spergillosi broncopolmonare allergica riassumendo i principali aspet-ti della diagnosi e del trattamento di questa patologia, con un riferi-mento particolare ai pazienti affetti da fibrosi cistica.Fernando de Benedictis e collaboratori presentano un elegante
articolo sui meccanismi coinvolti nella regolazione dell’apoptosi. Contrattazione scientificamente rigorosa gli autori descrivono il ruolodell’alterata apoptosi nella patogenesi dell’infiammazione cronica edel rimodellamento delle vie aeree nei soggetti asmatici.Infine Nicola Fuiano presenta un ampio excursus sulle malattie
respiratorie ad eosinofili, analizzando in modo esauriente ed accura-to tutte le principali patologie di interesse pediatrico nelle quali que-ste cellule hanno un ruolo centrale.In conclusione un numero che ci da l’opportunità di allargare i
confini delle nostre conoscenze ed arricchire il nostro bagaglio cul-turale sul doppio fronte dell’immuno-allergologia e della pneumolo-gia pediatrica.A tutti un augurio di buona lettura!
Eugenio Baraldi
EditorialeView point
Pneumologia Pediatrica 2009; 35: 21 21

Introduzione
Il passaggio dallo stato di semplice infezione aquello di malattia, la severità di quest’ultima e l’out-come finale dipendono tanto da fattori legati alpatogeno (quali la carica e virulenza) quanto dafattori legati all’ospite. Negli ultimi anni si è resosempre più evidente che questi ultimi hanno unruolo prevalente rispetto ai fattori microbici. Unamalattia infettiva può infatti manifestarsi con unventaglio di quadri clinici, di maggiore o minoreseverità, in rapporto alla capacità dell’ospite diprovvedere ad un’efficace clearance dell’agentemicrobico. Come immaginabile, quest’ultima è asua volta in stretta dipendenza dalle caratteristichegenetiche dell’individuo. Nell’ambito delle patolo-gie infettive respiratorie è acclarato infatti che,accanto ai fattori di ordine anatomico (malforma-zioni bronchiali, sindromi da broncoinalazione,etc.), un ruolo determinante è svolto da varianti digeni implicati nella risposta immune.
Le dimostrazioni del peso che hanno i geni nel-l’outcome finale delle malattie infettive sono ormainumerose. È noto ad esempio che la mortalità perpatologie infettive in individui adottati, nati da geni-tori deceduti per complicanze settiche, è maggio-re rispetto a quella della popolazione generale, edè invece sovrapponibile a quella dei genitori natu-rali (1). Inoltre, il rischio per un paziente ospeda-lizzato per polmonite pneumococcica di riamma-larsi della stessa patologia è significativamentemaggiore rispetto a quello di individui ricoveratiper altre cause (2). Studi di popolazione hannoanche dimostrato un’alta percentuale di concor-danza tra gemelli monocoriali per patologie comela tubercolosi, la lebbra, la malaria e l’infezione daHelicobacter pylori (3).Un esempio paradigmatico di suscettibilità geneti-ca alle infezioni è rappresentato dalle sindromi daimmunodeficienza primitiva (primary immune
Fabio Cardinale, Fernanda Cristofori, Iolanda Chinellato, Francesca Di Domenico, Annarita Cappiello,Francesca Carella, Paola Piccarreta, Maria Felicia Mastrototaro
Dipartimento di Biomedicina dell’Età evolutiva, Clinica Pediatrica “S. Maggiore”, A.O. Policlinico,Università di Bari
22
La suscettibilità genetica alle infezionirespiratorie
Genetic susceptibility to airway infections
Parole chiave: genetica, immunità innata, infezioni respiratorie, polmonite
Keywords: genetics, innate immunity, airways infections, pneumonia
Riassunto. Numerose evidenze della letteratura indicano che fattori legati all’ospite rivestono un peso determinante nellasuscettibilità alle infezioni. Un esempio paradigmatico di questo rapporto è rappresentato dalle sindromi da immunodeficit pri-mitivo e da patologie multisistemiche come la fibrosi cistica e la discinesia ciliare primitiva. Negli ultimi anni, comunque, sonostate descritte numerose mutazioni e varianti alleliche di geni dell’immunità innata in grado di condizionare l’evoluzione clinicadi alcuni tipi di infezione, soprattutto sistemiche e respiratorie. Una maggiore conoscenza di questi fenomeni è probabile chein un prossimo futuro conduca a sviluppare nuove terapie e ad identificare individui a rischio di manifestare malattie infettivesevere e/o loro complicanze.
Accettato per la pubblicazione il 31 agosto 2009.
Corrispondenza: Fabio Cardinale, Clinica Pediatrica “S. Maggiore”, Università di Bari, P.zza G. Cesare 11, 70125 Bari;e-mail: [email protected]
Pneumologia Pediatrica 2009; 35: 22-33

La suscettibilità genetica alle infezioni respiratorie 23
defects, PID), patologie in cui un disturbo maggio-re a carico di uno o più pathway del sistemaimmune comporta un’accresciuta suscettibilità neiconfronti di patogeni comuni o opportunisti.Tuttavia risulta oggi chiaro che queste rare e seve-re sindromi da immunodeficienza ricoprono solouna minima parte dei difetti immunitari primitiviad oggi riconosciuti.Negli ultimi anni, infatti, grande attenzione è statadedicata all’esistenza, accanto alle forme “classi-che” di PID, di forme nuove, “non convenzionali”,interessanti soprattutto il sistema immune innato.Queste ultime sono caratterizzate da uno spettrodi suscettibilità microbica molto più ristrettorispetto alle forme classiche (un solo patogeno opochi patogeni e talvolta anche una sola infezionesevera nell’arco di tutta la vita), da un esordio aqualunque età (anche nell’adulto), dal non espri-mersi esclusivamente nelle cellule ematopoietichee dall’assenza di alterazioni immunologiche evi-denti in periferia (4). Esempi di PID “selettive”sono rappresentati dalle sindromi da suscettibilitàmendeliana ai micobatteri (MSMD), dal deficit diIRAK4/MyD88 (vedi oltre), e dalla sindrome linfo-proliferativa X-linked, che predispongono rispetti-vamente a infezioni severe da micobatteri, polmo-niti gravi e/o sepsi pneumococciche e mononu-cleosi complicata (5).Peraltro, accanto ai deficit immunitari maggiori nonconvenzionali, sono state identificate negli ultimianni numerose varianti polimorfiche di geni del-l’immunità innata e adattativa in grado di condizio-nare la risposta immune dell’ospite nei confronti diuno o più microrganismi.La forma più comune di variante genica è rappre-sentata da singole sostituzioni nucleotidiche (singlenucleotide polymorphism, SNPs) a carico di undeterminato gene. Si stima che SNPs siano pre-senti in almeno 1%-2% dei soggetti normali ogni1200-1300 paia di basi (pb) (6). Ne consegue chel’intero genoma ne può contenere oltre 10 milio-ni. Tuttavia solo alcuni di questi polimorfismi rive-stono un significato funzionale, mentre la maggiorparte è priva di significato clinico.Un esempio di variabilità genetica del sistemaimmune è rappresentato dal complesso maggio-re di istocompatibilità (HLA), localizzato sul cro-mosoma 6. Da tempo è nota una correlazione traspecifici aplotipi HLA e suscettibilità nei confron-ti di malattie come la malaria (7), la tubercolosi(8), la lebbra (9), l’infezione da HIV (10), l’epatite
B (11) e l’infezione da virus di Epstein-Barr (12).Accanto all’HLA, comunque, grande importanzaavrebbero i polimorfismi di altri geni, questa voltaa carico dell’immunità innata, nel determinismo delfenotipo clinico delle infezioni, specialmente di tiposistemico o respiratorio. Gran parte della lettera-tura in quest’ambito riguarda l’adulto ma nonmancano studi pediatrici sull’argomento.
Generalità sull’immunità innata
È noto che nell’uomo e negli animali superiori ilsistema immunitario può essere schematicamenteripartito in due compartimenti: quello dell’immuni-tà innata e quello dell’immunità adattativa o acqui-sita (13). Quest’ultima è organizzata principalmen-te attorno a due classi di cellule specializzate, i lin-fociti T ed i B, ed è caratterizzata rispetto quellainnata da una maggior potenza e selettività d’azio-ne ma da tempi di attivazione di svariati giorni.Il sistema innato, a sua volta, trova i suoi strumen-ti effettori nei fagociti, nelle cellule natural killer NK,nella via alterna di attivazione del complemento(C) e in un complesso di peptidi con attività anti-microbica, presenti nel citoplasma, nell’ambienteextracellulare e nelle secrezioni (Figura 1).I tempi brevissimi occorrenti per la sua attivazionefanno sì che la funzione preminente del sistemaimmune innato sia quella di assicurare una lineadifensiva di tipo “pronto” nei confronti di un qual-sivoglia agente infettivo, in attesa dell’intervento dimeccanismi più raffinati e selettivi quali quelli T e Blinfocito-mediati. Caratteristiche fondamentali diquesto sistema sono infatti la mancanza di memo-ria immunologica e la non clonalità. Esso quindi, adifferenza di quello adattativo, non richiede unaprecedente esposizione ai patogeni, sebbene alcu-ni dei suoi componenti possano essere inducibili.Tutto il sistema immune innato è organizzatoattorno a recettori codificati allo stato germ-linedenominati pathogen-recognition receptors (PRR),espressi su numerosi tipi cellulari, ma soprattuttosulle antigen presenting cells (APC) professionali.Il funzionamento del sistema immune innato sibasa sul riconoscimento, attraverso i PRR, di uncomplesso di molecole, fortemente conservatenella filogenesi e presenti in grandi gruppi dimicrorganismi, definite pathogen-associated molecu-lar patterns (PAMP). Ne sono esempi il peptidogli-cano batterico, l’RNA virale a doppia elica o ilipoarabinomannani dei micobatteri. Sebbenedistinti tra di loro sotto il profilo chimico-strutturale,

Cardinale, et al.24
tutti i PAMP hanno in comune il fatto di esserepresenti solo nel mondo microbico, di rappresen-tare strutture essenziali per la sopravvivenza deimicroorganismi e di essere condivisi da intere clas-si di patogeni.Dal punto di vista funzionale, questi recettori pos-sono essere distinti in tre classi: secretivi, endociti-ci e di segnale.I PRR secretivi agiscono soprattutto come opsoni-ne, legandosi a strutture molecolari della paretemicrobica, sì da attivare il complemento (C) e pro-muoverne la fagocitosi. A questa classe apparten-gono le defensine, la mannose binding lectin (MBL)e le proteine surfattanti A e D (SP-A, SP-D). Ledefensine sono peptidi cationici a basso pesomolecolare dotati di potente attività microbicida.La MBL rappresenta invece una proteina della faseacuta appartenente alla famiglia delle collectine,con un ruolo importante nei processi di opsoniz-zazione e di attivazione del C, rappresentando ilprincipale artefice dell’innesco della via lectinica(Figura 2). Le SP-A e SP-D, oltre a entrare nell’ar-chitettura del surfattante polmonare, intervengo-no nelle difese immuni respiratorie grazie allacapacità di opsonizzare numerosi agenti infettivi, dipromuovere l’attivazione dei macrofagi e il killingmicrobico a livello degli spazi alveolari e delle vieaeree superiori.
I PRR endocitici sono localizzati sulla superficie deifagociti e intervengono nell’uptake e nell’ingressodei patogeni nei lisosomi dove vengono sottopo-sti a digestione enzimatica. Esempi di questa classedi recettori sono gli scavenger receptors e i recet-tori lectinici di tipo C, tra cui i meglio studiati sonoil macrophage mannose receptor e il recettore DC-SIGN. Quest’ultimo viene espresso sulle celluledendritiche e sui macrofagi alveolari, ed intervienenell’internalizzazione dei micobatteri (14).I PRR di segnale sono così denominati in quanto, aseguito del riconoscimento delle sequenze mole-colari associate al patogeno, attivano pathway spe-cifici di trasduzione del segnale all’interno delle cel-lule, che conducono all’espressione di diversi geni,codificanti per molecole coinvolte nella rispostaimmunitaria e nell’infiammazione (15).I più importanti recettori di segnale identificatinegli ultimi anni sono senza dubbio i toll-like recep-tors (TLRs), principali sensori della presenza dimicrorganismi da parte del sistema immune inna-to, con un ruolo chiave anche nella regolazionedella risposta immune adattativa. Sono stati identi-ficati almeno dieci TLRs nei mammiferi, alcuniespressi sulla membrana cellulare, altri nel com-partimento endosomico, ognuno provvisto di unamultispecificità di classe (ad es, i TLR4 per l’LPSbatterico e la proteina di fusione del VRS, i TLR5
Figura 1 Strumenti effettori del sistema immune innato.
Strumenti effettori dell’immunità innata
• C (via alterna e lectinica)• Fagociti, DC, NK, MC, eosinofili, cell. epiteliali• Peptidi antimicrobici (lisozima, collectine, pentraxine, defensine, … )• Altre molecole solubili (NOx etc.)
Macrofago Batterio
PRR (Pattern Recognition Receptors)
• Secretivi (es. collectine, ficoline, SP-A, …)• Endocitici (es. scavenger receptors,
C-type lectin receptors …)• Di segnale (es. toll-like receptors, NLRs…)
PAMP(Pathogen-AssociatedMolecular Patterns)
LPSPeptidoglicanoRNA/DNA viraleDNA battericoMannani, GlucaniAltro

La suscettibilità genetica alle infezioni respiratorie 25
per la flagellina dei batteri e così via) (15).L’attivazione dei TLRs condurrebbe al reclutamen-to a cascata di altre molecole di segnale (IRAK1-4,MyD88, TRAF6 e molte altre) e, in ultima analisi,all’attivazione dell’NF-κB che, una volta traslocatonel nucleo, darebbe luogo alla trascrizione di unamoltitudine di geni implicati della risposta immune(Figura 3).
Geni di suscettibilità per la bronchiolite
La presentazione e il decorso clinico dell’infezionedaVRS è estremamente variabile e può andare dasfumati sintomi a carico delle vie aeree superiori oinferiori fino a forme gravi di bronchiolite con dis-tress respiratorio. Fattori di rischio noti da tempoper bronchiolite severa sono la prematurità, ilbasso peso alla nascita, la broncodisplasia e le mal-formazioni cardiache. Negli ultimi anni, tuttavia,diversi studi hanno dimostrato l’importanza deideficit minori dell’immunità innata nel condiziona-re il decorso di questa patologia (Tabella 1).È stato dimostrato, ad esempio, che mutazioni delTLR-4 sono fortemente “over-rappresentate” in
bambini ospedalizzati per bronchiolite severa(20,2%) rispetto a soggetti di controllo costituitida adulti sani (4,9%) o bambini con sintomi bron-chiolitici di tipo lieve (5,6%) (16).Alcuni lavori indicano il ruolo anche di variantigenetiche delle SP-A e SP-D nel decorso clinicodella bronchiolite. È noto infatti che le proteinesurfattanti intervengono nella clearance del VRSlegandosi a componenti del virus e inducendonel’aggregazione (17).Alcuni Autori hanno dimostra-to livelli ridotti di SP-A, SP-B ed SP-D nel BAL dibambini con bronchiolite severa, ventilati meccani-camente, rispetto a pazienti di controllo (18). Sullascorta di queste premesse, l’importanza di varian-ti genetiche della SP-A nel condizionare la severi-tà della bronchiolite è stata dimostrata da uno stu-dio in cui veniva osservata una maggior frequenzadi specifici aplotipi del gene SP-A2 in bambiniaffetti da bronchiolite grave rispetto ad una popo-lazione controllo (19).Un altro lavoro ha dimostrato l’importanza diSNPs a carico del gene che codifica per l’IL-10 nel-l’outcome della bronchiolite da VRS. Gli Autoriinfatti hanno dimostrato, in un gruppo di lattanti
Figura 2 Attivazione della cascata complementare attraverso la via lectinica mediata dalla Mannose-binding lectin (MBL).
Via classica
C1q
C4bC2a
C1rC1s MBL
C3b
D
B
C3
C3a
C5
C5a C5b
C3b
C3bBb
C3 convertasiclassica
C3 convertasialterna
C5 convertasi
Complesso litico
C4 e C2 MASP1MASP2
Via lectinica Via alterna
BatterioBatterio
Batterio

Cardinale, et al.26
ospedalizzati per bronchiolite da VRS, una over-rappresentazione di due polimorfismi dell’IL-10 trai pazienti che avevano necessitato di ventilazionemeccanica (20). Un ruolo importante nel condi-zionare il decorso della bronchiolite sembra esse-re svolto comunque anche da geni codificanti peraltre citochine di tipo Th-2, come l’IL-4 e l’IL-18,insieme a quello per alcuni recettori delle chemo-chine, per il recettore della vitamina D e per laNOS (ossido nitrico sintetasi) di tipo 2 (21-24).
Geni di suscettibilità per le otiti
L’otite media acuta (OMA) rappresenta una dellepiù frequenti patologie infettive in età pediatrica.Studi su gemelli monozigoti hanno dimostratoche, accanto a fattori di rischio di ordine anatomi-co o ambientale (anomalie cranio-facciali, esposi-zione al fumo, allattamento artificiale, etc.) anchefattori genetici contribuiscono alla vulnerabilità neiconfronti di questa patologia.Alcuni Autori hanno dimostrato ad esempio cheSNPs a carico di geni che codificano per alcune
citochine proflogogene, come il TNF-α e l’IL-6,sono associati ad un rischio da 2 a 3 volte supe-riore di OMA ricorrente e miringostomia (25).Nello stesso lavoro, peraltro, veniva osservato uneffetto di protezione per un polimorfismo deiTLR4.Un altro gene implicato nell’OMA sembra esserequello che codifica per la SP-A, proteina espressaoltre che negli spazi alveolari anche a livello tuba-rico e delle vie aree superiori, in grado di pro-muovere la fagocitosi dello Streptococcus pneumo-niae e dell’Hemophilus influenzae. È stato infattidimostrato che la frequenza di alcuni aplotipi egenotipi della SP-A in bambini con OMA ricor-rente o precoce (cioè con esordio nei primi 6mesi) è significativamente differente da quella dibambini sani (26).Un ruolo importante nella genetica dell’OMA èprobabilmente svolto anche da varianti allelichedella MBL. Infatti, la presenza di varianti allelichedella MBL si associa ad un maggior rischio di OMAricorrente nel bambino, limitatamente però allafascia di età inferiore ai 24 mesi (27).
Figura 3 Pathway di trasduzione del segnale a valle dei Toll-like receptors che porta, in ultima analisi, all’attivazionedell’NF-κB. Quest’ultimo, una volta traslocato nel nucleo, induce la trascrizione di una moltitudine di geni implicatinella risposta immune.
Citoplasma
Nucleo
Membrana citoplasmaticaTLR
Myd88
IRAK4
TRAF6
IκκB
NF-κκB
IKK
NEMO/IKKλλ
αα ββ
TAK1
MAP Kinasi
Induzione di geni infiammatori e della risposta immune
IRAK1
IL-1R

La suscettibilità genetica alle infezioni respiratorie 27
Geni di suscettibilità per le pleuropolmoniti
Le maggiori evidenze sulla genetica delle polmoni-ti sono state raccolte sulle forme da legionella.Alcuni lavori hanno infatti dimostrato che uno SNPa carico del ligand binding domain del TLR5, coneffetto “stop” sulla trascrizione, si associa negli adul-ti ad un rischio aumentato di polmoniti da legio-nella mentre, al contrario, polimorfismi del TLR4sembrano essere associati a maggior resistenza neiconfronti dello stesso patogeno (28, 29).Per numerosi altri geni è stato dimostrato unruolo nell’outcome delle polmoniti acquisite incomunità (community-acquired pneumonia, CAP)da parte di svariati agenti infettivi. Uno studiorecente su una grossa casistica di adulti ospedaliz-zati per CAP ha dimostrato che varianti allelichedella MBL correlano con una maggior frequenza dicomplicanze (sepsi e insufficienza respiratoriaacuta) ed una prognosi nel complesso peggiorerispetto ai carriers di alleli wild-type (30). Un altrostudio, pur non confermando un’associazione travarianti della MBL e decorso clinico nelle CAP, hacomunque evidenziato un ruolo di queste nella
possibilità di sviluppare una coinfezione virale (31).Altri studi hanno dimostrato che SNPs a caricodel promoter dell’IL-10, in grado di aumentare lasecrezione di questa citochina antiinfiammatoria,predispongono in pazienti adulti con CAP ad unamaggior frequenza di complicanze ed ad un’au-mentata mortalità (32).Di contro, polimorfismi a carico del promoterdell’IL-6, associati ad un’upregulation della sintesi diquesta citochina, avrebbero un effetto protettivocontro le disseminazioni ematogene in corso dipolmonite da pneumococco (33).Infine, tra le complicanze delle polmoniti, lo shocksettico è stato associato ad un genotipo “high secre-tor” del TNF-α, mentre l’insufficienza respiratoria inassenza di shock sembra essere più frequente neipazienti con genotipo “low secretor” (34).
Geni di suscettibilità per le infezioniinvasive da pneumococco
Fattori di rischio ereditari per le infezioni invasiveda Streptococcus pneumoniae noti da molto tempocomprendono l’anemia a cellule falciformi e le PID“classiche”. Tra queste ultime vanno ricordati i
Tabella 1 Principali SNPs di geni di suscettibilità per la bronchiolite.
Gene Effetto Riferimento bibliografico
TLR4 ↑ Severità Tal, J Infect Dis 2004; 189: 2057.
TLR4 ↑ Severità Mandelberg, Clin Exp Immunol 2006; 144: 48.
IL-8 ↑ Rischio wheezing Goetghebuer, Clin Exp Allergy 2004; 34: 801.
IL-8 ↑ Severità infezione RSV Thorax 2000; 55: 1023.
IL4 e IL4r ↑ Severità (ospedalizzazione) Hoebee, J Infect Dis 2003; 187: 2.
IL-10 ↑ Severità (ospedalizzazione) Hoebee, J Infect Dis 2004; 189: 239.
IL-10 ↑ Severità Wilson, J Infect Dis 2005; 191: 1705.(ricovero Unità Terpia Intensiva)
IL-18 ↑ Suscettibilità Puthothu, Ped Infect Dis J 2007; 26: 1094.
VDR, JUN, IFNA5, NOS2 ↑ Suscettibilità Janssen, J Infect Dis 2007; 196: 826.
CD14 ↑ Suscettibilità Inoue, J Infect Dis 2007; 195: 1618.
RANTES ↑ Suscettibilità Amanatidou, Ped Infect Dis J 2008; 27: 38.
CCR5 ↑ Suscettibilità Hull, J Infect Dis 2003; 188: 904.
CX3CR1 ↑ Suscettibilità Amanatidou, Ped Infect Dis J 2006; 25: 410.
SP-A ↑ Suscettibilità Lofgren, J Infect Dis 2002; 185: 283.
SP-D ↑ Suscettibilità Lahti, Pediatric Res 2002; 51: 696.

Cardinale, et al.28
deficit anticorpali e del complemento, le neutro-penie e l’asplenia congenita. Più recentementesono stati descritti come causa di batteriemiericorrenti da pneumococco o altri Gram+ i difettia carico di alcuni fattori intracellulari (IRAK4,MyD88, NEMO, IκB) implicati nel signaling a valledei TLRs (35, 36) (Figura 3). Questi difetti com-portano un’alterazione della risposta infiammato-ria acuta dovuta alla mancata attivazione delnuclear factor-κB (NF-κB). Il gruppo francese diCasanova ha descritto negli ultimi anni numerosicasi di bambini con infezioni invasive da pneumo-cocco e da Stafilococcus aureus (meningite, artrite,cellulite, osteomielite, ascessi), precedute o menoda polmonite, per effetto di un deficit di IRAK4trasmesso con modalità AR. Caratteristica peculia-re di questi pazienti sembra essere l’attenuazionedei segni clinici e biologici di flogosi durante gli epi-sodi infettivi (37, 38).A parte queste, che rappresentano delle vere eproprie PID, alcuni SNPs a carico di geni dell’im-munità innata avrebbero un ruolo importante nellasuscettibilità alle infezioni sistemiche da pneumo-cocco. Un importante studio caso-controllo hadimostrato su una grossa casistica di adulti consepsi pneumococciche che lo status di omozigosiper varianti alleliche della MBL comporta un rischioaumentato (circa 3 volte superiore) di malattiarispetto a soggetti provvisti degli alleli wild-type(39). Altri SNPs di geni dell’immunità innata sem-brerebbero avere un effetto protettivo sulle infe-zioni invasive da pneumococco. Tra questi, va citatoin particolare il ruolo protettivo di alcuni polimor-fismi di MAL (o TIRAP), una proteina di segnale avalle dei TLR2 e TLR4 con attività pleiotropica neiconfronti di molti agenti infettivi (vedi oltre) (40).
Geni di suscettibilità alle infezioni damicobatteri
Come prima riportato, la MSMD rappresenta ilmiglior esempio di PID “non convenzionale” inquanto caratterizzata da una predisposizione a svi-luppare infezioni sistemiche da micobatteri nor-malmente poco virulenti (come il bacillo diCalmette–Guerin, o i micobatteri atipici) e, menofrequentemente, anche da Mycobacterium tuberco-losis, in assenza di sensibilità ad altri patogeni. Nellecasistiche di pazienti con MSMD compaiono spes-so, nell’ambito della stessa fratria, bambini con TBCpolmonare severa e bambini con infezioni dissemi-nate da micobatteri atipici. Ad oggi si conoscono
almeno 13 diversi genotipi di questa condizione, lecui basi molecolari sono da ascriversi a mutazionia carico di geni del circuito IFN-γ/IL-12, trasmessecon modalità AR, AD o X-linked (41).Quest’associazione è facilmente comprensibileove si pensi che l’interferone gamma (IFN-γ) è unpotente attivatore del macrofago e che quest’ulti-mo occupa un ruolo chiave nella difesa nei con-fronti dei micobatteri e nella formazione del gra-nuloma. L’IL-12, a sua volta prodotta dalle APCprofessionali, costituisce il principale induttoredella produzione di IFN-γ (Figura 4). Sulla scorta diquesta letteratura qualificati Autori ritengono chela genetica della TBC nel bambino sia differente daquella dell’adulto (v. oltre) e che ciò apra in alcunicasi delle prospettive per una terapia sostitutivacon IFN-γ.Ancora una volta, anche SNPs a carico di geni del-l’immunità innata svolgono un ruolo nel determi-nare la suscettibilità ai micobatteri maggiori. In unostudio recente, su una coorte di 1.262 pazientiadulti affetti da tubercolosi (TBC) polmonare, èstato riscontrato un effetto protettivo da parte diun polimorfismo del gene CD209 (che codificaper il recettore DC-SIGN), in particolare nei con-fronti delle forme di TBC cavitaria. Questo recet-tore, appartenente alla famiglia dei recettori lecti-nici di tipo C, è espresso su cellule dendritiche emacrofagi alveolari, e interviene nei processi diinternalizzazione dei micobatteri, con un effettofinale di sopprimere la trasduzione del segnale avalle dei TLRs (14). Studi recenti indicano che anche uno SNP in ete-rozigosi codificante per una variante funzionaledi MAL (conosciuta anche come TIRAP), mole-cola di segnale in grado di attivare l’NFκB, puòsvolgere un ruolo protettivo nei confronti dellaTBC polmonare. Tale effetto, peraltro, si estende-rebbe anche ad altre infezioni, quali la malaria, lesepsi in generale e le infezioni disseminate dapneumococco (40).Infine, un ruolo importante nella suscettibilità allaTBC sembra svolto dal recettore per la vitamina D(VDR). Attraverso l’interazione con la vitamina De con i VDRE (Vitamin D Response Elements) ilVDR riveste un’importante funzione immunomo-dulante, che include la fagocitosi dei micobatteri,l’attivazione dei monociti e della risposta cellulo-mediata e la soppressione della proliferazione lin-focitaria. Alcuni Autori hanno quindi dimostratol’importanza di varianti genetiche del VDR, in uno

La suscettibilità genetica alle infezioni respiratorie 29
studio su 382 famiglie, in cui hanno riscontratoun’associazione tra peculiari aplotipi del gene VDRe aumentata frequenza di TBC (42).
Geni di suscettibilità per le infezionirespiratorie ricorrenti
Le migliori evidenze del ruolo della genetica nelleinfezioni ricorrenti indifferenziate delle vie aereesuperiori (IRR) vengono dalla MBL. Uno studiodanese compiuto su una coorte di 252 bambini dietà inferiore a 2 anni ha dimostrato una frequen-za di IRR più di due volte superiore nei bambinicon varianti alleliche della MBL rispetto a soggettiportatori degli alleli wild-type. In particolare, que-st’associazione sembra essere importante neibambini tra i 6 e i 18 mesi, epoca in cui le risposteIgG2 verso gli antigeni polisaccaridici risultanodifettive (43).
Geni di suscettibilità per la sinusite
Oltre alle PID, anche la fibrosi cistica (FC) e la dis-cinesia ciliare primitiva (DCP) predispongono ainfezioni ricorrenti e/o croniche a carico delle vieaeree superiori e inferiori. Per quanto concerne laFC, trattandosi di una malattia a carattere AR,mutazioni del CFTR sono per definizione conside-rate espresse soltanto in condizioni di omozigosi o
eterozigosi composta. Alcune osservazioni hannoperò sottoposto a challenge questo concetto. Inparticolare, uno studio eseguito su 147 adulti consinusite cronica ha fatto osservare una frequenzadi mutazioni in eterozigosi (∆F508) e di polimorfi-smi (M470V) del CFTR significativamente superio-re in questa patologia rispetto alla popolazionegenerale (44), a dimostrazione del fatto che ilCFTR costituisce probabilmente un gene maggio-re di suscettibilità alle sinusopatie.
Geni dell’immunità innata come modifierdel fenotipo polmonare nella fibrosicistica
È noto come la correlazione genotipo/fenotiponella fibrosi cistica (FC) sia modesta e come, anchein pazienti con genotipi identici di CFTR, vi sia unampio spettro di severità nella malattia polmona-re. Nel tempo si è andata rafforzando la teoria chevarianti polimorfiche di altri geni possano agirecome modifier del quadro polmonare. Ad oggisono stati descritti più di 20 geni modifiers per ilfenotipo polmonare nella FC, tra cui il TNF, la SP-A, la NOS, l’α1-antitripsina e molti altri (45-48).Un ruolo importante sembra svolto ancora unavolta dalla MBL, essendo stata dimostrata un’asso-ciazione tra varianti alleliche di questo gene e seve-rità del danno polmonare in termini di funzionalità
Figura 4 Circuito IFNγ-IL-12 implicato nella sindrome da aumentata predisposizione alle infezioni invasive da mico-batteri (MSMD).
Micobatterio IL-12 IL-12R
NEMO
STAT
IFN-γγIFN-γγ R
CD40 CD40L
Fagocito/cellula dendritica T linfocito

Cardinale, et al.30
respiratoria, colonizzazione da Pseudomonas aerugi-nosa e tempo di sopravvivenza (49, 50).Specifiche varianti genetiche della β-defensina 1,un peptide antimicrobico sensibile alla concentra-zione salina, sembrano avere un ruolo nel condi-zionare la colonizzazione polmonare da pseudo-monas nei pazienti affetti da FC (51).Infine, specifici polimorfismi del promoter del genedell’IL-10 associati ad un aumento di produzionedella suddetta citochina, sembra possano incre-mentare la suscettibilità dei pazienti FC all’infezio-ne polmonare da pseudomonas (52).
Suscettibilità alle infezioni e asma
È noto che l’infezione da rhinovirus rappresentauno dei trigger più frequenti dell’asma e che ipazienti asmatici risultano maggiormente suscetti-bili nei confronti di questo tipo di infezione.Recenti studi hanno dimostrato che questa corre-lazione può essere spiegata da un deficit specificonella risposta immune innata verso questi virus. Inparticolare, in asmatici adulti è stata osservata unaproduzione difettiva di IFN-β e λ nei confronti deirhinovirus da parte dell’epitelio bronchiale e deimacrofagi alveolari, avente come effetto un’altera-zione dell’apoptosi cellulare e un aumento della
replicazione virale (53-55). Se questo difetto virus-specifico dell’immunità innata sia comunque primi-tivo, e cioè geneticamente determinato, o acquisi-to, non è ancora noto.
Conclusioni e prospettive future
L’aumentata resistenza agli antibiotici ha indotto afocalizzare l’attenzione sulla necessità di svilupparenuove strategie terapeutiche nella cura dellemalattie infettive (farmaci immunomodulatori,terapie sostitutive). Alla luce delle più recenticonoscenze, appare sempre più evidente chemolti individui “normali”, forse la totalità, possonodi fatto presentare uno o, probabilmente, più difet-ti immunitari minori, la cui espressione clinicadipende da fattori ambientali, di natura microbicao legati all’intervento umano (5). Dalla somma diquesti difetti deriva la suscettibilità dell’individuoalle infezioni. Un precoce riconoscimento dei sog-getti maggiormente a rischio potrà permettere infuturo di intervenire con misure preventive ad hoce/o terapie sostitutive o immunomodulanti. Perchéquesto processo si compia è necessaria comun-que una migliore conoscenza della patogenesidelle malattie infettive e delle differenze immuno-logiche interindividuali.

La suscettibilità genetica alle infezioni respiratorie 31
1. Sørensen TI, Nielsen GC, Andrersen PK,Teasdale TW. Genetic and environmental influenceson premature death in adult adoptees. N Engl JMed 1988; 318: 727-732.
2. Hedlund JU, Ortqvist AB, Kalin M, et al. Risk ofpneumonia in patients previously treated in hospitalfor pneumonia. Lancet 1992; 340: 396-397.
3. Kwiatkowski D. Science, medicine and thefuture: susceptibility to infection. BMJ 2000; 321:1061-1065.
4. Casanova JL, Fieschi C, Zhang SY, Abel L.Revisiting human primary immunodeficencies. JIntern Med 2008; 264: 115-127.
5. Casanova JL, Abel L. Human genetics of infec-tious diseases: a unified theory. EMBO J 2007; 26:915-922.
6. Bochud PY, Bochud M, Talenti A, Calandra T.Innate immunogenetics: a tool for exploring newfrontiers of host defence. Lancet Infect Dis 2007; 7:531-542.
7. Hill AV, Allsopp CE, Kwiatkowski D, et al.Common west African HLA antigens are associatedwith protection from severe malaria. Nature 1991;352: 595-600.
8. Singh SP, Mehra NK, Dingley HB, et al. Humanleukocyte antigen (HLA)-linked control of suscepti-bility to pulmonary tubercolosis and association withHLA-DR types. J Infect Dis 1983; 148: 676-681.
9.Todd JR, West BC, McDonald JC. Human leuko-cyte antigen and leprosy: study in northern Louisianaand review. Rev Infect Dis 1990; 12: 63-74.
10. Carrington M, Nelson GW, Martin MP, et al.HLA and HIV-1: heterozygote advantage andB*35-Cw*04 disadvantage. Science 1999; 283:1748-1752.
11. Thursz MR, Thomas HC, Greenwood BM,Hill AV. Heterozygote advantage for HLA class IIin Hepatitis virus infection. Nat Genet 1997; 17:11-12.
12. McAulay KA, Higgins CD, Macsween KF, et al.HLA class I polymorphisms are associated with devel-opment of infectious mononucleosis upon primaryEBV infection. J Clin Invest 2007; 117: 3042-3048.
13. Medzhitov R, Janeway C Jr. Innate immunity. NEngl J Med 2000; 343: 338-344.
14.Vannberg FO, Chapman SJ, Khor , et al. CD209genetic polymorphism and tuberculosis disease.PLoS One 2008; 3(1): e1388.
15. Akira S, Takeda K. Toll-like receptor signalling.Nat Rev Immunol 2004; 4: 499-511.
16. Tal G, Mandelberg A, Dalal I, et al. Associationbetween common Toll-Like receptor 4 mutations andsevere respiratory syncytial virus disease. J Infect Dis2004; 189: 2057-2063.
17. Le Vine AM, Gwozdz J, Stark J, et al.Surfactant protein-A enhances respiratory syncytialvirus clearance in vivo. J Clin Invest 1999; 103:1015-1021.
18. Kerr MH, Paton JY. Surfactant protein levels insevere respiratory syncytial virus infection. Am JRespir Crit Care Med 1999; 159: 1115-1118.
19. Löfgren J, Rämet M, Renko M, et al. Associationbetween surfactant protein A gene locus and severerespiratory syncytial virus infection in infant. J InfectDis. 2002; 185: 283-289.
20.Wilson J, Rowlands K, Rockett K, et al. Geneticvariation at the IL-10 gene locus is associated withseverity of respiratory syncytial virus bronchiolitis. JInfect Dis 2005; 191: 1705-1709.
21. Hoebee B, Rietveld E, Bont L, et al. Associationof severe respiratory syncytial virus bronchiolitis withinterleukin-4 and interleukin-4 receptor alpha poly-morphisms. J Infect Dis 2003; 187: 2-11.
22. Hoebee B, Bont L, Rietveld E, et al. Influenceof promoter variants of interleukin-10, interleukin-9,and tumor necrosis factor-alpha genes on respirato-ry syncytial virus bronchiolitis. J Infect Dis 2004; 189:239-247.
23. Puthothu B, Krueger M, Forster J, et al.Interleukin (IL)-18 polymorphism 133C/G is associ-ated with severe respiratory syncytial virus infection.Pediatr Infect Dis J 2007; 26: 1094-1098.
24. Janssen R, Bont L, Siezen CL, et al. Genetic sus-ceptibility to respiratory syncytial virus bronchiolitis ispredominantly associated with innate immunegenes. J Infect Dis 2007; 196: 826-834.
BIBLIOGRAFIA
Bibliografia

Cardinale, et al.32
25. Patel JA, Nair S, Revai K, et al. Association ofproinflammatory cytokine gene polymorphisms withsusceptibility to otitis media. Pediatrics 2006; 118:2273-2279.
26. Rämet M, Löfgren J, Alho OP, Hallman M.Surfactant protein-A gene locus associated withrecurrent otitis media. J Pediatr 2001;138: 266-268.
27. Wiertsema SP, Herpers BL, Veenhoven RH, etal. Functional polymorphisms in the mannan-bindinglectin 2 gene: effect on MBL levels and otitis media.JACI 2006; 117: 1344-1350.
28. Hawn TR, Verbon A, Lettinga KD, et al. A com-mon dominant TLR5 stop codon polymorphism abol-ishes flagellin signaling and is associated with sus-ceptibility to legionnaires’disease. J Exp Med 2003;198: 1563-1572.
29. Hawn TR, Verbon A, Janer M, et al. Toll-likereceptor 4 polymorphisms are associated with resist-ance to Legionnaires’disease. Proc Natl Acad Sci US A 2005; 102: 2487-2489.
30. Garcia-Laorden M, Sole-Violan J, Rodriguezde Castro F, et al. Mannose-binding lectin andmannose-binding lectin-associated serine protease2 in susceptibility, severity, and outcome of pneu-monia in adults. J Allergy Clin Immunol 2008; 122:368-374.
31. Endeman H, Herpers BL, de Jong BA, et al.Mannose-binding lectin genotypes in susceptibility tocommunity-acquired pneumonia. Chest 2008;134:1135-1140.
32. Gallangher PM, Lowe G, Fitzgerald T, et al.Association of IL-10 polymorphism with severity ofillness in community acquired pneumonia. Thorax2003; 58: 154-156.
33. Schaaf B, Rupp J, Müller-Steinhardt M, et al.The interleukin-6 -174 promoter polymorphism isassociated with extrapulmonary bacterial dissemi-nation in Streptococcus pneumoniae infection.Cytokine 2005; 31: 324-328.
34.Waterer G, Quasney M, Cantor R, WunderinkR. Septic shock and respiratory failure in communi-ty-acquired pneumonia have different TNF polymor-phism associations. Am J Respir Crit Care Med2001; 163: 1599-1604.
35. Picard C, Puel A, Bonnet M, et al. Pyogenicbacterial infections in humans with IRAK-4 deficien-cy. Science 2003; 299: 2076-2079.
36. Chapman SJ, Khor CC, Vannberg FO, et al.I� B genetic polymorphism and invasive pneumo-coccal disease. Am J Resp Crit Care Med 2007;176: 181-187.
37. Cardens M, von Bernuth H, García-SaavedraA, et al. Autosomal recessive interleukin-1 receptor-associated kinase 4 deficiency in fourth-degree rela-tives. J Pediatr 2006; 148: 549-551.
38. Ku CL, Picard C, Erdös M, et al. IRAK4 andNEMO mutations in otherwise healthy children withrecurrent invasive pneumococcal disease. J MedGenet 2007; 44: 16-23.
39. Roy S, Knox K, Segal S, et al. MBL genotype andrisk of invasive pneumococcal disease: a case-controlstudy. Lancet 2002; 359: 1569-1573.
40. Khor CC, Chapman SJ, Vannberg FO, et al. A Malfunctional variant is associated with protection againstinvasive pneumococcal disease, bacteremia, malariaand tuberculosis. Nat Genet 2007; 3: 523-527.
41. Casanova JL. Mendelian susceptibility tomycobacterial infection in man. Swiss Med Wkly2001; 131: 445-454.
42. Bornman L, Campbell SJ, Fielding K et al.Vitamin D receptor polymorphisms and susceptibili-ty to tuberculosis in West Africa: a case-control andfamily study. J Infect Dis 2004; 190: 1631-1641.
43. Koch A, Melbye M, Sørensen P, el al. Acute res-piratory tract infections and mannose-binding lectininsufficiency during early childhood. JAMA 2001;285: 1316-1321.
44. Wang X, Moylan B, Leopold DA, et al.Mutation in the gene responsible for cystic fibrosisand predisposition to chronic rhinosinusitis in thegeneral population. JAMA 2000; 284: 1814-1819.
45. Hull J, Thomson AH. Contribution of geneticfactors other than CFTR to disease severity in cysticfibrosis.Thorax 1998; 53: 1018-1021.
46. Von Bredow C, Birrer P, Griese M. Surfactantprotein A and other bronchoalveolar lavage fluid pro-teins are altered in cystic fibrosis. Eur Respir J 2001;17: 716-722.
BIBLIOGRAFIA

La suscettibilità genetica alle infezioni respiratorie 33
47. Choi E, Ehrmantraut M, Foster CB, et al.Association of common haplotypes of surfactantprotein A1 and A2 (SFTPA1 and SFTPA2) genes withseverity of lung disease in cystic fibrosis. PediatrPulmonol 2006; 41: 255-262.
48. Grasemann H, Knauer N, Büscher R, et al.Airway nitric oxide levels in cystic fibrosis patientsare related to a polymorphism in the neuronal nitricoxide synthase gene. Am J Respir Crit Care Med2000; 162: 2172-2176.
49. Gabolde M, Guilloud-Bataille M, Feingold J,Besmond C. Association of variant alleles of man-nose binding lectin with severity of pulmonary dis-ease in cystic fibrosis: cohort study. BMJ 1999; 319:1166-1167.
50. Garred P, Pressler T, Madsen HO, et al.Association of mannose-binding lectin gene hetero-geneity with severity of lung disease and survival incystic fibrosis. J Clin Invest 1999; 104: 431-437.
51. Cardinale F, Tesse R, Santostasi T, et al.Association of beta-defensin-1 gene polymorphismswith Pseudomonas aeruginosa airway colonization incystic fibrosis. Genes Immun 2008; 9: 57-60.
52. Tesse R, Cardinale F, Santostasi T, et al.Association of interleukin-10 gene haplotypes withPseudomonas aeruginosa airway colonization in cys-tic fibrosis. J Cyst Fibros 2008; 7: 329-332.
53. Contoli M, Message SD, Laza-Stanca V, et al.Role of deficient type III interferon-lambda produc-tion in asthma exacerbations. Nat Med 2006; 12:1023-1026.
54. Mallia P, Johnston SL. How viral infections causeexacerbation of airway diseases. Chest 2006; 130:1203-1210.
55. Wark P, Johnston SL, Bucchieri F, et al.Asthmatic bronchial epithelial cells have a deficientinnate immune response to infection with rhinovirus.J Exp Med 2005; 201; 937-947.
BIBLIOGRAFIA

Introduzione
Attualmente l’asma rappresenta la più comunemalattia cronica dell’infanzia con una prevalenzanei bambini italiani tra i 6-7 anni del 6,4% nellefemmine e dell’11% nei maschi (1-3). Anche leallergie alimentari sono particolarmente frequentinell’età pediatrica con una prevalenza stimata del6%-8% (4). Entrambe sviluppano nel primo trien-nio le fasi più critiche per espressione clinica emomenti patogenetici. In questo articolo puntua-lizzeremo da una parte come l’asma può esseredovuta anche ad allergia alimentare, dall’altracome la allergia alimentare predice, prepara e faci-lita lo sviluppo di asma nel bambino.
Quando l’allergia alimentare si esprimecon l’asma bronchiale
Negli ultimi 20 anni numerosi studi hanno specifi-camente valutato i rapporti tra allergia alimentareed asma dimostrando come la presenza di sintomirespiratori anche in assenza di altre manifestazioni(cutanee, gastrointestinali, anafilattiche) sia unevento non così raro e da tenere nella dovuta con-siderazione nel processo diagnostico di un bambino
con asma. Si tratta di lavori delicati da valutare,perché la delicatezza diagnostica su entrambi ifronti costituisce un moltiplicatore di difficoltà.Nell’analizzarli, un occhio deve essere tenuto allacompletezza della diagnosi sia di allergia alimenta-re (che andrebbe posta sempre con un DBPCFC -double blind, placebo-controlled food challenges), che diasma bronchiale (in un’età in cui vi possono esseredifficoltà anche solo nel rilevare la bronco-ostruzio-ne in un bambino raramente collaborante). Un’altraconsiderazione è che ci troviamo davanti ad un cro-giuolo di situazioni differenti. Orientativamente, tra ibambini con allergia alimentare sono il 33% quelliche presentano asma come unica manifestazione eun altro 33% manifesta asma e dermatite atopica(DA) (5). Alimenti ed asma sono strettamente asso-ciati anche nella pollen food syndrome dove la sensi-bilizzazione a proteine dei pollini inalanti può indur-re asma dopo ingestione di alcuni tipi di frutta evegetali a causa della presenza di proteine omologhe(6). Anche gli additivi alimentari possono esserecausa di asma bronchiale, con una prevalenza infe-riore al 5% nei pazienti asmatici (Tabella 1) (7, 8).
Alberto Martelli, Alessandra Caddeo, Sara El Oksha, Elena Calcinai, Luigi Terracciano, Alessandro Fiocchi
Melloni Pediatria, Milano
34
Allergia alimentare e asmaFood allergy and asthma
Parole chiave: asma, allergia alimentare
Keywords: asthma, food allergy
Riassunto. Asma ed allergia alimentare si interfacciano in tre situazioni. 1. L’asma può essere determinato da alimenti; talvol-ta capita agli stessi genitori di individuarne la causa, ma più spesso si tratta di forme persistenti, non risolte, forme cioè di asmadifficile. 2. L’asma si può manifestare come sintomo di anafilassi durante le reazioni immediate ad alimenti. 3. L’asma può esse-re preceduto e predetto dall’allergia alimentare, soprattutto in presenza di eczema. A questo terzo tema dedichiamo una rifles-sione in ottica possibilmente preventiva.
Accettato per la pubblicazione il 22 settembre 2009.
Corrispondenza: Dott. Alberto Martelli, Melloni Pediatria, Via Macedonio Melloni, 52, Milano; e-mail: [email protected]
Pneumologia Pediatrica 2009; 35: 34-41

Allergia alimentare e asma 35
Le caratteristiche cliniche del bambinocon allergia alimentare e asma
L’asma indotta da allergia alimentare generalmentecondiziona segni e sintomi più severi (9), tanto che iricercatori di Tucson hanno inserito la sensibilizzazio-ne ad arachidi, latte o uova tra i fattori di rischio perla persistenza di asma nell’età adulta (10). È quindi inquelle situazioni in cui la malattia condiziona negati-vamente la qualità di vita del bambino e della suafamiglia (11) che l’allergia alimentare va cercata conmaggiore determinazione.Gli alimenti possono scatenare asma non solo dopoingestione ma anche mediante inalazione, eventuali-tà riportata in letteratura per le arachidi, le carni, ilpesce, i molluschi, il latte, con crisi che possonoanche essere particolarmente severe (12, 13).Queste osservazioni rendono ragione delle grossedifficoltà che si incontrano nell’identificazione dell’al-lergene responsabile della sintomatologia.Il bambino con asma da allergia alimentare presentacaratteristiche piuttosto uniformi in tutte le osserva-zioni che comprendono la sensibilizzazione prevalen-te per alcuni allergeni (14) (arachide, uovo, pesce,latte, molluschi, noci e soia) anche se nessun alimentopuò essere escluso, come dimostrato dalle numero-sissime segnalazioni sporadiche di reazioni allergicheasmatiche a una grande varietà di cibi. È da segnalarecome specialmente nel caso dell’asma da arachidi lecrisi possano essere di assoluta gravità ed evolveresovente nel quadro di una reazione anafilattica (15).Secondo la position paper della European Academy ofAllergology and Clinical Immunology (EAACI) del2007, la compresenza di asma persistente ed allergiaalimentare rappresenta peraltro un’indicazione asso-luta alla prescrizione dell’adrenalina (16).
La diagnosi di asma da allergia alimentare
James (17) ha puntualizzato quali criteri ricercare perpoter sospettare un asma da allergia alimentare.
Criteri anamnestici per sospettare asma da allergiaalimentare
- i sintomi respiratori acuti da allergia alimentaresono normalmente accompagnati da sintomi cuta-nei e/o intestinali- sintomi respiratori cronici isolati da allergia ali-mentare sono rari- sintomi di asma da additivi alimentari possonoverificarsi molto raramente- reazioni allergiche ad alimenti possono indurreasma favorendo l’instaurarsi di iperreattività bron-chiale anche senza provocare sintomi acuti dopol’assunzione dell’alimento- i sintomi respiratori su base alimentare, in parti-colare l’asma, sono considerati fattori di rischio perreazioni gravi o fatali ad alimenti - l’uovo, il latte, le arachidi, la soia, il pesce e i mol-luschi sono i più comuni allergeni alimentari colle-gati a sintomi respiratori.
Criteri clinici all’esame obiettivo per sospettareasma da allergia alimentare
- L’asma da alimenti è più frequente nel bambinodi pochi anni di età che nel bambino più grande enell’adulto.- Bambini con dermatite atopica da alimenti sono arischio maggiore di asma da alimenti
Criteri di laboratorio per sospettare asma da aller-gia alimentare
- Una sensibilizzazione precoce verso alimentidurante l’infanzia è predittiva per il successivo svi-luppo di allergie respiratorie e di asma.- La presenza di livelli elevati di IgE totali in assenzadi sensibilizzazioni respiratorie orienta verso la pos-sibile presenza di una allergia alimentare.- Se è vero che nell’asma severo è più probabiletrovare allergia alimentare, per converso nei bam-bini con allergia alle proteine del latte vaccino (PLY)la presenza di asma o rinite all’esordio è un fattore
Tabella 1 Prevalenza dell’asma indotta da alimenti (17).
Popolazione pediatrica considerata Prevalenza stimata
Bambini con asma 5,7%
Bambini con APLV 29%
Wheezing indotto da alimento nelle reazioni acute 2%-24%
Wheezing indotto da additivi alimentari <5%
Bambini con dermatite atopica 17%-27%

Martelli, et al.36
predittivo di più tardiva tolleranza alle PLV (18).- Scattato il sospetto sulla base dell’anamnesi, del-l’esame obiettivo e della sensibilizzazione ai piùcomuni allergeni alimentari, il test di provocazioneorale in doppio cieco contro placebo rimane ilcardine per una diagnosi accurata.Fu proprio nei bambini con asma bronchiale cheper la prima volta venne condotta un’indaginesistematica per le reazioni allergiche ad alimentiutilizzando i DBPCFC (19), che rappresentanooggi il gold standard per la diagnosi di allergie ali-mentari (20, 21). Con questa metodica, Oehlig(22) prima e Rancé (23) poi, osservarono che ilbroncospasmo indotto da alimento era presentenell’8,5% dei bambini asmatici e che la maggiorparte delle sensibilizzazioni alimentari si verificavanel primo anno di vita, specialmente nei confrontidi uovo, arachidi, latte, merluzzo e senape. Anchela sensibilizzazione alle proteine del pesce puòessere un fattore rischio per asma. Infatti i bambi-ni sensibilizzati alle proteine del pesce nei primianni di vita, oltre che come già detto all’uovo,manifestano più spesso asma e broncoreattivitànella successiva età scolare (24).Benché in assenza di studi comparativi per alcuniparametri specifici (come ad esempio l’intervallodi tempo fra due challenge successivi o l’incre-mento delle dosi proposte) non sia possibile stila-re linee guida che possano uniformare ogni pas-saggio nelle procedure dei test di provocazioneorale con alimento (TPO), la position paperdell’EAACI ha contribuito a definire e standardiz-zare alcune caratteristiche dei TPO valide ancheper lo studio dell’asma da alimenti (25). In realtàmentre pare discretamente definita la modalità dipreparazione degli alimenti e la metodologia per lasomministrazione, più difficile sembra l’utilizzo delo dei test (a seconda dell’età del bambino) pervalutare l’effetto offendente dell’alimento sullebasse vie dell’apparato respiratorio. Nel caso delbambino collaborante, ma facciamo perlopiù rife-rimento al bambino di età superiore ai 6 anni, laspirometria basale e la spirometria ripetuta dopochallenge alimentare può essere diagnostica per leforme immediate. Più difficile la valutazione dellareazione ritardata non IgE mediata che può esserefatta sia attraverso l’osservazione clinica con l’a-scoltazione del torace, sia attraverso la ripetizione,in ospedale, di un’altra spirometria a distanza dialcuni giorni. Ma entro quanto tempo può esseredefinita una reazione ritardata dopo l’assunzione
di un alimento? In casi dubbi, per quanto concer-ne le reazioni ritardate, la ripetizione del challen-ge e l’analoga ricomparsa dei segni respiratoripossono confermare la diagnosi. Non sembramai sicura la possibilità di affidare la diagnosi aigenitori attraverso il monitoraggio domiciliare delpicco di flusso espiratorio con gli apparecchi por-tatili in dotazione. Anche la saturimetria pre epost challenge non è un parametro validato, siaperché non è stabilito di quanto dovrebbe ridur-si per assegnare la positività di un challenge, siaperché non dobbiamo dimenticare che la dimi-nuzione della saturimetria compare sempre intempi successivi rispetto alla broncostruzione (neè l’effetto). L’osservazione clinica attraverso lacomparsa di tosse, la polipnea, i rientramentitoracici indotti dall’attività della muscolaturaaccessoria del torace e l’auscultazione del torace,con la ricerca dei segni di broncostenosi, riman-gono il cardine per l’assegnazione della positivitàdel challenge. In virtù di tale considerazione èmolto importante che il bambino che si devesottoporre al challenge non solo sia in ottimecondizioni generali e senza un concomitante epi-sodio respiratorio ma che abbia sospeso, ovepossibile, la terapia farmacologia di fondo dell’a-sma da almeno 1 settimana. Una sospensione far-macologica più tardiva (troppo prossima all’ese-cuzione del challenge) potrebbe infatti indurre uneffetto “rebound” con un peggioramento dell’a-sma non indotta dall’alimento testato ma dallasospensione troppo recente della terapia farma-cologica. Pertanto anche il momento ottimaledell’esecuzione del challenge deve essere attenta-mente valutato.La valutazione clinica rimane il cardine della dia-gnosi anche perché, nel bambino asmatico, lo stu-dio delle variazioni sieriche di alcune citochinedurante l’esecuzione del test di provocazioneorale con alimento non è risultato significativo(26). Nel sospetto di asma secondario ad inalazio-ne di alimento il challenge bronchiale è lo stru-mento diagnostico più fine (27).Uno strumento promettente per la valutazioneobiettiva dei primissimi segni di broncostenosi incorso di challenge bronchiale in bambini di etàprescolare è l’uso di sensori acustici più fini del-l’orecchio umano. Esso è in grado di rivelare ilprimo wheeze già nei primi 2 minuti di nebulizza-zione, ancora non percepibile dall’orecchio delpediatra, nel 31% dei bambini risultati positivi al

Allergia alimentare e asma 37
challenge (28). Una precoce sospensione delchallenge positivo evita il procrastinare di un’inu-tile stimolazione asmogena. È auspicabile chenuove tecniche, come l’interrupter resistance(Rint) (Figura 1), adatte allo studio delle resisten-ze respiratorie nel bambino più piccolo, possanoessere utilizzate, nel TPO, per il monitoraggio del-l’effetto asmogeno dell’alimento sulle basse vierespiratorie; tuttavia la mancanza di parametrivalidati in tal senso pone la valutazione di questemetodiche solo su una base sperimentale.
Asma e additivi alimentari
Gli additivi alimentari possono essere causa diasma bronchiale, con una prevalenza inferiore al5% nei pazienti asmatici (29, 30). È anche contro-verso se i pazienti con asma possano presentare,rispetto alla popolazione generale, più frequentireazioni dopo assunzione di glutammato monoso-dico (MSG). Woods et al. in uno studio in doppiocieco su 12 pazienti che riferivano, in anamnesi, unpeggioramento dell’asma dopo assunzione diMSG non hanno evidenziato né asma dopo chal-lenge con MSG né modificazione di alcuni para-metri proteici di attivazione (ad esempio triptasi eproteina cationica dell’eosinofilo) (31).
La marcia atopica
L’altro aspetto di interesse per la relazione traasma ed allergia alimentare è lo studio della com-parsa di malattia respiratoria nel contesto di quel-la che viene comunemente identificata come“marcia atopica”. Con questo termine intendiamoun decorso della malattia atopica nel quale, in lineagenerale, i sintomi ed i segni delle diverse malattieallergiche si manifestano in sequenza con delle fasidi compresenza delle diverse malattie e altre in cuile stesse si susseguono in una definita sequenzatemporale (32). Di solito la dermatite atopica e lasensibilizzazione ad allergeni alimentari precedonoquella ad allergeni inalatori con il successivo svi-luppo di rinite e di asma bronchiale (33). Talvolta larinite precede l’asma, senza che siano evidenziabi-li nella storia clinica segni o sintomi di eczema osensibilizzazione agli alimenti.Recentemente è stato anche descritto che nonsolo esiste associazione fra allergia alimentare easma ma che questa associazione è tanto piùstretta quanto più il bambino presenta poliallergiaalimentare o allergia alimentare severa (34).
Secondo il modello classico di marcia atopica, circala metà dei bambini con dermatite atopica svilup-pa i segni cutanei nei primi 6 mesi di vita, e l’85%dei pazienti con eczema ha avuto i primi segni dellamalattia cutanea nei primi 5 anni di vita (35).Parallelamente si manifesta l’allergia alimentare cheha un picco di incidenza ad 1 anno di vita, quandocolpisce il 6%-8% della popolazione, per poi decli-nare gradatamente fino ad una prevalenza dell’1%-2% nella popolazione adulta. La sensibilizzazioneagli inalanti inizia intorno ai 3 anni di vita, nelle etàimmediatamente successive all’allergia alimentare esi incrementa rapidamente, tanto che il 60% degliasmatici ha l’esordio della malattia entro i primi 6anni di vita (36). Come é noto, la sensibilizzazionead alimenti è un potente fattore predittivo dellosviluppo successivo di sensibilizzazione allergica peri comuni aeroallergeni dell’infanzia. In particolare èstato documentato che la presenza di IgE specifi-che per l’albume (> 0.35 kU/l) correla con lo svi-luppo di asma. In combinazione con una anamnesifamiliare positiva per atopia, questo fattore è alta-mente predittivo di allergia ad inalanti all’età di 3anni con una specificità del 99% ed un valore pre-dittivo positivo del 78% (37, 38).Del tutto recentemente il concetto di marcia atopi-ca è stato in parte rivisitato: ecco gli aspetti consoli-dati e le nuove considerazioni su questo argomento.In base ai dati osservati nei bambini della coorteMAS con dermatite atopica seguiti per 7 anni èstato osservato che, almeno in un gruppo di bam-bini, la DA non precede l’asma in quanto moltimanifestano broncospasmo contemporaneamen-te alla DA (39). In questa coorte il 35,6% dei bam-bini ha avuto broncospasmo entro il terzo anno di
Figura 1 Interrupter resistance (Rint): un metodo applicabile allostudio della funzione respiratoria nel test da carico.

Martelli, et al.38
vita. I bambini con dermatite atopica più precoceo più severa avevano una maggiore prevalenza dibroncospasmo precoce rispetto a quelli senza DAo con DA lieve, rispettivamente il 46,3 contro il32,1% (p= 0,001). Questo gruppo di bambiniaveva un pattern di sensibilizzazione caratteristicoe distinto dagli altri, includente sensibilizzazioni ali-mentari, ed inoltre all’età di 7 anni avevano unafunzionalità respiratoria significativamente ridottarispetto agli altri bambini. Questi fattori suggeri-scono l’ipotesi che ci si trovi dinanzi ad un fenoti-po di malattia diverso, nel quale la progressionedella marcia allergica non si verifica, in quanto DAed asma coesistono fino dai primi momenti diespressione della malattia atopica.Peraltro vi sono alcuni aspetti di questo studio chesi conciliano bene con il modello della marcia aller-gica: in particolare negli 87 pazienti con DA preco-ce e broncospasmo precoce, la DA precedeva l’a-sma nel 56% dei casi, era concomitante nell’11% einvece seguiva il broncospasmo nel 33%, dati chenon sono completamente conciliabili con l’ipotesi diun fenotipo che esprime coesistenza delle malattie.Inoltre i bambini con DA e sensibilizzazione preco-ce, ma senza broncospasmo precoce eranocomunque quelli a maggior rischio di sviluppare l’a-sma, dato questo assolutamente coerente con ilmodello della marcia atopica. Se aggiungiamo che ilbroncospasmo precoce è sovente indotto da virused ha caratteristiche transitorie, possiamo conclu-dere che anche se questo studio getta alcuneombre sul modello della marcia allergica, sononecessarie conferme per indurre ad una sostanzia-le revisione degli attuali concetti.Ancora uno studio (40) condotto sulla coorte dibambini arruolata nell’isola di Wight mette in lucealcuni aspetti del decorso naturale dell’atopia e dellemalattie allergiche che si inquadrano difficilmente nelconcetto di marcia allergica. Sono stati arruolati1.456 bambini, che hanno eseguito dosaggio delleIgE, prick test a 4 e 10 anni, valutazione della funzio-nalità polmonare e challenge bronchiale. I fenotipiatopici sono stati definiti come: mai atopici (68%) sei prick erano negativi a 4 e 10 anni, atopici precoci(4,3%) se i prick erano positivi solo a 4 anni, atopicitardivi (11,2%) se erano positivi solo a 10 anni e ato-pici cronici (16,5%) se erano positivi a 4 e 10 anni.I pazienti del fenotipo “mai atopico” avevanocomunque una piccola ma ben valutabile preva-lenza di malattie allergiche persistenti quali asma(8,3%), rinite (0,5%) ed eczema (3,9%).
Oltre a questo dato vi sono delle peculiarità nelpattern di acquisizione della sensibilizzazione per-ché quella agli alimenti non precede sempre quel-la ad inalanti. Un’osservazione è stata condotta,recentemente, anche osservando il periodo miglio-re per eventuali strategie di prevenzione: il primoanno di vita. Infatti anche la sensibilizzazione adallergeni respiratori ed alimentari è riportata, spe-cie nel primo anno di vita, come fattore predittivodi sviluppo di successive manifestazioni atopicheall’età di 6 anni in bambini con familiarità atopica(41). È noto però che la procrastinata esposizionead allergeni alimentari, peraltro mai dimostratacome efficace nella prevenzione della sensibilizza-zione e dell’asma, è spesso incompleta e difficile darealizzare nella maggior parte dei casi (42). Presiinsieme, i dati di questi studi e quelli dello studioMiCMAC suggeriscono la possibilità che durante losviluppo della malattia allergica vi siano dei bambi-ni per i quali il trattamento dell’allergia alimentarepossa cogliere una finestra di opportunità per laprevenzione dell’asma; in altri casi, invece, lo svilup-po concomitante delle patologie allergiche rendedifficile pensare ad una tale strategia.L’importanza della sensibilizzazione allergica nellapredizione dell’asma è particolarmente evidentenei bambini geneticamente predisposti, quali quel-li con mutazione dei geni della filaggrina, in cui pre-disposizione genetica e sensibilizzazione ad aller-geni alimentari si esaltano a vicenda rappresentan-do due differenti meccanismi che interagiscononella patogenesi dell’asma. Infatti nei bambini coneczema e sensibilizzazione ad allergeni alimentarilo studio delle mutazioni della filaggrina potrebbe-ro predire l’asma prima ancora dell’esordio deisegni clinici, identificando così sottogruppi specificidi bambini dove poter strategicamente mirareinterventi di prevenzione nella progressione dal-l’eczema all’asma.
Conclusioni
L’associazione tra allergia alimentare ed asma èprobabilmente sottostimata a causa dell’ampiavariabilità delle manifestazioni cliniche che talvoltaconduce a diagnosi errate e della scarsa collabo-razione dei bambini all’esecuzione dei test di fun-zionalità respiratoria. Inoltre, ad oggi, manca unesame dotato contemporaneamente di sensibilitàe di specificità elevate per la diagnosi di asmasecondario ad allergia alimentare. I numerosi studirealizzati hanno dimostrato che i primi anni di vita

Allergia alimentare e asma 39
di ogni bambino sono predittivi del rischio di insor-genza e di persistenza di asma allergico: contribui-scono la storia familiare, la presenza di dermatiteatopica e la sensibilizzazione agli allergeni alimentaripiù comuni (43). Nell’asma, come in numerose altremalattie croniche, la prevenzione è di primariaimportanza, ma purtroppo le conoscenze attualinon solo non sono sufficienti ad elaborare delle
linee guida di profilassi primaria, ma nemmenosappiamo se tale profilassi sia possibile. Tuttavia,dato che gli studi epidemiologici lasciano intravve-dere qualche possibilità in tal senso, vista la gravitàdella patologia ci pare che sia opportuno investirenella realizzazione di programmi di screening perl’individuazione dei soggetti a rischio, cui far segui-re gli opportuni interventi di profilassi e terapia.
1.Woolcock AJ, Peat JK. Evidence for the increasein asthma worldwide. Ciba Found Symp 1997; 206:122-134.
2. International Study of Asthma and Allergies inChildhood Steering Committee. Worldwide varia-tions in the prevalence of atopic disease: theInternational Study of Asthma and Allergies inChildhood (ISAAC). Lancet 1998; 351: 1225-1232.
3. SIDRIA (Italian Studies on RespiratoryDisorders in Childhood and the Environment)Asthma and respiratory symptoms in 6-7 yr old Italianchildren: gender, latitude, urbanization and socioeco-nomic factors. Eur Respir J 1997; 10: 1780-1786.
4. Sampson HA. Food allergy - accurately identifyingclinical reactivity. Allergy 2005; 60 (suppl): 19-24.
5. Sicherer SH, Noone SA, Munoz-Furlong. A Theimpact of childhood food allergy on quality of life.Ann Allergy Asthma Immunol 2001; 87: 461-464.
6. Kanny G, Moneret-Vautrin DA, Flabbee J, et al.Population study of food allergy in France. J AllergyClin Immunol 2001; 108: 133-140.
7. Onorato J, Merland N, Terral C, et al. Placebo-controlled double-blind food challenges in asthma. JAllergy Clin Immunol 1986; 78: 1139-1146.
8. James JM, Crespo JF. Allergic reactions to foodsby inhalation. Curr Allergy Asthma Rep 2007; 7:167-174.
9.Wang J, Visness CM, Sampson HA. Food allergensensitization in inner-city children with asthma. JAllergy Clin Immunol. 2005;115: 1076-1080.
10. Guilbert TW, Morgan WJ, Geiger RS, et al.Atopic characteristics of children with recurrentwheezing at high risk for the development of child-hood asthma. J Allergy Clin Immunol 2004; 114:1282-1287.
11. Everhart RS, Fiese BH. Asthma severity andchild quality of life in pediatric asthma: a systematicreview. Patient Educ Couns 2009; 75: 162-168.
12. Bahna SL. Exquisite food allergy without eating.Allergy 1994; 49: 129-130.
13. Roberts G, Patel N, Levi-Schaffer F, et al. Foodallergy as a risk factor for life-threatening asthma inchildhood: a case-controlled study. J Allergy ClinImmunol 2003; 112: 168-174.
14. Rancé F, Kanny G, Dutau G, Moneret-VautrinDA. Food hypersensitivity in children: clinical aspectsand distribution of allergens. Pediatr AllergyImmunol 1999; 10: 33-38.
15. Sicherer SH, Furlong TJ, Munoz-Furlong A, etal. A voluntary registry for peanut and tree nut aller-gy: characteristics of the first 5149 registrants. JAllergy Clin Immunol 2001; 108: 128-132.
16. Muraro A, Roberts G, Clark A, et al. for theEAACI Task Force on Anaphylaxis in Children.The management of anaphylaxis in childhood:position paper of the European academy of aller-gology and clinical immunology. Allergy 2007; 62:857-871.
17. James J. Respiratory manifestations of food aller-gy. Pediatrics 2003; 6: 1625-1630.
Bibliografia

Martelli, et al.40
18. Fiocchi A, Terracciano L, Bouygue GR, et al.Incremental prognostic factors associated with cow’smilk allergy outcomes in infant and child referrals:the Milan Cow’s Milk Allergy Cohort study. AnnAllergy Asthma Immunol 2008; 101: 166-173.
19. May CD. Objective clinical and laboratory stud-ies of immediate hypersensivity reactions to foods inasthmatic children. J Allergy Clin Immunol 1976;58: 500-515.
20. Bock SA, Sampson HA, Atkins FM, et al.Double-blind, placebo-controlled food challenge(DBPCFC) as an official procedure: A manual. JAllergy Clin Immunol 1988; 82: 986-997.
21. Sicherer SH. Food allergy: when and how toperform oral food challenges. Pediatr AllergyImmunol 1999; 10: 226-234.
22. Oehling A, Baena Cagnani CE. Food allergyand child asthma. Allergol Immunopathol 1980; 8:7-14.
23. Rancé F, Kanny G, Dutau G, Moneret-VautrinDA. Food hypersensitivity in children: clinical aspectsand distribution of allergens. Pediatr AllergyImmunol 1999; 10: 33-38.
24. Priftis KN, Mermiri D, Papadopoulou A, et al.Asthma symptoms and bronchial reactivity in schoolchildren sensitized to food allergens in infancy. JAsthma 2008; 45: 590-595.
25. Bindslev-Jensen C, Ballmer-Weber BK,Bengtsson U, et al. Standardization of food chal-lenges in patients with immediate reactions to food- position paper from the European Academy ofAllergology and Clinical Immunology. Allergy 2004;59: 690-697.
26. Krogulska A, Wasowska-Królikowska K,Polakowska E, Chrul S. Cytokine profile in childrenwith asthma undergoing food challenges. J InvestigAllergol Clin Immunol 2009; 19: 43-48.
27. Roberts G, Golder N, Lack G. Bronchial chal-lenges with aerosolized food in asthmatic, food-aller-gic children. Allergy 2002; 57: 713-717.
28. Godfrey S, Cohen S, Avital A, Springer C.Timing and nature of wheezing at the endpoint of abronchial challenge in preschool children. PediatrPulmonol 2005; 39: 262-267.
29. Onorato J, Merland N, Terral C, et al. Placebo-controlled double-blind food challenges in asthma. JAllergy Clin Immunol 1986; 78: 1139-1146.
30. Cardinale F, Mangini F, Berardi M, et al.Intolerance to food additives: an update. MinervaPediatr 2008; 60: 1401-1409.
31. Woods RK, Weiner JM, Thien F, et al. The effectsof monosodium glutamate in adults with asthma whoperceive themselves to be monosodium glutamate-intolerant. J Allergy Clin Immunol 1998; 101: 762-771.
32. Kjellman NM, Nilsson L. From food allergy andatopic dermatitis to respiratory allergy. PediatrAllergy Immunol 1998; (s11): 13-17.
33. Bergmann RL, Edenharter G, Bergmann KE, etal.Atopic dermatitis in early infancy predicts allergicairways disease at 5 years. Clin Exp Allergy 1998;28: 965-970.
34. Schroeder A, Kumar R, Pongracic JA, et al.Food allergy is associated with an increased risk ofasthma. Clin Exp Allergy 2009; 39: 261-270.
35. Kay J, Gawkrodger DJ, Mortimer MJ, et al. Theprevalence of childhood atopic eczema in a generalpopulation. J Am Acad Dermatol 1994; 30: 35-39.
36. Rhodes HL, Thomas P, Sporik R, et al. A birthcohort study of subjects at risk of atopy: twentytwoyear follow-up of wheeze and atopic status. Am JRespir Crit Care Med 2002; 165: 176-180.
37. Nickel R, Kulig M, Forster J. Sensitization tohen’s egg at the age of twelve months is predictivefor allergic sensitization to common indoor and out-door allergens at the age of three years. J AllergyClin Immunol, 1997; 99: 613-617.
38.Wahn U, Bergmann R, Kulig M, et al. The nat-ural course of sensitisation and atopic disease ininfancy and childhood. Pediatr Allergy Immunol1997; 8 (suppl): 16-20.
39. Illi S, Von Mutius E, Lau S, et al. The naturalcourse of atopic dermatitis from birth to age 7 yearsand the association with asthma. J Allergy ClinImmunol 2004; 113: 925-931.
40. R. J. Kurukulaaratchy, S. Matthews,S. H. Arshad.Defining childhood atopic phenotypes to investigatethe association of atopic sensitization with allergicdisease. Allergy 2005; 60: 1280-1286.

Allergia alimentare e asma 41
41. Brockow I, Zutavern A, Hoffmann U, et al.;GINIplus StudyGroup. Early allergic sensitizationsand their relevance to atopic diseases in childrenaged 6 years: results of the GINI study. J InvestigAllergol Clin Immunol 2009; 19:180-187.
42. Vlieg-Boerstra BJ, van der Heide S, BijleveldCM, Kukler J, et al. Dietary assessment in children
adhering to a food allergen avoidance diet for aller-gy prevention. Eur J Clin Nutr 2006; 60: 1384-1390.
43. Marenholz I, Kerscher T, Bauerfeind A, et al. Aninteraction between filaggrin mutations and earlyfood sensitization improves the prediction of child-hood asthma. J Allergy Clin Immunol 2009; 123:911-916.

Introduzione
Una serie di evidenze epidemiologiche, fisiopatolo-giche e cliniche dimostra che asma e rinite sonospesso associate tra loro e che, specie nei bambinie negli adolescenti, la rinite costituisce un fattore dirischio per il successivo sviluppo di asma (1, 2, 3).Inoltre, nei soggetti affetti da rinite è frequenteriscontrare una condizione di iperreattività bron-chiale non specifica, che correla con l’infiammazione
eosinofila delle vie aeree superiori (4). Infine, iltrattamento di bambini affetti da rinite allergicacon antistaminici di terza generazione o steroidinasali è in grado di indurre un significativo miglio-ramento dei sintomi asmatici (5, 6, 7). Evidenze dicarattere anatomo-patologico, fisiopatologico eclinico hanno ampiamente dimostrato la strettacorrelazione esistente tra vie aeree superiori ed
Mariangela Tosca, Giorgio Ciprandi
U.O. Pneumologia ed Allergologia, Istituto “G. Gaslini”; Dipartimento di Medicina Interna, Universitàdi Genova
42
Rinite allergica ed asmain età pediatrica
Allergic rhinitis and asthma in childhood
Parole chiave: rinite allergica, asma
Keywords: allergic rhinitis, asthma
Riassunto. Evidenze di carattere anatomo-patologico, fisiopatologico e clinico hanno ampiamente dimostrato la stretta cor-relazione esistente tra vie aeree superiori ed inferiori, per cui nel caso di asma e rinite allergica è possibile parlare di un’unicaentità clinica.La rinite allergica costituisce un importante fattore di rischio per lo sviluppo di asma e per tale motivo, soprattutto in campo pedia-trico, va opportunamente indagata e trattata. Per quanto concerne i dati italiani, mediamente la prevalenza di sintomi nasali, dinatura allergica, si attesta sul 17,6 % nella fascia 6-7 anni e sul 31,3% nella fascia 13-14 anni. La valutazione della comorbidità asma-rinite in Europa si attesta sull’1,9% nei bambini di età scolare e sul 3,7% negli adolescenti. Pur essendo l’asma molto frequenteanche in età pediatrica, risulta spesso difficile stabilirne con precisione la reale prevalenza nelle diverse fasce di età. La definizionedi asma in età pediatrica non è infatti affatto scontata, anche se molte evidenze anatomopatologiche suggeriscono una quasi com-pleta sovrapposizione delle alterazioni immuno-flogistiche a livello bronchiale dell’adulto e del bambino con patologia asmatica.Pertanto, l’individuazione di fenotipi di “respiro sibilante” o di asma, anche al di sotto dei cinque anni di età, è notevolmente utilenella gestione pratica del paziente, come proposto da documenti recentemente pubblicati da diverse società scientifiche. La rini-te allergica inoltre rappresenta spesso la “spia” di una patologia asmatica, non opportunamente indagata e la sua durata appareessere una causa determinante per il progressivo deterioramento sia della funzione nasale che di quella bronchiale. Quindi ognipaziente con rinite allergica, anche laddove questa appaia come un quadro isolato, dovrebbe sempre essere accuratamente valu-tato eseguendo quando possibile uno studio della funzionalità respiratoria, ed avviando un trattamento adeguato che controlli lasintomatologia nasale e conseguentemente prevenga l’evoluzione asmatica o migliori la funzionalità bronchiale.Attualmente obbiettivo primario di diverse società scientifiche è individuare nuove strategie farmacologiche preventive, possi-bilmente in grado di modificare la storia naturale della malattia allergica, in considerazione dell’alto impatto che l’asma e la rini-te hanno sulle risorse finanziarie dei singoli paesi: l’elevato numero di pubblicazioni ne rimarca infatti il notevole interesse scien-tifico e socio-economico.
Accettato per la pubblicazione il 31 agosto 2009.
Corrispondenza: Dott.ssa Mariangela Tosca, Centro Malattie Allergiche, U.O. Pneumologia, Istituto “G. Gaslini”,Largo G. Gaslini 5, 16147 Genova; e-mail: [email protected]
Pneumologia Pediatrica 2009; 35: 42-49

Rinite allergica ed asma in età pediatrica 43
inferiori, per cui nel caso di asma e rinite allergicaè possibile parlare di un’unica entità clinica (unitedairway disease) (1, 8).
Epidemiologia
La prevalenza della rinite allergica nella popolazionegenerale è in aumento anche nel nostro Paese. Unaricerca multicentrica condotta in Italia settentriona-le nell’ambito della European Community RespiratoryHealth Survey (ECRHS) ha infatti riportato una pre-valenza della rinite allergica pari al 18,5% (9).Confrontando tali dati con quelli relativi ad alcunidecenni fa, si evidenzia chiaramente come la preva-lenza di questa manifestazione clinica sia progressi-vamente aumentata di oltre il 50%, con importantiripercussioni di ordine socio-economico.Analizzando la conoscenza dei dati epidemiologici,appare inconfutabile il concetto che anche l’asma,oltre alla rinite allergica costituisca, anche in etàpediatrica, una realtà con un elevato impatto epi-demiologico. Al riguardo, le informazioni più preci-se ed attendibili sono fornite dallo studio ISAAC(International Study of Allergy and Asthma inChildhood) giunto già alla terza fase (10).La prima fase dello studio dimostrava che esisteuna notevole variazione della prevalenza dei sinto-mi di asma e di rinite nelle varie aree geografiche,così come nelle due fasce di età studiate (bambinidi 6-7 anni e di 13-14 anni). Le prevalenze mag-giori erano osservate nei paesi cosiddetti occiden-tali. Analisi più approfondite avevano individuatouna serie di fattori associati con queste variazioni:lo stato socio-economico, l’alimentazione, il clima,le infezioni e le caratteristiche dei pollini (11).La seconda fase dello studio era rivolta ad appro-fondire i possibili fattori etiologici coinvolti, impie-gando una serie di test più approfonditi. La com-ponente allergica appariva essere predominantesempre nei paesi che disponevano di maggioririsorse economiche (12).La terza fase dello studio ISAAC si basa sullaripetizione delle prime osservazioni a distanza di5-10 anni per esaminare il trend in funzione deltempo trascorso, includendo un numero maggio-re di centri. Per quanto concerne i dati italiani,mediamente la prevalenza di sintomi nasali siattesta sul 17,6 % nella fascia 6-7 anni e sul 31,3%nella fascia 13-14 anni (10). La valutazione dellacomorbidità asma-rinite è stata considerata percontinenti: nell’Europa si attesta sull’1,9% nei
bambini più piccoli e sul 3,7% in quelli più grandi.Pur essendo l’asma una patologia molto frequenteanche in età pediatrica, risulta spesso difficile stabi-lirne con precisione la reale prevalenza, per ragio-ni differenti, nelle diverse fasce di età.Nell’adolescente, infatti, specie nei soggetti sensibi-lizzati agli acari, la coesistenza di asma e rinite èquasi una costante e la presenza dei sintomi pro-vocati dall’interessamento delle vie aeree inferiorifa talvolta sottostimare i sintomi dall’ostruzionenasale, spesso non inquadrata come una formaallergica e pertanto sottostimata, anche per lamancanza del corteo sintomatologico classico(rinorrea, starnutazioni, prurito nasale) tipico delleforme pollinosiche e più caratteristico delle etàsuccessive (10). In questi bambini, di età scolare eprescolare, l’esacerbazione asmatica rappresentauna complicanza comune, soprattutto nel pazien-te con malattia allergica respiratoria: ricordiamoinfatti che ben il 90% delle esacerbazioni asmati-che sono scatenate da un’infezione virale respira-toria, di cui quasi il 60% imputabile a rinovirus. Laflogosi allergica favorisce l’innesco del circolo vizio-so esistente tra infezioni respiratorie ed esacerba-zioni asmatiche, dal momento che il paziente aller-gico esposto all’allergene appare particolarmentesuscettibile alle virosi respiratorie, in particolare darinovirus, in quanto presenta, a livello mucosale,un’aumentata espressione della molecola di ade-sione ICAM-1 che è il principale recettore deirinovirus umani (14).Per quanto concerne l’asma, vi è un ampio con-senso sul fatto che vada considerata non comesingola patologia ma come una sindrome chericonosce molti meccanismi fisiopatologici che sipossono sovrapporre pur rimanendo distinti; unastoria naturale che può variare significativamenteda un bambino all’altro e una risposta ai tratta-menti alquanto variabile (15-17). In favore di uninquadramento unitario dei diversi fenotipi in unasingola entità nosografica vi sono recenti studieffettuati su biopsie bronchiali che dimostranouna sostanziale analogia delle alterazioni tissutaliimmuno-flogistiche nell’asma dell’adulto e delbambino. Infatti il tessuto bronchiale del bambinoasmatico presenta le stesse alterazioni strutturalidel “bronco” dell’adulto (danno epiteliale, infiltra-zione tissutale di cellule infiammatorie, prevalen-temente eosinofili, inspessimento della membranabasale, neoangiogenesi, remodeling) (18, 19). Talialterazioni appaiono presenti precocemente

Tosca, et al.44
anche in bambini di età pre-scolare (19) a sup-porto dell’importanza di un trattamento preco-ce, preventivo ed antiinfiammatorio, possibilmen-te in grado di modificare la storia naturale dellamalattia.Tuttavia, sia le linee guida GINA (Global Initiativefor Asthma) sia il consensus document PRACTALL(Practicing Allergology) sottolineano l’importanzadell’età nel considerare i diversi fenotipi di asmain età pediatrica ed identificano tre sottogruppiben distinti: (a) i bambini della prima infanzia; (b)quelli dell’età prescolare; e (c) quelli della terzainfanzia ed adolescenti (16, 17). Non vi è alcundubbio che l’individuazione dei diversi fenotipi siaclinicamente rilevante e utile nella gestione deibambini asmatici, concetto ripreso anche daldocumento PRACTALL, che propone l’adozionedi una classificazione basata sull’identificazione deifattori scatenanti (17).Non vi è dubbio che nel bambino in età presco-lare i sintomi siano variabili e non specifici e che,nella pratica clinica, non sia possibile studiareadeguatamente le principali “caratteristiche” del-l’asma, ovvero la flogosi delle vie aeree e la fun-zionalità respiratoria. Partendo da queste osser-vazioni, nel recente statement della Task Force delEuropean Respiratory Society (20) e nel nuovodocumento GINA “pediatrico” (21) è stato stabi-lito un principio importante: in età prescolare ladefinizione di asma deve essere fatta con unapproccio prettamente descrittivo, ovvero basan-dosi esclusivamente sulla individuazione di sinto-mi (20, 21). La Task Force della EuropeanRespiratory Society riconosce fondamentalmen-te due tipi di preschool wheezers: (a) coloro chepresentano wheezing in maniera episodica (episo-dic wheezers) generalmente a seguito di infezionivirali, e (b) coloro che “fischiano” dopo stimoli divaria natura (multiple trigger wheezers). Gli episo-dic wheezers non presentano sintomi tra un epi-sodio e l’altro, e tendono generalmente a miglio-rare nel tempo sino a “guarire” intorno ai sei annidi età. Tuttavia, un sottogruppo di episodic whee-zers può gradualmente trasformarsi in multipletriggers wheeze, bambini che vanno incontro afenomeni broncospastici indotti dall’esposizionea vari trigger, inclusi gli allergeni.Al contrario dell’asma, la definizione di riniteallergica, anche in età pediatrica, è più stringentee si basa, secondo il documento ARIA (AllergicRhinitis and its Impact on Asthma), sulla presenza
di sintomi tipici, quali la rinorrea, le starnutazioni,il prurito, l’ostruzione nasale, la dimostrazionedella produzione di IgE e l’esposizione a fattoriscatenanti specifici: gli allergeni (22). Si può quin-di concordemente considerare che la rinite aller-gica costituisca la controparte dell’asma a livellodelle vie aeree superiori. I sintomi della rinite quali rinorrea, starnutazioni, eprurito nasale, dipendono essenzialmente daglieffetti conseguenti al rilascio di istamina e sonoanche definiti “sintomi irritativi”: infatti, recedonospesso in maniera significativa in seguito alla som-ministrazione di un antistaminico. Mentre l’ostru-zione nasale è un sintomo più complesso, in quan-to dipende dal concorso di più eventi fisiopatologi-ci (l’edema mucosale, la congestione vascolare e l’i-perproduzione di muco), l’ostruzione è il sintomoche maggiormente esprime la presenza della flogo-si allergica. Infatti, recenti studi hanno dimostratocome il grado di gravità dell’ostruzione nasale siacorrelato con l’intensità dell’infiltrato infiammatorioa livello mucosale (23). Proprio per questo motivo,l’ostruzione nasale risente maggiormente di untrattamento antiinfiammatorio, in particolare del-l’impiego di corticosteroidi topici nasali.La comparsa dei sintomi è stretta conseguenzadell’esposizione all’allergene causale, che incon-trandosi con le IgE specifiche adese sulla superficiedelle cellule effettrici primarie (i mastociti) innescauna cascata di reazioni biochimiche che, da unaparte si traduce nel rilascio di mediatori prefor-mati (soprattutto l’istamina) che causano la com-parsa immediata dei sintomi irritativi, dall’altradetermina il rilascio di citochine che danno l’avvioalla cascata infiammatoria, responsabile a sua voltadella comparsa della sintomatologia ostruttiva.La rinite allergica si basa quindi sulla presenza disintomi nasali tipici conseguenti ad una rispostaflogistica IgE-mediata, e deve essere pertanto con-siderata una patologia infiammatoria delle vieaeree superiori, così come lo è l’asma delle vieaeree inferiori.
Rapporto naso-bronchi: meccanismipatogenetici
È stato ampiamente dimostrato che la rinite aller-gica costituisce uno dei più importanti fattori dirischio per l’esordio della patologia asmatica edinoltre una volta esordita l’asma ne costituisce unimportante fattore di aggravamento.

Rinite allergica ed asma in età pediatrica 45
I meccanismi fisiopatologici attraverso i quali la rini-te allergica può indurre o peggiorare l’asma sonodiversi: la progressione per contiguità della flogosiallergica dalle vie aeree superiori a quelle inferiori,il rilascio sistemico di mediatori e citochine, larespirazione orale, il riflesso rino-bronchiale, il post-nasal drip e la ridotta sintesi rino-sinusale di NO.La flogosi allergica è caratterizzata da un tipicoassetto della risposta immunitaria: la cosiddettapolarizzazione dei linfociti T helper 2 (Th2) (24).Nel soggetto allergico infatti prevalgono i Th2rispetto ai Th1, che fisiologicamente sono predo-minanti nel soggetto normale. L’abbondanza di Th2si traduce in un’aumentata produzione di citochi-ne Th2-dipendenti, tra cui spiccano l’interleuchina4 (IL-4) e l’IL-13: entrambe sono fondamentali perindirizzare la sintesi di IgE specifiche e per avviare,mantenere ed amplificare l’infiammazione mucosa-le, caratterizzata da un infiltrato eosinofilo. Sonoquindi tre gli stipiti cellulari caratteristici della rea-zione allergica: i linfociti Th2 che “orchestrano” lareazione allergica, i mastociti che la scatenanodopo l’esposizione allergenica, e gli eosinofili chesono responsabili del danno tissutale. A questoriguardo, diversi studi hanno evidenziato che ilgrado di infiltrazione eosinofila (e quindi l’intensitàdella reazione infiammatoria) è significativamentecorrelato sia con il quadro clinico che con il datofunzionale respiratorio. Inoltre, da alcuni anni sonostate scoperte nuove sottopopolazioni di linfocitiT che svolgono una funzione regolatoria: i cosid-detti T regolatori (Treg) (25). È stato dimostratoche nel soggetto allergico esiste un difetto funzio-nale di cellule Treg allergene-specifiche, difetto chepuò essere colmato dopo un trattamento conimmunoterapia specifica (26). Il difetto di Treg sitraduce in una ridotta produzione di alcune cito-chine: IL-10 e TGF-β, che, tra le numerose funzio-ni che esercitano, sono anche responsabili del con-trollo della sintesi di IgG e IgA allergene-specifiche.A questo proposito va ricordato che un allergeneper definizione è comunque un antigene: ciò impli-ca che anche un soggetto normale presenti unarisposta anticorpale nei confronti di qualsiasi aller-gene cui è esposto, ma le classi sintetizzate sonoIgG e IgA, cioè non patogene, ma espressione ditolleranza. Nell’allergico invece, essendo scarsa-mente efficienti le cellule Treg allergene-specifichee quindi le citochine che orientano verso le classiIgG e IgA, la risposta anticorpale viene indirizzataverso la classe E. Ancora più recentemente è stato
suggerito che un’altra sottopopolazione T potreb-be essere coinvolta nella genesi della reazioneallergica: i Th17; infatti i livelli sierici di IL-17 sonoaumentati nei soggetti maggiormente allergici (27).D’altro canto sono stati individuati alcuni meccani-smi ambientali, quali l’esposizione ad animali e adendotossine che, nella prima infanzia, favorirebbe-ro lo sviluppo di tolleranza e ridurrebbero ilrischio di sensibilizzazione ad allergeni e malattiaallergica. La cosiddetta ipotesi “igienica” (hygienehypotesis), si basa appunto sulla dimostrazione cheuna ridotta esposizione a stimoli antigenici duran-te l’infanzia, come accade nello stile di vita “occi-dentale”, favorisca la persistenza della polarizza-zione Th2, che è fisiologica nel feto, in quanto con-sente di evitare il rigetto immunologico materno(il feto è ovviamente un non-self per la madre).L’igiene, a volte eccessiva, dello stile di vita occi-dentale non permetterebbe quindi un’adeguatamaturazione del sistema immunitario in senso Th1o T regolatorio, favorendo lo sviluppo delle malat-tie allergiche (28). Ma esistono anche (e più com-plessi) meccanismi genetici, o di interazione tragene ed ambiente, che sono ad oggi in via di defi-nizione. È stato infatti dimostrato che sia l’asmache la dermatite atopica sono collegati al polimor-fismo della “filaggrina” (FLG), un gene, che contri-buisce all’integrità epiteliale (29). Questo aspettosuggerisce una base genetica della cosiddetta“marcia allergica”. Contribuiscono poi allo svilup-po delle malattie allergiche svariati fattori ambien-tali, quali infezioni virali, esposizione ad allergeni edinquinanti, che agiscono precocemente sul sistemaimmunitario e sulle vie aeree in via di sviluppo(30). Ma la risposta a tali fattori è in parte “misu-rata” da specifici pattern genetici. Ad esempio spe-cifici aplotipi del gene che codifica per il CD14,possono essere associati con un alto o bassorischio di sviluppare asma in relazione alla quanti-tà di endotossina presente nell’ambiente (31).Ritornando all’analisi dei meccanismi fisiopatologiciche sono coinvolti nel rapporto naso-bronchi, la flo-gosi allergica nasale si può quindi propagare versole vie aeree inferiori attraverso meccanismi di pas-saggio per continuità e contiguità, non esistendobarriere anatomiche nell’ambito dell’albero respira-torio. Inoltre, durante la complessa cascata di even-ti infiammatori si assiste ad un rilascio di numerosimediatori e citochine che, oltre ad esercitare uneffetto locale, giungono al torrente ematico per dif-fondere poi anche a livello del distretto bronchiale.

Tosca, et al.46
Un altro meccanismo fisiopatologico molto impor-tante è costituito dal venir meno della funzionenasale durante la rinite allergica. Ricordiamo perinciso che il naso esercita una funzione di “clima-tizzazione” dell’aria inalata: cioè riscalda, umidifica efiltra l’aria dell’ambiente. Ciò avviene attraverso iturbinati: queste strutture molto vascolarizzate,grazie alla loro conformazione anatomica, genera-no dei moti turbolenti che consentono un’ottima-le distribuzione del flusso aereo sulla maggiorsuperficie mucosale disponibile. Così l’aria può almeglio essere riscaldata ed umidificata, grazie allacontinua produzione di muco. Inoltre, il gel cheriveste la mucosa nasale viene continuamenterimosso attraverso un movimento metacronaledelle ciglia vibratili presenti sulla superficie epitelia-le, movimento che favorisce la clearance dellesostanze potenzialmente nocive.L’ostruzione nasale determina l’attivazione riflessadella respirazione orale: viene così ad essere sop-pressa la funzione nasale, pertanto ai bronchi giun-gerà aria relativamente fredda e secca, che si rive-lerà essere irritante, scatenando così l’iperreattivi-tà bronchiale. Va ricordato che la respirazioneorale è il principale meccanismo coinvolto nellagenesi dell’asma da sforzo: aumentando l’intensitàdello sforzo fisico la respirazione nasale diventainsufficiente a garantire l’ematosi e quindi subentrala respirazione orale che però induce la comparsadi iperreattività bronchiale.Un altro meccanismo fisiopatologico è rappresen-tato dal cosiddetto post-nasal drip: cioè il pasaggioretro-nasale (o rinorrea posteriore) di muco.L’iperproduzione di muco da una parte prende lavia anteriore e giunge alle coane, dall’altra scendein rino-faringe fino a stimolare gli irritant receptorselicitando il riflesso tussigeno.Ancora, la rinite allergica induce la stimolazione diun riflesso naso-bronchiale mediato dal nervovago, che si traduce in un riflesso broncospastico.Infine, le vie aeree superiori costituiscono un’ab-bondante fonte di produzione di ossido nitrico(NO) che svolge anche un’attività broncodilatatri-ce, la rinite allergica si caratterizza per una ridottaproduzione di NO, che potrebbe quindi contribui-re ad aggravare la patologia asmatica.Alla luce di queste premesse appaiono quindi benchiari due importanti concetti fisiopatologici: lostretto rapporto naso-bronchi e la persistenza dellaflogosi allergica. Questo secondo aspetto presentaun’immediata implicazione di ordine cronologico:
maggiore è la durata dell’esposizione all’allergene,maggiore è la cronicità della rinite allergica.In termini generali, è nozione risaputa ed ovvia cheuna patologia cronica col tempo, soprattutto senon adeguatamente trattata, possa presentareun’evoluzione solitamente peggiorativa: progressi-vo deterioramento funzionale e danno anatomico,aggravamento dei sintomi e comparsa di sequelee complicanze.
Studi clinici inerenti la durata della rini-te ed il suo impatto sull’asma
Peraltro, a fronte di tale considerazione, ovveroche la patologia cronica nasale può indurre unprogressivo danno funzionale delle vie aeree intoto, in letteratura sono pochi gli studi che hannoaffrontato questa problematica: cioè la valutazionedelle possibili conseguenze della durata della riniteallergica sulla funzionalità bronchiale.Un primo studio preliminare condotto su giovaniadulti ha avuto come obbiettivo la valutazione del-l’andamento nel tempo, in funzione della duratadella rinite allergica, dell’indagine spirometrica,valutata mediante un particolare parametro: il FEF25-75 (32). Questo parametro merita una parti-colare considerazione: infatti, sebbene non rivestaun ruolo diagnostico per l’asma, rappresentando iflussi aerei distali è stato dimostrato essere unmarcatore precoce di coinvolgimento bronchialein pazienti con sola rinite allergica e “spia” di unacondizione di iperreattività bronchiale. Questaindagine è stata condotta in un ampio gruppo dipazienti affetti da rinite allergica persistente chepercepivano solo sintomi nasali. I risultati hannoevidenziato in maniera molto significativa che isoggetti che presentavano valori di FEF 25-75<80% del predetto avevano una durata della rini-te allergica significativamente maggiore rispetto aisoggetti con normali valori di FEF 25-75.Per confermare tale dato è quindi condotto unsuccessivo studio su una casistica analoga, avendocome obiettivo primario la valutazione del FEV1 infunzione della durata della rinite allergica (33).Questo studio confermava il precedente, in quan-to aumentando la durata della rinite si assisteva adun progressivo deterioramento della funzionebronchiale, valutata mediante i valori di FEV1.Inoltre, è stata dimostrata una progressione deldeterioramento della funzione bronchiale stretta-mente correlata alla durata della rinite: infatti la

Rinite allergica ed asma in età pediatrica 47
contestuale alterazione di 3 parametri spirometri-ci (FEV1, FVC e FEF 25-75) si osservava neipazienti con maggiore durata, mentre la singolaalterazione del solo FEF 25-75 era tipico dei sog-getti con minor durata.Questo studio ha evidenziato due importanti fat-tori che possono svolgere un ruolo favorente l’e-voluzione della rinite allergica in asma: anzitutto ladurata della rinite ed il tipo di sensibilizzazione.Infatti chi è sensibilizzato agli acari presenta un ele-vato rischio (OR= 11,7) di avere un FEV1 <80%.Inoltre per ogni anno di durata della rinite allergi-ca, il fattore di rischio, relativo allo sviluppo diasma, sale di 2,3 volte. Sono entrambi valori moltosignificativi e rilevanti. Quindi nella pratica clinicaquotidiana, di fronte ad un paziente con riniteallergica quando questa duri da tempo e/o si asso-ci ad un’allergia agli acari bisogna sempre sospet-tare un possibile coinvolgimento bronchiale.Dato che la caratteristica fisiopatologica principa-le dell’asma è la presenza di iperreattività bron-chiale, è stato condotto un altro studio per valu-tare l’effetto della durata della rinite allergica sullapossibile comparsa di iperreattività bronchiale(34). Ricordiamo che anche nel soggetto con solarinite allergica può essere documentata la pre-senza di iperreattività bronchiale, che in questocaso può rivestire il significato di una possibileevoluzione verso l’asma. Lo studio pertanto havalutato, in un campione di pazienti con riniteallergica persistente, proprio la possibile presenzadi iperreattività bronchiale in funzione della dura-ta della rinite stessa. Tutti i pazienti rinitici sonostati sottoposti al test di provocazione bronchialecon metacolina al fine di definire la presenza omeno di iperreattività bronchiale e stabilirne ilgrado, valutando la PD20.Questo studio ha evidenziato che una notevolepercentuale di soggetti rinitici presentano iper-reattività bronchiale. Correlando la presenza ed ilgrado di iperattività bronchiale con la duratadella rinite, si è confermato che l’iperreattivitàgrave è significativamente associata ad una mag-gior durata della rinite (34). Un altro studio,appena pubblicato, ha valutato in un ampio grup-po di pazienti pediatrici con rinite allergica isola-ta, la risposta al test di broncodilatazione, effet-tuato dopo una spirometria basale, dimostrandoche più del 20% di essi presentavano una rispo-sta positiva al test. Si trattava dei pazienti conFEV1 più basso, una durata della rinite maggiore
e con allergia perenne. Questo trial sottolineaulteriormente l’importanza dello studio della fun-zionalità respiratoria nei soggetti con sola rinite,anche nei pazienti pediatrici (35).In conclusione, la rinite allergica è un importantefattore di rischio per l’asma e la sua durata appareessere un aspetto determinante verso questa evo-luzione. Un non adeguato trattamento della flogo-si allergica nasale causa infatti un rilevante deterio-ramento sia della funzione nasale che di quellabronchiale, che si rivelano essere progressivi neltempo. Quindi ogni paziente con rinite allergica,anche laddove questa appaia come un quadro iso-lato, e non associato all’asma, deve sempre essereaccuratamente indagato eseguendo sempre, sepossibile, uno studio della funzionalità respiratoria,ed avviando un trattamento che controlli la flogo-si allergica sottostante.
Conclusioni
Senza dubbio la rinite e l’asma allergica, rappresen-tano le forme più frequenti di malattia allergica, esono, a tutt’oggi, un argomento di estremo interes-se nel campo della ricerca. I dati epidemiologici sot-tolineano infatti l’elevata prevalenza di queste duepatologie, e la loro stretta sovrapposizione ci con-duce a considerarle come un’unica patologia.Inoltre un’attenta analisi socio-economica consen-te di rilevare il loro alto impatto sulle risorse finan-ziarie e l’elevato numero di pubblicazioni scientifi-che ne rimarca il notevole interesse socio-econo-mico. D’altro canto le attuali conoscenze sulle aller-gie respiratorie, soprattutto in ambito pediatrico,rimangono ancora carenti. In particolare, alcuniimportanti quesiti rimangono ancora insoluti: esistela possibilità di una reale prevenzione nei bambinipredisposti? Possiamo agire modificando la pro-gressione della malattia? L’immunomodulazione èrealmente efficace? Un tentativo di affrontarerazionalmente l’argomento è l’obbiettivo delConsorzio Internazionale sulle Allergie e l’Asma inPediatria (iPAC) (36), che affronterà temi quali: 1. migliorare la caratterizzazione dell’asma inpediatria, determinando quali fattori genetici oambientali indirizzano verso specifici fenotipi.2. Identificare marker specifici dei singoli fenotipi.3. Prevenire il remodeling, utilizzando strategiecombinate (farmaci ed immunoterapia).4. Identificare precocemente i bambini a rischio.5. Sviluppare farmaci preventivi “anti-virali”, chepossano prevenire lo sviluppo di asma.

Tosca, et al.48
6. Condurre studi prospettici sullo sviluppo delsistema immune e l’esordio di malattia allergica.7. Misurare con specifici parametri lo sviluppo delsistema immune a livello delle vie aeree.8. Studiare le interazioni gene-ambiente e come ipolimorfismi genetici interagiscono con specificifattori ambientali nel modificare il rischio di malat-tia allergica cronica.9. Studiare i meccanismi con cui l’esposizione aspecifici allergeni influenzi l’eziopatogenesi dellemalattie allergiche.
10. Valutare il rapporto costo-efficacia dei singolitrattamenti.Tutto ciò dovrebbe aiutarci a migliorare la com-prensione dei meccanismi patogenetici delle malat-tie allergiche, favorendone la prognosi. Tali ricerchedovrebbero inoltre contribuire all’individuazione dinuove strategie farmacologiche preventive, prima-rie e secondarie, specifiche per i singoli fenotipi,individualizzando la terapia per “categorie” dipazienti e riducendone in tal modo i costi.
1. Bousquet J, Khaltaev N, Cruz AA, et al. AllergicRhinitis and its Impact on Asthma (ARIA) 2008update (in collaboration with the World HealthOrganization, GA(2)LEN and AllerGen). Allergy2008; 63 (suppl) 86: 8-160.
2. Brozek JL, Baena-Cagnani CE, Bonini S, et al.Methodology for development of the Allergic Rhinitisand its Impact on Asthma guideline 2008 update.Allergy 2008; 63: 38-46. Review.
3. Leynaert B, Bousquet J, Neukirch C, et al.Perennial rhinitis: An independent risk factor for asth-ma in nonatopic subjects: results from the EuropeanCommunity Respiratory Health Survey. J Allergy ClinImmunol 1999; 104: 301-304.
4. Sale R, Silvestri M, Battistini E, et al. Nasal inflam-mation and bronchial reactivity to methacholine inatopic children with respiratory symptoms. Allergy2003; 58: 1171-1175.
5. Rossi GA, Tosca MA, Passalacqua G, et al.Evidence of desloratadine syrup efficacy and tolera-bility in children with pollen-induced allergic rhinitis.Allergy 2005; 60: 416-417.
6. Agondi RC, Machado ML, Kalil J, et al. Intranasalcorticosteroid administration reduces nonspecificbronchial hyperresponsiveness and improves asthmasymptoms. J Asthma 2008; 45: 754-775.
7. Camargos P, Ibiapina C, Lasmar L, et al.Obtaining concomitant control of allergic rhinitis and
asthma with a nasally inhaled corticosteroid. Allergy2007; 62: 213-215.
8. Bousquet J, Van Cauwenberge P, Khaltaev N;ARIA Workshop Group; World HealthOrganization. Allergic rhinitis and its impact onasthma. J Allergy Clin Immunol 2001; 108: S147-S334.
9. Verlato G, Corsico A, Villani S, et al. Is the pre-valence of adult asthma and allergic rhinitis stillincreasing? Results of an Italian study. J Allergy ClinImmunol 2003; 111: 1232-1238.
10. Ait-Khaled N, Pearce N, Anderson HR, et al.Global map of the prevalence of symptoms of rhi-noconjunctivitis in children: The International Study ofAsthma and Allergies in Childhood (ISAAC) PhaseThree. Allergy 2009; 64: 123-148.
11. The International Study of Asthma andAllergies in Childhood (ISAAC) SteeringCommittee. Worldwide variation in prevalence ofsymptoms of asthma, allergic rhinoconjunctivitis, andatopic eczema. Lancet 1998; 351: 1225-1232.
12. Asher I. ISAAC International Study of Asthmaand Allergies in Childhood. Pediatr Pulmonol 2007;42: 100.
13. Silvestri M, Oddera S, Rossi GA, et al.Sensitization to airborne allergens in children withrespiratory symptoms. Ann Allergy AsthmaImmunol 1996; 76: 239-244.
Bibliografia

Rinite allergica ed asma in età pediatrica 49
14. Ciprandi G, Buscaglia S, Pesce GP, et al.Minimal persistent inflammation is present at muco-sal level in asymptomatic rhinitic patients withallergy due to mites. J. Allergy Clin Immunol 1995;96: 971-979.
15. Beydon N, Davis SD, Lombardi E, et al. An offi-cial American Thoracic Society/European RespiratorySociety statement: pulmonary function testing in pre-school children. Am J Respir Crit Care Med 2007;175: 1304-1345.
16. Bateman ED, Hurd SS, Barnes PJ, et al. Globalstrategy for asthma management and prevention:GINA executive summary. Eur Respir J 2008; 31:143-178.
17. Bacharier LB, Boner A, Carlsen KH, et al.Diagnosis and treatment of asthma in childhood: aPRACTALL consensus report.Allergy 2008; 63: 5-34.
18. Barbato A, Turato G, Baraldo S, et al. Epithelialdamage and angiogenesis in the airways of childrenwith asthma. AJRCCM 2006; 174: 975-981.
19. Saglani S. Payne DN, Zhu J, et al. Early detec-tion of airway wall remodeling and eosinophilicinflammation in preschool wheezers. AJRCCM2007; 176; 858-864.
20. Brand PL, Baraldi E, Bisgaard H, et al.Definition, assessment and treatment of wheezingdisorders in preschool children: an evidence-basedapproach. Eur Respir J 2008; 32: 1096-1111.
21. The Global Strategy for the Diagnosis andManagement of Asthma in Children 5 Years andYounger. Global Initiative for Asthma (GINA) 2009.Disponibile su: http://www.ginasthma.org.
22. Bousquet J, Bodez T, Gehano P, et al.Implementation of Guidelines for Allergic Rhinitis inSpecialist Practices. A Randomized PragmaticControlled Trial. Int Arch Allergy Immunol 2009;150: 75-82.
23. Ciprandi G, Cirillo I, Pistorio A. Relationshipbetween severity of rhinitis symptoms and nasal air-flow. Rhinology 2008 Sep; 46: 209-212.
24. Christodoulopoulos P, Cameron L, Durham S,et al. Molecular pathology of allergic disease. IIUpper airway disease. J Allergy Clin Immunol 2000;105: 211-223.
25. Jutel M, Akdis M, Blaser K, et al. Mechanism ofallergen specific immunotherapy – T cell toleranceand more. Allergy 2006; 61: 796-807.
26. Ciprandi G, Fenoglio D, Tosca MA, et al.Induction of interleukin 10 by sublingual immuno-therapy for house dust mites: a preliminary report.Ann Allergy Asthma Immunol 2005; 95: 38-44.
27. Ciprandi G, Fenoglio D, De Amici M, et al. SerumIL-17 in allergic rhinitis. JACI 2008; 122: 650-651.
28. Kramer MS, Matush L, Bogdanovich N, et al.The low prevalence of allergic disease in EasternEurope: are risk factors consistent with the hygienehypothesis? Clin Exp Allergy 2009; 39: 708-716. E-pub 2009 Mar 3.
29. Palmer CN, Irvine AD, Terron-Kwiatkowski A,et al. Common loss-of-function variants of the epi-dermal barrier protein filaggrin are a major predi-sposing factor for atopic dermatitis. Nat Genet2006; 38: 441-446. Epub 2006 Mar 19
30. Jackson DJ, Gangnon RE, Evans MD, et al.Wheezing rhinovirus illnesses in early life predictasthma development in high-risk children. Am JRespir Crit Care Med 2008; 178: 667-672. E-pub2008 Jun 19.
31. Martinez FD. CD14, endotoxin, and asthmarisk: actions and interactions. Proc Am Thorac Soc2007; 4: 221-225.
32. Ciprandi G, Cirillo I, Pistorio A. Impact of aller-gic rhinitis on asthma. JACI 2007; 119: 1558-1559.
33. Ciprandi G, Cirillo I, Pistorio A. Impact of aller-gic rhinitis on asthma: effects on spirometric para-meters. Allergy 2008; 63: 255-260.
34. Ciprandi G, Cirillo I, Tosca MA, et al. Impact ofallergic rhinitis on asthma: effects on bronchial hyper-reactivity. Annals Allergy Asthma Immunol 2008;101: 42-46.
35. Capasso M, Varricchio A, Ciprandi G. Impact ofallergic rhinitis on asthma in children: effects on bron-chodilation test. Allergy 2009. Epub ahead of print.
36. Papadopoulos NG, Borres M, Gern J, et al.New visions in respiratory allergy (asthma and aller-gic rhinitis: An iPAC summary and future trends.Pediatr Allergy Immunol 2008; 19: 51-59.

Introduzione
Le aspergillosi sono infezioni causate da varie spe-cie di aspergillus (in particolare Aspergillus fumiga-tus, A. flavus, A. niger e A. terreus).Rappresentano un’importante causa di mortalità emorbidità in pazienti immunodepressi con neutro-penia cronica, infezione da HIV in stadio avanzato,immunodeficienza congenita e in pazienti sottopo-sti a trapianto allogenico di cellule staminali ema-topoietiche e/o trapianto di polmone. Soggetticon asma possono essere maggiormente predi-sposti ad aspergillosi broncopolmonare.
Patogenesi
L’inalazione di spore (2-5µm) dall’ambiente circo-stante seguito dalla proliferazione di ife nel mucodell’albero bronchiale stimola una risposta immuneTh2 CD4+ T cellule, con conseguente produzionedi anticorpi di classe IgG, IgE ed IgA (Figura 1) (1).
Fattori predisponenti
- fattori genetici: HLA-DR2/DR5; DR4/DR7: ↑ predisposizione; HLA DQ2: ruolo protettivo;
- fattori ambientali: eccessiva produzione di mucovie aeree → germinazione e rilascio Ag → attiva-zione Th2 → ↑ IgE, ↑ eosinofili.
Diagnosi
Esistono tre forme principali di aspergillosi (2):- Invasiva: comprende infezioni delle basse vierespiratorie, dei seni mascellari e della cute comevia d’ingresso del germe. Il sistema nervoso cen-trale (SNC), l’apparato cardiovascolare ed even-tuali altri tessuti possono essere infettati per disse-minazione ematogena o diretta estensione da siticontigui di infezione;- Cronica (saprofitica): otomicosi e aspergillomapolmonare;- Allergica: comprende prevalentemente sinusiti ebroncopolmoniti.Per la aspergillosi invasiva la diagnosi si definisce intre categorie:- Certa: quando l’infezione è stata documentatamediante esame istopatologico (ife tipiche dellevarie specie di Aspergillus) e colturale effettuati sucampioni prelevati da sedi normalmente sterili;
Valeria Raia, Fabiola De Gregorio, Paolo Buonpensiero, Angela Sepe
Dipartimento di Pediatria, Università Federico II Napoli
50
Aspergillosi broncopolmonareBronchopulmonary aspergillosis
Parole chiave: aspergillosi, fibrosi cistica, terapia antifungina
Keywords: aspergillosis, cystic fibrosis, antifungal therapy
Riassunto. L’aspergillosi broncopolmonare è una rara complicanza nei pazienti immunocompromessi, asmatici e più frequen-te nei soggetti affetti da fibrosi cistica. È correlata ad una risposta IgE-mediata in presenza di aspergillus che colonizza le vie respi-ratorie, e in grado di condizionare la prognosi. Diversi scenari clinici sono descritti in età pediatrica, con diversa gravità e pro-gnosi. Asma ricorrente, infiltrati polmonari e bronchiectasie, dal punto di vista clinico, ed eosinofilia, alti livelli di IgE totali, anticorpianti Aspergillus di tipo IgE e IgG, immunoprecipitine, prick test positivi per aspergillus, dal punto di vista di laboratorio, rappre-sentano i principali criteri di diagnosi. Corticosteroidi per via sistemica, eventualmente associati od in alternativa ad antimicoticirappresentano il trattamento di scelta, con durata e dosaggio correlati alla sede di infezione e alla gravità della patologia.
Accettato per la pubblicazione il 31 agosto 2009.
Corrispondenza: Dott.ssa Valeria Raia, Dipartimento di Pediatria Università “Federico II” Napoli, Via S. Pansini 5,80131 Napoli; e-mail: [email protected]
Pneumologia Pediatrica 2009; 35: 50-54

Aspergillosi broncopolmonare 51
- Probabile: quando sono soddisfatti almeno trecriteri quali condizione dell’ospite, manifestazionicliniche (sintomi, segni, quadro radiologico) e risul-tati microbiologici (positività dell’esame colturale edi quello microscopico diretto per la valutazionedi funghi filamentosi);- Possibile: quando sono presenti manifestazionicliniche suggestive in condizioni a rischio dell’ospi-te, in assenza di isolamento del microrganismopatogeno, ma positività di markers antigenici (2).La diagnosi di aspergillosi broncopolmonare sibasa sulla presenza dei seguenti parametri clinici elaboratoristici:a. Asmab. Infiltrati polmonari radiografici o tomografiacomputerizzata (TC) (lobo superiore e medio)c. Reattività cutanea immediata ad aspergillus (pre-valenza reattività cutanea per aspergillo, 23%-28%nei pazienti con asma)d. IgE totali sieriche elevate (>417 IU/mL o >1000ng/mL) e. Anticorpi sierici specifici vs A. fumigatus presenti f. Bronchiectasie centrali (2/3 superiori) alla TCpolmonare g. Eosinofilia periferica (>1.0x109/l)h. IgE o IgG anti aspergillus sierici elevati.
Esami diagnostici
A. Esami colturale, istopatologico e microscopico- sono effettuati su lavaggio broncoalveolare, agoaspirato o biopsia polmonare;
- sono il gold standard per la diagnosi diAspergillosi polmonare invasiva;- possono essere falsamente negativi in un pazien-te già in trattamento antimicotico;- sono di difficile esecuzione in pazienti in condi-zioni critiche;B. Emocoltura: di limitata utilità perché spesso nega-tiva anche in presenza di infezione disseminata;C. Tomografia computerizzata: segni radiologiciquali halo sign e air-crescent sign sono suggestivi manon caratteristici di aspergillosi polmonare invasivaD.Titolazione sierica dell’antigene galattomannano(EIA): rappresenta un buon marker di sensibilitàper la diagnosi di aspergillosi polmonare invasivase associata a segni radiologici. Il suo utilizzo èancora in fase di valutazione.E. Ricerca (1 >3)-β-D-glucani: non specifici peraspergillus; frequenti falsi positiviF. PCR: test ancora non standardizzato e non dis-ponibile in commercio.
Trattamento farmacologico
Numerosi sono gli antimicotici attivi sulle varie spe-cie di aspergillus: amfotericina B desossicolato enelle forme lipidiche, itraconazolo, e, più recente-mente, voriconazolo (3, 4), e caspofungina, ma solovoriconazolo e amfotericina B sono approvaticome terapia di prima linea dell’aspergillosi invasiva(5). Itraconazolo, voriconazolo per via orale eamfotericina B per aerosol sono raccomandati peril mantenimento delle concentrazioni terapeutiche.
Figura 1 Patogenesi dell’infezione da Aspergillo. Modificata da [1].
Spore AF
Micelle
CD4 TCR
HLA-DR2-/DR5
CD28
CD86
CD40L
CD40
IL-10
CCR3
IL-4
IL-13
CD23CD21
β cellula a riposo
IL-3
IL-5
CD23
CCR3
IL-4
IL-4
IL-4IL-5
CD23
NK cellule sCD23DR2/DR5
Immunoblasta IgE
IgC IgAAf
IgE
IgE
MBPVLA-4
eosinofiloipodenso
Af
Mastcellule
IL-5
CD23 VLA-4
Eotassina
Istamina
VCAM-1
CCR3
IFN-γ
Th0
APC
Th1
Th2
Asp f2
f4
f6
Tessuto linfoidebroncheoalveolare
Midolloosseo
Eosinofiloipodenso
Infiammazione bronchiale

Raia, et al.52
La notevole variabilità di risposta individuale, l’inte-razione con altri farmaci quali corticosteroidi edantibiotici, la necessità di monitoraggio farmacolo-gico e la scarsa disponibilità di studi clinici rando-mizzati controllati in età pediatrica non consento-no al momento attuale di definire protocolli ditrattamento standard.Promettenti risultati del voriconazolo per via ina-latoria sono stati dimostrati in modelli animali perla profilassi dell’aspergillosi polmonare invasiva (6).
Aspergillosi e fibrosi cistica
L’isolamento di specie di aspergillus (prevalente-mente A. fumigatus) nelle secrezioni bronchiali dipazienti affetti da fibrosi cistica (FC) è un eventoabbastanza frequente. Le spore di A. fumigatus sonoubiquitarie e mostrano una particolare predisposi-zione a colonizzare le vie aeree dei soggetti con FC.Ciò è dovuto prevalentemente sia alle dimensionidelle spore (diametro di 2-7 micron) che favorisco-no l’inalazione e la penetrazione nelle vie aeree, chea fattori locali a livello del polmone, principalmenterappresentati da un’alterazione della struttura del-l’albero bronchiale (cisti, cavità e presenza di bron-chiectasie), particolarmente compromesso in FC.L’inalazione di spore di aspergillus è quindi un’eve-nienza frequente, ma lo sviluppo di malattia a causadell’aspergillo è raro. Nel 10% circa dei soggetti conFC si verifica una particolare reazione allergica ad A.fumigatus (nota con l’acronimo ABPA, cioè aspergil-losi broncopolmonare allergica), il cui quadro clinicoè caratterizzato da episodi di bronco-ostruzionecon infiltrati polmonari radiograficamente documen-tabili, cutipositività ad allergeni del fungo ed elevati
livelli sierici di IgE totali e specifiche. Spesso il quadroclinico dell’ABPA si sovrappone a quello della malat-tia di base, pertanto la diagnosi di questa complican-za può non essere semplice. Raramente le ife delfungo tendono a riempire le cavità polmonari, for-mando una lesione nota come aspergilloma.
Epidemiologia
Circa il 50% dei pazienti FC hanno le vie aereecolonizzate da Aspergillus fumigatus (1). Questofungo (inalato come spore) per lo più rimane alungo come saprofita, ma in un certo numero dipazienti può indurre una risposta immunitaria IgE-mediata, ma anche di tipo IgG, e da attivazione dirisposte specifiche da parte dei globuli bianchi dellaserie linfocitica. Questa complessa risposta immu-nitaria può indurre danno a livello broncopolmo-nare, con comparsa di infiltrati polmonari, bronchi-te e bronchiectasie. Tale complicanza viene definita“Aspergillosi broncopolmonare allergica” o ABPA.L’APBA interessa dall’1% al 15% di tutti i pazientiFC (1).In uno studio epidemiologico europeo condottosu oltre 12.000 pazienti con FC (7) la frequenza diABPA è risultata in media di circa l’8%: assai raranei pazienti fino ai 6 anni di età, essa si assestaintorno al 10% nelle età successive.Alcuni fattori predispongono a questa complicanza,quale infezione cronica da Pseudomonas aeruginosa,una funzionalità polmonare ed uno stato nutrizio-nale a lungo compromessi (7, 8) e, forse, qualchetratto genetico predisponente, oggi allo studio.Diversi sono i quadri clinici di Aspergillosi che pos-sono associarsi alla fibrosi cistica (Tabella 1).
Tabella 1 Classificazione dei quadri clinici di Aspergillosi in Fibrosi Cistica.
• ABPA: presente nell’ l'8-10% dei pazienti con Fibrosi Cistica. Complicanza allergica.
• Aspergillosi allergica dei seni paranasali
• Aspergillosi invasiva: complicanza severa da Aspergillus fumigatus, con disseminazione in vari organi e tessuti.Rara in FC, prevalentemente in soggetti sottoposti a trapianto di organi.
• Bronchite da Aspergillo: presenza costante di Aspergillus nelle colture di escreato, associata a manifestazionipolmonari non rispondenti al trattamento antibiotico mirato ai batteri isolati, in assenza di altri segni di ABPA,responsivo al trattamento con farmaci antifungini, con esito assai favorevole (4).
• Aspergillosi saprofitica: colonizzazione delle vie aeree, senza coinvolgimento polmonare;
• Aspergilloma: massa fungina rotondeggiante che contiene ife del fungo, fibrina, cellule infiammatorie e cellulebronchiali di desquamazione. Processo benigno che, in rari casi, può essere asportato chirurgicamente con successo

Aspergillosi broncopolmonare 53
L’ABPA è una complicanza che può far deteriora-re il decorso della malattia polmonare in FC, ma,se trattata precocemente e adeguatamente, speciein giovane età, può risolversi senza sostanziali con-seguenze. Nello studio europeo (7), nel periododi studio di 4 anni, non è stato registrato un decli-no di funzione respiratoria nei pazienti con ABPAdiverso rispetto agli altri pazienti.La diagnosi di ABPA si basa:- sul rilievo di un deterioramento respiratorio,talora con sintomi di tipo asmatico;- comparsa di nuovi infiltrati polmonari che nonrispondono al trattamento antibatterico, even-tualmente bronchiectasie e impatto di muco neibronchi; - elevati livelli di IgE totali nel siero (oltre 1.000unità);
- immunoglobuline IgE e IgG specifiche anti-Aspergillus fumigatus (1);- positività ai test cutanei per antigeni di A. fumiga-tus (circa il 40%-60% di tutti i pazienti con FC pos-sono presentare positività ai test cutanei - iper-sensibilità, che non consente da sola di far diagno-si di ABPA e non obbligatoriamente prelude alquadro di aspergillosi broncopolmonare allergica.È consigliato uno screening annuale dell’ABPA neipazienti di età >6 aa: - in presenza di segni clinici di sospetto è utiletestare le IgE totali;- se i livelli sierici di IgE sono >500 κU/l è utileeffettuare prick/RAST per aspergillo;- se i livelli sierici di IgE sono <500 κU/l, in pre-senza di persistenti sintomi di sospetto di ABPA ènecessario monitorare frequentemente le IgE.
Tabella 2 Terapia dell’Aspergillosi in Fibrosi Cistica.
Raccomandazioni nel trattamento dell’Aspergillosi in Fibrosi Cistica
Patologia Terapia
Primaria Seconda Scelta Commenti
Aspergillosi Cortisone per via orale Itraconazolo, Il dosaggio e la durata dibroncopolmonare a dosaggio sostenuto e voriconazolo trattamento non sono mai
allergica per periodi protratti (mesi). stati standardizzati emancano studi clinici
prospettici e controllati.Il cortisone non previene
eventuali recidive e presentagli effetti collaterali tipicidei cortisonici. Non c’èevidenza di efficacia diterapia con cortisonici
somministrati per aerosol.Per quanto riguarda gli
antifungini mancano studiclinici prospettici randomiz-
zati controllati (6)
Aspergillosi Nessuna o Pochi dati sugliallergica itraconazolo altri antimicotici
dei seni paranasali
Aspergilloma Terapia chirurgica Itraconazolo oper casi selezionati Voriconazolo
Aspergillosi Voriconazolo Amfotericina B, Non è raccomandatainvasiva per via parenterale caspofungina. una terapia primaria di
Itraconazolo o associazione.voriconazolo per os come In età pediatrica
farmaci di mantenimento e mancano dati clinicicome terapia di prevenzione
nei soggetti a rischio (7)

Raia, et al.54
Trattamento farmacologicodell’Aspergillosi in FC
- Il trattamento (Tabella 2) si basa essenzialmentesulla somministrazione protratta di cortisonici (perparecchie settimane ma talora anche per mesi);- La durata del trattamento dipende dalla rispostaclinica e dal comportamento dei livelli di immuno-globuline E (devono abbassarsi sotto una certa
soglia), il cui dosaggio periodico nel sangue servea monitorare l’efficacia del trattamento. - Oggi viene quasi sempre associato anche un trat-tamento con antibiotici antifungini (itraconazolo inparticolare, ma anche voriconazolo, quest’ultimomeno sperimentato), anche se questo interventosuscita ancora qualche controversia (10).
1. Stevens DA, Moss RB, Kurup VP, et al. AllergicBronchopulmonary Aspergillosis in Cystic Fibrosis-State of the Art: Cystic Fibrosis Foundation ConsensusConference. Clin Infect Dis 2003; 37 (Suppl 3):S225-S264.
2.De Pauw B, Walsh TJ, Donnelly JP, et al. Revised def-initions of invasive fungal disease EORT/MSG ConsensusGroup. Clin Infect Dis 2008; 46: 1813-1821.
3.Walsh TJ, Lutsar I, Driscoll, T, et al. Voriconazole inthe treatment of aspergillus, scedosporios and otherinvasive fungal infections in children. Pediatr InfectDis 2002; 21: 240-248.
4. Scott LJ, Simpson D. Voriconazole: a review of itsuse in the management of invasive fungal infections.Drugs 2007; 67: 269-298.
5. Walsh TJ, Anaissie EJ, Denning DW, et al.Treatment of aspergillosis: clinical practice guidelinesof the infectious diseases society of America. ClinInfect Dis 2008; 46: 327-360.
6. Tolman JA, Wiederhold NP, McConville JT, et al.Inhaled voriconazole for prevention of invasive pul-monary aspergillosis. Antimicrob AgentsChemother 2009; 53: 2613-2615.
7. Mastella G, Rainisio M, Harms HK, et al. Allergicbronchopulmonary aspergillosis in cystic fibrosis. AEuropean epidemiological study. EpidemiologicRegistry of Cystic Fibrosis. Eur Respir J 2000; 16:464-471.
8. Ritz N, Ammann RA, Casaulta Aebischer C, etal. Risk factors for allergic bronchopulmonaryaspergillosis and sensitisation to Aspergillus fumigatusin patients with cystic fibrosis. Eur J Pediatr 2005;164: 577-582.
9. Shoseyov D, Brownlee KG, Conway SP, KeremE. Aspergillus Bronchitis in Cystic Fibrosis. Chest2006; 130: 222-226.
10. Hilliard T, Edwards S, Buchdahl R, Francis J, et al.Voriconazole therapy in children with cystic fibrosis. JCyst Fibrosis 2005; 4: 215-220.
11. Schoeni MH. To treat or not to treat with anti-fungal agents? J Cystic Fibrosis 2005; 4: 213-214.
12. Mantadakis E, Samonis G. Novel preventativestrategies against invasive aspergillosis. Med Mycol2006; 44 (Suppl): 327-327.
Bibliografia

L’asma allergico è caratterizzato da infiammazionecronica delle vie aeree, dominata da T linfocitiCD4+ specifici che producono citochine di tipo Th2(IL-4, IL-5, IL-9, IL-13, GM-CSF) (1). Queste citochi-ne orchestrano la risposta infiammatoria polmona-re, aumentando il reclutamento locale e la sopravvi-venza di diverse cellule infiammatorie come mast-cellule, eosinofili e neutrofili (Figura 1). La morte cel-lulare è necessaria per mantenere l’omeostasiimmunologica, e la mancata eliminazione delle cellu-le attivate prolunga la risposta immune e promuovel’infiammazione cronica delle vie aeree (2).Un’alterazione dell’apoptosi (una sequenza con-trollata di eventi che porta alla morte cellularesenza il rilascio di sostanze dannose nell’ambientecircostante) è stata implicata in diverse malattiecome tumori, malattie neurodegenerative, malattieautoimmuni e asma allergico (3).In questo articolo sono discussi i meccanismi del-l’apoptosi che probabilmente giocano un ruolo
nella patogenesi dell’asma ed analizzate le attivitàproapoptotiche dei farmaci antiasmatici tradizio-nali e delle nuove terapie “biologiche”.
Omeostasi del sistema immunitario emeccanismi dell’apoptosi
Nonostante la costante esposizione ad antigeniesterni e ad autoantigeni, il sistema immunitario èripristinato al livello di pre-immunizzazione, man-tenendo in tal modo uno stato di equilibrio(omeostasi) tale da garantire una risposta efficacein seguito al contatto con nuovi stimoli antigenici.La risposta immune termina con l’apoptosi dellepopolazioni linfoidi espanse e non più necessarie,come le cellule T e B e le cellule che presentanol’antigene (4) (Figura 2).L’apoptosi interviene quando una cellula è dan-neggiata oltre la possibilità di riparazione; da unpunto di vista filogenetico ha il fine di prevenire
55Pneumologia Pediatrica 2009; 35: 55-66
Fernando Maria de Benedictis1, Diletta de Benedictis2, Carmelo Gabriele1
1Dipartimento di Pediatria, A.O.U. “Ospedali Riuniti”, Ancona; 2Dipartimento di Pediatria, Università diPerugia
Asma e apoptosiAsthma and apoptosis
Parole chiave: asma, apoptosi, infiammazione
Keywords: asthma, apoptosis, inflammation
Riassunto. I linfociti T giocano un ruolo cruciale nel riconoscimento di antigeni esterni e nell’orchestrare la risposta immune.L’apoptosi dei linfociti T attivati è essenziale per il mantenimento dell’omeostasi del sistema immune, in quanto la mancata eli-minazione delle cellule attivate prolunga la risposta immune e promuove l’infiammazione cronica. Risultati sperimentali sugge-riscono che i meccanismi di controllo dell’apoptosi possono essere alterati nell’asma. L’aumento del reclutamento e dellasopravvivenza cellulare e la comunicazione tra linfociti T ed il microambiente circostante giocano un ruolo fondamentale nellosviluppo dell’infiammazione bronchiale, del danno della mucosa e del rimodellamento tissutale nell’asma. La ricerca su model-li animali ed umani ha concentrato l’attenzione prevalentemente sulle citochine derivate dalle cellule T come causa dominan-te dell’infiammazione bronchiale. I nuovi agenti “biologici” antiasma che bloccano specifici meccanismi patogenetici non hannodimostrato particolare efficacia, nonostante sopprimano l’infiltrazione eosinofilica nelle vie aeree. Farmaci antiasmatici tradizio-nali come i corticosteroidi o gli antagonisti recettoriali dei leucotrieni, che possono indurre l’apoptosi dei linfociti T, sembranoessere associati a migliori risultati clinici. Studi futuri dovranno chiarire se è possibile modulare l’apoptosi combinando nuovi evecchi farmaci antinfiammatori.
Accettato per la pubblicazione il 31 agosto 2009.
Corrispondenza: Prof. Fernando Maria de Benedictis, Dipartimento di Pediatria, Ospedale “G. Salesi”, Via Corridoni 11,60123 Ancona; e-mail: [email protected]

de Benedictis, et al.56
l’utilizzazione di nutrienti dell’organismo o di evita-re, nel caso si tratti di una cellula infettata, l’ulte-riore diffusione dell’infezione. A differenza dellanecrosi, l’apoptosi comporta una progressiva alte-razione strutturale della cellula con protrusionisulla membrana, contrazione della cellula, conden-sazione della cromatina nucleare e frammentazio-ne del DNA. Come atto finale, i frammenti cellu-lari sono inglobati dai fagociti senza che venganorilasciati all’esterno enzimi intracellulari potenzial-mente pericolosi, e dunque senza indurre unarisposta infiammatoria.Il processo dell’apoptosi è controllato da diversi tipidi segnale, di origine extracellulare e intracellulare. I
segnali extracellulari (tossine, ormoni, fattori dicrescita, ossido nitrico, citochine) per esercitare illoro effetto devono attraversare la membrana cel-lulare oppure agire tramite trasduzione. Il segnaleintracellulare può indurre il suicidio cellulare inrisposta a stress (radiazioni, infezioni virali, ipossia)(5) (Figura 3).Sono stati identificati due principali meccanismi diregolazione dell’apoptosi: il sistema mitocondrialee la trasduzione diretta del segnale (Figura 4).- Regolazione mitocondriale: Le proteine mitocon-driali conosciute come SMACs sono rilasciate nelcitosol in seguito a un aumento della permeabili-tà. Le SMAC si legano alle IAPs e le inattivano,
Figura 1 Meccanismi patogenetici dell’asma. Gli aspetti moderni della patogenesi dell’asma includono le interazionitra gli allergeni, le cellule immuni mucosali e le terminazioni vascolari/nervose. Queste ultime possono influenzare lareattività bronchiale e le fasi precoci dell’infiammazione (edema, perdita di plasma e vasodilatazione), mentre le inte-razioni cellulari possono influenzare l’infiltrato infiammatorio, il rimodellamento e la fibrosi (ristampato con il permes-so di Nature Publishing Group).
Allergene
Eosinofilo
Tappo mucoso
Ipersecrezionedi mucoIperplasia Vasodilatazione
Nuovi vasi(angiogenesi)
Versamento plasmaticoEdema
Neutrofilo
Sfaldamento epiteliale
Fibroblasto
Epitelio dellevie aeree
Fibrosisubepiteliale
Attivazione del nervo sensorio
Riflessocolinergico
Broncocostrizione Ipertrofia/iperplasia
Strato sottocutaneo
Muscolaturaliscia dellevie aeree
Attivazionedel nervo
Mast-cellula
TH2
Macrofago / cellula dendritica

Asma e apoptosi 57
Figura 2 Omeostasi del sistema immunitario nelle malattie allergiche. Stadi dello sviluppo e dell’omeostasi della rispo-sta immune. I segnali che inducono la proliferazione e la differenziazione linfocitaria sono indicati in blu e quelli cheterminano la risposta immune e mantengono l’omeostasi sono mostrati in nero. Modificata da [4].
Receptorengagement
?
Receptorengagement
Feedbackmechanism
Memory cell
Activatedcell
Differentiation
Loss of survival signals
Apoptosis
Inibition ofimmune response
Antigen and second signals
Loss of receptorengagement
Apoptosis
Naivecell
Effectorcell
Maintenanceof memory cells
Maintenanceof naive cells
Immune response
Maintenanceof survival signals
Migrationfrom
generative lymphoid
organs
Before antigen explosure After antigen explosure
permettendo in tal modo l’attivazione delle caspa-si, una famiglia di cistein proteasi, e la progressionedell’apoptosi.- Trasduzione diretta del segnale: Il recettore Fas(conosciuto anche come Apo-1 o CD95) si legaal FasL, una proteina transmembrana apparte-nente alla famiglia del Tumor Necrosis Factor(TNF). L’interazione tra Fas e FasL induce la for-mazione di molecole come FADD (Fas-associa-ting protein with death domain) e del segnale dimorte DISC (death-inducing signaling complex), ingrado sia di favorire il rilascio di fattori proapop-totici dai mitocondri sia di attivare la cascatadelle caspasi.Nel primo caso la caspasi-8 attiva la proteina cheinteragisce con il BCL-2 interacting domain (BID)inducendo il rilascio del citocromo C dai mitocon-dri attraverso il canale di membrana MAC. Unavolta rilasciato, il citocromo C si lega al apoplecticprotease activating factor-1 (APAF-1) insieme
all’ATP formando un complesso proteico deno-minato apoptosoma. L’apoptosoma attiva lacaspasi-9 innescando un meccanismo di attiva-zione a cascata delle altre caspasi. La famigliadelle proteine Bcl-2 prende parte alla regolazio-ne del processo mitocondriale tramite un bilan-ciamento tra fattori proapoptotici (Bax, Bak, Bad,Bid, Bim) e antiapoptotici (Bcl-xL, Bcl-2 Bcl-xS,Bcl-w). La proporzione di Bcl-2 e Bax presentinel mitocondrio regola il processo apoptotico: lapredominanza dell’espressione di Bcl-2 bloccal’apoptosi, mentre la predominanza dell’espres-sione di Bax promuove l’implementazione deisegnali di morte.Nel secondo caso, la caspasi-8 attiva direttamentela caspasi-3 che a sua volta attiva il DNAFragmentation Factor (DFF) con conseguente fram-mentazione del DNA e condensazione della cro-matina nucleare, elemento caratteristico dellaapoptosi.

de Benedictis, et al.58
Alterata apoptosi, allergia ed infiammazione bronchiale
Esistono diverse dimostrazioni a sostegno di un’al-terazione del processo di morte cellulare pro-grammata nell’asma. L’incapacità di eliminare le cel-lule attivate prolunga la risposta immune e puòessere responsabile dell’infiammazione cronicadelle vie aeree.Gli eosinofili rappresentano le cellule preminentinell’infiammazione allergica e diversi studi suggeri-scono che l’alterata apoptosi degli eosinofili puòavere un ruolo nell’asma. Nonostante l’apoptosidegli eosinofili non sia facile da dimostrare nei pol-moni, l’ipotesi che queste cellule siano eliminatetramite apoptosi seguita da fagocitosi macrofagicanon può essere esclusa, in quanto le cellule apop-totiche potrebbero essere rimosse così rapida-mente da non essere più riscontrabili nei tessuti(6). Dopo challenge allergenico su cavie, gli eosi-nofili possono essere eliminati dalle vie aeree tra-mite meccanismi alternativi, come la progressionenel lume e la successiva clearance mucociliare (7).È stato dimostrato che il numero di eosinofili non-apoptotici in biopsie bronchiali è significativamente
superiore negli asmatici rispetto ai controlli e chegli eosinofili degli asmatici esprimono una maggio-re quantità di Bcl-2 (3). Inoltre, studi su biopsiebronchiali (3) e sullo sputo indotto (8) evidenzia-no una stretta correlazione tra gravità dell’asma enumero di eosinofili apoptotici. Alla luce di questistudi si può ipotizzare che l’accumulo di eosinofilinelle vie aeree degli asmatici sia dovuto all’aumen-tato reclutamento locale di queste cellule e all’ele-vata inibizione della morte cellulare programmata.È verosimile che la ridotta apoptosi degli eosinofi-li nell’asma sia indotta dalle citochine proinfiam-matorie espresse in eccesso. Tali citochine infattisono in grado di indurre sia una ridotta espressio-ne dei recettori di morte (FAS, TNF-r, TRAIL: TNF-Related Apoptosis-Inducing Ligand) sia un’alterazio-ne dei meccanismi mitocondriali dell’apoptosi,come conseguenza di una up-regulation di geniche codificano per proteine ad azione antiapop-totica e di una down-regulation di geni che codifi-cano per proteine ad azione proapoptotica (9).L’espressione del Fas mRNA è infatti ridotta inmaniera dose-dipendente nelle cellule T trattatecon IL-4 (10) e negli eosinofili trattati con IL-5 (11).
Figura 3 Eventi metabolici nell’apoptosi. Diversi eventi (fattori di crescita, citochine) agiscono su specifici recettori.Farmaci, radiazioni e/o agenti tossici possono agire direttamente sulla trascrizione di particolari geni. Modificata da [5].
Withdrawal oftrophic factors
GF receptors
Gene transcription Activation of Ca++ dependentenzymes
2nd messengersCycl-cCeramide
Caspase activation
RadiationsAnti-cancer drugsDioxin
New cellsurface marker Cell phagocytosis without
inflammation
GC hormonesThyroid hormonesRetinoids
TNF-αTGF-βHIV gp 120CD95 Ligand
Endonuclease activationCalpain: distruption ofcytoskeletonTGT: cross-linking ofcytosolic proteins

Asma e apoptosi 59
Inoltre, topi transgenici che esprimono IL-4, IL-5,IL-9, IL-11 ed IL-13 nei polmoni accumulano cellu-le infiammatorie nel BAL (bronchoalveolar lavage) emostrano iperreattività bronchiale e deposizionedi collagene nelle vie aeree, indicando che l’espo-sizione cronica a citochine di tipo Th2 induce lecaratteristiche tipiche dell’asma allergico (12).L’allontanamento delle citochine al termine dellarisposta proliferativa è il meccanismo sottostantel’apoptosi delle cellule T e che determina la down-
regulation della risposta immune (13). Il numerodei linfociti nei polmoni dipende dall’equilibrio trala loro entrata, l’uscita, la proliferazione e la morte.Un meccanismo di regolazione centrale potrebberisiedere nelle cellule dendritiche intraepiteliali enei macrofagi alveolari commissionati alla fagocito-si dei corpi apoptotici.Si ritiene che i linfociti T giochino un ruolo centralenel controllo dell’infiammazione allergica dell’asma.L’aumentato numero di cellule T nella mucosa degli
Figura 4 Meccanismi di regolazione dell’apoptosi. Sistema mitocondriale e trasduzione diretta del segnale.
Fas ligand
Cell membrane
Apaf-1Fas receptor
Bax, Bad, Bim
Bid
Caspase-8
Caspase-3
DFF
DFF45
Nucleus
Oligomerof DFF40
DNAfragmentation
Caspase-9
Procaspase-3
Procaspase-9
Bid C terminal
Procaspase 8 Bcl-2, Bcl-XI
Citochromo c
Mitochondrion
dATP/ATP
Apoptosome
Apoptotic stimuli
FAD

de Benedictis, et al.60
asmatici è il risultato di un maggiore reclutamentodi queste cellule nelle vie aeree, ma può anchedipendere da un’aumentata sopravvivenza. Nellebiopsie bronchiali di soggetti asmatici la maggiorparte dei linfociti T appaiono infatti in fase non-apoptotica ed esprimono elevate quantità di Bcl-2(3). Le cellule T degli asmatici non raggiungono ilnormale grado di apoptosi che segue il legamecon il recettore Fas, inducendo ad ipotizzare un’i-nefficiente attivazione della trasduzione del segna-le. Inoltre, i linfociti prelevati dal BAL di asmaticisono più resistenti all’apoptosi dopo inalazione diallergeni (14).Nelle vie aeree degli asmatici il microambiente checirconda le cellule T è alterato. Questa caratteristi-ca potrebbe modificare il comportamento dellecellule T e fornire alle cellule un complesso di cito-chine e fattori di crescita tale da innescare unacascata di eventi capaci di promuovere la soprav-vivenza cellulare (15). La comunicazione (“cross-talk”) tra i linfociti T ed il microambiente tessutalepuò essere uno dei meccanismi coinvolti nella per-sistenza e nella sopravvivenza delle cellule T attiva-te. Recenti studi sperimentali evidenziano che lacooperazione tra cellule strutturali aiuta a creareuno specifico microambiente che sostiene l’attiva-zione e la sopravvivenza delle cellule immunitarienei tessuti infiammati (16). D’altro canto, altre citochine come Interferon γ(IFN-γ), IL-2 o TGF-β, sostengono il meccanismoapoptotico e mantengono l’omeostasi immunologi-ca del polmone tramite una up-regulation del FasmRNA e del recettore sulla superficie dei linfociti Te di altre cellule infiammatorie (17, 18). Tuttavia ilTGF-β1, prodotto abbondantemente da cellulestrutturali ed infiammatorie degli asmatici, può inibi-re l’espressione del FasL nelle cellule T e la conse-guente morte cellulare indotta dall’attivazione (19).Una forte evidenza scientifica indica che lo sbilan-ciamento a favore della risposta Th2 presente nel-l’asma è dovuto soprattutto ad una aumentataapoptosi delle cellule Th1 (20). In particolare, il difet-to della produzione di IFN-γ caratteristico dellarisposta immuno-allergica potrebbe essere respon-sabile della ridotta apoptosi dei linfociti T attivati dal-l’allergene, tramite la modulazione del Fas e FasLsulla superficie delle cellule T attivate (21).La ridotta apoptosi delle cellule T di memoria èassociata ad un’aumentata espressione di Bcl-2 epuò essere un meccanismo importante per l’accu-mulo e/o la persistenza di queste cellule nelle vie
aeree degli asmatici (22). La soppressione locale ela rimozione dei linfociti T di memoria potrebberidurre il rilascio di citochine proinfiammatorie con-tribuendo all’iperreattività bronchiale, infiammazio-ne e rimodellamento dell’asma. È stato suggeritoche l’identificazione dei meccanismi che modulanole popolazioni cellulari T di memoria potrebbe esse-re utilizzata come strategia terapeutica per ridurrela risposta infiammatoria nell’asma allergico.Il ruolo dei neutrofili nell’asma rimane poco chia-ro. Tuttavia l’infiammazione neutrofilica delle vieaeree, verosimilmente mediata da IL-8 e GM-CSFderivati dall’epitelio, è fortemente associata all’a-sma grave steroide-resistente e all’asma fatale adesordio improvviso (23). È verosimile che sia l’IL-8sia il GM-CSF contribuiscano ad un aumentatoreclutamento di neutrofili dal torrente circolatorioverso la mucosa bronchiale, ma queste citochinepotrebbero anche giocare un ruolo nell’alterataregolazione dell’apoptosi dei neutrofili in soggetticon asma grave (3).Ci sono alcune evidenze sul ruolo del NO nellaregolazione dell’apoptosi dei neutrofili. Negliasmatici le citochine proinfiammatorie aumentanol’espressione della forma inducibile della NO-sin-tasi nelle cellule epiteliali delle vie aeree, giustifi-cando gli elevati livelli di NO nell’esalato. La coltu-ra di neutrofili umani con donatori di NO causauna drammatica induzione dell’apoptosi, concen-trazione-dipendente (24).Infine, è stato dimostrato che la frammentazione ela desquamazione dell’epitelio bronchiale, quadroistologico tipico dell’asma, possono essere dovutea un’aumentata apoptosi di cellule epiteliali media-te dal TGF-β espresso in elevata quantità negliasmatici (25).
Alterata apoptosi, rimodellamento e fibrosi
La persistenza di T linfociti e di granulociti, in par-ticolar modo gli eosinofili, nei siti dell’infiammazio-ne allergica è accompagnata dal rimodellamentotissutale. Il rimodellamento delle vie aeree è carat-terizzato da un’aumentata deposizione di proteinedella matrice extracellulare nella membrana reti-colare basale e nella mucosa bronchiale, da unaumento della muscolatura liscia, da iperplasiadelle cellule caliciformi e dalla neoformazione divasi ematici (Figura 5). Gli eosinofili sono una fontedi diverse molecole implicate in questo processodi rimodellamento, incluse TGF-α, TGF-β1, FGF-2,VEGF, MMP-9 (matrix metallo proteinase), TIMP-1

Asma e apoptosi 61
(tissue inhibitor of metalloproteinase), IL-13 e IL-17(26). Gli eosinofili ed altre cellule infiammatoriecome i macrofagi, le cellule T, le mastcellule ed ifibroblasti secernono TGF-β1, il quale aumenta lapresenza di procollageno 3, proteoglicani, tenasci-na e lumicano nella membrana basale e favoriscel’infiltrazione da parte di precursori di miofibrobla-sti derivati dai fibrociti circolanti (27). Anche i lin-fociti CD4+ attivati possono contribuire al rimo-dellamento della muscolatura liscia delle vie aeree.Nell’asma sperimentale il contatto diretto tra lecellule T CD4+ e i miociti aumenta la proliferazio-ne ed inibisce l’apoptosi dei miociti delle vie aeree,suggerendo che l’interazione tra cellule partecipaalla regolazione del turnover dei miociti ed all’indu-zione del rimodellamento (28).Le cellule epiteliali bronchiali possono giocare unruolo attivo nello sviluppo dell’infiammazione dellevie aeree e del rimodellamento dell’asma. Le cel-lule epiteliali esprimono diversi marker di mem-brana, incluse le molecole di adesione, e rilasciano
un ampio spettro di molecole che partecipano allariparazione nelle vie aeree, incluse la fibronectina, ifattori di crescita, le citochine, le chemochine el’eotassina (27). Nell’asma il rimodellamento dellevie aeree potrebbe essere la conseguenza dieccessivi tentativi di riparazione che fanno seguitoal danno infiammatorio cronico. Tuttavia l’apoptosidelle cellule epiteliali può giocare un ruolo addi-zionale (3, 25, 29).Il rimodellamento delle vie aeree è stato osserva-to in diverse sindromi fibrotiche, quali le malattiepolmonari interstiziali, dove le citochine Th2 pos-sono giocare un ruolo proinfiammatorio (30).Studi in modelli umani ed animali di polmoniteinterstiziale mostrano che il danno alveolare sipresenta per primo ed è seguito da infiltrazionedei linfociti B e T, di leucociti polimorfonucleati, pla-smacellule e macrofagi, tale da portare all’organiz-zazione finale dell’essudato alveolare. In pazienticon sclerosi sistemica, l’infiltrato linfocitario segue ildanno epiteliale ed è probabilmente sostenuto
TNF-αTGF-βPDGF
VEGFFGF2
TIMPsMMP9
Dysregulatedcommunication
Alveolar epithelial cellsIncreased apoptosisDecreased proliferationDecreased migration
Fibroblasts/MyofibroblastsDecreased apoptosisIncreased ECM productionIncreased migrationIncreased proliferation
Th2 Citokines(IL-4, IL-13)
Fibrin
Figura 5 Fibrosi delle vie aeree e rimodellamento. Il rimodellamento delle vie aeree è favorito dall’alterata regolazio-ne della comunicazione tra cellule epiteliali e mesenchimali. Diverse citochine sono implicate nel favorire la morte dellecellule epiteliali (TNF-alfa, TGF-beta, PDGF) e nell’aumentare la sopravvivenza dei fibroblasti o dei miofibroblasti (IL-4, IL-13, FGF-2, VEGF), mentre l’aumentata deposizione delle proteine della matrice extracellulare, l’ipertrofia dellamuscolatura liscia, l’iperplasia delle cellule ghiandolari caliciformi e la neoformazione di vasi ematici contribuiscono alriarrangiamento tessutale.
Apoptotic signal

de Benedictis, et al.62
dalla produzione di citochine Th2 attraverso laproduzione oligoclonale delle cellule CD8+ (31).La bleomicina, le tossine che danneggiano il DNA,i virus e le reazioni immuni sono agenti scatenantidell’apoptosi di cellule epiteliali, sia direttamentesia tramite citochine quali il TNF-α ed il TGF-β.Infatti, in topi lpr (Fas-/-) e gld (Fas-L-/-) l’iniezionedi bleomicina attenua l’apoptosi delle cellule epite-liali e protegge dalla fibrosi (30). Risultati similisono stati osservati in topi normali quando unantagonista solubile del recettore Fas o un anti-corpo anti-FasL era somministrato per via endo-venosa o tramite inalazione (30). Questi dati chia-riscono i meccanismi tramite i quali le citochineTh2 possono interferire con la risoluzione del pro-cesso infiammatorio. Il loro meccanismo di azioneantiapoptotico aumenta la sopravvivenza di linfoci-ti, granulociti, plasmacellule e macrofagi, ma l’attivi-tà profibrotica dovuta alla prolungata sopravviven-za dei fibroblasti dovrebbe anche essere conside-rata come un fattore importante dell’infiammazio-ne e del rimodellamento delle vie aeree.
Orchestrare l’apoptosi per controllarel’infiammazione delle vie aeree
La terapia bersaglio (target therapy) è comune-mente definita come un trattamento farmacologi-co che agisce specificamente su un meccanismobiologico ben definito che, quando inattivato,porta alla regressione del processo patologico. Glianticorpi monoclonali diretti contro molecole disuperficie o recettori delle citochine rappresenta-no alcuni dei nuovi approcci “biologici” nel tratta-mento del cancro e delle malattie autoimmuni.Negli anni passati la complessa cornice delle inte-razioni biochimiche e cellulari che caratterizzanol’asma ha favorito lo sviluppo di strategie terapeu-tiche rivolte verso specifici aspetti dell’infiamma-zione allergica (ad es., IgE elevate, IL-4, IL-5). Taliinterventi immunologici sono stati basati principal-mente su studi animali (ad es. modelli di topiknock-out e sul ben noto paradigma delle citochi-ne Th2 nell’asma allergico) (32). Tuttavia, la maggiorparte di questi trattamenti non sono stati associa-ti con importanti risultati nei trial clinici.
Terapie biologiche
Le IgE giocano un ruolo centrale nelle malattie aller-giche. In individui sensibilizzati, le IgE si legano a recet-tori presenti sulla superficie delle mastcellule e dei
basofili e rilasciano mediatori preformati e prodottide novo, che iniziano una cascata immunologicaresponsabile dell’infiammazione. Nonostante itopi knock-out per IgE mostrino infiltrati eosinofi-lici dopo stimolazione allergenica, un anticorpomonoclonale umanizzato anti-IgE (recombinanthumanized monoclonal antibody rhu mAb-E25,omalizumab) è stato somministrato in trial con-trollati per il trattamento delle malattie allergiche.In pazienti con asma allergico moderato-grave, ilfarmaco ha diminuito i livelli di IgE circolanti, mala riduzione della dose di corticosteroidi inalato-ri è stata modesta e il controllo dell’asma non èdurato nel tempo (33). In pazienti con asma scar-samente controllato nonostante alte dosi di cor-ticosteroidi inalatori, l’omalizumab ha ridotto ladose di corticosteroidi ed ha migliorato la quali-tà della vita (34). Il meccanismo d’azione deglianticorpi anti-IgE non è ancora chiaro, ma è statoipotizzato che possa interferire con i linfociti B dimemoria che esprimono recettori transmembra-na per le IgE (mIgE). L’apoptosi di linfociti BmIgE+ inibisce la loro differenziazione in plasma-cellule IgE secernenti riducendo la produzione diIgE totali (35).È stato ipotizzato che gli anticorpi monoclonaliumanizzati contro IL-5 antagonizzano il pattern diproduzione di citochine di tipo Th2 nei polmoni,particolarmente l’infiltrazione eosinofilica dipen-dente da IL-5. Nei pazienti con asma moderato,questo tipo di trattamento ha determinato unadeplezione degli eosinofili nel sangue periferico,ma non è stato evidenziato alcun effetto sullarisposta asmatica tardiva o sull’iperreattività bron-chiale (36). Un anticorpo anti-IL-5 (SCH55700) haridotto l’eosinofilia periferica in maniera dose-dipendente nei pazienti con asma grave inducen-do un miglioramento della funzionalità polmonare(37). Infine, in biopsie bronchiali di asmatici tratta-ti con mAb anti-IL5, mepolizumab, è stata riscon-trata una riduzione significativa dell’espressione ditenascina, lumicano e procollageno-3 nella mem-brana reticolare basale della mucosa bronchiale,così come del TGF-α1 nel BAL (38). Poiché IL-12 prodotta dalle cellule dendritiche èrisultata capace di stimolare i linfociti T a secerne-re citochine di tipo Th1 in vitro e di aumentare ilnumero di cellule apoptotiche CD4+ in unmodello murino di asma (39), è stato condotto untrial clinico con IL-12 umana ricombinante sotto-cute in pazienti con asma lieve. Dopo l’inalazione

Asma e apoptosi 63
dell’allergene, è stata osservata una riduzionesignificativa degli eosinofili nel sangue e nell’espet-torato, ma il trattamento non ha mostrato alcuneffetto sull’iperreattività bronchiale all’istamina osulla risposta asmatica tardiva (40).Infine, poiché l’espressione del TNF-α è aumenta-ta nelle vie aeree di pazienti con asma grave refrat-tario alla terapia, è stato somministrato a questotipo di pazienti il recettore solubile ricombinanteper il TNF-α. Sono stati ottenuti significativi miglio-ramenti sui sintomi, sulla qualità di vita, sulla fun-zionalità polmonare e sull’iperreattività bronchiale,ma sono stati riscontrati scarsi effetti sugli indici diinfiammazione delle vie aeree (41, 42).Altre terapie “biologiche” promettenti nell’asmagrave, includono l’uso di IFN-γ, anti-IL-13 ed anti-corpi monoclonali anti-CD25 (daclizumab) (43,44, 45). Il numero sostanziale di pazienti in cui l’a-sma non è adeguatamente controllato con terapiaconvenzionale giustifica gli sforzi per cercare diidentificare nuovi meccanismi molecolari verso cuipossono essere diretti gli agenti biologici (46).Queste terapie costose sono ancora più impor-tanti per i nuovi sub-fenotipi di asma, in modo dapoter identificare coloro che hanno maggiore pro-babilità di rispondere a specifici trattamenti.
Farmaci antiasmatici tradizionali
Uno degli obiettivi principali della terapia antia-smatica è recuperare la sensibilità all’apoptosi daparte delle cellule che infiltrano le vie aeree. Pocoè noto relativamente alla potenziale attività proa-poptotica dei farmaci antiasmatici tradizionali.Dalla prima descrizione di apoptosi indotta dai cor-ticosteroidi nei timociti corticali, numerosi studihanno dimostrato l’effetto dei corticosteroidi sullecellule T periferiche attivate (47). Nonostante i lin-fociti T del sangue periferico siano relativamenteresistenti all’apoptosi indotta dal desametazone, icloni di cellule T attivate o di cellule T allergene-specifiche derivate dalla mucosa mostrano un’au-mentata suscettibilità all’apoptosi indotta da varisteroidi, inclusi desametazone, beclometasone edeflazacort per via orale, e fluticasone per via ina-latoria (48-51). Il salmeterolo da solo non hainfluenzato l’apoptosi delle cellule T periferichenegli asmatici, ma l’associazione con il fluticasone hamostrato un effetto sinergico sull’apoptosi (52).Prove indirette dell’attività degli steroidi nell’asmavengono anche da studi sul BAL o su biopsie bron-chiali di soggetti asmatici. Inoltre, la riduzione dei
linfociti T infiltrati può persistere per settimaneanche dopo la riduzione degli steroidi (49).Solo pochi studi hanno valutato l’effetto deglisteroidi sull’apoptosi degli eosinofili (53). Il miglio-ramento clinico indotto dai corticosteroidi negliasmatici è stato associato ad un elevato numerodi eosinofili apoptotici nel BAL e nelle biopsiebronchiali (54). Inoltre, l’entità dell’apoptosi deglieosinofili in pazienti che assumono corticosteroi-di è maggiore di quello dei pazienti che nonusano steroidi e non differente da quello di sog-getti sani (55).La teofillina, un broncodilatatore a lungo utilizzatoin passato per il trattamento dell’asma, è statarecentemente rivalutata per il suo meccanismo diazione antinfiammatoria (56). In vitro, la teofillinaha indotto l’apoptosi di eosinofili attivati dalla IL-3,probabilmente attraverso una diminuzione dell’e-spressione del Bcl-2 (57). Inoltre, l’incubazione conla teofillina ha ridotto la sopravvivenza degli eosi-nofili stimolata dalla IL-3, come verosimile conse-guenza dell’attivazione della caspasi-3 (58). La teo-fillina riduce la sopravvivenza dei neutrofili tramiteun meccanismo che sembra indipendente dall’ini-bizione delle fosfodiesterasi.Nonostante l’efficacia clinica degli antagonisti recet-toriali dei leucotrieni e degli inibitori della sintesidei leucotrieni sia stata attribuita principalmenteall’inibizione della broncocostrizione e della che-miotassi dei monociti, studi recenti suggerisconoche questi farmaci possono anche accelerare l’a-poptosi degli eosinofili (59). L’inibizione dei leuco-trieni riduce il numero degli eosinofili nel sanguecircolante, nello sputo e nel BAL dopo challengeallergenico (60). In una serie di studi in vitro, il mon-telukast (antagonista dei recettori dei cisteinil-leu-cotrieni, Cys-LT-R) ha indotto l’apoptosi di diversicloni di cellule T (61). In vitro, l’interazione del mon-telukast con i suoi recettori, che sono espressi dallecellule T attivate, inibisce la risposta proliferativa edinduce parziale perdita di vitalità. Colture di celluleT anti-CD3 attivate con differenti quantità di mon-telukast non modificano la produzione di IL-4,mentre la produzione di IFN-γ aumenta in manie-ra lineare con l’incremento delle concentrazioni delmontelukast. L’apoptosi delle cellule T attivateaumenta in maniera significativa in modo dose-dipendente dopo esposizione al montelukast epuò essere dovuta all’inibizione di geni associati allasopravvivenza come Bcl-2 e/o all’attivazione di geniproapoptotici come il p53, Fas e TRAIL.

de Benedictis, et al.64
Conclusioni
L’infiammazione, il danno tissutale e il rimodella-mento sostengono il complesso processo che siverifica nelle vie aeree degli asmatici. La riduzionedell’apoptosi dei linfociti T sembra essere la carat-teristica dell’infiammazione persistente. Ulterioristudi sono necessari per definire come la cono-scenza sperimentale dell’apoptosi delle cellule Tpossa essere tradotta in vivo. Poiché gli obiettivi deltrattamento dell’asma sono l’ottimizzazione delcontrollo della malattia, il prevenire la riduzione
della funzionalità polmonare, la minimizzazionedella necessità di farmaci al bisogno e il raggiungi-mento di una buona qualità della vita (GlobalInitiative for Asthma. Global strategy for asthmamanagement and prevention. Update December2008. Disponibile su: www.ginasthma.com), biso-gnerebbe compiere ogni sforzo per evitare il trat-tamento in difetto della malattia. Per ottenere unbuon controllo dell’asma potrebbe essere neces-saria in futuro la combinazione di opzioni tera-peutiche biologiche e tradizionali (62).
1. Busse WW, Lemanske RF. Asthma. N Engl JMed 2001; 344: 350-362.
2. Cohen JJ. Apoptosis: mechanisms of life anddeath in the immune system. J Allergy ClinImmunol 1999; 103: 548-554.
3. Vignola AM, Chiappara G, Gagliardo R, et al.Apoptosis and airway inflammation in asthma.Apoptosis 2000; 5: 473-485.
4.Van Parijs L, Abbas AK. Homeostasis and self-tol-erance in the immune system: turning lymphocytesoff. Science 1998; 280: 243-248.
5. Spinozzi F, de Benedictis D, de Benedictis FM.Apoptosis, airway inflammation and anti-asthma ther-apy. Pediatr Allergy Immunol 2008; 19: 287-295.
6. O’Sullivan MP, Tyner JW, Holtzman MJ.Apoptosis in the airways: another balancing act inthe epithelial program. Am J Respir Cell Mol Biol2003; 29: 3-7.
7. Erjefalt JS, Uller L, Malm-Erjefalt M, Persson CG.Rapid and efficient clearance of airway tissue gran-ulocytes through transepithelial migration. Thorax2004; 59: 136-143.
8.Duncan CJ, Lawrie AS, Blaylock MG, et al. Reducedeosinophil apoptosis in induced sputum correlates withasthma severity. Eur Respir J 2003; 22: 484-490.
9. Segal M, Niazi S, Simons MP, et al. Bid activationduring induction of extrinsic and intrinsic apoptosis ineosinophils. Immunol Cell Biol 2007; 85: 518-524.
10. Spinozzi F, Fizzotti M, Agea E, et al. Defectiveexpression of Fas mRNA and Fas receptor on pul-monary T cells from patients with asthma. AnnIntern Med 1998; 128: 363-369.
11. Robinson DS, Kay AB, Wardlaw AJ. Eosinophils.Clin Allergy Immunol 2002; 16: 43-75.
12. Spinozzi F, Agea E, Bertotto A. Bronchial aller-gic inflammation: how it begins and why it persists.Current Trends Immunol 1998; 1: 149-157.
13. Strasser A, Pellegrini M. T lymphocyte deathduring shutdown of an immune response. TrendsImmunol 2004; 25: 610-615.
14. Muller M, Grunewald J, Olgart Hoglund C, etal. Altered apoptosis in bronchoalveolar lymphocytesafter allergen exposure of atopic asthmatic subjects.Eur Respir J 2006; 28: 513-522.
15. Cormican L, O’Sullivan S, Burke CM, PoulterLW. IFN-� but not IL-4 T cells of the asthmaticbronchial wall show increased incidence of apopto-sis. Clin Exp Allergy 2001; 31:731-739.
16. Darveau ME, Jacques E, Rouabhia M, et al.Increased T-cell survival by structural bronchial cellsderived from asthmatic subjects cultured in an engi-neered human mucosa. J Allergy Clin Immunol2008; 121: 692-699.
17. Spinozzi F, Fizzotti M, Agea E, et al. Defectiveexpression of Fas mRNA and Fas receptor on pul-monary T cells from patients with asthma. AnnIntern Med 1998: 128: 363-369.
Bibliografia

Asma e apoptosi 65
18. Spinozzi F, Agea E, Fizzotti M, et al. Role of T-helpertype 2 cytokines in down-modulation of Fas mRNA andreceptor on the surface of activated CD4+ T cells:molecular basis for the persistence of the allergicimmune response. FASEB J 1998: 12: 1747-1753.
19. Genestier L, Kasibhatla S, Brunner T, GreenDR. Transforming growth factor beta1 inhibits Fas lig-and expression and subsequent activation-inducedcell death in T cells via downregulation of c-Myc. J ExpMed 1999; 189: 231-239.
20. Akkoc T, de Koning PJA, Ruckert B, et al.Increased activation-induced cell death of high IFN� -producing TH1 cells as a mechanism of TH2 pre-dominance in atopic diseases. J Allergy ClinImmunol 2008; 121: 652-658.
21. De Rose V, Cappello P, Sorbello V, et al. IFN-�inhibits the proliferation of allergen-activated T lym-phocytes from atopic, asthmatic patients by inducingFas/FasL-mediated apoptosis. J Leukoc Biol 2004;76: 423-432.
22. Lamb JP, James A, Carroll N, et al. Reducedapoptosis of memory T-cells in the inner airway wallof mild and severe asthma. Eur Respir J 2005; 26:265-270.
23. Kamath AV, Pavord ID, Ruparelia PR, ChilversER. Is the neutrophil the key effector cell in severeasthma?Thorax 2005; 60: 529-530.
24. Ward C, Wong TH, Murray J, et al. Induction ofhuman neutrophil apoptosis by nitric oxide donors:evidence for a caspase-dependent, cyclic-GMP-inde-pendent, mechanism. Biochem Pharmacol 2000; 59:305-314.
25. Pelaia G, Cuda G, Vatrella A, et al. Effects oftransforming growth factor-[beta] and budesonide onmitogen-activated protein kinase activation andapoptosis in airway epithelial cells. Am J Respir CellMol Biol 2003; 29: 12-18.
26. Kay AB, Phipps S, Robinson DS. A role foreosinophils in airway remodelling in asthma. TrendsImmunol 2004; 25: 477-482.
27. Fedorov IA, Wilson SJ, Davies DE, Holgate ST.Epithelial stress and structural remodelling in child-hood asthma. Thorax 2005; 60: 389-394.
28. Ramos-Barbòn B, Presley JF, Hamid QA, et al.Antigen-specific CD4+ T cells drive airway smoothmuscle remodelling in experimental asthma. J ClinInvest 2005; 115: 1580-1589.
29. O’Sullivan MP, Tyner JW, Holtzman MJ.Apoptosis in the airways: another balancing act in theepithelial program. Am J Respir Cell Mol Biol 2003;29: 3-7.
30. Kuwano K, Hagimoto N, Kawasaki M, et al.Essential role of the Fas-Fas ligand pathway in thedevelopment of pulmonary fibrosis. J Clin Invest1999; 104: 13-19.
31. Atamas SP, Yurovsky VV, Wise R, et al.Production of type-2 cytokines by CD8+ lung cells isassociated with greater decline in pulmonary functionin patients with systemic sclerosis. Arthritis Rheum1999; 42: 1168-1178.
32. Romagnani S. The Th1/Th2 paradigm and aller-gic disorders. Allergy 1998; 53: 12-15.
33. Milgrom H, Fick RB Jr, Su JQ, et al. Treatment ofallergic asthma with monoclonal anti-IgE antibody. NEngl J Med 1999; 341: 1966-1973.
34.Holgate ST, Djukanovic R, Casale T, Busquet J. Anti-immunoglobulin E treatment with omalizumab in aller-gic diseases: an update on anti-inflammatory activity andclinical efficacy. Clin Exp Allergy 2005; 35: 408-446.
35. Infürh D, Crameri R, Lamers R, Achatz G.Molecular and cellular targets of anti-IgE antibodies.Allergy 2005; 60: 977-985.
36. Leckie MJ, ten Brinke A, Khan J, et al. Effects ofan interleukin-5 blocking monoclonal antibody oneosinophils, airway hyperresponsiveness and lateasthmatic response. Lancet 2000; 356: 2144-2148.
37. Kips JC, O’Connor BJ, Langley SJ, et al. Effect ofSCH55700, a humanized anti-human interleukin-5antibody in severe persistent asthma: a pilot study. AmJ Respir Crit Care Med 2003; 167: 1655-1659.
38. Flood-Page P, Menzies-Gow A, Phipps, S et al. Anti-IL-5 treatment reduces deposition of ECM proteins inthe bronchial subepithelial basement membrane of mildatopic asthmatics. J Clin Invest 2003; 112: 1029-1036.
39. Kodama T, Kuribayashi K, Nakamura H, et al.Role of interleukin-12 in the regulation of CD4+ Tcell apoptosis in a mouse model of asthma. Clin ExpImmunol 2003; 131: 199-205.
40. Bryan SA, O’Connor BJ, Matti S, et al. Effects ofrecombinant human interleukin-12 on eosinophils,airway hyperresponsiveness and the late asthmaticresponse. Lancet 2000; 356: 2149-2153.

de Benedictis, et al.66
41. Howarth PH, Babu KS, Arshad Hs, et al.Tumour necrosis factor (TNFalpha) as a novel thera-peutic target in symptomatic corticosteroid-depend-ent asthma. Thorax 2005; 60: 1012-1018.
42. Berry MA, Hargadon B, Shelley M, et al.Evidence of a role of tumor necrosis factor alpha inrefractory asthma. N Engl J Med 2006; 354: 697-708.
43. Simon HU. Cytokine and anti-cytokine therapy forasthma. Curr Allergy Asthma Rep 2006: 6:117-121.
44. Kroegel C, Bergmenn N, Foerster M, et al.Interferon-alphacon-1 treatment of three patientswith severe corticosteroid asthma. Effect on diseasecontrol and systemic glucocorticosteroid dose.Respiration 2006; 73: 566-570.
45. Busse WW, Baker JW, Charous BL, et al.Preliminary safety and efficacy of daclizumab in thetreatment of patients with moderate to severe per-sistent asthma. J Allergy Clin Immunol 2004; 113:S286-S287.
46. Holgate ST, Polosa R. The mechanisms, diag-nosis, and management of severe asthma in adults.Lancet 2006; 368: 780-793.
47. Spinozzi F, Agea E, Bistoni O, et al. T lympho-cytes bearing the γδ T cell receptor are susceptibleto steroid-induced programmed cell death. Scand JImmunol 1995; 41: 504-508.
48.Watanabe K, Shirasaki H, Kanaizumi E, Himi T.Effects of glucocorticoids on infiltrating cells andepithelial cells of nasal polyps. Ann Otol RhinolLaryngol 2004; 113: 465-473.
49. Spinozzi F, Agea E, Bistoni O, et al. Increasedallergen-specific, steroid-sensitive � � T cells in bron-choalveolar lavage fluid from patients with asthma.Ann Intern Med 1996; 124: 223-227.
50. Melis M, Siena L, Pace E, et al. Fluticasoneinduces apoptosis in peripheral T-lymphocytes: acomparison between asthmatic and normal sub-jects. Eur Respir J 2003; 219: 257-266.
51. O’Sullivan S, Cormican L, Burke CM, PoulterLW. Fluticasone induces T cell apoptosis in thebronchial wall of mild to moderate asthmatics.Thorax 2004; 59: 657-661.
52. Pace E, Gagliardo R, Melis M, et al. Synergisticeffects of fluticasone propionate and salmeterol onin vitro T-cell activation and apoptosis in asthma. JAllergy Clin Immunol 2004; 114: 1216-1223.
53. Druilhe A, Létuvé S, Pretolani M. Glucocorticoid-induced apoptosis in human eosinophils: mechanism ofaction. Apoptosis 2003; 8: 481-495.
54. Uller L, Persson GC, Kallstrom L, Erjefalt JF.Lung tissue eosinophils may be cleared throughluminal entry rather than apoptosis: effects of steroidtreatment. Am J Respir Crit Care Med 2001; 164:1948-1956.
55. Kankaanranta H, Lindsay MA, Giembycz MA,et al. Delayed eosinophil apoptosis in asthma. JAllergy Clin Immunol 2000; 106: 77-83.
56. Markham A, Faulds D. Theophylline. A reviewof its potential steroid sparing effects in asthma.Drugs 1998; 56: 1081-1091.
57. Takeuchi M, Hayakawa A, Takagi K, et al.Theophylline induces apoptosis of the IL-3-activatedeosinophils of patients with bronchial asthma.Apoptosis 1999; 4: 461-468.
58. Sullivan P, Bekir S, Jaffar Z, et al. Anti-inflamma-tory effects of low-dose oral theophylline in atopicasthma. Lancet 1994; 343: 1006-1008.
59. Lee E, Robertson T, Smith J, Kilfeather S.Leukotriene receptor antagonists and synthesisinhibitors reverse survival in eosinophils of asthmaticindividuals. Am J Respir Crit Care Med 2000;161:1881-1886.
60. Calhoun WJ, Lavins BJ, Minkwitz MC, et al.Effect of zafirlukast on cellular mediators of inflam-mation. Bronchoalveolar lavage fluids findings aftersegmental antigen challenge. Am J Respir Crit CareMed 1998; 157: 1381-1389.
61. Spinozzi F, Russano AM, Piattoni S, et al.Biological effects of montelukast, a cysteinyl-leukotriene receptor-antagonist, on T lymphocytes.Clin Exp Allergy 2004; 34: 1876-1882.
62. de Benedictis FM, de Benedictis D, Spinozzi F.Apoptosis in asthma. In: Modern insight into dis-ease from molecule to man: Apoptosis. Ed: PreedyVR 2009 (in press).

67Pneumologia Pediatrica 2009; 35: 67-78
Nicola Fuiano
Pediatria ed Allergologia Pediatrica, Presidio Polispecialistico, Distretto Sanitario n. 1 San Severo,ASL della Provincia di Foggia
Le malattie respiratorie ad eosinofilinel bambino
Eosinophil-associated respiratory diseasein children
Parole chiave: eosinofilo, NARES, polmoniti eosinofile, sindrome ipereosinofila, sindrome di Churg-Strauss
Keywords: eosinophil, NARES, eosinophilic pneumonitis, hypereosinophilic syndrome, Churg-Strauss syndrome
Riassunto. Gli eosinofili sono cellule leucocitarie particolarmente importanti nella flogosi allergica, coinvolte anche in unaserie di patologie non allergiche, spesso a carico delle vie respiratorie. Per quanto riguarda le riniti, la forma più nota è la rini-te non allergica eosinofila (“NARES” nella letteratura internazionale), mentre le vie respiratorie inferiori possono essere inte-ressate da un’ampia serie di disordini che comprendono bronchite eosinofila e polmoniti eosinofile. Queste ultime si distin-guono in “idiopatiche” (primitive, intrinseche), che includono polmonite eosinofila acuta e cronica, sindrome di Churg- Strass,sindrome ipereosinofilica (HES) e granuloma eosinofilo o istiocitosi polmonare X, e “secondarie o estrinseche”, cioè provoca-te da fattori esterni quali farmaci, parassiti, funghi e altri microrganismi, che causano il reclutamento e l’attivazione degli eosi-nofili e il successivo danno a carico dell’apparato respiratorio.L’età pediatrica è interessata dalle riniti eosinofile e in particolare dalla NARES, che costituisce una quota piuttosto rilevantedelle riniti non allergiche. Per quanto riguarda le vie respiratorie inferiori, bronchite e polmoniti eosinofile, sindrome ipereosi-nofilica e granuloma eosinofilo sono da considerare rare nel bambino, sebbene soprattutto per le polmoniti si disponga di unaserie di case report. La sindrome di Churg-Strauss dispone anche di revisioni sistematiche che hanno permesso di delinearnele similarità e gli aspetti contrastanti tra bambino e adulto, che riguardano soprattutto il coinvolgimento polmonare e cardia-co (superiore nel bambino) e la mortalità.
Accettato per la pubblicazione il 31 agosto 2009.
Corrispondenza: Dott. Nicola Fuiano, Pediatria e Allergologia Pediatrica, Presidio Polispecialistico, Distretto Sanitario n. 1,Via Turati 44, 71016 San Severo Foggia; e-mail: [email protected]
Introduzione
Gli eosinofili hanno rappresentato una scopertafondamentale nel campo della immuno-allergolo-gia e, grazie alle acquisizioni degli ultimi anni, meri-tano un ruolo di primo piano nella patogenesidella flogosi allergica (1).Gli eosinofili costituiscono le cellule maggiormen-te responsabili delle alterazioni strutturali conse-guenti alla flogosi allergica, al punto da meritarsi ladefinizione di “braccio armato” di questo tipo diinfiammazione (2).Descritti per la prima volta nel 1879 da PaulEhrlich, furono successivamente riscontrati nelsangue periferico e a livello tissutale in soggetti
con asma bronchiale, ad opera di Ellis. Nel 1922un fondamentale articolo sulla patogenesi dell’a-sma, a firma di Huber e Koessler, evidenziava l’as-sociazione tra eosinofilia ematica e tissutale neipolmoni di soggetti deceduti per asma (3).Nel 1975 Horn e collaboratori dimostravanocome in pazienti affetti da asma si registrasse unpiù elevato numero di eosinofili nel sangue perife-rico in presenza di più bassi livelli di VEMS, a dimo-strazione del ruolo degli eosinofili nella patogene-si dell’asma (4).In realtà, la principale caratteristica della patologiadell’asma è rappresentata dalla massiva infiltrazione

del parenchima polmonare da parte degli eosino-fili e dal danneggiamento della mucosa (5).Nel corso di un attacco d’asma si calcola che piùdi mezzo miliardo di eosinofili possano accumular-si nel tessuto polmonare rendendosi responsabilidel danno tissutale (6).Gli eosinofili devono il loro nome alla peculiarecolorazione che, in vir tù del metodoRomanowsky, in presenza di eosina, evidenzia inumerosi granuli intracitoplasmatici all’interno deiquali è presente un elevato numero di mediatori,proteine, citochine e chemochine (Tabella 1).Anche la membrana cellulare presenta costituentitra i quali la proteina di Charcot-Leyden ad attivi-tà lisofosfolipasica, frequentemente presente nel-l’escreato di pazienti con asma bronchiale esoprattutto nella sindrome di Churg-Strauss (7).Sulla membrana plasmatica degli eosinofili sonopresenti recettori ad alta affinità per le IgE (FcεRI)e, in piccola percentuale, anche recettori a bassaaffinità per le IgE (FcεRII).Gli eosinofili attivati esprimono varie molecole diadesione, soprattutto integrine e antigeni di isto-compatibilità di classe I e II (7).Gli eosinofili sono leucociti post-mitotici, dellalinea mieloide, prodotti nel midollo osseo graziealla stimolazione di GM-CSF, IL-1, IL-3, IL-5 (8).Queste stesse citochine sono responsabili dellamorte programmata degli eosinofili.Della grandezza di 10 µ-12 µ, in condizioni nor-mali di riposo, dopo aver stazionato nel midolloper circa 4 giorni, gli eosinofili si trasferiscono nelsangue periferico per brevi periodi (6-12 ore)
rappresentando l’1%-6% del pool totale dei leu-cociti, in concentrazione inferiore a 350/µl, conlivelli diurni che variano in misura inversa al livellodel cortisolo plasmatico. Il picco si registra nelcorso della notte; il minimo, invece, al mattino.Successivamente, con la cooperazione delle che-mochine (eotaxin-1, eotaxin-2, RANTES), gli eosi-nofili attraversano le giunzioni intercellulari endo-teliali per diapedesi e migrano nei tessuti (primevie respiratorie, tratto gastrointestinale, cute,utero) con emivita di 1-5 giorni (9).Dal punto di vista funzionale, si distinguono trediverse popolazioni di eosinofili: 1. eosinofili nor-modensi a riposo; 2. eosinofili normodensi attivati;3. eosinofili ipodensi attivati. Gli eosinofili dei grup-pi 2 e 3 presentano maggior numero di recettoridi membrana e sono pertanto dotati di attivitàfunzionale aumentata (7).L’attivazione degli eosinofili e la conseguentedegranulazione si traducono in una perdita di den-sità della cellula. Il numero degli eosinofili ipodensisi correla con la gravità della reazione (2).
Le eosinofilie
Sono oltre 30 le malattie che possono esserecausa di un numero di eosinofili superiore a350/µl, sia per aumentata produzione sia per rila-scio dai tessuti al sangue periferico (10).Un incremento di eosinofili può riscontrarsi anchein altri fluidi corporei (per esempio, liquido cerebro-spinale, urine) e anche nei tessuti (cute, polmone,cuore, fegato, intestino, vescica, midollo osseo (11).
Tabella 1 Mediatori prodotti dagli eosinofili. Modificata da [2, 7].
mediatori presenti nei granuli citoplasmatici
• CitochineIL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, IL-16, GM-CSF, TGF-α, TGF-β, TNF-α
• ChemochineRANTES, MIP-1α
• Enzimiarilsulfatasi, catalasi, collagenesi, esterasi non specifiche, fosfatasi acide, fosfolipasi-D, istaminasi
• Proteine basicheperossidasi eosinofila (EPO), proteina basica maggiore (MBP), proteina cationica eosinofila (ECP), proteina X degli eosinofili (EPX), neurotossina eosinofila (EDN)
mediatori derivati dalla membrana
• LCT4, PAF, 15HETE, PGD2, PGE1, PGE2, TXB2
Fuiano68

Le malattie respiratorie ad eosinofili nel bambino 69
Negli Stati Uniti e nei paesi occidentali la causa piùfrequente di eosinofilia resta la malattia allergica.Meno frequenti invece sono le infezioni parassitarie,l’ipersensibilità a farmaci, le malattie polmonari, legastroenteriti, le malattie autoimmuni e i tumorimaligni (12, 13). Non si registrano differenze signifi-cative di prevalenza per età, sesso, etnia (14).Talvolta molto comuni e di facile approccio dia-gnostico, talaltra le eosinofilie necessitano di unapproccio multidisciplinare e specialistico.Le eosinofilie (15) si distinguono in idiopatiche esecondarie, ovviamente definendo idiopatiche(idiopatic hypereosinophilic syndrome, HES) quellecon eosinofilia prolungata (>6 mesi) con coinvol-gimento di più organi ma senza anormalità citoge-netiche né molecolari (16).In funzione del numero degli eosinofili presenti nelsangue periferico (Tabella 2), si distinguono: a. eosi-nofilie lievi; b. eosinofilie moderate; c. eosinofiliesevere (17).Più elevato è il numero di eosinofili circolanti, mag-giore è il rischio di complicanze direttamente cor-relate alla presenza di eosinofili nei tessuti. Gliorgani più frequentemente coinvolti sono la cute,i polmoni, l’apparato digerente, il sistema nervoso,il cuore (12).
Riniti eosinofile
La corretta diagnosi di rinite (o meglio rinosinusi-te) (18) resta un autentico banco di prova. Unconsiderevole contributo (Tabella 3) viene daldocumento ARIA (Allergic rhinitis and its impact on
Asthma) (19) e dalla successiva classificazione(Tabella 4) proposta da Settipane e Lieberman (20).La più nota delle riniti eosinofile è la NARES (nonallergic rhinitis with eosinophilia syndrome), la cuiprima segnalazione risale al 1981 (21) ed era rela-tiva a 52 pazienti con sintomi nasali perenni distarnuti parossistici e rinorrea acquosa profusasenza poliposi, sinusite, otite media o asma, contest allergologici negativi e con eosinofilia nasale.La diagnosi di NARES è quindi una diagnosi diesclusione che si fonda sulla presenza dei segni esintomi della rinite allergica in soggetti non atopici(con prick test e determinazione delle IgE serichenegativi) con marcata presenza di eosinofili all’esa-me citologico della mucosa nasale) (18, 20-24).Numerosi studi hanno indicato nel 20% la presen-za di eosinofili alla citologia nasale (18, 20-24)anche se altri suggeriscono un range compreso tra5% e 25% (25, 26).I soggetti con NARES tendono a presentare sinto-mi nasali più intensi rispetto ai pazienti con riniteallergica o rinite vasomotoria. Frequente è il riscon-tro di anosmia (21, 22), molto comune lo sviluppodi poliposi e micropoliposi nasale (20-24).Kramer e collaboratori (27) in uno studio con 26soggetti affetti da apnee notturne (10 affetti daNARES e 16 senza segni e sintomi nasali) ha con-cluso che la NARES costituisce un fattore di rischioper lo sviluppo di sindrome delle apnee notturne.Più frequentemente a carico dell’adulto e del sessofemminile (28), la NARES non risparmia l’età pedia-trica (29). È presente nel 13%-33% dei soggetti con
Tabella 2 Cause comuni e rare di eosinofilia. Modificata da [17].
Cause comuni Cause meno comuni o rare
Eosinofilia lieve (351-1,500/µL)
Rinite allergica Malattie autoimmuniAsma Infezioni parassitarie
Reazioni a farmaci NeoplasieDermatite atopica Malattia polmonare occupazionale
Eosinofilia moderata (1,501-5,000/µL)
Asma NeoplasieReazioni a farmaci Malatti autoimmuni
Parassiti Sindrome ipereosinofilica
Eosinofilia severa (>5,000/µL)
Sindrome eosinofilica polmonare Reazioni a farmaciSindrome ipereosinofila Neoplasie
Parassitosi severe

rinite nonallergica (22). Non ancora sufficientemen-te definita da punto di vista etiologico e fisiopatolo-gico (23, 24), la NARES può costituire il precurso-re clinico della caratteristica triade: poliposi nasale,asma intrinseco e intolleranza all’aspirina (30).Frequentemente si registra familiarità positiva perNARES e si riconoscono quali fattori scatenanti
eventi aspecifici, come variazioni metereologiche eodori forti (31).Una chiave di lettura in funzione della fisiopatolo-gia delle NARES è suggerita da Ellis e Keith (24)che enfatizzano il ruolo dell’infiammazione cronicaeosinofilica che, insieme allo sviluppo della polipo-si e micropoliposi, conduce all’auto-perpetuazionedella flogosi stessa.Un ruolo trovano anche i mastociti (24, 32) la cuiinfiltrazione dà vita alla rinite non allergica masto-citaria (NARMA), meno frequente della NARES,per la quale è necessaria la citologia ai fini delladiagnosi. Non infrequentemente nel rinocitogram-ma di soggetti con NARMA è possibile ritrovareneutrofili, eosinofili e linfociti (32). Di recenteGelardi e collaboratori hanno individuato nuoveentità di riniti non allergiche: “NARNE” laddovel’infiltrato è costituito da neutrofili, e “NARESMA”quando sia eosinofili sia mastociti partecipano allacostituzione dell’infiltrato. La variante NARESMArappresenta un quadro clinico particolarmentesevero caratterizzato da poliposi nasale importan-te facilmente associata ad asma. La varianteNARNE è più frequentemente presente nei lavo-ratori dell’industria, dell’artigianato, oltre che neifumatori cronici (32).Un modello particolarmente efficace per com-prendere lo sviluppo e la cronicizzazione nelleNARES è stato proposto da Moneret-Vautrin ecollaboratori (33) che hanno individuato nellamigrazione degli eosinofili dal letto vascolare allesecrezioni nasali la prima tappa. Successivamentegli eosinofili penetrano nella mucosa dando luogo
Fuiano70
Tabella 3 Classificazione della rinite secondo il documentoARIA. Modificata da [19].
• InfettivaViraleBatterica
• AllergicaIntermittentePersistente
• Occupazionale (allergica/non allergica)IntermittentePersistente
• Indotta da farmaciAspirinaAltri farmaci
• Ormonale
• Da altre causeRinite non allergica con sindrome eosinofilica (NARES)Da irritantiDa alimenti Da emozioniAtroficaDa reflusso gastroesofageo
• Idiopatica
Tabella 4 Classificazione delle riniti non allergiche (in base a caratteristiche immunologiche e citologiche). NARES, nonallergic rhinitis with eosinophilia syndrome; BENARS, blood eosinophilia nonallergic syndrome. Modificata da [20].
Rinite non allergicaperenne (infiammatoria)
• Malattia nasale eosinofilica(NARES, BENARS)
• Malattia nasale basofilicametacromatica
• Infettiva
• Poliposi nasale
• Rinite atrofica
• Malattia nasaleimmunologica (non-IgE mediata)
Rinite non infiammatorianon allergica
• Rinite medicamentosa
• Rinite neurovegetativa(luce intensa o altri stimoli fisici)
• Rinite vasomotoria- Rinite irritativa- Rinite da aria fredda- Rinite gustatoria
• Rinite secca
• Rinite metabolica(da estrogeni o da ipotiroidismo)
Rinite da alterazioni strutturali
• Deviazione del setto, deformazioni deiturbinati, disfunzione nasale
• Neoplasie e masse non neoplastiche
• Miscellanea (atresia coanale, trauma dacorpo estraneo, malformazioni,palatoschisi, ipertrofia adenoidea)

Le malattie respiratorie ad eosinofili nel bambino 71
a poliposi e micropoliposi (33). Con gli eosinofilipartecipa alla flogosi la ECP responsabile di effet-to citotossico a carico dell’epitelio, di ciliostasi e lisidell’epitelio (34). Alla flogosi partecipa anche latriptasi, proteina presente nei mastociti, che -unita-mente all’istamina rilasciata per effetto della degra-nulazione- esalta la permeabilità vascolare.La “BENARS” (blood eosinophilia nonallergic rhinitissyndrome) è un sottogruppo delle NARES, carat-terizzato da ipereosinofilia nel sangue perifericospesso non associata a sinusite, con negatività deiprick test, normalità delle IgE seriche, incrementataeosinofilia nelle secrezioni nasali. È presente inibi-zione dell’attività delle cilia della mucosa nasalecon conseguente sviluppo di infezioni e di polipo-si nasale (28).Per quanto riguarda la terapia, lo steroide intrana-sale costituisce la prima opzione: riduce la sinto-matologia rinitica, migliora la respirazione nasale,ridimensiona la grandezza delle poliposi (21), nonmigliora però l’olfatto (35). Utile l’impiego di ste-roide intranasale associato a loratadina che sorti-sce un effetto migliore rispetto a quello dei far-maci adoprati singolarmente (24). Anche i cromo-ni (36) e gli antagonisti dei leucotrieni (37) sonostati suggeriti nella terapia delle NARES.
Bronchite eosinofila
La tosse cronica, definita da una durata superiorea 8 settimane, è frequente e costituisce un impor-tante problema di salute (38, 39). Le sue causeincludono patologie con coinvolgimento deglieosinofili, quali asma, tosse asthma-like, tosse ato-pica e bronchite eosinofila non asmatica (39, 40).Quest’ultima si segnala sempre più frequentemen-te come causa di tosse cronica: dal 10% al 30% deisoggetti con tosse cronica hanno bronchite eosino-fila (41) che si caratterizza, come l’asma, per la pre-senza di eosinofili nello sputo indotto (42) ma che,a differenza dell’asma, non presenta broncocostri-zione reversibile né iperreattività bronchiale (43).In realtà l’iperreattività bronchiale nel soggettoasmatico resta una delle più importanti conse-guenze dell’infiammazione e del rimodellamento.Un contributo utile a capire le differenze tra asmae bronchite eosinofila viene da Park e collabora-tori che hanno condotto uno studio su pazienticon asma, pazienti con bronchite eosinofila, e sog-getti sani (44), riscontrando una percentuale diarea di parete delle vie aeree significativamentemaggiore negli asmatici rispetto ai pazienti con
bronchite eosinofila e ai soggetti sani, mentreambedue i gruppi di pazienti presentavano unaprominenza centrolobulare e un intrappolamentodi aria significativamente superiore rispetto ai sog-getti sani. Tali osservazioni portavano gli Autori aipotizzare che l’elemento maggiormente correlatoalla presenza di iperreattività bronchiale sia la per-centuale di area delle vie respiratorie interessata.Una ulteriore chiave di lettura nel comprendere lediversità che si registrano tra asma e bronchiteeosinofila, viene proposta da Berry e collaboratoriche hanno riscontrato livelli significativamente ele-vati di espressione di IL-13 nell’espettorato indottonei soggetti con asma vs soggetti con bronchiteeosinofila e soggetti sani (45). Inoltre, il reclutamen-to dei mastociti sulla superficie delle vie respirato-rie e la loro attivazione appaiono essere una carat-teristica della bronchite eosinofila, a confronto conl’infiltrazione mastocitaria nel muscolo liscio signifi-cativamente più elevata nell’asma; nella bronchiteeosinofila, l’assenza di alta espressione di IL-13 puòcontribuire alla normale reattività bronchiale (46).Esiste infine una recente ipotesi relativa al fatto chela diversa funzionalità delle vie respiratorie neipazienti con asma e con bronchite eosinofila possaesser causata da uno squilibrio nella produzione dibroncocostrittori quali leucotriene LTC4 e bron-coprotettori quali prostaglandine PGE2. Ciò è statosuggerito dai risultati di uno studio su 13 pazienticon asma, 13 con bronchite eosinofila e 11 sogget-ti sani, che ha dimostrato, in presenza di livelli para-gonabili di eosinofili, citochine proinfiammatorie eleucotrieni nell’espettorato dei due gruppi di mala-ti, livelli significativamente più elevati di PGE2 neipazienti con bronchite eosinofila (47).L’esito più comune della bronchite eosinofila è lacronicizzazione, mentre la risoluzione completarisulta piuttosto rara. Una analisi mediante regres-sione multipla lineare ha identificato nel fumo disigaretta, nel sesso femminile e nell’area sotto lacurva del conteggio di eosinofili nel tempo, gli indi-ci maggiormente predittivi di declino del VEMS (48).La terapia della bronchite eosinofila non può pre-scindere dall’impiego dei corticosteroidi inalatori(CSI) pur con i limiti attuali che non ci consento-no di stimare dosaggi e tempi di trattamento(49). In realtà il dosaggio e i tempi di trattamen-to differiscono da paziente a paziente. I pazienticon bronchite eosinofila possono anche necessi-tare di corticosteroidi per via sistemica orale alungo termine (46).

Polmoniti eosinofile
Le polmoniti eosinofile (EP) si caratterizzano perl’incrementato numero di eosinofili nel sangueperiferico, nel tessuto polmonare, nell’espettoratoe nel liquido di lavaggio broncoalveolare (BAL). LeEP si presentano con sintomatologia respiratoria,evidenza di anomalie radiografiche, rischio di mani-festazioni sistemiche (50).In chiave di fisiopatologia le EP rappresentano ilrisultato dell’infiltrazione del tessuto polmonare daparte degli eosinofili, che costituiscono meno del2% della popolazione cellulare nel BAL di sogget-ti normali, mentre la percentuale può raggiungereil 5%-25% delle cellule nel BAL in numerose con-dizioni tra cui le EP (51). Una popolazione di eosi-nofili superiore al 25% nel BAL è sicuro indice dipolmonite eosinofila (51-54).Le EP si distinguono in due gruppi:a. EP idiopatiche, primitive, intrinseche (51-54)- Polmonite eosinofila acuta (AEP)- Polmonite eosinofila cronica (CEP)- Sindrome di Churg- Strass (CSS)- Sindrome ipereosinofilica (HES)- Granuloma eosinofilo (EG) noto anche comeistiocitosi polmonare Xb. EP secondarie, estrinseche (52, 54, 55) provo-cate da fattori esterni quali polveri, farmaci,parassiti, funghi e altri microrganismi, fattori checausano il reclutamento e l’attivazione degli eosi-nofili e il successivo danno a carico dell’apparatorespiratorio. Tra queste EP “secondarie” vi è lasindrome di Loeffler.
Polmonite eosinofila acuta e cronica
Le EP sono il risultato del reclutamento e dell’atti-vazione degli eosinofili da parte di IL-5 a sua voltaattivata dai linfociti Th2. IL-5 e IL-18 sono presenti inelevate concentrazioni nel BAL. IL-5 peraltro inibi-sce l’apoptosi degli eosinofili (53, 55). Anche IL-6 eIL-10 sono implicate nella patogenesi delle EP (52).Gli eosinofili reclutati degranulano nell’interstizio enegli alveoli con rilascio di proteina basica maggio-re, proteina cationica, neurotossine e conseguentedanno tissutale; il successivo rilascio di leucotrieniporta a broncospasmo che unitamente alle secre-zioni bronchiali presenti nelle vie respiratorie con-duce all’insufficienza respiratoria (56). Le EP pos-sono essere secondarie, le possibili cause sonoelencate nelle Tabelle 5 e 6.La polmonite eosinofila acuta (AEP nella nomen-clatura internazionale) è stata descritta la primavolta nel 1989 e ha poi ricevuto numerose segna-lazioni, più spesso in soggetti di giovane età (etàmedia 29 anni), prevalentemente di sesso maschi-le, e sovente (circa il 40%) con storia di fumo disigaretta (51, 57). Un recente articolo ha descrittoun caso di AEP in un bambino di 12 anni, con eosi-nofilia periferica corrispondente a 33.800/mm3
con il 90% di eosinofili nel BAL, trattato con suc-cesso con ventilazione meccanica e terapia corti-costeroidea (58).In diagnosi differenziale, un rash cutaneo puòorientare verso una reazione da farmaci, infezioneparassitaria, Churg-Strauss o HES. La rino-sinusiteè anche presente nella sindrome di Churg-Strauss.
Fuiano72
Tabella 5 Cause di AEP secondaria. AEP, polmonite eosinofila acuta. Modificata da [51, 53].
Fattori Cause
Vaccinazioni e farmaci Vaccinazione BCG, minociclina, fludarabina, progesterone, sertralina
Infezioni Aspergillus, Coccidioidomycosis
Ambiente Fumo, polveri ambientali, benzina
Tabella 6 Cause di CEP secondaria. CEP, polmonite eosinofila cronica. Modificata da [56].
Fattori Cause
Farmaci FANS, salicilati, minociclina, cotrimossazolo, fludarabina, progesterone, sertralina
Infezioni Aspergillus (Aspergillosi broncopolmonare allergica), parassiti
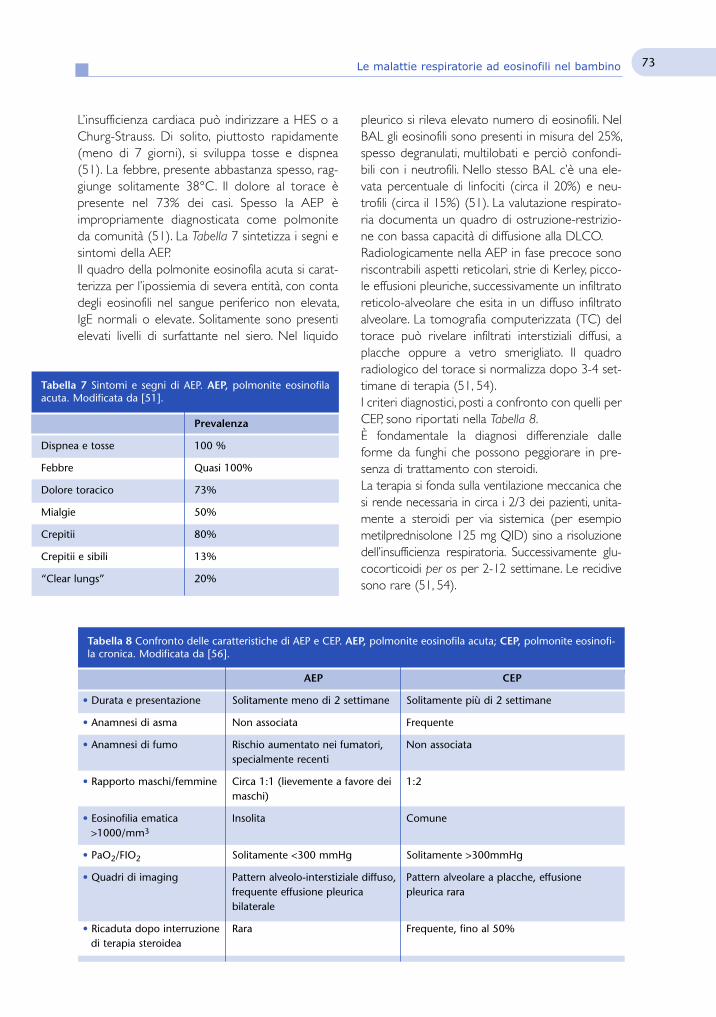
Le malattie respiratorie ad eosinofili nel bambino 73
L’insufficienza cardiaca può indirizzare a HES o aChurg-Strauss. Di solito, piuttosto rapidamente(meno di 7 giorni), si sviluppa tosse e dispnea(51). La febbre, presente abbastanza spesso, rag-giunge solitamente 38°C. Il dolore al torace èpresente nel 73% dei casi. Spesso la AEP èimpropriamente diagnosticata come polmoniteda comunità (51). La Tabella 7 sintetizza i segni esintomi della AEP.Il quadro della polmonite eosinofila acuta si carat-terizza per l’ipossiemia di severa entità, con contadegli eosinofili nel sangue periferico non elevata,IgE normali o elevate. Solitamente sono presentielevati livelli di surfattante nel siero. Nel liquido
pleurico si rileva elevato numero di eosinofili. NelBAL gli eosinofili sono presenti in misura del 25%,spesso degranulati, multilobati e perciò confondi-bili con i neutrofili. Nello stesso BAL c’è una ele-vata percentuale di linfociti (circa il 20%) e neu-trofili (circa il 15%) (51). La valutazione respirato-ria documenta un quadro di ostruzione-restrizio-ne con bassa capacità di diffusione alla DLCO.Radiologicamente nella AEP in fase precoce sonoriscontrabili aspetti reticolari, strie di Kerley, picco-le effusioni pleuriche, successivamente un infiltratoreticolo-alveolare che esita in un diffuso infiltratoalveolare. La tomografia computerizzata (TC) deltorace può rivelare infiltrati interstiziali diffusi, aplacche oppure a vetro smerigliato. Il quadroradiologico del torace si normalizza dopo 3-4 set-timane di terapia (51, 54).I criteri diagnostici, posti a confronto con quelli perCEP, sono riportati nella Tabella 8.È fondamentale la diagnosi differenziale dalleforme da funghi che possono peggiorare in pre-senza di trattamento con steroidi.La terapia si fonda sulla ventilazione meccanica chesi rende necessaria in circa i 2/3 dei pazienti, unita-mente a steroidi per via sistemica (per esempiometilprednisolone 125 mg QID) sino a risoluzionedell’insufficienza respiratoria. Successivamente glu-cocorticoidi per os per 2-12 settimane. Le recidivesono rare (51, 54).
Tabella 7 Sintomi e segni di AEP. AEP, polmonite eosinofilaacuta. Modificata da [51].
Prevalenza
Dispnea e tosse 100 %
Febbre Quasi 100%
Dolore toracico 73%
Mialgie 50%
Crepitii 80%
Crepitii e sibili 13%
“Clear lungs” 20%
Tabella 8 Confronto delle caratteristiche di AEP e CEP. AEP, polmonite eosinofila acuta; CEP, polmonite eosinofi-la cronica. Modificata da [56].
• Durata e presentazione
• Anamnesi di asma
• Anamnesi di fumo
• Rapporto maschi/femmine
• Eosinofilia ematica >1000/mm3
• PaO2/FIO2
• Quadri di imaging
• Ricaduta dopo interruzione di terapia steroidea
AEP
Solitamente meno di 2 settimane
Non associata
Rischio aumentato nei fumatori,specialmente recenti
Circa 1:1 (lievemente a favore dei maschi)
Insolita
Solitamente <300 mmHg
Pattern alveolo-interstiziale diffuso, frequente effusione pleurica bilaterale
Rara
CEP
Solitamente più di 2 settimane
Frequente
Non associata
1:2
Comune
Solitamente >300mmHg
Pattern alveolare a placche, effusione pleurica rara
Frequente, fino al 50%

La AEP può portare a decesso per insufficienzarespiratoria laddove non venga diagnosticata etrattata prontamente. Un netto miglioramento,con diagnosi e trattamento corretti e tempestivi, siregistra dopo 24-48 ore. La guarigione solitamen-te è completa, possono registrarsi come esitomodesti quadri di tipo restrittivo (51, 54).La polmonite eosinofila cronica (CEP) può coin-volgere pazienti di ogni età, più spesso soggettiintorno ai 45 anni circa (56, 57). Si stima che laCEP corrisponda al 2,5% dei casi di interstiziopa-tie. È più frequente nel sesso femminile, dal 50% al67% dei pazienti con CEP hanno storia di asmaalla diagnosi (56). Anamnesticamente spesso sitratta di atopici. Meno del 10% sono fumatori. Leforme di CEP secondarie sono riportate nellaTabella 6.Spesso si tratta di un problema di salute checoinvolge il paziente da un mese o più (anche 20settimane) con tosse di tipo produttivo, dispneasolo eccezionalmente tanto severa da meritarela ventilazione meccanica. Dolori al torace eemottisi sono rari. Sibili espiratori sono presentinel 50% dei pazienti; crepitii inspiratori solooccasionalmente (57).Perdita di peso, sudorazione notturna, febbre, aste-nia devono far considerare una patologia tumorale.La CEP può raramente avere una presentazionecon dolori al torace, artralgie, sintomi neurologici,coinvolgimento della cute, fegato, intestino così dasimulare sindrome di Churg-Strauss o HES (56).L’ipossiemia è di tipo lieve-moderato, l’eosinofilianel sangue periferico è marcata (più di 1.000 eosi-nofili/mm³). Gli indici di flogosi (VES e PCR) sonoelevati, elevati anche i livelli di IgE specialmentenegli asmatici (56). Il BAL si caratterizza per eleva-ta presenza di eosinofili (12%-95% della compo-nente cellulare). La percentuale di eosinofili spessoè più elevata di quella dei linfociti. È inoltre pre-sente degranulazione degli eosinofili (possibile ilcontrollo valutando la neurotossina nelle urine). Ilquadro respiratorio è in 1/3 dei pazienti di tipoostruttivo, ma è possibile registrare anche quadridi tipo restrittivo. Nella CEP il 25% dei pazientipresenta lieve riduzione della capacità di diffusionealla DLCO. E’importante cercare una possibilepatologia autoimmune associata come poliartritenodosa, artrite reumatoide, sclerodermia (54, 56).I quadri radiologici evidenziano infiltrati alveolariche comprendono aspetti a vetro smerigliato odi consolidamento, generalmente bilaterali e
periferici, con i lobi superiori come sedi predo-minanti. Gli infiltrati sono caratteristicamentemigranti, come si può rilevare con monitoraggioradiologico, la TC può essere utile per rilevare lepiccole lesioni a vetro smerigliato non visibili allaradiografia standard (54).I criteri diagnostici, posti a confronto con quelliper AEP, sono riportati nella Tabella 8.Il trattamento richiede solo raramente la ventila-zione assistita. I glicocorticoidi rappresentano laterapia di scelta con dosaggi da 0,3-1 mg kg/die diprednisolone (56) da ridurre gradualmente nell’ar-co di 6-12 mesi.Circa il 50% dei soggetti con CEP presentanorecrudescenza dopo la sospensione degli steroidie un numero significativo di soggetti sviluppa asmae necessita di terapia corticosteroidea cronica.
Sindrome di Churg-Strauss
Questa sindrome è espressione della possibilecorrelazione tra una condizione di atopia e l’insor-genza di vasculite. Malattia sistemica, con prevalen-za stimata in 0,75-1,3/100mila, più frequente insoggetti asmatici, molto rara in età pediatrica, conetà d’esordio tra i 15 e 70 anni. Può coinvolgerepolmoni, seni paranasali, tratto intestinale, reni,cuore (59).L’American College of Rheumatology richiede perla diagnosi di Churg-Strauss (CSS) la presenza dialmeno quattro delle seguenti caratteristiche:asma, eosinofilia, neuropatia, infiltrati polmonari,anomalie dei seni paranasali, vasculite eosinofila.L’asma è costante, l’eosinofilia periferica elevata(dal 10% al 75% della formula leucocitaria). La CSSè stata suddivisa in tre fasi: a. la prima con asmacon/senza rinite allergica; b. la seconda con eosi-nofilia nel sangue periferico e infiltrazioni eosinofi-le nei tessuti con quadri simili alla sindrome diLoeffler, alla polmonite eosinofila cronica, allagastroenterite eosinofila; c. la terza, quella vasculiti-ca, con coinvolgimento di polmoni, cuore, sistemanervoso periferico, fegato, linfonodi, muscoli, cute.Il 40% dei soggetti con CSS presenta anticorpi anti-citoplasmatici antineutrofili p-ANCA/MPO-ANCA.Considerati gli anticorpi ANCA, possiamo distin-guere due fenotipi: a. ANCA-positivi: con coinvol-gimento renale, neuropatia periferica, biopsie posi-tive per vasculite. La positività degli ANCA indicalo stato di flogosi e necrosi dei vasi di piccolo cali-bro di tipo vasculitico; b. ANCA-negativi, con feb-bre e malattia cardiaca (60).
Fuiano74

Le malattie respiratorie ad eosinofili nel bambino 75
Di rilievo la revisione sistematica effettuata daZwerina e collaboratori dal 2003 al 2007 sono statisegnalati 33 soggetti pediatrici, età media 12 anni,M:F ratio 0,74, tutti con eosinofilia ed asma. In tuttii soggetti studiati era presente eosinofilia e/o vascu-lite all’istologia, gli ANCA erano positivi nel 25% deltotale. Questi dati indicano le differenze della CSSin età adulta ed in età pediatrica: quest’ultima sicaratterizza per la prevalenza di manifestazioni car-diopolmonari, minore invece il coinvolgimento delsistema nervoso periferico, la mortalità è peraltroelevata (61).La diagnosi differenziale si pone con la granuloma-tosi di Wegener, la sindrome ipereosinofilica, la sin-drome di Langherans, le reazioni a farmaci, le infe-zioni fungine e parassitarie, i tumori maligni.I glucocorticoidi in monoterapia e gli immunosop-pressori (per es., ciclofosfamide) nelle forme piùaggressive, costituiscono la base del trattamento;restiamo in attesa di maggiori evidenze in meritoall’impiego di anticorpi monoclonali anti-interleuki-na-5 e anti-immunoglobulina E e di interferone α.
Sindrome ipereosinofilica
La sindrome ipereosinofilica (HES) è un disordinemieloproliferativo caratterizzato da persistenteeosinofilia associato a danneggiamento multiorga-no. L’eosinofilia periferica in soggetti con patologieorganiche è stata notata da più di 80 anni, ma ilriconoscimento della sindrome è dovuto a Hardy eAnderson nel 1968 (62). Le tre caratteristicherichieste per la diagnosi di HES sono: una contaassoluta superiore a 1.500 per più di sei mesi, l’as-senza di cause identificabili di eosinofilia e la pre-senza di segni e sintomi di interessamento di orga-ni interni (63). La malattia colpisce in prevalenza ilsesso maschile (rapporto M:F 9:1., solitamente inun’età compresa tra 20 e 50 anni, è rara nel bam-bino. Indolente nella maggior parte dei casi, lamalattia può risultare fatale. Uno studio condottoin Francia ha notato una sopravvivenza dell’80% a5 anni e del 42% a 15 anni (64).Il trattamento si basa su corticosteroidi e immuno-soppressori, ma anche su chemioterapici qualichlorambucil, etoposide, vincristina e cytarabina esulla terapia specifica per le localizzazioni d’organo.
Granuloma eosinofilo (EG)
Questa forma viene definita anche come granulo-matosi X di Langerhans o granulomatosi polmonarea cellule di Langerhans o istiocitosi polmonare X,
dove il termine istiocitosi descrive disordini prolifera-tivi degli istiociti o macrofagi caratterizzati da prolife-razione delle cellule di Langherans, che possono coin-volgere polmoni, ossa, ipofisi. Idiopatica, rappresenta il2,8% delle malattie interstiziali polmonari; più fre-quente nella 3a e 4a decade di vita; il 75% dei soggetticolpiti sono fumatori (65). Può decorrere in modoasintomatico, altrimenti ha un esordio con tosse, disp-nea; il 37,5% dei pazienti presenta pneumotorace(66), può verificarsi insufficienza respiratoria.Alla spirometria si osservano pattern variabili, men-tre alla DLCO si rilevano alterazioni significative. Lostudio radiografico può rivelare lesioni osteoliticheper accumulo di cellule di Langherans a effetto ero-sivo. Alla Rxgrafia del torace possono osservarsiimmagini di aspetto nodulare o cistico e infiltratireticolari o reticolo-nodulari (65), che possonoessere meglio valutati mediante TC ad alta risolu-zione (67). La presenza di cellule di Langherans nelBAL in soggetti con malattia in fase attiva confermala diagnosi. La diagnosi differenziale si pone con sar-coidosi, TBC, linfomi, polmoniti eosinofile, fibrosiinterstiziali polmonari. Il decorso è variabile ma lamalattia tende al peggioramento. La terapia concorticosteroidi e immunosoppressori non ha effet-to determinante e in circa il 25% dei pazienti ènecessario prendere in considerazione il trapiantodi polmone (65).
Polmoniti eosinofile secondarie o estrinseche
La più nota è la sindrome di Loeffler, presentatanella sua prima descrizione come espressione dirisposta immunitaria alla migrazione di larve diparassiti (più spesso ascaridi). nel parenchima pol-monare. Si distingue una forma asintomatica, rever-sibile, transitoria, che si autolimita senza necessità diterapia e una forma sintomatica (52).Occasionalmente associata ad asma, con febbrenon elevata; la sintomatologia respiratoria (tosse,dispnea, facile affaticabilità) può essere minima oassente. E’raro l’interessamento sistemico.L’eosinofilia ematica periferica è spiccata (60-80%dei leucociti), può essere riscontrata ipergamma-globulinemia e presenza di parassiti nelle feci. LaRxgrafia del torace dimostra solitamente infiltratiunilaterali che si sviluppano e scompaiono rapida-mente in diversi lobi oppure interessamento diffu-so con lesioni miliari o nodulari (52).La risoluzione è generalmente rapida e la prognosieccellente. La terapia, se necessaria, si basa su corti-sonici e controllo dell’infestazione parassitaria.

Conclusioni
Le patologie respiratorie con coinvolgimentodegli eosinofili comprendono un gruppo eteroge-neo di malattie unificate proprio dalla presenza diquantità elevate di tali leucociti. L’età pediatrica èinteressata dalle riniti eosinofile e in particolaredalla NARES, che costituisce una quota piuttostorilevante delle riniti non allergiche. Per quantoriguarda le vie respiratorie inferiori, bronchiti epolmoniti eosinofile, sindrome ipereosinofilica e
granuloma eosinofilo o istiocitosi X sono daconsiderare rare nel bambino, sebbene soprat-tutto per le polmoniti si disponga di una serie di“case report”. La sindrome di Churg-Straussdispone anche di revisioni sistematiche chehanno permesso di delinearne le similarità e gliaspetti contrastanti tra bambino e adulto, cheriguardano soprattutto il coinvolgimento pol-monare e cardiaco (superiore nel bambino) e lamortalità (61).
Fuiano76
1. Martin LB, Kita H, Leiferman KM, Gleich GJ.Eosinophils in allergy: role in disease, degranulationand cytokines. Int Arch Allergy Immunol 1996;109: 207.
2. Boner AL. Disregolazione del sistema immune:iperreattività allergica. In: Plebani A. ImmunologiaPediatrica. Mc Graw-Hill Italia, 1998: 96.
3. Frigas E, Gleich GJ. The eosinophil and the patho-physiology of asthma. J Allergy Clin Immunol 1986;77: 527-537.
4. Horn BR, Robin ED, Theodore J, Van Kessel A.Total eosinophil counts in the management ofbronchial asthma. N Engl J Med 1975; 292: 1152.
5. Naylor B. The shedding of the mucosa of thebronchial tree in asthma. Thorax 1962; 17: 69.
6. Filley WV, Holley KE, Kephart GM, Gleich GJ.Identification by immunofluorescence of eosinophilgranule major basic protein in lung tissues ofpatients with bronchial asthma. Lancet 1982; 2: 11.
7. Errigo E. Malattie Allergiche. Lombardo Editore,Roma, 1999, vol. I, pagg. 142-147.
8. Wardlaw AJ. Eosinophils in the 1990s: new per-spectives on their role in health and disease.Postgrad Med J 1994; 70: 536-552.
9. Jose PJ, Griffiths-Johnson DA, Collins PD, et al.Eotaxin: a potent eosinophil chemoattractant cytokinedetected in a guinea pig model of allergic airwaysinflammation. J Exp Med 1994; 179: 881-887.
10. Rothenberg ME. Eosinophilia. N Eng J Med1998; 338: 1592-1600.
11. Spry CJF. Eosinophils: a comprensive review andguide to the scientific and medical literature. Oxford,England: Oxford Medical Publications, 1988.
12. Roufosse F, Cogan E. Practical approach tohypereosinophilia. Rev Med Brux 2008; 29: 400-408.
13. Seifert M, Gerth J, Gajda M, et al. Eosinophilia– a challenging differential diagnosis. Med Klin2008; 103: 591-597.
14. Teffari A. Modern diagnosis and treatment ofprimary eosinophilia. Acta Haematologica 2005;114: 52-60.
15. Roufosse F, Cogan E, Goldman M. Recentadvances in pathogenesis and management of hyper-eosinophilic syndromes. Allergy 2004; 59: 673-863.
16. Hardy WR, Anderson RE. The hypere-osinophilic syndromes. Ann Intern Med 1968; 68:1220-1229.
17. Brigden ML. A practical workup for eosinophil-ia. Postgrad Med 1999; 105: 193-210.
18. Fokkens W,Lund V, Bachert C, et al. EAACIposition paper on rhinosinusitis and nasal polypsexecutive summary. Allergy 2005; 60: 583-601.
19. Bousquet J, van Cauwenberge P. World HealthOrganisation Iniziative, allergic rhinitis and its impacton Asthma (ARIA). Geneva: WHO, 2000.
Bibliografia

Le malattie respiratorie ad eosinofili nel bambino 77
20. Settipane RA, Lieberman P. Update on nonal-lergic rhinitis. Ann Allergy asthma Immunol 2001;86: 494-507.
21. Jacobs RL, Freedman PM, Boswell RN.Nonallergic rhinitis with eosinophilia (NARES syn-drome). Clinical and Immunologic presentation. JAllergy Clin Immunol 1981; 67: 253-262.
22. Schiavino D, Nucera E, Milani A, et al. Nasallavage cytometry in the diagnosis of non allergicrhinitis with eosinophilia syndrome (NARES). AllergyAsthma Proc 1997; 18: 363-366.
23. Fokkens WJ. Thoughts on the pathophysiologyof nonallergic rhinitis. Curr Allergy Asthma Rep2002; 3: 203-209.
24. Ellis AK, Keith PK. Nonallergic rhinitis witheosinophilia syndrome and related disorders. ClinAllergy Immunol 2007; 19: 87-100.
25. Mygind N, Dirkasen A, Johnsen NJ, Weeke B.Perennial rhinitis: an analysis of skin testing serumIgE and blood and smear eosinophilia in 201patients. Clin Otolaryngol 1978; 3: 189-196.
26. Crobach M, Hermans J,Kaptein A et al. Nasalsmear eosinophilia for the diagnosis of allergic rhini-tis and eosinophilic nonallergic rhinitis. Scand J PrimHealth Care 1996; 14: 116-121.
27. Kramer MF, de la Chaux R, Fintelmann R, RaspG. NARES: a risk factor for obstructive sleep apnea?Am J Otolaryngol 2004; 25: 173-177.
28. Settipane GA, Klein DE. Nonallergic rhinitis:demography of eosinophils in nasal smear, bloodtotal eosinophil counts, and IgE levels. N Engl RegAllergy Proc 1985; 6: 363-366.
29. Gelardi M,Russo C, Fiorella ML. Fisiopatologiadella mucosa nasale: dalla tipologia cellulare alripristino terapeutico della “Integrità di barriera”. It JAllergy Clin Immunol 2009; 19: 1-9.
30. Moneret-Vautrin DA, Hsieh V, Waioff M,Nonallergic rhinitis with eosinophilia syndrome a pre-cursor of the triad: nasal polyposis, intrinsic asthma,and intolerance to aspirin. Ann Allergy 1990; 64:513-518.
31. Swierczynska M, Strek P, Skladzien J, et al.Nonallergic rhinitis with eosinophilia syndrome: stateof knowledge. Otolaryngol Pol 2003; 57: 81-84.
32. Gelardi M, Maselli del Giudice A, Fiorella ML,et al. Non-allergic rhinitis with eosinophils and mastcells constitutes a new several disorder. Int JImmunopathol Pharmacol 2008; 21: 325-331.
33. Moneret-Vautrin DA, Jankowski R, Wayoff M.Clinical and pathogenetic aspects of NARES (nonal-lergic rhinitis with eosinophilic syndrome. RevLaryngol Otol Rhinol 1991; 112: 41-44.
34. Kramer MF, Burow G, Pfrogner E, Rasp G. Invitro diagnosis of chronic nasal inflammation. ClinExp Allergy 2004; 34: 1086-1092.
35. Mygind N. Effects of corticosteroid therapy innon-allergic rhinosinusitis. Acta Otolaryngol 1996;116: 164-166.
36. Nelson BL, Jacobs RL. Response of nonallergicrhinitis with eosinophilia (NARES) syndrome to 4%cromolyn sodium nasal solution. J Allergy ClinImmunol 1982; 70: 125-128.
37. Peters-Golden M, Henderson WR Jr. The roleof leukotrienes in allergic rhinitis. Ann AllergyAsthma Immunol 2005; 94: 609-618.
38. Scott KA, Wardlaw J. Eosinophilic airway disease.Semin Respire Crit Care Med 2006; 27: 128-131.
39. Chung KF, Pavord ID. Prevalence, pathogenesis,and causes of chronic cough. Lancet 2008; 371:1364-1374.
40. Niimi A, Matsumoto H, Mishima M.Eosinophilic airway disorders associated with chron-ic cough. Pulm Pharmacol Ther 2009; 22: 114-120.
41. Brightling CE. Chronic cough due to nonasthmat-ic eosinophilic bronchitis: ACCP evidence- based clinicalpractice guidelines. Chest 2006; 129: 116S-121S.
42. Gibson PG, Dolovich J, Denburg J, et al.Cronich cough eosinophilic bronchitis without
asthma. Lancet 1989; 1: 1346-1348.
43. Brightling CE, Ward R, Goh KL,et al.Eosinophilic bronchitis is an important cause ofchronic cough. Am J Respir Crit Care Med 1999;160: 406-410.
44. Park SW, Park JS, Lee YM, et al. Differences inradiological/HRCT findings in eosinophilic bronchitisand asthma: implication for bronchial responsive-ness. Thorax 2006; 61: 41-47.

Fuiano78
45. Berry MA, Parker D, Neale N, et al. Sputumand bronchial submucosal IL-13 expression in asth-ma and eosinophils bronchitis. J Allergy ClinImmunol 2004; 114: 1106-1109.
46. Gonlugur U, Gonlugur TE. Eosinophilic bron-chitis without asthma. Int Arch Allergy Immunol2008; 147: 1-5.
47. Sastre B, Fernàndez-Nieto M, Mollà R, et al.Increased prostaglandin E2 levels in the airway ofpatients with eosinophilic bronchitis. Allergy 2008;63: 58-66.
48. Berry MA, Hargadon B, McKenna S, et al.Observational study on the natural history ofeosinophil bronchitis. Clin Exp Allergy 2005; 35:598-601.
49. Gibson PG, Fujimura M, Niimi A. Eosinophilicbronchitis: clinical manifestations and implications fortreatment. Thorax 2002; 57: 178-182.
50. Wechsler ME. Pulmonary eosinophilic syn-dromes. Immunol Allergy Clin North Am 2007;27: 477-492.
51. Allen J. Acute eosinophilic pneumonia. SeminRespir Crit Care Med 2006; 27: 142-147.
52. Alberts WM. Eosinophilic interstitial lung dis-ease. Curr Opin Pulm Med 2004; 10: 419-424.
53. Cottin V, Cordier JF. Eosinophilic pneumonias.Allergy 2005; 60: 841-857.
54. Katz U, Shoenfeld Y. Pulmonary eosinophilia.Clin Rev Allergy Immunol 2008; 34: 367-371
55. Tefferi A, Patnaik MM, Pardanani A.Eosinophilia: secondary, clonal and idiopathic. Br JHaematol 2006; 133: 468-492.
56. Marchand E, Cordier JF. Idiopatic chroniceosinophilic pneumonia. Semin Respir Crit CareMed 2006; 27: 134-141.
57. Voss M, Allewelt M, Lode H. Chroniceosinophilic pneumonia. Dtsch Med Wochenschr2004; 29: 1858-1860.
58. Khèmiri M, Quederni M, Ben Mansour F et al.Acute respiratory failure revealing an idiophaticacute eosinophilic pneumonia: report of a pediatriccase. Ann Fr Anesth Reanim 2008; 27: 502-504.
59. Katzenstein AL. Diagnostic features and dif-ferential diagnosis of Churg-Strauss syndrome inthe lung. A Review. Am J Clin Pathol 2000; 114:767-772.
60. Pagnoux C, Guilpain P, Guillevin L. Churg-Strauss syndrome. Curr Opin Rheumatol 2007; 19:25-32.
61. Zwerina J, Eger G, Englbrecht M, et al. Churg-Strauss syndrome in childhood: a systematic litera-ture review and clinical comparison with adultpatients. Semin Arthritis Rheum 2008; 16. E-pubahead of print.
62. Hardy WR, Anderson RE. The hypere-osinophilic syndromes. Ann Inter Med 1968; 68:1220-1229.
63. Klion AD, Bochner BS, Gleich GJ, et al, for theHypereosinophilic Syndromes Working Group.Approaches to the treatment of hypereosinophilicsyndromes: a workshop summary report. J AllergyClin Immunol 2006;117: 1292-1302.
64. Lefebvre C, Bletry O, Degoulet P, et al.Prognostic factors of hypereosinophilic syndrome.Study of 40 cases. Ann Med Interne 1989; 140:253-257.
65. Callebaut W, Demedts M, Verleden G.Pulmonary Langherans’cell granulomatosis (histiocy-tosis X): clinical analysis of 8 cases. Acta Clin Belg1998; 53: 337-343.
66. Bianchi M, Cataldi M. Pneumothorax secondaryto pulmonary histiocytosis X. Minerva Chir 1999;54: 361-366.
67. Lynch DA, Hay T, Newell JD, et al. Pediatric dif-fuse lung disease: diagnosis and classification usinghigh-resolution CT. AJR Am J Roentgenol 1999;173:713-718.

Il wheezing episodico virale è una condizione assaifrequente in età prescolare che si presenta consintomatologia respiratoria tipo broncospasmo diseverità variabile, che spesso richiede un interven-to diagnostico-terapeutico da parte del pediatra dibase od in ospedale. Di solito questi bambini sonoasintomatici fra gli episodi di broncospasmo (1).L’impiego di una terapia corticosteroidea (per viainalatoria o per via sistemica) nel trattamento degliepisodi di wheezing episodico virale è una praticaclinica largamente adottata dalla maggior parte deipediatri, ma che in realtà sembra basata su evi-denze provenienti da studi condotti su bambini inetà scolare e con caratteristiche di asma. (2, 3).
L’efficacia di tale strategia terapeutica, in bambini dietà prescolare, in cui una diagnosi di asma non èspesso possibile né chiara, è stata posta in discus-sione dal gruppo inglese coordinato da JonathanGrigg, in un trial randomizzato controllato pubblica-to su the Lancet nel 2003 (4): un ciclo breve di cor-ticosteroidi (prednisolone 1 mg/kg/die per via oraleper 5 giorni) somministrato dai genitori in bambinidi età prescolare che a domicilio presentavano sin-tomi compatibili con wheezing virale non si è dimo-strato efficace nel prevenire la ospedalizzazione oridurre la gravità clinica dei sintomi respiratori acuti.Il gruppo di Vuillermin et al, nel 2007, hanno pubbli-cato con la Cochrane Collaboration una revisione
Enrico Opocher1, Marco Balzani1, Marta Zambolin1, Luca Bartolini1, Eugenio Baraldi1
1Gruppo Redazione Journal Club, Dipartimento di Pediatria, Università degli Studi di Padova
Corticosteroidi per via orale in bambinidi età pre-scolare con wheezing
episodico virale:cosa resta di questa terapia?
Corticosteroids treatment in pre-schoolchildren with viral induced wheezing:
where does this leave us?
Parole chiave:Wheezing virale, corticosteroidi, prednisolone, età prescolare
Keywords: viral wheezing, corticosteroids, prednisolone, preschool age
Riassunto. Il trattamento con steroidi per via orale in bambini di età prescolare con broncospasmo indotto da infezioni respi-ratorie virali rappresenta una pratica consolidata nella pediatria. Tuttavia tale approccio terapeutico è mutuato dall’efficacia chegli steroidi hanno nei bambini con asma acuto. Un recente trial clinico randomizzato controllato pubblicato sul New EnglandJournal of Medicine nel2009, mostra invece come, in bambini condotti in ospedale, un breve ciclo di prednisolone per via oralenon migliora i sintomi di broncospasmo lieve-moderato indotto da virus, né accorcia l’ospedalizzazione. Da questo studio losteroide per os sembra non essere efficace neppure in un sottogruppo di pazienti che risulta ad alto rischio di sviluppare asmain età scolare. Il trial clinico è metodologicamente corretto con una buona validità interna, pertanto non può essere ignoratoe invita a ripensare alla nostra pratica medica quotidiana di fronte ad un bambino con wheezing episodico virale.
Accettato per la pubblicazione il 31 agosto 2009.
Corrispondenza: Dott. Enrico Opocher, Dipartimento di Pediatria, Università di Padova,Via Giustiniani 3, 35128 Padova; e-mail: [email protected]
Pneumologia Pediatrica 2009; 35: 79-82 R U B R I C A : j o u r n a l c l u b 79

sistematica di due trial randomizzati controllati,dimostrando che non vi è evidenza di efficacia diun trattamento precoce con corticosteroidi pervia orale (intrapreso a domicilio dai familiari)rispetto al placebo in termini di necessità di ospe-dalizzazione, miglioramento dei sintomi respirato-ri e utilizzo di broncodilatatori per via inalatorianei bambini con “wheezing” virale (5).Più di recente, Panickar et al (6), dello stesso grup-po londinese, hanno pubblicato sul New EnglandJournal of Medicine (NEJM) a Gennaio 2009 un trialrandomizzato controllato con il medesimo schemadi trattamento con prednisolone orale, sommini-strato in bambini di età prescolare con wheezingvirale che venivano ricoverati in ospedale per la per-sistenza di sintomi respiratori acuti dopo un’ inizialeterapia con broncodilatatore per via inalatoria (sal-butamolo). I risultati di questo trial indicano chiara-mente che la terapia corticosteroidea per via oralenon è efficace nel ridurre la durata della ospedaliz-zazione né l’entità dei sintomi respiratori acuti.Questi studi, in modo complementare, sembranoaffermare che questa sia una pratica clinica quan-tomeno “imperfetta” e debba quindi essere ridi-scussa ed eventualmente modificata (7).In particolare, lo studio di Panickar et al. è un trialrandomizzato controllato, in cui sono stati arruo-lati lattanti e bambini di età compresa tra i 10 mesied i 6 anni con segni e sintomi di un’infezione vira-le delle alte vie respiratorie, diagnosticata clinica-mente e che si presentavano in uno degli Ospedaliinglesi partecipanti allo studio.I bambini eleggibili, arruolati nello studio, venivanotrattenuti in ospedale se, nonostante la sommini-strazione di broncodilatatore (10 puff diSalbutamolo tramite spray + distanziatore oppuredi 2,5 mg in soluzione se <3 anni o 5 mg se > 3anni mediante nebulizzatore) non vi era migliora-mento dei sintomi respiratori. I bambini ricoverativenivano quindi randomizzati ad assumere 1 cap-sula per os di prednisolone 10 mg se età ≤24 mesi,20 mg se età > 24 mesi in polvere, oppure 1 cap-sula per os contenente placebo in polvere, entram-bi miscelati con 10 ml di una bevanda aromatizza-ta, una volta al giorno per 5 giorni. Sono statiesclusi bambini con patologie croniche cardiacheo polmonari, in terapia immunosoppressiva oaffetti da qualche immuno-deficienza congenita equelli esposti di recente a casi di varicella.Nel corso dell’ospedalizzazione, oltre al farmaco instudio poteva essere somministrato salbutamolo
al bisogno per aerosol e ossigeno tramite masche-ra facciale, in caso di ipossia, a discrezione delmedico curante.Ai pazienti che erano stati assegnati ai 2 gruppi,subito dopo il ricovero, a 5’ dalla somministrazio-ne del broncodilatatore, veniva assegnato unoscore clinico basale, PRAM (Preschool RespiratoryAssessment Measure), che prevedeva un punteggiovariabile crescente da 0 a 12, dipendente dalla gra-vità del distress respiratorio e che veniva registra-to, anche dopo 4 , 12 e 24 ore. Al momento delladimissione (decisa dal pediatra del reparto in basealle stesse variabili utilizzate per ricoverare) venivafornito ai genitori un diario sul quale registrare acasa i sintomi respiratori del figlio, per una duratadi 7 giorni e le capsule residue per continuare iltrattamento per i 5 giorni previsti.Nello studio sono stati valutati un outcome princi-pale (durata dell’ospedalizzazione) e alcuni outco-me minori (PRAM score a 4, 12, 24 ore; dosi tota-li di salbutamolo durante l’ospedalizzazione; mediadello score dei sintomi su 7 giorni; media delledosi di salbutamolo a casa; tempo per il “ritornoalla normale attività”; nuova ospedalizzazione perwheezing entro 1 mese dalla dimissione).Sono stati valutati 1180 pazienti per l’arruolamen-to; di questi, 700 sono stati randomizzati. Dei 480scartati, 162 non soddisfacevano i criteri di eleggi-bilità, mentre di 318 i genitori non hanno dato ilconsenso. Dei 700 bambini randomizzati, a 343 èstato somministrato prednisolone ai dosaggi sopracitati. L’altro gruppo (344 pazienti) è stato trattatocon placebo. I due gruppi possedevano caratteri-stiche cliniche omogenee e paragonabili.Nei risultati viene sottolineato come la differenzadella durata dell’ospedalizzazione, tra gruppo trat-tato con prednisolone e gruppo trattato con pla-cebo, per quanto presente (-1.9 ore di differenzamediana a favore dei trattati con cortisonici) nonrisulti significativa (p= 0,16) e non sia comunquesignificativa dal punto di vista clinico (una differen-za di meno di 2 ore di durata di ospedalizzazione).Gli Autori concludono quindi che non vi sono dif-ferenze tra i 2 gruppi analizzati per quanto riguar-da gli outcome considerati. Gli stessi outcome inoltre sono stati valutati ancheper un sottogruppo di pazienti (124 bambini-58 nelgruppo placebo e 66 nel gruppo prednisolone)considerati ad “alto rischio” di sviluppare asma inetà scolare (secondo i criteri di Castro-Rodriguez)(8). Tuttavia, neppure in questo sottogruppo, sono
80 Opocher, et al.

Rubrica: journal club - Corticosteroidi per via orale in bambini di età pre-scolare ... 81
state rilevate differenze statisticamente significativetra i pazienti trattati con prednisolone e quelli trat-tati con placebo.Viene quindi suggerito da Panickar et al. di nonusare il prednisolone per os “di routine” nei pazien-ti in età prescolare che vengono condotti in ospe-dale con un “acute, mild-to-moderate virus-inducedwheezing”.La validità metodologica (validità interna) dellostudio di Panicker et al. è ben dimostrata e lasciaveramente pochi dubbi od “ombre”.Lo studio è prospettico randomizzato-controllato,a carattere multicentrico (4 centri dell’Inghilterra),con una numerosità campionaria soddisfacente, èstato condotto a doppio cieco, con partecipantirandomizzati a blocchi di 10, con una lista di ran-domizzazione inaccessibile prima della conclusionedello studio. L’analisi è stata condotta secondol’Intention-To-Treat (ITT) (Tabella 1) ed il follow-upè di durata adeguata e include praticamente tuttigli arruolati. Tra le limitazioni metodologiche “minori” vi è lascelta di includere partecipanti con limite inferioredi età pari a 10 mesi, una situazione che non esclu-de la possibilità che siano stati inclusi casi di bron-chiolite, condizione quest’ultima in cui non vi è evi-denza di alcun effetto della terapia corticosteroi-dea; un altro limite, che nella pratica clinica ha peròscarsa applicabilità e rilevanza è il non aver esegui-to alcun esame colturale per confermare la pre-senza di un’infezione virale respiratoria concomi-tante, la cui diagnosi era esclusivamente clinica. Dei318 bambini eleggibili ma non arruolati nello studioper rifiuto da parte dei genitori, resta inoltre il dub-bio sulle loro condizioni cliniche (“più gravi? menogravi?”) e la possibile influenza sui dati di outcome.
In un numero successivo del NEJM, SalmanMroueh commenta come la gravità del wheezing(score PRAM) prima della somministrazione di sal-butamolo è assente nello studio di Panicker et al.ciò non permette di determinare chiaramente l’ef-ficacia della terapia con broncodilatatore (primadella randomizzazione con prednisolone), tale dapoter eventualmente selezionare chi ha più biso-gno di una terapia cortisonica (9).Questo limite, insieme al fatto che i dati sono pro-venienti da una realtà dove la pediatria sul territo-rio è assente ed i criteri di ricovero discutibili (sin-tomatologia spesso lieve-moderata con scorePRAM medio pari a 4,3) sebbene non mettano indiscussione la “forza” del messaggio finale dellostudio di Panickar et al., non permettono di dimo-strarne con certezza la sua generalizzabilità (validi-tà esterna).Emerge, come sottolineato dagli stessi autori, chela gravità dei sintomi nei bambini effettivamenterandomizzati era “lieve-moderata”. I dati di questostudio non escludono quindi l’efficacia della terapiacorticosteroidea in bambini con wheezing virale“severo”.Vi sono in letteratura alcuni studi che cercano didistinguere un sottogruppo di bambini che potreb-be beneficiare più di altri della terapia cortisonicanel wheezing virale.Jartti et al. (10) dimostrerebbe infatti come unciclo di 3 giorni di prednisolone per os in bambinidi un anno di età, ospedalizzati per un episodio dibroncospasmo (venivano esclusi, tra gli altri, ipazienti che avevano presentato in passato più diun episodio di wheezing), pur non riuscendo aridurre la durata del ricovero, diminuiva però inmaniera statisticamente significativa, rispetto al
Tabella 1 Metodologia in pillole.
Cosa è l’Intention to treat (ITT) analysis?
L’ITT è un metodo di analisi per gli studi clinici con-trollati in cui per ogni individuo l’outcome di interesseviene valutato sulla base del gruppo di trattamentocui la randomizzazione ha assegnato l’individuo stessoe non sulla base del trattamento che ha effettivamen-te ricevuto.
ITT è la valutazione dei risultati di tutti i pazienti,secondo il loro gruppo di appartenenza, indipenden-temente dal fatto che abbiano o meno completato lostudio e che abbiano o meno assunto la terapia.
Che cosa implica per la validità di uno studio?
Una analisi non condotta secondo ITT potrebbe por-tare ad un analisi dei risultati influenzata dalle modifi-che di trattamento dei pazienti in base ad un decorsodifferente (sfavorevole?)

placebo, le ricadute nei 2 mesi successivi. La sceltadi includere nello studio di Panickar anche bambi-ni al primo episodio di wheezing, rischierebbequindi di confondere (almeno in parte) i dati fina-li. Sarebbe utile ed interessante, a nostro modo divedere, confrontare gli effetti della terapia cortiso-nica su un first episode of wheezing rispetto ad unbambino con episodi ripetuti pregressi.Lo stesso autore, in altri due interessanti studi (10,11) dimostra in una post-hoc analysis, come bam-bini con alcuni tipi di infezione virale (rhinovirus)oppure con associate caratteristiche di atopia(sensibilizzazione per allergeni respiratori, livelloIgE totale, numero di allergeni, eczema, eosinofilia,livelli di ossido nitrico esalato), avrebbero unamigliore risposta al cortisone in termini di duratadi ospedalizzazione e di minori ricadute.Tuttavia, tra i risultati dello studio (anche se nonviene particolarmente sottolineato dagli autori)sembra non esserci alcuna risposta alla terapia ste-roidea nemmeno nei cosiddetti “atopic wheezers”,cioè soggetti con predisposizione allergico-atopica.
Conclusioni e “take home message”
In due studi simili (3, 5) che complessivamentehanno arruolato più di 900 bambini, è stato dimo-strato che iniziare una terapia orale con cortico-steroidi nella maggior parte dei bambini di età pre-scolare affetti da wheezing virale episodico, non ènecessario, in quanto non riduce in modo signifi-cativo la gravità dei sintomi né previene la neces-sità o la durata dell’ ospedalizzazione.Vista la scarsa efficacia della terapia orale cortico-steroidea nei bambini in età prescolare, i possibilieffetti collaterali di dosi ripetute di corticosteroidi(particolarmente rilevanti se consideriamo gli epi-sodi ripetuti e frequenti di wheezing virale), questoarticolo apre un importante discussione e costrin-ge a ripensare sulla nostra pratica medica quoti-diana perché l’uso routinario della terapia oralecorticosteroidea nel wheezing virale lieve-modera-to nei bambini in età prescolare sembrerebbe nonpiù giustificato, sia a domicilio che in ospedale ed ilsuo utilizzo dovrebbe essere riservato ai bambiniospedalizzati con wheezing severo.
Opocher, et al.82
1. Brand PLP, Baraldi E, Bisgaard H, et al. Definition,assessment and treatment of wheezing disorders inpreschool children: an evidence-based approach. EurRespir J 2008; 32: 1096-1110.
2. Smith M, Iqbal S, Elliott TM, et al. Cortico-steroidsfor hospitalised children with acute asthma.Cochrane Database Syst Rev 2003;1: CD002886.
3. Weinberger M. Consensus statement from aconference on treatment of viral respiratory infec-tion-induced asthma in young children. J Pediatr2003; 142 (suppl): S45-S46.
4. Oommen A, Lambert PC, Grigg J. Efficacy of ashort course of parent-initiated oral prednisolone forviral wheeze in children aged 1-5 years: randomisedcontrolled trial. Lancet 2003; 362: 1433-1438.
5.Vuillermin PJ, Robertson CF, South M. Parent-ini-tiated oral corticosteroid therapy for intermittentwheezing illnesses in children: systematic review.Paediatr Child Health 2007; 43: 438-442.
6. Panickar J, Lakhanpaul M, Lambert PC, et al. Oralprednisolone for preschool children with acute virus-induced wheezing. N Engl J Med 2009; 360: 329-338.
7. Bush A. Practice imperfect - treatment for wheezingin pre-schoolers. N Engl J Med 2009; 360: 409-410.
8. Castro-Rodriguez JA, Wright AL, Taussig LM,Martinez FD. A clinical index to define risk of asth-ma in young children with recurrent wheezing. Am JRespir Crit Care Med 2000;162: 1403-1406.
9. Weinberger M. Oral corticosteroids in childrenwith wheezing. N Engl J Med 2009; 16; 360: 1673-1674; author reply 1675-1676.
10. Jartti T, Lehtinen P, Vanto T, et al. Efficacy of pred-nisolone in children hospitalized for recurrent wheez-ing. Pediatr Allergy Immunol 2007; 18: 326-334.
11. Jartti T, Lehtinen P, Vanto T et Al. Atopic charac-teristics of wheezing children and responses to pred-nisolone Pediatr Pulmonol. 2007; 42: 1125-1133.
Bibliografia

83
CongressiCongresses
CongressiCongresses
SETTEMBRE 2009
Il test da sforzo cardio-polmonare(CPET): basi e applicazioniParma 24 - 25 settembre 2009Organizzato da:Azienda Ospedaliero-Universitaria di ParmaSegreteria organizzativa:Planning congressi SrlTel. 051. 300100E-mail: [email protected]: www.planning.it
OTTOBRE 2009
XIII Convegno della Società Italiana perle Malattie Respiratorie InfantiliNapoli 15 - 17 ottobre 2009Organizzato da:Società Italiana per le Malattie Respiratorie InfantiliSegreteria organizzativa:iDea congress SrLTel. 06. 36381573E-mail: [email protected]
Pneumomeeting 2009La gestione integrata in MedicinaRespiratoriaTaormina 16 - 17 ottobre 2009Organizzato da:iDea congress SrLTel. 06. 36381573E-mail: [email protected]
CHEST 2009San Diego (Stati Uniti) 31 ottobre - 5 novembre2009Organizzato da:American College of Chest PhysiciansTel. +39. 847. 498. 1400Info: http://www.chestnet.org/
NOVEMBRE 2009
Medical aerosols: ins and outs of inhalation therapyAmsterdam (Olanda) 12 - 14 novembre 2009Organizzato da:European Respiratory SocietyTel. +41. 21. 213 0101E-mail: [email protected]: http://dev.ersnet.org/
Bambino e attività sportiva. Stili di vita, prevenzione e terapia.Corso teorico pratico sul test da sforzocardiopolmonare e corso PBLS-D(Pediatric Basic Life Support and earlyDefibrillation)Roma 20 - 21 novembre 2009Segreteria organizzativa:Center Comunicazione e CongressiTel. 081 19578490Fax 081 19578071Info: www.centercongressi.com/basp
Empiema pleurico e pleuriti infettiveBrescia 28 novembre - 2 dicembre 2009Organizzato da:U.O. Pneumologia, A.O. “Spedali Civili”Segreteria organizzativa:SP.i.cTel. 030. 382336E-mail: [email protected]
DICEMBRE 2009
Certezze scientifiche e criticità organizzative in PneumologiaMilano 2 - 5 dicembre 2009Organizzato da:Associazione Italiano Pneumologi Ospedalieri eUnione Italiana per la PneumologiaSegreteria organizzativa:iDea congressTel. +39 06 36381573E-mail: [email protected]: www.ideacpa.com

Informazioni per gli autoricomprese le norme per la preparazione dei manoscritti
La Rivista pubblica contributi redatti in forma di editoriali,articoli d’aggiornamento, articoli originali, articoli originalibrevi, casi clinici, lettere al Direttore, recensioni (da libri,lavori, congressi), relativi a problemi pneumologici e aller-gologici del bambino.I contributi devono essere inediti, non sottoposti contempo-raneamente ad altra Rivista, ed il loro contenuto conformealla legislazione vigente in materia di etica della ricerca.Gli Autori sono gli unici responsabili delle affermazioni con-tenute nell’articolo e sono tenuti a dichiarare di aver otte-nuto il consenso informato per la sperimentazione e per lariproduzione delle immagini.La redazione accoglie solo i testi conformi alle norme edi-toriali generali e specifiche per le singole rubriche.La loro accettazione è subordinata alla revisione critica diesperti, all’esecuzione di eventuali modifiche richieste ed alparere conclusivo del Direttore.
NORME GENERALITesto: in lingua italiana o inglese, in triplice copia, dattilo-scritto, con ampio margine, con interlinea doppia, massimo25 righe per pagina, con numerazione delle pagine a parti-re dalla prima, e corredato di:1) titolo del lavoro in italiano, in inglese;2) parola chiave in italiano, in inglese;3) riassunto in italiano, (la somma delle battute, spazi inclusi,non deve superare le 2.500);4) titolo e didascalie delle tabelle e delle figure.Si prega di allegare al manoscritto anche il testo memoriz-zato su dischetto di computer, purché scritto con pro-gramma Microsoft Word versione 4 e succ. (per Dos eApple Macintosh).Nella prima pagina devono comparire: il titolo (conciso);i nomi degli Autori e l’istituto o Ente di appartenenza; larubrica cui si intende destinare il lavoro (decisione che ècomunque subordinata al giudizio del Direttore); il nome,l’indirizzo e l’e-mail dell’Autore cui sono destinate la corri-spondenza e le bozze.Il manoscritto va preparato secondo le norme interna-zionali (Vancouver system) per garantire la uniformità dipresentazione (BMJ 1991; 302: 338-341). È dunque indi-spensabile dopo una introduzione, descrivere i materiali ei metodi, indagine statistica utilizzata, risultati, e discussio-ne con una conclusione finale. Gli stessi punti vannoriportati nel riassunto.Nelle ultime pagine compariranno la bibliografia, le dida-scalie di tabelle e figure.Tabelle (3 copie): devono essere contenute nel numero(evitando di presentare lo stesso dato in più forme), dattilo-scritte una per pagina e numerate progressivamente.Figure (3 copie): vanno riprodotte in foto e numerate sulretro. I grafici ed i disegni possono essere in fotocopia, pur-ché di buona qualità.Si accettano immagini su supporto digitale (floppy disk, zip,cd) purché salvate in uno dei seguenti formati: tif, jpg, eps econ una risoluzione adeguata alla riproduzione in stampa(300 dpi); oppure immagini generate da applicazioni pergrafica vettoriale (Macromedia Freehand, Adobe Illustratorper Macintosh). Sono riproducibili, benché con bassa resaqualitativa, anche documenti generati da Power Point. Alcontrario, non sono utilizzabili in alcun modo le immaginiinserite in documenti Word o generate da Corel Draw.La redazione si riserva di rifiutare il materiale ritenuto tec-nicamente non idoneo.
Bibliografia: va limitata alle voci essenziali identificate neltesto con numeri arabi ed elencate al termine del mano-scritto nell’ordine in cui sono state citate. Se gli autori sonofino a quattro si riportano tutti, se sono cinque o più siriportano solo i primi tre seguiti da “et al.”.Esempi di corretta citazione bibliografica per:articoli e riviste:Zonana J, Sarfarazi M, Thomas NST, et al. Improved definitionof carrier status in X-linked hypohydrotic ectodermal dysplasiaby use of restriction fragment lenght polymorphism-based linka-ge analysis. J Pediatr 1989; 114: 392-395.libri:Smith DW. Recognizable patterns of human malformation.Third Edition. Philadelphia: WB Saunders Co. 1982.capitoli di libri o atti di Congressi:Krmpotic-Nemanic J, Kostovis I, Rudan P. Aging changes of theform and infrastructure of the external nose and its importancein rhinoplasty. In: Conly J, Dickinson JT, (eds). “Plastic and recon-structive surgery of the face and neck”. New York, NY:Grune and Stratton 1972: 84-95.
Ringraziamenti, indicazioni di grants o borse di studio, vannocitati al termine della bibliografia.Le note, contraddistinte da asterischi o simboli equivalenti,compariranno nel testo a piè di pagina.Termini matematici, formule, abbreviazioni, unità e misuredevono conformarsi agli standard riportati in Scienze 1954;120: 1078.I farmaci vanno indicati col nome chimico.
Per la corrispondenza scientifica:
Prof. Eugenio BaraldiDipartimento di PediatriaUniversità di PadovaVia Giustiniani 335128 [email protected]
RICHIESTA ESTRATTIGli estratti devono essere richiesti all’Editore contestual-mente alle bozze corrette.Gli estratti sono disponibili in blocchi da 25.Il costo relativo, comprese le spese di spedizione in con-trassegno, è il seguente:25 estratti (fino a 4 pagine): h 60,0025 estratti (fino a 8 pagine): h 80,0025 estratti (fino a 12 pagine): h 100,00Si applicano i seguenti sconti in funzione del numero dicopie degli estratti:- per 50 copie, sconto del 5% sul totale- per 75 copie, sconto del 10% sul totale- per 100 copie, sconto del 15% sul totale
ABBONAMENTIPneumologia Pediatrica è trimestrale. Viene inviata gratuita-mente a tutti i soci della Società Italiana per le MalattieRespiratorie Infantili; i prezzi di abbonamento annuo per inon soci sono i seguenti:Italia ed Estero: h 72,00; singolo fascicolo: h 20,00.Le richieste di abbonamento e ogni altra corrispondenzarelativa agli abbonamenti vanno indirizzate a:
Primula Multimedia S.r.L.Via G. Ravizza, 22/b56121 Pisa - Loc. Ospedaletto

85Articoli del prossimo numero
Articoli del prossimo numero
SIMRI e Ricerca
1
2
4
3
5
9
La ricerca italiana in pneumologia pediatrica:il presente con uno sguardo sul futuroA. Rubino
Il fumo di sigaretta altera la funzione meccanica di barriera dell’epitelio bronchiale attraverso la disorganizzazione delle giunzioni strette tra le cellule epitelialiL. Petecchia, et al.
Fisiologia e patologia del tessuto adenoideoG.L. Marseglia, et al.
Variazioni circadiane di citochine e cortisolo in bambini con OSASL. Nosetti, et al.
L’asma in bambini in età prescolare in Italia:prevalenza, fattori di rischio e utilizzo delle risorse sanitarieD.G. Peroni, et al.
Funzionalità respiratoria nella displasia broncopolmonare: il follow-up del grippo di PadovaE. Baraldi, et al.
Markers nel condensato dell'aria espirata e fenotipi dell'asmaS. Carraro, et al.
Il lavaggio broncoalveolare e la biopsia bronchiale nei bambini asmatici e negli atopici non asmaticiD. Snijders, et al.
Discinesia ciliare primaria: problemi d’interpretazione diagnosticaF. Saretta, et al.
Un nuovo strumento per l’imaging polmonare: vibration responseimaging (VRI). Esperienza in una casistica pediatricaF. Saretta, et al.
7
8
6
10

Articoli del prossimo numero86
Infiammazione delle vie aeree e declino della funzione respiratorianella bronchiolite obliterante post-infettiva del bambinoS. Cazzato, et al.
Desensibilizzazione orale al latte: come scegliere la dose iniziale?C.O. Battaglini, et al.
Impedenza respiratoria e risposta al broncodilatatore in bambini saniin età prescolareC. Calogero, et al.
Sintomi respiratori, funzionalità polmonare e markers di infiammazionebronchiale in bambini che vivono in un’area sottoposta ad inquinamentoindustrialeF. Rusconi, et al.
Le colture cellulari da brushing nasale: un nuovo metodo per semplifi-care l’iter diagnostico nei casi dubbi di Discinesia Ciliare PrimariaM. Pifferi, et al.
Sensibilizzazione allergica in bambini con dermatite atopica: confronto tra studi internazionaliF. Franceschini, et al.
Sensibilizzazione per allergeni respiratori ed elevati livelli di IgE totalisono associati ad iperresponsività bronchiale in bambini con wheezingin età prescolareM. Verini, et al.
FeNO come marker di infiammazione delle vie aeree: possibili impli-cazioni nella gestione dell'asma nel bambinoM. Verini, et al.
Deossiribonucleasi umana ricombinante nel trattamento di bambinicon bronchiolite moderata-severaF. Midulla, et al.
Virus respiratorio sinciziale, bocavirus umano e rinovirus in bambinicon bronchioliteF. Midulla, et al.
La capacità funzionale e la valutazione della dispnea in bambinioperati per correzione di ernia diaframmatica congenitaA. Turchetta, et al.
11
12
14
13
15
19
17
18
16
21
20

Articoli del prossimo numero 87
24
26
29
28
30
31
Caratteristiche del sonno in bambini con sindrome di Prader-Willi ed effetti della terapia con GHE. Verrillo, et al.
La presenza di anomalie parossistiche all’elettroencefalogramma in unapopolazione di bambini con sindrome delle apnee ostruttive in sonnoS. Miano, et al.
Basi biochimiche dell’infiammazione nei bambini con sindrome delleapnee ostruttive notturne e nei bambini con obesitàM.P. Villa, et al.
Flogosi ed iperreattività bronchiale aspecifica in bambini con asmabronchialeC. Capristo, et al.
Valutazione del danno polmonare mediante risonanza magnetica adalto campo nella malattia polmonare cronica non dovuta a fibrosi cis-tica: confronto con la tomografia computerizzata ad alta risoluzione ecorrelazione con la funzionalità respiratoriaS. Montella, et al.
Disturbi respiratori nel sonno in bambini obesi del sud ItaliaL. Brunetti, et al.
Markers di flogosi nel condensato di aria espirata: recenti applicazionidiagnostioche nel bambino con wheezing/asma e dermatite atopicaL. Brunetti, et al.
Correlazione tra ossido nitrico nell’esalato (FeNO) e consumo di lipidi e antiossidanti nella dieta di bambini con asmaF. Cardinale, et al.
I determinanti che influenzano il livello di FeNO: studio su un campione di adolescenti italianiS. La Grutta
Efficacia a lungo termine dell’immunoterapia sublinguale: follow-up a 8 anniS. Leonardi, et al.
Efficacia a lungo termine dell’immunoterapia sublinguale: follow-up a 8 anniS. Leonardi, et al.
23
25
27
22
32





























