Embed Size (px)
Citation preview

The Expanded Universe of Prokaryotic Argonaute Proteins
Sergei Ryazansky,a Andrey Kulbachinskiy,a Alexei A. Aravina,b
aInstitute of Molecular Genetics, Russian Academy of Sciences, Moscow, RussiabDivision of Biology and Biological Engineering, California Institute of Technology, Pasadena, California, USA
ABSTRACT Members of the ancient family of Argonaute (Ago) proteins are presentin all domains of life. The common feature of Ago proteins is the ability to bindsmall nucleic acid guides and use them for sequence-specific recognition—andsometimes cleavage— of complementary targets. While eukaryotic Ago (eAgo) pro-teins are key players in RNA interference and related pathways, the properties andfunctions of these proteins in archaeal and bacterial species have just started toemerge. We undertook comprehensive exploration of prokaryotic Ago (pAgo) pro-teins in sequenced genomes and revealed their striking diversity in comparison witheAgos. Many pAgos contain divergent variants of the conserved domains involved ininteractions with nucleic acids, while having extra domains that are absent in eAgos,suggesting that they might have unusual specificities in the nucleic acid recognitionand cleavage. Many pAgos are associated with putative nucleases, helicases, andDNA binding proteins in the same gene or operon, suggesting that they are in-volved in target processing. The great variability of pAgos revealed by our analysisopens new ways for exploration of their functions in host cells and for their use aspotential tools in genome editing.
IMPORTANCE The eukaryotic Ago proteins and the RNA interference pathways theyare involved in are widely used as a powerful tool in research and as potential ther-apeutics. In contrast, the properties and functions of prokaryotic Ago (pAgo) pro-teins have remained poorly understood. Understanding the diversity and functionsof pAgos holds a huge potential for discovery of new cellular pathways and noveltools for genome manipulations. Only few pAgos have been characterized by struc-tural or biochemical approaches, while previous genomic studies discovered about300 proteins in archaeal and eubacterial genomes. Since that time the number ofbacterial strains with sequenced genomes has greatly expanded, and many previ-ously sequenced genomes have been revised. We undertook comprehensive analysisof pAgo proteins in sequenced genomes and almost tripled the number of knowngenes of this family. Our research thus forms a foundation for further experimentalcharacterization of pAgo functions that will be important for understanding of thebasic biology of these proteins and their adoption as a potential tool for genomeengineering in the future.
KEYWORDS Ago, RNA interference, genome editing, horizontal gene transfer,prokaryotic Argonaute proteins
Argonaute (Ago) proteins play the key role in the RNA interference (RNAi) pathwaysin eukaryotes. All known eukaryotic Agos (eAgos) bind small RNA molecules and
use them as guides for sequence-specific recognition of long RNA targets. Uponrecognition of the target, it can be cleaved by the intrinsic endonuclease activity of theAgo protein (1–4). Alternatively, Ago proteins, especially the members of the family thatlack nuclease activity, can recruit partner proteins to the target RNA, leading to itsdegradation and/or repression of its translation (5–7). Recognition of nascent RNA by
Received 3 September 2018 Accepted 20November 2018 Published 18 December2018
Citation Ryazansky S, Kulbachinskiy A, AravinAA. 2018. The expanded universe ofprokaryotic Argonaute proteins. mBio9:e01935-18. https://doi.org/10.1128/mBio.01935-18.
Editor Eleftherios T. Papoutsakis, University ofDelaware
Copyright © 2018 Ryazansky et al. This is anopen-access article distributed under the termsof the Creative Commons Attribution 4.0International license.
Address correspondence to Sergei Ryazansky,[email protected], or Alexei A. Aravin,[email protected].
RESEARCH ARTICLEMolecular Biology and Physiology
crossm
November/December 2018 Volume 9 Issue 6 e01935-18 ® mbio.asm.org 1
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

some eAgos can also lead to modification of the chromatin structure—DNA andhistone methylation— of the target locus (8–10).
The proteins that belong to the Ago family are also present in the genomes of manybacterial and archaeal species (11, 12). Structural and biochemical studies of prokaryoticAgos (pAgos) have provided key insights into the mechanisms of RNAi in eukaryotesand revealed that Ago proteins directly bind short nucleic acid guides and can cleavecomplementary targets (13–21). The same studies showed that pAgos can associatewith short DNA guides and preferentially recognize DNA targets, in contrast to allknown eAgos (17, 20, 22–28). Despite these differences, solved structures of severalpAgos and eAgos combined with their sequence alignments revealed a conserveddomain organization of these proteins (reviewed in references 12 and 59). All eAgosand all pAgos except one that were experimentally characterized to date possess fourdomains that are organized in a bilobal structure, with N- and PAZ (PIWI-Argonaute-Zwille) domains forming one lobe and MID (Middle) and PIWI (P-element InducedWimpy Testis) domains forming another lobe (16, 18, 20, 29–34). The nucleic acids arebound between the lobes; the MID and PAZ domains interact with the 5= and 3= endsof the small nucleic acid guide, respectively, and the PIWI domain contains an RNaseH-like fold with a catalytic tetrad of conserved amino acid residues involved in thetarget cleavage (18, 19, 23–25, 27, 28). The N-domain is the least conserved in Agoproteins; it was proposed to facilitate RNA duplex unwinding during guide loading byeAgos (35) and prevent extended duplex formation during target recognition by somepAgos (23).
Until now only a few pAgos were characterized by structural or biochemicalapproaches (see Fig. 1B). At the same time, earlier genomic studies revealed that up to32% of the Archaea and 9% of the Eubacteria with sequenced genomes contain genesencoding proteins from the Ago superfamily and showed that the diversity of pAgos isfar greater than that of eAgos (11, 12). Indeed, many pAgos contain substitutions of keycatalytic residues in the PIWI domain (and are probably inactive), and a large class ofpAgos lack the N- and PAZ domains (“short pAgos”), while many pAgos containadditional domains absent in eAgos, either in the same protein or as a part of putativeoperons. Furthermore, previous studies of the genomic context of pAgo genes revealedthat catalytically inactive pAgos are usually associated with several types of nucleasesfrom the SIR2, TIR, PLD, Mrr (which include proteins with novel RecB-like and RecB2domains), or Cas4 family (11, 12). Mrr and Cas4 are related to a highly divergent familyof PD-(D/E)XK nucleases (36, 37). Furthermore, a subclass of pAgos was found inassociation with Cas1 and Cas2 nucleases within the CRISPR loci (24). The diversity ofpAgos revealed by genomic studies together with few examples of biochemicallycharacterized proteins suggested new cellular functions for these proteins (38–41).Beyond understanding functions of pAgos in their host cells, analysis of pAgos mayyield new proteins that can be potentially harnessed for biotechnology, in particular asan alternative to the CRISPR/Cas genome editing tools (42).
Previous genomic studies identified a nonredundant set of 261 pAgos (11, 12).However, since their discovery the number of bacterial strains with sequenced ge-nomes has more than tripled, and many previously sequenced genomes have beenrevised, resulting in the removal of some pAgo genes from the databases. Therefore, wehave undertaken comprehensive exploration of the diversity of pAgos using the latestversion of the RefSeq protein database for bioinformatic search. The results of thesearch almost tripled the number of identified pAgo genes. Our analysis supports thepreviously reported separation of pAgos into short pAgos and long pAgos (which bythemselves can be separated into two distinct clades) and reveals several subclasseswith significant differences in the domain organization. These include variations in theMID and PAZ domains involved in guide binding and substitutions of catalytic residuesin the PIWI domain. A large fraction of pAgos, including those that lack intrinsicendonuclease activity, are associated with putative nucleases and other DNA bindingproteins. Overall, the great diversity of pAgos revealed by our analysis opens a way for
Ryazansky et al. ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 2
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

exploration of their biochemical properties and cellular functions, as well as their use aspotential tools in genome engineering.
RESULTSThe expanded set of prokaryotic Argonaute proteins. To reveal the diversity of
pAgos and to get new insights into their evolution, we performed comprehensiveanalysis of proteins that belong to the Ago family in sequenced genomes of prokaryoticspecies. For this, we searched 116 million proteins fetched from the RefSeq NCBIprotein database using known pAgos as queries by PSI-BLAST. We found 1,010 pAgosencoded in 1,385 completely and partially sequenced genomes of 1,248 eubacterialand archaeal strains (Fig. 1; see also Table S1 in the supplemental material). In total,pAgos were found in 17% of eubacterial and 25% of archaeal genera (Fig. 1A). Themajority (1,186, or 95%) of archaeal and eubacterial strains carry only one pAgo gene(876 pAgos, or 86.7% of all proteins). However, the genomes of 57 strains carry twopAgo genes, four strains carry three pAgo genes, and one strain encodes four pAgos.pAgos are in general randomly distributed in different prokaryotic clades as the number
1/71/71/21/61/51/32/51/3
2/252/111/61/72/22/112/44/103/82/45/82/44/233/154/874/129/18
2/714/25
11/1024/6023/20119/14721/246
27/4617/55
42/9333/156
43/8760/343
88/325
ChlorobiaDeferribacteresRhodothermia
SaprospiriaThermoleophilia
ZetaproteobacteriaBalneolia
DehalococcoidiaMethanomicrobia
NitrososphaeriaNitrospiraOpitutae
RubrobacteriaAquificae
ArchaeoglobiAnaerolineae
VerrucomicrobiaeMethanococciAcidobacteriia
ThermococciThermoproteiSpirochaetia
DeltaproteobacteriaThermotogaeChitinophagia
DeinococciPlanctomycetia
SphingobacteriiaCytophagia
ClostridiaBacilli
ActinobacteriaHalobacteria
BacteroidiaFlavobacteriia
BetaproteobacteriaCyanobacteria
GammaproteobacteriaAlphaproteobacteria
long pAgo short pAgo
Found pAgo proteins
Cla
sses
0 100 200
20
FIG 1 Phylogenetic analysis of pAgo proteins. (A) The numbers of identified short and long pAgo proteins in the classes of Eubacteria and Archaea. Pinkasterisks near the clade names indicate the archaeal classes. The numbers on the right of each bar depict the number of genera encoding pAgos versus thetotal number of genera with the complete or partially sequenced genomes in the corresponding class. (B) The circular phylogenetic tree of the nonredundantset of pAgos constructed based on the multiple alignment of the MID-PIWI domains. The pAgo proteins were annotated as follows, from the inner to the outercircles: the type of protein, in which long-A pAgos are colored in green (truncated variants without the PAZ domain, light green), long-B pAgos are light green(truncated variants without PAZ, green), and short pAgos are orange; the type of the PIWI domain, depending on the presence of the catalytic tetrad DEDX;the type of the 5=-end guide binding motif in the MID domain (the first two conserved residues are indicated); the presence and the type of the PAZ/APAZdomains in pAgos; and the superkingdom to which the corresponding pAgo belongs. Biochemically characterized pAgos (or the most similar redundanthomologs for RsAgo and TtAgo) with resolved structures are highlighted in red. The scale bar represents the evolutionary rate calculated under the JTT�CATevolutionary model.
The Diverse World of Prokaryotic Argonautes ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 3
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

of genera in each class that encode pAgos in their genomes correlates with the totalnumber of genera in the class (Fig. 1A; Fig. S1). The nonredundant set of pAgos withthe level of similarity of less than 90% comprises 721 proteins, which is almost triple thenumber of previously identified pAgo proteins (11, 12).
As described below, the vast majority of pAgos have the MID and PIWI domains, butmany proteins lack the N- and PAZ domains conserved in eAgos. Therefore, we usedmultiple alignments of the MID-PIWI domains to construct a phylogenetic tree of pAgos(Fig. 1B; Fig. S2). The tree revealed three large clades of pAgos, while two proteins fromthermophilic archaea Thermoproteus uzoniensis and Vulcanisaeta moutnovskia could notbe unambiguously classified as belonging to any of these clades due to their highdivergence. Following Makarova and coauthors (11), we call one of these clades “shortpAgos” (Fig. 1B), as all proteins in this clade lack the N- and PAZ domains present ineAgos. The majority of pAgos that belong to the other two clades include the PAZdomain, and we therefore designate them as long-A and long-B. The diversity of pAgosvastly exceeds the diversity of eAgos that were previously shown to form a singlebranch on the phylogenetic tree of long pAgos (as illustrated in Fig. 1B) (11, 12).
The three clades of pAgos can be found in both archaeal and eubacterial species.Furthermore, the majority of prokaryotic classes that include large numbers of generaencode both short and long pAgos. However, some classes have a clear bias towardshort or long pAgo types; thus, genomes of Alphaproteobacteria predominantly encodeshort pAgos, while Cyanobacteria predominantly encode long pAgos (Fig. 1A). pAgosthat belong to different clades can be found in the same genome (for the strains thatencode several proteins). For example, Enhydrobacter aerosaccus ATCC 27094 encodesthree short and one long-A pAgo, Parvularcula bermudensis HTCC2503 and Halomi-cronema hongdechloris C2206 encode two short and one long-B pAgos, and Methylo-microbium agile ATCC 35068 contains one short and one long-B pAgo. Generally, thephylogenetic tree of pAgos has little similarity with the phylogenetic tree of hostspecies built using classic molecular markers such as rRNA genes, thus confirmingpreviously published observations (11, 12) and suggesting that pAgos have mostlyspread through horizontal gene transfer (HGT). Notably, a few pAgos that have beenbiochemically studied almost all belong to the long-A clade, while only two pAgos fromthe long-B clade and no proteins from the short pAgo clade have been characterizedto date (indicated in Fig. 1B).
Domain architecture of pAgos. To gain further insight into the structural diversityof pAgo, we analyzed their domain architecture. The exploration of multiple alignmentsof PIWI, MID, and PAZ domains was used to identify the main structural and functionalfeatures of pAgos. We did not include the N-domain in our analysis because it has thelowest level of conservation, which impedes its identification in diverse pAgos. UsingInterProScan and CDD-batch programs and the Pfam and Superfamily databases, wealso searched for other domains in pAgo proteins. This analysis allowed us to proposea comprehensive classification of the pAgo domain organization (Fig. 2).
As justified by their name, we found that all members of the short pAgo clade lackthe PAZ domain implicated in binding of the 3= end of a guide molecule. The majorityof short pAgos (470, or 81%) have only two domains, MID and PIWI. Six short pAgos(�1%) are further truncated and lack the MID domain, suggesting that they may beunable to interact with a guide. In addition to the MID and PIWI domains, 106 shortpAgos (18%) that all belong to a single monophyletic branch encompass the APAZ(“analog of PAZ”) domain (Fig. 1B and 2A). The APAZ domain has no sequence similarityto PAZ and was previously described as a domain always associated with short pAgoseither as a part of the same protein or present in the same operon with short pAgos(11). Almost all (102 out of 106) APAZ-containing short pAgos also carry domainsrelated to the SIR2 family (SIR2_1 subfamily) (Fig. 1B and 2A), which are considered tobe bona fide nucleases (11, 12).
In contrast to short pAgos, the majority (378, or 90%) of pAgos that belong to thelong pAgo clades have the domain architecture similar to eAgos, including PAZ, MID,
Ryazansky et al. ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 4
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

and PIWI domains (the N-domain was not included in the analysis). However, 31 pAgosthat undoubtedly belong to long pAgos based on alignments of their MID-PIWIdomains lack the PAZ domain, including previously characterized AfAgo from Archaeo-globus fulgidus (13–15). These truncated long pAgos are scattered at different positionson the phylogenetic tree, suggesting multiple independent cases of the loss of thisdomain during evolution of long pAgos. One of these truncated proteins also lacks theMID domain. On the other hand, 15 long pAgos that have the PAZ domain also carryadditional putative nucleic acid binding domains at their N termini: 7 pAgos containDNA-binding domains (Schlafen, PF04326, or lambda-repressor-like, SSF47413) and8 pAgos contain a nucleic acid binding domain with the OB-fold (SSF50249) (Fig. 1Band 2B).
Analysis of the diversity of MID, PIWI, and PAZ domains revealed significant varia-tions in their structures characteristic for different groups of pAgos (Fig. 1B and 2), asdescribed below. In particular, subsets of proteins from both clades of long pAgoscontain MID* domains with substitutions of key residues involved in interactions withthe guide 5= end; many long-A pAgos and all long-B and short pAgos contain PIWI*variants with substitutions of essential catalytic residues; and most long-B pAgoscontain incomplete variants of the PAZ* domain involved in the 3=-guide interactions(Fig. 2).
Binding of nucleic acid guides in the MID domain. The vast majority (1,001, or99% of all proteins) of pAgos contain the MID domain that has been implicated inbinding of the 5= end of guide molecules. The resolved 3D structures of pAgos,including AfAgo (pAgo of A. fulgidus) (13–15, 22), RsAgo (Rhodobacter sphaeroides) (25,28), TtAgo (Thermus thermophilus) (17, 19, 23), and MjAgo (Methanocaldococcus jann-aschii) (27), have shown that the MID domain anchors the phosphorylated 5=-guidenucleotide by a set of conserved amino acid residues (Fig. 3). These interactions arecomplemented by the PIWI domain that provides additional residues for 5=-guide anddivalent cation binding (13, 15, 19, 23, 25, 27, 28).
247
218
102
5
3
1
(HK)-MID
(HK)-MID
FIG 2 The diversity of domain architecture of pAgo proteins. Various types of domain organization of short (A) and long (B) pAgo proteins are schematicallyshown. The numbers on the right of each type are the frequency of its occurrence in the redundant set of proteins. Based on the presence of the DEDX tetradin the PIWI domain and the putative 5=-P guide binding motif in the MID domain, all long pAgos can be separated into four types shown here. Green PIWIdomains carry the active DEDX catalytic tetrad while turquoise PIWI* domains do not contain the canonical tetrad. Yellow MID domains contain different typesof the 5=-end guide binding motif as indicated, while light-yellow MID* domains carry substitutions of the crucial amino acid residues in this motif. Orange PAZdomains have the full-sized pocket responsible for 3=-end guide binding, while light-brown PAZ* variants do not have the second subdomain. The OB-fold isthe nucleic acid-binding domain SSF50249; the DNA-binding domains are Schlafen domain with AlbA_2 (PF04326) or lambda-repressor-like domain (SSF47413).
The Diverse World of Prokaryotic Argonautes ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 5
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from
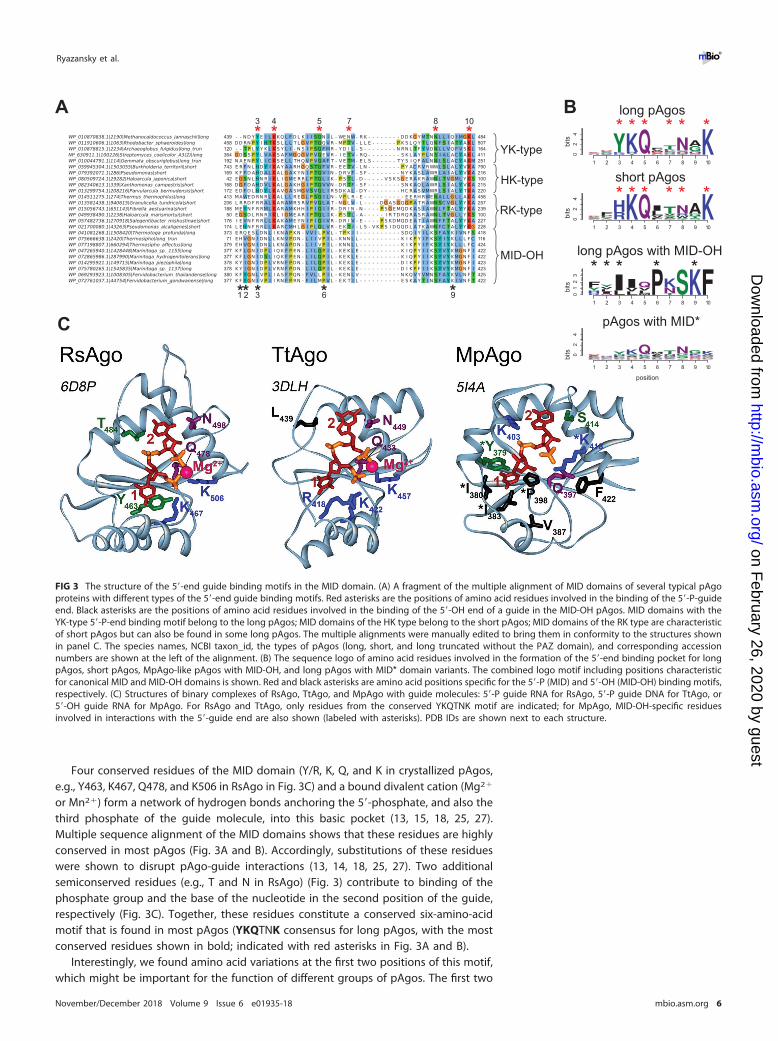
Four conserved residues of the MID domain (Y/R, K, Q, and K in crystallized pAgos,e.g., Y463, K467, Q478, and K506 in RsAgo in Fig. 3C) and a bound divalent cation (Mg2�
or Mn2�) form a network of hydrogen bonds anchoring the 5=-phosphate, and also thethird phosphate of the guide molecule, into this basic pocket (13, 15, 18, 25, 27).Multiple sequence alignment of the MID domains shows that these residues are highlyconserved in most pAgos (Fig. 3A and B). Accordingly, substitutions of these residueswere shown to disrupt pAgo-guide interactions (13, 14, 18, 25, 27). Two additionalsemiconserved residues (e.g., T and N in RsAgo) (Fig. 3) contribute to binding of thephosphate group and the base of the nucleotide in the second position of the guide,respectively (Fig. 3C). Together, these residues constitute a conserved six-amino-acidmotif that is found in most pAgos (YKQTNK consensus for long pAgos, with the mostconserved residues shown in bold; indicated with red asterisks in Fig. 3A and B).
Interestingly, we found amino acid variations at the first two positions of this motif,which might be important for the function of different groups of pAgos. The first two
* * * * * *
** * * *
439458120364192743169
42168172413206188
50176174373
71379377377378378380377
484507164411251790216100215220458257239100227228418116424422422423423425422
1 2 3 4 5 6 7 8 9 10
02
4
position
bits
bits
bits
bits
1 2 3 4 5 6 7 8 9 10
02
4
1 2 3 4 5 6 7 8 9 10
01
23
1 2 3 4 5 6 7 8 9 10
02
4
long pAgos
short pAgos
long pAgos with MID-OH
pAgos with MID*
BA* * * * **
* * * * **
* * * **
C
YK-type
HK-type
MID-OH
RK-type
1 2 3
4 5
6
7 8
9
3 10
FIG 3 The structure of the 5=-end guide binding motifs in the MID domain. (A) A fragment of the multiple alignment of MID domains of several typical pAgoproteins with different types of the 5=-end guide binding motifs. Red asterisks are the positions of amino acid residues involved in the binding of the 5=-P-guideend. Black asterisks are the positions of amino acid residues involved in the binding of the 5=-OH end of a guide in the MID-OH pAgos. MID domains with theYK-type 5=-P-end binding motif belong to the long pAgos; MID domains of the HK type belong to the short pAgos; MID domains of the RK type are characteristicof short pAgos but can also be found in some long pAgos. The multiple alignments were manually edited to bring them in conformity to the structures shownin panel C. The species names, NCBI taxon_id, the types of pAgos (long, short, and long truncated without the PAZ domain), and corresponding accessionnumbers are shown at the left of the alignment. (B) The sequence logo of amino acid residues involved in the formation of the 5=-end binding pocket for longpAgos, short pAgos, MpAgo-like pAgos with MID-OH, and long pAgos with MID* domain variants. The combined logo motif including positions characteristicfor canonical MID and MID-OH domains is shown. Red and black asterisks are amino acid positions specific for the 5=-P (MID) and 5=-OH (MID-OH) binding motifs,respectively. (C) Structures of binary complexes of RsAgo, TtAgo, and MpAgo with guide molecules: 5=-P guide RNA for RsAgo, 5=-P guide DNA for TtAgo, or5=-OH guide RNA for MpAgo. For RsAgo and TtAgo, only residues from the conserved YKQTNK motif are indicated; for MpAgo, MID-OH-specific residuesinvolved in interactions with the 5=-guide end are also shown (labeled with asterisks). PDB IDs are shown next to each structure.
Ryazansky et al. ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 6
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

residues in the motif are usually YK in long pAgos; subsets of long-A pAgos contain KR,RK, HK, or YY combinations, while long-B pAgos are almost exclusively YK variants. Thearomatic ring of the first Y stacks on the first base at the 5= end of a guide, therebycontributing to the binding of the first nucleotide within the MID pocket (13, 15, 25, 27,28). In contrast to tyrosine, R is positively charged and not aromatic and so unable tostack the first base of the guide, as exemplified in the structure of TtAgo (Fig. 3C) (18,23); the structure of any pAgo with histidine at the first position of the motif remainsunknown.
The conserved residues implicated in binding of the 5=-phosphorylated guide endare present in the majority (80%) of long pAgos (Fig. 1B). However, 84 (20%) of longpAgos have at least one amino acid substitution at the most conserved positions in the5= binding motif in their MID* domain; namely, they do not contain Y/H/R at the firstposition of the YKQTNK motif and/or K/R/Y at the second position, and/or Q at the thirdposition, and/or K at the last position. A separate subgroup of these proteins (MID-OH),which form a tight clade on the phylogenetic tree (Fig. 1B), includes pAgo fromMarinitoga piezophila (MpAgo) that was recently shown to bind guide molecules with5=-OH instead of 5=-P ends (24). The multiple sequence alignment showed that thisgroup includes 9 pAgos (compared to 3 proteins identified in the previous study [24])and that positions of amino acid residues involved in 5=-guide interactions in theseproteins overlap the positions of the 5=-P-end binding motif in other pAgos (Fig. 3A andB). However, in comparison with most pAgos, these residues form a more hydrophobicpocket for 5=-OH binding (Fig. 3C). In addition, the MID pocket of MpAgo does not bindMg2� ions (Fig. 3C) (24). The absence of guide contacts with the Mg2� ion is in partcompensated by interactions between the phosphates of second and third nucleotidesof guide RNA with lysine residues specific for this group of pAgos (K403 and K418 inMpAgo; Fig. 3B and C). While the first tyrosine of the YKQTNK motif that stacks withthe first nucleotide base in the guide molecule in other pAgos is replaced with ahydrophobic residue in the MID-OH domain, its role is taken by a precedingaromatic residue (Y or F) that stacks from the other side of the same base (Y379 inMpAgo; Fig. 3B and C).
Another 75 pAgos with noncanonical MID* variants, none of which is characterizedto date, belong to several distinct branches on the phylogenetic tree of pAgos, andsome of them are located close to the MID-OH pAgos (Fig. 1B). The alignment of thesesequences showed that in general they share the same YKQTNK motif, although theconservation of individual positions is much lower than for canonical pAgos (see logoin Fig. 3B; alignment in Data Set S1). It remains to be established whether these proteinsinclude any additional subgroups with noncanonical MID* domains of different spec-ificities, as already revealed for MpAgo, or whether they just represent divergentvariants of the same canonical motif.
In contrast to long pAgos, all short pAgos have the canonical 5=-P-end binding motif(Fig. 3B). Interestingly, almost all of them contain HK and RK residues at the first twopositions of this motif, instead of YK in long pAgos, further supporting their separationinto a distinct clade from long pAgos (Fig. 1, 2B, and 3A and B). Though none of shortpAgos was biochemically characterized to date, the conservation of key residues in theMID domain strongly suggests that these proteins bind 5=-phosphorylated guidenucleic acids similarly to most long pAgos and eAgos.
Endonuclease activity of the PIWI domain. Structural and biochemical studies ofseveral pAgos and eAgos demonstrated that the conserved tetrad of amino acidresidues in the PIWI domain (DEDX, where X is D, H, or K) is responsible for endonu-cleolytic cleavage of the target upon its recognition by the Ago-guide complex (18–20,24, 40). Similarly to previous reports (11, 12), we found that all pAgos that belong to theshort clade lack a canonical DEDX catalytic tetrad in the PIWI domain (PIWI* variants)(Fig. 2A). Similarly to short pAgos, all proteins in the long-B clade also lack the catalytictetrad. In contrast, the majority of long-A pAgos (79%) have the canonical DEDXcatalytic tetrad in the PIWI domain, suggesting that they possess endonucleolytic
The Diverse World of Prokaryotic Argonautes ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 7
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

activity. However, this branch also harbors pAgos with substitutions in the catalytictetrad, suggesting several independent events of the loss of endonuclease activity inthis branch.
Binding of the guide 3= end in the PAZ domain. It was previously shown that theamino acid sequences of the PAZ domains of the pAgo proteins are divergent, but theirstructural folds are similar. The full-length PAZ domain has two subdomains, eachconsisting of two nucleic acid binding regions, which are oriented to form a hydro-phobic pocket that anchors the 3= end of the guide strand in the binary pAgo-guidecomplex (16, 18, 20, 28); a typical structure of PAZ is illustrated for TtAgo in Fig. 4A. Thefirst subdomain consists of an oligosaccharide binding fold (OB-fold)-like structure withone or two helices on one side and includes nucleic acid binding regions R1 and R4. Thesecond subdomain consists of an �-helix and a �-hairpin or loop structure, sometimesfollowed by another �-helix, and includes nucleic acid binding regions R2 and R3(Fig. 4A) (16, 18, 20). In TtAgo, all four regions contribute to anchoring of the guide 3=end in the PAZ pocket (Fig. 4A) (18). However, some pAgos, such as RsAgo and MpAgo,contain incomplete variants of the PAZ domain that lack the second subdomain,including regions R2 and R3, and therefore do not possess the PAZ pocket (24, 25, 28).Nevertheless, structural analysis of the binary guide-MpAgo complex revealed that theguide 3= end can still be bound in the incomplete PAZ domain, although with adifferent orientation relative to TtAgo (24). These differences may possibly affect thekinetics of guide binding, the stability of binary guide pAgo complexes, or their abilityto recognize complementary nucleic acid targets (see Discussion).
We determined how often the PAZ domain without the second subdomain (referredto here as PAZ*) is present among long pAgos. As noted above, the structural elementsof PAZ involved in nucleic acid binding are organized by the four poorly conservedregions, R1 to R4 (Fig. S3, as described in reference 28), with the DNA-binding �-helixin the second subdomain formed by stretches of 7 to 15 amino acids from region R3(illustrated for TtAgo and Pyrococcus furiosus PfAgo in Fig. 4). Thus, we analyzed thepresence of this region as a signature of the second subdomain of PAZ in pAgos. Usingmultiple sequence alignments of the identified PAZ domains, we found that R3 is found
FIG 4 PAZ domains in pAgo proteins. (A) The structure of the 3=-guide binding pocket in TtAgo (PDBID: 3DLH). Structural regions R1 to R4 forming the pocket are shown in surface representation. (B) Ageneral scheme of the PAZ and PAZ* domain structures in long pAgos. All PAZ domains can have R1, R2,R3, and R4 regions. PAZ* is defined as variants of PAZ lacking full-length region R3. No regions R1, R2,and/or R4 could be detected in multiple alignments of some PAZ or PAZ* variants (shown by brokenboxes), but their actual presence needed to be tested due to their low conservation. The numbers showhow many long pAgos have corresponding domain structures. The pAgos with resolved structureshaving PAZ and PAZ* domains are indicated. (C) The three-dimensional structures of PAZ (TtAgo andPfAgo) and PAZ* (MpAgo and RsAgo) variants in crystallized pAgos and their binary complexes with DNA(for TtAgo) or RNA (for MpAgo) guide molecules. Tyrosine and lysine/arginine residues from R3 in TtAgoand PfAgo, probably involved in guide binding, are indicated. PDB IDs are shown next to each structure.
Ryazansky et al. ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 8
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

in more than half of the PAZ domains in long pAgos, including most long-A pAgos(Fig. 1B and 4A; alignment in Data Set S2), indicating their canonical two-lobe structure.The rest of PAZ* domains are smaller and do not have R3, suggesting that they havethe one-subdomain organization without the nucleic acid binding pocket, as previouslyobserved for RsAgo and MpAgo. Interestingly, the PAZ* domain is found in almost alllong-B pAgos (Fig. 1B), suggesting that it was present in the common ancestor of thisgroup.
Phylogenetic analysis of APAZ domain-containing proteins. Previous analysesidentified the so-called APAZ domain that is present in some short pAgos as well as ingenes contained in putative operons containing short pAgos (11). The APAZ domainwas proposed to be the functional analog (but not homolog) of the PAZ domain (11)or the N-domain (43) that is absent in short pAgos. To gain insight into the diversity andphylogeny of APAZ-containing proteins, we iteratively searched the RefSeq proteindatabase by DELTA-BLAST using the sequences of known APAZ domains as queries. Intotal, we found 5,385 protein hits. However, the vast majority (4,753, or 88%) of thefound proteins belonged to the HisG (PRK01686) and EIIB families (TIGR01996). Wetherefore performed the reverse DELTA-BLAST search using them as queries to test ifthese protein families were indeed related to the other APAZ proteins; however, we didnot find any APAZ-containing proteins within obtained hits. Moreover, the hisG pro-teins were found only among BLAST hits of the SIR2-APAZ protein from Sphingomonaswittichii RW1, while EIIB proteins were found only among BLAST hits of the TIR-APAZprotein from Chlorobium phaeobacteroides BS1. Thus, we concluded that HisG and EIIBfamilies were revealed artificially and are not related to the APAZ proteins, and weexcluded them all from the further phylogenetic analysis. The remaining set of APAZdomains included 632 proteins.
The multiple alignments of APAZ domains were used to build their phylogenetictree, which revealed that the APAZ proteins can be separated into four large groupsdesignated Ia, Ib, IIa, and IIb and a fifth group III of 18 proteins from several archaealspecies that have remote similarity to groups I and II (Fig. 5; alignment in Data Set S3).All five groups are present in putative operons with pAgos. We further analyzed thedomain architectures of APAZ-containing proteins using InterProScan and the Pfamand Superfamily databases. The first group of APAZ proteins designated Ia includes 117proteins that in addition to APAZ contain the SIR2 domain (the “SIR2-APAZ” type). Themajority of proteins in this group are short pAgos that also contain the MID-PIWIdomains; however, 31 proteins from this group lack the MID-PIWI domains. Since thesevariants are scattered along different branches in this group, they might have inde-pendently lost their MID-PIWI parts. In contrast, while a significant part of APAZproteins from the four other groups are associated with pAgos, only one of them ispAgo itself. Most proteins that belong to group Ib (150) are related to group Ia and alsocontain SIR2 besides APAZ, but do not have MID and PIWI. There are also severalproteins in both groups Ia and Ib that lack SIR2 or any other domains (the “APAZ” type;4 and 6 proteins for Ia and Ib, correspondingly). Most proteins of group IIa (122) arecharacterized by the presence of the TIR domain on their N termini (“the TIR-APAZ”type); one of them is short pAgo (also Fig. 2); 16 proteins in this group lack TIR domains.Group IIb, which encompasses 225 proteins, is diverse: 112 proteins contain unchar-acterized domain DUF4365 (“DUF4365-APAZ”), and 13 have the SIR2 domain, while 100do not have any extra domains (“APAZ”). Our analysis of the DUF4365 domain hasshown that it corresponds to the recently identified RecB-like domain of the Mrrsubfamily of PD-(D/E)XK nucleases (see below) (11). Finally, the proteins from group III,similarly to a major part of proteins from group IIb, do not have any other domainsexcept APAZ (the “APAZ” type).
APAZ-containing proteins are found almost exclusively in eubacterial species. Theanalysis revealed tight reciprocal association between APAZ-containing proteins andshort pAgos. Indeed, although only one branch of short pAgos contains the APAZdomain, the majority of other short pAgos that do not have APAZ themselves contain
The Diverse World of Prokaryotic Argonautes ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 9
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

APAZ-containing genes in their operons (Fig. 1B and 2). Reciprocally, 397, or 63%, of allgenes with the APAZ domain are positioned close (within 10 genes on the samegenomic strand) to short pAgos, likely in the same operons (Fig. 5) (see below). Thus,APAZ proteins are likely an integral component of functional pathways mediated byshort pAgos.
WP
_082
8220
67.1
|439
92|B
rady
rhiz
obiu
m li
aoni
ngen
se
WP_093992232.1|1819565|Loktanella m
arina
WP
_081
8527
74.1
|112
5973
|Bos
ea s
p. 1
17
WP_091963848.1|1761772|Bradyrhizobium sp. err11
WP
_093
0391
41.1
|128
7727
|Ros
eova
rius
azor
ensi
s
WP
_090
0726
29.1
|655
353|
Coh
aesi
bact
er m
aris
flavi
WP_062680196.1|222|Achrom
obacter
WP
_014
1327
57.1
|108
2930
|Pel
agib
acte
rium
WP_
0975
4471
4.1|
1184
720|
Rhi
zobi
um a
nhui
ense
WP_018016394.1|2426|Teredinibacter turnerae
WP_081730962.1|68287|Mesorhizobium
WP
_097
0827
96.1
|570
013|
Rho
doba
cter
sp.
JA
431
WP_0
2919
3119
.1|5
9839
|Pae
niba
cillu
s al
gino
lyticu
s
WP_053571900.1|1353886|Caballeronia cordobensis
WP_0
8729
2483
.1|1
9655
88|A
naer
ofilu
m s
p. A
n201
WP_007215639.1|246787|B
acteroides c
ellulosil
yticu
s
WP_087738802.1|1701573|Paraburkholderia piptadeniae
WP
_006
7462
22.1
|108
010|
Thio
alka
livib
rio p
arad
oxus
WP
_056186121.1|1736259|Methylobacterium
sp. Leaf113
WP_038943875.1|375|Bradyrhizobium
japonicum
WP_
0788
1466
9.1|
4846
7|Pr
osth
ecob
acte
r deb
ontii
WP_039858603.1|205844|Novosphingobium pentaromativorans
WP_093081846.1|1884369|Sphingobium sp. AP50
WP_
0756
3405
0.1|
1672
749|
Rhi
zobi
um rh
izos
phae
rae
WP_
1002
3990
5.1|
1983
755|
Sphi
ngob
ium
sp.
LB1
26
WP
_051044811.1|91459|Methylobacterium
sp. B1
WP_036051529.1|999627|Sedimentitalea nanhaiensis WP_089318528.1|1610492|Pontibacter ummariensis
WP
_078056299.1|1234595|Pacificim
onas flava
WP_006685036.1|133448|Citrobacter youngae
WP_038205789.1|151755|Xenophilus azovorans
WP_008781127.1|469592|Bacteroides sp. 3_1_19
WP
_067969880.1|83263|Am
inobacter aminovorans
WP
_094291737.1|2004485|Acidovorax sp. K
ND
SW
-TSA
6
NP
_951611.1|243231|Geobacter sulfurreducens
WP_061958301.1|82633|Cupriavidus pauculus
WP_018273545.1|29448|Bradyrhizobium elkanii
WP
_050
0385
98.1
|223
7|H
aloa
rcul
a
WP_0
7309
9040
.1|98
8|Lee
uwen
hoek
iella
marino
flava
WP_096483475.1|223967|Methylobacterium populi
WP_096462202.1|1675686|Sulfurifustis variabilis
WP
_082
7381
20.1
|136
87|S
phin
gom
onas
WP_
0917
8116
4.1|
1761
778|
Burk
hold
eria
sp.
yr2
81
WP_065998852.1|1886027|Mucilaginibacter sp. PPCGB 2223
WP_063002581.1|508464|Nocardia mikamii
WP_075285887.1|339|Xanthomonas campestris
WP
_082731713.1|13687|Sphingom
onas
WP_037187103.1|1500259|Rhizobium
sp. YR519
WP
_087
2528
36.1
|196
5647
|Dra
ncou
rtel
la s
p. A
n57
WP
_071
1456
52.1
|185
2362
|Bac
tero
ides
ihua
e
WP_
0676
9636
7.1|
4996
56|E
ryth
roba
cter
sp.
AP2
3
WP_079605750.1|1437360|Bradyrhizobium erythrophlei
WP_
0507
8387
7.1|
5164
66|B
urkh
olde
ria s
p. H
160
WP
_056152149.1|1736266|Duganella sp. Leaf126
WP_003807800.1|518|Bordetella bronchiseptica
WP
_013632722.1|151895|Pseudopedobacter saltans
WP_092021235.1|488535|Marinobacter zhejiangensis
WP_037435767.1|380|Sinorhizobium
fredii
WP_037716570.1|68239|Streptomyce
s mira
bilis
WP_013299755.1|208216|Parvularcula bermudensis
WP_062419304.1|229921|Levilinea sa
ccharolyt
ica
WP_035838021.1|765699|Defluviim
onas indica
WP
_059
6229
37.1
|605
52|B
urkh
olde
ria v
ietn
amie
nsis
siral
ucis
evsa
nomi
dnuv
erB|
6721
4|1.
1525
7348
0_P
W
WP
_026932797.1|279359|Gram
ella echinicola
WP
_092684840.1|1513892|Rhodopseudom
onas pseudopalustris
WP
_098
0634
74.1
|146
9170
|Lon
gim
onas
hal
ophi
la
WP_028849834.1|37926|Thermocrispum municipale
WP_052117900.1|205844|Novosphingobium pentaromativorans
WP_062785679.1|13688|Novosphingobium
capsulatum
WP_050399989.1|630921|Bradyrhizobium embrapense
WP
_052
3032
32.1
|605
48|P
arab
urkh
olde
ria g
ram
inis
WP_0
5603
4460
.1|17
3636
6|Chr
yseo
bacte
rium sp
. Lea
f404
WP_004271609.1|28077|Nitrospirillum amazonense
WP
_052114702.1|172045|Elizabethkingia m
iricola
WP_092996169.1|1566262|Rhizobium sp. NFR07
WP
_081215336.1|13690|Sphingobium
yanoikuyae
WP_029005631.1|281091|Azorhizobium doebereinerae
WP
_089
6991
20.1
|660
521|
Hal
ogra
num
gel
atin
ilytic
um
WP_050771919.1|404236|Maritimibacter alkaliphilus
WP
_059401471.1|1603291|Alicycliphilus sp. B
1
WP_0
2815
3625
.1|3
75|B
rady
rhizo
bium
japo
nicu
m
WP_026042451.1|1225184|Pantoea sp. A4
WP_056209640.1|1736485|Flavobacterium sp. Root186
WP_100788469.1|817|Bacteroides fragilis
WP_0
4756
4307
.1|1
5744
09|M
esor
hizo
bium
sp.
F7
WP
_082442752.1|311410|Labrenzia alba
WP
_083
7167
55.1
|125
0539
|Pel
agib
aca
abys
si
WP
_090
2706
87.1
|657
014|
Lito
rimic
robi
um ta
eane
nse
WP_064816696.1|1703960|Rhizobium sp. N113
WP_093164034.1|1882828|Variovorax sp. YR216
00B082
CJSL.ps
muibozihroseM|6337821|1.660086320_
PW
WP
_073035593.1|337701|Roseovarius pacificus
WP_082576376.1|1736568|Lysobacter sp. R
oot604
WP_054211403.1|1526658|Bosea vaviloviae
WP_089334214.1|1411120|Hymenobacter mucosus
WP_016393047.1|70775|Pseudomonas plecoglossicida
WP_
1000
8505
1.1|
1524
80|B
urkh
olde
ria a
mbi
faria
WP_068222132.1|333140|Roseivirga spongicola
WP
_044
0159
94.1
|153
9298
|Tre
pone
ma
sp. O
MZ
838
WP_092637302.1|1884352|Rhizobiales bacterium GAS113
WP_056160041.1|1839|Nocardioides
WP
_033317603.1|279369|Sphingopyxis baekryungensis
WP_045057112.1|1827278|Aliterella atlantica
WP_027412346.1|279356|Aquimarin
a muelle
ri
WP
_008140723.1|387090|Bacteroides coprophilus
WP_008615787.1|453852|Joostella marina
WP
_081
6247
27.1
|330
35|B
laut
ia p
rodu
cta
WP_093590751.1|235987|Streptomyces jietaisiensis
WP
_085937812.1|225324|Enhydrobacter aerosaccus
WP_036017302.1|102129|Leptolyn
gbya sp
. PCC 7375
WP_081903350.1|58117|Microbispora rosea
WP_068185907.1|1857568|M
acellib
acteroides s
p. HH-Z
S
WP_072017557.1|371042|Erwinia typographi
WP_0
0256
6035
.1|2
0847
9|[C
lost
ridiu
m] b
olte
ae
WP_0
8209
6051
.1|1
6035
55|C
andid
atus
Nitr
osot
enuis
cloa
cae
WP
_074129929.1|1680160|Bradyrhizobium
sp. NA
S96.2
WP
_026782276.1|257440|Pleom
orphomonas koreensis
NP_952413.1|243231|Geobacter sulfurreducens
WP_092119516.1|1855396|Porphyromonadaceae bacterium KH3CP3RA
WP_082722381.1|1385591|Burkholderia sp. BDU6
WP_031452536.1|1232437|Desu
lfobacu
la sp. T
S
WP_083808461.1|940615|Granulicella tundricola
esne
parb
memu
iboz
ihry
dar
B|12
9036
|1.1
8919
9380
_P
W
WP_0
2716
0975
.1|1
0409
84|M
esor
hizob
ium sp
. WSM
1293
WP_024810211.1|1305735|Oceanicola sp. HL-35
WP_009507644.1|794903|Opitutaceae bacterium TAV5
WP
_011
0210
17.1
|221
4|M
etha
nosa
rcin
a ac
etiv
oran
s
WP_036212047.1|1415754|Marinobacter sp. MCTG268
WP_044447509.1|589873|Alteromonas australica
WP
_025
4931
97.1
|123
2455
|Lac
hnos
pira
ceae
bac
teriu
m V
E20
2-12
WP_042565353.1|1587524|Flavo
bacteriu
m sp. M
EB061
WP_097031611.1|439529|Rhodobacter ovatus
WP_010424308.1|177400|Anaerophaga thermohalophila
WP_077684787.1|1332264|Tessaracoccus sp. NSG39
WP_084600957.1|80878|Acidovorax tem
perans
WP_080811272.1|1209493|Halom
icronema hongdechloris
WP_082469977.1|1736242|M
ethylobacte
rium sp
. Leaf86
WP_091133619.1|549386|Microvirga guangxiensis
WP_077230992.1|29448|Bradyrhizobium
elkanii
WP_077293451.1|187304|Labrenzia aggregata
WP_020809957.1|357|Agrobacterium
WP_071240727.1|1882733|Thalassospira sp. MIT1004
WP_083942961.1|266127|Sphingom
onas soli
WP_080801769.1|1183401|Agrobacterium genomosp. 1
WP_101603479.1|817|Bacteroides fragilis
WP_068818385.1|268141|Phormidesmis priestleyi
WP_051821414.1|1463855|Streptomyces sp. N
RRL F-5065
WP_0
8547
4560
.1|5
6106
1|Sph
ingob
acte
rium
psy
chro
aqua
ticum
WP
_023727136.1|1287285|Mesorhizobium
sp. LSH
C412B
00
WP
_052
0271
61.1
|118
7851
|Rho
dovu
lum
sp.
PH
10
WP_050631536.1|1654716|Bradyrhizobium viridifuturi
WP_032541222.1|816|Bacteroides
WP_011831497.1|105560|Methylibium petroleiphilum
WP_064102550.1|1805633|Acinetobacter sp. SFA
WP
_041864518.1|40324|Stenotrophom
onas maltophilia
WP_012486312.1|155077|Cellvibrio japonicus
WP
_029134085.1|191960|Sedim
enticola selenatireducens
WP_005783348.1|47883|Pseudomonas synxantha
WP_017505568.1|1229204|alpha proteobacterium L41A
WP_
0854
8289
2.1|
1515
439|
Para
burk
hold
eria
sus
onge
nsis
WP_090227846.1|1007099|Pseudomonas kuykendallii
WP_024973836.1|329|Ralstonia pickettii
WP
_019
0156
71.1
|457
934|
Elio
raea
tepi
diph
ila
WP_016278428.1|671267|Bacteroides sartorii
WP
_014020246.1|104|Cyclobacterium
marinum
WP_048755578.1|1035|Afipia felis
WP_008983418.1|553384|Alishewanella agri
WP_062365449.1|34073|Variovorax paradoxus
WP_100221482.1|80842|Herbaspirillum rubrisubalbicans
WP
_080826259.1|1183411|Agrobacterium
genomosp. 6
WP
_093
2808
27.1
|489
703|
Sol
imon
as a
quat
ica
WP_091979223.1|1502776|Methylobacterium sp. 13MFTsu3.1M2
WP
_013
6370
54.1
|861
208|
Agr
obac
teriu
m s
p. H
13-3
WP
_028
0811
00.1
|413
479|
Sol
imon
as s
oli
WP_082795034.1|33051|Sphingomonas sanguinis
WP
_044394680.1|80878|Acidovorax tem
perans
WP_011767948.1|189918|Mycobacterium sp. KMS
WP
_064683281.1|1138189|Rhizobium
bangladeshense
WP_085933358.1|225324|Enhydrobacter aerosaccus
WP
_046846704.1|113574|Hyphom
icrobium sp. G
J21
WP_049803085.1|375|Bradyrhizobium japonicum
WP
_081
2956
19.1
|384
|Rhi
zobi
um le
gum
inos
arum
WP
_096
3917
61.1
|104
8396
|Hal
open
itus
pers
icus
WP
_027
1015
16.1
|129
8913
|Com
amon
adac
eae
bact
eriu
m U
RH
A00
28
WP
_082657619.1|165697|Sphingopyxis
WP_052530012.1|36809|Mycobacterium abscessus
WP
_068847673.1|1841610|Planctopirus sp. JC
280
WP_1
0099
1967
.1|2
0570
25|S
piros
oma
sp. H
A7
WP_015056745.1|651143|Fibrella aestuarina
WP_083473000.1|269536|Frankia sp. R43
WP
_062
4869
46.1
|171
5989
|Can
dida
tus
Nitr
ospi
ra in
opin
ata
WP
_012
4894
04.1
|379
|Rhi
zobi
um
WP_035240078.1|1380348|Acidobacteriaceae bacterium URHE0068
WP_036267940.1|73780|Methylocaldum szegedienseWP_013685229.1|191579|Fluviicola taffensis
WP_092821445.1|1380357|Rhodospirillales bacterium
URHD0017
WP
_072559535.1|1913578|Sphingopyxis sp. LP
B0140
WP_081684175.1|266854|Granulicoccus phenolivorans
WP_004264290.1|587|Providencia rettgeri
WP_045167303.1|28131|Prevotella in
termedia
WP
_048
0768
61.1
|117
3487
|Hal
orub
rum
sp.
AJ6
7
WP_028337809.1|29448|Bradyrhizobium elkanii
WP_013654325.1|991904|Polymorphum gilvum
WP_093153036.1|420996|Thalassobaculum litoreum
WP_088492884.1|837|Porphyromonas gingivalis
WP_008657175.1|816|B
acteroides
WP
_083
5097
81.1
|121
290|
Hyp
hom
icro
bium
sul
foni
vora
ns
WP
_064304386.1|303|Pseudom
onas putida
WP
_092459739.1|1393122|Crenotalea therm
ophila
WP_0
7306
7332
.1|11
9409
0|Aliif
odini
bius r
oseu
s
WP
_037165528.1|1500300|Rhizobium
sp. CF
258
WP
_055143318.1|39492|[Eubacterium
] siraeum
WP
_097
2007
09.1
|188
4383
|Var
iovo
rax
sp. Y
R75
2
WP_003280119.1|316|Pseudomonas stutzeri
WP_053381025.1|42253|Nitrospira moscoviensis
WP_
0757
9371
1.1|
1141
883|
Mas
silia
put
ida
WP_068094658.1|76978|Novosphingobium rosa
WP
_066
8029
57.1
|406
81|S
phin
gom
onas
asa
ccha
roly
tica
WP_081000780.1|87883|Burkholderia multivorans
WP_048500689.1|232216|Chryseobacterium koreense
WP_084218942.1|328301|Syntrophorhabdus aromaticivorans
WP_071980017.1|1917158|Alteromonas sp. RW2A1
WP_050631537.1|1654716|Bradyrhizobium viridifuturi
WP_078813596.1|48467|Prosthecobacter debontii
WP
_092
0336
21.1
|185
5398
|Por
phyr
omon
adac
eae
bact
eriu
m K
HP
3R9
WP_0
7968
5319
.1|6
8886
7|Oht
aekw
angia
kore
ensis
esneiranacmuibozihrydar
B|540552|1.463053580_P
W
WP
_080902475.1|294|Pseudom
onas fluorescens
WP
_014200913.1|253245|Ow
enweeksia hongkongensis
WP
_047468669.1|1339242|Delftia sp. ZN
C0008
WP_005372458.1|39773|Methylomicrobium
WP_018005262.1|164546|Cupriavidus taiwanensis
WP
_071
9700
84.1
|191
7485
|Sul
fitob
acte
r sp
. AM
1-D
1
WP
_008510818.1|1218315|Brucella inopinata
WP_097674963.1|142585|Bradyrhizobium sp. C9
WP
_049
9067
81.1
|119
434|
Hal
orub
rum
tebe
nqui
chen
se
WP_
0599
4530
4.1|
1503
055|
Burk
hold
eria
terri
torii
WP_010683362.1|39956|Methylobacterium mesophilicum
WP_051880056.1|421072|Chryseobacterium halperniae
WP_044237100.1|122|G
imesia
maris
WP_004308218.1|816|Bacteroides
WP_051600234.1|1280944|Hyphomonas sp. CY54-11-8
WP_013078838.1|88688|Caulobacter segnis
WP_058745238.1|172044|Sphingom
onas yabuuchiae
WP_091379109.1|1881103|Geodermatophilus sp. DSM 45219
WP_012473795.1|1096|Chlorobium phaeobacteroides
WP_072995904.1|192903|Pseudozobellia thermophila
WP_042419826.1|405782|Streptacidiphilus anmyonensis
WP_002993155.1|28453|Sphingobacterium
WP
_090997603.1|414048|Pedobacter insulae
WP_091126757.1|574651|Nocardioides terrae
WP_022545094.1|1400053|Bacteroidales bacterium CF
WP_093736217.1|1839759|Streptomyces sp. D
valAA-14
esneruhtam
muibognihpsovoN|099824|1.245137970_
PW
WP_101944482.1|2067453|Uliginosibacterium sp. TH139
WP_011923975.1|114615|Bradyrhizobium sp. ORS 278
WP_0
9062
4110
.1|14
7743
7|Par
aped
obac
ter i
ndicu
s
WP_102206007.1|372787|Fischerella thermalis
WP_056695931.1|414371|Aureim
onas
WP
_052
5648
96.1
|795
830|
Can
dida
tus
Bro
cadi
a si
nica
WP_099104184.1|1206980|Nostoc sp. 'Peltigera m
alacea cyanobiont' DB3992
WP_016263566.1|286|Pseudomonas
WP_0
8423
4542
.1|1
0017
6|Pap
illiba
cter
cin
nam
ivora
ns
WP
_070666720.1|838|Prevotella
WP_071098581.1|1172030|M
icromonosp
ora sp. W
MMB235
WP_053326746.1|1324352|Chryseobacterium gallinarum
WP
_049
8970
37.1
|689
11|N
atria
lba
chah
anna
oens
is
WP_081705202.1|1416614|G
loeobacter kilaueensis
WP_
0174
5467
8.1|
8084
2|H
erba
spiri
llum
rubr
isub
albi
cans
WP_073904454.1|1490482|Mycobacterium sp. SWH-M3
WP
_074121857.1|1680155|Bradyrhizobium
sp. AS
23.2
WP_069020256.1|1818881|Candidatus Thiodiazotropha endoloripes
WP_099463435.1|1944636|Parabacteroides sp. Marseille
-P3668
WP
_077292092.1|187304|Labrenzia aggregataWP_082670027.1|33051|Sphingomonas sanguinis
WP_083637046.1|1922337|Leptolyngbya
sp. 'h
ensonii'
WP_004748890.1|29498|Vibrio tubiashii
WP
_088664285.1|1317112|Roseovarius sp. 22II1-1F6A
WP_0
8728
7470
.1|1
9655
76|P
seud
oflav
onifr
acto
r sp.
An1
84
WP
_022
7873
70.1
|877
414|
Clo
strid
iale
s ba
cter
ium
NK
3B98
WP
_083779632.1|314267|Sulfitobacter sp. N
AS
-14.1
WP
_015
4491
54.1
|753
09|R
hoda
noba
cter
WP
_092
8388
21.1
|100
5928
|Ros
eova
rius
lutim
aris
WP_023692463.1|1287325|M
esorhizobium sp. LSJC268A00
WP_069707909.1|158500|Novosphingobium resinovorum
WP_090828645.1|1233|Nitrosovibrio tenuis
WP
_079392070.1|286|Pseudom
onas
WP_041534907.1|208216|Parvularcula bermudensis
WP_013833356.1|165696|Novosphingobium
WP_098195322.1|2029983|Chitinophagaceae bacterium 13
WP_0
0887
7326
.1|4
8972
2|M
esor
hizo
bium
met
allid
uran
s
WP
_085984230.1|1131813|Methylobacterium
sp. 88A
WP
_081
4351
15.1
|160
791|
Sph
ingo
mon
as w
ittic
hii
WP
_026
8086
85.1
|861
04|A
reni
bact
er la
teric
ius
WP_0
4809
0856
.1|1
3929
98|C
andid
atus
Met
hano
pere
dens
nitr
ored
ucen
s
WP
_053
0581
40.1
|909
625|
Rub
roba
cter
apl
ysin
ae
WP
_006270760.1|76891|Asticcacaulis biprosthecium
WP_010123961.1|1030157|Sphingom
onas sp. KC8
WP_
0564
9544
3.1|
1735
692|
Sphi
ngom
onas
sp.
Lea
f25
WP_
0476
1820
2.1|
398|
Rhi
zobi
um tr
opic
iW
P_065664090.1|1841655|Agrobacterium sp. B131/95
WP_065010823.1|68287|Mesorhizobium
WP_0
1197
1023
.1|1
1032
1|Sin
orhi
zobi
um m
edica
e
WP_
0599
8250
1.1|
1015
71|B
urkh
olde
ria u
bone
nsis
WP_099503355.1|69665|Caulobacter sp. FWC26
WP_091516911.1|998|Flexibacter flexilis
WP
_081
7432
00.1
|288
965|
[Clo
strid
ium
] cla
rifla
vum
WP_099463422.1|1944636|Parabacteroides sp. Marseille-P3668
WP_0
2503
0983
.1|2
0479
9|Nitr
atire
duct
or a
quib
iodo
mus
WP_083657918.1|1892903|Herbaspirillum sp. W
T00C
WP
_092
6623
01.1
|136
7881
|Hal
orie
ntal
is p
ersi
cus
WP_019960613.1|240237|Woodsholea maritima
WP_092048392.1|1576369|Planctomicrobium piriforme
WP
_089355802.1|447679|Ekhidna lutea
WP_085773725.1|655015|Methylocystis bryophila
WP_094530879.1|1980935|Pseudanabaena sp. SR411
WP_008706033.1|1265734|Rhodopirellula maiorica
WP_073409929.1|986|Flavo
bacteriu
m johnso
niae
WP_0
4747
9138
.1|15
0028
3|Chr
yseo
bacte
rium sp
. CF35
6
WP_082340617.1|339|Xanthomonas campestris
WP_035638686.1|566679|Bradyrhizobium
sp. OR
S 375
WP
_092
9101
36.1
|188
2830
|Rhi
zobi
ales
bac
teriu
m G
AS
191
WP
_073365442.1|1124188|Flavobacterium
fontis
WP_013109336.1|120|Planctopirus limnophila
WP
_009301608.1|665942|Desulfovibrio sp. 6_1_46A
FAA
WP_084467687.1|103730|Actinokineospora inagensis
WP_065006787.1|1854057|M
esorhizobium sp. AA22
WP_052744930.1|1538294|Micr
omonospora sp
. HK10
WP_022842741.1|278963|Acidobacteriaceae bacterium TAA166
WP
_082
5495
97.1
|173
6555
|Mes
orhi
zobi
um s
p. R
oot5
52
WP_083869695.1|477641|Modestobacter marinus
WP_083592922.1|1317114|Aurantim
onas sp. 22II-16-19i
WP_049284968.1|286|Pseudomonas
WP_0
0462
4807
.1|2
9371
|[Clo
strid
ium
] ter
mitid
is
WP_095840540.1|2033437|Chitinophaga sp. MD30
WP_046756489.1|1348245|Kordia jejudonensis
WP
_042
6191
22.1
|358
|Agr
obac
teriu
m tu
mef
acie
ns
WP
_008
3879
95.1
|411
361|
Hal
ogeo
met
ricum
pal
lidum
WP_066752989.1|1685010|Chryseobacterium sp. IHBB 10212
WP
_095
4474
52.1
|271
865|
Och
roba
ctru
m s
p. A
44
WP_075061401.1|11
34406|Orn
atilinea apprim
a
WP
_082
7559
86.1
|135
5477
|Bra
dyrh
izob
ium
dia
zoef
ficie
ns
WP
_009043888.1|587753|Pseudom
onas chlororaphis
WP
_095387429.1|2012632|Sphingopyxis sp. G
W247-27LB
WP
_089
7614
43.1
|408
074|
Chi
tinop
haga
terr
ae K
im a
nd J
ung
2007
WP_061535229.1|279058|Collimonas arenae
WP_052259141.1|56459|Xanthomonas vasicola
WP
_052
4942
27.1
|581
33|N
itros
ospi
ra s
p. N
pAV
WP_095565911.1|2024580|Plantactinospora sp. KBS50
WP_092226358.1|1855318|Bradyrhizobium sp. Gha
WP
_010913851.1|2066070|Mesorhizobium
japonicum
WP_099588558.1|40324|Stenotrophomonas maltophilia
WP_075628770.1|464029|Rhizobium oryzae
WP_0
1997
7020
.1|2
47|E
mpe
doba
cter
bre
vis
WP
_074487307.1|1302689|Mucilaginibacter polytrichastri
WP_084522028.1|53432|Nocardia uniformis
WP_044272248.1|1470345|Bacteroides timonensis
WP_015664541.1|44255|Bradyrhizobium oligotrophicum
WP_084625517.1|1207055|Sphingobium sp. C100
WP_087337819.1|357276|Bacteroides dorei
WP_
0122
2225
8.1|
3399
6|G
luco
nace
toba
cter
dia
zotro
phic
us
WP_068133588.1|980254|R
oseim
aritima ulva
e
WP
_064835082.1|396|Rhizobium
phaseoli
WP_071507486.1|305|Ralstonia solanacearum
WP_028095445.1|337702|Pseudodonghicola xiamenensis
sano
mogn
ihp
S|78
631|
1.65
7804
640_
PW
WP_
0236
9906
8.1|
1287
322|
Mes
orhi
zobi
um s
p. L
SJC
265A
00
WP
_036284031.1|622637|Methylocystis sp. ATC
C 49242
WP_068768974.1|1184151|Opitutaceae bacterium TSB47
WP
_068017268.1|674703|Rhodoplanes sp. Z
2-YC
6860
WP_051443965.1|1429916|Afipia sp. P52-10
WP_084182446.1|39687|Mycobacterium austroafricanum
WP_062341847.1|1768752|Novosphingobium
sp. CCH12-A3
WP
_036632782.1|823|Parabacteroides distasonis
WP
_092416776.1|1801620|Collim
onas sp. OK
307
WP
_034679768.1|236814|Chryseobacterium
formosense
WP_061048384.1|221822|Phaeobacter inhibens
WP_073132933.1|947013|Chryseolinea serpens
WP_005988489.1|56460|Xanthomonas vesicatoria
WP_028057923.1|1298857|Solirubrobacter s
p. URHD0082
WP
_098195162.1|2029983|Chitinophagaceae bacterium
13
WP_004290502.1|28111|Bacte
roides eggerth
ii
WP
_081
4449
39.1
|124
|Bla
stop
irellu
la m
arin
a
WP_090603244.1|332977|Parapedobacter koreensis
WP_095589297.1|1590614|Actibacterium
ureilyticum
11rr
e.ps
muib
ozih
ryda
rB|
2771
671|
1.09
7369
190_
PW
WP
_093307966.1|34072|Variovorax
WP_024740054.1|107401|Te
nacibacu
lum mariti
mum
WP_065137571.1|1778|Mycobacterium gordonae
WP
_078
3669
26.1
|280
66|R
hodo
fera
x fe
rmen
tans
WP
_083
2407
32.1
|177
4968
|Met
hylo
cean
ibac
ter
met
hani
cus
WP
_030539670.1|1303256|Sphingobium
sp. DC
-2
WP_059000176.1|1552121|Leptolyngbya sp. NIES-2104
WP_082932868.1|190721|Ralstonia insidiosa
WP_093896586.1|1839774|Streptomyces sp. Ncost-T10-10d
WP
_067673142.1|1822215|Erythrobacter sp. H
I00D59
WP_050591131.1|68287|Mesorhizobium
WP
_004
5941
99.1
|292
82|H
aloa
rcul
a ja
poni
ca
WP_085936213.1|225324|Enhydrobacter aerosaccus
WP_051882337.1|1247963|Parvularcula oceani
WP_083960493.1|980561|Methylomonas lenta
WP_005801097.1|817|Bacteroides fragilis
WP
_096683443.1|1973475|Nostoc sp. N
IES
-2111
WP
_052
1921
92.1
|156
4113
|Sph
ingo
mon
as s
p. A
nt H
11
WP_0
9117
0676
.1|5
5199
6|M
ucila
giniba
cter g
ossy
pii
WP
_005
2771
91.1
|121
7695
|Aci
neto
bact
er s
p. N
IPH
185
9
WP_034728376.1|1233950|Chryseobacterium sp. JM1
WP_062547850.1|1727164|Pedobacter sp. PACM 27299
WP_042087938.1|1492281|alpha proteobacterium Q-1
WP_072766083.1|558155|Arenibacter n
anhaiticus
WP_096614338.1|862134|Sphingomonas ginsenosidimutans
WP_085870580.1|1839|Nocardioides
WP
_097
6138
65.1
|203
5448
|Rhi
zobi
um s
p. C
5
WP
_006019181.1|56946|Afipia broom
eae
WP_064665416.1|182141|Pseudoalteromonas prydzensis
WP
_065656395.1|358|Agrobacterium
tumefaciens
WP_084685017.1|244365|Methylohalobius crimeensis
WP_027853030.1|404770|Marinobacterium litorale
WP
_049858702.1|1681194|Pseudom
onas sp. RIT-P
I-a
WP
_015596988.1|53399|Hyphom
icrobiumdenitrificans
WP_074343878.1|36809|Mycobacterium abscessus
WP_102073885.1|2045210|Pusillimonas sp. JR1/69-3-13
WP_080700186.1|103855|Bordetella hinzii
WP_079791392.1|28901|Salmonella enterica
WP_085809851.1|1917973|Sphingomonas sp. TZW2008
WP
_022727544.1|1121832|Fodinicurvata sedim
inis
WP_080810443.1|1209493|H
alomicronema hongdech
loris
WP_036945621.1|118173|Pseudanabaena sp. PCC 6802
WP
_080
5747
28.1
|294
49|R
hizo
bium
etli
WP
_091535647.1|1780376|Alistipes sp. C
HK
CI003
WP_083871010.1|1288121|Alistipes senegalensis
WP_087406053.1|1965620|Bacteroides sp. An279
WP_051677674.1|1380355|Bradyrhizobium sp. URHD0069
WP
_081912819.1|1522072|Sphingobium
sp. ba1
WP_011940822.1|351604|Geobacter uraniireducens
WP_013801384.1|742013|Delftia sp. Cs1-4
WP_091639649.1|145854|Micromonospora pallida
WP
_081
4419
52.1
|314
344|
Mar
ipro
fund
us fe
rroo
xyda
ns
WP
_064790849.1|502049|Thalassospira profundim
aris
WP
_051294653.1|220343|Gem
mobacter nectariphilus
WP_013596009.1|80867|Acidovorax avenae
WP_0
1438
7263
.1|31
2277
|Flav
obac
terium
indic
um
WP_099076851.1|92942|Nostoc linckia
WP_008137934.1|1144344|Bradyrhizobium
sp. YR681
WP
_051
6025
27.1
|85|
Hyp
hom
onas
WP_
0894
5754
7.1|
2015
357|
Burk
hold
eria
sp.
AU33
803
WP_080522385.1|1420851|Methyloprofundus sedimenti
sirtsulapsano
moduespodohR|6701|1.120405110_
PW
WP_051634474.1|358|Agrobacterium
tumefaciens
WP_057482737.1|270918|Salegentibacter mishustinae
murasonimugel
muibozihR|483|1.748615050_
PW
WP
_092
2827
52.1
|937
334|
Cal
dico
prob
acte
r fa
ecal
is
02S
D.pssano
midnuverB|5552351|1.472567450_
PW
WP_042571950.1|321846|Pseudomonas simiae
WP_083808707.1|1032527|Pedosphaera parvula
WP_
0882
0156
1.1|
382|
Sino
rhizo
bium
mel
iloti
WP
_015
8958
34.1
|330
75|A
cido
bact
eriu
m c
apsu
latu
m
WP
_061934617.1|1638162|Aureim
onas sp. AU
22
WP_057026385.1|108015|Bradyrhizobium yuanmingense
WP_009860218.1|328812|Parabacteroides goldsteinii
WP
_084
4627
63.1
|580
166|
Oce
anib
acul
um p
acifi
cum
WP
_089
7698
62.1
|660
517|
Hal
obel
lus
clav
atus
WP_065578027.1|1196095|Gilliamella apicola
WP_091471579.1|492660|Methylophilus rhizosphaerae
WP
_081147616.1|1703345|Niastella vici
WP
_101
2375
71.1
|177
6741
|Aci
neto
bact
er p
rote
olyt
icus
WP_066514273.1|120107|Sphingobium cloacae
WP_0
6732
5608
.1|1
8540
58|M
esor
hizo
bium
sp.
AA23
WP_073243758.1|983|Flavobacterium flevense
WP_
0101
6514
8.1|
1112
216|
Sphi
ngom
onas
sp.
PAM
C 2
6617
WP
_078
7925
95.1
|238
|Eliz
abet
hkin
gia
men
ingo
sept
ica
WP_057221537.1|106591|Ensifer
WP_016736429.1|396|Rhizobium phaseoli
WP_088892393.1|1962290|Leptolyngbya ohadii
WP_057834160.1|280332|Bradyrhizobium jicamae
WP
_074
7935
83.1
|357
99|N
itros
ospi
ra b
riens
is
WP_060847086.1|270351|M
ethylobacterium aquaticum
WP_099746952.1|48296|Acinetobacter pittii
WP
_014
7461
85.1
|171
437|
Tist
rella
mob
ilis
Tree scale: 2
pAgo operons pAgo fusion
short pAgo
superkingdom
Archaea
Bacteria
APAZ in pAgo operon
Ia
Ib
IIa
IIb
operonpAgo fusionkingdom
proteintype
MID
APAZ
APAZ
APAZ
APAZ
APAZDUF4365
III
FIG 5 Phylogenetic analysis of APAZ domains. The circular phylogenetic tree of the five groups of APAZ domains. The phylogenetic tree was annotated asfollows, from the inner to the outer circles: in the “protein type” circle the APAZ-containing proteins are colored according to their phylogenetic groups anddomain compositions, where each type of protein structure is exemplified by the callouts with corresponding domain schemes; isolated APAZ domains in allgroups are shown in light blue; the “operon” circle indicates whether the APAZ-containing protein is found in a pAgo coding operon; the “pAgo fusion” circleshows APAZ domains which are fused with short pAgo proteins; and the “kingdom” circle indicates the superkingdom to which the correspondingAPAZ-containing protein belongs.
Ryazansky et al. ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 10
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

Functional classification of proteins enriched in the genomic context of pAgos.Analysis of the functions of proteins encoded in the same operons with pAgos mightshed light on the molecular pathways involving pAgos. We therefore analyzed thegenomic context of pAgos encoded in the 1,385 genomes of 1,248 strains and exploredthe proteins encoded in their proximity. We defined a window centered on pAgoencompassing 20 genes (i.e., 10 genes upstream and 10 genes downstream of pAgo)in each genome. The 16,274 proteins encoded in these windows were clustered intoorthogroups based on the sequence similarities that resulted in 1,892 orthogroups thathad at least 2 proteins (see Materials and Methods). The orthogroups contained 12,668proteins, and the largest one comprised 298 related proteins (Table S2). We furtherdefined pAgo operons that included only genes that are continuously located on thesame genomic strand as pAgo, which resulted in a set of operons with a mean size of6.4 genes. We separately considered operons of five different groups of pAgos (Fig. 6):
RecB(52%), APAZ(96%)
PD
-(D
/E)X
K p
redi
ctio
n
1. w
/o A
PA
Z2.
with
SIR
2-A
PA
Z3.
long
-A4.
long
-B5.
long
-A w
ith M
ID-O
H
Fraction of predicted PD(E/D)XK proteins in orthogroup
Percentage of operonswith protein(-s) fromthe orthogroup
BLAST Pfam
Annotation of orthogroups (% of proteins)
Prim-pol (SSF56747(82%), SSF48452(73%))
***
***
**
*
*
*
**
*
*Cas4Cas4
SIR2_1-APAZ
Putative DNA-binding domain (Schlafen, AlbA_2), SIR2_1, APAZ
SIR2_2
Repeat Associated Mysterious Proteins (RAMP)
0
Cas6
Ort
hogr
oups
of p
rote
ins
?
?
?
?
RecB2
*
DNA-binding domain
* Nuclease activity
P-loop NTPase domainHelicase activity
Hydrolase/phosphataseSAM-dependent methyltransferases
efflux membrane transport
?
FIG 6 Functional annotations of proteins encoded in pAgo operons. The heat map on the left shows the percentage of operons for each pAgo group(columns) that have at least one protein from the particular orthogroup (rows). The orthogroups are ordered according to the number of proteins in eachgroup, from the largest to the smallest. Since many operons do not contain any proteins included in the orthogroups analyzed here, the total number ofoperons with orthogroup-specific proteins may be less than 100%. The PD-(D/E)XK column shows the fraction of proteins from each orthogroup that arepredicted to be nucleases from the PD-(D/E)XK superfamily. The annotation of proteins was performed by BLAST analysis for identification of APAZ, RecB-like,and RecB2 domains and by InterProScan using the Pfam and Superfamily databases. Only the Pfam annotation of domains is shown (with the exception ofog_209, for which the Superfamily annotation is shown). The percentage in the brackets after the domain name shows the fraction of proteins in thecorresponding orthogroup having this type of domain. Since proteins may contain several domains, the sum of percentages in each row may be greater than100%. Only Pfam or Superfamily domains found in at least 30% of orthogroup proteins are shown. The descriptions of domain functions are shown on theright of the panel. Proteins from several orthogroups do not have any known domains and are marked by “?.” After the manual curation of domain functions,several of the most common functional features were selected, which are shown as colored symbols and described below the table.
The Diverse World of Prokaryotic Argonautes ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 11
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

(i) short pAgos that are not fused to the APAZ domain, (ii) short pAgos that contain theSIR2-APAZ domains, (iii) all long-A pAgos except the MID-OH variants, (iv) long-B pAgos,and (v) long-A pAgos with the MID-OH domain. Proteins from 47 orthogroups that werepresent in at least 5% of operons in at least one of the five pAgo groups were furtheranalyzed in detail. In Fig. 6, the orthogroups are sorted in descending order of thenumber of proteins in each orthogroup.
We defined the domain structure of proteins in each orthogroup using InterProScanwith Pfam, Superfamily, and CDD databases containing known domain annotations. Foridentification of the APAZ, RecB-like, and RecB2 domains, previously described byMakarova et al. (11) and absent from these databases, we performed PSI-BLAST searchusing previously identified proteins as queries. In addition, we proposed that there maybe other pAgo-associated proteins belonging to the PD-(D/E)XK superfamily of nu-cleases, besides previously identified Mrr (RecB-like and RecB2) and Cas4 proteins (11,12). To check this, we have searched for the characteristic PD-(D/E)XK sequence motifin the orthogroup proteins. The identified domains found in each orthogroup ofproteins and the putative functional types of pAgo-associated proteins are shown inFig. 6.
The proteins with APAZ domains are present in the orthogroups og_3, og_4, og_9,and og_40. The proteins from these orthogroups also contain other types of domains,including DUF4365 (og_3), SIR2_1 subtype of SIR2 (og_4), TIR (og_9), and SIR2_1 fusedwith Schlafen domain (og_40). Most of the revealed APAZ-containing proteins arelocated in group 1 operons of short pAgos that do not have their own APAZ domain.In total, 91% of group 1 operons contain proteins with APAZ. In contrast, only 6% ofgroup 2 operons of short pAgo fused with APAZ and SIR2 encode APAZ-containingproteins. Taken together, 93% of short pAgos are associated with the APAZ domain asa part of the same protein or present in the same operon with short pAgos. In contrast,long pAgos are only rarely coencoded with APAZ proteins (2%, 3%, and 0% of operonsfrom groups 3, 4, and 5, respectively).
We found several different types of nucleases and DNA-binding proteins abundantin the pAgo operons (Fig. 6). In total, 17 out of 47 orthogroups (36%) encode proteinswith putative nuclease activity. These proteins are found in 68% of pAgo operons.Ninety-two percent of operons of short pAgos that themselves do not have the APAZand SIR2 domains (group 1 of pAgos) encode different types of nucleases. In contrast,only 32% of operons of short pAgos fused with the SIR2 and APAZ domains (group 2)contain nucleases. For group 3 of long-A pAgos, 45% of pAgos carrying the catalytictetrad have nucleases in their operons, while this number is increased to 53% foroperons of long-A lacking the DEDX tetrad. This number is further increased to 67% forgroup 4 of long-B pAgos, which are all catalytically inactive. The nucleases found in thepAgo operons are quite diverse and include proteins with SIR2 (og_4, og_40, andog_44), TIR (og_9), and RecB2 (og_15) domains; the DUF4365 domain that belongs toRecB-like nucleases (og_3); Cas4 proteins (og_56 and og_60), and phospholipase Dproteins with putative nuclease activity (og_136). A set of pAgo operons also containproteins with putative nucleases of the HNH type with DUF252 and DUF1254 domains(og_28).
Two pAgos from Marinitoga piezophila and Thermotoga profunda, which are repre-sentatives of the small group of long-A pAgos containing the MID-OH domain (Fig. 1B,2B, and 3A), were previously reported to be encoded in the CRISPR loci between theCas1 and Cas2 genes (24). We found that all pAgos containing the MID-OH domainfrom 9 prokaryotic strains (group 5 of pAgos) are encoded within CRISPR loci with Cas1(og_154) and Cas2 (og_209) genes nearby (Fig. 6). Other proteins specific for thesubtype III-B CRISPR locus are also observed in association with MpAgo-like pAgos: Cas6(og_190), primase (og_209), and Cmr (Cas module RAMP) proteins (og_455).
In addition, proteins in several other orthogroups have motifs characteristic for thePD-(D/E)XK nucleases [Fig. 6, the PD-(D/E)XK column]. These proteins include the UvrDhelicase and P-loop NTPase (AAA� subfamily) domains (og_6), SAM-dependent meth-yltransferase domain (og_13), restriction endonuclease of type III (og_21, og_34), DEAD
Ryazansky et al. ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 12
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

box helicase and P-loop NTPase (AAA� subfamily) (og_34 and og_37), YhgA-typeputative transposases (og_72), and proteins without known domains (og_257 andog_603). Notably, proteins from three of these orthogroups have predicted helicaseactivity (domains related to the UvrD helicase, og_6, and DEAD box helicase, og_34 andog_37).
Proteins with DNA-binding domains are abundant in operons of short pAgos fusedto the APAZ and SIR2 domains (33%) and of long-B pAgos (33%). In addition, proteinswith DNA-binding domains are present in 19% and 13% of operons of long-A pAgosand short pAgos without APAZ-SIR2 domains, respectively. The DNA-binding domainsin pAgo-associated proteins include the lambda repressor-like DNA-binding domain(og_2), helix-turn-helix DNA-binding domains (og_18, og_49, og_73, and og_79), theSchlafen domain with an AlbA_2 subdomain (og_37), and DNA-binding components ofthe type I restriction-modification system (HsdS) (og_11). Interestingly, the lambdarepressor-like DNA-binding domain (PF01381) from highly represented orthogroupog_2 and the Schlafen DNA-binding domain AlbA_2 (PF04326) from og_37 were foundto be fused with several long-B pAgos (Fig. 2B).
Overall, analysis of proteins in pAgo operons strongly indicates the involvement ofnuclease, helicase, and DNA-binding activities in cellular pathways dependent onpAgos (see Discussion). Beyond nucleases and DNA-binding proteins, 10% of all longpAgo operons encode hydrolases and phosphatases (og_47, og_62, and og_55). Finally,14% of long-B pAgo operons encode multidrug efflux transporters of the resistance-nodulation-cell division (RND) superfamily (og_14 and og_31), but their possible rolesin the functional pathways involving pAgos remain unknown.
In the present analysis, we could not reveal any defined operon compositions forspecific pAgo groups. While the median number of all genes associated with pAgo onthe same strand is 6.4, these numbers are much lower if we consider only proteinsincluded in the orthogoups analyzed here: 2.0 for all short pAgos, 3.0 for each long-Aand long-B pAgo, and 5.0 for the MID-OH long-A pAgos. The small sizes of operons onthe one hand and the large number of orthogroups on the other result in numerouspossible operon configurations, making the construction of the consensus operonstructure impossible. The only exception is the MID-OH long-A pAgos, for which thestructure can be presented as Cas1-Cas2-Cas6-pAgo-Cmr. The same operon schemewas reported for MpAgo previously (24). The list of all observed configurations oforthogroup proteins in pAgo operons is presented in Table S3.
The genomes encoding pAgos do not show decreased transposon copy num-ber. Several eukaryotic Ago proteins were shown to prevent propagation of trans-posons and viruses in the genomes (reviewed in references 44 and 45). Similarly, a fewstudied pAgos were proposed to protect prokaryotic cells from foreign DNA, includingplasmids, transposons, and phages (38, 40, 41). In particular, pAgos may be involved inthe protection of the prokaryotic genome from the invasion of mobile genetic elementsor suppress the activity of genomic copies of such elements. If pAgos are able to reducethe expansion of transposons in their host genomes, then the number of transposoninsertions should be lower in strains that encode pAgo. To test this hypothesis, wecounted the number of transposon insertions in the genomes of strains encoding ornot encoding pAgos from different groups (short pAgos, short pAgos fused to theSIR2-APAZ domains, long-A with PIWI and PIWI* domains, long-B pAgos, and long-ApAgos with the MID-OH domain). We found that the genomes of strains encodingpAgos do not have lower copy numbers of transposons than strains without pAgogenes (Fig. 7A). Moreover, we observed a moderate but significant increase in the meannumber of transposon copies per genome in pAgo-contaning strains for both short andlong classes of pAgos. For the MID-OH pAgos, the number of transposon copies incorresponding genomes was smaller than in pAgo-minus strains, but this differencewas statistically insignificant due to the small number of pAgos in this group. Sincedifferent phylogenetic groups may contain different transposon numbers, we com-pared the transposon content within several genera (Fig. 7B). For this, we selected 32prokaryotic genera having at least 10 strains with sequenced genomes encoding and
The Diverse World of Prokaryotic Argonautes ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 13
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

not encoding pAgos. For the strains of each genus, the transposon copy numbers weresubjected to log2 transformation and z-scaling. If a strain has a z-score of ��2, thenthere is only a 2.3% chance that its genome has the transposon content typical for thisgenus, and so it can be considered the one that has a significantly lower transposonnumber than average. We have found that only one strain among 108 pAgo-encodingstrains from 32 genera (703 strains in total) has a z-score of ��2 (Fig. 7B). The genomesof all other strains with pAgo have the transposon copy numbers that are typical of thecorresponding genera. Overall, our results indicate that pAgos present in the genomesmay not be able to stop transposon insertions; however, this does not exclude apossibility that pAgos can repress transposon expression.
DISCUSSION
Eukaryotic systems of RNA interference have evolved from several diverse prokary-otic components, including RNase III-like enzymes, RNA-dependent RNA polymerases,helicases, and pAgos, which gave rise to various types of eAgos and RNAi pathways(46). While being highly variable and involved in different interference pathways, alleAgos share the same modus operandi: they always bind small RNA guides to locate andinactivate cRNA targets (or attract other components to modify corresponding codingDNA loci). Perhaps not surprisingly, pAgos are much more diverse, with significantsequence and domain variations (11, 12). Moreover, a few published functional studiesof pAgos demonstrated that, while retaining the same basic principle of guided nucleicacid recognition, they may significantly differ in their modes of action in comparisonwith eukaryotic counterparts (38–41, 47). We have now revealed even a larger diversityof pAgos that allows us to suggest their updated classification and propose possibleevolutionary pathways and cellular functions for this conserved family of nucleicacid-targeting proteins.
*******
*****
193 37 69 30 75 4 5914
2
64
2048
shor
t pA
gos
shor
t pA
gos
with
SIR
2-A
PAZ
long
-A p
Ago
s w
ith P
IWI
long
-A p
Ago
s w
ith P
IWI*
long
-B p
Ago
s
long
-A p
Ago
s w
ith M
ID-O
H
no p
Ago
Tn
copi
es in
gen
omes
(lo
g2)
A B
1/11
11/2
7
3/11
2/17
8/19
15/5
4
2/11
3/31
3/11
1/19
4/13
2/11
10/1
5
1/13
3/12
3/62
5/13
2/40
1/10
1/11
5/12
1/22
1/17
2/23
3/14
2/93
5/29
1/10
1/11
1/28
4/19
1/14
-2
0
2
Aci
dovo
rax
Aci
neto
bact
er
Alte
rom
onas
Bor
dete
lla
Bra
dyrh
izob
ium
Bur
khol
deria
Cal
othr
ix
Clo
strid
ium
Cup
riavi
dus
Ent
erob
acte
r
Geo
bact
er
Kle
bsie
lla
Mes
orhi
zobi
um
Met
hano
sarc
ina
Met
hylo
bact
eriu
m
Myc
obac
teriu
m
Nos
toc
Pae
niba
cillu
s
Par
abur
khol
deria
Pla
noco
ccus
Pre
vote
lla
Pse
udoa
ltero
mon
as
Ser
ratia
She
wan
ella
Sph
ingo
bium
Stre
ptom
yces
Ther
moc
occu
s
Ther
mot
oga
Trep
onem
a
Vibr
io
Xan
thom
onas
Yers
inia
Genus
Tn c
opy
num
ber i
n ge
nom
es (l
og2,
z-s
cale
d)
short pAgos
short pAgos with SIR2-APAZ
long-A pAgos with PIWI
long-A pAgos with PIWI*
long-B pAgos
long-A pAgos with MID-OH
FIG 7 The number of transposon copies in genomes with and without pAgo genes. (A) The violin plot shows the log2-transformed distribution of copy numbersof transposons in the genomes of prokaryotic strains encoding or not encoding pAgos. The black dots with whiskers indicate the means and standarddeviations. The numbers of analyzed genomes for each group of pAgos are shown below each violin. The P values shown above the plot were calculated withthe Wilcoxon two-sided rank test: ****, P value � 1 � 10�4; ***, P value � 1 � 10�3; **, P values � 0.05. (B) The violin plot shows log2-transformed and z-scaleddistribution of copy numbers of transposons in the genomes of prokaryotic strains encoding or not encoding pAgos for selected genera. The colored dotsindicate the transposon content of strain genomes encoding pAgos; the correspondence of pAgo type and dot color is specified above the plot. The numbersof analyzed genomes for each genus are shown above each violin (strains with pAgo/total number of strains).
Ryazansky et al. ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 14
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

The diversity of pAgos. Our results revealed that pAgos are widespread in archaealand eubacterial species. The availability of new sequenced genomes allowed identifi-cation of an almost 3-fold-larger set of pAgos than previous analysis. Analysis of thisexpanded set demonstrated that pAgos are highly diverse and can be separated intothree large clades, short, long-A and long-B, which diverged early in evolution (Fig. 1B;see also Fig. S1 in the supplemental material). While this division is based solely onsequence alignments of the MID-PIWI domains, these groups also reveal characteristicvariations in specific structural elements involved in nucleic acid binding and process-ing. Thus, the members of the long-A clade mostly contain both catalytically active PIWIdomains and PAZ domains of normal size. In contrast, long-B pAgos contain inactivePIWI* domains and also usually shortened PAZ* domains, although other combinationswere also observed for both clades (Fig. 1 and 2). Furthermore, proteins from the long-Aclade contain several variants of the 5=-P-guide binding motif while long-B pAgosalmost always contain the canonical YK motif. Short pAgos always contain substitutionsin the active site (PIWI*) and a subset of canonical MID motifs (HK/RK).
The exact relationship between the three clades is ambiguous, with two possiblealternative scenarios (12). In one scenario, short and long pAgos diversified first,followed by a further split of long pAgos into two clades. Alternatively, catalyticallyinactive long-B pAgos and short pAgos may have originated from a common ancestorcontaining substitutions in the active site. The phylogenetic analysis of the MID-PIWIdomains has shown that the evolutionary distance between combined short pAgo andlong-B pAgo clades on the one side and the long-A clade on the other is slightly largerthan between short and combined long-A and long-B clades (Fig. S1). This argue infavor of the second scenario of the evolutionary origin of the three clades of pAgos.
Binding of nucleic acids by pAgos. Tight association with small nucleic acid guidesseems to be a defining feature of pAgos and eAgos alike (reviewed in references 12 and59). Analysis of the nucleic acid binding pockets of the MID domain revealed that themajority of pAgos that belong to all three clades (short, long-A, and long-B) likely bindsmall nucleic acid guides with monophosphorylated 5= ends. At the same time, wediscovered significant variations in the conserved 5=-guide binding motifs in the long-ApAgos, including substitutions of key residues involved in the 5=-phosphate interac-tions (i.e., YK, YY, KR, YR, MID*, and MID-OH variants). While most these variants likelyinteract with 5=-P-guides, some proteins might bind guides with an alternative 5=-endstructure, as already shown for the MID-OH pAgos, or not use nucleic acid guides at all.Interestingly, almost all members of the short pAgo clade have MID variants that areclose to the consensus (HK/RK), suggesting that the canonical MID pocket structure isessential for their function in the absence of the PAZ domain. Future biochemicalcomparisons of the long and short pAgos with various noncanonical MID motifs shouldfind out if the observed differences in the 5=-end pockets may influence the specificity,affinity, and kinetics of their interactions with guide and target molecules.
While all known eAgos use small RNA guides to recognize RNA targets, the majorityof proteins in a relatively small sample of pAgos characterized so far prefer to bindsmall DNA as a guide, except for RsAgo (38) and MpAgo (24), which associate with smallRNAs. Furthermore, all studied pAgos preferably interact with DNA targets. Few aminoacid residues involved in the binding of ribose and deoxyribose backbone of the guideand target molecules were identified in the structures of experimentally characterizedpAgos (17, 23–25, 28); however, these residues are not well conserved. Therefore,sequence analysis is not able to predict preference for DNA versus RNA binding, andthis information has to be obtained experimentally.
The PAZ domain of most studied eAgo and pAgo proteins is composed of twosubdomains that together form the 3=-end guide binding pocket (16, 18, 20, 28).However, our results revealed different organizations of the PAZ domain among longpAgos, indicating that a single conserved structure of PAZ may not be required forguide binding. The structure is not conserved even among the few experimentallycharacterized pAgos. While AaAgo and TtAgo contain both subdomains (18, 20), the
The Diverse World of Prokaryotic Argonautes ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 15
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

PAZ* domains of MpAgo and RsAgo lack the second subdomain and therefore do notpossess the PAZ pocket (24, 25, 28). While no three-dimensional structure of the binarycomplex is available for RsAgo, the guide 3= end still interacts with PAZ in the binarycomplex of MpAgo but is differently oriented (Fig. 4) (24). Formation of the extendedguide-target base-pairing during target recognition is accompanied by guide releasefrom PAZ (19, 23, 24, 26, 48). Thus, structural characteristics of PAZ may have animportant role in the recognition and binding of nucleic acid targets by pAgo proteins,for example, by influencing the kinetics of target recognition and cleavage (19, 23, 24,26, 48, 49). Interestingly, the incomplete PAZ* domain is characteristic for the long-Bclade of catalytically inactive pAgos, suggesting that the stepwise guide-target anneal-ing controlled by full-length PAZ may be more important in the active long-A pAgos,e.g., for prevention of premature nonspecific target cleavage.
All short pAgos and a small but significant fraction of long pAgos lack the PAZdomain entirely, raising the question of how nucleic acid guides can be bound by theseproteins. Structural and functional analysis of the truncated long-A AfAgo lacking thePAZ domain demonstrated that it can nevertheless bind and stabilize short guide-target duplexes (14, 15, 22). Furthermore, since the guide 3= end is released from thePAZ domain upon target recognition, PAZ may be important for protection of the guidefrom cellular nucleases until the target is found—and the protection may be lessimportant for short pAgos or truncated long pAgos that use DNA guides. However, itremains to be established whether any accessory proteins may facilitate guide andtarget binding by such pAgos. The finding of association between short pAgos and theAPAZ domain led to the suggestion that APAZ might play the role of PAZ in shortpAgos (11). Recently, it was proposed that APAZ may be instead homologous to theN-domain of full-length Agos (43). However, both proposals await experimental vali-dation.
Possible functional activities of pAgo-associated proteins. In contrast to theirability to bind nucleic acids, the endonuclease activity toward the target is not auniversal feature of pAgos: the proteins that are predicted to have the catalytic tetradin the PIWI domain are found in only the long-A clade of pAgos (�18% of all pAgos).However, the lack of catalytic activity in PIWI* does not rule out the possibility that suchpAgos are involved in processing of target nucleic acids. Indeed, our analysis confirmedand expanded previous observations on the association of catalytically inactive pAgowith proteins that might have nuclease activity (11, 12). Markedly, the number ofputative nuclease genes in operons containing catalytically inactive pAgo variants issignificantly higher than for the active pAgos.
The proteins in one branch of short pAgos have the SIR2 domain with potentialnuclease function, while many more pAgos that belong to the short and long-B cladesof catalytically inactive pAgos harbor in their operons separate proteins with predictednuclease activities. The absence of the catalytic tetrad in the PIWI* domain in suchpAgos and their tight association with proteins containing SIR2, TIR2, DUF4365 (novelRecB-like nuclease of the Mrr subfamily), RecB2, Cas4, PLD, and HNH nuclease domainssuggest that these domains might replace the intrinsic endonuclease activity of theAgo protein. Although the APAZ domain is not related to any nuclease, its strongassociation with short pAgos and the proteins with nuclease domains suggests thatAPAZ might also be involved in the nuclease activity. Thus, the functions of targetrecognition and its further processing seem to be separated, with the first stepperformed by pAgos and the second by nucleases encoded in the pAgo operons.Notably, these operons often include predicted helicases and DNA-binding proteins,suggesting their involvement in DNA processing. Future analysis of the functionalactivities of these proteins will be required to uncover the molecular details of pAgoaction.
Possible functions of pAgos in prokaryotic cells. eAgos and their small RNApartners play diverse functions, from the repression of selfish genomic elements suchas transposons to the regulation of host gene expression. Furthermore, eAgos can
Ryazansky et al. ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 16
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

direct changes of chromatin marks and even DNA elimination (44, 45). Compared toeAgos, experimental data about pAgo functions are very scarce, as activities of only afew proteins were characterized in vitro and even smaller numbers of proteins (TtAgoand RsAgo) were studied in their host cells in vivo. Available data suggest that pAgosmight participate in protection of prokaryotic genomes against foreign elements suchas plasmids, transposons, and phages (38, 39). However, the only direct results obtainedso far were the ability of TtAgo and RsAgo (and also PfAgo and MjAgo in theheterologous Escherichia coli system) to decrease the amount of plasmid DNA in the cell(38–41). RsAgo was also demonstrated to decrease plasmid transcription and prefer-entially associate with genomic DNA sequences corresponding to transposons and ISelements (38). While the molecular mechanisms of this specific targeting remain to beestablished, we infer that other types of pAgos discovered in our analysis may also beinvolved in host defense against certain types of genetic elements.
Intriguingly, we observed a positive, not negative, correlation between the presenceof various types of pAgos and the number of transposon copies in sequenced genomes.This observation suggests several possibilities. First, transposons may not be the maintargets of all pAgos, most of which may instead target phages and plasmids. Second,the presence of pAgos in genomes with increased transposon numbers might bebeneficial for these strains because pAgos suppress the transposon expression or theirfurther detrimental transpositions. Third, it can be speculated that pAgos might per-form a plethora of other functions in prokaryotic cells, including genetic regulation andDNA repair. Indeed, association of pAgos with multiple DNA-binding and DNA-processing proteins suggests their active involvement in DNA metabolism in prokary-otic cells.
As reported previously (11, 12) and in this work, the pattern of distribution of pAgosamong eubacterial and archaeal species does not correspond to the phylogeny of hostspecies, suggesting their spread by horizontal gene transfer. While we could not builda conserved operon structure for any group of pAgo proteins, their strong associationwith genes encoding proteins involved in DNA processing suggests their commonfunction and joint horizontal transfer. Statistical analysis of the genomic neighborhoodsof pAgos also revealed a significant link to phage resistance systems (11). The functionalroles of such associations should be investigated experimentally. Other genes spreadby HGT include various adaptation and resistance systems such as CRISPR, restriction/modification, drug resistance, and toxin/antitoxin modules that have common featuresof cell protection in the highly diverse and developing environments. While it isattractive to suggest that most pAgos use associated nucleic acid guides for sequence-specific recognition of foreign DNA targets in order to repress them, other scenarios oftheir action are possible as well. For example, pAgos together with their proteinpartners might constitute a suicidal system that is activated under stress conditions orupon viral infections and triggers processing of host DNA unless its activity is sup-pressed. Our study opens new possibilities for detailed analysis of various groups ofpAgos and associated factors, which can reveal their new cellular functions andactivities and possibly adopt pAgos as an efficient tool for genomic manipulations.
MATERIALS AND METHODSProkaryotic protein and genome databases. The set of proteins was downloaded from the NCBI
FTP site in January 2018. Altogether, the database included more than 116 million RefSeq proteins from4,364 taxons with completely sequenced and assembled genomes (“Complete Genome” and “Chromo-some” statuses) and 15,505 taxons with unassembled genomic sequences (“Scaffold” and “Contig”statuses) of Archaea and Eubacteria. The genomic sequences and genome annotations of prokaryoteswere fetched from the NCBI FTP site in January 2018.
Identification of pAgo- and APAZ-containing proteins and analysis of their domain structure.The identification of homologs of already-known pAgo- and APAZ domain-containing proteins wascarried out using the PSI-BLAST and DELTA-BLAST program from the NCBI-BLAST� package, v.2.6.0(-num_descriptions 1000 -num_alignments 1000 -evalue 10E�5 -num_iterations 5) (50). The search wasperformed with five iterations, sufficient for the full convergence of the results. For the search, sequencesof only the PIWI-MID domains of already-known pAgos (12) or APAZ domains (11) were used as queries.The domain architecture of the found proteins was analyzed by the CDD-batch program (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi) for the Pfam and CDD databases as well as InterProScan
The Diverse World of Prokaryotic Argonautes ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 17
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

(v.5.28-67) for the Pfam and Superfamily databases. The proteins were aligned by the MAFFT program,v.7.3 (-ep 0 – genafpair –maxiterate 1000) (51); multiple alignments were manually curated and used forthe extraction of domain borders and features.
Phylogenetic analysis. To construct a nonredundant, representative sequence set for the phyloge-netic analysis, sequences of the PIWI-MID and APAZ domains were clustered using the UCLUST 4.2program (52) with the sequence identity threshold of 90%. The longest sequence was selected torepresent each cluster. The multiple alignment of PIWI-MID domains was carried out by the MAFFTprogram, v. 7.3 (-ep 0 – genafpair –maxiterate 1000) (51). Positions including greater than �0.5 gaps wereremoved from the alignment by trimAl, v.1.4 (53). Phylogenetic analysis was performed using theFastTree program (54) with default parameters, with the WAG evolutionary model and the discretegamma model with 20 rate categories. The tree structure was validated with bootstrap analysis (n � 100).
Analysis of the composition of pAgo operons. The protein-coding sequences of 16,264 genesneighboring all pAgo genes (10 upstream and 10 downstream genes for each pAgo) were clusteredbased on sequence homology. The pairwise similarity was calculated by using the profile-profile metricwith HHblits and HHsearch from the HH-suite (55, 56), as suggested in reference 57. In contrast to themore common sequence-sequence comparison method with using all-against-all BLAST analysis, thecomparison of HMM profiles represents a more sensitive strategy for detecting distant evolutionaryrelationships among diverged proteins. For this, a profile for each sequence was constructed with HHblits(-n 1 -E 1E�3) and the uniclast30_2017_10 HMM database (http://wwwuser.gwdg.de/~compbiol/uniclust/2017_10/). Then, all-against-all profile–profile search was conducted using HHsearch (-b 1 -b1000 -z 1 -Z 1000 -E 1E�5), which provided E values similar to what BLAST does. Clustering of genes intoorthogroups was done with the MCL algorithm implemented in the clusterMaker2 plug-in of Cytoscape3.6.1 (inflation factor � 2).
The annotation of obtained 1,892 protein orthogroups was performed with InterProScan (v.5.28-67)for the Pfam and Superfamily databases. Also, we have performed the PSI-BLAST search of APAZ,RecB-like, and RecB2 domains in the orthogroup proteins. The prediction of nucleases belonging to thePD-(D/E)XK superfamily was carried out with an SVM-based approach using the standalone version of the“PDEXK recognition” program kindly provided by Mindaugas Margelevicius (58). For each pAgo operon,we then determined the orthogroups to which operon genes belonged to. If a pAgo was encoded inseveral genomes, only the genome with the largest operon was selected for final counting. Onlyorthogroups that were present in at least 5% of operons for each pAgo type (minimum three operonsof any type of pAgo operon) were kept.
Counting the transposon copy number in genomes. The transposon insertions in genomes werecounted using genomic annotations of prokaryotic strains fetched from NCBI. Only genomes withstatuses “Complete Genome” and “Chromosome” were taken into account. For each genome, wecounted the number of occurrences of keyword “insertion sequence” or “transposase” in the genomicannotation file. If there were several completed genomes for a given strain, then the median number oflog2-transformed transposon insertions was calculated. If a pAgo gene was found in different strainsof the same species (having the same taxon_id in the NCBI annotation), then the median number oftransposon insertions among all strains was taken. For the comparison of transposon contents of strainsencoding or not encoding pAgos within a genus, the additional z-scaling was performed.
SUPPLEMENTAL MATERIALSupplemental material for this article may be found at https://doi.org/10.1128/mBio
.01935-18.FIG S1, PDF file, 0.01 MB.FIG S2, PDF file, 0.04 MB.FIG S3, PDF file, 0.5 MB.TABLE S1, XLS file, 0.3 MB.TABLE S2, XLSX file, 0.3 MB.TABLE S3, XLS file, 0.03 MB.DATA SET S1, TXT file, 0.02 MB.DATA SET S2, TXT file, 0.1 MB.DATA SET S3, TXT file, 0.1 MB.
ACKNOWLEDGMENTSWe thank Mindaugas Margelevicius for the help with the prediction of PD-(D/E)XK
motifs in proteins.This work was supported by the grant of the Ministry of Education and Science of
the Russian Federation 14.W03.31.0007.
REFERENCES1. Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song J-J, Ham-
mond SM, Joshua-Tor L, Hannon GJ. 2004. Argonaute2 is the catalyticengine of mammalian RNAi. Science 305:1437–1441. https://doi.org/10.1126/science.1102513.
Ryazansky et al. ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 18
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

2. Miyoshi K, Tsukumo H, Nagami T, Siomi H, Siomi MC. 2005. Slicerfunction of Drosophila Argonautes and its involvement in RISC forma-tion. Genes Dev 19:2837–2848. https://doi.org/10.1101/gad.1370605.
3. Rivas FV, Tolia NH, Song JJ, Aragon JP, Liu J, Hannon GJ, Joshua-Tor L.2005. Purified Argonaute2 and an siRNA form recombinant human RISC.Nat Struct Mol Biol 12:340 –349. https://doi.org/10.1038/nsmb918.
4. Rand TA, Ginalski K, Grishin NV, Wang X. 2004. Biochemical identificationof Argonaute 2 as the sole protein required for RNA-induced silencingcomplex activity. Proc Natl Acad Sci U S A 101:14385–14389. https://doi.org/10.1073/pnas.0405913101.
5. Djuranovic S, Nahvi A, Green R. 2012. miRNA-mediated gene silencing bytranslational repression followed by mRNA deadenylation and decay.Science 336:237–240. https://doi.org/10.1126/science.1215691.
6. Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E.2006. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev20:1885–1898. https://doi.org/10.1101/gad.1424106.
7. Rehwinkel J, Behm-Ansmant I, Gatfield D, Izaurralde E. 2005. A crucialrole for GW182 and the DCP1:DCP2 decapping complex in miRNA-mediated gene silencing. RNA 11:1640 –1647. https://doi.org/10.1261/rna.2191905.
8. Kuramochi-Miyagawa S, Watanabe T, Gotoh K, Totoki Y, Toyoda A, IkawaM, Asada N, Kojima K, Yamaguchi Y, Ijiri TW, Hata K, Li E, Matsuda Y,Kimura T, Okabe M, Sakaki Y, Sasaki H, Nakano T. 2008. DNA methylationof retrotransposon genes is regulated by Piwi family members MILI andMIWI2 in murine fetal testes. Genes Dev 22:908 –917. https://doi.org/10.1101/gad.1640708.
9. Sienski G, Dönertas D, Brennecke J. 2012. Transcriptional silencing oftransposons by Piwi and maelstrom and its impact on chromatin stateand gene expression. Cell 151:964 –980. https://doi.org/10.1016/j.cell.2012.10.040.
10. Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, Moazed D. 2004.RNAi-mediated targeting of heterochromatin by the RITS complex. Sci-ence 303:672– 676. https://doi.org/10.1126/science.1093686.
11. Makarova KS, Wolf YI, van der Oost J, Koonin EV. 2009. Prokaryotichomologs of Argonaute proteins are predicted to function as key com-ponents of a novel system of defense against mobile genetic elements.Biol Direct 4:29. https://doi.org/10.1186/1745-6150-4-29.
12. Swarts DC, Makarova K, Wang Y, Nakanishi K, Ketting RF, Koonin EV,Patel DJ, van der Oost J. 2014. The evolutionary journey of Argonauteproteins. Nat Struct Mol Biol 21:743–753. https://doi.org/10.1038/nsmb.2879.
13. Ma J-B, Yuan Y-R, Meister G, Pei Y, Tuschl T, Patel DJ. 2005. Structuralbasis for 5=-end-specific recognition of guide RNA by the A. fulgidus Piwiprotein. Nature 434:666 – 670. https://doi.org/10.1038/nature03514.
14. Parker JS, Roe SM, Barford D. 2004. Crystal structure of a PIWI proteinsuggests mechanisms for siRNA recognition and slicer activity. EMBO J23:4727– 4737. https://doi.org/10.1038/sj.emboj.7600488.
15. Parker JS, Roe SM, Barford D. 2005. Structural insights into mRNArecognition from a PIWI domain–siRNA guide complex. Nature 434:663– 666. https://doi.org/10.1038/nature03462.
16. Song JJ, Smith SK, Hannon GJ, Joshua-Tor L. 2004. Crystal structure ofArgonaute and its implications for RISC slicer activity. Science 305:1434 –1437. https://doi.org/10.1126/science.1102514.
17. Wang Y, Juranek S, Li H, Sheng G, Tuschl T, Patel DJ. 2008. Structureof an argonaute silencing complex with a seed-containing guide DNAand target RNA duplex. Nature 456:921–926. https://doi.org/10.1038/nature07666.
18. Wang Y, Sheng G, Juranek S, Tuschl T, Patel DJ. 2008. Structure of theguide-strand-containing argonaute silencing complex. Nature 456:209 –213. https://doi.org/10.1038/nature07315.
19. Wang Y, Juranek S, Li H, Sheng G, Wardle GS, Tuschl T, Patel DJ. 2009.Nucleation, propagation and cleavage of target RNAs in Ago silencingcomplexes. Nature 461:754 –761. https://doi.org/10.1038/nature08434.
20. Yuan Y-R, Pei Y, Ma J-B, Kuryavyi V, Zhadina M, Meister G, Chen H-Y,Dauter Z, Tuschl T, Patel DJ. 2005. Crystal structure of A. aeolicusargonaute, a site-specific DNA-guided endoribonuclease, provides in-sights into RISC-mediated mRNA cleavage. Mol Cell 19:405– 419. https://doi.org/10.1016/j.molcel.2005.07.011.
21. Yuan YR, Pei Y, Chen HY, Tuschl T, Patel DJ. 2006. A potential protein-RNA recognition event along the RISC-loading pathway from the struc-ture of A. aeolicus Argonaute with externally bound siRNA. Structure14:1557–1565. https://doi.org/10.1016/j.str.2006.08.009.
22. Parker JS, Parizotto EA, Wang M, Roe SM, Barford D. 2009. Enhancement
of the seed-target recognition step in RNA silencing by a PIWI/MIDdomain protein. Mol Cell 33:204 –214. https://doi.org/10.1016/j.molcel.2008.12.012.
23. Sheng G, Zhao H, Wang J, Rao Y, Tian W, Swarts DC, van der Oost J, PatelDJ, Wang Y. 2014. Structure-based cleavage mechanism of Thermusthermophilus Argonaute DNA guide strand-mediated DNA target cleav-age. Proc Natl Acad Sci U S A 111:652– 657. https://doi.org/10.1073/pnas.1321032111.
24. Kaya E, Doxzen KW, Knoll KR, Wilson RC, Strutt SC, Kranzusch PJ, DoudnaJA. 2016. A bacterial Argonaute with noncanonical guide RNA specificity.Proc Natl Acad Sci U S A 113:4057– 4062. https://doi.org/10.1073/pnas.1524385113.
25. Miyoshi T, Ito K, Murakami R, Uchiumi T. 2016. Structural basis for therecognition of guide RNA and target DNA heteroduplex by Argonaute.Nat Commun 7:11846. https://doi.org/10.1038/ncomms11846.
26. Doxzen KW, Doudna JA. 2017. DNA recognition by an RNA-guidedbacterial Argonaute. PLoS One 12:e0177097. https://doi.org/10.1371/journal.pone.0177097.
27. Willkomm S, Oellig CA, Zander A, Restle T, Keegan R, Grohmann D,Schneider S. 2017. Structural and mechanistic insights into an archaealDNA-guided Argonaute protein. Nat Microbiol 2:17035. https://doi.org/10.1038/nmicrobiol.2017.35.
28. Liu Y, Esyunina D, Olovnikov I, Teplova M, Kulbachinskiy A, Aravin AA,Patel DJ. 2018. Accommodation of helical imperfections in Rhodobactersphaeroides Argonaute ternary complexes with guide RNA and targetDNA. Cell Rep 24:453– 462. https://doi.org/10.1016/j.celrep.2018.06.021.
29. Elkayam E, Kuhn CD, Tocilj A, Haase AD, Greene EM, Hannon GJ, Joshua-Tor L. 2012. The structure of human argonaute-2 in complex withmiR-20a. Cell 150:100 –110. https://doi.org/10.1016/j.cell.2012.05.017.
30. Nakanishi K, Weinberg DE, Bartel DP, Patel DJ. 2012. Structure of yeastArgonaute with guide RNA. Nature 486:368 –374. https://doi.org/10.1038/nature11211.
31. Faehnle CR, Elkayam E, Haase AD, Hannon GJ, Joshua-Tor L. 2013. Themaking of a slicer: activation of human Argonaute-1. Cell Rep3:1901–1909. https://doi.org/10.1016/j.celrep.2013.05.033.
32. Schirle NT, Sheu-Gruttadauria J, MacRae IJ. 2014. Structural basis formicroRNA targeting. Science 346:608 – 613. https://doi.org/10.1126/science.1258040.
33. Matsumoto N, Nishimasu H, Sakakibara K, Nishida KM, Hirano T, IshitaniR, Siomi H, Siomi MC, Nureki O. 2016. Crystal structure of silkwormPIWI-clade Argonaute Siwi bound to piRNA. Cell 167:484 – 497.e9.https://doi.org/10.1016/j.cell.2016.09.002.
34. Park MS, Phan HD, Busch F, Hinckley SH, Brackbill JA, Wysocki VH,Nakanishi K. 2017. Human Argonaute3 has slicer activity. Nucleic AcidsRes 45:11867–11877. https://doi.org/10.1093/nar/gkx916.
35. Kwak PB, Tomari Y. 2012. The N domain of Argonaute drives duplexunwinding during RISC assembly. Nat Struct Mol Biol 19:145–151.https://doi.org/10.1038/nsmb.2232.
36. Bujnicki JM, Rychlewski L. 2001. Identification of a PD-(D/E)XK-like do-main with a novel configuration of the endonuclease active site in themethyl-directed restriction enzyme Mrr and its homologs. Gene 267:183–191. https://doi.org/10.1016/S0378-1119(01)00405-X.
37. Kinch LN, Ginalski K, Rychlewski L, Grishin NV. 2005. Identification ofnovel restriction endonuclease-like fold families among hypotheticalproteins. Nucleic Acids Res 33:3598 –3605. https://doi.org/10.1093/nar/gki676.
38. Olovnikov I, Chan K, Sachidanandam R, Newman DK, Aravin AA. 2013.Bacterial argonaute samples the transcriptome to identify foreign DNA.Mol Cell 51:594 – 605. https://doi.org/10.1016/j.molcel.2013.08.014.
39. Swarts DC, Jore MM, Westra ER, Zhu Y, Janssen JH, Snijders AP, Wang Y,Patel DJ, Berenguer J, Brouns SJJ, van der Oost J. 2014. DNA-guided DNAinterference by a prokaryotic Argonaute. Nature 507:258 –261. https://doi.org/10.1038/nature12971.
40. Swarts DC, Hegge JW, Hinojo I, Shiimori M, Ellis MA, Dumrongkulraksa J,Terns RM, Terns MP, van der Oost J. 2015. Argonaute of the archaeonPyrococcus furiosus is a DNA-guided nuclease that targets cognate DNA.Nucleic Acids Res 43:5120 –5129. https://doi.org/10.1093/nar/gkv415.
41. Zander A, Willkomm S, Ofer S, van Wolferen M, Egert L, Buchmeier S,Stockl S, Tinnefeld P, Schneider S, Klingl A, Albers SV, Werner F, Grohm-ann D. 2017. Guide-independent DNA cleavage by archaeal Argonautefrom Methanocaldococcus jannaschii. Nat Microbiol 2:17034. https://doi.org/10.1038/nmicrobiol.2017.34.
42. Hegge JW, Swarts DC, van der Oost J. 2018. Prokaryotic Argonaute
The Diverse World of Prokaryotic Argonautes ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 19
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from

proteins: novel genome-editing tools? Nat Rev Microbiol 16:5–11.https://doi.org/10.1038/nrmicro.2017.73.
43. Willkomm S, Makarova KS, Grohmann D. 2018. DNA silencing by pro-karyotic Argonaute proteins adds a new layer of defense against invad-ing nucleic acids. FEMS Microbiol Rev 42:376 –387. https://doi.org/10.1093/femsre/fuy010.
44. Juliano C, Wang J, Lin H. 2011. Uniting germline and stem cells: thefunction of Piwi proteins and the piRNA pathway in diverse organisms.Annu Rev Genet 45:447– 469. https://doi.org/10.1146/annurev-genet-110410-132541.
45. Hutvagner G, Simard MJ. 2008. Argonaute proteins: key players inRNA silencing. Nat Rev Mol Cell Biol 9:22–32. https://doi.org/10.1038/nrm2321.
46. Koonin EV. 2017. Evolution of RNA- and DNA-guided antivirus defensesystems in prokaryotes and eukaryotes: common ancestry vs conver-gence. Biol Direct 12:5. https://doi.org/10.1186/s13062-017-0177-2.
47. Swarts DC, Koehorst JJ, Westra ER, Schaap PJ, van der Oost J. 2015.Effects of Argonaute on gene expression in Thermus thermophilus. PLoSOne 10:e0124880. https://doi.org/10.1371/journal.pone.0124880.
48. Zander A, Holzmeister P, Klose D, Tinnefeld P, Grohmann D. 2014.Single-molecule FRET supports the two-state model of Argonaute ac-tion. RNA Biol 11:45–56. https://doi.org/10.4161/rna.27446.
49. Jung SR, Kim E, Hwang W, Shin S, Song JJ, Hohng S. 2013. Dynamicanchoring of the 3=-end of the guide strand controls the target disso-ciation of Argonaute-guide complex. J Am Chem Soc 135:16865–16871.https://doi.org/10.1021/ja403138d.
50. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, LipmanDJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of proteindatabase search programs. Nucleic Acids Res 25:3389 –3402. [9254694]https://doi.org/10.1093/nar/25.17.3389.
51. Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment soft-ware version 7: improvements in performance and usability. Mol BiolEvol 30:772–780. https://doi.org/10.1093/molbev/mst010.
52. Edgar RC. 2010. Search and clustering orders of magnitude faster thanBLAST. Bioinformatics 26:2460–2461. https://doi.org/10.1093/bioinformatics/btq461.
53. Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: atool for automated alignment trimming in large-scale phylogeneticanalyses. Bioinformatics 25:1972–1973. https://doi.org/10.1093/bioinformatics/btp348.
54. Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximatelymaximum-likelihood trees for large alignments. PLoS One 5:e9490.https://doi.org/10.1371/journal.pone.0009490.
55. Söding J. 2005. Protein homology detection by HMM-HMM comparison.Bioinformatics 21:951–960. https://doi.org/10.1093/bioinformatics/bti125.
56. Remmert M, Biegert A, Hauser A, Söding J. 2011. HHblits: lightning-fastiterative protein sequence searching by HMM-HMM alignment. NatMethods 9:173–175. https://doi.org/10.1038/nmeth.1818.
57. Bernardes JS, Vieira FR, Costa LM, Zaverucha G. 2015. Evaluation andimprovements of clustering algorithms for detecting remote homolo-gous protein families. BMC Bioinformatics 16:34. https://doi.org/10.1186/s12859-014-0445-4.
58. Laganeckas M, Margelevicius M, Venclovas C. 2011. Identification of newhomologs of PD-(D/E)XK nucleases by support vector machines trainedon data derived from profile-profile alignments. Nucleic Acids Res 39:1187–1196. https://doi.org/10.1093/nar/gkq958.
59. Lisitskaya L, Aravin AA, Kulbachinskiy A. DNA interference and beyond:structure and functions of prokaryotic Argonaute proteins. Nat Com-mun, in press.
Ryazansky et al. ®
November/December 2018 Volume 9 Issue 6 e01935-18 mbio.asm.org 20
on February 26, 2020 by guest
http://mbio.asm
.org/D
ownloaded from





























