Embed Size (px)
Citation preview

523
Abstract. – OBJECTIVE: MiR-26a is involved in regulating myocardial remodeling and it is al-so related to organ fibrosis. Its role in myocar-dial fibrosis process is still controversy. As a definite target gene of miR-26a, phosphatase and tensin homology does on chromosome ten (PTEN) plays a role in regulating PTEN-PI3K/AKT signaling pathway. This study explored the function of miR-26a in regulating PTEN-PI3K/AKT signaling pathway, MMP-9 expression, and myocardial fibrosis after acute myocardial in-farction (AMI).
MATERIALS AND METHODS: AMI model was established on Sprague-Dawley (SD) rats. Hy-droxyproline, COL1A1, miR-26a, PTEN, p-AKT, and MMP-9 expressions in myocardial tissue at 1 week, 2 weeks, and 4 weeks after modeling were detected. Human cardiac fibroblasts (HCF) were cultured in vitro to detect miR-26a, PTEN, p-AKT, MMP-9, COL1A1, and α-SMA expressions in the process of myofibroblast differentiation (P1, P3, P5). HCF in P5 were transfected with miR-26a mimics or inhibitor to test miR-26a, PTEN, p-AKT, MMP-9, COL1A1, and α-SMA expressions.
RESULTS: Hydroxyproline, COL1A1, miR-26a, p-AKT, and MMP-9 overexpressed, while PTEN downregulated in myocardial tissue during the process of myocardial fibrosis after AMI. MiR-26a, PTEN, p-AKT, MMP-9, COL1A1, and α-SMA expression gradually enhanced, while PTEN de-clined, following the process of HCF differenti-ating into myofibroblast. MiR-26a elevation sup-pressed PTEN expression, and increased p-AKT, MMP-9, COL1A1, and α-SMA levels. MiR-26a re-duction significantly upregulated PTEN level, weakened PI3K/AKT signaling pathway activity, and declined MMP-9, COL1A1, and α-SMA pro-tein expression.
CONCLUSIONS: MiR-26a upregulation may play a role in myocardial fibrosis after AMI by suppressing PTEN, enhancing PI3K/AKT signal-ing pathway and MMP-9 levels.
Key Words:miR-26a, PTEN, PI3K/AKT, AMI, Myocardial fibrosis.
Introduction
Myocardial infarction (MI) refers to myocar-dial necrosis caused by coronary artery acute or persistent hypoxia-ischemia, featured as se-vere and persistent retrosternal pain in clinic, complicated with cardiac arrhythmia, shock, or heart failure. It is one of the important reasons of cardiac death1,2. MI caused by myocardial hypoxia-ischemia is a type of cardiovascular disease which seriously harms human health and causes heart failure. It is characterized as complex pathogenesis and critical dangerous; thus, not timely treatment may lead to high dis-ability and lethality rates3-5. Insufficient blood supply induced MI is easy to cause oxidative stress, leading to myocardial tissue necrosis, inflammatory reaction6, and pathological myo-cardial remodeling7. Myocardial fibrosis is an important pathological change after MI. Cardiac physiological structures are destroyed, and myo-cardial interstitial extracellular matrix excessive proliferation and accumulation under the effect of multiple pro-fibrogenic factors after MI, lead-ing to increased heart tissue stiffness reduced myocardial systolic and diastolic function ab-normality of reserve, and eventually resulting in abnormal cardiac electrophysiological function, malignant arrhythmia, and sudden death8,9. Ex-tracellular matrix protein abnormal proliferation and accumulation is related to the enhancement of cardiac fibroblasts activation, proliferation, differentiation to myofibroblasts, and collagen secretion10. MicroRNAs is a kind of endogenous single non-coding RNAs at the length of 22-25 nucleotides. They are involved in all types of bi-ological processes including energy metabolism, cell growth, survival, and differentiation by identifying the 3’-UTR sequence of target gene
European Review for Medical and Pharmacological Sciences 2018; 22: 523-531
S. ZHANG, R. CUI
Department of Cardiology, Heilongjiang provincial Hospital,Harbin, Heilongjiang, P.R. China
Shu Zhang and Rong Cui contributed equally to this work
Corresponding Author: Rong Cui, MD; e-mail: [email protected]
The targeted regulation of miR-26a on PTEN-PI3K/AKT signaling pathway in myocardial fibrosis after myocardial infarction

S. Zhang, R. Cui
524
mRNA to suppress mRNA translation or direct degrade mRNA11. Numerous studies12-14 showed that myocardial fibrosis process was regulated by a series of signaling pathways and miRNAs. It was found that miR-26a was involved in the regulation of myocardial remodeling15, and al-so associated with organ fibrosis16,17. However, whether it participates in myocardial fibrosis process is still unclear. Matrix metalloproteinas-es (MMPs) play an important role in the degra-dation of extracellular matrix and organ fibrosis. As one of important members, MMP-9 activity elevated throughout the course of myocardial fi-brosis18,19. It was showed that PI3K/AKT signal-ing pathway played a critical role in regulating MMP-9 expression20. Abnormal activation of PI3K/AKT signaling pathway is closely relat-ed to the occurrence of myocardial infarction after MI21,22. Phosphatidylinositol (3,4,5)-tris-phosphate (PIP3) is the most important substrate of phosphatase and tensin homology located on chromosome ten (PTEN). PTEN suppresses PI3K/AKT signaling pathway activation through dephosphorylating PIP323. Downregulation of PTEN and excessive activation of PI3K/AKT signaling pathway are confirmed to participate in the occurrence of myocardial fibrosis24. As the definite target gene of miR-26a, PTEN ex-pression and function is negatively regulated by miR-26a25. The role of miR-26a in regulating PTEN-PI3K/AKT signaling pathway, MMP-9 expression, and myocardial fibrosis is lack of investigation.
Materials and Methods
Main Reagents and MaterialsHealthy male Sprague Dawley (SD) rats
weighted 220-250 g and at 6-7 weeks old were purchased from Heilongjiang Laboratory Animal Center, Academy of Military Medical Sciences (Heilongjiang, China). Human cardiac fibroblasts (HCF) and HCF specific medium Fibroblast Me-dium-2 were bought from ScienCell (Carlsbad, CA, USA). Opti-MEM I medium was got from Gibco (Rockville, MD, USA). RNA extraction kit was acquired from Omega (Norcross, GA, USA). Reverse transcription kit and Real-time PCR kit were from TaKaRa (Dalian, Liaoning, China). Oligonucleotide for transfection and PCR primers were designed and synthetized by Gene Phar-ma (Shanghai, China). PTEN primary antibody was got from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). MMP-9 and COL1A1 primary antibodies were from Abcam (Cambridge, MA, USA). P-AKT and α-SMA antibodies were from Cell Signaling Technology (CST, Danvers, MA, USA). Hydroxyproline ELISA kit was from ML-BIO (Shanghai, China). Rats were used for all experiments, and all procedures were approved by the Animal Ethics Committee of Heilongjiang Hospital.
Rat MI Model EstablishmentThe experimental rats were randomly equal-
ly divided into Sham group and AMI group with 15 in each group. Each group was further divided into postoperative 1 week, 2 weeks, and 4 weeks subgroups with 5 in each subgroup. SD rats were anesthetized by 10% chloral hydrate (3 mg/g) intraperitoneal injection and fixed on the plastic foam board after no righting reac-tion. The limbs were connected with, electro-cardiogram (ECG) monitor. The neck skin was incised under aseptic condition and the muscle was separated to expose the trachea. An animal breathing machine was connected for assisted respiration after endotracheal intubation, with the breathing ratio at 1:2, respiratory frequency at 70 bpm, and tidal volume at 10-12 mL. The chest was opened between 3rd and 4th left ribs to expose the heart. The left anterior descend-ing coronary artery was identified between the pulmonary arterial cone and aorta, and ligated using 6-0 no damage stitches. ECG monitor exhibited ST segment arch lift for 0.1 mV or T wave high, pale myocardium, and abate pulse were applied to confirm the MI model success. Then, the incision was closed and penicillin was adopted to prevent infection. The rats in sham group received the same operation with-out left anterior descending coronary artery ligation. The rats were anesthetized by 10% chloral hydrate intraperitoneal injection and received cardiac ultrasonography at 1 week, 2 weeks, and 4 weeks after surgery, respectively. Left ventricular end systolic diameter and left ventricular diastolic diameter were recorded at the level of papillary muscle prior to mi-tral valve through the left ventricular short axis view. Left ventricular ejection fraction (LVEF) and left ventricular fractional short-ening (LVFS) were automatically calculated. The tissue samples at MI region were extracted from the rats at 1 week, 2 weeks, and 4 weeks after modeling, and stored in liquid nitrogen and then in -80°C cryogenic refrigerator.

MiR-26a regulates PTEN in myocardial fibrosis
525
Cell Culture and GroupingThe cell culture dish was coated by poly-L-ly-
sine at 2 μg/cm2 and incubated at 37°C overnight.
HCF cells were seeded into the dish at 5000 cells/cm2 and maintained in Fibroblast Medium-2. The cells were digested by enzyme when the fusion reached 70-80% and named P1. The cells were then passaged or used for experiments. HCF cells in P5 were divided into four groups, including NC mimics, miR-26a mimics, NC inhibitor, and miR-26a inhibitor. Lipofectamine 2000 and oli-gonucleotide were diluted in Opti-MEM I and transfected to the cells for 6 h at 37°C and 5% CO2. The cells were further cultured for 48 h for the following experiments.
qRT-PCRTotal RNA was extracted using the kit
from OMEGA. The cells were added with 350 μl TRK buffer and 350 μl 70% ethanol. After blending, the solution was moved to the fil-tration column and centrifuged at 10,000 rpm for 1 min. After washed by 500 μl Wash buf-fer I for 1 time and 500 μl Wash buffer II for 2 times, the membrane was air dried at room temperature and added with 40 μl RNase free water for 2 min. The solved RNA was moved to the EP tube after centrifuged at 10,000 rpm for 1 min. The reverse transcription system in 20 μl contained 2 μg total RNA, 1 μl dNTP (10 mmol/L), 4 μl RT buffer (5 ×), 2 μl RT primer (1 μmol/L), 2 μl reverse transcriptase, 0.5 μl RNase inhibitor, and RNase free H2O. The reverse transcription reaction was per-formed at 16°C for 30 min, 42°C for 15 min, and 85°C for 5 min. The obtained cDNA was stored at -20°C refrigerator. PCR amplifica-tion was performed using cDNA as template under the effect of TaqDNA polymerase. The primers sequences used were as follows. miR-26aPF: 5′-TTGGATCCGTCAGAAATTCTCTC-CCGAGG-3′, miR-26aPR: 5′-GGTCTAGAT-GTGAACTCTGGTGTTGGTGC-3′; U6PF: 5′-CTCGCTTCGGCAGCACA-3′, U6PR: 5′-AAC-GCTTCACGAATTTGCGT-3′; PTENPF: 5′-CT-GGTCTGCCAGCTAAAGGT-3′, PTENPR: 5′-TCACCACACACAGGTAACGG-3′; MMP9PF: 5′-TGTACCGCTATGGTTACACTCG-3′, MMP-9PR: 5′-GGCAGGGACAGTTGCTTCT-3′; CO-L1A1PF: 5′-GTGCGATGACGTGATCTGTGA-3′, COL1A1PR: 5′-CGGTGGTTTCTTGGTCGGT-3′; α-SMAPF: 5′-AAAAGACAGCTACGTGGGT-GA-3′, α-SMAPR: 5′-GCCATGTTCTATCGGG-TACTTCT-3′; β-actinPF: 5′-GCACTCTTCCAG-
CCTTCC-3′, β-actinPR: 5′-AGAAAGGGTGTA-ACGCAACTAAG-3′. PCR reaction system in 10 μl volume was composed of 4.5 μl 2 × SYBR Green Mixture, 0.5 μl primer (5 μmol/L), 1 μl cD-NA, and 3.5 μl ddH2O. The reaction was started at 95°C for 5 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min.
Western BlotThe cell lysis in SDS was boiled for 5 min
and quantified using BCA kit. A total of 60 μg sample was separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membrane. After blocked by 5% skim milk at room temperature for 1 h, the membrane was incubated in primary antibody at 4°C overnight and washed by phosphate-buff-ered saline Tween (PBST) for three times. Next, the membrane was incubated in horseradish peroxidase (HRP) labeled secondary antibody at room temperature for 60 min and washed by PBST for three times. At last, the membrane was treated by ECL reagent and developed. The data was analyzed using Quantity One software (Bio-Rad).
Myocardial Hydroxyproline Content Measurement
A total of 100 mg myocardial tissue was di-gested and hydrolyzed to obtain the supernatant. Hydroxyproline content was determined by ELI-SA. In brief, 50 μl standard substrate or diluted sample were added to the plate and incubated at 37°C for 30 min. After washed by washing buffer for 5 times, 50 μl enzyme-labeled reagent were added to each well at 37°C for 30 min. The plate was washed by washing buffer for 5 times and treated by 50 μl color developing agent A and 50 μl color developing agent B at 37°C for 15 min. Next, the plate was added with 50 μl stop buffer and read at 450 nm.
Statistical AnalysisAll data analysis was performed on SPSS
18.0 software (SPSSS Inc., Chicago, IL, USA). The measurement data was presented as mean ± standard deviation, while the enumeration data was depicted as percentage. Data between groups were compared by x2-test or one-way ANOVA when necessary. LSD was performed for post-hoc test. p < 0.05 was considered as statistical significance.
S. Zhang, R. Cui
526
Results
The degree of Myocardial Fibrosis Gradually Increased After MI
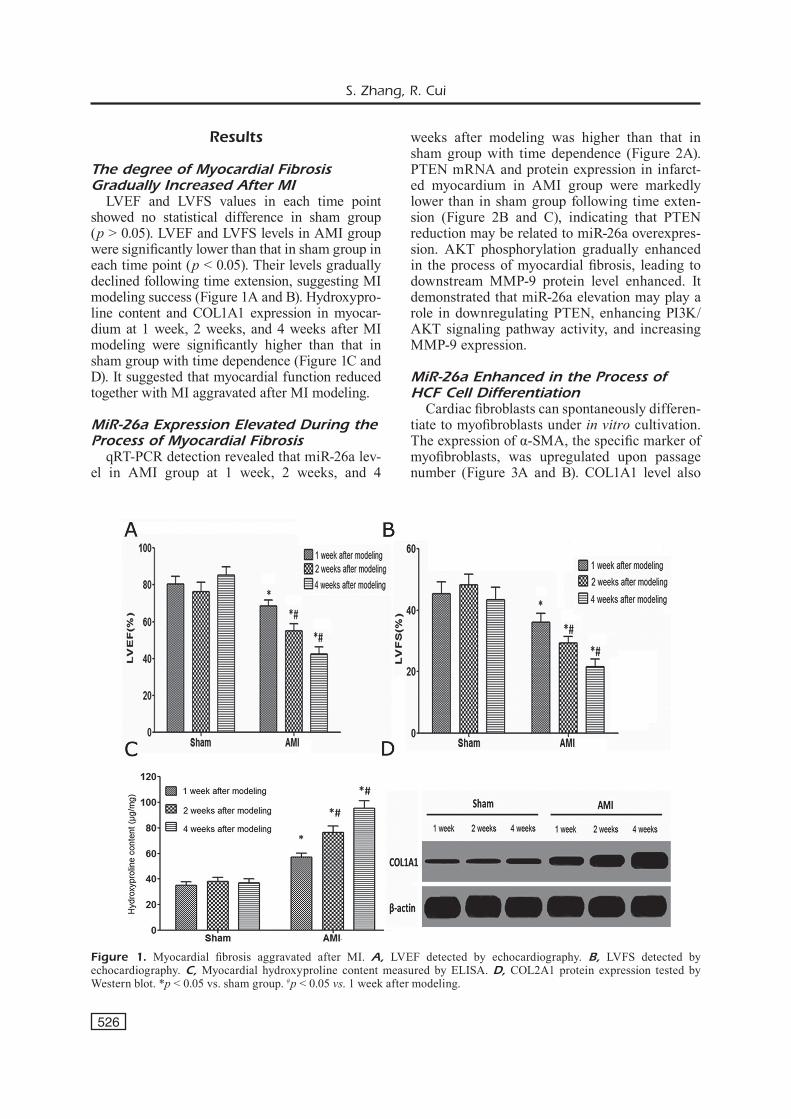
LVEF and LVFS values in each time point showed no statistical difference in sham group (p > 0.05). LVEF and LVFS levels in AMI group were significantly lower than that in sham group in each time point (p < 0.05). Their levels gradually declined following time extension, suggesting MI modeling success (Figure 1A and B). Hydroxypro-line content and COL1A1 expression in myocar-dium at 1 week, 2 weeks, and 4 weeks after MI modeling were significantly higher than that in sham group with time dependence (Figure 1C and D). It suggested that myocardial function reduced together with MI aggravated after MI modeling.
MiR-26a Expression Elevated During the Process of Myocardial Fibrosis
qRT-PCR detection revealed that miR-26a lev-el in AMI group at 1 week, 2 weeks, and 4
weeks after modeling was higher than that in sham group with time dependence (Figure 2A). PTEN mRNA and protein expression in infarct-ed myocardium in AMI group were markedly lower than in sham group following time exten-sion (Figure 2B and C), indicating that PTEN reduction may be related to miR-26a overexpres-sion. AKT phosphorylation gradually enhanced in the process of myocardial fibrosis, leading to downstream MMP-9 protein level enhanced. It demonstrated that miR-26a elevation may play a role in downregulating PTEN, enhancing PI3K/AKT signaling pathway activity, and increasing MMP-9 expression.
MiR-26a Enhanced in the Process of HCF Cell Differentiation
Cardiac fibroblasts can spontaneously differen-tiate to myofibroblasts under in vitro cultivation. The expression of α-SMA, the specific marker of myofibroblasts, was upregulated upon passage number (Figure 3A and B). COL1A1 level also
Figure 1. Myocardial fibrosis aggravated after MI. A, LVEF detected by echocardiography. B, LVFS detected by echocardiography. C, Myocardial hydroxyproline content measured by ELISA. D, COL2A1 protein expression tested by Western blot. *p < 0.05 vs. sham group. #p < 0.05 vs. 1 week after modeling.

MiR-26a regulates PTEN in myocardial fibrosis
527
elevated according to passage number, revealing that the ability of synthetizing collagen gradually enhanced in the process of cardiac fibroblasts differentiate to myofibroblasts (Figure 3A and B). Further experiments showed that PTEN reduced, while p-AKT and MMP-9 upregulated following miR-26a increase in the differentiation process (Figure 3A and B).
MiR-26a Regulated PI3K/AKT Activity and MMP-9 Expression in HCF Cells
HCF cells in P5 were applied for miR-26a mimic or miR-26a inhibitor transfection. It was demonstrated that miR-26a upregulation sig-nificantly suppressed PTEN expression, and in-creased p-AKT, MMP-9, COL1A1, and α-SMA levels (Figure 4A and B). MiR-26a reduction
upregulated PTEN level, weakened PI3K/AKT signaling pathway activity, and declined MMP-9, COL1A1, and α-SMA protein expression (Figure 4A and B).
Discussion
Myocardial fibrosis, resulting from collagen excessive deposition, characterized by elevation of collagen concentration and volume various collagen imbalance and disorder, and myocardial interstitial structure abnormality, is the common pathological feature of various cardiovascular diseases, such as atherosclerosis, hypertension, viral myocarditis, cardiomyopathy, arrhythmia, MI. Myocardial fibrosis and AMI caused ventric-
Figure 2. MiR-26a expression elevated during the process of myocardial fibrosis. A, miR-26a expression detected by qRT-PCR. B, PTEN mRNA expression detected by qRT-PCR. C, Protein expression tested by Western blot. *p < 0.05 vs. sham group. #p < 0.05 vs. 1 week after modeling.
Figure 3. MiR-26a enhanced in the process of HCF cell differentiation. A, mRNA expression detected by qRT-PCR. B, Protein expression detected by Western blot. *p < 0.05 vs. P1. #p < 0.05 vs. P3.

S. Zhang, R. Cui
528
ular remodeling are important reasons of heart failure, malignant arrhythmia, and cardiac death. There are numerous types of collagen in myocar-dial interstitium, while type I collagen accounts for more than 80%. It plays a key role in maintain-ing the intensity of ventricular wall because of its thick fiber, stiffness, and anti-traction. Type I collagen content or proportion excessive increase will reduce the compliance of the ventricular wall; thus, it is reported to participate in the process of myocardial fibrosis26. This study used animal model to observe the myocardial fibrosis process after MI. It was showed that the expression of type I collagen protein in myocardium from AMI group was significantly higher than the sham group with time dependence, which was similar to the results of Hookana et al26. Hydroxyproline is the main component of collagen, while almost all hydroxyproline in the animal tissue is derived from collagen. As a result, hydroxyproline con-tent in myocardial tissues can represent collagen level to reflect the degree of myocardial fibrosis27. Our results demonstrated that hydroxyproline content was higher in the AMI group, confirming myocardial fibrosis after MI. MMPs are a kind of Zn2+ dependent extracellular matrix protein hy-drolysis enzyme family that can degrade almost all components of extracellular matrix. MMPs exist in normal myocardial tissue in inactive state and may be quickly activated under the stimulus of ischemia-hypoxia or inflammation28. The lev-els of MMPs with enzyme activity significantly
increase in the myocardial tissue and circulating blood during myocardial fibrosis, while MMP-9 is the leading type29. Extracellular collagen is degraded by elevated MMP-9, leading to abnor-mal collagen reticular formation promoting the occurrence of myocardial fibers30. This study observed that MMP-9 level in myocardium from AMI group increased after infarction, suggesting that MMP-9 elevation participated in the process of myocardial fibrosis. It was found that miR-26a was involved in the regulation of myocardial re-modeling15 and associated with organ fibrosis16,17. However, whether it participates in myocardial fibrosis is controversy. This study showed that miR-26a upregulated in the myocardium after infarction. It also gradually increased following the degree of myocardial fibrosis, suggesting that miR-26a may promote myocardial fibrosis. As a member of PTP gene family, PTEN locates on 10q23.3 with a transcription product at 515 kb31. PTEN can dephosphorylate PIP3 to antago-nize the phosphorylation effect of PI3K on PIP2, thus preventing AKT and downstream signaling pathway activation23. Several studies demonstrat-ed that PI3K/AKT signaling pathway activation played an important role in upregulating MMP-932-34 and promoting myocardial fibrosis35. As a negative regulatory factor of PI3K/AKT signaling pathway, PTEN downregulation plays a promot-ing role in myocardial fibrosis36. The expression and function of PTEN is negatively regulated by miR-26a25. This study investigated the role of
Figure 4. MiR-26a regulated PI3K/AKT activity and MMP-9 expression in HCF cells. A, mRNA expression detected by qRT-PCR. B, Protein expression detected by Western blot. *p < 0.05 vs. mimic NC. #p < 0.05 vs. inhibitor NC.

MiR-26a regulates PTEN in myocardial fibrosis
529
miR-26a in regulating PTEN-PI3K/AKT signal-ing pathway, MMP-9 expression, and myocardial fibrosis. The results exhibited that PTEN level reduced in myocardial tissue after infarction, which was similar to Gao et al36 findings; PTEN expression declined in infarcted myocardial tis-sue following time extension. Furthermore, AKT phosphorylation also increased in infarcted myo-cardial tissue, which was in accordance with the upregulation trend of MMP-9. It suggested that miR-26a abnormal elevation may play a role in reducing PTEN, enhancing PI3K/AKT signal-ing pathway activity, and upregulating MMP-9, which may be a mechanism of its participating in myocardial fibrosis. Cardiac fibroblast is an im-portant component in myocardial tissue that can regulate extracellular matrix synthesis and deg-radation. MMPs in myocardial tissue are mainly synthetized and secreted by cardiac fibroblasts. Cardiac fibroblasts abnormally activate, increase proliferation and migration, differentiate to se-cretory myofibroblasts, and enhance the ability of collagen synthesis and secretion, thus playing a critical role in promoting myocardial fibrosis10. Upon in vitro HCF spontaneous differentiate to myofibroblasts model, this study observed miR-26a expression gradually increased, while PTEN reduced, and PI3K/AKT signaling pathway activ-ity, MMP-9 and COL1A1 expression elevated in the differentiation process, which was in accor-dance with the animal model. Zhong et al37 found that PTEN downregulation was associated with cardiac fibroblasts proliferation and migration abilities enhancement. Lorenzen et al24 reported that reducing PTEN level can promote cardiac fibroblasts survival and proliferation. They sug-gested that PTEN abnormal expression may be involved in accelerating myocardial fibrosis. This study observed that PTEN level declined in car-diac fibroblasts in the process of differentiation to myofibroblasts, indicating that PTEN down-regulation may participate in myocardial fibrosis. This may have the common theoretical basis and mechanism found by Zhong et al37 and Lorenzen et al24. In addition, cardiac fibroblasts differ-entiate into muscle fibers in the process of cell differentiation, not only resulting in enhanced collagen synthesis, but also overexpressed MMP-9 in the process of cardiac fibroblasts differentiate to myofibroblasts, revealing collagen degradation is strengthened. Vadla et al38 showed that except collagen synthesis elevation, MMP-2 and MMP-9 with the function of collagen degradation also significantly upregulated in myocardial fibrosis,
suggesting the myocardial fibrosis process was the result of collagen synthesis abnormal increase and degradation disorder. To further clarify the influence of miR-26a on myocardial fibrosis, this study transfected miR-26a mimic or miR-26a inhibitor to change miR-26a level in HCF, re-spectively. It was confirmed that miR-26a had the ability to restrain PTEN expression, enhance PI3K/AKT signaling pathway activity, and pro-mote MMP-9 expression.
Conclusions
MiR-26a abnormal elevation inhibited PTEN, strengthened PI3K/AKT signaling pathway ac-tivity, and upregulated MMP-9 level, thus play-ing a part in facilitating cardiac fibrosis after AMI.
AcknowledgementsThis project was supported by the National Natural Science Foundation of China Youth Foud (NO. 81400392).
Conflict of InterestThe Authors declare that they have no conflict of interests.
References
1) Lisowska a, Makarewicz-wujec M, FiLipiak kj. Risk factors, prognosis and secondary prevention of myocardial infarction in young adults in Poland. Kardiol Pol 2016; 74: 1148-1153.
2) Davies N. Treating ST-elevation myocardial infarc-tion. Emerg Nurse 2016; 24: 20-25.
3) YaNg H, suN w, QuaN N, waNg L, cHu D, cates c, Liu Q, zHeNg Y, Li j. Cardioprotective ac-tions of Notch1 against myocardial infarction via LKB1-dependent AMPK signaling pathway. Bio-chem Pharmacol 2016; 108: 47-57.
4) DeNg F, Xia Y, Fu M, Hu Y, jia F, raHarDjo Y, DuaN Y, He L, cHaNg j. Influence of heart failure on the prognosis of patients with acute myocardial in-farction in southwestern China. Exp Ther Med 2016; 11: 2127-2138.
5) sHibata t, kawakaMi s, NogucHi t, taNaka t, asauMi Y, kaNaYa t, Nagai t, Nakao k, FujiNo M, Nagatsuka k, isHibasHi-ueDa H, NisHiMura k, MiYaMoto Y, kusaNo k, aNzai t, goto Y, ogawa H, YasuDa s. Prevalence, clinical features, and prognosis of acute myocar-dial infarction attributable to coronary artery em-bolism. Circulation 2015; 132: 241-250.
6) westMaN pc, LipiNski Mj, Luger D, waksMaN r, boNow ro, wu e, epsteiN se. Inflammation as a

S. Zhang, R. Cui
530
driver of adverse left ventricular remodeling af-ter acute myocardial infarction. J Am Coll Cardiol 2016; 67: 2050-2060.
7) LiNDseY ML, iYer rp, juNg M, DeLeoN-peNNeLL kY, Ma Y. Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling. J Mol Cell Cardiol 2016; 91: 134-140.
8) FraNcis stuart sD, De jesus NM, LiNDseY ML, rip-pLiNger cM. The crossroads of inflammation, fibro-sis, and arrhythmia following myocardial infarc-tion. J Mol Cell Cardiol 2016; 91: 114-122.
9) zHu F, Li Y, zHaNg j, piao c, Liu t, Li HH, Du j. Senescent cardiac fibroblast is critical for cardi-ac fibrosis after myocardial infarction. PLoS One 2013; 8: e74535.
10) vaN NieuweNHoveN Fa, turNer Na. The role of car-diac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vascul Pharmacol 2013; 58: 182-188.
11) NoLLet e, HoYMaNs vY, vaN craeNeNbroeck aH, vriNts cj, vaN craeNeNbroeck eM. Improving stem cell therapy in cardiovascular diseases: the po-tential role of microRNA. Am J Physiol Heart Circ Physiol 2016; 311: H207-218.
12) tao H, YaNg jj, Hu w, sHi kH, DeNg zY, Li j. Non-coding RNA as regulators of cardiac fibrosis: cur-rent insight and the road ahead. Pflugers Arch 2016; 468: 1103-1111.
13) piccoLi Mt, bar c, tHuM t. Non-coding RNAs as modulators of the cardiac fibroblast phenotype. J Mol Cell Cardiol 2016; 92: 75-81.
14) waNg j, Liew ow, ricHarDs aM, cHeN Yt. Over-view of MicroRNAs in cardiac hypertrophy, fibro-sis, and apoptosis. Int J Mol Sci 2016; 17: pii: E749.
15) zHaNg zH, Li j, Liu br, Luo cF, DoNg Q, zHao LN, zHoNg Y, cHeN wY, cHeN Ms, Liu sM. MicroRNA-26 was decreased in rat cardiac hypertrophy model and may be a promising therapeutic target. J Car-diovasc Pharmacol 2013; 62: 312-319.
16) Li X, Liu L, sHeN Y, waNg t, cHeN L, Xu D, weN F. MicroRNA-26a modulates transforming growth factor beta-1-induced proliferation in human fetal lung fibroblasts. Biochem Biophys Res Commun 2014; 454: 512-517.
17) LiaNg H, Xu c, paN z, zHaNg Y, Xu z, cHeN Y, Li t, Li X, Liu Y, HuaNgFu L, Lu Y, zHaNg z, YaNg b, gitau s, Lu Y, sHaN H, Du z. The antifibrotic effects and mech-anisms of microRNA-26a action in idiopathic pul-monary fibrosis. Mol Ther 2014; 22: 1122-1133.
18) cHeN r, Xue j, Xie M. Osthole regulates TGF-be-ta1 and MMP-2/9 expressions via activation of PPARalpha/gamma in cultured mouse cardiac fi-broblasts stimulated with angiotensin II. J Pharm Pharm Sci 2013; 16: 732-741.
19) HeYMaNs s, Lupu F, tercLavers s, vaNwetswiNkeL b, Herbert jM, baker a, coLLeN D, carMeLiet p, MooNs L. Loss or inhibition of uPA or MMP-9 attenuates LV remodeling and dysfunction after acute pres-sure overload in mice. Am J Pathol 2005; 166: 15-25.
20) guaN bz, YaN rL, HuaNg jw, Li FL, zHoNg YX, cHeN Y, Liu FN, Hu b, HuaNg sb, YiN LH. Activation of G Protein Coupled Estrogen Receptor (GPER) promotes the migration of renal cell carcinoma via the PI3K/AKT/MMP-9 Signals. Cell Adh Migr 2015 Jan 14:0. [Epub ahead of print]
21) FaN H, Ma L, FaN b, wu j, YaNg z, waNg L. Role of PDGFR-beta/PI3K/AKT signaling pathway in PDGF-BB induced myocardial fibrosis in rats. Am J Transl Res 2014; 6: 714-723.
22) worou Me, beLMokHtar k, boNNet p, vourc’H p, MacHet Mc, kHaMis g, eDer v. Hemin decreases cardiac oxidative stress and fibrosis in a rat mod-el of systemic hypertension via PI3K/Akt signal-ling. Cardiovasc Res 2011; 91: 320-329.
23) bassi c, Mak tw. Regulation of the phosphatidy-linositide 3-kinase pathway by the lipid phospha-tase PTEN. Clin Chem 2016; 62: 884-885.
24) LoreNzeN jM, scHauerte c, HubNer a, koLLiNg M, Mar-tiNo F, scHerF k, batkai s, ziMMer k, FoiNQuiNos a, kaucsar t, FieDLer j, kuMarswaMY r, baNg c, Hart-MaNN D, gupta sk, kieLsteiN j, juNgMaNN a, katus Ha, weiDeMaNN F, MuLLer oj, HaLLer H, tHuM t. Osteo-pontin is indispensible for AP1-mediated angioten-sin II-related miR-21 transcription during cardiac fi-brosis. Eur Heart J 2015; 36: 2184-2196.
25) cui c, Xu g, Qiu j, FaN X. Up-regulation of miR-26a promotes neurite outgrowth and ameliorates apoptosis by inhibiting PTEN in bupivacaine in-jured mouse dorsal root ganglia. Cell Biol Int 2015; 39: 933-942.
26) HookaNa e, juNttiLa Mj, kaikkoNeN ks, porvari k, kaija H, risteLi j, korteLaiNeN ML, Huikuri Hv. In-creased type I collagen synthesis in victims of sudden cardiac death due to idiopathic myocardi-al fibrosis. Ann Med 2014; 46: 318-323.
27) wu X, Qi X, Lu Y, LiN c, YuaN Y, zHu Q, YiN Q, Li w, Li Y, biaN H. Liguzinediol protects against cardiac fibrosis in rats in vivo and in vitro. Biomed Phar-macother 2016; 80: 260-267.
28) Xu X, DiNg F, paNg j, gao X, Xu rk, Hao w, cao jM, cHeN c. Chronic administration of hexarelin atten-uates cardiac fibrosis in the spontaneously hyper-tensive rat. Am J Physiol Heart Circ Physiol 2012; 303: H703-711.
29) Li j, Li L, cHu H, suN X, ge z. Oral sophocarpine protects rat heart against pressure overload-in-duced cardiac fibrosis. Pharm Biol 2014; 52: 1045-1051.
30) waNg X, Lv H, gu Y, waNg X, cao H, taNg Y, cHeN H, HuaNg c. Protective effect of lycopene on car-diac function and myocardial fibrosis after acute myocardial infarction in rats via the modulation of p38 and MMP-9. J Mol Histol 2014; 45: 113-120.
31) rizvi Na, cHaN ta. Immunotherapy and oncogenic pathways: the PTEN connection. Cancer Discov 2016; 6: 128-129.
32) Liu t, zHou w, cai b, cHu j, sHi g, teNg H, Xu j, Xiao j, waNg Y. IRX2-mediated upregulation of MMP-9 and VEGF in a PI3K/AKT-dependent manner. Mol Med Rep 2015; 12: 4346-4351.

MiR-26a regulates PTEN in myocardial fibrosis
531
33) cHeN s, cHeN w, zHaNg X, LiN s, cHeN z. Overex-pression of KiSS-1 reduces colorectal cancer cell invasion by downregulating MMP-9 via blocking PI3K/Akt/NF-kappaB signal pathway. Int J Oncol 2016; 48: 1391-1398.
34) kiM D, kiM s, koH H, YooN so, cHuNg as, cHo ks, cHuNg j. Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase pro-duction. FASEB J 2001; 15: 1953-1962.
35) zHaNg k, He X, zHou Y, gao L, Qi z, cHeN j, gao X. Atorvastatin ameliorates radiation-induced cardiac fibrosis in rats. Radiat Res 2015; 184: 611-620.
36) gao Y, cHu M, HoNg j, sHaNg j, Xu D. Hypoxia in-duces cardiac fibroblast proliferation and pheno-
typic switch: a role for caveolae and caveolin-1/PTEN mediated pathway. J Thorac Dis 2014; 6: 1458-1468.
37) zHoNg c, waNg k, Liu Y, Lv D, zHeNg b, zHou Q, suN Q, cHeN p, DiNg s, Xu Y, HuaNg H. miR-19b controls cardiac fibroblast proliferation and migration. J Cell Mol Med 2016; 20: 1191-1197.
38) vaDLa gp, veLLaicHaMY e. Anti-fibrotic cardio protective efficacy of aminoguanidine against streptozotocin induced cardiac fibrosis and high glucose induced collagen up regulation in car-diac fibroblasts. Chem Biol Interact 2012; 197: 119-128.





























