Embed Size (px)
Citation preview

METABOLIC DISORDERS

METABOLISM
METABOLISM IS THE SUM TOTAL OF ALL
THE CHEMICAL REACTIONS IN THE
PROCESS OF BREAKDOWN AND
RENEWAL OF THE BODY TISSUES

METABOLISM
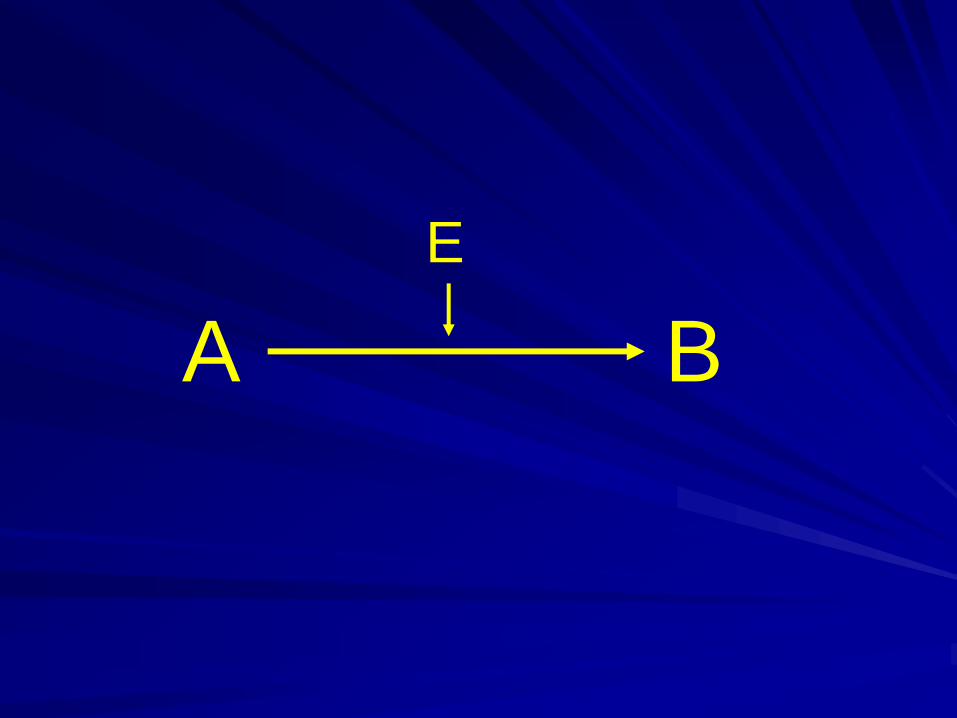
Enzymes play the main role in the process
of converting one chemical metabolite to
another
Enzyme – protein.
Mutations may affect enzyme activity –
amount- defect enzyme production, rapid
breakdown,impaired activity
A B
E
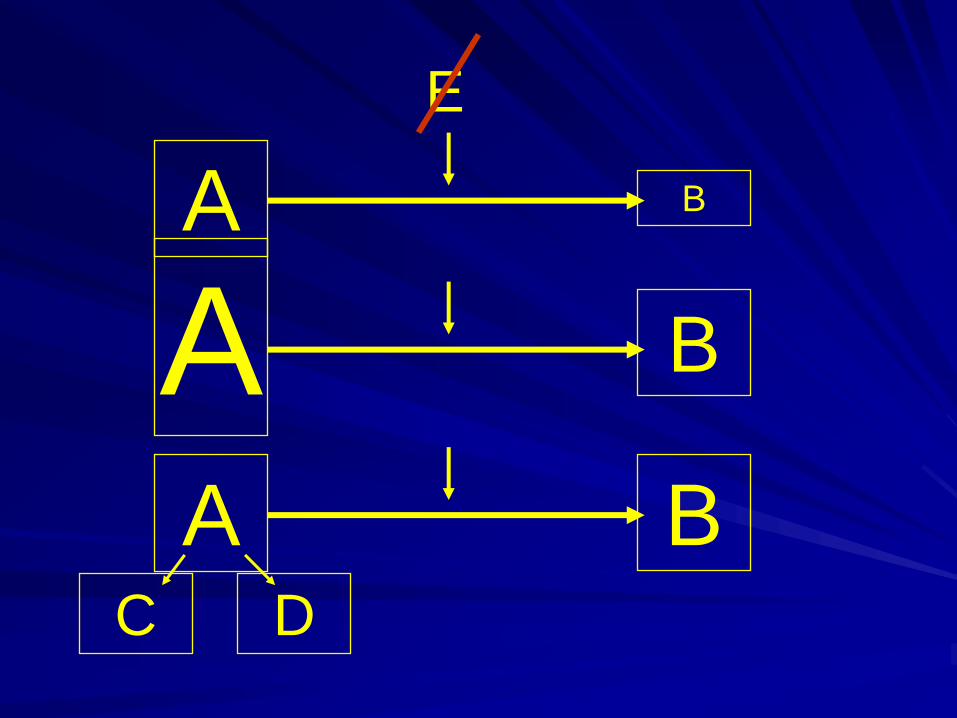
A B
E
A B
A BC D

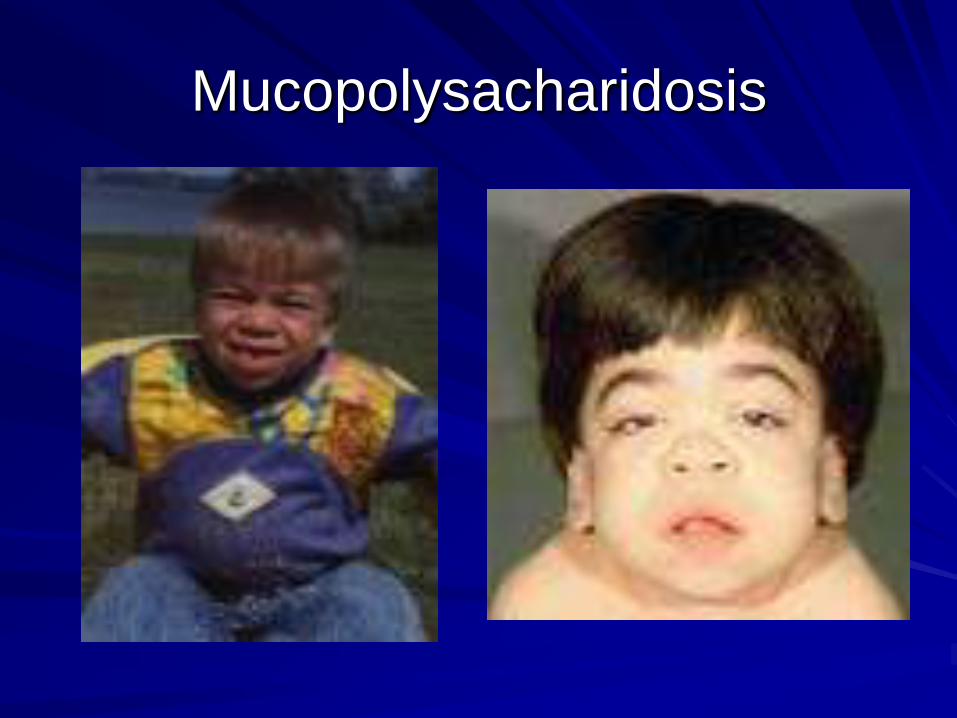
Phenylketonuria


Metabolic disorders
Mutation – protein synthesis defect
Changes in enzymes, receptors, transporters, Na-k pump
Complete vs partial changes – severity of the disorder : mild to death
Crisis during stress : starvation, dehydration, fever , vomiting, etc.
Inheritance: dominant, recessive, x-linked, mithochondrial

Metabolic disorders
Metabolite deficiency
Metabolite accumulation or storage
Toxic metabolites
Transport defects

Metabolic disorders
Neurological anomalies
Neonatal catastrophe – crisis
Liver dysfunction
Acid-base disorders
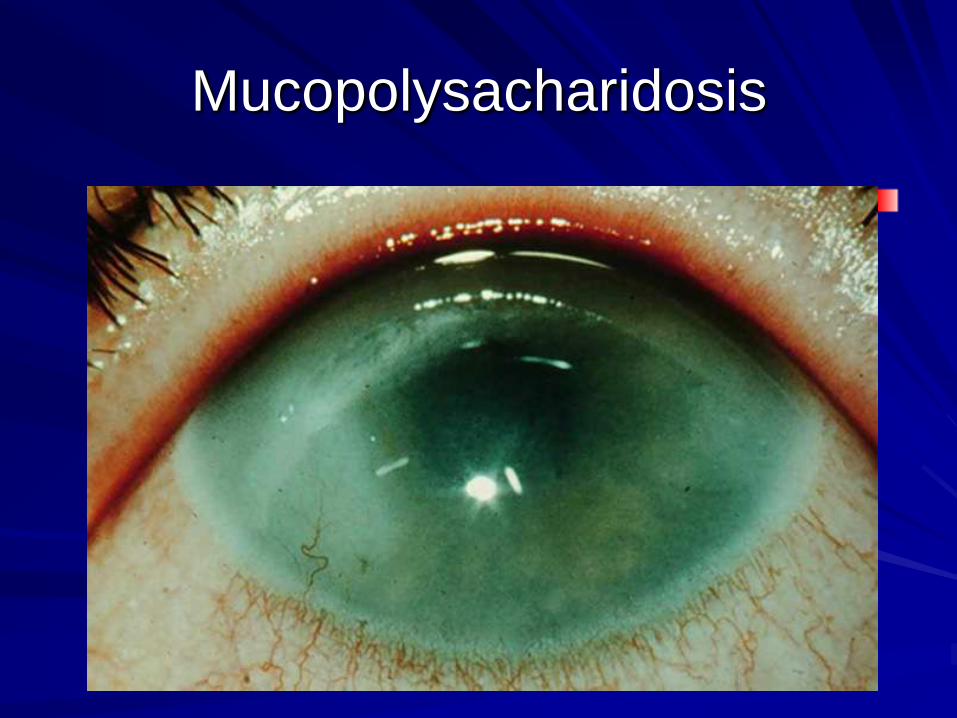
Storage disorders- dysmorphism

Neurological syndrome
Chronic encephalopathy
Acute encephalopathy
Myopathy
Movement disorder
Psychiatric or behavioral abnormalities Neurologic disorders

Neurological syndromeCHRONIC
Global developmental delay
Severe irritability, hyperactivity,
aggressiveness impulsivity
Progressive deterioration
Associated with other neurological
dysfunction- seizures, extra pyramidal
deficits,

Neurological syndromeacute encephalopathy
Emergency
Previous healthy infant/child
Deterioration of consciousness

Neurological syndromeacute encephalopathy

Neurological syndromeacute encephalopathy
Urea cycle disorders
Organic acidopathies(MMA, PA, IVA,GA)
MSUD
Fatty acid oxidation defects
NKHG
Energy defects

Neurologic disorders
Represents toxic effect of the brain
Neurological deterioration
Progressive muscle weakness – hypotonic
Myoglobinuria - elevated cpk
Hyper/hyporeflexia
Convulsions generalized, focal, myoclonic
Movement disorders- dystonia, ataxia choeoathetosis,
Apathy- lethargy

Neurologic disorders
Learning disorders
Behavioral changes – hyperactivity
Psychiatric changes
Vascular events
MRI/CT changes
Sensory neural deafness
Peripheral neuropathy
Optic atrophy, ophtalmoplegia, retinitis pigmentosa

Neonatal crisis
Usually normal newborn- sometimes
intrauterine disease
Maternal-placental fetal unit normal
metabolites delivery with toxic metabolites
evacuation
After delivery – infant self metabolism
Normal infant in the first few days

Neonatal crisis
Poor feeding and sucking
Vomiting
Apathy irritability lethargy
Muscle tone changes – hypotonic
Respiratory changes – tachypnea – apnea
Convulsions, loss of conscious- coma

Neonatal crisis
Differential diagnosis :
sepsis,
cyanotic heart disease
Milk allergy
Pyloric stenosis
Endocrinological disorder

Neonatal crisis
Hypoglycemia
• Glycogen storage disease
• Gluconeogenesis disorder
• Fatty oxidation disorders
• Organic acidurias
• Mitochondrial disorders
• Hyperamonemia
• Acidosis

Neonatal crisis
Jaundice
• Galactosemia
• Tyrosinemia
Dysmorphic features
Peroxisomal disorders
Cholesterol synthesis disorders

Acid base balance disorders
Acidosis
Tachypnea - bradypnea - apnea
Palor – cyanosis
decreased perfusion – cutis marmorata
Increased anion gap (Na+k) – (Cl+Hco3)
Organic metabolites - lactate

Acid base balance disorders
Hyperamonemia
Urea cycle disorders – organic acidurias
Stimulatory effect on CNS
Respiratory alkalosis

Chronic metabolic acidosis
Failure to thrive
Feeding difficulties
Recurrent vomiting
Developmental delay
During infancy or late childhood
Partial enzymatic deficiency

Acid base balance disorders
Amino acid metabolism disorders – MSUD
Organic acidurias – isovaleric acidemia,
methylmalonic acidemia
Glycogen storage diseases
Gluconegenesis disorders
Fatty acid oxidation defects
Lactate-pyruvate metabolism defects
respiratory chain defects

Liver diseases
Hepatomegaly
Direct hyperbilirubinemia
Liver function disorders
Coagulation defects
Hypoglycemia- utilization of liver glycogen

Table 3

Liver diseases
Chronic
Storage disease
Mitochondrial
disorders
Galactosemia
tyrosinemia
Acute
Urea cycle disorders
Fatty acid oxidation
Galactosemia
Fructosemia
Tyrosinemia
Mitochondrial liver
disease

Storage diseases
Cherry red spots
Deafness
Mental retardation
Developmental delay
Neurological
deterioration
Hepatosplenomegaly
Dysmorphic features
Coarse face
Skeletal disorders
Corneal opacity
cardiomyopathy

Storage diseases
Lysosomal
Peroxisomal
Cholesterol metabolism
Glycolisation defects

Dysmorphism
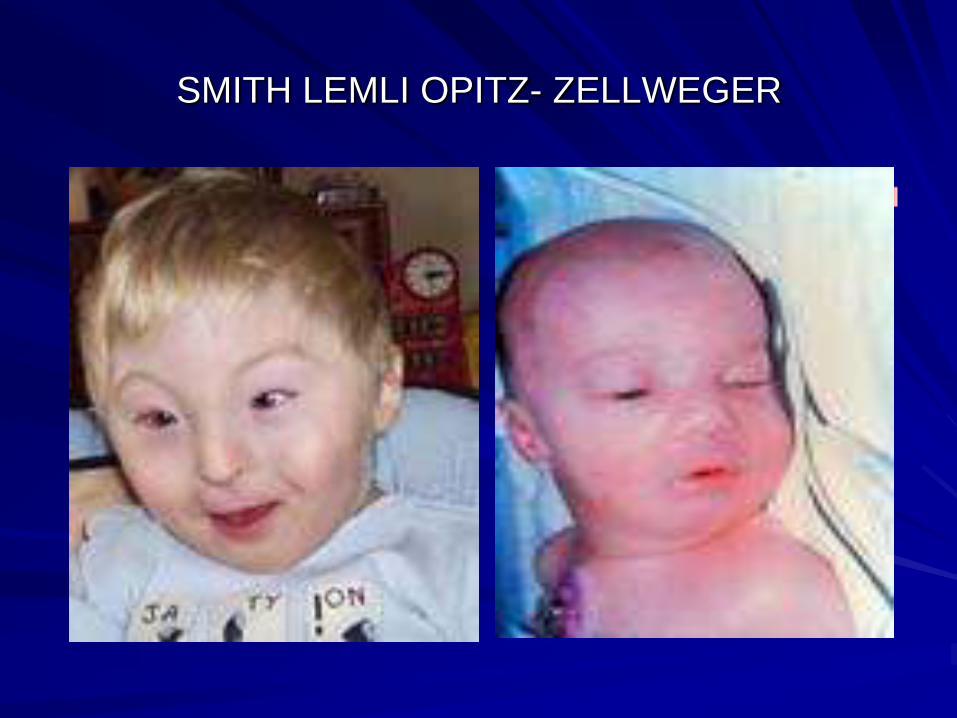
Zellweger syn

הפרוקסיזום
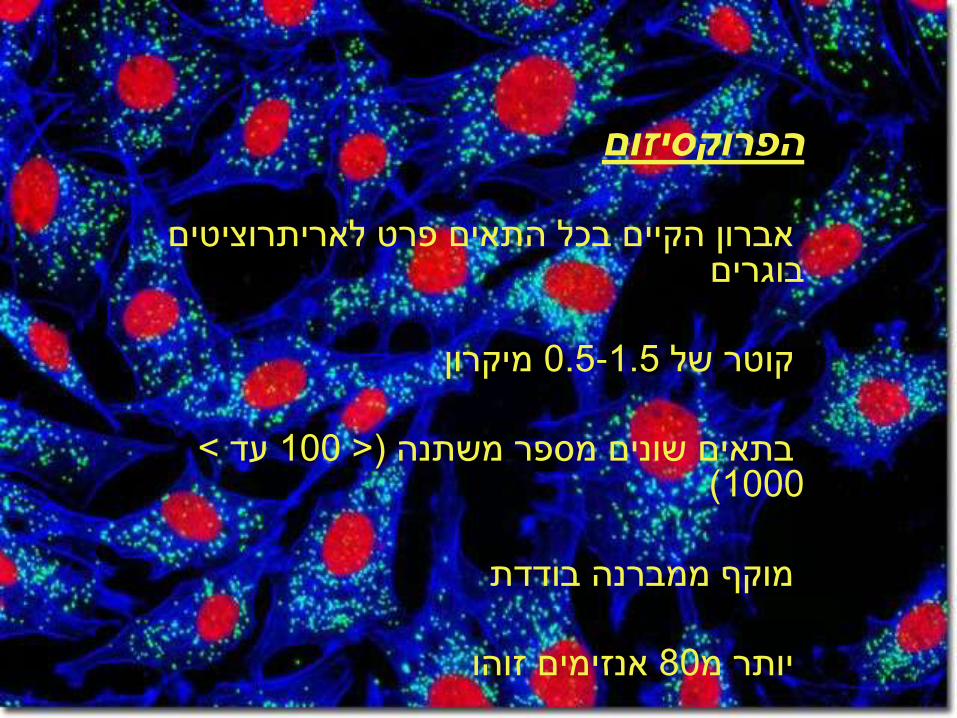
הפרוקסיזום
אברון הקיים בכל התאים פרט לאריתרוציטים בוגרים
מיקרון 0.5-1.5קוטר של
< עד 100> )בתאים שונים מספר משתנה 1000 )
מוקף ממברנה בודדת
אנזימים זוהו 80יותר מ

תפקידים
β אוקסידציה של חומצות שומן ארוכות מאד
α אוקסידציה של חומצות שומן
(פלסמלוגנים)ייצור אתרפוספוליפידים
מטבוליזם של פרוקסיד החמצן
כולסטרול , מטבוליזם חומצות מרה
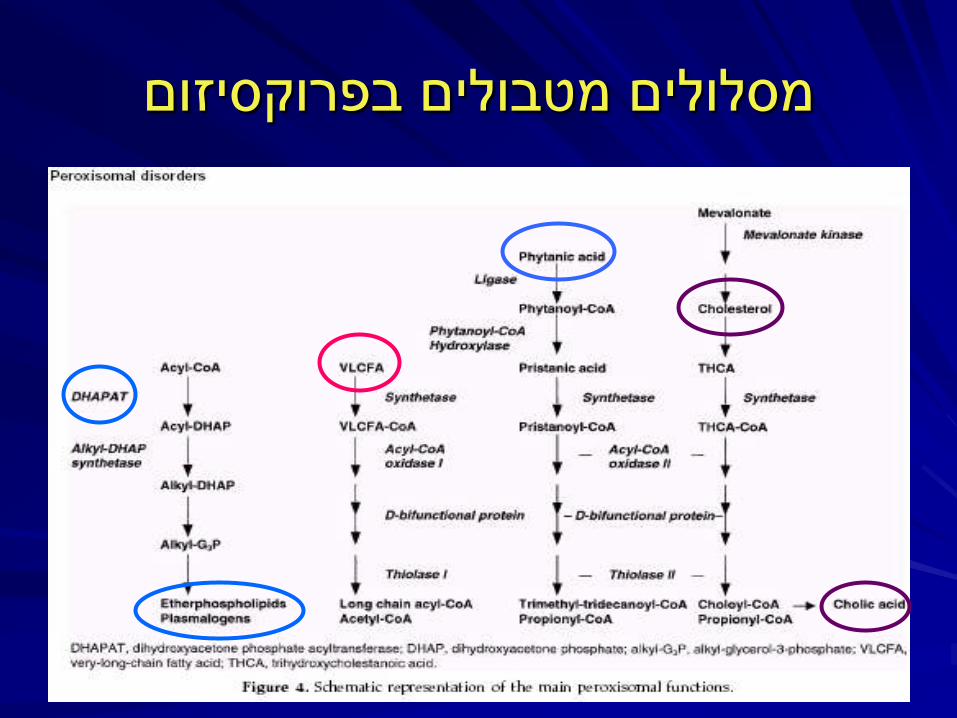
מסלולים מטבולים בפרוקסיזום

PEROXISOMAL BIOGENESIS
רוב האנזימים הפרוקסיזומלים מיוצרים בריבוזום ומכוונים לפרוקסיזום
הביוגנזה היא התהליך בו נוצרות ממברנות הפרוקסיזום וחלבוני
המטריקס עוברים מהציטוזול לתוך האורגנלה
Peroxinsהחלבונים הקשורים בביוגנזה של הפרוקסיזום נקראים
PEX genesי "מקודדים ע -Peroxinsה

PEX GENES

Smith lemli opitz

Abnormal odors
Glutaric aciduria sweaty feet
Isovaleric acidemia sweaty feet
Maple syrup urine disease maple syrup
Phenylketonuria mousy
Trimethylaminuria rotten fish
Tyrosinemia cabbage
Methionine malabsorption cabbage like

Other systems disorders
Cardiomyopathy
Renal tubular acidosis
Diarrhea – villous atrophy
Diabetes mellitus, dwarfism,
hypoparathyroidism, diabetes insipidus.
Neutropenia,
thrombocytopenia,sideroblastic anemia
Pancreatic insufficiency
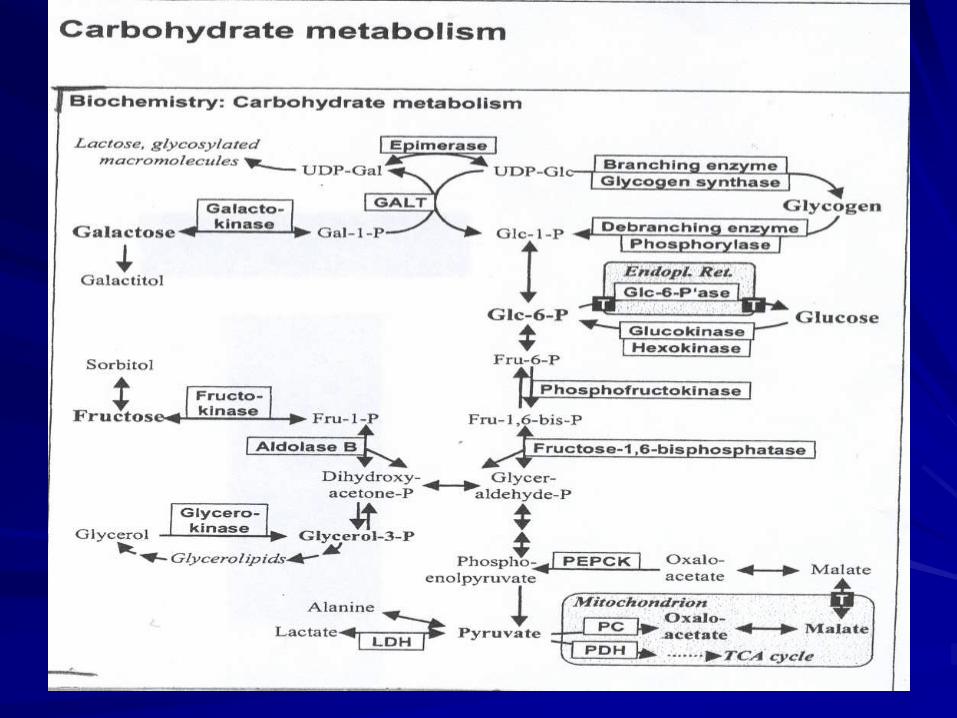


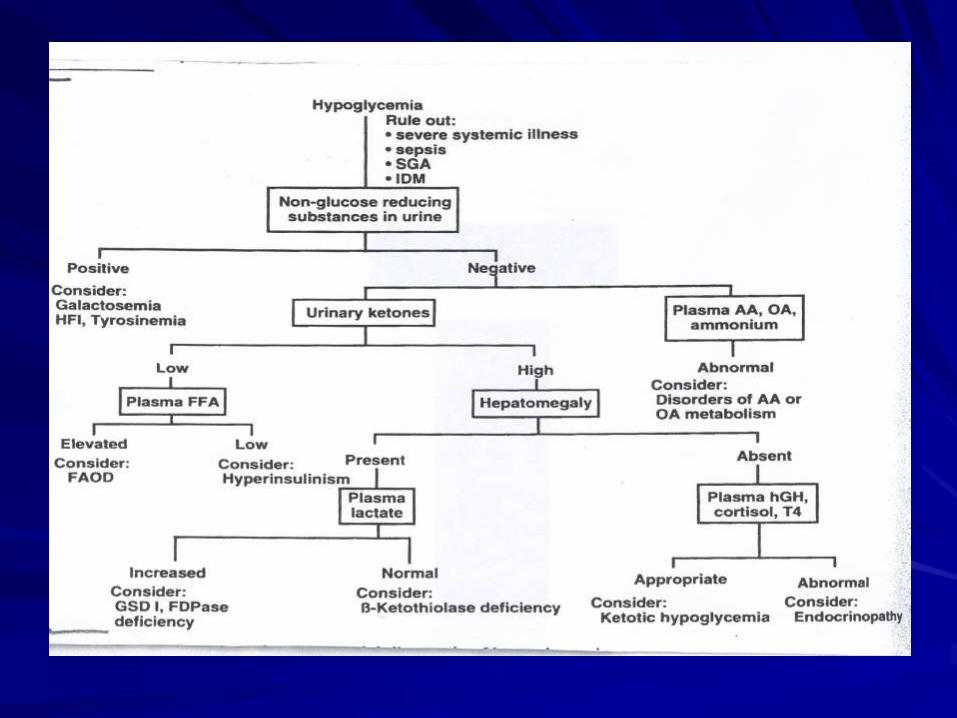
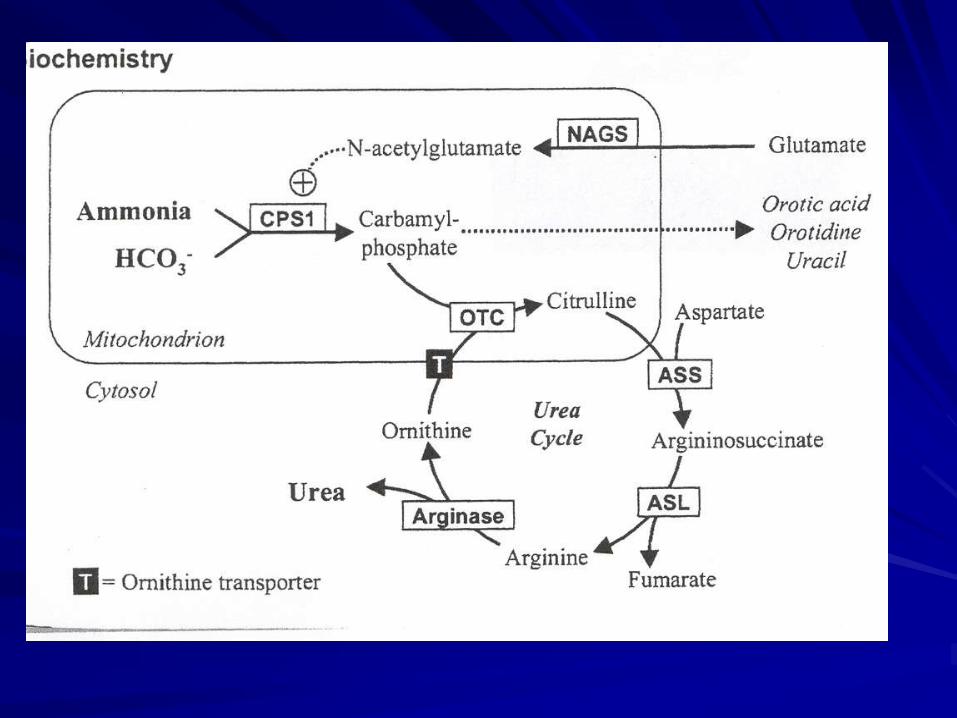
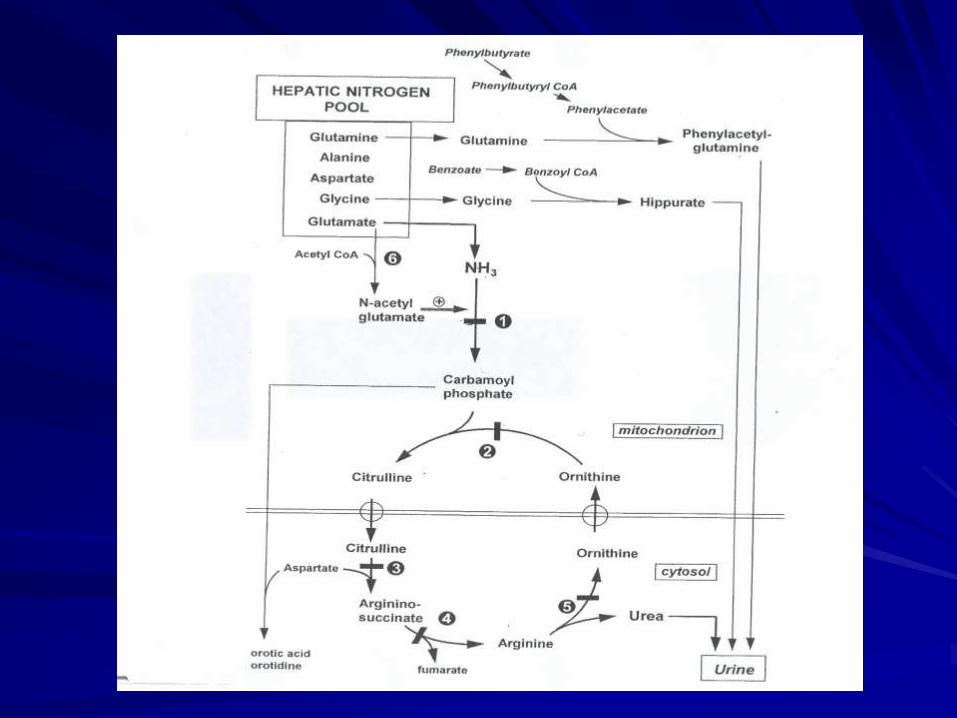


Fig 1

Metabolic acidosis

Metabolic acidosis
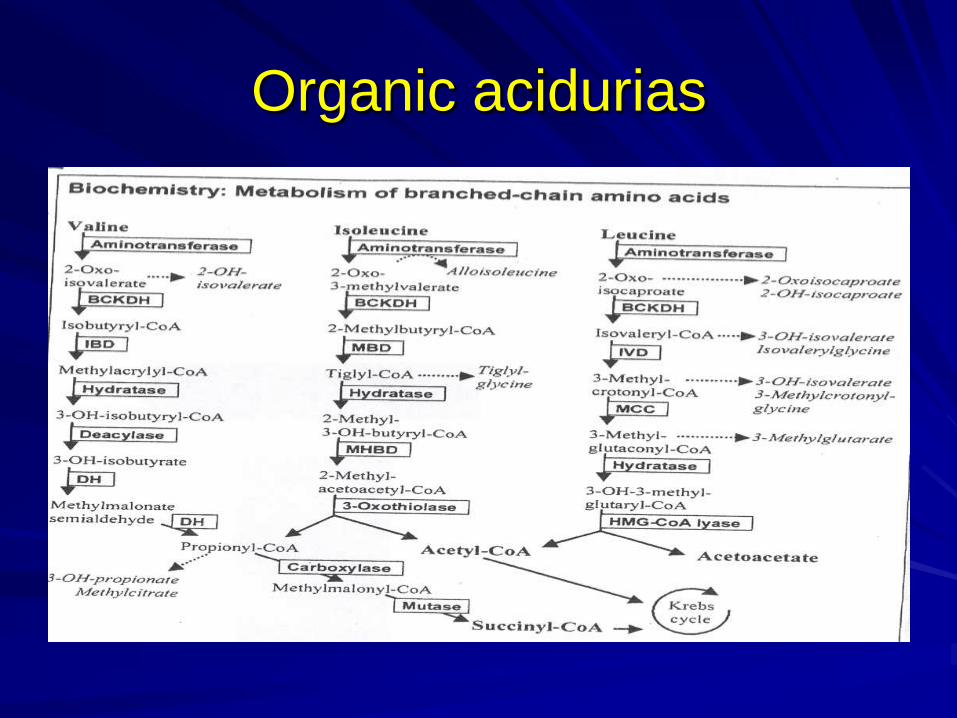
Organic acidurias

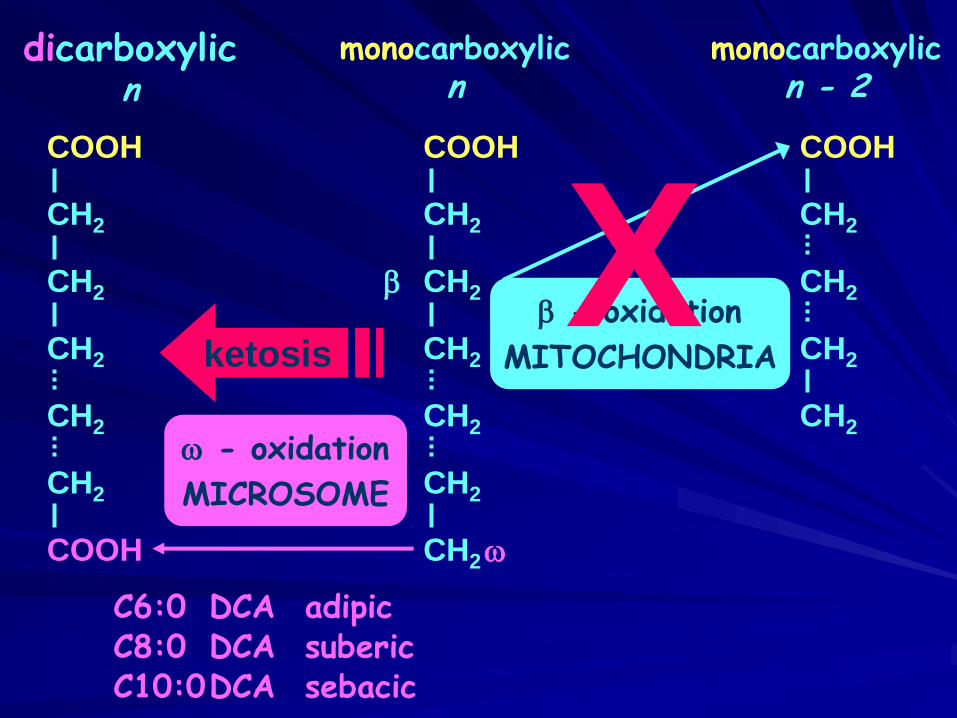
COOH
CH2
CH2
CH2
CH2
COOH
CH2
w - oxidation
MICROSOME
dicarboxylicn
monocarboxylicn
monocarboxylicn - 2
COOH
CH2
CH2
CH2
CH2
CH2
CH2
COOH
CH2
CH2
CH2
CH2b - oxidation
MITOCHONDRIA
b Xw
C6:0 DCA adipicC8:0 DCA subericC10:0DCA sebacic
ketosis
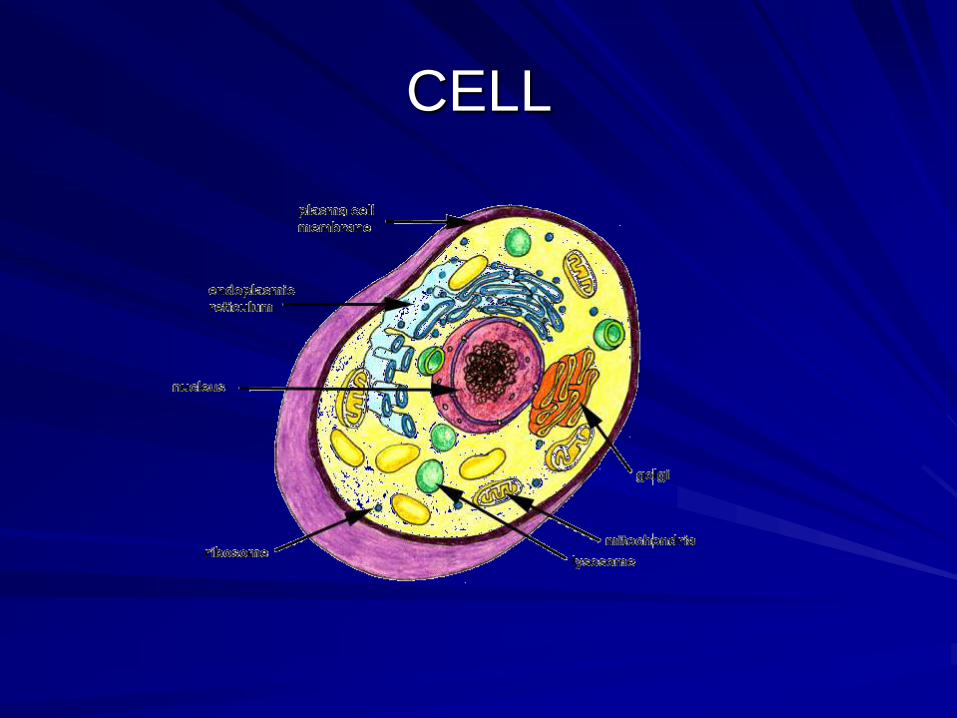
CELL


MITOCHONDRIA
MITOCHONDRIA ARE INTRACELLULAR
ORGANELLES MAINLY CONCERNED
WITH ENERGY PRODUCTION USED TO
SUPPORT THE CELL’S BIOCHEMICAL
AND MECHANICAL FUNCTIONS
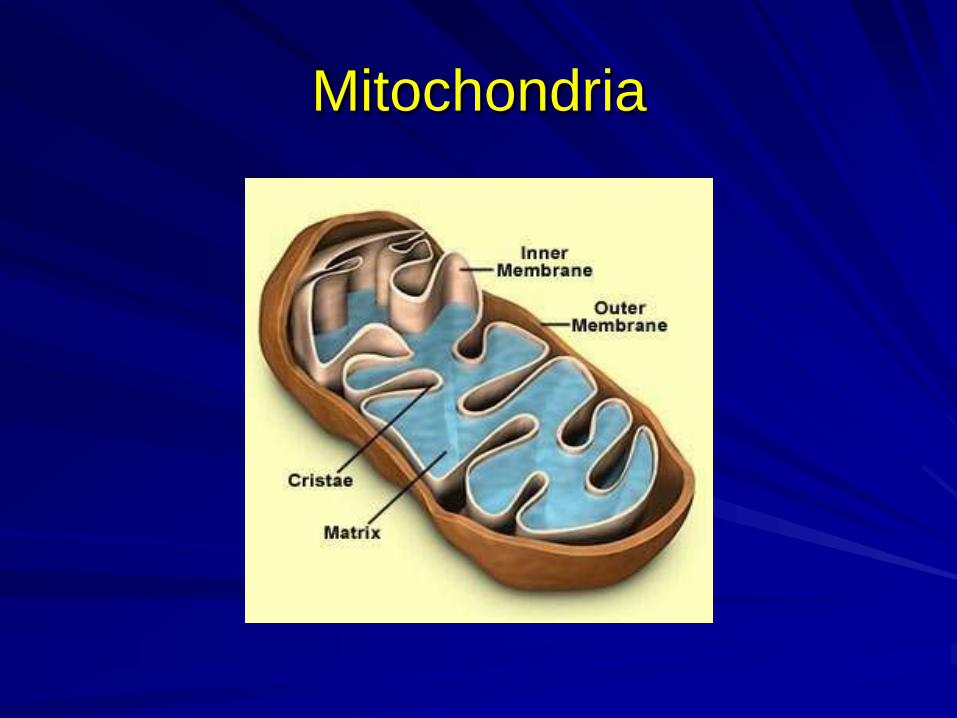
Mitochondria

Mitochondria

Mitochondria
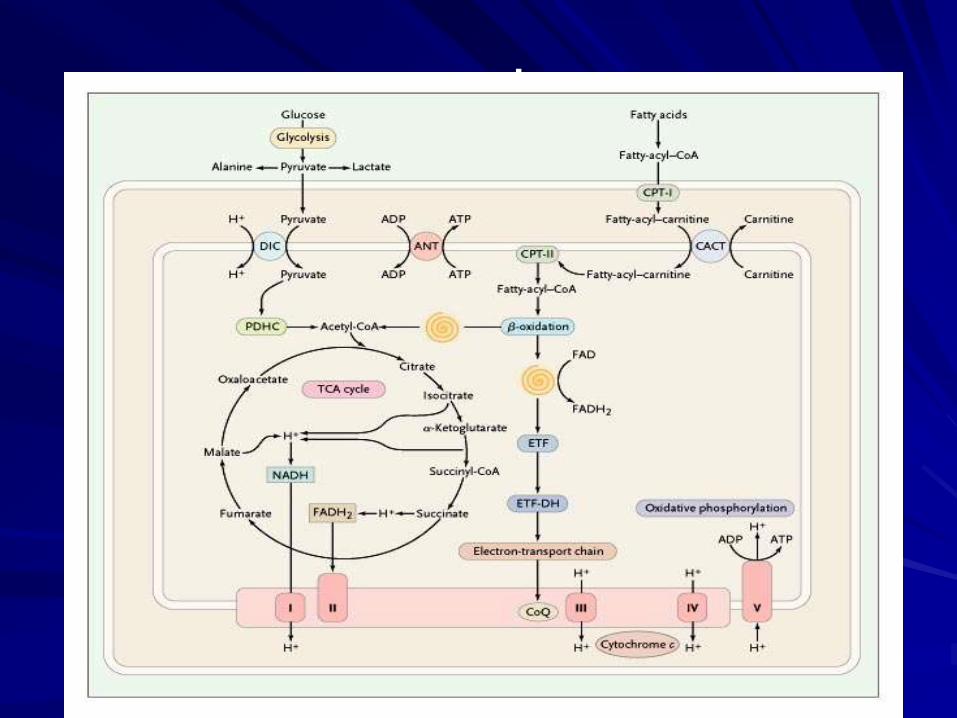
מעגלי אנרגיה



ENERGY
1Mol GLUCOSE 38Mol ATP






Mitochondria
200-500 mitochondria in every cell
Eye -80%, heart - 40% of cell volume
according to energy needs

Mitochondrial Disorders
The mitochondria are self duplicating and
contain their own DNA and RNA
Mitochondrial proteins are encoded by
both nuclear and mitochondrial DNA

Inheritance Pattern
Mitochondrial DNA is the only extranuclear
DNA in the cell
A circular double stranded molecule
containing 16569 base pairs
Contains only 37 genes
These genes code for: 13 proteins, 2
ribosomal RNA’s, 22 transfer RNA’s

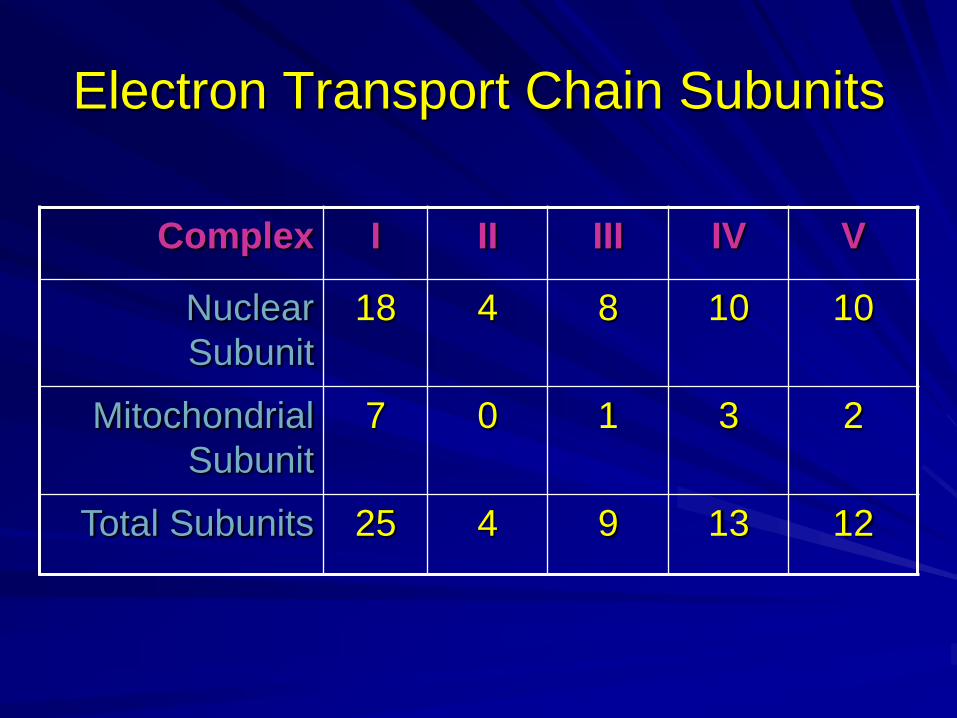
Electron Transport Chain Subunits
VIVIIIIIIComplex
10108418Nuclear
Subunit
23107Mitochondrial
Subunit
12139425Total Subunits

Maternal Inheritance
The condition will be transmitted from the
mother to all her children
Only the females will pass the condition to
succeeding generations

MATERNAL INHERITANCE

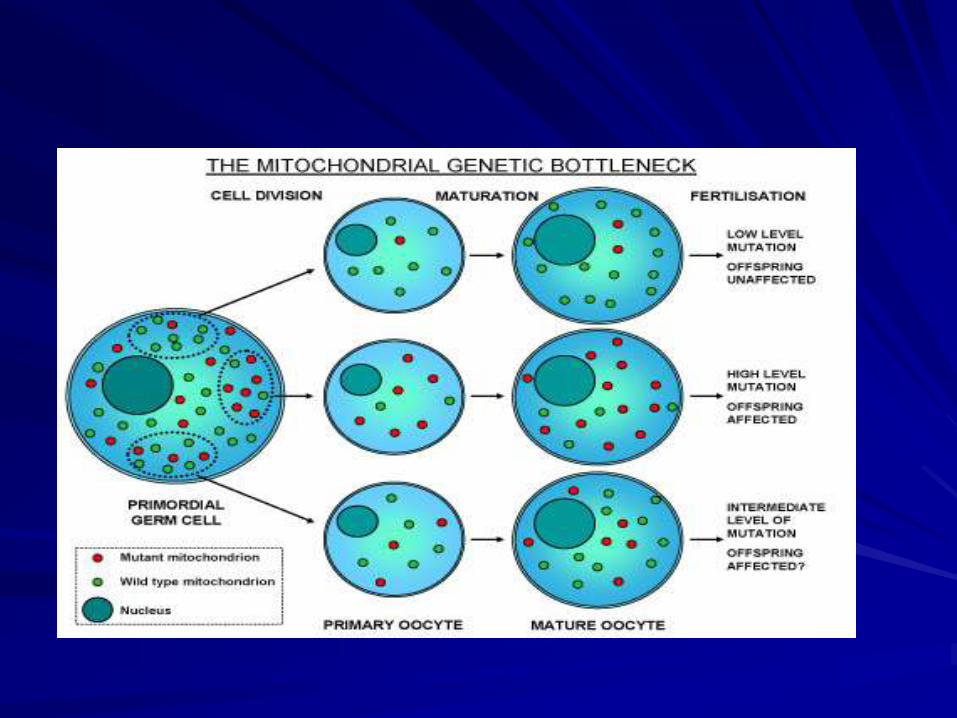
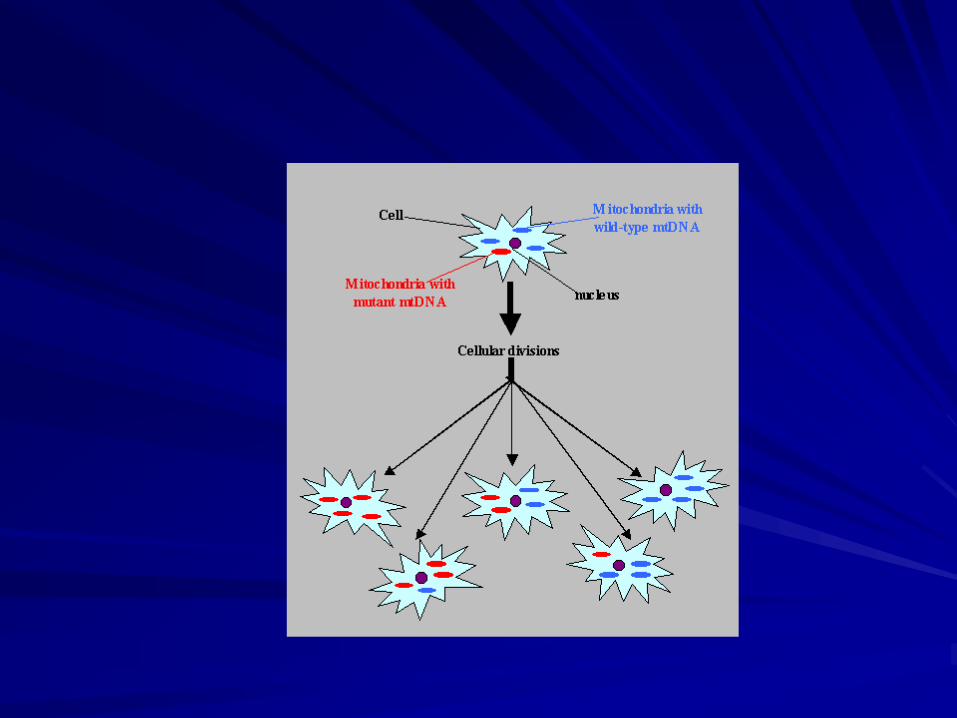
Mitochondrial heteroplasmy – each cell
contains a mixture of normal (wild type)
mtDNA and mutated mtDNA
Replicative segregation – during cell division
some cells may accumulate a greater or lesser
number of wild type mtDNA
Threshold effect – the cellular phenotype will
be determined by the more abundant type of
mtDNA


A Mutation in the mtDNA is
Pathological if:
Does not exist in normals
It shows heteroplasmy
It produces an abnormal protein
There are affected maternal relatives
There is a correlation between the
percentage of the mutation and the
severity of the disease

Mitochondrial disease
Mitochondrial DNA point mutation
Mitochondrial DNA deletion
Mitochondrial DNA depletion
DNA nuclear mutation

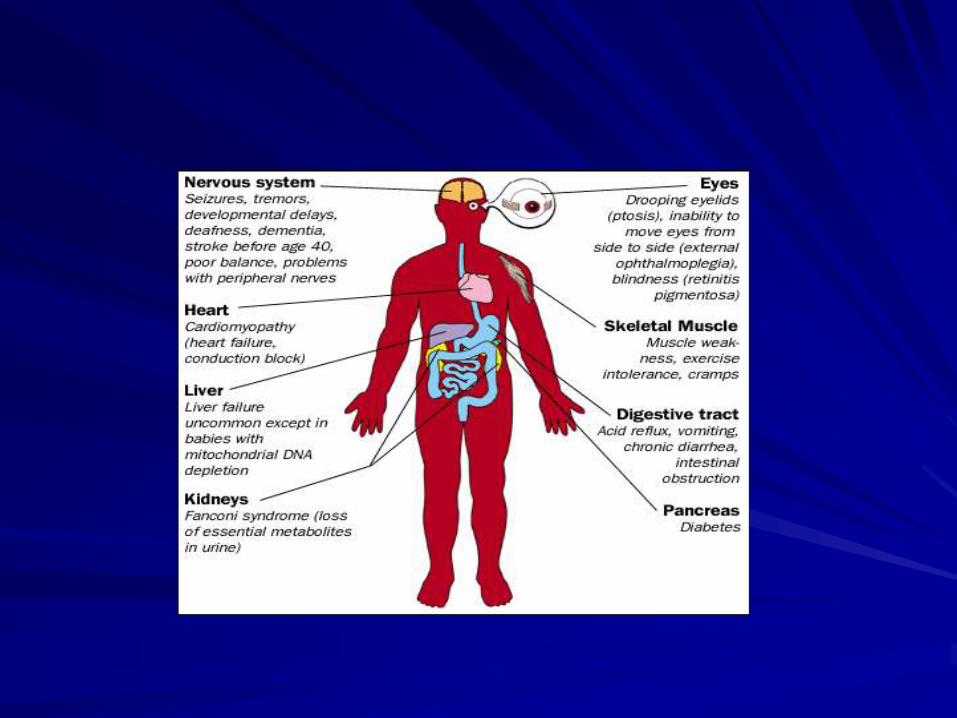
Mitochondrial diseases derive from dysfunction of
organ systems that are highly dependent on aerobic
metabolism:
Nervous system: psychomotor retardation, deceleration of head growth, convulsions, impairment of hearing and vision, ataxia, myopathy, neuropathy
Heart: cardiomyopathy, conduction block
Kidney: renal tubular acidosis
Endocrine: short stature, diabetes mellitus, hypothyroidism, hypoparathyroidism
Hematology: sideroblastic anemia, neutropenia
Hepatic gastrointestinal

Diagnostic criteria
Clinical
– multisystemic symptoms at least 3
– Family history
– Progressive course
Histology
Enzymology
Functional –ATP synthesis rate
Molecular

Biochemistry
Increased serum lactate
Increased CSF lactate
Pyruvate
Alanine – serum
Urinary Organic acids- lactate, krebs cycle
metabolites , specific- methylglutaconic
acids
MRI -leukodystrophies

Therapy
PDH – Ketogenic diet
Cofactors-
– Thiamine,riboflavin,biotin
Enzyme activators – dichloroacetate
Electron transporters –CoQ10 Idebenone , uridine
Radical scavengers –Vitamin C, E, K
L-carnitine
Creatine - high energy phosphate compounds

Metabolic evaluation
PRIMARY
Blood bicarbonate
Glucose
Electrolytes, CPK
Liver functions – coagulation
urinary reducing substances, ketones
Blood amonia, lactate,

Metabolic evaluation
Blood and urinary amino acids
CSF lactate and amino acids
Carnitine
pyruvate
Urinary organic acids
Acylcarnitines
VLCFA
Muscle, liver , skin fibroblasts
Genetics- DNA

Treatment
IV glucose 10% -hydration catabolism
prevention
Improving hypoglycemia
Treatment of acidosis
Hyperamonemia – sodium benzoate ,
phenylbutitate
Treatment of the crisis precipitator-
infection, dehydration etc.

Treatment
Cofactors – carnitine,riboflavin thiamin etc.
Peritoneal dialysis
Hemodialysis
Diagnosis and treatment of the dying child

Metabolic disorders
Mitochondrial
disorders
Urea cycle
Amino acids
Carbohydrates
disorders
Purines and
pyrimidines
Neurotransmiters
Lysosomes
Peroxisomal
disorders
Cholesterol synthesis
disorders
Glycoproteins
porphyria

Summary
Acute / chronic presentation
Multisystemic expression
Metabolic presentation – hypoglycemia ,
acidosis, lactic acidemia,
hyperammonemia
Neurologic disorder
To think about

THANKS

Galactosemia
חד סוכר שיחד עם גלוקוז יוצר את –גלקטוז
לקטוז–סוכר החלב
כשלון בהעברת גלקטוז לגלוקוז
Galactose-1-Phosphateחסר ב
Uridyltransferase
פוספט-1-הצטברות גלקטוז

Galactosemia
FTTסרוב לאכול
ישנוניות ואפטיה
צהבת
ascites-הפטוספלנומגליה
מחלה כליתית טובולרית
היפוגליקמיה
קטרקט
חמרים מחזרים בשתן
פיגור-במחלה כרונית

Galactosemia
איבחון
חמרים מחזרים בשתן–
ניר גטרי–חסר באנזים בכדוריות אדומות –
איבחון גנטי –
טיפול
אלימינציה של חלב ומוצריו–
הוצאת גלקטוז מהכלכלה–
ייעוץ גנטי–

Galactosemia
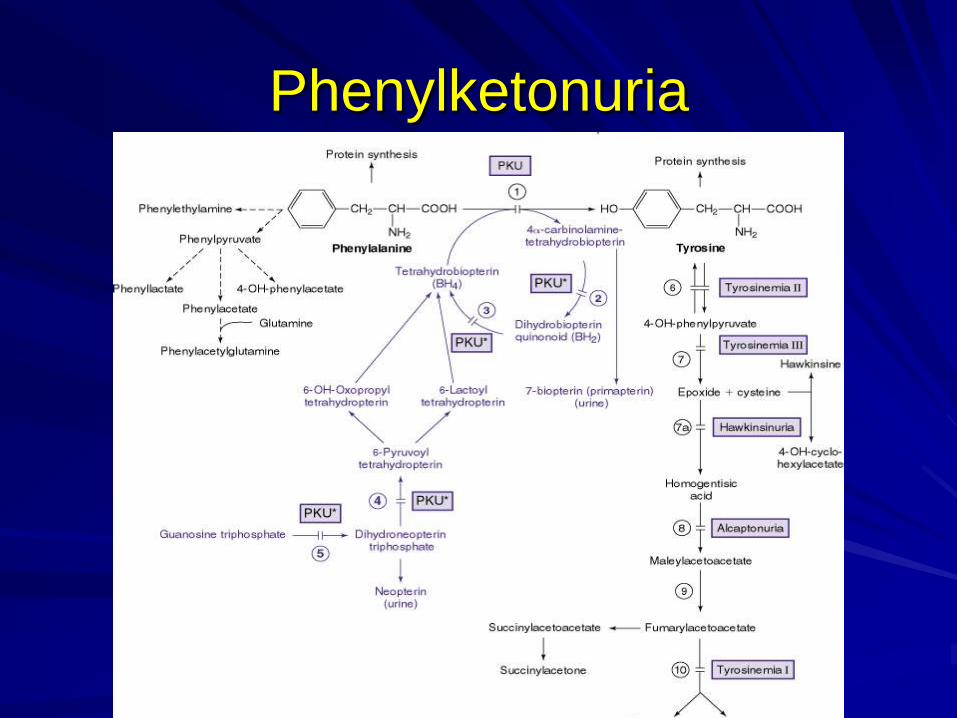
Phenylketonuria

Phenylketonuria
הפרעה במטבוליזם פניל אלנין
בעיקר בגלל חסר בphenylalanine hydroxylase
400 12מחלה אוטוסומלית רצסיבית כרומוסום ,מוטציות
הצטברות פניל אלנין בנוזלי הגוף ובCNS
של האנזים ורמת חומרת המחלה לפי מידת החסרהפנילאלנין בדם
1:14000-1:20,000שכיחות

Phenylketonuria
קליניקה
תינוק נורמלי בלידה
פיגור שכלי מתפתח בהדרגה בחדשים הראשונים
לחיים
ללא טיפול הפיגור עמוק
הקאה
תנועות לא , היפראקטיביות–ילדים גדולים יותר
.מכוונות ואתטוזיס

Phenylketonuria
קליניקה
"תימנים בלונדיניים"–ילדים בהירים
פריחה סבוראית
ריח רע מהגוף
פירכוסים 25%
EEGהפרעות ב 50%מעל
טונוס גוף מגבר
מיקרוצפליה בולטת
הפרעה בגדילה

Phenylketonuria
בכל הארצות screenאיבחון נאונטלי כ
המפותחות בניר גטרי
טיפול בפורמולה שמגבילה פניל אלנין
%מג 2-6ניטור רמת הפנילאלנין בדם בין
הגבלת דיאטה כדי –נשים עם פנילאלנין גבהה
למנע פגיעה מהעובר

Isovaleric acidemia

Isovaleric acidemia
organic aciduria
Isovaleryl CoA dehydrogenaseחסר ב
הצטברות חומצה איזוולרית
מחלה אוטוסומלית רצסיבית
15הגן מופה ונמצא על כרומוסום

Isovaleric acidemia
קליניקה
מחלה חריפה עם חמצת קשה הקאה 50%
אפטיה לטרגיה
comaפירכוסים
Sweaty feet odor
מחלה קלה יותר עם ביטוי מאוחר יותר וביטויים
חריפים קלים יותר
אנמיה לאוקופניה, טרומבוציטופניה

Isovaleric acidemia
במעבדה–
ANION GAP>20חמצת מטבולית עם
בשתן רמה גבהה של חומצה איזוולרית
ומטבוליטים נוספים כמו איזוולרילגליצין

Isovaleric acidemia
טיפול:
מניעת מצב קטאבולי
הידרציה
תיקון חמצת
טיפול בקרניטין וגליצין
דיאליזה
לאחר מצב חריף
דיאטה דלת לאוצין
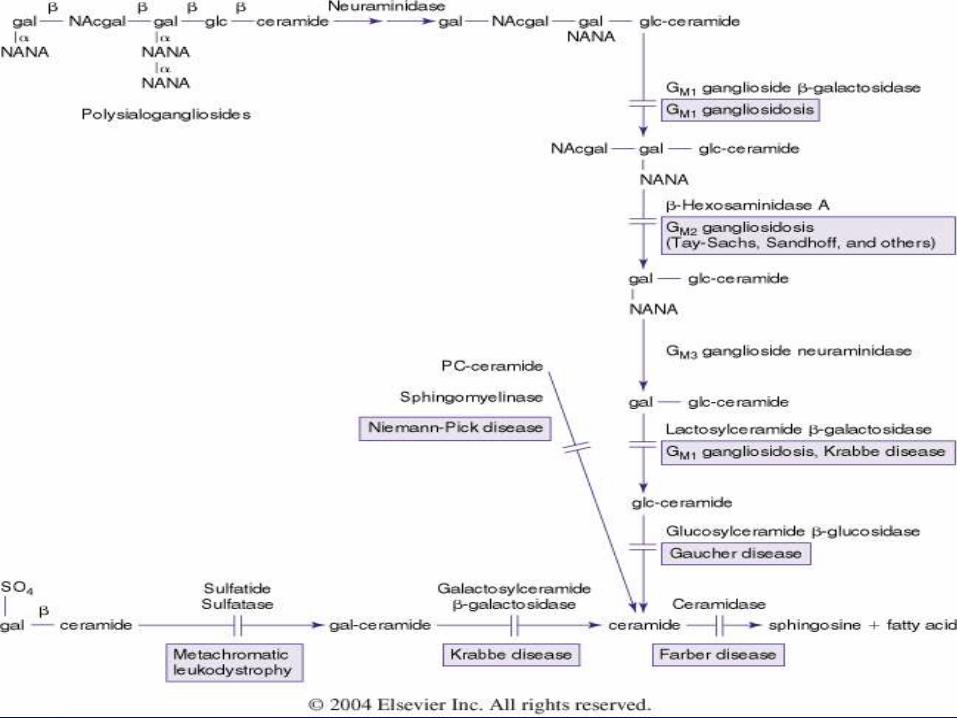


מחלות אגירה
אגירה של סובסטרטים של שומנים בליזוזומים
מחלה נאורודגנרטיבית - CNSאגירה ב
הפרעות , הפטוספלנומגליה–אגירה ויסצרלית
.תסנינים ריאתיים, בשלד
, איבחון הדפקט האנזימתי בלאוקוציטים
פיברובלסטים
איבחון גנטי

מחלות אגירה
Niemann-pick
בלידה נורמליים
צהבת ישירה ממושכת
חסר שיגשוג בינקות
הפטוספלנומגליה מסיבית
חדשים 6הדרדרות נוארדגנרטיבית מתקדמת מגיל
מות עד גיל שנתיים
acid sphingomyelinaseחסר פעילות ב
הצטברות ספינגומיאלין
11מחלה אוטוסומלית רצסיבית כרומוסום

הפרעות במעגל האוריאה

מעגלי אנרגיה

METABOLIC DISORDERS

Fig 1

חמצת מטבולית

Dysmorphism
Zellweger syn


סיכום ביניים