Embed Size (px)
Citation preview

American Journal of Medical Genetics 124A:148–157 (2004)
Longterm Follow-Up in ChondrodysplasiaPunctata, Tibia–Metacarpal Type,Demonstrating Natural History
Ravi Savarirayan,1,3* Robert J. Boyle,1 John Masel,2 John G. Rogers,1,3 and Leslie J. Sheffield1,3
1Genetic Health Services Victoria and Murdoch Childrens Research Institute, Royal Children’s Hospital,Parkville, Victoria, Australia2Department of Radiology, The Royal Children’s Hospital Brisbane, Brisbane, Queensland, Australia3Department of Paediatrics, University of Melbourne, Parkville, Victoria, Australia
We report the longterm clinical and radiolo-gical progression in three unrelated patientswith the tibia–metacarpal form of chondro-dysplasia punctata (CDP-TM). The patientswere followed for 37, 25, and 32 years,respectively. At follow-up intellectual func-tion was normal, and physical function waswell preserved. There was also markedresolution of several significant early radio-graphic features. The patients attained adultheights of 152, 138, and 148 cm. Two patientshad chronic serous otitis media requiringtympanostomy tubes during childhood. Onepatient suffered persisting back pain relatedto spinal stenosis and required lumbar lami-nectomy at the age of 26 years. One patienthad hip dysplasia requiring orthopedic sur-gical intervention. All patients had recur-rent patella dislocation. Sterol and verylong chain fatty acid profiles, FISH analysisfor SHOX gene deletions, blood lymphocytekaryotype, and phytanic acid levels werenormal in those tested, and no mutations inarylsulfatase D and E genes were detected.These data suggest that the longterm clinicaland functional prognosis in this conditionappears to be better than that expectedbased on initial clinical and radiologicalfindings. � 2003 Wiley-Liss, Inc.
KEY WORDS: calcific stippling; punctateepiphyseal dysplasia; osteo-chondrodysplasia; mesome-lia
INTRODUCTION
The tibia–metacarpal type of chondrodysplasia punc-tata (CDP-TM) has now been described in at least 11children [Haynes and Wangner, 1951; Asanti andHeikel, 1963; Burck, 1982; Rogers, 1989; Rittler et al.,1990; Matsui et al., 1994; Mayden Argo et al., 1996;Jansen et al., 2000]. This form of CDP is characterized byshortening and bowing of the tibiae, shortened meta-carpals, shortened ulnae with radial bowing, and verte-bral clefts or delayed vertebral ossification. There isstippled calcification, typically in the sacral, tarsal, andcarpal regions and most marked in the newborn period.Patients may display rhizomelia, mesomelia, or micro-melia [Mayden Argo et al., 1996]. The disorder isthought to be inherited as an autosomal dominanttrait, although no cases of familial recurrence have beenreported. The underlying etiology of CDP-TM isunknown. The longterm prognosis and natural historyof this condition has not been previously describedbeyond one child followed to the age of 11 years and8 months [Rittler et al., 1990; Matsui et al., 1994]. Wereport the clinical and radiological features of threepatients with CDP-TM followed into adulthood andreview previously reported cases.
CLINICAL REPORTS
Patient One (F.K.)
F.K.: Figure 1a–i. Rogers [1989] previously reportedthis boy. He was born to healthy, non-consanguineousAustralian parents, mother aged 35 and father aged 47,after a normal pregnancy. He has three healthy siblingsof normal stature (191 cm male, 168 cm female, and162 cm female). His mother also had two spontaneousearly miscarriages. One sister has four healthy childrenof normal stature. He was born at term with birth weight2,700 g (<3rd centile) and length 38 cm (�3rd centile,50th centile for 28 weeks of gestation). His mother is168 cm tall (90th centile) and father 173 cm tall (50thcentile). As a neonate, he was noted to be hypotonic andhad occasional cyanotic spells of unknown origin. At theage of 3 months, he was noted to have short and bowed
*Correspondence to: Dr. Ravi Savarirayan, Genetic HealthServices Victoria, 10th Floor, Royal Children’s Hospital, Parkville3052, Australia. E-mail: [email protected]
Received 23 January 2003; Accepted 22 April 2003
DOI 10.1002/ajmg.a.20383
� 2003 Wiley-Liss, Inc.

limbs and radiographs revealed calcific stippling of thesacrum, carpal and tarsal regions with other featuressuggesting a primary osteochondrodysplasia (see Radio-graphic data). At the age of 9 months, length was 58 cm(�1st centile, 50th centile for the age of 2 months),weight 6.75 kg (1st centile), and head circumference45 cm (50th centile). During childhood he developedchronic serous otitis media which required placement oftympanostomy tubes. He was noted to have a hypoplas-tic left optic disc with reduced visual acuity, partial leftsided hearing loss, and left sided upper motor neuronefacial nerve palsy. His developmental progress wasnormal for age throughout childhood. He subsequentlyattended college, attained a higher degree, and cur-rently works as a librarian. His height, at the age of 37, is152 cm (<1st centile), weight 71 kg (50th centile), andhead circumference 56 cm (50th centile). Upper segmentto lower segment ratio was 1.05. He has short forearms(upper arm length 33 cm, 50th centile, forearm length
22 cm, <3rd centile) and short shanks (upper leg length44 cm, 75th centile, lower leg length 33 cm,�1st centile)bilaterally. Clinically, pes planus, reduced internal rota-tion of his hips and 20 degree fixed flexion deformities atthe elbows were present with dislocated radial heads.
He had recurrent subluxation of the left patella thathas resolved in adulthood without surgery. He cycles,jogs, and plays tennis, cricket and basketball. He com-plains of occasional back stiffness and knee pain afterexercise.
Patient Two (C.C.)
C.C.: Figure 2a–h. A female was born to healthy, non-consanguineous parents of Australian/English origin,father aged 31 and mother aged 30. During pregnancyher mother had a ‘‘flu-like’’ illness at 8 weeks of gestationand was treated with amoxycillin for a possible urinarytract infection at 4 and 26 weeks of gestation. She was
Fig. 1. Clinical photographs of patient 1 (F.K.) from age 3 years to age37 years. a: Facial appearance at the age of 3. Note short columella. b: Age3 years. Note short limbs with marked shortening of the shanks and normalsize hands. c: Age 9 years. Normal hand. d: Age 9 years. Note relativelengthening of the shanks, which remain short. e: Age 15 years. Note shortstature, mesomelia, and development of fixed flexion contractures at the
elbows with dislocated radial heads. f: Age 24 years. Note increase bowing ofradii. g: Age 24 years. Short shanks with distal fibula overgrowth and mildtibial bowing. h: Age 37 years. Note short columella and flat midface. i: Age37 years. Note disproportionate short stature with mildly short shanks.[Color figure can be viewed in the online issue, which is available atwww.interscience.wiley.com.]
Tibia–Metacarpal Type CDP 149

born by normal vaginal delivery at term with birthweight of 2,500 g (<3rd centile) and length 42 cm (<3rdcentile, 50th centile for 32 weeks). At the age of 3 monthsher length was 51 cm (<3rd centile), weight 4.75 kg (3rd
centile), and head circumference 39.5 cm (50th centile).Her parents are of normal stature. She has a healthybrother and sister each with healthy children and allare of normal stature. She was referred at the age of
Fig. 2. Clinical photographs of patient 2 (C.C.) from age 3 months to age25 years. a: Age 3 months. Note short columella with upturned nasal tip andlong philtrum. b: Age 3 months. Note short limbs. c: Age 3 months. Noteshort ring finger and small hand. d: Age 10 years. Short columella with flatmidface. e: Age 18 years. Note high position of right patella and relatively
normal shanks. f: Age 25 years. Note short stature with short limbs. g: Age25 years. Note flat midface and upturned nasal tip. h: Age 25 years. Noteshort 4th, 5th metacarpals. [Color figure can be viewed in the online issue,which is available at www.interscience.wiley.com.]
150 Savarirayan et al.

3 months for evaluation of short stature with a flat facialappearance. Clinically she exhibited short limbs and aflat midface with short upturned nose and columella.Radiographs during the first year of life showed punc-tate calcification in the tarsal, coccygeal, metacarpal,and thyroid areas with other features consistent with ageneralized skeletal dysplasia (see Radiographic data).She had two episodes of acute otitis media during thefirst 2 years of life and had tympanostomy tubes insertedin childhood for chronic serous otitis media. She was alsonoted to have an absent left kidney and psoriasis. At theage of 6 years she had bilateral distal femoral osteo-tomies for valgus deformity and secondary fixed flexiondeformity at the knees. She had right hip surgery at theage of 8 years for hip dysplasia and suffered recurrentpatella dislocations during childhood and adolescence,most recently at the age of 20 years. Growth duringchildhood and adolescence remained just below, butparallel to the 3rd centile. She required speech therapyfor speech delay during early childhood but otherwisedevelopmental progress was normal.
Currently, at the age of 25 years, she has neck painwith upper limb parasthesiae and a minor cervical spinekyphosis radiographically. She works as a chef’s assis-tant and has no intellectual difficulties. She swimsregularly and reports no restrictions on her physicalactivity. She has one child, a girl, aged 3 years, who hasno clinical or radiographic signs of CDP-TM. Her finaladult height is 138 cm (�1st centile), head circumfer-ence 55 cm (50th centile), and weight 52 kg (25thcentile). Upper arm and forearm lengths are normal(29 cm, 25th centile and 23 cm, 50th centile, respec-tively) as were lower limb measurements. Apart fromshort stature, short 4th and 5th metacarpals, and mobilepatellas, examination is normal.
Patient Three (P.W.)
P.W.: Figure 3a–d. This boy was born after a normalpregnancy and delivery to non-consanguineous, healthyparents. He has one healthy brother and no familyhistory of short stature. He was noted to be short at
Fig. 3. Clinical photographs of patient 3 (P.W.), age 32 years. a: Note short stature, short limbs, and fixed flexion at elbows. b: Note flat midface and shortcolumella. c: Note short arm with flexion at elbow and loss of antecubital fossa. d: Brachydactyly. [Color figure can be viewed in the online issue, which isavailable at www.interscience.wiley.com.]
Tibia–Metacarpal Type CDP 151

birth and radiographs revealed changes consistent withCDP-TM. He had normal developmental progress,completed school by the age of 18 years, and workedinitially as a clerical officer. He had recurrent patelladislocation and underwent tendon transfer proceduresaged 14 and 17 years for this. He has a lumbar lordosis,has suffered chronic back pain and underwent lumbarlaminectomy with fusion at L4/5 aged 26 for manage-ment of spinal stenosis. At follow-up, aged 32, he is148 cm tall (�1st centile) with an arm span of 140 cm. Hehas a flattened columella and midface. He has restrictedelbow movement due to radial head dislocation with 30degrees fixed flexion deformity bilaterally. He continuesto suffer chronic low back pain, works as a video repairerand is able to function without hindrance in thiscapacity. His does not engage in sports due to back pain.Radiographic features of CDP-TM showed markedimprovement during childhood and adolescence (seeRadiographic data).
MATERIALS AND METHODS
Karyotype, FISH for SHOX Deletion,Sterol Analysis
Karyotype was analyzed using venous blood lymp-hocyte cultures. Molecular cytogenetic studies usedfluorescent in situ hybridization (FISH) with thechromosome X and Y probe c34F5 (DXYS233) to lookfor an X chromosome deletion involving the SHOXregion. Sterols were determined using gas chromato-graphy–mass spectrometry as previously described[Kelley, 1995] with minor modifications. Selected ionchromatograms for 351 m/z and 229 m/z were used todetermine 8-dehydrocholesterol and cholest-8(9)-en-3beta-ol, respectively.
DNA Extraction and Analysisfor Arylsulfatase D and E Mutations
DNA extraction from blood and cultured fibroblastswas carried out as published by Blok et al. [1995].Individual exons were PCR amplified by use of intronspecific primers [Franco et al., 1995; G. Meroni andcolleagues, unpublished data]. Single strand conforma-tion polymorphism (SSCP) detection was performed onthe amplified exon fragments as described by Orita et al.[1989]. Fragments were labeled internally with a33P-dATP during PCR amplification [Dean and Greenwald,1995]. PCR fragments were purified from 3% NuSieveagarose gels using a 200 ml plugged tip purificationmethod [Dean and Greenwald, 1995]. The DNA wassequenced as described by Blok et al. [1995], using aSequenase version 2.0 kit (USB, Cleveland, OH).
RESULTS
Radiographic Findings
Patient 1. Radiographs were taken at the age of3 months. These revealed calcific stippling at the wristsand ankles as well as in the sacral area. There washypoplasia of several thoracic vertebrae and sagittal
clefting of cervical and thoracic vertebrae. The humeriwere short and the distal ulnae were hypoplastic withrelatively long, bowed radii (Fig. 4a). The hands wererelatively normal at this age. The femora were bowedand the tibiae markedly shortened with relatively longfibulae (Fig. 4b).
Radiographic appearance by midchildhood had im-proved with growth of the long bones but continuingmarked distal ulnar hypoplasia. Tibial growth wasimpressive with only mild relative fibular overgrowth(proximally and distally) remaining by age 6.5 years(Fig. 4c). Tarsal and carpal bone development wasrelatively normal with no residua of stippling. Thevertebrae were abnormal in shape with anterior wed-ging and posterior scalloping (Fig. 4d). At the age of37 years, radiographs of the upper limbs revealed signi-ficant resolution of abnormal findings compared withthe initial studies. Despite this, the ulnae remainedshort distally, with relatively long radii with dislocatedheads (Fig. 4e). The distal radius was angulated giving a‘‘Madelung’’ type appearance (Fig. 4e). The lower limbsshowed remarkable resolution of radiographic changewith almost normal appearing tibiae and only minimalproximal fibular overgrowth (Fig. 4f ). Hand radio-graphs remained normal (Fig. 4g).
Patient 2. Initial radiographs at the age of3 months revealed similar features to patient 1, in-cluding short tibiae and tarsal, sacral, and metacarpalstippling (Fig. 5a). In addition, there was shorteningof the 4th metacarpal present (Fig. 5b). The distalulnae were not hypoplastic (Fig. 5b). By age 8 yearsthe tibiae were almost normal with mild relativefibular overgrowth (Fig. 5c). At the age of 25 yearsupper and lower limb radiographs were relativelynormal. The hand radiographs revealed hypoplastic4th and 5th metacarpals but normal carpal devel-opment and distal ulnae and radii (Fig. 5d). Thecervical spine showed loss of normal lordosis with minorkyphos at the C4/C5 level, but no other abnormalities(Fig. 5e).
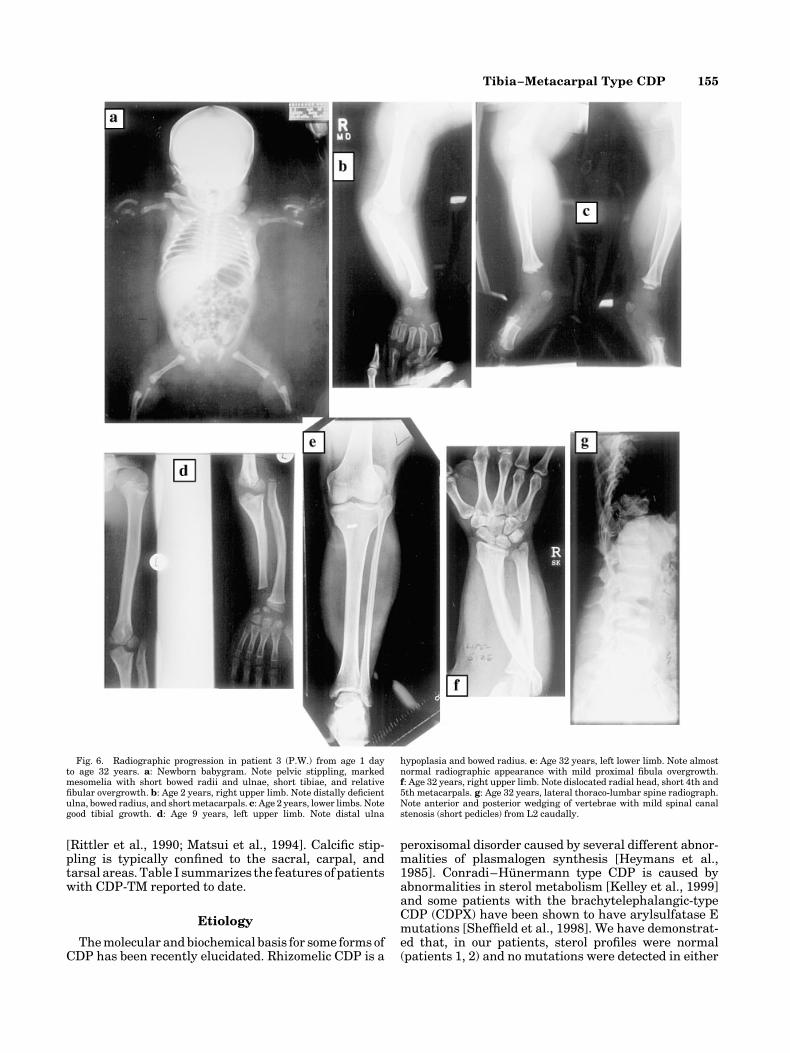
Patient 3. Newborn radiographs revealed stipplingof the proximal humeri, proximal femora, and sacralarea with short ulnae and bowed radii and short tibiaeand relatively long fibulae (Fig. 6a). The tibial dispro-portion lessened over time but the distal ulnar hypopla-sia remained significant during childhood (Fig. 6b–d).By age 32 years there had been almost completeresolution of lower limb radiographic changes with gooddistal ulnar growth and short 4th and 5th metacarpals(Fig. 6e,f ). Lateral thoraco-lumbar spine radiographsshowed anterior wedging with posterior scalloping andevidence of mild residual spinal canal stenosis from L2caudally (Fig. 6g).
Cytogenetic and Biochemical Analysis
Patient 1. Karyotype at the age of 2 years and6 months showed a normal male, 46,XY. Plasma sterolprofile is normal and the patient was unwilling toundergo further blood tests when recently reviewed.
Patient 2. Karyotype aged 24 was normal female,46,XX and FISH analysis showed no deletion at the
152 Savarirayan et al.

SHOX locus. Plasma sterol analysis (7-dehydrocho-lesterol 1 micromol/L, cholesterol 3.7 micromol/L, 8-dehydrocholesterol, and cholest-8(9)en-ol not detected)was normal. SCCP analysis and sequencing of the exonsof arylsulfatase D and E did not detect a mutation.
Patient 3. Karyotype was normal male, 46,XY.Very long chain fatty acid profile and blood phytanicacid levels were normal.
DISCUSSION
CDP and CDP-TM
Stippled calcifications at sites of endochondral ossifi-cation occur secondary to a number of congenitalconditions and in the various forms of CDP as recentlyclassified by the International Skeletal DysplasiaSociety [Hall, 2002]. An alternate classification has
Fig. 4. Radiographic progression in patient 1 (F.K.) from age 3 months toage 37 years documenting marked resolution of early features. a: Age 3months, left upper limb. Note short humerus, distal hypoplasia of ulna,stippling at wrist, and relatively long, bowed radius with relatively normalhands. b: Age 9 months, lower limbs. Note bowed femora, short, bowed tibiaewith relatively long fibulae. c: Age 6.5 years, lower limbs. Note impressivetibial growth with mild residual proximal and distal fibular overgrowth.
d: Age 9 years, lateral spine radiograph. Note abnormal vertebral morphol-ogy with anterior rounding and posterior scalloping. e: Age 37 years, leftupper limb. Note short, distally shortened ulna with relatively long, bowedradius with dislocated radial head. f: Age 37 years, right lower limb. Notealmost complete resolution of early radiographic features with residual(relative) proximal fibular overgrowth. g: Age 37 years. Normal left handradiograph.
Tibia–Metacarpal Type CDP 153

been proposed [Dahl et al., 1999], which includes thecommon (or ‘‘symmetrical’’ form) of CDP and also dis-orders where the etiology is, as yet, unknown.
It has been suggested that a rare ‘Humero-Metacar-pal’ form of CDP may represent a distinct entity[Borochowitz, 1991; Fryburg and Kelly, 1996]. Recentlya clinical form similar to rhizomelic CDP but without thebiochemical features has been described [Kumada et al.,2001]. The patients reported by Leroy of ‘‘dominantmesomelic dwarfism’’ appear to have a condition that isdistinct from CDP-TM, as there was no recorded calcificstippling, normal spinal radiographs, no metacarpal orphalangeal abnormalities, and tibial pseudarthrosespresent [Leroy et al., 1975].
CDP-TM is distinguished by short tibiae, short 4thmetacarpals, and vertebral abnormalities (coronal cleftsand deficient ossification) most marked at birth. Origin-ally termed mesomelic dysplasia [Burck et al., 1980;Burck, 1982], a number of patients have since beendescribed with rhizomelia [Haynes and Wangner, 1951;Rittler et al., 1990; Mayden Argo et al., 1996] ormicromelia [Rittler et al., 1990]. Although a shortenedand bowed tibia in the newborn is a constant feature ofthis condition there may also be marked shortening ofthe humerus and femur with or without ulnar short-ening. General shortening of the metacarpals andphalanges particularly the proximal 2nd phalanx andthe 3rd and 4th metacarpals are also common features
Fig. 5. Radiographic progression in patient 2 (C.C.) from age 3 months toage 25 years. a: Age 3 months, lower limbs. Note short tibiae, relatively longfibulae, and tarsal stippling. b: Age 3 months, right hand. Note short 4thmetacarpal and normal distal radius and ulna. c: Age 8 years, lower limbs.
Note good tibial ‘‘catch-up’’ growth with mild relative proximal fibulaovergrowth. d: Age 25 years, right hand. Note short 4th and 5thmetacarpals. e: Age 25 years, lateral cervical spine radiograph. Note lossof normal lordosis.
154 Savarirayan et al.
[Rittler et al., 1990; Matsui et al., 1994]. Calcific stip-pling is typically confined to the sacral, carpal, andtarsal areas. Table I summarizes the features of patientswith CDP-TM reported to date.
Etiology
The molecular and biochemical basis for some forms ofCDP has been recently elucidated. Rhizomelic CDP is a
peroxisomal disorder caused by several different abnor-malities of plasmalogen synthesis [Heymans et al.,1985]. Conradi–Hunermann type CDP is caused byabnormalities in sterol metabolism [Kelley et al., 1999]and some patients with the brachytelephalangic-typeCDP (CDPX) have been shown to have arylsulfatase Emutations [Sheffield et al., 1998]. We have demonstrat-ed that, in our patients, sterol profiles were normal(patients 1, 2) and no mutations were detected in either
Fig. 6. Radiographic progression in patient 3 (P.W.) from age 1 dayto age 32 years. a: Newborn babygram. Note pelvic stippling, markedmesomelia with short bowed radii and ulnae, short tibiae, and relativefibular overgrowth. b: Age 2 years, right upper limb. Note distally deficientulna, bowed radius, and short metacarpals. c: Age 2 years, lower limbs. Notegood tibial growth. d: Age 9 years, left upper limb. Note distal ulna
hypoplasia and bowed radius. e: Age 32 years, left lower limb. Note almostnormal radiographic appearance with mild proximal fibula overgrowth.f: Age 32 years, right upper limb. Note dislocated radial head, short 4th and5th metacarpals. g: Age 32 years, lateral thoraco-lumbar spine radiograph.Note anterior and posterior wedging of vertebrae with mild spinal canalstenosis (short pedicles) from L2 caudally.
Tibia–Metacarpal Type CDP 155

the arylsulfatase D or E genes (patient 2). The etiology ofCDP-TM therefore remains unknown at this time.
Inheritance
It has been suggested that CDP-TM may be inheritedas an autosomal dominant trait due to the involvementof both males and females, advanced paternal age in onecase, and the absence of consanguinity [Rittler et al.,1990]. There are no reported instances of transmissionof CDP-TM from parent to child or of familial recurrence.Our patient 2, who has a healthy unaffected daughter(age 3) is the only recorded instance of child-bearing inthis condition. The mode of transmission in CDP-TMremains debatable, and although autosomal dominantinheritance remains the most likely mechanism, X-linked inheritance cannot be excluded given the overallpedigree data.
Natural History
It has previously been suggested that the calcificstippling and vertebral clefts of CDP-TM disappear withtime, but there are few data [Rittler et al., 1990] re-garding longterm developmental progress and func-tional outcome. This is the first report of patients withCDP-TM followed into adulthood. All three patients hadnormal development and school progress and aregainfully employed. Only one patient with CDP-TMhas been reported with a suggestion of motor delay at theage of 9 months, which is most likely secondary to shortstature [Matsui et al., 1994]. All our patients hadrecurrent patellar dislocation, a problem also noted byBurck [1982]. Two of our patients required tympanost-omy tube insertion during childhood for chronic serousotitis media. One patient had symptomatic spinal steno-sis requiring laminectomy, and one patient has devel-oped a cervical and upper limb pain that may be relatedto vertebral abnormalities. The final adult heights ofall three patients are well below the 3rd centile. Tworemain physically active while one has physical limita-tions related to ongoing back pain.
Patients reported by several authors [Haynes andWangner, 1951: case 2; Mosekilde, 1952: case 1;Yakovac, 1954: case 1] were all stillborn or died in in-fancy, and two of the patients reported by Rittler et al.[1990] cases 1 and 2 had severe respiratory problemsduring infancy. It may be that these patients reflectgenetic heterogeneity within CDP-TM or that theyrepresent the most severe end of the phenotypicspectrum.
One of our patients (patient 1) had unilateral opticatrophy from birth, and Savignac [1952] also reported apatient with many of the features of CDP-TM andunilateral optic atrophy. This patient also had bilateralcongenital cataracts that may have been related to theoptic atrophy. In the absence of further reported asso-ciations, a firm causal connection between optic atrophyand CDP-TM cannot be made at this time.
The longterm prognosis in CDP is very variable, andrelates most often to the primary cause underlyingthe skeletal changes. The rhizomelic form of CDP likeother peroxisomal disorders has a poor prognosis,usually leading to death in early childhood [Raymond,1999; White et al., 2003]. The brachytelephalangic (orsymmetrical)-type CDP has a good prognosis and indeedmay be difficult to recognize in adulthood as the charac-teristic punctate epiphyseal changes usually resolve inthe first 2 years of life [Sheffield et al., 1976]. A risk ofspinal stenosis and myelopathy, however, is well re-cognized in some forms of CDP including the Conradi–Hunermann form and rhizomelic CDP [Goodman andDominguez, 1990; Takano et al., 1998; Khanna et al.,2001]. It is clear from this report that spinal stenosismay also be a feature of CDP-TM and should be moni-tored for clinically and radiographically.
It is clear from the longterm follow-up of thesepatients that the marked early radiological features ofCDP-TM improve significantly during childhood andadolescence. Affected patients have normal intellect,short stature (with final adult height below the 3rdcentile) and a range of potential medical complications.These include recurrent otitis media, recurrent patellardislocation, genu valgum, and radial head dislocation
TABLE I. Summary of Medical Complications in Previously Reported Patients With CDP-TM
Case Sex Age (yrs) Ht (cm) Medical complications
Our case 1 M 37 152 Patella subluxationOur case 2 F 25 138 Patella subluxationOur case 3 M 32 148 Patella subluxation, symptomatic spinal stenosisRittler 1 M 3 mo NR Cleft palate, micrognathia, death from pneumonia at 3/12Rittler 2 F 1 NR Frequent respiratory tract infections. Small at birthRittler 3 F 6 101 Normal developmentRittler 6 M 2 79 NilRittler 7 M 12 133 NilBurck 1 (Rittler 5) F 8 yrs 8 mo 115 Patella dislocationBurck 2 (Rittler 4) F 6 80 Patella dislocationAsanti case 3 F 1 mo NR NRMayden Argo M 3 NR Small at birthMatsui M 9 mo NR Delayed motor developmentHaynes M 3 hr NR Died at 3 hr respiratory distressYakovac M Birth 34 cm Stillborn
M, male; F, female; NR, not recorded/unknown.
156 Savarirayan et al.

with secondary fixed flexion deformity at the elbow.Most importantly, as in other forms of CDP, there is arisk of symptomatic spinal stenosis. These data suggestthat individuals with CDP-TM warrant careful ongoingclinical and radiographic surveillance and appro-priate education and anticipatory guidance regardingthe complications of the condition and its anticipatednatural history.
ACKNOWLEDGMENTS
We thank Dr. David Danks, Dr. Christine Oley, andDr. Simon Latham for clinical management of thepatients, Dr. Fred Jensen for radiographic opinion,and Amelia Osborn and Wendy Hutchison for DNAanalysis of the arylsulfatase D and E genes.
REFERENCES
Asanti R, Heikel P-E. 1963. Chondroangiopathia calcarea or punctata.Ann Paediatr Fenn 9:280–289.
Blok RB, Thorburn DR, Thompson GN, Dahl HH. 1995. A topoisomerase IIcleavage site is associated with a novel mitochondrial DNA deletion.Hum Genet 95:75–81.
Borochowitz Z. 1991. Generalized chondrodysplasia punctata with short-ness of humeri and brachymetacarpy: Humero-metacarpal (HM) type:Variation or heterogeneity? Am J Med Genet 41:417–422.
Burck U. 1982. Mesomelic dysplasia with punctata epiphyseal calcifica-tions—A new entity of chondrodysplasia punctata? Eur J Pediatr 138:67–72.
Burck U, Schaefer E, Held KR. 1980. Mesomelic dysplasia with shortulna, long fibula, brachymetacarpy, and micrognathia. Clinical andradiological differential diagnostic features. Pediatr Radiol 9:161–165.
Dahl H-HM, Osborn AH, Hutchison WM, Thorburn DR, Sheffield LJ. 1999.Late diagnosis of maternal PKU in a family segregating an arylsulfataseE mutation causing symmetrical chondrodysplasia punctata. Mol GenMetab 68:503–506.
Dean AD, Greenwald JE. 1995. Use of filtered pipette tips to elute DNA fromagarose gels. BioTechniques 18:980.
Franco B, Meroni G, Parenti G, Levilliers J, Bernard L, Gebbid M, Cox L,Maroteaux P, Sheffield L, Rappoid GA, Andria G, Petit C, Ballabio A.1995. A cluster of sulfatase genes on xp 22.3: mutations in chondrodys-plasia punctata (CDPX) and implications for warfarin embryopathy.Cell 81:15–25.
Fryburg JS, Kelly TE. 1996. Chondrodysplasia punctata, humero-metacar-pal type: A second case. Am J Med Genet 64:493–496.
Goodman P, Dominguez R. 1990. Cervical myelopathy in Conradi–Hunermann disease: MRI diagnosis. Magn Reson Imaging 8:647–650.
Hall CM. 2002. International Working Group on the Classification ofConstitutional Disorders of Bone. International Nosology and classifica-tion of constitutional disorders of bone (2001). Am J Med Genet 113:65–77.
Haynes ER, Wangner WMF. 1951. Chondroangioapathia calcarea seupunctata. Review and case report. Radiology 57:547–550.
Heymans HS, Oorthuys JW, Nelck G, Wanders RJ, Schutgens RB. 1985.Rhizomelic chondrodysplasia punctata: Another peroxisomal disorder[letter]. N Engl J Med 313:187–188.
Jansen V, Sarafoglou K, Rebarber A, Greco A, Genieser NB, Wallerstein R.2000. Chondrodysplasia punctata, tibia–metacarpal type in a 16 weekfetus. J Ultrasound Med 19(10):719–722.
Kelley RI. 1995. Diagnosis of Smith–Lemli–Opitz syndrome by gaschromatography/mass spectrometry of 7-dehydrocholesterol in plasma,amniotic fluid, and cultured skin fibroblasts. Clin Chimica Acta 236:45–58.
Kelley RI, Wilcox WG, Smith M, Kratz LE, Moser A, Rimoin DS. 1999.Abnormal sterol metabolism in patients with Conradi–Hunermann–Happle syndrome and sporadic lethal chondrodysplasia punctata. Am JMed Genet 83:213–219.
Khanna AJ, Braverman NE, Valle D, Sponseller PD. 2001. Cervical stenosissecondary to rhizomelic chondrodysplasia punctata. Am J Med Genet99:63–66.
Kumada S, Hayashi M, Kenmochi J, Kurosawa S, Shimozawa N, Kratz LE,Kelley RI, Taki K, Okaniwa M. 2001. Lethal form of chondrodysplasiapunctata with normal plasmalogen and cholesterol biosynthesis. Am JMed Genet 98:250–255.
Leroy JG, de Vos J, Timmermans J. 1975. Dominant mesomelic dwarfism ofthe hypoplastic tibia, radius type. Clin Genet 7:280.
Matsui M, Honma Y, Oguro N, Shiraishi H, Kobayashi S, Yanagisawa M,Nakamura K. 1994. Case report: A newborn case of chondrodysplasiapunctata, tibia–metacarpal type. Br J Radiol 67:97–99.
Mayden Argo K, Toriello HV, Jelsema RD, Zuidema LJ. 1996. Prenatalfindings in chondrodysplasia punctata, tibia–metacarpal type. Ultra-sound Obstet Gynecol 8:350–354.
Mosekilde E. 1952. Stippled epiphyses in the newborn and in infants. ActaRadiol [Diagn] (Stockh) 37:297–307.
Orita M, Suzuki Y, Sekiya T, Hayashi K. 1989. Rapid and sensitive detectionof point mutations and DNA polymorphisms using the polymerase chainreaction. Genomics 5:874–879.
Raymond GV. 1999. Peroxisomal disorders. Curr Opin Pediatr 11:572–576.
Rittler M, Menger H, Spranger J. 1990. Chondrodysplasia punctata, tibia–metacarpal (MT) type. Am J Med Genet 37:200–208.
Rogers J. 1989. Mesomelic dysplasia. Confirmation of a new clinical type ofchondrodysplasia punctata. Clinical Genetics Conference Abstracts. AmJ Med Genet 34:135–147, A36.
Savignac EM. 1952. Chondrodystrophia calcificans congenita: Report of acase. Radiology 58:415–420.
Sheffield LJ, Danks DM, Mayne V, Hutchinson AL. 1976. Chondrodysplasiapunctata—23 Cases of a mild and relatively common variety. J Pediatr89:916–923.
Sheffield LJ, Osborn AH, Hutchison WM, Sillence DO, Forrest SM, WhiteSJ, Dahl H-H. 1998. Segregation of mutations in arylsulfatase E andcorrelation with the clinical presentation of chondrodysplasia punctata.J Med Genet 35:1004–1008.
Takano H, Smith WL, Sato Y, Kao SC. 1998. Cervical spine abnormalitiesand instability with myelopathy in warfarin-related chondrodysplasia:17-Year follow-up. Pediatr Radiol 28:497–499.
White AL, Modaff P, Holland-Morris F, Pauli RM. 2003. Natural history ofrhizomelic chondrodysplasia punctata. Am J Med Genet, publishedonline 27th Jan. DOI: 10.1002/ajmg.a.20009
Yakovac WC. 1954. Calcareous chondropathies in the newborn infant.Arch Pathol Lab Med 57:62–79.
Tibia–Metacarpal Type CDP 157





























