Embed Size (px)
Citation preview

Lecture 7.1 1
The Ensembl Database
Erin Pleasance
Steven Jones
Canada’s Michael Smith Genome Sciences Centre, Vancouver
Lecture 7.1 2
www.ensembl.org

Lecture 7.1 3
What is Ensembl?
• Public annotation of mammalian and other genomes
• Open source software• Relational database system• The future of genomic bioinformatics?

Lecture 7.1 4
The Ensembl Project
“Ensembl is a joint project between EMBL European Bioinformatics Institute and the Sanger Institute to develop a software system which produces and maintains automatic annotation on eukaryotic genomes. Ensembl is primarily funded by the Wellcome Trust”

Lecture 7.1 5
The Ensembl Project
“The main aim of this campaign is to encourage scientists across the world - in academia, pharmaceutical companies, and the biotechnology and computer industries - to use this free information.”
- Dr. Mike Dexter, Director of the Wellcome Trust
Lecture 7.1 6
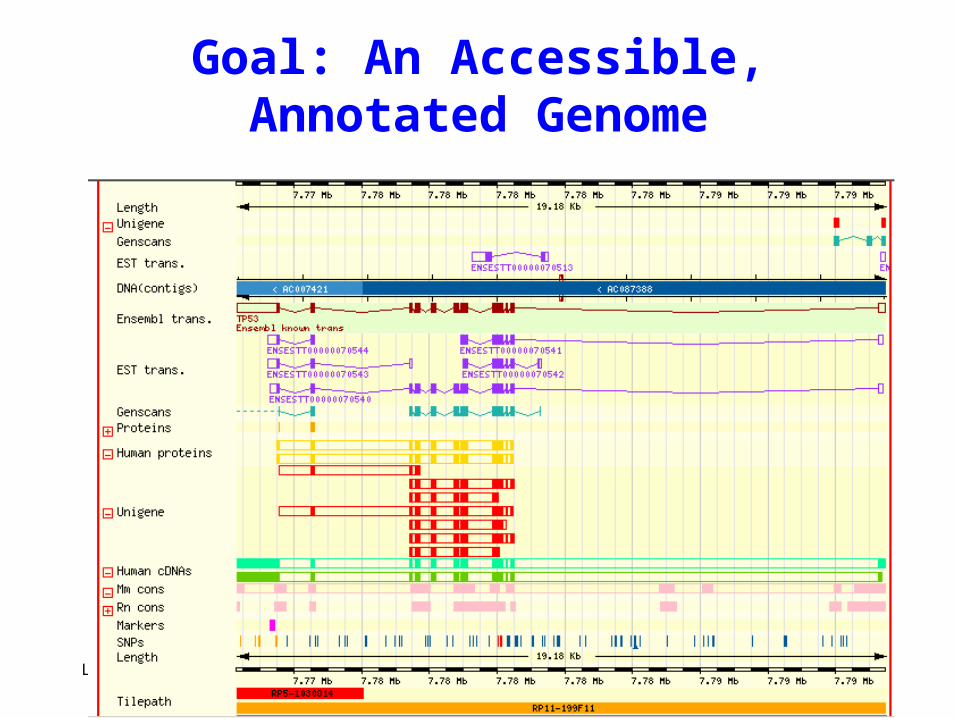
Diagram of contigview as “what we want in the end”
Goal: An Accessible, Annotated Genome

Lecture 7.1 7
Ensembl Software System
• Uses extensively BioPerl (www.bioperl.org)• The free mySQL database• Entire Ensembl code base is freely available
under Apache open source license. • Mainly written in Perl, extensions in C. Some
viewers have been written in Java (e.g. Apollo).

Lecture 7.1 8
Ensembl Genome Annotation
• Utilizes raw DNA sequence data from public sources
• Creates a tracking database (The “Ensembl database”)
• Joins the sequences - based on a sequence scaffold or “Golden Path”
• Automatically finds genes and other features of the sequence
• Associates sequence and features with data from other sources
• Provides a publicly accessible web based interface to the database

Lecture 7.1 9
The Genome Problem
• The problem with the genome (particularly human) is that it is “large, complicated, and opaque to analysis” (Ewan Birney, Ensembl)
• Genome features to identify include:– Genes: protein coding, RNA, pseudogenes– Regulatory elements– SNPs, repeats, etc….

Lecture 7.1 10
DNA sequence in Ensembl
• Sequences are determined in fragments (contigs)• Features cross boundaries between fragments
• Entire sequence too large and changes too much (constantly updated and reassembled) to be stored as one long database entry

Lecture 7.1 11
DNA sequence in Ensembl
• Core design feature is the “virtual contig” object
• Allows genome sequence to be accessed as a single large contiguous sequence even though it is stored as a collection of fragments
• VC object handles reading and writing features to the DNA sequence

Lecture 7.1 12
Ensembl Gene Build System
• Three-part gene build system– “Best in genome” matches for known genes– Alignment of homologous genes– Ab initio gene finding
• Genes predicted on repeat-masked DNA• All genes predicted based on experimental
(available sequence) evidence

Lecture 7.1 13
“Best in genome” predictions
• Find known proteins from SPTREMBL on genome using pmatch
• Incorporate cDNAs using exonerate and EST_genome– Align with gaps placed preferentially at splice
consensus sites– Allows prediction of 5’ and 3’ UTRs
• Refine predictions using genewise
Lecture 7.1 14
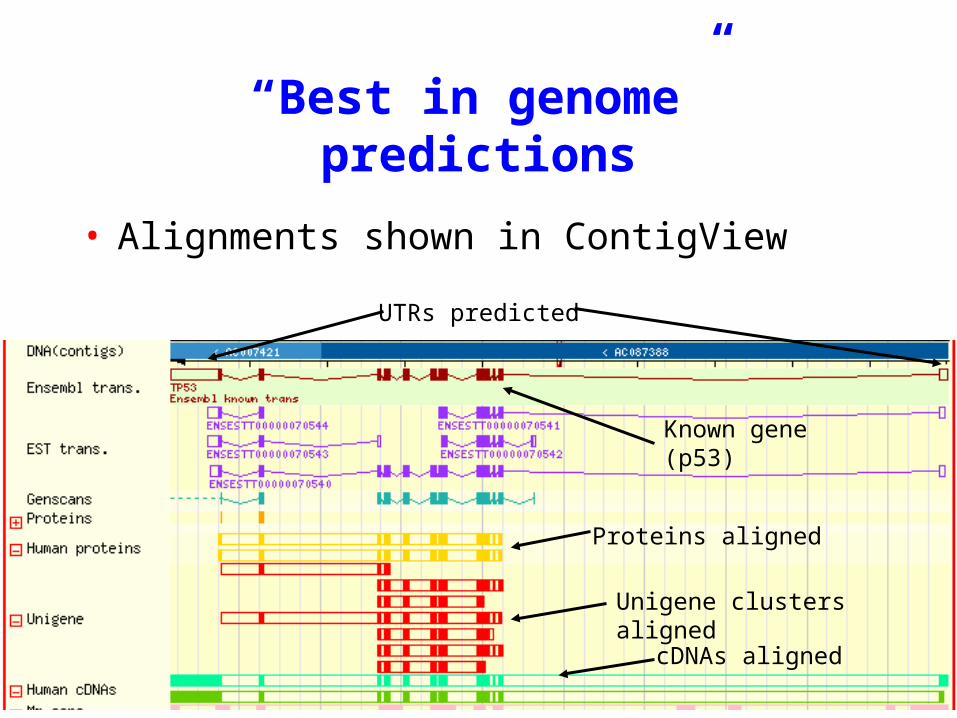
“Best in genome” predictions
ContigView of best in genome gene with associated evidence
Known gene (p53)
Proteins aligned
cDNAs aligned
UTRs predicted
Unigene clusters aligned
• Alignments shown in ContigView

Lecture 7.1 15
Homology predictions
• Align homologous proteins using BLAST, genewise– Paralogs (from same organism)– Orthologs (from closely related organisms)
• Assemble novel genes

Lecture 7.1 16
Ab initio gene predictions
• Use Genscan to identify novel exons• Confirm exons by BLAST to known proteins, mRNAs,
UniGene clusters• Based on ab initio predictions but require homology
evidence
ContigView of homology gene with associated evidence
Novel geneGenScan predictions
Proteins aligned
Unigene clusters aligned
Lecture 7.1 17
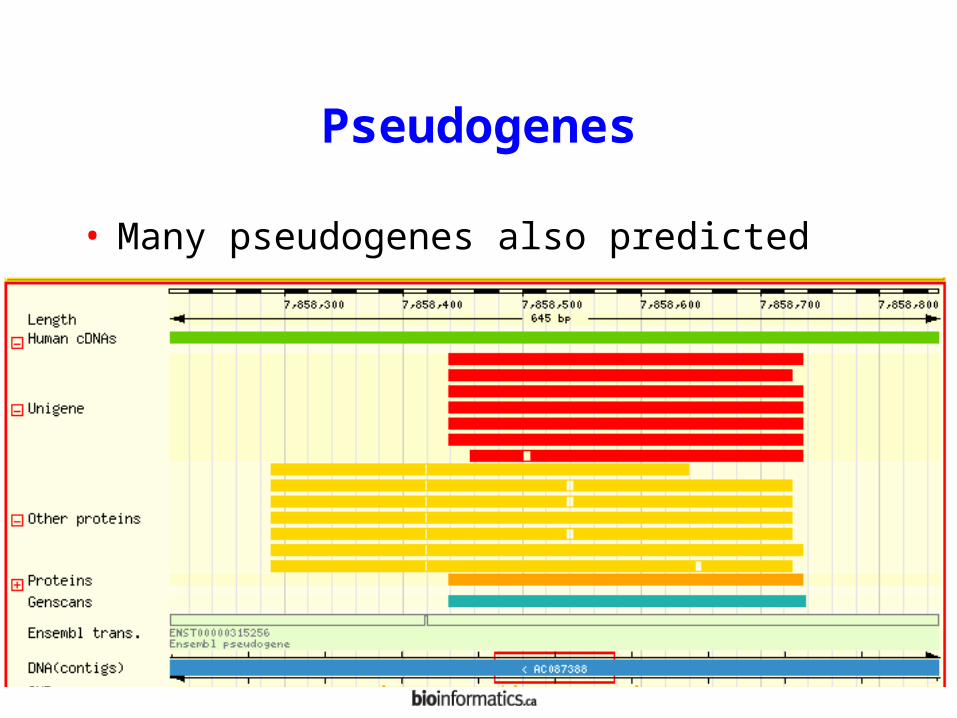
Pseudogenes
• Many pseudogenes also predicted

Lecture 7.1 18
Ensembl Gene Build System
• Resulting “Ensembl genes” are highly accurate with low false positive rates
• Ensembl human gene identifiers are 95% stable between builds
Snapshot or stats on genes

Lecture 7.1 19
Ensembl EST genes
• ESTs not accurate enough to produce Ensembl genes, but important especially for identifying alternative transcripts
• Create an independent set of “EST genes”
Known gene
Unigene clusters aligned
EST genes

Lecture 7.1 20
Ensembl EST genes
• Map ESTs to genome using Exonerate, BLAST, and EST2Genome
• Define transcripts by merging redundant ends, setting splice sites to common ends– Finds splice sites and defines UTRs– Alternative transcript predicted if at least one
alternatively spliced EST exists
• Process transcripts with Genomewise to find longest ORF for each

Lecture 7.1 21
Ensembl EST genes
• Evidence for genes shown (ExonView)

Lecture 7.1 22
Manual gene annotation: Otter
• Manual annotation done with applications eg. Apollo
• Otter database/server allows manual annotations to be integrated with automated annotations

Lecture 7.1 23
Manually curated genes: VEGA
• Chromosomes 6,7,13,14, 20 and 22 contain manually curated genes from VEGA database
Lecture 7.1 24
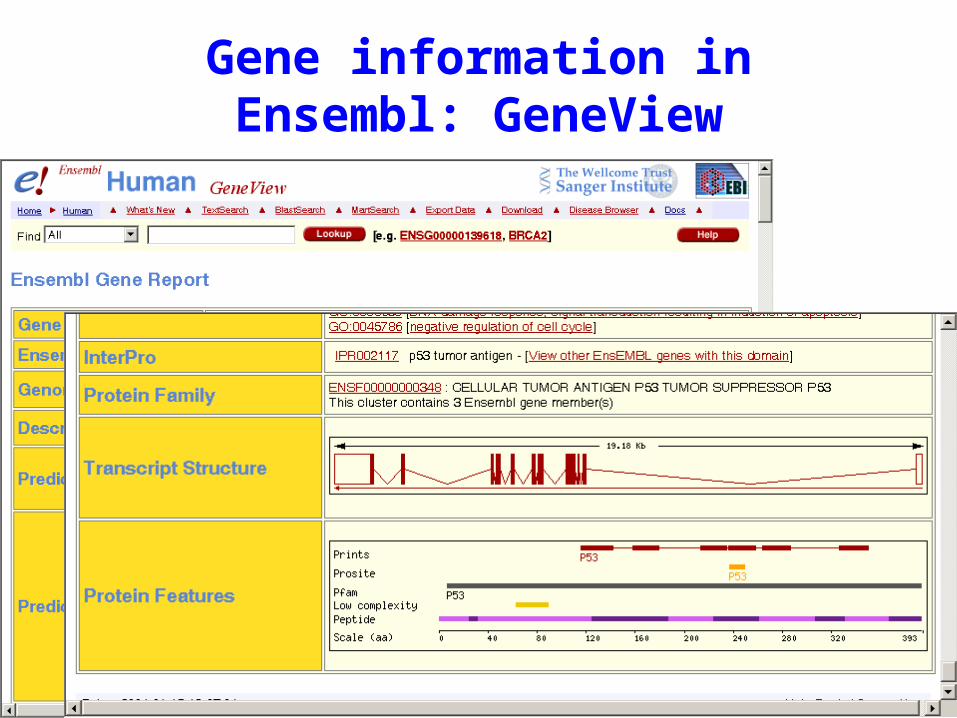
Gene information in Ensembl: GeneView
Lecture 7.1 25
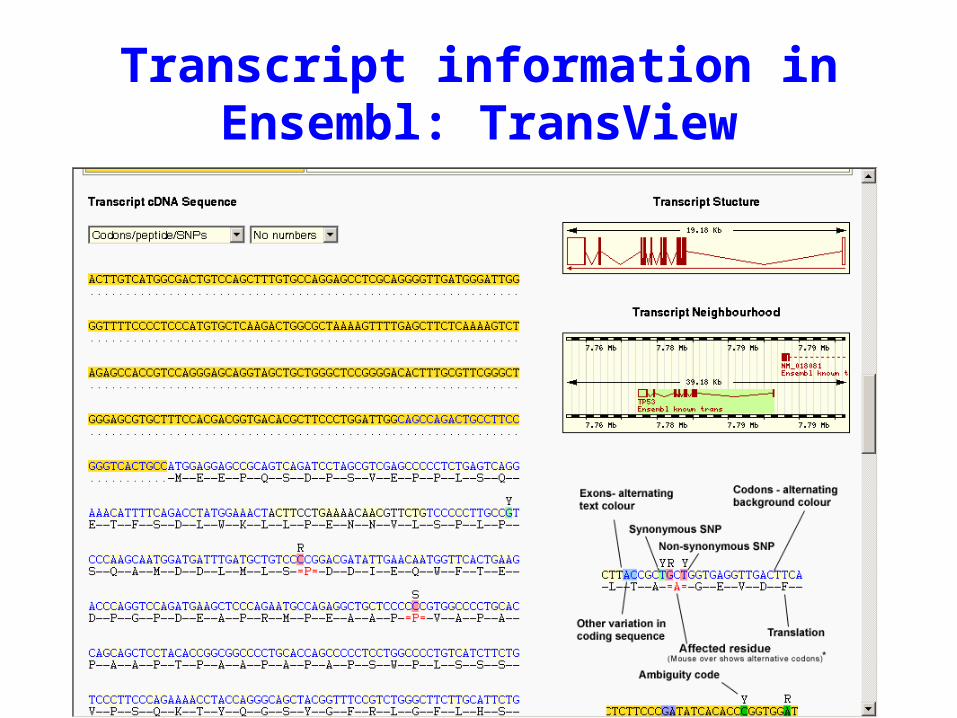
Transcript information in Ensembl: TransView

Lecture 7.1 26
Protein information in Ensembl: ProteinView
Lecture 7.1 27
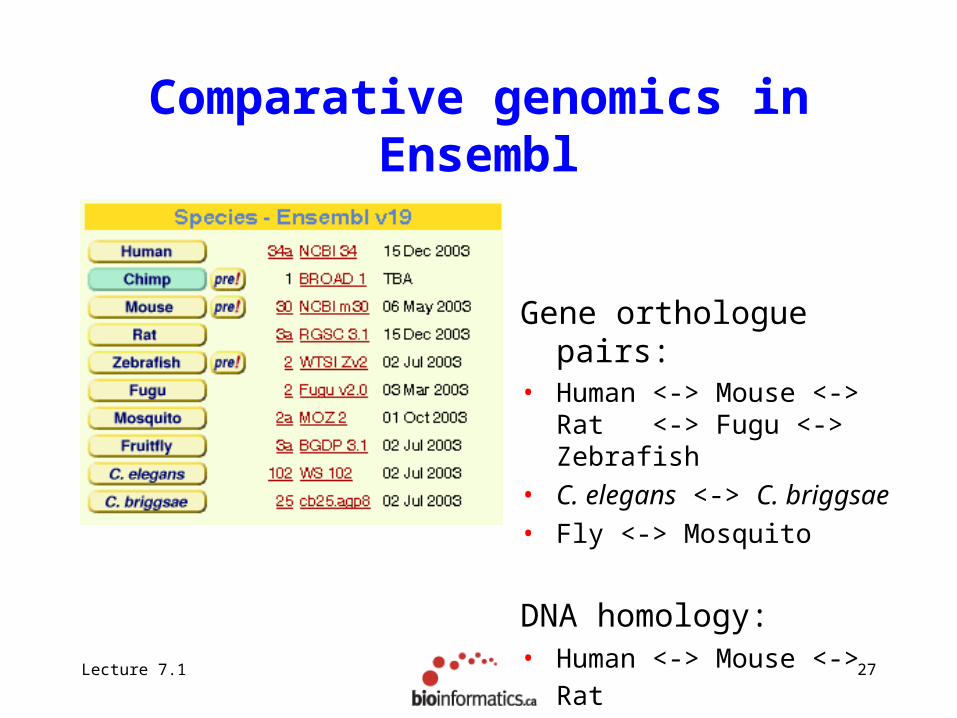
Comparative genomics in Ensembl
Gene orthologue pairs:• Human <-> Mouse <-> Rat
<-> Fugu <-> Zebrafish• C. elegans <-> C. briggsae• Fly <-> Mosquito
DNA homology:• Human <-> Mouse <-> Rat

Lecture 7.1 28
Comparative genomics in Ensembl: Gene orthologs
• Gene ortholog pairs shown in GeneView• Calculated by BLAST (reciprocal best BLAST hits, or
BLAST + synteny)• dN/dS = nonsynonymous/synonymous change
(measure of selection)
Lecture 7.1 29
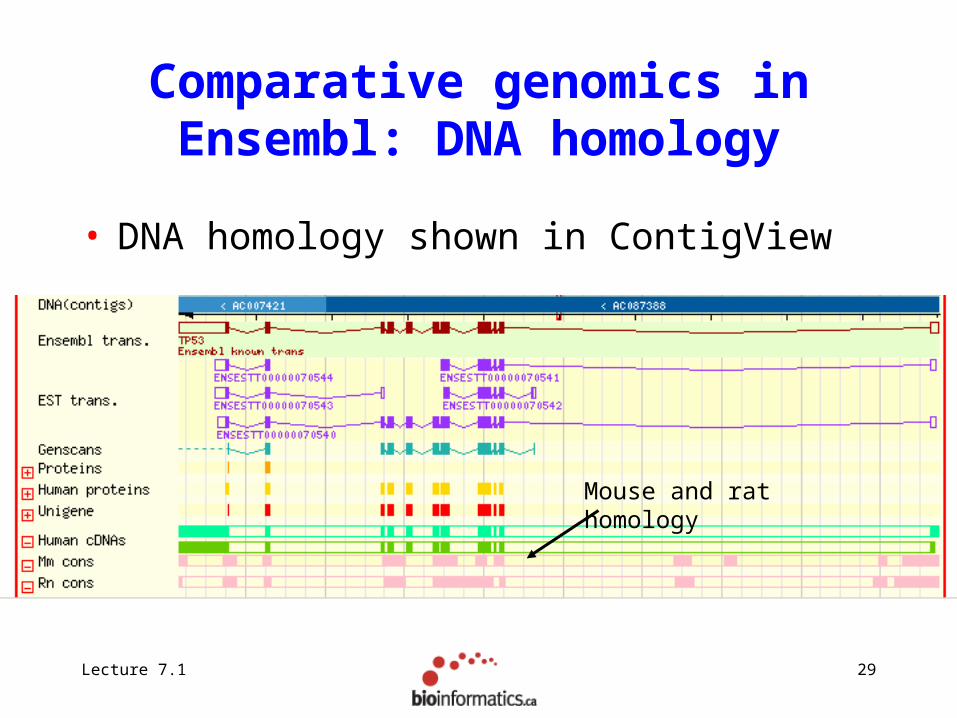
Comparative genomics in Ensembl: DNA homology
• DNA homology shown in ContigView
Mouse and rat homology
Lecture 7.1 30
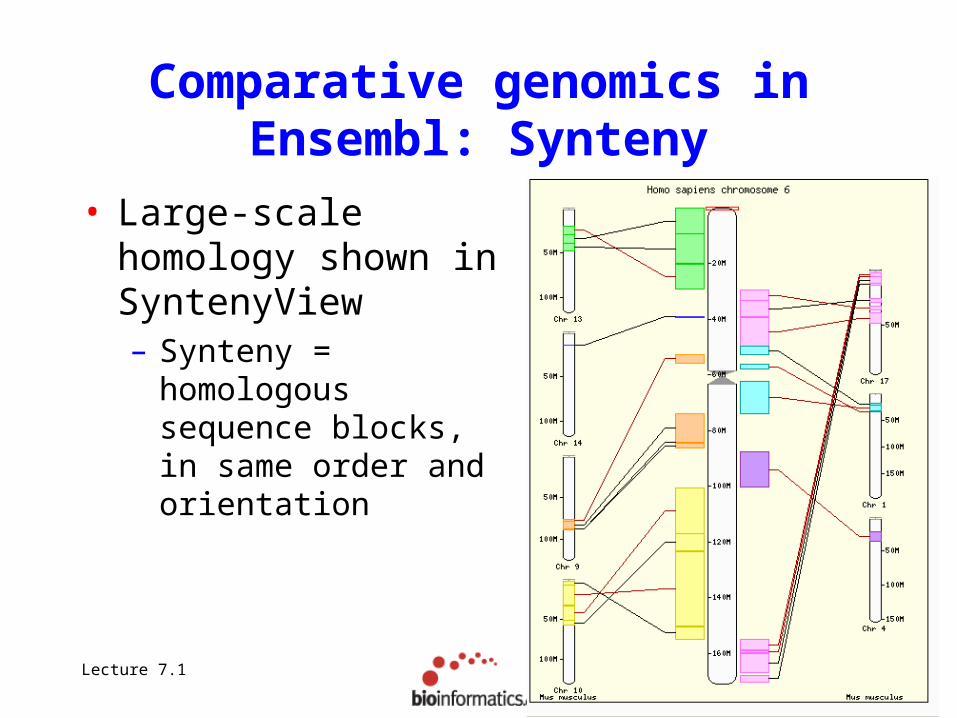
Comparative genomics in Ensembl: Synteny
• Large-scale homology shown in SyntenyView– Synteny = homologous
sequence blocks, in same order and orientation

Lecture 7.1 31
Other features in Ensembl
• Menus provide other feature options
• Features eg. SNPs and markers have special views

Lecture 7.1 32
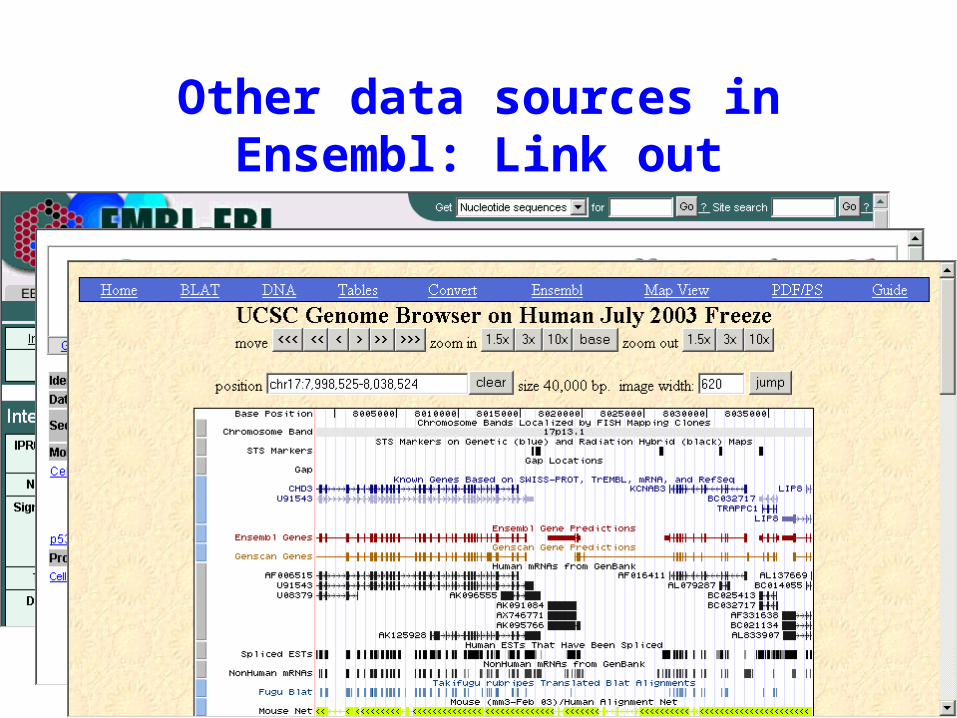
Other data sources in Ensembl
• Ensembl incorporates gene and feature info from many other datasources
OMIM
SwissProt
Lecture 7.1 33
Other data sources in Ensembl: Link out

Lecture 7.1 34
The Distributed Annotation System
• Allows viewing third-party annotation of the genomic scaffold
• Users can choose the annotation they are interested in
• Features are viewed in consistent user interface/display
• Allows specialized feature annotation and the comparison of different methodologies

Lecture 7.1 35
DAS: Selecting data

Lecture 7.1 36
GeneDAS
• GeneDAS allows exchange of annotations on gene level– eg. access to SwissProt annotations from GeneView

Lecture 7.1 37
DAS: Add your own annotations
• Anyone can add data and upload it to DAS server for others to view

Lecture 7.1 38
Sequence similarity searching
• Two search methods– SSAHA: very fast, good for identifying near-exact
DNA-DNA matches – BLAST: slower but more accurate, can do DNA or
protein searches
• Can search against any species• Can search against genomic sequence,
cDNAs (Ensembl or Genscan), or protein sequences
Lecture 7.1 39
Lecture 7.1 40
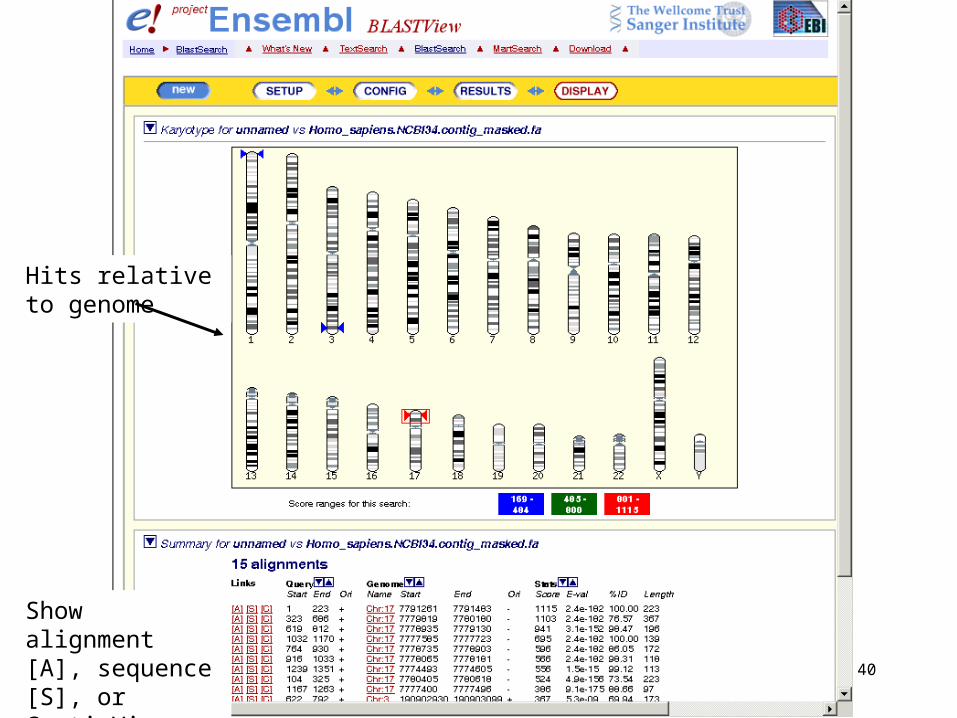
Show alignment [A], sequence [S], or ContigView [C]
Hits relative to genome
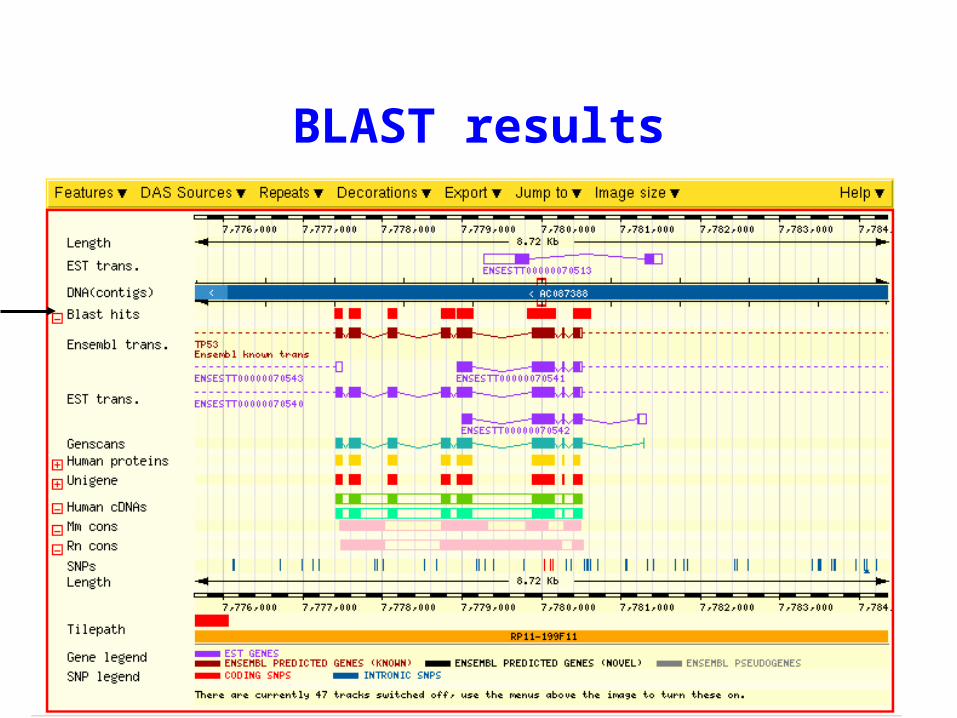
Lecture 7.1 41
BLAST results

Lecture 7.1 42
Data Mining with EnsMart
• EnsMart - organizes data from Ensembl into a query-optimized database
• Allows very fast, cross-data source querying• Accessible from:
– Ensembl website (MartView)– Stand-alone application (MartExplorer)– Command-line interface (MartShell)
• Extremely powerful for data mining

Lecture 7.1 43
Dataming with Ensmart
•Mouse homologues for human disease genes.
•Coding SNPs for all novel kinases.
•Genes on chromosome 1 expressed in liver.
•Ensembl genes mapped to RefSeq identifiers.
•Upstream sequence for all Ensembl genes mapped to U95A chip.
•Disease related genes between markers (eg D10S255 and D10S259).
•Transmembrane proteins with an Ig-MHC domain (IPR003006) on chromosome 2.
•Genes with associated coding SNPs on chromosomal band 5q35.3
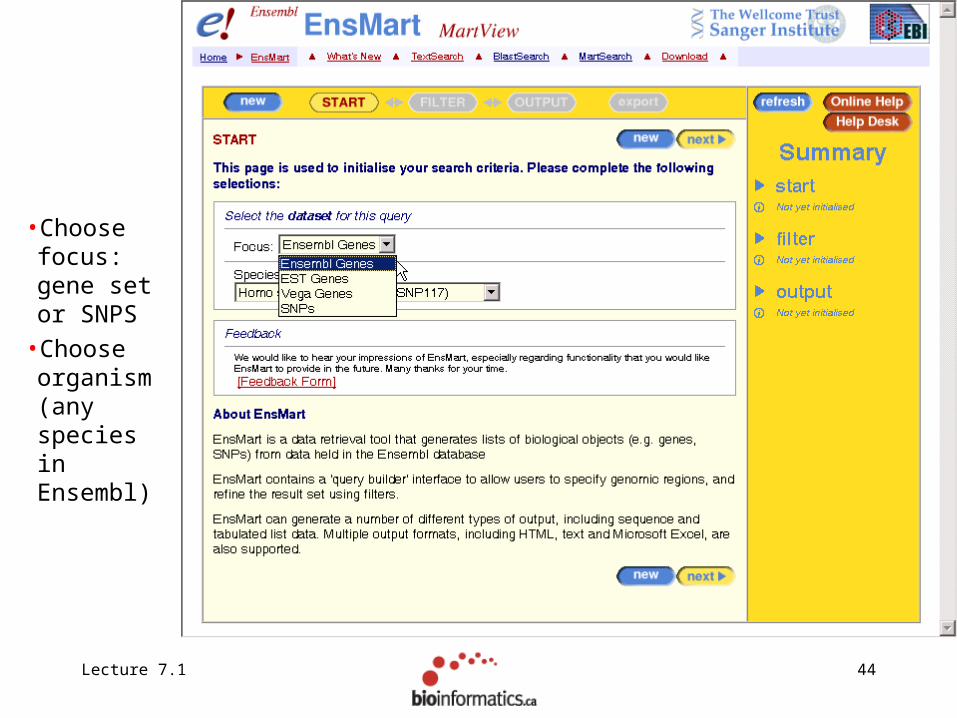
Lecture 7.1 44
•Choose focus: gene set or SNPS
•Choose organism (any species in Ensembl)
Lecture 7.1 45
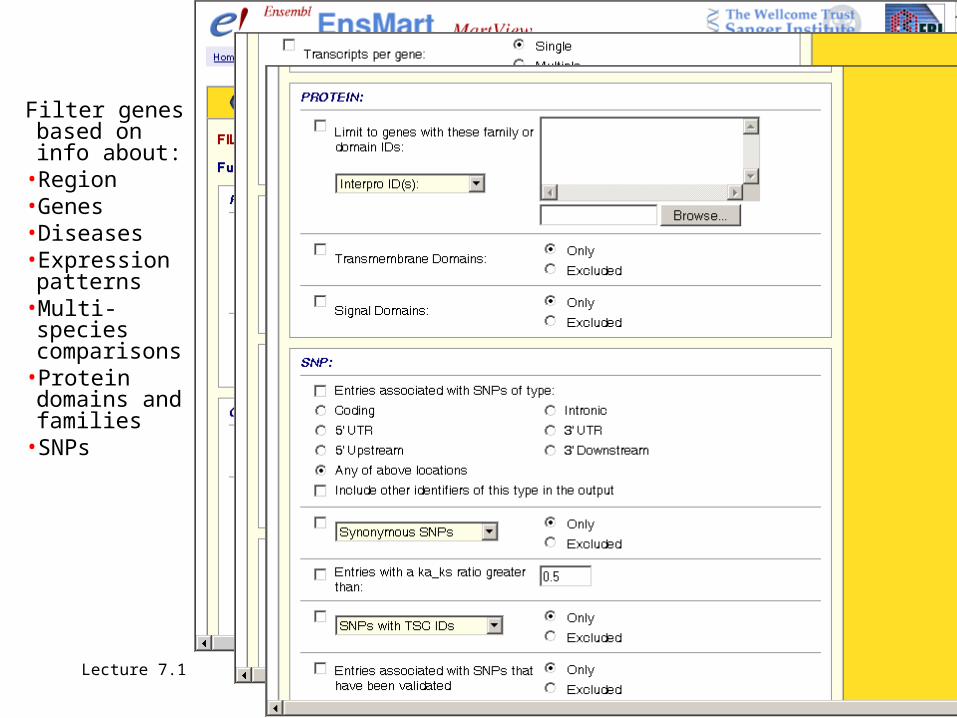
Filter genes based on info about:
•Region•Genes•Diseases•Expression patterns
•Multi-species comparisons
•Protein domains and families
•SNPs
Lecture 7.1 46
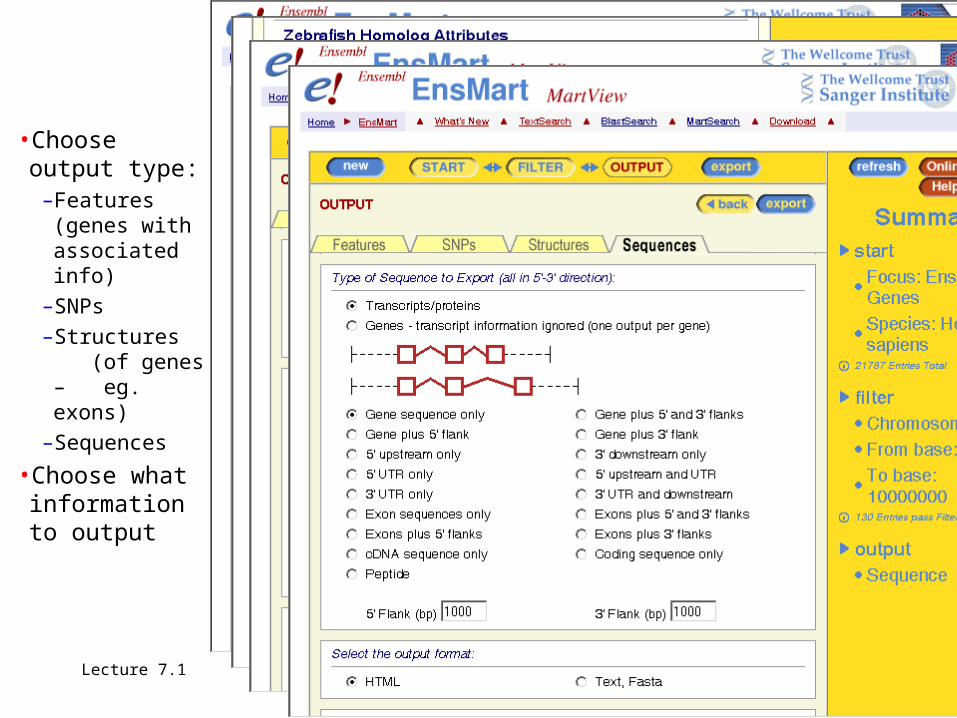
•Choose output type:
–Features (genes with associated info)
–SNPs–Structures
(of genes – eg. exons)
–Sequences
•Choose what information to output

Lecture 7.1 47
Multiple Programming Interfaces now exist for Ensembl
Lecture 7.1 48
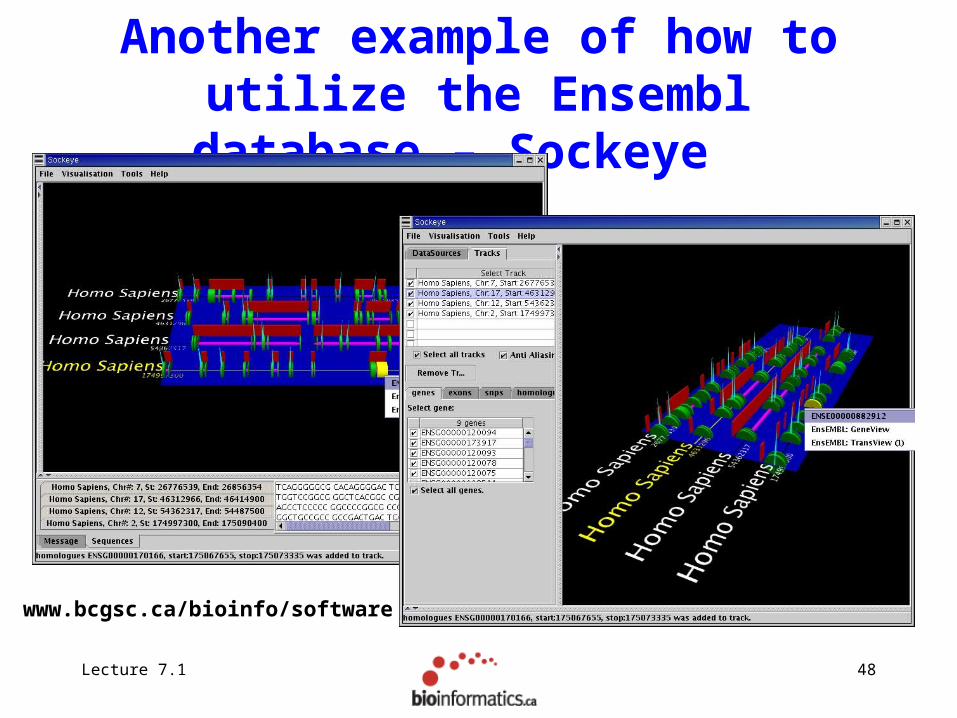
Another example of how to utilize the Ensembl database – Sockeye
www.bcgsc.ca/bioinfo/software
Lecture 7.1 49
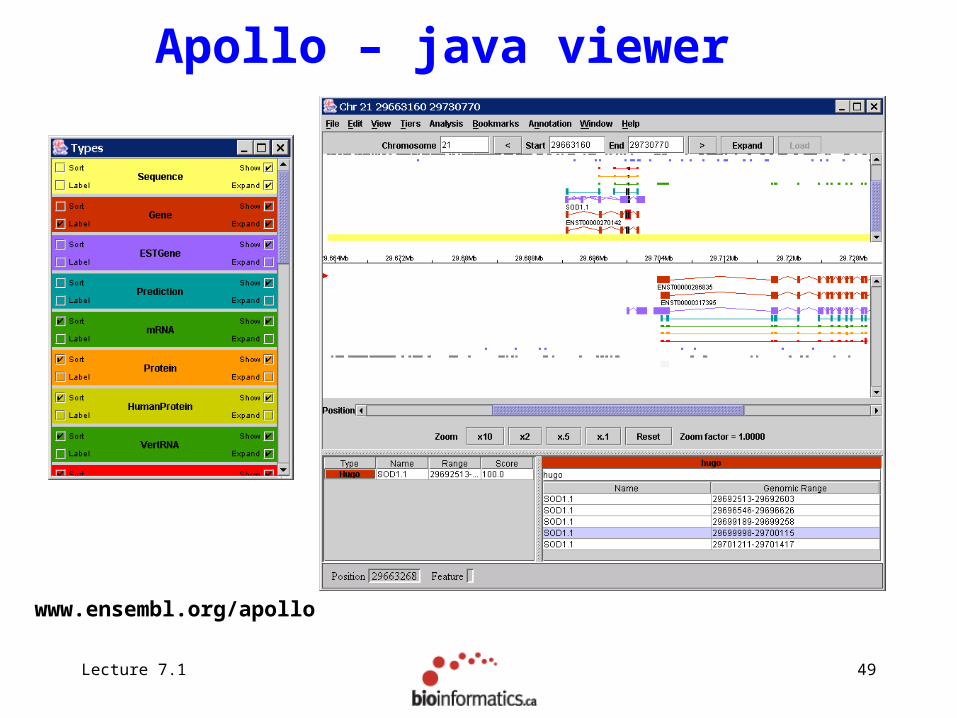
Apollo – java viewer
www.ensembl.org/apollo

Lecture 7.1 50
Ensembl updates
• Monthly• Include:
– Changes in genome builds (with new annotations)– Changes in code or database schema– Additional views and tools on website

Lecture 7.1 51
Pre-Ensembl
• Full annotation can take weeks• Pre-Ensembl site provides in-progress annotation
– Placement of known proteins– Ab initio gene predictions– Repeat masking– BLAST and SSAHA searching

Lecture 7.1 52
Ensembl Software System
• Software can be accessed by FTP• Can also be accessed through CVS
(concurrent versions system)• Possible to set up a mirror of the entire
Ensembl system.

Lecture 7.1 53
Further Information
• The Ensembl Project: www.ensembl.org• VEGA: vega.sanger.ac.uk• EnsMart: www.ensembl.org/EnsMart/• Distribributed Annotation System: www.biodas.org• Human Genome Central Resources:
www.ensembl.org/genome/central• References:
– Ensembl:• Hubbard et al, 2002. NAR 30 (1), 38-41.• Clamp et al, 2003. NAR 31 (1), 38-42.• Birney et al, 2004. NAR 32, D468-D470.
– EnsMart: Birney et al, 2004. Genome Res. 14, 160-169.





























