Embed Size (px)
Citation preview

Kopplungsanalyse und Kandidatengenansatz auf
Chromosom 14 zur Identifikation des ursächlichen Gens in
einer Familie mit Leberscher kongenitaler Amaurose (LCA)
Inauguraldissertation
zur Erlangung des Grades eines Doktors der Medizin
des Fachbereichs Medizin
der Justus-Liebig-Universität Gießen
vorgelegt von Kreutz, Annette, geb. Schick
aus Amöneburg
Gießen, 2016

Aus dem Labor für Molekulare Ophthalmologie der Klinik und Poliklinik für Augenheilkunde,
Universitätsklinikum Gießen und Marburg GmbH, Standort Gießen.
Gutachter: Prof. Dr. Müller
Gutachter: PD Dr. Preising
Tag der Disputation: 05.08.2016

Inhaltsverzeichnis
1 Einleitung ............................................................................................................. 1
1.1 Lebersche kongenitale Amaurose ................................................................. 1
1.1.1 Definition .................................................................................................. 1
1.1.2 Phänotyp.................................................................................................. 1
1.1.3 Vererbungsmuster ................................................................................... 2
1.1.4 Genmutationen und Untertypen der LCA ................................................. 3
1.1.5 Differentialdiagnosen ............................................................................... 5
1.1.6 Genotyp Phänotyp Korrelation ................................................................. 6
1.1.7 Therapie................................................................................................... 8
1.2 Zielsetzung .................................................................................................. 10
2 Material und Methoden ...................................................................................... 11
2.1 Patient und Familie ...................................................................................... 11
2.2 Molekulargenetische Methoden ................................................................... 12
2.2.1 Geräte .................................................................................................... 12
2.2.2 Reagenzien, Enzyme, Chemikalien, Puffer............................................ 13
2.2.3 Phenolextraktion von DNA ..................................................................... 14
2.3 DNA-Konzentrationsbestimmung und Ansetzen der Gebrauchslösungen aus
den DNA-Stammlösungen .................................................................................... 15
2.4 Amplifikation definierter Sequenzen durch die Polymerasekettenreaktion
(PCR) .................................................................................................................... 15
2.5 Gelelektrophorese ....................................................................................... 17
2.5.1 Herstellung des Agarosegels ................................................................. 18
2.5.2 Auftrennung der PCR-Produkte ............................................................. 18
2.6 QIAxcel ........................................................................................................ 19
2.7 Gensequenzierung ...................................................................................... 19
2.8 Kopplungsanalyse ....................................................................................... 19
2.8.1 Definition ................................................................................................ 19
2.8.2 Das menschliche Genom ....................................................................... 20
2.8.3 Identifizierung von pathologischen Genveränderungen ......................... 20

2.8.4 Single Nucleotide Polymorphism (SNP) ................................................ 21
2.8.5 Mikrosatellitenmarker ............................................................................. 21
2.8.6 Logarithmic Odds Ratio (LOD)-Score .................................................... 22
2.8.7 SNP-Genotyping Microarrays ................................................................ 23
2.8.8 Parametrische Analyse .......................................................................... 23
2.9 Ermittlung geeigneter Mikrosatellitenmarker ............................................... 24
2.10 Auswahl möglicher Kandidatengene ........................................................ 28
3 Ergebnisse ........................................................................................................ 29
3.1 Anamnese der Familie ................................................................................. 29
3.2 SNP Genotypsierung und Kopplungsanalyse .............................................. 31
3.3 Optimierung der Mikrosatellitenamplifikation ............................................... 33
3.3.1 Amplifikation der Mikrosatellitenmarker mit den DNA-Proben der Familie
35
3.4 Kopplungsanalyse mit den Mikrosatellitenmarkern ..................................... 35
3.4.1 Haplotypen der Mikrosatellitenmarker auf Chromosom 7 ...................... 35
3.4.2 Haplotypen der Mikrosatellitenmarker auf Chromosom 8 ...................... 37
3.4.3 Haplotypen der Mikrosatellitenmarker auf Chromosom 14 .................... 37
3.4.4 Ergebnis der Haplotypenanalyse ........................................................... 38
3.5 Berechnung der Kopplungswahrscheinlichkeit mit MERLIN ........................ 39
3.6 Evaluation möglicher Kandidatengene auf Chromosom 14 ......................... 46
3.6.1 Tubulin Polymerisation-Promoting Protein family member 2 (TPPP2) ... 47
3.6.2 Defender Against Cell death (DAD1) ..................................................... 47
3.6.3 Retinitis pigmentosa GTPase regulator-interacting Protein (RPGRIP1) 48
3.6.4 Neural Retina Leucine Zipper (NRL) ...................................................... 49
3.7 Ergebnis der Kandidatengenanalyse ........................................................... 49
4 Diskussion ......................................................................................................... 50
4.1 Bewertung des Patientenmaterials .............................................................. 50
4.2 Bewertung der Untersuchungstechnik ......................................................... 50
4.3 Bewertung der Kandidatengene .................................................................. 52
4.4 Die RPGRIP1-Mutation ............................................................................... 53
4.5 Aussichten und Nutzen für die Familie ........................................................ 54

4.6 Ausblick ....................................................................................................... 55
5 Zusammenfassung ............................................................................................ 56
6 Summary ........................................................................................................... 57
7 Abbildungsverzeichnis ....................................................................................... 58
8 Tabellenverzeichnis ........................................................................................... 59
9 Abkürzungsverzeichnis ...................................................................................... 60
10 Literaturverzeichnis ........................................................................................... 60
11 Ehrenwörtliche Erklärung .................................................................................. 76
12 Danksagung ...................................................................................................... 77

1
1 Einleitung
1.1 Lebersche kongenitale Amaurose
1.1.1 Definition
Die nach dem Heidelberger Ophthalmologen Theodor Carl Gustav von Leber benannte
Lebersche kongenitale Amaurose (LCA) beschreibt eine heterogene Gruppe von
Aderhaut- und Netzhautdystrophien mit schweren Sehbeeinträchtigungen bis hin zur
Erblindung [9]. Die bei 5 % der retinalen Dystrophien nachgewiesene LCA ist die am
frühesten einsetzende und am schwersten verlaufende Form der erblichen retinalen
Dystrophien. Die Inzidenz der LCA liegt bei 1–9 : 100.000 [50, 77, 103].
Die Diagnose LCA wird meist nach der Geburt oder in den ersten Lebensmonaten
gestellt und weist eine hohe phänotypische und genotypische Heterogenität auf [71,
103]. Hauptmerkmal des Krankheitsbildes ist eine sich im frühen Lebensalter
manifestierende Einschränkung des Sehvermögens bis hin zur Erblindung [77]. Die
LCA ist die früheste Form der schweren frühkindlichen Netzhautdegenerationen (engl.
early onset severe retinal dystrophy, EOSRD oder engl. severe early childhood onset
retinal dystrophy, SECORD) [102].
1.1.2 Phänotyp
Der Fundus oculi ist bei den meisten Patienten zunächst unauffällig. Im Verlauf der
Erkrankung treten Fundusveränderungen, wie Hyper- und Hypopigmentierungen,
sowie eine unterschiedlich stark ausgeprägte Netzhautatrophie auf [9, 50]. Im weiteren
Verlauf zeigen sich häufig generalisierende Gesichtsfeldeinschränkungen sowie ein
deutlich reduzierter Visus. Die Patienten sind zudem meist hyperob [18, 34, 51, 71].
Auch das Farbensehen ist in der Regel eingeschränkt [71].
Erkrankte sind nicht in der Lage, Gegenstände zu fixieren. Man beobachtet wandernde
Augenbewegungen oder einen sensorischen Nystagmus. Weitere Symptome sind ein
Strabismus oder das okulodigitale Zeichen [9, 17].
Die Pupillenreaktion ist in einigen Fällen träge, die Pupillenreflexe erscheinen meist
schon kurz nach der Geburt abgeschwächt.
Die Gefäße des Augenhintergrundes sind häufig eng und die Papille blass. Außerdem
treten Pseudopapillenödeme oder Papillenödeme zum Teil schon in den ersten
Lebensmonaten auf [9].

2
Schon bei der Geburt ist der Visus der Betroffenen meist stark eingeschränkt bis hin zu
einer vollständigen Erblindung. In einigen Fällen bleibt ein Visus von 0,05 bis zum
zehnten Lebensjahr erhalten.
Die Diagnostik erfolgt über ein Elektroretinogramm (ERG), bei dem über die von der
Netzhaut gebildeten elektrischen Potentiale die elektrischen Aktivitäten der Stäbchen,
Zapfen und der nachgeschalteten Bipolarzellen bestimmt werden. Bei LCA-Patienten
sind die Signale im ERG bereits in den ersten Lebensjahren meist nicht mehr
nachweisbar [35, 71].
Eine ortsauflösende Analyse der Stäbchen- und Zapfenfunktion gelingt mit der
Zweifarben-Schwellenperimetrie (2FSP). Mit Hilfe dieser psychophysikalischen
Methode kann die Funktion bzw. deren Einschränkung beider Systeme an einem
definierten Ort unterschieden werden [71].
Zur Untersuchung des Augenhintergrundes nutzt man die Autofluoreszenz des
retinalen Pigmentepithels (RPE, Fundus-Autofluoreszenz). Dabei werden
Ablagerungen von Lipofuszin im RPE zur Fluoreszenz angeregt. Bei der Untersuchung
kann sowohl eine Hyperfluoreszenz (bei Läsionen im RPE) als auch eine
Hypofluoreszenz (aufgrund von abgestorbenen Photorezeptoren) beobachtet werden
[71, 84]. Eine potentiell auftretende, pathologisch veränderte Schichtung der
Neuroretina wird mit Hilfe der optischen Kohärenztomographie (OCT), mit der die
Neuroretina, das RPE und die Aderhaut darstellt werden kann, diagnostiziert [71].
Die Skiaskopie dient der objektiven Refraktionsbestimmung. Bei dieser Methode dient
ein virtuelles Objekt als Lichtquelle, dessen Bild aus einer gleichmäßigen Beleuchtung
des Augenhintergrundes besteht. Betroffene zeigen meist eine Hyperopie > 3 dpt, nur
selten treten Myopien auf [9].
1.1.3 Vererbungsmuster
Das Vererbungsmuster der LCA ist in den meisten Fällen autosomal-rezessiv [73].
Patienten mit autosomal-dominanter Vererbung wurden im Zusammenhang mit
Mutationen im Cone rod homeobox-(CRX)-, Inosine-5'-monophosphate-
Dehydrogenase-1-(IMPDH1)- und Guanylate-cyclase-2D-(GUCY2D)-Gen beschrieben
[8, 32, 67].
Bei dem horizontalen Vererbungsmuster der autosomal-rezessiven Vererbung der LCA
sind häufig nur einzelne Personen in der Familie erkrankt. Sowohl Eltern als auch
Kinder der betroffenen Person sind in der Regel gesund. Haben Eltern bereits ein
erkranktes Kind, besteht für weitere Kinder unabhängig vom Geschlecht eine
Wahrscheinlichkeit von 25 % ebenfalls zu erkranken, da beide Elternteile heterozygote
Träger der Genmutation sind.

3
Bei einer Konsanguinität der Eltern erhöht sich das Erkrankungsrisiko für die direkten
Nachkommen, da die Wahrscheinlichkeit für das Zusammentreffen zweier genetisch
veränderter Gene deutlich erhöht ist. In Familien mit mehreren blutsverwandten Paaren
kommen verglichen mit der Normalbevölkerung in verschiedenen Generationen
gehäuft erkrankte Individuen vor [73].
1.1.4 Genmutationen und Untertypen der LCA
Ursächlich für die LCA sind Mutationen in unterschiedlichen Genen (siehe Tabelle 1).
Der Erbgang ist in den meisten Fällen autosomal-rezessiv.
Die am häufigsten auftretende Genmutation ist die Centrosomal protein 290 kDa-
(CEP290)-Mutation c.2991+1655A>G, die bei bis zu 30 % der LCA-Patienten
ursächlich für die Erkrankung ist [16].
Nicotinamide-nucleotide-adenylyltransferase-1-(NMNAT1)-Mutationen sind in 5 % und
Retinol-Dehydrogenase-12-(RDH12)-Mutationen bei < 4 % aller LCA-Fälle beschrieben
[25, 66].
Autosomal-rezessive Mutationen im GUCY2D-Gen führen zur LCA1 [67]. Heterozygote
Mutationen im CRX-Gen wurden als Ursache einer autosomal-dominanten LCA
beschrieben [26, 65, 67]. Die Mutation des Gens IMPDH1, die zur LCA11 führt, wird
ebenfalls autosomal-dominant vererbt [8].

4
Tabelle 1: Bekannte Gene mit ursächlichen Mutationen der LCA
Gen OMIM Form Funktion
GUCY2D Guanylate-cyclase-2D
600179 LCA1 Phototransduktion [67]
RPE65 Retinal pigment epithelium-
specific 65 kDa protein
180069 LCA2 Retinozyklus [59, 69]
SPATA7 Spermatogenesis associated 7
609868 LCA3 Zilienbildung/-funktion [68]
AIPL1 Aryl hydrocarbon receptor
interacting protein like 1
604392 LCA4 Proteinverteilung/-synthese
[78]
C6ORF152
Lebercilin
611408 LCA5 Zilienbildung/-funktion [20]
RPGRIP1 Retinitis pigmentosa GTPase
regulator interacting protein 1
605446 LCA6 Zilienbildung/-funktion [23,
31]
CRX Cone-rod homeobox
602225 LCA7 Retinale Entwicklung [27, 40,
81]
CRB1 Crumbs 1, cell polarity complex
component
604210 LCA8 Retinale Entwicklung [19]
NMNAT1 Nicotinamide nucleotide
adenylyltransferase 1
608700 LCA9 zellulärer Metabolismus,
beteiligt an der
Nikotinsäureamid-
Adenindinukleotid-(NAD)+-
Synthese [25]
CEP290 (=NPHP6)
Centrosomal protein 290
610142 LCA10 Zilienbildung/-funktion [21]
IMPDH1 Inosine monophosphate-
dehydrogenase 1
146690 LCA11 Zilienbildung/-funktion [8]
C1ORF36 (=RD3) Retinal degeneration 3
610612 LCA12 Transport der
Guanylatcyclase [4, 28]
RDH12 Retinol dehydrogenase 12
608830 LCA 13 Vitamin A Metabolismus [42]

5
LRAT Lecithin retinol acyltransferase
604863 LCA14 Vitamin A Metabolismus [82]
TULP1 Tubby like protein 1
602280 LCA15 Zilienbildung/-funktion [33]
KCNJ13 Potassium voltage-gated channel subfamily J member 13 channel, inwardly rectifying subfamily J, member 13
603208 LCA16 Reizleitung [76]
GDF6 Growth differentiation factor 6
601147 LCA17 Retinale Entwicklung [2]
PRPH2 Peripherin 2
179605 LCA18 Zilienbildung/-funktion [85]
IQCB1 (=NPHP5) IQ motif containing B1
609237 LCA Zilienbildung/-funktion
[12,86]
MERTK MER proto-oncogene, tyrosine kinase
604705 LCA Phagozytose [29]
ALMS1 Centrosome and basal body associated protein
606844 LCA Zilienbildung/-funktion [86]
CNGA3 Cyclic nucleotide gated channel alpha 3
600053 LCA Phototransduktion [86]
PDE6C Phosphodiesterase 6C
600827 LCA Phototransduktion [86]
ADAMTS18 ADAM metallopeptidase with thrombospondin type 1 motif 18
607512 LCA retinale Entwicklung [64]
MyoVIIa Myosin VIIA
600060 LCA Zilienbildung/-funktion [86]
1.1.5 Differentialdiagnosen
Die LCA hat eine Reihe von Differentialdiagnosen, weil retinale Dystrophien einen
ähnlichen Phänotyp aufweisen. Teilweise können Mutationen der beschriebenen Gene
auch zu anderen Erkrankungen (Zapfen-Stäbchen-Dystrophie, Stäbchen-Zapfen-
Dystrophie, Retinitis pigmentosa (RP) simplex, autosomal-rezessive RP) führen.
Anders als bei der autosomal-rezessiven Form der RP ist bei der RP simplex innerhalb
einer Familie häufig nur ein Individuum - meist aufgrund einer Neumutation - erkrankt
[71].
Die juvenile RP ähnelt der LCA in ihrem klinischen Bild, manifestiert sich allerdings erst
später. Auffällige Befunde, wie Hyperpigmentierungen und Knochenbälkchen, treten
häufig erst ab dem sechsten Lebensjahr auf. Die Sehschärfe liegt meist unter 0,2; die

6
Erkrankten sind leicht myop oder hyperop. Die Pupillenreaktion bei Patienten mit
juveniler RP ist im Vergleich zu der Mehrzahl der LCA-Patienten unauffällig [3].
Die Zapfen-Stäbchen-Dystrophie ist durch einen früheren Verlust der Zapfen- und
Stäbchenfunktion charakterisiert. Die Funktion der Stäbchen nimmt entweder
gleichzeitig oder zeitlich nach hinten versetzt mit der Funktion der Zapfen ab.
Betroffene zeigen einen stark eingeschränkten Visus und zentrale Gesichtsfeldausfälle
mit Nystagmus. In den ersten Lebensjahren imponiert eine hohe
Blendungsempfindlichkeit. Der Fundus ist zu Beginn der Erkrankung meist unauffällig
[53].
Die autosomal-dominante GUCY2D-Mutation folgt in ihrem klinischen Bild der Zapfen-
Stäbchen-Dystrophie [32].
Auch Mutationen des Gens AIPL1 wurden mit einer autosomal-dominanten Zapfen-
Stäbchen-Dystrophie in Verbindung gebracht. Diese Befunde ließen sich in folgenden
Untersuchungen allerdings nicht wiederholen [78].
Die Stäbchen-Zapfen-Dystrophie zeigt im Gegensatz zur Zapfen-Stäbchen-Dystrophie
zuerst eine Einschränkung der Stäbchenfunktion. Das Sehvermögen der Patienten ist
deutlich weniger eingeschränkt. Vor allem der Phänotyp der LCA2, die durch Retinal
pigment epithelium-specific 65 kDa protein-(RPE65)-Mutationen hervorgerufen wird,
folgt dem Verlauf einer Stäbchen-Zapfen-Dystrophie [53].
Darüber hinaus führen Mutationen der Gene LRAT und RPGRIP1 zu einem klinischen
Bild einer frühen Stäbchen-Zapfen-Dystrophie [23, 82].
Differentialdiagnostisch sind diese Dystrophien von systemischen oder metabolischen
Erkrankungen (z.B. Periarteriitis nodosa, Retinopathie bei Syphilis,
Rötelnembryopathie, Vitamin-A Mangel), die aufgrund von Pigmentretinopathien zu
ähnlichen klinischen Bildern führen, zu trennen. Darüber hinaus besteht auch die
Möglichkeit einer Pigmentretinopathie durch Medikamente (z.B. Resochin,
Phosphodiesterase-Hemmer, Tamoxifen) [9].
1.1.6 Genotyp Phänotyp Korrelation
Bei der durch Mutationen des GUCY2D-Gens hervorgerufenen LCA1 haben die
Betroffenen von Beginn an ein stark eingeschränktes Sehvermögen. Typisch ist die
Diskrepanz zwischen der Sehqualität und der Funduspathologie, die in den ersten
Jahren kaum Auffälligkeiten zeigt. Kennzeichnend für die Klinik ist eine von Beginn an
hohe Blendungsempfindlichkeit der Patienten. Im Verlauf erinnert die Erkrankung an
eine Zapfen-Stäbchen-Dystrophie [53].
Die pathologischen Veränderungen des Fundus bei der durch Mutationen des RPE65-
Gens hervorgerufenen LCA2 erinnern an eine RP. Allerdings sind die

7
Hyperpigmentierungen weniger stark ausgeprägt und die für eine RP typischen
Knochenbälkchen fehlen meist [54]. Analysen der Fundus-Autofluoreszenz sind nicht
durchführbar, weil im RPE kein Lipofuszin gebildet wird [55]. Die sich im Verlauf
entwickelnden Funduspathologien erinnern an eine Stäbchen-Zapfen-Dystrophie.
Klinisch weisen die Patienten eine deutlich geringere Seheinschränkung und
Blendungsempfindlichkeit auf als LCA1 Patienten [54, 72].
Im Gegensatz zu von anderen Genmutationen Betroffenen haben Erkrankte mit einer
RPE65-Mutation einen relativ guten Visus, der abhängig von der Beleuchtung bei ca.
0,3 liegt. Häufig tritt eine absolute Hemeralopie auf [54].
Die autosomal-dominanten Mutationen des CRX-Gens korrelieren mit einer Zapfen-
Stäbchen-Dystrophie. In der Funduskopie sind viele kleinere Läsionen, die
unregelmäßig über die gesamte Netzhaut verteilt sind, zu erkennen. In der
Fundusautofluoreszenz zeigt sich ein entsprechendes Muster mit reduzierten oder
erhöhten Signalen. Die Veränderungen erstrecken sich ausgehend von der Makula
über den gesamten Fundus [71].
Bei den autosomal-dominanten Mutationen des GUCY2D-Gens kommt es zu einer
starken Zapfenschädigung und Veränderungen der Makula [32].
Die autosomal-rezessiven Mutationen des GUCY2D-Gens führen im Gegensatz zur
autosomal-dominanten Mutation zu einer sehr frühen Form der Erkrankung. Der
Augenhintergrund zeigt über lange Zeit hinweg keine Veränderungen. Gleichzeitig
haben die Betroffenen einen Visus unter 0,1 und eine träge Pupillenreaktion.
Ursächlich für das klinische Bild der Erkrankung ist ein erniedrigter retinaler cyclischer-
Guanosinmonophosphat-(cGMP)-Spiegel, wodurch Lichtreize auf retinaler Ebene nicht
weitergeleitet werden und die Bildung des Rezeptorpotentials unterbleibt [22]. Bei den
Mutationen des GUCY2D-Gens handelt es sich entweder um Missense-Mutationen
oder Mutationen, die zu Translationsabbrüchen führen. Erkrankte mit Missense-
Mutationen, die sich auf die extrazelluläre Domäne des Proteins auswirken, zeigen
eine verzögerte Krankheitsprogression [62].
Mutationen des CRB1-Gens führen zu einer charakteristischen Verdickung der
Neuroretina, die mit Hilfe der optischen Kohärenztomographie (OCT) nachweisbar ist.
Bei anderen Mutationen (z.B. RPE65 oder GUCY2D) ist die Neuroretina meist
verdünnt [41].
Mutationen im Gen CEP290, welches für das Protein Nephrocystin 6 kodiert, gehören
zu den häufigsten LCA-assoziierten Mutationen. In einer Studie von den Hollander et
al. wurden bei 16 von 76 nicht konsanguinen Patienten Mutationen in diesem Gen
gefunden [21]. Coppieters et al. fanden in einer Studie mit 91 LCA-Patienten bei 30 %
der Erkrankten eine Beteiligung des CEP290-Gens [16].

8
Mutationen im CEP290- und IQCB1-Gen führen zur Ziliopathie der Photorezeptoren.
Auch die frühe und schnell fortschreitende Degeneration der Stäbchen ist
charakteristisch; dabei sind hauptsächlich die Außensegmente betroffen. In der Fovea
centralis bleiben die Zapfen meist noch eine Weile erhalten, verlieren aber – wie für die
LCA typisch – schnell ihre Funktion [49]. Klinisch erinnert das Bild daher an eine
Stäbchen-Zapfen-Dystrophie. Zilien kommen nicht nur in den Zellen des Auges,
sondern in fast allen Körperzellen des Menschen vor. In der Niere sind sie für die
Signalweiterleitung und den Flüssigkeitstransport von elementarer Bedeutung. Je nach
Ausprägung der Ziliopathie kann eine CEP290-Mutation neben den
ophthalmologischen Dysfunktionen auch respiratorische und nephrologische
Funktionsstörungen zur Folge haben [61].
1.1.7 Therapie
Die LCA ist eine genetische Erkrankung. Eine Therapie der Betroffenen gestaltet sich
daher als schwierig.
Im Fokus des ärztlichen Handelns steht die Beratung von Erkrankten und
Mutationsträgern im Sinne einer Primärprophylaxe. Dabei steht die Aufklärung über die
Wahrscheinlichkeit erkrankte Kinder zu bekommen im Mittelpunkt.
Häufig bleibt bei LCA-Patienten ein sehr geringes Sehvermögen für einige Jahre
bestehen. In diesen Fällen ist es wichtig eine Refraktionskorrektur durchzuführen, um
eine zusätzliche Sehminderung durch eine Deprivationsamblyopie zu verhindern [3].
Zudem sollte bei der Untersuchung von LCA-Patienten auf Begleiterkrankungen
geachtet werden, weil Netzhautdystrophien auch im Zusammenhang mit
verschiedenen Syndromen (z. B. Joubert-Syndrom, Senior-Loken-Syndrom) auftreten
können [74]. Bei der atypischen LCA sollte ebenso an ein cerebro-hepato-renales
Syndrom oder an eine juvenile Nephronophthise gedacht werden [3]. Im
Zusammenhang mit CEP290- oder IQCB1-Mutationen stehende Ziliopathien können
zu schwerwiegenden Nieren- und Lungenfunktionsstörungen mit Superinfektionen
führen [61].
Um weitere Folgeschäden in der Entwicklung zu minimieren, sollte eine allgemeine
pädiatrische und neuropädiatrische Diagnostik erfolgen. Durch dieses Vorgehen lassen
sich Nierenfehlbildungen, angeborene Epilepsien oder geistige Retardierungen
frühzeitig erkennen und therapieren [3].
In jüngster Zeit gewinnt die Gentherapie in der Medizin zunehmend an Bedeutung und
gibt von Mutationen Betroffenen neue Hoffnung. Die Entwicklung einer
Gensubstitutionstherapie bei retinalen Dystrophien infolge von RPE65-Mutationen hat
in den letzten Jahren große Fortschritte gemacht [58, 79]. Bei einer spezifischen

9
Gentherapie werden gesunde Allele der mutierten Gene mithilfe von rekombinanten
Adeno-assoziierten viralen-(AAV)-Vektoren transferiert [5]. Diese Vektoren werden in
der Gentherapie häufig genutzt, weil sie als nicht humanpathogen gelten [57]. Zudem
tritt nach einer Behandlung mit AAV-Vektoren nur eine geringe Immunreaktion auf [14].
Der limitierende Faktor der Gentherapie mit AAV-Vektoren ist die begrenzte Länge
(maximal 4,8 kb) des transferierbaren Gens. Eine Möglichkeit, diese Einschränkung zu
umgehen, ist die Verteilung längerer Sequenzen auf verschiedene AAV-Vektoren [6].
In klinischen Versuchen zur Gentherapie von LCA2-Patienten mit RPE65-Mutationen
konnte eine geringe Verbesserung der Netzhautfunktion nachgewiesen werden [58].
Die subretinale Applikation von rAAV2/2.RPE65 (AAV-Vektor mit RPE65-Gen) ergab
bei LCA2-Patienten eine signifikante Verbesserung der Sehfähigkeit bei Dämmerung.
Ein Fortschreiten der Netzhautdegeneration konnte mit der Therapie nicht aufgehalten
werden [11, 39].
In einem Tierversuch mit RPE65-/--Hunden konnte durch eine intravitreale Injektion von
9-cis-Retinol die Sehkraft bei den therapierten Hunden wiederhergestellt werden. Nach
der Injektion konnte im ERG eine höhere Aktivität der Stäbchen und Zapfen
nachgewiesen werden [30]. In einer Studie mit 14 LCA-Patienten mit LRAT- oder
RPE65-Mutationen wurde eine orale Therapie über sieben Tage mit zwei
verschiedenen Dosen eines synthetischen 9-cis-Retinols (QLT091001) durchgeführt.
Drei Patienten zeigten nach sieben Tagen eine klinisch relevante Verbesserung der
visuellen Funktionen (Gesichtsfeld, Sehschärfe und ERG), die bis zu vier Monate nach
Behandlungsende anhielt [47].
In einem weiteren Versuch wurden Stäbchen und Zapfen mithilfe von CRX-positiven
Spenderzellen in CRB1rd8/rd8- (Deletion einer Base mit nachfolgender Verschiebung des
Leserasters) - und GUCY2E-/--Mäusen transplantiert.
Dazu wurden CRX-exprimierende Zellen aus CRX-green-fluorescent-protein-(GFP)
transgenen Mäusen entnommen und in die Empfängerretina verpflanzt. Mit Hilfe des
GFP lassen sich die Transgen-tragenden Zellen lokalisieren. In dem Versuch
integrierten sich die embryonalen Spenderzellen in die äußere Körnerschicht der
Empfängerretinae. Obwohl die Spenderzellen hauptsächlich aus Zapfenvorläuferzellen
bestanden, entwickelten sie sich zu Zapfen und Stäbchen in einem ähnlichen
Verhältnis wie in der Empfängerretina (1 : 35) weiter. Offensichtlich beeinflusste das
Milieu der Empfängerretina die Differenzierung der embryonalen Vorläuferzellen.
Bei den GUCY2E-/--Mäusen konnte ein höherer Anteil an Zapfen nachgewiesen
werden als bei den CRB1rd8/rd8-Mäusen. Diese Befunde deuten darauf hin, dass ein
starker Verlust der Zapfen (z. B. durch eine GUCY2E-Mutation) bessere

10
Voraussetzungen für eine Zapfentransplantation bietet als eine weniger stark
ausgeprägte Zapfendegeneration (z. B. durch eine CRB1-Mutation).
Die Daten belegen weiter, dass embryonale CRX-positive Zellen das Potential haben,
verlorengegangene Photorezeptoren zu ersetzen. Diese Befunde stellen damit einen
wichtigen Ansatzpunkt für weitere Studien zur Therapie von erblich bedingten, retinalen
Degenerationen dar [49].
2011 wurde ein Mausmodell entwickelt, das das LCA-Krankheitsbild bei CEP290- bzw.
IQCB1-Mutationen imitiert. Wie auch beim Menschen zeigte sich bei den Mäusen eine
frühe Degeneration der Stäbchen. Meist deutete nur noch der Nachweis von Lipofuszin
im RPE auf die Existenz dieser Zellen hin [11]. Dieses Mausmodell eignet sich daher
für präklinische Studien für Gentherapien der LCA auf der Basis von CEP290- bzw.
IQCB1-Mutationen.
Aktuell werden im Tiermodell neue Gentransfermöglichkeiten erforscht. Im Fokus
dieser Forschung steht der Transfer von Genen in Nanopartikeln mithilfe der
Elektroporation [15, 44]. Dazu werden mit plasmid-desoxyribonucleic-acid-(DNA)
beladene Nanopartikel subretinal in Mausaugen injiziert. Durch die Elektroporation
werden die Zellmembranen vorübergehend permeabilisiert, so dass die Nanopartikel in
die Zielzellen gelangen können [44].
Die Vorteile der Nanopartikel sind die geringe Immunreaktion, der einfache Transfer
durch die Zellmembran und die kostensparende Herstellung. Mithilfe von
Modifikationen der Nanopartikel ist es möglich, den Transport so zu steuern, dass die
DNA-Fragmente nur in gewünschten Zielzellen freigesetzt werden. Damit werden
mögliche systemische Effekte beim Übertreten der Blut-Retina-Schranke vermieden
[43].
1.2 Zielsetzung
Ziel dieser Arbeit war die Identifikation der ursächlichen Mutation(en) eines Gens in
einer Familie mit einem LCA-Patienten.
Mithilfe von Kopplungsanalysen über Single Nucleotide Polymorphism-(SNP)-Marker
konnten im Vorfeld drei chromosomale Kandidatenregionen eingegrenzt werden. Über
Mikrosatellitenmarker sollten diese drei Regionen auf eine einzelne Kandidatenregion
mit einem Minimum an Kandidatengenen reduziert werden. Über eine Analyse der
Funktion und der Lokalisation der Gene sollte eine Auswahl von Kandidatengenen
getroffen werden. Das ursächliche Gen sollte dann über eine direkte Sequenzierung
identifiziert werden.

11
2 Material und Methoden
2.1 Patient und Familie
Die in der vorliegenden Arbeit untersuchten DNA-Proben stammten von einer
konsanguinen Familie türkischer Herkunft. Für die Analyse lagen Blutproben von
beiden Elternteilen und deren fünf Kindern, von denen der jüngste Sohn an LCA
erkrankt war, vor (Abbildung 1).
Der Patient wurde im Kinderzentrum in Bielefeld, einem akademischen
Lehrkrankenhaus der Universitätsklinik in Münster, untersucht und diagnostiziert.
Die Familie wurde über die geplanten genetischen Untersuchungen aufgeklärt und das
Einverständnis wurde eingeholt. Die Studie wurde von der Ethikkommission der
Universität Regensburg (Antrag 00/106) und der Ethikkommission der Universität
Gießen (Antrag 149/07) befürwortet.
Abbildung 1: Stammbaum der Familie 967
12
2.2 Molekulargenetische Methoden
2.2.1 Geräte
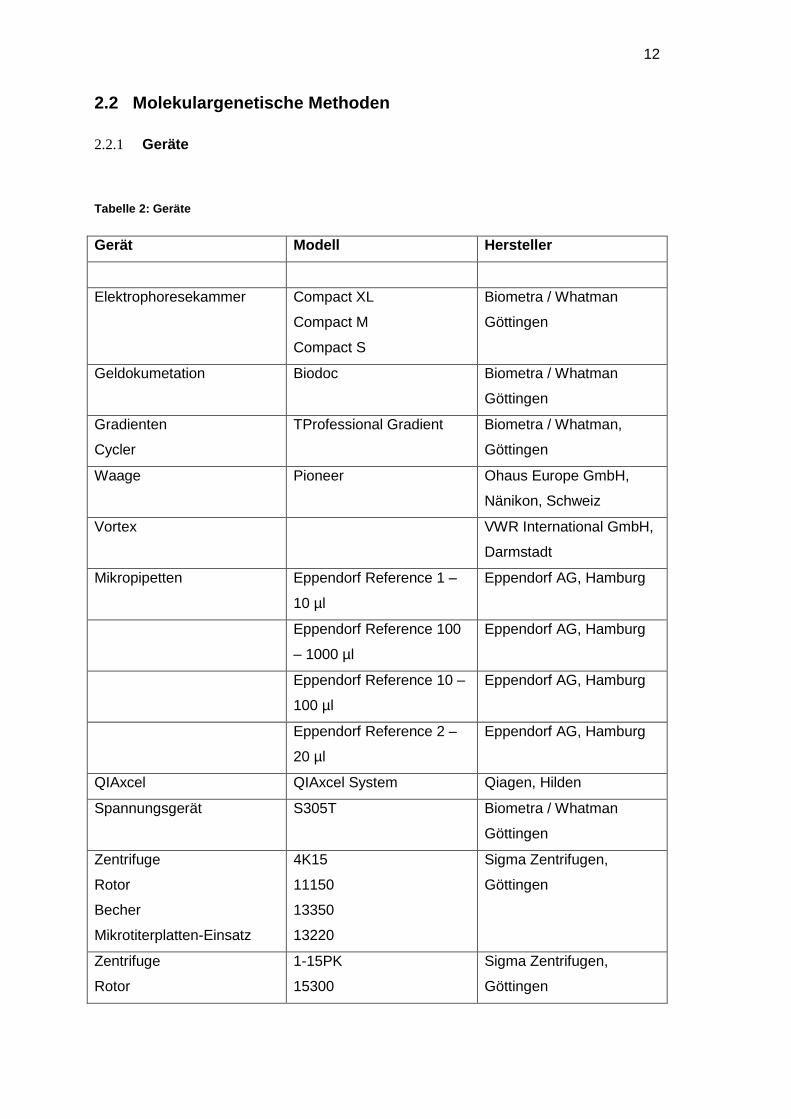
Tabelle 2: Geräte
Gerät Modell Hersteller
Elektrophoresekammer Compact XL
Compact M
Compact S
Biometra / Whatman
Göttingen
Geldokumetation Biodoc Biometra / Whatman
Göttingen
Gradienten
Cycler
TProfessional Gradient Biometra / Whatman,
Göttingen
Waage Pioneer Ohaus Europe GmbH,
Nänikon, Schweiz
Vortex VWR International GmbH,
Darmstadt
Mikropipetten Eppendorf Reference 1 –
10 µl
Eppendorf AG, Hamburg
Eppendorf Reference 100
– 1000 µl
Eppendorf AG, Hamburg
Eppendorf Reference 10 –
100 µl
Eppendorf AG, Hamburg
Eppendorf Reference 2 –
20 µl
Eppendorf AG, Hamburg
QIAxcel QIAxcel System Qiagen, Hilden
Spannungsgerät S305T Biometra / Whatman
Göttingen
Zentrifuge
Rotor
Becher
Mikrotiterplatten-Einsatz
4K15
11150
13350
13220
Sigma Zentrifugen,
Göttingen
Zentrifuge
Rotor
1-15PK
15300
Sigma Zentrifugen,
Göttingen

13
2.2.2 Reagenzien, Enzyme, Chemikalien, Puffer
Tabelle 3: Chemikalien
Reagenz Beschreibung Hersteller
Agarose, Seakem LE C12H18O9 VWR International GmbH,
Darmstadt
Borsäure H3BO3 Merck, Darmstadt
Bromphenolblau Indikator C19H10Br4O5S Merck, Darmstadt
Dimethylsulfoxid (DMSO) C2H6OS Merck, Darmstadt
Ethidiumbromid C21H20BrN3 Sigma-Aldrich
Ethanol (EtOH) C2H6OH Carl Roth GmbH & Co KG,
Karlsruhe
Magnesiumchlorid MgCl2 Merck, Darmstadt
Trishydroxy-methyl-aminoethan (TRIS)
C4H11NO3 Merck, Darmstadt
Mineral Oil QIAGEN, Hilden
QX DNA Alignment Marker 15–500 bp
QIAGEN, Hilden
Tabelle 4: Molekularbiologische Reagenzien
Chemikalien Bestellnummer Hersteller
Desoxyribonukleosid-Triphosphat-(dNTP)-Set (jeweils 100 mM)
R0186 Fermentas, St. Leon-Roth
DNA-Ladder 100 bp (0,5 µg/µl) SM321 Fermentas, St. Leon-Roth
Go Taq DNA-Polymerase (5 U/µl)
M3005 Promega, Mannheim

14
2 x Stammlösungen:
10 × TBE-Puffer
2 mM EDTA-2 Na 0,7 g
89 mM TRIS 10,8 g
89 mM Borsäure 5,5 g
ad 1 l Aqua Dest
Gelladepuffer:
20 % w/v Sucrose 10 g
0,005 % w/v Bromphenolblau 0,025 g
0,005 % w/v Xylene Cyanol 0,025 g
ad 50 ml 1x TBE
2.2.3 Phenolextraktion von DNA
Für die Phenolextraktion wurden für jede DNA-Probe vier Eppendorfgefäße mit der
Probennummer beschriftet. Anschließend wurde die DNA-Lösung in ein
Eppendorfgefäß pipettiert und dabei das Volumen der DNA-Probe bestimmt. Danach
wurde die gleiche Menge TE-Puffer gesättigtes Phenol hinzugefügt. Durch Schütteln
des Eppendorfgefäßes entstand eine homogene, trübe Flüssigkeit, die anschließend
10 min bei 17968 g zentrifugiert wurde.
Nach der Zentrifugation wurde die obere wässrige Phase sorgfältig abpipettiert und in
ein weiteres Eppendorfgefäß überführt. Die untere Phase (Phenol) wurde verworfen.
Zu der oberen Phase wurde das gleiche Volumen an Chloroform hinzugefügt. Nach
dem Schütteln erfolgte eine weitere Zentrifugation (10 min bei 17968 g).
Danach wurde die obere Phase in das dritte Eppendorfgefäß pipettiert. Erneut wurde
das gleiche Volumen Chloroform zugegeben. Das Eppendorfgefäß wurde wiederum
nach dem Schütteln 10 min in zentrifugiert. Danach wurde die obere Phase in das
vierte Eppendorfgefäß pipettiert. Die untere Phase wurde aufgehoben, da aufgrund der
hohen Salzkonzentration eine Phasenumkehr möglich war.
Zur Fällung der DNA wurden ein Zehntel des Volumens 3 M Natriumacetatlösung und
zwei Volumenanteile eiskaltes EtOH (absolut) hinzugefügt. Das Eppendorfgefäß wurde
für eine halbe Stunde bei -20 °C aufbewahrt.
Anschließend erfolgte eine Zentrifugation (20 min, 4 °C, 17968 g). Nach dem Abgießen
des EtOH wurde das Pellet anschließend mit 1 ml 70 % EtOH gewaschen. Nach einer
weiteren Zentrifugation (5 min) wurde das EtOH wieder abgegossen. Anschließend

15
wurde das Eppendorfgefäß mit dem Pelltet für 15 min bei 37 °C im Trockenschrank
gelagert. Danach wurde die gewaschene DNA in der gleichen Menge sterilem 1x TE-
Puffer für mehrere Minuten gelöst.
Zur Reinheitsprüfung wurde die OD230, OD260 und OD280 bestimmt (siehe Kapitel 2.3).
2.3 DNA-Konzentrationsbestimmung und Ansetzen der
Gebrauchslösungen aus den DNA-Stammlösungen
Die Konzentration einer DNA-haltigen Lösung lässt sich photometrisch bei einer
Wellenlänge von 260 nm bestimmen. Eine Optische Dichte (OD)260 = 1 entspricht bei
einem pH von 7,0 einer Konzentration von 50 µg/ml doppelsträngige DNA. Die Proben
wurden mit Wasser verdünnt.
Durch die Isolationsmethode kann die DNA-Lösung mit Proteinen, ribonucleic-acid
(RNA) oder Phenolresten kontaminiert werden. Eine Verunreinigung mit Proteinen
kann über eine photometrische Messung bei 280 nm bestimmt werden. Bei dieser
Wellenlänge liegt das Absorptionsmaximum der aromatischen Aminosäurereste.
Die Reinheit einer DNA-Lösung kann durch das Verhältnis der OD260 und OD280
bestimmt werden. Ein OD260/OD280-Quotient = 1,8 spricht für eine reine DNA-Lösung.
Bei einer Kontamination mit Proteinen ist dieser Wert signifikant kleiner.
Um eine Verunreinigung mit Polysacchariden nachzuweisen, wird zusätzlich der OD230-
Wert bestimmt. Der OD230/OD260-Quotient sollte im Idealfall bei 0,45 liegen [89].
Aus der DNA-Stammlösung wurden Gebrauchslösungen mit einer Konzentration von
20 ng/µl mit einem Volumen von 500 µl angesetzt. Dazu musste zunächst das
einzusetzende Volumen der Stammlösung berechnet werden:
Endkonzentration x Endvolumen = einzusetzende Volumen der
Stammlösung Konzentration der Stammlösung
Das berechnete Volumen der einzusetzenden Stammlösung wurde mit doppelt
destilliertem Wasser (ddH2O) auf das Endvolumen eingestellt.
2.4 Amplifikation definierter Sequenzen durch die
Polymerasekettenreaktion (PCR)
Die Standardansatzgröße für die PCR-Ansätze betrug 10 µl. Tabelle 5 zeigt die
genaue Zusammensetzung für einen PCR-Ansatz.

16
Tabelle 5: Zusammensetzung des PCR-Standardansatzes
Reagenz Menge (µl)
ddH2O 2,4
Puffer (5x) 2
dNTP (1,25 mM) 1,6
MgCl2 ( 25 mM) 1,2
Primer forward (10 mM) 0,4
Primer reverse (10 mM) 0,4
Polymerase (5 U/µl) 0,08
DNA (20 ng/µl) 2
Gesamt 10
Um die optimalen PCR-Bedingungen für jeden Marker zu finden, wurde jeweils ein
Mastermix (62,5 µl, Tabelle 6) hergestellt, der in sechs Ansätze mit 10 µl zur Testung
unterschiedlicher Temperaturen aufgeteilt wurde.
Tabelle 6: Zusammensetzung eines PCR-Mastermix mit einer Ansatzgröße von 62,5 µl
Mastermix: Einheit: µl
ddH2O 14,75
Puffer (5x) 12,5
dNTP (1,25 mM) 10
MgCl2 ( 25 mM) 7,5
Primer forward (10 mM) 2,5
Primer reverse (10 mM) 2,5
Polymerase GoTaq (5 U/µl) 0,25
DNA (20 ng/µl) 12,5
Gesamt 62,5
Für die Amplifikation der Marker wurde zunächst die optimale PCR-
Annealingtemperatur für jeden Marker mit einer Kontroll-DNA-Probe ermittelt. Mit dem
PCR-Gradienten-Cycler wurden PCRs mit zwölf verschiedenen Annealingtemperaturen

17
zwischen 53 und 65 C durchgeführt. Da die Temperaturdifferenzen maximal 1,6 °C
betrugen, war es ausreichend, jeden Marker nur bei jeder zweiten Temperaturstufe
(entweder Temperaturstufe 1, 3, 5, 7, 9 und 11 oder Temperaturstufe 2, 4, 6, 8, 10 und
12, Tabelle 7) zu testen, weil kleine Temperaturunterschiede keine Auswirkung auf die
Amplifikationseffizienz hatten.
Tabelle 7: Temperaturstufen der Gradienten PCR
Well 1 2 3 4 5 6 7 8 9 10 11 12
T [°C] 53 53,3 54,2 55,4 56,8 58,2 59,8 61,2 62,6 63,8 64,7 65
In jedem PCR-Durchlauf wurden 16 verschiedene Marker bei jeweils sechs
unterschiedlichen Annealingtemperaturen amplifiziert. Die PCR wurde mit dem in
Tabelle 8 beschrieben Zyklus durchgeführt.
Tabelle 8: Parameter der Gradienten PCR
Hot Start: 94 °C 5 min
Annealing: 53 - 66 °C 30 - 60 s
Extension: 72 °C 30 s
Denaturierung: 94 °C 30 s
Annealing: 53 - 65 °C 30 - 60 s
Extension: 72 °C 5 min
Konnten im ersten Durchlauf keine zufriedenstellenden Amplifikationsergebnisse erzielt
werden, wurde die PCR für diese DNA-Proben mit einer Zykluszeit von 60 Sekunden je
Schritt wiederholt. Wenn dadurch keine Verbesserung der Amplifikation erzielt werden
konnte, wurde die MgCl2-Konzentration im Ansatz verändert.
Nach der Identifikation der optimalen PCR-Bedingungen wurde jeder Marker unter den
ermittelten Parametern mit einem 10 µl Standardansatz getestet.
2.5 Gelelektrophorese
Die Gelelektrophorese dient zur Auftrennung verschieden großer DNA-Fragmente. Das
Trägergel besteht aus Agarose, TBE-Puffer und Ethidiumbromid. Die Größe der Poren
ist abhängig vom Vernetzungsgrad des Agarosegels und damit von der
Agarosekonzentration. Die Proben werden in den Geltaschen, die sich an der einen

18
Seite befinden, aufgetragen. Nach Anlegen einer elektrischen Spannung wandern die
DNA-Fragmente aufgrund ihrer negativ geladenen Phosphatgruppen von der Kathode
zur Anode. Die Wanderungsgeschwindigkeit eines DNA-Fragments ist umgekehrt
proportional zum dekadischen Logarithmus seines Molekulargewichts, das heißt,
kleinere Fragmente bewegen sich schneller als größere Fragmente [88].
Zur Längenbestimmung der DNA-Fragmente wurde ein DNA-Größenstandard mit
Fragmenten bekannter Größe parallel zu den zu analysierenden DNA-Fragmenten in
der Gelelektrophorese aufgetrennt [52].
2.5.1 Herstellung des Agarosegels
Da die erwartete Länge der PCR-Produkte zwischen 62 und 282 Basenpaaren (bp)
lag, wurde für die Gelelektrophorese ein zweiprozentiges Gel (5 g Agarose in 250 ml 1
x TBE) verwendet, das für die Auftrennung kleiner Fragmente (0,2 – 1 kb) geeignet ist.
Der Puffer mit dem Agarosepulver wurde bis zur vollständigen Lösung der Agarose
aufgekocht.
Anschließend wurde der verdunstete Teil des Puffers durch Zugabe von
Ionenaustauscher ausgeglichen.
Nach dem Abkühlen auf ca. 42 °C wurden 20 µl 1 % Ethidiumbromidlösung
hinzugegeben und das Gel gegossen.
Nach dem Festwerden des Gels wurde es in die Elektrophoresekammer gelegt und mit
1 x TBE-Puffer vollständig bedeckt. Dann wurden die Kämme für die Geltaschen
entfernt.
Nach Verfestigung des Gels wurde dieses in die Elektrophoresekammer gelegt und mit
1 x TBE-Puffer vollständig bedeckt. Anschließend wurden die Kämme entfernt. Die
Slots mussten dabei mit der Öffnung nach oben an der Kathodenseite der
Elektrophorese liegen, damit die DNA Fragmente in Richtung der positiv geladenen
Anode wanderten.
2.5.2 Auftrennung der PCR-Produkte
Von jedem PCR-Produkt wurden 5 µl in eine Geltasche pipettiert. In eine randständige
Geltasche wurden 7 µl des DNA-Größenstandards pipettiert. Nach einer Stunde bei
100 Volt waren die DNA-Fragmente soweit gewandert, dass das Gel unter
ultraviolettem Licht ausgewertet werden konnte.
Das im Agarosegel enthaltene Ethidiumbromid hatte sich zwischen die Doppelstränge
der DNA–Fragmente eingelagert und fluoreszierte dadurch. Anhand der bekannten
Fragmentgrößen des DNA-Größenstandards konnte man über die Position der Banden
die Größen der PCR-Produkte abschätzen.

19
2.6 QIAxcel
Das QIAxcel-System von Qiagen ermöglicht die parallele Auftrennung von zwölf PCR-
Produkten in matrixgefüllten Kapillaren. Die Vorteile dieser Methode gegenüber der
Gelelektrophorese sind ein geringerer Zeitaufwand, die Durchführbarkeit mit geringerer
Probenmenge und eine exaktere Bestimmung der PCR-Fragmentgrößen. Bei DNA-
Fragmenten mit weniger als 500 bp ist eine Auflösung von 2–3 bp möglich. Die
Datenerfassung und -analyse erfolgte standardisiert mit einer geräteeigenen
Auswertesoftware. Die Daten wurden graphisch als Elektropherogramme dargestellt.
Anhand der Peaks konnten die Fragmentgrößen und Mengen der PCR-Produkte
ermittelt werden [91]. Ein Alignment-Marker mit Fragmenten zwischen 15 und 500 bp
diente als Referenzgröße zur Berechnung der PCR-Produktlänge. Die PCR-
Bedingungen wurden soweit optimiert, dass mindestens eine und maximal zwei
Banden pro Marker vorlagen.
2.7 Gensequenzierung
Bei der Sequenzierung nach Sanger (Kettenabbruch-Methode) bewirkt der Einbau von
Didesoxynukleotiden (ddNTP) den Abbruch des synthetisierten DNA-Stranges. Die
Methode wird für jedes zu sequenzierende DNA-Fragment in vier parallelen Ansätzen
mit Zusatz jeweils eines anderen ddNTPs durchgeführt. Dadurch liegen am Ende der
entstehenden DNA-Fragmente definierte Nukleotide. Durch die Auftrennung der
Fragmente nach ihrer Größe kann die Abfolge der Basensequenz ermittelt werden [46].
Die Cycle-Sequenzierung ist eine Weiterentwicklung der Sanger-Methode. Dazu führt
man eine PCR, in der neben den dNTPs die vier verschiedenen, mit unterschiedlichen
Farbstoffen markierten ddNTPs enthalten sind, durch. Die Basensequenz kann in einer
nachfolgenden Kapillarelektrophorese mithilfe eines Fluoreszenz-Detektors direkt
bestimmt werden.
Die Sequenzierungen der Kandidatengene wurden bei Seqlab in Göttingen
durchgeführt.
2.8 Kopplungsanalyse
2.8.1 Definition
Bei der Kopplungsanalyse wird untersucht, ob ein potentiell krankheitsverursachendes
Gen mit einer bestimmten chromosomalen Region bzw. einem Marker gekoppelt
vererbt wird. Ziel der Kopplungsanalyse ist es, genetische Loci zu ermitteln, die

20
statistisch gesehen häufiger miteinander vererbt werden, als es aufgrund der
meiotischen Teilung zu erwarten ist [71].
2.8.2 Das menschliche Genom
Das menschliche Genom besteht aus 46 Chromosomen, die sich in 44 Autosomen und
zwei Gonosomen unterteilen. Das gesamte Genom hat eine Länge von etwa 3
Milliarden bp. Bei der Transkription werden die kodierenden Regionen der Gene in
ribonucleic-acid (RNA) umgeschrieben. Aus diesem primären Transkript werden DNA-
Abschnitte (Introns) ausgeschnitten. Die verbleibenden Exons (durchschnittliche Länge
122 bp), die nur etwa 2 % des menschlichen Genoms ausmachen, bilden die
messenger RNA (mRNA). Die Länge der Introns übertrifft die der Exons meist um ein
Vielfaches [10].
2.8.3 Identifizierung von pathologischen Genveränderungen
Bei der Identifikation von Genen, deren Veränderungen zu erblichen Krankheiten
führen, werden vier Strategien unterschieden: die funktionsspezifische Klonierung, die
positionelle Klonierung, sowie positionsunabhängige und positionsabhängige
Kandidatengenverfahren [10].
Bei der funktionsspezifischen Klonierung (engl. functional cloning) ist zwar die
Genfunktion, nicht aber der Gen-Lokus bzw. die DNA-Sequenz bekannt. Die
Identifizierung des Gens erfolgt über die Genfunktion mithilfe spezieller
Proteinantikörper oder bereits bekannter Proteinsequenzen [56].
Wenn die chromosomale Region eines Gens bekannt ist, kann dessen genaue Position
mithilfe einer positionellen Klonierung ermittelt werden. Die chromosomale Region wird
über eine Kopplungsanalyse bestimmt. Dieses zeitaufwändige Verfahren verliert seit
dem Humangenomprojekt und mit der Weiterentwicklung von Sequenziertechniken
(Next Generation Sequencing, NGS) zunehmend an Bedeutung.
Mit dem positionsunabhängigen Kandidatengenverfahren werden potentielle
krankheitsauslösende Gene ohne eine vorgeschaltete Kopplungsanalyse bestimmt.
Das Verfahren basiert auf einem Vergleich der zu untersuchenden Erkrankung mit
Phänotypen von Menschen oder Tieren mit bekannten Mutationen. Eine weitere
Möglichkeit ist die Zuordnung des unbekannten Gens zu einer bereits bekannten
Genfamilie aufgrund der Krankheitsbefunde.
Die zurzeit vielversprechendste Methode zur Identifizierung krankheitsauslösender
Gene ist das positionelle Kandidatengenverfahren. Wie bei der positionellen Klonierung
muss für dieses Verfahren eine chromosomale Region, in der das
krankheitsverursachende Gen liegt, bekannt sein. Diese Region wird im Vorfeld durch
eine Kopplungsanalyse ermittelt. Über Datenbanken des Humangenomprojekts

21
(http://www.ebi.ac.uk/embl/ oder http://www.ncbi.nlm.nih.gov), die eine Vielzahl von
Informationen über verschiedene Gene enthalten, lassen sich die Gene bestimmen, die
in der identifizierten Region lokalisiert sind [10].
Nach Ermittlung der Kandidatengene werden diese sequenziert und potentielle
Genmutationen identifiziert [56].
2.8.4 Single Nucleotide Polymorphism (SNP)
SNPs werden auch als „erfolgreiche Punktmutationen“ bezeichnet, weil sie bei
mindestens 1 % der Population vorkommen und sich daher im Genpool durchgesetzt
haben. Unter einer Punktmutation versteht man den Austausch einer Base der DNA
gegen eine andere. Die Folge einer Punktmutation kann ein verändertes Genprodukt
sein, wenn die betroffene Position innerhalb von kodierenden Sequenzen liegt.
Da sich rein statistisch gesehen menschliche Genome an jeder tausendsten Stelle
voneinander unterscheiden, enthält ein individuelles Genom rein rechnerisch etwa drei
Millionen SNPs [46].
SNPs führen nur selten zu einer Erkrankung, da sie auch außerhalb von Genen
vorkommen. Im Vergleich zu Mikrosatellitenmarkern ist mithilfe von SNPs aufgrund
ihrer Häufigkeit eine höhere Feinauflösung genetischer Karten möglich.
SNPs bestehen aus maximal zwei Allelen, sind wenig polymorph und eignen sich
daher gut zur automatisierten Typisierung. Diese Eigenschaften prädestinieren SNPs
als Marker zur Lokalisation krankheitsverursachender Gene in definierten
chromosomalen Regionen [70].
In der Genomanalyse werden aktuell Oligonukleotid-Arrays angewandt, bei denen
SNPs durch die Hybridisierung mit genomischer DNA charakterisiert werden [56].
2.8.5 Mikrosatellitenmarker
Auf den Chromosomen befinden sich durchschnittlich alle 30 kb sogenannte repetitive
DNA-Sequenzen. Dabei handelt es sich um kurze Sequenzen, die in
Tandemwiederholungen aufeinanderfolgen [46]. Nach Größe der Wiederholungseinheit
und Anzahl der Wiederholungen unterscheidet man Satelliten-DNA, Minisatelliten-DNA
und Mikrosatelliten-DNA. Mikrosatelliten machen ca. 2 % des Genoms aus. Am
häufigsten handelt es sich bei den wiederholten Einheiten um Dinukleotide, meist
CA/TG- oder AC/GT-Repeats. Tri- und Tetranukleotide sind wesentlich seltener. Da
Mikrosatelliten polymorph sind, werden sie gerne als genetische Marker in
Kopplungsanalysen eingesetzt. Eine noch höhere Feinauflösung genetischer Karten
erreicht man mit Einzelnukleotid-Polymorphismen, die sogenannten SNPs [10].
Mit SNPs kann man zwar eine höhere Feinauflösung genetischer Karten erzielen, sie
haben aber den Nachteil, dass sie nur als zwei Allele vorkommen [10]. Mikrosatelliten

22
liefern aufgrund ihrer Variabilität einen höheren Informationsgehalt, weil die Genome
beider Elternteile bis zu vier verschiedene Markerallele (zwei je Elternteil) aufweisen.
DNA-Sequenzen, die in einer SNP-Analyse homozygot sind, können deshalb durch
Mikrosatellitenmarker weiter differenziert werden. Aus diesem Grund werden nach der
Kopplungsanalyse über SNPs Mikrosatellitenmarker bestimmt, die auf den durch die
SNP-Analyse bestimmten Chromosomenbereichen liegen.
2.8.6 Logarithmic Odds Ratio (LOD)-Score
Beim Erstellen der genetischen Karten untersucht man mit Hilfe der Kopplungsanalyse,
mit welcher Wahrscheinlichkeit zwei Merkmale auf einem Chromosom voneinander
getrennt, bzw. gekoppelt vererbt werden [10]. Für eine Familienuntersuchung muss ein
Elternteil sowohl für die Genmutation als auch für den Marker heterozygot sein [73].
Über die Rate meiotischer Rekombination zwischen zwei Loci wird die Position eines
Quantitative Trait Locus (QTL = Phänotyp) relativ zu einem definierten Locus bestimmt.
Dabei ist ein Locus durch eine Sequenzposition im Genom definiert, die in
verschiedenen Allelen vorkommt. Liegen zwei Loci auf demselben Chromosom, ist
eine meiotische Rekombination nur bei einem Crossing Over zwischen den homologen
Chromosomen des diploiden Chromosomensatzes im Rahmen der Keimzellbildung
möglich [105]. Je größer der Abstand zweier Loci auf einem Chromosom ist, desto
häufiger werden diese getrennt vererbt, weil die Wahrscheinlichkeit für ein Crossing-
Over über die Distanz zunimmt. Dicht beieinander liegende Loci werden dagegen
häufiger gekoppelt vererbt.
Der Abstand zwischen zwei Genen wird in Centi-Morgan (cM) angegeben. Ein Centi-
Morgan entspricht einer Rekombinationshäufigkeit von 1 % und umfasst eine Sequenz
von 1 Mbp.
Die Rekombinationswahrscheinlichkeit beträgt Null, wenn eine vollständige Kopplung
vorliegt. Bei freier Rekombination beträgt sie 0,5. In diesem Fall liegen die beiden Loci
auf verschiedenen Chromosomen oder sehr weit voneinander entfernt auf demselben
Chromosom.
Die statistische Bewertung von Kopplungsanalysen erfolgt über den LOD-Score [10].
Dieser ist definiert als der Logarithmus zur Basis 10 des Quotienten aus der
Wahrscheinlichkeit für eine Kopplung durch die Wahrscheinlichkeit für eine
Nichtkopplung. Eine signifikante Kopplung liegt vor, wenn der LOD-Score über 3 liegt
(d.h. der Quotient beträgt 1000 : 1). In diesem Fall ist die Wahrscheinlichkeit einer
Kopplung beider Loci um einen Faktor 1000 höher als die Wahrscheinlichkeit einer
Nichtkopplung. Die Wahrscheinlichkeit für ein falsch positives Ergebnis liegt somit bei
0,001. Ein Wert unter -2 steht für eine Nichtkopplung. Die Wahrscheinlichkeit für eine

23
Nichtkopplung ist dann mindestens 100 mal höher als die Wahrscheinlichkeit einer
Kopplung [10, 56].
Mit einer genomweiten Kopplungsanalyse lassen sich chromosomale Regionen
ausfindig machen, auf denen potentielle Kandidatengene liegen.
2.8.7 SNP-Genotyping Microarrays
Die DNA-Proben der sieben Familienmitglieder wurden im Gene Mapping Center des
Max Delbrück Center for Molecular Medicine (MDC), Berlin-Buch, mit dem Human
Mapping 50K Array Xba 240 (Version 01-2007) von Affymetrix hybridisiert und
detektiert. Danach wurden die Daten von Dr. Franz Rüschendorf mit Hilfe der Software
ALOHOMORA (V0.29) für die weitere Analyse in MERLIN ausgewertet (Annotation
File: NA21, Xba240 map file vom 02.01.2007, Reference NCBI 36.3, Built HG18 von
03/2006 für dbSNP vom 23.10.2007) und zur Verfügung gestellt. ALOHOMORA ist
eine Software, die die Kopplungsanalyse mit dem Mapping-Array kombiniert, indem sie
die SNP-Daten aufbereitet und Eingabedateien für verschiedene Kopplungsanalyse-
und Auswerteprogramme erstellt.
Der bei der Untersuchung verwendete Human Mapping 50K Array Xba 240 von
Affymetrix ermöglicht die Genotypisierung von 50.000 SNPs.
Voraussetzung für die Durchführung der Analysen war eine DNA-Ausgangsmenge von
250 ng pro Array und eine Mindestkonzentration von 50 ng/µl. Weiter musste die DNA
doppelsträngig und frei von PCR-Inhibitoren vorliegen.
2.8.8 Parametrische Analyse
Die statistische Einschätzung für die Wahrscheinlichkeit einer Genkopplung gelingt mit
einer parametrischen oder nicht-parametrischen Analyse. Anders als bei der nicht-
parametrischen Analyse ist bei der parametrischen Analyse ein Krankheitsmodell
vorgegeben, das bestimmte Komponenten wie ein Vererbungsmuster, die
Genfrequenz, die Häufigkeit und die Penetranz des Genotyps enthält [1]. Die Struktur
der in der vorliegenden Analyse untersuchten Familie erlaubte die Anwendung eines
parametrischen Verfahrens.
Bei Vererbungsmustern unterscheidet man zwischen autosomal-dominant, autosomal-
rezessiv, X-chromosomal dominant, X-chromosomal rezessiv, Y-chromosomal und
mitochondrial [73].
Die dritte Mendelsche Regel (Unabhängigkeitsregel) beschreibt die Vererbung zweier
verschiedener Merkmale mit je zwei Allelen, die auf unterschiedlichen Chromosomen
lokalisiert sind. In diesem Fall werden beide Merkmale unabhängig voneinander
vererbt und jedes Merkmal wird für sich nach der ersten und zweiten Mendelsche
Regel vererbt [70]. Die Wahrscheinlichkeit, dass zwei bestimmte Allele zweier

24
verschiedener Marker auf zwei unterschiedliche Tochterzellen verteilt werden, beträgt
50 %.
Liegen zwei Loci hingegen auf einem Chromosom, beträgt die Wahrscheinlichkeit einer
Trennung bei der Meiose durch Rekombination unter 50 %, d.h. < 0,5. Je näher die
beiden Loci auf einem Chromosom aneinander liegen, desto unwahrscheinlicher ist
eine Rekombination durch Crossing Over. Bei einer chromosomalen Kopplung kann
die Rekombinationsrate zwischen 0 und < 0,5 liegen [56].
Unter Penetranz versteht man die Wahrscheinlichkeit, mit der sich ein bestimmter
Genotyp auch phänotypisch auswirkt. Trägt eine Person eine Genveränderung,
erkrankt aber nicht, hat der betreffende Genotyp eine unvollständige Penetranz [56,
73].
Für die Durchführung von parametrischen Kopplungsanalysen stehen verschiedene
Softwareprogramme, wie beispielsweise MERLIN (Multipoint Engine for Rapid
Likelihood Inference), zur Verfügung [1].
MERLIN errechnet die Kopplungswahrscheinlichkeiten anhand von Kopplungsdaten
und verschiedenen Parametern wie Familienstruktur, Vererbungsmodel und Prävalenz
der Erkrankung. Für die Durchführung der Kopplungsanalyse muss der Stammbaum
der Familie des Patienten bekannt sein. Die zur Untersuchung verwendeten Marker
(Mikrosatellitenmarker oder SNPs) müssen für alle Familienmitglieder genotypisiert
werden.
Die Geno- und Phänotypen der Familienmitglieder wurden in einer Stammbaumdatei
(pedigree file), einer Stammbaum spezifischen Parameterdatei (data file) und einer
Kartierungsdatei mit den chromosomalen Positionen (map file) beschrieben.
Der Stammbaum lieferte Informationen über die Geschlechterverteilung,
Familienmitglieder und Generationen. Neben der Anzahl der Erkrankten wurde auch
die Anzahl der betroffenen Generationen berücksichtigt. Von Bedeutung war außerdem
die Anzahl der Krankheitsüberträger.
Die berechneten LOD-Scores (siehe Kapitel 2.8.6) für jeden Marker wurden gegen die
genomische Position aufgetragen. Anschließend wurde der DNA-Abschnitt mit der
höchsten Wahrscheinlichkeit für die Lokalisation des Kandidatengens bestimmt [104].
2.9 Ermittlung geeigneter Mikrosatellitenmarker
Gekoppelte Loci, die in kosanguinen Familien über SNP-Arrays ermittelt werden,
umfassen meist einen weiten Bereich. Für die Suche nach Kandidatengenen müssen
diese Regionen weiter eingegrenzt werden. Dazu eignen sich Mikrosatellitenmarker, da
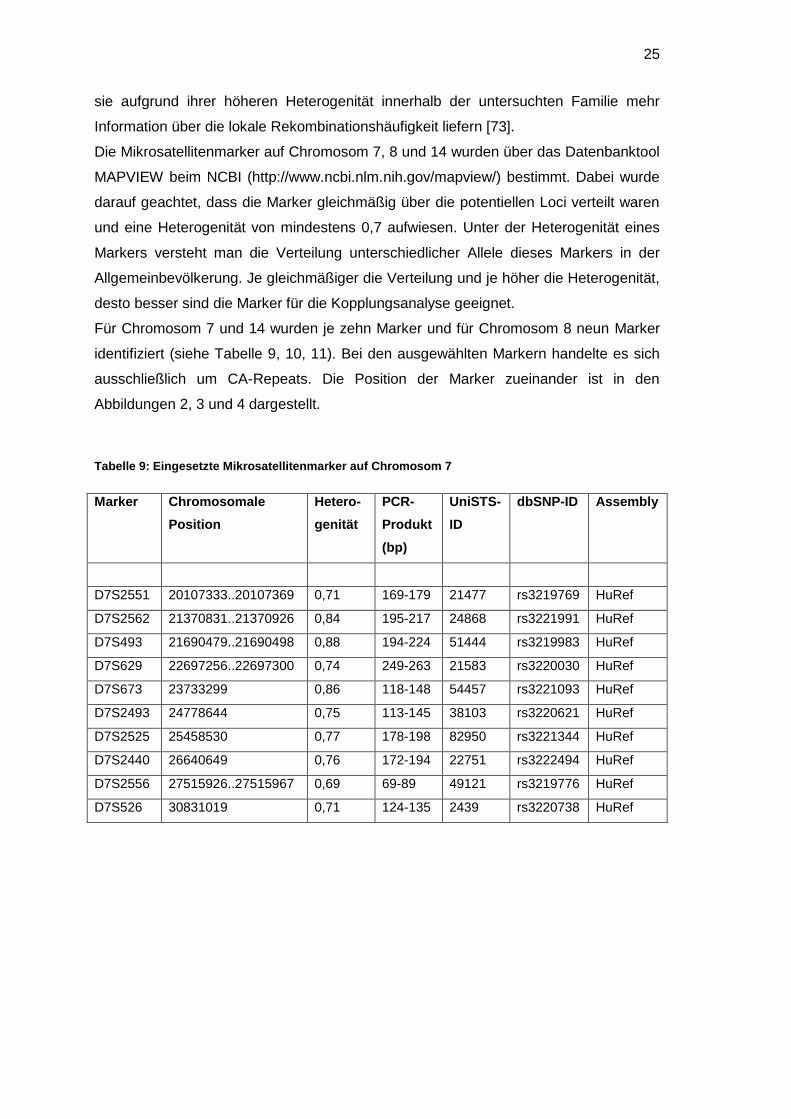
25
sie aufgrund ihrer höheren Heterogenität innerhalb der untersuchten Familie mehr
Information über die lokale Rekombinationshäufigkeit liefern [73].
Die Mikrosatellitenmarker auf Chromosom 7, 8 und 14 wurden über das Datenbanktool
MAPVIEW beim NCBI (http://www.ncbi.nlm.nih.gov/mapview/) bestimmt. Dabei wurde
darauf geachtet, dass die Marker gleichmäßig über die potentiellen Loci verteilt waren
und eine Heterogenität von mindestens 0,7 aufwiesen. Unter der Heterogenität eines
Markers versteht man die Verteilung unterschiedlicher Allele dieses Markers in der
Allgemeinbevölkerung. Je gleichmäßiger die Verteilung und je höher die Heterogenität,
desto besser sind die Marker für die Kopplungsanalyse geeignet.
Für Chromosom 7 und 14 wurden je zehn Marker und für Chromosom 8 neun Marker
identifiziert (siehe Tabelle 9, 10, 11). Bei den ausgewählten Markern handelte es sich
ausschließlich um CA-Repeats. Die Position der Marker zueinander ist in den
Abbildungen 2, 3 und 4 dargestellt.
Tabelle 9: Eingesetzte Mikrosatellitenmarker auf Chromosom 7
Marker Chromosomale
Position
Hetero-
genität
PCR-
Produkt
(bp)
UniSTS-
ID
dbSNP-ID Assembly
D7S2551 20107333..20107369 0,71 169-179 21477 rs3219769 HuRef
D7S2562 21370831..21370926 0,84 195-217 24868 rs3221991 HuRef
D7S493 21690479..21690498 0,88 194-224 51444 rs3219983 HuRef
D7S629 22697256..22697300 0,74 249-263 21583 rs3220030 HuRef
D7S673 23733299 0,86 118-148 54457 rs3221093 HuRef
D7S2493 24778644 0,75 113-145 38103 rs3220621 HuRef
D7S2525 25458530 0,77 178-198 82950 rs3221344 HuRef
D7S2440 26640649 0,76 172-194 22751 rs3222494 HuRef
D7S2556 27515926..27515967 0,69 69-89 49121 rs3219776 HuRef
D7S526 30831019 0,71 124-135 2439 rs3220738 HuRef
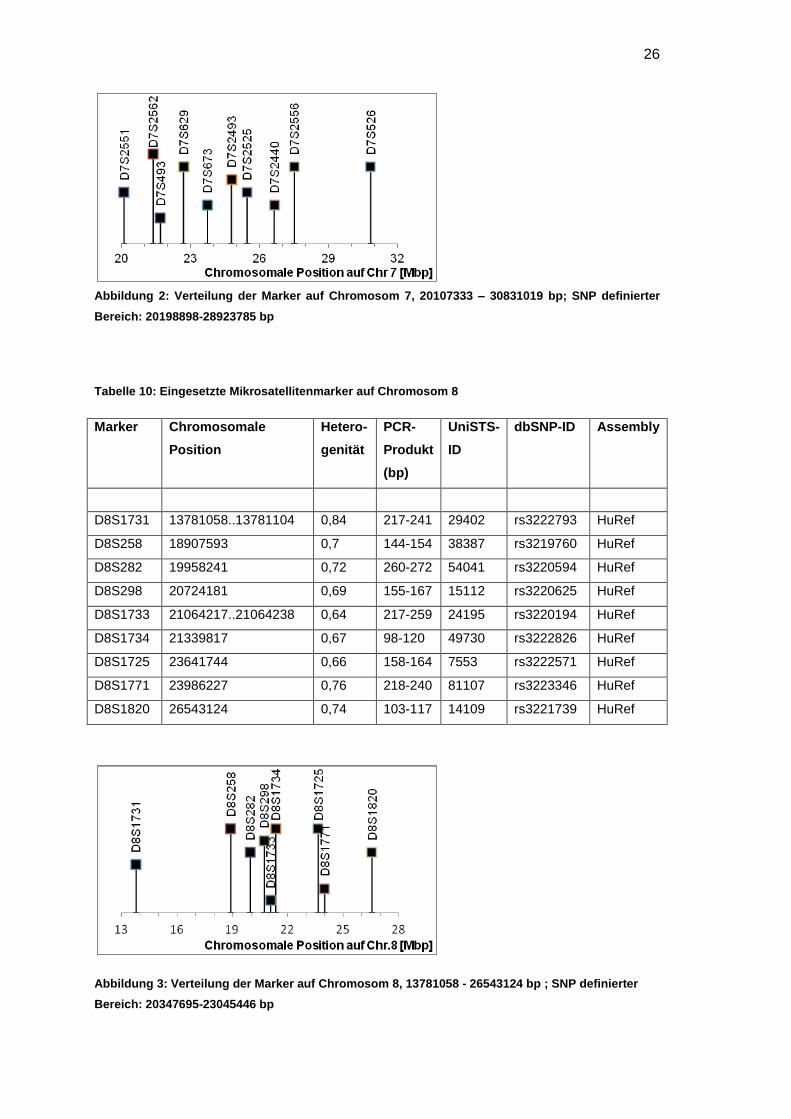
26
Abbildung 2: Verteilung der Marker auf Chromosom 7, 20107333 – 30831019 bp; SNP definierter
Bereich: 20198898-28923785 bp
Tabelle 10: Eingesetzte Mikrosatellitenmarker auf Chromosom 8
Marker Chromosomale
Position
Hetero-
genität
PCR-
Produkt
(bp)
UniSTS-
ID
dbSNP-ID Assembly
D8S1731 13781058..13781104 0,84 217-241 29402 rs3222793 HuRef
D8S258 18907593 0,7 144-154 38387 rs3219760 HuRef
D8S282 19958241 0,72 260-272 54041 rs3220594 HuRef
D8S298 20724181 0,69 155-167 15112 rs3220625 HuRef
D8S1733 21064217..21064238 0,64 217-259 24195 rs3220194 HuRef
D8S1734 21339817 0,67 98-120 49730 rs3222826 HuRef
D8S1725 23641744 0,66 158-164 7553 rs3222571 HuRef
D8S1771 23986227 0,76 218-240 81107 rs3223346 HuRef
D8S1820 26543124 0,74 103-117 14109 rs3221739 HuRef
Abbildung 3: Verteilung der Marker auf Chromosom 8, 13781058 - 26543124 bp ; SNP definierter
Bereich: 20347695-23045446 bp

27
Tabelle 11: Eingesetzte Mikrosatellitenmarker auf Chromosom 14
Marker Chromosomale
Position
Hetero-
genität
PCR-
Produkt
(bp)
UniSTS-
ID
dbSNP-ID Assembly
D14S72 21371146 0,82 257-271 479968 rs3220241 GCH37
D14S1023 21442117 0,82 93-109 71727 rs3223415 GCH37
D14S283 22687564-226875 0,82 125-153 53734 rs3221322 GCH37
D14S990 23586459 0,85 135-161 57093 rs3222520 GCH37
D14S972 24347835-
24347870
0,75 201-211 76329 rs3222058 GCH37
D14S64 24560330 0,75 126-136 54008 rs3219681 GCH37
D14S264 25280072-
25280107
0,72 216-234 31557 rs3219695 GCH37
D14S1032 26211356 0,73 268-282 73907 rs4915421 GCH37
D14S275 26696825-
26696868
0,71 194-205 62366 rs3221112 GCH37
D14S80 27724479 0,83 132-156 40831 rs3220706 GCH37
Abbildung 4: Verteilung der Marker auf Chromosom 14, 21371146 - 27724479 bp; SNP definierter
Bereich: 21452992 – 26740590 bp
Nach Auswahl der Mikrosatellitenmarker wurden die Sequenzen für die PCR-Primer
aus der UniSTS- und dbSNP-Datenbank entnommen. Die Primer wurden bei Metabion
International GmbH (Martinsried) nach Angaben aus den Internetdatenbanken
(http://www.ncbi.nlm.nih.gov/SNP/) synthetisiert.

28
2.10 Auswahl möglicher Kandidatengene
Bei der Auswahl der Kandidatengene wurden verschiedene Kriterien beachtet, um die
Wahrscheinlichkeit für einen Zusammenhang mit der untersuchten Erkrankung zu
erhöhen.
Zuerst wurde die Funktion des Gens betrachtet und hinterfragt, ob eine Mutation in
diesem Gen mit dem Phänotyp der Erkrankung in Verbindung gebracht werden konnte.
Dabei wurde berücksichtigt, dass biochemische Prozesse häufig nicht unmittelbar mit
den Symptomen einer Erkrankung in Verbindung gebracht werden können. Weiter
wurden die phänotypischen Auswirkungen von Mutationen in homologen Genen
berücksichtigt.
Anhand des Expressionsmusters wurde untersucht, ob die Kandidatengene in von der
Erkrankung betroffenen Geweben exprimiert werden.
Gene, deren Mutation bei Tieren zu Erkrankungen mit einem ähnlichen Phänotyp
führen, galten ebenfalls als vielversprechende Kandidaten.
Bei der Untersuchung größerer Genregionen wurde die Größe der zu untersuchenden
Gene berücksichtigt. Kamen zwei Gene in Frage, die sich nur anhand ihrer Exon-
Anzahl unterschieden, wurde zunächst das kleinere Gen untersucht, da der Aufwand
der Analyse direkt mit der Größe des Genes korreliert [73].

29
3 Ergebnisse
3.1 Anamnese der Familie
Von der Familie des an LCA erkrankten Kindes wurden DNA Proben aus zwei
Generationen (Eltern, zwei Töchter, drei Söhne) analysiert. In der zweiten Generation
war der jüngste Sohn an LCA erkrankt. In der Familie war kein weiterer Krankheitsfall
bekannt. Die eineinhalb Jahre ältere Schwester litt an Neurodermitis, die anderen drei
Geschwister waren, soweit berichtet, gesund. Es bestand eine Konsanguinität, da
Mutter und Vater Cousin und Cousine waren. Ein Onkel mütterlicherseits war im
fortgeschrittenen Alter an Diabetes mellitus Typ II erkrankt und aufgrund dieser
Erkrankung erblindet.
Die Blutproben der sieben Familienangehörigen (967.01-07, Abbildung 1) wurden in
Form von EDTA-Blut für die Untersuchung an das molekulargenetische Labor der
Abteilung für Pädiatrische Ophthalmologie, Strabismologie und Ophthalmogenetik
(KSO) des Universitätsklinikums Regensburg gesandt. Die DNA-Proben wurden aus
jeweils 4 ml EDTA-Blut der Geschwister und jeweils 10 ml EDTA-Blut der Eltern
extrahiert.
Die extrahierte DNA wurde zunächst im molekulargenetischen Labor der KSO auf
Mutationen damals bekannter LCA-Gene untersucht. Die Untersuchungen wurden
anschließend im Labor für molekulare Ophthalmologie der Klinik und Poliklinik für
Augenheilkunde der Justus-Liebig-Universität in Gießen mit einer Kopplungsanalyse
(die vorliegende Arbeit) fortgeführt.
Der Patient 967.01 wurde 2002 im Kinderzentrum Bielefeld, Lehrkrankenhaus der
Universitätsklinik Münster, untersucht (Tabelle 12). Aus der Krankenakte ging hervor,
dass den Eltern bei ihrem Sohn eine schlechte Kopfkontrolle und ein Unvermögen,
Gegenstände richtig zu fixieren, aufgefallen war. Bei der Aufnahme war der vier
Monate alte Säugling in einem guten Allgemein- und Ernährungszustand. Er zeigte
eine auffällige Augenmotorik mit Nystagmus in wechselnder Richtung. Die Pupillen
waren weit, die Pupillenreaktion stark verlangsamt und es bestand eine leichte Miosis.
Erst bei längerer Lichtexposition verstärkte sich die Miosis. Eine Fixation der Augen
war nicht sicher nachweisbar. Der Patient blinzelte oder erschrak nicht, wenn
Gegenstände direkt auf ihn zu bewegt wurden.
Auffällig waren weiterhin die unkoordinierten Bewegungsmuster der Arme und Beine.
Die Muskeleigenreflexe waren seitengleich auslösbar. Weiter zeigte der Säugling einen
Moro-Reflex.

30
Bei einem Traktionsversuch, bei dem der Säugling an den Armen hochgezogen wurde,
fiel eine schlechte Kopfkontrolle auf. Um eine Muskelerkrankung auszuschließen,
wurde die gesamte Muskulatur untersucht. Der generalisierte Muskeltonus erwies sich
als nicht pathologisch. Die schlechte Kopfhaltung wurde durch die mangelnde
Fähigkeit, die Motorik visuell zu koordinieren, erklärt.
Auch die weitere körperliche Untersuchung ergab keine pathologischen Befunde. Das
Blutbild des Patienten war unauffällig. In der Blutgasanalyse ließ sich eine leichte
kompensierte metabolische Azidose nachweisen. Serologisch gab es keinen Hinweis
auf eine abgelaufene Cytomegalievirus- (CMV), Röteln- oder Toxoplasmose-Infektion.
Im Alter von vier Monaten wurde der Säugling einem niedergelassenen Augenarzt
vorgestellt. Die augenärztliche Untersuchung ergab normal brechende Medien und
eine unauffällige Darstellung des Nervus opticus. Eine Fixation der Augen war nicht
detektierbar. Gelegentlich bestand der Eindruck, dass eine starke Lichtquelle mit den
Augen verfolgt wurde. Die Pupillenreaktion war stark eingeschränkt, da erst nach
längerer Lichtexposition eine deutliche Miosis einsetzte. Der Fundus war nur schwer
beurteilbar. Eine Makulahypoplasie, Optikusatrophie oder retinale Degenerationen
waren nicht erkennbar. Aufgrund des klinischen Bildes wurde eine primäre
Optikusatrophie angenommen.
Die Familie des Patienten stand laut Angaben des einsendenden Kinderarztes für
weitere Untersuchungen nicht mehr zur Verfügung.

31
Tabelle 12: Zusammenfassung der klinischen Untersuchungsergebnisse des Patienten
Untersuchung Ergebnis
Augenärztliche Untersuchung
- Keine Fixation möglich
- Stark eingeschränkte
Pupillenreaktion
- Fundus schwer beurteilbar
Visuell evozierte Potentiale (VEP)
- Keine reproduzierbaren VEPs,
weder seitengetrennt noch
binokulär
- Deutlicher Hinweis auf eine
Netzhauterkrankung oder
Leitungsstörung im Verlauf der
Sehbahn
Traktionsversuch - Schlechte Kopfbalance
Blutgasanalyse - Leicht kompensierte metabolische
Azidose
Serologie - CMV / Toxoplasmose / Röteln
negativ
Urinstatus - Ohne pathologischen Befund
Qualitative Urintests: Phenylketonurie-
Screening, Reduktionsprobe, Ninhydrin,
Brand´sche Probe
- Ohne pathologischen Befund
Sonographie Schädel - Unauffällig
Nuclear magnetic resonance-(NMR)-
Spektroskopie des Neurocraniums - Normalbefund
Blutbild (inklusive Leber und Nierenwerte) - Unauffällig
3.2 SNP Genotypsierung und Kopplungsanalyse
Mit den sieben vorliegenden DNA-Proben wurde im Gene Mapping Center des Max
Delbrück Center for Molecular Medicine, wie in Kapitel 2.3.4 beschrieben, mithilfe des
Human Mapping 50K Array Xba 240 von Affymetrix, eine Genotypisierung der SNPs
durchgeführt. Im Anschluss wurden die Daten mit ALOHOMORA aufbereitet und einer
parametrischen Kopplungsanalyse in MERLIN unterzogen. Dazu wurde eine kurze
Konsanguinitätsschleife in den Stammbaum eingeführt. Durch die genomweite
Kopplungsanalyse konnten auffällige LOD-Scores an drei Loci auf Chromosom 7, 8
und 14 (grüne Pfeile) ermittelt werden (Abbildung 5, Tabelle 13).

32
Abbildung 5: Auswertung des Human Mapping 50K Array Xba 240 von Affymetrix mit MERLIN
Tabelle 13: LOD-Scores der Kopplungsanalyse mittels SNP-Marker
Chromosom LOD
Score Chromosomale Position Assembly
7 1,8 20198898 – 28923785 bp HuREF [89, 99]
8 1,7 20347695 – 23045446 bp HuREF [100, 101]
14 1,8 21452992 – 26740590 bp GCH37 [96, 97]
Höhere LOD-Scores waren nicht zu erwarten, da nur eine begrenzte Anzahl von
Meiosen (DNA-Proben aus zwei Generationen) zur Verfügung stand, die in die
Berechnung mit einbezogen werden konnten. Die Ergebnisse konnten nicht als
signifikant gewertet werden, da keine LOD-Scores > 3 auftraten. Daher musste eine
Feinkartierung der Loci durchgeführt werden, um die möglichen Kandidatengene näher
einschränken zu können.
Für die weitere Untersuchung der Chromosomenbereiche wurden
Mikrosatellitenmarker benutzt, die über das MAPVIEW Tool des National Center for
Biotechnology Information (http://www.ncbi.nlm.nih.gov/mapvieww//) bestimmt wurden.

33
3.3 Optimierung der Mikrosatellitenamplifikation
Zunächst wurden mit einer Kontroll-DNA (DNA 0005) die optimalen Bedingungen zur
Amplifikation der Mikrosatellitenmarker bestimmt (siehe Kapitel 2.5). Variiert wurden
dazu die Annealingzeit und -temperatur der PCR und die Zusammensetzung des
Mastermixstandards.
Für jeden Mikrosatellitenmarker wurde eine PCR mit sechs verschiedenen
Annealingtemperaturen zwischen 53 – 65 °C durchgeführt. Die optimalen Bedingungen
für jeden Marker sind in der Tabelle 14 zusammengefasst.
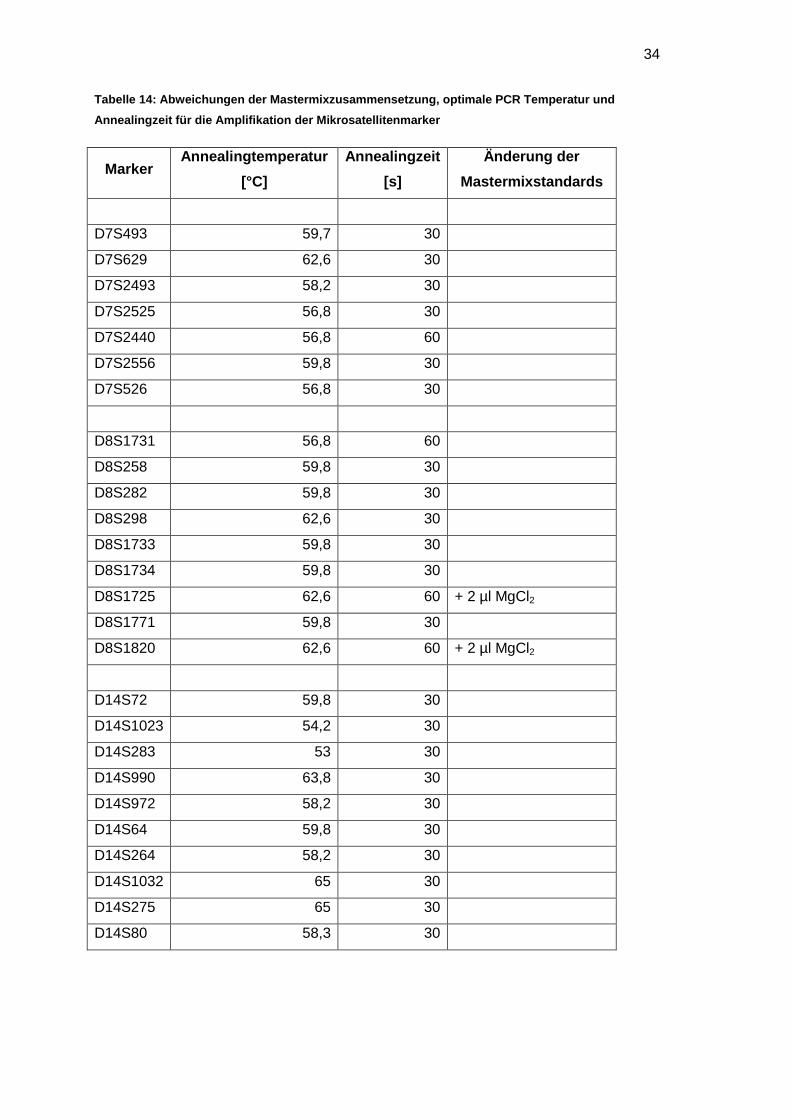
34
Tabelle 14: Abweichungen der Mastermixzusammensetzung, optimale PCR Temperatur und
Annealingzeit für die Amplifikation der Mikrosatellitenmarker
Marker Annealingtemperatur
[°C]
Annealingzeit
[s]
Änderung der
Mastermixstandards
D7S493 59,7 30
D7S629 62,6 30
D7S2493 58,2 30
D7S2525 56,8 30
D7S2440 56,8 60
D7S2556 59,8 30
D7S526 56,8 30
D8S1731 56,8 60
D8S258 59,8 30
D8S282 59,8 30
D8S298 62,6 30
D8S1733 59,8 30
D8S1734 59,8 30
D8S1725 62,6 60 + 2 µl MgCl2
D8S1771 59,8 30
D8S1820 62,6 60 + 2 µl MgCl2
D14S72 59,8 30
D14S1023 54,2 30
D14S283 53 30
D14S990 63,8 30
D14S972 58,2 30
D14S64 59,8 30
D14S264 58,2 30
D14S1032 65 30
D14S275 65 30
D14S80 58,3 30

35
3.3.1 Amplifikation der Mikrosatellitenmarker mit den DNA-Proben der Familie
Unter den optimierten PCR-Bedingungen wurde die PCR mit den sieben zu
untersuchenden DNA-Proben durchgeführt.
Die DNA-Proben wurden mit einer Konzentration von 20 ng/µl angesetzt.
Für jeden Marker wurde jeweils ein PCR Ansatz für jedes Familienmitglied hergestellt.
Nach Auftrennung des PCR Produkte in der Agarosegelelektrophorese fiel auf, dass
die Banden bei allen PCR Ansätzen der DNA Proben 967.06 und 967.07 sehr schwach
waren. Grund dafür war ein zu hoher Proteinanteil in den DNA-Proben, der mit einer
Phenolextraktion der DNA verringert wurde. Diese wurde, wie in Kapitel 2.5
beschrieben, durchgeführt. Die aufgereinigten DNA Proben wurden erneut auf 20 ng/µl
konzentriert und die PCRs mit anschließender Gelelektrophorese bei beiden DNA
Proben für alle Marker wiederholt.
3.4 Kopplungsanalyse mit den Mikrosatellitenmarkern
Nach der Amplifikation wurde die Allelgröße der Mikrosatellitenmarker mit der QIAxcel
bestimmt.
Nach Typisierung aller Allele der untersuchten Mikrosatellitenmarker der zur Verfügung
stehenden DNA-Proben wurden die Haplotypen bestimmt.
Die Genotypisierungen wurden graphisch mit CYRILLIC 2.1 dargestellt. CYRILLIC 2.1
erlaubt auch eine Haplotypisierung, die für jedes Chromosom separat durchgeführt
wurde (Abbildungen 6 – 8). Nicht informative Marker wurden nicht dargestellt.
Der Haplotyp des an LCA erkrankten Kindes wurde mit den Haplotypen der anderen
Familienmitglieder verglichen. Dabei wurde analysiert bei welchen
Mikrosatellitenmarkern Unterschiede bestanden.
3.4.1 Haplotypen der Mikrosatellitenmarker auf Chromosom 7
Abbildung 6 zeigt den Stammbaum der Familie und die Mikrosatellitenmarkeranalyse
für Chromosom 7.

36
Abbildung 6: Stammbaum der Familie 967 mit den Haplotypen der Mikrosatellitenmarker auf
Chromosom 7
Die Haplotypen der Mikrosatellitenmarker der Kinder waren verschiedene
Konstellationen aus je einem Haplotyp der entsprechenden Marker des Vaters und der
Mutter. Die Auswertung der Mikrosatellitenmarker D7S493 und D7S2556 ergab keine
brauchbaren Informationen, da beide Eltern homozygote Träger des gleichen
Haplotyps waren und daher keine Rückschlüsse auf das Verteilungsmuster bei den
Kindern möglich war.
Sowohl der erkrankte Sohn, als auch ein gesunder Bruder und eine gesunde
Schwester wiesen für den Mikrosatellitenmarker D7S2440 den gleichen Haplotyp auf,
so dass eine Lage des Kandidatengens in diesem Bereich ausgeschlossen werden
konnte.
Die Mikrosatellitenmarker D7S629, D7S2493, D7S2525 und D7S526 wurden weiter
untersucht.
Der Vergleich der Haplotypkonstellationen der Geschwister und der Eltern ergab keine
auffälligen Unterschiede zwischen dem erkrankten Kind und den gesunden
Geschwisterkindern bzw. den phänotypisch gesunden Eltern.

37
3.4.2 Haplotypen der Mikrosatellitenmarker auf Chromosom 8
Abbildung 7 zeigt den Stammbaum der Familie und die Mikrosatellitenmarkeranalyse
für Chromosom 8.
Abbildung 7: Stammbaum der Familie 967 mit den Haplotypen der Mikrosatellitenmarker auf
Chromosom 8
Auf Chromosom 8 wurden neun Mikrosatellitenmarker untersucht. Die Marker
D8S1733 und D8S298 lieferten keine auswertbaren Informationen, weil die gesunden
Geschwister und das erkrankte Kind identische Haplotypen besaßen. Auffällig war,
dass das erkrankte Kind bezüglich des Mikrosatellitenmarkers D8S1820 ein
homozygoter Allelträger war, während sowohl die Eltern als auch die gesunden
Geschwisterkinder heterozygote Allele besaßen.
3.4.3 Haplotypen der Mikrosatellitenmarker auf Chromosom 14
Abbildung 8 zeigt den Stammbaum der Familie und die Mikrosatellitenmarkeranalyse
für Chromosom 14.

38
Abbildung 8: Stammbaum der Familie 967 mit den Haplotypen der Mikrosatellitenmarker auf
Chromosom 14
Auf Chromosom 14 wurden zehn Mikrosatellitenmarker untersucht. Die Haplotypen des
Markers D14S275 waren bei allen Geschwisterkindern identisch. Alle fünf Geschwister
waren homozygot für diesen Haplotyp. Die Haplotypenanalyse auf Chromosom 14
zeigte bei sechs Mikrosatellitenmarkern signifikante Unterschiede zwischen der
Allelkonstellation des erkrankten Kindes und der Allelkonstellation der vier gesunden
Geschwister. Das erkrankte Kind war bezüglich der Marker D14S283, D14S990,
D14S972, D14S64, D14S264 und D14S1032 homozygot, während sowohl die Eltern
als auch die gesunden Geschwistern entweder heterozygot oder homozygot für ein
anderes Allel waren.
3.4.4 Ergebnis der Haplotypenanalyse
Die Haplotypenanalyse auf Chromosom 7 ergab keinen Hinweis für eine mögliche
Kopplung, da sich kein Haplotyp des erkrankten Kindes bei nicht mindestens einem
gesunden Geschwisterkind wiederfand.

39
Auf Chromosom 8 zeigte sich nur bei dem Mikrosattelitenmarker D8S1820 ein
Unterschied bezüglich der Allelverteilung zwischen dem erkrankten Kind und den nicht
erkrankten Geschwistern.
Die Haplotypenanalyse auf Chromosom 14 ergab für sechs Mikrosatellitenmarker
(D14S283, D14S990, D14S972, D14S64 D14S264 und D14S1032) einige interessante
Unterschiede bei der Allelkonstellation des erkrankten Kindes im Vergleich zu den
Haplotypen der vier Geschwister. Der Chromosomenbereich, der durch diese
Mikrosatellitenmarker abgedeckt wurde, erstreckte sich von 21.442.117 bp bis
26.211.356 bp auf Chromosom 14.
Eine mögliche Genkopplung auf Chromosom 7 und 8 war unwahrscheinlich, allerdings
nicht ausgeschlossen.
Um eine mögliche Kopplungsregion genauer zu definieren, wurde ergänzend eine
Kopplungsanalyse der Mikrosatellitenmarker mit MERLIN durchgeführt.
3.5 Berechnung der Kopplungswahrscheinlichkeit mit MERLIN
Im Anschluss an die Haplotypenanalyse wurden die Daten der Genotypisierung für die
Kopplungsanalyse mit MERLIN aufgearbeitet, um die Kopplung zu quantifizieren und
die Kopplungsregion darüber näher einzugrenzen. Es wurden für jeden Locus vier
Dateien erstellt:
1. eine Stammbaumdatei (967-xx.ped) (Tabellen 15 - 17)
2. eine Stammbaum spezifische Parameterdatei (967-xx.dat) (Tabelle 18)
3. eine Kartierungsdatei mit den chromosomalen Positionen jeden Markers (967-
xx.map) (Tabelle 19)
4. eine Allefrequenzdatei mit den Häufigkeiten der untersuchten Markerallele in
der Normalbevölkerung (967-xx.fq) (Tabelle 20).

40
Tabelle 15: Stammbaumdatei zur Kopplungsanalyse am Chromosom 7
967 11 0 0 1 1 x x x x x x x x
967 12 0 0 2 1 x x x x x x x x
967 21 0 0 1 1 x x x x x x x x
967 22 11 12 2 1 x x x x x x x x
967 23 11 12 1 1 x x x x x x x x
967 24 0 0 2 1 x x x x x x x x
967 7 21 22 1 1 249 249 129 141 186 186 124 132
967 8 23 24 2 1 249 253 129 133 186 190 132 124
967 2 7 8 2 1 249 253 129 133 186 190 132 132
967 3 7 8 1 1 249 253 141 129 186 190 132 132
967 4 7 8 2 1 249 253 129 129 186 190 124 124
967 5 7 8 1 1 249 253 129 129 186 190 124 124
967 1 7 8 1 2 249 249 129 129 186 186 132 132
Tabelle 16: Stammbaumdatei zur Kopplungsanalyse am Chromosom 8
967 11 0 0 1 1 x x x x x x x x x x x x x x
967 12 0 0 2 1 x x x x x x x x x x x x x x
967 21 0 0 1 1 x x x x x x x x x x x x x x
967 22 11 12 2 1 x x x x x x x x x x x x x x
967 23 11 12 1 1 x x x x x x x x x x x x x x
967 24 0 0 2 1 x x x x x x x x x x x x x x
967 7 21 22 1 1 221
239
148
148
262
262
104
98
162
162
232
236
103
111
967 8 23 24 2 1 235
237
154
144
264
264
104
98
162
160
230
232
103
111
967 2 7 8 2 1 221
237
148
154
262
264
104
104
162
160
232
230
103
103
967 3 7 8 1 1 221
237
148
154
262
264
104
104
162
162
232
232
103
111
967 4 7 8 2 1 221
237
148
154
262
264
104
98
162
162
232
232
103
111
967 5 7 8 1 1 221
237
148
154
262
264
104
98
162
162
236
232
103
111
967 1 7 8 1 2 221
237
148
144
262
264
104
104
162
160
236
232
111
111
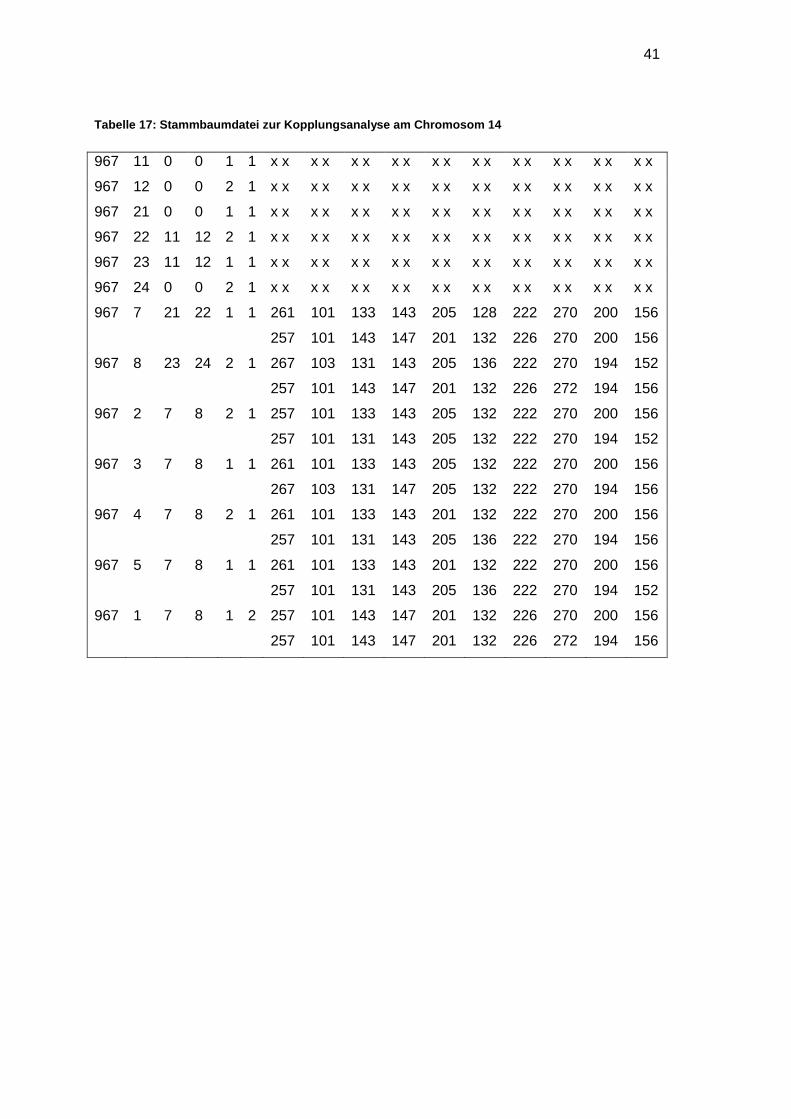
41
Tabelle 17: Stammbaumdatei zur Kopplungsanalyse am Chromosom 14
967 11 0 0 1 1 x x x x x x x x x x x x x x x x x x x x
967 12 0 0 2 1 x x x x x x x x x x x x x x x x x x x x
967 21 0 0 1 1 x x x x x x x x x x x x x x x x x x x x
967 22 11 12 2 1 x x x x x x x x x x x x x x x x x x x x
967 23 11 12 1 1 x x x x x x x x x x x x x x x x x x x x
967 24 0 0 2 1 x x x x x x x x x x x x x x x x x x x x
967 7 21 22 1 1 261
257
101
101
133
143
143
147
205
201
128
132
222
226
270
270
200
200
156
156
967 8 23 24 2 1 267
257
103
101
131
143
143
147
205
201
136
132
222
226
270
272
194
194
152
156
967 2 7 8 2 1 257
257
101
101
133
131
143
143
205
205
132
132
222
222
270
270
200
194
156
152
967 3 7 8 1 1 261
267
101
103
133
131
143
147
205
205
132
132
222
222
270
270
200
194
156
156
967 4 7 8 2 1 261
257
101
101
133
131
143
143
201
205
132
136
222
222
270
270
200
194
156
156
967 5 7 8 1 1 261
257
101
101
133
131
143
143
201
205
132
136
222
222
270
270
200
194
156
152
967 1 7 8 1 2 257
257
101
101
143
143
147
147
201
201
132
132
226
226
270
272
200
194
156
156
42
Tabelle 18: Parameterdatei zur Kopplungsanalyse am A) Chromosom 7, B) Chromosom 8, C)
Chromosom 14
A A LCA967 B A LCA967 C A LCA967
M D7S629 M D8S1731 M D14S72
M D7S2493 M D8S258 M D14S1023
M D7S2525 M D8S282 M D14S283
M D7S526 M D8S1734 M D14S990
M D8S1725 M D14S972
M D8S1771 M D14S64
M D8S1820 M D14S264
M D14S1032
M D14S275
M D14S80
Tabelle 19: Kartierungsdatei zur Kopplungsanalyse am A) Chromosom 7, B) Chromosom 8, C)
Chromosom 14
A 7 D7S629 22697.078 B 8 D8S173
1
13780.985 C 1
4
D14S72 1492.157
7 D7S249
3
2477.8583 8 D8S258 18907.500 1
4
D14S102
3
1563.201
7 D7S252
5
2537.9498 8 D8S282 19958.427 1
4
D14S283 2805.161
7 D7S526 30830.954 8 D8S173
4
21339.754 1
4
D14S990 3703.431
8 D8S172
5
23641.673 1
4
D14S972 4464.891
8 D8S177
1
23986.152 1
4
D14S64 4675.199
8 D8S182
0
26543.094 1
4
D14S264 5394.043
1
4
D14S103
2
6326.397
1
4
D14S275 6812.039
1
4
D14S80 7840.875
Die dritte Spalte gibt die chromosomale Position in Kilobasen an.

43
Zu jeder Kartierungsdatei wurde eine korrespondierende Datei mit den Allelfrequenzen
der gelisteten Mikrosatellitenmarker angelegt. Die Allelfrequenzen wurden der dbSNP-
Datenbank am NCBI entnommen. Die Datei hatte das in Tabelle 20 dargestellte
Grundformat.
Tabelle 20: Allelfrequenzdatei
M Markername
A Allel 1 Allelfrequenz
A Allel 2 Allelfrequenz
...
A Allel n Allelfrequenz
Wegen des Umfangs der Dateien wird hier auf eine detaillierte Wiedergabe verzichtet.
Letztlich wurde eine Modelldatei angelegt, die die Parameter des Modells für die
parametrische Kopplungsanalyse definierte. Diese waren:
Prävalenz 1 : 100.000
Penetranz: vollständig
Erbgang: autosomal-rezessiv (nur Träger mit zwei mutierten Allelen sind
betroffen)
In den Abbildungen 9 – 11 sind die Ergebnisse der Kopplungsanalyse mit den
Mikrosatellitenmarkern (blaue Punkte) gegen die Kopplungsanalyse mit den SNP-
Markern (schwarze Kreise) als LOD-Scores an einer definierten chromosomalen
Position aufgetragen. Alle drei Abbildungen decken eine vergleichbare
Chromosomenlänge ab (14 Mbp), um die Größe der Kopplungsbereiche besser
vergleichen zu können.

44
Abbildung 9: Ergebnisse der Kopplungsanalyse mit den SNP- und Mikrosatellitenmarkern auf
Chromosom 7
Abbildung 9 stellt die Kopplungsdaten für Chromosom 7 dar. Keiner der untersuchten
Mikrosatellitenmarker (blaue Punkte) erreichte einen LOD-Score (> 3,0), der auf eine
signifikante Kopplung in dieser Region hinwies. Die LOD-Scores für D7S2493 und
D7S2525 schlossen eine Kopplung nicht grundsätzlich aus. Die Mikrosatellitenmarker
D7S829 und D7S526 erzielten einen LOD-Score von -∞.
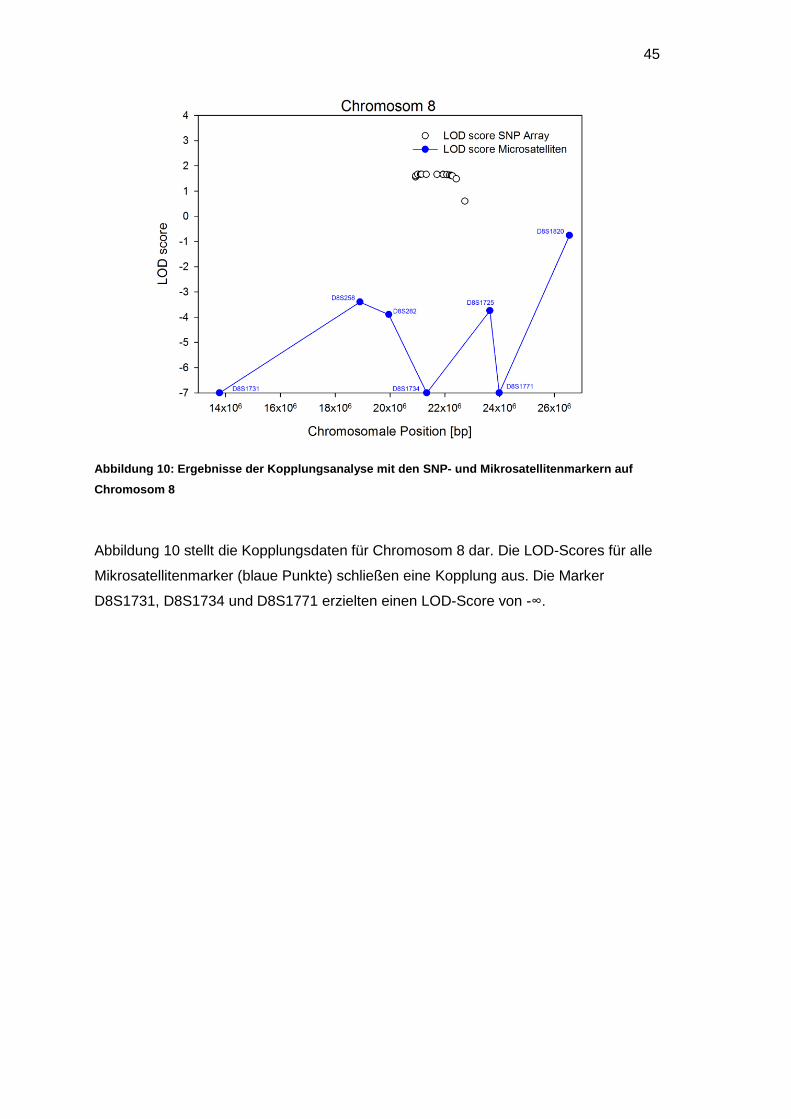
45
Abbildung 10: Ergebnisse der Kopplungsanalyse mit den SNP- und Mikrosatellitenmarkern auf
Chromosom 8
Abbildung 10 stellt die Kopplungsdaten für Chromosom 8 dar. Die LOD-Scores für alle
Mikrosatellitenmarker (blaue Punkte) schließen eine Kopplung aus. Die Marker
D8S1731, D8S1734 und D8S1771 erzielten einen LOD-Score von -∞.

46
Abbildung 11: Ergebnisse der Kopplungsanalyse mit den SNP- und Mikrosatellitenmarkern auf
Chromosom 14
Abbildung 11 stellt die Kopplungsdaten für Chromosom 14 dar. Keiner der
Mikrosatellitenmarker (blaue Punkte) erreichte einen LOD-Score, der eine Kopplung in
dieser Region sicher bestätigte. Die LOD-Scores für D14S283, D14S990 und D14S972
schlossen eine Kopplung in der Region nicht grundsätzlich aus. Die Marker D14S72
und D14S64 erzielten einen LOD-Score von -∞.
Die Auswertung der Haplotypenanalyse ergab, dass eine Kopplung auf Chromosom 14
am wahrscheinlichsten war. Die Kopplungsanalyse zeigte vergleichbar gute LOD-
Scores für verschiedene der Mikrosatellitenmarker auf Chromosom 7 und 14. Die damit
abgedeckte Region auf Chromosom 14 war mit > 3 Mbp deutlich größer als auf
Chromosom 7 mit > 1 Mbp. Da aufgrund der Haplotypenanalyse eine Genkopplung auf
Chromosom 7 als unwahrscheinlich zu bewerten war, wurde die Region auf
Chromosom 14 als Kandidatengenregion definiert.
3.6 Evaluation möglicher Kandidatengene auf Chromosom 14
Der über die SNP-Kopplungsanalyse identifizierte und anschließend über die
Haplotypen- und Mikrosatellitenmarkeranalyse eingegrenzte Locus auf Chromosom 14
enthielt einschließlich der flankierenden Chromosomenbereiche 294 Gene. Mithilfe des
Ensembl Genome Browsers wurde eine Liste dieser Gene erstellt. Anschließend
erfolgte eine weitere Eingrenzung nach den in Kapitel 2.10 beschriebenen Kriterien.
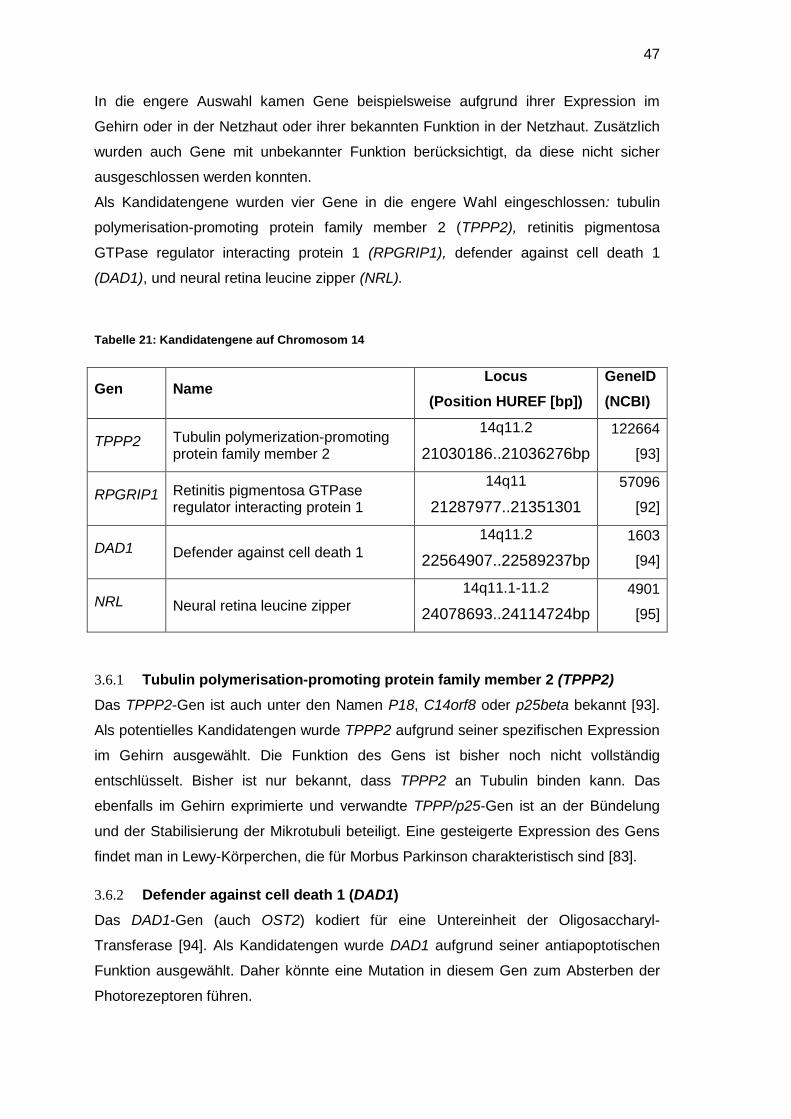
47
In die engere Auswahl kamen Gene beispielsweise aufgrund ihrer Expression im
Gehirn oder in der Netzhaut oder ihrer bekannten Funktion in der Netzhaut. Zusätzlich
wurden auch Gene mit unbekannter Funktion berücksichtigt, da diese nicht sicher
ausgeschlossen werden konnten.
Als Kandidatengene wurden vier Gene in die engere Wahl eingeschlossen: tubulin
polymerisation-promoting protein family member 2 (TPPP2), retinitis pigmentosa
GTPase regulator interacting protein 1 (RPGRIP1), defender against cell death 1
(DAD1), und neural retina leucine zipper (NRL).
Tabelle 21: Kandidatengene auf Chromosom 14
Gen Name Locus
(Position HUREF [bp])
GeneID
(NCBI)
TPPP2 Tubulin polymerization-promoting protein family member 2
14q11.2
21030186..21036276bp
122664
[93]
RPGRIP1 Retinitis pigmentosa GTPase regulator interacting protein 1
14q11
21287977..21351301
57096
[92]
DAD1 Defender against cell death 1
14q11.2
22564907..22589237bp
1603
[94]
NRL Neural retina leucine zipper
14q11.1-11.2
24078693..24114724bp
4901
[95]
3.6.1 Tubulin polymerisation-promoting protein family member 2 (TPPP2)
Das TPPP2-Gen ist auch unter den Namen P18, C14orf8 oder p25beta bekannt [93].
Als potentielles Kandidatengen wurde TPPP2 aufgrund seiner spezifischen Expression
im Gehirn ausgewählt. Die Funktion des Gens ist bisher noch nicht vollständig
entschlüsselt. Bisher ist nur bekannt, dass TPPP2 an Tubulin binden kann. Das
ebenfalls im Gehirn exprimierte und verwandte TPPP/p25-Gen ist an der Bündelung
und der Stabilisierung der Mikrotubuli beteiligt. Eine gesteigerte Expression des Gens
findet man in Lewy-Körperchen, die für Morbus Parkinson charakteristisch sind [83].
3.6.2 Defender against cell death 1 (DAD1)
Das DAD1-Gen (auch OST2) kodiert für eine Untereinheit der Oligosaccharyl-
Transferase [94]. Als Kandidatengen wurde DAD1 aufgrund seiner antiapoptotischen
Funktion ausgewählt. Daher könnte eine Mutation in diesem Gen zum Absterben der
Photorezeptoren führen.

48
Die Oligosaccharyl-Transferase spielt eine wesentliche Rolle bei der N-Glykosylierung
in Eukaryonten [94]. Die N-Glykosylierung findet im glatten endoplasmatischen
Retikulum und abschließend im Golgi-Apparat statt. Dieser Prozess besteht aus einer
Bindung von Oligosacchariden an die NH2-Gruppe des Asparagins [52]. Aufgrund der
Expression in Dünndarmtumoren wird diesem Gen eine Rolle bei der Tumorgenese
zugeschrieben [48].
3.6.3 Retinitis pigmentosa GTPase regulator-interacting Protein (RPGRIP1)
Das Gen RPGRIP1 kodiert für ein Photorezeptor-Protein, das im verbindenden Zilium
(engl. connecting cilium, CC) zwischen Photorezeptor-Außensegment und -
Innensegment lokalisiert ist. Das RPGRIP-Protein besteht aus einer amino-terminalen
Domäne, die über eine coiled-coil Struktur mit der Carboxy-(C)-terminalen Domäne
verbunden ist [37]. RPGRIP spielt bei der Entwicklung der Photorezeptoren eine Rolle,
wobei die genaue Funktion bis heute nicht geklärt ist. RPGRIP beeinflusst die
Zilienbildung und -funktion und ist wahrscheinlich an der Disc-Morphogenese beteiligt
[23, 31, 87]. In den Photorezeptoren ist RPGRIP an der Verankerung eines weiteren
Photorezeptor-Proteins, des RP GTPase regulator (RPGR) im CC beteiligt [63]. Die CC
sind hochspezialisierte Organellen, die für den intraflagellaren Transport (IFT) in den
Photorezeptoren zuständig sind und das Außen- mit dem Innensegment verbinden
[38].
RPGRIP1-Mutationen führen zur Degeneration der Photorezeptor-Außensegmente und
zur Dislokation von Opsin-Proteinen in Stäbchen und Zapfen [63]. Mäusen, denen
RPGRIP fehlt, weisen vergrößerte Außensegmente der Photorezeptoren auf und sind
stärker erkrankt als Mäuse ohne RPGR. Der Verlust beider Photorezeptor-Proteine
unterscheidet sich phänotypisch nicht von dem Verlust von RPGRIP alleine. Die C-
terminale Domäne von RPGRIP weist Homologien zur entsprechenden Domäne der
Aktin-Fragmin-Kinase auf. Dieser Befund ist möglicherweise eine Erklärung dafür, dass
RPGRIP nicht nur bei der Zilienentwicklung eine Rolle spielt, sondern auch über die
Regulierung des Aktin-Zytoskeletts die Morphologie der Disks der Photorezeptoren
beeinflusst [87].
Klinisch äußern sich Mutationen im RPGRIP1-Gen in autosomal-rezessiven
kongenitalen Dystrophien [92]. Meist handelt es sich um Missense-Mutationen, die in
den für die C-terminalen C2-Domänen kodierenden Bereichen lokalisiert sind. An
diesem Ende des RPGRIP-Proteins befindet sich auch eine Ca2+-Bindungsstelle.
Modellversuche legen nahe, dass diese Ca2+-Bindungsstelle durch die Missense-
Mutationen von RPGRIP1 zerstört wird. RPGRIP steht in enger Wechselwirkung mit
einem weiteren Zilienprotein namens Nephrocystin-4. D. Nephrocystin-4 bindet

49
spezifisch an die C2-Domäne von RPGRIP. Mutationen des NPHP4-Gens sind für
Nephronophthisen oder das Senior-Loken Syndrom (SLSN), einer Kombination aus RP
und Nierendysplasie, verantwortlich. Die Interaktion von NPHP4 und RPGRIP1 kann
durch Mutationen in einem der beiden Gene gestört werden [75].
Eine weitere Wechselwirkung besteht zwischen RPGRIP1 und der Nek4-
Serin/Threonin-Kinase. In den Flimmerzellen kommt diese Kinase hauptsächlich in den
Basalkörperchen vor und ist Teil sowohl des RPGRIP1- als auch des RPGRIP1L-
Protein-Komplexes. Mutationen im RPGRIP1L-Gen führen zum Meckel-Gruber oder
Joubert-Syndrom. Beides sind schwere Ziliopathien. In einer Studie zur Funktion von
RPGRIP1 und RPGRIP1L und zu ihrer Interaktion mit der Nek4-Serin/Threonin-Kinase
führte eine verminderte Aktivität der Nek4-Serin/Threonin-Kinase zur Abnahme der
Ziliensynthese. Es wurde die Hypothese aufgestellt, dass RPGRIP1 und RPGRIP1L
die Funktion der zilienstabilisierenden Nek4-Serin/Threonin-Kinase beeinflussen [13].
Bis heute sind mehr als 70 unterschiedliche RPGRIP1-Mutationen bekannt [90].
Hauptsächlich handelt es sich um Basenaustausch-Mutationen in den kodierenden
Sequenzen. 2014 wurde erstmals eine homozygote Deletion von Exon 17 in RPGRIP1
beschrieben [80].
3.6.4 Neural Retina Leucine Zipper (NRL)
Das NRL-Gen kodiert für einen Transkriptionsfaktor, der die Entwicklung der Stäbchen
steuert [95]. Mutationen dieses Gens wurden bereits bei der autosomal-dominanten
und der autosomal-rezessiven RP beschrieben [7, 36]. Bei Betroffenen wird eine stark
reduzierte Funktion der Stäbchen diagnostiziert; die Patienten sind seit frühester
Kindheit nachtblind. In der Peripherie des Fundus auf der Ebene des RPE lassen sich
große, aggregierte Pigmentablagerungen nachweisen [60].
3.7 Ergebnis der Kandidatengenanalyse
Bei der DNA-Probe des erkrankten Kindes wurde zunächst die RPGRIP1-Sequenz
durch Seqlab (Sequence Laborities, Göttingen) bestimmt, da Mutationen des RPGRIP1
als Ursache der autosomal-rezessiven LCA bereits beschrieben wurden.
Die Sequenzierung ergab zwei Sequenzveränderungen des RPGRIP1-Gens: einen
homozygoten c.1111C>T-Austausch in Exon 9, der für eine Nonsense-Mutation
(p.R371*) kodiert und einen heterozygoten Austausch c.3097G>C in Exon 18, der für
eine Missense-Mutation (p.Q1033E) kodiert. Die Segregationsanalyse in der Familie
des Patienten ergab, dass der c.1111C>T Austausch bei den Eltern und bei dreien der
vier nicht betroffenen Geschwister in heterozygotem Zustand vorlag.

50
4 Diskussion
4.1 Bewertung des Patientenmaterials
In dieser Arbeit wurden sieben Mitglieder einer Familie mit einem an LCA erkrankten
Kind untersucht. Da sowohl beide Eltern als auch die vier Geschwister nicht erkrankt
waren, standen ausreichend Referenzproben für eine Kopplungsuntersuchung zur
Verfügung, deren Genotypen mit dem Genotyp des Patienten verglichen werden
konnten. Da nur DNA-Proben aus zwei Generationen und nur eines Betroffenen zur
Verfügung standen, lag der theoretische erreichbare LOD-Score zu niedrig, um mit
Hilfe einer Kopplungsanalyse allein das ursächliche Gen eindeutig zu identifizieren.
Phänotypisch zeigte das erkrankte Kind Symptome einer degenerativen
Netzhauterkrankung. Aufgrund der Konsanguinität der Eltern war ein Gendefekt als
Ursache der Erkrankung wahrscheinlich. Da degenerative Netzhautdegenerationen ein
ähnliches klinisches Bild besitzen, war eine genaue Diagnose aufgrund des Phänotyps
zum Zeitpunkt der Untersuchung nicht möglich. Typisch für eine LCA war vor allem das
frühe Auftreten der Symptome. Sowohl die Konsanguinität der phänotypisch gesunden
Eltern als auch die Tatsache, dass nur eines der fünf Kinder erkrankt war, deutete auf
eine autosomal-rezessive Erkrankung hin.
Die klinischen Untersuchungen wurden zudem dadurch erschwert, dass es sich bei
dem erkranken Kind um einen Säugling handelte. Da der Patient seine Beschwerden
nicht beschreiben konnte, waren die behandelnden Ärzte bei der Diagnose auf
fremdanamnestische Aussagen der Eltern bzw. auf objektive Untersuchungsmethoden
angewiesen. Laut Rücksprache mit dem Kinderarzt war eine erneute Untersuchung
des Kindes im fortgeschrittenen Alter nicht möglich, da die Familie für weitere
Untersuchungen nicht zur Verfügung stand.
4.2 Bewertung der Untersuchungstechnik
Da für die Analyse nur die DNA-Proben des betroffenen Kindes, der Eltern und der vier
nicht betroffenen Geschwistern und keine DNA-Proben weiterer Generationen
zugänglich waren, konnten nur eine begrenzte Anzahl von Meiosen in die LOD-Score-
Berechnung mit einfließen. Zudem gab es nur einen Betroffenen in der Familie. Die
berechneten LOD-Scores konnten daher nur Hinweise auf mögliche
Kopplungsregionen auf Chromosom 7, 8 und 14 liefern.
Die Werte betrugen:
Chromosom 7: LOD-Score 1,8

51
Chromosom 8: LOD-Score 1,7
Chromosom 14: LOD-Score 1,8.
Das initiale Ergebnis der Kopplungsanalyse mit SNP-Markern war als nicht signifikant
zu werten, da kein LOD-Score > 3 vorlag.
Ein Nachteil von SNP-Markern ist ihre im Vergleich zu Mikrosatelliten-Markern
geringere Heterogenität. Um die potentiellen Kopplungsbereiche genauer eingrenzen
zu können, wurden anschließend Kopplungsanalysen mit Mikrosatellitenmarkern
durchgeführt.
Zunächst wurden zur Untersuchung der Kandidatenregionen je zehn
Mikrosatellitenmarker für die Chromosomen 7, 8 und 14 ausgewählt. Da für
Chromosom 7 drei Mikrosatellitenmarker und für Chromosom 8 ein
Mikrosatellitenmarker aufgrund einer unzureichenden Qualität der Amplifikation nicht
verwendet werden konnten, wurden für Chromosom 7 sieben Mikrosatellitenmarker, für
Chromosom 8 neun und für Chromosom 14 zehn Mikrosatellitenmarker typisiert.
Die Anzahl der zur Eingrenzung des Bereichs notwendigen Mikrosatellitenmarker
hängt von der Größe der Kandidatengenregion und der Qualität der
Mikrosatellitenmarker ab. In der vorliegenden Arbeit wurden zunächst zehn Marker
definiert, die die jeweilige Kandidatenregion gleichmäßig abdeckten. Die Anzahl der
Mikrosatellitenmarker erwies sich für Chromosom 8 als ausreichend, da die Lage des
krankheitsverursachenden Gens auf diesem Chromosom klar ausgeschlossen werden
konnte.
Für den zu untersuchenden Bereich auf Chromosom 14 konnte mithilfe der zehn
Mikrosatellitenmarker die Kandidatenregion deutlich eingegrenzt werden.
Lediglich für Chromosom 7 war die Anzahl der ausgewählten Mikrosatellitenmarker zu
gering, da drei Marker unter den publizierten Bedingungen nicht ausreichend zu
amplifizieren waren und drei weitere Marker für die folgende Typisierung keine
zusätzlichen Informationen lieferten. Die verbleibenden vier Marker lagen so verteilt,
dass eine engere Eingrenzung der Kandidatenregion vorgenommen werden konnte.
Ein sicherer Ausschluss konnte nur im direkten Vergleich mit den anderen
Kandidatenregionen geführt werden.
Im Laufe der Kopplungsanalyse ergaben sich Schwierigkeiten, die eng im
Zusammenhang mit der Konsanguinität der Familie standen. Aufgrund der
Verwandtschaft der Eltern bestand nur eine eingeschränkte Heterogenität zwischen
den nachweisbaren Allelen der polymorphen Mikrosatellitenmarker (siehe Chromosom
7). Darüber hinaus waren die Eltern für einige Mikrosatellitenmarker zwar heterozygot,
trugen aber die gleichen Allel-Kombinationen. Daher ließ sich bei den Kindern häufig
nicht nachvollziehen, welches Allel vom Vater bzw. von der Mutter vererbt wurde. Die

52
Kinder zeigten ebenfalls eine eingeschränkte Variabilität der
Mikrosatellitenmarkerallele. Diese sogenannten „nicht-informativen“
Mikrosatellitenmarker erschwerten die Kopplungsanalyse.
Dennoch erwies sich die Nutzung von Mikrosatellitenmarkern im Vergleich zu der
Kopplungsanalyse mit SNP-Markern als vorteilhaft, da bei den multiallelen
Mikrosatellitenmarkern, trotz der Verwandtschaft Unterschiede zu identifizieren waren.
Durch die Kopplungsanalyse mit den Mikrosatellitenmarkern konnte die potentielle
Kopplungsregion von ca. 5 Mbp (Ergebnis der SNP-Markeranalyse) auf ca. 3 Mbp
eingegrenzt werden.
Aufgrund der rasant fortgeschrittenen Entwicklung neuer Sequenzierungstechnologien
ist es aktuell möglich, zahlreiche Sequenzen in einem einzigen Lauf zu bestimmen.
NGS-Techniken wurden zunächst in der Genomforschung genutzt. Wegen der hohen
Qualität der NGS-Daten können diese Technologien auch in der humangenetischen
Diagnostik eingesetzt werden. Durch die erfolgte Automatisierung der Techniken
konnten sowohl die Untersuchungskosten als auch der Zeitaufwand deutlich reduziert
werden [106]. Die Untersuchungsmethode eignet sich besonders für genotypisch und
phänotypisch heterogene Erkrankungen wie die LCA.
Auf der Basis der ständigen Weiterentwicklung der Diagnostik und Forschung auf dem
Gebiet der LCA wurden in den letzten Jahren zahlreiche Mutationen als Ursache der
Erkrankung identifiziert. In einer Arbeit von Suzuki et al. wurden 2014 mithilfe von zwei
Microarrays (Affymetrix resequencing microarrays) 33 Gene sequenziert, die im
Zusammenhang mit der LCA, der autosomal-rezessiven RP und der Stargardt disease
beschrieben wurden [80]. Differentialdiagnostisch werden durch dieses Verfahren
phänotypisch ähnliche Augenerkrankungen mit eingeschlossen.
4.3 Bewertung der Kandidatengene
Die Kopplungsanalysen konnten keine eindeutige Kandidatenregion identifizieren. Die
Region von 21442117 bp bis 26211356 bp auf Chromosom 14 war die
erfolgversprechendste unter den potentiellen Kandidatenregionen, da hier die
gekoppelten Mikrosatellitenmarker den größten zusammenhängenden Abschnitt
abbildeten. In dieser Region wurden auf der Basis der Expressionsmuster und
bekannten Funktionen TPPP2, RPGRIP1, DAD1 und NRL als potentielle
Kandidatengene ausgewählt.
Eine Mutation der Gene DAD1 und TPPP2 erschien eher unwahrscheinlich, da diese
beiden Gene bisher nicht im Zusammenhang mit Netzhautdegenerationen beschrieben
wurden. Dennoch wurde die beiden Gene aufgrund der spezifischen Expression von

53
TPPP2 im ZNS bzw. der funktionellen Beteiligung von DAD1 an der Apoptose mit in
die Auswahl aufgenommen. Bei den Genen RPGRIP1 und NRL ist ein Zusammenhang
mit degenerativen Netzhauterkrankungen wie LCA oder RP bereits bekannt.
Aufgrund der Ergebnisse der Kopplungsanalyse der Mikrosatellitenmarker war eine
Genmutation von TPPP2 beziehungsweise NRL eher unwahrscheinlich. Der Marker
D14S72, der auf dem Chromosom in der Nähe des TPPP2-Gens lag, erzielte einen
LOD-Score von -∞. Bei dem Marker D14S64, der im Bereich des NRL-Gens lag, betrug
der LOD-Score ebenfalls -∞. Gänzlich konnte eine Kopplung aber nicht
ausgeschlossen werden, da sowohl TPPP2 als auch NRL in der Nähe der
Mikrosatellitenmarker D14S283, D14S990 und D14S972 lagen, deren LOD-Scores auf
eine potentielle Kopplung hinwiesen.
Aufgrund ihrer Lokalisation war eine Genmutation von RPGRIP1 oder DAD1
wahrscheinlicher, weil beide Gene nicht im Randbereich der identifizierten
Kopplungsregion, sondern zentral in der Nähe der Mikrosatellitenmarker D14S283,
D14S990 und D14S972 lagen, deren LOD-Score auf eine Kopplung hindeutete.
Da bereits Mutationen im RPGRIP1-Gen als Ursache der autosomal-rezessiven LCA
beschrieben worden waren, wurde dieses Gen als wahrscheinlichster Kandidat
eingestuft.
4.4 Die RPGRIP1-Mutation
Die Sequenzierung des RPGRIP1-Gens ergab bei dem erkrankten Kind einen
homozygoten Basenaustausch, der für eine Nonsense-Mutation in Exon 9 kodiert
[c.1111C>T (p.R371X)]. Der Austausch lag bei beiden Elternteilen und bei drei von vier
Geschwistern im heterozygoten Zustand vor. Zusätzlich wurde ein heterozygoter
Basenaustausch in Exon 18 bei dem erkrankten Kind nachgewiesen [c.3097G>C
(p.Q1033E)]. Dieser Austausch kodiert für eine Missense-Mutation.
Daher stellte sich die Frage, ob die Erkrankung des betroffenen Kindes durch die
homozygote Nonsense-Mutation in Exon 9 oder durch die heterozygote Missense-
Mutation in Exon 18 ausgelöst wurde. Der c.1111C>T Basenaustausch ist zwar in der
dbSNP des NCBI (http://www.ncbi.nlm.nih.gov/SNP/) erfasst (rs375859404), allerdings
spricht eine Allelfrequenz von 0,0002 gegen einen Polymorphismus. Der c.3097G>C
Basenaustausch ist mit einer Allelfrequenz von 0,2953 (rs3748361) deutlich häufiger.
Daher ist der c.1111C>T Basenaustausch als ursächlich anzusehen.
Ein weiteres Indiz war die Segregation des c.1111C>T Basenaustausches, der mit den
phänotypischen Befunden der Familienmitglieder übereinstimmte, auch wenn kein
weiterer Betroffener zur endgültigen Bestätigung herangezogen werden konnte. Bisher

54
bekannte RPGRIP1-Mutationen sind Frameshift-Mutationen (Insertionen, Deletionen),
Splice-site-Mutationen, Nonsense-Mutationen in der RPGR-interacting Domäne und
Missense-Mutationen in der coiled-coil Domäne [23, 34, 80, 90, 92].
Eine Beziehung zwischen der Art der Mutation und einem bestimmten Phänotyp
konnte bisher nicht gezeigt werden [45].
4.5 Aussichten und Nutzen für die Familie
Da es sich bei der Erkrankung des Kindes um einen autosomal-rezessiven Erbgang
handelte, waren beide Allele des RPGRIP1-Gens von der Mutation betroffen. Damit
sind alle potentiellen Nachkommen des Erkrankten heterozygote RPGRIP1-
Mutationsträger. Der Patient sollte daher eine Heirat in der Familie meiden, weil dann
die Wahrscheinlichkeit eine Überträgerin zu ehelichen deutlich erhöht wäre. Die
zukünftige Lebenspartnerin sollte sich aber generell genetisch untersuchen lassen, um
eine heterozygote und damit phänotypisch unauffällige RPGRIP1-Mutation
auszuschließen. Eine solche Untersuchung ist vor dem Hintergrund, dass RPGRIP1-
Mutationen bei bis zu 12,5 % der LCA-Fälle ursächlich sind, von besonderer
Bedeutung [24].
Ausgehend von dem Befund, dass nur die homozygote Stopp-Mutation im RPGRIP1-
Gen eine LCA auslöste, läge die Wahrscheinlichkeit ein erkranktes Kind zu bekommen,
bei einer heterozygoten Stopp-Mutation im RPGRIP1-Gen der Partnerin, unabhängig
vom Geschlecht, bei 50 % (die anderen 50 % wären phänotypisch gesunde
Überträger). Bei einer Partnerin ohne Mutation im RPGRIP1-Gen wären alle Kinder
phänotypisch gesund, aber heterozygote Mutationsträger.
Darüber hinaus sollten auch die heterozygoten Mutationsträger der Familie beraten
werden.
Die heterozygote Nonsense-Mutation wird mit einer Wahrscheinlichkeit von 50 %
weitervererbt. Sollte der Partner ebenfalls Träger einer Mutation im RPGRIP1-Gen
sein, bestände die Möglichkeit ein an LCA erkranktes Kind zu bekommen. Zudem
sollte eine Aufklärung über das vermehrte Auftreten von erblichen Erkrankungen in
konsanguinen Familien stattfinden.
Aktuell ist einer Heilung der LCA noch nicht möglich. Neuere gentherapeutische
Ansätze (Kapitel 1.1.7) haben aber gezeigt, dass möglicherweise in Zukunft mutierte
krankheitsauslösende Gene ersetzt werden können. Eine erfolgversprechende
Therapie für LCA-Patienten mit einer RPGRIP1-Mutation ist aber aktuell noch nicht in
der Entwicklung.

55
4.6 Ausblick
Die Kopplungsanalyse ist eine altbewährte und effektive Methode zur Identifizierung
von ursächlich veränderten Genen bei monogenen Erkrankungen. Die Entwicklung
dichter Markerkarten mit einfach zu typisierenden Markern hat dazu beigetragen die
Identifikation von ursächlichen Genen zu beschleunigen. Dennoch tritt die
Kopplungsanalyse seit der ersten Veröffentlichung des humanen Genoms und der
Entwicklung der Hochdurchsatzsequenzierung hinter die direkte Sequenzierung des
Exoms oder des gesamten Genoms zurück. Der Vorteil der
Hochdurchsatzsequenzierung liegt in der Vielfältigkeit der Daten, die sowohl speziell
auf Veränderungen in bekannten ursächlichen Genen für erbliche
Netzhauterkrankungen untersucht werden können (Paneldiagnostik), als auch auf
Veränderungen in kodierenden Sequenzen des Genoms (Whole Exome Sequencing,
WES), die bislang nicht mit erblichen Netzhauterkrankungen assoziiert waren. Die
Auswertung der so erhobenen Daten führt in den meisten Fällen bereits bei
Einzelpersonen zur ursächlichen Veränderung [24]. So kann inzwischen je nach Form
der erblichen Netzhautdystrophie in bis zu 90 % die genetische Ursache trotz der
genetischen Heterogenität kostengünstig identifiziert werden.
Letztlich kann die Hochdurchsatzsequenzierung für das gesamte Genom (Whole
Genom Sequenzierung, WGS) konfiguriert werden, so dass die Daten auch verwendet
werden können, um Kopplungsanalysen zu initiieren, wenn der direkte Zugang zu
keinem schlüssigen Ergebnis führt. Immerhin bleiben nach Paneldiagnostik und WES
immer noch etwa 10 % der Familien mit erblichen frühkindlichen
Netzhauterkrankungen, wo die ursächlichen Veränderungen nicht zu identifizieren sind.
Die Kosten für die WGS-gestützte Kopplungsanalyse liegen derzeit allerdings noch
höher, als für den in dieser Arbeit gegangenen Weg, sodass eine Kopplungsanalyse
über SNP- und Mikrosatellitenmarker nach wie vor in einigen Spezialfällen der Weg der
Wahl bleibt.

56
5 Zusammenfassung
Ziel der vorliegenden Arbeit war es, die Mutation, die in einer siebenköpfigen
konsanguinen Familie (Eltern, drei Söhne, zwei Töchter) zur Erkrankung des jüngsten
Sohnes an einer Leberschen kongenitalen Amaurose (LCA) geführt hatte, mithilfe einer
genomweiten Kopplungsanalyse zu identifizieren.
Durch Single Nucleotide Polymorphism-(SNP)-Kopplungsanalysen wurden mögliche
Kopplungsregionen auf Chromosom 7, 8 und 14 identifiziert. Da in der Familie nur ein
Kind betroffen war und nur DNA-Proben des Betroffenen, der Geschwister und der
Eltern verfügbar waren, konnte nur eine verhältnismäßig geringe Zahl der Meiosen in
die Logarithmic Odds Ratio-(LOD)-Score-Berechnung für diese Regionen mit
einbezogen werden. Daher wurden diese Bereiche mithilfe von Mikrosattelitenmarker
genauer untersucht.
Durch die SNP-Kopplungsanalyse sowie durch die Kopplungs- und Haplotypenanalyse
der Mikrosatellitenmarker konnte auf Chromosom 14 ein Kandidatenlocus für den
gesuchten Gendefekt eingegrenzt werden.
Die anhand ihres Expressionsmusters bzw. ihrer Funktion ausgewählten
Kandidatengene tubulin polymerization-promoting protein family member 2 (TPPP2),
retinitis pigmentosa GTPase regulator interacting protein 1 (RPGRIP1), defender
against cell death 1 (DAD1) und neural retina leucine zipper (NRL) wurden genauer
betrachtet. Die Sequenzierung von RPGRIP1 führte bei dem erkrankten Kind zur
Identifizierung zweier bekannter Mutationen. Hierbei handelte es sich um eine
homozygote Nonsense-Mutation in Exon 9 (c.1111C>T; p.R371X) und um eine
heterozygote Missense-Mutation in Exon 18 (c.3097G>C; p.Q1033E).
Da in der Familie von einem autosomal-rezessiven Erbgang auszugehen war,
bestätigte das Segregationsverhalten der Nonsense-Mutation in Exon 9 (beide Eltern
und drei Geschwister waren heterozygote Träger, phänotypisch aber gesund) diese als
Ursache der Erkrankung.
Die Kopplungsanalyse ist beim Vorliegen eines ausreichend großen Stammbaums und
bei der Verfügbarkeit von DNA-Proben der Familienmitglieder eine valide Technik zur
Identifikation von krankheitsassoziierten Allelen. Allerdings ist die in der vorliegenden
Arbeit verwendete Strategie (SNP- und nachfolgende Mikrosatellitenanalyse) in Zeiten
der Hochdurchsatzsequenzierung und des Whole Exom Sequencings (WES) veraltet.
Das WES liefert valide Ergebnisse und kann auch beim alleinigen Vorliegen der
Patienten-DNA eingesetzt werden. Auch eine kostengünstige Paneldiagnostik, die
gezielt Gene mit bekannter Assoziation zu Netzhautdegenerationen untersucht, hat im
Fall der LCA eine Nachweisquote von mindestens 80 %.

57
6 Summary
The fundamental idea of this work was to identify the mutation, which inside a seven-
headed consanguine family (parents, three sons, two daughters) was the cause of
Leber's congenital amaurosis (LCA) disease of the youngest son, using a genome-
wide linkage-analysis.
Possible linkage regions were identified through Single nucleotide polymorphism-
(SNP)-based linkage-analysis on chromosome 7, 8 and 14.
Because only one child was affected and only the DNA-samples of the affected child,
of the siblings and of the parents were available, only a comparatively low number of
meiosis could be included in the logarithm odds ratio-(LOD)-score calculation.
Therefore these regions were testet with a microsatellite marker-based linkage-
analysis.
The combination of SNP-based linkage-analysis and microsatellite marker-based
haplotype- and linkage-analysis specified a region on chromosome 14 as the candidate
locus for the gene defect.
Bases on their cell-type specific expression and/or on their function tubulin
polymerization-promoting protein family member 2 (TPPP2), retinitis pigmentosa
GTPase regulator interacting protein 1 RPGRIP1, defender against cell death 1 (DAD1)
and neural retina leucine zipper (NRL) were selected as candidate genes. The
sequencing of RPGRIP1 identified two well-known mutations inside the genome of the
affected child: one homozygous nonsense-mutation in exon 9 (c.1111C>T; p.R371X)
and one heterozygous missense-mutation in exon 18 (c.3097G>C; p.Q1033E).
Because of the assumption of an autosomal recessive succession inside the family, the
segregation of the nonsense-mutation in exon 9 (both parents and three siblings were
heterozygous carriers but phenotypic unaffected) confirmed this mutation as the cause
of the disease.
The linkage analysis is a valid technology for the identification of disease-related
alleles, if an adequate large family-tree and DNA-samples of family-members are
available. However the strategy used in this work (an SNP- followed by microsatellite-
analysis) is rather outdated in times of high-throughput-sequencing and whole exome
sequencing (WES)
The WES provides valid results while being more cost efficient and can be used, if only
the DNA of the patient is available. Moreover a cost efficient panel-diagnosis,
examining genes with a well-know association to retinal degeneration, shows in case of
LCA at least an 80 % detection-rate.

58
7 Abbildungsverzeichnis
Abbildung 1: Stammbaum der Familie 967 ..................................................... 11
Abbildung 2: Verteilung der Marker auf Chromosom 7, 20107333 – 30831019
bp; SNP definierter Bereich: 20198898-28923785 bp ...................................... 26
Abbildung 3: Verteilung der Marker auf Chromosom 8, 13781058 - 26543124
bp; SNP definierter Bereich: 20347695 - 23045446 bp .................................... 26
Abbildung 4: Verteilung der Marker auf Chromosom 14, 21371146 - 27724479
bp; SNP definierter Bereich: 21452992 – 26740590 bp ................................... 27
Abbildung 5: Auswertung des Human Mapping 50K Array Xba 240 von
Affymetrix mit MERLIN ..................................................................................... 32
Abbildung 6: Stammbaum der Familie 967 mit den Haplotypen der
Mikrosatellitenmarker auf Chromosom 7 .......................................................... 36
Abbildung 7: Stammbaum der Familie 967 mit den Haplotypen der
Mikrosatellitenmarker auf Chromosom 8 .......................................................... 37
Abbildung 8: Stammbaum der Familie 967 mit den Haplotypen der
Mikrosatellitenmarker auf Chromosom 14 ........................................................ 38
Abbildung 9: Ergebnisse der Kopplungsanalyse mit den SNP- und
Mikrosatellitenmarkern auf Chromosom 7 ........................................................ 44
Abbildung 10: Ergebnisse der Kopplungsanalyse mit den SNP- und
Mikrosatellitenmarkern auf Chromosom 8 ........................................................ 45
Abbildung 11: Ergebnisse der Kopplungsanalyse mit den SNP- und
Mikrosatellitenmarkern auf Chromosom 14 ...................................................... 46

59
8 Tabellenverzeichnis
Tabelle 1: Bekannte Gene mit ursächlichen Mutationen der LCA ...................... 4
Tabelle 2: Geräte ............................................................................................. 12
Tabelle 3: Chemikalien .................................................................................... 13
Tabelle 4: Molekularbiologische Reagenzien ................................................... 13
Tabelle 5: Zusammensetzung des PCR-Standardansatzes ............................ 16
Tabelle 6: Zusammensetzung eines PCR-Mastermix mit einer Ansatzgröße von
62,5 µl ............................................................................................................... 16
Tabelle 7: Temperaturstufen der Gradienten PCR .......................................... 17
Tabelle 8: Parameter der Gradienten PCR ...................................................... 17
Tabelle 9: Eingesetzte Mikrosatellitenmarker auf Chromosom 7 ..................... 25
Tabelle 10: Eingesetzte Mikrosatellitenmarker auf Chromosom 8 ................... 26
Tabelle 11: Eingesetzte Mikrosatellitenmarker auf Chromosom 14 ................. 27
Tabelle 12: Zusammenfassung der klinischen Untersuchungsergebnisse des
Patienten .......................................................................................................... 31
Tabelle 13: LOD-Scores der Kopplungsanalyse mittels SNP-Marker .............. 32
Tabelle 14: Abweichungen der Mastermixzusammensetzung, optimale PCR
Temperatur und Annealingzeit für die Amplifikation der Mikrosatellitenmarker 34
Tabelle 15: Stammbaumdatei zur Kopplungsanalyse am Chromosom 7 ....... 40
Tabelle 16: Stammbaumdatei zur Kopplungsanalyse am Chromosom 8 ....... 40
Tabelle 17: Stammbaumdatei zur Kopplungsanalyse am Chromosom 14 ..... 41
Tabelle 18: Parameterdatei zur Kopplungsanalyse am A) Chromosom 7,
B) Chromosom 8, C) Chromosom 14 ............................................................... 42
Tabelle 19: Kartierungsdatei zur Kopplungsanalyse am A) Chromosom 7,
B) Chromosom 8, C) Chromosom 14 .............................................................. 42
Tabelle 20: Allelfrequenzdatei .......................................................................... 43
Tabelle 21: Kandidatengene auf Chromosom 14 ............................................. 47

60
9 Abkürzungsverzeichnis
°C Grad Celsius
µg Mikrogramm
µl Mikroliter
2FSP Zweifarbenschwellenperimetrie
A Adenin
AAV-Vektor Adeno-assoziierter viraler Vektor
ADAMTS1 ADAM metallopeptidase with thrombospondin type 1 motif, 18
AIPL1 Aryl Hydrocarbon Receptor Interacting Protein-Like 1
ALMS1 Alstrom syndrome protein 1
bp Basenpaare
bzw. beziehungsweise
C Cytosin
CC Connecting cilium
C1ORF36 Retinal degeneration 3
CEP290 Centrosomal protein 290kDa
cM centi-Morgan
CNGA3 Cyclic nucleotide gated channel alpha 3
CRB1 Crumbs family member 1
CRX Cone rod homeobox
C-terminal Carboxy-terminal
ddH2O doppeltdestilliertes Wasser
ddNTP Didesoxyribonukleosid-Triphosphat
dNTP Desoxyribonukleosid-Triphosphat
DNA Desoxyribonucleic-acid
EDTA Ethylendiamintetraacetat
EOSRD Early-Onset Severe Retinal Dystrophie
ERG Elektroretinogramm
EtBr Ethidiumbromid
EtOH Ethanol
FAF Fundusautofluoreszenz
G Guanin
g Gramm
GDF6 Growth differentiation factor 6
GUCY2D Guanylate-cyclase-2D

61
IFT intraflagellarer Transport
IMPDH1 Inosine-5'-monophosphate-Dehydrogenase-1
IQCB1 IQ motif containing B1
kb Kilobase
KCNJ13 Potassium channel, inwardly rectifying subfamily J, member13
LCA Lebersche kongenitale Amaurose
LOD Logarithmic Odds Ratio
LRAT Lecithin retinol acyltransferase
Mbp Megabasenpaare
MDC Max-Delbrück-Center for Molecular Medicine
MERTK MER proto-oncogene, tyrosine kinase
min Minute(n)
ml Milliliter
mM Millimol
mRNA messenger Ribonucleic-acid
NGS Next Generation Sequencing
nm Nanometer
NMR Nuclear Magnetic Resonance
NPHP4 Nephrocystin-4
NRL Neural retina leucine zipper
OCT optische Kohärenztomographie
OD optische Dichte
PCR Polymerase Chain Reaction
PDE6C Phosphodiesterase 6C, cGMP-specific, cone, alpha prime
pH potentia hydrogenii
PKU Phenylketonurie
PRPH2 peripherin 2 (retinal degeneration, slow)
QTL Quantitative Trait Locus
RDH12 retinol dehydrogenase 12
RP Retinitis Pigmentosa
RPE retinales Pigmentepithel
RPE65 Retinal pigment epithelium-specific 65 kDa protein
RPG RP GTPase regulator
RPGRIP1 Retinitis Pigmentosa GTPase Regulator Interacting Protein 1
SECORD Severe Early Childhood Onset Retinal Dystrophy
s Sekunde(n)
SNP Single Nucleotide Polymorphism

62
SPATA7 Spermatogenesis Associated 7
T Thymin
TBE TRIS-Borat-EDTA
TPPP2 Tubulin polymerization-promoting protein family member 2
TRIS Tris(hydroxymethyl)-aminomethan
TULP1 tubby like protein 1
u.a. unter anderem
w/v Weight/Volume
WES Whole Exome Sequencing
WGS Whole Genom Sequencing
z.B. zum Beispiel

63
10 Literaturverzeichnis
[1] Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin-rapid
analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 30:97-
101
[2] Asai-Coakwell M, March L, Dai XH, Duval M, Lopez I, French CR, Famulski J,
De Baere E, Francis PJ, Sundaresan P, Sauvé Y, Koenekoop RK, Berry FB, Allison
WT, Waskiewicz AJ, Lehmann OJ (2013) Contribution of growth differentiation
factor 6-dependent cell survival to early-onset retinal dystrophies. Hum Mol Genet.
22:1432-1442
[3] Augustin, AJ, Collins JF (2001) Augenheilkunde. 2. Auflage, Berlin, Heidelberg,
New York: Springer Verlag, 389-394
[4] Azadi S, Molday LL, Molday RS (2010) RD3, the protein associated with Leber
congenital amaurosis type 12, is required for guanylate cyclase trafficking in
photoreceptor cells. Proc Natl Acad Sci U S A. 107:21158-21163
[5] Bainbridge JW, Tan MH, Ali RR (2006) Gene therapy progress and prospects:
the eye. Gene Ther. 13:1191-1197
[6] Bellingrath JS, Fischer MD (2015) Gentherapie als Behandlungskonzeot für
erbliche Netzhauterkrankungen. Der Ophtalmologe 112:720-727
[7] Bessant DA, Payne AM, Mitton KP, Wang QL, Swain PK, Plant C, Bird AC, Zack
DJ, Swaroop A, Bhattacharya SS (1999) A mutation in NRL is associated with
autosomal dominant retinitis pigmentosa. Nat Genet. 21:355-356
[8] Bowne SJ, Liu Q, Sullivan LS, Zhu J, Spellicy CJ, Rickman CB, Pierce EA,
Daiger SP (2006) Why do mutations in the ubiquitously expressed housekeeping
gene IMPDH1 cause retina-specific photoreceptor degeneration? Invest
Ophthalmol Vis Sci. 47:3754-3765
[9] Burk A, Burk R (1996) Checkliste Augenheilkunde. 3. überarbeitete und
erweiterte Auflage, Stuttgart, New York: Georg Thieme Verlag, 41, 192, 202, 304-
305, 365

64
[10] Buselmaier WT, Tariverdian G (2006) Humangenetik. 4.Auflage, Heidelberg:
Springer Verlag, 6-11, 56-57
[11] Cideciyan AV, Jacobson SG, Beltran WA, Sumaroka A, Swider M, Iwabe S,
Roman AJ, Olivares MB, Schwartz SB, Komáromy AM, Hauswirth WW, Aguirre GD
(2013) Human retinal gene therapy for Leber congenital amaurosis shows
advancing retinal degeneration despite enduring visual improvement. Proc Natl
Acad Sci U S A. 110:E517-E525
[12] Cideciyan AV, Rachel RA, Aleman TS, Swider M, Schwartz SB, Sumaroka A,
Roman AJ, Stone EM, Jacobson SG, Swaroop A (2011) Cone photoreceptors are
the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290)
blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the
human retinal ciliopathy. Hum Mol Genet. 20:1411-1423
[13] Coene KL, Mans DA, Boldt K, Gloeckner CJ, van Reeuwijk J, Bolat E, Roosing
S, Letteboer SJ, Peters TA, Cremers FP, Ueffing M, Roepman R (2011) The
ciliopathy-associated protein homologs RPGRIP1 and RPGRIP1L are linked to
cilium integrity through interaction with Nek4 serine/threonine kinase. Hum Mol
Genet. 20:3592-3605
[14] Colella P, Auricchio A (2012) Gene therapy of inherited retinopathies: a long
and successful road from viral vectors to patients. Hum Gene Ther. 23: 796-807
[15] Conley SM, Naash MI (2010) Nanoparticles for retinal gene therapy. Prog
Retin Eye Res. 29:376-397
[16] Coppieters F, Casteels I, Meire F, De Jaegere S, Hooghe S, van Regemorter
N, Van Esch H, Matuleviciene A, Nunes L, Meersschaut V, Walraedt S, Standaert
L, Coucke P, Hoeben H, Kroes HY, Vande Walle J, de Ravel T, Leroy BP, De
Baere E (2010) Genetic screening of LCA in Belgium: predominance of CEP290
and identification of potential modifier alleles in AHI1 of CEP290-related
phenotypes. Hum Mutat. 31:E1709-1766
[17] Cremers FP, van den Hurk JA, den Hollander AI (2002) Molecular genetics of
Leber congenital amaurosis. Hum Mol Genet. 11:1169-1176
[18] den Hollander AI, Roepman R, Koenekoop RK, Cremers FP (2008) Leber
congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye
Res. 27:391-419

65
[19] den Hollander AI, Heckenlively JR, van den Born LI, de Kok YJ, van der Velde-
Visser SD, Kellner U, Jurklies B, van Schooneveld MJ, Blankenagel A,
Rohrschneider K, Wissinger B, Cruysberg JR, Deutman AF, Brunner HG,
Apfelstedt-Sylla E, Hoyng CB, Cremers FP (2001) Leber congenital amaurosis and
retinitis pigmentosa with Coats-like exudative vasculopathy are associated with
mutations in the crumbs homologue 1 (CRB1) gene. Am J Hum Genet. 69:198-203
[20] den Hollander AI, Koenekoop RK, Mohamed MD, Arts HH, Boldt K, Towns KV,
Sedmak T, Beer M, Nagel-Wolfrum K, McKibbin M, Dharmaraj S, Lopez I, Ivings L,
Williams GA, Springell K, Woods CG, Jafri H, Rashid Y, Strom TM, van der Zwaag
B, Gosens I, Kersten FF, van Wijk E, Veltman JA, Zonneveld MN, van Beersum
SE, Maumenee IH, Wolfrum U, Cheetham ME, Ueffing M, Cremers FP, Inglehearn
CF, Roepman R (2007) Mutations in LCA5, encoding the ciliary protein lebercillin,
cause Leber congenital amaurosis. Nat Genet. 39:889-895
[21] den Holllander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE,
Zonneveld MN, Strom TM, Meitinger T, Brunner HG, Hoyng CB, van den Born LI,
Rohrschneider K, Cremers FP (2006) Mutations in the CEP290 (NPHP6) gene are
a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 79:556-561
[22] Dharmaraj SR, Silva ER, Pina AL, Li YY, Yang JM, Carter CR, Loyer MK, El-
Hilali HK, Traboulsi EK, Sundin OK, Zhu DK, Koenekoop RK, Maumenee IH (2000)
Mutational analysis and clinical correlation in Leber congenital amaurosis.
Ophthalmic Genet. 21:135-150
[23] Dryja TP, Adams SM, Grimsby JL, McGee TL, Hong DH, Li T, Andréasson S,
Berson EL (2001) Null RPGRIP1 alleles in patients with Leber congenital
amaurosis. Am J Hum Genet. 68:1295-1298
[24] Eisenberger T, Neuhaus C, Khan AO, Decker C, Preising MN, Friedburg C,
Bieg A, Gliem M, Charbel Issa P, Holz FG, Baig SM, Hellenbroich Y, Galvez A,
Platzer K, Wollnik B, Laddach N, Ghaffari SR, Rafati M, Botzenhart E, Tinschert S,
Börger D, Bohring A, Schreml J, Körtge-Jung S, Schell-Apacik C, Bakur K, Al-
Aama JY, Neuhann T, Herkenrath P, Nürnberg G, Nürnberg P, Davis JS, Gal A,
Bergmann C, Lorenz B, Bolz HJ (2013) Increasing the yield in targeted next-
generation sequencing by implicating CNV analysis, non-coding exons and the
overall variant load: the example of retinal dystrophies. PLoS One. 8:e78496
[25] Falk MJ, Zhang Q, Nakamaru-Ogiso E, Kannabiran C, Fonseca-Kelly Z,
Chakarova C, Audo I, Mackay DS, Zeitz C, Borman AD, Staniszewska M, Shukla

66
R, Palavalli L, Mohand-Said S, Waseem NH, Jalali S, Perin JC, Place E, Ostrovsky
J, Xiao R, Bhattacharya SS, Consugar M, Webster AR, Sahel JA, Moore AT,
Berson EL, Liu Q, Gai X, Pierce EA (2012) NMNAT1 mutations cause Leber
congenital amaurosis. Nat.Genet. 44:1040-1045
[26] Freund CL, Gregory-Evans CY, Furukawa T, Papaioannou M, Looser J, Ploder
L, Bellingham J, Ng D, Herbrick JA, Duncan A, Scherer SW, Tsui LC, Loutradis-
Anagnostou A, Jacobson SG, Cepko CL, Bhattacharya SS, McInnes RR (1997)
Cone-rod dystrophy due to mutations in a novel photoreceptor - spezific homebox
gene (CRX) essential for maintenance of the photoreceptor. Cell 91:543-553
[27] Freund CL, Wang QL, Chen S, Muskat BL, Wiles CD, Sheffield VC, Jacobson
SG, McInnes RR, Zack DJ, Stone EM (1998) De novo mutations in the CRX
homeobox gene accociated with Leber congenital amaurosis. Nat Genet. 18:311-
312
[28] Friedman JS, Chang B, Kannabiran C, Chakarova C, Singh HP, Jalali S,
Hawes NL, Branham K, Othman M, Filippova E, Thompson DA, Webster AR,
Andréasson S, Jacobson SG, Bhattacharya SS, Heckenlively JR, Swaroop A
(2006) Premature truncation of an novel protein, RD3, exhibiting subnuclear
localizaion is associated with retinal degeneration. Am J Hum Genet. 79:1059-1070
[29] Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG, Apfelstedt-Sylla E,
Vollrath D (2000) Mutations in MERTK, the human orthologue of the RCS rat retinal
dystrophy gene, cause retinitis pigmentosa. Nat Genet. 26:270–271
[30] Gearhart PM, Gearhart C, Thompson DA, Petersen-Jones SM (2010)
Improvement of visual performance with intravitreal administration of 9-cis-retinal in
Rpe65-mutant dogs. Arch Ophthalmol. 128:1442-1448
[31] Gerber S, Perrault I, Hanein S, Barbet F, Ducroq D, Ghazi I, Martin-Coignard
D, Leowski C, Homfray T, Dufier JL, Munnich A, Kaplan J, Rozet JM (2001)
Complete exon-intron structure of the RPGR-in-teracting protein (RPGRIP1) gene
allows the identification of mutations underlying Leber congenital amaurosis. Eur J
Hum Genet. 9:561-571
[32] Gregory-Evans K, Kelsell RE, Gregory-Evans CY, Downes SM, Fitzke FW,
Holder GE, Simunovic M, Mollon JD, Taylor R, Hunt DM, Bird AC, Moore AT (2000)
Autosomal dominant cone-rod retinal dystrophy (CORD6) from heterozygous

67
mutation of GUCY2D, which encodes retinal guanylate cyclase. Ophthalmology.
107:55-61
[33] Hagstrom SA, North MA, Nishina PL, Berson EL, Dryja TP (1998) Recessive
mutations in the gene encoding the tubby-like protein TULP1 in the patients with
retinitis pigmentosa. Nat Genet. 18:174-176
[34] Hanein S, Perrault I, Gerber S, Tanguy G, Barbet F, Ducroq D, Calvas P,
Dollfus H, Hamel C, Lopponen T, Munier F, Santos L, Shalev S, Zafeiriou D, Dufier
JL, Munnich A, Rozet JM, Kaplan J (2004) Leber congenital amaurosis:
comprehensive survey of the genetic heterogeneity, refinement of the clinical
definition, and genotype-phenotype correlations as a strategy for molecular
diagnosis. Hum Mutat. 23:306-317
[35] Heckenlively JR, Yoser SL, Friedman LH, Oversier JJ (1988) Clinical Findings
and common symptoms in retinitis pigmentosa. Am J Ophthalmol. 105:504-511
[36] Hernan I, Gamundi MJ, Borràs E, Maseras M, García-Sandoval B, Blanco-
Kelly F, Ayuso C, Carballo M (2012) Novel p.M96T variant of NRL and shRNA-
based suppression and replacement of NRL mutants associated with autosomal
dominant retinitis pigmentosa. Clin Genet. 82:446-452
[37] Hong DH, Yue G, Adamian M, Li T (2001) Retinitis pigmentosa GTPase
regulator (RPGRr)-interacting protein is stably associated with the photoreceptor
ciliary axoneme and anchors RPGR to the connecting cilium. J Biol Chem.
276:12091-12099
[38] Hosch J, Lorenz B, Stieger K (2010) RPGR: Role in the photoreceptor cilium,
human retinal disease, and gene therapy. Ophthalmic Genet. 32:1-11
[39] Jacobson SG, Cideciyan AV, Roman AJ, Sumaroka A, Schwartz SB, Heon E,
Hauswirth WW (2015). Improvement and decline in vision with gene therapy in
childhood blindness. N Engl J Med. 372:1920-1926
[40] Jacobson SG, Cideciyan AV, Huang Y, Hanna DB, Freund CL, Affatigato LM,
Carr RE, Zack DJ, Stone EM, McInnes RR (1998) Renital degenerations with
truncation mutations in the cone-rod homeobox (CRX) gene. Invest Ophthalmol Vis
Sci. 39:2417-2426
[41] Jacobson SG, Cideciyan AV, Aleman TS, Pianta MJ, Sumaroka A, Schwartz
SB, Smilko EE, Milam AH, Sheffield VC, Stone EM (2003) Crumbs homolog 1

68
(CRB1) mutations result in a thick human retina with abnormal lamination. Hum Mol
Genet. 12:1073-1078
[42] Janecke AR, Thompson DA, Utermann G, Becker C, Hübner CA, Schmid E,
McHenry CL, Nair AR, Rüschendorf F, Heckenlively J, Wissinger B, Nürnberg P,
Gal A (2004) Mutations in RDH12 encoding a photoreceptor cell retinol
dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet.
36:850-854
[43] Jo DH, Lee TG, Kim JH (2011) Nanotechnology and nanotoxicology in
retinopathy. Int J Mol Sci. 12:8288-8301
[44] Johnson CJ, Berglin L, Chrenek MA, Redmond TM, Boatright JH, Nickerson
JM (2008) Technicel brief: subretinal injection and electroporation into adult mouse
eyes. Mol Vis. 14:2211-2226
[45] Khan AO, Abu-Safieh L, Eisenberger T, Bolz HJ, Alkuraya FS (2013) The
RPGRIP1-related retinal phenotype in children. Br J Ophthalmol 97:760-764
[46] Knippers R (2006) Molekulare Genetik. 9. Auflage, Stuttgart, New York: Georg
Thieme Verlag, 312-314, 505-514
[47] Koenekoop RK, Sui R, Sallum J, van den Born LI, Ajlan R, Khan A, den
Hollander AI, Cremers FP, Mendola JD, Bittner AK, Dagnelie G, Schuchard RA,
Saperstein DA (2014) Oral 9-cis retinoid for childhood blindness due to Leber
congenital amaurosis caused by RPE65 or LRAT mutations: an open-label phase
1b trial. Lancet 384:1513-1520
[48] Kulke MH, Freed E, Chiang DY, Philips J, Zahrieh D, Glickman JN, Shivdasani
RA (2008) High-resolution analysis of genetic alterations in small bowel carcinoid
tumors reveals areas of recurrent amplification and loss. Genes Chromosomes
Cancer. 47:591-603
[49] Lakowski J, Baron M, Bainbridge J, Barber AC, Pearson RA, Ali RR, Sowden
JC (2010) Cone and rod photoreceptor transplantation in models of the retinopathy
Leber congenital amaurosis using flow-sorted Crx-positive donor cells. Hum Mol
Genet. 19:4545-4559
[50] Leber T (1869) Über Retinitis pigmentosa und angeborene Amaurose. Albrecht
von Graefes Arch Ophtalmol. 15:1-25

69
[51] Leber T (1871) Über anormale Formen der Retinitis pigmentosa. Albrecht von
Graefes Arch Ophthalmol. 17:314-341
[52] Lewin B (1998) Molekularbiologie der Gene. Heidelberg, Berlin: Spektrum
Akademischer Verlag GmbH, 59, 173-174, 831-834
[53] Lorenz B, Preising M (2003) Frühkindliche schwere Netzhautdystrophien.
Aktuelle Aspekte zur Leberschen kongenitalen Amaurose. Zeitschrift für praktische
Augenheilkunde & augenärztliche Fortbildung. 24:367-372
[54] Lorenz B, Gyürüs P, Preising M, Bremser D, Gu S, Andrassi M, Gerth C, Gal A
(2000) Early-onset severe rod-cone dystrophy in young children with RPE65
mutations. Invest Ophtalmol Vis Sci 41:2735-2742
[55] Lorenz B, Wabbels B, Wegscheider E, Hamel CP, Drexler W, Preising MN
(2004) Lack of fundus autofluorescence to 488 nanometers from childhood on in
patients with early-onset severe retinal dystrophie accociated with mutations ins
RPE65. Ophtalmologiy 111:1585-1594
[56] Lottspeich FE, Engels JW (2006) Bioanalytik. 2. Auflage, München: Spektrum
Akademischer Verlag, 952
[57] Louis Jeune V, Joergensen JA, Hajjar RJ, Weber T (2013). Pre-existing anti-
adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum Gene
Ther Methods 24:59-67
[58] Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr, Mingozzi F, Bennicelli J,
Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR,
Konkle B, Stone E, Sun J, Jacobs J, Dell'Osso L, Hertle R, Ma JX, Redmond TM,
Zhu X, Hauck B, Zelenaia O, Shindler KS, Maguire MG, Wright JF, Volpe NJ,
McDonnell JW, Auricchio A, High KA, Bennett J (2008). Safety and efficacy of gene
transfer for Leber´s congenital. N Engl J Med 358:2240-2248
[59] Marlhens F, Bareil C, Griffoin JM, Zrenner E, Amalric P, Eliaou C, Liu SY,
Harris E, Redmond TM, Arnaud B, Claustres M, Hamel CP (1997) Mutations in
RPE65 cause Leber´s congenital amaurosis. Nat Genet. 17:139-141
[60] Nishiguchi KM, Friedman JS, Sandberg MA, Swaroop A, Berson EL, Dryja TP
(2004) Recessive NRL mutations in patients with clumped pigmentary retinal
degeneration and relative preservation of blue cone function. Proc Natl Acad Sci U
S A. 101:17819-17824

70
[61] Papon JF, Perrault I, Coste A, Louis B, Gérard X, Hanein S, Fares-Taie L,
Gerber S, Defoort-Dhellemmes S, Vojtek AM, Kaplan J, Rozet JM, Escudier E
(2010). Abnormal respiratory cilia in non-syndromic Leber congenital amaurosis
with CEP290 mutations. J Med Genet. 47:829-834
[62] Paunescu K, Preising MN, Friedburg C, Rosenberg T, Lorenz B (2006)
Variation of phenotype in patients with compound heterozygous mutations of
RetGC1 depending on the affected domains. Invest Ophtalmol Vis Sci. 47:5802
[63] Pawlyk BS, Smith AJ, Buch PK, Adamian M, Hong DH, Sandberg MA, Ali RR,
Li T (2005). Gene replacement therapy rescues photoreceptor degeneration in a
murine model of Leber congenital amaurosis lacking RPGRIP. Invest Ophthalmol
Vis Sci. 46:3039-3045
[64] Peluso I, Conte I, Testa F, Dharmalingam G, Pizzo M, Collin RW, Meola N,
Barbato S, Mutarelli M, Ziviello C, Barbarulo AM, Nigro V, Melone MA; European
Retinal Disease Consortium, Simonelli F, Banfi S (2013). The ADAMTS18 gene is
responsible for autosomal recessive early onset severe retinal dystrophy. Orphanet
J Rare Dis. 8:16
[65] Perrault I, Hanein S, Gerber S, Barbet F, Dufier JL, Munnich A, Rozet JM,
Kaplan J (2003) Evidence of autosomal dominant Leber congenital amaurosis
(LCA) underlain by a CRX heterozygous null allele. J Med Genet. 40: e90
[66] Perrault I, Hanein S, Gerber S, Barbet F, Ducroq D, Dollfus H, Hamel C, Dufier
JL, Munnich A, Kaplan J, Rozet JM (2004) Retinal Dehydrogenase 12 /RDH12
mutations in Leber congenital amaurosis. Am J Hum Genet. 75:639-646
[67] Perrault I, Rozet JM, Gerber S, Kelsell RE, Souied E, Cabot A, Hunt DM,
Munnich A, Kaplan J (1998) A retGC-1 mutation in autosomal dominant cone-rod
dystrophy. Am J Hum Genet. 63:651-654
[68] Perrault, I. Hanein S, Gerard X, Delphin N, Fares-Taie L, Gerber S,
Pelletier V, Mercé E, Dollfus H, Puech B, Defoort-Dhellemmes S, Petersen
MD, Zafeiriou D, Munnich A, Kaplan J, Roche O, Rozet JM (2010) Spectrum
of SPATA7 mutations in Leber congenital amaurosis and delineation of the
associated phenotype. Hum Mutat. 31E1241-E1250
[69] Perrault I, Rozet JM, Ghazi I, Leowski C, Bonnemaison M, Gerber S, Ducroq
D, Cabot A, Souied E, Dufier JL, Munnich A, Kaplan J (1999) Different functional

71
outcome of RetGC1 and RPE65 gene mutatinos in Leber congenital amaurosis.
Am J Hum Genet 64: 1225-1228
[70] Poeggel G, Hammelehle R (2005) Kurzlehrbuch Biologie. Stuttgart: Georg
Thieme Verlag
[71] Preising MN, Paunescu K, Friedburg C, Lorenz B (2007) Genetische und
klinische Heterogenität bei LCA-Patienten: Das Ende der Einheitlichkeit. Der
Ophtalmologe. 104:490-498
[72] Preising, MN, Rosenberg, T., Kellner, U., Meschede, D., Brauer, U., Lorenz, B.
(2001). Mutation Screening of RetGC1 and RPE65 in Patients with LCA and RP.
Invest. Ophtalmol. Vis. Sci. 42:644
[73] Read A, Donnai D (2008). Angewandte Humangenetik. 1. Auflage, Berlin, New
York: Walter de Gruyter, 13-17, 90-91, 234, 228-229, 317-319
[74] Robert B,H. , Ahmed ZM, Corrêa ZM, Sisk RA (2012) Gene therapy for Leber
congenital amaurosis: advances and future directions. Graefes Arch Clin Exp
Ophthalmol. 250:1117-1128
[75] Roepman R, Letteboer SJ, Arts HH, van Beersum SE, Lu X, Krieger E,
Ferreira PA, Cremers FP (2005) Interaction of nephrocystin-4 and RPGRIP1 is
disrupted by nephronophthisis or Leber congenital amaurosis-associated
mutations. Proc Natl Acad Sci U S A. 102:18520-18525
[76] Sergouniotis PI, Davidson AE, Mackay DS, Li Z, Yang X, Plagnol V, Moore AT,
Webster AR (2011) Recessive mutations in KCNJ13, encoding an inwardly
rectifying potassium channel subunit, cause leber congenital amaurosis. Am J Hum
Genet. 89:183-190
[77] Sohocki MM, Bowne SJ, Sullivan LS, Blackshaw S, Cepko CL, Payne AM,
Bhattacharya SS, Khaliq S, Qasim Mehdi S, Birch DG, Harrison WR, Elder FF,
Heckenlively JR, Daiger SP (2000) Mutations in a new photoreceptor-pineal gene
on 17p cause Leber congenital amaurosis. Nat Genet. 24:79-83
[78] Sohocki MM, Perrault I, Leroy BP, Payne AM, Dharmaraj S, Bhattacharya SS,
Kaplan J, Maumenee IH, Koenekoop R, Meire FM, Birch DG, Heckenlively JR,
Daiger SP (2000) Prevalence of AIPL1 mutations in in herited retinal degenerative
diesease. Mol Genet Metab. 70:142-150

72
[79] Stein L, Roy K, Lei L, Kaushal S (2011) Clinical gene therapy for the treatment
of RPE65-associated Leber congenital amaurosis. Expert Opin Biol Ther. 11:429-
439
[80] Suzuki T, Fujimaki T, Yanagawa A, Arai E, Fujiki K, Wada Y, Murakami A
(2014) A novel exon 17 deletion mutation of RPGRIP1 gene in two siblings with
Leber congenital amaurosis. Jpn J Ophthalmol. 58:528-535
[81] Swaroop A, Wang QL, Wu W, Cook J, Coats C, Xu S, Chen S, Zack DJ,
Sieving PA (1999) Leber congenital amaurosis caused by a homozygous mutation
(R90W) in the homeodomain of the retinal transcription factor CRX: direct evidence
for the involvement of CRX in the development of photoreceptor function. Hum Mol
Genet. 8:299-305
[82] Thompson DA, Li Y, McHenry CL, Carlson TJ, Ding X, Sieving PA, Apfelstedt-
Sylla E, Gal A (2001) Mutations in the gene encoding lecithin retinol acyltransferase
are associated with early-onset severe retinal dystrophy. Nat Genet. 28:123-124
[83] Tirian L, Hlavanda E, Oláh J, Horváth I, Orosz F, Szabó B, Kovács J, Szabad
J, Ovádi J (2003) TPPP/p25 promotes tubulin assemblies and blocks mitotic
spindle formation. Proc Natl Acad Sci U S A. 100:13976-13981
[84] von Rückmann A, Fitzke FW, Bird AC (1997) Fundus autofluorescence in age-
related macular disease imaged with a laser scanning ophthalmoscope. Invest
Ophthalmol Vis Sci. 38:478-486
[85] Wang X, Wang H, Sun V, Tuan HF, Keser V, Wang K, Ren H, Lopez I,
Zaneveld JE, Siddiqui S, Bowles S, Khan A, Salvo J, Jacobson SG, Iannaccone A,
Wang F, Birch D, Heckenlively JR, Fishman GA, Traboulsi EI, Li Y, Wheaton D,
Koenekoop RK, Chen R (2013) Comprehensive molecular diagnosis of 179 Leber
congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next
generation sequencing. J Med Genet. 50:674-688
[86] Wang X, Wang H, Cao M, Li Z, Chen X, Patenia C, Gore A, Abboud EB, Al-
Rajhi AA, Lewis RA, Lupski JR, Mardon G, Zhang K, Muzny D, Gibbs RA, Chen R
(2011) Whole-exome sequencing identifies ALMS1, IQCB1, CNGA3, and MYO7A
mutations in patients with Leber congenital amaurosis. Hum Mutat. 32:1450-1459
[87] Zhao Y, Hong DH, Pawlyk B, Yue G, Adamian M, Grynberg M, Godzik A, Li T
(2003) The retinitis pigmentosa GTPase regulator (RPGR)- interacting protein:

73
subserving RPGR function and participating in disk morphogenesis. Proc Natl Acad
Sci U S A. 100:3965-3970
[88]
http://campus.doccheck.com/uploads/tx_dcmedstudscripts/7260_agarosegel_elektr
ophorese_folien_.pdf (abgerufen am 29. 04.2010)
[89]
http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/5/bc/vlus/gentechnik/prak_dna
_isolierung.vlu/Page/vsc/de/ch/5/bc/gentechnik/methoden/reinheit_ns/reinheit_ns.v
scml.html (abgerufen am 29.04.2010)
[90]
http://www.hgmd.cf.ac.uk/ac/gene.php?gene=RPGRIP1 (abgerufen am
29.10.2015)
[91]
http://www.laborpraxis.vogel.de/analytik/bioanalytik/elektrophorese/articles/121197.
(abgerufen am 21.05.2010)
[92]
http://www.ncbi.nlm.nih.gov/mapview/maps.cgi?taxid=9606&chr=14|HuRef&MAPS
=ugHs,genes-r&cmd=focus&fill=40&query=uid%2816330499%29&QSTR=RPGRIP
(abgerufen am 05.05.2010)
[93]
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=retrieve&dopt=full_report&
list_uids=122664 (abgerufen am 05.05.2010)
[94]
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=retrieve&dopt=full_report&
list_uids=1603 (abgerufen am 05.05.2010)
[95]
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=retrieve&dopt=full_report&
list_uids=4901 (abgerufen am 05.05.2010)
[96]
http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=10483315 (abgerufen am
12.04.2014)

74
[97]
http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=1952517 (abgerufen am
12.04.2014)
[98]
http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=2390287 (abgerufen am
12.04.2014)
[99]
http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=719675 (abgerufen am
12.04.2014)
[100]
http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=7816898 (abgerufen am
12.04.2014)
[101]
http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=9314269 (abgerufen am
12.04.2014)
[102]
http://www.orpha.net/consor/cgi-
bin/Disease_Search.php?lng=EN&data_id=22365&Disease_Disease_Search_dise
aseGroup=Severe-early-childhood-onset-retinal-
dystrophy&Disease_Disease_Search_diseaseType=Pat&Disease%28s%29/group
%20of%20diseases=Severe-early-childhood-onset-retinal-dystrophy&title=Severe-
early-childhood-onset-retinal-dystrophy&search=Disease_Search_Simple
(abgerufen am 10.11.2015)
[103]
http://www.orpha.net/consor/cgi-
bin/Disease_Search.php?lng=EN&data_id=3243&Disease_Disease_Search_disea
seGroup=Leber-congenital-
amaurosis&Disease_Disease_Search_diseaseType=Pat&Disease%28s%29/group
%20of%20diseases=Leber-congenital-amaurosis&title=Leber-congenital-
amaurosis&search=Disease_Search_Simple (abgerufen am 10.11.2015)
[104]
http://www.sph.umich.edu/csg/abecasis/merlin/tour/parametric.html (abgerufen am
22.05.2010)

75
[105]
http://www.zmb.uni-kiel.de/institute/imis/genet_epidem (abgerufen am 21.05.2010)
[106]
https://www.genetikum.de/de/genetikum/Aktuell/meldungen_detail.php?oid=254&dtl
=Diagnostik+mit+Gen-Panels+am+genetikum (abgerufen am 31.10.2015)

76
11 Ehrenwörtliche Erklärung
„Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig und ohne unzulässige
Hilfe oder Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe. Alle
Textstellen, die wörtlich oder sinngemäß aus veröffentlichten oder nichtveröffentlichten
Schriften entnommen sind, und alle Angaben, die auf mündlichen Auskünften beruhen,
sind als solche kenntlich gemacht. Bei den von mir durchgeführten und in der
Dissertation erwähnten Untersuchungen habe ich die Grundsätze guter
wissenschaftlicher Praxis, wie sie in der „Satzung der Justus-Liebig-Universität Gießen
zur Sicherung guter wissenschaftlicher Praxis“ niedergelegt sind, eingehalten sowie
ethische, datenschutzrechtliche und tierschutzrechtliche Grundsätze befolgt. Ich
versichere, dass Dritte von mir weder unmittelbar noch mittelbar geldwerte Leistungen
für Arbeiten erhalten haben, die im Zusammenhang mit dem Inhalt der vorgelegten
Dissertation stehen, oder habe diese nachstehend spezifiziert. Die vorgelegte Arbeit
wurde weder im Inland noch im Ausland in gleicher oder ähnlicher Form einer anderen
Prüfungsbehörde zum Zweck einer Promotion oder eines anderen Prüfungsverfahrens
vorgelegt. Alles aus anderen Quellen und von anderen Personen übernommene
Material, das in der Arbeit verwendet wurde oder auf das direkt Bezug genommen wird,
wurde als solches kenntlich gemacht. Insbesondere wurden alle Personen genannt, die
direkt und indirekt an der Entstehung der vorliegenden Arbeit beteiligt waren. Mit der
Überprüfung meiner Arbeit durch eine Plagiatserkennungssoftware bzw. ein
internetbasiertes Softwareprogramm erkläre ich mich einverstanden.“
_____________________ _______________________
Ort, Datum Unterschrift

77
12 Danksagung
Ein großer Dank gilt Herrn PD Dr. rer. medic. Markus Preising für die Stellung des
Themas und stetige Unterstützung, Hilfsbereitschaft und Betreuung während der
gesamten Zeit.
Ich danke Frau Prof. Dr. med. Birgit Lorenz für die Bereitstellung des Arbeitsplatzes
und der Materialen.
Ich danke meinem Ehemann Marc, der mich immer wieder aufs Neue motiviert hat und
mir auch in schwierigen Momenten zur Seite stand.
Ich danke meinem Bruder Martin und meiner lieben Freundin Annika für ihre Hilfe.
Zuletzt danke ich meinen Eltern, die mir durch ihre Unterstützung ein erfolgreiches
Studium ermöglicht haben.





























