Embed Size (px)
Citation preview

J. MOE. Biol. (1972) 63, 21-39
Kinetic Complexity of RNA Molecules
MAX L. BIRNSTIEL, BRUCE H. SE&s? AND IAN F. PURDOM
Medical Research Council Epigenetics Research Group. Department of Genetics, University of Edinburgh, West Mains Road,
Edinburgh EH9 3JN, Scotland
(Received 7 June 1971 and in revised form 20 September 1971)
A procedure has been developed for calculating the base sequence complexity of polyribonucleotides using RNA-DNA hybridization on membrane filters. Optimal rate temperature ( TOPt) in hybridization media containing 6 x SSC (SSC is 0.15 M-Nacl, 0.015 M-sodium citrate) (pH 7.2), 50% formamide or 1.15 M-sodium acetate (pH 5), 60% formamide were determined for a variety of RNA species.
The To,, in formamide were found to be unexpectedly high for all RNA species investigated. It is suggested that the high T,,, are caused by increased stability of RNA-DNA hybrids in these incubation mixtures.
It is demonstrated that RNA-DNA reactions with homogeneous RNA species present in excess are most easily characterized by C,t+ where C, is the molar concentration of RNA nucleotides in solution and t& is the time taken to reach the 50yo saturation value. Within certain limits C,t+ is independent of the amount of DNA on filter disks and the degree of fragmentation of the RNA. C,tk increases linearly with the base sequence complexity of RNA molecules. Using comple- mentary RNA derived from a balanced synthetic RNA-@X DNA hybrid wo show that the slope of the straight linear standard curve which links C,4& to base sequence complexity is 9.3 x 10e9 moles.sec/l. dalton for 6 x SSC, 50% forms,- mide. In 1.15 M-sodium acetate (pH 5), 60% formamide, the slope is 2.3 x 10-a moles.sec/l. daltons.
Applications of the kinetic standard curve for evaluation of purity and concentration of isolated messenger RNA’s are discussed. We show that transfer RNA of both Escherichia coli and Xenopw laevis is made up from 30 to 40 basic nucleotide sequences distinguishable by RNA-DNA hybridization. In Xenopus there are some 6500 cistrons for tRNA and so each tRNA sequence is, on average, 160-fold reiterated. 5 s RNA of Xenopus, coded by some 9000 cistrons, and ribosomal RNA of Bacillus subtilis, rabbit and Xenopus coded by 8, 250 and 610 cistrons, respectively, each contain a single family of nucleotide sequences and are therefore highly conserved in these species.
1. Introduction The object of the experiments described in this paper is to develop a simple procedure for determining the base sequence complexity of polyribonucleotides using RNA-DNA
hybridization techniques and by this method to establish the complexity of a few selected RNA species by comparison to standards of known genetic complexity.
Previous workers have established a relationship between the rate of renaturation
and base sequence complexity of DNA (Britten & Kohne, 1968; Wetmur & Davidson, 1968). DNA renaturation results from the collision of complementary strands and
t On leave from St. Jude Children’s Research Hospital, Memphis, Tennessee, U.S.A.
21

22 M. L. BIRNSTIEL, B. II. SELLS AND I. F. PURDOM
follows second-order kinetics. Thus, the half period of reassociation is inversely proportional to the molar concentrations of the DNA complements. Cot?, the product of the initial molar concentration C, (expressed as moles deoxynucleotide/l.) and half time of reassociation t, (see) is a measure of the base sequence complexity of the renaturing DNA (Britten & Kohne, 1968).
In RNA-DNA hybridization experiments, where the RNA is well in excess of the complementary DNA, the hybridization reaction can conveniently be presented by the straight linear curve of a double reciprocal plot, l/hybridization versus l/time (Bishop, 1969). We provide further examples in this paper that this method of presenting the hybridization reactions, although thermodynamically not understood, is very useful for the evaluation of kinetic data and we further show that it yields saturation values comparable to those obtained by standard saturation experiments. At a given R;“U’A input the rate of hybridization is directly proportional to concentration of the RNA and inversely proportional, roughly, to the genetic complexity of the RNA (Bishop, 1969), thus as in the case of DNA renaturation, the reaction is governed by the molar concentration of nucleotide sequence in solution.
From our experiments we have determined conditions of optimal rate for RNA-Dh‘A hybridization by the Gillespie & Spiegelman (1965) procedure for which we have adapted the low-temperature formamide technique of McConaughy, Laird & McCarthy (1969). First we show that an RNA of given base sequence is most simply characterized by C& the product of molar concentration of RNA sequences and the time necessary to reach half saturation; second, we developed a standard curve which correlates, with some precision, the rate of hybridization with base sequence complexity of RNA; third, we discuss possible applications of the kinetic analysis of RNA-DNA hybridiza- tion in determining the purity of messenger RNA, of messenger concentrations in RNA mixtures and the degree of base sequence divergence within RNA species derived from repetitive DNA sequences.
2. Materials and Methods (a) Materials
n-[2-3-3H]Valine (29 Ci/m-mole), L-[3H]leucine (34 Ci/m-mole), [5-3H]uridinc ( > 24 Ci/m-mole) and 3H-labelled nucleoside triphosphates were obtained from R’adiochcmical Centre, Amersham, England. E. coli tRNA used in charging experiments was purchased from General Biochemicals (Chagrin Falls, Ohio, U.S.A.). Frozen E. coli MRE 600 were kindly provided by the Microbiological Research Establishment, Porton, England. @X 174 virus particles and E. co.% RNApolymerase were purchased from Miles-Seravac, Maidenhead, England. A kidney (Rafferty) cell line of Xenopus Zaewti was a gift from Dr J. Gall (New Haven, Corm.). Rabbit kidney cells RK13 were purchased from Flow Laboratories, Irvine, Scotland and Falcon tissue culture flasks from Falcon Plastics, Oxnard, Calif., U.S.A.
(b) Preparation of DNA Xenopus DNA was prepared from nucleated blood cells. Toads were decapitated and
the blood collected in 0.15 M-N&I, 0.1 M-EDTA, lye /I-mercaptoethanol, pH 8.0 (100 to 200 ml./toad), and lysed with 1% SDS? (final concn). The DNA was prepared according to a modified Marmur procedure (1961) using repeatedly RNase (O.O1o!o) and pronaso (O.OS’$h). For the preparation of human placental or rabbit liver DNA, the tissue was first homo- genized in an Omnimix blender with 3 vol. of a. solution containing 0.4 M-sucrose, 0.001 ar- &Cl,, 0.05 M-Tris, pH 7.2. The homogenate was filtered through cheese cloth, the filtrate treated with 10 vol. saline-EDTA (Marmur, 1961) lysed by addition of 1 y. SDS and deproteinized as above.
t Abbreviation used: SDS, sodium dodecyl sulphate.

KINETICS OF RNA-DNA HYBRIDIZATION 23
E. co& cells were lysed in saline-EDTA, 1% SDS (Marmur, 1961). B. subtilis cells were washed in 0.1 M-NaCl, 0.01 M-EDTA, 0.01 M-Tris, pH 8, and resuspended in 20 vol. of the same medium. Lysozyme to a concentration of 0.5 mg/ml. was added and left at 0°C for 5 min. SDS was then added to a final concentration of 1%. The cell suspension was heated to 37°C and 5 min later, pronase (0.5 mg/ml.) was added. After 15 min the lysate was cooled to room temperature. Both E. coli and B. subtilis DNA were deproteinized as above..
(c) Preparation of 3H-labelled ribosomal RNA
Pormanent tissue culture lines from Xen0pu.s Zaevis and rabbit kidney were grown in. Falcon 75-cm2 bottles in Eagles MEM, supplemented with O.ll”,& NaHCO,, 10% foetal. bovine strum (Flow Laboratory) and antibiotics (streptomycin 130 i.u./ml.; penicillin 100 i.u./ml.). For Xenopus cells the medium was diluted by addition of 15% (vol.) water.. The cells were labelled by incubation for 36 to 72 hr with 50 to 100 &i/ml. [3H]uridino (2 24 Ci/m-mole) and chased for 36 hr with 4 changes of medium containing a loo-fold. excess of unlabelled uridinc. The incubation medium was removed, the cells detached by addition of the RNA extraction medium (containing 0.15 M-NaCl, 0.01 M-Tris (pH 7.5)., 0.05% sodium dodecyl sulphate (Sigma, London) ) and by scraping the flask with a rubber t,ipped spatula. The lysate was homogenized gently in a glass-Teflon homogenizer and dcproteinized by shaking with an ecpral vol. of water-saturated phenol added at room temperature. After centrifugation, the supernatant was re-extracted with phenol. This procedure yields RNA free from DNA. To the supernatant, 0.1 vol. of a 3 M-NaCl solution was added and the RNA precipitated by addition of 2.5 vol. of ethanol. After storage: overnight at -2O”C, the RNA was collected, washed 3 times in ethanol, dissolved in 0.02 M-sodium acetate pH 5, and fractionated on a 5 to 4096 sucrose gradient (0.1 M-NaCl, 0.02 M-sodium acetate, 0.001 M-EDTA, pH 5.0) in a Spinco SW 25.2 rotor at 15’C, 24,000 rev./min for 18 hr. To the 28 s and 18 s peak fractions, 0.1 vol. of 3 M-NaCl was added and the RNA recovered by precipitation with 2.5 vol. of ethanol.
The specific activities of rRNA preparations ranged from 4 x lo5 to 3.1 x lo6 disint.,l min/pg RNA. The rRNA was shown to be free from other hybridizable material by hybridization with a complete range of DNA fractions taken from a CsCl density gradient (Birnstiol, Speirs, Purdom, Jones & Loening, 1968). In all cases hybridization was confined to the high density rDNA, leaving the main band DNA u&belled (Fig. l(a) and (c) ). 1,abelled B. subtilis and Proteus mirabilis rRNA had a specific activity of 3.2 x lo* and 3.8 ;< lo5 disint./min/pg, and were the gift of Dr J. Bishop (Edinburgh). The bacterial rRNA was shown to hybridize exclusively to high density bacterial DNA (cf. Purdom, Bishop 8; Birnstiel, 1970); (Fig. l(d)) and hence was free from contaminating messenger RNA.
(d) Preparation of labelled and unlabelled 5 s RNA amZ transfer RNA from Xenopus laevis
Tissue culture cells were labelled for 3 days with 100 &i [3H]uridine/ml. RNA from labclled cells or livers from Xenopus toads were extracted as above. The RNA was passed through a 200-cm Sephadex CT100 column (bed vol, 630 ml.) which had been equilibrated with 0.01 M-sodium acetate, pH 5; 0.0001 M-EDTA according to Brown & Littna (1966). ‘rho flow rate was 12 ml./hr. The relevant peak fractions were collected, concentrated, if necessary, by lyophilization and used for hybridization. The 3H-labelled 5 s and 3H- lnbelled tRNA’s had a specific activity in excess of 2.8 x lo6 disint./min/pg. When annealed t,o a complete range of CsCl density gradient fractions, hybridization of 3H-labolled 5 s RNA was found to be exclusively with the lighter fractions of the gradient (Fig. l(a) ). 3H- labclled tRNA, in the presence of an excess of unlaballed 5 S, 18 s and 28 s rRNA, annealed to DNA of slightly heavier than average density (Fig. l(b) ). Both these findings are in accordance with the observation of Brown & Weber (1968) and attest to the hybridiz&ion specificity of the preparation.
(e) Preparation of sH-labelled complementary RNA from @X 174
Approximately 6 to 15 x 1013 @X particlesweresuspended in 0.1 M-NaCl, 0.025 M-EDTA , 0.035 nl-borate buffer (pH 9.1), 0.5% SDS and incubated with 0.05% pronase at 37°C for 3 hr. The solution was made 1% Sarcosyl and the DNA purified by banding in a 5.5ml. CsCl density gradient (25”C, 42,000 rev./min pav = 1.720 g/cm3) in c2 Spinco no. 50 angle

24 M. L. BIRNSTIEL, B. H. SELLS AND I. F. PURDOM
( a ) L M fufeu.5 lb) -I
I I
! ! u!’ \ ‘/RNA / ” -
Rotor centre ----+
FIG. 1. Hybridization of Xenopus 4 s, 5 s and rRNA rabbit and B. subtdis rRNA to C&l during gradient fractions of homologous DNA.
50 pg DNA in 4 ml. of 0.1 x SSC, together with M. Euteus DNA as a density marker, were added to 5.2 g CsCl and centrifuged at 42,000 rev./min at 25°C in an MSE 10 x 10 ml. rotor for 36 hr. Ten-drop fractions were collected and challenged at T,,, for 10 half-lives (tt) with (a) Xenpous 5 s RNA (0.014 pg/ml.) or 28 s + 18 s rRNA (3 pgglml.); (b) Xenopus tRNA (0.42 pg/ml.); (c) rabbit 28 s + 18 s rRNA (3 ,ug/ml.) and (d) B. subtilis 23 s + 16 s rRNA (2 pg/ml.).
rotor (Flamm, Bond & Burr, 1966), the fraction containing the O.D. peak dilutod 4-fold by addition of water and the DNA pelleted overnight in a Spinco no. 50 rotor at 45,000 rev./min and 15°C. The DNA pellet was dissolved in a small vol. of 0.01 M-Tris, pH 7.5. The @X RNA was transcribed in vitro according to the protocol of Chamberlin & Berg (1964). The incubation mixture contained in 1 ml., 40 mM-Tris buffer, pH 8,1 mM-ATP, CTP, GTP and UTP, each at a specific activity of N 50 mCi/m-mole, 4 mM-MgCl,, 1 mM-MnCl,, 12 m&r-%mercaptoethanol, 50 units of E. coli polymerase and 25 pg @X DNA. After incubation for 50 min at 37”C, the solution was chilled, made up to 2.5% Sarcosyl and deproteinized (twice) by shaking with an equal vol. of phenol. The supernatant was passed through a Sephadox G75 column (bed vol., 100 ml.) which had been equilibrated with 0.5 x SSC and the excluded fraction collected. The RNA : DNA ratio of the product was 1.06 by radioactivity. When examined in the Model E at 44,000 and 56,000 rev./min the product banded in C&SO, at 1.503 g/cm3 (Fig. 2) with only traces of U.V. absorbing material at 1.64 g/cm3 (where RNA is expected to band). Both these findings show that the RNA-@X DNA is a balanced hybrid (cf. Chamberlin & Berg, 1964). The RNA was dissociated from the DNA by a brief exposure (45 set) of the hybrid to 0.1 M-NaOH at O”C, quickly neutralized by addition of Tris buffer and 0.1 M-HCl and freed from DNA by centrifugation in a preparative C&SO1 gradient in a Spinco SW39 rotor (Chamberlin & Berg, 1964). The RNA was recovered, dialysed against 0.1 x SSC and lyophilized. The specific activity of the RNS was 8.5 x lo5 disint./min/pg.
(f) Preparation of aminoacyl symthetases
The synthotases were prepared from frozen E. coli MRE 600. The cells (30 g) were ground with 60 g of Al,O, and extracted with 150 ml. of Tris, 0.05 M, pH 7.5. The mixture was fnst centrifuged at 3000 g for 10 min and then at 6000 g for 10 min to remove Al,O,

KINETICS OF RNA-DNA HYBRIDIZATION 25
--Rotor centre
FIG. 2. Isopycnic centrifugation of @X RNA-DNA hybrid in CSZSOI. Xenopua DNA (1.422 g/em-3) was added as a density marker (see Text).
and cell debris. The supernatant was adjusted to 0.01 M-MgCl, and centrifuged for 4 hr at 27,500 rev./min in the no. 30 rotor of a Spinco ulbracentrifuge to remove ribosome particles. The supernatant was removed and adjusted to 0.33 ~-Kc1 and passed through a DEAE cellulose column (50 ml. bed vol. pre-equilibrated with 0.05 M-Tris. HCl in 0.33 M-KCl, pH 7,5) to remove tRNA (Wong, Mustard & Herbert, 1969). The peak fractions were collected, pooled in small fractions and frozen. The enzymes were stable for at least one month in the frozen state.
(g) Preparation of charged transfer RNA The hybridization studies with tRNA were performed by using tRNA charged with a
labelled amino acid. The charged tRNA was prepared at 37°C in a l-ml. reaction mixture (Waters & Novelli, 1968) containing the following components: 100 pmole /3-mercapto- ethanol, 1.2 mg tRNA and [3H]leucine (2 pg and 0.5 mCi) or [3H]valine (2.4 mg and 1 mCi). The incubation time was 15 min and was initiated by the addition of 0.3 mg of crude synthetase preparation. Following charging, the reaction mixture was chilled and an equal vol. of 0.1 x SSC-saturated phenol was added. The mixture was shaken at room temperature for 10 to 15 min, centrifuged, the water layer removed, and passed through a coarse G25 Sephadex column to remove small molecular weight components. The fractions containing the bulk of the radioactivity were combined and stored for use in hybridization studies.
(h) Preliminary procedure before charging To ensure that the tRNA’s specific for valine were saturated with amino acid, the
subsequent steps were followed: a series of incubations were performed in the charging medium with a standard amount of tRNA and varying concentrations of the amino acid under consideration but a constant amount of radioactivity. The number of counts obtained in the charged tRNA were determined at the different amino-acid concentrations. The lowest concentration of amino acid necessary to saturate the standard amount of RNA was obtained by measuring the specific activity (cts/min/mg RNA) at the various concen- trations of amino acid, then determining the concentration which produced a drop in specific activity proportional to the amount of ammo acid added. The amount of enzyme used was sufficient to complete the charging reaction within 10 to 15 min at 37°C. The rate of charging was determined by removing samples at intervals after enzyme addition and measuring the number of 10% trichloroacetic acid-precipitable counts.

26 &!I. L. BIRNSTIEL, B. II. SELLS AND I. F. PURDOJI
(i) Preparation of 3H-labelled transfer RNA from E. coli
To prepare high specific activity tRNA, E. coli (pyr-, Zm-, and vit B-) was used. A suspension of exponentially growing cells with an absorbance of 0.08 (at 660 nm) wns incubated in a minimal medium containing 5 mCi E3H]uridine (6 mg/ml.) until tho supply of uridino was exhausted (usually at an absorbance of 0.35 to 0.5). After a further 30 min, unlebellod uridine was added to a final concentration of 300 pg/ml. and the incubation continued until the absorbance reached 0.6 to 0.7.
To isolate the 3H-labelled tRNA the following procedure (Morell, Smith, Dubn:rkl & Marmur, 1967) was used. The cells were harvested by centrifugation and disrupted by grinding with Al,O,. The mixture was then extracted at 0 to 4°C with buffer (0.005 al-Tris and 0.01 M-MgCl,, pH 7.5). The extract was centrifuged at 5000 g for 10 min t,o rcmovo A1,03 and then at 24,000 g for 20 min to remove debris. To the supcrnat.ant DNtLsc W~LS added a final concentration of 5 pg/ml. and dextran sulphate 500 (60 pg/ml.) t#o inhibit. RNase. The preparation was then centrifuged at 38,000 rev./min for 3 hr in a Spinco no. 40 rotor to sediment ribosome particles. Following centrifugation the upper 3/4 of the supcr- natant was removed and added to an equal vol. of phenol (saturated with 0.1 ): SSC). The RNA was extracted by shaking t#he mixture for 15 min. The water layer containing the RNA was separated by centrifugation and the RNA precipitated by the addition of 0.1 vol. of 30yb potassium acetate and 2.5 vol. of absolute ethyl alcohol. The RNA was dissolved in 0.1 x SSC re-precipitated and washed with absolute ethyl alcohol and finally dissolved in 0.1 x ssc.
(j) Hybridization Focedure
13 mm HAWP Millipore filters were washed overnight in 2 x SSC. They were pl~~ced in stainless steel Swinney filtration apparatus (Millipore, U.K. Ltd.) fittod with lo-ml. Perspex tubes and washed with 5 ml. of 2 x SSC. DNA, usually in 0.5 ml. of 0.1 >: SSC, was denatured by addition of an equal vol. of 1 N-NaOH, which was neutralized 15 to 20 min later, by the addition of 4 vol of a medium containing 3 RI-N&I, 1 N-HCl and 1 M-Tris (pH 8.0) in a ratio of 2 : 1 : 1. The solution was filtered at a rate of 0.2 ml./min through t,he prepared HAWP disks, which were then washed with 5 ml. of 6 x SSC. The filters were heated at 80°C for 2 hr in a vacuum oven, marked with a pencil and stored at --20°C (Gillespie & Spiegelman, 1965).
Before use, the DNA containing filters were soaked in 6 x SSC, 50: formnmidr. Fol kinetic studies the hybridization medium was brought to optimum temperature (soo below) and wetted-filters rapidly immersed and agitated at the appropriate temperature. Thz volume of incubation medium was sufficient to allow the filters to move around freely in tho solution. For sampling, the filters were removed at various time intervals, and placed in a large vol. of ice cold 6 x SSC. The filters were washed by the batch method (Birnstiel et al., 1968) by placing them in a 2-1. beaker with 11. of 2 x SSC at room temperature and keeping them under continuous agitation for 30 min for each exchange (3 times). The filters then were incubat,ed with 10 pg RNase/ml. for 20 min in 2 x SSC at room temperature, rinsed with 2 x SSC and vacuum dried before counting in the scintillation counter. Tho back- ground controls using heterologous bacterial DNA instead of eukaryotic DNlZ were less than 0.5% of the homologous saturation values.
3H-labelled aminoacyl-tRNA’s were incubated in 600,; formamide, 1.15 ,Ir-sotlium (acetate), pH 5 at 46°C. Incubation for 2 hr in this medium led to only a minor discharge ( < 10%) of the [3H]valine-tRNA [3H]leucine-tRNA (Fig. 3). The washing procedure was carried out ss above except that there was no RN&se treatment. When aminoacyl-t RNA from E. co& was annealed to Xenopus DNA, the background was < 7 y0 of the homologous reaction. Both background noise and hybridization levels were unaffected by the add&ion of 10 or 100 units of T,-RNase/ml. in the final wash, nor was the hybridization : noise ratio improved (Table 1).
(k) Dissociation of RNA-DNA hybrids
rRNA-DNA hybrids from Xenopus and B. subtilis and cRNA-@X DNA hybrids wcrc: formed at optimal temperatures as described above. The filters were washed by the batch method and treated with RNase 8s described above. The filters were then incubated in

KINETICS OF RNA-DNA HYBRIDIZATION 27
FIG. 3. Stability of [3H]leucine-tRNA and [3H]valine-tRNA during hybridization. E. coli tRNA charged with [3H]leucine or [3H]valine were incubated in 1.15 M-sodium acet,atc
(pH 5), 60% formamide at 46°C. At times indicated a sample was withdrawn and injected intu 2 ml. of water, 100 pg yeast RNA (Sigma, London) was added and the RNA precipitated with a,n equal vol. of 5% trichloroacetic acid in the cold. The precipitate was recovered on hfillipore filters, washed 3 times with 5% trichloroacetic acid, dried, and counted.
TABLE 1
Effects of T, RNase on the stability of [3H]leucine-transfer RNA-DNA hybrids
Counts hybridized
No T1 RNase treatment
T, RNase treatment
10 units/ml. 100 units/ml.
Xenopus DNA 73 83 63 E. cobi DNA 1237 1180 1133
0.1 x SSC, 0.1% diethylpyrocarbonate at 20°C for 30 mm to remove the nuclease activity, rinsed exhaustively in 0.1 x SSC and dried at room temperature. To determine the tem- perature at which 50% of the RNA was released from the filter (T,), the filters were placed in 1 ml. of 0.1 x SSC or 6 x SSC; 50% formamide and heated in temperature increments of approximately 5°C. After 6 mm at each temperature, the released RNA was recovered by trichloroacetic acid precipitation and counted.
3. Results
In all experiments to be described here, the concentration of RNA was well in excess of the complementary DNA, usually by a factor of 50 to 100. The time-course of each reaction is presented conveniently as a double reciprocal plot, since this allows simple determination of both reaction half time and saturation value (cf. Bishop, 1969).
(a) Effects of temperature on rate of RNA-DNA hybridization
We have established the optimal temperatures for maximal initial rates of reaction in 6 x SSC (pH 7*2), 50% formamide or in 1.15 M-sodium acetate (pH EGO), GOT/, formamide for a variety of RNA components. Up to 25 pg DNA bound to 13 mm millipore HAWP filters were challenged with O-1 to 3 pg homologous RNAjml. A time-interval of incubation was selected appropriate for each RNA concentration to allow hybridization to approximately 20% of the saturation value. The counts hybridized were taken as a measure of initial rate reaction.
Figure 4 gives a selection of temperature-dependent rate curves. The temperature at which the reaction rates are maximal are well defined. From Table 2 it may be seen

28 M. L. BIRNSTIEL, B. H. SELLS AND I. F. PURDOM
3oc
2oc
IOC
C
IOC
5C 2 -5 5 .2 0 .s :: .e 2 a
100
0
400
200
(a) (b)
I
Cd)
(hl
200
100
0
200
100
-c c \
0 g
100 :$ $7 4 B
50
0 100
50
Temperature
FIG. 4. Initial rate of RNA-DNA hybridization as a function of temperature. Filters contained approximately 25 pg DNA. The RNA concentration and time of incubation
were chosen in such a way that about 20% of the complementary DNA reacted. (a) to (f) 6 x SSC, 50% formamide; (g) and (h) 1.15 sodium acetate (pH 5), 60% formamide;
(a) Xenopw 5 s RNA, (b) Xenopus 28 s + 18 s (upper) and 18 s rRNA (lower), (c) HeLa rRNA, (d) rabbit 18 s rRNA, (e) E. coli 16 s RNA, (f) B. subtilis rRNA, (g) Xenopus 28 s RNA, (h) E. coli tRNA. Unlabelled rRNA and 5 s RNA were added in 20 and 2 times excess.
that the temperature optima (T,,,) are high, especially for the ribosomal RNA species. The T, in 0.1 x SSC and 6 x SSC; 50% formamide are also listed for a selection of RNA-DNA hybrids (discussed below).
(b) Effects of fragment size of RNA molecules
We have determined the effect of fragment size of ribosomal RNA molecules on the rate of hybridization reaction as follows: 28 s rRNA of Xenopus Eaevis in 0.1 x SSC was exposed to 0.1 N-NaoH, at 20°C for 0, 3, 10 and 30 minutes to yield RNA with

TABL
E 2
Opt
imal
ra
te a
nd
mel
ting
tem
pera
ture
s fo
r RN
A-DN
A hy
brid
s
RNA
GSCt
T
opt
17,
(hyb
rid)
Tm
(DNA
) T,
(h
ybrid
) -
T,
(DNA
)-
cont
ent
6 x
SSC,
50
%
6 x
SSC,
50
%
6 x
SSC,
50
%
T,
(hyb
rid)
Tm
(DNA
) To
,, (h
ybrid
) To
,, (h
ybrid
)
(%I
form
amide
fo
rmam
ide
form
amide
o-
1 x
ssc
0.1
x ss
c 6
x SS
C,
500/
6 6
x SS
C,
50%
fo
rmam
ide
form
amide
In
6 x
SSC,
50
%
form
amide
16 8
E.
cdi
rRNA
23
+
16 s
B.
subt
iZis
23
+ 16
s P
. m
drab
iZis
(PX
cRNA
4
s Xe
nopu
s tR
NA
6 s
Xeno
pua
rRNA
18
s X
enop
wr
rRNA
28
+
18 s
Xen
opus
rR
NA
28
+ 18
s H
eLa
18 s
Rab
bit
28
+ 18
s R
abbit
In
1.15
M-
N&
+ 60
%
form
amide
4 s
E.
coli
tRNA
23
+
16 s
B.
subt
ilis
28
s Xe
nopw
rR
NA
53
60
69
9 53
58
75
69
74
76
18
11
53
55
69
14
42
45
66
64
63
70
20
19
60
54
73
19
57
61
71
10
54
62
70
8
59
64
78
73
80
80
16
9 62
65
75
10
56
61
71
10
62
62
75
77
81
13
50
46
53
47
63
52
1 y0
G
+ C
arc
taken
fro
m
F.
Amal
di
(196
9);
Birn
sticl
et
al.
(196
8);
Brow
n &
Web
er
(196
8);
Brow
n 85
Littn
a (1
966)

30 M. L. BIRNSTIEL, B. H. SELLS AND I. F. PURDOM
TABLE 3
Effect of fragment size on hybridization
Sample no. S Cts/min hybridized
in 20 min
1 28 415 2 8 408 3 5 376 4 3.5 401
S-values of 28, 9, 5 and 3.5 s RNA. When samples of these RNA preparations were hybridized to Xenopus DNA on disks the rate of hybridization was similar for all preparations, irrespective of initial molecular weight before incubation (Table 3). This means, in practice, that for the purpose of kinetic comparison the various RNA mole- cules do not have to be brought to similar size.
(c) Effects of DNA concentrations
Gillespie & Spiegelman (1965) noted that the hybridization reaction is strongly influenced by RNA concentration, but that the concentration of DNA was less important. To study the effects of the amount of DNA bound to membrane filters more quantitatively, we prepared disks containing heterologous bacterial DNA (10 pg) with the addition of 0.01 to 1.0 pug Xenopus rDNA (Birnstiel et al., 1968). When these filters were challenged with radioactive Xenopus rRNA the absolute initial rate of hybridization (at 20 min reaction time) was proportional to the rDNA input (Table 4). When the initial rate is divided by the hybridization saturation value it is evident that the relative rates (i.e. percentages of the rDNA hybridized at a given time) are similar for the different concentrations, at least at low rDNA input. At low input of comple- mentary DNA, therefore, the relative rate of hybridizat)ion is unaffected by t’he amount of DNA on the disks.
The important practical consequence of this finding is that when studying specific DNA sequences under condition of RNA excess, the degree of reiteration of these sequences (i.e. the amount of complementary DNA) has little effect on the relative rate of hybridization. Consequently, when investigating the rate of hybridization of cellular RNA’s which are encoded by DNA segments of disparate multiplicities, a comparison of the rate for a given RNA concentration can be obtained simply by measuring the time required to reach 50% saturation of those sequences (see below).
TABLE 4
Hybridization of ribosomal RNA to Xenopus ribowmal DNA
Amount of rDNAt bLg/dW
RNA hybridized (cts/min) ratio
at 20 min at t = co cts/min, t = 20
cts/min, t = co
0.01 65 200 0.32 0.10 730 2020 0.36 0.33 2031 6940 0.28 1.0 3496 20100 0.17
t Disks also contained 10 pg of Proteus mirabilis DNA.
KINETICS OF RNA-DNA HYBRIDIZATION
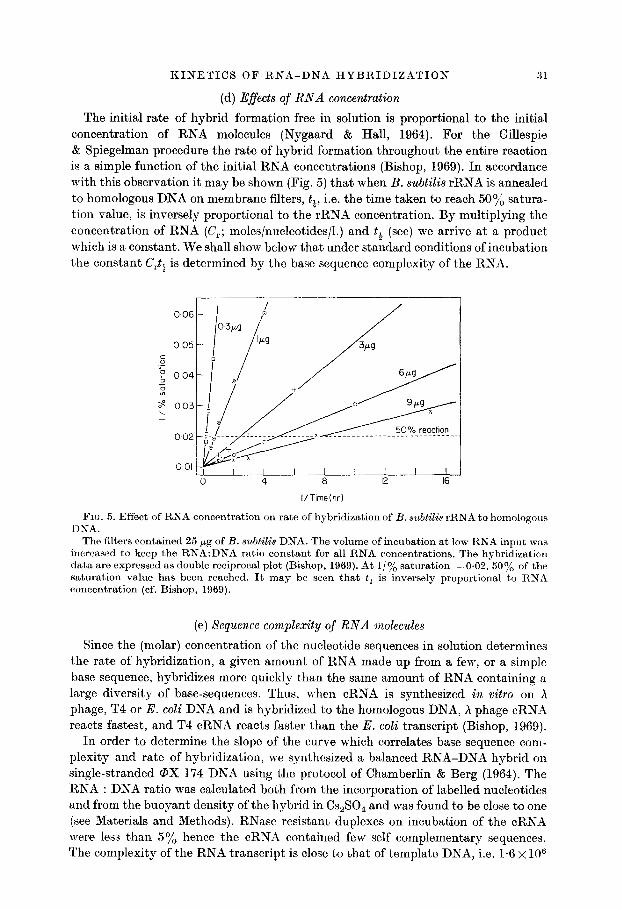
(d) Effects of RNA concentration
31
The initial rate of hybrid formation free in solution is proportional to the initiad concentration of RNA molecules (Nygaard & Hall, 1964). For the Gillespie & Spiegelman procedure the rate of hybrid formation throughout the entire reaction is a simple function of the initial RNA concentrations (Bishop, 1969). In accordance with this observation it may be shown (Fig. 5) that when B. subtilis rRNA is annealed to homologous DNA on membrane filters, tt, i.e. the time taken to reach 50% satura.- tion value, is inversely proportional to the rRNA concentration. By multiplying the concentration of RNA (C,; moles/nucleotides/l.) and tt (set) we arrive at a product which is a constant. We shall show below that under standard conditions of incubation the constant C,t, is determined by the base sequence complexity of the RNA.
006
I/Time(hr)
FIG. 5. Effect of RNA concentration on rate of hybridization of B. subtilis rRNA to homologous DNA.
The filters contained 25 pg of B. subtilis DNA. The volume of incubation at low RNA input was increased to keep the RNA:DNA ratio constant for all RNA concentrations. The hybridization data are expressed as double reciprocal plot (Bishop, 1969). At l/ye saturation =0.02, 50% of the saturation value has been reached. It may be seen that t+ is inversely proportional to RNA concentration (cf. Bishop, 1969).
(e) Xequence complexity qf RNA molecules
Since the (molar) concentration of the nucleotide sequences in solution determines the rate of hybridization, a given amount of RNA made up from a few, or a simple base sequence, hybridizes more quickly than the same amount of RNA containing a large diversity of base-sequences. Thus, when cRNA is synthesized in vitro on A phage, T4 or E. coli DNA and is hybridized to the homologous DNA, h phage cRN.4 reacts fastest, and T4 cRNA reacts faster than the E. coli transcript (Bishop, 1969).
In order to determine the slope of the curve which correlates base sequence com- plexity and rate of hybridization, we synthesized a balanced RNA-DNA hybrid on single-stranded @X 174 DNB using the protocol of Chamberlin & Berg (1964). The RNA : DNA ratio was calculated both from the incorporation of labelled nucleotides and from the buoyant density of the hybrid in C&30, and was found to be close to one (see Materials and Methods). RNase resistant duplexes on incubation of t.he cRNA were less than 5% hence the cRNA contained few self complementary sequencej. The complexity of the RNA transcript is close to that of template DNA, i.e. 1.6 x 106

32 M. L. BIRNSTIEL, B. H. SELLS AND I. F. PURDOM
I I
(b)
' -)
Ii-L---- I I I I 33 I 2 4 6 8 IO
I/hour
FIG. 6. Double reciprocal plots of RNA-DNA hybridization (a) for 6 x SSC, 50% formamide; (b) 1.15 M-sodium aoetate, 60% formamide.
Filters contained 0.01 pg @X DNA with 10 pg Xenopus DNA added as a carrier. For hybridiza- tion of bacterial RNA, the filters contained 10 to 20 pg DNA, for hybridization of eukaryotic DNA, 25 pg filters were used. 3H-labelled tRNA preparations were supplemented by addition of S-fold excess of m&belled homologous rRNA and an equal amount of unlabelled 5 s RNA, separated by G 100 gel filtration (see Materials and Methods). The amounts of RNA shown in Fig. 6 are pg/ml.

TABL
E 5
Anal
ytica
l an
d kin
etic
com
plex
ity
of R
NA
mol
ecul
es
RNA
In
6 x
SW;
50%
fo
rmam
ide
OX
174
cRNA
4
s E.
co
li tR
NA
23
x 16
s
B.
subt
ilis
rRNA
23
x
16 s
P.
wv
irabil
is rR
NA
4 s
Xeno
pus
tRNA
5
s Xe
nopu
s I-R
NA
18 s
Xen
opus
rR
NA
28
s Xe
nopu
s rR
NA
28
+ 18
s
Xeno
pus
rKNA
28
+
18 s
Rab
bit
rRNA
In
1.15
.\r-
Na+
; 60
:/,
form
amide
4 s
E.
coli
valyl
-tR,N
At
4 s
E.
coli
tRNA
(to
tal,
char
ged
0.02
5 0.
0032
0.
025
- wi
t,h
valin
e)
4 s
h’.
cold
tR
S-1
(tota
l, ch
arge
d 0.
025
with
leu
cine)
4
s E.
eo
li 3H
-labe
lled
tRNA
0.
025
4 s
Xeno
pus
tRNA
0.
025
23
+ 16
s
B.
subti
lis rR
NA
1.6
28
s Xe
nopu
s rR
NA
1.5
Analy
tical
com
plexit
y (d
alton
s x
10-s)
Com
plem
entar
y DN
A
(o/o)
Geno
mic
redu
ndan
cy
t, (a
t 3
PLg)
(m
in)
@we
)
1.6
95
1 24
-27
0.02
5 0.
06
60
9-14
1.
6 0.
64
8 19
-24
1.6
0.41
6
24
0.02
5 0.
009
6.5
x lo3
12
-17
0.03
8 0.
020
9x10
3 04
-0.8
0.7
0.02
4 6.
1 x
1Oa
7-12
1.
5 0.
051
6.1
x lo2
16
-20
2.2
0.07
5 6.
1 x
1Oa
24-3
0 2.
5 0.
03
2.5~1
0~
24-3
4
-
0.06
0.
009
0.64
0.
051
3 0.
7*
0.4
0.01
8 -
29
17
0.71
- 25
17
0.
45
60
39
18-2
4 0.
95
6.5
x lo3
42
24
1.
05
6 66
37
Ki
netic
sta
ndar
d 6.
1 x
lo2
74
42
l-7
c&t*
x 10
3 (m
oles.s
ec/l.)
(a
vera
ge)
Kine
tic
com
plexit
y (d
alton
s x
10T6
) (a
vera
ge)
15
Kine
tic
stand
ard
6.7
0.78
12
1.
4 13
1.
5 7.
8 0.
90
0.37
iz
0.04
3 5.
3 0.
61
11
1.3
16
1.9
17
2.0
*t Th
e cC
,nce
ntra
tion
of
3H-la
bello
d va
lyl-tE
NA
was
calcu
late
d fro
m
the
level
of
incor
pora
tion
of
[3H]
vslin
e int
o tR
NS.

34 MM. L. BIRNSTIEL, B. H. SELLS AND I. F. PURDOM
daltons (Sinsheimer, 1959). We have determined the C,t, for this RNA species at optimal temperature. From Figure 6(a), it may be seen that at 0.94 pg @X cRNA ml. saturates half of the DNA complements in 80 minutes.
A variety of cellular RNA’s have also been investigated at T,,,. Since 5 s RNA hybridizes very fast, the concentrations of this RNA was kept between0.1 to 0.5 pg/ml. For the other RNA species a range of 0.5 to 6 pg was employed. The exact composition of the reaction mixture is given in Figure 6(a) and (b). These Figures show the double reciprocal plots for the various RNA species in 6 x SSC, 50% formamide (pH 7.2), or 1.15 M-sodium acetate (pH 5), 60% f ormamide. The reaction half times (tJ were determined for all RNA species. Using the inverse relationships between t+ and concen- tration (Bishop, 1969; Fig. 5) we have calculated t+ for an RNA concentration of 3 pg/ml. for each species and tabulated the observed ranges of values, together with the corrcs- ponding average Crt+ in Table 5.
4. Discussion
(a) Optimal rate temperatures for hybridization of RNA to DNA in the presence of formamide
Nygaard & Hall (1964) demonstrated that in O-3 M-Nacl, 0.05 M-!lh’iS, pH 7.5, the initial rate of hybridization of T4 RNA to homologous DNA was optimal at 67”C, a temperature some 16°C lower than the T, of the T4 RNA-DNA hybrid. This tempera- ture differential is noticeably smaller than that for DNA renaturation where the rat’e of reassociation has been shown to be maximal about 25°C below the mean denaturation temperature (Wetmur & Davidson, 1968). McCarthy & Church (1970) have recently reviewed the effect of reaction temperatures on the fidelity of base pairing. Generally, little is known about the temperature dependence of the kinetics of RNA-DNA reactions.
Hybridization of rRNA is routinely performed at 67°C in media containing 2 or 6 x SSC. In initial experiments we observed that under these conditions, t’emperatures in excess of 80°C were required to permit maximal rate of hybridization of eukaryotic rRNA. Since these temperatures were deleterious to the stability of the RNA we have consequently supplemented these media with high concenhrations of formamide. Addition of this compound allows maximal rate of annealing at relatively low temperatures (McConaughy et al., 1969). We find that a solution composed of 6 x SSC and 50% formamide provides conditions allowing a reasonable rate of hybridizat,ion while eliminating largely the problems of RNA instability.
Table 2 lists the T,,, for various RNA species. The values are compared t’o the T, of RNA-DNA hybrids determined by dissociation of RNase treated hybrids in either 6 x SSC, 50% formamide or O-1 x SSC (Fig. 7). We also list the T, of the DNA complements as predicted from the G + C content of the RNA. For this purpose we determined the optical melting curves for Xenopus (40% G -k C), E. coli (50% G + C) and 01. Zuteus (71% G + C) DNA in 6 x SSC, 50yo formamide and in 0.1 x SSC (not shown) and calculated the T, according to the predicted G + C content of the DNA complements by linear interpolation. It should be noted that the T, of RNA-DNA hybrids are those obtained for undegraded RNA. Reduction of rRNA to 3 to 4 s for instance, reduces the T, by approximately 5°C.
An unexpected finding is that RNA-DNA hybrids appear to be more stable in 6 x SSC; 50% formamide, by some 2 to 6”C, than native DNA of comparable G+C

KINETICS OF RNA-DNA HYBRIDIZATION 35
80
60
20 40 60 80 100 40 60 80 100
Temperature (deg. C)
FIG. 7. Melting temperature of RNA-DNA hybrids. DNA was challenged with saturating amounts of homologous RNA and dissociated as described
in the Materials and Methods section. (a) Dissociation in 6 x SSC, 50% formamide; (b) dissociation in 0.1 x SSC; -O--O--,
@X RNA-DNA hybrid; -O-O-, B. subtilis rRNA-DNA hybrid; -A--A--, Xenopzcs rRNA-DNA hybrid.
content (cf. Table 2), and by up to 8°C when compared to reassociated DNA duplexes (Grunstein, unpublished observations). By contrast the T, of the same RNA-DNA hybrids dissociated in O-1 x SSC are equal to or somewhat lower than, those of DNA complements and in this they conform to the generally accepted rule that RNA-DNA hybrids are less stable than corresponding native DNA segments (cf. Walker, 1969).
For RNA of high molecular weight T,,, for RNA hybridization in 6 x SSC; 507/o formamide is approximately 10 deg. C below the T, of corresponding DNA comple- ments. The differential for low molecular weight RNA is larger, but here the T,,, is likely to be lower because of the reduced stability of short RNA-DNA hybrids. When the T, of the hybrid and T,,, are both determined at high formamide concentrations, a temperature differential as high as 20°C is obtained (see Table 2) and in this the reac- tion resembles the hybridization of mRNA to T4 DNA (Nygaard & Hall, 1964). Our combined data suggest that the high concentration of formamide imparts high stability to RNA-DNA hybrids and consequently produces high optimal rate temperatures and high T,.
We have also measured T,,, in an incubation mixture similar to that of Weiss, Hsu, Foft & Scherberg (1968) and of Daniel, Sarid & Littauer (1970) which allows the measurement of the kinetics of 3H-labelled aminoacyl-tRNA hybridization with little hydrolysis of the aminoacyl ester bond (see Materials and Methods; Fig. 3). In this medium, the formamide concentration is increased to 60% and the pH lowered to pi. T,,, are listed in Table 2.
There are probably at least four characteristics of the RNA which influence T,,,. These are: fragment size, fidelity of base pairing, G $ C content and secondary structure of the RNA. Amongst the RNA species the To,, of rRNA is highest of all,
and T, (DNA)-To,, (hybrid) smallest. In rRNA a large proportion of the nucleotide sequences are present in double-stranded pin-loop form. This could lead to a severe sequestration of nucleotide sequences and thus slow down the reaction by lowering of the effective nucleotide concentration in solution. It is possible that in this case a large

36 31. L. BIRNSTIEL, B. H. SELLS AND I. F. PURDOM
portion of the base sequence must first be made available by melting of the double- stranded RNA region. Consistent with this idea it is observed that B. subtilis and Xenopus rRNA when heated to optimal rate temperatures in 6 x SSC, 50% formamide appear to be largely melted but still maintain 15% of their hyperchromicity (Speirs, unpublished observations). The optimal rate temperatures observed probably represent a compromise between the melting of RNA molecules and the stability of the initial RNA-DNA hybrids around the nucleation sites.
From the high optimal rate temperatures it would appear that previous annealing experiments with formamide, especially those utilizing rRNA, mere carried out ~\ell below the optimal discriminating temperatures of annealing. It’is noteworthy that, the saturation values for RNA’s such as Xenopus and B. subtilis rRNA are identical at optimal and sub-optimal temperatures, while at higher temperatures the value is reduced. Saturation levels were determined at t = co from all double reciprocal plots and these are listed in Table 5. They fall well within the ranges published by other laboratories. The hybridization saturation value of @X cRNA represents an interest)ing case. Here, apparenbly most of the @X DNL4 bound to filters is available for hybridiza- tion and no large “hybridization factor” (Bishop & Robertson, 1969) exists for this RNA species.
(b) Choice of standards for the determination of rates of hybridization
Because of the relatively well-defined temperature optima it is essential to stan- dardize the reaction condition rigorously. Once this has been achieved, RNA-DNA hybridization may be used to analyse the base sequence complexity of RNA molecules. The procedure is relatively simple because within the limits stated above, the degree of fragmentation of t,he RNA and t,he mass of the complement’ary DNS on membrane filters have little influence on the relative rate of RXA-DNA hybridization. d small molecule such as Xenopus 5 s R,NA encoded for by a multiplicity of some 9000 DNS segments (cf. Table 5) can be compared without difficulty to the relatively large bacterial ribosomal RNA, which is complementary to only a few DNA segments per genome.
In choosing kinetic standards we have avoided self-complement’ary cRNA since here neither t’he true complexity nor the concentration of freely react,ive RKA is adequately known because of self annealing of the RNA during the hybridiza- tion reaction. Standards provided by in vivo t’ranscripts from repetitive sequences could always be open to the objection that minor heterogeneity amongst redundant genes might measurably influence the rate of hybridization. Other problems with cellular RNA’s might arise from poor locu s specificity of hybridization reactions (McCa~rthy & Church, 1970). For example, the base sequences in E. coli rRPLTA are similar enough to cross-hybridize between 23 s and 16 s cistrons (Attardi, Huang & Kabat, 1965; Mangiarotti, Apirion, Schlessinger & Silengo, 1968) so that the kinetic complexity of this RNA is not clearly predictable. In our hands, E. coli rRNA has so far provided the only exception to the hybridization series of Table 5, in that it appears to react more slowly by a factor of 1.7 than B. subtilis rRNA. But we have not been able to test this RNA for its hybridization purity and so this observation must be further investigated.
For these reasons we have used cRNA from a balanced (PX RNA-DNA hybrid for the prime kinetic standard, while we consider RNA species such as 5 s RNA of Xenopus laevis and 23 s and 16 s rRNA of B. subtilis (which do not cross-hybridize) as

KINETICS OF RNA-DNA HYBRIDIZATION 3’7
secondary kinetic standards. When the kinetic complexity of these secondary standards (and eukaryotic rRNA’s) are calculated by comparison to the 0X cRNA (Table 5), it is clear that in all cases the kinetic complexity closely approximates the molecular weight of the RNA’s. For most cellular RNA’s the complexity is in fact slightly less than anticipated and this may be caused by the faster reaction of G + C rich RNA molecules compared to the low G + C standard. For 6 x SSC, 50% formamide the slope which links C& and complexity is 9,3 x 10es moles.sec/l. daltons. From Table 5 it may also be deduced that the slope of the standard curve for 1.15 M-sodium acetate (pH 5); 60% formamide is 2.3 x lows’ moles.sec/l. daltons.
(c) Kinetic complexity of RNA molecules
In this discussion the analytical complexity of an RNA is defined as its molecular weight, while the kinetic complexity, also measured in daltons is defined as the rate at which RNA-DNA hybrids form and is determined by comparison to the above mentioned kinetic standards. These concepts have already been usefully employed when dealing with DNA renaturation experiments (Wetmur & Davidson, 1968).
At its simplest the relationship between C,t, and base-sequence complexity under- lines the long-recognized fact that a given amount of RNA made up from simple sequences supports rapid hybridization while an equal amount of more complex RNA molecules anneals more slowly. Under our condition of incubation, saturation is closely approximated in 6.5 minutes for 3 pg 5 s RNA/ml. Longer incubation only ma,gnifies the contribution of contaminants in the RNA preparation. Conversely, the reaction of bacterial ribosomal RNA requires a period of 240 minutes to achieve the same degree of saturation.
Where a messenger RNA fraction has been isolated which is suspected to code for a single protein, the purity of this RNA can be quickly established. From the mole.. cular weight of this RNA (analytical complexity) a C,t+ can be calculated. By comparison to the experimentally obtained Crtt it may be established whether analytical and kinetic complexity coincide, i.e. whether the mRNA in question is a single species or a mixture of different RNA species. In hybridization experiments, using messenger RNA coding for globin (Williamson, Morrison & Paul, 1970) and, for histone (Kedes & Birnstiel, 1971) it was noted that hybridization was rapid, but no quantitative analyses of the kinetics have yet been made. As pointed out by Britten (1968), the kinetics of hybridization should be helpful in the determination of the concentration of messenger RNA’s Wherever the base-sequence complexity of the messenger is known, measurement of t; will yield C,.
Finally, RNA species such as tRNA, 5 s and rRNA are encoded by highly redundant genes, and kinetic analysis can be used to determine whether the gene transcripts form one, two or more families of base sequences. Five examples are given below.
(1) There are 5 to 8 ribosomal cistrons in the genome of B. subtilis (Oishi & Sueoka,, 1965, cf. Table 5). In agreement with the renaturation kinetics of isolated bacterial ribosomal DNA complements (Kohne, 1969) we show that the 23 s and the 16 s bacterial rRNA possesses a kinetic complexity, which is that of a single family of base sequence of w 1.4 x lo6 daltons.
(2) Hybridization experiments indicate that of the order of 0.02% of Xenopus DNA are complementary to 5 s RNA (Brown & Weber, 1968, cf. Table 5). From this it may be calculated that 5 s cistrons are some 9000 times repetitive in the haploid genome of

38 M. L. BIRNSTIEL, B. H. SELLS AND I. F. PURDOkT
Xenopus laevis. Fingerprint analysis of this RNA species (Williamson, personal communication) has shown this species t’o be largely homogeneous, Consistent with this we find that 5 s RNA anneals some 40 times faster than @X cRNA. 5 s RNA, although encoded by highly repetitive cistrons, has a kinetic complexity close t,o that of a single family of RNA molecules.
(3) The ribosomal cistrons from Xenopus laevis and rabbit are approximately 450 to 800 and 250 to 700-fold redundant (cf. Table 5, Birnstiel et al., 1966; Brown & Weber, 1968; Moore & McCarthy, 1968). The analytical complexity of amphibian and mammalian 28 s and 18 s rRNA molecules combined are 2.2 and 2.5 x 106 daltons, respectively (Loening, 1968). The Crts values at optimal rate temperatures are listed in Table 5. By comparison to the kinetic standards it may be seen that the kinetic complexities of these eukaryotic rRNA’s are closely similar to the analytical complexity. It follows that most, if not all, rRNA molecules belong to a single family of nucleotide sequences. In both Xenopus and rabbit the ribosomal sequences are highly conserved.
(4) In our hands, O*O58o/o of the E. coli genome is complementary to tRNA. This compares to values of 0.04 to O.O6o/o found by Morel1 et al. (1967) and Zehavi-Willner & Comb (1966) for E. coli and 0*07% for B. s&ilk DNA by Oishi & Sueoka (1965). For all 60 tRNA cistrons combined we anticipate an analytical complexity of approxi- mately 1.5 x 106. As may be seen from Table 5 [3H]uridine labelled E. coli tRNA exhibits a complexity of only O-7 to 0.8 x lo6 daltons. tRNA labelled by acylation with [3H]leucine or [3H]valine exhibits a composite complexity of 0.60 and 0.71 x 10E daltons. The kinetic complexity of unfractionated E. coli tRNA, uniformly labelletl with [3H]uridine or acylated with [3H]valine or [3H]leucine is about half the value of the predicted analytical complexity. This can be explained in two ways. 1. The over-all reaction may be accelerated by the presence of a few tRNA species in disproport,ion- ately high concentrations. The rapid annealing rate of tRNA labelled with [3H]leucine may reflect this aspect of the hybridization. 2. Some sequences amongst the tRNA species may be similar enough that they are not resolved from one another by hybridization experiments. This is perhaps documented by the finding that [3H]valine- tRNA anneals to a multiplicity of DNA segments but behaves kinetically as if it were homogeneous.
(5) The results presented in Table 4 indicate that Xenopus tRNA is encoded by approximately 6500 cistrons. Its kinetic complexity, however, is similar to that of E. coli tRNA which is coded for by 60 cistrons. In spite of this great difference in redundancy between the bacterial and amphibian species the base-sequence complexity is similar. Thus, the Xenopus tRNA genome contains N 40 families of tRNA base sequences each of which is reiterated about 160-fold. These data are interesting in terms of what is known about the heterogeneity of tRNA’s examined by various column procedures. Novelli and his colleagues (Novelli, 1969) using reverse-phase chromatography have demonstrated a number of subspecies of tRNA for each amino acid in both animal and bacterial cells. Yang & Novelli (1971) have demonstrated in rat, mouse and man that the iso-accepting species of tRNA for the 20 amino acids are separable into some 60 peaks. Our kinetic studies are in agreement with these studies in that they suggest limited base sequence heterogeneity despite the high degree of genetic redundancy of the tRNA cistrons.
The technical assistance of Mrs Joan O’Neil is greatly appreciated. Wo wish to thank Dr J. 0. Bishop for supplying us with labelled B. subtilis and P. mirabilis rRNA. These studies were supported in part by the Damon Runyon Memorial Fund.

KINETICS OF RNA-DNA HYBRIDIZATION 39
REFERENCES
Amaldi, F. (1969). Nature, 221, 95. Attardi, G., Huang, P. C. & Kabat, S. (1965). Proc. Nat. Acud. Sci., Wash. 53, 1490. Birnstiel, M. L., Speirs, J., Purdom, I., Jones, K. & Loening, U. E. (1968). Nature, 219,
454. Birnstiel, M. L., Wallace, H., Sirlin, J. L. & Fischberg, M. (1966). Nut. Cancer Inst. Monogr..
23, 431. Bishop, J. 0. (1969). Biochem. ,T. 113, 805. Bishop, J. 0. & Robertson, F. W. (1969). Biochem. J. 115, 253. Britten, R. J. (1968). Carnegie Inst. Yrb. 67, 335. Britten, R. J. & Kohne, D. E. (1968). Science, 161, 529. Brown, D. D. & Littna, E. (1966). J. ~%foZ. Biol. 20, 95. Brown, D. D. & Weber, C. S. (1968). J. Mol. Biol. 34, 661. Chnmberlin, M. & Berg, P. (1964). J. Mol. BioZ. 8, 297. Daniel, V., Sarid, S. & Littauer, U. Z. (1970). Science, 167, 1682. Flamm, W. G., Bond, H. E. & Burr, H. E. (1966). Biochim. biophys. Acta, 129, 310. Gillespie, D.& Spiegelman, S. (1965). J. Mol. BioZ. 12, 829. Kedes, L. & Birnstiel, M. L. (1971). Nature, 230, 165. Kohne, D. E. (1969). Biophys. J. 8, 1104. Loening, U. E. (1968). J. Mol. BioZ. 38, 355. McCarthy, B. J. & Church, R. B. (1970). Ann. Rev. Biochem. 39, 131. McConaughy, B. L., Laird, C. D. & McCarthy, B. J. (1969). Biochemistry, 8, 3289. Mangiarotti, G., Apirion, D., Schlessinger, D. & Silengo, L. (1968). Biochemistry, 7, 456. Marmur, J. (1961). J. iqToZ. BioZ. 3, 208. Moore, R. L. & McCarthy, B. J. (1968). Biochem. Genetics, 2, 75. Morell, P., Smith, I., Dubnau, D. & Marmur, J. (1967). Biochemistry, 6, 258. No\relli, C;. D. (1969). J. Cell. Physiol. Suppl. 1, 74. Nygaard, A. P. & Hall, B. D. (1964). J. Mol. BioZ. 9, 125. Oishi, M. & Sueoka, N. (1965). Proc. Nat. Acad. Sci., Wash. 54, 483. Purdom, I., Bishop, J. 0. & Birnstiel, M. (1970). Nature, 227, 239. Sinsheimor, R. L. (1959). J. Mol. BioZ. 1, 37. Walker, P. M. B. (1969). Progress in Nucleic Acid Research, 9, 301. Waters, L. C. & Novelli, G. D. (1968). Biochem. Biophys. Res. Comm. 32, 971. U’eiss, S. B., Hsu, W., Foft, J. W. & Scherberg, N. H. (1968). Proc. Nat. Acad. Sci., Wash.
61, 114. Wotmur, J. G. & Davidson, N. (1968). J. Mol. BioZ. 31, 349. Williamson, R., Morrison, M. & Paul, J. (1970). Biochem. Biophys. Res. Comm. 40, 740. Wong, J. T., Mustard, M. & Herbert, E. (1969). Biochim. biophys. Acta, 174, 531. Yang, W. K. & Novelli, G. D. (1971). Methods in Embryology XII, Part C. Methods in
Nucleic Acids, ed. by Grossman & Moldavo. Zehavi-Willner, T. & Comb, D. G. (1966). J. Mol. Biol. 16, 250.
Note added i-n proof: Yaniv & Barr&l (1971, Nature New Biol., 223, 113) have recently shown that valine-tRNA from E. coli is separable into three subspecies. The sequences of valine,, and valinez,-tRNA were shown to be identical but for six residues, while valine,- tRNA shared 55 out of 77 bases with valine,-tRNA. This is consistent with our hybridiza- tion data (Table 5) which reveal the presence of three closely similar genes for valine-tRNA in the E. coli genome.














![Understanding the Functions of Long Non-Coding …...Two types of RNA molecules exist [1]: messenger RNA (mRNA) molecules, which possess the ability to encode the amino acid sequence](https://img.dokumen.tips/doc/110x75/5f0a5b967e708231d42b3fa6/understanding-the-functions-of-long-non-coding-two-types-of-rna-molecules-exist.jpg)














