Embed Size (px)
Citation preview

CARE OF THE ADULT WITH SICKLE CELL
ANEMIA
Julie Overbey DNP(c), FNP-C, ACNP-BC, OCNNurse PractitionerAdult Sickle Cell ProgramBanner Health

Objectives Overview of sickle cell disease Not to be missed sequelae!
Secondary Pulmonary Hypertension Acute Chest Syndrome Cardiovascular Aplastic Crisis Splenic Sequestration Exchange versus simple transfusion
Legal obligation to treat Aberrant Behaviors My role Final Thoughts

Overview Most common in African Americans but
can be seen in Mediterranean, Saudi Arabia and South America
Interracial marriages have increased the incidence of sickle cell disease in Native Americans and Hispanics Hgb SC/Beta Thal more common in
NA/Hispanics Protective properties against malaria Mutant gene was discovered in individuals
who didn’t contract malaria during outbreaks or who relatively mild cases

Genetics Autosomal recessive genetic disease
Most common genetically inherited hemoglobinopathy Both mother and father must pass on genetic mutation. Parents may not have sickle cell disease – only trait
Trait is NOT sickle cell disease Trait is usually asymptomatic Hematuria – rare No one is the 1/100,000 individual who is “different”
Parents with trait/disease have: 25 percent chance of having an unaffected child with
normal hemoglobin A 50 percent chance of having a child who also is a
carrier/trait A 25 percent chance of having a child with sickle cell
anemia

Prevalence SCD affects 90,000 to 100,000
Americans. SCD occurs in about 1 out of every 500
Black or African-American births. SCD occurs in about 1 out of every
36,000 Hispanic-American births. SCT occurs in about 1 in 12 Blacks or
African Americans.

Phenotype Overview Hgb SS (6-9 g/dl)
Most severe form Profound anemia Most common Increased incidence of the sequelae of disease Life expectancy 45-48 years
Hgb SC (8-11 g/dl) Fewer painful crisis Higher incidence of bone infarcts Minimal need for transfusion Increased risk of blindness Life expectancy >70 years

Pathophysiology of Sickling Cells Average RBC is in circulation approximately 120
days Sickle cell RBC’s last approximately 14-21 days Sickle hemoglobin has
Low solubility Rapid polymerization with deoxygenation Rapid turnover of cells leads to chronic anemia Reduced nitric oxide
Cells clump and stick in vessel bifurcations Sickling causes cell hypoxia which leads to painful
crisis Sickling leads to decreased oxygen carrying
capacity which can lead to end-organ damage and bony infarcts

Common Causes of Pain Stress
Physical psychosocial
Dehydration Changes in weather
Extremes in weather Rain or impending storms
Infection Precipitous drop in hemoglobin from baseline Illicit drug use
Any drug that increases metabolism – cocaine, methamphetamines
Bony Infarcts

Common Issues Renal failure
Caused by sticky RBC’s clogging tubules Inability to concentrate urine
Cardiovascular Accident More common in children
Blindness More common in Hgb SC
Skin Ulcerations Sticky RBC’s provide inadequate circulation to periphery
Gallstones increased bilirubin collection in gallbladder
Infection Usually as a result of an autosplenectomy

Secondary Pulmonary Hypertension
Mechanism in SCD is unknown Possible causes:
Fat embolism Sequestered erythrocytes causing a
vasculopathy Recurrent infection
asplenic Chronic hypoxia causing remodeling of the
vasculature with smooth muscle proliferation and fibrosis
Pulmonary scarring from repeated episodes of ACS

Secondary Pulmonary Hypertension As PH worsens, patients complain of chest
pain and dyspnea, and have hypoxemia at rest High risk of
right sided heart failure (cor pulmonale) syncope sudden death from pulmonary thromboembolism systemic hypotension cardiac arrhythmias
Unless an echocardiogram shows tricuspid regurgitation with increased pulmonary artery pressure, the diagnosis requires right sided cardiac catheterization.

Acute Chest Syndrome Second most common cause of
hospitalization Occurs in 15-43% of all patients Recurrent episodes occur approximately 80% of
patients who have had a previous episode Of those 80% mortality increases 50% with
each subsequent episode Responsible for up to 25% of deaths
Components New infiltrate in at least one lobe Fever Cough/Tachypnea/Chest Pain Increased leukocytosis

Acute Chest Syndrome Some thought that aggressive hydration
may contribute to ACS but that is not supported
Etiology is unclear Infection Vaso-occlusion Combination of both Reduction of nitric oxide as a result of rapid
hemolysis Organisms
Chlamydia pneumoniae Mycoplasma pneumoniae RSV

Treatment Recommendations Identify and treat all underlying precipitating factors
Macrolide and quinolone (coverages for atypicals) Transfuse if indicated Hydration
Supplemental oxygen to treat hypoxia and maintain arterial oxygen tension above 70 mm Hg
Optimal pain control and fluid management
Ongoing respiratory therapy Bronchodilators Incentive spirometry Supplemental oxygen
Simple or exchange transfusion to reduce hemoglobin S concentration and to enhance oxygen carrying capacity Multiple lobe involvement Increased oxygen needs
Miscellaneous: NO inhalation, systemic steroids, mechanical ventilation, and extracorporeal membrane oxygenation


Cardiovascular Cardiac exam findings are rarely normal in
sickle cell disease The heart is usually enlarged and the
precordium hyperactive, systolic murmurs and premature contractions are often present in adults
Physical work capacity is reduced to about half in adults with sickle cell anemia and 60 to 70 percent in children
This is related to the severity of the anemia Sudden unexpected and unexplained death
is common in adults with sickle cell anemia

Cardiovascular Patients with SCD can have autonomic nervous
system dysfunction that may contribute to sudden death.
Chest pain, a common complaint and often leads to patients being told they have had a heart attack Ask if the pain is consistent with their usual sickle
cell pain or does it feel different MI is usually from other comorbidity
Obvious myocardial infarction is unusual Paradoxically, coronary artery occlusion is not
common suggesting that small vessel disease is responsible for the cardiac damage

Splenic Sequestion Acute splenic sequestration complication (ASSC) is
caused by intrasplenic trapping of red cells causing a precipitous drop in
hemoglobin level potential for hypoxic/hypovolemic shock
ASSC remains a leading cause of death in children with SCD
ASSC may be defined by a decrease of at least 2 g/dL from the steady-state
hemoglobin concentration evidence of increased erythropoiesis such as a markedly
elevated reticulocyte count an acutely enlarging spleen
The attacks are often associated with viral or bacterial infections
Acute chest syndrome occurs in 20 percent

Splenic Sequestration The usual clinical manifestations are
sudden weakness pallor tachycardia tachypnea abdominal fullness
The immediate treatment of acute splenic sequestration is directed toward correction of hypovolemia with red blood cell transfusion (simple transfusion)
Severe ASSC can be fatal within a few hours so emergent transfusion is required
Once transfusion is employed, red cells sequestered in the spleen are remobilized, splenomegaly regresses, and the hemoglobin level increases, often to a level greater than predicted

Aplastic Crisis Aplastic: “unable to form” Temporary cessation of red blood cell
proliferation Absence of reticulocytosis Temporary (usually 5-10 days) Often associated with
Parvo B19 virus (fifth’s disease) Strep EBV

Aplastic Crisis Symptoms:
Pallor Fatigue Activity intolerance Shortness of breath Pancytopenia rarely occurs
More prevalent in children Treatment
Simple transfusion with Hgb S- PRBC’s Supportive symptom management

Red Cell Exchange Transfusion Clinical Indication
Acute infarctive stroke (mostly in children – STOP 1 and STOP 2 clinical trials)
Acute chest syndrome Preoperative for Hgb SC patients undergoing
medium to high risk surgery under general anesthesia
Priapism Multi-system organ dysfunction/failure Reduction in circulating Hgb S levels Reduce viscosity Not proven to be helpful in resolution of vaso-
occlusive crisis
Red Cell Exchange Works by:
Decreasing rate of hemolysis Decreasing liver processing of bilirubin
Severe cholestasis Right upper quadrant syndrome
Damage to renal tubular cells Scavenging of nitric oxide by free
hemoglobin released from sickling cells

Red Cell Exchange Transfusion Benefits:
Iron neutrality Decreased fluid burden
Risks Line failure Blood reaction Vasculature collapse
Goals Keep Hgb close to 10 g/dl – do not exceed Hgb S % <35 Approximately 8-9 units of Hgb S- PRBC’s

Simple Transfusion The transfusion of a single unit of Hgb S- red
cells Phlebotomy vs simple transfusion All PRBC’s need to be Hgb S- (unless emergent) No clear evidence to support a specific Hgb level
Useful in patients with: Chronic pain syndrome Antepartum (for fetus – not mother) Preoperatively to maintain Hgb of 10 g/dl
Benefits Increased oxygenation Decreased % of Hgb S (sickling cells)

Simple TransfusionRisks
Delayed transfusion reaction TRALI Iron overload Alloimmunization Autoimmunization Hemolysis Hyperviscosity

Indication How To Transfuse
Hepatic sequestration Exchange or simple transfusion
Intrahepatic cholestasis Exchange or simple transfusion
Multisystem organ failure (MSOF) Exchange or simple transfusion
Aplastic crisis Simple transfusion
Symptomatic anemia (see page 43 in the “Managing Acute Complications of Sickle Cell Disease” chapter)
Simple transfusion
Indication How To Transfuse Quality of Evidence Strength of
Recommendation
Symptomatic acute chest syndrome (ACS) combined with a decreased Hb of 1 g/dL below baseline
Simple transfusion Low Weak
Symptomatic severe ACS (as defined by an oxygen saturation less than 90% despite supplemental oxygen)
Exchange transfusion Low Strong
Acute splenic sequestration plus severe anemia
Simple transfusion Low Strong
Stroke Simple or exchange transfusion
Low Moderate

Legal Implications Appropriate triage Appropriate treatment for symptoms
Pain Does not require IV analgesia May use oral or IM Does not require opioids
Could use NSAID’s Fluids
Hypotonic preferred Prescriptions
Review CSPMP No legal obligation to provide prescriptions

Admission Criteria Every facility is different No legal obligation to admit for primary
pain control Immediate or foreseeable need to
transfuse Hgb <7 or with hydration will drop to this
level Symptomatic anemia
Infection Concern for “symptoms not to miss”
sequelae Risk to self or others

Example from CSPMP
08/28/2015 OXYCODONE AND ACETAMINOPHEN, 325 MG;5 MG, TABLET 30.00 (10) FG1983205 (MIHS)
08/19/2015 OXYCODONE AND ACETAMINOPHEN, 325 MG;5 MG, TABLET 20.00 (5) FB2883139 (BDMC)
07/07/2015 OXYCODONE HYDROCHLORIDE, 30 MG, TABLET 60.00 (10) AB2187905 (Hematologist)
06/29/2015 OXYCODONE HYDROCHLORIDE, 30 MG, TABLET 30.00 (7) BK2676558 (Phoenix St. Luke’s)
06/19/2015 OXYCODONE HYDROCHLORIDE, 15 MG, TABLET 20.00 (5) BC7786140 (St. Joe’s)
06/10/2015 OXYCODONE HYDROCHLORIDE, 15 MG, TABLET 15.00 (3) MG2395398 (St. Joe’s)
06/02/2015 MORPHINE SULFATE, 30 MG, TABLET, EXTENDED RELEASE 15.00 (5)FA0933108 (Scottsdale Osborne)
05/20/2015 OXYCODONE HYDROCHLORIDE, 30 MG, TABLET 24.00 (4)FA0933108 (Scottsdale Osborne)
05/18/2015 ALPRAZOLAM, 2 MG, TABLET 8.00 (4)FA0933108 (Scottsdale Osborne)
04/20/2015 MORPHINE SULFATE, 30 MG, TABLET, EXTENDED RELEASE 40.00 (20)BS9877842 (Arizona Oncology)
All but 1 of these were cash pay
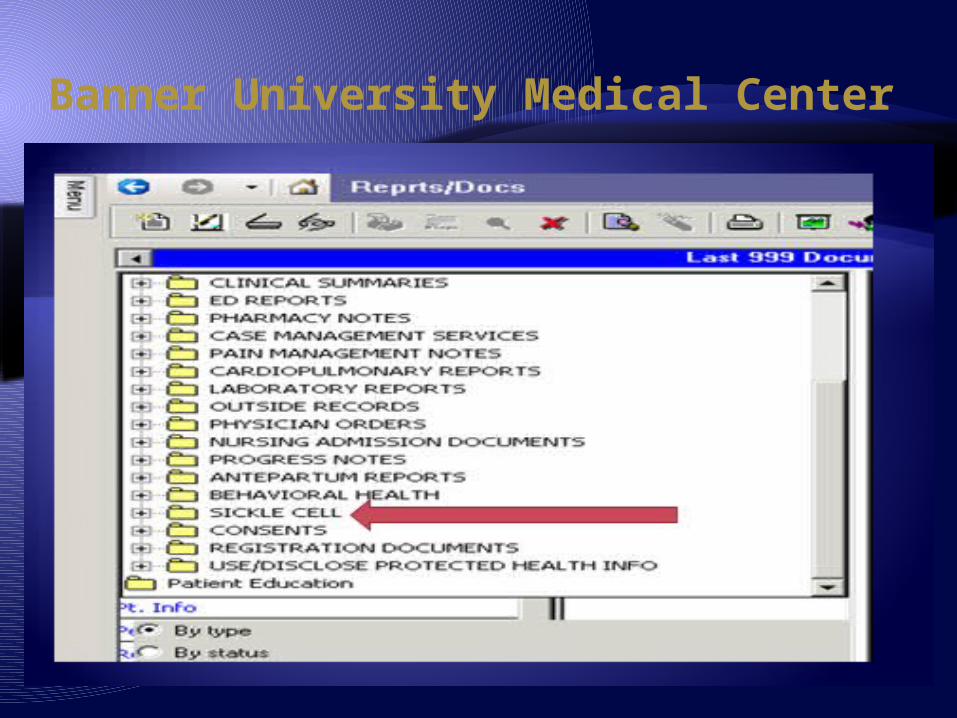
Banner University Medical Center

Aberrant behaviors This is a population full of challenges Development of aberrant behaviors
To get what they want In response to the quality of care Treated as drug addicts or drug seeking
Doses of IV opioids less than what the current home regimen is
Lack of trust in healthcare – adversarial relationship
Lack of discharge follow through No PCP No outside pain provider or hematologist

Behavior Hospital hopping High tolerance Will ask for the “cocktail”
Dilaudid, Phenergan and Benadryl IVP Okay to refuse – no obligation to provide
together Threats
File a complaint about care Racial discrimination Harm to you or your family

My role Was created in response to:
Nursing and physician dissatisfaction Aberrant behaviors Develop standardized care
What is offered: 4.5 years ago development of the outpatient
infusion clinic dedicated to adults with sickle cell Only program of its kind in Arizona
Hydration/pain management Transfusions 2 visits per week - 4 hours per visit

Final Thoughts Patients will try to treat their pain at
home for 3-5 days before coming in Pain is what the patient says it is
Until proven otherwise Stereotyping will eventually lead to
missed signs and symptoms of more serious illnesses associated with SCD Acute chest syndrome MI - rare Subtle presentation of a stroke – rare in
adults

Resources National Heart, Lung and Blood Institute Evidence-based management of sickle
cell disease (2014) https://www.nhlbi.nih.gov/sites/www.nhlbi.nih.gov/files/sickle-cell-disease-report.pdf

How to contact me Cell
Personal Cell – please do not give to patients
Office (602) 839-4766








![Lezione3 acnp 2010-2011 [modalità compatibilità]](https://img.dokumen.tips/doc/110x75/54b3ce8b4a7959dc618b456b/lezione3-acnp-2010-2011-modalita-compatibilita.jpg)




















