Embed Size (px)
Citation preview

J Neurosurg Volume 122 • April 2015
case reportJ Neurosurg 122:773–777, 2015
Desmoplastic small round cell tumors (DSRCTs) are rare primitive neoplasms that have an aggressive clinical course. Despite multiple modalities of
treatment, these tumors remain incurable and have poor long-term survival rates. They were first described in 1989 by Gerald and Rosai.3 These tumors are more common in males and usually present in adolescence or early adulthood. There are no reliable biomarkers or specific radiological signs for their diagnosis; however, recent advances in molecular biology and immunohistochemistry allow reliable diagnosis through examination of a biopsy specimen in most cases. Cytogenetic analysis in DSRCTs has shown a specific chromosomal abnormality, t(11;22)(p13;q12), as a result of gene translocation and rearrangement of the
genes for Ewing’s sarcoma (EWS) and Wilms tumor 1 (WT1). The gene fusion of EWS-WT1 is described to be specific to DSRCTs.4
DSRCTs typically arise from abdominal and pelvic peritoneum and spread over serosal surfaces, leaving multiple small peritoneal implants. There have been a small number of reports of DSRCTs affecting other organs, such as lymph nodes, ovaries, kidneys, pancreas, tunica vaginalis, testis, liver, and pleura. Such tumors arising within the cranium are extremely rare. Three definite and two possible cases of intracranial DSRCTs (one tentorial, one cerebellopontine angle, one temporal lobe, and two cerebellar tumors) have been previously reported.1,7,10,11 Here we report the first case of intracranial DSRCT presenting as
abbreviatioNs DSRCT = desmoplastic small round cell tumor; EMA = epithelial membrane antigen; EWS = Ewing’s sarcoma; FISH = fluorescent in situ hybridization; PLAP = placental alkaline phosphatase; RT-PCR = reverse transcription polymerase chain reaction; WT1 = Wilms tumor 1.submitted November 12, 2013. accepted October 16, 2014.iNclude wheN citiNg Published online December 5, 2014; DOI: 10.3171/2014.10.JNS132490.disclosure The authors report no conflict of interest concerning the materials or methods used in this study or the findings specified in this paper.
Intracranial desmoplastic small round cell tumor presenting as a suprasellar masssravan K. thondam, mrcp,1 daniel du plessis, Frcpath,2 daniel J. cuthbertson, phd,1 Kumar s. v. das, Frcr,3 mohsen Javadpour, Frcs(sN),3 ian a. macFarlane, Frcp,1 James leggate, Frcs,2 brian haylock, Frcr,4 and christina daousi, Frcp1
1Department of Endocrinology, University Hospital Aintree, Liverpool; 2Neuropathology Unit and Clinical Neurosciences Center, Salford Royal Hospital, Salford, Greater Manchester; 3Walton Center for Neurology & Neurosurgery, Liverpool; and 4Clatterbridge Cancer Center, Liverpool, United Kingdom
Desmoplastic small round cell tumors (DSRCTs) are rare, aggressive neoplasms that typically arise from abdominal and pelvic peritoneum in young adults. Other primary sites are uncommon, and an intracranial origin is exceptionally rare. Here the authors report the first case of a DSRCT presenting as a primary suprasellar tumor causing panhypopituitarism and severe bitemporal hemianopia in a young man. Macroscopic debulking of the tumor was undertaken, and histology revealed features of DSRCT. Reverse transcription polymerase chain reaction confirmed the presence of Ewing’s sar-coma–Wilms tumor 1 (EWS-WT1) gene rearrangement specific to DSRCT. Postoperative whole-body imaging showed no primary malignancy elsewhere. The tumor recurred 4 months after surgery, and this was followed by cervical and mediastinal lymph node metastases. The patient died 20 months after initial presentation of rapidly progressive disease. DSRCTs should be included in the differential diagnosis of an unusual suprasellar mass in young adults. Early diagnosis is essential, and once the tumor is identified histologically, gross-total resection and radical postoperative treatment involving radiotherapy, chemotherapy, and close surveillance are required because of the lesion’s potential for rapidly progressive malignancy.http://thejns.org/doi/abs/10.3171/2014.10.JNS132490Key words desmoplastic small round cell tumor; suprasellar mass; EWS-WT1 gene fusion; intracranial malignant tumor; oncology
773©AANS, 2015
Unauthenticated | Downloaded 04/01/22 01:46 PM UTC
s. K. thondam et al.
J Neurosurg Volume 122 • April 2015
a suprasellar mass. The tumor caused panhypopituitarism and visual field defects as a result of optic chiasm compression in a young man and had a fatal outcome because of progressive disease.
case reportHistory and Examination
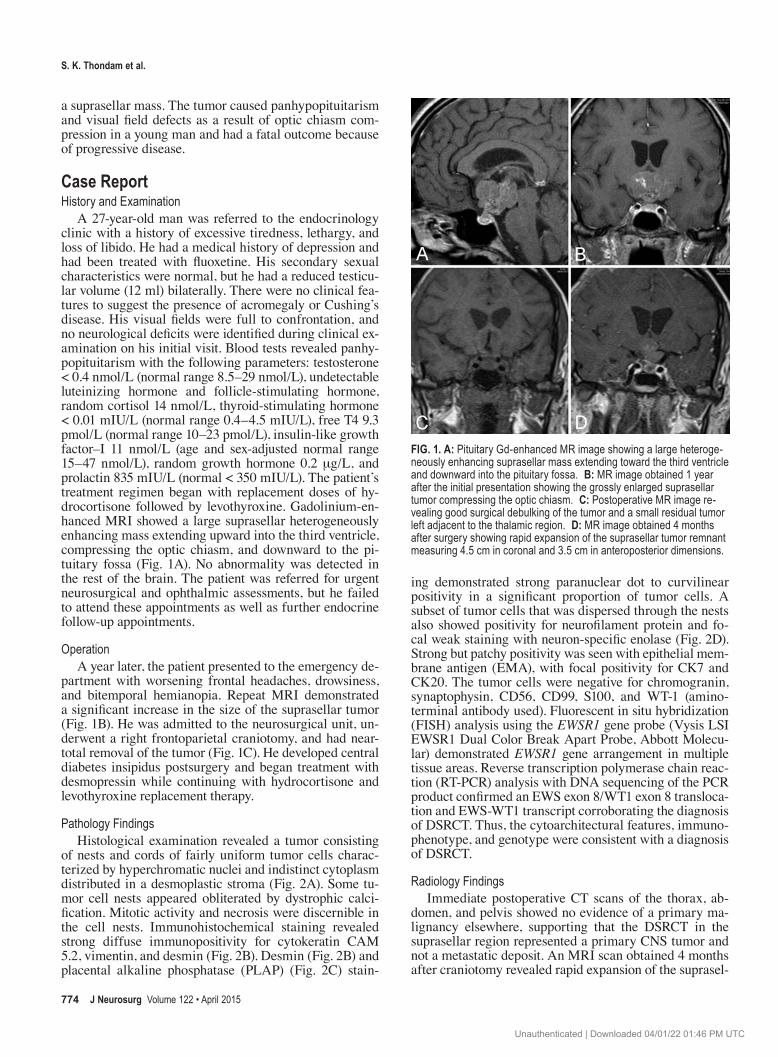
A 27-year-old man was referred to the endocrinology clinic with a history of excessive tiredness, lethargy, and loss of libido. He had a medical history of depression and had been treated with fluoxetine. His secondary sexual characteristics were normal, but he had a reduced testicular volume (12 ml) bilaterally. There were no clinical features to suggest the presence of acromegaly or Cushing’s disease. His visual fields were full to confrontation, and no neurological deficits were identified during clinical examination on his initial visit. Blood tests revealed panhypopituitarism with the following parameters: testosterone < 0.4 nmol/L (normal range 8.5–29 nmol/L), undetectable luteinizing hormone and folliclestimulating hormone, random cortisol 14 nmol/L, thyroid-stimulating hormone < 0.01 mIU/L (normal range 0.4–4.5 mIU/L), free T4 9.3 pmol/L (normal range 10–23 pmol/L), insulin-like growth factor–I 11 nmol/L (age and sex-adjusted normal range 15–47 nmol/L), random growth hormone 0.2 μg/L, and prolactin 835 mIU/L (normal < 350 mIU/L). The patient’s treatment regimen began with replacement doses of hydrocortisone followed by levothyroxine. Gadoliniumenhanced MRI showed a large suprasellar heterogeneously enhancing mass extending upward into the third ventricle, compressing the optic chiasm, and downward to the pituitary fossa (Fig. 1A). No abnormality was detected in the rest of the brain. The patient was referred for urgent neurosurgical and ophthalmic assessments, but he failed to attend these appointments as well as further endocrine followup appointments.
OperationA year later, the patient presented to the emergency de
partment with worsening frontal headaches, drowsiness, and bitemporal hemianopia. Repeat MRI demonstrated a significant increase in the size of the suprasellar tumor (Fig. 1B). He was admitted to the neurosurgical unit, underwent a right frontoparietal craniotomy, and had neartotal removal of the tumor (Fig. 1C). He developed central diabetes insipidus postsurgery and began treatment with desmopressin while continuing with hydrocortisone and levothyroxine replacement therapy.
Pathology FindingsHistological examination revealed a tumor consisting
of nests and cords of fairly uniform tumor cells characterized by hyperchromatic nuclei and indistinct cytoplasm distributed in a desmoplastic stroma (Fig. 2A). Some tumor cell nests appeared obliterated by dystrophic calcification. Mitotic activity and necrosis were discernible in the cell nests. Immunohistochemical staining revealed strong diffuse immunopositivity for cytokeratin CAM 5.2, vimentin, and desmin (Fig. 2B). Desmin (Fig. 2B) and placental alkaline phosphatase (PLAP) (Fig. 2C) stain
ing demonstrated strong paranuclear dot to curvilinear positivity in a significant proportion of tumor cells. A subset of tumor cells that was dispersed through the nests also showed positivity for neurofilament protein and focal weak staining with neuron-specific enolase (Fig. 2D). Strong but patchy positivity was seen with epithelial membrane antigen (EMA), with focal positivity for CK7 and CK20. The tumor cells were negative for chromogranin, synaptophysin, CD56, CD99, S100, and WT-1 (amino-terminal antibody used). Fluorescent in situ hybridization (FISH) analysis using the EWSR1 gene probe (Vysis LSI EWSR1 Dual Color Break Apart Probe, Abbott Molecular) demonstrated EWSR1 gene arrangement in multiple tissue areas. Reverse transcription polymerase chain reaction (RT-PCR) analysis with DNA sequencing of the PCR product confirmed an EWS exon 8/WT1 exon 8 translocation and EWS-WT1 transcript corroborating the diagnosis of DSRCT. Thus, the cytoarchitectural features, immunophenotype, and genotype were consistent with a diagnosis of DSRCT.
Radiology FindingsImmediate postoperative CT scans of the thorax, ab
domen, and pelvis showed no evidence of a primary malignancy elsewhere, supporting that the DSRCT in the suprasellar region represented a primary CNS tumor and not a metastatic deposit. An MRI scan obtained 4 months after craniotomy revealed rapid expansion of the suprasel
Fig. 1. a: Pituitary Gd-enhanced MR image showing a large heteroge-neously enhancing suprasellar mass extending toward the third ventricle and downward into the pituitary fossa. b: MR image obtained 1 year after the initial presentation showing the grossly enlarged suprasellar tumor compressing the optic chiasm. c: Postoperative MR image re-vealing good surgical debulking of the tumor and a small residual tumor left adjacent to the thalamic region. d: MR image obtained 4 months after surgery showing rapid expansion of the suprasellar tumor remnant measuring 4.5 cm in coronal and 3.5 cm in anteroposterior dimensions.
774
Unauthenticated | Downloaded 04/01/22 01:46 PM UTC

suprasellar desmoplastic small round cell tumor
J Neurosurg Volume 122 • April 2015
lar tumor remnant measuring 4.5 × 3.5 cm (Fig. 1D). The patient started receiving fractionated conformal radiotherapy, but treatment was discontinued after 1 week because of general deterioration in his condition. A repeat CT scan of the thorax, abdomen, and pelvis showed extensive metastatic disease involving mediastinal, subcarinal, and hilar lymph nodes, causing occlusion of the right main bronchus and right pulmonary infiltrates. No intraabdominal masses or lymph nodes were identified, and the appearances of the liver, spleen, and adrenal glands were normal. The histological results of a fine-needle aspirate obtained from a cervical lymph node were once again consistent with DSRCT. Palliative chemotherapy was considered; however, the patient’s clinical condition deteriorated rapidly and he died 20 months after his initial presentation.
discussionAn intracranial DSRCT presenting as a suprasellar
mass poses a major challenge in diagnosis and management. A tissue biopsy is essential to differentiate this lesion from other suprasellar tumors, such as invasive pituitary adenomas, craniopharyngiomas, gliomas, meningiomas, and metastases. Although the majority of DSRCTs have characteristic histological features, a significant proportion can exhibit a wide range of morphological features.8 The main differential diagnoses for these tumors presenting in the CNS based on histological findings should include medulloblastomas, atypical teratoid/rhabdoid tumors, and primitive neuroectodermal tumors.7 Immunocytochemistry and molecular genetics help to distinguish DSRCTs reliably from other tumors.
Cytogenetic analysis in DSRCTs has shown a specific chromosomal abnormality, t(11;22)(p13;q12), as a result of gene translocation and rearrangement of the genes for
EWS and WT1.4 This can be demonstrated by RTPCR or by FISH. The authors of a large series of DSRCTs reported a significant variation in clinical features, cytology, and immunoreactivity, but the gene fusion EWS-WT1 was consistently distinctive to this tumor.2 Histological examination, immunocytochemistry, and genotyping in our case demonstrated features typical of a DSRCT, which are quite distinct from the other tumors mentioned above.
The rare occurrence of these tumors makes the study of different treatment modalities and their impact on survival very difficult. Kushner et al.5 showed that intense alkylator-based therapy with the P6 protocol (cyclophosphamide, doxorubicin, vincristine, ifosfamide, and etoposide) improved remission rates in patients with intraabdominal or pelvic DSRCTs. In that study, 7 of 12 patients achieved a shortterm remission, and 4 had complete remission for over 1 year. Quaglia and Brennan9 studied 40 patients, 27 of whom had intraabdominal tumors. Overall survival at 3 years from diagnosis was 29%, and induction chemotherapy with P6 protocol and gross-total resection of the tumor were associated with improved survival. Lal et al.6 showed that multimodality treatment improved the survival rate in a study of 66 patients with predominantly intraabdominal tumors. The 3-year survival rate was 55% in patients receiving induction chemotherapy (P6 protocol) combined with surgical debulking and radiotherapy.
Systematic review of the literature yielded only 5 cases of intracranial DSRCTs. Evaluation of presenting symptoms, pathological findings, management, and outcome of these limited cases shows the aggressive nature of these tumors despite multiple modalities of treatment (Table 1). Tison et al.10 reported on a 24-year-old man with posterior cranial fossa tumor adherent to the tentorium. This patient underwent a partial excision of the tumor and received 3 cycles of chemotherapy (PCNU cisplatin and V16) combined with radiotherapy and was receiving intracranial methotrexate at the time the study was published. There are no details published on longterm survival for this patient. Neder et al.7 reported on 2 cases of DSRCT. The first patient was a 37-year-old man with a cerebellopontine angle tumor. He underwent subtotal resection and stereotactic irradiation but the disease progressed to spinal metastases 6 months later. He died 2 years after diagnosis despite receiving wholebrain and wholespine radiotherapy and chemotherapy. The second patient was a 39-year-old man, with multiple lesions in the left cerebellar hemisphere and spinal leptomeninges, who presented with cauda equina syndrome. He underwent surgical decompression of the spinal cord and conus medullaris followed by radiotherapy and 3 courses of chemotherapy. He developed further posterior fossa and spinal axis lesions 27 months after diagnosis.
Yachnis et al.11 described a possible DSRCT in a 9-month-old girl with an aggressive posterior cranial fossa tumor requiring the placement of a ventriculoperitoneal shunt for obstructive hydrocephalus. She required a second craniotomy for tumor debulking surgery 18 months later despite intensive chemotherapy. Long-term survival details of this patient are not available. Bouchireb et al.1 described a 6-year-old girl with a right temporal lobe tumor in whom histological and immunochemistry findings
Fig. 2. a: Nests of tumor cells with hyperchromatic nuclei and indistinct cytoplasm surrounded by desmoplastic stroma. H & E stain, original magnification ×100. b: Paranuclear dot and curvilinear desmin im-munopositivity of tumor cells. Desmin, original magnification ×630. c: Distinctive paranuclear curvilinear and dot positivity for PLAP. Original magnification ×400. d: Subset of tumor cells showing neurofilament immunostaining. Original magnification ×200. Figure is available in color online only.
775
Unauthenticated | Downloaded 04/01/22 01:46 PM UTC

s. K. thondam et al.
J Neurosurg Volume 122 • April 2015
tabl
e 1.
lite
ratu
re re
view
of r
epor
ted
case
s of i
ntra
cran
ial d
srct
s
Authors &
Year
Age, Se
x
Clinical Featur
es
on Initia
l Presentation
MRI Find
ings
Histo
logy, Immu
nocytochem
istry,
& Cy
togenetics
Site of
Metastasis
Treatment
Outco
me
Yachnis
et
al., 1992
9 mos, F
Proje
ctile vomiting
& inc
reased
head circum
fer-
ence
Enhancing
rt ce
rebellar m
ass
comp
ressing
& displac
ing
brain
stem
Marked o
bstru
ctive hy
dro-
cephalu
s
Dema
rcate
d nests of tum
or ce
lls
separated
by mesenchym
al strom
aNe
st cell p
ositiv
e for GFA
P, cyto-
keratin, &
vime
ntin
Negative for de
smin immu
nore-
activity
None at tim
e of
publication
Ventr
iculop
eritoneal shunt
Chem
o w/ cisp
latin, cy
clophospham
ide,
vincristine
, & high-dose m
ethotr
exate
Second craniotom
y for de
bulking su
rgery
1.5 yrs a
fter o
rigina
l presentation
Alive
at tim
e of
publication
Tison e
t al.,
1996
24 yrs, M
Headache, vom
it-ing
, vertigo, &
impaired h
earing
Poste
rior crania
l fossa tum
or
(3 × 4 × 3.5 c
m) ad
herent
to ten
torium
& pe
trous pa
rt of tem
poral bone, dis
plac-
ing lt cerebellar h
emi-
sphere
Nests
of sm
all un
iform
round &
oval tum
or ce
lls se
parated
by
desm
oplas
tic stroma
Strong im
munoreactivity for kera-
tin, vimentin, desmin, & EM
ASo
uthern blot for g
enom
ic DN
A extra
ction, P
CR an
alysis
of
EWS-WT1 fusio
n gene
None at tim
e of
publication
Partial excis
ion of tumo
rTh
ree c
ycles
of ch
emo c
onsis
ting o
f PCN
U cis
platin & V16; in
tracranial meth
otrex-
ate ev
ery 4
0 days; radio
therapy
Alive
& no
clini cal
signs of re
-lap
se at tim
e of
publication
Bouchireb et
al., 2008
6 yrs, F
3-wk
histo
ry of
headaches,
comp
lex pa
rtial
seizu
res, &
dysphasia
Well-dem
arcated
heter
oge-
neously
enhancing
mass in
rt tem
poral lo
be
Small malignant cells w
/ hyper-
chroma
tic nu
clei & eo
sino-
philic
cytop
lasm em
bedded in
fibromy
xoid strom
aPo
sitive
stain
ing for vimentin,
desm
in, & sy
napto
physin
EWS-WT1 translo
cation b
y FISH
None at tim
e of
publication
Comp
lete e
xcision of tumo
rSe
ven c
ourses of ch
emo w
/ P6 p
rotocol
(cyclo
phospham
ide, doxorubicin, vin
-cristine
, ifosfamide, &
etoposide
); focal
confo
rmal radio
therapy to tum
or be
d at
54 Gy
Alive
& disease
free a
fter 18
mos o
f follow-
up
Neder et al.,
2009
37 yrs, M
5-mo
histo
ry of lt-
sid
e hearing
loss &
tinnitus
Heter
ogeneously enhancing
ma
ss in lt CP
A w/ mild
mass effec
t
Intracranial & sp
inal biop
sies:
round to o
val hyperchroma
tic
nucle
i w/ in
conspic
uous nu
cle-
oli in ad
dition
to de
smoplas
tic
strom
a Po
sitive
stain
ing for E
MA, CAM
5.2, desm
in, & nu
clear IN
I-1EW
S-WT1 translo
cation b
y RT-
PCR & FISH
Spina
l lepto
me-
ninges 6
mos
after
diagno-
sis
Initia
l subtotal resection of CPA
mass &
ste
reotactic ra
diation to tum
or be
dDe
bulking of intra
dural spin
al nodules
&
spina
l radia
tion (4500 ra
d); radios
urgery
to CP
A (10
0 rad) &
whole brain
(360
0 rad)
Chem
o (carboplatin tem
ozolo
mide)
Died 2 yrs a
fter
diagnosis w/
progressive
dis
ease
39 yrs, M
4-mo
histo
ry of ga
it imbalan
ce &
bilat low
er lim
b we
akness; la
ter
develop
ed
urina
ry & fecal
incontinence
Multiple e
nhancin
g lesion
s in
lt cerebellar he
misphere &
metasta
sis in sp
inal le
pto-
menin
geal space
Laminecto
my sa
mples
: tumo
r cells w/ oval to
irregular nu
clei
w/ co
arse ch
roma
tin & sc
anty
cytop
lasm; po
sitive
stain
ing for
EMA, CAM
5.2, desm
in, &
nucle
ar IN
I-1; E
WS-WT1
transloc
ation by
RT-PC
R
Metastasis
to
spina
l lep to
-me
ningeal
space a
t presentation
Decomp
ression
of sp
inal cord &
conus
medullaris followe
d by radiotherapy
Laminecto
my of T1
2–L5 sh
owed ca
uda
equin
a nerve ro
ots studded w
/ gray
tumor no
dules
Three c
ourses of ch
emo (cis
platin, etop
o-sid
e, & Ho
loxan)
Increase in po
s-
terior fossa
tumor; further
spread in sp
i-nal axis
after
27 mos of
follow-up
chem
o = ch
emotherapy; C
PA = ce
rebellopontine
angle
.
776
Unauthenticated | Downloaded 04/01/22 01:46 PM UTC

suprasellar desmoplastic small round cell tumor
J Neurosurg Volume 122 • April 2015
were not diagnostic of DSRCT but had the characteristic EWS-WT1 translocation. This patient underwent excision of the mass followed by P6 protocol chemotherapy5 and focal conformal irradiation of tumor bed at 54 Gy. She was reported to be free of the disease 18 months after diagnosis.
In summary, we have described the first case of an intracranial DSRCT presenting as a suprasellar mass, causing panhypopituitarism and bitemporal hemianopia in a young man who died of progressive disease. Despite multimodality treatment, the aggressive behavior of this tumor was similar to DSRCTs arising from the peritoneal cavity and the few rare intracranial cases. Failure to attend followup outpatient appointments and possible noncompliance with treatment may have also contributed to the poor prognosis in our patient. It is important to include DSRCTs in the differential diagnosis of an unusual suprasellar mass in young adults. Once the tumor is identified histologically, grosstotal resection and radical postoperative treatment involving radiotherapy, chemotherapy, and close surveillance are required because of its potential for an aggressive clinical course.
references 1. Bouchireb K, Auger N, Bhangoo R, Di Rocco F, Brousse N,
Delattre O, et al: Intracerebral small round cell tumor: an unu sual case with EWS-WT1 translocation. Pediatr Blood Cancer 51:545–548, 2008
2. Gerald WL, Ladanyi M, de Alava E, Cuatrecasas M, Kushner BH, LaQuaglia MP, et al: Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small roundcell tumor and its variants. J Clin Oncol 16:3028–3036, 1998
3. Gerald WL, Rosai J: Case 2. Desmoplastic small cell tumor with divergent differentiation. Pediatr Pathol 9:177–183, 1989
4. Gerald WL, Rosai J, Ladanyi M: Characterization of the genomic breakpoint and chimeric transcripts in the EWS-WT1 gene fusion of desmoplastic small round cell tumor. Proc Natl Acad Sci U S A 92:1028–1032, 1995
5. Kushner BH, LaQuaglia MP, Wollner N, Meyers PA, Linds-ley KL, Ghavimi F, et al: Desmoplastic small round-cell tumor: prolonged progressionfree survival with aggressive multimodality therapy. J Clin Oncol 14:1526–1531, 1996
6. Lal DR, Su WT, Wolden SL, Loh KC, Modak S, La Quaglia MP: Results of multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg 40:251–255, 2005
7. Neder L, Scheithauer BW, Turel KE, Arnesen MA, Ketterling RP, Jin L, et al: Desmoplastic small round cell tumor of the central nervous system: report of two cases and review of the literature. Virchows Arch 454:431–439, 2009
8. Ordóñez NG: Desmoplastic small round cell tumor: I: a histopathologic study of 39 cases with emphasis on unusual histological patterns. Am J Surg Pathol 22:1303–1313, 1998
9. Quaglia MPL, Brennan MF: The clinical approach to desmoplastic small round cell tumor. Surg Oncol 9:77–81, 2000
10. Tison V, Cerasoli S, Morigi F, Ladanyi M, Gerald WL, Rosai J: Intracranial desmoplastic small-cell tumor. Report of a case. Am J Surg Pathol 20:112–117, 1996
11. Yachnis AT, Rorke LB, Biegel JA, Perilongo G, Zimmerman RA, Sutton LN: Desmoplastic primitive neuroectodermal tumor with divergent differentiation. Broadening the spectrum of desmoplastic infantile neuroepithelial tumors. Am J Surg Pathol 16:998–1006, 1992
author contributionsConception and design: Thondam, Daousi. Acquisition of data: Thondam, du Plessis, Das, Haylock, Daousi. Drafting the article: Thondam, du Plessis, Cuthbertson, Daousi. Critically revising the article: all authors. Reviewed submitted version of manuscript: all authors. Approved the final version of the manuscript on behalf of all authors: Thondam.
correspondenceSravan Kumar Thondam, University Hospital Aintree, Department of Endocrinology, Clinical Sciences Centre, 3rd Fl., Lower Ln., Liverpool L9 7AL, United Kingdom. email: [email protected].
777
Unauthenticated | Downloaded 04/01/22 01:46 PM UTC


















![Case Report Desmoplastic non-infantile ganglioglioma: A low ...cells.[1] The first report of DIGG, earlier known as desmoplastic supratentorial neuroepithelial tumor, was by Vandenberg](https://img.dokumen.tips/doc/110x75/611dcd70c65a242b5322953a/case-report-desmoplastic-non-infantile-ganglioglioma-a-low-cells1-the-first.jpg)










