Embed Size (px)
Citation preview

Intermetallic Compound AlPd As a Selective Hydrogenation Catalyst:A DFT StudyM. Krajcí*,† and J. Hafner‡
†Institute of Physics, Slovak Academy of Sciences, Bratislava SK-84511, Slovakia‡Faculty for Physics, Center for Computational Materials Science, Vienna University, Vienna A-1090, Austria
ABSTRACT: Recently, it has been demonstrated that intermetallic compounds composed of Pdand Ga or Co and Al provide excellent selectivity for the hydrogenation of acetylene to ethylene.Motivated by experimental works on GaPd catalysts, we have performed a detailed ab initio study ofacetylene hydrogenation by the pseudo 5-fold (120) surface of the isostructural and isoelectronicAlPd compound crystallizing in the B20-type structure. The structure of the surface can bedescribed by a triangle−rectangle tiling, and we demonstrate that the most active sites forhydrogenation are triangular arrangements of two Al and one Pd atom. Acetylene is bound to bridgesites between two Al atoms. The most favorable adsorption site for ethylene is on top of the moststrongly protruding Pd atom. Activation energies for all steps of the reaction have been calculated.We demonstrate that the activation energies for the rate-controlling steps are comparable to thoseon reference catalysts (Pd, Pd−Ag, and Al13Co4) and that a desorption energy for ethylene that islower than the activation energy of ethylene to ethyl provides thus good selectivity. We show thatthe decisive factors for the activity and selectivity of the catalyst are the same on both intermetalliccompounds AlPd and Al13Co4.
■ INTRODUCTIONThe hydrogenation of acetylene to ethylene, C2H2+H2 →C2H4, is a chemical reaction of industrial importance. Ethyleneproduced by a steam cracking process contains a small fractionof acetylene. In the ethylene feedstock used for the productionof polyethylene, any contamination by acetylene has to beremoved to avoid poisoning of the polymerization catalyst.1
Usually Pd-based hydrogenation catalysts are used for thehydrogenation of acetylene to ethylene, and because furtherhydrogenation of ethylene to ethane is undesired, the selectivityof the catalyst plays a significant role. A typical hydrogenationcatalyst contains metallic palladium dispersed on an inert oxidicsupport, e.g., Al2O3. Dispersed palladium exhibits high activitybut unsatisfactory selectivity.The properties of Pd as catalyst for the hydrogenation of
alkynes to alkenes have been studied experimentally andtheoretically for many years. It has been demonstrated that purePd catalysts are selective if the reaction is performed underconditions where subsurface carbon is formed.2,3 However, thesubsurface hydrogen promotes further hydrogenation to analkane and hence reduces selectivity.4 It was also found thatalloying Pd with other metals such as Ag, Au, Pb, or Ga cansignificantly improve selectivity.Osswald et al.5,6 compared the performance of various
catalysts for acetylene hydrogenation under laboratoryconditions. The relevant parameters are the rate of conversion(expressing the concentration of acetylene before and after thereaction) and the selectivity (measuring the concentration ofthe desired product, ethylene, after the reaction). For pure Pd
on an Al2O3 support, a conversion rate of 43% and a lowselectivity of only 17% were reported. For a Pd20Ag80 alloycatalyst, the conversion rate increased to 83% and the selectivityto 49%. Alloying Pd with Ag can be considered as the currentindustrial solution for acetylene hydrogenation.7
Recently, a novel concept for the design of selective andstable catalysts for the hydrogenation of alkynes has beenannounced by Kovnir et al.:8 the isolation of the active sites onthe surface of a complex intermetallic compound. On thesurface of pure metals, every atom is a potential active site. AsPd atoms react strongly with alkynes and alkenes, the reactantsare fully hydrogenated to alkanes and the catalysts have poorselectivity. The coordination of Pd atoms by less reactive atomssuch as Ag in a Pd−Ag alloy catalyst reduces the adsorptionenergies of alkenes and favors their desorption over furtherhydrogenation to alkanes. Detailed ab initio density functionalcalculations, combined with kinetic Monte Carlo studies ofNeurock et al.9−11 have demonstrated that electronic effects(changes in the local electronic density of states) are far lessimportant than geometric effects (the number of Ag atomsoccupying nearest neighbor sites around a Pd atom). Insubstitutional alloys, both components are randomly distrib-uted, and the optimal local coordination is realized only arounda fraction of the Pd atoms present on the alloy surface. Incontrast, the surfaces of intermetallic compounds are ordered,
Received: December 21, 2011Revised: February 15, 2012Published: February 21, 2012
Article
pubs.acs.org/JPCC
© 2012 American Chemical Society 6307 dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−6319

and by appropriately choosing the composition and the crystalstructure of the compound, the environment of the active sitescan be optimized. The isolation of the active sites avoidsundesired interactions and permits to control the activity,selectivity, and stability of the catalyst.Osswald et al.5,6 suggested to use intermetallic Ga−Pd
compounds as a promising new class of highly active andselective catalysts for acetylene hydrogenation. They inves-tigated the activity, selectivity, and long-term stability of GaPdand Ga7Pd3 under different reaction conditions. For a GaPdcatalyst, both conversion and selectivity increased by 91% and56%, respectively, in comparison with the reference catalystsPd/Al2O3 and Pd20Ag80. The initially observed drawback of alow active surface area was overcome by milling and chemicaletching.12 The superior properties of Ga−Pd catalysts areattributed to the isolation of the active Pd sites according to theactive-site isolation concept. In the GaPd compoundcrystallizing in the B20 (FeSi-type) structure,13 each Pd atomis surrounded by 7 Ga atoms only. In the more complexstructure of the Ga7Pd3 compound, Pd atoms are coordinatedto 8 Ga atoms.However, so far the atomistic scenario of the catalytic
reactions on GaPd surfaces is unknown. Enhanced covalency ofGa−Pd bonding prevents the formation of subsurface hydridsand reduces hydrogen supply for unselective hydrogenation,and this contributes to enhance selectivity.14 A GaPdcompound exhibits also a significantly reduced electron densityat the Fermi level and a shift of the Pd 4d band to higherbinding energies compared to elemental Pd.In this work, ab initio DFT methods have been used to
explore a complete atomistic scenario for the complex multistephydrogenation process (coadsorption of hydrocarbons andmolecular hydrogen, dissociation of hydrogen, and reaction ofatomic hydrogen with hydrocarbon molecules and intermedi-ates) on the surface of an AlPd compound. AlPd and GaPd areisostructural and isoelectronic; both crystallize in the B20structure. The chemical properties of Al and Ga in thecompounds with Pd are almost identical. The reason why westarted our investigations with AlPd was the desire to comparethe results with those available from our previous works on thecomplex intermetallic compound Al13Co4 as a selectivehydrogenation catalyst.15,16 These studies have been motivatedby preliminary reports on experiments demonstrating asuperior performance and selectivity of this catalyst.17,18 Thetheoretical results demonstrated that the active sites forhydrogenation are Al atoms forming pentagons around isolatedCo atoms. Because of the high stability of large pentagonalbipyramids forming the constituent building blocks of thecomplex orthorhombic structure of Al13Co4, the surface of thecompound is strongly corrugated, and the active sites areexposed at the edges of stripes formed by Al5Co pentagons. It ishence possible that geometry effects are as important inpromoting the catalytic activity of the Al atoms as theirinteraction with the Co atoms (to whom the role of active siteshad originally been attributed). The comparison with acatalytically active Al-rich compound with a simpler crystalstructure will thus provide further insight into the relativeimportance of structural, chemical, and electronic effects inpromoting an improved catalytic performance.DFT methods allow to identify reaction centers, to
determine adsorption and activation energies, and to searchfor optimal reaction paths, providing information not directlyaccessible to experiment. A prerequisite for any study is a
reliable structural model of the surface. Although the B20structure is not too complex (8 atoms per elementary cell),little is known about the atomic structure of the surfaces. TheB20 structure has a certain hidden relation to the structure oficosahedral Al−transition metal quasicrystals. We shalldemonstrate that it is just the pseudo 5-fold (p5f) surfacecomparable to the 5-fold surface of quasicrystals that isresponsible for the superior catalytic properties of Al(Ga)Pdcompounds.Since DFT does not provide absolute values of adsorption
and activation energies with chemical accuracy (i.e., within ±1−2 kJ/mol), it is important to compare the results with those fora well-studied reference system. For acetylene hydrogenation,such a reference is the (111) surface of Pd. Ab initio DFTstudies of acetylene hydrogenation over Pd(111) have beenpresented, e.g., by Sheth et al.9 For the C2H2+H → C2H3reaction, activation energies of 66 and 50 kJ/mol are reportedfor surface coverages of 25% and 33%, respectively. At thesehigh coverages, lateral interactions between the reactantscannot be neglected so that the comparison with a reactionat the surface of a complex intermetallic compound (where thedistances between active sites are much larger) is notstraightforward. We have calculated the energetics of acetylenehydrogenation over Pd(111) at a coverage of 4% and foundthat the activation energy for this reaction increases to 75 kJ/mol. Calculations for lower coverages require large models, andtherefore, they are computationally very demanding.Hydrogenation of acetylene to ethylene over Pd(111)
proceeds through a Horiuti−Polanyi mechanism scheme via avinyl intermediate. While the hydrogenation of acetylene tovinyl is almost thermo-neutral, the following hydrogenation ofvinyl to ethylene is strongly exothermic and irreversible.Reactants, intermediates, and products are bound to the samePd surface atoms. On the p5f surface of an AlPd compound, thehydrogenation reaction also follows the Horiuti−Polanyischeme, but the active site is not the same at all steps of thereaction. Acetylene adsorbs preferentially in a bridge positionbetween two Al atoms close to a Pd atom, and this is the activesite for the hydrogenation to vinyl. During the secondhydrogenation step from vinyl to ethylene, the molecularcomplex shifts from the bridge position to a position on top ofa nearby Pd atom. Selectivity is than determined by thecompetition between further hydrogenation of Pd-boundethylene to ethyl and desorption of ethylene. This scenario isvery similar to that identified and reported for the Al13Co4catalyst,16 where the active sites for the initial steps of thereaction are also Al atoms close to the transition metal (TM)atom and where selectivity is determined by the competitionbetween desorption and reaction of transition metal boundethylene.
■ COMPUTATIONAL METHODS AND MODELSElectronic structure calculations have been performed using theVienna ab initio simulation package (VASP).19,20 VASPproduces an iterative solution of the Kohn−Sham equationsof density functional theory (DFT) within a plane wave basis.We used the semilocal exchange-correlation functional in thegeneralized gradient approximation (GGA) proposed byPerdew et al.21 The basis set contained plane waves with akinetic energy up to Ecut‑off = 700 eV. The self-consistencyiterations were stopped when total energies are converged towithin 10−6 eV. The optimized geometries of the surface and ofthe adsorbate−substrate complexes were determined using
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−63196308

static relaxations using a quasi-Newton method and theHellmann−Feynman forces acting on the atoms. Transitionstates were determined using the nudged elastic band method(NEB).22 In structural optimizations and transition statesearches, convergence criteria of 10−4 eV for total energiesand 0.1 eV/Å for forces acting on the atoms were applied.The surface of AlPd is represented by a slab cut from the
bulk structure. The surface area x × y of the orthorhombiccomputational cell is 10.97 Å × 9.82 Å. It consists of theelementary surface cell doubled in the y direction. Thethickness of the slab should be large enough to stabilize thesurface. We have found that a thickness of four layers, withfixed bulk-like coordinates of atoms in the bottom layer issufficient. Neighboring slabs are separated by a vacuum layer of12 Å. The large lateral dimensions of the computational cellallowed to perform many exploratory calculations using the Γ-point, but all final results were calculated using a 2 × 2 × 1 k-point mesh for Brillouin-zone integration. The accuracy ofcalculated adsorption and activation energies was verified forseveral configurations by comparison with calculations at a finer3 × 3 × 1 k-point mesh.Crystal Structure of the B20 Compound. The B20
(FeSi-type) structure has space group P213 (No. 198). TheBravais lattice is simple cubic, but the overall point symmetry istetrahedral. The space group consists of four 3-fold rotationalaxes oriented along the ⟨111⟩ directions and three 2-fold screw21 axes consisting of a 180° rotation around a cubic axis,followed by a nonprimitive translation by (1/2,1/2,0)a andcombinations thereof. The B20 structure has 8 atoms perelementary cell; the Pearson symbol is cP8. In FeSi, both Feand Si are located at Wyckoff positions (4a) with coordinates(u,u,u), (u + 0.5,0.5 − u,−u), (− u,0.5 + u,0.5 −u), and (0.5 −u,−u,0.5 + u) where u(Fe) = 0.1358 and u(Si) = 0.844. Thepoint symmetry of the Fe and Si sites is C3, i.e., a 3-foldrotation. In the AlPd compound, Al occupies Si sites, and Fe isreplaced by Pd. The values of the experimental internalcoordinates are23 u(Pd) = 0.143 and u(Al) = 0.847. The latticeparameter is a = 4.859 Å. The calculated values are a = 4.908 Å,and u(Pd) = 0.147 and u(Al) = 0.844.The FeSi structure may be considered as derived from the B1
(NaCl) structure by a distortion involving a displacement of Feor Si atoms along a ⟨111⟩ direction to sites with u(Fe) = 0.25and u(Si) = 1 − u(Fe) = 0.75 These distortions are rather largeand reduce the space group symmetry from Fm3m to P213.Another interesting view of the B20 structure is in terms of theideal B20 structure proposed by Vocadlo et al.24 For u(Fe) = 1/(4τ) = 0.1545 and u(Si) = 1− 1/(4τ) = 0.8455 (where τ = (1 +√ 5)/2 is the golden mean), each atom has exactly sevennearest neighbors of the opposite kind at a distance of a√3/(2τ). Note that the positions of atoms in the AlPd compoundare close to the ideal ones. The seven nearest neighbors occupyseven of the twenty vertices of a pentagonal dodecahedroncentered at the atom. The 7-fold coordination and thearrangement of the coordinating atoms have led Dmitrienko25
to interpret the B20 structure as a low-order crystallineapproximant to an icosahedral Al−TM quasicrystal (e.g., i−Al−Pd−Mn). The quasiperiodicity of icosahedral quasicrystal isclosely related to the golden mean τ number. A systematicmethod to construct periodic approximants to the infinitequasicrystal consists of replacing this irrational number by afraction of two subsequent Fibonacci numbers. One gets asequence of periodic approximants with increasing size of the
elementary cell. The B2 and B20 structures are the lowestapproximants in this sequence.The electronic structure of FeSi and of isoelectronic
compounds such as RuSi and AlRh is characterized by anarrow band gap in the Fe-d−Si-p bands around the Fermienergy. In AlPd with one electron more per formula unit, thegap reduces to a pseudogap located about 1 eV below theFermi energy.26 The formation of a band gap is closely relatedto the topology of the lattice. In the undistorted B1 structure,the bands are multiply connected at the high symmetry pointsΓ and X. The distortion leading to the formation of the idealB20 structure lifts the degeneracy and permits the opening of agap. The further distortion from the ideal to the real B20structure splits the shell of seven equidistant nearest Pdneighbors around each Al at 2.63 Å into 1 + 3 + 3 neighborslocated at increasing distances of 2.58, 2.60, and 3.04 Å. Thedistortion correspond to the formation of −Al−Pd−Al−Pd−chains with alternating short (2.60 Å) and long (3.04 Å)distances. It leads to a stronger hybridization between Pd-d andAl-p states and contributes to a partially covalent character ofthe Al−Pd bonds resulting in the stabilization of the B20 overthe B1 structure.26
Structure of the Pseudo 5-Fold Surface of B20. Thedetermination of the stable surfaces of intermetallic compoundsis a rather complex task. The still rather limited experience withthe surface physics and chemistry of intermetallic compoundsand quasicrystals has established a few simple rules:27−34 (i) thecrystal cleaves preferentially along dense atomic planesseparated by wide density gaps. (ii) Because the surface energyof Al is substantially lower than that of transition metals, forAl−TM compounds, Al-rich surfaces generally have lowersurface energies. (iii) The sites on surfaces with a mixed Al−TM composition binding adsorbates most strongly are TMatoms or Al atoms close to TM atoms.In closely packed cubic crystals, the low-index surfaces (100),
(110), and (111) fulfill these rules. For the cubic B20 structure,all atomic planes perpendicular to the [100] direction areidentical, but they are relatively sparsely populated, and thedistance between neighboring planes is only 1.49 Å.Perpendicular to the [110] direction, one finds many sparseatomic planes at small distances. Perpendicular to the [111]direction in one period of a√3, there are 9 atomic planes: threeflat planes occupied by Pd only, three flat planes occupied by Alatoms only, and three slightly puckered Al−Pd planes. BecauseAl-rich surfaces have lower surface energies than terminationswhere many TM atoms are exposed at the surface, one canexpect that the (111) surface will be Al-rich and that most Pdatoms will be buried ∼0.9 Å below the surface. Details of ourstudy of the structural properties of the (111) surface and othersurface terminations of the B20 compounds will be publishedelsewhere.In the B20 structure, one finds perpendicular to the (120)
direction a stacking sequence of slightly puckered planescontaining Pd and Al atoms in equal numbers, separated by agap of 2.19 Å; see Figure 1. The atomic arrangement in theseplanes has pseudo 5-fold (p5f) symmetry, the angle betweenthe pseudo 5-fold direction, and the [100] direction is β, sin β =τ/(τ + 2)1/2, β = 58.28°, where τ is the golden mean. This anglediffers only slightly from the 60° between the [100] and [120]directions. Upon relaxation, the distance between the first Al−Pd layer, and the surface layer is reduced to 2.15 Å. Relaxationalso reduces the puckering of the top layer by 9% but hardly
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−63196309

affects the lateral distances between the atoms in the surfacelayer.Figure 2 shows a top view at the p5f surface. The ordering of
atoms in the surface layer can be described by a tiling consistingof golden thick rhombi and squashed hexagons (RH tiling) andthe vertices being occupied alternatingly by Pd and Al atoms.The acute angles in the rhombi and the hexagons are 2π/5. Wenote that if each hexagon is decomposed into one golden thickrhombus and two golden thin rhombi (see the dashed lines inFigure 2), one gets a Penrose tiling. In the Penrose tiling,vertices linked by a short diagonal of a thin rhombus are leftvacant. Alternatively, if each thick rhombus is split into twotriangles and each hexagon is decomposed into two trianglesand one rectangle (see full red lines in Figure 2), one gets arectangle−triangle (RT) tiling with atoms at all vertices; seealso Figure 3b.If the hexagons are split into two irregular quadrangles (see
the dotted line in Figure 2), one creates a tiling that allows tosee the relations with the (110) surface of the B2 (CsCl)structure. If the positions of the atoms in the surface layer areshifted a little such that all quadrangular tiles becomerectangular, the structure becomes identical to that of the(110) B2 surface. The p5f surface of the B20 structure thusplays a role analogous to the intensively studied (110) surfacesof the B2-type Al−TM compounds.35,36 It is possible to assigna parity to each vertex in the RH tiling. The Al atoms are in sayodd vertices, while Pd atoms occupy vertices with even parity.Moreover, there are two kinds of Al sites and two kinds of Pdsites. Pd sites of the first kind have 3 Al neighbors; Pd sites ofthe second kind have 4 Al neighbors (note the number of edgesof the RH tiling with a common vertex). Each Pd site of thesecond kind is at the center of an incomplete pentagon of Alatoms: four Al atoms occupy the vertices of a regular pentagon,and the fifth is replaced by an Al−Pd pair occupying one edgeof a rectangular tile. This configuration resembles to the
pentagonal configurations of atoms on the catalytically active(100) surface of Al13Co4.
16
The p5f surface is slightly corrugated (see Figure 1). InFigure 3, the heights of all atoms in the relaxed surface relativeto the average surface plane are given in angstroms. The heightof the Pd atom in site P2 is at least by ∼0.3 Å greater than thatof the surrounding Al atoms A1, A3, A1′, and A2′. However, thePd atom at site P4 is more than ∼0.3 Å deeper than thesurrounding Al atoms. A similar situation had been found onthe puckered P plane of the (100) surface of the Al13Co4compound.16
In the following, we shall use the p5f surface of B20-typeAlPd to examine the catalytic properties of this compound.
■ RESULTS
The first step toward the exploration of an atomistic scenariofor the multistep hydrogenation reaction must be thedetermination of the energetically most favorable adsorptionsites of reactants, intermediates, and products, via thecalculation of the binding energies at specific adsorption sites.Binding energies are defined by the energy difference betweenthe adsorbate/substrate complex and the energies of theadsorbed species in the gas phase and of the clean substrate.Figure 3a shows a top view of the computational cell used in
our simulations. The surface cell contains eight Al and eight Pdatoms. The relaxed surface is appreciably corrugated. Theheight of the Pd atoms relative to the average surface plane
Figure 1. Side view at the layered structure of AlPd projected onto the(001) plane. Positions of atoms are shown by circles: Al, light gray; Pd,dark gray. The blue dashed line shows the position of the cleavageplane determining the pseudo 5-fold (120) surface and the red dashedline the cleavage plane for the formation of a (100) surfaceperpendicular to the 2-fold screw axis. The dotted square marks oneunit cell.
Figure 2. Top view of the pseudo 5-fold (120) surface of AlPd. Asurface area of 3 unit cells is shown. Positions of atoms are shown bycircles: Al, light gray; Pd, dark gray. The arrangement of atoms in thesurface layer can be described by different planar tilings: a tilingconsisting of hexagons and thick golden rhombi (full blue lines), or atriangle−rectangle tiling if all hexagons and rhombi are split asindicated by full red lines. The hexagonal tile can also be divided intoone thick and two thin rhombi (as shown by the dashed lines),producing a Penrose tiling. If the hexagons are split into two irregularquadrangles, as shown by the dotted line, the tiling can be described asarising by a distortion of tiling describing the (110) surface of the B2structure.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−63196310

varies between +0.36 Å for the most strongly protruding P2sites and −0.32 Å for the P4 buried rather deeply below. Forthe Al atoms, the height varies between +0.29 Å and −0.27 Å.Part b of the surface shows a contour plot of the valenceelectron density distribution in the average surface plane,superposed by the rectangle−triangle tiling. Note that the tilesare not flat, but skewed relative to the surface plane. Because ofthe corrugation of the surface, we have to distinguish betweentwo types of rectangles (P1−P2−A1−A2 and P4′−P3′−A4−A3), and the triangles are also inequivalent. There are triangleswith two Al and one Pd atoms (A1−A3−P2, A3−A1′−P2, A1′−A2′−P2, A4−A3−P4, A3−A1−P4, etc.) and triangles with oneAl and two Pd atoms (A4−P4−P3, A2′−P1−P2, etc.), differingalso in their inclinations relative to the surface plane.Molecular and Dissociative Adsorption of Hydrogen.
Besides the active sites binding the hydrocarbon species, otheractive sites must be present, which promote the dissociativeadsorption of hydrogen. Atomic hydrogen must also besufficiently mobile to approach the reactants and intermediates.In this section, we determine the energetics of hydrogendissociation and diffusion.On Pd(111) surface, hydrogen molecules are preferentially
adsorbed on top of Pd atoms, 1.82 Å above the surface plane.On the p5f surface of AlPd, a H2 molecule is bonded veryweakly; at most surface sites, it is not chemisorbed at all. Thesite promoting the strongest adsorption of the H2 molecule,with an adsorption energy of Eb = −7.6 kJ/mol, is on top of themost protruding P2 atom. On the other Pd atoms lying deeperin the surface, a H2 molecule does not bind at all.We have calculated hydrogen dissociation over the P2 and P4
sites. For each site, several dissociation paths are possible.Dissociated H atoms from the hydrogen molecule on top of theP2 site can find their final positions in, e.g., H2 + H4 or H4 +H3′ sites (for the definition of the adsorption sites, see Figure4). The adsorption energies of H atoms coadsorbed at thesesites are −21 and −26 kJ/mol, respectively. The adsorptionenergies of coadsorbed atoms are not a superposition of theadsorption energies of single H atoms presented in Table 1.The difference originates from repulsive interaction of thecoadsorbates. For the P2 and P4 sites, we found activationenergies for dissociation of Ea = 54 and 58 kJ/mol, respectively.The dissociated H atoms are located at the sites H3′ and H4after dissociation over P2 and approximately at the sites H7 andH8 after dissociation over site P4. Hydrogen dissociation is an
exothermic process; for the P2 site, we found a heat of reactionof ΔEreact = −18 kJ/mol. The activation energy for therecombinative desorption of molecular hydrogen is thus 72 kJ/mol. These values can be compared with the data for thedissociative adsorption of hydrogen on the Pd(111) surface37
where activation energies of 55 kJ/mol for H2 dissociation and67 kJ/mol for the recombinative desorption were found. The
Figure 3. (a) Atomic sites on the surface in the computational cell. The violet circles mark positions of Pd atoms; the light-blue circles are Al atoms.The atoms below the top surface layer are darker. On the p5f surface, there are four inequivalent sites for Al (labeled A1−A4) and four for Pd(labeled P1−P4). The surface is appreciably corrugated, with a difference of 0.68 Å in the heights of the most strongly protruding atoms and thosebelow the average. The numbers inside the circles indicate the height of the atoms (in Å) with respect to the average height. (b) Valence electrondensity distribution in the surface layer, superposed by the rectangle−triangle tiling.
Figure 4. Adsorption sites of H atoms Hi, i = 1−9, on the pseudo 5-fold surface of AlPd. Two examples of diffusion paths are shown, onein x direction ([21 0]) and one in y direction ([001]).
Table 1. Adsorption Energies Eb (in kJ/mol) and Height hb(in Å) above the Surface of Molecular and Atomic HydrogenAdsorbed at Different Sites on the p5f Surface of AlPd (seeFigure 4)a
pseudo 5-fold surface of AlPd
adsorbate site description Eb hb
H2 P2 top −7.6 2.24H H1 hollow −2.3 0.90
H2 hollow −12.1 1.13H3 hollow −5.0 1.18H4 bridge −23.5 1.24H5 bridge 5.6 1.42H6 bridge 6.5 1.03H7 hollow −0.5 1.13H8 hollow −0.8 0.90H9 hollow −21.8 1.06
aEnergies are calculated relative to the energy of the H2 molecule inthe gas phase; heights are relative to the average surface plane.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−63196311

difference of ΔEreact = −12 kJ/mol corresponds to theadsorption energy of the coadsorbed atoms. All energiescalculated for Pd(111) are thus comparable to those calculatedfor the p5f surface of the AlPd compound.On the p5f surface of AlPd, dissociated hydrogen atoms
occupy preferably bridge or hollow sites formed by Pd and Alatoms. Figure 4 shows that the positions are all (meta)stableadsorption sites for H atoms. Adsorption energies are listed inTable 1. The binding energies were defined with respect to thegas phase, i.e., one-half of the binding energy of a H2 moleculehas been subtracted. Small values around zero mean that thebinding of atomic hydrogen to the surface is of comparablestrength to the binding in the hydrogen molecule. Around theprotruding P2 site, there are four stable H sites, while aroundthe other Pd sites, only three H sites were found. If such sitesare simultaneously occupied, the interaction between thecoadsorbed H atoms is mostly repulsive.Diffusion of Hydrogen. Dissociated H atoms on the p5f
surface can move via thermally activated diffusion jumps bylocally stable adsorption sites. Figure 4 shows two possible
diffusion paths: one roughly along the x direction [210],connecting the sites H1, H2, H4, H5, H7, and H8, and one inthe y direction [001] through the sites H2′, H4′, H3′, H2, H4,and H3. Figure 5 presents the energy profiles for both diffusionpaths; activation energies for each step have been determinedusing the nudged elastic band method.22 The optimizeddiffusion paths show that H atoms tend to move around Pdatoms, following a region of lower electron density rather thanto go over the top of the atoms. In contrast, H atoms jump overAl atoms, following almost a straight line between initial andfinal locations.It is evident that jumps over Al atoms require much higher
activation energies than jumps of H around Pd atoms. Thehighest activation energies of 45 and 41 kJ/mol have beencalculated for the jumps from H3 to H4 (across the Al atom insite A1) and from H5 to H7 (across the Al atom in site A3). Allother activations energies are significantly lower. From thediffusion paths presented in Figure 5, it is obvious that adiffusion movement of H atoms along the [001] direction ismuch easier than that along the [210] direction. The important
Figure 5. Energy profiles of the diffusion paths of adsorbed H atoms (a) in x direction and (b) in y direction; cf. Figure 4. Activations energies aregiven in kJ/mol.
Table 2. Adsorption Energies Eb (in kJ/mol, Relative to the Molecular Species in the Gas Phase), Height hb (in Å) of the Centerof Gravity of the Molecule above the Average Surface Plane, Length dC−C (in Å) of the C−C Bond, Length dC−H (in Å) of theShortest C−H Bond, Length dC−Pd (in Å) of the Shortest C−Pd Bond, and Average C−C−H Bond Angle ϕC−C−H (in deg) forVarious C2Hx (x = 2−5) Species Adsorbed at Different Sites on the p5f Surface of AlPd (see Figure 6)
pseudo 5-fold surface of AlPd
adsorbate site description Eb hb dC−C dC−H dC−Pd ϕC−C−H
C2H2 P2 top −37 2.72 1.24 1.07 2.24 20.1P1−P2 bridge −47 2.56 1.30 1.09 2.09 47.6A1−A3−P2 T center −148 2.21 1.39 1.10 2.25 60.4A3−A1′−P2 T center −131 2.20 1.39 1.10 2.32 60.5A3−A4−P4 T center −137 2.14 1.38 1.10 2.41 60.7A1′−A2′−P1′−P2′ R center −150 1.96 1.40 1.10 2.18 62.2A3−A4−P3′−P4′ R center −109 1.99 1.40 1.10 2.17 63.0
C2H3 A1−A3−P2 T center −294 2.38 1.38 1.10 2.29 60.5A3−A1−P2 T center −282 2.42 1.39 1.09 2.32 60.5A1′−A2′−P1′−P2′ R center −255 2.05 1.42 1.10 2.28 62.6
C2H4 P1 top −27 2.66 1.37 1.09 2.44 58.7C2H4 P2 top −45 2.73 1.38 1.09 2.33 59.0C2H4 P3 top −4 2.57 1.38 1.09 2.43 59.0C2H4 P4 top − 2.37 1.39 1.09 − 59.9C2H5 P2 top −173 3.04 1.51 1.09 2.16 68.2
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−63196312

conclusion is that the activation energies for dissociation anddiffusion of hydrogen are lower than that of the activationenergies for the following hydrogenation steps, so that thecatalytic activity is not controlled by the dissociation andmobility of hydrogen.Adsorption of Hydrocarbons. The adsorption energies of
different hydrocarbon species (acetylene, vinyl, ethylene, andethyl) at the most stable sites are listed in Table 2, togetherwith the height of the center of gravity of the molecule abovethe average surface plane, the length of the C−C bond, lengthdC−H of the shortest C−H bond, length dC−Pd of the shortestC−Pd bond, and the average angle of the C−H bonds withrespect to the C−C axis. All energies are calculated relative tothe total energy of the same species in the gas phase and theclean AlPd surface. An acetylene molecule can bind, e.g., on topof Pd atoms, Eb(P2) = −37 kJ/mol or in a bridge positionbetween two Pd atoms Eb(P1−P2) = −47 kJ/mol. However, onthe p5f surface, the acetylene molecule is the most stronglyadsorbed at special positions that are very close to bridgepositions between two Al atoms, above the center of atriangular (T) tile, whose corners are occupied by two Al andone Pd atom, or of a rectangular (R) tile with two Al and twoPd atoms at its corners (see Figure 6). Because of the puckeringof the surface layer, the T sites are not all equivalent; the
binding energies of the C2H2 molecule vary between −131 to−148 kJ/mol. Triangular tiles with two Pd and only one Alatoms are less favorable for acetylene adsorption. For instance,the position of the C2H2 molecule in the triangular tile A2′−P1−P2 is unstable. The molecule shifts to the bridge positionP1−P2 already listed in Table 2. From the A1′−A2′−P2triangle, the acetylene molecule is displaced into the A1′−A2′−P1′−P2′ rectangle.For two inequivalent R sites at the p5f surface, we have found
the adsorption energies of −109 and −150 kJ/mol. The lessfavorable R sites are characterized by a larger difference of 0.7 Åin the heights of the atoms occupying the corners. All theseadsorption energies are substantially larger than the bindingenergies at other sites.The strong adsorption at the T and R tiles also affects the
molecular geometry of the adsorbates. While gas phaseacetylene has a linear geometry, the C−H bonds of theadsorbate are tilted by about 60° relative to the C−C axis, awayfrom the surface. The length of the C−C bond is about 1.39 Å,increased by 0.19 Å relative to the free molecule and almostequal to the length of a CC double bond in gas phase C2H4.The elongation of the C−C bond reflects the activation of the
molecule, the T and R sites can be considered as reactioncenters for the hydrogenation process.There is a significant difference in the character of bonding of
acetylene on top of a Pd atom or in a bridge position betweentwo atoms occupying the corner of a T or R tile. On top of Pdacetylene is bonded via the hybridization between the ppπ-states of the molecule with d-orbitals of the transition metal, inthe bridge position the bonding has di-σ character. In gas phaseacetylene, the triple CC bond is formed by one sp hybridorbital and two p orbitals. Upon adsorption, the intramolecularhybridization changes to sp2, the three sp2 hybrid promoting (i)the C−H bond (which is now tilted relative to the molecularaxis), (ii) a C−C bond, and (iii) the bonding to the substrate.The second C−C bond is a ppπ bond; the elongation of theC−C distance reflects the change from a triple- to a double-bond.Vinyl (C2H3) preferably binds with the H−C end to one of
the Al atoms from a T or R tile. In the nomenclature used inTable 2, the bonding Al atom is listed first, i.e., in the T(A1−A3−P2) reaction center, the C−H group of the C2H3 moleculebinds to the A1 atom. Alternatively, the C−H group can alsobind to the Al atom in the A3 site. The two configurationsdiffer by 12 kJ/mol in the adsorption energy. The CH2 isshifted toward the Pd atom in site P2. In the gas phase, themolecule is planar; on the carbon atom of the C−H group, theorbitals form sp hybrids, and those of the C−H2 group form sp2
hybrids. Upon adsorption, the hybridization changes to sp2 inthe C−H and to sp3 in the C−H2 group. All three C−H bondsare tilted away from the surface. On the C−H end, vinyl bindsstrongly to Al through one of the sp2 hybrids and the unpairedp-electron; on the C−H2 end, there is only a weak binding viaone of the sp3 hybrids to the Pd atom. The C atom in the C−H2 group is by 0.86 Å higher above the surface than the other Catom, but the C−C distance is only slightly elongatedcompared to adsorbed acetylene from 1.38 to 1.42 Å.Ethylene (C2H4) is adsorbed rather weakly. It binds
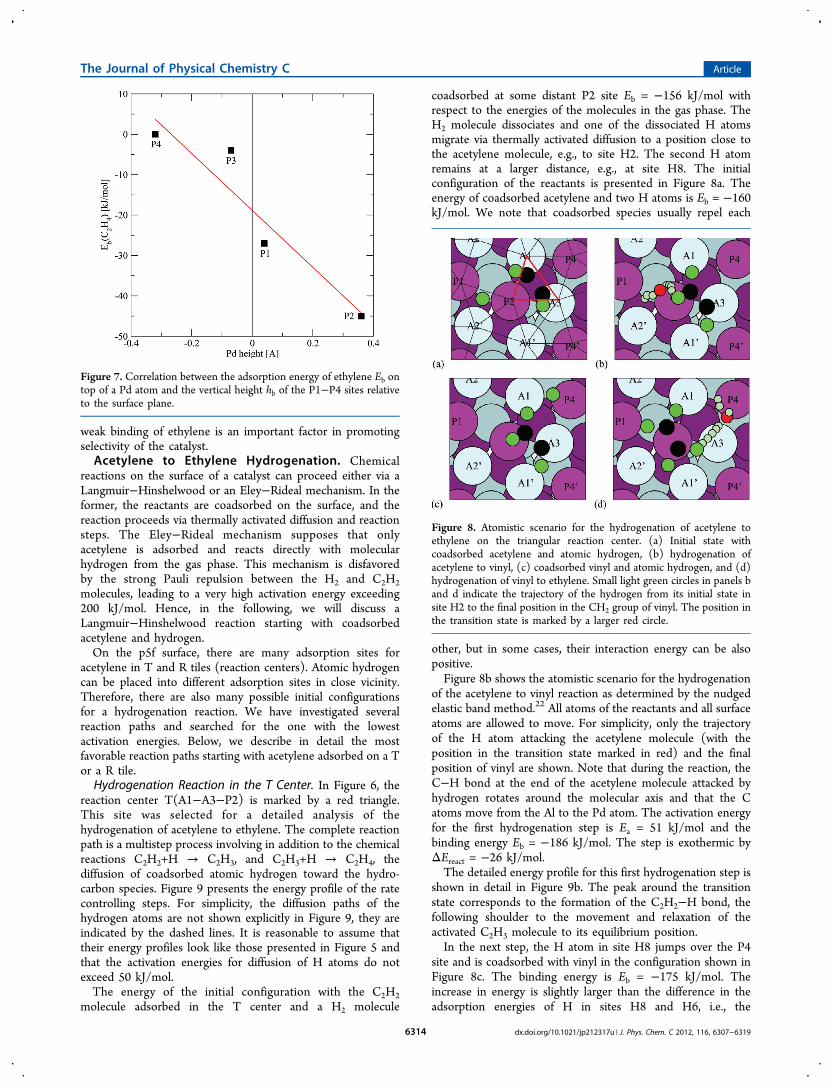
preferably on top of the protruding Pd at the P2 site. Thereis clear correlation between the height of a Pd atom above thesurface plane and the adsorption energy; see Figure 7. On theP4 site buried in the surface, ethylene is not bound at all.Binding is promoted by the interaction of one of the sp3
hybrids on each C atom with the d states of Pd. For a moleculeadsorbed at the P2 site, the center of gravity of the molecule islocated 2.73 Å above the surface. The length of the C−C is 1.38Å as in the gas phase, and the C−H bonds are tilted away fromthe surface. We shall see that the vertical position of the Pdatom above the surrounding Al atoms and consequently thelarge C-surface distance is an important factor determiningboth the activity and selectivity of the catalyst. Ethyl is morestrongly bound, also on top of the protruding Pd atom at aheight of about 3 Å.It is remarkable that the adsorption of hydrocarbons on the
p5f surface of AlPd is very similar to that on the (100) surfaceof orthorhombic Al13Co4: binding of acetylene at Al−Al bridgesites close to TM atoms, shifting to adsorption on top of a TMatom. In the most stable sites, adsorption energies on Al13Co4are higher by about 30 kJ/mol for acetylene, vinyl, and ethyleneand by 140 kJ/mol for ethyl.The adsorption energy of Eb(P2) = −45 kJ/mol of the
ethylene molecule on top of the protruding P2 atoms issignificantly lower than the adsorption energy of −71 kJ/molon top of protruding Co atoms in the case of Al13Co4 surface.On the buried P4 site, the ethylene does not bind at all. The
Figure 6. Preferred adsorption geometries of acetylene over triangular(T) and rectangular (R) tiles on the pseudo 5-fold surface of AlPd.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−63196313
weak binding of ethylene is an important factor in promotingselectivity of the catalyst.Acetylene to Ethylene Hydrogenation. Chemical
reactions on the surface of a catalyst can proceed either via aLangmuir−Hinshelwood or an Eley−Rideal mechanism. In theformer, the reactants are coadsorbed on the surface, and thereaction proceeds via thermally activated diffusion and reactionsteps. The Eley−Rideal mechanism supposes that onlyacetylene is adsorbed and reacts directly with molecularhydrogen from the gas phase. This mechanism is disfavoredby the strong Pauli repulsion between the H2 and C2H2molecules, leading to a very high activation energy exceeding200 kJ/mol. Hence, in the following, we will discuss aLangmuir−Hinshelwood reaction starting with coadsorbedacetylene and hydrogen.On the p5f surface, there are many adsorption sites for
acetylene in T and R tiles (reaction centers). Atomic hydrogencan be placed into different adsorption sites in close vicinity.Therefore, there are also many possible initial configurationsfor a hydrogenation reaction. We have investigated severalreaction paths and searched for the one with the lowestactivation energies. Below, we describe in detail the mostfavorable reaction paths starting with acetylene adsorbed on a Tor a R tile.Hydrogenation Reaction in the T Center. In Figure 6, the
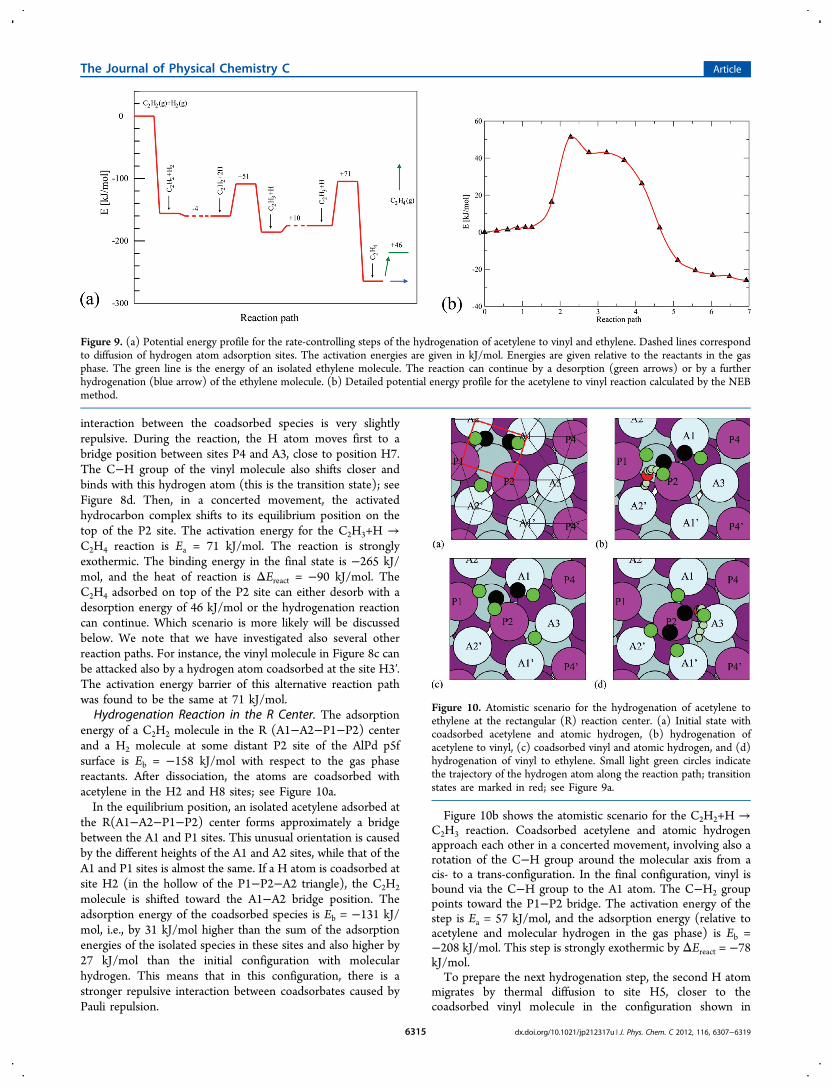
reaction center T(A1−A3−P2) is marked by a red triangle.This site was selected for a detailed analysis of thehydrogenation of acetylene to ethylene. The complete reactionpath is a multistep process involving in addition to the chemicalreactions C2H2+H → C2H3, and C2H3+H → C2H4, thediffusion of coadsorbed atomic hydrogen toward the hydro-carbon species. Figure 9 presents the energy profile of the ratecontrolling steps. For simplicity, the diffusion paths of thehydrogen atoms are not shown explicitly in Figure 9, they areindicated by the dashed lines. It is reasonable to assume thattheir energy profiles look like those presented in Figure 5 andthat the activation energies for diffusion of H atoms do notexceed 50 kJ/mol.The energy of the initial configuration with the C2H2
molecule adsorbed in the T center and a H2 molecule
coadsorbed at some distant P2 site Eb = −156 kJ/mol withrespect to the energies of the molecules in the gas phase. TheH2 molecule dissociates and one of the dissociated H atomsmigrate via thermally activated diffusion to a position close tothe acetylene molecule, e.g., to site H2. The second H atomremains at a larger distance, e.g., at site H8. The initialconfiguration of the reactants is presented in Figure 8a. Theenergy of coadsorbed acetylene and two H atoms is Eb = −160kJ/mol. We note that coadsorbed species usually repel each
other, but in some cases, their interaction energy can be alsopositive.Figure 8b shows the atomistic scenario for the hydrogenation
of the acetylene to vinyl reaction as determined by the nudgedelastic band method.22 All atoms of the reactants and all surfaceatoms are allowed to move. For simplicity, only the trajectoryof the H atom attacking the acetylene molecule (with theposition in the transition state marked in red) and the finalposition of vinyl are shown. Note that during the reaction, theC−H bond at the end of the acetylene molecule attacked byhydrogen rotates around the molecular axis and that the Catoms move from the Al to the Pd atom. The activation energyfor the first hydrogenation step is Ea = 51 kJ/mol and thebinding energy Eb = −186 kJ/mol. The step is exothermic byΔEreact = −26 kJ/mol.The detailed energy profile for this first hydrogenation step is
shown in detail in Figure 9b. The peak around the transitionstate corresponds to the formation of the C2H2−H bond, thefollowing shoulder to the movement and relaxation of theactivated C2H3 molecule to its equilibrium position.In the next step, the H atom in site H8 jumps over the P4
site and is coadsorbed with vinyl in the configuration shown inFigure 8c. The binding energy is Eb = −175 kJ/mol. Theincrease in energy is slightly larger than the difference in theadsorption energies of H in sites H8 and H6, i.e., the
Figure 7. Correlation between the adsorption energy of ethylene Eb ontop of a Pd atom and the vertical height hb of the P1−P4 sites relativeto the surface plane.
Figure 8. Atomistic scenario for the hydrogenation of acetylene toethylene on the triangular reaction center. (a) Initial state withcoadsorbed acetylene and atomic hydrogen, (b) hydrogenation ofacetylene to vinyl, (c) coadsorbed vinyl and atomic hydrogen, and (d)hydrogenation of vinyl to ethylene. Small light green circles in panels band d indicate the trajectory of the hydrogen from its initial state insite H2 to the final position in the CH2 group of vinyl. The position inthe transition state is marked by a larger red circle.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−63196314
interaction between the coadsorbed species is very slightlyrepulsive. During the reaction, the H atom moves first to abridge position between sites P4 and A3, close to position H7.The C−H group of the vinyl molecule also shifts closer andbinds with this hydrogen atom (this is the transition state); seeFigure 8d. Then, in a concerted movement, the activatedhydrocarbon complex shifts to its equilibrium position on thetop of the P2 site. The activation energy for the C2H3+H →C2H4 reaction is Ea = 71 kJ/mol. The reaction is stronglyexothermic. The binding energy in the final state is −265 kJ/mol, and the heat of reaction is ΔEreact = −90 kJ/mol. TheC2H4 adsorbed on top of the P2 site can either desorb with adesorption energy of 46 kJ/mol or the hydrogenation reactioncan continue. Which scenario is more likely will be discussedbelow. We note that we have investigated also several otherreaction paths. For instance, the vinyl molecule in Figure 8c canbe attacked also by a hydrogen atom coadsorbed at the site H3′.The activation energy barrier of this alternative reaction pathwas found to be the same at 71 kJ/mol.Hydrogenation Reaction in the R Center. The adsorption
energy of a C2H2 molecule in the R (A1−A2−P1−P2) centerand a H2 molecule at some distant P2 site of the AlPd p5fsurface is Eb = −158 kJ/mol with respect to the gas phasereactants. After dissociation, the atoms are coadsorbed withacetylene in the H2 and H8 sites; see Figure 10a.In the equilibrium position, an isolated acetylene adsorbed at
the R(A1−A2−P1−P2) center forms approximately a bridgebetween the A1 and P1 sites. This unusual orientation is causedby the different heights of the A1 and A2 sites, while that of theA1 and P1 sites is almost the same. If a H atom is coadsorbed atsite H2 (in the hollow of the P1−P2−A2 triangle), the C2H2
molecule is shifted toward the A1−A2 bridge position. Theadsorption energy of the coadsorbed species is Eb = −131 kJ/mol, i.e., by 31 kJ/mol higher than the sum of the adsorptionenergies of the isolated species in these sites and also higher by27 kJ/mol than the initial configuration with molecularhydrogen. This means that in this configuration, there is astronger repulsive interaction between coadsorbates caused byPauli repulsion.
Figure 10b shows the atomistic scenario for the C2H2+H →C2H3 reaction. Coadsorbed acetylene and atomic hydrogenapproach each other in a concerted movement, involving also arotation of the C−H group around the molecular axis from acis- to a trans-configuration. In the final configuration, vinyl isbound via the C−H group to the A1 atom. The C−H2 grouppoints toward the P1−P2 bridge. The activation energy of thestep is Ea = 57 kJ/mol, and the adsorption energy (relative toacetylene and molecular hydrogen in the gas phase) is Eb =−208 kJ/mol. This step is strongly exothermic by ΔEreact = −78kJ/mol.To prepare the next hydrogenation step, the second H atom
migrates by thermal diffusion to site H5, closer to thecoadsorbed vinyl molecule in the configuration shown in
Figure 9. (a) Potential energy profile for the rate-controlling steps of the hydrogenation of acetylene to vinyl and ethylene. Dashed lines correspondto diffusion of hydrogen atom adsorption sites. The activation energies are given in kJ/mol. Energies are given relative to the reactants in the gasphase. The green line is the energy of an isolated ethylene molecule. The reaction can continue by a desorption (green arrows) or by a furtherhydrogenation (blue arrow) of the ethylene molecule. (b) Detailed potential energy profile for the acetylene to vinyl reaction calculated by the NEBmethod.
Figure 10. Atomistic scenario for the hydrogenation of acetylene toethylene at the rectangular (R) reaction center. (a) Initial state withcoadsorbed acetylene and atomic hydrogen, (b) hydrogenation ofacetylene to vinyl, (c) coadsorbed vinyl and atomic hydrogen, and (d)hydrogenation of vinyl to ethylene. Small light green circles indicatethe trajectory of the hydrogen atom along the reaction path; transitionstates are marked in red; see Figure 9a.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−63196315

Figure 10c. The energy of the coadsorbed species is Eb = −192kJ/mol. Hydrogenation occurs by a concerted movement of thereactants: vinyl moves to a position on top of site P2.Simultaneously, the C−H group rotates around the molecularaxis to allow the hydrogen atoms to bind to the C atom. Theactivation energy for the C2H3+H → C2H4 reaction is Ea = 76kJ/mol. The reaction is also strongly exothermic, the bindingenergy in the final state is −264 kJ/mol, and the heat ofreaction is ΔEreact = −71 kJ/mol. Figure 10d shows the positionof the C2H4 molecule after the reaction. From the trajectory ofthe attacking hydrogen atom and its position in the transitionstate, it is evident that in this case, a large shift of the vinylmolecule is required before the second hydrogenation can takeplace. The energy profile of the whole reaction path is shown inFigure 11a.For both reaction paths explored above, the rate-controlling
step for the transformation of acetylene to ethylene is thesecond hydrogenation step from vinyl to ethylene, withactivation energies of 71 and 76 kJ/mol for a reaction at theT and R center, respectively. All earlier diffusion and reactionsteps have activation energies of 50 kJ/mol or lower. This hasto be compared with the energy profile for the hydrogenationover the puckered surface of Al13Co4 where we have foundactivation energies of 61 ± 2 kJ/mol for all steps.16
It is remarkable that both reactions starting from differentinitial states at the R(A1−A2−P1−P2) and T(A1−A3−P2)centers finish in the same final configuration: C2H4 adsorbedon top of the most protruding P2 site. They differ only in arotational orientation. The ethylene molecule can either desorbwith a desorption energy of 45 kJ/mol or the hydrogenationreaction can continue. Which scenario is more likely is nowanalyzed.Ethylene Hydrogenation and Selectivity. The ethylene
molecule is weakly bonded at the P2 site. Its equilibriumposition is 2.33 Å above the Pd atom. A direct attack by atomicH coadsorbed at a nearby site is difficult because of thedifferent heights of the coadsorbed species. The H atomessentially has to break the bond to the surface to approach theethylene molecule high above the surface. This leads to a very
high activation energy comparable to that for an Eley−Ridealreaction.Nevertheless, the reaction is possible via a two-step
mechanism. Similar to the reaction on the Al13Co4(100)surface, we have found that a reaction of ethylene withhydrogen is possible only if the H atom is first moved to a sitejust below the ethylene molecule. On Al13Co4, this first steprequires a very high activation energy of 80 kJ/mol, and theAl13Co4 catalyst has a high selectivity because this energy ishigher than the desorption energy of ethylene of 70 kJ/mol.16
The following reaction of H with C2H4 requires only anactivation energy of 15 kJ/mol.On the p5f surface of AlPd, the reaction C2H4+H → C2H5 is
also a two step process. The atomistic scenario is presented inFigure 12, the energy profile in Figure 11b. The activationenergies for the first and second steps are 41 and 31 kJ/mol,respectively. In the initial step, ethylene is located on top of a
Pd atom at site P2 and coadsorbed with hydrogen in site H9.The coadsorption energy is Eb = −274 kJ/mol. The repulsionbetween the coadsorbates costs an energy of 12 kJ/mol. Thefirst diffusion step bringing the H atom into position H2(moving around a Pd atom in site P1; see Figure 12a) requiresan activation energy of 41 kJ/mol. This step is endothermic byΔEreact = 34 kJ/mol because site H2 is energetically less
Figure 11. (a) Potential energy profile for the rate-controlling steps of the hydrogenation of (a) acetylene to vinyl and ethylene (red line), followingthe atomistic scenario shown in Figure 10. (b) Potential energy profile for the hydrogenation of ethylene to ethyl (blue line), according to theatomistic scenario shown in Figure 12. The green line in panel a is the energy of an isolated ethylene molecule. The green rectangle in panel b showsthe range of desorption energies for ethylene in the presence of coadsorbed H atoms. The activation energies are given in kJ/mol. The energy scale iswith respect to the reactants in the gas phase.
Figure 12. Atomistic scenario for the hydrogenation of ethylene toethyl. (a) A diffusion jump of hydrogen atom toward ethylene and (b)reaction of the hydrogen atom with ethylene. Small light green circlesindicate the trajectory of the hydrogen atoms along the reaction path;transition states are marked in red; see Figure 11b.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−63196316

favorable than site H9 and because the repulsive interactionbecomes stronger. The binding of the H atom with coadsorbedethylene requires a rotational movement of the C−H2 grouparound the molecular axis such that a C−H3 group can beformed with the H atom approaching ethylene from below (seeFigure 12a,b).Compared to Al13Co4, the activation energy is much lower
for the first and higher for the second step. The first barrier islower because diffusion of the H atom around the Pd atom atthe P1 site requires less energy than the jump over an Al atomon the Al13Co4 surface. The second barrier is higher because arotation of the C−H2 is required to make space for theadditional H atom, while this is not the case on Al13Co4 wherethe configuration of ethylene remains unchanged.The selectivity of the catalyst is determined by the
relationship between the activation energy for the hydro-genation of ethylene to ethyl and the desorption energy ofethylene. The hydrogenation is a two-step process, with atomichydrogen coadsorbed very close to ethylene as an intermediate.The energy of the barrier for the second reaction step is 56 kJ/mol higher than the initial state: this by 11 kJ/mol than thedesorption energy of an isolated ethylene molecule. Hence,desorption is favored over hydrogenation, and therefore, thecatalyst is selective. Selectivity is further enhanced by (i) adesorption energy for ethylene coadsorbed with hydrogen,which is considerably lower than the 45 kJ/mol calculated forisolated ethylene, and (ii) an activation energy for the backwardreaction from the intermediate to the initial state, which ismuch lower than the barrier for the forward reaction (seeFigure 11b). For the coadsorbed configuration shown in Figure12b, the desorption energy is only 10 kJ/mol. In dependenceon the position of coadsorbed hydrogen, the desorption energyvaries in a 35 kJ/mol wide interval, between 10 and 45 kJ/molas indicated in Figure 11b by a green rectangle.
■ DISCUSSION AND CONCLUSIONSAb initio DFT calculations have been used to construct anatomistic scenario for the selective hydrogenation of acetyleneto ethylene catalyzed by the p5f surface of intermetallic AlPdcrystallizing in the B20-type structure. The atomic order of thesurface can be described by a tiling of golden rhombi andhexagons,or, for our purpose, more appropriately by a triangle−rectangle tiling. The surface is rather strongly corrugated. Theheight of atoms is differing by up to 0.68 Å. The dissociativeadsorption of molecular hydrogen is an activated process withan activation energy of 54−58 kJ/mol. Atomic hydrogen israther mobile on this surface; activation energies for diffusivemotion between neighboring adsorption sites vary widely,between 2 and 45 kJ/mol as expected for such a corrugatedsurface. Two different reaction centers with acetylene adsorbedin Al−Al bridge positions in a triangular (T) or a rectangular(R) tile have been identified. For both reaction paths, the rate-controlling step is the second hydrogenation from vinyl toethylene with activation energies of 71 and 76 kJ/mol for the Tand R center, respectively. In contrast to acetylene and vinyl,ethylene is bound to the surface not via an Al atom but ratheron top of a Pd atom protruding from the surface. Thehydrogenation of ethylene to ethyl is a two-step reaction, withan intermediate in the form of atomic hydrogen coadsorbedvery close to but at a significantly lower height than ethylene.The height of the combined barrier for this reaction is with 56kJ/mol, significantly higher than the desorption energy ofethylene, varying between 10 and 45 kJ/mol depending on the
presence and location of coadsorbed hydrogen. We also notethat the strong exothermicity of the vinyl to ethylene reaction isimportant. It helps to enhance selectivity because theprobability of a backwards reaction will be very low; but ofcourse, despite our effort, we cannot claim that the reactionpathway we have found is the most favorable. This is alsounnecessary: if the reaction is efficient and selective along thepath we have found, this is sufficient to support our claim thatAlPd is a good catalyst. Difficulties could arise only if there is areaction path for ethylene to ethyl with a lower barrier, but thisis unlikely because ethylene binds preferentially on top of P2,reducing strongly the number of possible reaction scenarios. Inthis configuration, the C−C bond of ethylene is very close tothe length of a CC double bond in gas phase ethyleneindicating the absence of any activation for further hydro-genation. Thus, our simulations parallel the experimentalresults5,6,8,12 for the isostructural and isoelectronic compoundGaPd: both intermetallic compounds are efficient and selectivecatalysts for the hydrogenation of acetylene to ethylene.It is interesting to compare our results with the reaction path
and its energetic profile constructed for other hydrogenationcatalysts. For a Pd(111) surface, Mei et al.10,11 and Studt et al.3
reported activations energies of 66/74/72 kJ/mol for thehydrogenation of acetylene to vinyl, vinyl to ethylene, andethylene to ethyl, compared to a desorption energy of 82 kJ/mol for ethylene. The difference of 10 kJ/mol in favor ofcontinued hydrogenation explains the poor selectivity of purePd. For a Pd−Ag alloy catalyst, the activation energies arelowered to 66/10/61 kJ/mol, but the activation energy for theformation of ethyl remains lower by 10 kJ/mol than thedesorption energy of ethylene. It was argued that selectivitydoes not result from a low desorption barrier of ethylene butfrom the fact that the activation energy for dissociation ofhydrogen from ethyl (and formation of ethylene) is lower thanthe activation energy for the hydrogenation of ethyl to ethane(and also much lower than the desorption energy of ethyl).9−11
For acetylene hydrogenation on Al13Co4, we have calculatedactivation energies of 63/61/80 kJ/mol for the successivehydrogenation steps, and a desorption energy of ethylenevarying between 54 and 71 kJ/mol, depending on coadsorbates.Thus, the activation energy for the rate-controlling step of thehydrogenation of acetylene to ethylene is lower for the Al−Cocompound, but the difference between the activation energy forthe formation of ethyl and the desorption of ethylene issomewhat larger for the AlPd compound. Thus, the activity ofthe Al13Co4 catalyst is expected to be better; however, theselectivity of AlPd is expected to be somewhat better than thatof Al13Co4.It is remarkable that only for the two intermetallic
compounds selectivity results from a desorption energy forethylene that is lower than the activation energy for completehydrogenation. It is remarkable that the adsorption config-urations of the reactants on the Al13Co4(100) surface and onthe pseudo 5-fold surface of AlPd are quite similar; see Figure13. In both cases the catalytically active sites are formed by apentagonal arrangement of Al atoms around a slightlyprotruding transition metal atom. The most active reactioncenters in both cases are triangular configurations of two Alatoms and one TM atom. The preferable adsorption geometryfor acetylene is the Al−Al bridge position, forming a di-σ bond.Vinyl is adsorbed in a rather asymmetric configuration, with thestrongest bond between the C−H group and an Al atom. Anethylene molecule is adsorbed via a π-bond on top of the TM
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−63196317

atom. During the hydrogenation reaction, the molecularcomplex shifts from the bridge position between the Al sitesto the top position on top of the TM site where it is onlyweakly bound, favoring desorption. This is in contrast to theadsorption configurations on Pd or Pd−Ag surfaces where bothacetylene and ethylene are bound via di-σ bonds to two Pdatoms, irrespective of surface composition.Another important factor with a significant influence on both
activity and selectivity is the relative height of the Pd atomabove the Al atoms in the reactive center. The Pd atom in theP2 site lies 0.36 Å above the average surface plane; see Figure 3.For comparison, the Co atom in the center of the Al-pentagonof Al13Co4 is 0.45 Å above surrounding Al atoms. On one hand,the binding energy of ethylene increases with the height of thePd atom. This makes desorption more difficult, but a highbinding energy of ethylene also contributes to reduce theactivation energy for the second and rate controlling hydro-genation step.The location of adsorbed ethylene on top of a protruding Pd
atom also increases the activation energy for the formation ofethyl. Similarly, as on Al13Co4, on the p5f surface of AlPd, theethylene hydrogenation, C2H4+H→ C2H5, is possible only if ina two-step reaction first, the H atom is brought to a positionvery close but significantly lower than the position of theethylene molecule. In the second step, H is attached to thehydrocarbon molecule: this requires the loosening of thehydrogen-surface bond. The combined activation energy forthis process is higher than for the desorption of ethylene.Hence, both the isolation of the TM atom (preventing theformation of a di-σ bond of ethylene with the surface) and itsheight in a corrugated surface are important factors influencingboth activity and the selectivity of the catalytic reaction. Weassume that the TM−Al2 triplet configurations that weidentified as the catalytically active sites for acetylenehydrogenation can exist also at selected surfaces of other Al−TM intermetallics. However, our results suggest that the (111)or (100) surfaces of AlPd are presumably catalytically inactive.The top layer of the (111) surface is occupied by Al atoms; Pdatoms are buried 0.9 Å below the top plane and cannotcontribute to the catalytic process.Our results demonstrate that a B20-type AlPd compound
should be an active as well as selective hydrogenation catalyst. A
theoretical investigation of catalytic properties of theisostructural and isoelectronic GaPd compound is under way.
■ AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis work has been supported by the Austrian Ministry forEducation, Science and Art through the Center for Computa-tional Materials Science. M.K. also thanks the support from theGrant Agency for Science of Slovakia (No. 2/0111/11), fromCEX FUN-MAT, and from the Slovak Research and Develop-ment Agency (Grant No. APVV-0647-10).
■ REFERENCES(1) Schbib, N. S.; Garcia, M. A.; Gigola, C. E.; Errazu, A. F. Ind. Eng.Chem. Res. 1996, 35, 1496−1505.(2) Teschner, D.; Borsodi, J.; Wootsch, A.; Revay, Z.; Havecker, M.;Knop-Gericke, A.; Jackson, S. D.; Schogl, R. Science 2008, 320, 86−89.(3) Studt, F.; Abild-Petersen, F.; Bligaard, T.; Sørensen, R. Z.;Christensen, C. H.; Nørskov, J. K. Angew. Chem., Int. Ed. 2008, 47,9299−9302.(4) Doyle, A. M.; Shaikhudinov, S. K.; Freund, H.-J. J. Catal. 2004,223, 444−453.(5) Osswald, J.; Giedigkeit, R.; Jentoft, R. E.; Armbruster, M.;Girgsdies, F.; Kovnir, K.; Ressler, T.; Grin, Y.; Schlogl, R. J. Catal.2008, 258, 210−218.(6) Osswald, J.; Kovnir, K.; Armbruster, M.; Giedigkeit, R.; Jentoft, R.E.; Wild, U.; Grin, Y.; Schlogl, R. J. Catal. 2008, 258, 219−227.(7) Collins, B. M. Selective hydrogenation of highly unsaturatedhydrocarbons in the presence of less unsaturated hydrocarbons. U.S.Patent 4,126,645, 1978.(8) Kovnir, K.; Armbruster, M.; Teschner, D.; Venkov, T. V.; Jentoft,F. C.; Knop-Gericke, A.; Grin, Y.; Schlogl, R. Sci. Technol. Adv. Mater.2007, 8, 420−427.(9) Sheth, P. A.; Neurock, M.; Smith, C. M. J. Phys. Chem. B 2003,107, 2009−2017.(10) Mei, D.; Sheth, P. A.; Neurock, M.; Smith, C. M. J. Catal. 2006,242, 1−15.(11) Mei, D.; Neurock, M.; Smith, C. M. J. Catal. 2009, 268, 181−195.(12) Kovnir, K.; Osswald, J.; Armbruster, M.; Teschner, D.;Weinberg, G.; Wild, U.; Knop-Gericke, A.; Ressler, T.; Grin, Y.;Schlogl, R. J. Catal. 2009, 264, 93−103.(13) Bhargava, M. K.; Gadalla, A. A.; Schubert, K. J. Less-CommonMet. 1975, 42, 69−76.(14) Kovnir, K.; Armbruster, M.; Teschner, D.; Venkov, T. V.;Szentmiklosi, L.; Jentoft, F. C.; Knop-Gericke, A.; Grin, Y.; Schlogl, R.Surf. Sci. 2009, 603, 1784−1792.(15) Krajcí, M.; Hafner, J. Philos. Mag. 2011, 91, 2904−2913.(16) Krajcí, M.; Hafner, J. J. Catal. 2011, 278, 200−207.(17) Armbruster, M.; Kovnir, K.; Grin, J.; Schlogl, R.; Gille, P.;Heggen, M.; Feuerbacher, M. Ordered Cobalt-Aluminum and Iron-Aluminum Intermetallic Compounds as Hydrogenation Catalysts.European Patent 09157875.7, 2009.(18) Armbruster, M.; Kovnir, K.; Grin, Y.; Schlogl, R. In ComplexMetallic Alloys: Fundamentals and Applications; Dubois, J.-M., Belin-Ferre, E., Eds.; Wiley-VCH: Berlin, Germany, 2010; pp 385−399.(19) Kresse, G.; Furthmuller, J. Phys. Rev. B 1996, 54, 11169−11186.(20) Kresse, G.; Joubert, D. Phys. Rev. B 1999, 59, 1758−1775.(21) Perdew, J.; Wang, Y. Phys. Rev. B 1992, 45, 13244−13249.(22) Henkelman, G.; Jonsson, H. J. Chem. Phys. 1999, 111, 7010−7022.
Figure 13. Comparison of the catalytically active surfaces of Al13Co4(a) and AlPd (b). The violet circles represent the TM atoms. The redtriangles mark the most active reaction centers. On both surfaces, themost preferable adsorption site for the acetylene molecule is a bridgeposition between two Al atoms. The vertical position of theneighboring TM atom is an important parameter controlling activityand selectivity of the catalyst.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−63196318

(23) Villars, P.; Calvert, L. D. Pearson’s Handbook of CrystallographicData for Intermetallic Phases; American Society for Metals: Metals Park,OH, 1985.(24) Vocadlo, L.; Price, G. D.; Wood, I. G. Acta Crystallogr., Sect. B:Struct. Sci. 1999, 55, 484−493.(25) Dmitrienko, V. E. Acta Crystallogr., Sect. A: Found. Crystallogr.1994, 50, 515−526.(26) Krajcí, M.; Hafner, J. Phys. Rev. B 2007, 75, 024116.(27) Krajcí, M.; Hafner, J. Phys. Rev. B 2005, 71, 054202.(28) Krajcí, M.; Hafner, J.; Mihalkovic, M. Phys. Rev. B 2006, 73,134203.(29) Krajcí, M.; Hafner, J. Surf. Sci. 2008, 602, 182−197.(30) Deniozou, T.; Addou, R.; Shukla, A. K.; Heggen, M.;Feuerbacher, M.; Krajcí, M.; Hafner, J.; Widmer, R.; Groning, O.;Fournee, V.; et al. Phys. Rev. B 2010, 81, 125418.(31) Gaudry, E.; Shukla, A. K.; Duguet, T.; Ledieu, J.; de Weerd, M.-C.; Dubois, J.-M.; Fournee, V. Phys. Rev. B 2010, 82, 085411.(32) Addou, R.; Gaudry, E.; Deniozou, T.; Heggen, M.; Feuerbacher,M.; Gille, P.; Grin, Y.; Widmer, R.; Groning, O.; Fournee, V.; et al.Phys. Rev. B 2009, 80, 014203.(33) Ledieu, J.; Krajcí, M.; Hafner, J.; Leung, L.; Wearing, L. H.;McGrath, R.; Lograsso, T. A.; Wu, D.; Fournee, V. Phys. Rev. B 2009,79, 165430.(34) Krajcí, M.; Hafner, J. Phys. Rev. B 2011, 84, 115410.(35) Blum, V.; Hammer, L.; Schmidt, C.; Meier, W.; Wieckhorst, O.;Muller, S.; Heinz, K. Phys. Rev. Lett. 2002, 89, 266102.(36) Hammer, L.; Blum, V.; Schmidt, C.; Wieckhorst, O.; Meier, W.;Muller, S.; Heinz, K. Phys. Rev. B 2005, 71, 075413.(37) Dong, W.; Kresse, G.; Hafner, J. J. Mol. Catal. A: Chem. 1997,119, 69−76.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp212317u | J. Phys. Chem. C 2012, 116, 6307−63196319





























