Embed Size (px)
Citation preview

ORIGINAL ARTICLE
Erlend Moldrheim Æ Michael J. Hannon
Isabelle Meistermann Æ Alison Rodger Æ Einar Sletten
Interaction between a DNA oligonucleotide and a dinuclear iron(II)supramolecular cylinder; an NMR and molecular dynamics study
Received: 8 November 2001 /Accepted: 8 February 2002 / Published online: 6 April 2002� SBIC 2002
Abstract A tetracationic supramolecular cylinder, [Fe2L3]
4+ (L=C25H20N4), with a triple-helical architecture,is just the right size to fit into the major groove of DNAbut too big to fit into the minor groove. A detailedNMR spectroscopic analysis supported by moleculardynamics (MD) calculations shows unambiguously theclose fit between the cylinder and a duplex oligonucle-otide, [d(GACGGCCGTC)2]. Furthermore, only theleft-handed enantiomer of the cylinder seems to fit thegroove geometry. With both free and complexed speciesof [Fe2L3]
4+ and DNA in solution, the NMR spectra aretoo complicated for a detailed structure determination.Based on differences in chemical shifts and extensiveMD calculations, a realistic qualitative picture of theDNA-cylinder adduct is presented. Several sets ofchemical shifts assigned to the protons of the three li-gand strands in the cylinder indicate that the ironcomplex situated in the major groove exhibits restrictedrotation on the NMR timescale around the cylindricalaxis. The NMR NOE data support a model where thecylinder undergoes both a translational and rotationaloscillation in the major groove. The results of an NOErestrained MD calculation indicates that the cylinderinduces a 40� bend of the double helix, in accordancewith linear dichroism measurements. Other distinct
features to be noticed are the very low value of the he-lical twist (16�) induced at the G4C5 step. Electronicsupplementary material to this paper, comprisingFigs. S1–S4 and Tables S1–S4, can be obtained by usingthe Springer Link server located at http://dx.doi.org/10.1007/s00775-002-0354-2.
Keywords Metallo-supramolecular structure ÆDNA oligonucleotide Æ Nuclear magneticresonance Æ Docking Æ Molecular dynamics
Abbreviations CHELPG: charges from electrostaticpotentials – grid based Æ ESP: electrostatic poten-tial Æ MD: molecular dynamics Æ NOESY: nuclearOverhauser enhancement spectroscopy Æ PME: particlemesh Ewald Æ ROESY: rotating frame Overhauserenhancement spectroscopy Æ TOCSY: total correlationspectroscopy Æ VMD: visual molecular dynamics
Introduction
While DNA encodes the essential blueprint for life,within biological systems its structure and function areregulated by proteins. These proteins generally achievesequence-specific code recognition through surface mo-tifs which interact with either the minor or, more com-monly, the major groove of DNA in a non-covalentfashion. In the post-genomic environment, the ability toartificially stimulate or prevent the processing of thegenetic code is an important goal and offers new op-portunities for disease prevention or control. To achievethis, synthetic agents are required that recognize thegenetic code in a sequence-selective fashion. Syntheticagents that target the major groove of DNA with rec-ognition through non-covalent surface motifs thereforehave the potential to be a powerful new tool. However,to date, little progress has been made in this direction,owing in large part to the size of the molecular surfacesrequired to achieve this. Indeed, synthetic molecules thatdo achieve sequence selectivity, such as amide-linked
J Biol Inorg Chem (2002) 7: 770–780DOI 10.1007/s00775-002-0354-2
Electronic supplementary material to this paper, comprisingFigs. S1–S4 and Tables S1–S4, can be obtained by using theSpringer Link server located at http://dx.doi.org/10.1007/s00775-002-0354-2
E. Moldrheim Æ E. Sletten (&)Department of Chemistry,University of Bergen, Allegt. 41,5007 Bergen, NorwayE-mail: [email protected].: +47-5-5583352Fax: +47-5-5589490
M.J. Hannon Æ I. Meistermann Æ A. RodgerDepartment of Chemistry,University of Warwick,Coventry CV4 7AL, UK

imidazole/pyrrole oligomers which bind in the minorgroove [2], are rare. Oligonucleotides (synthetic andnatural) can selectively recognize DNA by forming tri-plexes through binding in the major groove [3] andneutral oligonucleotide analogues (e.g. PNAs) canachieve similar effects, although more commonly theyachieve sequence selectivity through strand displacement[4].
Metal complexes are particularly attractive vectorsfor non-covalent DNA recognition because of the cat-ionic charge that the metallo centres can impart, whichaffords a substantial energetic contribution to the non-covalent binding to anionic DNA. Covalent DNAbinding by metal complexes (e.g. cisplatin) has beenextensively studied and usually focuses on binding to N7of G and A residues [5]. Non-covalent binding of metalcomplexes to DNA is a less well-developed area and hasprimarily centered around spherical ruthenium poly-pyridyl complexes and complexes bearing planar inter-calating units (or combinations thereof) ([6, 7, 8] andreferences therein). Their small size often results in theirlocation in the minor rather than the major groove andalso means that they cannot target more than two basepairs. Consequently, they do not afford surfaces bestsuited to act as scaffolds for sequence-specific recogni-tion. Supramolecular chemistry provides methodologyfor the design of large synthetic arrays and consequentlyallows us synthetically to bridge the size-gap betweentraditional small molecule and larger biomolecule DNA-recognition motifs. In particular, we have developed an‘‘inexpensive approach’’ to generate sophisticated su-pramolecular architectures based on the interaction ofmetal ions with imine-based ligands [9, 10, 11, 12]. Ofparticular importance to this work, a synthetic proce-dure has been described for making tetracationic su-pramolecular cylinders with a triple helical architecture(Fig. 1) which are just the right size to fit into the majorgroove of DNA but too big to fit into the minor groove
[1, 12, 13]. We describe herein the binding of such asupramolecular metallo cylinder to a DNA oligonucle-otide. A preliminary report on this work has recentlybeen published [1]. The present paper describes in detailthe results of NMR spectroscopic work and moleculardynamics (MD) calculations for the DNA-cylinder sys-tem.
Materials and methods
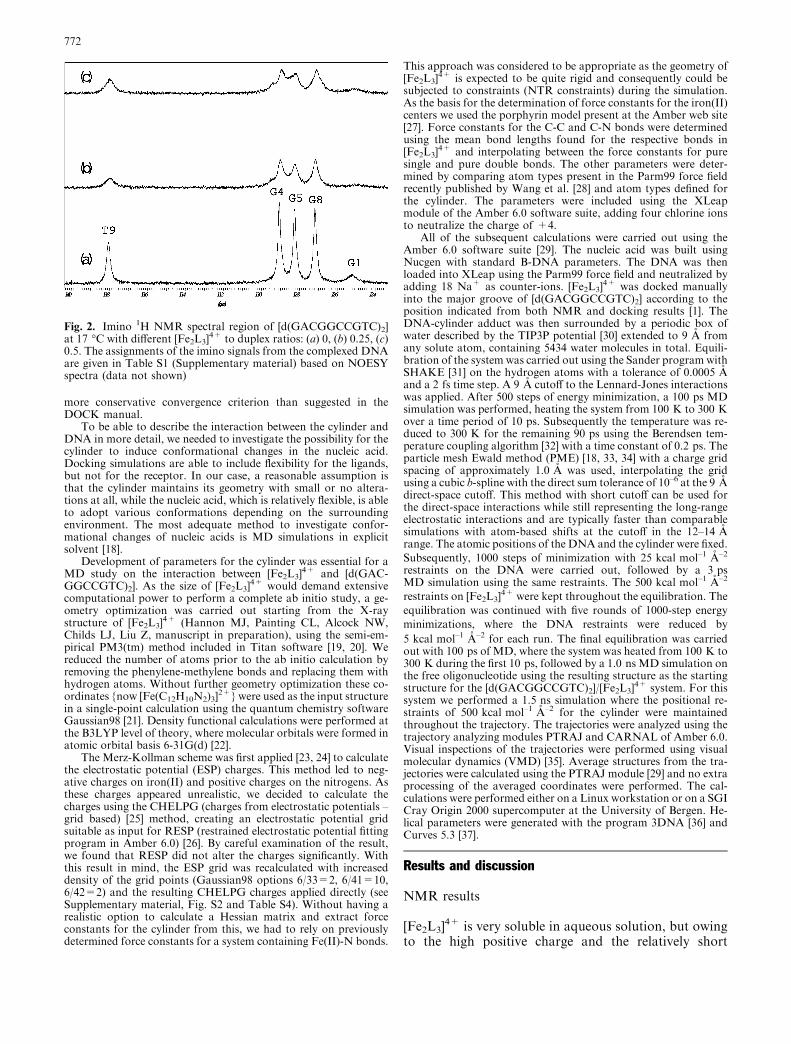
The synthesis of [Fe2L3]Cl4 (L=C25H20N4) (Fig. 1) has been de-scribed previously [12, 13]. The sodium salt of the deoxyoligonu-cleotide [d(G1A2C3G4G5C6C7G8T9C10)2] was purchased fromOswel DNA Service and purified by ion-exchange chromatogra-phy, desalted and paramagnetic impurities removed using a Chelex100 column (Sigma-Aldrich). The oligonucleotide was then dis-solved in 90% H2O/10% D2O containing 50 mM phosphate bufferand 40 mM NaCl, with a final concentration of 2.8 mM in duplex.Aliquots of [Fe2L3]Cl4 were added to the duplex in five steps toreach a cylinder:duplex ratio of 1:2. This ratio was selected so thatno precipitation should occur. NMR spectra were recorded on aBruker DRX 600 MHz instrument. A combination of through-space nuclear Overhauser effect spectroscopy (NOESY) andthrough-bond total correlation spectroscopy (TOCSY) two-di-mensional spectra were recorded at 17 �C using double pulsed fieldgradients spin echo water suppression (dpfgsew5) [14, 15]. The datawere processed using XWIN-NMR (Bruker) and analyzed usingSparky [16]. All five imino protons involved in Watson-Crick hy-drogen bonding were clearly observed in the 1D spectrum for thefree duplex (Fig. 2). The assignments for the imino resonances inthe adduct were obtained from a NOESY spectrum.
Based on the fact that the interaction between [Fe2L3]4+ and
DNA is expected to be purely electrostatic, we decided to investi-gate this with molecular docking calculations using the DOCK4.0.1 suite of programs (University of California, San Francisco)[17]. The duplex geometry of the oligonucleotide was built usingstandard B-DNA parameters in InsightII (Biosym). Based onNOESY cross-peaks between the cylinder and the duplex, a se-lected cluster was generated in the major groove. A box with di-mensions 14.2·14.1·14.1 (A) was constructed around these clustersand a grid calculation were carried out with a grid spacing 0.15 A.Initially the docking was performed with a sampling size of 10,000structures. The 15 best results were then energy minimized using a
Fig. 1. The molecular structureof the ligand (with numbering)and the tetracationic triplehelical supramolecular cylinder[Fe2(C25H20N4)3]Cl4. A sche-matic route of synthesis is alsoshown
771
more conservative convergence criterion than suggested in theDOCK manual.
To be able to describe the interaction between the cylinder andDNA in more detail, we needed to investigate the possibility for thecylinder to induce conformational changes in the nucleic acid.Docking simulations are able to include flexibility for the ligands,but not for the receptor. In our case, a reasonable assumption isthat the cylinder maintains its geometry with small or no altera-tions at all, while the nucleic acid, which is relatively flexible, is ableto adopt various conformations depending on the surroundingenvironment. The most adequate method to investigate confor-mational changes of nucleic acids is MD simulations in explicitsolvent [18].
Development of parameters for the cylinder was essential for aMD study on the interaction between [Fe2L3]
4+ and [d(GAC-GGCCGTC)2]. As the size of [Fe2L3]
4+ would demand extensivecomputational power to perform a complete ab initio study, a ge-ometry optimization was carried out starting from the X-raystructure of [Fe2L3]
4+ (Hannon MJ, Painting CL, Alcock NW,Childs LJ, Liu Z, manuscript in preparation), using the semi-em-pirical PM3(tm) method included in Titan software [19, 20]. Wereduced the number of atoms prior to the ab initio calculation byremoving the phenylene-methylene bonds and replacing them withhydrogen atoms. Without further geometry optimization these co-ordinates {now [Fe(C12H10N2)3]
2+} were used as the input structurein a single-point calculation using the quantum chemistry softwareGaussian98 [21]. Density functional calculations were performed atthe B3LYP level of theory, where molecular orbitals were formed inatomic orbital basis 6-31G(d) [22].
The Merz-Kollman scheme was first applied [23, 24] to calculatethe electrostatic potential (ESP) charges. This method led to neg-ative charges on iron(II) and positive charges on the nitrogens. Asthese charges appeared unrealistic, we decided to calculate thecharges using the CHELPG (charges from electrostatic potentials –grid based) [25] method, creating an electrostatic potential gridsuitable as input for RESP (restrained electrostatic potential fittingprogram in Amber 6.0) [26]. By careful examination of the result,we found that RESP did not alter the charges significantly. Withthis result in mind, the ESP grid was recalculated with increaseddensity of the grid points (Gaussian98 options 6/33=2, 6/41=10,6/42=2) and the resulting CHELPG charges applied directly (seeSupplementary material, Fig. S2 and Table S4). Without having arealistic option to calculate a Hessian matrix and extract forceconstants for the cylinder from this, we had to rely on previouslydetermined force constants for a system containing Fe(II)-N bonds.
This approach was considered to be appropriate as the geometry of[Fe2L3]
4+ is expected to be quite rigid and consequently could besubjected to constraints (NTR constraints) during the simulation.As the basis for the determination of force constants for the iron(II)centers we used the porphyrin model present at the Amber web site[27]. Force constants for the C-C and C-N bonds were determinedusing the mean bond lengths found for the respective bonds in[Fe2L3]
4+ and interpolating between the force constants for puresingle and pure double bonds. The other parameters were deter-mined by comparing atom types present in the Parm99 force fieldrecently published by Wang et al. [28] and atom types defined forthe cylinder. The parameters were included using the XLeapmodule of the Amber 6.0 software suite, adding four chlorine ionsto neutralize the charge of +4.
All of the subsequent calculations were carried out using theAmber 6.0 software suite [29]. The nucleic acid was built usingNucgen with standard B-DNA parameters. The DNA was thenloaded into XLeap using the Parm99 force field and neutralized byadding 18 Na+ as counter-ions. [Fe2L3]
4+ was docked manuallyinto the major groove of [d(GACGGCCGTC)2] according to theposition indicated from both NMR and docking results [1]. TheDNA-cylinder adduct was then surrounded by a periodic box ofwater described by the TIP3P potential [30] extended to 9 A fromany solute atom, containing 5434 water molecules in total. Equili-bration of the system was carried out using the Sander programwithSHAKE [31] on the hydrogen atoms with a tolerance of 0.0005 Aand a 2 fs time step. A 9 A cutoff to the Lennard-Jones interactionswas applied. After 500 steps of energy minimization, a 100 ps MDsimulation was performed, heating the system from 100 K to 300 Kover a time period of 10 ps. Subsequently the temperature was re-duced to 300 K for the remaining 90 ps using the Berendsen tem-perature coupling algorithm [32] with a time constant of 0.2 ps. Theparticle mesh Ewald method (PME) [18, 33, 34] with a charge gridspacing of approximately 1.0 A was used, interpolating the gridusing a cubic b-spline with the direct sum tolerance of 10–6 at the 9 Adirect-space cutoff. This method with short cutoff can be used forthe direct-space interactions while still representing the long-rangeelectrostatic interactions and are typically faster than comparablesimulations with atom-based shifts at the cutoff in the 12–14 Arange. The atomic positions of the DNA and the cylinder were fixed.
Subsequently, 1000 steps of minimization with 25 kcal mol–1 A–2
restraints on the DNA were carried out, followed by a 3 psMD simulation using the same restraints. The 500 kcal mol–1 A–2
restraints on [Fe2L3]4+ were kept throughout the equilibration. The
equilibration was continued with five rounds of 1000-step energy
minimizations, where the DNA restraints were reduced by
5 kcal mol–1 A–2 for each run. The final equilibration was carriedout with 100 ps of MD, where the system was heated from 100 K to300 K during the first 10 ps, followed by a 1.0 ns MD simulation onthe free oligonucleotide using the resulting structure as the startingstructure for the [d(GACGGCCGTC)2]/[Fe2L3]
4+ system. For thissystem we performed a 1.5 ns simulation where the positional re-straints of 500 kcal mol–1 A–2 for the cylinder were maintainedthroughout the trajectory. The trajectories were analyzed using thetrajectory analyzing modules PTRAJ and CARNAL of Amber 6.0.Visual inspections of the trajectories were performed using visualmolecular dynamics (VMD) [35]. Average structures from the tra-jectories were calculated using the PTRAJ module [29] and no extraprocessing of the averaged coordinates were performed. The cal-culations were performed either on a Linux workstation or on a SGICray Origin 2000 supercomputer at the University of Bergen. He-lical parameters were generated with the program 3DNA [36] andCurves 5.3 [37].
Results and discussion
NMR results
[Fe2L3]4+ is very soluble in aqueous solution, but owing
to the high positive charge and the relatively short
Fig. 2. Imino 1H NMR spectral region of [d(GACGGCCGTC)2]at 17 �C with different [Fe2L3]
4+ to duplex ratios: (a) 0, (b) 0.25, (c)0.5. The assignments of the imino signals from the complexed DNAare given in Table S1 (Supplementary material) based on NOESYspectra (data not shown)
772

oligonucleotide used in this study, we observed precipi-tation beyond a [Fe2L3]
4+/DNA ratio of 1:2. Evidentlythis precipitation is caused by charge neutralization,which has been observed with ditercalinium (a positivelycharged bisintercalating molecule) complexed to shortoligonucleotides [38] and also with binding of simpledivalent metal ions (e.g. Hg2+) to duplexes [39]. Thestepwise addition of [Fe2L3]
4+ to the duplex solution toreach the 1:2 ratio was followed by 1D 1H NMR. Thecolor of the solution was now deep purple (the color ofthe cylinder) and there was no sign of precipitation overa period of several months. The base pairing of theoligonucleotide was maintained, as confirmed by thepresence of Watson-Crick imino protons both in the freeand complexed form of the duplex (Fig. 2; see alsoSupplementary material, Fig. S1 and Table S1). Itshould be noted that several attempts were made toprepare samples with higher concentrations of thecylinder involving extensive stirring, the use of variousco-solvents and different salt concentrations. However,these attempts were not successful.
Since the solution contains both free and complexedDNA, the resulting NMR spectra are quite complex anddifficult to analyze. However, we were able to assign themajority of the signals belonging to both the free andbound DNA. The two-fold symmetry of the central partof the palindrome is lifted upon coordination with[Fe2L3]
4+. This is clearly seen in the TOCSY spectrum(Fig. 3), where the number of cross-peaks originatingfrom the coupling between H5 and H6 on the cytosinesC3, C6 and C7 increased from three to six (C3II is
overlapped with C3). Thus, we were able to assign adouble set of nucleotide resonances, which are labeled Iand II. This change in symmetry has also been observedfor other groove binding and intercalating molecules [40,41, 42]. The fact that relatively narrow linewidths areobserved for the resonances of the DNA-cylinder indi-cates that this adduct is quite stable. This is in agreementwith an ethidium bromide (EB) fluorescence competitionbinding assay previously performed by Rodger et al.[13], showing that [Fe2L3]
4+ has a binding constant to-wards DNA that is higher than EB (in the order of107 M–1).
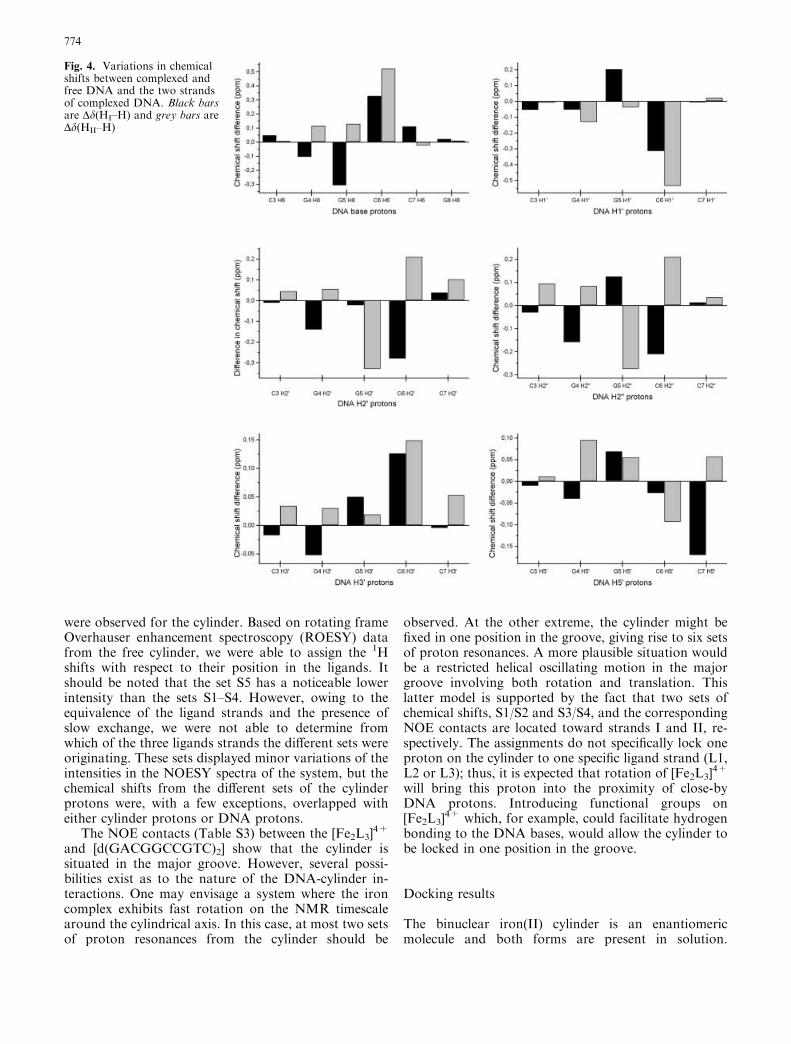
Variations in chemical shift between free and boundDNA (Table S1 and Fig. 4) indicate significant confor-mational changes in [d(GACGGCCGTC)2] upon com-plexation with the cylinder. The largest changes inchemical shift are found in the central G-G-C-C quartet.As the interaction between the cylinder and DNA isprimarily electrostatic, it is likely that the chemical shifteffects found for the nucleotide protons are induced ei-ther by conformational changes to the DNA and/orinfluence by ring current effects generated by the prox-imity of the six phenyl groups of the [Fe2L3]
4+ ligands.In a double helical environment the position of sugarproton resonances from H2¢ is mainly affected by thering current and magnetic anisotropy of the base at-tached to this sugar [43]. Furthermore, it has been sug-gested that the H2¢ signal should be rather sensitive todeviations from a regular double helical conformation[43, 44, 45]. For example, a large upfield shift of H2¢ in acis-platinated duplex was attributed to shielding by thedestacked adenine base of the kinked structure [45]. It isnow well established that Y-R steps (Y=pyrimidine,R=purine) like CpG are geometrically flexible [46] andseveral studies have shown that CpG steps are involvedin DNA bending [47, 48]. Since linear dichroism showsthat very low loading of [Fe2L3]
4+ has a dramaticbending effect on the DNA [1] and atomic force mi-croscopy images show that this is an intramolecular ef-fect resulting in coils of DNA [1], it is interesting to lookfor similar bending patterns in the NMR data. The H2¢resonances of the central residues G5II and C6I of theadduct display significant upfield shifts, in accordancewith a bent structure found by other methods (vide su-pra). However, similar upfield shifts are also observedfor the corresponding H2¢¢ resonances. Thus, the prox-imity of the cylinder ligand containing six ring-current-producing phenyl groups prevents a quantitativeconformational analysis based on chemical shifts.
The sequential walk between base protons and the H1¢protons are shown in Fig. 5 for the free duplex and forthe two strands I and II in the adduct. Every expectedcross-peak is present (though partially overlapped), butthe intensity of the cross-peaks in the central region isdecreased. This is most noticeable for the cross-peaksG5I H1¢ to C6I H5 and C6II H1¢ to C7II H6.
The chemical shift data and intramolecular NOEsoriginating from [Fe2L3]
4+ are presented in Tables S2and S3, respectively. Five different sets of signals (S1–S5)
Fig. 3. 1H 2D-TOCSY spectrum of the [d(GACGGCCGTC)2]/[Fe2L3]
4+ sample showing the cytosine H5-H6 cross-peaks. TheTOCSY spectrum was recorded with 80 ms mixing time at 25 �C
773
were observed for the cylinder. Based on rotating frameOverhauser enhancement spectroscopy (ROESY) datafrom the free cylinder, we were able to assign the 1Hshifts with respect to their position in the ligands. Itshould be noted that the set S5 has a noticeable lowerintensity than the sets S1–S4. However, owing to theequivalence of the ligand strands and the presence ofslow exchange, we were not able to determine fromwhich of the three ligands strands the different sets wereoriginating. These sets displayed minor variations of theintensities in the NOESY spectra of the system, but thechemical shifts from the different sets of the cylinderprotons were, with a few exceptions, overlapped witheither cylinder protons or DNA protons.
The NOE contacts (Table S3) between the [Fe2L3]4+
and [d(GACGGCCGTC)2] show that the cylinder issituated in the major groove. However, several possi-bilities exist as to the nature of the DNA-cylinder in-teractions. One may envisage a system where the ironcomplex exhibits fast rotation on the NMR timescalearound the cylindrical axis. In this case, at most two setsof proton resonances from the cylinder should be
observed. At the other extreme, the cylinder might befixed in one position in the groove, giving rise to six setsof proton resonances. A more plausible situation wouldbe a restricted helical oscillating motion in the majorgroove involving both rotation and translation. Thislatter model is supported by the fact that two sets ofchemical shifts, S1/S2 and S3/S4, and the correspondingNOE contacts are located toward strands I and II, re-spectively. The assignments do not specifically lock oneproton on the cylinder to one specific ligand strand (L1,L2 or L3); thus, it is expected that rotation of [Fe2L3]
4+
will bring this proton into the proximity of close-byDNA protons. Introducing functional groups on[Fe2L3]
4+ which, for example, could facilitate hydrogenbonding to the DNA bases, would allow the cylinder tobe locked in one position in the groove.
Docking results
The binuclear iron(II) cylinder is an enantiomericmolecule and both forms are present in solution.
Fig. 4. Variations in chemicalshifts between complexed andfree DNA and the two strandsof complexed DNA. Black barsare Dd(HI–H) and grey bars areDd(HII–H)
774

Recently we were able to separate the two enantiomers[49] and tests against DNA using linear dichroism (LD)show that the left-handed enantiomer displays thehighest activity towards DNA. By utilizing simplemolecular modeling, the left-handed enantiomer wasfound to fit the major groove of a right-handedB-DNA much better than the right-handed form. Tofurther test whether the major groove of DNA is alikely coordination site for [Fe2L3]
4+ or not, we per-formed molecular docking calculations using DOCK4.0 [17]. At this stage the canonical B-DNA version of[d(GACGGCCGTC)2] as the receptor for the cylinderwas used. The docking calculations were performedusing two kinds of scoring functions: contact score(molecular shape complementarity) and energy score[50] (based on non-bonded terms of the Amber force
field). Among the 15 top-scoring results with respect tocontact score, 10 complexes were almost superimposedin the middle of the major groove (spanning C3 to G8).The other five were spanning G4 to T9. The resultscalculated with respect to energy score were not thathomogeneous. Here, only three out of 15 complexeswere located in the middle of the groove. The six top-scoring complexes were all located between G4 and T9.A closer inspection of the results showed that the in-termolecular van der Waals energy was the dominatingfactor with respect to the energy score. The threecomplexes in the middle of the groove all have favor-able values for the intermolecular electrostatic energybut correspondingly higher values for the intermolec-ular van der Waals energy. The left-handed enantiomerof [Fe2L3]
4+ displays a remarkable fit in the groovewhere two of the pyridine rings of ligand strands L1and L2 ‘‘follow’’ the base geometry of the two nucle-otide strands (see Fig. S3).
MD calculations: free DNA and DNA-cylinder adduct
Initially, MD calculations were carried out for the freeDNA duplex in order to check any sequence-specificstructural deviations from regular B-form DNA. Theaveraged structure from the last 200 ps of the 1.0 nstrajectory of the free duplex displays a number of in-teresting features (Fig. S4). The structure is character-ized by a wide major groove in the central part and amoderate bend (10.3�). Other features of the structureare negative x-displacement values for all base pairsteps, except at the ends, and positive tip and roll valuesfor the two CpG steps.
The parameters developed for [Fe2L3]4+ were pri-
marily prepared to account for the charge of thecomplex. Reasonable force constants were extracted bycomparing the geometry for atom types included in thelatest force field published for Amber [28] and with theatoms in [Fe2L3]
4+. The results of a relatively shortMD test run (150 ps) showed that the coordinationgeometry around the two irons changed from thepseudo-octahedral geometry found in the X-ray struc-ture (Hannon MJ, Painting CL, Alcock NW, ChildsLJ, Liu Z, manuscript in preparation) to a geometrybest described as a compressed octahedron. However,these differences are hardly significant and further MDcalculations were carried out using positional restraintsfor the cylinder to maintain the X-ray structure duringthe simulation. We also know from spectroscopy (e.g.UV-vis and CD) that any changes in the helicatestructure are at best minimal.
Snapshots of the 1.5 ns trajectory from the MDsimulation performed on the [Fe2L3]
4+/[d(GAC-GGCCGTC)2] system clearly show that the cylinder hasa dramatic influence on the conformation of the oli-gonucleotide (Fig. 6). A more detailed analysis of thesimulation process is given in Figs. 7, 8, 9. The mostremarkable feature is the bending of DNA, which was
Fig. 5.1H 2D-NOESY spectrum of the [d(GACGGCCGTC)2]/
[Fe2L3]4+ adduct showing the sequential pathway for the aromatic
H1¢/H5 region: a free DNA; b the two strands in complexed DNA.The NOESY spectrum was recorded at 25 �C using a mixing timeof 200 ms. The solvent is 90% H2O/10% D2O. The sequentialpathways for free DNA and the adduct are separated for clarity
775

measured using the distance between the center of massof the first base-pair and the center of mass of the lastbase-pair (Fig. 7). The bending reaches a maximum of38.9� at 496 ps, but decreases significantly over the next150 ps. During the following �700 ps the DNA displaysa distinct stretched structure (Fig. 6). With no anchoringrestraints imposed, the cylinder is seen to move awayfrom the initial coordination site during the long tra-jectory. The animation of the trajectory using VMDshows that the cylinder moves down the major groove,with one end aligning along the phosphate backboneand the other end facing out of the major groove duringthe last �200 ps. The variation in RMS deviation duringthe simulation run (Fig. 8) was calculated with theequilibrated structure as a reference and follows thechange in curvature, which apparently is the majorcontributor to the RMS deviation.
NOE-restrained MD calculations
Based on NOESY data alone, it was not possible tocarry out a complete NMR structure determination ofthe DNA-cylinder adduct owing to excessive overlap inthe 2D spectra. An alternative approach was to use in-termolecular NOE contacts as anchors in a MD simu-lation. To test this possibility we used a fewunambiguous NOE contacts as anchors in an explicitwater MD simulation. The same protocol for equili-bration as previously described was used, except that thenumber of minimization steps was increased from 1000to 2000. The system was left to equilibrate over a periodof 50 ps. In this period the system was heated from100 K to 380 K over a period of 5 ps and then cooledslowly to 300 K over a period of 5 ps. In the remain-ing 40 ps the system was equilibrated to constant
Fig. 6. Three snapshots takenfrom the trajectory and theaverage structure(1360–1500 ps). [d(GAC-GGCCGTC)2] displays aremarkable flexibility duringthe course of the trajectory. Thefigure was generated with VMD[35] and Raster3D [51]
776

temperature (300 K). During this simulation the systemappeared to reach an equilibrated state. A relativelyshort production run of 400 ps was then performed. The
Fig. 7. Variation in DNA bending during the simulation cycles asmeasured by the distance between the centers of mass of the endbase-pairs in [d(GACGGCCGTC)2]
Fig. 8. Plot of the RMS deviation between the equilibratedstructure and the frames in the simulation trajectory
Fig. 9. Variation in distances between the center of mass in[Fe2L3]
4+ and the center of mass of the two central base pairsG5-C16 and C6-G15 during the simulation cycles
Fig. 11. Close-up of NOE contacts between S3 H7 and G4II H8,G5II H8 and C6II H6
Fig. 10. Averaged structure over the last 200 ps from the 400 psMD simulation including NOE restraints on the distances betweenS3 H7 and G4II H8, G5II H8 and C6II H6
Fig. 12. A comparison of the structures from the unrestrained andNMR-restrained MD simulations. The structure from the NMR-restrained simulation is shown in thick lines. Hydrogens areomitted for clarity. The most notable differences in the twostructures are the position of the cylinder and the displacements ofthe central base-pairs. The total RMS deviation between thestructures without hydrogens is 2.35 A (calculated with VMD)
777
entire system and a close-up of the central part of theadduct based on coordinates averaged over 200–400 psfrom the restrained MD calculations are shown inFigs. 10 and 11. A comparison of these coordinates and
the coordinates from the 1.5 ns trajectory is shown inFig. 12.
Helical parameters
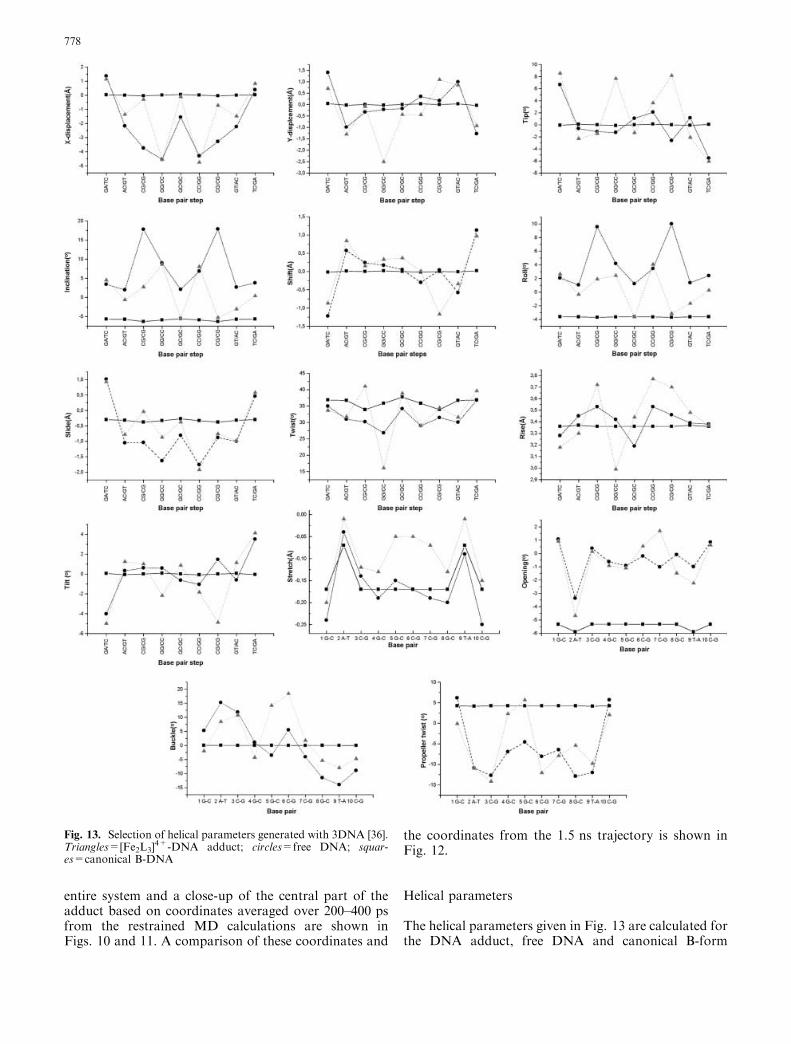
The helical parameters given in Fig. 13 are calculated forthe DNA adduct, free DNA and canonical B-form
Fig. 13. Selection of helical parameters generated with 3DNA [36].Triangles=[Fe2L3]
4+-DNA adduct; circles=free DNA; squar-es=canonical B-DNA
778

DNA. The coordinates for free DNA are those based onthe averaged structures over the last 200 ps from the1.0 ns trajectory, whereas the averaged structure for theDNA adduct was calculated using the coordinates from1360 to 1500 ps. The results show several structuralchanges, particularly in the central part of [d(GAC-GGCCGTC)2] where the [Fe2L3]
4+ induces a noticeableshift of symmetry in the duplex. Thus, the MD simula-tion supports the lift of symmetry found in the NMRspectra of the system. The wide major groove found inthe free DNA is extended even more to accommodatethe cylinder (Fig. 14). Among other distinct features ofthe structure, which differ from the free DNA, is the lowvalue for the helical twist (16.05�) found for the G4pG5
step. This step also displays a low value for the y-dis-placement, a high value for the inclination and a verysmall rise. The G5pC6 step follows the base-pair stepparameters of canonical B-DNA with surprising accu-racy, but clearly deviates from the results found for thefree DNA. Furthermore, the parameters of the G5-C16
and C6-G15 base pairs exhibit distinct differences forstretch, buckle and propeller twist. The C6pC7 step ischaracterized by a large value for the rise parameter,where the distance between the base pairs is 3.77 A.During parts of the trajectory, one of the pyridine ringson [Fe2L3]
4+ is partially intercalated between C6 andC7. NMR data were not able to confirm this mode ofinteraction. High values for inclination and rise char-acterize the C7pG8 step.
Conclusions
We have presented a detailed analysis of the interactionbetween a binuclear iron(II) supramolecular cylinderand the DNA decamer [d(GACGGCCGTC)2] with theuse of NMR, molecular docking and MD calculations,which reveal the potential of this cylinder to act as amajor groove binder. The interaction mode of thecylinder is characterized by its ability to induce
conformational changes in DNA. This has been shownby a combination of NMR spectroscopy and MD cal-culations. The development of synthetic major groovebinding agents, which recognize the DNA through non-covalent interactions inducing dramatic structural ef-fects, has important implications for genome recognitionand we are currently extending our studies to function-alized systems and enantiopure cylinders. Based on thepresent results, one way to proceed is to attach polargroups to the cylinder to act as either hydrogen-bonddonors or acceptors in the major groove involving basenitrogen atoms and/or carbonyl groups. In this way thecylinder could be locked to dsDNA in a sequence-specific manner, depending on the position of the polargroups.
Acknowledgements This work was supported by The NorwegianResearch Council (135055/410), the Leverhulme Trust (F/215/BC),the EPSRC life sciences network (GR/M91105) and the Universityof Warwick. Dr. Knut Børve (Department of Chemistry, Univer-sity of Bergen) and Dr. Ulf Ryde (Department of TheoreticalChemistry, University of Lund, Sweden) have been of great help inthe preparation of force field parameters for [Fe2L3]
4+. Computertime from the Parallab High Performance Computer Center at theUniversity of Bergen is gratefully acknowledged.
References
1. Hannon MJ, Moreno V, Prieto MJ, Moldrheim E, Sletten E,Meistermann I, Isaac CJ, Sanders KJ, Rodger A (2001) AngewChem Int Ed 40:879–884
2. White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB(1998) Nature 391:468–471
3. Thuong NT, Helene C (1993) Angew Chem Int Ed Engl 32:666–690
4. Nielsen PE, Haaima G (1997) Chem Soc Rev 26:73–785. Lippert B (ed) (1999) Cisplatin, a leading anti-cancer drug.VCH, Weinheim
6. Coggan DZM, Haworth IS, Bates PJ, Robinson A, Rodger A(1999) Inorg Chem 38:4486–4497
7. Onfelt B, Lincoln P, Norden B (1999) J Am Chem Soc121:10846–10847
8. Erkkila KE, Odom DT, Barton JK (1999) Chem Rev 99:2777–2795
9. Childs LZ, Alcock NW, Hannon MJ (2001) Angew Chem IntEd 40:1079–1081
10. Hannon MJ, Painting CL, Alcock NW (1999) Chem Commun2023–2024
11. Hannon MJ, Bunce S, Clarke AJ, Alcock NW (1999) AngewChem Int Ed 38:1277–1278
12. Hannon MJ, Painting CL, Jackson A, Hamblin J, Errington W(1997) Chem Commun 1807–1808
13. Rodger A, Sanders KJ, Hannon MJ, Meistermann I, ParkinsonA, Vidler DS, Haworth IS (2000) Chirality 12:221–236
14. Liu AM, Mao X, Ye C, Huang H, Nicholson JK, Lindon JC(1998) J Magn Reson 132:125–129
15. Hwang T-L, Shaka AJ (1995) J Magn Reson Ser A 112:275–27916. Goddard TD, Kneller DG () SPARKY3. University of Cali-
fornia, San Francisco17. Ewing TJA, Kuntz ID (1997) J Comput Chem 18:1175–118918. Cheatham TE III, Kollman PA (2000) Annu Rev Phys Chem
51:435–47119. Stewart JJP (1989) J Comput Chem 10:209–22020. Wavefunction () Titan. Wavefunction, Irvine, Calif21. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA,
Cheeseman JR, Zakrzewski VG, Montgomery Jr JA,
Fig. 14. Refined major groove widths generated with 3DNA [36]
779

Stratmann RE, Burant JC, Dapprich S, Millam JM, DanielsAD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V,Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, CliffordS, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K,Malick DK, Rabuck AD, Raghavachari K, Foresman JB,Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G,Liashenko A, Piskorz P, Komaromi I, Gomperts R, MartinRL, Fox DJ, Keith T, Al-Laham MA, Peng CY, NanayakkaraA, Challacombe M, Gill PMW, Johnson B, Chen W, WongMW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES,Pople JA (1998) Gaussian 98, revision A9. Gaussian, Pittsburgh
22. Rassolov VA, Pople JA, Ratner MA, Windus TL (1998)J Chem Phys 109:1223–1229
23. Singh UC, Kollman PA (1984) J Comput Chem 5:129–14524. Besler BH, Merz KM, Kollman PA (1990) J Comput Chem
11:431–43925. Breneman CM, Wiberg KB (1990) J Comput Chem 11:361–37326. Bayly CI, Cieplak P, Cornell WD, Kollman PA (1993) J Phys
Chem 97:10269–1028027. http://www.amber.ucsf.edu/amber/ff94/contrib/heme/frcmod
hemall28. Wang J, Cieplak P, Kollman PA (2000) J Comput Chem
21:1049–107429. Case DA, Pearlman DA, Caldwell JW, Cheatham TE III, Ross
WS, Simmerling CL, Darden TA, Merz KM, Stanton RV,Cheng AL, Vincent JJ, Crowley M, Tsui V, Radmer RJ, DuanY, Pitera J, Massova I, Seibel GL, Singh UC, Weiner PK,Kollman PA (1999) Amber 6. University of California, SanFrancisco
30. Jorgensen WL, Chandrasekhar J, Madura J, Impey RW, KleinML (1983) J Chem Phys 79:926–937
31. Ryckaert JP, Ciccotti G, Berendsen HJC (1997) J Comput Phys23:327–341
32. Berendsen HJC, Postma JPM, van Guensteren WF, DiNola A,Haak JR (1984) J Chem Phys 81:3684–3690
33. Essmann U, Perera L, Berkowitz ML, Darden T, Lee H,Pedersen LG (1995) J Chem Phys 103:8577–8593
34. Spakova N, Berger I, Sponer J (2001) J Am Chem Soc123:3295–3307
35. Humphrey W, Dalke A, Schulten K (1996) J Mol Graph 14:33–38
36. Lu X-J, Shakked Z, Olson WK (2000) J Mol Biol 300:819–84037. Lavery R, Sklenar H () Curves 5.3 (http://apex.ibpc.fr/
UPR9080/CurForm_bis.html)38. Delbarre A, Delepierre M, Langlois d’Estaintot B, Igolen J,
Roques BP (1987) Biopolymers 26:1001–1033
39. Frøystein NA, Sletten E (1994) J Am Chem Soc 116:3240–325040. Bostock-Smith CE, Harris SA, Laughton CA, Searle MS (2001)
Nucleic Acids Res 29:693–70241. Kumar RA, Ikemoto N, Patel DJ (1997) J Mol Biol 265:173–
18642. Chen H, Patel DJ (1995) J Mol Biol 246:164–17943. Wijmenga SS, Kruithof M, Hilbers CW (1997) J Biomol NMR
10:337–35044. Fouchet MH, Guittet E, Cognet JAH, Kozelka J, Gauthier C,
Bret ML, Zimmermann K, Chottard JC (1997) J Biol InorgChem 2:83–92
45. Marzilli LG, Saad JS, Kuklenyik Z, Keating KA, Xu Y (2001)J Am Chem Soc 123:2764–2770
46. Packer MJ, Dauncey MP, Hunter CA (2000) J Mol Biol295:71–83
47. Dickerson RE (1998) Nucleic Acids Res 26:1906–192648. Bolshoy A, McNamara P, Harrington RE, Trifonov EN (1991)
Proc Natl Acad Sci USA 88:2312–231649. Hannon MJ, Meistermann I, Isaac CJ, Blomme C, Aldrich-
Wright JR, Rodger A (2001) Chem Commun 1078–107950. Meng EC, Shoichet BK, Kuntz ID (1992) J Comput Chem
13:504–52451. Merrit EA, Bacon DJ (1997) Methods Enzymol 277:505–524
780





























