Embed Size (px)
Citation preview

REVIEW
Inherited genetic susceptibility to multiple myelomaGJ Morgan1, DC Johnson1, N Weinhold2, H Goldschmidt2, O Landgren3, HT Lynch4, K Hemminki5,6 and RS Houlston7
Although the familial clustering of multiple myeloma (MM) supports the role of inherited susceptibility, only recently has directevidence for genetic predisposition been demonstrated. A meta-analysis of two genome-wide association (GWA) studies hasidentified single-nucleotide polymorphisms (SNPs) localising to a number of genomic regions that are robustly associated with MMrisk. In this review, we provide an overview of the evidence supporting a genetic contribution to the predisposition to MM andMGUS (monoclonal gammopathy of unknown significance), and the insight this gives into the biological basis of disease aetiology.We also highlight the promise of future approaches to identify further specific risk factors and their potential clinical utility.
Leukemia (2014) 28, 518–524; doi:10.1038/leu.2013.344
Keywords: myeloma; SNP; MGUS
EPIDEMIOLOGICAL STUDIESEpidemiological studies have established increasing age, malegender, familial background and a past history of MGUS as beingrisk factors for MM.1 MGUS is detectable in 3–5% of individualsaged 50 years or older in European populations2 and is typicallyassociated with an annual risk of progression to MM of around 1%.As MGUS plasma cells have many of the genetic characteristics ofMM plasma cells, it has been suggested that MM is alwayspreceded by a MGUS phase. This is supported by long-termfollow-up studies in a sample set of 77 469 healthy adults who hadbanked serum samples3 and a military cohort.2,4 Observationstherefore indicate that susceptibility to MM is either shared withMGUS risk or related to the transition between the twopathological states.
A number of lifestyle and environmental risk factors have beenproposed to increase MM risk, including obesity,5 immunedysfunction, agricultural, chemical6 and radiation exposure.1 Theresults from these purported associations are, however, notconcordant between independent studies possibly reflectingissues with study design.1 Obesity is probably the mostconsistently reported association and various mechanisms havebeen proposed to explain the relationship including increasedoxidative stress, alterations in immunologic response, metabolicresponse and the levels of endogenous hormones (for example,sex steroids, insulin and insulin-like growth factor-1). A number ofstudies have also suggested that the risk of MM may be related toautoimmunity or its treatment.7
FAMILIAL EPIDEMIOLOGICAL STUDIESCase reports of the familial clustering of MM provide support forthe role of inherited factors in disease aetiology. Epidemiologicalcase–control and cohort studies have consistently shown anincreased risk of MM or MGUS in first-degree relatives of patientswith MM or MGUS.8–11 The largest study to date, involving
11 752 MM patients diagnosed in Sweden between 1958 and2002, showed that the risk of MM was increased 4.25-fold in first-degree relatives (95% CI: 1.81–8.41).8 A survey carried out by theUtah Cancer Registry, making use of family data from 1354 cases,confirms this association but also demonstrated a relationshipwith risk of prostate and melanoma,12 a finding supported by twoother reports.13–15 Two studies have shown the risk of MM andMGUS is increased threefold in relatives of individuals withMGUS.16,17 In addition, it was observed that there was also anincreased risk of developing NHL and CLL8,9,18,19 Collectively, thesedata are consistent with an inherited genetic susceptibility to MM,which is somewhere in the order of two- to fourfold. In addition tothis epidemiological data, over 100 MM families (Table 1) havebeen described in the literature, recent reports coming fromUS,20,21 France,22,23 Sweden8 and Iceland.10 As well as providingevidence for inherited predisposition to MM some of thesefamilies show MM present in three generations (Figure 1)compatible with Mendelian transmission of the risk.23–26
RACIAL DIFFERENCESRacial differences in the risk of developing MM are wellrecognised, with a greater prevalence of MGUS and MM beingseen in African Americans as compared with those with EuropeanAncestry.4,27,28 A study based on US Veterans found a two tothreefold higher prevalence of MGUS among African Americanscompared with Caucasians, while the risk of transformation fromMGUS to MM was the same.29 An increased prevalence of MGUSrelative to whites has also been reported from Ghana,4,30 whereasrates of MGUS and MM in American Chinese, Japanese andMexicans populations are lower.31,32 Recent data from the UnitedStates has found that the mean age at diagnosis of myeloma is65.8 and 69.8 years for African Americans and Caucasians,respectively.33 Although inequalities and confounding effectsdue to environment and behavioural factors cannot beexcluded, both the higher rates and earlier age of onset of MM
1Haemato-Oncology Research Unit, Division of Molecular Pathology, Institute of Cancer Research, Surrey, UK; 2Department of Internal Medicine V, University of Heidelberg,Heidelberg, Germany; 3Multiple Myeloma Section, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA; 4Department of Preventive Medicine, Creighton’sHereditary Cancer Center, Omaha, NE, USA; 5Division of Molecular Genetic Epidemiology, German Cancer Research Centre (DKFZ), Heidelberg, Germany; 6Center for PrimaryHealth Care Research, Lund University, Malmo, Sweden and 7Division of Genetics and Epidemiology, Institute of Cancer Research, Surrey, UK. Correspondence: ProfessorGJ Morgan, Haemato-Oncology Research Unit, Division of Molecular Pathology, The Institute of Cancer Research, 15 Cotswold Road, Sutton SM2 5NG, UK.E-mail: [email protected] 30 July 2013; revised 15 October 2013; accepted 17 October 2013; accepted article preview online 19 November 2013; advance online publication, 20 December 2013
Leukemia (2014) 28, 518–524& 2014 Macmillan Publishers Limited All rights reserved 0887-6924/14
www.nature.com/leu

in African Americans is supportive of a genetic contribution to theaetiology of MM.33
GENETIC ASSOCIATIONSUntil recently, the search for genetic variants influencing MM riskhas almost exclusively been based on analyses of polymorphismsin candidate genes. In reviewing the published literature ongenetic association studies in MM catalogued by PUBMED since2000, six major hypotheses have been examined, including therole of cytokines and immune response, DNA repair, folatemetabolism, ADME (absorption, distribution, metabolism andexcretion), insulin-like growth factors and apoptosis.29,47–52
Studies have reported positive associations with MM risk, butnone of these studies report associations lower than the stringentthreshold of genome-wide significance of 5� 10� 8 withindependent replication. Most of these associations have beenbased on small sample sizes with limited accountancy forpopulation substructure or cryptic-relatedness, and thereforehave limited power to robustly demonstrate a relationship
between genotype and MM risk. The nature and design of thesestudies have been reviewed previously.52–54 Importantly, withouta clear understanding of the biological basis of MM developmentdeciding what represents a candidate gene is inherently difficult.Although future association studies with greater cases andcontrols may be powered to replicate some of these previouslydescribed candidate gene study associations.
GENOME-WIDE ANALYSESCataloguing the haplotype structure of the human genome hasled to the development of a comprehensive set of tagging SNPsthat capture common genetic variation. The analysis of around500 000 of these tagSNPs allows for the search of commonvariants influencing cancer risk to be conducted on a genome-wide basis. Such studies are agnostic and are not based onpreconceptions of biology such as those used in candidate genestudies. Genome-wide association studies (GWAS) have beenconducted on most common cancers and multiple risk lociidentified. As well as vindicating the long-held hypothesis of
Table 1. A summary of MM families with 42MM cases that have been described in the literature
Author Year No. offamilies
No. of MMindividuals
No. of MGUSindividuals
Relationship of another MMindividual with proband
Relationship of another MGUSindividual with proband
MM Pedigrees/case reportsShoenfeld et al.34 1982 37 94 NA 4—Parent–child
26—sibling pairs3—larger sibling group
NA
McCrea and Morris35 1986 6 13 NA 3—parent–child7—sibling pairs3—larger sibling group
NA
Roddie et al.36 1998 1 3 NA 2—Sibling pairs NAGrosbois et al.22 1999 15 30 3 6—Parent–child
10—Sibling pairs1—Larger sibling group
3—Larger sibling group
Lynch et al.37 2001 1 3 2 2—Sibling pairs 2—Sibling pairsSobol et al.23 2001 1 6 NA 2—Parent-child
2—Sibling pairs1—Larger sibling group
NA
Lynch et al.21 2005 39 74 4 20—Parent–child12—Sibling pairs2—Larger sibling group
NA
Ogmundsdottir et al.11 2005 4 8 6 2—Sibling pairs2—Larger sibling group
1—parent–child1—sibling pairs4—larger sibling group
Gerkes et al.38 2007 2 5 NA 3—Parent–child1—Sibling pairs
NA
Lynch et al.26 2008 1 5 1 2—Sibling pairs2—Larger sibling group
3—sibling pairs1—larger sibling group
Jain et al.24 2009 8 11 8 2—Parent–child3—Sibling pairs
2—parent–child5—sibling pairs
Grass et al.39 2011 4 6 4 1—Parent–child2—Sibling pairs
1—Parent–child2—sibling pairs1—larger sibling group
MM TwinsOgawa et al.40 1970 1 2 NA Monozygotic twin NAJudson et al.41 1985 1 2 NA Monozygotic twin NAMcCrea and Morris35 1986 1 2 NA Monozygotic twin NAComotti et al.42 1987 1 2 NA Monozygotic twin NASnowden and Greaves43 1995 1 2 NA Monozygotic twin NAOlujohungbe et al.44 2006 1 2 NA Monozygotic twin NA
MM families within cancer registriesLichtenstein et al.45 2000 1 2 NA Monozygotic twin NAHemminki46 2002 8 16 NA 8—Parent-child NAAltieri et al.8 2006 32 NA NA NA NA
Abbreviation: NA, not applicable.
Inherited genetic susceptibility to MMGJ Morgan et al
519
& 2014 Macmillan Publishers Limited Leukemia (2014) 518 – 524

polygenic inheritance to cancer, the loci identified haveprovided novel insights into the genes influencing risk. Twogenome-wide association studies of MM have been recentlyreported.55,56 The initial meta-analysis of UK and German GWASstudies comprises 1675 MM cases and 5903 controls;55 thesestudies were then extended in a second meta-analysis combining4692 MM cases and 10 990 controls.56 Cases and controls weredrawn from populations of Northern and Western Europeandescent. The studies identified SNPs at chromosomes 2p23.3,3p22.1, 3q26.2, 6p21.33, 7p15.3, 17p11.2 and 22q13.1 robustlyassociated with risk of MM, Table 2. Importantly none of the genesannotated by SNPs at these loci have previously been evaluated incandidate gene studies.
The 2p23.3 association annotates a gene, DNA (cytosine-5)-methyltransferase 3 alpha (DNMT3A) that encodes a DNAmethyltransferase.57–59 DNMT3A is highly expressed gene in MM-regulating cytokines such as IFN-g, TNFa and IL-4.60,61 In addition,DNMT3A directly interacts with MYC, a key myeloma related gene,as well as acting as a co-repressor through methylation ofpromoters at genes including the cyclin-dependent kinaseinhibitors p21Cip1 and p15INK4B.62
The 3p22.1 association is associated with the A542T poly-morphism in the serine/threonine-protein kinase unc-51-like kinase4 (ULK4) gene. ULK4 is highly expressed in MM, and the Atg1/ULKcomplex is a key regulator of mTOR-mediated autophagy. Theregion of LD to which ULK1 A542T maps also encompasses theB-catenin gene (CTNNB1), which is known to activate transcriptionfactors including MYC.63 A correlation between the expression ofthe ULK4 and CTNNB1 exists, suggesting the possibility of theshared regulation of this region in MM.64,65
The 3q26.2 association is defined by a SNP mapping 50 to thetelomerase RNA component gene (TERC). TERC is the ncRNAcomponent of telomerase that together with telomerase reversetranscriptase (TERT) form part of a ribonuclear protein complex thathelps maintains telomere ends. Interestingly, telomerase reactivationand telomerase-mediated elongation of shorter telomeres is afeature of MM66 and the major allele is associated with longertelomeres; thus there is strong a priori evidence for genetic variationat this locus having a role in MM.67,68 Variants at this locus have alsobeen associated with prostate cancer69 and colorectal cancer.70
The 6p21.33 SNP association localises to intron 5 of a putativepsoriasis susceptibility gene (PSORS1C1), but it is entirely possiblebecause of the LD structure across this region, that the associationreflects variation within the extended MHC region and hence HLAgenotype.
The 7p15.3 SNP association encompasses the 30 part of theCDCA7L (cell division cycle associated 7-like) gene and theDNAH11 (dynein, axonemal, heavy chain 11 (MIM 603339)) gene.DNAH11 has exonic mutations and is also affected by translocationin cases of MM.71 CDCA7L is expressed at relatively high levels andincreases on the progression from ‘MGUS’ to ‘MM’ in mice.72
Interestingly, in the context of the mechanism underlying MM risk,CDCA7L is a cell cycle-related gene that directly interacts with MYC(c-Myc).73–75 Expression of a set of conserved miRNAs in the 30-UTRof CDCA7L is higher in MM cell lines compared with a normalplasma cell control.76 hsa-miR-25 and hsa-miR-32 are among agroup miRNA that are upregulated in MM and predicted toregulate CDCA7L.77,78 Thus, there is a plausible hypothesissupporting a role for CDCA7L in mediating MM risk throughactivation of downstream growth control genes.79
Figure 1. Pedigree showing family members with multiple myeloma or monoclonal gammopathy of undetermined significance (MGUS). Thisis an updated pedigree first published, Lynch et al.26
Inherited genetic susceptibility to MMGJ Morgan et al
520
Leukemia (2014) 518 – 524 & 2014 Macmillan Publishers Limited
The 17p11.2 SNP association maps within intron 2 of tumournecrosis factor receptor superfamily member 13B (TNFRSF13B).TNFRSF13B also known as TACI (transmembrane activator andcalcium modulator and cyclophilin ligand interactor) has a key rolein promoting differentiation of Ig-secreting cells. Variation at thischromosome region has been implicated as a determinant ofcirculating immunoglobulin levels.80,81 TNFRSF13B is preferentiallyexpressed on marginal zone B cells, CD27þ memory B-cellsubsets and plasma cells.82 TNFRSF13B is receptor for key MMgrowth factors BAFF and APRIL,83 and mutations identified inTNFRSF13B can activate the classical NFkB pathway.84
The 22q13.1 SNP association localises to intron 2 of the geneencoding chromobox homologue 7 (CBX7). CBX7 encodes apolycomb group protein (PcG) known to be downregulated inmany cancers.85–88 It is thought to be a tumour suppressornegatively regulating CCNE1 expression.89 These proteins formpart of a gene regulatory mechanism that determines cell fateduring development as well as contributing to the control of cellgrowth and differentiation.90 High levels of CBX7 are seen ingerminal center lymphocytes and in follicular lymphoma.88 CBX7can cooperate with MYC to promote lymphomagenesis as well asaffecting cellular lifespan through regulation of both the p16Ink4a/Rb and the Arf/p53 pathways.91,92
ASSOCIATIONS WITH MOLECULAR SUBGROUPSIt is being increasingly recognised that genetic susceptibilitiesare subtype specific reflecting differences in biology andthat analysis by specific molecular features can reveal keyrisk relationships. Myeloma displays considerable molecularheterogeneity, which is reflected in clinical outcome. Thesubgroups defined by t(11;14)(q13;q32), t(4;14)(p16;q32),t(6;14)(p21;q32), t(14;16)(q32;q23) and t(14;20)(q32;q11) transloca-tions93 show transcriptional activation of CCND1, FGFR3/MMSET,CCND3, MAF and MAFB, respectively, which directly contribute tothe development of MM.93 The other major subgroup of myelomaare patients without a primary translocation, who commonlydisplay hyperdiploidy.
A case-only analysis of the identified MM risk loci in theoriginal GWAS data sets provided little evidence forsubtype-specific associations, apart from the CBX7 SNP rs877529that showed evidence of an association driven by non-t(11;14)MM.56 As subset-only analyses are powered by a sectionof the available cases, larger samples sets will be required todefinitively determine whether these genetic risk loci arerestricted to subsets or that these regions have a generic effecton MM risk.
Through a stratified analysis of GWAS data, it has been shownthat the G870A polymorphism in cyclin D1 is a determinant of therisk of developing t(11;14) MM.94 Cyclin D1 is a component of thecore cell cycle machinery and increased levels are seen in manycancers including MM. In addition to activating cyclin-dependent
kinases CDKN4 and CDK6, cyclin D1 has kinase-independentfunctions in DNA repair, notably directly binding RAD51, arecombinase that drives homologous recombination.95,96
Overproduction of a D group cyclin is common biologicalfeature of MM and in the t(11;14) subtype it is CCND1, which isupregulated through its close proximity to the powerful Emenhancer of the IgH locus as a result of a reciprocaltranslocation.93,97
The G870 allele creates an optimal splice donor site at theintron 4/exon 5 boundary resulting in the cyclin D1a transcript. Bycontrast, A870 hinders splicing allowing for read-through intointron 4 and production of the variant cyclin D1b transcript.Although 870A is preferentially associated with D1b production,the A allele is not fully penetrant.96 D1a/D1b transcript ratios havebeen shown to be enhanced in some cancers, suggesting thatdifferential alternative splicing can influence tumourigenesis.96
Recent data have demonstrated that while cyclin D1b is a potentoncogene operating through aberrant kinase activity cyclin D1arecruits RAD51 to local chromatin in response to DNAdamage.95,98 However, although the association between G870Aand t(11;14) MM represents the first report of germline variationbeing a determinant of risk for a specific chromosometranslocation, the biological basis remains to be established.G870A cannot operate through a general DNA repair mechanismas increased risk in other translocation groups of MM is not seen.As an association with t(11;14) MGUS is seen but no associationshown for Mantle cell lymphoma, it suggests the impact of G870Aoperates through a as yet unidentified mechanism early on in thedevelopment of t(11;14) MM.94,99
DEFINING INDIVIDUAL RISKAlthough the risk of MM associated with each of the variantsidentified through GWAS is individually modest, they are commonin the population; therefore, they contribute significantly to theoverall disease burden. Moreover, alleles can act additivelyconferring substantive risks to those carrying multiple risk alleles.Currently the seven known risk alleles account for B13% of thefamilial risk of MM;56 therefore, the discriminatory value ofgenotyping individuals based on such a panel is very limitedand we have a long way to go before we can predict high riskindividuals confidently. If we are to effectively identify high-riskindividuals with the sensitivity and specificity required in a clinicalsetting, it has been reasoned that we would need to identifyupwards of 150 independent risk loci at OR¼ 1.5 or 250 atOR¼ 1.25 with minor allele frequencies greater than 10%.100 Tofacilitate the identification of additional risk alleles throughassociation-based and analyses, it requires access to largesample series something only achievable through multi-centrecollaborative efforts such as the ‘MyelomA Genetics InternationalConsortium’ (MAGIC).101
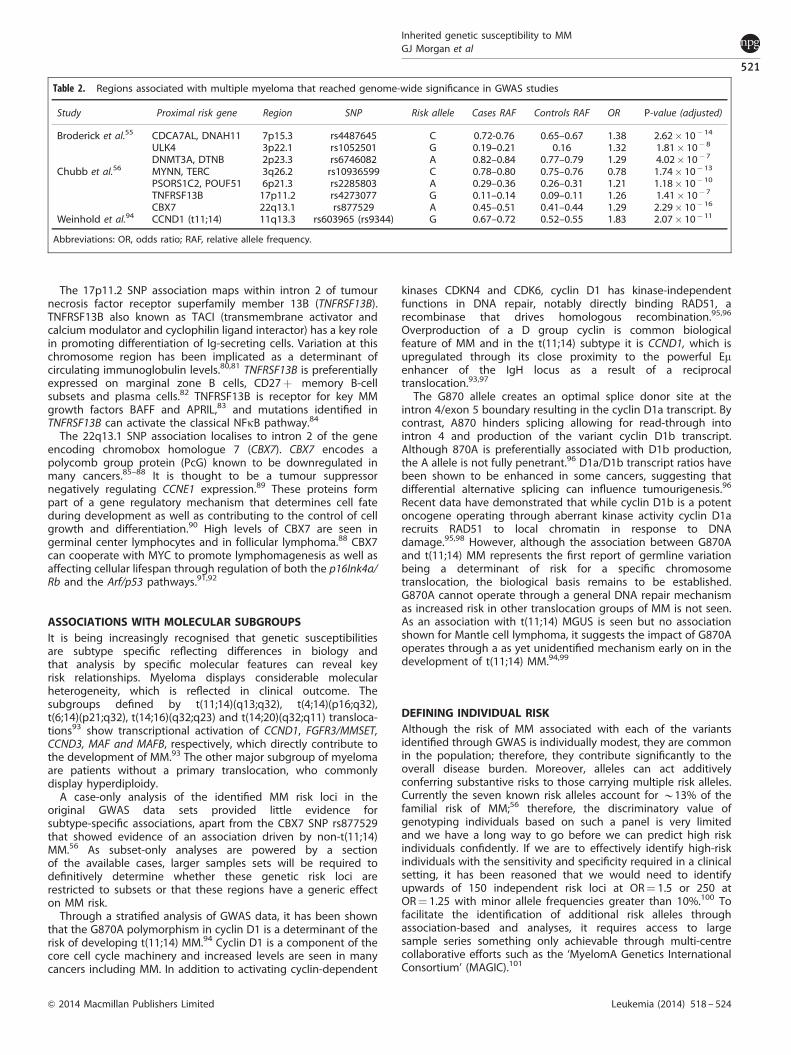
Table 2. Regions associated with multiple myeloma that reached genome-wide significance in GWAS studies
Study Proximal risk gene Region SNP Risk allele Cases RAF Controls RAF OR P-value (adjusted)
Broderick et al.55 CDCA7AL, DNAH11 7p15.3 rs4487645 C 0.72-0.76 0.65–0.67 1.38 2.62� 10� 14
ULK4 3p22.1 rs1052501 G 0.19–0.21 0.16 1.32 1.81� 10� 8
DNMT3A, DTNB 2p23.3 rs6746082 A 0.82–0.84 0.77–0.79 1.29 4.02� 10� 7
Chubb et al.56 MYNN, TERC 3q26.2 rs10936599 C 0.78–0.80 0.75–0.76 0.78 1.74� 10� 13
PSORS1C2, POUF51 6p21.3 rs2285803 A 0.29–0.36 0.26–0.31 1.21 1.18� 10� 10
TNFRSF13B 17p11.2 rs4273077 G 0.11–0.14 0.09–0.11 1.26 1.41� 10� 7
CBX7 22q13.1 rs877529 A 0.45–0.51 0.41–0.44 1.29 2.29� 10� 16
Weinhold et al.94 CCND1 (t11;14) 11q13.3 rs603965 (rs9344) G 0.67–0.72 0.52–0.55 1.83 2.07� 10� 11
Abbreviations: OR, odds ratio; RAF, relative allele frequency.
Inherited genetic susceptibility to MMGJ Morgan et al
521
& 2014 Macmillan Publishers Limited Leukemia (2014) 518 – 524

BIOLOGICAL INSIGHTS BASED ON THESE VARIANTSUntil now it has been possible to gain important insights into thelater stages of myeloma pathogenesis by the study of tumour-acquired genetic lesions such as copy-number variants andmutations. These studies have defined important pathologicallyderegulated genes and pathways that re-wire the biologicalfunction of a normal plasma cell into one that has the ‘molecularhallmarks’ of a myelomatous plasma cell. Early events in thismalignant transformation process, however, have been moredifficult to study and largely it has only been the class switchdriven translocations that have given relevant insights. Theassociation studies we describe here are important because theycan give insights into the potential mechanisms relevant early inthe pathogenesis of MM as well as highlighting how inheritedgenetic variants might interact with tumour-acquired variants toenhance progression towards myeloma.
Interestingly, we find few abnormalities, in this first pass of thedata, that would have been predicted to be associated withmyeloma based on a priori hypotheses and that have beenincluded in previous candidate gene studies. However, we seederegulation of pathways involved in DNA methylation, telomerelength, differentiation and autophagy, which is consistent withMM risk being governed by pathways important in the longevityof plasma cells and the adaptation to paraprotein production. Wesee an association with TNFRSF13B, the receptor for BAFF andAPRIL that activates the NFkB pathway, a key growth and survivalpathway in plasma cells. This association argues that constitutiveactivation of the NFkB pathway may be an important early eventincreasing the likelihood that a cell, bearing this variant, may bedirected down a pathway eventually leading to MM.
MYC deregulation has been reported to be critical to myelomapathogenesis and is deregulated in the majority of cell lines aswell as in a significant proportion of clinical cases. Thederegulation of MYC, late in the natural history of myeloma, isbrought about by a range of molecular mechanisms includingtumour-acquired DNA insertion, deletion and translocation. Themyeloma risk genes CDCA7L, DNMT3A and CBX7 are all reported tohave interactions with MYC and indicate that the deregulation ofMYC early in the pathogenesis of myeloma is, in some part,mediated by the inheritance of risk variants. This observation, ofinherited variation in MYC function and predisposition tomyeloma, seems to be a common mechanism in a range of B-celltumours. Associations with MYC are seen in the results of GWAstudies in other B-cell entities including non-Hodgkin lym-phoma,102 Hodgkin’s lymphoma103 and chronic lymphocyticleukaemia.104 The mechanisms deregulating MYC are, however,different in these tumour types involving inherited variants in agene desert upstream of MYC.
Cell cycle abnormalities are critical features of myelomapathogenesis and a number of MM risk genes have functionsthat may influence the cell cycle pathway early in the naturalhistory of the disease. The expression of checkpoint genesp16Ink4a/Rb, Arf/p53 and CCNE1 can be altered by activity of theMM risk gene CBX7. Whereas DNMT3A can repress p21Cip1 andp15INK4B, and CDCA7L can influence the expression of CCND1. It isof course fascinating that an inherited variant in CCND1, a criticalcell cycle gene deregulated in a significant proportion ofmyeloma, seemingly predisposes patients to develop transloca-tions into that locus. This observation further informs us of thecritical nature of deregulation of the G1S cell cycle checkpoint inthe transformation of a normal to a malignant plasma cell.
The key conclusions of these initial GWAS analyses are that thepathways deregulated by the acquisition of genomic changesgiving rise to the ‘molecular hallmarks of myeloma’ are alsoimportant early in the disease natural history where their functionis impacted by inherited variants. Analogous to tumour-acquiredvariants such inherited variants can also be considered as ‘genetic
hits’ pushing a normal cell along the pathway to a malignantplasma cell. Thus, a person with an inherited variant is either onestep further along the pathway and so more likely to develop MMor alternatively they may have an increased likelihood ofdeveloping MM-specific variants.
RARE MODERATE-HIGH PENETRANCE SUSCEPTIBILITY TO MMIn addition to common variation influencing MM risk, it is likelythat other classes of susceptibility exist, an assertion supported byfamilies in which the inheritance of MM is compatible with a majordisease locus. An example of a rare low-penetrance susceptibilityallele in MM is provided by the germ-line mutations observed inCDKN2A (p16INK4A).13 Current GWA-based strategies are notoptimally configured to identify such low frequency variants;however, genotyping strategies using increased density arraysinformed by sequence studies such as the 1000 genomes willallow rarer variants important to the pathogenesis of MM to beidentified.
Despite this current deficiency in array-based approaches,recent advances in molecular-based technologies make theutilisation of sequencing approaches a feasible proposition.Association testing using either exome or whole-genomesequence heralds a fundamental development that will allowinvestigation of the full spectrum of inherited variation impactingthe risk developing MM. In addition, although the computingchallenges of processing the large data sets for sequence-basedassociation testing are considerable, the bioinformatic tools arebeginning to be developed to undertake such analyses.105–107
Confirming rarer variants predisposing to MM will require evenlarger case series and is even stronger rationale for collaborativestudies across the MM community.
CONCLUSIONAssociation testing offers hypothesis-free testing of cases thatcombined with powerful sequence-based technologies will lead toa deeper understanding of genes and interactions underlying thepathogenesis of MM. Recent developments in the functionalannotation by the ENCODE projects will no doubt accelerate thefunctional characterisation of these associations. Ultimately, thistype of strategy will highlight novel therapeutic molecular targetsfor the development of new therapeutic strategies. In addition, theidentification of variants that predisposes individuals to distinctgenetic pathogenetic subtypes hold the promise of providingclinicians with screening tools to allow greater personalisation oftherapies and clinical management. To date, MM GWAS studieshave only considered patients with a Northern and WesternEuropean descent (CEU), it is important to follow-up these findingsin other populations. In addition to new therapies and improvedmanagement of MM patients, further elucidation of MM andMGUS risk factors may eventually allow related family members ofMM patients or individuals with MGUS to be more informed oftheir potential predisposing risk of developing MM.
CONFLICT OF INTERESTThe authors declare no conflict of interest.
ACKNOWLEDGEMENTSLaboratory work of GJM is supported by funding from Myeloma UK. The work ofRSH is supported by grants from Leukaemia Lymphoma Research and MyelomaUK. HG and KH are supported by funding was provided to Dietmar-Hopp-Stiftungin Walldorf, The German Ministry of Education and Science (Gliomics 01ZX1309B),the German Cancer Aid, Deursche Krebshilfe and the University HospitalHeidelberg.
Inherited genetic susceptibility to MMGJ Morgan et al
522
Leukemia (2014) 518 – 524 & 2014 Macmillan Publishers Limited

REFERENCES1 Morgan GJ, Davies FE, Linet M. Myeloma aetiology and epidemiology. Biomed
Pharmacother 2002; 56: 223–234.2 Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy
precedes multiple myeloma in most patients. Blood 2009; 113: 5418–5422.3 Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB et al.
Monoclonal gammopathy of undetermined significance (MGUS) consistentlyprecedes multiple myeloma: a prospective study. Blood 2009; 113: 5412–5417.
4 Landgren O, Weiss BM. Patterns of monoclonal gammopathy of undeterminedsignificance and multiple myeloma in various ethnic/racial groups: support forgenetic factors in pathogenesis. Leukemia 2009; 23: 1691–1697.
5 Soderberg KC, Kaprio J, Verkasalo PK, Pukkala E, Koskenvuo M, Lundqvist E et al.Overweight, obesity and risk of haematological malignancies: a cohort study ofSwedish and Finnish twins. Eur J Cancer 2009; 45: 1232–1238.
6 Ruder AM, Hein MJ, Hopf NB, Waters MA. Mortality among 24 865 workersexposed to polychlorinated biphenyls (PCBs) in three electrical capacitormanufacturing plants: A ten-year update. Int J Hyg Environ Health 2013; 13:pii: S1438-4639: 00060–00066.
7 Lindqvist EK, Goldin LR, Landgren O, Blimark C, Mellqvist UH, Turesson I et al.Personal and family history of immune-related conditions increase therisk of plasma cell disorders: a population-based study. Blood 2011; 118:6284–6291.
8 Altieri A, Chen B, Bermejo JL, Castro F, Hemminki K. Familial risks and temporalincidence trends of multiple myeloma. Eur J Cancer 2006; 42: 1661–1670.
9 Bourguet CC, Grufferman S, Delzell E, DeLong ER, Cohen HJ. Multiple myelomaand family history of cancer. A case-control study. Cancer 1985; 56: 2133–2139.
10 Ogmundsdottir HM, Einarsdottir HK, Steingrimsdottir H, Haraldsdottir V. Familialpredisposition to monoclonal gammopathy of unknown significance, Walden-strom’s macroglobulinemia, and multiple myeloma. Clin Lymphoma Myeloma2009; 9: 27–29.
11 Ogmundsdottir HM, Haraldsdottirm V, Johannesson GM, Olafsdottir G, Bjarna-dottir K, Sigvaldason H et al. Familiality of benign and malignant para-proteinemias. A population-based cancer-registry study of multiple myelomafamilies. Haematologica 2005; 90: 66–71.
12 Camp NJ, Werner TL, Cannon-Albright LA. Familial myeloma. N Engl J Med 2008;359: 1734–1735author reply 1735.
13 Dilworth D, Liu L, Stewart AK, Berenson JR, Lassam N, Hogg D. Germline CDKN2Amutation implicated in predisposition to multiple myeloma. Blood 2000; 95:1869–1871.
14 Eriksson M, Karlsson M. Occupational and other environmental factors andmultiple myeloma: a population based case-control study. Br J Ind Med 1992; 49:95–103.
15 Eriksson M, Hallberg B. Familial occurrence of hematologic malignancies andother diseases in multiple myeloma: a case-control study. Cancer Causes Control1992; 3: 63–67.
16 Landgren O, Kristinsson SY, Goldin LR, Caporaso NE, Blimark C, Mellqvist UH et al.Risk of plasma cell and lymphoproliferative disorders among 14621 first-degreerelatives of 4458 patients with monoclonal gammopathy of undeterminedsignificance in Sweden. Blood 2009; 114: 791–795.
17 Vachon CM, Kyle RA, Therneau TM, Foreman BJ, Larson DR, Colby CL et al.Increased risk of monoclonal gammopathy in first-degree relatives of patientswith multiple myeloma or monoclonal gammopathy of undetermined sig-nificance. Blood 2009; 114: 785–790.
18 Brown LM, Linet MS, Greenberg RS, Silverman DT, Hayes RB, Swanson GM et al.Multiple myeloma and family history of cancer among blacks and whites in theUS. Cancer 1999; 85: 2385–2390.
19 Linet MS, McLaughlin JK, Harlow SD, Fraumeni JF. Family history of autoimmunedisorders and cancer in multiple myeloma. Int J Epidemiol 1988; 17: 512–513.
20 Jain M, Cook GM, Davis FG, Grace MG, Howe GR, Miller AB. A case-control studyof diet and colo-rectal cancer. Int J Cancer 1980; 26: 757–768.
21 Lynch HT, Watson P, Tarantolo S, Wiernik PH, Quinn-Laquer B, Isgur Bergsagel Ket al. Phenotypic heterogeneity in multiple myeloma families. J Clin Oncol 2005;23: 685–693.
22 Grosbois B, Jego P, Attal M, Payen C, Rapp MJ, Fuzibet JG et al. Familial multiplemyeloma: report of fifteen families. Br J Haematol 1999; 105: 768–770.
23 Sobol H, Vey N, Sauvan R, Philip N, Noguchi T, Eisinger F. Re: familial multiplemyeloma: a family study and review of the literature. J Natl Cancer Inst 2002; 94:461–462; author reply 463.
24 Jain M, Ascensao J, Schechter GP. Familial myeloma and monoclonal gammo-pathy: a report of eight African American families. Am J Hematol 2009; 84: 34–38.
25 Lynch HT, Thome SD. Familial multiple myeloma. Blood 2009; 114: 749–750.26 Lynch HT, Ferrara K, Barlogie B, Coleman EA, Lynch JF, Weisenburger D et al.
Familial myeloma. N Engl J Med 2008; 359: 152–157.27 Landgren O, Rajkumar SV, Pfeiffer RM, Kyle RA, Katzmann JA, Dispenzieri A et al.
Obesity is associated with an increased risk of monoclonal gammopathy of
undetermined significance among black and white women. Blood 2010; 116:1056–1059.
28 Greenberg AJ, Vachon CM, Rajkumar SV. Disparities in the prevalence, pathogen-esis and progression of monoclonal gammopathy of undetermined significanceand multiple myeloma between blacks and whites. Leukemia 2011; 26: 609–614.
29 Spink CF, Gray LC, Davies FE, Morgan GJ, Bidwell JL. Haplotypic structure acrossthe I kappa B alpha gene (NFKBIA) and association with multiple myeloma.Cancer Lett 2007; 246: 92–99.
30 Landgren O, Katzmann JA, Hsing AW, Pfeiffer RM, Kyle RA, Yeboah ED et al.Prevalence of monoclonal gammopathy of undetermined significance amongmen in Ghana. Mayo Clin Proc 2007; 82: 1468–1473.
31 Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF et al. Along-term study of prognosis in monoclonal gammopathy of undeterminedsignificance. N Engl J Med 2002; 346: 564–569.
32 Landgren O, Gridley G, Turesson I, Caporaso NE, Goldin LR, Baris D et al. Risk ofmonoclonal gammopathy of undetermined significance (MGUS) and sub-sequent multiple myeloma among African American and white veterans in theUnited States. Blood 2006; 107: 904–906.
33 Waxman AJ, Mink PJ, Devesa SS, Anderson WF, Weiss BM, Kristinsson SY et al.Racial disparities in incidence and outcome in multiple myeloma: a population-based study. Blood 2010; 116: 5501–5506.
34 Shoenfeld Y, Berliner S, Shaklai M, Gallant LA, Pinkhas J. Familial multiple mye-loma. A review of thirty-seven families. Postgrad Med J 1982; 58: 12–16.
35 McCrea AP, Morris TC. Concurrent familial myeloma in Northern Ireland. Cancer1986; 58: 394–396.
36 Roddie PH, Dang R, Parker AC. Multiple myeloma in three siblings. Clin LabHaematol 1998; 20: 191–193.
37 Lynch HT, Sanger WG, Pirruccello S, Quinn-Laquer B, Weisenburger DD. Familialmultiple myeloma: a family study and review of the literature. J Natl Cancer Inst2001; 93: 1479–1483.
38 Gerkes EH, de Jong MM, Sijmons RH, Vellenga E. Familial multiple myeloma:report on two families and discussion of screening options. Hered Cancer ClinPract 2007; 5: 72–78.
39 Grass S, Preuss KD, Thome S, Weisenburger DD, Witt V, Lynch J et al. Paraproteinsof familial MGUS/multiple myeloma target family-typical antigens: hyperpho-sphorylation of autoantigens is a consistent finding in familial and sporadicMGUS/MM. Blood 2011; 118: 635–637.
40 Ogawa M, Wurster DH, McIntyre OR. Multiple myeloma in one of a pair ofmonozygotic twins. Acta Haematol 1970; 44: 295–304.
41 Judson IR, Wiltshaw E, Newland AC. Multiple myeloma in a pair of monozygotictwins: the first reported case. Br J Haematol 1985; 60: 551–554.
42 Comotti B, Bassan R, Buzzetti M, Finazzi G, Barbui T. Multiple myeloma in a pairof twins. Br J Haematol 1987; 65: 123–124.
43 Snowden JA, Greaves M. IgA lambda myeloma presenting concurrently inidentical twins with subsequent transformation to ’aggressive phase’ in one. ClinLab Haematol 1995; 17: 95–96.
44 Olujohungbe AB, Gledhill T, Satchithananathan G. Temporal development ofmyeloma in a syngeneic twin pair. Br J Haematol 2006; 133: 211–212.
45 Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M et al.Environmental and heritable factors in the causation of cancer—analyses of cohortsof twins from Sweden, Denmark, and Finland. N Engl J Med 2000; 343: 78–85.
46 Hemminki K. Re: familial multiple myeloma: a family study and review of theliterature. J Natl Cancer Inst 2002; 94: 462–463; author reply 463.
47 Hayden PJ, Tewari P, Morris DW, Staines A, Crowley D, Nieters A et al. Variation inDNA repair genes XRCC3, XRCC4, XRCC5 and susceptibility to myeloma. Hum MolGenet 2007; 16: 3117–3127.
48 Pratt G, Fenton JA, Allsup D, Fegan C, Morgan GJ, Jackson G et al. A poly-morphism in the 3’ UTR of IRF4 linked to susceptibility and pathogenesis inchronic lymphocytic leukaemia and Hodgkin lymphoma has limited impact inmultiple myeloma. Br J Haematol 2010; 150: 371–373.
49 Morgan GJ, Adamson PJ, Mensah FK, Spink CF, Law GR, Keen LJ et al. Haplotypes inthe tumour necrosis factor region and myeloma. Br J Haematol 2005; 129: 358–365.
50 Roddam PL, Rollinson S, O’Driscoll M, Jeggo PA, Jack A, Morgan GJ. Geneticvariants of NHEJ DNA ligase IV can affect the risk of developing multiple mye-loma, a tumour characterised by aberrant class switch recombination. J MedGenet 2002; 39: 900–905.
51 Davies FE, Rollinson SJ, Rawstron AC, Roman E, Richards S, Drayson M et al. High-producer haplotypes of tumor necrosis factor alpha and lymphotoxin alpha areassociated with an increased risk of myeloma and have an improved progres-sion-free survival after treatment. J Clin Oncol 2000; 18: 2843–2851.
52 Greenberg AJ, Rajkumar SV, Vachon CM. Familial monoclonal gammopathy ofundetermined significance and multiple myeloma: epidemiology, risk factors,and biological characteristics. Blood 2012; 119: 5359–5366.
53 Vangsted A, Klausen TW, Vogel U. Genetic variations in multiple myeloma I:effect on risk of multiple myeloma. Eur J Haematol 2012; 88: 8–30.
Inherited genetic susceptibility to MMGJ Morgan et al
523
& 2014 Macmillan Publishers Limited Leukemia (2014) 518 – 524

54 Vangsted A, Klausen TW, Vogel U. Genetic variations in multiple myeloma II:association with effect of treatment. Eur J Haematol 2012; 88: 93–117.
55 Broderick P, Chubb D, Johnson DC, Weinhold N, Forsti A, Lloyd A et al. Commonvariation at 3p22.1 and 7p15.3 influences multiple myeloma risk. Nat Genet 2012;44: 58–61.
56 Chubb D, Weinhold N, Broderick P, Chen B, Johnson DC, Forsti A et al. Commonvariation at 3q26.2, 6p21.33, 17p11.2 and 22q13.1 influences multiple myelomarisk. Nat Genet 2013; 45: 1221–1225.
57 Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem2005; 74: 481–514.
58 Kangaspeska S, Stride B, Metivier R, Polycarpou-Schwarz M, Ibberson D, Car-mouche RP et al. Transient cyclical methylation of promoter DNA. Nature 2008;452: 112–115.
59 Metivier R, Gallais R, Tiffoche C, Le Peron C, Jurkowska RZ, Carmouche RP et al.Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008;452: 45–50.
60 El Gazzar M, Yoza BK, Chen X, Hu J, Hawkins GA, McCall CE. G9a and HP1 couplehistone and DNA methylation to TNFalpha transcription silencing duringendotoxin tolerance. J Biol Chem 2008; 283: 32198–32208.
61 Gamper CJ, Agoston AT, Nelson WG, Powell JD. Identification of DNA methyl-transferase 3a as a T cell receptor-induced regulator of Th1 and Th2 differ-entiation. J Immunol 2009; 183: 2267–2276.
62 Brenner C, Deplus R, Didelot C, Loriot A, Vire E, De Smet C et al. Myc repressestranscription through recruitment of DNA methyltransferase corepressor. EMBO J2005; 24: 336–346.
63 Davies FE, Dring AM, Li C, Rawstron AC, Shammas MA, O’Connor SM et al.Insights into the multistep transformation of MGUS to myeloma using micro-array expression analysis. Blood 2003; 102: 4504–4511.
64 Walker BA, Leone PE, Jenner MW, Li C, Gonzalez D, Johnson DC et al. Integrationof global SNP-based mapping and expression arrays reveals key regions,mechanisms, and genes important in the pathogenesis of multiple myeloma.Blood 2006; 108: 1733–1743.
65 Broyl A, Hose D, Lokhorst H, de Knegt Y, Peeters J, Jauch A et al. Gene expressionprofiling for molecular classification of multiple myeloma in newly diagnosedpatients. Blood 2010 2010; 116: 2543–2553.
66 Jones CH, Pepper C, Baird DM. Telomere dysfunction and its role in haemato-logical cancer. Br J Haematol 2012; 156: 573–587.
67 Codd V, Mangino M, van der Harst P, Braund PS, Kaiser M, Beveridge AJ et al.Common variants near TERC are associated with mean telomere length. NatGenet 2010; 42: 197–199.
68 Jones AM, Beggs AD, Carvajal-Carmona L, Farrington S, Tenesa A, Walker M et al.TERC polymorphisms are associated both with susceptibility to colorectal cancerand with longer telomeres. Gut 2012; 61: 248–254.
69 Kote-Jarai Z, Olama AA, Giles GG, Severi G, Schleutker J, Weischer M et al. Sevenprostate cancer susceptibility loci identified by a multi-stage genome-wideassociation study. Nat Genet 2011; 43: 785–791.
70 Houlston RS, Cheadle J, Dobbins SE, Tenesa A, Jones AM, Howarth K et al. Meta-analysis of three genome-wide association studies identifies susceptibility locifor colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet 2010;42: 973–977.
71 Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC et al.Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471:467–472.
72 Carrasco DR, Sukhdeo K, Protopopova M, Sinha R, Enos M, Carrasco DE et al. Thedifferentiation and stress response factor XBP-1 drives multiple myelomapathogenesis. Cancer Cell 2007; 11: 349–360.
73 Taddesse-Heath L, Meloni-Ehrig A, Scheerle J, Kelly JC, Jaffe ES. Plasmablasticlymphoma with MYC translocation: evidence for a common pathway in thegeneration of plasmablastic features. Mod Pathol 2010; 23: 991–999.
74 Dib A, Gabrea A, Glebov OK, Bergsagel PL, Kuehl WM. Characterization of MYC trans-locations in multiple myeloma cell lines. J Natl Cancer Inst Monogr 2008; 39: 25–31.
75 Chng WJ, Huang GF, Chung TH, Ng SB, Gonzalez-Paz N, Troska-Price T et al.Clinical and biological implications of MYC activation: a common differencebetween MGUS and newly diagnosed multiple myeloma. Leukemia 2011; 25:1026–1035.
76 Jima DD, Zhang J, Jacobs C, Richards KL, Dunphy CH, Choi WW et al. Deepsequencing of the small RNA transcriptome of normal and malignant human Bcells identifies hundreds of novel microRNAs. Blood 2010; 116: e118–e127.
77 Pichiorri F, Suh SS, Ladetto M, Kuehl M, Palumbo T, Drandi D et al. MicroRNAsregulate critical genes associated with multiple myeloma pathogenesis. Proc NatlAcad Sci USA 2008; 105: 12885–12890.
78 Pichiorri F, Suh SS, Rocci A, De Luca L, Taccioli C, Santhanam R et al. Down-regulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2autoregulatory loop in multiple myeloma development. Cancer Cell 2010; 18:367–381.
79 Yang L, Lin C, Liu W, Zhang J, Ohgi KA, Grinstein JD et al. ncRNA- and Pc2methylation-dependent gene relocation between nuclear structures mediatesgene activation programs. Cell 2011; 147: 773–788.
80 Osman W, Okada Y, Kamatani Y, Kubo M, Matsuda K, Nakamura Y. Association ofcommon variants in TNFRSF13B, TNFSF13, and ANXA3 with serum levels of non-albumin protein and immunoglobulin isotypes in Japanese. PLoS One 2012; 7:e32683.
81 Kamatani Y, Matsuda K, Okada Y, Kubo M, Hosono N, Daigo Y et al. Genome-wideassociation study of hematological and biochemical traits in a Japanese popu-lation. Nat Genet 2010; 42: 210–215.
82 Darce JR, Arendt BK, Wu X, Jelinek DF. Regulated expression of BAFF-bindingreceptors during human B cell differentiation. J Immunol 2007; 179: 7276–7286.
83 Novak AJ, Darce JR, Arendt BK, Harder B, Henderson K, Kindsvogel W et al.Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism forgrowth and survival. Blood 2004; 103: 689–694.
84 Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ et al. Promiscuousmutations activate the noncanonical NF-kappaB pathway in multiple myeloma.Cancer Cell 2007; 12: 131–144.
85 Forzati F, Federico A, Pallante P, Fedele M, Fusco A. Tumor suppressor activity ofCBX7 in lung carcinogenesis. Cell Cycle 2012; 11: 1888–1891.
86 Zhang XW, Zhang L, Qin W, Yao XH, Zheng LZ, Liu X et al. Oncogenic role of thechromobox protein CBX7 in gastric cancer. J Exp Clin Cancer Res 2010; 29: 114.
87 Karamitopoulou E, Pallante P, Zlobec I, Tornillo L, Carafa V, Schaffner T et al. Lossof the CBX7 protein expression correlates with a more aggressive phenotype inpancreatic cancer. Eur J Cancer 2010; 46: 1438–1444.
88 Scott CL, Gil J, Hernando E, Teruya-Feldstein J, Narita M, Martinez D et al. Role ofthe chromobox protein CBX7 in lymphomagenesis. Proc Natl Acad Sci USA 2007;104: 5389–5394.
89 Forzati F, Federico A, Pallante P, Abbate A, Esposito F, Malapelle U et al. CBX7 is atumor suppressor in mice and humans. J Clin Invest 2012; 122: 612–623.
90 Gil J, Bernard D, Peters G. Role of polycomb group proteins in stem cell self-renewal and cancer. DNA Cell Biol 2005; 24: 117–125.
91 Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S et al. Molecularinterplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 bypolycomb CBX7 in transcriptional silencing of INK4a. Mol Cell 2010; 38: 662–674.
92 Gil J, Bernard D, Martinez D, Beach D. Polycomb CBX7 has a unifying role incellular lifespan. Nat Cell Biol 2004; 6: 67–72.
93 Sawyer J. The prognostic significance of cytogenetics and molecular profiling inmultiple myeloma. Cancer Genet 2011; 204: 3–12.
94 Weinhold N, Johnson DC, Chubb D, Chen B, Forsti A, Hosking FJ et al. The CCND1c.870G4A polymorphism is a risk factor for t(11;14)(q13;q32) multiple myeloma.Nat Genet 2013; 45: 522–525.
95 Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F et al. A functionfor cyclin D1 in DNA repair uncovered by protein interactome analyses in humancancers. Nature 2012; 474: 230–234.
96 Knudsen KE, Diehl JA, Haiman CA, Knudsen ES. Cyclin D1: polymorphism,aberrant splicing and cancer risk. Oncogene 2006; 25: 1620–1628.
97 Chng WJ, Glebov O, Bergsagel PL, Kuehl WM. Genetic events in the pathogenesisof multiple myeloma. Best Pract Res Clin Haematol 2007; 20: 571–596.
98 Li Z, Jiao X, Wang C, Shirley LA, Elsaleh H, Dahl O et al. Alternative cyclin D1splice forms differentially regulate the DNA damage response. Cancer Res 2010;70: 8802–8811.
99 Johnson DC, Ross FM, Dickens NJ, Davies FE, Child JA, Durie B et al. Associationof Genetic Variants with FISH-Based Karyotyping Status in Multiple MyelomaPatients. Clin Lymphoma Myelom 2009; 9: S132–S133.
100 Pepe MS, Gu JW, Morris DE. The potential of genes and other markers to informabout risk. Cancer Epidemiol Biomarkers Prev 2010; 19: 655–665.
101 Morgan G, Johnsen HE, Goldschmidt H, Palumbo A, Cavo M, Sonneveld P et al.MAGIC (MyelomA Genetics International Consortium). Leuk Lymphoma 2011; 53:796–800.
102 Conde L, Halperin E, Akers NK, Brown KM, Smedby KE, Rothman N et al. Genome-wide association study of follicular lymphoma identifies a risk locus at 6p21.32.Nat Genet 2010; 42: 661–664.
103 Enciso-Mora V, Broderick P, Ma Y, Jarrett RF, Hjalgrim H, Hemminki K et al. A genome-wide association study of Hodgkin’s lymphoma identifies new susceptibility loci at2p16.1 (REL), 8q24.21 and 10p14 (GATA3). Nat Genet 2010; 42: 1126–1130.
104 Crowther-Swanepoel D, Broderick P, Di Bernardo MC, Dobbins SE, Torres M,Mansouri M et al. Common variants at 2q37.3, 8q24.21, 15q21.3 and 16q24.1influence chronic lymphocytic leukemia risk. Nat Genet 42: 132–136.
105 Asimit J, Zeggini E. Testing for rare variant associations in complex diseases.Genome Med 2011; 3: 24.
106 Asimit J, Zeggini E. Rare variant association analysis methods for complex traits.Annu Rev Genet 2010; 44: 293–308.
107 Morris AP, Zeggini E. An evaluation of statistical approaches to rare variantanalysis in genetic association studies. Genet Epidemiol 2010; 34: 188–193.
Inherited genetic susceptibility to MMGJ Morgan et al
524
Leukemia (2014) 518 – 524 & 2014 Macmillan Publishers Limited





























