Embed Size (px)
Citation preview

CHAPTER 8
Inherited Tumour Syndromes
Inherited cancer susceptibility is now recognized as a signifi-cant risk for cancer of the breast and female genitals organs.For many inherited tumour syndromes, the underlying germlinemutations have been identified. This allows genetic testing andcounseling of at risk family members and to estimate the asso -ciated disease burden. The genetic basis involves mutationalinactivation of tumour supressor and DNA repair genes. Suchgermline mutations follow a mendelian inheritance pattern andusually confer substantial cancer risks, with breast and ovaryas most frequent target organs. Additional familial aggrega-tions have been observed but the responsible genes have notyet been identified and may involve multigenic traits.

336 Inherited tumour syndromes
Evidence of familial aggregation ofb reast, ovarian, and other tumours of thefemale genital organs derived from anec-dotal observation of large families andf rom systematic analyses of cancer inci-dence in relatives of cancer cases.Although there are a number of potentialm e a s u res of familial aggregation, themost commonly used is the familial re l a-tive risk (FRR) or standardized incidencerate (SlR). This is defined as the ratio ofthe incidence of disease among re l a t i v e sof an individual with disease compare dwith the incidence in the population as awhole. The FRR is most often estimatedt h rough comparison of family history databetween cases and controls, with theresulting odds ratio used as an estimatorof the familial risk. Using genealogicalre s o u rces linked to cancer registries hasa number of advantages; the number ofcases is usually large compared to case-c o n t rol studies and, more import a n t l y, allcancers found among relatives are con-f i rmed in the cancer re g i s t ry.
Breast cancerEvidence that women with a positive fam-ily history of breast cancer are ati n c reased risk for developing the diseasehas been accumulating for over 50 years;v i rtually every study has found signifi-cantly elevated relative risks to female re l-atives of breast cancer patients. Moststudies have found relative risks between2 and 3 for first-degree relatives of bre a s tcancer patients selected without re g a rdto age at diagnosis or laterality. A re c e n treview of 74 published studies {2238} cal-culated familial relative risks of 2.1 (95%CI 2.0, 2.2) for breast cancer in any firstd e g ree relative, 2.3 for a sister aff e c t e d ,and 2.0 for an affected mother, and a re l-ative risk of 3.6 if an individual had both amother and sister affected. For individu-als with a first degree relative diagnosedwith breast cancer under age 50, the re l-ative risk to develop breast cancer beforeage 50 was 3.3 (CI 2.8, 3.9). In a population-based study of familialcancer using the Utah PopulationDatabase, Goldgar et al. {1029} studied
the incidence of breast and other can-cers among 49 202 first-degree relativesof 5559 breast cancer probands diag-nosed before age 80. This study estimat-ed a relative risk of 1.8 in first degree rel-atives of these breast cancer probands.When restricted to early-onset cancer(diagnosed before age 50), the relativerisk of breast cancer among first-degreerelatives increased to 2.6 and the risk forearly-onset breast cancer among theserelatives was 3.7 (95% CI. 2.8–4.6). TheSwedish family cancer database {715}contains >9.6 million individuals, withdata on nearly 700,000 invasive cancersand consists of individuals born inSweden after 1934 and their parents.Analyzing cancers diagnosed betweenthe years 1958 to 1996, the standardizedincidence ratio for breast cancer was1.85 (95% CI 1.74–1.96) for having anaffected mother, 1.98 (1.79–2.18) for hav-ing an affected sister, and 2.4 (1.72–3.23) if both mother and sister werea ffected. Other studies found largerfamilial effects among relatives of youngbilateral probands compared with youngprobands with unilateral breast cancer{700,1246,2129}. The issue of relationship of histology tofamilial breast cancer is less clear {375,500,1724,2441,2989}. Some studiesfound that lobular carcinoma is moreoften associated with a positive familyh i s t o ry {791} while others {1566}observed that cases with tubular carci-noma were more frequently associatedwith a positive family history. Multi-centricity was also found to be positivelyassociated with family history {1564}.Occurrence of breast cancer in a maleconveys a two to three fold increased riskof breast cancer in female re l a t i v e s{94,2449}.
Ovarian cancerIn a population-based case-control studyof families of 493 ovarian cancer casesand 2465 controls, Schildkraut andThompson {2557} reported an odds ratiofor ovarian cancer in first degree rela-tives of 3.6 (95% CI 1.8–7.1). A compre-
hensive study of first-degree relatives of883 ovarian cancer probands from theUtah Population Database estimated arelative risk of 2.1 (1.0–3.4) for ovariancancer in the relatives {1029}. Analysis ofthe Swedish family cancer database{715} found a standardized incidenceratio for ovarian cancer of 2.81 (95% CI2.21–3.51) for having an affected mother,1.94 (0.99–3.41) for having an affectedsister, and 25.5 (6.6–66.0) if both motherand sister were affected. A meta-analysisof all case-control and cohort studiespublished before 1998 estimates the riskto first degree relatives at 3.1, with a95%CI of 2.6-3.7 {2801}.
Endometrial cancerGruber and Thompson {1071} in a studyof first-degree relatives of 455 cases ofp r i m a ry epithelial carcinoma of theendometrium and 3216 controls, reportan odds ratio (OR) of 2.8 (CI 1.9 – 4.2) forhaving one or more relatives affectedwith endometrial cancer. In a similar sizestudy (726 cases and 2123 controls)Parrazini et al. {2173} found a smallereffect, with an OR of 1.5 (CI 1.0–2.3). Thismay partly be explained by the fact thatin the former study, cases were restrictedto ages 20-54, while in the latter, themedian age at diagnosis was 61. ADanish case-control study of 237 casesof endometrial cancer diagnosed underage 50 and 538 population contro l sreported an OR for family history of 2.1(1.1–3.8). In contrast to most other sites,the two registry/geneaology based stud-ies of endometrial cancer produced con-flicting results, with the Utah study find-ing a FRR of 1.32 and the Swedish fami-ly cancer database reporting a SIR of2.85. The reason for this discrepancy isunclear, but may to some extent reflectdifferences in the age distribution of thetwo populations.
Cervical cancerIn the Utah Population Database {1029},a FRR to first degree relatives of 999 cer-vical cancer cases of 1.74 was obtained(95% CI 1.03-2.53) while in the Swedish
D. GoldgarM.R. StrattonFamilial aggregation of cancers of the
breast and female genital organs

337Familial aggregation of cancers of the breast and female genital organs
family cancer database {715}, a slightlyhigher risk of 1.93 (1.52-2.42) in mothersof invasive cervical cancer cases and2.39 (1.59-3.46) in sisters. Unlike manyother cancers, there did not appear to bea significant effect of age at diagnosis infamilial risk of cervical cancer, althoughthe risks to mothers did depend on thenumber of affected daughters. In thiss t u d y, significant familial aggre g a t i o nwas also found for in situ carcinoma ofthe cervix (FRR 1.79, (1.75-1.84).
Multiple cancer sitesIn most but not all studies, a familialassociation between cancers of thebreast and ovary have been found, par-ticularly when the breast cancer caseshave been diagnosed at a young age.Undoubtedly, the majority of the associa-tion between breast and ovarian cancerdetected in these population studies isdue to the BRCA1 gene, which is knownto be involved in a large proportion ofextended kindreds with clearly inheritedsusceptibility to breast and ovarian can-cer. It is likely that some of the discrepantresults are linked to the frequency ofBRCA1 deleterious alleles in the respec-tive populations in these studies.For breast cancer, the most consistentfinding has been a small (FRR/SIR ~ 1.2)but highly significant familial associationwith prostate cancer. Other sites found tobe associated in at least two studies withbreast cancer in the familial context havebeen thyroid cancer and other endo-crine-related tumours.For endometrial cancer, there is a familialassociation with colorectal cancer whichis consistently found in a number of stud-ies with statistically significant OR/SIRsranging from from 1.3 to 1.9. Some, butnot all studies have also reported associ-ations with ovarian cancer, particularlyamong relatives of younger patients. The strongest and most consistent famil-ial association between cervical andother sites is for lung cancer with statisti -cally significant SIRs of 1.8 and 1.64found in the Swedish FCDB and the UtahUPDB, respectively. Other cancers withpossible associations in both studies are
lip/skin (SIR 2.4 and 1.83) and bladdercancer (SIR=1.6), though the latter wasnot statistically significant in the UPDBstudy.In addition to this statistical and observa-tional evidence for the role of genetic fac-tors in the development of these cancers,a number of specific genes have beenidentified. Of these, the most important interms of both risk and frequency are thebreast cancer susceptibility loci BRCA1and BRCA2, and the mismatch repairgenes MSH2, MLH1, and MSH6 in thecontext of the hereditary non-polyposiscolorectal cancer (HNPCC).
Search for additional genesWhile some of the familial clustering maybe due to shared environmental factors,it seems likely that a number of additionalloci remain to be identified for cancers ofthe breast and female genital tract. Somestudies have shown that only about one-fifth of the familial aggregation of breast
cancer is attributable to the BRCA1 andBRCA2 genes {107,592,2230} and thatthese genes only explain less than half ofall high risk site-specific breast cancerfamilies {898,2631}. Whether the remain-ing familial aggregation is due to addi-tional moderate to high risk loci or to thecombined effects of a number of morecommon, but lower risk, susceptibilityalleles is unknown {2236}. In contrast, itappears that almost all of the familialclustering in ovarian cancer can beascribed to the effects of the BRCA1/2and HNPCC loci {2802}. Although nosystematic studies have been done forendometrial cancer, it is also likely thatthe HNPCC loci account for a substantialfraction of familial aggregation in thiscancer as well.
Table 8.01Specific inherited syndromes involving cancers of the breast and female genital organs.
Syndrome MIM Gene Location Associated sites / tumours
BRCA1 syndrome 113705 BRCA1 17q Breast, ovary, colon, liver,endometrium, cervix,fallopian tube, peritoneum
BRCA2 syndrome 600185 BRCA2 13q Breast (female and male), ovary, fallopian tube, prostate, pancreas, gallbladder, stomach, melanoma
Li-Fraumeni 151623 TP53 17p Breast, sarcoma, brain,adrenal, leukaemia
Cowden 158350 PTEN 10q Skin, thyroid, breast, cerebellum, colon
HNPCC 114500 MLH1 3p Colon, endometrium, small intestine,MSH2 2p ovary, ureter/renal pelvis,MSH6 2p hepatobiliary tract, brain, skin
Muir Torre 158320 MLH1 3p HNPCC sites plus sebaceous glandsMSH2 2p
Peutz-Jeghers 175200 STK11 19p Small intestine, ovary, cervix, testis,pancreas, breast
Ataxia Telangiectasia 208900 ATM 11q Breast (heterozygotes)

338 Inherited tumour syndromes
DefinitionInherited tumour syndrome with autoso-mal dominant trait and markedlyi n c reased susceptibility to breast andovarian tumours, due to germline muta-tions in the B R C A 1 gene. Additional organsites include colon, liver, endometrium,cervix, fallopian tube, and peritoneum.
MIM No. 113705 {1835}
SynonymsBreast cancer 1, early onset breast ova-rian cancer syndrome.
IncidenceThe prevalence of BRCA1 mutations inmost Caucasian populations is estimatedto be 1 in 883 {897}. However, in certainpopulations, this is higher, e.g. 1% inAshkenazi Jews {3065}. Using recombi-nation techniques, B R C A 1 m u t a t i o n shave been dated to the early Romantimes {1997}. De novo mutations are rare.
Diagnostic criteriaA definitive diagnosis is only possible bygenetic testing. BRCA1 mutations arecommon in certain populations and infamilies with numerous early onset breastcancer cases (≥4 cases of breast cancerat <60 years) or in those with ovariancancer at any age in addition to earlyonset breast cancer. The chance of amutation in either BRCA1 or BRCA2 islower (<30%) when only two or threebreast cancer cases are present in af a m i l y. The main diff e rence betweenBRCA1 and BRCA2 is the increased riskof male breast cancer in BRCA2. TheAmerican Society of Clinical Oncology(ASCO) guidelines suggest offering test-ing at a probability of mutation of >10%but many other countries will only offertesting to those with a chance >30%because of the need to concentrateresources.
Breast tumoursPenetranceAnalyses of worldwide data submitted tothe Breast Cancer Linkage Consortium
(BCLC) have provided general estimatesof penetrance {8}. Estimates for specificpopulations have shown that theAshkenazim have a lower than averagelifetime breast cancer penetrance ofabout 50-60% {3065}. Population basedstudies in UK breast cancer patients alsorevealed a lower penetrance and indi-cate that the presence of a mutation with-in a familial breast cancer cluster doesconfer a higher penetrance {2230}. Thismay be due to an association with othergenes or epidemiological factors that arepresent in the family. There are alsoreports of variable penetrance depend-ent on the position of the mutation withinthe BRCA1 gene {2914}.
Clinical features Breast cancer in BRCA1 mutation carri-ers occurs more often at a younger age,typically before age 40 {1687}. It tends top ro g ress directly to invasive diseasewithout a precancerous DCIS component{8,1574}. Accordingly, there appears tobe a lower chance of early detection bymammographic screening and a higherproportion of invasive cancers {1025}.There is an almost linear increase in thelifetime risk of contralateral breast cancerfrom the age of 35 years, reaching a levelof 64% by the age of 80 {742}.
PathologyCertain morphological types of breastcancer, including medullary carcinoma,tubular carcinoma, lobular carcinoma insitu, and invasive lobular carc i n o m a ,
have been reported more commonly inpatients with a positive family history ofb reast cancer {191,1566,1684,1724,2441}. Patients with BRCA1 germline mutationshave an excess of medullary or atypicalmedullary carcinoma compared to con-trols {8,764,1767}. Tumours in BRCA1mutation carriers are generally of a high-er grade than their sporadic counterparts{8,764,1767}. Ductal carcinoma in situ(DCIS) adjacent to invasive cancer isobserved less frequently while the fre-quency of lobular neoplasia in situ is sim-ilar in both groups {8}. However, in a mul-tifactorial analysis of the BCLC database,the only features significantly associatedwith BRCA1 were total mitotic count, con-tinuous pushing margins, and lympho-cytic infiltrate. All other features, includ-ing the diagnosis of medullary and atyp-ical medullary carcinoma, were not foundto be significant {1572}.B R C A 1-associated tumours are morelikely to be estrogen (ER) and proges-t e rone receptor (PgR) negative {766,1352,1574,2121}. Data on ERBB2 arelimited but BRCA1-linked tumours aremore likely to be negative than controls{1352,1574}. B R C A 1-linked tumoursshow a higher frequency of TP53 muta-tions and p53 expression than sporadicb reast cancer {580,581,765,1574}.BRCA1-associated tumours show verylow expression of Cyclin D1 in both theinvasive and in situ components {2122}.The absence of Cyclin D1 in thesetumours could be an additional evidence
BRCA1 syndrome
Table 8.02Probability of BRCA1/2 mutation in women with breast/ovarian cancer.
Chance of mutation Clinical criteria
<10% Single breast cancer / ovarian cancer case at <40 years in non Ashkenazim10-30% 2-3 female breast cancers <60 years (no ovarian / male breast cancer)
30% One female breast cancer <60 and one ovarian cancerFemale breast cancer <40 in Ashkenazi
>60% Four cases of female breast cancer at <60 years≥2 cases female breast cancer <60 and ovarian cancer any age ≥2 cases female breast cancer <60 and male breast cancer any age
__________________From R.A. Eeles {749}.
D. Goldgar R.H.M. VerheijenR. Eeles C. SzaboD. Easton A.N. MonteiroS.R. Lakhani P. DevileeS.Piver S. NarodJ.M. Piek E.H. Meijers-HeijboerP.J. van Diest N. Sodha

of hormone independence of BRCA1-associated breast cancers.
Prognosis and prognostic factorsStudies on the prognosis of breast can-cer associated with BRCA1 range frompoorer prognosis, to no difference, to abetter prognosis {441}. There is a poten-tial survival bias since at least one patientin each family must have survived inorder to have blood taken for gene test-ing. The most optimal studies are there-fore those which have taken this intoconsideration, either by discounting theproband in a family who has presentedfor testing {3022} or by testing specificfounder mutations in archival tumour tis-sue material from all cases in a specificpopulation (for example, see Foulkes etal. {904}).
Ovarian tumoursAge distribution and penetranceAbout 7-10% of ovarian carcinomas aredue to inherited B R C A 1 (or B R C A 2)mutations; as these are on autosomes,they can be inherited from either themother or the father. Although ovariancancer can occur earlier in BRCA1 (andindeed BRCA2) carriers, the presence ofan older onset ovarian cancer still canindicate an underlying mutation in eitherof these genes. The penetrance for ovar-ian cancer in BRCA1 mutation carriers isshown in Fig. 8.02; it starts to rise at anearlier age than the curve for BRCA2,which starts to rise at about 50 years.The penetrance is 44-60% by age 70.This is markedly higher than the lifetimerisk of 1.8% (1 in 55) for sporadic ovariancancer in women living in developedcountries.
Clinical featuresIn a retrospective cohort study of Jewishsubjects, women with advanced-stageovarian cancer and a BRCA1 or BRCA2founder mutation had a longer survivalthan women with non-hereditary ovariancancer (P = 0.004) and a longer mediantime to recurrence (14 months versus 7months) (P< 0.001) {329}.B R C A 1 / 2 h e t e rozygotes had higherresponse rates to primary therapy com-pared with patients who had sporadicdisease (P = 0.01), and those withadvance-stage disease had impro v e dsurvival compared with patients who hadadvanced stage sporadic carc i n o m a{422}.
PathologyIn patients with B R C A 1 g e rmline muta-tions, epithelial tumours (carcinomas) arethe most common histological diagnosis.All subtypes of malignant epithelial ovarianneoplasms have been re p o rted, includingthe very rare entity of malignant transitionalcell carcinoma {3102}. Interobserver varia-tion in typing of ovarian carcinoma is likelyto account, at least in part, for the diff e re n tresults re p o rted to date {572,1716,2513}.Some studies indicate that papillaryserous adenocarcinoma is the predomi-nant ovarian cancer that occurs in famil-ial ovarian cancer syndromes {229,2479,
2800} while others report that they occurwith similar frequency in BRCA1/2 muta-tion carriers and sporadic cases {329,2239,3102}. The large majority of studieshave shown mucinous carcinoma to beunder-represented in BCRA1 mutationcarriers {50,229,1974,2239,2479,2800,3102}.The frequency of endometrioid and clearcell carcinoma occurring in B R C A 1mutation carriers is similar to that of spo-radic cases {50,229,1353,2239,2479,2800,3102,3272}.The current data suggest that germlinemutations in BRCA1/2 genes do not pre-
Fig. 8.02 The ovarian cancer penetrance of BRCA1and BRCA2 from the BCLC data.
Fig. 8.01 The breast cancer penetrance of BRCA1and BRCA2 from the BCLC data.
Table 8.03Lifetime cancer risks of BRCA1 carriers.
Cancer site Relative risk Cumulative risk by age 70, %(95% CI) (95% CI)
Breast Age-dependent 87
Ovary Age-dependent 44
Colon 4.11 (2.36-7.15) {896} -2.03 (1.45-2.85) {2915} 1
Cervix 3.72 (2.26-6.10) 3.57 (3.16-4.04)
Uterus 2.65 (1.69-4.16) 2.47 (2.02-3.04)
Pancreas 2.26 (1.26-4.06) 1.2 (0.9-1.7)
Prostate 3.33 (1.78-6.20) {896} 2.64 (1.95-3.57) (Europe)1.82 (1.01-3.29) {2915} 2 7.67 (4.77-12.20) (North America)
All cancers3 – male 0.95 (0.81-1.12) 16.89 (14.52-19.81)
All cancers3 – female 2.30 (1.93-2.75) 23.27 (21.73-24.89)
________From D. Ford et al. {896} and D. Thompson et al. {2915}.
1 When considered together with rectal cancer, the relative risk was no longer significantly elevatedabove 1.0; no excess risk was noted among men.
2 For men under the age of 65.3 All cancers other than nonmelanoma skin cancer, breast cancer, or ovarian cancer.
339BRCA1 syndrome

340 Inherited tumour syndromes
dispose individuals to the developmentof borderline neoplasms {1044,1704}.H o w e v e r, occasional invasive {2479,3272} and borderline {50} mucinous neo-plasms have been reported. Stromal tumours and malignant germ cellovarian neoplasms appear not to beassociated with BRCA1/2 germline muta-tions. However, several families in whichmore than one relative had been diag-nosed with a malignant ovarian germ celltumour have been published {2790}.Single cases of dysgerminoma {3103}and transitional cell ovarian carcinoma{3101} have been observed in BRCA1carriers with a family history of breastand ovarian cancer. The development ofthese lesions may be unrelated to theg e rmline B R C A 1 mutations in thesecases. The first report on BRCA1-associatedovarian carcinoma found that overall thetumours were of higher grade and higherstage than their historic age-matchedc o n t rols {2479}. These findings havebeen largely reproduced by a number ofother groups {50,229,2239,3102,3272}.In contrast, Berchuck et al. {229} foundthat although the BRCA1 cases in theirstudy were all of advanced stage (III/IV),they were half as likely to be as poorlyd i ff e rentiated as cases without muta-tions. Johannsson et al. {1353} did notidentify a difference in grade betweenthe ovarian cancers in their BRCA1 muta-tion carriers and the control population-based cancer registry group.
Prognosis and prognostic factorsThe majority of BRCA1 ovarian cancersare serous cystadenocarcinomas whichhave a poor prognosis generally if diag-nosed when they have spread outsidethe ovary. Studies of ovarian canceroccurring in BRCA1 carriers have report-ed a somewhat better prognosis {213},but it is uncertain whether this is becauseof the bias in carrier detection in thispopulation or whether they are more sen-sitive to treatment. If the latter were true,this would refer to platinum treatments asthese data have been reported prior tothe use of taxanes.
Tumours of the fallopian tubeDefinitionHereditary fallopian tube carcinoma aris-es from epithelium overlying the laminapropria of the endosalpinx in women athigh hereditary risk to develop ovarian
carcinoma, typically due to loss of thewild-type allele of BRCA1 or BRCA2. Thetumour has to fulfill the clinical and histo-logical criteria for tubal carcinoma {1256}as well as clinical genetic criteria shownin Table 8.02.
IncidenceFrom 1997 to 2002, a total of 15 heredi-tary breast/ovarian family related tubaltumours have been reported in literature.In 8 cases a B R C A 1 mutation wasdetected. However, the true incidence ofboth hereditary and sporadic tubal carci-noma is probably much higher. This iscaused by the fact that primary tubaltumours are often mistaken for primaryovarian carcinomas {3150}. More o v e r,some primary ovarian carcinomas mightactually derive from inclusion cysts linedby tubal epithelial cells included into theovarian stroma {2247}.
Age distributionIn general the age of onset is younger inhereditary cases when compared to spo-radic cases.
Diagnostic criteriaThe criteria of Hu et. al. {1256} as modi-fied by Sedlis {2614} and Yo o n e s s i{3185} are applied to differentiate hered-itary tubal carcinomas from ovarian- ande n d o m e t r i a l - c a rcinoma. These criteriarequire that: a) the main tumour is in thefallopian tube and arises from the endo-salpinx, b) the histological feature sresemble a tubal pattern, c) if the tubalwall is involved, the transition betweenmalignant and benign tubal epitheliumshould be detectable, d) the fallopiantube contains more tumour than theovary or endometrium.
Clinical featuresSymptoms and signs. To date, there isno indication that clinical hereditary tubalc a rcinoma features are diff e rent fro mthose of its sporadic counterpart. Inaddition to occasional abdominal dis-comfort, the classical triad of symptomsinclude: (i) prominent watery vaginal dis-charge, (ii) pelvic pain and (iii) a pelvicmass {158}. Cervical cytology revealsa d e n o c a rcinomatous cells in appro x i-mately 10% of patients {3185}.
Tumour marker. As in ovarian carcinoma,elevation of serum CA125 levels arefound in approximately 80% of cases{1173}.
Imaging. CT/MRI are inconclusive withrespect to the differential diagnosis oftubal or ovarian carcinomas. However,these techniques can be helpful in deter-mining the extent of disease. Likewise,ultrasonography can not distinguishtubal from ovarian disease {2720}.
Histopathology and gradingSerous papillary carcinoma is the mostcommon form of hereditary tubal carci-noma. Grading is of limited value in thesetumours and, if used, is based on thepapillary architecture, nuclear atypia andmitotic activity. Grade I cancers showpapillary growth with well differentiatedcolumnar cells and low mitotic rate.Grade II cancers are papillary with evi-
Table 8.04BRCA1 mutation status in relation to histopatholo-gy of 200 malignant ovarian epithelial tumours.
Histologic BRCA1- BRCA1-type negative positive
f a m i l i e s f a m i l i e s
Serous 80 (59%) 44 (67%)Mucinous 12 (9%)* 0 (0%)*Endometrioid 10 (7%) 5 (8%)Clear cell 13 (10%) 3 (4%)Undifferentiated 10 (7%) 9 (14%)MMMTa 3 (2%) 1 (2%) Transitional cell 2 (2%) 1 (2%)Mixedb 5 (4%) 2 (3%)Total 135 (100%) 65 (100%) ________Excludes borderline tumours. Adapted from B.A. Werness et al. {3102}.a Malignant müllerian mixed tumour.b > 10% minor histologic type. * P = 0.01 for the difference in prevalence betweenBRCA1-positive and BRCA1-negative families.
Fig. 8.03 Average age of onset of BRCA1 and BRCA2related carcinomas.
341BRCA1 syndrome
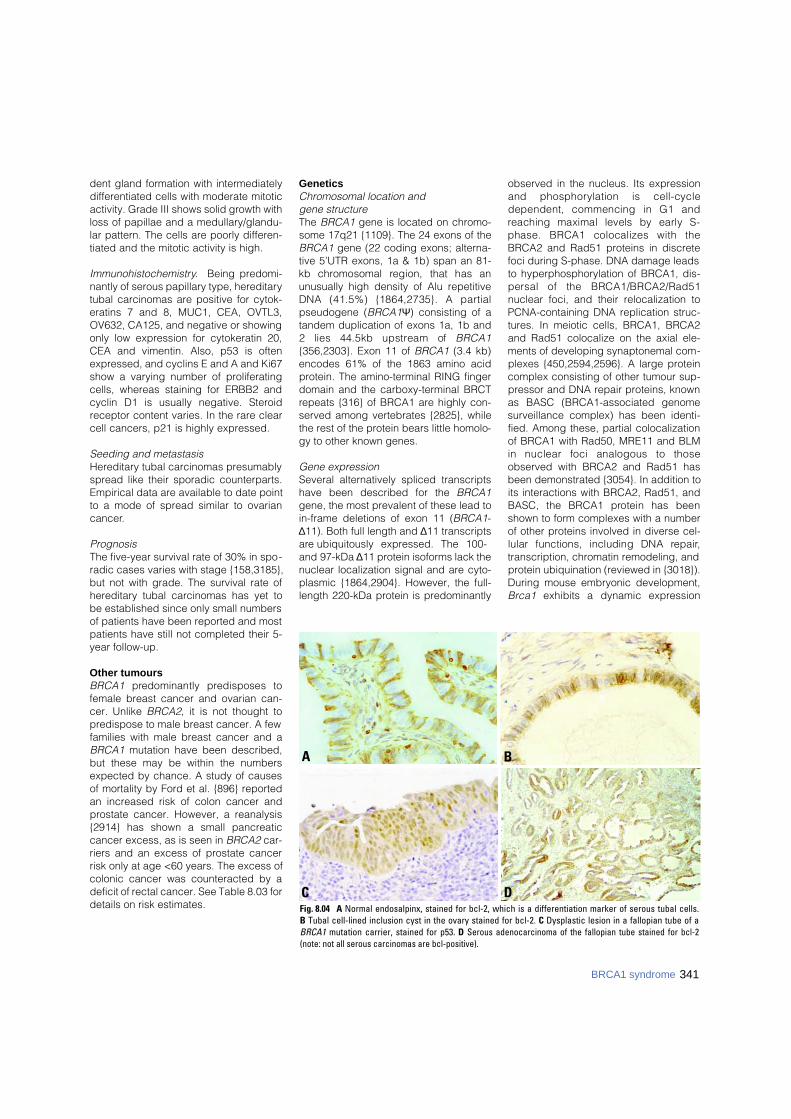
dent gland formation with intermediatelydifferentiated cells with moderate mitoticactivity. Grade III shows solid growth withloss of papillae and a medullary/glandu-lar pattern. The cells are poorly differen-tiated and the mitotic activity is high.
Immunohistochemistry. Being predomi-nantly of serous papillary type, hereditarytubal carcinomas are positive for cytok-eratins 7 and 8, MUC1, CEA, OVTL3,OV632, CA125, and negative or showingonly low expression for cytokeratin 20,CEA and vimentin. Also, p53 is oftenexpressed, and cyclins E and A and Ki67show a varying number of proliferatingcells, whereas staining for ERBB2 andcyclin D1 is usually negative. Steroidreceptor content varies. In the rare clearcell cancers, p21 is highly expressed.
Seeding and metastasisHereditary tubal carcinomas presumablyspread like their sporadic counterparts.Empirical data are available to date pointto a mode of spread similar to ovariancancer.
PrognosisThe five-year survival rate of 30% in spo-radic cases varies with stage {158,3185},but not with grade. The survival rate ofhereditary tubal carcinomas has yet tobe established since only small numbersof patients have been reported and mostpatients have still not completed their 5-year follow-up.
Other tumoursB R C A 1 p redominantly predisposes tofemale breast cancer and ovarian can-cer. Unlike BRCA2, it is not thought topredispose to male breast cancer. A fewfamilies with male breast cancer and aBRCA1 mutation have been described,but these may be within the numbersexpected by chance. A study of causesof mortality by Ford et al. {896} reportedan increased risk of colon cancer andprostate cancer. However, a reanalysis{2914} has shown a small pancreaticcancer excess, as is seen in BRCA2 car-riers and an excess of prostate cancerrisk only at age <60 years. The excess ofcolonic cancer was counteracted by adeficit of rectal cancer. See Table 8.03 fordetails on risk estimates.
GeneticsChromosomal location and gene structureThe BRCA1 gene is located on chromo-some 17q21 {1109}. The 24 exons of theBRCA1 gene (22 coding exons; alterna-tive 5’UTR exons, 1a & 1b) span an 81-kb chromosomal region, that has anunusually high density of Alu repetitiveDNA (41.5%) {1864,2735}. A part i a lpseudogene (BRCA1Ψ) consisting of atandem duplication of exons 1a, 1b and2 lies 44.5kb upstream of B R C A 1{356,2303}. Exon 11 of BRCA1 (3.4 kb)encodes 61% of the 1863 amino acidprotein. The amino-terminal RING fingerdomain and the carboxy-terminal BRCTrepeats {316} of BRCA1 are highly con-served among vertebrates {2825}, whilethe rest of the protein bears little homolo-gy to other known genes.
Gene expressionSeveral alternatively spliced transcriptshave been described for the BRCA1gene, the most prevalent of these lead toin-frame deletions of exon 11 (BRCA1-∆11). Both full length and ∆11 transcriptsa re ubiquitously expressed. The 100-and 97-kDa ∆11 protein isoforms lack thenuclear localization signal and are cyto-plasmic {1864,2904}. However, the full-length 220-kDa protein is predominantly
observed in the nucleus. Its expressionand phosphorylation is cell-cycledependent, commencing in G1 andreaching maximal levels by early S-phase. BRCA1 colocalizes with theBRCA2 and Rad51 proteins in discretefoci during S-phase. DNA damage leadsto hyperphosphorylation of BRCA1, dis-persal of the BRCA1/BRCA2/Rad51nuclear foci, and their relocalization toPCNA-containing DNA replication struc-tures. In meiotic cells, BRCA1, BRCA2and Rad51 colocalize on the axial ele-ments of developing synaptonemal com-plexes {450,2594,2596}. A large proteincomplex consisting of other tumour sup-pressor and DNA repair proteins, knownas BASC (BRCA1-associated genomesurveillance complex) has been identi-fied. Among these, partial colocalizationof BRCA1 with Rad50, MRE11 and BLMin nuclear foci analogous to thoseobserved with BRCA2 and Rad51 hasbeen demonstrated {3054}. In addition toits interactions with BRCA2, Rad51, andBASC, the BRCA1 protein has beenshown to form complexes with a numberof other proteins involved in diverse cel-lular functions, including DNA re p a i r,transcription, chromatin remodeling, andprotein ubiquination (reviewed in {3018}). During mouse embryonic development,B rc a 1 exhibits a dynamic expre s s i o n
BA
DCFig. 8.04 A Normal endosalpinx, stained for bcl-2, which is a differentiation marker of serous tubal cells. B Tubal cell-lined inclusion cyst in the ovary stained for bcl-2. C Dysplastic lesion in a fallopian tube of aBRCA1 mutation carrier, stained for p53. D Serous adenocarcinoma of the fallopian tube stained for bcl-2(note: not all serous carcinomas are bcl-positive).

342 Inherited tumour syndromes
pattern, which parallels Brca2 expres-sion in that the highest expression levelsoccur in epithelial tissues undergoingconcurrent proliferation and differentia-tion. In adult mice, Brca1 and Brca2expression is induced during mammarygland ductal proliferation, morphogene-sis and differentiation occuring at puber-ty and again during proliferation of themammary epithelium during pregnancy{1582,1769,2323}.Consistent with its role as a tumour-sup-p ressor gene, the wild-type allele ofBRCA1 is lost in the majority of tumoursof individuals with inherited mutations,presumably leading to absence of nor-mal protein {560}. In sporadic cancer,BRCA1 protein expression is absent orreduced in the majority of high gradebreast carcinomas and sporadic ovariantumours {2493,3130}. Although fewsomatic mutations in the BRCA1 codingsequence have been identified {1846},somatic inactivation of protein expres-sion may occur through several mecha-nisms, including gross chro m o s o m a lrearrangements – approximately 50% ofprimary breast tumours show loss of het-erozygosity of chromosome 17q21 {559,1134}, or epigenetic inactivation ofexpression, such as promoter hyperme-thylation {426}.
Gene functionThe BRCT domain of BRCA1 is a protein-protein interaction module found in pro-teins involved in DNA repair and cellcycle control {316}. The RING domainmediates the interaction with BARD1 andthe dimer displays ubiquitin ligase (E3)activity {159}. The physiologic substratesof this activity remain unknown althoughthe Fanconi anaemia D2 protein is a like-ly candidate {958}. The integrity of theRING and BRCT domains is indispensa-ble for the functions of BRCA1 as demon-strated by the presence of cancer-asso-ciated mutations in these regions.A number of different mutations havebeen introduced into mouse Brca1, allresulting in embryos with γ- i r r a d i a t i o nhypersensitivity and genetic instability.Mice with a conditional mutation of Brca1in the mammary gland developed tumori-genesis associated with genetic instabil-ity, providing an important link to humandisease {3167}. Intere s t i n g l y, mousecells lacking Brca1 are deficient in repairof chromosomal double-strand breaks(DSB) by homologous re c o m b i n a t i o n{1931}. Taken together, these results sug-gest a role for BRCA1 in the DNA dam-age response.Expression of wild type but not disease-associated B R C A 1 alleles in B R C A 1-
deficient human cells restores resistanceto DNA-damaging agents {2595} andseveral BRCA1-containing complexesinvolved in DNA repair have been identi-fied. These include S-phase nuclear focicontaining BRCA2 and Rad51 {450}, theh R a d 5 0 - h M re 1 1 - N B S 1p 9 5 (R/M/N) com-plex, involved in a wide variety of DNArepair processes {3258}, and the BASCcomplex which contains ATM, the BLMhelicase, mismatch repair pro t e i n sMSH2, MSH6, MLH1 and the R/M/N com-plex {3054}. DNA damaging agentsinduce BRCA1 hyperphosphory l a t i o n ,which is likely to modulate the associa-tion of BRCA1 with these different proteincomplexes {2597}. These biochemicalapproaches corroborate the notion thatBRCA1 participates in the cellularresponse to promote DNA break recogni-tion and repair, as shown in Fig. 8.08. The involvement of BRCA1 in a variety ofDNA repair processes suggests that itmay be an upstream effector common tovarious responses to DNA damage{3018}. In line with the idea of BRCA1’spleiotropic role, it also acts as a negativeregulator of cell growth. Ectopic expres-sion of BRCA1 causes cell cycle arrest atG1 via the induction of the cdk inhibitorp21Waf1/CiP1 {2745}. Conversely, inhibitionof B R C A 1 e x p ression with antisenseoligonucleotides results in the accelerat-ed growth of mammary epithelial celllines {2917}. Also, BRCA1 seems to berequired for efficient radiation-inducedG2/M and S-phase checkpoints pointingto a broad involvement of BRCA1 incheckpoint control {3166,3178}.Several lines of evidence suggest thatone of the molecular functions of BRCA1is the regulation of transcription. TheBRCA1 C-terminus acts as a transactiva-tion domain and germline mutationsfound in B R C A 1 abolish this activity{1899}. BRCA1 can be copurified withRNA polymerase II and upon replicationblockage, a novel complex containingBRCA1 and BARD1 is formed, suggest-ing that BRCA1 protein redistributes todifferent complexes in response to repli-cation stress {476,2593}. BRCA1 alsoassociates and, in some cases, modu-lates the activity of several pro t e i n sinvolved in the regulation of geneexpression such as transcription factors,coactivators, core p ressors and chro-matin remodeling complexes {297,1247,1899,3255}. A recent exciting develop-ment, of yet unknown physiologic signifi-
Fig. 8.05 To assess whether wild-type and/or mutated BRCA1 alleles are lost in dysplastic tubal epithelium ofa BRCA1 mutation carrier, light-cycler polymerase chain reaction (PCR) melting curve analysis is performed.This technique utilizes the properties of probes to anneal less stringent to mutated DNA than to wild-type DNA,resulting in a lower denaturation temperature for mutated DNA. Two peaks, indicating different denaturing tem-peratures, are detected in non-dysplastic epithelium, indicating the presence of both wild-type and mutatedBRCA1 DNA. One clear peak at the melting temperature for the mutated BRCA1 DNA in the dysplastic epitheli-um indicates loss of wild-type BRCA1 DNA. From J.M. Piek et al. {2246}.

cance, was the discovery of direct DNAbinding by BRCA1 in vitro which may beimportant for its function in transcriptionand DNA repair {2198}.Putative BRCA1 transcriptional targetgenes identified so far play a role insome aspect of the DNA damageresponse. BRCA1 induces the transacti-vation of p21WAF1/CiP1 in p53-dependentand independent manners, insuring apotent cell cycle arrest, reinforcing theconnection between cell cycle check-point control and transcription regulation{2130,2745}. Experiments using cDNAarrays identified the DNA-damage-responsive gene GADD45 as a major tar-get of BRCA1-mediated transcription{1138,1727}. These results, coupled withstudies showing that disruption of p53partially rescues embryonic lethality inBrca1-/- mouse, link the p53 pathway andBRCA1 function {1108,1710}. Import -antly, the majority of tumours derivedf rom B R C A 1-linked patients or fro mBrca1-/- mice present mutations in p53{581,3167}.
Mutation spectrumGermline mutations in BRCA1 have beendetected in 15-20% of clinic-basedbreast cancer families, and in 40-50% ofb reast-ovarian cancer families {2657,3023}. Mutations occur throughout thee n t i re coding region, and hence themutation spectrum has taught us rela-tively little about the gene’s function. Themajority of the mutations are predicted tolead to a prematurely truncated proteinwhen translated. In conjunction with theobserved loss of the wildtype allele intumours arising in mutation carriers{560}, this indicates that inactivation ofthe gene is an important step in tumori-genesis. Despite the strong variability inmutations detected in families, foundereffects have led to some mutations beingvery prevalent in certain populations ofdefined geographical or ethnic back-ground. An example is the 185delAGmutation, which is present in approxi-mately 1% of all individuals of AshkenaziJewish descent {1151}. As a result, muta-tion spectra may vary according to eth-nic background of the sampled popula-tion {2824}. In some populations, specif-ic large interstitial deletions or insertions,which are difficult to detect by conven-tional PCR-based mutation scanningtechnologies, have been observed to beparticularly frequent. They may comprise
between 10 and 20% of the total muta-tion spectrum {944,1229}.In recent years, an increasing numberof missense changes are being detect-ed in B R C A 1, of which the clinical sig-nificance is uncertain. These alre a d ycomprise up to 40% of all knownsequence changes in B R C A 1. TheB reast Cancer Information Core (BIC)maintains a website providing a centralre p o s i t o ry for information re g a rd i n gmutations and polymorphisms{ h t t p : / / re s e a rc h . n h g r i . n i h . g o v / b i c / } .
Genotype-phenotype correlationsInitially, the breast and ovarian cancercancer risks conferred by mutations inB R C A 1 w e re estimated from B R C A 1-linked, multiple-case families (see Figs.8.01 and 8.02) {896,898}. More recently,estimates from specific populations havecome up with lower estimates{106,3065}. This could point to 1) theexistence of mutation-specific risks(because different populations have dif-ferent mutation spectra, the overall can-cer risks would differ), 2) the existence ofgenetic variants in other genes, particu-larly prevalent in certain populations,which might modify the BRCA1-relatedcancer risks, 3) population-specific dif-ferences in environmental risk modifiers.
BRCA1 mutation positionOne report observed a significant corre-lation between the location of the muta-tion in the gene and the ratio of breast toovarian cancer incidence within eachfamily {974}, suggesting a transition inrisk such that mutations in the 3' third ofthe gene were associated with a lowerproportion of ovarian cancer. It wasn’tclear, however, whether this was due tohigher breast cancer risks, or lower ovar-ian cancer risks. A much larger study of
356 BRCA1-linked families {2914} foundthe breast cancer risk associated withmutations in the central region to be sig-nificantly lower than for other mutations(relative risk, 0.71), and the ovarian can-cer risk associated with mutations 3' tonucleotide 4191 to be significantlyreduced relative to the rest of the gene(relative risk, 0.81). Recent work sug-gests that the risk to ovarian cancermight also be influenced by genetic vari-ation in the wildtype BRCA1 copy inBRCA1 carriers {1009}.
Genetic risk modifiersOne study showed that the risk for ovari-an cancer was 2.11 times greater forBRCA1 carriers harbouring one or tworare HRAS1 alleles, compared to carrierswith only common alleles (P = 0.015).Susceptibility to breast cancer did notappear to be affected by the presence ofrare HRAS1 alleles {2240}. Likewise, alength-variation of the polyglutaminerepeats in the estrogen receptor co-acti-vator NCOA3 and the androgen receptorinfluences breast cancer risk in carriersof BRCA1 and BRCA2 {2342,2345}. Thevariant pro g e s t e rone receptor allelenamed PROGINS was associated withan odds ratio of 2.4 for ovarian canceramong 214 BRCA1/2 carriers with nopast exposure to oral contraceptives,c o m p a red to women without ovariancancer and with no PROGINS allele{2487}. These results support the hypothesis thatpathways involving endocrine signallingmay have a substantial effect onB R C A 1 / 2-associated cancer risk.Genetic variation in the genes constitut-ing the DNA repair pathways might alsobe involved. A C/G polymorphism in the5' untranslated region of RAD51 wasfound to modify both breast and ovarian
Fig. 8.06 Functional domains in BRCA1. The RING domain contains a C3HC4 motif that interacts with otherproteins. NLS = nuclear localization signal. BRCT = BRCA1-related C-terminal. The proportion encoded byexon 11 is indicated.
343BRCA1 syndrome

cancer risk, initially only in carriers ofBRCA2 {1328,1644,3053}.
Hormonal factors as risk modifiersOral contraceptivesBecause of the observed pro t e c t i v eeffects of oophorectomy and tamoxifen, itis of concern that supplemental estro-gen, in the form of oral contraceptives orh o rmone replacement therapy, mayincrease the risk of breast cancer. In theOxford overview analysis, current use ofbirth control pills was associated with arelative risk of 1.2 {539}. However, in arecent large American case-contro ls t u d y, no adverse effect was noted{2607}. In a large international case-con-t rol study of oral contraceptives andhereditary breast cancer {1977} a mildincrease in risk was seen among BRCA1carriers (relative risk 1.2) but not amongBRCA2 carriers (relative risk 0.89). Theoverall result was not significant, but riskincreases were found for women whofirst took a contraceptive before age 30,for women who developed breast cancerbefore age 40, for women with five ormore years of pill use, and for womenwho first took an oral contraceptive priorto 1975. It appears that short-term use ofm o d e rn contraceptives poses noincrease in risk, but further studies areneeded in this regard. No studies havebeen conducted yet regarding whetheror not HRT increases the risk of breastcancer in BRCA1/2 mutation carriers.
It is important to establish whether oralcontraceptives are hazardous to thebreast, because their use has been pro-posed as a preventive measure againstovarian cancer. A protective effect of oralcontraceptives on ovarian cancer riskhas been observed in three case-controlstudies of BRCA1/2 mutation carriers{1976,1979,1980} but there has beenone conflicting report {1886}. In a recentstudy of 232 ovarian cancer cases and232 controls, oral contraceptive use wasassociated with a 56% reduction in therisk of ovarian cancer (p = 0.002) {1976}.Tubal ligation has been found to be pro-tective against ovarian cancer in the gen-eral population {1126} and amongB R C A 1 mutation carriers {1980}. Anadjusted relative risk of 0.39 was report-ed for tubal ligation and subsequentovarian cancer (a risk reduction of 61%).The mechanism of risk reduction isunclear.
PregnancyHormonal levels rise dramatically duringpregnancy and two groups found preg-nancy to be a risk factor for early breastcancer in BRCA1/2 mutation carriers.Johannsson et al. reported ten pregnan-cy-related breast cancers in 37 BRCA1/2mutation carriers, versus the expected3.7 {1351}. Jernstrom et al. reported thatthe risk of breast cancer increased witheach pregnancy in BRCA1/2 c a r r i e r sbefore the age of 40 {1348}. This was
found for BRCA1 and BRCA2 mutationcarriers, but was only significant for theformer group. In the general population,p regnancy offers protection againstbreast cancer after the age of 40, butappears to increase the risk for veryearly-onset breast cancer {227}. This isconsistent with the hypothesis that theovarian hormones produced duringpregnancy are mitogenic, and acceler-ate the growth of existing tumours.During pregnancy breast differentiationoccurs and thereafter the population ofsusceptible cells is reduced. This mayexplain why pregnancy prevents breastcancers at a later age. In the generalpopulation, only a small proportion ofbreast cancers occur before age 40, andpregnancy confers an overall advantage.Early-onset breast cancers are typicalamong BRCA1 mutation carriers, how-ever, and a high proportion of cancersoccur before age 40. A case-contro lstudy of breast-feeding and breast can-cer in BRCA1/2 mutation carriers report-ed a protective effect in women withBRCA1 mutations, but not with BRCA2mutations {1347}. BRCA1 mutation carri-ers who breast-fed for more than oneyear were 40% less likely to have breastcancer than those who breast-fed for ashorter period (p = 0.01). The observedprotective effect among BRCA1 carrierswas greater than that observed for mem-bers of the general population {224}.
Prognosis and preventive optionsThe overall life expectancy of unaffectedwomen with a BRCA1 or BRCA2 mutationclearly is decreased due to their high riskof developing breast cancer and ovariancancer, in particular at young ages. Theoverall mortality from breast and ovariancancer within 10 years of diagnosis ofcancer is still significant, 40% and 60%respectively.C u r rently the following avenues arebeing explored to improve the prognosisof women with a BRCA1 or BRCA2 muta-tion, all aiming for either early detectionor prevention of breast cancer and/orovarian cancer: i) regular surveillance, ii)prophylactic surgery, and iii) chemopre-vention.
Preventive surveillanceNo evidence exists that regular breastsurveillance using mammography leadsto earlier detection of cancers in mutationcarriers {1442}. Preliminary results on
344 Inherited tumour syndromes
Fig. 8.07 Factors that modify risk of breast or ovarian cancer. Most of these proposed factors are based onresults of a single study and require confirmation. From S.A. Narod {1975}.
breast surveillance using MRI suggestthat there is an increased frequency ofearly detection of tumours, but definiteconclusions cannot yet be made {1875,2835}. Also, no evidence exists that reg-ular ovarian surveillance detects ovariancancer at curable stages.
Prophylactic surgeryProphylactic bilateral mastectomy lowersthe risk of breast cancer in mutation car-riers by more than 90%, also on the long-term {178,1407}. Prophylactic bilateralsalpingo-oophorectomy prevents ovariancancer, though a minimum long-term riskof 4% of peritoneal cancer remains afterthis procedure {2344}. The incidence of breast cancer inBRCA1 carriers is maximal in the agegroup 40 to 55 and then declines slightlythereafter {1978}. This observation sug-gests that ovarian hormones may have apromoting role in breast carcinogenesis.In support of this, oophorectomy hasbeen found to be protective againstbreast cancer in BRCA1/2 mutation carri-ers in several studies {1976,2504}.Rebbeck et al. compared the breast can-cer risk in a historical cohort of BRCA1mutation carriers, some of whom hadundergone an oophorectomy and someof whom had both ovaries intact{2343,2344}. The estimated relative riskof breast cancer associated withoophorectomy was approximately one-half. This was confirmed in a case-con-trol study {763} and in a prospective fol-low-up study of 170 women {1413}.Among BRCA1 mutation carriers; the riskof breast cancer among women who hadan oophorectomy was decreased by61% (odds ratio 0.39; 95% CI 0.20 to0.75). These studies suggest thatoophorectomy might be used as a strat-egy to decrease the risk of breast canceramong B R C A 1 mutation carriers.However, in young women the procedureis associated with acute and long-termside effects. Members of a BRCA1-linked family are atrisk also to develop tubal carc i n o m a{3271}. Piek et al. studied prophylactical-ly removed fallopian tubes of 12 womenwith a predisposition for ovarian cancer,in 7 of whom a BRCA1 mutation wasdetected {2246}. Six showed dysplasia,including one case of severe dysplasia.Five harboured hyperplastic lesions, andin one woman no histological aberrationswere found. Therefore, it is recommend-
ed to perform a complete adnexectomyin women harbouring a BRCA1 mutation.Whether an abdominal hystere c t o m yshould be performed to dissect the intra-uterine part of the tube, is still in debate.However, most studies indicate that tubalcarcinomas in fact predominantly arise indistal parts of the tube.The interest of women with a BRCA1 orBRCA2 mutation in the various optionsdiffers greatly between countries {1425},and may also change over time when theefficacy of surveillance, chemopreven-tion, or treatment improves. However, atpresent in some countries up to 50-60%of unaffected women chose to have pro-phylactic bilateral mastectomy, and 65%p rophylactic bilateral salpingo-oophorectomy {1012,2285}.
ChemopreventionTamoxifen is an anti-estrogenic drug thatis routinely used in the treatment of estro-gen-receptor positive breast cancer thathas also been demonstrated to be of
value in reducing the risk of primary inva-sive and pre-malignant breast cancer inhigh risk women {865,1464,1976} and ofcontralateral breast cancer in unselectedwomen {10}. Narod, et al. {1976} studiedtamoxifen and contralateral breast can-cer in a case-control study of BRCA1 andBRCA2 mutation carriers. Tamoxifen usewas equivalent to a 62% risk reduction inBRCA1 carriers. A reduction in risk ofcontralateral cancer was also seen withoophorectomy and chemotherapy. Thisresult implies that the combination oftamoxifen and oophorectomy may bem o re effective than either tre a t m e n talone, and that the two prevention strate-gies may be complementary. Until moredefinitive guidlines are established, theinterest in participation in chemopreven-tion trials is likely to remain small {2285}.
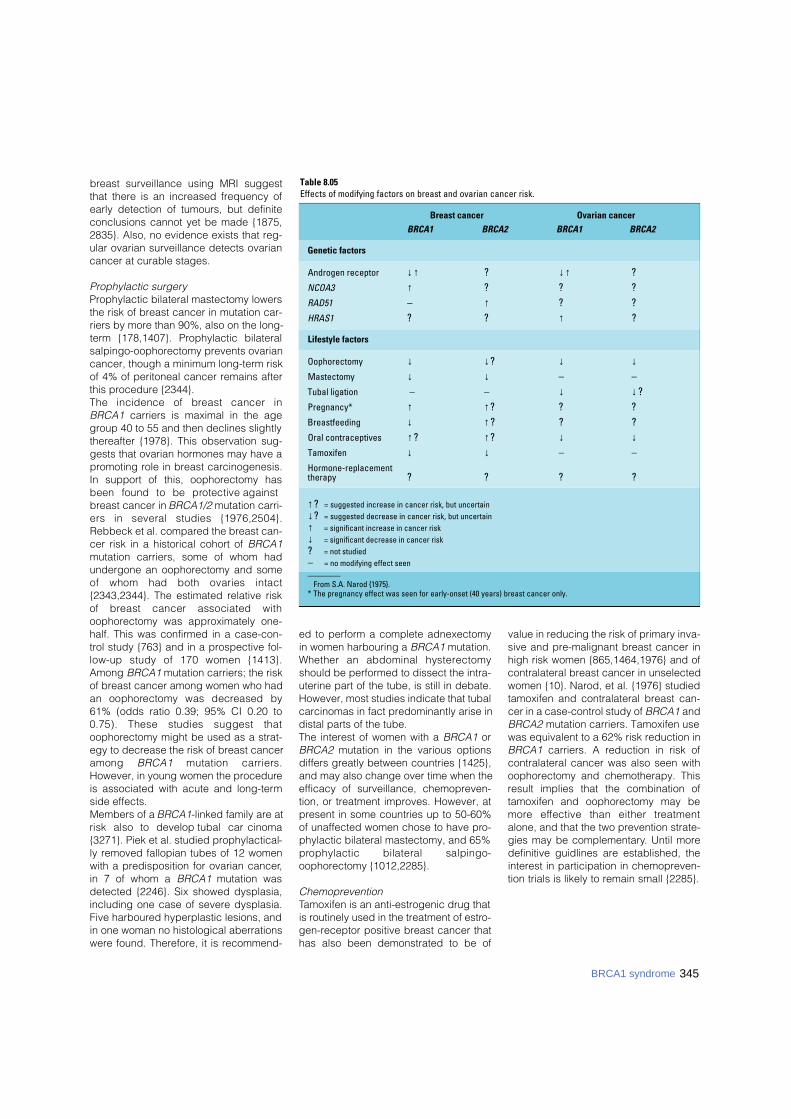
Table 8.05Effects of modifying factors on breast and ovarian cancer risk.
Breast cancer Ovarian cancer
BRCA1 BRCA2 BRCA1 BRCA2
Genetic factors
Androgen receptor ↓↑ ? ↓↑ ?NCOA3 ↑ ? ? ?RAD51 – ↑ ? ?HRAS1 ? ? ↑ ?
Lifestyle factors
Oophorectomy ↓ ↓? ↓ ↓Mastectomy ↓ ↓ – –Tubal ligation – – ↓ ↓?Pregnancy* ↑ ↑? ? ?Breastfeeding ↓ ↑? ? ?Oral contraceptives ↑? ↑? ↓ ↓Tamoxifen ↓ ↓ – –Hormone-replacementtherapy ? ? ? ?
↑? = suggested increase in cancer risk, but uncertain ↓? = suggested decrease in cancer risk, but uncertain ↑ = significant increase in cancer risk ↓ = significant decrease in cancer risk ? = not studied – = no modifying effect seen________
From S.A. Narod {1975}.* The pregnancy effect was seen for early-onset (40 years) breast cancer only.
345BRCA1 syndrome

DefinitionInherited tumour syndrome with autoso-mal dominant trait and markedlyincreased susceptibility to early onsetbreast cancer and an additional risk forthe development of male breast cancerand, less fre q u e n t l y, pancreatic andovarian cancer. Occasionally, carriers ofa BRCA2 germline mutation present withskin melanoma, gall bladder and bileduct tumours, and cancer of the fallopiantube.
MIM No. 600185 {1835}
SynonymsSite specific early onset breast cancersyndrome, breast cancer 2, FANCD1.
IncidenceThe B R C A 2 s y n d rome is generallyuncommon (about 1 in 1000 individuals),but in certain populations, it is moreprevalent. For example, a specific muta-tion (6174delT) is present in 1.5% of the
Ashkenazim and another (999del5) in0.6% of Icelanders, due to foundereffects {2382,2921}.
Diagnostic criteriaBRCA2 mutations are more often presentin families with multiple female breastcancer (>4 cases of early onset at <60years) and male breast cancer. The riskof ovarian cancer is lower than in BRCA1families. The definitive diagnosis relieson the identification of a BRCA2 germlinemutation.
Breast tumoursPenetrance and age distributionAnalyses of the worldwide data submit-ted to the Breast Cancer LinkageConsortium (BCLC) studies have beenused to provide general estimates ofpenetrance (see Fig. 8.01) {8}. Popula-tion based studies of mutations in breastcancer patients from the UK have showna lower penetrance than the BCLC, indi-cating that the presence of a mutation
within a familial breast cancer clusterdoes confer a higher penetrance {2230}.This may be due to association with othergenes or exposure and lifestyle factorsthat are present in the family. Specificestimates for different populations haveshown that the Ashkenazim have asomewhat lower lifetime breast cancerpenetrance of about 50-60% {3065}.There are also reports of variable pene-trance, dependent upon mutation posi-tion {2914}. There is an increased risk ofcontralateral breast cancer of about 56%lifetime after a diagnosis of a first breastprimary. Breast cancer in BRCA2 carriersoccurs more often at younger ages thanin the general population, but at olderages than in BRCA1 carriers.
PathologyAlthough lobular and tubulo-lobular car-cinoma has been reported to be associ-ated with BRCA2 germline mutation inone study {1767}, this has not been con-firmed in a larger study and no specifichistological type is thought to be associ-ated with BRCA2 {8,1572}. In a multifac-torial analysis, the only factors found tobe significant for BRCA2 were tubules c o re, fewer mitoses and continuouspushing margins. All other features werenot found to be significant {1572}.BRCA2 tumours are overall higher gradethan sporadic cancers {8,43,1767}.Ductal carcinoma in situ (DCIS) isobserved less frequently in B R C A 1cases than in controls, but this is not thecase for BRCA2. Lobular carcinoma insitu shows no difference between thegroups {8}.Invasive lobular carcinoma clearly doeshave a familial association and a trendhas been identified in familial breast can-cer not linked to BRCA1 or BRCA2 (i.e.BRCAX) {1571}. BRCA2 tumours are similar to sporadiccancers in steroid receptor (ER, PgR)e x p ression {766,1574,2121}. Data onERBB2 are limited but B R C A 1 a n dBRCA2 tumours are more likely to benegative than controls {1574}. BRCA2tumours do not show a higher frequency
BRCA2 syndrome R. EelesS. PiverS.R. LakhaniJ.M. PiekA. Ashworth
P. DevileeS. Narod
E.H. Meijers-HeijboerA.R. Venkitaraman
Fig. 8.08 Several genes (ATM, CHEK2, BRCA1 and BRCA2) whose inactivation predisposes people to breastand other cancers participate in the error-free repair of breaks in double-stranded DNA by homologousrecombination. Genes for another chromosome instability disorder named Fanconi anaemia have been con-nected to this DNA repair pathway. Ub denotes mono-ubiquitin. From A.R. Venkitaraman {3019}.
346 Inherited tumour syndromes

of TP53 mutation and p53 expressionc o m p a red to sporadic breast cancer{580,581,1574}.
Prognosis and prognostic factorsSince the breast cancers associated withBRCA2 mutations are more often estro-gen receptor positive and are associatedwith DCIS, they would be expected tohave a better prognosis. The most sys-tematic study to investigate pro g n o s i shas analysed the survival of Ashkenaziwomen with breast cancer who havemutations as tested from paraff i n - s t o re dtissue. This is possible because theyhave a single 6174delT founder mutation.T h e re was no diff e rence in survivalbetween carriers and non-carriers {441}.
Risk modifiers and preventionThe preventive effect of oophorectomyand tamoxifen, mastectomy, and the pos-sible hazard associated with oral contra-ceptives are similar in both BRCA syn-dromes have been dealt with in the pre-ceding section on BRCA1.
Ovarian tumoursPenetrance and age distributionAbout 7-10% of ovarian carcinomas aredue to inherited BRCA1 or BRCA2 muta-tions; as these are on autosomes, theycan be inherited from either the mother orthe father. Although ovarian cancer canoccur earlier in B R C A 1 and indeedBRCA2 carriers, the presence of an olderonset ovarian cancer still can indicate anunderlying mutation in either of thesegenes. The penetrance of ovarian cancerin BRCA2 carriers is shown in Fig. 8.02;the risk of developing ovarian cancer byage 70 in BRCA2 families is approxi-mately 27% {898}. It should be noted thatthe penetrance curve starts to rise laterthan for BRCA1 which could have impli-cations for the timing of prophylacticoophorectomy.
PathologyCompared with the information on thepathology of BRCA1-associated ovariancancers, little is re p o rted on B R C A 2m u t a t i o n - related ovarian tumours. Thepaucity of information is accounted forby the low incidence of this diseasec o m p a red with that of B R C A 1- l i n k e dcases {329,973}. Some recent studiesindicate that the histological phenotypeof these ovarian neoplasms is similar tothat of BRCA1-associated carcinomas
and are predominantly of papillaryserous type {329,2239,3272}. A singlecase of an ovarian malignant mixed mül-lerian tumour (carc i n o s a rcoma), hasbeen reported as occurring in a BCRA2mutation carrier {2748}.The data on grade are similar to those ofBRCA1 ovarian cancers with an associa-tion with higher grade but limited num-bers in study and interobserver variation{329,2239,2479,3102,3272} in the scor-ing of grade should be taken intoaccount when considering the evidenceThere are no data to support a role ofB R C A 2 in borderline ovarian lesions{1044,1704} nor are there germ cell orsex cord stromal tumours.
Prevention by oral contraceptivesAlthough it has been long known that oralcontraceptives can decrease the risk ofdeveloping ovarian cancer in the generalpopulation {2}, recently there is evidencethat this may also be true for hereditaryovarian cancer {1976,1979,1980}. Seethe preceding section on BRCA1 syn-drome for further details.
Prognosis and prognostic factorsIn a retrospective cohort study, womenwith BRCA1 or BRCA2 founder mutationadvanced-stage ovarian cancer had alonger survival compared with womenwith non-hereditary ovarian cancer (P =0.004) and a longer median time torecurrence (14 months versus 7 months)(P< 0.001) {329}.Studies of ovarian cancer occurring inBRCA2 carriers have reported a betterp rognosis {329}, but it is uncert a i nwhether this is because of the bias incarrier detection in this population orwhether they are more sensitive to treat-ment. If the latter is true, this would beplatinum treatments as these data areprior to the use of taxanes.
Tumours of the fallopian tubeHereditary fallopian tube carcinoma aris-es from epithelium overlying the laminapropria of the endosalpinx in women athigh hereditary risk to develop ovariancarcinoma. Loss of the wild-type breastcancer 1 or 2 gene (BRCA1/2) allele ismost likely pivotal in carcinogenesis ofthese tumours. To be unequivocally iden-tified, the tumour has to fulfill the clinicaland histological criteria for tubal carcino-ma {1256} as well as clinical genetic cri-teria.
Incidence F rom 1997 to 2002, a total of 15 here d i t a ryb reast/ovarian family related tubaltumours have been re p o rted in literature .In 4 cases, a B R C A 2 mutation was detec-ted. However, the true incidence of here d i-t a ry tubal carcinoma is probably muchh i g h e r, as is suggested for its sporadicc o u n t e r p a rt. This is caused by the fact thatp r i m a ry tubal tumours are often mistakenfor primary ovarian carcinomas {3150}.M o re o v e r, some primary ovarian carc i n o-mas might actually derive from inclusioncysts lined by tubal epithelial cells inclu-ded into the ovarian stroma {2247}.
Age distributionIn general the age of onset is younger inhereditary cases when compared to spo-radic cases.
Diagnostic criteriaThe criteria of Hu et. al. {1256} as modi-fied by Sedlis {2614} and Yo o n e s s i{3185} are applied to differentiate hered-itary tubal carcinomas from ovarian andendometrial carcinoma. These criteriarequire that: (i) the main tumour is in thefallopian tube and arises from the endo-salpinx, (ii) the histological feature sresemble a tubal pattern, (iii) if the tubalwall is involved, the transition betweenmalignant and benign tubal epitheliumshould be detectable, (iv) the fallopiantube contains more tumour than theovary or endometrium.
Clinical features
Symptoms and signs. To date, there isno indication that clinical hereditary tubalc a rcinoma features are diff e rent fro mthose of its sporadic counterpart ;abdominal discomfort is more or lesscommon, but an atypical complaint. Theclassical but rare triad of symptomsinclude: (i) prominent watery vaginal dis -charge, (ii) pelvic pain and (iii) pelvicmass {158}. It has been reported thatapproximately 10% of patients will havea d e n o c a rcinomatous cells in cervicalcytology {3185}.
Tumour markers. As in ovarian carcino-ma elevation of serum CA125 levels canbe found in approximately 80% of cases{1173}.
Imaging. CT / MRI are inconclusive withrespect to the differential diagnosis of
347BRCA2 syndrome

348 Inherited tumour syndromes
tubal or ovarian carcinomas. However,these techniques can be helpful in deter-mining the extent of disease. Likewise,ultrasonography can not distinguishtubal from ovarian disease {2720}.
Pathology
Histopathology and grading. S e ro u spapillary carcinoma is the most commonf o rm of here d i t a ry tubal carc i n o m a .Grading is of limited value in thesetumours and, if used, is based on thepapillary architecture, nuclear atypia andmitotic activity. Grade I cancers showpapillary growth with well differentiatedcolumnar cells and low mitotic rate.Grade II cancers are papillary with evi-dent gland formation with intermediatelydifferentiated cells with moderate mitoticactivity. Grade III shows solid growth withloss of papillae and a medullary/glandu-lar pattern. The cells are poorly differen-tiated and the mitotic activity is high.
Immunoprofile. Being predominantly ofserous papillary type, hereditary tubalcarcinomas are positive for cytokeratins7 and 8, MUC1, CEA, OVTL3, OV632,CA125, and negative or showing only lowexpression for cytokeratin 20, CEA andvimentin. Also, p53 is often expressed,and cyclins E and A and Ki67 show av a rying number of proliferating cells,w h e reas staining for HER-2/neu andcyclin D1 is usually negative. Steroidreceptor content varies. In the rare clearcell cancers, p21 is highly expressed.
Seeding and metastasisHereditary tubal carcinomas presumablyspread like their sporadic counterparts.However, only empirical data are avail-able to date, pointing to a mode ofspread similar to ovarian cancer.
SurvivalThe five-year survival rate of 30% in spo-radic cases varies with stage {158,3185},but not with grade. The survival rate ofhereditary tubal carcinomas has yet tobe established since only small numbersof patients have been reported and mostpatients have still not completed their 5-year follow-up.
Prophylactic interventionsIn one study, 30 women with either a doc-umented deleterious BRCA1 or BRCA2mutation or a suggestive family history
underwent prophylactic oophore c t o m y{1617}. Five of these (17%) were found tohave clinically occult malignancy, 3 ofwhich involved a primary fallopian tubemalignancy. Three of the five were knownBRCA1 mutation carriers, one had a doc-umented BRCA2 mutation. Therefore, it isrecommended to perform a completeadnexectomy in women carrying aBRCA1 or a BRCA2 mutation. Whetheran abdominal hysterectomy should bep e rf o rmed to dissect the intra-uterinep a rt of the tube, is still in debate.However, most studies indicate that tubalcarcinomas in fact predominantly arise indistal parts of the tube.
Other tumoursBRCA2 confers an increased risk of ovar-ian cancer, but not as high as that forBRCA1. Statistically significant increasesin risk were observed for a number ofother tumour types, including prostate,pancreatic and stomach cancer. The riskfor prostate cancer is probably not suffi-ciently high to cause an appreciablefraction of early-onset prostate cancercases. The risk for male breast cancer,although the hallmark of BRCA2 muta-tions, is based on only four observedcases and hence is very imprecise.
GeneticsChromosomal location and genestructureB R C A 2 is located on chro m o s o m e13q12.3. It consists of 27 exons, of whichexon 11 is remarkably large (4.9 kb). Theopen reading frame is 10,254 basepairs,encoding a protein of 3,418 aminoacidsthat has no significant similarity to anyknown protein. Exon 11 encodes a struc-tural motif consisting of eight ‘BRC’repeats, through which BRCA2 controlsthe function of RAD51, a recombinaseenzyme, in pathways for DNA repair byhomologous recombination.
Gene expression A wide range of human tissues expressBRCA2 mRNA, in a pattern very similar tothat of BRCA1, but the highest levelswere observed in breast and thymus,with slightly lower levels in lung, ovary,and spleen {2891}. In normal cells,BRCA2 is a nuclear protein, preferential-ly expressed during the late-G1/early-Sphase of the cell cycle {258,480,3012}. Inmice, Brca1 and Brca2 are coordinatelyupregulated during ductal proliferation,morphogenesis and diff e rentiation ofbreast epithelial cells occurring at puber-ty, pregnancy and lactation {1582,1769,
Table 8.06Cancer risks of BRCA2 carriers.
Cancer site Relative risk Cumulative Risk By Age 70, %or type (95% CI) (95% CI)
Breast (female) Age-dependent 84 (43 – 95)
Breast (male) 150 6.3 (1.4 – 25.6)
Ovary Age-dependent 27 (0 – 47)
Gall bladder and bile ducts 4.97 (1.50 – 16.5) -
Prostate 4.65 (3.48 – 6.22) 7.5 (5.7 – 9.3)
Prostate before age 65 7.33 (4.66 – 11.52) -
Pancreas1 3.51 (1.87 – 6.58) Males: 2.1 (1.2 – 3.0))Females: 1.5 (0.9 – 2.1)
Stomach1 2.59 (1.46 - 4.61) -
Malignant melanoma 1 2.58 (1.28 – 5.17) -
All cancers2 2.45 (2.15 – 2.78) -________
From D. Ford et al. 1998 {898}, D.F. Easton et al. 1997 {744} and the Breast Cancer Linkage Consortium 1999 {11}.1 Relative risks were slightly higher for individuals aged 65 or under.2 All cancers other than nonmelanoma skin cancer, breast cancer, or ovarian cancer.

2323}. Both proteins co-exist with RAD51in subnuclear foci during S phase, whichredistribute following DNA damage{450,2193}.Exon 12 of the messenger is alternative-ly spliced, and there is some suggestionthat this splice variant is expressed athigher levels in about a third of sporadicbreast tumour when compared to normalepithelial cells {266}. In sporadic breasttumours, BRCA2 mRNA-expression washigher than that in normal surroundingtissues in 20% of the cases, and lower in11% {267}. In agreement with this, nohypermethylation of the BRCA2 promotorregion has been detected in breast andovarian cancer {541}.
Gene functionLoss, or mutational inactivation, of thesingle wild-type allele in heterozygouscarriers of mutations in the BRCA2 geneis a key step in tumourigenesis. Themechanism by which the encoded pro-tein contributes to disease progression isnot yet completely understood but isthought to be related, at least in part, tothe proposed role of BRCA2 in the repairof damaged DNA. B R C A 2 encodes a very large (3,418amino acids in humans) protein that isexpressed during S phase of the cellcycle when it is present in the cell nucle-us. Although the amino acid sequence ofthe BRCA2 protein presents few directclues as to its normal cellular role, somefunctional domains have been defined.The C-terminal region of BRCA2 containsa functional nuclear localization se-quence; many pathogenic truncatingmutations in human BRCA2 are proximalto this domain and would therefore bepredicted to encode cytoplasmic pro-teins. The central part of the proteinencoded by the large exon 11 containseight copies of a novel sequence (theBRC repeat) that has been shown to becapable of binding RAD51 pro t e i n .RAD51 is a key protein involved in dou-ble-strand DNA break repair and homol-ogous recombination and the interactionwith BRCA2 was the first evidence impli-cating the protein in these processes.BRCA2-deficient cells and tumours char-acteristically accumulate aberrations inc h romosome structure {3018}. Theselesions include breaks involving one ofthe two sister chromatids, as well as tri-radial and quadri-radial chromosomestypical of Bloom syndrome and Fanconi
anaemia. Thus, BRCA2 deficiency maybe similar in its pathogenesis to othergenetic diseases in which unstable chro-mosome structure is linked to cancerpredisposition.C h romatid-type breaks, tri-radial andquadri-radial chromosomes are thoughtto arise from defects in the repair of DNAdouble-strand breaks (DSBs) during theS phase of cell cycle. During S phase,DSB repair proceeds pre f e re n t i a l l ythrough mechanisms involving homolo-gous recombination. These mechanismsenable error-free repair of broken DNA,taking advantage of the availability of thereplicated sister chromatid as a sub-strate for recombination reactions. InBRCA2-deficient cells, DSB repair byhomologous recombination is defective.However, alternative – but error-prone -
mechanisms for DSB repair such as end-joining or strand-annealing are still pres-ent. The end result is that DSBs inBRCA2-deficient cells are mis-repaired,giving rise to mutations and chromoso-mal rearrangements including transloca-tions or deletions. The resulting geneticinstability is believed to potentiate theacquisition of mutations that transform anormal cell into a cancer cell. Thus,BRCA2 works as a tumour suppressorindirectly through its ‘caretaker’ role inprotecting chromosomal stability.BRCA2 is essential for homologousrecombination because it controls theintra-cellular transport and activity ofRAD51. In BRCA2-deficient cells, RAD51fails to efficiently enter the nucleus. Aftere x p o s u re of BRCA2-deficient cells to DNAdamaging agents, RAD51 fails to localize
Fig. 8.10 Aberrations in chromosome structure reminiscent of Bloom syndrome and Fanconi anaemia accu-mulate during the division of BRCA2-deficient cells in culture. Enlargements of characteristic aberrations areshown in the panels on the right hand-side (ctb, chromatid break, tr, tri-radial and qr, quadri-radial).Reproduced from K.J. Patel et al. {2193}.
349BRCA2 syndrome
Fig. 8.09 Functional domains in BRCA2. There are 8 BRC repeats in the central region of the protein whichinteract with RAD51. NLS = nuclear localization signal. The proportion encoded by exon 11 is indicated.

350 Inherited tumour syndromes
in typical nuclear foci that may re p re s e n tsites for DNA damage pro c e s s i n g .M o re o v e r, BRCA2 controls the assemblyof RAD51 into a nucleoprotein filamentthat coats DNA, a critical interm e d i a t es t r u c t u re in recombination re a c t i o n s .Unexpected and potentially inform a t i v einsight into the role of BRCA1/2 genes inDNA repair in humans in vivo has comef rom recent studies on Fanconi anaemia( FA), a complex disorder characterizedby congenital abnormalities, pro g re s s i v ebone marrow failure and cancer suscepti-b i l i t y. FA is a recessively inherited disor-der which can result from mutation in atleast 8 individual genes. It has re c e n t l ybeen suggested that one of the pre v i o u s-ly unidentified FA genes, FANCD1, is infact BRCA2 {1251}. The cellular conse-quences of homozygosity for B R C A 2mutation, including spontaneous chro m o-some instability and hypersensitivity toDNA cross-linking agents, are rather sim-ilar to those observed in cells derivedf rom FA patients. This is not the only linkbetween FA and breast cancer suscepti-bility genes. Another FA gene pro d u c t ,FANCD2, can interact and co-localizewith BRCA1 {958}. Thus it seems that thepathways disrupted in FA and breast can-cer susceptibility are intimately connect-ed. Only a small pro p o rtion of FA, which initself is rare, is caused by B R C A 2 m u t a-tion but the importance of this finding isthat it connects together two pre v i o u s l yd i ff e rent bodies of work on DNA re p a i r. A current simplified model on howBRCA2 and several other genes involvedin breast cancer predisposition act coor-dinately to repair DNA damage is indi-cated in Fig. 8.08. ATM and CHEK2 pro-tein kinases signal the presence of dou-ble-stranded DNA breaks and phospho-rylate (red arrows) a number of down-s t ream effector proteins, includingBRCA1. This induces their migration tosites where DNA is repaired. BRCA2 car-ries the DNA-recombination enzymeRAD51 to the same sites, guided thereby the DNA-binding structures formedbetween its C-terminal domain and Dss1protein. A complex of Fanconi anaemiaproteins – termed A, C, D2, E, F, and G –triggers the ubiquitination of the D2 pro-tein alone and its colocalization withBRCA1.Other roles for BRCA2 have been sug-gested in chromatin remodelling andgene transcription {1442}. Such func-tions – which remain very poorly charac-
terized – may help to explain why cancerpredisposition associated with BRCA2mutations should be specific to tissuessuch as the breast and ovary. However,notwithstanding these other potentialfunctions, it seems likely that loss ofBRCA2 function engenders genomicinstability leading to oncogene activationand tumour suppressor loss that culmi-nates in tumourigenic pro g ression. Amajor challenge for future work will be tounderstand how this basic pathogenicmechanism plays out in the complex tis-sue environments of the breast, ovary orp rostate, giving rise to site-specificepithelial malignancies.
Mutation spectrumG e rmline mutations in B R C A 2 have beendetected in 5-10% of clinic-based bre a s tcancer families, and in similar fre q u e n-cies of breast-ovarian cancer families{2657,3023}. Somatic mutations in spo-radic breast and ovarian tumours aree x t remely rare. Mutations occur thro u g h-out the entire coding region, and hencethe mutation spectrum did not pro v i d eimmediate clues to functional genedomains. The majority of the mutationsa re predicted to lead to a pre m a t u re l ytruncated protein when translated. Inconjunction with the observed loss of thewildtype allele in tumours arising in muta-tion carriers {560}, this indicates thei m p o rtance of gene inactivation fortumourigenesis to occur. Despite thes t rong variability in mutations detected infamilies, founder effects have led to somemutations being very prevalent in cert a i npopulations of defined geographical orethnic background. Examples are the999del5 mutation, which is present ina p p roximately 0.6% of all Icelandic indi-viduals {2920}, and the 6174delT muta-tion found in an equal pro p o rtion ofAshkenazi Jews {2083}. As a result, muta-tion spectra may vary according to ethnicb a c k g round of the sampled population{2824}. In recent years, an incre a s i n gnumber of missense changes are beingdetected in B R C A 2 of which the clinicalsignificance is uncertain in the absenceof a functional assay. These already com-prise up to 50% of all known sequencechanges in B R C A 2. Although many ofthem are expected to be rare neutralpolymorphisms, some might be associat-ed with elevated levels of breast cancerrisk. An example is the arginine for histi-dine substitution at codon 372 {1167}.
Many known deleterious B R C A 1 a n dB R C A 2 mutations affect splicing, andthese typically lie near intron/exon bound-aries. However, there are also potentiali n t e rnal exonic mutations that disruptfunctional exonic splicing enhancer (ESE)sequences, resulting in exon skipping. AT2722R mutation segregated with aff e c t-ed individuals in a family with breast can-cer and disrupted 3 potential ESE sites{816}. The mutation caused deleteriousp rotein truncation and suggested apotentially useful method for determ i n i n gthe clinical significance of a subset of themany unclassified variants of B R C A 1 a n dB R C A 2. As more functional and structur-al information on the BRCA1 and BRCA2p roteins accumulates, our understandingof genetic variation in these genes willi m p rove. The Breast Cancer Inform a t i o nC o re (BIC) maintains a website pro v i d i n ga central re p o s i t o ry for inform a t i o nre g a rding mutations and polymorphisms( h t t p : / / re s e a rc h . n h g r i . n i h . g o v / b i c / ) .
Genotype-phenotype correlationsEvidence is accumulating that the risksconferred by pathogenic BRCA2 muta-tions are dependent on the position ofthe mutation in the gene, genetic varia-tion in other genes, and environmental orlifestyle factors.
BRCA2 mutation positionTruncating mutations in families with thehighest risk of ovarian cancer relative tobreast cancer are clustered in a region ofapproximately 3.3 kb in exon 11 {972}.This region of B R C A 2, bounded bynucleotides 3035 and 6629, was dubbedthe 'ovarian cancer cluster region,' orOCCR. Notably, this region coincideswith the BRC repeats that are critical forthe functional interaction with the RAD51protein. A much larger study of 164 fam-ilies confirmed that OCCR mutations areassociated with a lower risk of breastcancer and with a higher risk of ovariancancer {2913}. The extent of risk modifi-cation is too moderate, however, to beused in genetic counseling.
Genetic risk-modifiersA length-variation of the polyglutaminerepeats in the estrogen receptor co-acti-vator NCOA3 influences breast cancerrisk in carriers of B R C A 1 and B R C A 2{2345}. Although it should be noted thatmost of the carriers in these studies areB R C A 1 carriers, and there was insuff i-

cient power to determine the effect inB R C A 2 carriers alone. Similarly, the vari-ant pro g e s t e rone receptor allele namedPROGINS was associated with an oddsratio of 2.4 for ovarian cancer among214 B R C A 1 / 2 carriers with no paste x p o s u re to oral contraceptives, com-p a red to women without ovarian cancerand with no PROGINS allele {2487}. AC/G polymorphism in the 5' untranslatedregion of R A D 5 1 was found to modify
both breast and ovarian cancer risk incarriers of B R C A 2 {1644,3053}. Theseresults support the hypothesis thatgenetic variation in the genes constitut-ing endocrine signalling and DNA re p a i rpathways may modify B R C A 2- a s s o c i a t-ed cancer risk.
Hormonal risk modifiersAs in the BRCA1 syndrome, the breastcancer risk of BRCA2 carriers is influ-
enced by hormonal factors, includingoral contraceptives and pregnancy (seepage 56).
Prognosis and preventionLife expectancy and preventive strate-gies are similar to those discussed forBRCA1 carriers (see page 56) .
Li-Fraumeni syndrome P. HainautR. EelesH. OhgakiM. Olivier
DefinitionLi-Fraumeni syndrome (LFS) is aninherited neoplastic disease with auto-somal dominant trait. It is character-ized by multiple primary neoplasms inc h i l d ren and young adults, with a pre-dominance of soft tissue sarc o m a s ,o s t e o s a rcomas, breast cancer, and ani n c reased incidence of brain tumours,leukaemia and adre n o c o rtical carc i n o-ma. The majority of Li-Fraumeni casesis caused by a T P 5 3 g e rmline muta-t i o n .
MIM Nos. {1835}Li-Fraumeni syndrome 151623TP53 mutations
(germline and sporadic) 191170CHEK2 mutations 604373
SynonymS a rcoma family syndrome of Li andFraumeni.
IncidenceF rom 1990 to 1998, 143 families with aT P 5 3 g e rmline mutations were re p o rt-
ed {2086}. The IARC Database { w w w.i a rc . f r / p 5 3 / g e rmline.html} c u r rently con-tains 223 families {2104a}.
Diagnostic criteriaThe criteria used to identify an affectedindividual in a Li-Fraumeni family are: (i)occurrence of sarcoma before the age of45 and (ii) at least one first degree rela-tive with any tumour before age 45 and(iii) a second (or first) degree relativewith cancer before age 45 or a sarcomaat any age {273,957,1650}.
BAFig. 8.11 A A fraction of tumours in families with a TP53 germline mutation. B Mean age of patients with tumours caused by a TP53 germline mutation, accordingto organ site.
351BRCA2 syndrome / Li-Fraumeni syndrome

Breast tumoursFrequency Breast cancers are the most frequentneoplasms developed in families with aTP53 germline mutation. Thirty-seven %of these families are defined as Li-Fraumeni syndrome and 30% as Li-Fraumeni-like syndrome. In the 219 fami-lies with a TP53 germline mutation report-ed in 1990–2001 (IARC TP53 database:www.iarc.fr/p53), a total of 562 tumoursdeveloped in individuals with a con-firmed TP53 germline mutation. Of these,158 (28%) were breast tumours. Eighty-three (38%) families with a TP53 mutationhad at least one family member with abreast tumour. Among the families inwhich at least one case of breast cancerdeveloped, the mean number of breasttumours per family was 1.9.
Age and sex distributionBreast cancers associated with a TP53germline mutation develop earlier thantheir sporadic counterparts, with a meanage of 35+10 years (range 14-67 yearsold). The mean age of women with Li-Fraumeni-like syndrome (LFL) is approx-imately 8 years higher {2104a} However,b reast cancers associated with T P 5 3germline mutations never developed inyoung children, suggesting that hormon-al stimulation of the mammary glandsconstitutes an important co-factor.Sporadic breast tumours occur approxi-mately 100 times more frequently infemales than in males {1475}, and noneoccurred in males among the 158 report-ed breast cancer with TP53 germlinemutations.
PathologyOf the 158 breast tumours recorded, themajority (146 cases, 92%) have not beenclassified histologically, but recorded asjust breast cancers. Histologically classi-fied cases included carcinoma in situ (4cases), adenocarcinoma (1 case), Pagetdisease (2 cases), malignant phyllodestumour (2 cases), comedocarcinoma (1case), spindle cell sarcoma (1 case),and stromal sarcoma (1 case).
Prognosis and prognostic factorsThe breast cancers that occur in LFS areof younger onset and so may have ap o o rer prognosis due to this early age atdiagnosis. In mice, there is re l a t i v er a d i o resistance in p 5 3 mutants, how-e v e r, radioresistance due to germ l i n e
mutation has not been convincinglyshown in man.
Other tumoursFrequencyFollowing breast cancer, brain tumoursand sarcomas (osteosarcomas and softtissue sarcomas) are the next most fre-quent manifestations. The sporadicc o u n t e r p a rts of these tumours alsoshow somatic T P 5 3 mutations, suggest-ing that in these neoplasms, T P 5 3 m u t a-tions are capable of initiating thep rocess of malignant transform a t i o n{ 1 4 7 5 , 2 0 8 7 } .
Age distributionIn general, tumours associated with aTP53 germline mutation develop earlierthan their sporadic counterparts, butthere are marked organ-specific differ-ences. As with sporadic brain tumours,the age of patients with nervous systemneoplasms associated with TP53 germ-line mutations shows a bimodal distribu-tion. The first peak of incidence (repre-senting medulloblastomas and relatedprimitive neuroectodermal tumours) is inchildren, and the second (mainly astro-cytic brain tumours) in the third andfourth decades of life {2087}. Adreno-cortical carcinomas associated with aTP53 germline mutation develop almostexclusively in children, in contrast to spo-radic adrenocortical carcinomas, whichhave a broad age distribution with a peakbeyond age 40 {1475}.
Genetics – TP53Chromosomal locationThe TP53 gene encompasses 20 kilo-bases on chromosome 17p13.1. TP53belongs to a family of growth suppres-sors that also comprises two other mem-bers, TP73 and TP63. Whereas the twolatter genes are mostly involved in theregulation of differentiation and develop-ment, TP53 plays specialized functionsas a tumour suppressor {1643}.
Gene structureThe gene contains 11 exons, the first onenon-coding. The first intron is particularlylarge (10 kilobases). The codingsequence is concentrated over 1.3 kilo-bases. TP53 is ubiquitously expressed,mostly as a single mRNA species(although rare alternatively spliced vari-ants have been reported). The promoterdoes not contain a classical TATA boxbut shows binding elements for severalcommon transcription factors, includingc-Jun and NF-kappaB {1107}.
Gene expressionThe p53 protein is constitutivelye x p ressed in most cell types but, in nor-mal circumstances, does not accumu-late to significant level due to rapiddegradation by the proteasome machin-e ry. In response to various types of cel-lular stress, the p53 protein undergoes anumber of post-translational modifica-tions that release p53 from the negativec o n t rol of MDM2, a protein that binds top53 and mediates its degradation.
352 Inherited tumour syndromes
Fig. 8.12 Age distribution of patients with tumourscaused by a T P 5 3 germline mutation.

353Li-Fraumeni syndrome
These modifications result in the intranu-clear accumulation of p53 and in its acti-vation as a transcription factors. Tw omajor signaling pathways can triggerT P 5 3 activation. The first, and best char-acterized, is the pathway of response toDNA damage, including large kinases ofthe phosphoinositol-3 kinase family suchas AT M (ataxia telangiectasia mutated)and the cell-cycle re g u l a t o ry kinaseC H E K 2. Both of these kinases phospho-rylate p53 in the extreme N-term i n u s(serines 15, 20 and 37), within the re g i o nthat binds to MDM2. The second is acti-vated in response to the constitutivestimulation of gro w t h - p romoting signal-ing cascades. The central regulator inthis pathway is p14ARF, the altern a t i v ep roduct of the locus encoding thecyclin-kinase inhibitor p 1 6 / C D K N 2 a.p14ARF expression is activated by E2Ftranscription factors, and binds toMDM2, thus neutralizing its capacity toinduce p53 degradation. This pathwaymay be part of a normal feedback con-t rol loop in which p53 is activated as acell-cycle brake in cells exposed toh y p e r p roliferative stimuli {2267}.
Gene functionAfter accumulation, the p53 protein actsas a transcriptional regulator for a panelof genes that differ according to thenature of the stimulus, its intensity andthe cell type considered. Broadly speak-ing, the genes controlled by p53 fall intot h ree main categories, including cell-cycle regulatory genes (WAF1, GADD45,14-3-3S, CYCLING), pro - a p o p t o t i cgenes (FAS/APO1/CD95, KILLER/DR5,AIF1, PUMA, BAX) and genes involved inDNA repair (O6MGMT, MLH2). The p53protein also binds to compoments of thetranscription, replication and re p a i rmachineries and may exert additionalcontrols on DNA stability through themodulation of these mechanisms.Collectively, the p53 target genes medi-ate two type of cellular responses: cell-cycle arrest, followed by DNA repair incells exposed to light forms of genotoxicstress, and apoptosis, in cells exposedto levels of damage that cannot be effi-ciently repaired. Both responses con-tribute to the transient or permanent sup-pression of cells that contain damaged,potentially oncogenic DNA. In themouse, inactivation of Tp53 by homolo-gous recombination does not preventnormal growth but results in a strong pre-
disposition to early, multiple cancers,illustrating the crucial role of this gene asa tumour suppressor {714}.
Mutation spectrumThe T P 5 3 gene is frequently mutated inmost forms of sporadic cancers, withp revalences that range from a few per-cents in cervical cancers and in malignantmelanomas to over 50% in invasive carc i-nomas of the aero-digestive tract. Over75% of the mutations are single base sub-stitutions (missense or nonsense), cluster-ing in exons 5 to 8 that encode the DNA-binding domain of the protein. Codons175, 245, 248, 273 and 282 are majormutation hotspots in almost all types ofcancers. To g e t h e r, these codons containover 25% of all known T P 5 3 m u t a t i o n s .Other codons are mutation hotspots inonly specific tumour types, such as codon249 in hepatocellular carcinoma andcodon 157 in bronchial cancer. Mutationp a t t e rns can differ significantly betweenbetween diff e rent types cancers orbetween geographic areas for the samecancer type (as for example hepatocellu-lar carcinoma). These observations haveled to the concept that mutation pattern smay reveal clues on the cellular or envi-ronmental mechanisms that have causedthe mutations {1107}. In sporadic bre a s tcancers, T P 5 3 is mutated in about 25% ofthe cases. However, several studies havere p o rted accumulation of the p53 pro t e i nwithout mutation in up to 30-40% of inva-
sive ductal carcinoma in situ. The mutationp a t t e rn is similar to that of many other can-cers and does not provide information onpossible mutagenic events. There is limit-ed evidence that the mutation pre v a l e n c eis higher in BRCA1 mutation carriers. G e rmline T P 5 3 mutations have beenidentified in 223 families. Of these fami-lies, 83 match the strict LFS criteria, 67c o r respond to the extended, LFL defini-tion, 37 have a family history of cancerthat does not fit within LFS or LFL defini-tions and 36 have germline mutationswithout documented familial history ofcancer (IARC TP53 mutation database,w w w. i a rc.fr/p53). The codon distributionof germline T P 5 3 mutations show thesame mutational hotspots as somaticmutations {1475}. The distribution ofinherited mutations that predisposes tob reast cancer are scattered along exons5 to 8 with relative "hotspots" at codons245, 248 and 273, which are also com-monly mutated in somatic breast cancer.In contrast, a total of 16 breast cancershave been detected in 5 families with ag e rmline mutation at codon 133, a posi-tion which is not a common mutationhotspot in somatic breast cancer. Itremains to be established whether thismutant has particular functional pro p e r-ties that predispose to breast cancer.
Genotype-phenotype correlationsBrain tumours appear to be associatedwith missense TP53 mutations in the
Fig. 8.13 The p53 signaling pathway. In normal cells the p53 protein is kept in a latent state by MDM2.Oncogenic and genotoxic stresses release p53 from the negative control of MDM2, resulting in p53 accu-mulation and activation. Active p53 acts as transcription factor for genes involved cell cycle control, DNArepair and apoptosis, thus exerting a broad range of antiproliferative effects.

354 Inherited tumour syndromes
DNA-binding loop that contacts the minorg roove, while early onset brain tumoursw e re associated with mutations likely toresult in absence of protein or loss offunction {2104a}. Adre n o c o rtical carc i n o-mas were associated with missensemutations in the loops opposing the pro-tein-DNA contact surface {2104a}.
Genetics – CHEK2Chromosomal locationCHEK2 is on chromosome 22q12.1.
Gene structureCHEK2 has 14 exons and there are sev-eral homologous loci, which encompassexons 10-14 of the gene, scattere dt h roughout the genome. These genefragment copies can present problemswhen analysing C H E K 2 for germ l i n emutations in genomic DNA, and it isimportant to ensure that the correct copyis being amplified {2742}. This problemcan be overcome by amplifying exons10-14 by the use of a long range PCRusing primers located in the non-dupli-cated region of the gene {2741}. Theindividual exons can then be subse-quently amplified using the product ofthe long range PCR as a template.
Gene expressionCHEK2 is expressed in nonpro l i f e r a t i n gand terminally diff e rentiated cells. It is
homogenously expressed in renewing cellpopulations such as epidermis, esopha-gus, rectum, bladder, stomach, intestineand colon, and heterogenously in condi-tionally renewing tissues such as lung,b reast kidney, salivary, thyroid, parathy-roid, adrenal glands, pancreas, pro s t a t e ,epididymis, sweat glands, endometruim,s t romal mesenchymal cells, blood ves-sels, lymphoid tissues, smooth and car-diac muscle tissues and pre i p h e r a lnerves. It is absent or cytoplasmic in stat-ic tissues such as muscle and brain.CHEK2 remains expressed and can beactivated in all phases of the cell cycle inresponse to DNA damage {1714}.
Gene functionHuman CHEK2 is a homolog of the yeastG2-checkpoint kinases C D S 1 a n dRAD53 {1791}. In response to DNA dam-age, CHEK2 propagates the checkpointsignal along several pathways, whicheventually causes cell-cycle arrest in G1,S and G2/M phases {449,820}; activationof DNA repair {1609}, and in apoptoticcell death {1315}. Four of the down-s t ream checkpoint effectors that areestablished as substrates of CHEK2 invivo include p53, BRCA1 and Cdc25Aand Cdc25C.
Mutation spectrumRecently, heterozygous germline muta-
tions in CHEK2 have been identified inthree of a subset of individuals with thedominantly inherited Li-Fraumeni syn-drome which do not harbour TP53 muta-tions {209}. However, one of these wasfound to be in a pseudogene copy of theCHEK2 gene. Another one appeared tobe neutral polymorphism in the Finnishpopulation. The third was a protein-trun-cating mutation, 1100delC in exon 10,which abolishes the kinase function ofCHEK2. The possibility that this gene isonly contributing to the breast cancercases within LFS families rather than LFSper se has been raised {2740}. The frequency of 1100delC has beenestimated in healthy control populations,and was found to vary between 0.3%and 1.7% {1840,2084,2984}. This wouldalso suggest that the 1100delC is a poly-morphism, rather than a disease-causingmutation. Yet among unselected patientswith breast cancer, its prevalence wasfound to be approximately 1.5-fold high-er than in controls. Significantly elevatedfrequencies were found among patientswith a positive family history and amongpatients with bilateral breast cancer{2984}. The strongest enrichment of1100delC carriers (approximately 5-fold)was found among familial breast cancerpatients in whom the presence of BRCA1or B R C A 2 mutations were excluded{1840,2984}. However, in families withthe 1100delC mutation, it appears to co-segregate poorly with breast cancer. Theresults suggest that CHEK2*1100delC isa low risk breast cancer susceptibilityallele which may make a significant con-tribution to familial clustering of breastcancer, including families with smallernumbers of affected cases. As it isenriched among multiple-case families,but unable to explain all breast cancer infamilies with at least one carrier case, itmay interact with other, as yet unknownbreast cancer susceptibility alleles.
Search for additional LFS genesThe paucity of large LFS kindreds makesclassical linkage methodology difficult. Acandidate approach is there f o re beingused. Candidate genes are those involvedin cell cycle pathways, those commonlymutated in multiple tumour types and theb reast cancer genes, as this site is com-monly affected in LFS kindreds. Usingthese approaches, the genes P 1 6 a n dP T E N {379} have been analysed and nog e rmline mutations found.
Fig. 8.14 Codon distribution of somatic (top) or germline (bottom) TP53 mutations associated with breastcancers. Hotspot mutations are indicated. Mutation at codon 133 has been reported in 5 Li-Fraumeni fami-lies with breast cancers, but is not a frequent site for somatic mutation in breast cancer in the general pop-ulation. Compiled from: IARC TP53 database, www.iarc.fr/p53.

355Cowden syndrome
Cowden syndrome C. Eng
D e f i n i t i o nCowden syndrome (CS) is an autoso-mal dominant disorder caused byg e rmline mutaions of the P T E N gene. Itis characterized by multiple hamar-tomas involving organs derived from allt h ree germ cell layers and a high risk ofb reast, uterine and non-medullary thy-roid cancer. The classic hamartoma isthe trichilemmoma and is pathogno-monic for CS.
MIM No. 158350 {1835}
S y n o n y m sCowden disease, multiple hamart o m as y n d ro m e .
I n c i d e n c eThe single most comprehensive clinicalepidemiologic study before the CS sus-ceptibility gene was identified estimat-ed the prevalence to be 1:1 000 000{1990,2776}. Once the gene was iden-tified {1654}, a molecular-based esti-mate of prevalence in the same popu-lation was 1:300 000 {1989}. Becauseof the difficulty in recognizing this syn-d rome, prevalence figures are likelyu n d e re s t i m a t e d .
Diagnostic criteriaBecause of the variable and bro a de x p ression of CS and the lack of uni-f o rm diagnostic criteria prior to 1996,the International Cowden Consort i u m{1990} compiled operational diagnosticcriteria for CS, based on the publishedl i t e r a t u re and their own clinical experi-ence {785}. These criteria have beenrecently revised in light of new data,and have been adopted by the US-based National Compre h e n s i v eCancer Network Practice Guidelines{786,1299}. Trichilemommas and papil-lomatous papules are part i c u l a r l yi m p o rtant to recognize. CS usuallyp resents by the late 20's. It has vari-able expression and, pro b a b l y, an age-related penetrance although the exactpenetrance is unknown. By the thirddecade, 99% of affected individuals
would have developed the mucocuta-neous stigmata although any of the fea-t u res could be present alre a d y.Because the clinical literature on CSconsists mostly of re p o rts of the mostflorid and unusual families or casere p o rts by subspecialists interested intheir respective organ systems, thespectrum of component signs isunknown. Despite this, the most commonlyre p o rted manifestations are mucocuta-
neous lesions, thyroid abnorm a l i t i e s ,f i b rocystic disease and carcinoma ofthe breast, gastrointestinal hamar-tomas, multiple, early-onset uterineleiomyoma, macrocephaly (specifical-l y, megencephaly) and mental re t a rd a-tion {1133, 1693,1748,2776}. Recentdata have suggested that endometrialc a rcinoma should be a componentcancer of CS {657,786,1772}. What itsf requency is in mutation carriers is asyet unknown.
Table 8.07International Cowden Syndrome Consortium Operational Criteria for the Diagnosis of Cowden Syndrome(Ver. 2000)*.
Pathognomonic Mucocutanous lesions:criteria Trichilemmomas, facial
Acral keratosesPapillomatous papulesMucosal lesions
Major criteria Breast carcinomaThyroid carcinoma (non-medullary), esp. follicular thyroid carcinomaMacrocephaly (Megalencephaly) (say, >97%ile)Lhermitte-Duclos disease (LDD)Endometrial carcinoma
Minor criteria Other thyroid lesions (e.g. adenoma or multinodular goiter)Mental retardation (say, IQ < 75)GI hamartomasFibrocystic disease of the breastLipomasFibromasGU tumours (e.g. renal cell carcinoma, uterine fibroids) or malformation
Operational diagnosis 1. Mucocutanous lesions alone if:in an individual a) there are 6 or more facial papules, of which 3 or more
must be trichilemmoma, orb) cutaneous facial papules and oral mucosal papillomatosis, orc) oral mucosal papillomatosis and acral keratoses, ord) palmoplantar keratoses, 6 or more
2. Two major criteria but one must include macrocephaly or LDD3. One major and 3 minor criteria4. Four minor criteria
Operational diagnosis in a 1. The pathognomonic criterion/iafamily where one individual 2. Any one major criterion with or without minor criteriais diagnostic for Cowden 3. Two minor criteria
*Operational diagnostic criteria are reviewed and revised on a continuous basis as new clinical and genetic informa-tion becomes available. The 1995 version and 2000 version have been accepted by the US-based NationalComprehensive Cancer Network High Risk/Genetics Panel.

Breast tumoursAge distribution and penetranceInvasive carcinomas of the breast havebeen diagnosed as early as the age of14 years and as late as in the 60’s{1693}. However, the majority of CS-related breast cancers occur after theage of 30-35 years {786,788}. A singlepopulation-based clinical study, with-out the benefit of genetic analysis, sug-gested that benign breast disease canoccur in two-thirds of affected womenwhile CS females have a 25-50% life-time risk of developing invasive bre a s tcancer {786,2776}. Male breast cancercan occur in CS as well but the fre-quency is unknown {817,1771}.
Clinical featuresIt is believed that the clinical pre s e n t a-tion of breast cancer in CS is no diff e r-ent from that of the general population.H o w e v e r, no formal data is curre n t l ya v a i l a b l e .
P a t h o l o g yLike other inherited cancer syndro m e s ,multifocality and bilateral involvementis the rule. With re g a rd to the individualcancers, even of the breast and thy-roid, as of mid 1997, there has yet to bea systematic study published. Thereexists, however, one study which hasattempted to look at benign and malig-nant breast pathology in CS patients.Although these are pre l i m i n a ry studies,without true matched controls, it is, todate, the only study that examinesb reast pathology in a series of CScases. Breast histopathology from 59cases belonging to 19 CS women wassystematically analysed {2578}. Thirt y -five specimens had some form ofmalignant pathology. Of these, 31(90%) had ductal adenocarc i n o m a ,one tubular carcinoma and one lobularc a rcinoma-in-situ. Sixteen of the 31had both invasive and in situ (DCIS)components of ductal carcinoma while12 had DCIS only and two only invasivea d e n o c a rcinoma. Intere s t i n g l y, it wasnoted that 19 of these carc i n o m a sa p p e a red to have arisen in the midst ofdensely fibrotic hamartomatous tissue.Benign breast disease is more commonthan malignant, with the form e rbelieved to occur in 75% of aff e c t e dfemales. Fibrocystic disease of theb reast, breast hamartomas, andf i b roadenomas are commonly seen.
Uterine tumoursAge distribution and penetranceSince endometrial carcinomas haveonly recently been suggested to be aminor component of CS {786}, it isunknown what the true frequency isamong mutation carriers or what theage distribution is. Anecdotal casessuggest that the frequency could be 6-10% in affected women.Benign tumours of the uterus are com-mon in CS. Uterine leiomyomas arebelieved to occur in almost half ofa ffected women {1693}. They are usu-ally multi-focal and occur at a youngage, even in the 20’s. Other benignuterine pathologies such as polyps andhyperplasias have been found in CSpatients but are of unknown fre q u e n c y.
Clinical featuresT h e re have been no systematic studiesof uterine tumours in CS. Clinical obser-vation and anecdotal re p o rts suggestthat the leiomyomas can become quitesymptomatic, presenting with bleedingand pain. It is unclear if the clinicalp resentation of the endometrial carc i n o-mas is diff e rent from that of sporadicc a s e s .
P a t h o l o g yT h e re have been no systematic studiesof uterine tumours in CS although it isbelieved that the histopathology is nod i ff e rent from that of typical sporadicc a s e s .
Prognosis and prognostic factorsWhether the prognosis differs from spo-radic cases is unknown.
Thyroid tumoursAge of distribution and penetranceA p a rt from breast cancer, the othermajor component cancer in CS is non-m e d u l l a ry thyroid cancer. Nonmedullaryt h y roid carcinomas occur at a fre q u e n c yof 3-10% of affected individuals, re g a rd-less of sex, in non-systematic clinicalseries {1693,2776}. It is unclear, howev-e r, whether the age of onset is earlierthan that of sporadic cases.Benign thyroid disease occurs ina p p roximately 70% of affected individu-als. Component features include multin-odular goitre and follicular adenomas.These benign tumours can occur at anyage and can even manifest in teen-a g e r s .
Clinical featuresMany of the benign tumours in CS indi-viduals remain asymptomatic. However,the most common presenting sign orsymptom would be a neck mass. Likemany inherited syndromes, CS thyro i dlesions can be multifocal and bilobar.
P a t h o l o g yNo systematic studies have been per-f o rmed to examine the thyroid in CS.H o w e v e r, clinical observations and clin-ical re p o rts suggest that the histology ofthe nonmedullary thyroid carcinoma isp redominantly of the follicular type {786,2 7 7 6 } .
Other tumoursDysplastic gangliocytoma of the cere-bellum (Lhermitte-Duclos disease) is themajor manifestation in the central nerv-ous system. Peripheral lesions includeverrucous skin changes, cobblestone-like papules, fibromas of the oralmucosa, multiple facial trichilemmomasand hamartomatous polyps of the colon.
G e n e t i c sChromosomal location and mode oft r a n s m i s s i o nCS is an autosomal dominant disord e r,with age related penetrance and vari-able expression {787}. The CS suscepti-bility gene, P T E N, resides on 10q23.3{1651, 1654,1990}.
Gene structureP T E N / M M A C 1 / T E P 1 is comprised of 9exons spanning 120-150 kb of genomicdistance {1649,1651,1654,2777}. It isbelieved that intron 1 occupies much ofthis (approximately 100 kb). P T E Nencodes a transcript of 1.2 kb.
Gene expressionP T E N is expressed almost ubiquitouslyin the adult human. In normal humane m b ryonic and foetal development,PTEN protein is expressed ubiquitouslyas well, although levels might changet h roughout development {1008}. P T E Nis very highly expressed in the develop-ing central nervous system as well asneural crest and its derivatives, e.g.enteric ganglia {1008}.
Gene functionPTEN encodes a dual specificity lipidand protein phosphatase [reviewed in{3043}]. It is the major 3-phosphatase
356 Inherited tumour syndromes

acting in the phosphoinositol-3-kinase(PI3K)/Akt apoptotic pathway {1730,2774}. To date, virtually all naturallyoccurring missense mutations testeda b rogate both lipid and protein phos-phatase activity, and one mutant,G129E, affects only lipid phosphataseactivity [reviewed in {3043}]. Over-e x p ression of PTEN results, for the mostp a rt, in phosphatase-dependent cellcycle arrest at G1 and/or apoptosis,depending on cell type [reviewed in{3043}]. There is also growing evidencethat PTEN can mediate growth arre s tindependent of the PI3K/Akt pathwayand perhaps independent of the lipidphosphatase activity {3096-3098}[ reviewed in {3042}].Murine models null for Pten result inearly embryonic death {688,2268,2817}.Hemizygous knock-out of Pten result invarious neoplasias, and the spectra ared i ff e rent depending on the part i c u l a rmodel. While the neoplasias are re m i n i s-cent of the component tumours found inthe human syndrome, none of the thre emodels are similar to CS.
Mutation spectrumAs with most other tumour suppre s s o rgenes, the mutations found in P T E N a res c a t t e red throughout all 9 exons. Theycomprise loss-of-function mutationsincluding missense, nonsense,frameshift and splice site mutations{309, 1771}. Approximately 30-40% ofg e rmline P T E N mutations are found inexon 5, although exon 5 re p resents 20%of the coding sequence. Furt h e r,a p p roximately 65% of all mutations can
be found in one of exons 5, 7 or 8 {309,1 7 7 1 } .Although P T E N is the major susceptibli-ty gene for CS, one CS family, withoutP T E N mutations, was found to have ag e rmline mutation in B M P R 1 A, which isone of the susceptibility genes for juve-nile polyposis syndrome {1250,3262}.Whether B M P R 1 A is a minor CS sus-ceptiblity gene or whether this familywith CS features actually has occultjuvenile polyposis is as yet unknown.
Genotype-phenotype correlationsA p p roximately 70-80% of CS cases, asstrictly defined by the Consortium cri-tieria, have a germline P T E N m u t a t i o n{1654,1771}. If the diagnostic criteriaa re relaxed, then mutation fre q u e n c i e sd rop to 10-50% {1723,1991,2959}. Af o rmal study which ascertained 64 unre-lated CS-like cases revealed a mutationf requency of 2% if the criteria are notmet, even if the diagnosis is made shortof one criterion {1772}.A single re s e a rch centre study involving37 unrelated CS families, ascert a i n e da c c o rding to the strict diagnostic criteriaof the Consortium, revealed a mutationf requency of 80% {1771}. Exploratorygenotype-phenotype analyses re v e a l e dthat the presence of a germline mutationwas associated with a familial risk ofdeveloping malignant breast disease{1771}. Furt h e r, missense mutationsand/or mutations 5’ of the phosphatasec o re motif seem to be associated with as u r rogate for disease severity (multi-organ involvement). One other smallstudy comprising 13 families, with 8
P T E N mutation positive, could not findany genotype-phenotype associations{1989}. However, it should be noted thatthis small sample size is not suitable forstatistical analyses and no conclusionsshould be drawn.P reviously thought to be clinically dis-tinct, Bannayan-Riley-Ruvalcaba syn-d rome (BRR, MIM 153480), which ischaracterized by macro c e p h a l y, lipo-matosis, haemangiomatosis and speck-led penis, is likely allelic to CS {1773}.A p p roximately 60% of BRR families andisolated cases combined carry ag e rmline P T E N mutation {1774}. Intere s -t i n g l y, there were 11 cases classified astrue CS-BRR overlap families in thisc o h o rt, and 10 of the 11 had a PTENmutation. The overlapping mutationspectrum, the existence of true overlapfamilies and the genotype-phenotypeassociations which suggest that thep resence of germline P T E N mutation isassociated with cancer strongly suggestthat CS and BRR are allelic and arealong a single spectrum at the molecu-lar level. The aggregate term of PTENh a m a rtoma tumour syndrome (PHTS)has been suggested {1774}.R e c e n t l y, the clinical spectrum of PHTShas expanded to include subsets ofP roteus syndrome and Pro t e u s - l i k e(non-CS, non-BRR) syndromes {3260}.G e rmline P T E N mutations in one case ofm a c rocephaly and autism and hydro-cephaly associated with VATER associa-tion have been re p o rted {625,2341}.
357Cowden syndrome

DefinitionHereditary nonpolyposis colorectal can-cer (HNPCC) is an autosomal dominantdisorder characterized by the develop-ment of colorectal cancer, endometrialcancer and other cancers due to inherit-ed mutations in one of the DNA mismatchrepair (MMR) genes {1725}.
MIM Nos. {1835}Familial nonpolyposis colon cancer, type 1 120435Familial nonpolyposis colon cancer, type 2 120436
SynonymsLynch syndrome, hereditary colorectalendometrial cancer syndrome {3007},h e re d i t a ry defective mismatch re p a i rsyndrome {595}.
IncidenceA p p roximately 2-5% of all cases of col-o rectal cancer are due to HNPCC {12}.The estimated frequency of carriers ofa DNA mismatch repair gene mutationin the general population is one in1000.
Diagnostic criteriaThe International Collaborative Group onHNPCC (ICG-HNPCC) proposed a set ofdiagnostic criteria (Revised AmsterdamCriteria) to provide uniformity in clinicalstudies {3010}. These criteria identifyfamilies that are very likely to representHNPCC. Other widely used criteria arethe Bethesda Criteria that can be used toidentify families suspected of HNPCCthat need testing for microsatellite insta-bility {2398}.
Endometrial tumoursP redisposed individuals from HNPCCfamilies have a high risk (30-80%) ofdeveloping colorectal cancer. The mostfrequent extracolonic cancer is endome-trial cancer. The lifetime risk of develop-ing this cancer is 30-60% by age 70 {14,731,3009,3071}. HNPCC-associatedendometrial cancer is diagnosed approx.10 years earlier than in the general pop-
ulation. The mean age at diagnosis is 50years. Patients with colorectal cancerassociated with HNPCC have a betterprognosis than patients with commonsporadic colorectal cancer {2526,3070}.In contrast, a recent study showed thatthe survival of endometrial cancer asso-ciated with HNPCC does not differ signif-icantly from endometrial cancer in thegeneral population {305}.
Pathology of endometrial tumoursIn patients from families with pro v e ngermline mutations in the MMR genes,MLH1, MSH2, MSH6, or from (suspect-ed) HNPCC families, the majority ofendometrial tumours were reported to beof the endometrioid type with diversegrading and staging {650,2174}. Certainhistopathologic features such as muci-nous diff e rentiation, solid-cribriformgrowth pattern, high grade and possiblenecrosis might suggest that a tumour isdue to a mismatch repair defect {1481,2174,2206}. Loss of MLH1 protein expression occursin endometrial cancer associated withHNPCC {235,650,1276, 1768,2174,2264}but also in 15-30 % of sporadic cancerswith somatic inactivation of MLH1{2518,2772}. Abrogation of MSH2 and/or MSH6 pro-tein expression, especially at a youngage seems to be a more specific indica-tor for HNPCC {235, 650, 2174,2264}.A l ready in the hyperplastic pre c u r s o rlesions such loss of expression can beencountered {235, 650}.
Other cancersMany other cancers have been reportedin HNPCC {13,14,3009,3010}. The fre-quency of specific cancers depends onthe prevalence of the cancer in the back-ground population {2178}. Cancer of thestomach for example is fre q u e n t l yobserved in families from Finland andJapan, both countries with a high preva-lence of stomach cancer in population.The ages at diagnosis of most cancersreported are earlier than their sporadiccounterparts.
358 Inherited tumour syndromes
H.F.A. VasenH. Moreau P. PeltomakiR. Fodde
Hereditary non-polyposis colon cancer(HNPCC)
1. Individuals with two HNPCC-related can-cers, including synchronous and metachro-nous colorectal cancers or associatedextracolonic cancers (endometrial, ovarian,gastric, hepatobiliary, small bowel canceror transitional cell carcinoma of the renalpelvis or ureter)
2. Individuals with colorectal cancer and afirst degree relative with colorectal cancerand/or HNPCC-related extracolonic cancerand/or colorectal adenoma; one of the can -cers diagnosed at age <45 y, and the ade-noma diagnosed at age <40 y
3. Individuals with colorectal cancer orendometrial cancer diagnosed at age <45 y
4. Individuals with right-sided colorectal can-cer with an undifferentiated pattern onhistopathology diagnosed at age <45 y
5. Individuals with signet-ring-cell-type col-orectal cancer diagnosed at age <45 y
6. Individuals with adenomas diagnosed atage <40
Table 8.09Bethesda Criteria.
There should be at least three relatives withcolorectal cancer (CRC) or with an HNPCC-associated cancer: cancer of the endometri-um, small bowel, ureter or renal pelvis.
– one relative should be a first degree relativeof the other two,
– at least two successive generations shouldbe affected,
– at least one tumour should be diagnosedbefore age 50,
– familial adenomatous polyposis should beexcluded in the CRC case if any,
– tumours should be verified by histopatholog-ical examination.
Table 8.08Revised Amsterdam Criteria.

Genetics of MLH1, MSH2, MSH6Chromosomal location and structure HNPCC is associated with germ l i n emutations in five genes with verified orputative DNA mismatch repair function,viz. MSH2 (MutS homologue 2), MLH1(MutL homologue 1), PMS2 (Postmeioticsegregation 2), MSH6 (MutS homologue6), and possibly MLH3 (MutL homologue3). Structural characteristics of thesegenes are given in Table 8.11. Endo-metrial cancer appears to be part of thesyndrome in families with mutations inany one of these genes, but is particular-ly associated with M S H 2 and M S H 6germline mutations {236,3011,3114}.
Gene productHNPCC genes show ubiquitous, nucleare x p ression in adult human tissues, andthe expression is particularly prominent inthe epithelium of the digestive tract aswell as in testis and ovary {860,1602,3132}. These genes are also expressed inn o rmal endometrium, and loss of pro t e i ne x p ression is an early change in endome-trial tumorigenesis. Studies of M S H 2 o rM L H 1 mutation carriers have shown thatthese proteins may be lost already in atyp-ical hyperplasia (precursor lesion ofendometrial cancer) or even in endometri-al hyperplasia without atypia in severalmonths before the diagnosis of endome-trial cancer, suggesting that immunohisto-chemical analysis of MSH2 and MLH1p roteins may be useful for pre - s c re e n i n gpurposes in HNPCC patients {235,1277}.
Gene functionThe protein products of HNPCC genesare key players in the correction of mis-
matches that arise during DNA replica-tion {1496}. Two different MutS-relatedheterodimeric complexes are responsi-ble for mismatch recognition: MSH2-MSH3 and MSH2-MSH6. While the pres-ence of MSH2 in the complex is manda-tory, MSH3 can replace MSH6 in the cor-rection of insertion-deletion mismatches,but not single-base mispairs. Followingmismatch binding, a heterodimeric com-plex of MutL-related proteins, MLH1-PMS2 or MLH1-MLH3, is recruited, andthis larger complex, together with numer-ous other proteins, accomplishes mis-match repair. The observed functionalredundancy in the DNA mismatch repairp rotein family may help explain whymutations in MSH2 and MLH1 are preva-lent in HNPCC families, while those inMSH6, PMS2 and MLH3 are less fre-quent (and MSH3 mutations completelyabsent), although alternative hypotheses(e.g. based on the differential participa-tion of the DNA mismatch repair proteinsin apoptosis signaling {863}) have alsobeen proposed.It is not known why some female HNPCCpatients develop endometrial cancer,while others develop colon cancer.Comparison of these two tumour typesoriginating from identical germline muta-tion carriers suggests the existence ofsome important tissue-specific diff e r-ences that may indicate different patho-genetic mechanisms. For example,a c q u i red loss of M S H 2 and M S H 6appears to characterize endometrial, butnot colon carcinomas developing inpatients with inherited mutations of MLH1{2589}. Moreover, the general MSI pat-terns and target genes for MSI seem dif-
ferent in endometrial and colorectal can-cers from HNPCC patients {1527}. Earlyinactivation of PTEN characterizes mostendometrial cancers from HNPCCpatients {3261} and tumorigenesis medi-ated by PTEN inactivation is acceleratedby mismatch repair deficiency {3052}.Apart from biosynthetic errors, the DNAmismatch repair proteins also recognizeand eliminate various types of endoge-nous and exogenous DNA damage, anddifferential exposure to such agents orvariable capacity to correct lesionsinduced by them may also play a role inthe organ-specific cancer susceptibilityin HNPCC {655}.
Gene mutationsThe International Collaborative Group onHNPCC maintains a database forHNPCC-associated mutations and poly-morphisms (http://www.nfdht.nl). To date(May 2002), there are 155 different MSH2mutations (comprising 39% of all muta-tions) and 200 (50%) MLH1 mutationsreported to the database, together with30 (8%), 5 (1%) and 10 (3%) mutations inMSH6, PMS2, and MLH3, respectively.Most MSH2 and MLH1 mutations aretruncating {2214}. However, 30-40% ofMLH1 and MSH6 mutations are of themissense type (leading only to an aminoacid substitution), which constitutes adiagnostic problem concerning theirpathogenicity. Besides commonly usedtheoretical predictions (evolutionary con-servation status of the amino acid, con-servativeness of the amino acid change,occurrence of the variant in the normalpopulation, co-segregation with diseasephenotype) functional tests may be nec-
359Hereditary non-polyposis colon cancer (HNPCC)
Table 8.10Extracolonic cancer in 144 HNPCC families knownat the Dutch HNPCC Registry.
Cancer Number Mean Range site age (yrs) (yrs)
Endometrium 87 49 24-78
Stomach 26 51 23-82
Ureter/pyelum 24 55 37-72
Small bowel 22 51 25-69
Ovarian cancer 28 48 19-75
Brain 18 42 2-78
Table 8.11Characteristics of HNPCC-associated human DNA mismatch repair genes.
Gene Chromosomal Length of Number Genomic Referenceslocation cDNA (kb) of exons size (kb)
MSH2 2p21 2.8 16 73 {1495,16802213,2563}
MLH1 3p21-p23 2.3 19 58-100 {353,1121,14941666,1679,2167}
PMS2 7p22 2.6 15 16 {2008,2010}
MSH6 2p21 4.2 10 20 {30,2009. 2163,2563}
MLH3 14q24.3 4.3 12 37 {1674}

essary in the evaluation of the patho-genicity of missense changes.
Microsatellite instability Microsatellite instability (MSI) is the hall-mark of tumours that arise in carriers ofM L H 1, M S H 2, of M S H 6 m u t a t i o n s .Overall, MSI is detected in approximate-ly 15% of all colorectal cancers. It ismeasured as alterations in the length ofsimple repetitive genomic sequences,usually dinucleotide repeats, or mononu-cleotide runs. As these repeats have atendency to form mismatches duringDNA replication, a mismatch re p a i rdefect is expected to increase theirmutation frequency. Because the defini-tion of instability applied has been vari-able, in 1998 an international workingg roup recommended the use of fivemarkers to assess MSI {306}. Tumoursa re characterized as having high-fre-quency MSI (MSI-H) if two or more of thefive markers show instability (i.e. haveinsertion/deletion mutations), or as hav-ing low-frequency MSI (MSI-L) if only oneof the five markers shows instability. Thedistinction between microsatellite stable(MSS) and low frequency MSI (MSI-L)can only be accomplished if a greaternumber of markers is utilized. MSI analy-sis, in conjunction with immunohisto-chemistry, can greatly improve the effica-cy of the molecular screening for HNPCC{650,2516}.In one study, all 12 endometrial carcino-mas from carriers of MLH1 and MSH2g e rmline mutations demonstrated anMSI-high phenotype involving all types ofrepeat markers, while this was found inonly 4 out of 11 (36%) endometrial carci -nomas from M S H 6 mutation carriers{650}. In another study, MSI-patterns inendometrial cancers differed from thosein colorectal cancers, even though thepatients had identical pre d i s p o s i n gmutations in the MMR genes MLH1 orMSH2 {1527}. In endometrial cancers,the pattern was more heterogeneous andinvolved a lower proportion of unstablemarkers per tumour and shorter allelicshifts for BAT markers. These resultsmight point to gene-specific and/ororgan-specific differences that may beimportant determinants of the HNPCCtumour spectrum.
Mutation spectrum Hereditary non polyposis colorectal can-cer (HNPCC) is caused by germ l i n e
mutations in one of 5 DNA mismatchrepair genes (MMR): MSH2 {864}, MLH1{353}, PMS1 {2010}, PMS2 {2010}, andMSH6 (formerly GTBP) {53,1884}. Othergenes like E X O 1 {3161}, M L H 3{1674,3162} and TGFbRII {1705} havebeen re p o rted to possibly causeHNPCC-like syndromes, although nodefinitive evidence has been deliveredyet, both in terms of pathogenicity and/orcosegregation with the disease of thealleged germline mutations in affectedfamilies. To date, more than 300 different predis-posing mutations have been identified,most in MSH2 and MLH1 and in familiescomplying with the clinical Amsterdamcriteria (AMS+) {2214}. Many HNPCCfamilies, however, do not fully complywith these criteria, and in most of thesecases the disease-causing mutations areyet unknown. Mutations in MSH6 havebeen found in atypical HNPCC families(see below).In general, MMR mutations are scatteredalong the coding sequence of MSH2 andMLH1 and predict either the truncation ofthe corresponding protein products, or asubtler amino acid substitutions. Thesemutations appear evenly distributedthroughout the coding regions of themain MMR genes, with some clusteringin MSH2 exon 12 {2214} and MLH1 exon16 {3115}. While most of the MSH2 muta-tions consist of frameshift or nonsensechanges, MLH1 is mainly affected byframeshift or missense alterations. Mostof the mutations found to date areunique, with a few common recurringones {2214}. Genomic deletions havealso been found at both loci {442,2070,3116}. MSH2 deletions appear to be avery frequent cause of HNPCC, con-tributing for up to a quarter of the familiesselected by Amsterdam criteria {3116}.MLH1 deletions are less frequent than inM S H 2 {1793,2070}. Southern analysisand/or other PCR-based methods todetect larger rearrangements at thegenomic level {443} should be routinelyemployed when approaching the muta-tion analyses of these major mismatchrepair genes.
Genotype-phenotype correlationsThe combination of clinical (number andtype of tumours, age of onset, clinicalcourse of the disease, etc.) and genetic(different mismatch repair genes, trun-cating and missense mutations) hetero-
geneity in HNPCC represents an idealopportunity to attempt the establishmentof genotype-phenotype corre l a t i o n s .Unfortunately, and notwithstanding thelarge number of mutations and clinicaldata collected to date, no clear-cut cor-relations have been observed betweenspecific MMR gene mutations and theirclinical outcome. For example, the iden-tification of identical mutations both inHNPCC and in Muir-Torre or Turcot syn-drome does not support the existence ofconsistent genotype-phenotype correla-tions {179,1115,1494}. The most reliable correlation found todate is the association between clear-cutpathogenic mutations at MSH2, MLH1and MSH6, and the resulting spectrum ofc o l o rectal and extracolonic tumours.HNPCC kindreds due to MSH2 or MLH1germline mutations are characterized byhigh penetrance and early onset of col-o rectal and endometrial cancer. Thediagnostic criteria, Amsterdam I and II,established by the Intern a t i o n a lCollaborative Group on HNPCC {3008,3010} well serve the purpose of selectingfamilies with a high likelihood to carryMSH2 and MLH1 mutations {3117}. Inaddition to the fulfillment of the above cri-teria, other factors represent valid pre-dictors of the presence of germ l i n eMSH2 and MLH1 mutations in HNPCCfamilies. These include 1. young age atdiagnosis of colorectal cancer, and 2. theoccurrence of at least one patient with anextra-colonic cancer, such as those ofthe endometrium, small intestine, brain,and stomach, within an AMS+ HNPCCk i n d red. The frequency of mutationsidentified in these families increased toabout 70% {3117}. Moreover, the occur-rence of at least one patient with multiplesynchronous or metachronous colorectalcancers, and the combined occurrenceof colorectal cancer with endometrialcancer in one patient are very good pre-dictors of M S H 2 or M L H 1 m u t a t i o n s{3117}.The first reports on MSH6 germline muta-tions already indicated that the clinicalphenotype differed from the "classical"HNPCC caused by MSH2 and MLH1mutations {53,1884}. More re c e n t l y,M S H 6 g e rmline mutations have beendemonstrated in a considerable numberof the atypical HNPCC families, i.e. notcomplying with the Amsterdam criteria(ACI and II) {1497,3039,3114,3160}. Ingeneral, the penetrance of colore c t a l
360 Inherited tumour syndromes

cancer seemed to be reduced whileendometrial cancer seems to represent am o re important clinical manifestationamong female MSH6 mutation carriers.Also, the mean age of onset of colorectaland endometrial cancer appeared to bedelayed in families with MSH6 germlinemutations {3011,3039,3114}. Notably,
MSI analysis of tumours from M S H 6mutation carriers suggests a reducedpenetrance of the MSI-H phenotype andpreferential instability at mononucleotiderepeats {650,1497,3114,3160}.An additional MSH6-associated clinicalphenotype is the papillary transitionalcell carcinoma of the ureter and renal
pelvis, observed in approx. 10% of thecarriers from an extended MSH6 kindred{3039}. Notably, the lifetime cumulativerisk of this tumour type in MLH1 or MSH2mutation carriers is only 2.6% {2673}.
361Hereditary non-polyposis colon cancer (HNPCC)
DefinitionAtaxia telangiectasia syndrome (A-T) is arare, progressive neurological disorderthat manifests at the toddler stage. Thedisease is characterized by cerebellardegeneration (ataxia), dilated blood ves-sels in the eyes and skin (telangiectasia),immunodeficiency, chromosomal insta-b i l i t y, increased sensitivity to ionizingradiation and a predisposition to cancer,in particular leukaemias and lymphomas.Germline mutations in the ATM gene(ataxia telangiectasia mutated), homozy-gous or compound heterozygous, arethe cause of this autosomal recessivedisorder. Heterozygous carriers are phe-notypically unaffected but exhibit anincreased risk to develop breast cancerand often display a variety of age relateddisorders which may result in reducedlife expectancy {2808}.
MIM No. 208900 {1835}
Synonyms Louis-Bar Syndrome, A-T complementa-tion group A (ATA), group C (ATC), groupD (ATD) and group E (ATE). The differentcomplementation groups are all linked tothe ATM gene.
Incidence The rare A-T disease occurs in both gen-ders and world wide among all races. Thedisease has an estimated incidence of oneper 40,000 to one per 300,000 live birt h s .
A p p roximately 0.2-1% of the general pop-ulation has been estimated to be hetero z y-gous carriers of a type of germline muta-tion in the AT M gene that in homozygousstate causes the A-T syndro m e .
Tumours in A-T patients Individuals with A-T have a 50 to 150 foldexcess risk of cancer, with approximate-ly 70% being lymphomas and T cellleukaemias. In younger patients, anacute lymphoblastic leukaemia is mostoften of T-cell origin, although the pre-Bcommon ALL of childhood has also beenseen in A-T patients. When leukaemiadevelops in older A-T patients it is usual-ly an aggressive T-cell leukaemia (T-PLL,T cell prolymphocytic leukaemia).Lymphomas are usually B cell types. Awide range of solid tumours makes upthe remainder of the tumours seen in A-Tpatients and includes cancers of thebreast, stomach, ovary and melanoma. The presence of missense mutations inA-T patients has been associated with amilder clinical phenotype and alteredcancer predisposition. In two British A-Tfamilies a T>G tranversion at base pair7271 was found to be associated with amilder clinical phenotype, lowerradiosensitivity but an increased risk ofbreast cancer. This increased risk wasobserved in both the homozygote andheterozygotes carriers of this modifica-tion (RR 12.7 p=0.0025) {2775}. Thissequence alteration has subsequently
been found in multiple-case breast can-cer families. The expression and activityanalyses of the ATM protein in heterozy-gous cell lines carrying this sequencechange indicated that this mutation wasdominant negative {462}.
Breast cancer in ATM heterozygotes Heterozygous carriers of ATM mutationshave a higher mortality rate and an earli -er age at death from cancer andischemic heart disease than non-carriers{2808}. A-T heterozygotes have beenreported to have a 3 to 8 fold increasedrisk of breast cancer. The associationbetween ATM heterozygosity and breastcancer risk was initially found amongblood relatives of A-T patients {2820},and in almost every study of A-T relativessince an increased breast cancer riskhas been detected {318,741,981,1291,1334,2105,2257,2819}. Paradoxically, inthe years following the cloning of theATM gene {2546}, several studies inves-tigating large breast cancer cohort sfailed to find an increased incidence ofATM mutations of the type found in A-Tpatients, and a controversy aro s eregarding the role of ATM in breast can -cer susceptibility {194,281,884}.However, a number of recent studies,analysing the frequency of all type ofAT M mutations, did confirm pre v i o u sfindings of an elevated breast cancerrisk in ATM mutation carriers {129,351,462,2592,2775}.
A. Broeks L.J. van’t Veer A.L. Borresen-DaleJ. Hall
Ataxia telangiectasia syndrome

Age distribution and penetranceMost of the studies finding an incre a s e drisk of breast cancer in A-T relatives pointto an early onset of the disease. The pen-etrance has been difficult to estimatesince most of the studies are small, andd i ff e rent mutations may have diff e re n te ffects. In several studies of A-T re l a t i v e s ,the elevated risk is restricted to obligatecarriers (mothers), and is not increased inother relatives according to their pro b a-bility of being a mutation carrier. This maypoint to an interaction with enviro n m e n t a land/or other genetic factors contributingto the elevated breast cancer risk.
Clinical and pathological features No typical clinical or pathological fea-tures are so far known for ATM heterozy-gous breast cancer patients, other thanearly age at onset (before age 50) andfrequent bilateral occurrence {351}.
Response to therapy and prognosisATM heterozygotes with breast cancerdo not seem to exhibit acute radiationsensitivity as A-T patients do, and exces-sive toxicity has not been observed afterradiotherapy {115,2331,3088}. It hashowever been speculated whether ATMh e t e rozygous breast cancer patientshave an increased risk of developing asecond breast cancer after radiationtreatment, and large multi-center studiesare ongoing to answer this question.There are only few studies evaluating theprognosis of A-T carriers with breastcancer, pointing to a long-term survival.This may be due to their tumours beingmore susceptible to cell killing by ioniz-ing radiation than tumour cells in non-carriers {2809}.
ATM expression in breast cancerNormal breast tissue shows a distinctpattern of ATM expression, the proteinbeing found in the nucleus of the ductalepithelial cells and to a lesser extent inthe surrounding myoepithelial cells.D e c reased ATM expression is oftenobserved in breast carcinomas {102,1384} and ATM mRNA levels have alsobeen found to be lower in invasive breastcarcinomas than in normal tissues orbenign lesions {3041}.Significant loss of heterozygosity in spo-radic breast tumours across chro m o-some 11q22-23 where the ATM gene islocated has been reported {1118,1439,1553,1754,2375}.
GeneticsChromosomal locationThe ATM gene is located on human chro-mosome 11q22-23.
Gene structureThe ATM gene has 66 exons scanning150 kilobase of genomic DNA and isexpressed in a wide range of tissues asan approximately 12-kilobase messen-ger RNA encoding a 350 kD serine/thre-onine protein kinase. The initiation codonfalls within exon 4. The last exon is 3.8kband contains the stop codon and a 3’-untranslated region of about 3600nucleotides {2983}.
Gene expressionThe major 13 kb AT M transcript isobserved in every tissue tested to date.N o rt h e rn blots and RT-PCR pro d u c t sfrom various tissues failed to discloseany evidence of alternative forms withinthe coding region. However the first fourexons, which fall within the 5’-untranslat-ed region (UTR), undergo extensivea l t e rnative splicing. Diff e rential poly-adenylation results in 3’UTRs of varyinglengths. These structural features sug-gest that ATM expression might be sub-ject to complex post-transcriptional regu-lation {2547}.
Gene functionThe ATM protein plays a central role insensing and signalling the presence ofDNA double-strand breaks (DSBs)f o rmed in cells as a result of normal DNAmetabolism (e.g. meiotic or V(D)J re c o m-bination) or damage caused by extern a lagents. The kinase domain in the car-b o x y - t e rminal region of the protein con-tains the signature motifs of phos-
phatidylinositol 3-kinases. AT M ’s kinaseactivity is itself enhanced in response toDNA double-strand breaks resulting in ap h o s p h o rylation cascade activatingmany proteins each of which in turna ffects a specific signalling pathway.These substrates include the pro t e i np roducts of a number of well character-ized tumour- s u p p ressor genes includingT P 5 3, B R C A 1 and C H E K 2 which playi m p o rtant roles in triggering cell cyclea r rest, DNA repair or apoptosis (re v i e w e din Shiloh et al. {2660}). Additional DSB-induced responses that are ATM depend-ent include the activation of transcriptionfactors such as AP-1, p73 and NFKB, anddeacetylation of chromatin pro t e i n s( reviewed in Barzilai et al. {189}).
Mutation spectrum in A-T patientsSince the AT M gene was cloned {2546}m o re than 300 diff e rent A-T disease-causing mutations have been re p o rt e d .The profile of these has revealed thatmost are unique and uniformly distributedalong the length of the gene, no muta-tional hotspots have been detected. Themajority of A-T patients are compoundh e t e rozygotes having two diff e rent AT Mmutations and patients homozygous forthe same AT M mutation are rare. The pre-dominate type of mutation found in theAT M gene in A-T patients results in a trun-cated and unstable ATM protein. Some A-T patients have a milder phenotype (vari-ant A-T) that may be related to the pre s-ence of missense mutations or mutationsp roducing an ATM protein retaining somen o rmal function {1825,2592}.
Genotype-phenotype correlationsGatti et al. {971}, in distinguishingbetween truncating mutations where no
362 Inherited tumour syndromes
Table 8.12Proposed ATM genotype / phenotype relationships {971}.
Genotype Phenotype
ATM wt/wt Normal
ATM trun/trun Ataxia telangiectasia, High cancer risk
ATM trun/mis Ataxia telangiectasia, Variant A-T?, High cancer risk?
ATM mis/mis Ataxia telangiectasia? Variant A-T?, High cancer risk?
ATM trun/wt A-T relatives, Elevated breast cancer risk, Increased age related disorders?
ATM mis/wt Few A-T relatives, Moderate breast cancer risk? Increased age related disorders?

363Ataxia telangiectasia syndrome
ATM protein is detected and missensesubstitutions where mutant protein ofvariable stability is observed, have sug-gested that this mutant protein could pro-duce a dominant negative effect in het-erozygotes, resulting in an altered phe-notype and an increased breast cancersusceptibility. The expected phenotypesthat might arise from having two types ofA-T carriers in the general population areshown in Table 8.12. The genotype ATM trun/trun with two truncat-ing mutations causes the classical A-Tdisorder. The genotype ATMmis/mis with two
missense mutations is also found insome children with the classical form ofthe disease, in particular when these arelocated within the ATM kinase domain(for instance Belzen et al. {2987}, Angeleet al. {101}) but may also be associatedwith a variant A-T phenotype with someneurological features and cancer sus-ceptibility. Two types of ATM heterozy-gotes exist and the phenotypes differ, i.e.those with truncating mutations thatmake no protein and those with missensemutations that make reduced amount orp a rtly defective protein {115,462,971,
1825,2331,2592,2775,2809,3088}, andthese two groups may have differentb reast cancer risks. If this pro p o s e dmodel is correct it necessitates a re-analyses of the epidemiological datastratifying for the two types of heterozy-gotes. The literature to date suggeststhat germline ATM missense mutationsare more frequent than the 0.2-1% fre-quency of A-T causing mutations andhence contribute to a larger fraction ofbreast cancer patients {351,462,2592}.




























