-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
1/25
Review
Immune and inflammatory mechanisms in neuropathic pain
Gila Moalem, David J. Tracey
School of Medical Sciences, University of New South Wales,
Sydney, NSW 2052, Australia
A R T I C L E I N F O A B S T R A C T
Article history:
Accepted 17 November 2005
Available online 4 January 2006
Tissue damage, inflammation or injury of the nervous system may
result in chronic
neuropathic pain characterised by increased sensitivity to
painful stimuli (hyperalgesia), the
perception of innocuous stimuli as painful (allodynia) and
spontaneous pain. Neuropathic
pain has been described in about 1% of the US population, is
often severely debilitating and
largely resistant to treatment. Animal models of peripheral
neuropathic pain are now
available in which the mechanisms underlying hyperalgesia and
allodynia due to nerve
injury or nerve inflammation can be analysed. Recently, it has
become clear that
inflammatory and immune mechanisms both in the periphery and the
central nervous
system play an important role in neuropathic pain.
Infiltrationof inflammatory cells, as well
as activation of resident immune cells in response to nervous
system damage, leads to
subsequent production and secretion of various inflammatory
mediators. These mediators
promote neuroimmune activation and can sensitise primary
afferent neurones and
contribute to pain hypersensitivity. Inflammatory cells such as
mast cells, neutrophils,macrophages and T lymphocytes have all been
implicated, as have immune-like glial cells
such as microglia and astrocytes. In addition, the immune
response plays an important role
in demyelinating neuropathies such as multiple sclerosis (MS),
in which pain is a common
symptom, and an animal model of MS-related pain has recently
been demonstrated. Here,
we will briefly review some of the milestones in research that
have led to an increased
awareness of the contribution of immune and inflammatory systems
to neuropathic pain
and then review in more detail the role of immune cells and
inflammatory mediators.
2005 Elsevier B.V. All rights reserved.
Keywords:
Neuropathic pain
Nerve injury
Immunity
Inflammation
Abbreviations:
5HT, 5-hydroxytryptamine,
serotonin8R, 15S-diHETE, (8R, 15S)-
dihydroxyeicosa-(5E-9,11,13Z)-
tetraenoic acid
ADP, adenosine diphosphate
AMP, adenosine monophosphate
ATP, adenosine triphosphate
BDNF, brain-derived neurotrophic
factor
CCR2, chemokine (CC motif)
receptor 2
CGRP, calcitonin-gene-related
peptide
COX-1, cyclooxygenase-1COX-2, cyclooxygenase-2
CX3CR1, G-protein-coupled
chemokine receptor for fractalkine
DRG, dorsal root ganglion
GABA, gamma aminobutyric acid
GBS, GuillainBarr syndrome
GDNF, glial-cell-line-derived
neurotrophic factor
B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4 0 2
6 4
Corresponding author. Fax: +61 2 9385 8016.E-mail address:
[email protected] (D.J. Tracey).
0165-0173/$ see front matter 2005 Elsevier B.V. All rights
reserved.doi:10.1016/j.brainresrev.2005.11.004
a v a i l a b l e a t w w w . s c i e n c e d i r e c t . c o
m
w w w . e l s e v i e r . c o m / l o c a t e / b r a i n r e s
r e v
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
2/25
IL-1, interleukin-1
L-NAME, NG-nitro-L-arginine methyl
ester
LTB4, leukotriene B4MCP-1, monocyte chemoattractant
protein-1
MHC, major histocompatibility
complex
MIP-1, macrophage inflammatory
protein-1
MRP-14, cytosolic-calcium-binding
protein of 14 kDa
MS, multiple sclerosis
NGF, nerve growth factor
NO, nitric oxide
NOS, nitric oxide synthase
NSAID, non-steroidal
anti-inflammatory drug
NT-3, Neurotrophin-3
NT-4/5, Neurotrophin-4/5
P2X, ATP-gated ion channels
P2Y, G-protein-coupled ion channels
activated by purine/pyrimidine
nucleotides
PAR-2, protease-activated receptor-2
PGE2, Prostaglandin E2PGI2, Prostaglandin I2TCA, tricyclic
antidepressant
TEMPOL, 4-hydroxy-2,2,6,6-
tetramethylpiperidine-1-oxyl
Th1 cells, T helper 1 cells
Th2 cells, T helper 2 cells
TLR4, Toll-like receptor 4
TNF, tumour necrosis factor
TrkA, tyrosine kinase receptor A
TrkB, tyrosine kinase receptor B
TrkC, tyrosine kinase receptor C
TRPV1, transient receptor potential
(receptor) vanilloid 1
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
242
1.1. Chemical environment of sensory neurones contributes to
neuropathic pain . . . . . . . . . . . . . . . . . 2421.2.
Inflammatory models of neuropathic pain. . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . 243
1.3. Neuropathic pain in autoimmune diseases of the nervous
system . . . . . . . . . . . . . . . . . . . . . . . 243
1.4. Direct and indirect actions of mediators released by immune
cells . . . . . . . . . . . . . . . . . . . . . . . 243
2. Immune cells . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
244
2.1. Mast cells . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
244
2.2. Neutrophils . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 245
2.3. Macrophages . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 247
2.4. Lymphocytes . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 247
2.5. Glia and Schwann cells . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 248
3. Mediators . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
249
3.1. Bradykinin . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 249
3.2. ATP and adenosine. . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 249
3.3. Serotonin . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 250
241B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4 0
2 6 4
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
3/25
3.4. Eicosanoids. . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
250
3.5. Cytokines. . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
251
3.5.1. Interleukin-1 . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . 2 52
3.5.2. Interleukin-6 . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . 2 52
3.5.3. Tumour necrosis factor . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . 252
3.5.4. Interleukin-10 . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . 2 53
3.6. Neurotrophins . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 253
3.7. Nitric oxide and reactive oxygen species . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2544.
Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 255
References. . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. 255
1. Introduction
Neuropathic pain is caused by lesion or inflammation of
the nervous system and is relatively common, with an
incidence estimated at 0.6% to 1.5% in the US population
(Warfield and Fausett, 2002). It is often severely
debilitating
and largely resistant to treatment (Harden and Cohen,
2003) mainly because the underlying mechanisms are still
poorly understood. Symptoms of neuropathic pain may
include allodynia (pain resulting from a stimulus that is
normally non-painful), hyperalgesia (an excessive response
to painful stimuli) and spontaneous pain. The study of
neuropathic pain can be traced back to Weir Mitchell and
his classic work on nerve injuries from the American Civil
War (Mitchell, 1872). Since that time, a great deal has been
written on neuropathic pain and its possible causes, but it
was only with the development of animals models of pain
due to nerve injury that some real progress was made in
understanding some of the mechanisms involved. For
recent reviews of these mechanisms, see Julius and
Basbaum (2001) and Scholz and Woolf (2002). The first
widely used animal model of neuropathic pain was chronic
constriction injury of the rat sciatic nerve (Bennett and
Xie,
1988), in which the sciatic nerve is encircled by four
ligatures of chromic gut. This leads to the cardinal
symptoms of neuropathic painhyperalgesia, allodynia
and apparent spontaneous pain. Other widely used animal
models include partial ligation of the sciatic nerve
(Seltzer
et al., 1990) and section of one or more of the spinal
nerves which contribute to the sciatic (Kim and Chung,
1992). Research on these animal models of peripheral
neuropathic pain (the pain syndrome resulting from
lesions of the peripheral nervous system) has made it
clear that a number of mechanisms are involved, including
ectopic excitability of sensory neurones, altered gene
expression of sensory neurones and sensitisation of
neurones in the dorsal horn of the spinal cord (Scholz
and Woolf, 2002; Woolf, 2004). However, there is increasing
evidence that inflammatory and immune mechanisms also
play a role, and we will begin by looking at a brief history
of how this evidence has accumulated. We will then look
in more detail at the evidence for involvement of immune
cells and inflammatory mediators in neuropathic pain.
However, the review does not provide exhaustive coverage
of all immune and inflammatory mechanisms that con-
tribute to pain following nerve injury. For readers who
would like to know more about these and other aspects of
neuropathic pain, several recent reviews are available,
dealing with immune and glial mechanisms (Watkins and
Maier, 2002, 2005; DeLeo et al., 2004; McMahon et al., 2005;
Tsuda et al., 2005), inflammatory aspects (Woolf and
Costigan, 1999; DeLeo and Yezierski, 2001; DeLeo et al.,
2004; Sommer and Kress, 2004) and cytokines (Sommer andKress,
2004).
1.1. Chemical environment of sensory neurones
contributes to neuropathic pain
At the time that the first animal models of pain due to
nerve injury were developed, it was widely believed that
injury or loss of myelinated and unmyelinated sensory
axons in the sciatic nerve was the key factor in producing
symptoms of neuropathic pain. However, some studies in
the early 1990s suggested that other factors might also be
involved. For example, Maves showed that chromic gut
alone placed next to the sciatic nerve induced thermal
hyperalgesia without any loss of myelinated axons (Maves
et al., 1993). Of course, it had long been known that C-
polymodal nociceptors are sensitised or activated by
chemical stimuli, including inflammatory mediators (Wood
and Docherty, 1997), but these effects were thought of as
being responsible for inflammatory pain, and little atten-
tion was given to the idea that immune or inflammatory
cells or their mediators might play a role in neuropathic
pain until the advent of useful animal models. It was also
known that nerve injury resulted in activation of mast cells
(Olsson, 1967) and recruitment of neutrophils and macro-
phages (Perry et al., 1987). However, it was not until 1993
that acute changes in the endoneurial microenvironment
were explicitly linked with the initial development of
hyperalgesia (Frisen et al., 1993; Sommer et al., 1993).
Further work confirmed suggestions that the factors
responsible for neuropathic hyperalgesia included Wallerian
degeneration and macrophage activation (Sommer et al.,
1995). At around the same time, Clatworthy followed up the
observations of Maves et al. and showed that suppression
of the inflammatory response in the injured sciatic nerve
blocked the development of hyperalgesia, while enhancing
the inflammatory response augmented the hyperalgesia
(Clatworthy et al., 1995). Evidence on the involvement of
immune cells and inflammatory mediators in hyperalgesia
was reviewed at this time, and it was suggested that these
242 B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4
0 2 6 4
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
4/25
mediators (and cytokines in particular) were a probable link
between the activation or recruitment of immune cells to
the injured nerve and the development of hyperalgesia and
neuropathic pain (Tracey and Walker, 1995; Watkins et al.,
1995). Glial cells supporting neurones in the spinal cord
(Meller et al., 1994) and Schwann cells supporting axons in
the peripheral nerve (Constable et al., 1994; Wagner and
Myers, 1996a) were added to the list of cell types whichmight
contribute to neuropathic pain. In fact, glial cells and
Schwann cells may also play an active role in the immune
process by releasing mediators, including cytokines, and
acting as antigen-presenting cells following nerve injury
(Argall et al., 1992; Bergsteinsdottir et al., 1992; Constable
et
al., 1994). These ideas were not widely accepted at the
time, but, gradually, evidence accumulated to give them
further support. For example, tumour necrosis factor (TNF)
and interleukins 1 and 6 (IL-1, IL-6) had already been
shown to induce acute or short-term hyperalgesia (Ferreira
et al., 1988; Cunha et al., 1992) but were now implicated
directly in neuropathic pain, involving chronic hyperalgesia
and allodynia (Wagner and Myers, 1996b; DeLeo et al., 1997;
Arruda et al., 1998).
Another example is the contribution of nerve growth
factor (NGF), which shares many attributes with typical
cytokines (Bonini et al., 2003). NGF is best known for its
role in protecting some neuronal types from cell death
during development, but Levine and his colleagues showed
that the N-terminal octapeptide of NGF elicited hyperalge-
sia in the rat when applied to injured tissue (Taiwo et al.,
1991). These experiments were provoked by apparent
similarities between the NGF fragment and bradykininan
inflammatory mediator known to activate or sensitise
nociceptors and to elicit hyperalgesia (Dray and Perkins,
1993; Dray, 1997). By this stage, it was known that the
level
of NGF rises substantially in inflamed tissue and in injured
nerve (Heumann et al., 1987; Weskamp and Otten, 1987),
and a causal relationship between tissue levels of NGF and
inflammatory hyperalgesia was confirmed by showing that
anti-NGF antibodies reduced inflammatory hyperalgesia
(Lewin et al., 1994; Woolf et al., 1994). NGF was later
shown to contribute to neuropathic hyperalgesia as well
(Herzberg et al., 1997; Theodosiou et al., 1999). The
underlying mechanism is not clear, but it is agreed that
NGF has a cytokine-like action on inflammatory cells
(Otten, 1991) including mast cells, basophils, neutrophils
and lymphocytes. Furthermore, NGF is produced and
released by leukocytes such as macrophages and T
lymphocytes (Vega et al., 2003). NGF is a potent degranu-
lator of mast cells (Pearce and Thompson, 1986), which
appear to play an important role in inflammatory (Woolf et
al., 1996) as well as neuropathic hyperalgesia (Zuo et al.,
2003). It seems likely that mediators released by activated
mast cells contribute to hyperalgesia following nerve injury
histamine has been strongly implicated (Zuo et al., 2003),
but several other mediators may also be involved.
1.2. Inflammatory models of neuropathic pain
It is now apparent that early animal models of neuropathic
pain suffer from the disadvantage that symptoms such as
hyperalgesia are most likely the combined result of nerve
injury itself (e.g. ectopic firing of injured sensory axons)
and
the release of algesic mediators by immune cells activated
near the site of injury. In fact, neuropathic pain may
develop
without any injury of sensory axons. This was shown in rats
by induction of neuropathic hyperalgesia by placing segments
of chromic gut next to the sciatic nerve (Maves et al., 1993)
or
by lesion of the ventral roots of spinal nerves (Li et al.,
2002;Sheth et al., 2002). In both cases, typical signs of
neuropathic
pain were elicited without damage to sensory axons, implying
that sensitisation or activation of sensory fibres by
inflam-
matory mediators is sufficient to cause neuropathic pain. As
a
result of observations like these, inflammatory models of
neuropathic pain have been developed in which carrageenan
or complete Freund's adjuvant (Eliav et al., 1999) or
zymosan
(Chacur et al., 2001) is applied around the intact sciatic
nerve
of the rat. These models should help us to understand the
role
of inflammatory and immune mechanisms in neuropathic
pain.
1.3. Neuropathic pain in autoimmune diseases of thenervous
system
Autoimmune diseases of the nervous system including
multiple sclerosis (MS) and GuillainBarr syndrome (GBS)
are among the most common disabling neurologic diseases,
which cause not only demyelination with impaired motor
function, but also abnormal sensory phenomena including
chronic neuropathic pain. MS is an autoimmune demyelinat-
ing disease of the central nervous system that causes
relapsing and chronic neurological impairment. Pain is
experienced by approximately 65% of patients with MS at
some time during the course of their disease (Kerns et al.,
2002). GBS is an acute inflammatory demyelinating neuropa-
thy caused by an autoimmune attack on the peripheral
nervous system and characterised by motor disorders such
as weakness or paralysis and variable sensory disturbances
(Hughes et al., 1999). Pain is a common symptom of GBS
occurring in 7090% of cases (Pentland and Donald, 1994;
Moulin et al., 1997). Immune cells including macrophages and
T cells are believed to play a critical role in the pathogenesis
of
both MS and GBS and may contribute to the related pain.
Studying neuropathic pain in these autoimmune syndromes
has been hampered by the lack of proper animal models for
autoimmunity-related pain. However, recent study in mice
has shown that transient demyelination of peripheral affer-
ents without axonal loss results in neuropathic pain behav-
iour (Wallace et al., 2003), and a suitable animal model of
MS-
related pain has recently been developed using both active
and passive induction of experimental autoimmune enceph-
alomyelitis (Aicher et al., 2004).
1.4. Direct and indirect actions of mediators released by
immune cells
While there is good evidence that inflammatory mediators
contribute to neuropathic pain, it is not clear just how or
where these mediators act. Do they act directly on receptors
located on the cell membrane of the neurone or do they
activate non-neuronal cells (such as mast cells or Schwann
243B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4 0
2 6 4
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
5/25
cells), which then release a secondary mediator that exerts
a
direct action on neuronal receptors? Are the neuronal
receptors for chemical mediators located on the nerve
terminals or cell bodies of sensory neurones, even though
these may be many centimetres from the lesion site? The
answer to the first question is that both direct and
indirect
mechanisms are likely to operate. Some mediators (such as
prostaglandin E2) probably sensitise nociceptors directly
byacting on receptors on the neuronal cell membrane (Pitchford
and Levine, 1991). While IL-1 may also have a direct action
on
nociceptors (Sommer and Kress, 2004), it has additional
indirect actions by eliciting the production of
prostaglandins
(Cunha et al., 1992) or even more indirectly via neural
pathways from vagal afferents to the brainstem and then to
the spinal cord (Watkins et al., 1995). Theanswer to
thesecond
question is that receptors for inflammatory mediators may be
located not only on the neuronal cell body and peripheral
nerve terminals, but also on the axon itself.
Recent work by Grafe and his colleagues makes it clear
that some of the neuronal receptors are located on the axon
itself (Irnich et al., 2002; Lang et al., 2003; Moalem et
al.,
2005). This makes it easier to understand how mediators
released by immune cells clustered around the nerve lesion
can influence the excitability of sensory neurones since
these mediators would be released close to axonal receptors
and would not need to diffuse distally to nerve terminals or
proximally to cell bodies. For example, it was shown that
ATP increased the excitability of unmyelinated C-fibres in
an
in vitro preparation of an isolated segment of the sural
nerve, in which no cell bodies or nerve terminals were
present. Tissue levels of ATP are raised in inflamed tissue,
and ATP activates nociceptors causing pain and hyperalgesia
(Sawynok and Liu, 2003); it also plays a role in neuropathic
pain (Barclay et al., 2002; Jarvis et al., 2002). It may well
be
that ATP contributes to neuropathic pain by its action on
axonal receptors.
2. Immune cells
Several inflammatory and immune-like glial cells have been
implicated in the pathogenesis of neuropathic pain. They
include mast cells, neutrophils, macrophages and T cells in
the peripheral nervous system (Fig. 1) and microglia (Fig.
2)
and astrocytes in the central nervous system. Table 1
summarises immune cells and the mediators they release
with their most significant actions.
2.1. Mast cells
Mast cells are not only critical effector cells in allergic
disorders but are also important initiators and effectors of
innate immunity (Galli et al., 2005). There is a resident
population of mast cells in the peripheral nerve (Olsson,
1968), and these mast cells are degranulated at the site of
a
nerve lesion (Olsson, 1967; Zuo et al., 2003). The granules
contain mediators such as histamine, proteases and cytokines
(Metcalfe et al., 1997; Galli et al., 2005), several of which
are
capable of sensitising or activating neurones. For example,
histamine sensitises visceral nociceptors (Koda and Mizu-
mura, 2002). Although it elicits itch in normal skin,
histamine
application to the skin of patients with neuropathic pain
did
not evoke an itch but instead induced a severe increase in
spontaneous burning pain (Baron et al., 2001). Activation of
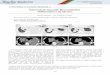
Fig. 1 Peripheral nerve injury induces activation of resident
immune cells as well as recruitment of inflammatory cells to
the
nerve. Injury of a peripheral nerveinitiates an inflammatory
cascade in whichmast cells residing in the nerve are the first to
be
activated. They release mediators such as histamine (hist) and
TNF, which sensitise nociceptors and contribute to the
recruitment of neutrophils and macrophages. Mediators released
by neutrophils (including the chemokine MIP-1 and the
cytokine IL-1) assist in recruitment of macrophages. Both
neutrophils and macrophages in the nerve produce and secrete
mediators such as TNF and PGE2 that can further sensitise
nociceptors. Nerve injury also initiates Schwann cell
de-differentiation and the release of several algesic mediators
such as pro-inflammatory cytokines, NGF, PGE2 and ATP. These
initial events promote the recruitment of T cells, which can
secrete a variety of cytokines depending on their subtype. This
cocktail of mediators serves as a mechanism of enhanced
inflammatory response in the injured nerve and contributes to
neuropathic pain.
244 B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4
0 2 6 4
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
6/25
mast cells may also cause secretion of mediators without
overt degranulation (Theoharides and Cochrane, 2004) by
synthesis of lipid mediators such as prostaglandin D2 or by
transcription, translation and secretion of a wide range of
cytokines and chemokines (Mekori and Metcalfe, 2000).
Activation and degranulation of mast cells therefore release
some mediators (such as serotonin in the rat) which can
elicit
hyperalgesia by a direct action on the nociceptor (Rueff and
Dray, 1993; Sommer, 2004). Some mast cell mediators exert a
broader range of actions by initiating an inflammatory
cascade (Fig. 1). For example, release of histamine, leuko-
trienes and chemokines contributes to recruitment of leuko-
cytes including neutrophils and macrophages (Gaboury et al.,
1995; Malaviya and Abraham, 2000). In fact, recent work in
our
laboratory suggests that degranulation of mast cells plays a
key role in the initiation of neuropathic pain since, in
rats
subjected to partial ligation of the sciatic nerve,
stabilisation of
mast cells with cromoglycate strongly suppressed the devel-
opment of neuropathic pain and reduced the numbers of
neutrophils and macrophages at the lesion site (Zuo et al.,
2003). Treatment of the nerve-injured rats with H1 and H2
histamine receptor antagonists also alleviated neuropathic
hyperalgesia, although to a lesser extent than stabilisation
with cromoglycate. This suggests that nerve injury induces
degranulation of mast cells and release of histamine, whichacts
on histamine receptors on endothelial cells to induce
recruitment of leukocytes such as neutrophils and macro-
phages(Yamaki et al., 1998). Both of these cell types
contribute
to neuropathic pain (Liu et al., 2000b; Perkins and Tracey,
2000). The question of how mast cells are activated by nerve
injury is still open, but it could be brought about by
increased
levels of adenosine (Sawynok et al., 2000) or bradykinin
(McLean et al., 2000). Other mast cell mediators are
probably
also involved in the initiation of neuropathic pain. One
example is tryptase, a trypsin-like serine protease that is
specific to mast cells. One of the most prominent mast cell
mediators, it is associated with the mast cell granules and
released by degranulation. Tryptase acts on the protease-
activated receptor-2 (PAR-2), which is known to be present
on
primary sensory neurones and to play a role in inflammation.
Recently, it has been shown to trigger inflammatory hyper-
algesia and nociceptive behaviour in rats (Kawabata et al.,
2001; Vergnolle et al., 2001).
2.2. Neutrophils
Neutrophils (or polymorphonuclear leukocytes) are an essen-
tial part of the innate immune system. Their cytoplasm
contains granules and secretory vesicles that can release a
wide range of microbicidal effector molecules including
bactericidal proteins and cytokines, proteinases and
reactive
oxygen species (Witko-Sarsat et al., 2000; Faurschou and
Borregaard, 2003). The first steps in migration towards an
inflammatory focus areadhesionto thesurface of thevascular
endothelium (mediated by selectins) followed by rolling and
transendothelial migration. Neutrophils then migrate towards
the inflammatory site up a gradient of chemoattractants.
These include formyl peptides released by bacteria or dying
cells and chemokines released by immune cells (Witko-Sarsat
et al., 2000). Neutrophils contribute to inflammatory hyper-
algesia (Levine et al., 1984, 1985; Bennett et al., 1998b)
by
release of the 15-lipoxygenase product 8R, 15S-diHETE
(Levine
et al., 1985; White et al., 1990); their release of cytokines
such
as TNF (Witko-Sarsat et al., 2000) may also contribute.
Neutrophils are not observed in normal nerves but are found
in significant numbers at the site of injury in injured
peripheral nerves (Perry et al., 1987; Clatworthy et al.,
1995;
Perkins and Tracey, 2000; Zuo et al., 2003). Work in our
laboratory showed that depletion of circulating neutrophils
at
the time of nerve injury significantly attenuated the
induction
of hyperalgesia. However, depletion of neutrophils at day
8 post-injury did not alleviate hyperalgesia after its
normal
induction since by this time neutrophil numbers at the site
of
nerve injury have declined to negligible levels(Zuo
etal.,2003).
It seems likely that nerve injury induces an inflammatory
cascade initiated by mast cell activation. Activated mast
cells
release mediators that promote migration of neutrophils from
Fig. 2 Peripheral nerve injury induces glial activation in
the
dorsal horn of the spinal cord. Injury to a peripheral nerve
initiates increased release of neurotransmitters such as
glutamate, substance P and possibly ATP from the central
terminals of primary afferents. These neurotransmitters can
activate both second order neurones and glia. For example,
ATP can activate glial cells through binding to their
P2X4receptors, and fractalkine released by second order
neurones
can activate glia through binding to their CX3CR1 receptors.
The TLR4 receptor is involved in glial activation, but the
nature of its specific ligand in the spinal cord is
unclear.These triggers cause production and release of
inflammatory
mediators including pro-inflammatory cytokines (such as
TNF and IL-1), glutamate (Glu), prostaglandins (PGs) and
nitric oxide (NO) from glial cells. These agents are then
capable of further enhancing glial activation and production
of inflammatory mediators that sensitise dorsal horn
neurones, thereby contributing to neuropathic pain.
245B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4 0
2 6 4
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
7/25
Table 1 Immune cells, mediators released and actions
Cell type Mediators Actions
Mast cells Cytokines and
chemokines (Galli
et al., 2005; Mekori
and Metcalfe, 2000)
Recruit leukocytes;
IL-1 facilitates the
release of CGRP from
nociceptors (Fukuoka
et al., 1994) and elicits
hyperalgesia (Ferreira
et al., 1988; Follenfant
et al., 1989; Sung et al.,
2004; Zelenka et al.,
2005); TNF sensitises
C-fibres ( Junger and
Sorkin, 2000; Sorkin
et al., 1997) and DRG
neurones (Schfers et
al., 2003a,b) and
produces hyperalgesia
(Cunha et al., 1992;
Schfers et al., 2003a,b;
Zelenka et al., 2005).
Chemokines produce
excitatory effects on
DRG neurons and
produce allodynia
(Oh et al., 2001)
Histamine
(Mekori and
Metcalfe, 2000)
Sensitises nociceptors
(Koda and Mizumura,
2002), recruits
leukocytes (Gaboury
et al., 1995; Yamaki
et al., 1998); enhances
neuropathic pain in
patients (Baron et al.,
2001)
Leukotriene B4
(Galli et al., 2005;
Mekori and
Metcalfe, 2000)
Recruits neutrophils
(Bennett et al.,
1998a,b; Levine et al.,
1984)
Mast cell protease
(e.g. tryptase)
(Metcalfe et al.,
1997)
Activates PAR-2
receptor (Kawabata
et al., 2001; Vergnolle
et al., 2001)
Nerve growth
factor (Bonini et
al., 2003)
Sensitises nociceptors
(Bennett et al.,
1998a,b; Bennett,
2001; Koltzenburg et
al., 1999a,b), indirectactions via immune
cells and sympathetic
neurones (Bennett et
al., 1998a,b; Lewin et
al., 1994; Woolf et al.,
1996)
Prostaglandins
(PGD2, PGE2) (Galli
et al., 2005; Mekori
and Metcalfe, 2000)
PGE2 sensitises
nociceptors (Taiwo
and Levine, 1989),
depolarises spinal
neurons (Baba et al.,
2001)
Table 1 (continued)
Cell type Mediators Actions
Serotonin (in
murine rodents)
(Galli et al., 2005)
Sensitises nociceptors
(Lang et al., 1990;
Rueff and Dray, 1993)
and sensory axons
(Moalem et al., 2005)
Neutrophils 8R, 15S-diHETE
(Levine et al., 1986)
Sensitises nociceptors
(White et al., 1990)
Cytokines
(Witko-Sarsat et al.,
2000) and chemokines
(Scapini et al., 2000)
See above entry for
cytokines and
chemokines
Defensins (Faurschou
and Borregaard, 2003)
Recruit monocytes,
T-cells (Faurschou and
Borregaard, 2003;
Welling et al., 1998)
Reactive oxygen
species (Witko-Sarsat
et al., 2000)
Sensitise neurons in
PNS and CNS (Aley et
al., 1998; Kim et al.,
2004; Liu et al., 2000a,b;
Meller and Gebhart,
1993; Twining et al.,
2004)
Opioid peptides
(Brack et al., 2004)
Anti-nociceptive (Brack
et al., 2004)
Macrophages Cytokines (Nathan,
1987) and
chemokines
See above entry for
cytokines and
chemokines
Prostaglandins
(PGE2, PGI2)
(Nathan, 1987)
PGE2, PGI2 sensitise
nociceptors (Taiwo and
Levine, 1989), PGE2 acts
on spinal neurons (Baba
et al., 2001)
Reactive oxygen
species (Nathan, 1987)
See above entry for
reactive oxygen species
T lymphocytes Pro-inflammatory
cytokines (Mosmann
et al., 1986; Sad et
al., 1995)
Increase
hyperalgesia and
allodynia (Moalem
et al., 2004)
Anti-inflammatory
cytokines (Mosmann
et al., 1986; Sad et al.,
1995)
Decrease hyperalgesia
and allodynia (Moalem
et al., 2004)
Glia and
Schwann
cells
Cytokines and
chemokines
(Hanisch, 2002)
See above entry for
cytokines and
chemokines
Excitatory amino
acids (Araque et al.,
2001; Watkins et al.,
2001)
Modulate synaptic
transmission and
neuronal excitability
(Araque et al., 2001)
246 B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4
0 2 6 4
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
8/25
small blood vessels into inflamed tissue (Gaboury et al.,
1995),
and neutrophils in turn release mediators such as chemokines
and defensins which are chemotactic for macrophages (Well-
ing et al., 1998; Scapini et al., 2000) (Fig. 1). Finally, it is
worth
noting that neutrophils may also have an anti-inflammatory
and anti-nociceptive role. MRP-14 is a calcium-binding
protein
that forms a significant proportion of the cytoplasmic
protein
in neutrophils. It de-activates macrophages in vitro and
suppresses inflammatory pain in mice (Giorgi et al., 1998).
Under inflammatory conditions, neutrophils may secrete
opioid peptides, which bind to opioid receptors on
peripheral
sensory neurones and mediate anti-nociception (Brack et al.,
2004). The evidence for an anti-nociceptive role for neutro-
phils appears to contradict the pro-nociceptive role
outlined
above. However, neuropathic pain and inflammatory pain
differ in some respects, even though they share some basic
mechanisms. In particular, a tertiary peripheral site of
opioid
analgesia is initiated by acute inflammation which may notbe
activated in neuropathic pain (Bridges et al., 2001). This
difference may help to explain why opioids are less
effective
in relieving neuropathic pain than inflammatory pain (Delle-
mijn, 1999).
2.3. Macrophages
Macrophages phagocytose foreign particles such as microbes
but also have an important role in removing injured or dead
tissue. In the context of this review, they play a vital part
in
phagocytosing the dead or dying remnants of injured
Schwann cells and axotomised axons in Wallerian degener-
ation (Brck, 1997). There is a resident population of
macrophages in the peripheral nerve and dorsal root ganglia;
their equivalent in the CNS is the microglia. In peripheral
nerve, the resident macrophages are assisted with clearance
of cellular debris by an influx of haematogenous macro-
phages (Bendszus and Stoll, 2003; Mueller et al., 2003).
Clearance of debris is a prerequisite to regeneration in
peripheral nerves (Perry and Brown, 1992; Correale and
Villa,
2004), and Wallerian degeneration is delayed in mice with a
genetic defect which delays the recruitment of macrophages
(Perry and Brown, 1992; Araki et al., 2004). Hyperalgesia is
also delayed in these mice, suggesting that macrophages
contribute to pain resulting from nerve injury (Myers et
al.,
1996). This suggestion was confirmed by the demonstration
that depletion of macrophages in nerve-injured rats allevi-
ated neuropathic hyperalgesia (Liu et al., 2000b). However,
another study found only a limited role of macrophages inthe
generation of mechanical allodynia following nerve
injury (Rutkowski et al., 2000). Furthermore, in a model of
neuropathic pain, the development of mechanical allodynia
was totally abrogated in mice with a knockout for the
chemokine receptor CCR2, involved in monocyte recruitment
(Abbadie et al., 2003). It is noteworthy that macrophages
also
invade the dorsal root ganglia following peripheral nerve
lesion and may contribute to neuropathic pain by release of
excitatory agents that generate ectopic activity in sensory
neurones (Hu and McLachlan, 2002).
What are the signals that recruit macrophages? Essential
roles are played by monocyte chemoattractant protein-1,
MCP-1; macrophage inflammatory protein-1, MIP-1; and
the IL-1 (Perrin et al., 2005); the latter two are known to
be
released by neutrophils (Witko-Sarsat et al., 2000). Release
of
defensin by neutrophils may also be involved (Welling et
al.,
1998). Which are the mediators that are released by macro-
phages to cause hyperalgesia? Macrophages secrete prosta-
glandins, including prostaglandin E2 and I2 (Nathan, 1987),
which sensitise primary afferents directly. Prostaglandin
release by macrophages is strongly implicated in neuropathic
pain since inhibition of cyclooxygenase, an enzyme respon-
sible for prostaglandin synthesis, relieves hyperalgesia in
nerve-injured rats (Syriatowicz et al., 1999), and
cyclooxygen-
ase-2 is upregulated in macrophages in the injured nerve (Ma
and Eisenach, 2002, 2003b). Other algesic mediators that are
released by macrophages (Nathan, 1987) and most likely
contribute to neuropathic pain include reactive oxygen
species and the cytokines TNF (Sommer et al., 1998b), IL-1
and IL-6 (Sommer and Kress, 2004) (Fig. 1).
2.4. Lymphocytes
Lymphocytes can be classed as B lymphocytes, responsible
for antibody production, T lymphocytes, which are the
mediators of cellular immunity, and natural killer cells.
Both T cells and natural killer cells are found at the site
of
the nerve lesion in rat models of neuropathic pain (Cui et
al., 2000). T cells are also found in increased numbers in
the dorsal root ganglia and spinal cord after lesions of the
peripheral nerve, leading to the suggestion that they may
play a role in generating neuropathic pain (Hu and
McLachlan, 2002; Sweitzer et al., 2002). This suggestion
has now been confirmed by the finding that congenitally
athymic nude rats, which lack mature T cells, develop
significantly less mechanical allodynia and thermal hyper-
algesia when subjected to sciatic nerve injury than their
heterozygous littermates (Moalem et al., 2004). T cells are
rather heterogeneous, they can be divided into CD4+
(helper) and CD8+ (cytotoxic) cells, and each of these
subpopulations can be further divided into type 1 and
type 2 subsets according to their cytokine secretion profile
Table 1 (continued)
Cell type Mediators Actions
Nitric oxide (Minghetti
and Levi, 1998)
Activates guanylyl
cyclase, sensitises
neurons in CNS (Luo
and Cizkova, 2000;
Minghetti and Levi,
1998)
Prostaglandins
(Minghetti and Levi,
1998; Muja and
DeVries, 2004)
See above entries
for prostaglandins
ATP (Anderson et al.,
2004; Liu et al., 2005)
Activates nociceptors
(Hamilton and
McMahon, 2000),
increases sensory
C-fibre excitability
(Irnich et al., 2002;
Moalem et al., 2005)
247B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4 0
2 6 4
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
9/25
(Mosmann et al., 1986; Sad et al., 1995). The functions of T
helper 1 (Th1) and Th2 cells correlate well with their
distinctive cytokines. Th1 cells produce IL-2 and
interferon-
gamma and are involved in cell-mediated inflammatory
reactions, while Th2 cells produce IL-4, IL-5, IL-6, IL-9,
IL-10
and IL-13, are involved in antibody and allergic responses
and inhibit synthesis of pro-inflammatory cytokines by Th1
cells (Mosmann and Sad, 1996). Th1 and Th2 cytokines aremutually
inhibitory for the functions of the reciprocal
phenotype. Recent work by Moalem et al. has demonstrated
that passive transfer of type 1 T cells, which produce pro-
inflammatory cytokines, into nude rats increased their
sensitivity to noxious stimuli to a level comparable with
that of heterozygous rats. By contrast, passive transfer of
type 2 T cells, which release anti-inflammatory cytokines,
into heterozygous rats reduced their pain sensitivity
(Moalem et al., 2004). These findings indicate that T cells
take part in the course of neuropathic pain (Fig. 1) and
suggest that Th1 and Th2 cells have opposite effects on
neuropathic pain as a result of the distinct sets of
cytokines they release. The role of B cells in neuropathic
pain remains to be elucidated.
2.5. Glia and Schwann cells
Glial cells surround and support neurones in the nervous
system and are 10 to 50 times as numerous as neurones. In
the last few years, it has become clear that glia are by no
means passive bystanders in the nervous system but have
important metabolic and immune functions. In the periph-
eral nervous system, glia are represented by Schwann cells,
while, in the CNS, there are three types of glial cell that
can
be distinguished the astrocytes and oligodendrocytes
(macroglia) and the microglia. Microglia form a population
of resident macrophages in the CNS, constituting about 5
10% of glial cells in the CNS. In the resting state, they have
a
small soma with fine processes, but, when activated by
stimuli such as trauma, ischemia or inflammation, they take
on an amoeboid shape and function as phagocytes ( Vilhardt,
2005). At this stage, they express the major
histocompatibil-
ity complex (MHC), which has a role in presenting antigen to
T lymphocytes, and release mediators that include pro-
inflammatory cytokines (Piehl and Lidman, 2001). Astrocytes
represent the largest cell population in the CNS. They
closely
interact with neurones to provide active maintenance of
homeostasis by regulating extracellular ion and neurotrans-
mitter concentrations and extracellular pH in their micro-
environment. The relative contribution of microglia and
astrocytes to neuropathic pain is still under investigation.
However, recent studies suggest that microglia are more
important for the initiation, while astrocytes are more
important for the maintenance of neuropathic pain (Colburn
et al., 1999; Raghavendra et al., 2003). In this review, we
will
use the term glia for both microglia and astrocytes in the
CNS. Evidence has been accumulating since the early 1990s
that spinal glia contribute to neuropathic pain (DeLeo and
Colburn, 1999; Watkins et al., 2001; McMahon et al., 2005;
Tsuda et al., 2005). Microglia are activated by peripheral
nerve injury (Colburn et al., 1999; Tanga et al., 2004), and
inhibition of microglial activation in the several models of
neuropathic pain inhibits the development of hyperalgesia
and allodynia (Raghavendra et al., 2003; Ledeboer et al.,
2005). Glia are activated by several mediators including
ATP,
bradykinin, substance P and prostaglandins (Hosli and Hosli,
1993; Watkins et al., 2001). ATP is known to activate
nociceptors in the periphery by an action on P2X3 receptors
(Cook et al., 1997), but, in the CNS, it activates P2X4
receptors
that are selectively expressed by activated microglia andappear
to be required for neuropathic pain (Tsuda et al.,
2005). Recent evidence suggests that activation of microglia
by fractalkine (Milligan et al., 2004; Verge et al., 2004) and
by
the Toll-like receptor 4 (TLR4) (Tanga et al., 2005) is also
involved in the initiation of neuropathic pain (Fig. 2).
Fractalkine is a chemokine expressed on the surface of
spinal neurones, which activates the CX3CR1 receptor on
microglia, while the Toll-like receptors recognise invariant
molecular structures of pathogens and specific ligands
released after nerve injury.
Although we have some idea of how glial cells are
activated, there is little concrete evidence on how
activated
glial cells regulate neuronal function to bring about
symptoms
of neuropathic pain. Activated glia release a number of
mediators that excite spinal neurones (Watkins et al.,
2001),
including prostanoids and nitric oxide (Minghetti and Levi,
1998), cytokines and chemokines (Hanisch, 2002), excitatory
amino acids (Araque et al., 2001) and ATP (Anderson et al.,
2004). However, the role and relative importance of these
glial
mediators in neuropathic pain still need to be established.
Interestingly, microglia have been recently implicated in
mirror-image pain, pain arising from sites contralateral to
the site of pathology (Koltzenburg et al., 1999b; Twining et
al.,
2004). The mechanisms underlying the expansion of pain to
the contralateral site are not clear. However, one possible
mechanism is through spinal gap junction activation. Recent
study has demonstrated that treatment with a gap junction
decoupler reverses neuropathy-induced mirror-image pain
(Spataro et al., 2004). Thus, strong activation of glia at one
site
can lead to gap junctional propagation of calcium waves that
activates distant glia, leading to release of pain-enhancing
substances.
Schwann cells in the peripheral nervous system provide
the myelin sheath in myelinated axons and also envelop
small bundles of unmyelinated axons (Remak bundles). The
primary role of Schwann cells was long thought to be the
provision of an insulating sheath around myelinated axons
(facilitating conduction), maintenance of suitable environ-
ment for peripheral axons, and guiding regenerating axons
back to their targets after injury. In recent years, an
immune
function for Schwann cells has received increasing recogni-
tion since they interact with the immune system in T-cell-
mediated immune responses by expressing MHC class II
molecules (Bergsteinsdottir et al., 1992). Schwann cells
release several algesic mediators, including TNF (Murwani
et al., 1996; Wagner and Myers, 1996a; Shamash et al., 2002
),
IL-1 (Bergsteinsdottir et al., 1991; Shamash et al., 2002),
IL-6
(Bolin et al., 1995), NGF (Matsuoka et al., 1991),
prostaglandin
E2 (Muja and DeVries, 2004) and ATP (Liu et al., 2005).
Schwann cells also express a number of ion channels and
receptors for mediators including glutamate (Liu and Ben-
nett, 2003), ATP (Grafe et al., 1999; Irnich et al., 2001; Liu
et
248 B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4
0 2 6 4
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
10/25
al., 2005) and IL-1 (Skundric et al., 1997). Given the close
relationship between Schwann cells and peripheral axons
and the presence of axonal receptors for mediators such as
ATP (Irnich et al., 2001, 2002), it seems very likely that
Schwann cells contribute to neuropathic pain by sensitising
or activating the axons of nociceptors (Fig. 1). However, it
has not yet been possible to obtain direct evidence for this
as selective inhibition or depletion of Schwann cells in
anintact animal has not yet been achieved.
3. Mediators
The mediators released by inflammatory and immune cells
may act directly to sensitise or activate neurones (usually
nociceptors in the periphery or dorsal horn neurones in the
spinal cord). Alternatively, they may act on a non-neuronal
cell, which on activation releases another mediator that
does
act directly on the neurone. These mediators form a long and
increasing list that includes bradykinin, ATP and
adenosine,serotonin, eicosanoids, cytokines, neurotrophins and
reactive
oxygen species (Dray, 1995). This list is not exhaustive,
and
some inflammatory mediators such as protons and histamine
(see the Mast cells section) are not reviewed here.
3.1. Bradykinin
Bradykinin and kallidin are peptides that are formed from
precursors in the blood and other tissues. In the blood,
bradykinin is formed from high molecular weight kininogen
as part of the clotting cascade, while in other tissues
kallidin
(lysyl-bradykinin) is formed from a low molecular weight
kininogen. At least two subtypes of bradykinin receptors (B1
and B2) have been characterised on the basis of in vivo and
in
vitro functional studies, using selective agonists and
antago-
nists of these receptors (Regoli et al., 1998). Bradykinin
produces pain hypersensitivity by sensitising the peripheral
terminals of nociceptors and by potentiating glutamatergic
synaptic transmission in the spinal cord (Wang et al.,
2005).
Bradykinin also sensitises nociceptors by disinhibition of
the
TRPV1 receptor (Stucky et al., 1998; Di Marzo et al., 2002).
B2
receptors are constitutive and are found on a number of cell
types including sensory neurones and microglia (Dray, 1997).
Increased expression of B1 receptors plays a prominent role
in
inflammatory hyperalgesia (Dray and Perkins, 1993; Dray,
1997), and this upregulation is facilitated by cytokines such
as
IL-1 (Marceau, 1995). Bradykinin acts mainly on the B2
receptor, while des-Arg9-bradykinin, the major metabolite of
bradykinin, has high affinity for the B1 receptor but little
affinity for the B2 receptor (Dray and Perkins, 1993; Khasar
et
al., 1995). Both B1 and B2 receptors have been shown to be
involved in peripheral inflammatory hyperalgesia (Dray and
Perkins, 1993; Dray, 1997). Bradykinin stimulates
macrophages
to release TNF and IL-1 (Tiffany and Burch, 1989), as well
as
factors that are chemotactic for neutrophils and monocytes
(Sato et al., 1996). Bradykinin also elicits the release of
histamine from mast cells but does so by non-specific
binding
tothe cell surface rather than toB1 or B2receptors
(Reissmann
et al., 2000). In fact, peptide antagonists of B1 or B2
receptors
may also bind non-specifically to the cell surface and
elicit
degranulation (Griesbacher and Rainer, 1999).
Injury of the sciatic nerve in rats led to upregulation of
B2
and B1 receptors on lumbar dorsal root ganglia (Eckert et
al.,
1999; Levy and Zochodne, 2000), and in rats with a chronic
constriction injury of the sciatic nerve, constant infusion of
B2
or B1 receptor antagonists reduced hyperalgesia (Yamaguchi-
Sase et al., 2003). A recent study in nerve-injured mice
foundthat B2 receptors were mainly expressed by the cell bodies
of
unmyelinated axons and that their expression was drastically
decreased after nerve injury. By contrast, B1 receptors were
newly expressed on the cell bodies of myelinated axons,
suggesting that nerve injury induced a switch in the type of
sensory neurone and the type of bradykinin receptor involved
in nociception (Rashid et al., 2004). Bradykinin and its B2
receptor have recently been implicated in central pain
transmission. This work showed that the B2 receptor is
expressed by dorsal horn neurones and is involved in central
sensitisation evoked by primary afferents (Wang et al.,
2005).
Activation of B2 receptors on glial cells in the CNS could play
a
role in neuropathic pain, butthere isno evidencefor this
asyet.
3.2. ATP and adenosine
Purines such as ATP and adenosine are well known for their
role in energy metabolism but also have widespread effects
on
neurones and immune cells. ATP is a classical neurotrans-
mitter but is also released by non-neuronal cells and from
disrupted cells in injured tissue (Cook and McCleskey,
2002).
Adenosine is a neuromodulator that is produced in the
extracellular space by degradation of extracellular ATP and
is also transported from the cytoplasm into the interstitial
space by transport proteins (Linden, 1994). Inosine is
produced
by deamination of adenosine. It mayreach millimolar levelsin
ischemic tissue and can degranulate mast cells ( Jin et al.,
1997).
In 1977, Bleehen and Keele reported that ATP, ADP, AMP
andadenosine allproducedpain when applied to a blister base
in man (Bleehen and Keele, 1977). Since then, it has been
found that adenyl compounds and other purines activate
nociceptors and elicit pain by their actions on purinergic
receptors (Burnstock and Wood, 1996; Sawynok, 1998; Hamil-
ton and McMahon, 2000). Receptors for purines have been
classified into adenosine (P1) receptors and nucleotide (P2)
receptors (Ralevic and Burnstock, 1998). P2 receptors can be
subdivided into P2X receptors, which are ATP-gated ion
channels (Chizh and Illes, 2001), and P2Y receptors, which
are G-protein coupled receptors activated by purine or
pyrimidine nucleotides such as ATP or UTP (von Kugelgen
and Wetter, 2000; Burnstock and Knight, 2004).
Initial experiments on the role of ATP in neuropathic pain
implicated P2X3 receptors, although results were apparently
contradictory. Immunocytochemical studies in rats and
humans found that P2X3 receptor expression was significantly
reduced after axotomy (Bradbury et al., 1998; Yiangou et
al.,
2000) or partial nerve ligation (Kage et al., 2002), while
the
number of P2X3 immunoreactive neurones in the DRG or
trigeminal ganglia was increased after chronic constriction
injury of peripheral nerves (Eriksson et al., 1998; Novakovic
et
al., 1999). This apparent discrepancy may be due to the
249B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4 0
2 6 4
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
11/25
different types of nerve injury, leading to downregulation
of
P2X3 receptors in injured cells but upregulation in intact
cells
(Tsuzuki et al., 2001; Kennedy et al., 2003).
Electrophysiological
recordings from DRG neurones supported the idea that
axotomy results in an increase in purinergic sensitivity
(Chen et al., 2001), most likely mediated by P2X receptors
(Zhou et al., 2001). While these results are suggestive, they
do
not directly address the issue of neuropathic pain. This wasdone
by reducing the level of expression of P2X3 subunits in
DRG cells with intrathecal administration of antisense
oligo-
nucleotides. In the partial nerve ligation model of
neuropathic
pain, this inhibited the initiation of mechanical
hyperalgesia
and reversed established hyperalgesia (Barclay et al.,
2002).
This result was confirmed using a novel non-nucleotide
antagonist of P2X3 and P2X2/3 activation. Blocking these
receptors in the chronic constriction model of neuropathic
pain attenuated both thermal and mechanical allodynia
(Jarvis et al., 2002).
P2X4 and P2X7 receptors have also been implicated in
neuropathic pain. P2X4 receptors are upregulated in spinal
microglia following nerve injury, and pharmacological block-
ade or inhibition of P2X4 receptors reversed the accompa-
nying allodynia (Tsuda et al., 2003; Inoue et al., 2004).
P2X7receptors are expressed by immune cells, including mast
cells, macrophages and T lymphocytes. They have a key role
in the secretion of IL-1. Recently, it was shown that
disruption of the P2X7 purinoreceptor gene abolishes neuro-
pathic pain and that the receptor is upregulated in human
DRGs and injured nerves from neuropathic pain patients
(Chessell et al., 2005).
P2Y1 and P2Y2 receptors are found in sensory neurones
including nociceptors (Molliver et al., 2002; Xiao et al.,
2002;
Stucky et al., 2004) and in Schwann cells (Mayer et al., 1998).
In
normal rats, P2Y1 mRNA is found in about 20% of DRG
neurones(large as well as small), butthis percentage
increases
to about 70% after section of the sciatic nerve, suggesting
a
possible role in neuropathic pain (Xiao et al., 2002).
Intrathecal
administration of P2Y receptor agonists appears to have an
inhibitory effect on neuropathic pain, acting via P2Y2
and/or
P2Y4 receptors and P2Y6 receptors (Okada et al., 2002).
Adenosine elicits cutaneous pain and hyperalgesia when
applied peripherally (Bleehen and Keele, 1977; Taiwo and
Levine, 1990a). It also activates axonal receptors on human
C-
fibres (Irnich et al., 2002; Lang et al., 2002). However, its
role in
the CNS is primarily inhibitory (see Sawynok and Liu, 2003
for
review). Thus, in rats with spinal nerve ligation, spinal
administration of adenosine resulted in a dose-dependent
reduction in tactile allodynia (Lavand'homme and Eisenach,
1999). Inhibition of adenosine kinase increases the
availability
of extracellular adenosine in the spinal cord, and spinal
administration of adenosine kinase inhibitors relieves
tactile
allodynia or suppresses neuronal responses in nerve-injured
rats (Suzuki et al., 2001; Zhu et al., 2001). These inhibitors
may
therefore hold some promise for the treatment of patients
with neuropathic pain.
3.3. Serotonin
Serotonin (5-hydroxytryptamine, 5HT) is a neurotransmitter
synthesised and released by neurones in the CNS. Messenger
RNAs for 5HT1B, 5HT1D, 5HT2A,5HT2C, 5HT3 and 5HT7 receptors
have been found in dorsal root ganglia (Pierce et al., 1996),
and
5HT receptors are also found in the cytoplasm of Schwann
cells (Yoder et al., 1997).
There is a large projection of serotonergic neurones from
the raphe nuclei to laminae 1 and 2 of the spinal cord, and
these serotonergic axons contribute to the descending
inhibi-
tion of pain (Millan, 2002). While serotonin
suppressesnociception at the level of the spinal cord, it enhances
it in
the periphery, where serotonin sensitises nociceptors to
thermal stimuli and to bradykinin application (Lang et al.,
1990; Rueff and Dray, 1993). Serotonin also increases the
excitability of sensory C-fibres in isolated segments of
peripheral nerve (Moalem et al., 2005). In the periphery,
serotonin is not released as a neurotransmitter by the axon
terminals of serotonergic neurones but is an inflammatory
mediator released by platelets and murine mast cells
(seroto-
nin is not released by human mast cells). When injected into
the rat's paw, it induces a hyperalgesia thought to bedue to
an
action on 5HT1A receptors (Taiwo and Levine, 1992; Hong and
Abbott, 1994).
Work in our laboratory suggested that serotonin con-
tributes to established mechanical hyperalgesia in the
partial ligation model of neuropathic pain. Subcutaneous
injection of a 5HT2A receptor blocker or a 5HT3 receptor
blocker alleviated hyperalgesia in a dose-dependent fashion
when injected into the hindpaw of the nerve-injured limb.
Injection of the blockers into the contralateral hindpaw did
not relieve hyperalgesia, suggesting that the effect was
mediated locally rather than systemically (Theodosiou et
al., 1999). Other research on the involvement of serotonin
in
neuropathic pain has focussed on its role in the descending
inhibition of pain. Tricyclic antidepressants (TCAs) are
widely used to treat neuropathic pain in patients and
inhibit
reuptake of monoamines such as serotonin and noradren-
aline into nerve terminals. This is thought to increase the
efficacy of descending monoaminergic pathways that inhibit
pain. Some TCAs are effective in alleviating neuropathic
pain in animal models, but selective inhibitors of serotonin
reuptake were not effective in animal models or in patients
(Max et al., 1992; Jett et al., 1997). However,
fenfluramine,
which not only inhibits serotonin reuptake but also elicits
releases of serotonin from presynaptic terminals, produced a
significant reduction in mechanical allodynia in rats with
tight ligation of the L5/L6 spinal nerves (Wang et al.,
1999).
Serotonin reuptake is mediated by the serotonin transporter
(5HTT), and mice deficient for the serotonin transporter did
not develop thermal hyperalgesia following chronic constric-
tion of the sciatic nerve (Vogel et al., 2003). Whether this
was
due to altered tissue levels of serotonin or downregulation
of
5HT receptors was not clear.
3.4. Eicosanoids
Arachidonic acid is a polyunsaturated fatty acid that is
found
in the cell membrane. It is the major precursor of the
eicosanoids, and, once it has been freed from the cell
membrane by phospholipase A2, it is converted by enzymes
in the arachidonate cascade to compounds that include
prostaglandins, thromboxanes and leukotrienes.
250 B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4
0 2 6 4
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
12/25
Arachidonic acid is metabolised to prostanoids by the
cyclooxygenase pathway and to leukotrienes by the lipox-
ygenase pathway (Wolfe and Horrocks, 1994; Smith et al.,
2000). It may also be converted to isoprostanes by peroxida-
tion; the isoprostanes are inflammatory mediators that
augment nociception (Evans et al., 2000).
Prostaglandins PGE2 and PGI2 induce hyperalgesia in the
periphery (Taiwo and Levine, 1990b) where they act directlyon
the terminals of nociceptors (Taiwo and Levine, 1989)
and probably on macrophages as well (Ma and Eisenach,
2003a). More recently, it has been established that prosta-
glandins also contribute to nociception at the level of the
spinal cord (Malmberg and Yaksh, 1992; Vanegas and Schaible,
2001).
There are two primary isoforms of cyclooxygenase, COX-1
and COX-2. COX-1 is expressed constitutively in most tissues
and has a homeostatic or housekeeping role, while COX-2
expression is usually lowbut canbe induced by factors such
as
inflammatory cytokines in cells including mast cells, macro-
phages (Smith et al., 2000) and spinal neurones (Samad et
al.,
2002). In fact, COX-1 may also be induced in some conditions
such as spinal cord injury (Schwab et al., 2000; Samad et
al.,
2002). Prostaglandins exert their effects by actions on G-
protein-coupled receptors including the EP receptors (EP14
for
PGE2) and the IP receptor for PGI2 or prostacyclin (Hata and
Breyer, 2004).
Work in our laboratory showed that mechanical hyper-
algesia in nerve-injured rats wasalleviated for up to 10 days
by
subcutaneous injection of indomethacin (a classic inhibitor
of
cyclooxygenase) into the affected hindpaw. Subcutaneous
injection of selective COX-2 inhibitors or an EP1 receptor
blocker relieved thermal as well as mechanical hyperalgesia,
but with a shorter timecourse. We concluded that increased
expression of prostaglandins in the region of the nerve
lesion
contributed to neuropathic pain (Syriatowicz et al., 1999).
It
was then shown in several animal modelsof neuropathic pain
that the number of COX-2 immunoreactive cells was dramat-
ically increased in the region of the nerve lesion, that most
of
these cells were infiltrating macrophages (Ma and Eisenach,
2002, 2003b) and that this was the case in human patients as
well as nerve-injured rats (Durrenberger et al., 2004). In
fact,
the increase in COX-2 expression appears to be biphasic,
with
an early increase in Schwann cells (1 day after spinal nerve
injury) and an increase in macrophage expression at about 7
14 days (Takahashi et al., 2004). As one might expect,
increased levels of PGE2 are found in the injured nerves
(Schfers et al., 2004). Furthermore, cells immunoreactive
for
EP receptors are found in the injured nerve, but not in
normal
intact nerves. Once again, many of the immunoreactive cells
prove to be macrophages (Ma and Eisenach, 2003a). These
observations, based on several animal models of sciatic
nerve
injury, support the idea that upregulation of COX-2 and EP
receptorsin the injured nerve contributes to neuropathic
pain.
However, in the spared nerve injury model (Decosterd and
Woolf, 2000), where degenerating axons distal to the lesion
do
not mingle with intact axons, treatment with the COX-2
inhibitor rofecoxib had no effect on the development of
allodynia and hyperalgesia (Broom et al., 2004).
Increased expression of cyclooxygenase after nerve injury
is not restricted to the nerve itself. COX-2 is also
upregulated
in the dorsal horn of the spinal cord and thalamus following
peripheral nerve injury (Zhao et al., 2000), and the
resulting
allodynia is attenuated by intrathecal injection of ketorolac,
a
COX-1 preferring inhibitor (Ma et al., 2002). COX-1 is
expressed
constitutively in glial cells and motor neurones of normal
rats.
It is also upregulated after ligation of L5L6 spinal nerves
so
that the number of cells immunoreactive for COX-1 is
increased in the superficial laminae of the dorsal horn at 4days
after spinal nerve ligation and remains high for at least 2
weeks (Zhu and Eisenach, 2003). How do increased levels of
prostaglandins in the spinal cord mediate neuropathic pain?
There is evidence for at least twomechanisms. Thefirst is
that
PGE2 depolarises wide dynamic range neurones in the deep
dorsal horn (Baba et al., 2001), and the second is that PGE2
also
blocks glycinergic inhibition of neurones in the superficial
laminae of the dorsal horn (Ahmadi et al., 2002; Harvey et
al.,
2004).
Given the evidence for a role of cyclooxygenases in the
development of neuropathic pain and the efficacy of cycloox-
ygenase inhibitors in treating inflammatory pain, it is
surprising that COX inhibitors are relatively ineffective in
the
treatment of neuropathic pain in patients (Namaka et al.,
2004). This may be due to the predominant role of
prostaglan-
dins in the development rather than maintenance of neuro-
pathic pain or that higher effective concentrations of COX
inhibitors are achieved in experimental animals than in
patients.
There are three mammalian lipoxygenases, which catalyse
the insertion of oxygen at positions 5, 12 and 15 of
arachidonic
acid. These are therefore referred to as 5-, 12- and 15-
lipoxygenases. The 5-lipoxygenases form leukotrienes, and
leukotriene B4 (LTB4) produces hyperalgesia by eliciting the
release of a mediator from neutrophils (Levine et al., 1984,
1985), which was identified as the 15-lipoxygenase product
(8R, 15S)-dihydroxyeicosa-(5E-9,11,13Z)-tetraenoic acid (8R,
15S-diHETE) (Levine et al., 1986). This was shown to
sensitise
nociceptors in the rat and may contribute to the role of
neutrophils in neuropathic pain (see the Neutrophils
section).
Nerve growth factor elicits hyperalgesia when injected into
the rat paw, and inhibition of 5-lipoxygenase inhibits
release
of LTB4, accumulation of neutrophils and the associated
hyperalgesia (Amann et al., 1996; Bennett et al., 1998b). It
is
likely that NGF produces hyperalgesia by inducing the
release
of LTB4 from mast cells, leading in turn to recruitment of
neutrophils (Bennett et al., 1998b), which then release
algesic
mediators and chemokines. Neutrophils and nerve growth
factor have both been directly implicated in the genesis of
neuropathic pain (Herzberget al., 1997; Theodosiou et al.,
1999;
Perkins and Tracey, 2000), and so it is likely that LTB4 and
8R,
15S-diHETE are also involved.
3.5. Cytokines
Cytokines are small proteins that mediate interactions
between cells over relatively short distances. They are
mostly
involved in responses to disease or infection. Many of them
are referred to as interleukins, denoting a mediator
released
by one leukocyte and acting on another, but they are
synthesised by most cell types. Several are pro-inflammato-
ry, such as IL-1, IL-6 and TNF, while others such as IL-10
are
251B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4 0
2 6 4
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
13/25
anti-inflammatory. Exogenous administration of these pro-
inflammatory cytokines elicits pain and hyperalgesia (Som-
mer and Kress, 2004), but they do not appear to be involved
in normal pain (Watkins et al., 2001). In fact, these three
pro-inflammatory cytokines induce the production of each
other, and they act synergistically (Watkins et al., 1999).
This
leads to the possibility of positive feedback, which could
lead
to chronic pain if not appropriately regulated. Other cyto-kines
such as IL-4 and IL-10 are anti-inflammatory and
suppress genes that code for IL-1, TNF and chemokines. The
algesic effects of pro-inflammatory cytokines are often
indirect, so that they may not act directly on the
nociceptor
but instead induce the expression of agents (such as PGE2)
that themselves sensitise nociceptors. In inflammation and
Wallerian degeneration, cytokine production is organised
into a sequence with TNF playing a leading role (Shamash et
al., 2002; Cunha et al., 2005). This sequence appears to be
one
of the mechanisms underlying neuropathic pain (for reviews,
see DeLeo and Colburn, 1999; Sommer, 2001; Sorkin, 2002;
Sommer and Kress, 2004). Chemokines are small chemotactic
cytokines that are important for leukocyte migration and
recruitment to damaged sites. Recently, they have been
shown to contribute directly to nociception by producing
excitatory effects on DRG neurones and inducing allodynia
after injection into the rat's paw (Oh et al., 2001). The
contribution of chemokines to pain has been reviewed
recently (Abbadie, 2005; White et al., 2005).
3.5.1. Interleukin-1
Binding of IL-1 to its receptor IL1-RI on the cell surface
initiates several signalling events, such as translocation of
NF-
B into the nucleus (Dinarello, 1999). NF-B then upregulates
transcription of several genes, including COX-2, inducible
nitric oxide synthase, TNF, IL-1 and IL-6 (Pahl, 1999;
Tegeder
et al., 2004). IL-1 may act directly as well as indirectly
on
nociceptors. Thus, Fukuoka and colleagues found that IL-1
facilitated release of CGRP from nociceptors in an in vitro
nerve-skin preparation. The latency of the effectwas too
short
for upregulation of receptors or changes in gene expression,
and the cell bodies were not present, suggesting a direct
sensitisation of nociceptors (Fukuoka et al., 1994; Sommer
and
Kress, 2004). IL-1 elicits hyperalgesia when injected
periph-
erally into the rat paw (Ferreira et al., 1988; Follenfant et
al.,
1989), intraneurally into rat sciatic nerve (Zelenka et al.,
2005),
intrathecally in the rat spinal cord (Sung et al., 2004) or
centrally into various regions of the brain (see Bianchi et
al.,
1998 for review). In fact, Watkins and her colleagues have
shown that IL-1 may induce an illness hyperalgesia via a
kind of supraspinal loop involving sensory fibres in the
vagus nerve, the nucleus of the solitary tract, nucleus
raphe
magnus and projections to the spinal cord (Watkins et al.,
1994; Watkins and Maier, 1999). IL-1 is implicated in
neuropathic pain since IL-1 and IL-1 are both upregulated
in injured peripheral nerve (Gillen et al., 1998; Okamoto et
al., 2001; Shamash et al., 2002). Neuropathic pain is
alleviated in nerve-injured mice by administration of neu-
tralizing antibodies to the IL-1 receptor (Sommer et al.,
1999;
Schfers et al., 2001) and in nerve-injured rats by
intrathecal
IL-1 receptor antagonist in combination with soluble TNF
receptor (Sweitzer et al., 2001).
3.5.2. Interleukin-6
IL-6 is synthesised by many cell types, including mast
cells,
monocytes, lymphocytes, neurones and glial cells. Once IL-6
has bound to its receptor IL-6R, it initiates two major
intracellular cascadesone involving Janus kinases and
STAT factors, the other using the Ras-dependent MAP kinase
pathway (De Jongh et al., 2003). Injury of the sciatic nerve
induces upregulation of IL-6 and its receptor in the region
ofthe lesion, where it appears in macrophages and Schwann
cells (Bolin et al., 1995; Kurek et al., 1996; Ito et al., 1998;
Grothe
et al., 2000). Upregulation of IL-6 in these macrophages
appears to be induced by PGE2 (Ma and Quirion, 2005).
Sciatic
nerve injury also increases IL-6 levels in the DRG and
spinal
cord, particularly in the superficial laminae of the dorsal
horn
(Murphy et al., 1995; DeLeo et al., 1996; Lee et al., 2004).
Exogenous IL-6 induces thermal hyperalgesia when injected
into the lateral cerebral ventricles of the rat (Oka et al.,
1995)
and increases the heat-evoked release of CGRP from cutane-
ous nociceptors (Opree and Kress, 2000; Obreja et al.,
2002).
However, there is little evidence that IL-6 plays a role in
normal nociception. One study did report that intraplantar
injection of IL-6 induced mechanical hyperalgesia in
naiverats
(Cunha et al., 1992). However, this finding has not been
reproduced and later work found that intraplantar IL-6 had
no
effect on mechanical thresholds (Czlonkowski et al., 1993)
or
produced a thermal hypoalgesia (Flatters et al., 2004). In
fact,
peripheral IL-6 inhibitedheat responses of nociceptors in an
in
vitro preparation.
There is limited evidence that IL-6 contributes to the
development of neuropathic pain. In the in vivo spinal cord,
peripheral IL-6 inhibited the responses of dorsal horn neu-
rones to thermal and mechanical stimuli; after spinal nerve
ligation, peripheral IL-6 released the inhibition of
mechanical
responses (Flatters et al., 2004). In IL-6 knockout mice,
nociceptive responses to thermal and mechanical stimuli are
the same as in wild-type mice (Bianchi et al., 1999; Murphy
et
al., 1999), but the development of mechanical allodynia
after
spinalnervelesion is delayed in IL-6 knockoutsby comparison
with wild-type mice (Ramer et al., 1998). It is not clear
whether
thermal hyperalgesia induced by a spinal nerve lesion is
reduced in IL-6 knockouts (Murphy et al., 1999) or not
(Ramer
et al., 1998).
3.5.3. Tumour necrosis factor
Tumour necrosis factor (TNF, TNFSF2, formerly TNF) is a
member of a large superfamily of proteins, which have an
unusual trifold symmetry. There is an equally large super-
family of receptors; the receptors activated by TNF are the
constitutively expressed TNFR1 (TNFRSF1A, p55-R) and the
inducible TNFR2 (p75-R) (Locksley et al., 2001). TNFR1 is
linked
to pathways for cell death, whereas TNFR2 is not. However,
activation of either receptor results in p38 MAP kinase
signalling (Schfers et al., 2003b), translocation of NFB to
the nucleus and activation of COX-2-dependent prostanoid
release, as described above for IL-1 (Dinarello, 1999). TNF
is
constitutively expressed in cutaneous mast cells (Walsh et
al.,
1991), but, in injury or inflammation, it may be released by
other cells including neutrophils and macrophages.
Intraplan-
tar injection of TNF into the rat's paw induces mechanical
hyperalgesia (Cunha et al., 1992). This can be attributed to
252 B R A I N R E S E A R C H R E V I E W S 5 1 ( 2 0 0 6 ) 2 4
0 2 6 4
-
8/14/2019 Immune and inflammatory mechanism in neuropathic
pain
14/25
sensitisation of C-fibres since subcutaneous injection in
the
distribution of the sural nerve lowered the thresholds of
cutaneous C-fibres to mechanical stimulation and led to
ongoing activity in some of these fibres ( Junger and
Sorkin,
2000). In fact, topical application of TNF to a restricted
portion
of the sciatic nerve led to ectopic firing of A- and
C-fibres
(Sorkin et al., 1997), and intraneural injection of TNF into
rat
sciatic nerve at physiological doses induced thermal
hyper-algesia and mechanical allodynia (Zelenka et al., 2005),
suggesting the presence of TNF receptors on the axons. An
alternative possibility is insertion of the TNF trimer into
the
cell membrane of the axon (Baldwin et al., 1996), creating a
pore that is permeable to sodium ions and may be voltage-
dependent (Kagan et al., 1992).
Injury of the sciatic nerve leads to upregulation of TNF and
its receptors in the nerve (George et al., 1999; Shubayev
and
Myers, 2000; George et al., 2005); this upregulation is
found
mainly in Schwann cells and endothelial cells (Wagner and
Myers, 1996a). Nerve injury also leads to increased TNF
expression in the dorsal horn of the spinal cord and in the
locus coeruleus and hippocampus (Ignatowski et al., 1999).
An
important role for TNF in neuropathic pain is indicated by
reducing TNF levels in nerve-injured rats or mice (Sommer,
2001). Inhibiting TNF synthesis with thalidomide or
treatment
with anti-TNF neutralising antibodies at the time of nerve
injury blocked the development of hyperalgesia and allodynia
in these animal models (Sommer et al., 1998a,b, 2001a).
Treat-
ment with etanercept, a recombinant TNF receptor (p75)-Fc
fusion protein that acts as a TNF antagonist, reversed
estab-
lished hyperalgesia in mice with a chronic constriction
injury
of the sciatic nerve (Sommer et al., 2001b), and endoneurial
injection of neutralising antibodies against TNF receptors
showed that neuropathic hyperalgesia is dependent on the
TNFR1 receptor (Sommer et al., 1998b). Furthermore, applica-
tion of exogenous TNF to the DRG increased the sensitivity
of
injured and adjacent uninjured primary sensory neurones and
elicited faster onset of allodynia and spontaneous pain
behaviour after spinal nerve ligation (Schfers et al.,
2003a).
3.5.4. Interleukin-10
IL-10 is generally regarded as anti-inflammatory. Its
expres-
sion increases gradually over at least 6 weeks following
nerve
injury (Okamoto et al., 2001), and treatment with a single
dose
of IL-10 at the site of a chronic constriction injury
significantly
reduces hyperalgesia, probably due in part to suppression of
TNF expression and macrophage recruitment (Wagner et al.,
1998). These results are consistent with findings that
thalid-
omide (an inhibitor of TNF synthesis) decreases TNF levels,
increases endoneurial IL-10 levels, and alleviates
hyperalgesia
in rats with a CCI nerve lesion (Sommer et al., 1998a; George
et
al., 2000). Recent work confirms theefficacyof IL-10 in
alleviat-
ing neuropathic pain, using virally driven spinal production
of
IL-10 in neuropathic pain models (Milligan et al., 2005a,b).
3.6. Neurotrophins
The neurotrophins are dimeric proteins that are essential
for
the normal development of the vertebrate nervous system.
This family includes nerve growth factor (NGF),
brain-derived
neurotrophic factor (BDNF), neurotrophin (NT)-3 and NT-4/5.
Glial-cell-line-derived neurotrophic factor (GDNF) also has
neurotrophic properties but is structurally unrelated to the
dimeric neurotrophin family (Lewin and Barde, 1996).
The neurotrophins act on a trio of tyrosine kinase (Trk)
receptors U TrkA, TrkB and TrkC, which primarily bind NGF,
BDNF and NT-4/5, and NT-3 respectively. There is also a low
affinity or pan-neurotrophin receptor, p75NTR, which acti-
vates a separate set of signalling pathways that interact
withthose activated by Trk receptors (Huang and Reichardt,
2003;
Nykjaer et al., 2005). During embryonic development, all
small-diameter sensory neurones express TrkA, the high
affinity receptor for NGF. Most of these neurones will
become
nociceptors, with thinly myelinated axons (A fibres) or
unmyelinated axons (C-fibres). After birth, the expression
of
TrkA is downregulated so that TrkA is only expressed by
those
small sensory neurones which are peptidergic, whereas the
non-peptidergic remainder (IB4 binding neurones) now
expresses receptor components for another neurotrophin,
GDNF (Bennett, 2001).
Neurotrophins are synthesised and released by a several
cell types of immune cells, including mast cells and lympho-
cytes (Moalem et al., 2000; Bonini et al., 2003). BDNF is
constitutively expressed by peptidergic nociceptors (Thomp-
son et al., 1999).
Nerve injury produces marked changes in expression of
neurotrophins and their receptors, primarily in Schwann
cells
(Frostick et al., 1998). Schwann cells in the intact
peripheral
nerve synthesise NGF, BDNF and GDNF, and this synthesis is
dramatically upregulated by nerve injury (Lindholm et al.,
1987; Meyer et al., 1992; Hammarberg et al., 1996). BDNF
synthesis is also upregulated in sensory neurones following
axotomy (Tonra et al., 1998). In the intactsciatic nerve,
mRNAs
for the neurotrophin receptors TrkB and TrkC are expressed,
but TrkA and p75NTR mRNAs are undetectable. Levels of
p75NTR mRNA are markedly increased in Schwann cells after
nerve injury, but TrkA levels remain undetectable (Funakoshi
et al., 1993).
It is now recognised thatsome neurotrophins (in particular,
NGF and BDNF) play a significant role in nociception
(Bennett,
2001) so that NGF sensitises nociceptors in the periphery
(Koltzenburg et al., 1999a) while BDNF increases the respon-
siveness of dorsal horn neurones in the spinal cord (Pezet
et
al., 2002). Intraplantar or systemic injection of NGF
induces
hyperalgesia (Lewin et al., 1994; Andreev et al., 1995;
Theodo-
siou et al., 1999). Tissue levels of NGF are increased by
inflammation, and the hyperalgesia resulting from inflamma-
tion is prevented by administration of anti-NGF neutralising
antibodies or a TrkA-IgG fusion molecule (Woolf et al.,
1994;
McMahon et al., 1995). Some of the hyperalgesic action of
NGF
is exerted directly on sensory neurones, but indirect
actions
via mast cells, neutrophils and sympa