Embed Size (px)
Citation preview

Współczesna diagnostyka
i monitorowanie leczenia
zaburzeń hemostazy
Anna Raszeja-SpechtKatedra Biochemii Klinicznej
Zakład Medycyny Laboratoryjnej GUMedWarszawa 12. 10. 2016

Co nowego – pytania:
Czy obserwujemy istotne zmiany w
fizjologii/ patofizjologii krzepnięcia?
Czy zmieniły się schematy postępowania
diagnostycznego?
Czy pojawiły się nowe metody
diagnostyczne?
Czy, kiedy i jak monitorować leczenie
przeciwrzepliwe?
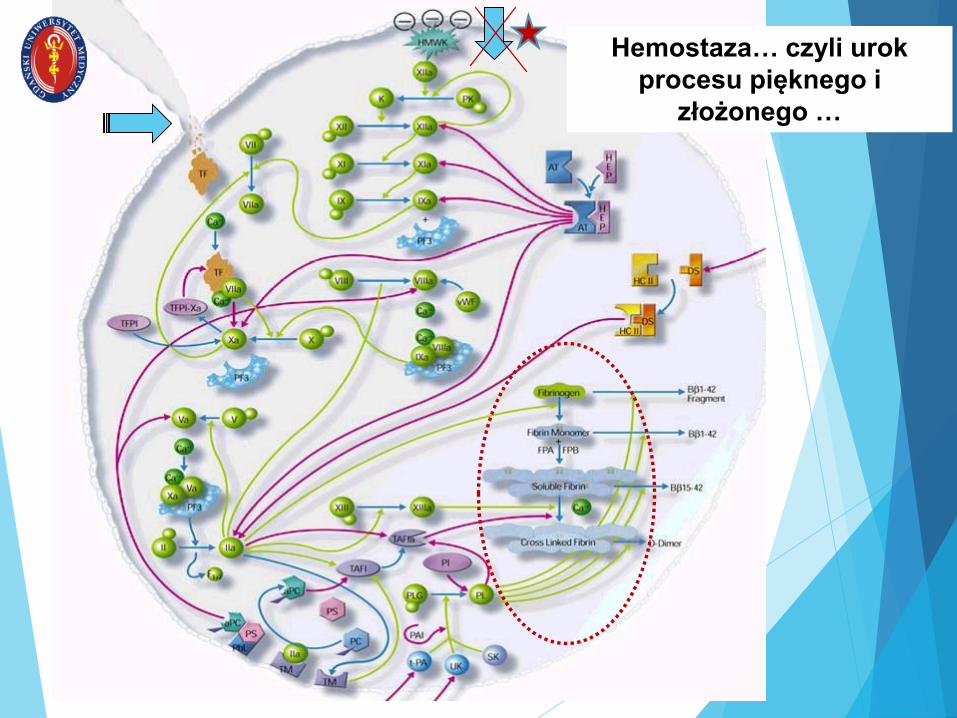
Hemostaza… czyli urok
procesu pięknego i
złożonego …

Skazy krwotoczne =
wrodzona lub nabyta skłonność do
krwawień
Skazy naczyniowe
•Skazy naczyniowe wrodzone: naczyniakowatość, choroby tkanki łącznej
•Skazy naczyniowe nabyte: zespół Schoenleina-Henocha, zaburzenia ściany naczyń i budowy naczyń (kruchość, przepuszczalność naczyń) - plamice
Skazy płytkowe
•Małopłytkowości, nadpłytkowości, zaburzenia funkcji płytek (trombocytopatie)
•Współistnienie zaburzeń jakościowych i ilościowych
Skazy osoczowe
•Skazy osoczowe wrodzone – ilościowe i jakościowe niedobory czynników
•Skazy osoczowe nabyte (awitaminoza K, choroby wątroby, obecność przeciwciał p-czynnikom krzepnięcia)
Zaburzenia mieszane
•Zwiększone zużycie/niedobory: DIC, hiperheparynemia, masywne przetoczenia krwi
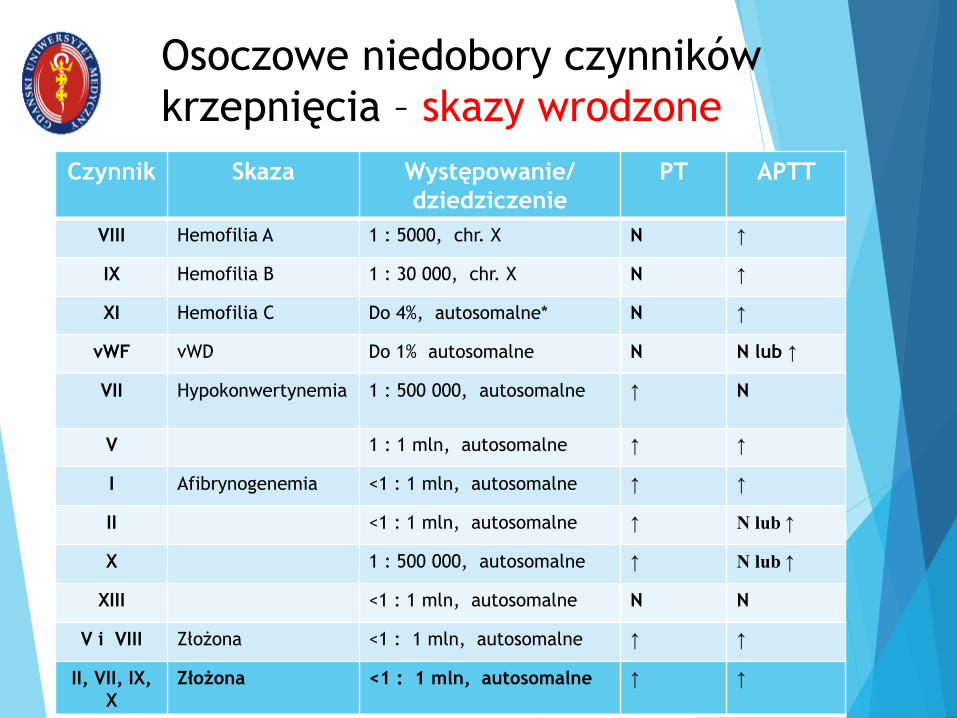
Osoczowe niedobory czynników
krzepnięcia – skazy wrodzone
Czynnik Skaza Występowanie/
dziedziczenie
PT APTT
VIII Hemofilia A 1 : 5000, chr. X N ↑
IX Hemofilia B 1 : 30 000, chr. X N ↑
XI Hemofilia C Do 4%, autosomalne* N ↑
vWF vWD Do 1% autosomalne N N lub ↑
VII Hypokonwertynemia 1 : 500 000, autosomalne ↑ N
V 1 : 1 mln, autosomalne ↑ ↑
I Afibrynogenemia <1 : 1 mln, autosomalne ↑ ↑
II <1 : 1 mln, autosomalne ↑ N lub ↑
X 1 : 500 000, autosomalne ↑ N lub ↑
XIII <1 : 1 mln, autosomalne N N
V i VIII Złożona <1 : 1 mln, autosomalne ↑ ↑
II, VII, IX,
X
Złożona <1 : 1 mln, autosomalne ↑ ↑

HEMOFILIA A
PODŁOŻE GENETYCZNE
Gen FVIII sklonowano w 1984 r., lokalizacja: długie ramię chromosomu X
(Xq28). Wielkość genu: 186 000 par zasad, 26 eksonów stanowi 4% genu
(96% to introny)
Inwersja intronu 22 (INT22) – ok. 50%: dystalna (83%), proksymalna (16%)
lub mieszana (1%), inwersja intronu 1 – ok. 2%
Inne duże rearanżacje w obrębie INT22 (Del22, Dup22) – rzadko
Delecje (duże/małe), rzadziej insercje
Liczne mutacje punktowe (> 1200), w obrębie całego genu FVIII
RYZYKO INHIBITORA CZ. VIII:
– największe w przypadku dużych delecji, potem INV22, mutacji
nonsensownych, mutacji splicingowych
– ale: również geny składników układu immunologicznego, rasa, czynniki
środowiskowe)

HEMOFILIA B
PODŁOŻE GENETYCZNE
Gen FIX sklonowano w 1982 r., lokalizacja: długie ramię
chromosomu X (Xq27), wielkość: 34 000 par zasad, 8
eksonów
Opisano ponad 2100 mutacji we wszystkich regionach
genu FIX:
mutacje punktowe (najczęstsze)
mutacje miejsc splicingowych
mutacje „przesunięcia ramki odczytu”
duże delecje/rearanżacje
dziedziczenie, obraz kliniczny, postacie – jak w hemofilii A
ok. 7 razy rzadsza od hemofilii A

CHOROBA VON WILLEBRANDA
epidemiologia
Defekty czynnika von Willebranda występują u
1% populacji.
Najczęstsza skaza, rozpoznawana zbyt rzadko
(postacie łagodne).
Gen VWF: 12p13.3, 52 eksony + 51 intronów
Pseudogen VWF: położony na chromosomie 22
(nieaktywna kopia genu, utrudniająca
diagnostykę genetyczną)
DIAGNOSTYKA MOLEKULARNA JEST
PRZYDATNA W ROZRÓŻNIENIU:
TYPU 1 OD 2
PODTYPÓW TYPU 2 (głównie A i M)
TYPU 2N OD ŁAGODNEJ HEMOFILII A
Erik von Willebrand
1926 r. (Finlandia)
„pseudohemofilia”

Klasyfikacja choroby von Willebrandawg..Medycyna Praktyczna, Zalecenia PTHiT 12, 2008, modyfikacja 2015
Typ Charakterystyka Postaćkliniczna (krwawienia)
1 Częściowy niedobór ilościowy, proporcjonalne obniżenie antygenu i aktywności
Łagodnalubumiarkowana
2A Niedobór jakościowy: upośledzona adhezyjna funkcja płytek, zmniejszenie ilości/brak dużych multimerów
Różna
2B Niedobór jakościowy: wzrost powinowactwa vWF do płytkowej GP Ib/IX,nieprawidłowe/brak dużych multimerów
Różna
2M Niedobór jakościowy: obniżenie powinowactwa vWF do płytkowej GP Ib/IX, brak zmian multimerowych
Różna
2N Niedobór jakościowy: spadek powinowactwa vWF do czynnika VIII
Różna
3 Całkowity niedobór ilościowy z dużym obniżeniem aktywności czynnika VIII
Ciężka

CHOROBA VON WILLEBRANDA
podłoże genetyczne(1)
Typ 1: Szerokie spektrum mutacji, głównie w regionie D4
domeny CK
Typ 2: Mutacje w określonych regionach genu, np. domena A1 VWF w
typie 2B, domena D` w typie 2N, domena A3 przy zaburzeniu
wiązania do kolagenu (2A)
Typ 3: heterozygoty mogą mieć objawy kliniczne! Duże/małe delecje, insercje, szereg mutacji punktowych
Najczęstsza mutacja w Europie (75% polskich rodzin): delecja
punktowa w eksonie 18
czasem dziedziczenie dwóch defektów VWD typu 1

CHOROBA VON WILLEBRANDA
podłoże genetyczne (2)
Zaburzenia modyfikacji potranslacyjnej:
Mutacje w obrębie domeny CK – zaburzenie dimeryzacji VWF
Mutacje w obrębie domen D1-D3 – zaburzenie multimeryzacji VWF
Warianty podtypu 2A
Homozygotyczne: fenotyp typu 3

Badania przesiewowe w kierunku
choroby von Willebranda (1)
TYP Czas krwawienia*
Czas okluzji(PFA-100 lub 200)
Liczba płytek APTT
1(75-80%) N lub Prawidłowa N lub
2A(15%) Prawidłowa N lub
2B Obniżona N lub
2M N lub Prawidłowa N lub
2N N N Prawidłowa
3 Prawidłowa
Med. Prakt. Zalecenia PTHiT, 2008 i 2014
Wg. Hayward, Int J Lab Hemat 2014, 36, 334-340

Badania celowane w przypadku
podejrzenia choroby von Willebranda
(2)TYP VIII:C vWF:Ag vWF:RCo RIPA
1 lub N lub N
2A lub N
2B* lub N lub N StymulacjaRIPA-LD
2M lub
2N N N N
3 lub brak
lub brak Brak (?)
Wg. Hayward, Int J Lab Hemat 2014, 36, 334-340
* vWF – wiązanie ze zmutowaną GPIbα met. ELISA

Schemat postępowania w podejrzeniu
choroby von Willebranda (zespołu vW)
Wywiad
Cechy skazy (częstość, rodzaj, lokalizacja)
Obciążenia rodzinne, choroby przewlekłe
Badanie fizykalne
Cechy i objawy skazy, lokalizacja, choroby predysponujące
Układ krzepnięcia
APTT
Morfologia
PLT
Czas okluzji
(PFA-100) lub inne
Badania podstawowe - celowane
vWF:Ag vWF:RCo
Badania dodatkowe
Multimery
vWF
Wiązanie: kolagen,
cz.VIII, GPIb
vWF w płytkach
Agregacja po botrocetynie
Przeciwciała anty-vWF
Analiza
DNA
PropeptydvWF
cz. VIII:C RIPA
Czas krwawienia (?)
ADAMTS-13 (?)

Zależność ADAMTS-13/ vWF
Prawidłowa hemostaza
Krwawienia
Stosunek:
ADAMTS-13/ vWF
O.30-1.5 μg/ml do 5-15 μg/ml
ADAMTS-13/ vWF
O,30-1,5 μg/ml do <5 μg/ml
Zakrzepica - TTP
ADAMTS-13/ vWF
<O,30μg/ml do <15 μg/ml

RZADKIE OSOCZOWE SKAZY
KRWOTOCZNE (1) dziedziczone w sposób autosomalny
podejrzenie: na podstawie badań podstawowych (APTT, PT, TT) – poza
niedoborem czynnika XIII i alfa2-antyplazminy (badania przesiewowe
prawidłowe, skaza ciężka klinicznie)
leczenie: FFP (poza koncentratem cz. XIII, cz. VII oraz fibrynogenu)
JEDNOSTKI CHOROBOWE: niedobór lub dysfunkcja fibrynogenu (afibrynogenemia, hipofibrynogenemia,
dysfibrynogenemia)
niedobór czynnika VII
niedobór czynnika XI (hemofilia C)
niedobór czynnika X, V, II, skojarzony niedobór czynnika V i VIII
skojarzone niedobory czynników zależnych od wit. K
niedobór czynnika XIII

RZADKIE OSOCZOWE SKAZY
KRWOTOCZNE (2)
dziedziczenie zwykle AR
częstość postaci homozygotycznych:
Niedobór FVII – 1/500 000
Pozostałe: < 1/1 000 000 (poza pewnymi populacjami (Żydzi, Śr. Wschód, Indie),
gdzie częstość niektórych skaz osoczowych jest znacznie większa (np. małżeństwa
osób spokrewnionych)
zwykle zaburzenie ilościowe (typ I), niekiedy jakościowe (typ II)
nosiciele (heterozygoty) – zmienny obraz kliniczny
Podłoże genetyczne: mutacja w genie danego czynnika krzepnięcia, za
wyjątkiem:
skojarzonego niedoboru cz. V i VIII (mutacja genu białka uczestniczącego w
wewnątrzkomórkowym transporcie czynników krzepnięcia)
skojarzonego niedoboru cz. II, VII, IX i X (mutacja genu enzymu uczestniczącego
w metabolizmie wit. K)

NIEDOBÓR FIBRYNOGENU
podłoże genetyczne
mutacje łańcuchów Aα, Bβ, γ
afibrynogenemia, ciężka hipofibrynogenemia – zwykle
mutacje null (delecje, nonsense, miejsc splicingowych),
niekiedy mutacje prowadzące do upośledzonej sekrecji
hipofibrynogenemia – dziedziczenie AD (często heterozygoty
pod względem mutacji null)
dysfibrynogenemia – zwykle dziedziczenie AD (w 90%
missense, czasem delecje/insercje)
„POLSKIE FIBRYNOGENY” – Poznań, Zabrze, Kraków, Gdańsk

Interpretacja wyników oznaczeń APTT, PT i
TT u pacjenta z objawami skazy krwotocznej
Rodzaj zaburzenia Badania uzupełniające APTT PT TT
Niedobór czynników VIII, IX, XI,
choroba von Willebranda
Oznaczenia w/w czynników,
próby korekcyjne (APTT)P N N
Niedobór czynnika VII, złożone
niedobory czynników K-zależnych
(niewielkie)
Oznaczenia czynników II, VII, X N P N
Niedobór II, V, X, złożone niedobory
czynników K-zależnych (znaczne),
zaburzenia po masywnych transfuzjach
Oznaczenia w/w czynników,
próby korekcyjne (APTT, PT)
P P N
Hypo i dys-fibrynogenemie, choroby
wątroby, DIC, obecność heparyny
Oznaczenia fibrynogenu,
dimeru D, FDP,
czas batroksobinowy
(reptylazowy),
testy parakoagulacji
P P P
Ważne – opracowanie schematów postępowania!

Osoczowe zaburzenia krzepnięcia
– skazy nabyte
Leczenie VKA (acenokumarol, warfaryna)
II, VII, IX, X – zależne od witaminy K
Niedobory pokarmowe, zespoły złego wchłaniania
II, VII, IX, X – zależne od witaminy K
Niewydolność wątroby Większość czynników
Amyloidoza Czynnik X
Nowotwory mieloproliferacyjne Czynnik V
DIC Większość czynników
Nabyty zespół von Willebranda vWF, VIII
Nabyta hemofilia A Czynnik VIII
Masywne przetoczenia krwi, leczenie heparyną Większość czynników (+ płytki)

Schemat diagnostyczny
w nabytej hemofilii
Izolowany przedłużony APTT
Potwierdzenie lub wykluczenie kontaminacji heparyną…
Test mieszania 1:1, PP:NPP, 37°C, czas 0 i 2h
Korekcja w czasie 0 i 2h
Podejrzenie niedoboru czynników –oznaczyć cz. VIII, IX, XI i XII
Niedobór pojedynczego czynnika
Brak korekcji (lub słaba)
Podejrzenie inhibitora: oznaczyć LA, cz. VIII, IX, XI
LA ujemny ORAZ izolowane obniżenie VIII (hamowanie zależne od czasu)
Miano inhibitora cz. VIII (j.Bethesda)
Nabyta hemofilia A
Konieczna korelacja z objawami klinicznymi!
LA dodatni
Antykoagulant tocznia
Konieczna korelacja z objawami klinicznymi
International Journal of Laboratory HematologyVolume 36, Issue 3, pages 398-407, 18 APR 2014 DOI: 10.1111/ijlh.12210

Schemat diagnostyczny
w podejrzeniu DIC
Wywiad: przyczyny DIC + cechy krwawienia
Badanie fizykalne: cechy krwawienia, choroby współistniejące…
Badania przesiewowe i uzupełniające
Przesiewowe
Liczba płytek
APTT, PT, TT, BtT
Fibrynogen
Uzupełniające I
AT, DD, FDP, MF, C
Uzupełniające II
TAT, PAP
Czynniki osoczowe i płytkowe *
• *Plazminogen, αAP, PAI-1, t-PA, czynniki V, VII, VIII, trombomodulina, TF, PF4, βTG, TXA2….
tromboelastografia
• Biomarkery molekularne: F1+2, FPA, Fib b-β 15-42 i 1-118, peptydy z cz. IX i X
Zmodyfikowane wg. Int.J.Lab.Hem 2014, 36, 228-236
oraz 2016, 38, 151-159

Badania laboratoryjne w
ostrym i przewlekłym DIC
Parametr Ostry DIC Przewlekły DIC
PLT <100 G/L ↓↑ lub N
PT ↑↑ ↑ lub N
APTT ↑↑ ↑ lub N
TT ↑↑ ↑ lub N
Fibrynogen ↓↓↓ ↓↑ lub N
Erytrocyty-morfologia Fragmentocyty N
Dimer D (FDP)* ↑↑ ↑ lub N
AT ↓ ↓
Białko C ↓ ↓ lub N
Zmodyfikowane wg. Int.J.Lab.Hem 2014, 36, 228-236
oraz 2016, 38, 151-159
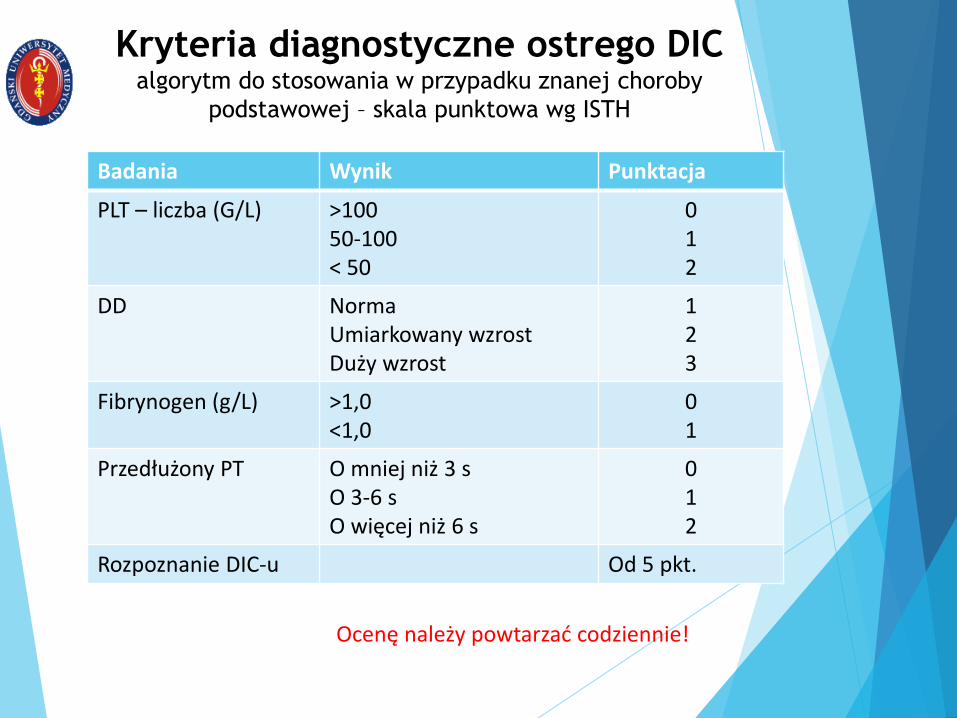
Kryteria diagnostyczne ostrego DICalgorytm do stosowania w przypadku znanej choroby
podstawowej – skala punktowa wg ISTH
Badania Wynik Punktacja
PLT – liczba (G/L) >10050-100< 50
012
DD NormaUmiarkowany wzrostDuży wzrost
123
Fibrynogen (g/L) >1,0<1,0
01
Przedłużony PT O mniej niż 3 sO 3-6 sO więcej niż 6 s
012
Rozpoznanie DIC-u Od 5 pkt.
Ocenę należy powtarzać codziennie!

Kryteria diagnostyczne przewlekłego DIC
Badania Wynik Punktacja
Czy pacjent ma schorzenie sprzyjające rozwojowi DIC?
TakNie
20
PLT – liczba (G/L)
Dynamika zmian PLT
>100<100Wzrost/Bez zmian /Spadek
01
-1/0/1
Przedłużony PT
Dynamika zmian PT
O mniej niż 3 sO więcej niż 3 sWzrost/ Bez zmian/Spadek
01
1/0/-1
DD
Dynamika zmian DD
NormaWzrostWzrost/Bez zmian/Spadek
01
1/0/-1
AT (stężenie/aktywność) NormaZmniejszenie
-10
Białko C NormaZmniejszenie
-10
TATF1+2 lub inne
NormaZmniejszone
-10
Rozpoznanie przewlekłego DIC-u Gorsze rokowanie Od 5 pkt

Różnicowanie stanów z
przedłużonym APTT - podsumowanie
Badania Ostry DIC Nabyta
hemofilia
Antykoagulant
tocznia
Heparyna
APTT ↑↑↑ ↑↑↑ ↑↑↑ ↑↑
PT ↑↑ N N N lub ↑
TT ↑↑↑ N N ↑↑↑
Fibrynogen ↓↓↓ N N N lub ↓ (met.)
Cz. VIII ↓ ↓↓↓ N lub ↓ N
Inne czynniki ↓↓↓ N lub ↓ N lub ↓ N
DD ↑↑↑ +/- 0 0
Krwawienie ++++ ++++ 0 +/-
Zakrzepica MODS 0 ++/+/- 0

Przypadek
65-letnia kobieta została skierowana na konsultację specjalistyczną przezstomatologa z powodu silnego krwawienia podczas ekstrakcji zęba. Wewczesnym dzieciństwie wykazywała skłonność do krwawień z nosa oraz doprzedłużających się krwawień z drobnych skaleczeń. Nie obserwowanowylewów do mięśni ani stawów.
Obecnie leczona z powodu szpiczaka; przyjmuje okresowo leki przeciwzapalne zpowodu bólów stawowych.
Zlecono badania przesiewowe układu krzepnięcia:
BT PLT APTT PT-INR Fib
min G/L s g/L
>15 220 42 1,1 3,3
Diagnoza?
Zaburzenia hemostazy pierwotnej?
Wrodzone czy nabyte?
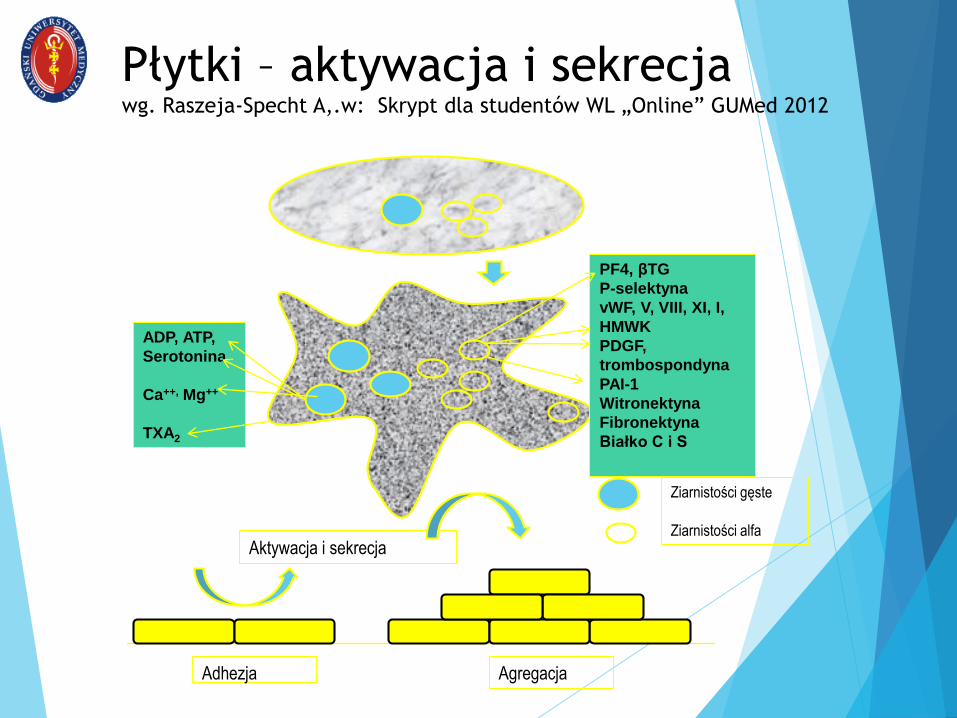
Płytki – aktywacja i sekrecjawg. Raszeja-Specht A,.w: Skrypt dla studentów WL „Online” GUMed 2012
ADP, ATP,
Serotonina
Ca++, Mg++
TXA2
PF4, βTG
P-selektyna
vWF, V, VIII, XI, I,
HMWK
PDGF,
trombospondyna
PAI-1
Witronektyna
Fibronektyna
Białko C i S
Adhezja Agregacja
Aktywacja i sekrecja
Ziarnistości gęste
Ziarnistości alfa

Badanie liczby płytek
krwi
Metoda: tradycyjna lub analizatory hematologiczne (np. metoda impedancyjna vs. optyczna)
Zaburzenia: małopłytkowości lub nadpłytkowości
Fałszywie zmniejszona liczba płytek: mikroskrzepy, tworzenie agregatów, małopłytkowość rzekoma, obecność tzw. płytek olbrzymich….
Fałszywie zwiększona liczba płytek: obecność fragmentów RBC lub WBC oraz zanieczyszczeń analizatora lub odczynników, znaczna mikrocytoza RBC…
Raszeja-Specht A., Topolewska I. Acta Hemat .Pol. 2013

Metody badania czynności
krwinek płytkowych
Testy ogólne - czas krwawienia, czas okluzji (PFA-100), czas tworzenia skrzepu (Xylum), tromboelastometria
Badania adhezji i agregacji płytek - w osoczu PRP lub w pełnej krwi (agregometria – impedancyjna, optyczna, przepływowa, szybkie testy funkcji -RPFA, badania reologiczne….
Pomiary markerów aktywacji:
β-tromboglobulina, czynnik płytkowy 4
metabolity TXA2 w moczu
rozpuszczalna P-selektyna (sCD62P) i serotonina
Badania metodami cytometrii przepływowej
wykrywanie krążących zaktywowanych płytek
wykrywanie mikropęcherzyków pochodzenia płytkowego
wykrywanie krążących agregatów płytkowych
Ocena aktywności prokoagulacyjnej
Badanie polimorfizmu glikoprotein i genów płytkowych

WRODZONE TROMBOCYTOPATIE
trombastenia Glanzmanna
zespół Bernarda-Souliera
zaburzenia wydzielania
ziarnistości płytkowych (choroby
puli magazynowej), np. zespół
szarych płytek
defekty receptorów płytkowych
(np. receptora dla kolagenu)
LICZBA PŁYTEK ZWYKLE
PRAWIDŁOWA lub
ŁAGODNA MAŁOPŁYTKOWOŚĆ

TROMBASTENIA GLANZMANNA
Mutacje genów kodujących glikoproteiny GPIIb (ITGA2B) i GPIIIa (ITGB3) na
chromosomie 17q21 (położone blisko siebie) – opisano > 100 mutacji,
wrodzony defekt syntezy kompleksu glikoprotein IIb/IIIa (receptor dla
fibrynogenu, trombospondyny, fibronektyny)
TYP 1: ekspresja GPIIb/IIIa < 5%
TYP 2: ekspresja GPIIb/IIIa 10-20%
WARIANT: ekspresja GPIIb/IIIa > 50%(funkcja kompleksu upośledzona)
obraz kliniczny: ciężka skaza skórno-śluzówkowa od wczesnego dzieciństwa
diagnostyka: przedłużony czas krwawienia, liczba płytek w normie, brak
agregacji pod wpływem ADP, kolagenu, kw. arachidonowego, adrenaliny,
agregacja pod wpływem rystocetyny prawidłowa
ZESPÓŁ BERNARDA-SOULIERA
mutacje genów kodujących glikoproteiny GP1BA, GP1BB, GP9
wrodzony defekt syntezy kompleksu Gp Ib/IX/V (receptor dla czynnika von
Willebranda)
obraz kliniczny: ciężka skaza skórno-śluzówkowa
diagnostyka: przedłużony czas krwawienia, umiarkowana małopłytkowość,
obecne płytki olbrzymie, brak agregacji pod wpływem rystocetyny (pod
wpływem pozostałych agonistów - prawidłowa)
Haemophilia
Volume 21, Issue 6, pages e510-e513, 31 JUL 2015 DOI: 10.1111/hae.12777
http://onlinelibrary.wiley.com/doi/10.1111/hae.12777/full#hae12777-fig-0001

Stare i nowe leki
przeciwkrzepliwe
Poziom „1” – TF/VIIa – TFPI, NAPc2
Poziom „2” – inhibitory Xa – pentasacharydy
(Fondaparinuks – Arixtra; Rywaroksaban –
Xarelto; Apiksaban - Eliquis)
Poziom „3” - bezpośrednie inhibitory
trombiny (Dabigatran- Pradaxa)

Schemat działania bezpośrednich i
pośrednich inhibitorów IIa i Xa
Raszeja-Specht A., Badanie i diagnoza 12, 2012

Działanie BDA - rywaroksabanu i dabigatranu
Rywaroksaban Dabigatran
Działanie antykoagulacyjne Anty-Xa Anty-IIa
Biodostępność 80% 6-7%
Początek działania Po 30 min (Tmax 2,5-4 h) Po 30 min (Tmax 0,5-3 h)
Czas działania
Czas połowicznej eliminacji
24 h
5-9 h (starsi do 13 h)
24-36 h
7-9 h (starsi do 14 h)
Droga eliminacji Nerkowa i wątrobowa* Nerkowa 80%, z żółcią 20%
Interakcja za dietą i alkoholem Niewielka lub brak
Interakcja z lekami Tak, inhibitory P450 i Gp-P Tak, inhibitory pompy H+ i
inh./akt. Gp-P
Dawkowanie Stałe
Monitorowanie leczenia Brak wskazań w warunkach standardowych *
Zastosowanie w ciąży i laktacji Przeciwwskazane
Zastosowanie w ciężkich chorobach
nerek
Ostrożne lub przeciwwskazane
Powrót do normy po odstawieniu leku 24 h 24-36 h
Antidotium Problematyczne, kosztowne (Idarucimab/Dabigatran)

Badania laboratoryjne w
monitorowaniu leczenia dabigatranem
PT - INR
APTT
TT (zmodyfikowany – w osoczu rozcieńczonym osoczem prawidłowym)
ECT
Materiał do badań:
osocze pacjentów leczonych
dawka terapeutyczna Pradaxa – 75-120-220 mg/dobę, p.o.

Badania laboratoryjne w monitorowaniu
leczenia rywaroksabanem
Aktywność anty-Xa
PT wyrażany jako INR-RIVA
Czas dRVVT (?)
APTT
Dawka terapeutyczna/profilaktyczna:
Xarelto 10-15-20 mg/dobę, p.o.

Interferencja nowych leków
przeciwkrzepliwych w badaniach układu
krzepnięcia
Dabigatran
(Pradaxa)
Rywaroksaban
(Xarelto)
PT ↑ ↑↑↑
APTT ↑↑↑ ↑↑
TT ↑↑↑↑ bz.
Fibrynogen (Clauss) Obniżony lub bz. (?) bz.
Czynniki – metody
koagulometryczne
Obniżone Obniżone
LA – dRVVT (APTT?) Fałszywie dodatni Fałszywie dodatni ↑ ↑
AT chromogenna (Xa)
AT chromogenna (IIa)
bz
↑
↑
bz
Białko C i S
(koagulometrycznie)
↑↑ ↑
APCR – koagulometrycznie
(APTT)
↑ ↑
ECT ↑↑ -
Int J Lab Hem 2013, 35, 262
Int J Lab Hem 2014, 36, 261-268

Wnioski: czego nie oznaczać -
ostrożna interpretacja (?)
AT – zawyżona, maskuje niedobór
Fibrynogen - zaniżony (met. Claussa)
APC-R – zawyżony, maskuje defekt
PC i PS – można, ale nie-koagulometrycznie…
LA i inne – silny wpływ, fałszywie dodatni?
Inne – czynniki krzepnięcia…

BDA
Rywaroksaban Dabigatran
Anty-Xa Anty-IIa
Apiksaban
PT
przesiew
Anty-Xa
monitorowanie
Anty-Xa
monitorowanieAPTT
przesiew
dTT lub ECT
monitorowanie
wg. Raszeja- Specht A., A. Michno,
Diagnostyka Laboratoryjna 2015, 51, 221-228
Schemat monitorowania leczenia - BDA
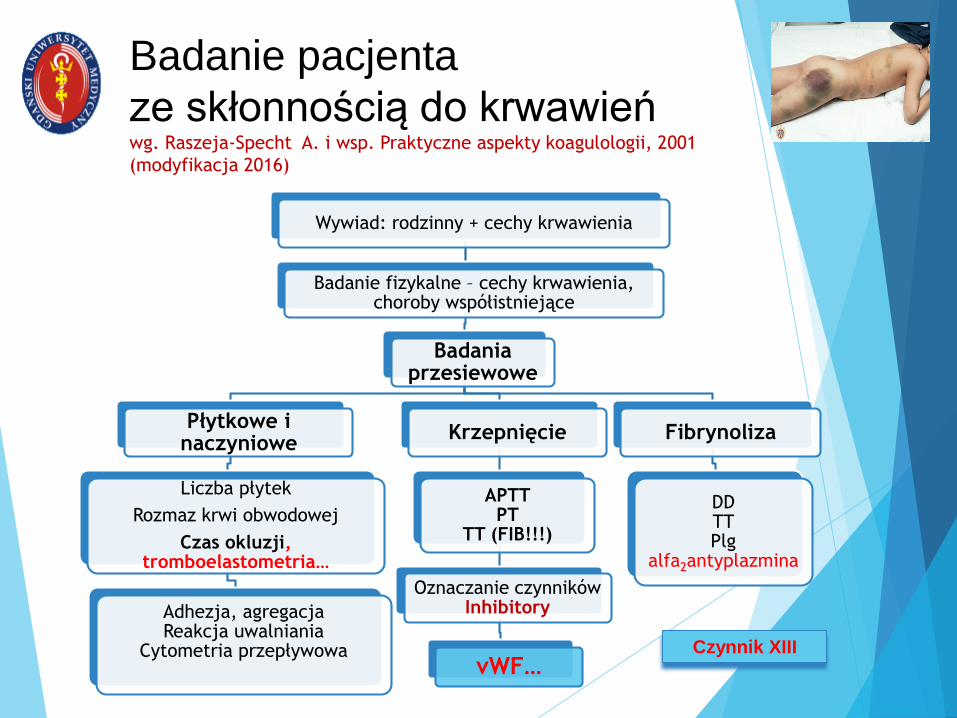
Badanie pacjenta
ze skłonnością do krwawieńwg. Raszeja-Specht A. i wsp. Praktyczne aspekty koagulologii, 2001
(modyfikacja 2016)
Wywiad: rodzinny + cechy krwawienia
Badanie fizykalne – cechy krwawienia, choroby współistniejące
Badania przesiewowe
Płytkowe i naczyniowe
Liczba płytek
Rozmaz krwi obwodowej
Czas okluzji, tromboelastometria…
Adhezja, agregacjaReakcja uwalniania
Cytometria przepływowa
Krzepnięcie
APTTPT
TT (FIB!!!)
Oznaczanie czynnikówInhibitory
vWF…
Fibrynoliza
DDTTPlg
alfa2antyplazmina
Czynnik XIII
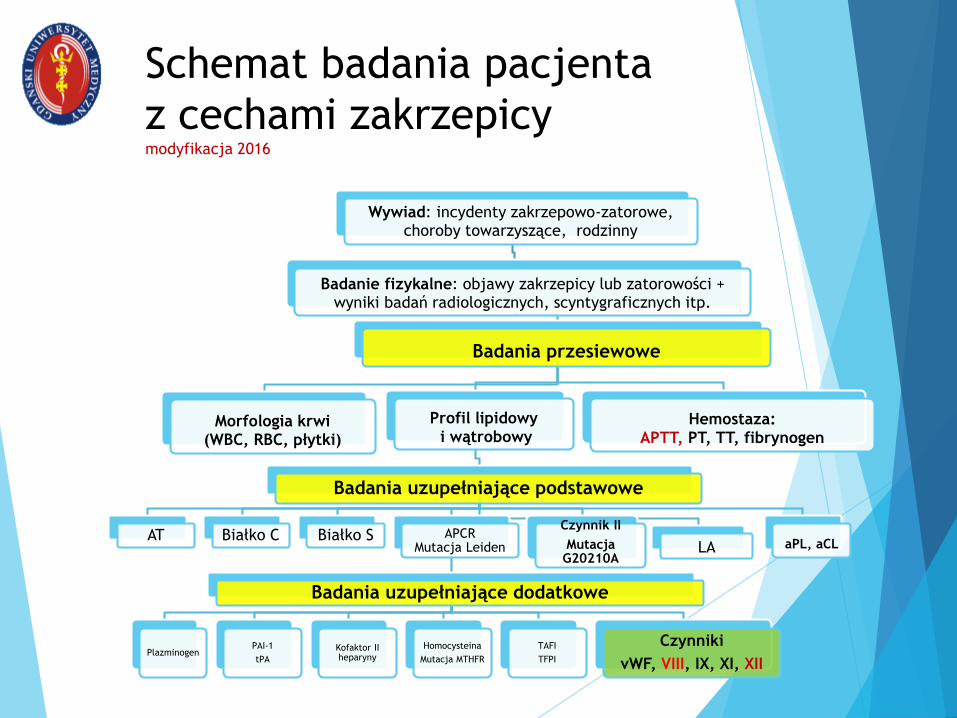
Schemat badania pacjenta
z cechami zakrzepicymodyfikacja 2016
Wywiad: incydenty zakrzepowo-zatorowe, choroby towarzyszące, rodzinny
Badanie fizykalne: objawy zakrzepicy lub zatorowości + wyniki badań radiologicznych, scyntygraficznych itp.
Badania przesiewowe
Morfologia krwi (WBC, RBC, płytki)
Profil lipidowyi wątrobowy
Badania uzupełniające podstawowe
AT Białko C Białko S APCRMutacja Leiden
Badania uzupełniające dodatkowe
PlazminogenPAI-1
tPA
Kofaktor II heparyny
Homocysteina
Mutacja MTHFR
TAFI
TFPI
Czynniki
vWF, VIII, IX, XI, XII
Czynnik II
Mutacja G20210A
LA aPL, aCL
Hemostaza:APTT, PT, TT, fibrynogen





























