Embed Size (px)
Citation preview

- ~v A - NOVO
/SETA~/pRESS/ r-.I 1\ P"
L_h j)
Environmental .r".i."t"gy and Chemi,'try, V"L 2\, N". 5,1'1'.980-983,2002" 2002 SETAC
I'rinted in the USA11730-72"'\102 $9.1)4) .t .1111
'-
OXlDATlON CHEMISTRY OF ACID-YOLATlLE SULFIDE DURING ANALYSIS
ADRIAN M. GONZALEZ*Amtcch Scrvi"cs In<:.,Dn lIannah Avcrmc, Knoxvillc, "Tcnncs.'<Cc:\7'>21,USA
(R.xâved 21 M{~y 2001; Accel'ted 21 Oct<)ner 2001)
Ab5tract.--The susccptibility of some componcnts of sediment add-volatile sul/ide (AVS) to chemical oxidation is a critical factorimpacting accurate measurcment of AVS in se<!iment samples. This well-documeutcd susccptibility tooxidatiau 100to Ih<:n:quircmentfor oxygen-free cou(ütions in lhe analytical method devcloped for AVS. lu light ofthis aL'"Utcpott.'t1tial tooxidi7.e, lhe sercnilipitousfinding that Rir can bc use<! in thc analysis of sOOiment AVS is countt.'Iintuitive and unexpccted. To dcmonstrate and investigatelhis interesting observation, extraction cxperiments were per{ormed using aqueous and solid-pbase sullide spcci<."S.Expcrimenlsusing Rir as lhe earrier gas showcd a mean percentage recovery of sul/ide matching that af traditional (nitrogen gíts) analysis (i.e.,>91"1..) and a time to completion ofless lhan 30 mio for aqueous sulfidc and less than (,() mio for scilimcnt samples. These resultsare consistenl with those of sul/ide oxidation studles rcported in lhe líterature. Usi~ mr asthe anaIytical carrier gas can provido:an interesting alternative for devclopiog aR analytical method to determine AVS paramcters in lhe field.
Keywords.--Acid-volalile sulfide Metal toxidty &-diment
INTROD1JCTION
Certain componenls of sediment acid-volatile sulfide (AVS;í.e., labile metal sulfides [primarily amorphous iron (11) andmanganese (fI) sulfideJ, FeS, and/or MnS)arequíte susceptíbleto chemical oxidalÍon [1-3). Detennining concentrations 01'simultaneously extmcled metaIs (SEM) and AVS is useful inscreening sediments for potential toxicity doe to elevated metalconcentrations. The accuracy of lhe SEM and AVS data, how-ever, is limiled by lhe slability of AVS in lhe sediment sample.Changes in AVS concentration, primarily through oxidationduring sampling, shipment, and manipulation, can lead to in-accurate SEM:AVS ratios and to inaccurate toxico}ogica} clas-sification of sedimcnts [4,5]. The current, /aboratory-basedmelhod foI determining SEM and se.iiment AVS concentra-tíons [6J altempts to do this by using ínert gas (e.g., nitrogen)10transfer hydrogen sulfide (B}S) released during sample acid-ífication fiom tJlC reaction vessel to a suUide-stabilizing me-diurno The method BIso specífies using flasks, water, acid rc-agent, and su/fide-stabilizíng solution purged of oxygen (i.e.,deaerated).
The goal of tbis papel" was to document lhe successful useof air as a carricr-gas matrí.1\ for AVS ana/ysis. AJthough ini-lially counterintuitive, this phenornenon is repcatable and re-producibJe and can be cxplained in light of sulfide oxidationIheory as reflected in lhe current lileralure. Thís I)henomenonmíght Jind some usefulncss ín lhe devclopmclll of analyticalmclhods tor dctemlining AVS paranlelers in lhe field.
MATERIALS ANO METIIOOS
To demollstrale. lhe negligihle effe.ct ~~fair on sulfode re-covery during AVS analysis, extraction cxpcrirnc1Its were pcr-formcd on sulllde species ín severa I malríces. Prcpared solu-lions ofsodiurn sulfidc, sampled aliquots oinatural {resh~atersedimenl, and prepared samples of a laboratory-iormulaledsedimellt (LrS) amcndcd wíth synthetic AVS were analyzed
* Tu whom cmrc~pm\<km:c may bc a_"lres","\accn 1117(l.1)hotmait.<:om.\
Oxidation Analysis
for sulfidc content. Surface-rinsed sodium sulfide crystals(Na1S'9H2O) were dissolved in deaerated, àistilled walcr tomake stoek solutions lhat were then standarrli1',ed iorlomeuí-
cal/y. Oue samp/e of a freshwater sediment was collected ftoma suburban pond (Rivcrdale, NY, USA) using a Petite-Ponardredge (Wildlife Suprir, Buffalo, NY, USA) a1ld passedthrough a I.OO-mm sieve to remove Jarge gravei and debris.PolentiaJ oxidation caused by sieving this se.iiment samplewas ilTelevant to this study, because the goal was to comparerclatíve AV3 concc1luations in spli\, hornogenized suhsamplesby two methods (i.e., aír.and nitrogen). Thc LFS batches wcrcmade by mixing equal volume\: 01' a natural \:ubmergcd clay(Oak Ridge, TN, USA; sieved to a particle size of < 1,000I-Lm) anã sand (from a local vendar, KnoxviJIe, TN, USA;sieved to a particle si2'.c of <500 ILm). ~nthetic AVS for- "
mu/ations [7Jwere made bycomhiningstoichiometricam~ijJ1ISrof iron (ll) sulfate (FeSO..7Hp) and sodium sulfid€' (both ,dissolved in deaemted, distilled walcr) directly within lhe LFS tmatrix to forro the iron (lI) sult,de precipitate. Su1f\de ana1)!s)s \in lhe ínitial Jnvestigations followed lhe mcthod described byAllen ct ai. [6}. wilh the following minor modifications.
The nitrogen-bascd sulfide analytical method (Nz-AVS)used laboratory-grade nitrogen anã Ihrec 125-ml glass Erlen-meye~ flash (Belko Glass, Vineland, NJ, USA) for lhe re-action vessels (i.e., the reaction vessel, pU 4 buffer trap, andsulftde stabili",ation trap). A cartridge 01'oxygen-stripping resin ' "j "
(OxiClear; Diamond Tool anã Die, CIe~'elalld, 011, USA) W3S( rinstalled 'o lhe nitrogen supply line, replacing \he vanadium Ich/oride scrubbing solution described by Allen et aI. [6]. The
reaction vesscl in eacb analytical traio was charged wilh SO\ i \\ml 01' 1 M hydrochloric acid (instead of water) that was de- " '-oxygenated wítb nitrogen for more than 30 min before sampleaddition. '-oss of sulfidc by adding samples díreclly 10 lhe acidwas negligible, because the rcaclion nask was sealed ímllle-diately « 1 s) al\cr addi\ion 01' \he sample. Two sult.de rc-
covery chccks iu this syslem (using lhe 1>1andardizedsodíurnsulfide solutioll) resultcd in 96.6% recovcry.
The air-ha"oo ..ult;.u, analytical m..thod (air-AV~) u"..duu-
')80
Oxidation chemistry 01' AYS during analysis Ellvimlt. Tt,x;.:..,t. Ch<,m. 21, 2002 981
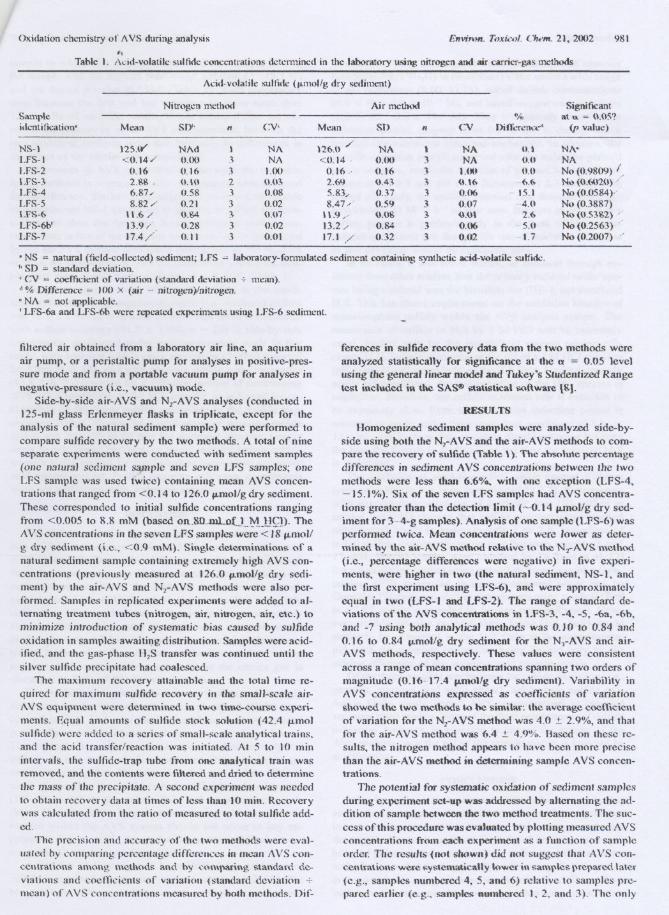
..Table I. Acid-volatilc sulfide conccntrations dctcnnined in lhe laboratory using nitrogen and air CarriL'T-gaSmetbods
.NS = natural (tield-collected) sediment; LFS ~ laboratory-fonnulated sedimcnt containing synthetic acid-volatile sullidc.h SD = standard dcviation.'CV = coefficient 01'variation (standard dcviation .:. mean).d % Difference = IO{) X (air - nitrogen)/nitrogen.'NA = not applicable.I LFS-6a and LFS-6b wcre repeated L"Xperinu..-ntsusing LFS-6 sedirncnt.
fillered air oblaincd from a laboratory air tine. an aquariumair pump. or a peristaltic pwnp for ana1yses in positive-pres-sure mode and from a portable vacuum pump for analyses inncgative-prcssurc (i.c.. vacuum) mode.
Side-by-side air-AVS and N,-AVS analyses (conducted in125-ml glass Erlenmcycr flasks in triplicatc. exccpt for tbeanalysis of Ibe natural sediment sample) were perfonned tocompare suJfide recovery by tbe two melhods. A total ofninescparate experiments were conducted witb scdiment samples(one natural sed imen I ~ple and seve" LFS samples; oneLFS samplc was used twiee) conlaining mcan AVS eoneen-tTalions tbal ranged from <0.14 to 126.0 jLmol/g dry sediment.These eorresponded to inilial sulfide eoncentrations rangingfTom <0.005 to 8.8 mM (based on.81l..mLofJ.M.J:!Ç!). TheAVS concenlrations in the seven LFS samples were < 18 !Lmol/g dry sediment (i.e.. <0.9 mM). Single detcnninations of anatural sediment sample containing extrernely bigb AVS con-centrations (previously measured aI 126.0 jLmol/g dry sedi-ment) by tbe air-AVS and N,-AVS rnetbods were BIso per-forrned. Samples in replícated experimenls were added to al-ternat1ng treatmenl tures (nitrogen, a1T.nittogcn. air, cte.) tomjnjmize introduction of syslernalic bias caused by sulfideoxidation in samples awaiti~g distribuliol1. Samples were aeid-ified. and lhe gas-pbase B,S transfer was contil1ued unlil lhesilver suICIde precipitale bad coalesced.
The maximllm recovery attainab1e and the tota1 time re-quired for maxímum sultide recovery in lhe small-scale air-AVS cquipmenl were detennined in tWQtirne-course experi-menls. Equal amollnts of sulfide stock solulion (42.4 p.molsulfide) were added to a series of small-scaJe analytkal trains.and tbe aeid transfer/reaclion was iniliated. AI 5 to tO min
ínlervals. lhe sulfidc-trap tllbe fcom ooe analylical traio wasremoved. and the contents were filtered and dTied to determinelhe mass 01' I.he predpitale. A secolld experimenl was neededto oblaín recovery data aI times of less tban 10 mino Recoverywas calculaled tTom Ibe ralio of measured to total sulfide add-ed.
The precision and accuracy of 111eIwo rnelhods were eval-ualed hy comparing percenlage diftcrenccs in mcan AVS con-ccntralions among mctho\1$ and by comparing standani de-viatíons and coenieienls of varialíon (standard deviation .;-mean) of AYS eonccnlrations measured by botb melbods. Dít:.
ferences in sulfide rccovery data florn the two rnetbods wcreana1Y7..ed statistiea11y for signiflCance at lhe a = 0.05 leve1
using Lhegeneral linear model and Tukey's Studentized Rangetest inctudcd iR lhe SAS~ statistical IOOftware{Si.
RESULTS
Bomogeni7..ed sediment samplcs were analyzed side-by-sitie using botb lhe N,-A VS and tlle air-A VS melhods to com-pare lhe recovery of sulfide (Table \). The absolute p<..-rccntagedifferences in sediment AVS coneenlTations belween lhe Iwo
metbods were less than 6.6'%, witb one exception (LFS-4.-15.1%). Six oftbe seven LFS samples bad AVS eoneenlra-lions grealer tban the detection limit (-O. t 4 fLIDol/gdry sed-iment for 3-4-g samp1es). Analysis of 000 samp1e (LFS-6) wasperfOlmed twiCd. Mean concentrations weriJ lower as deter-mined by lhe air-i\VS mcthod retative tQ lhe N,-i\YS method(i.e.. pereentage differences were negative) in tive experí-ments. were bigbcr in two (lhe natural sedimenl, NS-l. andtbe first experiment using LFS-6), and were approxirnatclycqual in two (LFS-I and LFS-2). The range of stanciard de-viations ofthe AVS concentrations in LFS-3. -4, -5. -6a. -6b.aRQ -7 usjng bolh ana)yJieaJ metborls was O.JO 10 0.84 and0.16 10 0.84 p.mol/g dry sedirnent for lhe N,-A VS and ai r-AVS melhods. respeetivcly. Tbcse values were consistentacross a range ofmean eoncentrations spanning two orders ofmagnitude (0.16-11.4 fLIDol/g dry sedimenl). Variabi1ity inAVS concentralíons exprcssed as coefficienls of variatíonshowed the two methods to be similar: ,!te average cocffidentof variation for lhe N[AVS rnctbod was 4.0 :t 2.9"/0, and tbalfor lhe aÍf-AVS mcthod was 6.4 ..!.:4.9%. Based on Ibese re-
sutis, tbe nittogen metbod appears 10 have becn more preciseIhan lhe air-A VS metllod in detennining sample AVS eoncen-tralÍons.
The potential for systematic oxidalion 01'sedirnent samplesduring. experiment set-up was addressed by altemating tbe ad-dition of sampte belwecn tlte two mclbod trealrnents. Tbe suc-cess 01'this procedure was evaluated by ptotting measured AVSe<mcentralions from eacb experirnent as a funelion of sampleorder. The resu/ts (oot shown) di<!nol suggest thal AVS con-ccntralions were "ystematkaUy Iowcr in sam\,lcs l\feparc<.-\lalcr(e.g.. samples numbercd 4, 5. and 6) relalive to samples pre-pared earlier (c.g.. samplcs nurnbcrcd I, 2. and 3). The only
Acid-volatile sulfide (,L111Ol/gdry sediment)
Nitrogen mcthod Air mcthod SigniflCant8a111l'le U,I" at fi O.OS'!identilication' Mean SOl' n CY' Mcan SI> n CY DiffL'Tene<." (p value)
NS-I 125.tr' NAd I NA 126.0 / NA I NA tU NA'LFS-I <0.14/ 0.00 3 NA <0.14, 0.00 3 NA 0.0 NALFS-2 0.16 0.16 3 1.00 0.16. 0.16 3 1.00 0.0 No (0.9809) .LFS-3 2.&&. 0.10 2 o.m 2.69 0.43 3 tU6 6.6 No (tH'Io.120)/LFS-4 6.87/ 0.58 3 0.08 5.83/ 0.37 3 (1.(16 . IS.I No (J.OS84)/LFS-5 8.82/ 0.21 3 0.02 8.47/ 0.S9 3 0.07 -4.0 No (0.3887)LFS-6 \ 1.6/ 0.84 3 0.07 11.9/ 0.08 3 0.0\ 2.6 No (05:W.2) ,LFS-6b' 13.9/ 0.28 3 0.02 13.2/ 0.84 3 0.(16 -- 5.0 No (0.2563)LrS- 7 17.4/ 0.11 3 0.01 17.1 / 0.32 3 0.02 -- 1.7 No (0.2007) /

982 Envimn. Tox;co[. Ch.'m. 21.2002
sample to which Ihis mar have occurrcd was sarople LFS-4.tbe sample with lhe bigbest percentage difference (- 15. I %)and lhe Iowesl p value (0.0584) (Table I). Total preparaliontime between tbe flrst and last sample was nev~ nmre than15 mino Bascd on Ihese results, il was assumcd Ihal any 01>-scrved differences in mcan AVS concentrations bctween the
two analytical methods were due primarily 10 difference.<; inlhe efTccl of lhe carrier-gas composition.
Differcnces in AVS collcelltrations bctwcen me two ana-
Iytical melhods in seven oflhe nine experiments were analyzedfor significance. StatisticaJ comparisons were not applicableto experiments NS-I (analy"..cd in singlet) or LFS-I (concen-tration Icss than lhe detection levei). Allhough sulfide con-centrations in five of lhe analyses were )ower wbcn deterrnincdby lhe air-AVS melhod Ihan when determined by lhe N,-A VSmethod, no diff~ences in AVS concentrations among lhe saro-pie pairs were significantly differcnt at o: = 0.05 (Table I).
The percentage recovery of aqucous sulfide in lhe small-scale apparatus using negative-pressure (i.e., vacuum) airflowwas 92. I 1: 2.9%(n = 4) afier 10 mino This result isconsistent
with sulfide rccovery (91.7 1: 7.0'Yo.n = 20) ill side-by-sideexperiments perforrned under positive-pressure mode in thisstudy, wilh aqueous su)fide spike recoveries (>90%) reportcdby Allcn et aJo[6). ano with aqueous sulfide spike recoveries(93.81: 6.7%) reported for a diffusion method ofdeterrniningAVS (9). Completion of tbe H,S transfer, as indicated by co-alescence of lhe silver sullide precipitale in lhe sullide traI',occuned within 20 min of acidification of &:\ucous sulfldespikes ano from 30 to 60 min for scdiment samples. Longertimes to prccipitate coalescence likely indicate longer sulfidetransfer times caused by inefficient or incomplete scdimelltlacid reagent mixing. Sulfide'recovery efficiellcy experimentswcre nol pcrforrncd wilh sedimcnt samples.
DlSCUSSION
The sulfide oxjdation literature was reviewcd for Ihree sul-
lide oxidation processes: Gascous H1S. aqucous sulfide. anosolid-phasc FeS. The results of Ihis review show Ihat. allhoughthese Ihree processes are bigbly complex ano dependent onexperimental condltions, lhe oxidation rale under lhe experi-mental condirions of AVS analysis (í.e.,low pH) is extremelyslow. The difference be\ween II,S oxidation ra1es ano extrac-tion and sequestration rales is wbat makes using air as thecarrier gas successful in this particular case. The relative po-tential impact of each oxidation OIcchanism on deterroiningaccurate AVS conccntrations using air as lhe c3JTÍer gas isdiscusscd in light of Ihis literature review.
The oxidation of gaseous 1-12Sin Rir is slow ano stronglydependent on tbe atmospberic' bydroxyl radical (011') con-centration [10). This mar partiaJly explain ils 311llOying(andI'otenlially toxic) pcrsistencc in lhe atmo\:\,here when relea~d.The residence time ofH1S in lhe Rir spacc wilhin the analyticaltraiu (-60 ml) at the Rir flow rale uscd (200 ml/min) is ap-proximately 60 ml + 200 rol/mia = 0.3 miR = 18 s. This is
much shorter than lhe residence lime of H2S in a doscd system( 102--IO' sI, wbcrc lhe only sulfide sink is almospheric oxi-dalion [ I IJ. Oxjd111ionby gas-pbase reaclion wiili almosphericoxygcn wilhin lhe AVS system should not oceur to any ap-preciable degrce within lhe analytieal systcm anã likely is nota sígnificant mcchanism for sulfidc loss.
A number of investígators bave studicd lhe oxidatíon 01'sulfide ill marine or freshwaler aqueous matriccs {12 181-Thebel<texample oflhcse I<ludicl<was one rerform,e,d by Chen 3nd
AM. GonzaIez
Morris [12]. They investigatcd oxidation kinetics of aqucoussulftde (as Na2S.Q\l20) in (nonsaline) water under a wide rangeof 1'11conditions (6.00-- I 1.75), inilial sulfade .:oocentralions(0.5 X 10'4..2.0 X 10.4 M), and initial OKygC4lo~cntrations(1.(, X \0-4 .8.0 x 10 . M). They consistenlly observcd aninduction períod, ranging fiom 0.2 to 6.0 h, during whichreactant concentrations remaincd uuchanged. In addition, lhespeciflC oxidation rale (k; observcd afier lhe induction period)was a cmnplex, nonlinear funclion of pH ano had maxrmumvalues aI pll 8.3 ano p/J J 1.2. Oelween pll .8.3 anel 6.0 (thelowest pH uscd), lhe spccifie rcachon rale dropped from ap-proximately 23 M-O9 h-I to ncar ".cm. Because at pU < 6 anysulfide prescnt is predominantly in lhe f0l11l of H2S, Iheseautbors spcculated tbat the spccific rale of oxidalion of su)f.dcin this 1'11range would be extremely slow.
In ali cases, it was ei~ shown, ar assumed through ev-ideare from other studies, thal lhe primary reduced sulfur spe-cies being oxidizcd was lhe bisulfide ion (HS--), not dissolved1-/2S.This has dircct implicatíons on lhe oxidation linetics ofaqueous-phase ~ulfide wilhin lhe AVS anaJysis system. Theconvcrsion of sulfidc to H2S by I M HCI will be exuernelyrapid, limitcd onJy by lhe samplelacid reagent mixing effi-ciency [12). Sulfide spcciation at lhe ,,1-1ofa I M HCI 8Olution(pH = O) l4) will strongly favor lhe 1-12Sfonu. Underextremely
seidic pH conditions, lhe concentration ofthe HS- spccies isnegligib'e; Iherefore, lhe sulflde oxidation rale is expeclcd tobc extremely slow. Even if lhe sborlest induclÍOII period isassumcd to apply under lheGe analytical condilions (0.2 b, or12 ruiu), significant sulfide oxidation should not occur betwecnlhe time lhe sample is acidificd ano lhe time the relcascd H,Sis stabilizcd.
The oxidation of solid-pbase FeS within sel/eral difTcrenllypcs of matrices ha.",been invcstigaled 1I,2,7}. Expcrrmenlswilh varying concentrations of synlhelic iron (11) sulfide inaeralcd waler [I) showed that sulfide concentrations'reacbcd .nondetectcd levels in approximalely I to 2 h, regardless ofinitial sulfide concentrations ranging bclween roughly 3 and17 ruM. Similar results were ohtaincd by Di Toro et aI. [2jduring oxidalion studies usíng natural scdiment samples (con-taining AVS) suspended in oxygen-saturated watcr and byGonzalez {7) during oxidaliOll cxperimcnts usíng synlhetic FeSiu a LFS matrix suspended in oKygen-salurated water. How-ever, sulfide samples in solid-phase forros (e.g.. FeS) wouldsimilarly be conl/ertcd to lhe 112Sforro at lhe experimentalpl I, with its complete conversion bcing limitcd only hy lhemixing ef11ciency of lhe scdimentlacid reagcnt matrix.
Oftbe tbrce possible mechanisms for suiCIdeoxidation (i.e.,gasoous, aqucous. and 8Olic:1phase), S<llid-phase sulfidc llXi-lUltion Iike1y would impacl AVS concentra\io"s in samplcdsediment most readily. None oflhethrcc mcchanisms ofsulfidcoxidalioll, however, is expccted to iml'3CI thc rcsults 01' IhcAVS analysis ollce lhe 1'11ofthe saml'lc matri is 10werC4.!hyacidiflCalion.
CONCUJSIONS
h lIas been shown lhal scdiment AVS can bc eJ<tracted usingair (as tlle carrier gas inlheacid extraction) willlout significanlimpacts on its quantitative (CCOl/cryand measuremcnt otlcethe samplc has becn acidiflCd. IR light of the acutc suscepti-hility ofreduccd sulfur sl'ccics (e.g.. scdiment AVS) to chclll-ical oxidation. this fmding is rdtbcr intercsting. Sullide rccov-cry cxpcrimcllts using Rir werc Rol statisticall) difTcrcnt ti'oll1I<ulfldecanccntratiolli: obtaincd usi,,!!. laboratory-gradc "i\(o-

Oxidation ChL'l1listryof AVS during anaJysis
gell. The fiean recovery efficiency for aqlleolls sodillffi slIlfidespikes, rallgíng from 10.2 to 42.4 v.mol, was 91.7 -'- 7J)% (n= 20), which ís colIsistelll wíth the resu/Cs of olher íllvesli-gations l4,9}. The successlul use 01' air as a carricr gas forAVS analysis works ill I1lis paI1icuJar applicalioll, howcver,bolh because the dominant form of sulfide present during anal-ysis is not the rapidly oxidized HS- but lhe slowly oxidizedH2S and because lhe rale of extraction/transfer of dissolvedH2S is much grcaler than the oxidation rale at acidie pll. Thcrelease, transler, and stabilízation 01'sulfide occur betore SIIb-
stantialloss via oxÍllation is realized. This obscrvation mightprove 10 he ali illtcresting alternative for developing an allll-Iytical method 01' detennining AVS pararnclers in the field.
Acknow/nigement...This study was pcrformed in part under Suocon-tract 18X-ST297C with CKY at Oak Ridb~ National Laboratory, man-aged by Lockheed Martin Encrgy Rescarch for the U.S. DcpartrnL"'!1taf Energy undi..-rcontract DE-ACO5-960R22464. Tbe l'Upt'artofl.ynnKszos and the assistance of Clint Rash are VL'TYmuch apprcciated.Critica! review of the manuscript by several anonY111ousrevicwcrs isalso apprecialed.
REFERF.NCES
I. Nelson MB. 1978. Kinctics and meehanism.s of the oxidation offelTOus suJtide. PhD thesis. SIanforo Vniversicy, Pala Alto, CA,USA.
2. Di Tara DM, Mahony JD, Gon7..atez AM. 1996. Partide oxidationmodcl of synthetic FeS and sedimcnt acid-voJatiJe sulfKle. En-vimn T<lxicot Cfwm 15:2156-2167.
3. Hanscn DJ, Mahony JD, Berry WJ, Benyi SJ, Ú}rbin JM, Pratt5D, AbJe MB. 19%. Chronic effeet of cadmium in sedimcnts oneoloni7.>\lion by b...>nlhicmarine arganisms: An eval""tion 01' lherole of interstitíal cadmimn and acid-volatile sultide in biologicalavailability. Envimn Sei Technotl5:2126 -2137.
4. U.8. Envirom11ental Protectioh AgL"'!1cy.1994. Melhods for mea-suring lhe toxicity anil bioaccumulation of sediment-associated
Envirml. ToxicoJ. Ch<m. 21, 2002 %3;,
contaminants witll IrL..mwater inwI1dmllL"S. EPA 600/R-<}4/024.W""hington, I>c.
5. Bccker DC, {iinn Te. IW5. EfI,..cts of stor~e time on toxicityof sediments from Puget Sound, Wa.<hington. Envimn ToxÜ:olChem 14:82'J-!BS.
6. Allen HE, Fu G, I.kng B. 1993. Analysís of acid-volalik suJjj,k(AVS) and simultaneousJy extraclelt metais (Sr;M) I..,r the esli-mation of po!<."'!1tia!toxicity in aquatic S(."dil1l4."'!1ts.Environ ToxicolCh.'m 12:144/145.3.
7. Gonzalez AM. 19%. A lahoratory-formuSated sediment incor-porating synlhetic acid-volatile sul/ide. ElI!vimn Toxico/ Ch"m/5:2W9- 22W.
8. SAS Institule. 1982. SAS~~ l!«>r'.' (;uide;' Sttltistic... V"r 6JJ4.Cary, NC, USA.
9. Brouwer 11, Murphy TP. 1994. Dil1'usion mt."'1bodfor lhe deter-""nalion af acid-volatile "ul f"k (AVI;) in ,..,.timent. Environ Tox-ico/ Chem 13:1273-1275.
10. Cux RA, Sheppard D. /980. Reactions ofOJl radicais with ga.<-"""" suJfm eampound.'l. Nalur., 2M::;'3<~.33\.
11. Stumm W, Morgan JJ. 1970. Aqudtic CIUJmi'try. Wilcy-lntcrsci-ence, New York. NY, USA.
U. Chcn KY, Morris JC. 1972. Kinetics of OXi.lalion of aqueoussulj;de by 01, Envimn Sei Techno/6:529 .537.
13. aine JD, Richards FA. 1%9. Oxygenation of hydrogcn sul/idein seawater at conslant salinity, temperature, and 1'11.Environ SÓTechno/ 3:838.-.843.
14. O'Bricn DJ, Birkner FO. 1977. Kineties of oxygenation ofrc-duced sulfur species in aqueous solution. Environ Sei Techno/11:1114..1120.
15. Hoffinann MR, Um Bc. 1979. Kinctics and mechanisms of lheoxidation of ""Ulfid,,by oxygen: Catalysis by bomageneous metal-phtha/ocyanine romp/exes. Environ Sei r.",hno/ 13..1406-1414.
\6. Millero FI. 1986. The thc<modynamic.. and kinc\ic.. af thc by-drogcn ~"UIfKleSystL-min natural walt.'TS.Mar Chem 18:121-147.
17. Wilmot PD, Cadee K, Katinic JJ, Kavanagh Ov. 1988. Kineticsof ,,"'lide oxidalion by dissolve" l">Xygen.Wol"r Envirl>nmt?nlR"."'Alrch JournaI60:1264-1270.
18. Millero FJ, Hubinger S, Femandcz M, Garnett S. 1987. Oxidatitmof BIS in scawaler as a function of K'!11(1Cralure,pU, and ionicstrenglh. Envinm Sâ T"chml/ 21 :439 443.





























