Embed Size (px)
Citation preview

КООРДИНАЦИОННАЯ ХИМИЯ, 2017, том 43, № 6, с. 357–368
357
HYDROTHERMAL SYNTHESIS, CRYSTAL STRUCTURE,
AND OPTICAL PROPERTIES OF FOUR SUPRAMOLECULAR
COORDINATION COMPOUNDS CONSTRUCTED
FROM 1,10-PHENANTHROLINE AND POLYCARBOXYLIC ACIDS
© 2017 J. Zhang1, 2, C. C. Wang1, 2, *, P. Wang1, 2, and S. J. Gao11Beijing Key Laboratory of Functional Materials for Building Structure and Environment Remediation,
Beijing University of Civil Engineering and Architecture, Beijing, 100044 P.R. China2Key Laboratory of Urban Stormwater System and Water Environment (Ministry of Education),
Beijing University of Civil Engineering and Architecture, Beijing, 100044 P.R. China
*e-mail: [email protected]
Received October 10, 2016
Four novel coordination compounds based on d10 metals, [Cd(Phen)2(H2Dczpb)] · H2O (I),[CdCl(Phen)2(HNda)] · 3H2O (II), [Mn2(Phen)4(H2Pma)] · (H3Pma)2 (III) and [Co2(Phen)6] · 2H3Pma ·· (H2Pma) · 2H2O (IV), where Phen = 1,10-phenanthroline, H4Dczpb = 3,4-dicarboxyl-(3',4'-dicarbox-ylazophenyl)benzene, H2Nda = 1,4-naphthalenedicarboxylic acid, H4Pma = pyromellitic acid), havebeen synthesized under hydrothermal conditions, and characterized by FT-IR spectroscopy, CNH elementalanalysis, single crystal X-ray diffraction (CIF files CCDC nos. 1439537–1439540 for I–IV, respectively) andUV-Vis diffuse reflectance spectra. Compounds I and III were built up of one-dimensional[Cd(Phen)2(H2Dczpb)] chains and [Mn2(Phen)4(H2Pma)] chains, respectively, while compounds II and IVwere composed of zero-dimensional discrete [CdCl(Phen)2(HNda)] unit and [Co2(Phen)6], being linked in-to 3D framework via hydrogen-bonding and π–π stacking interactions. The optical properties of I–IV havebeen studied in solid state at room temperature.
Keywords: supramolecular coordination compound, 1,10-phenanthroline, polycarboxylic acids, crystalstructure, optical energy gap
DOI: 10.7868/S0132344X17060135
INTRODUCTION
In the recent years, more attentions were paid tothe new solid-state structures involving the self-as-sembly of molecules into well-defined supramole-cules via non-covalent, multiple intermolecular in-teractions, like hydrogen-bonding, π–π stacking,and metal-ligand bond formation [1, 2]. Especially,the strong, selective, and directional hydrogenbonding interactions act as a most powerful orga-nizing force in supramolecular assembly, resultinginto 1D, 2D or 3D hydrogen-bonded architectures[3, 4]. Usually, the self-assembly of supramoleculesis also frequently inf luenced by several factors, likethe organic ligands, the geometric requirements ofthe metal atom, the anionic counter-ions, the reac-tion temperature, and the solvent system effect [5–7]. The key to construction of a desired architectureis the selection of organic ligands, and in some cas-es, a subtle alteration of organic ligands can lead toa new architecture [8, 9]. The combination withneutral N-donor ligands and anionic O-donor li-
gands can generate more interesting structureswhich cannot be obtained only via one type of li-gands [10–12].
In this regard, N-donor ligands are good candi-dates for the construction of supramolecules, as theappropriate aromatic N-donor chelating ligands arelikely via π–π interactions to produce fascinatingarchitecture [13]. As all known, 1,10-phenantroline(Phen) is the typical chelating N,N'-based second-ary ligand due to its excellent conjugated system andcoordinating ability which has been extensivelystudied to build novel supramolecular structures[14]. 3,4-Dicarboxyl-(3',4'-dicarboxylazophe-nyl)benzene (H4Dczpb) [15], 1,4-naphthalenedi-carboxylic acid (H2Nda) [16, 17], pyromellitic acid(H4Pma) [18, 19] (Scheme 1) had been employed asexo-multidentate ligands for the design and con-struction of novel coordination polymers owing totheir thermal stability and symmetry.
УДК 541.49

358
КООРДИНАЦИОННАЯ ХИМИЯ том 43 № 6 2017
ZHANG et al.
Scheme 1.
In this paper, four supramolecular coordinationcompounds, namely [Cd(Phen)2(H2Dczpb)] · H2O(I), [CdCl(Phen)2(HNda)] · 3H2O (II),[Mn2(Phen)4(H2Pma)] · (H3Pma)2 (III) and[Co2(Phen)6] · 2H3Pma · (H2Pma) · 2H2O (IV), con-structed by Phen, and H4Dczpb, H2Nda, H4Pma withdifferent transition metal ions (Cd2+, Mn2+, and Co2+)were presented. Further, their optical properties havealso been investigated in detail.
EXPERIMENTAL
Materials and methods. All chemicals used for thesyntheses were reagent grade, which were used with-out further purification. CHN elemental analyses wereobtained using an Elementar Vario EL-III instru-ment. FT-IR spectra, in the region 400–4000 cm–1,were recorded on a Nicolet 6700 Fourier Transforminfrared spectrophotometer. UV-Visible diffuse re-flectance spectra (UV-Vis DRS) of solid samples weremeasured from 200 to 800 nm by PerkinElmer Lamda650S spectrophotometer, in which BaSO4 was used asthe standard with 100% reflectance. Powder X-ray dif-fraction (PXRD) patterns were recorded using a Dan-donghaoyuan DX-2700B diffractometer employingCuKα radiation.
Synthesis of complex I. A mixture of CdCl2 · 2.5H2O(0.3 mmol, 0.0685 g), H4Dczpb (0.3 mmol, 0.1074 g)and Phen (0.6 mmol, 0.1189 g) with a molar ratio of1 : 1 : 2 was sealed in a 25 mL Tef lon-lined stainlesssteel Parr bomb containing deionized water (20 mL),heated at 160°C for 72 h, and then cooled down to roomtemperature. Dark red block-like crystals were isolatedand washed with deionized water and ethanol (yield72% based on CdCl2 · 2.5H2O).
FT-IR (KBr; ν, cm–1): 3446, 3049, 2923, 1707, 1622,1561, 1541, 1514, 1495, 1426, 1372, 1289, 1222, 1205,1172, 1143, 1100, 1065, 932, 862, 847, 825, 810, 777,729, 697, 682, 669, 637, 600.Synthesis of complex II. White block-like crystals of
II (yield 87% based on CdCl2 · 2.5H2O) were synthe-sized from a mixture of CdCl2 · 2.5H2O (0.3 mmol,0.0685 g), H2Nda (0.3 mmol, 0.0649 g) and Phen(0.6 mmol, 0.1189 g) with a molar ratio of 1 : 1 : 2 un-der the same conditions as for I.
FT-IR (KBr; ν, cm–1): 3449, 3049, 3007, 2651,2569, 1968, 1902, 1839, 1682, 1620, 1587, 1571, 1547,1510, 1494, 1465, 1424, 1384, 1342, 1301, 1261, 1201,1170, 1151, 1144, 1099, 1047, 1009, 998, 989, 975, 952,920, 858, 826, 812, 803, 786, 774, 746, 729, 720, 658,648, 634, 615, 581, 551, 456, 417.Synthesis of complex III. Yellow block-like crystals
of III (yield 87% based on MnCl2 · 4H2O) were syn-thesized from a mixture of MnCl2 · 4H2O (0.3 mmol,0.0594 g), H4Pma (0.3 mmol, 0.0762 g) and Phen(0.6 mmol, 0.1189 g) with a molar ratio of 1 : 1 : 2 un-der the same conditions as for I.
IR (KBr; ν, cm–1): 3441, 3053, 2922, 2563, 1919,1692, 1625, 1589, 1562, 1516, 1495, 1460, 1425, 1406,1343, 1277, 1246, 1198, 1150, 1122, 1102, 1034, 865,843, 818, 804, 785, 773, 726, 658, 638, 614, 548.Synthesis of complex IV. Dark red block-like crys-
tals of IV (yield 87% based on CoCl2 · 6H2O) were syn-thesized from a mixture of CoCl2 · 6H2O (0.3 mmol,
N NCOOH
COOH N
COOH
COOHN
COOH
COOH
COOH
COOH
COOH
COOH
Phen
H2Nda
H4Dczpb
H4Pma
For C40H26N6O9Cd
anal. calcd., %: C, 56.7; N, 9.9; H, 3.1.
Found, %: C, 57.0; N, 10.1; H, 3.2.
For C36H29N4O7ClCd
anal. calcd., %: C, 55.6; N, 7.2; H, 3.7.
Found, %: C, 55.5; N, 7.1; H, 3.9.
For C39H23N4O12Mn
anal. calcd., %: C, 58.9; N, 7.0; H, 2.9.
Found, %: C, 59.0; N, 6.9; H, 3.0.

КООРДИНАЦИОННАЯ ХИМИЯ том 43 № 6 2017
HYDROTHERMAL SYNTHESIS, CRYSTAL STRUCTURE 359
0.0714 g), H4Pma (0.3 mmol, 0.0762 g) and Phen(0.6 mmol, 0.1189 g) with a molar ratio of 1 : 1 : 2 un-der the same conditions as for I.
IR (KBr; ν, cm–1): 3649, 3439, 3072, 2922, 2613,1919, 1708, 1601, 1516, 1452, 1424, 1390, 1345, 1284,1242, 1143, 1125, 1112, 1103, 1050, 943, 924, 868, 849,819, 805, 775, 739, 728, 643, 607, 585, 458, 423.
X-ray crystallography. X-ray single-crystal datacollection for coordination compounds I–IV wereperformed with Bruker SMART 1000 CCD area de-tector diffractometer with a graphite-monochroma-tized MoKα radiation (λ = 0.71073 Å) using ϕ–ω modeat 298(2) K. The SMART software [20] was used for da-ta collection, and the SAINT software [21] was utilizedto carry out data extraction. Empirical absorption cor-rections were performed with the SADABS program[22]. The structure has been solved by direct methods
For C51H33N6O13Co
anal. calcd., %: C, 61.4; N, 8.4; H, 3.3.
Found, %: C, 61.5; N, 8.4; H, 3.5.
(SHELXS-97) [23], and refined by full-matrix-leastsquares techniques on F 2 with anisotropic thermal pa-rameters for all the non-hydrogen atoms (SHELXL-97)[23]. All hydrogen atoms were located by Fourier differ-ence synthesis and geometrical analysis. These hydrogenatoms were allowed to ride on their respective parentatoms. All structural calculations were performed us-ing the SHELX-97 program package [23]. Crystallo-graphic data and structural refinements for com-pounds I–IV are summarized in Table 1, and selectedbond lengths and angles are listed in Table 2.
Supplementary material for structures I–IV hasbeen deposited with the Cambridge CrystallographicData Centre (CCDC nos. 1439537–1439540; [email protected] or www.ccdc.cam.ac.uk/data_re-quest/cif).
RESULTS AND DISCUSSION
In compound I, the cadmium(II) center, in a dis-torted octahedral geometry, is coordinated by four ni-trogen atoms (N(3), N(4), N(5), and N(6)) from twodifferent phen ligands via a chelating mode, and two
Table 1. Crystallographic data and structure refinement for compounds I–IV
I II III IV
M 847.07 777.48 1589.11 1993.53
Crystal system Triclinic Monoclinic Triclinic Triclinic
Space group P1 P21/c P1 P1
a, Å 11.2030(9) 13.3665(12) 9.6710(9) 12.3910(11)
b, Å 12.0229(11) 13.1171(11) 13.0189(12) 12.9121(12)
c, Å 13.9701(12) 19.3681(18) 14.4541(14) 14.5519(13)
α, deg 91.5110(10) 90 75.6760(10) 96.1820(10)
β, deg 94.9620(10) 104.920(2) 85.657(2) 106.682(2)
γ, deg 105.849(2) 90 80.0650(10) 104.847(2)
V, Å3 1800.9(3) 3281.3(5) 1735.8(3) 2114.1(3)
Z 2 4 1 1
ρ(MoKα), mm–1 0.674 0.804 0.455 0.488
Total reflections 9214 16331 8936 10819
Unique reflections (Rint)
6261 (0.0278) 5785 (0.0768) 6030 (0.0482) 7333 (0.0233)
F(000) 856 1576 812 1024
Goodness-of-fit on F2 1.023 1.034 1.024 1.051
R1, wR2 (F2> 2σ(F2)) 0.0418, 0.1115 0.0488, 0.0841 0.0562, 0.0756 0.0509, 0.1272
R1, wR2 (all data) 0.0534, 0.1187 0.1092, 0.1008 0.1065, 0.0828 0.0755, 0.1374
Largest diff. peakand hole, e/Å3
0.900 and –0.657 0.869 and –0.563 0.452 and –0.404 0.628 and –0.615

360
КООРДИНАЦИОННАЯ ХИМИЯ том 43 № 6 2017
ZHANG et al.
Table 2. Selected bond lengths (Å) and angles (deg) for compounds I–IV
Symmetry transformations used to generate equivalent atoms: #1 –x, –y, –z + 2 (III).
Bond d, Å Bond d, Å Bond d, Å
I
Cd(1)–O(7) 2.281(3) Cd(1)–O(1) 2.300(3) Cd(1)–N(4) 2.386(3)Cd(1)–N(6) 2.405(3) Cd(1)–N(5) 2.409(3) Cd(1)–N(3) 2.422(3)
II
Cd(1)–N(4) 2.315(4) Cd(1)–N(2) 2.324(4) Cd(1)–O(1) 2.324(3)Cd(1)–N(3) 2.405(4) Cd(1)–N(1) 2.432(4) Cd(1)–Cl(1) 2.486(14)
III
Mn(1)–O(1) 2.122(3) Mn(1)–O(2)#1 2.126(2) Mn(1)–N(2) 2.231(3)Mn(1)–N(3) 2.245(3) Mn(1)–N(4) 2.285(3) Mn(1)–N(1) 2.287(3)
IV
Co(1)–N(5) 2.062(3) Co(1)–N(2) 2.077(3) Co(1)–N(4) 2.084(3)Co(1)–N(3) 2.093(3) Co(1)–N(6) 2.096(3) Co(1)–N(1) 2.123(3)
Angle ω, deg Angle ω, deg
I
O(7)Cd(1)O(1) 98.14(10) O(7)Cd(1)N(4) 152.53(11)O(1)Cd(1)N(4) 94.98(11) O(7)Cd(1)N(6) 117.67(11)O(1)Cd(1)N(6) 82.87(11) N(4)Cd(1)N(6) 87.79(11)O(7)Cd(1)N(5) 85.85(10) O(1)Cd(1)N(5) 150.17(11)N(4)Cd(1)N(5) 94.61(11) N(6)Cd(1)N(5) 69.35(11)O(7)Cd(1)N(3) 83.49(10) O(1)Cd(1)N(3) 123.18(10)N(4)Cd(1)N(3) 69.14(11) N(6)Cd(1)N(3) 145.40(11)N(5)Cd(1)N(3) 86.61(10)
II
N(4)Cd(1)N(2) 148.07(15) N(4)Cd(1)O(1) 107.79(13)N(2)Cd(1)O(1) 92.32(14) N(4)Cd(1)N(3) 70.58(15)N(2)Cd(1)N(3) 88.37(15) O(1)Cd(1)N(3) 82.56(13)N(4)Cd(1)N(1) 86.42(14) N(2)Cd(1)N(1) 70.23(15)O(1)Cd(1)N(1) 161.99(14) N(3)Cd(1)N(1) 92.23(13)N(4)Cd(1)Cl(1) 97.73(11) N(2)Cd(1)Cl(1) 106.07(11)O(1)Cd(1)Cl(1) 92.54(10) N(3)Cd(1)Cl(1) 164.98(11)N(1)Cd(1)Cl(1) 96.50(10)
III
O(1)Mn(1)O(2)#1 99.09(9) O(1)Mn(1)N(2) 88.90(11)
O(2)#1Mn(1)N(2) 92.34(10) O(1)Mn(1)N(3) 95.42(11)
O(2)#1Mn(1)N(3) 94.40(11) N(2)Mn(1)N(3) 171.32(11)O(1)Mn(1)N(4) 87.57(10) O(2)#1Mn(1)N(4) 166.57(10)N(2)Mn(1)N(4) 99.48(11) N(3)Mn(1)N(4) 73.25(11)O(1)Mn(1)N(1) 161.70(11) O(2)#1Mn(1)N(1) 85.92(10)N(2)Mn(1)N(1) 73.26(12) N(3)Mn(1)N(1) 101.76(12)N(4)Mn(1)N(1) 91.36(10)
IV
N(5)Co(1)N(2) 170.08(10) N(5)Co(1)N(4) 96.30(11)N(2)Co(1)N(4) 89.06(10) N(5)Co(1)N(3) 95.31(10)N(2)Co(1)N(3) 93.81(11) N(4)Co(1)N(3) 80.34(10)N(5)Co(1)N(6) 80.27(11) N(2)Co(1)(6) 95.68(11)N(4)Co(1)N(6) 170.46(10) N(3)Co(1)N(6) 91.06(10)N(5)Co(1)N(1) 91.56(10) N(2)Co(1)N(1) 79.47(10)N(4)Co(1)N(1) 97.01(10) N(3)Co(1)N(1) 172.86(10)N(6)Co(1)N(1) 92.00(10)
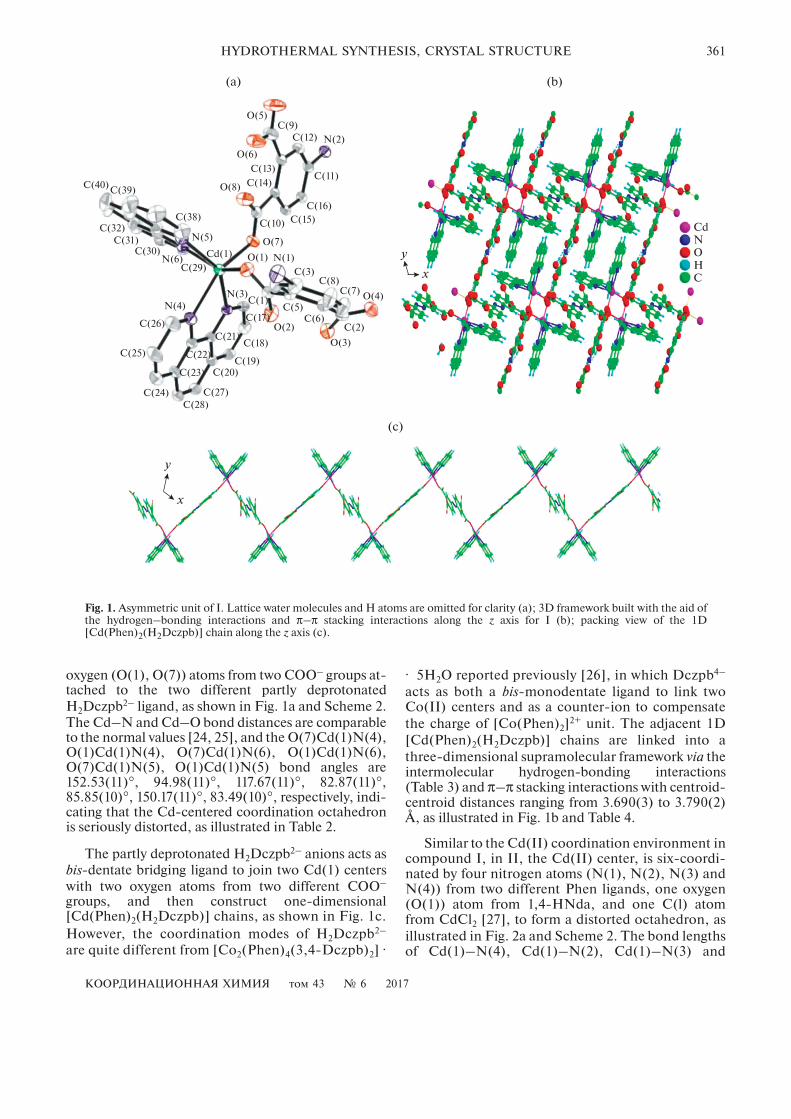
КООРДИНАЦИОННАЯ ХИМИЯ том 43 № 6 2017
HYDROTHERMAL SYNTHESIS, CRYSTAL STRUCTURE 361
oxygen (O(1), O(7)) atoms from two COO– groups at-tached to the two different partly deprotonatedH2Dczpb2– ligand, as shown in Fig. 1a and Scheme 2.The Cd–N and Cd–O bond distances are comparableto the normal values [24, 25], and the O(7)Cd(1)N(4),O(1)Cd(1)N(4), O(7)Cd(1)N(6), O(1)Cd(1)N(6),O(7)Cd(1)N(5), O(1)Cd(1)N(5) bond angles are152.53(11)°, 94.98(11)°, 117.67(11)°, 82.87(11)°,85.85(10)°, 150.17(11)°, 83.49(10)°, respectively, indi-cating that the Cd-centered coordination octahedronis seriously distorted, as illustrated in Table 2.
The partly deprotonated H2Dczpb2– anions acts asbis-dentate bridging ligand to join two Cd(1) centerswith two oxygen atoms from two different COO–
groups, and then construct one-dimensional[Cd(Phen)2(H2Dczpb)] chains, as shown in Fig. 1c.However, the coordination modes of H2Dczpb2–
are quite different from [Co2(Phen)4(3,4-Dczpb)2] ·
· 5H2O reported previously [26], in which Dczpb4–
acts as both a bis-monodentate ligand to link twoCo(II) centers and as a counter-ion to compensatethe charge of [Co(Phen)2]2+ unit. The adjacent 1D[Cd(Phen)2(H2Dczpb)] chains are linked into athree-dimensional supramolecular framework via theintermolecular hydrogen-bonding interactions(Table 3) and π–π stacking interactions with centroid-centroid distances ranging from 3.690(3) to 3.790(2)Å, as illustrated in Fig. 1b and Table 4.
Similar to the Cd(II) coordination environment incompound I, in II, the Cd(II) center, is six-coordi-nated by four nitrogen atoms (N(1), N(2), N(3) andN(4)) from two different Phen ligands, one oxygen(O(1)) atom from 1,4-HNda, and one C(l) atomfrom CdCl2 [27], to form a distorted octahedron, asillustrated in Fig. 2a and Scheme 2. The bond lengthsof Cd(1)–N(4), Cd(1)–N(2), Cd(1)–N(3) and
Fig. 1. Asymmetric unit of I. Lattice water molecules and H atoms are omitted for clarity (a); 3D framework built with the aid ofthe hydrogen–bonding interactions and π–π stacking interactions along the z axis for I (b); packing view of the 1D[Cd(Phen)2(H2Dczpb)] chain along the z axis (c).
(a) (b)
x
y
C(9)O(5)
O(6)
O(8)
O(7)
O(1)
O(4)
O(3)
O(2)
N(2)
N(1)
N(3)N(4)
N(6)
N(5)
Cd(1)
C(12)
C(11)
C(1)
C(17)C(5)
C(6)C(2)
C(7)C(8)
C(3)
C(18)
C(19)C(20)
C(27)C(28)
C(24)
C(25)
C(26)
C(29)
C(30)C(31)
C(32)
C(39)C(40)
C(38)
C(23)
C(22)
C(21)
C(16)C(15)
C(14)C(13)
C(10)
(c)
CdNOHCx
y

362
КООРДИНАЦИОННАЯ ХИМИЯ том 43 № 6 2017
ZHANG et al.
Table 3. Geometric parameters of hydrogen bonds for compounds I–IV
D–H⋅⋅⋅ODistance, Å
Angle DHA, deg Coordinates of A atomsD–H H⋅⋅⋅A D⋅⋅⋅A
I
O(3)–H(3)⋅⋅⋅O(2) 0.82 1.77 2.481(4) 144
O(6)–H(6)⋅⋅⋅O(8) 0.82 1.59 2.394(5) 168
O(9)–H(9C)⋅⋅⋅O(5) 0.85 1.97 2.818(7) 173 x + 1, y + 1, z
O(9)–H(9D)⋅⋅⋅O(6) 0.85 1.97 2.819(7) 173 –x + 1, –y + 1, –z
O(9)–H(9D)⋅⋅⋅O(5) 0.85 2.59 3.224(7) 132 –x + 1, –y + 1, –z
II
O(3)–H(3)⋅⋅⋅O(2) 0.82 1.75 2.560(5) 167 –x + 1, y + 1/2, –z + 3/2
O(5)–H(5C)⋅⋅⋅O(4) 0.85 2.15 2.973(7) 164
O(6)–H(6C)⋅⋅⋅O(2) 0.85 2.07 2.893(6) 163
O(6)–H(6C)⋅⋅⋅O(1) 0.85 2.42 3.116(6) 139
O(6)–H(6D)⋅⋅⋅O(5) 0.85 2.07 2.895(9) 163 –x + 1, y – 1/2, –z + 3/2
O(7)–H(7C)⋅⋅⋅O(6) 0.85 1.95 2.801(9) 177 –x + 1, –y + 1, –z + 1
III
O(4)–H(4)⋅⋅⋅O(8) 0.82 1.88 2.688(4) 167 –x + 1, –y, –z + 1
O(5)–H(5)⋅⋅⋅O(9) 0.82 1.79 2.601(4) 172 х – 1, y, z
O(5)–H(5)⋅⋅⋅O(10) 0.82 2.58 3.082(4) 121 x – 1, y, z
O(7)–H(7)⋅⋅⋅O(12) 0.82 1.75 2.573(3) 175 –x + 1, –y + 1, –z + 1
O(11)–H(11)⋅⋅⋅O(10) 0.82 1.59 2.381(4) 160
IV
O(2)–H(2)⋅⋅⋅O(13) 0.82 1.77 2.552(6) 158 x, y + 1, z
O(4)–H(4)⋅⋅⋅O(11) 0.82 1.71 2.499(3) 160
O(7)–H(7)⋅⋅⋅O(6) 0.82 1.64 2.450(4) 171
O(10)–H(10)⋅⋅⋅O(5) 0.82 1.76 2.569(3) 170 –x, –y + 1, –z + 1
O(13)–H(13C)⋅⋅⋅O(10) 0.85 2.26 3.029(6) 151 –x, –y + 1, –z + 1
O(13)–H(13D)⋅⋅⋅O(12) 0.85 1.99 2.760(6) 151 –x, –y + 1, –z + 1
Cd(1)–N(1) are 2.315(4), 2.324(4), 2.405(4) and2.432(4) Å, respectively, which are comparable witha typical bond length of Cd–N reported in the previousliterature [25]. The bond angles of N(4)Cd(1)O(1),N(2)Cd(1)O(1), N(4)Cd(1)N(1) and N(2)Cd(1)N(1)are 107.79(13)°, 92.32(14)°, 86.42(14)° and 70.23(15)°,respectively, implying CdN4OCl adopts a distortedoctahedral geometry. Thus, the crystal structure ofcompound II was built up from discrete and neutral[CdCl(Phen)2(HNda)] and three lattice water mole-cules, in which HNda– only coordinated with themetal atom Cd. However, in previously reportedcompounds, H2Nda ligand is bonded to metal atoms
and adopts a multidentate coordination mode [16].Finally compound II was strengthened by hydrogen-bonding interactions (Table 3) and π−π stacking in-teractions with centroid-centroid distances rangingfrom 3.465(3) to 3.926(3) Å, as illustrated in Fig. 2band Table 4.
The crystal structure analysis reveals that the com-pound III consists of the chain of the
and the anion H3Pma–.The Mn(II), in an octahedral geometry, is six-coordi-nated by four nitrogen atoms (N(1), N(2), N(3), andN(4)) from two different Phen ligands (Mn(1)–N(2)
( ) ( )[ ] +2 24
Mn Phen H Pman
n
КООРДИНАЦИОННАЯ ХИМИЯ том 43 № 6 2017
HYDROTHERMAL SYNTHESIS, CRYSTAL STRUCTURE 363
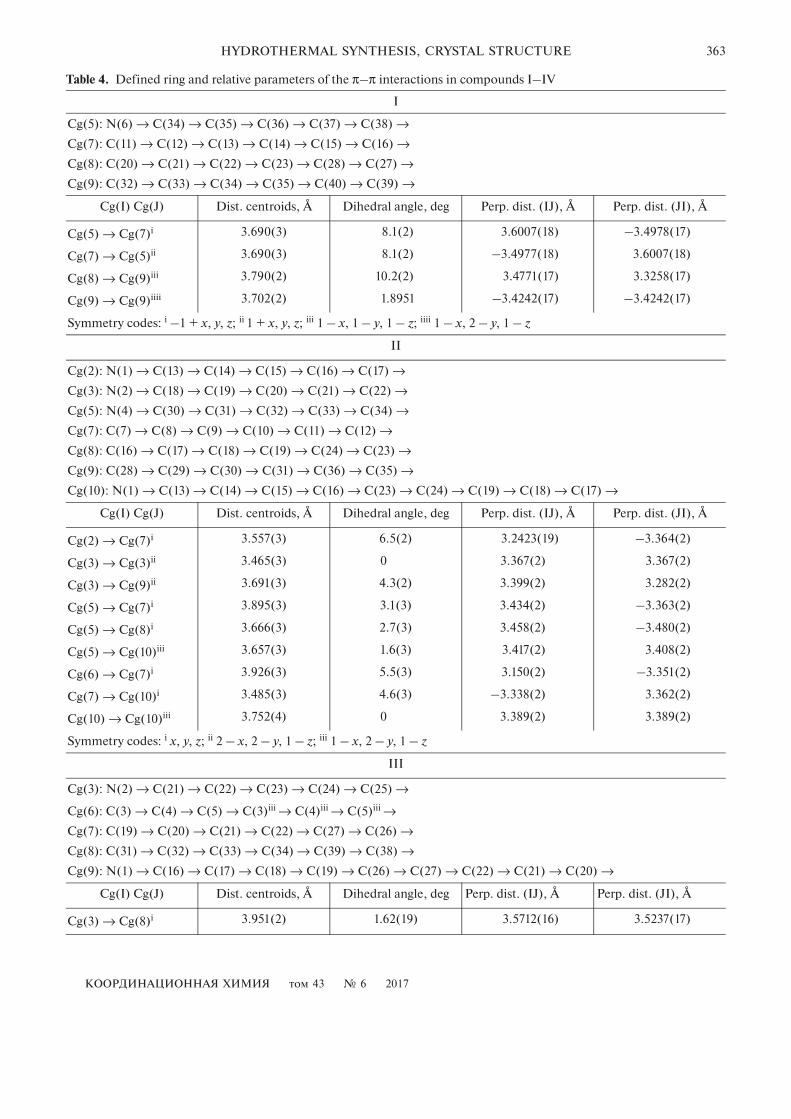
Table 4. Defined ring and relative parameters of the π–π interactions in compounds I–IV
I
Cg(5): N(6) → C(34) → C(35) → C(36) → C(37) → C(38) →
Cg(7): C(11) → C(12) → C(13) → C(14) → C(15) → C(16) →
Cg(8): C(20) → C(21) → C(22) → C(23) → C(28) → C(27) →
Cg(9): C(32) → C(33) → C(34) → C(35) → C(40) → C(39) →
Cg(I) Cg(J) Dist. centroids, Å Dihedral angle, deg Perp. dist. (IJ), Å Perp. dist. (JI), Å
Cg(5) → Cg(7)i 3.690(3) 8.1(2) 3.6007(18) –3.4978(17)
Cg(7) → Cg(5)ii 3.690(3) 8.1(2) –3.4977(18) 3.6007(18)
Cg(8) → Cg(9)iii 3.790(2) 10.2(2) 3.4771(17) 3.3258(17)
Cg(9) → Cg(9)iiii 3.702(2) 1.8951 –3.4242(17) –3.4242(17)
Symmetry codes: i –1 + x, y, z; ii 1 + x, y, z; iii 1 – x, 1 – y, 1 – z; iiii 1 – x, 2 – y, 1 – z
II
Cg(2): N(1) → C(13) → C(14) → C(15) → C(16) → C(17) →
Cg(3): N(2) → C(18) → C(19) → C(20) → C(21) → C(22) →
Cg(5): N(4) → C(30) → C(31) → C(32) → C(33) → C(34) →
Cg(7): C(7) → C(8) → C(9) → C(10) → C(11) → C(12) →
Cg(8): C(16) → C(17) → C(18) → C(19) → C(24) → C(23) →
Cg(9): C(28) → C(29) → C(30) → C(31) → C(36) → C(35) →
Cg(10): N(1) → C(13) → C(14) → C(15) → C(16) → C(23) → C(24) → C(19) → C(18) → C(17) →
Cg(I) Cg(J) Dist. centroids, Å Dihedral angle, deg Perp. dist. (IJ), Å Perp. dist. (JI), Å
Cg(2) → Cg(7)i 3.557(3) 6.5(2) 3.2423(19) –3.364(2)
Cg(3) → Cg(3)ii 3.465(3) 0 3.367(2) 3.367(2)
Cg(3) → Cg(9)ii 3.691(3) 4.3(2) 3.399(2) 3.282(2)
Cg(5) → Cg(7)i 3.895(3) 3.1(3) 3.434(2) –3.363(2)
Cg(5) → Cg(8)i 3.666(3) 2.7(3) 3.458(2) –3.480(2)
Cg(5) → Cg(10)iii 3.657(3) 1.6(3) 3.417(2) 3.408(2)
Cg(6) → Cg(7)i 3.926(3) 5.5(3) 3.150(2) –3.351(2)
Cg(7) → Cg(10)i 3.485(3) 4.6(3) –3.338(2) 3.362(2)
Cg(10) → Cg(10)iii 3.752(4) 0 3.389(2) 3.389(2)
Symmetry codes: i x, y, z; ii 2 – x, 2 – y, 1 – z; iii 1 – x, 2 – y, 1 – z
III
Cg(3): N(2) → C(21) → C(22) → C(23) → C(24) → C(25) →
Cg(6): C(3) → C(4) → C(5) → C(3)iii → C(4)iii → C(5)iii →
Cg(7): C(19) → C(20) → C(21) → C(22) → C(27) → C(26) →
Cg(8): C(31) → C(32) → C(33) → C(34) → C(39) → C(38) →
Cg(9): N(1) → C(16) → C(17) → C(18) → C(19) → C(26) → C(27) → C(22) → C(21) → C(20) →
Cg(I) Cg(J) Dist. centroids, Å Dihedral angle, deg Perp. dist. (IJ), Å Perp. dist. (JI), Å
Cg(3) → Cg(8)i 3.951(2) 1.62(19) 3.5712(16) 3.5237(17)

364
КООРДИНАЦИОННАЯ ХИМИЯ том 43 № 6 2017
ZHANG et al.
2.231(3), Mn(1)–N(3) 2.245(3), Mn(1)–N(4)2.285(3), Mn(1)–N(1) 2.287(3) Å), two oxygen at-om (O(1)) from two different H4Pma ligands(Mn(1)–O(1) 2.122(3), Mn(1)–O(2)#1 2.126(2) Å),
and the coordination environment of the Mn(II) issimilar to the previously reported compound [15].The bond angles of O(1)Mn(1)N(2) 88.90(11)°,O(1)Mn(1)N(3) 95.42(11)°, N(2)Mn(1)N(1)
Cg(6) → Cg(7)ii 3.617(2) 3.14(17) –3.4101(16) 3.3662(12)
Cg(I) Cg(J) Dist. centroids, Å Dihedral angle, deg Perp. dist. (IJ), Å Perp. dist. (JI), Å
Cg(6) → Cg(7)iii 3.617(2) 3.14(17) 3.4101(16) 3.3662(12)
Cg(7) → Cg(9)ii 3.604(2) 4.82(16) 3.3484(12) –3.3732(16)
Cg(7) → Cg(9)iii 3.604(2) 4.82(16) 3.3483(12) 3.3732(16)
Cg(9) → Cg(16)iiii 3.675(2) 9.88(17) –3.4210(16) –3.1544(12)
Symmetry codes: codes: i –x, –y, 1 – z; ii x, y, z; iii 1 – x, –y, 2 – z; iiii 1 – x, 1 – y, 1 – z
IV
Cg(9): C(19) → C(20) → C(21) → C(22) → C(27) → C(26) →
Cg(12): N(1) → C(16) → C(17) → C(18) → C(19) → C(26) → C(27) → C(22) → C(21) → C(20) →
Cg(I) Cg(J) Dist. centroids, Å Dihedral angle, deg Perp. dist. (IJ), Å Perp. dist. (JI), Å
Cg(9) → Cg(12)i 3.665(2) 2.34(17) –3.2968(15) –3.3489(15)
Symmetry codes: i 1 – x, –y, –z
Table 4. (Contd.)
Fig. 2. Asymmetric unit of II. Lattice water molecules and H atoms are omitted for clarity (a); 3D framework built with the aidof the hydrogen–bonding interactions and π–π stacking interactions along the z axis for II (b).
(a)
(b)
C(10)
C(11)
C(1)
C(3)
C(4)
C(5)
C(36)C(31)
C(32)C(33)
C(34)
C(30)
C(29)C(35)C(28)
C(27)
C(26)C(25)
C(22)
C(21)C(20)
C(19) C(24)
C(23)
C(18)
C(15)
C(14)C(13)
C(17)C(16)
N(4)
N(3)
N(1)
N(2)
Cl(1)
Cd(1)
C(2)
C(12)
C(9)
C(8)
C(7)
C(6)
O(2)
O(1)
O(3)
O(4)
CdClNOHC
x
y

КООРДИНАЦИОННАЯ ХИМИЯ том 43 № 6 2017
HYDROTHERMAL SYNTHESIS, CRYSTAL STRUCTURE 365
Fig. 3. Asymmetric unit of III. Lattice water molecules and H atoms are omitted for clarity (a); 3D framework built with the aidof the hydrogen–bonding interactions and π–π stacking interactions along the x axis for III(b); packing view of the chain of[Mn2(Phen)4(H2Pma)] along the y axis (c).
(a) (b)
(c)
x
z
MnNOHCz
y
O(4A)
O(3A)
O(2A)
O(1)
O(4)
O(3)
C(1)
C(4)
C(2) C(5)
C(37)
C(36)C(35)
C(34)C(39)
C(6)
C(11)
C(12) C(8)
C(13)
C(14)
C(9)C(15)
C(10)
C(7)O(7)
O(8)
O(9)
O(10)
O(11)
O(12)O(5)
O(6)
C(38)
C(17)
C(16)
C(30)
C(29)
C(33)
O(2)
C(1A)C(28)
N(3)
N(1)
N(2)
N(4)
Mn(1)
Mn(1A)
73.26(12)°, O(1)Mn(1)N(3) 95.42(11)°, respectively,showing MnN4O2 adopts a slightly distorted octahe-dral geometry. The partly deprotonated H2Pma2–
acts as a bis-monodentate ligand to link two Mn(II)centers via COO– group to form 1D
(Fig. 3c), and uncoordi-nated H3Pma– as a counter-ion to compensate the
charge of Finally, athree-dimensional framework is built up by hydro-gen-bonding and π–π stacking interactions with cen-troid-centroid distances ranging from 3.604(2) to3.951(2) Å, as listed in Fig. 3b and Table 4.
In compound IV, the Co2+ ion, is six-coordinated bysix nitrogen atoms (N(1), N(2), N(3), N(4), N(5) andN(6)) from three different Phen ligands (Scheme 2).Both the partly deprotonated HPma3– and H2Pma2–
act counter-ions to balance the cationic charge of[Co2(Phen)6]4+, without participating in coordinationto the Co(II) atoms, which is quite different from theones in compound III described-above and previously
( ) ( )[ ] +2 24
Mn Phen H Pman
n
( ) ( )[ ] +2 24
Mn Phen H Pma .n
n
reported coordination compounds [19, 28]. The twonitrogen atom (N(4) and N(6)) from chelating Phenligand occupy the axial positions, and the remainingfour nitrogen atoms (N(1), N(2), N(3) and N(5))from other Phen occupy the four sites of equatorialplane of the octahedron, as shown in Fig. 4a. And theN(5)Co(1)N(3) 95.31(10)°, N(2)Co(1)N(3) 93.81(11)°,N(5)Co(1)N(1) 91.56(10)°, N(2)Co(1)N(1) 79.47(10)°,respectively, show that the Co-centered coordination oc-tahedron is slightly distorted.
The adjacent [Co2(Phen)6] · 2H3Pma · (H2Pma) ·· 2H2O units is linked into a three-dimensionalframework via the intermolecular hydrogen-bondinginteractions, as shown in Table 3, and π–π stackinginteractions with centroid-centroid distances ofCg(9)–Cg(12) being 3.665(2) Å, in which Cg(9) isthe aromatic ring C(19), C(20), C(21), C(22), C(27),C(26) and Cg(12) is the centroid of aromatic ringN(1), C(16), C(17), C(18), C(19), C(26), C(27),C(22), C(21), C(20), as shown in Fig. 4b and Table 4.
Coordination environment of metal ions in com-pound I–IV are given in Scheme 2:

366
КООРДИНАЦИОННАЯ ХИМИЯ том 43 № 6 2017
ZHANG et al.
Scheme 2.
Broad bands observed at 3446, 3449, 3441 and3439 cm–1 for compounds I–IV, respectively, are as-signed to the carboxyl groups of the correspondingcarboxyl ligands. The C–H stretching mode for thePhen ring is relatively weak and observed at about3049, 3049, 3052 and 3072 cm–1 for I–IV, respective-
ly. Sharp bands at 1707 cm–1 for I, 1682 cm–1 for II,1692 cm–1 for III, 1708 cm–1 for IV, are attributed tothe asymmetric and symmetric vibrations of the carbox-ylate groups, reapectively. The characteristic absorptionpeaks of the Phen ligand are observed at 1426 cm–1 for I,1424 cm–1 for II, 1425 cm–1 for III, 1424 cm–1 for IV [26].
COOH
MOO
N N C O
N N
N NCOOH
OM
O
NNCOCd
Cl
N N
N N
COCd
O
CO
O
N
N N
NN N
Co
N
N
N N
Mn
N
N
NN
Mn
C O M
O
COOH
COOH
COO
O
O
(a)
(b)
(d)
(c)
Fig. 4. Asymmetric unit of IV. Lattice water molecules and H atoms are omitted for clarity (a); 3D framework built with the aidof the hydrogen–bonding interactions and π–π stacking interactions along the y axis for IV (b).
(a) (b)
CoNOHC
x
z
C(23) C(27)
C(26)C(19)C(22)
C(24)
C(25)C(37)
C(36)C(35)
C(34)
C(33)C(32)
C(28)C(29)C(40)
C(41)C(42)
C(43)C(50)
C(51)
C(44)
C(30)C(31)
C(38)C(3)
C(9)
C(10)C(5)
C(6)C(7)
C(2)
C(1)
C(8)
C(4)
O(5)
O(6)
O(7)O(1)O(3)
O(4)
O(9)
O(10)
O(11)
O(12)C(15)
C(12)C(14)C(13)
C(11)
O(2)
O(8)
C(39)
C(21)C(18)
C(20)C(17)
C(16)C(49)
C(48)
C(47)
C(46)
C(45)N(6)
N(5)
N(1)
N(2)
N(4)
N(3)Co(1)
КООРДИНАЦИОННАЯ ХИМИЯ том 43 № 6 2017
HYDROTHERMAL SYNTHESIS, CRYSTAL STRUCTURE 367
And the IR spectra contained four characteristic adsorp-tion peaks at 825 cm–1 for I , 826 cm–1 for II, which canbe assigned to Cd–N stretching vibrations [29], respec-tively, 658 cm–1 for III and 643 cm–1 for IV, which can beattributed to Mn–N and Co–N stretching vibrations.
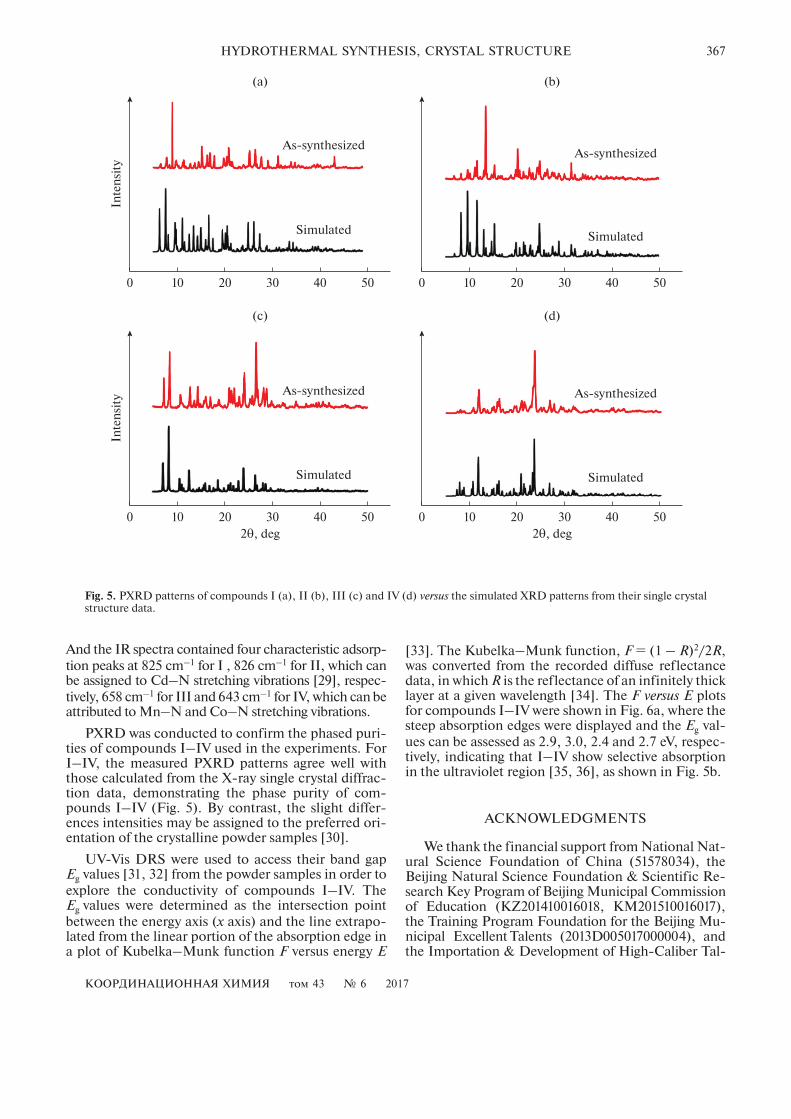
PXRD was conducted to confirm the phased puri-ties of compounds I–IV used in the experiments. ForI–IV, the measured PXRD patterns agree well withthose calculated from the X-ray single crystal diffrac-tion data, demonstrating the phase purity of com-pounds I–IV (Fig. 5). By contrast, the slight differ-ences intensities may be assigned to the preferred ori-entation of the crystalline powder samples [30].
UV-Vis DRS were used to access their band gapEg values [31, 32] from the powder samples in order toexplore the conductivity of compounds I–IV. TheEg values were determined as the intersection pointbetween the energy axis (x axis) and the line extrapo-lated from the linear portion of the absorption edge ina plot of Kubelka–Munk function F versus energy E
[33]. The Kubelka–Munk function, F = (1 – R)2/2R,was converted from the recorded diffuse reflectancedata, in which R is the reflectance of an infinitely thicklayer at a given wavelength [34]. The F versus E plotsfor compounds I–IV were shown in Fig. 6a, where thesteep absorption edges were displayed and the Eg val-ues can be assessed as 2.9, 3.0, 2.4 and 2.7 eV, respec-tively, indicating that I–IV show selective absorptionin the ultraviolet region [35, 36], as shown in Fig. 5b.
ACKNOWLEDGMENTS
We thank the financial support from National Nat-ural Science Foundation of China (51578034), theBeijing Natural Science Foundation & Scientific Re-search Key Program of Beijing Municipal Commissionof Education (KZ201410016018, KM201510016017),the Training Program Foundation for the Beijing Mu-nicipal ExcellentTalents (2013D005017000004), andthe Importation & Development of High-Caliber Tal-
Fig. 5. PXRD patterns of compounds I (a), II (b), III (c) and IV (d) versus the simulated XRD patterns from their single crystalstructure data.
0 10 20 30
Simulated
As-synthesized
(c)
40 502θ, deg
0 10 20 30
Simulated
As-synthesized
(d)
40 502θ, deg
Intensity
0 10 20 30
Simulated
As-synthesized
(a)
40 50 0 10 20 30
Simulated
As-synthesized
(b)
40 50
Intensity

368
КООРДИНАЦИОННАЯ ХИМИЯ том 43 № 6 2017
ZHANG et al.
ents Project of Beijing Municipal Institutions(CIT&CD201404076).
REFERENCES
1. Oh, M., Carpenter, G.B., and Sweigart, D.A., AccountsChem. Res., 2004, vol. 37, no. 1, p. 1.
2. Holliday, B.J. and Mirkin, C.A., Angew. Chem. Int. Ed.,2001, vol. 40, no. 11, p. 2022.
3. Wang, X.X., Huang, C.M., Dong, G.Y. and Cui, G.H.,Transition Met. Chem., 2015, vol. 40, no. 7, p. 733.
4. Wang, X.X., Zhang, S., Van Hecke, K., et al., Transi-tion Met. Chem., 2015, vol. 40, no. 5, p. 565.
5. Han, L.-J., Kong, Y.-J. and Sheng, N., Opt. Spectrosc.,2015, vol. 118, no. 1, p. 60.
6. Wang, X.-L., Sui, F.-F., Lin, H.-Y., et al., CrystEng-Comm, 2013, vol. 15, no. 36, p. 7274.
7. Hou, S., Liu, Q.-K., Ma, J.-P., and Dong, Y.-B., Inorg.Chem., 2013, vol. 52, no. 6, p. 3225.
8. Guo, H., Yan, Y., Wang, N., et al., CrystEngComm,2015, vol. 17, no. 34, p. 6512.
9. Eddaoudi, M., Kim, J., Rosi, N., et al., Science, 2002,vol. 295, no. 5554, p. 469.
10. Zhang, J., Wang, C.-C., Wang, P., and Gao, S.-J.,Transition Met. Chem., 2015, vol. 40, no. 8, p. 821.
11. Wang, C.-C., Guo, G.-L., and Wang, P., TransitionMet. Chem., 2013, vol. 38, no. 4, p. 455.
12. Wang, C.-C., Wang, P., and Guo, G.-L., TransitionMet. Chem., 2012, vol. 37, no. 4, p. 345.
13. Kong, Z.-G., Sun, X.-R., Li, L., et al., Transition Met.Chem., 2013, vol. 38, no. 4, p. 449.
14. Chen, X.M. and Liu, G.F., Chem. Eur. J., 2002, vol. 8,no. 20, p. 4811.
15. Wang, C.-C., Jing, H.-P., Wang, P., and Gao, S.-J.,J. Mol. Struct., 2015, vol. 1080, p. 44.
16. Raja, D.S., Luo, J.-H., Yeh, C.-T., et al., CrystEng-Comm, 2014, vol. 16, no. 10, p. 1985.
17. Huang-Fu, Y.-J., Chen, X.-Y., et al., Mater. Lett.,2015, vol. 155, p. 48.
18. Cao, R., Sun, D., Liang, Y., et al., Inorg. Chem., 2002,vol. 41, no. 8, p. 2087.
19. Hao, J.M., Li, Y.H., Li, H.H., and Cui, G., TransitionMet. Chem., 2014, vol. 39, no. 1, p. 1.
20. AXS SMART, Version 5.611, Madison (WI, USA):Bruker AXS, 2000.
21. AXS SAINT, Version 6.28, Madison (WI, USA): BrukerAXS, 2003.
22. SADABS, Version 2.03, Madison (WI, USA): BrukerAXS, 2000.
23. Sheldrick, G.M., SHELX-97, Göttingen (Germany):Univ. of Göttingen, 1997.
24. Yadav, P.K., Kumari, N., and Pachfule, P., Cryst.Growth Des., 2012, vol. 12, no. 11, p. 5311.
25. Chen, X.-L., Wu, Q.-R., Hu, H.-M., et al., Inorg.Chim. Acta, 2010, vol. 363, no. 2, p. 360.
26. Wang, C.-C., Jing, H.-P., Zhang, Y.-Q., et al., Transi-tion Met. Chem., 2015, vol. 40, no. 5, p. 573.
27. Li, Q.-Y., Yang, G.-W., Tang, X.-Y., et al., Inorg. Chem.Commun., 2010, vol. 13, no. 2, p. 254.
28. Luan, Y., Qi, Y., Jin, Z., et al., RSC. Adv., 2015, vol. 5,no. 25, p. 19273.
29. Warad, I., Al-Ali, M., and Hammouti, B., Res. Chem.Intermediat., 2013, vol. 39, no. 6, p. 2451.
30. Hao, J.-M., Yu, B.-Y., Van Hecke, K., and Cui, G.-H.,CrystEngComm, 2015, vol. 17, no. 11, p. 2279.
31. Ji, W.-J., Zhai, Q.-G., Li, S.-N., et al., Inorg. Chem.Commun., 2012, vol. 24, p. 209.
32. Du, P., Yang, Y., Yang, J., et al., Dalton Trans., 2013,vol. 42, no. 5, p. 1567.
33. Pankove, J.I., Optical Processes in Semiconductors,Courier Corporation, 2012.
34. Wendlandt, W.W. and Hecht, H.G., Reflectance Spec-troscopy, New York: Interscience, 1966.
35. Stylianou, K.C., Heck, R., Chong, S.Y., et al., J. Am.Chem. Soc., 2010, vol. 132, no. 12, p. 4119.
36. Laurier, K.G., Vermoortele, F., Ameloot, R., et al.,J. Am. Chem. Soc., 2013, vol. 135, no. 39, p. 14488.
Fig. 6. Kubelka–Munk-transformed DRS of compounds I–IV (a), UV–Visible DRS of compounds I–IV (b).
0
3
6
9
12
1 2
K–M fu
nction
3 4 5 6
III III IV
7Eg, eV
(a)
0
0.15
0.05
0.10
0.20
0.25
200 300
A
400 500 600 700
I II III IV
800Wavelength, nm
(b)






![Supramolecular anion recognition in water: synthesis of ... · Supramolecular anion recognition in water: synthesis of hydrogen-bonded supramolecular frameworks ... (TP) 2] n taken](https://img.dokumen.tips/doc/110x75/5b9ce37509d3f2321b8d8473/supramolecular-anion-recognition-in-water-synthesis-of-supramolecular-anion.jpg)






















