Embed Size (px)
Citation preview

1
Chelating Activity of Advanced Glycation End-Product (AGE) Inhibitors
by
David L. Price, Patricia M. Rhett, Suzanne R. Thorpe, and John W. Baynes
Department of Chemistry and Biochemistry
University of South Carolina
Columbia, SC 29208
Corresponding Author
John W. Baynes
Department of Chemistry and Biochemistry
University of South Carolina
Columbia, SC 29208
Phone/FAX: 803-777-7272
email: [email protected]
Keywords: advanced glycation end-product (AGE); aminoguanidine; ascorbic acid; autoxidation;
chelation; glycoxidation; oxidation; pyridoxamine.
Running Title: Chelating activity of AGE inhibitors
Abbreviations: AA, ascorbic acid; AG, aminoguanidine; AGE, advanced glycation end-product;
CML, Nε-(carboxymethyl)lysine; DAP, diaminophenazine; DTPA, diethylenetriaminepentaacetic
acid; HFBA, heptafluorobutyric acid; PM, pyridoxamine; PMTB, phenacyldimethylthiazolium
bromide; PTB, phenacylthiazolium bromide;.
Acknowledgement: This work was supported by Research Grant DK-19971 from the National
Institutes of Diabetes, Digestive and Kidney Diseases. The authors thank Mr. Shengzu Yang,
University of South Carolina, for the synthesis of phenacylthiazolium bromide and
phenacyldimethylthiazolium bromides.
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

2
ABSTRACT
The advanced glycation end-product (AGE) hypothesis proposes that accelerated chemical
modification of proteins by glucose during hyperglycemia contributes to the pathogenesis of
diabetic complications. The two most commonly measured AGEs, Nε-(carboxymethyl)lysine and
pentosidine, are glycoxidation products, formed from glucose by sequential glycation and
autoxidation reactions. Although several compounds have been developed as AGE inhibitors and
are being tested in animal models of diabetes and in clinical trials, the mechanism of action of these
inhibitors is poorly understood. In general, they are thought to function as nucleophilic traps for
reactive carbonyl intermediates in the formation of AGEs, however alternative mechanisms of
actions, such as chelation, have not been rigorously examined. In order to distinguish between the
carbonyl trapping and antioxidant activity of AGE inhibitors, we have measured the chelating
activity of the inhibitors by determining the concentration required for 50% inhibition of the rate of
copper-catalyzed autoxidation of ascorbic acid in phosphate buffer. All AGE inhibitors studied
were chelators of copper, as measured by inhibition of metal-catalyzed autoxidation of ascorbate.
Apparent binding constants for copper ranged from ~2 mM for aminoguanidine and pyridoxamine,
to 10-100 µM for carnosine, phenazinediamine, OPB-9195 and tenilsetam. The AGE-breakers,
phenacylthiazolium and phenacyldimethylthiazolium bromide, and their hydrolysis products, were
among the most potent inhibitors of ascorbate oxidation. We conclude that, at millimolar
concentrations of AGE inhibitors used in many in vitro studies, inhibition of AGE formation results
primarily from the chelating or antioxidant activity of the AGE inhibitors, rather than their carbonyl
trapping activity. Further, at therapeutic concentrations, the chelating activity of AGE inhibitors
and AGE breakers may contribute to their inhibition of AGE formation and protection against
development of diabetic complications.
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

3
INTRODUCTION
The Maillard or browning reaction between sugars and proteins leads to the formation of
chemical modifications and crosslinks in proteins, known as advanced glycation end-products
(AGEs) (1). These products contribute to the age-dependent chemical modification of long-lived
tissue proteins (2), and accelerated formation of AGEs during hyperglycemia is implicated in the
development of diabetic complications (1,2). Consistent with the AGE hypothesis, AGE inhibitors,
such as aminoguanidine (AG) (3) and pyridoxamine (PM) (4), inhibit the formation of AGEs in
various proteins in vitro and in collagen in vivo (5, 6), and retard the development of diabetic
complications in animal models. The two most commonly measured AGEs, Nε-
(carboxymethyl)lysine (CML) and pentosidine, are glycoxidation products, formed by sequential
glycation and oxidation reactions (7), and traces of transition metal ions in physiological buffers are
potent catalysts of the formation of these AGEs in vitro (8). AGE inhibitors are nucleophilic
compounds, designed to trap reactive carbonyl or dicarbonyl intermediates in AGE formation.
However, as shown here, some of these compounds also have potent chelating activity, making it
difficult to dissect the antioxidative, chelating activity from the carbonyl trapping activity of the
inhibitors in in vitro studies. In the present work, we measured the chelating activity of AGE
inhibitors by determining the concentration required for 50% inhibition of copper-catalyzed
autoxidation of ascorbic acid in phosphate buffer. Using this assay, we show that AGE inhibitors
have weak-to-potent metal chelating activity, and that, in fact, many experiments demonstrating the
activity of AGE inhibitors in vitro are likely to be measuring the chelating activity, rather than
carbonyl trapping activity, of these compounds. We also show that AGE-breakers and their
hydrolysis products are potent chelators of copper and conclude that both the carbonyl trapping and
chelation activity of AGE inhibitors and AGE breakers may be involved in their therapeutic
mechanism of action.
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

4
MATERIALS AND METHODS
Reagents - Unless otherwise noted, all chemicals were purchased from Sigma/Aldrich Chemical Co
(St. Louis, MO). Monobasic and dibasic sodium phosphate salts were purchased from EM Science
(Gibbstown, NJ); heptafluorobutyric acid (HFBA) from Acros (Pittsburgh, PA), and nitric acid and
HPLC grade acetonitrile from Fisher Scientific (Pittsburgh, PA). Tenilsetam ((±)-3-(2-thienyl)-2-
piperazinone) (9, 10) was a gift from Aventis Pharma, Germany, and OPB-9195 ((±)-2-
isopropylidenehydrazono-4-oxo-thiazolidin-5-ylacetanilide) (11, 12) from Otsuka Pharmaceuticals,
Japan. Phenacylthiazolium bromide (PTB) and phenacyldimethylthiazolium bromide (PMTB) were
synthesized according to the methods of Vasan et al. (13) and Wolfenbüttel et al., (14),
respectively, and purified to homogeneity by reverse-phase HPLC. 2,3-diaminophenzine (DAP)
was synthesized according to Soulis et al. (15). PTB, PMTB and DAP were homogeneous by
reverse phase HPLC analysis and by electrospray ionization liquid chromatography - mass
spectrometry. All water used in these experiments was distilled, and then deionized (15-18 MΩ)
using a mixed bed ion-exchanger with activated charcoal polisher. Chelex-100 (~80% water
content) was suspended and allowed to settle several times in water to remove soluble chelator and
fine particles (16). To remove metal ions from phosphate buffer, Chelex-100 resin (7 g/L, wet
weight) was added to 50 mM phosphate buffer, pH 7.4, in a 2 L plastic container, and rocked gently
overnight at room temperature. The buffer was stored at room temperature in the same container,
allowing the resin to settle to the bottom; aliquots were removed for experiments, as needed.
Measurement of the kinetics of oxidation of ascorbic acid - All incubations (1.5 ml total
volume) were conducted in chelex-treated 50 mM phosphate buffer, pH 7.4, in a water bath at 30oC;
reagents were pre-equilibrated at this temperature for at least 5 minutes. In preliminary
experiments, the kinetics of oxidation of AA was determined in quartz spectrophotometer cuvettes
by measuring the rate of decrease in absorbance at 265 nm, as described by Buettner (17); the
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

5
cuvettes were pre-cleaned by soaking overnight in 1% nitric acid. AA (500 µM) was stable (>99%
recovery) in chelex-treated 50 mM phosphate buffer, pH 7.4, for at least one hour. Addition of 500
nM CuCl2 yielded a half-life of approximately one-hour.
For studies with AGE inhibitors, the copper and inhibitor were pre-incubated in phosphate
buffer for 5 min, then the ascorbate was added from a concentrated solution (75 µl of 10 mM AA in
water) to initiate the reaction. Aliquots (150 µl) were removed at 0, 15, 30, 45 and 60 minutes and
transferred to autoinjector vials containing 15 µL of 10 mM diethylenetriaminepentaacetic acid
(DTPA) to quench the metal-catalyzed oxidation reaction. Samples were analyzed by reverse phase
HPLC (RP-HPLC), using a Supelcosil LC-18 column (25 cm x 4.6 mm, 5µm) (Supelco; Bellefonte,
PA) on a Shimadzu (Columbia, MD) LC-10Ai liquid chromatograph equipped with a SIL-10Ai
auto-injector, an SPD-10AV UV-Vis detector. Solvents for HPLC were: A (0.1% HFBA in water),
and B (50% acetonitrile). The gradient was: 100% A at 0 minutes, increase to 80% B from 0 to 4
minutes, return to 100% A from 4 to 4.1 minutes, then re-equilibrate in 100% A from 4.1 to 9
minutes. The absorbance of ascorbic acid was measured at 244 nm (Figure 1), and the peak area
was integrated to estimate the percent of AA remaining vs. time. The equations for the least-
squares-fits to these plots (triplicate analyses) were determined with SigmaPlot 2000 (Jandel
Scientific) and used to determine the concentration of each compound that inhibited the rate of AA
oxidation by 50% (IC50).
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

6
RESULTS
All assays with AGE inhibitors were performed in triplicate by HPLC, measuring residual
ascorbate by absorbance at 244 nm (Figure 1). None of the compounds tested co-eluted with, or
otherwise interfered with, the measurement of ascorbate by this RP-HPLC method. In preliminary
studies, we adjusted experimental conditions to obtain a concentration of cupric ion that would
catalyze the oxidation of AA (500 µM) with an approximate half-life of one hour. As shown in
Figure 2 (lower lines; without AG or PM), this was achieved by addition of 500 nM CuCl2 to 50
mM Chelex-treated phosphate buffer, pH 7.4, at 30°C. This half-life was comparable to the half-
life of ascorbate in 0.1 M phosphate buffers prepared in our laboratory, however buffers prepared
with different batches of phosphate salts varied in their catalytic activity because of batch-to-batch
variation in metal ion content.
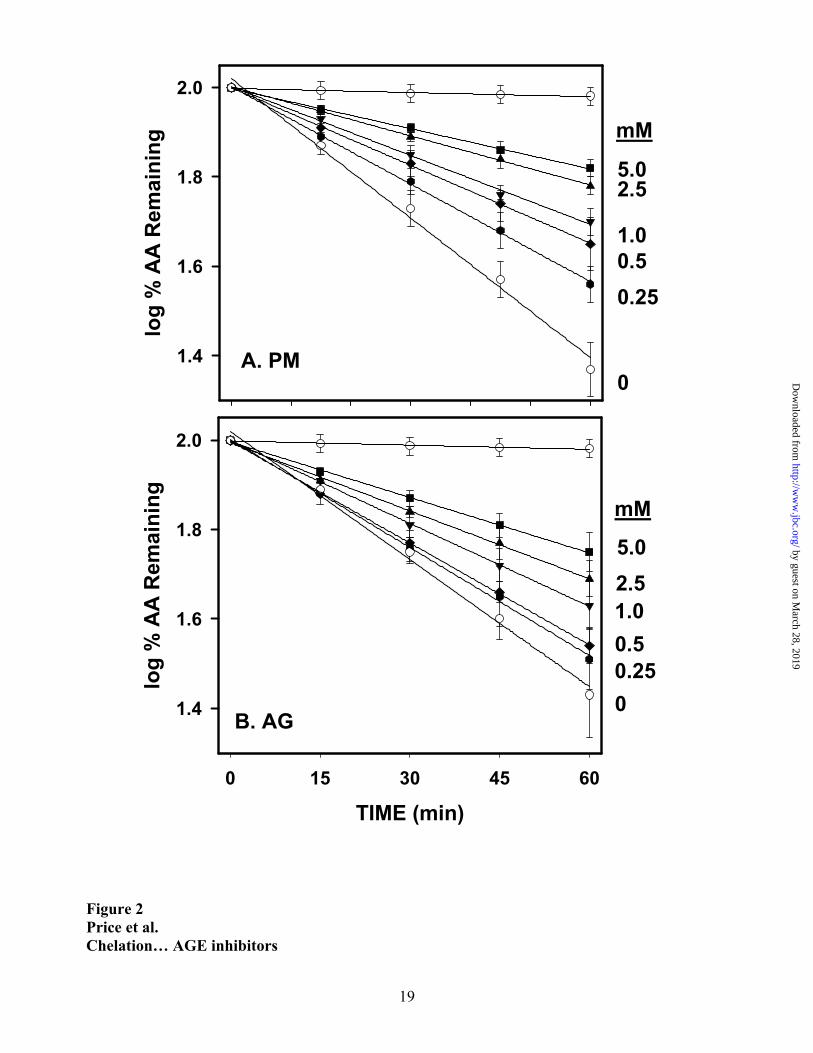
The AGE inhibitors, AG and PM, inhibited the oxidation of AA in a concentration-
dependent manner (Figure 2). The linearity of the semi-logarithmic plots illustrates that the AGE
inhibitors did not alter the pseudo-first order kinetics of the autoxidation reaction. Graphical
analysis of the rate of ascorbate oxidation vs. inhibitor concentration indicated that percent
inhibition was not a linear function of inhibitor concentration. The linear relationships between
percent inhibition and the square of the inhibitor concentration, shown in Figure 3, were consistent
with formation of 2:1 or higher valency complexes between AG or PM and Cu(II) ions. These data
yielded IC50 values of ~2.5 mM for AG and ~1 mM for PM. Amine nitrogens are common ligands
for transition metal ions, and pyridoxine (IC50 ~3.6 mM) and pyridoxal (IC50 >> 5mM), with a
hydroxymethyl or carboxaldehyde group, in lieu of the aminomethyl group of PM, were less
inhibitory than PM itself (Figure 3).
Carnosine and its the constituent amino acid histidine were potent inhibitors of AA
oxidation, with IC50 values ~4 and 12 µM, respectively (Figure 4A). The antioxidative effect of
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

7
carnosine and histidine was an asymptotic function of increasing inhibitor concentration, suggesting
that the chelate species may have some residual catalytic activity. Among other compounds tested,
taurine and ethanol had no effect on metal-catalyzed oxidation of AA. As shown in Figure 4B, the
AGE inhibitors, DAP and tenilsetam, had IC50 values of ~50 and 25 µM, respectively, while OPB-
9195 (Figure 4C) yielded a consistently non-linear response with a minimum at ~5 µM.
Both of the AGE breakers, PTB and PMTB, were also potent inhibitors of ascorbate
oxidation, with IC50 values of approximately 10 and 80 µM, respectively (Figure 5). Since the half-
life of PTB and PMTB were 2 and 8 hours, respectively, in phosphate buffer at 30°C (manuscript in
preparation), we also measured the activity of the hydrolysis products in the ascorbate oxidation
system. For these experiments, the AGE-breakers were incubated for 24 hours in buffer, prior to
addition of the ascorbate. As also shown in Figure 5, the hydrolysis products had IC50 values of < 2
µM and were among the most potent inhibitors of metal-catalyzed oxidation of ascorbate. The non-
linear response obtained with native PTB (Figure 5A) suggests that on-going hydrolysis,
approximately 30% during the course of the 1-hour incubation, may have contributed to the
increased inhibitory activity of this compound at higher concentrations.
In order to assess the potential role of AGE inhibitors and AGE-breakers as inhibitors of
metal-catalyzed oxidation chemistry in vivo, we also measured the effects of plasma albumin on the
kinetics of ascorbate oxidation. As shown in Figure 6, among all the compounds tested, bovine
serum albumin (BSA) was the most potent inhibitor of ascorbate oxidation on a molar basis. The
IC50 for BSA, ~80 nM (~5 µg/ml), was 16% of the concentration of Cu(II) in the reaction system,
suggesting at least 6 binding sites for copper.
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

8
DISCUSSION
Chelation activity of AGE inhibitors. The late Simon Wolff was a pioneer in focusing attention on
the metal chelating activity of drugs designed for other activities (18, 19). Aldose reductase
inhibitors and anti-inflammatory agents with anti-cataract activity were shown to have substantial
chelating activity, suggesting that their chelating activity might protect lens proteins from chemical
damage by inhibiting metal-catalyzed autoxidation of sugars or ascorbate (20, 21). In the present
study we have evaluated the chelating activity of various AGE inhibitors to determine the role of
metal chelation in the mechanism of action of AGE inhibitors. A typical reaction pathway for the
formation of AGEs from AA (and other carbohydrates) in vitro is shown in Figure 7. Metal-
catalyzed autoxidation chemistry accelerates the browning and crosslinking of protein by
carbohydrates and is essential for the formation of the glycoxidation products, CML and
pentosidine. Chelators such as DTPA are potent inhibitors of formation of these AGEs from
ascorbate (22) and glucose (23) (dotted arrows in Figure 7). In contrast, AGE inhibitors, such as
AG and PM, are thought to function as carbonyl traps (3, 4), reacting with electrophilic, carbonyl
intermediates formed on autoxidation of carbohydrates, thereby protecting against chemical
modification of nucleophilic residues on protein (solid arrow in Figure 7). Our experiments show,
however, that AGE inhibitors are also transition metal chelators and inhibit the autoxidation of
ascorbate, prior to formation of reactive carbonyl compounds. AGE inhibitors had IC50 values for
copper ranging from the millimolar range for AG and PM, to micromolar values for carnosine,
tenilsetam, DAP and OPB-9195. These results indicate that AGE inhibitors have two mechanisms
of action in vitro: first, inhibition of formation of reactive carbonyl intermediates; and second,
nucleophilic reaction with these intermediates.
AGE inhibitors in vitro. High concentrations of AG and other inhibitors have been employed in
many studies, including our own (23 – 26), on the mechanism of action of AGE inhibitors. These
high inhibitor concentrations have been justified because of the high concentrations of glucose used
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

9
to accelerate AGE formation in in vitro reactions. Based on the present work, however, it is more
likely that AG, at concentrations in the 5-25 mM range, i.e. 2-10 fold higher than its IC50, exerts its
primary effect by inhibiting autoxidation chemistry; this inhibition is observed not only with the
copper-supplemented Chelex-treated phosphate buffers used in our experiments, but also with
commercial phosphate buffers containing traces of mixed metal ions. Similar results were obtained
for AG and PM in the presence of iron salts, although iron was about 10-fold less effective as a
catalyst of ascorbate autoxidation reactions at neutral pH. Other AGE inhibitors, such as carnosine,
DAP, tenilsetam and OPB-9195, which have much stronger metal-binding activity (Table 1), are
likely to act primarily as chelators at the millimolar concentrations commonly employed for these
reagents in vitro. Chelation may, therefore, confound the identification of AGE inhibitors, e.g. in
two recent studies in which a large number of heterocyclic AGE inhibitors were described (27, 28),
but are more likely to inhibit AGE formation through their chelation, rather than carbonyl trapping
activity. For AG, and PM, however, triazines (29) and amides (30) have been identified as discrete
adducts to carbonyl compounds during glycoxidation and lipoxidation reactions, confirming that, in
addition to their chelating activity, they also act as true scavengers of carbonyl compounds. Similar
experiments will have to be performed with other AGE inhibitors in order to confirm their proposed
mechanism of action as carbonyl traps.
The estimate of the IC50 for inhibition of ascorbate oxidation is a useful, but comparative
measure of the chelation activity of AGE inhibitors. It is the empirical result of equilibrium
constants for metal binding by the multiple species of ascorbate (31), the AGE inhibitor and the
phosphate buffer ions in solution, rather than an actual Kd for the AGE inhibitor-metal complex. In
most cases, except for pyridoxamine (32) and carnosine (33), true metal binding constants have not
been measured. Because of the complexity of the equilibria involved, the IC50 for an AGE inhibitor
will vary with buffer, metal ion and ascorbate concentration, and even with the specific sugar under
study, e.g. glucose, which exists to a lesser extent in the enolate conformation, would bind transition
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

10
metal ions more weakly than ascorbate. Variations in the species and concentrations of metal ion
contaminants in buffers will also affect the apparent IC50 for specific inhibitors, depending on the
their respective binding constants with AGE inhibitors.
How can one avoid the complex and confounding effects of chelation in studies on AGE
inhibitors? One solution is to conduct these studies in the presence of a strong chelator, such as
DTPA, so that the contribution of the AGE inhibitor to chelation would be minimal. However,
DTPA itself is a potent inhibitor (>99% at 1 mM (23) of AGE formation from glucose and
ascorbate, so that reaction times would be excessive, even at high glucose concentration. We
suggest that it may be best to evaluate the efficacy of AGE inhibitors by studying the browning of
proteins by pentoses. Pentoses are 10 – 100 times more reactive than glucose with protein, so that
lower concentrations of sugar can be used. Glycation of protein by pentoses also leads to formation
of AGEs, crosslinks and fluorescence characteristic of the modification of protein by glucose (34).
Importantly, Maillard reactions of pentoses proceed readily under nitrogen and in the presence of
chelators (35). Experiments on the browning of proteins by pentoses can be conducted in the
presence of strong chelator such as DTPA, so that the chelating activity of the inhibitor would not
affect the kinetics of AGE formation in pentose model systems.
AGE inhibitors in vivo. The significance of the chelating activity of AGE inhibitors in vivo is
difficult to assess, considering the powerful inhibition of ascorbate oxidation observed with albumin
(Figure 6). Yet, albumin is subject to metal-catalyzed oxidation and formation of Maillard products
from glucose in vitro, and AGEs are present on albumin in vivo (36). It is likely that metals bound
to albumin are in equilibrium with metals bound to other proteins, and protein-bound metal ions,
even when bound to proteins such as albumin (37) or low density lipoprotein (38), may remain
redox-active and induce site-specific cleavage of the proteins in the presence of peroxide (39).
Despite lower binding constants, compared to albumin and chelators such as DTPA, AGE inhibitors
may gradually deplete excess chelatable iron in the body by promoting the excretion of metal ion
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

11
complexes in urine. Even a weak chelator, such as AG or PM, which might not inhibit autoxidation
reactions significantly at therapeutic concentrations (≤ 100 µM), could promote the gradual removal
of free metal ions from tissues and plasma for excretion in urine, decreasing overall metal-catalyzed
oxidative damage to proteins in vivo. Monnier and Eaton and colleagues (40,41) have proposed that
AGEs on proteins may serve to bind redox-active, transition metal ions, enhancing local oxidative
damage to proteins. If AGE inhibitors remove weakly bound metal ions from these "glycochelates,"
they may both inhibit AGE formation (by chelation and/or carbonyl trapping) and also reduce other
metal-catalyzed oxidative damage to tissues resulting from the presence of AGEs. Measurements
of chelatable iron and copper in control vs. AGE inhibitor-treated animals should clarify the
therapeutic significance of the weak chelating activity of AGE inhibitors. Notably, although
chelators are not used clinically for the treatment of diabetic complications, chelators have recently
proven effective in treatment of vascular disease (42) and neuropathy (43) in the STZ-diabetic rat.
AGE inhibitors vary widely in their IC50 values in the ascorbate model system, from the
millimolar range for AG and PM, to the micromolar range for carnosine, DAP, tenilsetam and OPB-
9195. At plasma concentrations of ≤ 100 µM, i.e. < 10% of their IC50, both AG and PM were
effective in inhibiting the formation of AGEs and development of complications in animal models
of diabetes (5). Among the stronger chelators, the concentration of carnosine in tissues (up to 20
mM) (44) far exceeds its IC50, while the therapeutic concentration of tenilsetam, ~10µM (45), is
comparable to its IC50 in the ascorbate system (25 µM; Figure 4B). The therapeutic concentrations
of DAP and OPB-9195 have not been reported, but are probably also in the micromolar range,
suggesting that chelation would contribute to their AGE inhibitory activity in vivo.
Chelating activity of AGE breakers. The AGE-breakers PTB and PMTB were among the most
potent inhibitors of copper-catalyzed oxidation of ascorbate, and the hydrolysis products of these
compounds exhibited even stronger chelating activity. Hydrolysis of PTB leads to opening of the
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

12
thiazolium ring, producing a sulfhydryl compound (46). Mercaptans are excellent ligands for
copper and other transition metals, which might explain the strong chelating activity of the
hydrolysis products of PTB and PMTB. Since PTB also protects E. coli against MGO toxicity,
almost as well as AG (47), it is also possible that the hydrolysis products may have carbonyl
trapping activity. The chelating properties of AGE breakers is sufficiently strong that they may also
have some lathyrogenic activity, inhibiting the enzymatic crosslinking of collagen, possibly leading
to decreased crosslinking and greater elasticity of newly synthesized collagen.
Summary. We have shown that AGE inhibitors have significant copper chelating activity. While
we have not studied other metals in detail, similar inhibitory effects were observable with phosphate
buffers containing a mixture of trace metal ions. The general metal chelating activity of AGE-
directed drugs containing nitrogen and thiol groups most certainly contributes to the chelating
activity of putative AGE inhibitors in vitro and could be important in their mechanism of action in
vivo. Screening assays for AGE inhibition should account for metal chelation by working with
inhibitor concentrations below their IC50 or by using pentoses as glycating agents. Identification
and characterization of products trapped by the AGE inhibitors is also critical for confirming their
mechanism of action. Defining the chelating vs. carbonyl trapping activity of AGE inhibitors is
essential for understanding the mechanism of action of these drugs and the possible benefits of
chelation therapy in diabetes, and for developing more effective, clinically useful inhibitors of
diabetic complications.
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

13
REFERENCES
1. Baynes, J.W., & Thorpe, S.R. (1999) in Diabetes in the New Millennium (Turtle, J.R., Kaneko,
T., and Osato, S., eds) pp. 337-350, Endocrinology and Diabetes Research Foundation, Sydney
2. Thorpe, S.R., and Baynes, J.W. (1996) Drugs & Aging 9, 69-77
3. Nilsson, B.O. (1999) Inflamm. Res. 48, 509-515
4. Khalifah, R.G., Baynes, J.W., and Hudson B.G. (1999) Biochem. Biophys. Res. Commun. 257,
251-258
5. Degenhardt, T.P., Alderson, N.L., Arrington, D.D., Beattie, R.J., Basgen, J.M., Steffes, M.W.,
Thorpe, S.R., and Baynes, J.W. Pyridoxamine inhibits early renal disease and dyslipidemia in
the streptozotocin-diabetic rat. Kidney Internat., in press.
6. Alderson, N.L., Metz, T.O., Chachich, M.E., Baynes, J.W., and Thorpe, S.R. (2001) Diabetes 50
(Suppl. 2), A172 (Abstr. #696P)
7. Baynes J.W., and Thorpe S.R. (1999) Diabetes 48, 1-9
8. Chace, K.V., Carubelli, R., and Nordquist, R.E. (1991) Arch. Biochem. Biophys. 288, 473-480
9. Munch, G., Taneli, Y., Schraven, E., Schindler, U., Schinzel, R., Palm, D., and Riederer, P.
(1994) J. Neural Transm. 8,193-208
10. Shoda, H., Miyata, S., Liu, B.F., Yamada, H., Ohara, T., Suzuki, K., Oimomi, M., and Kasuga,
M. (1997) Endocrinology 138, 1886-1892
11. Nakamura, S., Makita, Z., Ishikawa, S., Yasumura, K., Fujii, W., Yanagisawa, K., Kawata, T.,
and Koike, T. (1997) Diabetes 46, 895-899
12. Miyata, T., Ueda, Y., Asahi, K., Izuhara, Y., Inagi, R., Saito, A., Van Ypersele De Strihou, C.,
and Kurokawa, K. (2000) J. Am. Soc. Nephrol. 11, 1719-1725
13. Vasan, S., Zhang, X., Zhang, X., Kapurniotu, A., Bernhagen, J., Teichberg, S., Basgen, J.,
Wagle, D., Shih, D., Terlecky, I., Bucala, R., Cerami, A., Egan, J., and Ulrich, P. (1996) Nature
382, 211-212
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

14
14. Wolffenbüttel, B.H., Boulanger, C.M., Crijns, F.R., Huijberts, M.S., Poitevin, P., Swennen
G.N, Vasan, S., Egan, J.J., Ulrich, P., Cerami, A., Levy, B.I. (1998) Proc. Natl. Acad. Sci. USA
95, 4630-4634
15. Soulis, T., Sastra, S., Thallas, V., Mortensen, S.B., Wilken, M., Clausen, J.T., Bjerrum, O.J.,
Petersen, H., Lau, J., Jerums, G., Boel, E., and Cooper, M.E. (1999) Diabetologia 42, 472-479
16. Van Reyk, D.M., Brown, A.J., Jessup, W., and Dean, R.T. (1995) Free Radic. Res. 23, 533-535
17. Buettner, G.A. (1990) Meth. Enzymol. 186, 125-127
18. Woollard, A.C., Wolff, S.P., and Bascal, Z.A. (1990) Free Radic. Biol. Med. 9, 299-305
19. Jiang, Z.Y., Zhou, Q.L., Eaton, J.W., Koppenol, W.H., Hunt, J.V., and Wolff, S.P. (1991)
Biochem. Pharmacol. 42, 1273-1278
20. Wolff, S.P., Jiang, Z.Y., and Hunt, J.V. (1991) Free Radic. Biol. Med. 10, 339-352
21. Thornalley, P., Wolff, S., Crabbe, J., and Stern, A. (1984) Biochim. Biophys. Acta 797, 276-287
22. Dunn, J.A., Ahmed, M.U., Murtiashaw, M.H., Richardson, J.M., Walla, M.D., Thorpe, S.R.,
and Baynes, J.W. (1990) Biochemistry 29, 10964-10970
23. Fu, M.X., Wells-Knecht, K.J., Blackledge, J.A., Lyons, T.J., Thorpe, S.R., and Baynes, J.W.
(1994) Diabetes 43, 676-683
24. Brownlee, M., Vlassara, H., Kooney, A., Ulrich, P., and Cerami, A. (1986) Science 232, 1629-
1632
25. Glomb, M.A., and Monnier, V.M. (1995) J. Biol. Chem. 270, 10017-10026
26. Booth, A.A., Khalifah, R.G., Todd, P., and Hudson, B.G. (1997) J. Biol. Chem. 272, 5430-5437
27. Rahbar, S., Yernini, K.K., Scott, S., Gonzales, N., and Lalezari, I. (2000) Mol. Cell. Biol. Res.
Commun. 3, 360-366
28. Rahbar, S., Yernini, K.K., Scott, S., Gonzales, N., and Lalezari, I. (1999) Biochem. Biophys.
Res. Commun. 262, 651-656
29. Thornalley, P.J., Yurek-George, A., and Argirov, O.K. (2000) Biochem. Pharmacol. 60, 55-65
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

15
30. Onorato, J.M., Jenkins, A.J., Thorpe, S.R., and Baynes, J.W. (2000) J. Biol. Chem. 275, 21177-
21184
31. Hayakawa, K., and Hayashi, Y. (1977) J. Nutr. Sci. Vitaminol. (Tokyo) 23, 395-401
32. Gustafson, R.L., and Martell, A.E. (1957) Arch. Biochem. Biophys. 68, 485-498
33. Baran, E.J. (2000) Biochemistry (Moscow) 65, 789-797
34. Lee, K.W,. Simpson, G., and Ortwerth, B. (1999) Biochim. Biophys. Acta 1453, 141-151
35. Litchfield, J.E., Thorpe, S.R., Baynes, J.W. (1999) Int. J. Biochem. Cell. Biol. 31, 1297-1305
36. Miyata, T., Fu, M.X., Kurokawa, K., van Ypersele de Strihou, C., Thorpe, S.R., Baynes, J.W.
(1998) Kidney Int. 54, 1290-1295
37. Coussons, P.J., Jacoby, J., McKay, A., Kelly, S.M., Price, N.C., and Hunt, J.V. (1997) Free
Radic. Biol. Med. 22, 1217-1227
38. Lopes-Virella, M.F., Koskinen, S., Mironova, M., Horne, D., Klein, R., Chassereau, C.,
Enockson, C., and Virella, G. (2000) Atherosclerosis 152, 107-115
39. Hawkins, C.L., and Davies, M.J. (1997) Biochim. Biophys. Acta 1360, 84-96
40. Weiss, M.F., Saxena, A.K., and Monnier, V.M. (1999) Perit. Dial. Int. 19 (Suppl. 2), S62-S67
41. Qian, M., and Eaton, J. (2000) Free Radic. Biol. Med. 28, 652-656
42. Keegan, A., Cotter, M.A., Cameron, N.E. (1999) Free Radic. Biol. Med. 27, 536-543
43. Cameron, N.E., and Cotter, M.A. (2001) Diabetologia 44, 621-628
44. Quinn, P.R., Boldyrev, A.A., and Formazuyk, V.E. (1992) Mol. Aspects Med. 13, 379-444
45. Burrows, J.L., and Coppin, F.G. (1990) J. Chromatog. 529, 486-493
46. Thornalley, P.J., and Minhas, H.S. (1999) Biochem. Pharmacol. 57, 303-307
47. Furguson, G.P., VanPatten, S., Bucala, R., Al-Abed, Y. (1999) Chem. Res. Toxicol. 12, 617-
622
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

16
TABLE. Estimated IC50 for inhibition of copper-catalyzed oxidation of ascorbic acid by various
amines and AGE inhibitors.
Compound IC50 (µM)
Pyridoxamine 1000
pyridoxine 3600
pyridoxal >5000
aminoguanidine 2500
carnosine 4
histidine 12
taurine >5000
diaminophenazine 50
tenilsetam 25
OPB-9195 51
phenylthiazolium bromide 10 (<2)2
Phenyldimethylthiazolium bromide 80 (<2)2
1 Optimal inhibitory concentration. OPB-9195 was less effective at
higher and lower concentrations.
2 After 24-hr. incubation in metal-free phosphate buffer.
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

17
LEGENDS TO FIGURES
Figure 1. Typical data obtained from RP-HPLC analyses of the kinetics of oxidation of ascorbate.
These data were obtained for a reaction mixture containing 500 µM AA and 500 nM CuCl2.
Figure 2. Kinetics of oxidation of ascorbate in the absence and presence of various concentrations
of aminoguanidine (A) and pyridoxamine (B). Reaction mixtures contained 500µM AA, 500nM
CuCl2 and various concentrations of AG and PM. Data points represent means ± SD of triplicate
experiments.
Figure 3. Effect of aminoguanidine and B6 vitamer concentration on the kinetics of copper-
catalyzed oxidation of ascorbic acid. Data for AG and PM are from Figure 2. Inhibition is plotted
as a function of the square root of the concentration of AGE inhibitor. Dashed horizontal lines in
this and later figures indicate 50% loss of ascorbic acid
Figure 4. Effect of biological amines and AGE inhibitors on the kinetics of copper-catalyzed
oxidation of ascorbic acid. (top) carnosine and histidine; (middle) DAP and tenilsetam; (bottom)
OPB-9195.
Figure 5. Effect of AGE breakers and their hydrolysis products on the kinetics of copper-catalyzed
oxidation of ascorbic acid. (top) PTB ( ) and PTB hydrolysis products ( ) generated by
incubation in phosphate buffer for 24 hours. (bottom) PMTB ( ) and PMTB hydrolysis products
( ) generated by incubation in phosphate buffer for 24 hours.
Figure 6. Effect of BSA on the kinetics of copper-catalyzed oxidation of ascorbic acid.
Figure 7. Possible sites of action of AGE inhibitors in vitro. Carbonyl trapping (large arrow) is
considered the primary mechanism of action of AGE inhibitors. The present study illustrates that
many AGE inhibitors are potent metal chelators, effectively inhibiting AGE formation by inhibition
of metal-catalyzed oxidation reactions (dashed arrows).
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

18
Figure 1 Price et al. Chelation… AGE inhibitors
Time (min)0.0 3.0 3.5 4.0
Abs
orba
nce
(244
nm
)
t = 0
15 min
30 min
45 min
60 min
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from
19
Figure 2 Price et al. Chelation… AGE inhibitors
log
% A
A R
emai
ning
1.4
1.6
1.8
2.0
TIME (min)0 15 30 45 60
log
% A
A R
emai
ning
1.4
1.6
1.8
2.0
mM
5.02.51.00.50.250
A. PM
B. AG
mM
5.02.5
1.00.50.25
0
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

20
Figure 3 Price et al. Chelation… AGE inhibitors
(mM)1/20.0 0.5 1.0 1.5 2.0 2.5
Nor
mal
ized
Rat
e of
AA
Oxi
datio
n
0.2
0.4
0.6
0.8
1.0PL
AG
PN
PM
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

21
Figure 4 Price et al. Chelation… AGE inhibition
0 2 4 6 8 10 12 14 160.0
0.2
0.4
0.6
0.8
1.0
0 2 4 6 8 10 12 14 16
0.2
0.4
0.6
0.8
1.0
(uM)1/20 2 4 6 8 10 12 14 16
Rel
ativ
e R
ate
of A
A O
xida
tion
0.2
0.4
0.6
0.8
1.0
Histidine
Carnosine
OPB-9195
Tenilsetam
DAP
A.
B.
C.
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

22
Figure 5 Price et al. Chelation…. AGE inhibition
(uM)1/20 2 4 6 8 10 12 14 16
Nor
mal
ized
Rat
e of
AA
Oxi
datio
n
0.0
0.2
0.4
0.6
0.8
1.0
(uM)1/20 2 4 6 8 10 12 14 16
Nor
mal
ized
Rat
e of
AA
Oxi
datio
n
0.0
0.2
0.4
0.6
0.8
1.0 A.
B.
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

23
Figure 6 Price et al. Chelation…. AGE inhibition
BSA (nM)0 20 40 60 80 100 950 1000
Nor
mal
ized
Rat
e of
AA
Oxi
datio
n
0.0
0.2
0.4
0.6
0.8
1.0
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

24
Figure 7. Price et al. Chelation…. AGE inhibition
AGE-Inhibitors
Ascorbate DehydroascorbateReactiveCarbonylCompounds
Protein
Men+
[O2]
Men+
[O2]AGEs
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

David L. Price, Patricia M. Rhett, Suzanne R. Thorpe and John W. BaynesChelating activity of advanced glycation end-product (AGE) inhibitors
published online October 24, 2001J. Biol. Chem.
10.1074/jbc.M108196200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on March 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from





























