Embed Size (px)
Citation preview

1
F1 Praktikum Neurobiologie 2010
Block I Neurogenetik
Partho Halder, Mandy Jauch, Alexander Kapustjanskij, Juliane Klessen, Kirsa
Neuser, Nidhi Nuwal, Tulip Nuwal, Alice Schubert, Narendra Solanki, Erich
Buchner, Reinhard Wolf
„Nothing in neurobiology makes sense except in the light of behaviour“ (Martin Heisenber). In
diesem Kursteil wollen wir Ihnen die Möglichkeit bieten, an einem neurobiologischen
Modellorganismus ein breites Spektrum von Analysemethoden kennen zu lernen, das vom Gen
über das Protein über die Zelle, ihre Physiologie und Struktur, über das neuronale Netzwerk bis
hin zur systemischen Funktion reicht, wie sie sich im Verhalten manifestiert. Auf der
molekularen Ebene kam nach „Genomics“, dem Forschungsansatz, bei dem ganze Genome
sequenziert und analysiert werden, „Proteomics“, bei dem das „Proteom“, d.h. die Gesamtheit
aller Proteine aus einem Organismus betrachtet wird, und schließlich das „Transkriptom“, das die
Information über das zeitliche und räumliche Expressionsmuster für jedes Gen beinhaltet. Auf
der strukturellen Ebene spielt die confocale Mikroskopie eine herausragende Rolle, wobei die
Verbindung von Struktur und Funktion durch funktionelles Imaging und optogenetische
Stimulation hergestellt werden kann, Techniken, die im Kurs Anwendung finden. Zentrale
Techniken für den molekularen Ansatz sind Gel-Elektrophorese (1- und 2-dimensional), DNA-
Chips, RT-PCR und Antikörper. Antikörper spielen darüber hinaus auch eine essentielle Rolle
bei der Charakterisierung der Expression von Genen, da man mit ihrer Hilfe das Genprodukt
sowohl im Homogenat identifizieren als auch im Gewebe mit hoher Selektivität, Empfindlichkeit
und räumlicher Auflösung lokalisieren kann. Mit den beiden entsprechenden Techniken, dem
Western Blot und der Immunhistochemie, wollen wir in diesem Kursteil Fragen zur Abundanz
und Lokalisation von Genprodukten im Gehirn von Drosophila untersuchen.
Einführung: Funktionelle Anatomie des Fliegengehirns
Ein wesentlicher Aspekt der Funktion eines neuronalen Proteins ist sein spezifisches
Expressionsmuster im Gehirn. Um ein solches Expressionsmuster interpretieren zu können,
müssen wir uns mit der Anatomie des Drosophilagehirns vertraut machen. Der Grundtypus des
Zentralnervensystems von Articulata ist das Strickleiternervensystem. Ursprünglich ist jedem

2
Körpersegment ein Ganglienpaar zuzuordnen. Die Ganglien eines Segmentes sind durch eine
Kommissur (die "Sprossen der Strickleiter") miteinander verbunden. Die linken wie die rechten
Ganglien benachbarter Segmente verknüpfen Konnektive (die "Holme der Strickleiter").
Bei den rezenten Gliedertieren ist dieser Grundtypus mehr oder weniger stark
abgewandelt. Praktisch in allen Klassen, so auch bei den Insekten, hat eine starke Zentrierung der
Ganglienmassen stattgefunden. Am Vorderende ist dies Ausdruck der Cephalisation. Die
Anhäufung von Sinnesorganen am Kopf führt zu einer Verschmelzung der Kopfganglien
(Gehirnbildung). Als Gehirn im engeren Sinne wird bei vielen Insekten das Oberschlundganglion
bezeichnet, welches aus den paarigen Anlagen des Protocerebrums, des Deutocerebrums und des
Tritocerebrums entstanden ist. Bei Dipteren bilden Ober- und Unterschlundganglion jedoch eine
offensichtliche Einheit. Der Oesophagus zieht mitten durch das Gehirn. Die Reihenfolge
Protocerebrum, Deutocerebrum, Tritocerebrum, Unterschlundganglion - normalerweise ein
Hintereinander - bezeichnet bei Dipteren wie auch bei anderen Insekten ein Übereinander. Die
Hochachse des Gehirns entspricht entwicklungsbiologisch also seiner Längsachse. Dies ist dann
wichtig, wenn Homologisierungen mit dem Thoracalganglion versucht werden. In diesem sind
die verschmolzenen Pro-, Meso- und Metathoracalganglien hintereinander angeordnet.
Im Kurs wollen wir uns mit dem Gehirn von Drosophila melanogaster näher beschäftigen.
Die klassischen histologischen Methoden werden nur kurz besprochen. Die Golgi-Methode ist
beruht auf der zufälligen Kristallisation von metallischem Silber in einzelnen Neuronen, so dass
diese Neurone mit etwas Glück in ihrer gesamten Ausdehnung einen schwarzen Niederschlag
enthalten während der Rest des Gehirns transparent bleibt. Ihre Hauptstärke liegt in der
Darstellung einzelner Neuronengestalten. Semidünntechnik und Elektronenmikroskopie eignen
sich besonders gut für Drosophila. Aufgrund der Kleinheit der Fliege verliert man im
Elektronenmikroskop selten die Übersicht. Beispiele für Semi- und Ultradünn-Technik werden
gezeigt werden. Durch die Silberfärbung nach Blest werden so viele Neuronentypen angefärbt,
dass einzelne Zellen praktisch nicht mehr auflösbar sind, dafür werden aber die Grenzen
neuronaler Überstrukturen deutlich, welche häufig als "Körper" bezeichnet werden (z.B.
Pilzkörper, Zentralkörper, Ventralkörper etc.). Solche "Körper", die sich durch viele intrinsische
(in ihren Grenzen bleibende) Nervenzellen auszeichnen (was Golgi-Bilder zeigen), werden vom
sog. diffusen Neuropil umgeben. Diese Bereiche zeigen keine ins Auge springende Ordnung,
sondern erscheinen ungeordnet, diffus. Sehr viele verschiedene Zelltypen entsenden Seiten- oder
Hauptäste hierher. Funktionell dürfte das diffuse Neuropil nicht weniger bedeutsam sein als die
"Körper". Letzteren ist jedoch bisher die meiste wissenschaftliche Aufmerksamkeit zuteil
geworden. Dies hat z.T. vergleichende Gründe, da die "Körper" in den Gehirnen der
verschiedensten Insekten, ja Artikulaten zu finden sind, während die Bereiche des diffusen
Neuropils sich in der Evolution offensichtlich weit weniger konservativ verhalten. Strukturen des

3
Zentralhirns sollen im Kurs näher besprochen werden. Unser besonderes Interesse gilt jedoch
auch dem olfaktorischen System und den optischen Loben, die hier noch kurz diskutiert seien.
Düfte werden von Fliegen durch olfaktorische Rezeptorneurone auf den dritten antennalen
Segmenten und den Maxillarpalpen wahrgenommen. Diese Rezeptorneurone projizieren in den
Antennallobus, wo sie in spärischen Strukturen, den Glomeruli, terminieren. Lokale Interneurone
im Antennallobus prozessieren die eingehende Information durch laterale Inhibition und
Exzitation zwischen den Glomeruli. Desoxyglucose-Experimente (s.u.) an Drosophila und
Calcium-Imaging-Arbeiten an Honigbienen und Drosophila zeigen, dass Düfte im Antennallobus
durch die räumlich-zeitliche Aktivierung dieser Glomeruli repräsentiert werden.
„Projektionsneurone“ greifen die Information von den Glomeruli ab und projizieren einerseits in
das laterale Protocerebrum, andererseits in die Calyxregion des Pilzkörpers. Der Pilzkörper ist
eine für uns besonders interessante Struktur, weil er in unmittelbarem Zusammenhang mit
olfaktorischem Lernen steht.
Die optischen Loben sind seitliche Ausstülpungen des Oberschlundganglions, deren
Aufgabe darin besteht, aus den Meldungen der Lichtsinneszellen des Komplexauges
verhaltensrelevante Interpretationen des Umweltgeschehens zu gewinnen. Sie gliedern sich von
peripher nach zentral in Lamina, Medulla und Lobula-Komplex. Dieser ist bei Coleopteren,
Lepidopteren und Dipteren in eine posteriore Lobula-Platte und eine anteriore Lobula gespalten.
Eine der wichtigsten Struktureigenschaften der optischen Loben ist ihr isotoper Aufbau.
Jeder Bereich der Lamina entspricht einem Bereich der Medulla, der Lobula und der Lobula-
Platte, wobei die Nachbarschaftsbeziehungen erhalten bleiben. Die Neuromere sind aus
repetitiven Untereinheiten, den Säulen, aufgebaut, deren Zahl der Zahl der Ommatidien
entspricht. Tatsächlich sind die visuellen Säulen primär mit der Auswertung der Information
eines Sehpunktes beschäftigt. Bei dem neuralen Superpositionsauge der Dipteren bedeutet dies
jedoch nicht, dass eine Säule die Information eines Ommatidiums auswertet. Die 8 Sehzellen
eines Ommatidiums schauen auf 7 verschiedene Raumpunkte, und die Cartridges, die Säulen der
Lamina, erhalten dementsprechend die Axone von Sehzellen mit gleicher Blickrichtung aus 7
benachbarten Ommatidien. Obwohl die Säulenstruktur über die Medulla bis hinein in die Lobula
und Lobula-Platte zu verfolgen ist, wird in Lobula-Komplex und Medulla deutlich, dass die
Zusammenfassung der Information von verschiedenen Raumpunkten erst ein Bild ergibt. Die
tangentiale Streifung der Neuromere senkrecht zur Säulenrichtung verrät die Existenz von
großflächig abgreifenden Nervenzellen, die ins Zentralhirn projizieren. Dieses erhält jedoch
zusätzlich durch verschiedene Sätze von Säulenzellen der Lobula auch ein multiples
mosaikhaftes Bild der Umwelt.
In der funktionellen Neuroanatomie versucht man aus der Anordnung und Verbindung der
Bauelemente Rückschlüsse auf die Funktion der jeweiligen Gehirnteile zu ziehen. Was also lässt

4
sich aus dem Aufbau der optischen Loben aus Säulen und Schichten schließen? Eine
Arbeitshypothese ist, dass dort das Sehfeld "parametrisiert", d.h nach bestimmten Gesichtpunkten
ausgewertet wird. So wie in manchen Atlanten ein Erdteil nach seiner Oberfächenkontur, seinem
Klima, seinen Bodenschätzen und der Bevölkerungsdichte dargestellt ist, so könnte der Sehraum
in den verschiedenen tangentialen Schichten der optischen Loben z.B. hinsichtlich der mittleren
Helligkeit, seinen räumlichen Fourier-Komponenten, seinem mittleren Farbton oder hinsichtlich
gerichteter Bewegung analysiert werden. Selbst der Ort eines Ereignisses im Sehraum könnte
durch seine Koordinaten repräsentiert sein. Man kann sich leicht überzeugen, dass durch die
Relationen von vergleichsweise wenigen Parametern astronomisch viele Einzelzustände des
Sehraumes unterschieden werden können. Das Unterscheiden und Wiedererkennen von
Landmarken und Mustern ist allerdings nur eine von vielen Aufgaben des visuellen Systems.
Spekulative Ideen wie diese erhalten Nahrung durch elektrophysiologische
Untersuchungen in der Lobulaplatte. Die Großfeldneurone in den tangentialen Schichten dieser
Neuropilregion dienen der Kurskontrolle beim Flug und beim Laufen (siehe Kursteil
Optomotorik). Sie werten die Summe der Relativbewegungen Fliege - Umwelt für jeweils eine
bestimmte Bewegungsrichtung aus und ihre Signale werden dazu verwendet, das Bild der
Umwelt auf der Retina zu stabilisieren.
Im Gegensatz dazu weiß man fast nichts über die Funktionen der Sätze von
Kleinfeldzellen, die die Lobula mit dem Zentralhirn verbinden. Seit einigen Jahren ist es möglich,
Fragen nach der Funktionsspezialisierung von Zellpopulationen auch bei Drosophila direkt zu
untersuchen, obwohl elektrophysiologische Techniken hier wegen der Kleinheit der Zellen nur
begrenzt einsetzbar sind: Durch die sogenannte Desoxyglucosemethode wird physiologische
Aktivität im anatomischen Schnitt sichtbar gemacht. Das Verfahren beruht darauf, dass
physiologisch aktive Neurone mehr Zucker metabolisieren als inaktive. Wird neben der normalen
Glucose ein radioaktiv markiertes Zuckeranalog angeboten, das zwar von den Zellen
aufgenommen, aber nicht abgebaut werden kann, (3H-Deoxyglucose), so reichert sich in aktiven
Zellen radioaktives Material schneller an als in inaktiven. Dieser Unterschied kann im
histologischen Schnitt durch Autoradiographie sichtbar gemacht werden. Ein großer Nachteil der
Deoxyglucosemethode liegt jedoch darin, dass für eine sichtbare Erhöhung der Zuckeraufnahme
die Zellen für mindestens 30 min aktiv sein müssen. Reize im Zentralhirn habituieren jedoch
relativ rasch, wie man aus elektrophysiologischen Ableitungen an großen Insekten weiß. Daher
gibt es momentan zahlreiche Versuche, die Aktivität von Nervenzellen mit hoher zeitlicher
Auflösung über den Einstrom von Calcium, eine Änderung des pH-Werts oder direkt über die
Veränderung des Membranpotentials in vivo sichtbar zu machen. Für Tiere, für die Techniken
der Keimbahntransformation zur Verfügung stehen, wurden kürzlich sog. DNA-kodierte Sonden
entwickelt, bei denen der Calcium-, pH- oder Spannungs-Sensor ein Protein ist. Andere Proteine

5
erlauben die Depolarisation von Neuronen durch Licht (Channel-Rhodopsin, ChOP) oder die
Aktivierung des Enzyms Adenylatcyclase durch Licht (photoactivierte Adenylatcyclase PAC).
Ein kürzlich entwickelter DNA-kodierter Sensor, den Sie jetzt im Praktikum testen können,
ändert seine Farbe in Gegenwart von cAMP. Solche Sonden haben den immensen Vorteil, dass
sie mit genetischen Methoden, die wir für Drosophila ausführlich besprechen werden,
zellspezifisch und zeitlich geregelt exprimiert werden können. Die Vererbung der transformierten
Gene sorgt dafür, dass Versuche identisch wiederholt werden können, was bei Applikation z.B.
von klassischen Calcium- oder Spannungs-sensitiven Farbstoffen oder Pharmaka in der Regel
nicht ohne weiteres möglich ist.

6
Gruppe I: Visuelles Lernen im Flugsimulator (Narendra Solanki, Juliane Klessen und Reinhard Wolf)
Grundlagen Flugsimulator Ändert eine frei fliegende Fliege ihre Flugrichtung, so führt sie während dieser Richtungsänderung eine Rotation um ihre vertikale Körperachse aus. Physikalische Ursache für diese Rotation ist das Drehmoment, das die Fliege für die Dauer der Drehung mit einer entsprechenden Modulation der Schubkräfte beider Flügel erzeugt. Während dieser Drehung bewegt sich das visuelle Abbild der Fliegen-Umgebung (Panorama) entgegengesetzt zu der eigenen Rotationsrichtung über die Retina der Fliege. Im Flugsimulator werden diese Verhältnisse im stationären Flug simuliert. Das bedeutet, daß die (auf der Stelle fliegende) Fliege
sich bei einer Änderung der Flugrichtung nicht selbst um ihre eigene Achse dreht, sondern daß stattdessen das visuelle Panorama um die Fliege gedreht wird. Bezüglich des Zusammenhanges zwischen der selbst verursachten Drehrichtung und der Bewegungs-richtung des Panoramas auf der Retina ist (aus der Sicht der Fliege) kein Unterschied zwischen freiem Flug und dem Flug im Flugsimulator zu erkennen. Für den stationären Flug ist die Fliege mittels eines zwischen Kopf und Thorax befestigten Bügels aus Kupferdraht
(Präparation siehe Abb.6) an einem Drehmoment-Spannungs-Wandler (torquemeter) befestigt. Aus dem elektrischen Signal, das die Fliege während des Fluges mit ihrem Drehmoment generiert, berechnet ein Computer kontinuierlich die Änderungen der aktuellen Flugrichtung, welche der Fliege als bewegtes Bild in einem künstlichen Panorama in Echtzeit vorgespielt wird. Auf diese Weise kann die Fliege ihre Flugrichtung relativ zu künstlichen visuellen Landmarken im Panorama selbst bestimmen ("closed loop")1,2.
Abb. 1
Abb.2
Visuelles Lernen Während des closed loop Fluges kann man beobachten, daß die Fliegen häufiger Flugrichtungen zu den Landmarken hin einschlagen, als Flugrichtungen zwischen Landmarken hindurch ("Fixation")1. In Abb.1 und Abb.2a befinden sich 4

7
T-förmige Landmarken im Panorama, von denen 2 um 180° gedreht sind. Alle 4 Landmarken werden von Drosophila im langzeitlichen Mittel gleich häufig angeflogen. Mit Hilfe eines Hitze-Strahls (Infrarot-Laser, Abb.1) kann die Fliege konditioniert werden, Flugrichtungen zu einem bestimmten Mustertyp hin, z.B. in Richtung zu einem der aufrecht stehenden "T"s, zu vermeiden. Der "Performance Index" ( PI ) in Abb.2b ist definiert als PI = (tc - th)/(tc + th) in jeweils einem 2-Minuten-Intervall (tc = akkumulierte Flugzeit in Richtung zum "unbestraften" Muster; th = akkumulierte Flugzeit in Richtung zum "bestraften" Muster). Ein positiver PI bedeutet demnach überwiegende Flugrichtung zum "unbestraften" Muster während des Mess-Intervalles. Die dunklen Balken (orange) in Abb.3b kennzeichnen Trainings-Intervalle und die hellen Balken (gelb) stehen für 2-Minuten-Intervalle, in denen der Laserstrahl nicht eingeschaltet wird (Test). Das Histogramm in Abb.2b zeigt, daß die Fliegen nicht nur die Hitze während des Trainings, sondern darüber hinaus auch während der Test-Perioden die Flugrichtungen vermeiden, die während des Trainings mit der Hitze assoziiert waren3. Aus diesem Verhalten lassen sich folgende Schlüsse ziehen:
1. Die Fliegen können durch Auswahl der "richtigen" Flugrichtung lebensbedrohende Hitze vermeiden
2. Drosophila kann aufrecht stehende T's von umgekehrten T's unterscheiden 3. Die Fliegen können diesen Unterschied lernen (visuelles Gedächtnis)
Visuelles Gedächtnis Wie wir inzwischen festgestellt haben, werden im visuellen Gedächtnis keine Bilder, sondern lediglich unterschiedliche Musterparameter gespeichert, die aus den Bildern extrahiert werden4. Im hier gezeigten Beispiel mit den aufrechten und umgedrehten T's spielt die "T"-Form überhaupt keine Rolle, sondern es wird lediglich die unterschiedliche Höhe der Flächenschwerpunkte im visuellen Gedächtnis abgelegt. Bisher wurden bei Drosophila folgende visuell erinnerbare Parameter identifiziert (s. Abb.4)5:
1. unterschiedlich positionierte Höhe der Muster im Panorama 2. unterschiedliche Größe 3. unterschiedliche vertikale Ausdehnung (bei gleicher Schwärzung) 4. unterschiedliche Kantenorientierung 5. unterschiedliche Farbe (blau / grün)
Darüber hinaus können noch unterschiedliche vertikale Anordnungen gelernt werden ("relational parameters"), z.B. grün über blau vs. blau über grün, oder links geneigte über rechts geneigter Kante vs. rechts geneigte über links geneigter Kante.

8
"relational parameters"
Abb.3
Die verschiedenen Muster-parameter können unabhän-gig von der retinalen Trainingsposition wieder erkannt werden ("position invariance")5. Erst kürzlich ist es gelungen, den Ort des visuellen Gedächtnisses für zwei der Musterparameter (unterschiedliche Höhe des Musters und unterschiedliche Kantenorientierung, Abb.3) im Zentralkomplex des Fliegengehirnes zu lokalisieren6.
Molekulare Mechanismen der Klassischen Konditionierung Grundlage für Lern- und Gedächtnisprozesse ist auf zellulärer Ebene die Plastizität von Synapsen. Bei Drosophila sind mehrere Gene bekannt, die für die synaptische Plastizität benötigt werden. Eines davon, rutabaga, ist das Strukturgen für eine Ca2+/Calmodulin-abhängige
Adenylatcyclase, die cAMP aus ATP herstellt. Mutanten dieses Gens sind in allen, bisher untersuchten, assoziativen Lern- und Gedächtnisleistungen gestört. Man nimmt an, dass es sich bei der rutabaga-Adenylatcyclase um einen molekularen Koinzidenzdetektor handelt, der die neuronalen Aktivitäten verarbeitet, die durch den CS oder den US hervorgerufen werden. Neuronale Korrelate für rutabaga-abhängige, assoziative Plastizität wurden in
Abb. 4: Die zeitgleiche Ankunft des CS und des US erhöht die Konzentration von cAMP in der Zelle, was zu einer erhöhten Aktivität der PKA führt, die dann Proteine an der Synapse phosphoryliert (nach Heisenberg, 2003)
Abb.4

9
bestimmten Regionen des Insektengehirns identifiziert, wobei vor allem der Fächerförmige Körper im Zentralkomplex für die Bildung eines aversiven visuellen Gedächtnisses7 und die Pilzkörper für die Bildung eines aversiven olfaktorischen Gedächtnisses notwendig sind7. Während wir für das visuelle Gedächtnis bisher lediglich die beteiligten Zellgruppen im Fächerförmigen Körper identifiziert haben, gibt es für das olfaktorische Lernen bereits ein Funktionsmodell für die synaptische Plastizität in den Pilzkörpern8, an dessen Beispiel in Abb.4 die molekularen Vorgänge bei der klassischen Konditionierung eines Duftes (CS) mit Elektroschocks (US) gezeigt werden. Außer den Neuronen, die den Duft repräsentieren (Kenyonzellen), benötigen die Pilzkörper noch weitere neuronale Eingänge, die ihnen eine Wertung des Duftes mitteilen, wie 'positiv' oder 'negativ' (US-Neurone), und weiterhin Pilzkörperausgangs-neuronen, die mit allen Kenyon-zellsynapsen in Verbindung stehen und in Abhängigkeit von der Wertung ein entsprechendes Verhalten evozieren. Ohne jegliche Konditionierung sind diese Neuronen still und würden auf keinen gegebenen Stimulus reagieren. Bekommt die Fliege während eines Trainingsdurchlaufs einen Duft präsentiert, der koinzident mit einem Schock gepaart wird, werden die US-Neurone und die für den Duft spezifischen Kenyonzellen gleichzeitig antworten, was zu einer Hochregulierung der Transmitterfreisetzung in den Ausgangssynapsen der Kenyonzellen führt. Dies wiederum macht die Ausgangsneuronen des Pilzkörpers spezifisch empfindlich für den trainierten Duft. Obwohl bisher nicht gezeigt wurde, daß das oben beschriebene, rutabaga-abhängige Funktionsmodell für die synaptische Plastizität in den Pilzkörpern auch für das visuelle Lernen Anwendung finden könnte, so wurde immerhin gezeigt, daß das rutabaga-Gen zumindest bei den ersten vier (s. Abb.3) von den insgesamt fünf bisher bekannten, von Drosophila lernbaren Musterparametern benötigt wird. Nach bisher vorläufigen Ergebnissen scheint jedoch der fünfte Parameter, der Farb-Parameter, eine Ausnahme darzustellen: Fliegen mit einem defekten rutabaga-Gen scheinen den Farb-Parameter genauso gut lernen zu können, wie Wildtyp-Fliegen (pers. Mitteilung v. Li Liu, Inst. f. Biophysik, Peking). Versuch 1: Farb-Hitze-Konditionierung im Flugsimulator mit WT und rutabaga-Mutanten (Juliane Klessen, Reinhard Wolf)
Die Erklärung der in diesem Versuch verwendeten Apparatur und des Versuchsablaufes wird vor
Ort, im Labor, gegeben.
"Novelty-Lernen" Ein weiteres Paradigma, mit dessen Hilfe wir das visuelle Mustergedächtnis von Drosophila untersuchen können, benutzt den so genannten Novelty-Effekt9. Werden einer Fliege zunächst für eine bestimmte Dauer vier gleiche Muster (z.B. vier aufrechte T's) im Flugsimulator präsentiert,

10
so werden die entsprechenden Musterpositionen mit gleicher Häufigkeit angeflogen. Ersetzt man nach dieser Trainingsphase zwei gegenüberliegende Muster durch zwei andere Muster (in diesem Fall durch zwei umgedrehte T's), so werden die beiden neuen Muster häufiger in Flugrichtung gebracht, als die beiden alten Muster (Novelty-Effekt). Da diese Präferenz für die neuen Muster nur nach der vorhergegangenen Präsentation eines Panoramas mit 4 gleichen Mustern zu beobachten ist, ist anzunehmen, daß es sich hierbei um eine Gedächtnisleistung handelt, bei der das aktuelle visuelle Panorama mit der "Erinnerung" an das vorher gezeigte Panorama verglichen wird. Allerdings ist bisher nicht ganz klar, um welche Art des Lernens (assoziativ oder nicht-assoziativ) es sich hierbei handeln könnte. Das Training im Novelty-Experiment unterscheidet sich vom Training während des Standard-Experiments im Flugsimulator dadurch, daß kein offensichtlicher Bestrafungs- oder Belohnungsreiz beteiligt ist. Wir können jedoch nicht ausschließen, daß die künstlichen experimentellen Bedingungen im Flugsimulator (unnatürliches feedback, Fehlen der propriozeptiven Rückmeldungen, Unerreichbarkeit der angeflogenen Landmarken, ..etc) bei den Fliegen eine Art von unbehaglichem Stress oder Frustration erzeugen kann. Ob die Fliegen das während des Trainings präsentierte Muster mit diesen negativen Erfahrungen assoziieren, oder ob sie aus einer Art von 'Neugier' heraus das neue Muster bevorzugen, ist nicht bekannt. Als Alternative könnte man sich vorstellen, daß beim Novelty-Effekt eine Art von nicht-assoziativem Lernen (z.B. Habituation) beteiligt sein könnte.

11
Versuch 2: Novelty-Lernen im Flugsimulator mit operanter Abdomensteuerung (Narendra Solanki, Reinhard Wolf)
Versuchsaufbau (Abb.5):
Wie im Flugsimulator mit
Drehmoment-Steuerung (Abb.1),
fliegt die Versuchsfliege fliegt im
Zentrum der LED-Arena,
angeklebt an einen starren
Drahtbügel. Ihr Abdomen wird
von oben mit rotem Licht
beleuchtet und von einer
Videokamera (webcam) mit
Makrooptik auf einem Monitor
gegen einen dunklen Hintergrund
dargestellt. Zwei Solarzellen sind
mit Schlitzaperturen abgeklebt
und die rechteckigen Öffnungen
so auf dem Bildschirm befestigt,
daß sie senkrecht zu den
seitlichen linken bzw. rechten Konturlinien des Abdomens orientiert sind. Jede Solarzelle "sieht"
also ein Stück des hell dargestellten Abdomens und ansonsten den dunklen Bildhintergrund.
Wendereaktionen der angeklebt fliegenden Fliege gehen mit ruderartigen Verkrümmungen des
Abdomens in die intendierte Richtung einher. Die Solarzelle auf der Innenseite der intendierten
Kurve bekommt mehr von dem hellen Abdomen zu "sehen" und produziert daher mehr Strom,
die auf der Außenseite mehr vom Hintergrund und weniger vom hellen Abdomen; sie gibt
entsprechend weniger Strom ab. Über einen Differenzverstärker kann ein bipolares elektrisches
Signal erzeugt werden, das ungefähr proportional zum Abdomeneinschlag variiert. Dieses Signal
kann dazu benutzt werden, das Muster in der LED-Arena zu drehen (nach Strauss et al. 2001).
Mit der Stärke ihrer Steuerreaktion - hier also dem Grad der Abdomenverkrümmung - kann die
Fliege die Winkelgeschwindigkeit der Musterbewegung steuern und somit die Flugrichtung in
Bezug auf die Landmarken kontrollieren.
Abb.5
In früheren Versuchen wurde bereits gezeigt, daß die Fliegen in einer ähnlichen Anordnung in
der Lage sind, ihre Flugrichtung auf ein einzelnes visuelles Objekt (dunkler vertikaler Balken)

12
beizubehalten ("Fixation"). Interessanterweise können das die Fliegen auch mit invertiertem
Vorzeichen der Rückkoppelung, ohne erst lange trainieren zu müssen. Wird das Vorzeichen der
Kopplung invertiert, so wird z.B. bei einer Rechtswendung der Fliege visuell eine Linkswendung
vorgetäuscht. Die Situation erinnert an Fahrradlenken mit überkreuzten Armen (schon mal
probiert?). Dieses erstaunliche Verhalten deutet darauf hin, daß die Fliege vermutlich keine
vorbestimmte Erwartung darüber hat, in welche Richtung das Panorama reagieren wird, wenn das
Abdomen in eine bestimmte Richtung gekrümmt wird. Die Fliege bekommt durch Ausprobieren
heraus, daß und in welcher Weise sie ihre Flugrichtung mit dem Abdomen kontrollieren kann
(operantes Verhalten).
In dem Praktikums-Versuch soll erstmalig untersucht werden, ob die Fliegen auch in dieser
komplexen Situation in der Lage sind, den oben beschriebenen Novelty-Effekt zu zeigen.
Präparation der Fliegen für die Flugversuche Für die Flugversuche im stationären Flug müssen die Fliegen, möglichst schon einen Tag vor dem Versuch, an kleine Drahtbügel geklebt werden (bzw. die Drahtbügel an die Fliegen). Hierzu wird eine einzelne Fliege zunächst durch Abkühlung immobilisiert und dann auf eine ebenfalls gekühlte Metallplatte unter einem Binokular-Präpariermikroskop gelegt. Der Drahtbügel wird in die Halteklammer eines Mikromanipulators eingespannt. Mittels eines feinen Spatels (angeschliffene Präpariernadel) wird ein kleines Tröpfchen eines UV-aushärtenden Klebers an dem unteren Ende des Drahtbügels angebracht (Abb.4a). Mit dem Mikromanipulator wird dann der Bügel mit dem Kleber so positioniert, daß der Klebstofftropfen eine Brücke zwischen dem Hinterkopf der Fliege und dem Thorax bildet (Abb.4b). Hierbei sollte man besonders darauf achten, daß der Draht nicht zu tief zwischen Kopf und Thorax geschoben wird, damit das Hals-Konnektiv möglichst nicht mit Klebstoff benetzt wird. Die Fixierung des Kopfes ist notwendig für den Flug im Flugsimulator, um zusätzliche Bewegungsreize, die durch spontane Kopfbewegungen verursacht werden, zu verhindern. Zum Schluss wird der Klebstoff durch Bestrahlung mit ultraviolettem Licht ausgehärtet (ca 40 Sekunden aus größtmöglicher Nähe). Die so präparierten Fliegen (Abb.4c) werden dann einzeln in Plexiglas-Küvetten, mit einigen Körnchen Zucker versorgt, auf angefeuchtetem Filterpapier bis zum nächsten Versuchstag aufgehoben.

13
Abb. 6: Präparation der Fliegen für Flugversuche. (aus Heisenberg und Wolf 1984)
Die praktischen Anleitungen zur Präparation der Fliegen, sowie zur Durchführung und Auswertung der Experimente werden Ihnen vor Ort, im Labor und am Flugsimulator gegeben.
Referenzen
1Heisenberg, M., Wolf, R. (1984). Vision in Drosophila. Genetics of microbehavior. In: V. Braitenberg (Ed.), Studies of Brain Function, Vol. XII, Springer, Berlin, Heidelberg, New York.
2Heisenberg, M., Wolf, R. (1993). The sensory-motor link in motion-dependent flight control of flies. In: F.A. Miles and J. Wallman (Eds), Visual Motion and its Role in the Stabilization of Gaze, Elsevier
3Wolf, R., Heisenberg, M. (1991). Basic organization of operant behavior as revealed in Drosophila flight orientation. J. comp. Physiol. 169, 699-705
4Ernst, R., Heisenberg, M. (1999). The memory template in Drosophila pattern vision at the flight simulator. Vision Res. 39, 3920-3933.
5Tang, S.M., Wolf, R., Xu, S.P., Heisenberg, M. (2004). Visual pattern recognition in Drosophila is invariant for retinal position. Science 305, 1020-1022
6Liu, G., Seiler, H., Wen, A., Zars, T., Ito, K., Wolf, R., Heisenberg, M., Liu, L. (2006). Distinct memory traces for two visual features in the Drosophila brain. Nature 439, 551-556
7 Gerber, B., Tanimoto, H & Heisenberg, M. (2004). An engram found? Evaluating the evidence from fruit flies. Curr. Opin. Neurobiol. 14, 737-744.
8 Heisenberg, M. (2003). Mushroom body memoir: from maps to models. Nat. Rev. Neurosci. 4 (4): 266-275.
9Dill, M., Heisenberg, M (1995). Visual pattern memory without shape recognition. Phil. Trans. R. Soc. Lond. B. 349, 143-152
10Strauss R, Renner M, Götz KG (2001). Task-specific association of photoreceptor systems and steering parameters in Drosophila. J Comp Physiol A 187: 617-632.

14
Gruppe II: In-vivo Calcium und cAMP Imaging im olfaktorischen Pathway von
Drosophila melanogaster, Verhaltensversuche bei in-vivo Aktivierung
identfizierter Neurone, Expressionsanalyse durch real-time RT-PCR
(Alex, Benjamin, Nidhi) Zwei zentrale Methoden zum funktionellen Imaging basieren auf der Schlüsselrolle des Calcium-Ions bei der Aktivität von Nervenzellen bzw. des cAMP als zentraler sekundärer Botenstoff im Nervensystem. Erreicht z.B. ein Aktionspotential das synaptische Endköpfchen eines Neurons, so erfolgt Calcium-Einstrom ins Zellinnere und die Transmitterauschüttung in den synaptischen Spalt wird eingeleitet. Aber auch im Dendriten, im Zellkörper und im Axon beobachtet man einen Calcium-Einstrom bei Aktivität des Neurons. Calcium Imaging macht diesen Calcium-Einstrom sichtbar. Wir verwenden dazu als DNA-kodiertes Proteinkonstrukt das sog. Yellow Cameleon-2.1.
Das Molekül besteht aus zwei modifizierten Versionen des Green Fluorescent Protein (GFP),
welche über Calmodulin (Calcium-Bindungsdomäne) und einer Calmodulin-Bindungssequenz,
das sogenannte M13-Peptid, miteinander kovalent verbunden sind (Abb. 1). ECFP (Enhanced
Cyan Fluorescent Protein) kann durch Licht der Wellenlänge 436 nm angeregt werden. Ist die
Calciumkonzentration niedrig, so emittiert das angeregte ECFP Licht der Wellenlänge 485 nm.
Steigt die Calcium-Konzentration, bindet Calcium an Calmodulin, woraufhin das M13-Peptid an
das Calmodulin bindet. Dadurch ändert sich die Konformation des Proteins und ECFP gelangt in
räumliche Nähe zu EYFP (Enhanced Yellow Fluorescent Protein), wodurch ein strahlungsloser

15
Energietransfer (Fluorescence Resonance Energy Transfer, FRET) stattfinden kann. Das
bedeutet, dass das angeregte ECFP seine Energie auf das EYFP überträgt, d.h. ECFP emitiert nur
noch wenig Strahlung, das EYFP wird angeregt und emitiert Licht der Wellenlänge 535 nm. Das
Proteinkonstrukt Cameleon ist also in der Lage, unterschiedliche Calcium-Konzentrationen und
damit neuronale Aktivität optisch darzustellen: Eine niedrige Calciumkonzentration bedeutet
einen niedrigen Energietransfer und eine kleines EYFP/ECFP-Emissionsverhältnis. Eine hohe
Calciumkonzentration bedeutet einen hohen Energietransfer und eine großes EYFP/ECFP-
Emissionsverhältnis. Ein wichtiger Vorteil dieser Verhältnisänderung beim sog. „Ratio-Imaging“
ist, dass sie leicht von artefiziellen Absolutveränderungen z.B. durch Bleichen der Fluorophore
oder bei Bewegung der fluoreszierenden Strukturen zu unterscheiden ist.
cAMP ist an der Modulation der synaptischen Übertragung beteiligt und daher essentiell für
Lernen und Gedächtnis. Der Sensor besteht aus einer cyclic nucleotid binding domain (CNBD)
und, wie bei Cameleon, zwei modifizierten GFPs, CFP und YFP. Durch cAMP-Bindung and die
CNBD ändert sich deren Konformation, was den Abstand von CFP und YFP erhöht und dadurch
den FRET-Effekt abschwächt. Dies kann als Erniedrigung des YFP/CFP gemessen werden.

16
Die emittierte Fluoreszenz von ECFP und EYFP wird mittels eines Spektralteilers auf zwei
Hälften des Chips einer hochempfindlichen CCD-Kamera projiziert. Die beiden Halb-Bilder
werden im Rechner übereinandergelegt und Pixel für Pixel wird das CYFP/ECFP
Emissionsverhältnis berechnet. Während der Messung mit dem Cameleon Sensor bekommt die
Fliege Duftstoffe verabreicht, was zu einer gesteigerten neuronalen Aktivität führen sollte.
Dementsprechend erwartet man kurz nach der Duftgabe eine Erhöhung des EYFP/ECFP-
Emissionsverhältnisses. Zum Testen des cAMP Sensors verwenden wir bisher die Substanz
Forskolin, die alle Adenylatcyclasen aktiviert und somit in den Zellen den cAMP Spiegel erhöht.
Dies sollte zu einer Erniedrigung des YFP/GFP Verhältnisses führen.
Mit Hilfe des Gal4/UAS-Systems gelingt es, die Sensor-Konstrukte zum Beispiel selektiv
in den olfaktorischen Rezeptorneuronen (Gal4-Linie Or83b), in den olfaktorischen
Projektionsneuronen (Gal4-Linie GH146), in zwei kleinen Untergruppen von
Projektionsneuronen (Gal4-Linie MZ19), in den Neuronen des Pilzkörpers (Gal4-Linie 201y), in
den Neuronen mit dem Neurotransmitter Dopamin oder Octopamin (TH-Gal4 Linie/TDC-Gal4
Linie) oder in allen Neuronen (nrv-Gal4 Linie) zu exprimieren.
Im Rahmen dieses F1-Praktikums wollen wir Ihnen einen Einblick in die Methodik des
Funktions-Imagings vermitteln, die auf „Fluoreszenz Resonanz Energie Transfer“ (FRET) beruht,
einem physikalischen Prinzip, das generell häufig zur Analyse von molekularen
Wechselwirkungen eingesetzt wird. Hierzu werden Sie Untersuchungen mit Hilfe des cAMP-
Imagings in der olfaktorischen Bahn mit einer Pilzkörper-spezirischen Gal4 Linien durchführen.
Die Details des Versuchs werden im Praktikum besprochen.
In vivo optical imaging (Alex)
Visualizing cellular processes helps us to quantify, evaluate and finally better understand them. Utilizing several genetically encoded FRET based sensors, consisting of two flurophores fused to a binding domain for the molecule of interest, in combination with the technique of optical imaging allows us to take a closer look at various processes along the olfactory pathway of Drosophila melanogaster. Using an epifluorescent imaging setup coupled to a CCD Camera (Fig. 1), we will record Calcium and/or cAMP signals in various neuropiles (antennal lobes and mushroom body lobes Fig. 2) crucial for both odour represantation and odour learning.

17
In this practical course you will learn a special preparation and dissection procedure, which allows us to perform the imaging in vivo. To perform the recording, flies will be fastened in a pipette tip, with their head glued to a thin sheet of plastic foil. A small opening will be cut through the foil into the head cuticulla, in order to lay bare the neuronal structures of interest. To uphold physiological conditions and prevent dehydration of the tissue, the whole dissection procedure takes place under Ringer´s solution (Fig. 3)
Figure 1: Imaging setup
Data recording will be performed via the Metafluor software, with the intensity of both fluorescence channels as well as the ratio of the emission strength of both fluorophores recorded simultaneously. For the evaluation, the recorded pcitures will be transformed and aligned via ImageJ software in order to reduce the imact of various movement artifacts.
Figure 2: Major brain structures involved in the olfactory processing
ResinCover slip
Drop of Ringer
Objective
Figure 3: Fixation method
Pipette tip

18
Light induced neuronal activation
By generating transgenic flies we can incorporate proteins usually found in other species into the cells of our experimental animals. In this experiment we utilize the light-activated cation-selective ion channel Channelrhodopsin-2 (ChRh2) native to the green algae Chlamidomonas reinhardtii (Fig 4.). Cells featuring this channel can be depolarized by exposure to monochromatic light of 480nm wavelength. This is of particular use in the study of neuronal function and neuronal networks, as single neurons or neuronal populations can be activated individually by selective expression and directed light application.
Drop of Ringer
Objective
IR sensor
Walking ball
Figure 4: Channelrhodopsin-2 Under the control of the Gal4-UAS system, we can express the
channel in certain neurons of the central complex, a distinct part of the central brain composed of various neuronal structures putatively involved in spatial orientation and directional control of behavior. The impact of selective activation of central complex neurons on walking behavior can be studied in a specialized walking ball setup, featuring a styrofoam ball suspended by a laminar air current (Fig. 6). A fly prepared and fixated in a way similar to the imaging preparation can be placed in the walking ball apparatus, where it starts walking on the ball. The rotations of the ball and thus the
fly´s movement direction and speed can be tracked by an infrared sensor.
Figure 5: Central complex
Figure 5: Experimental setup
In order to direct the light to a distinct part of the central complex, the protocerebral bridge, we will utilize a fly line featuring GFP expression in the bridge neurons. Using the built-in apertures of the microscope either the full bridge, or only one half of the bride will be subjected to illumination.

19
Operant behavior in the “shock-box”
While pure associative learning is something relatively uncommon under natural conditions, goal oriented behavior most prominently features operant learning. In a simple paradigm we will introduce flies to a small chamber. Inside the chamber a single fly can pace back and forth. The
position of the fly will be constantly tracked by barcode scanner, while behavioral observations can be made via an IR-CCD camera. In the course of the experiments flies will be trained to avoid one side of the chamber, by application of electric shock while the fly
remains on the punished side and switching off the shock if the fly returns to the safe side. The experiment will consist of a pretest phase where normal fly behavior in the box will be recorded. After that an alternating series of training and test phases will be performed, a preference index for the time spent on both sides will be calculated for each phase of the experiment individually. The performance of a mutant called rut 2080 featuring a reduced function of the type I adenelyl
cyclase Rutabaga will be compared to the performance of the normal wild type flies. The Rutabaga adenelyl cyclase was previously reported to be involved in various learning types (olfactory, visual), therefore we will try to assess its necessity to the operant learning in the situation dictated by our experimental paradigm.

20
3D Segmentierung ausgewählter Gehirnabschnitte (Benjamin)
Am Konfokalmikroskop werden die Daten in Form von Stapel von 2D Bildern gespeichert. Diese Bilder können mit geeigneter Bildverarbeitungssoftware, z.B. ImageJ, weiterverarbeitet werden. Für ImageJ existieren dafür verschiedene Plugins. Anwendung finden hier der “ImageJ 3D Viewer” und das Plugin “Interactive 3D Segmentation”, die eigens zu diesem Zweck entwickelt wurden. Ersteres bietet die Möglichkeit, die Konfokalbilder dreidimensional darzustellen (in unterschiedlichen Modi, etwa als Volume renderings oder als Oberflächen). “Interactive 3D Segmentation” basiert auf dem 3D Viewer und wird im Folgenden verwendet, um semi-automatisch die Neuropile der optischen Loben zu segmentieren. Dafür werden jeweils ein Bild in ImageJ geöffnet und das Plugin gestartet. Es erscheint eine 3D Ansicht des Fliegengehirns, in der einzelne Punkte ausgewählt werden können: Ausgehend von diesen Startpunkten “wächst” eine Oberfläche um die entsprechenden Neuropile: Wie in der Abbildung ersichtlich ist, ist die entstandene Oberfläche unvollständig, bzw. enthält Löcher. Um
sie zu vervollständigen, werden vorhandene Referenz-Oberflächen von einem bereits segmentiertem Gehirn verwendet. Diese können von der Software an die bestehende Oberfläche aligniert und an das neue Bild angepasst werden. (In der folgenden Abbildung ist die alignierte Referenzoberfläche in grün dargestellt):

21
(Eine genauere Beschreibung des Plugins ist online unter http://3dviewer.neurofly.de/IntSeg_3D). Schließlich kann die Software die Volumina der einzelnen, angepassten Oberflächen berechnen. Der ganze Vorgang wird sowohl für den Wildtyp als auch für die white-Mutante durchgeführt, so dass die mittleren Volumina der Medulla, Lobula und Lobula-Platte von beiden Gruppen miteinander verglichen werden können.
Quantitative gene expression analysis (Nidhi):
Synapsin and Synapse Associated Protein of 47 KDa (Sap 47) are two synaptic proteins
involved in learning and memory. Mutants of genes coding for these proteins show a defect in
classical associative learning paradigm but show no other obvious phenotype. Therefore to
quantify the molecular changes occurring in these two mutants we have performed GeneChip
icroarray) experiments which have given information of gene expression modifications
ccurring in the genome of these mutants to cope with the mutation. The candidates selected by
we
the
bsence of these proteins. Since RNA code for proteins a measure of changes in protein levels
l time experiments would be
200 flies frozen in liquid nitrogen, heads (30 mg of tissue) isolated by vortexing.
RNeasy mini kit ion of total RNA. The place was
cleaned to b r sterile
tips were u
Steps:
(m
o
the microarray screen would be analysed during the course by real time PCR. In the course
would be analyzing the changes that occur at the transcript level (RNA) to compensate for
a
could be reflected by changes in the RNA copy number. Rea
performed for such quantification which would enable measurements of changes in transcript
number as the Polymerase Chain Reaction (PCR) proceeds.
Experimental procedure:
RNA extraction:
from QIAGEN was used for isolat
y RNase Zap from sigma to ma
sed at all steps.
ke the workplace RNase free. Filte

22
Fly heads were homogenised in 600 µl of RLT buffer and 6 µl of ß-
mercaptoethanol with glass homogenisers.
ple was centrifuged for 3 min at 13200 rpm.
et was discarded and supernatant was transferred into a new tube.
flow through was discarded and 700 µl of RW1 buffer was added to the
column and centrifuged at 12500 rpm for 15s.
ged for 15s at 12500 rpm. Once again
rpm.
min.
30 µl of RNase free H2O
in to elute the
ater bath for 10 min.
meter.
Verifi
e
apparatuses by
dissolv al concentration being 0.1% and left
ov and the rigs were treated
wit ated water. A
go d 260/230 at
2.0 ization of the gel 2
The homogenized sam
Pell
600 µl of 100% ethanol was added and gently mixed.
Take 600 µl of the mixture and add it to the columns of the kit.
600 µl of the sample was added to 2 ml RNeasy columns and centrifuged at 12500
rpm for 15s.
The
The columns were transferred to new collection tubes.
500 µl of RPE buffer was added and centrifu
500 µl was added to the columns and centrifuged for 2 min at 12500
The flow through was discarded and the column was centrifuged for 1
The column was transferred to new collection tube and
was added to the columns and centrifuged at 12500 rpm for 1 m
RNA. Then the columns were discarded.
1 µl of RNase-free DNase was added to the eluted RNA.
RNA was incubated at 37°C for 45 min to activate DNase function.
To inactivate DNase tube was placed in 95°C w
RNA concentration was measured using a spectrophoto
cation of RNA quality on a gel:
When working with RNA it is important to make sure that working space and all th
and reagents used are RNase free. Therefore water was made RNase free
ing DEPC (Di-Ethyl Pyro Carbonate) with fin
ernight for dissolving before autoclaving it. The gel running chamber
h 3% hydrogen peroxide and left overnight. They were rinsed with DEPC tre
od quality RNA should have Optical Density (OD) ratio 260/280 at least 1.8 an
. There should also be no degradation visible on the RNA gel. On visual

23
bands are observed at 2 Kb, one band corresponds to 18S RNA which should give a signal at 2Kb
nd the other band corresponds to 28S RNA which should ideally give signal at 4Kb, but in
Drosop
re was cooled to 65 °C and to it 2ml of 37% (12.3M) of formaldehyde was
dded under the hood.
ify.
aration:
To 4.5 µl of RNA sample 2 µl of 5X MOPS buffer, 3.5 µl formaldehyde (37%), and 10 µl
le.
RNA was extracted using procedure above. The
1 µl of oligo-dT (Invitrogen) primer and 1ul of dNTPs (Invitrogen) were added.
a
hila the 28S RNA is cleaved into two fragments which cannot be distinguished from each
other on the gel.
Steps for preparing Formaldehyde Agarose gel for RNA visualisation:
Gel preparation:
Preparation of 10X MOPS buffer: 200mM of 3-[morpholino] propanesulphonic acid,
MOPS with 50mM sodium acetate, 10mM EDTA and pH was set to 7.0 with NaOH
0.5 g of agarose was boiled in 50 ml of DEPC water and 6ml of 10X MOPS buffer
The mixtu
a
The mixture was poured into the casting tray and allowed to solid
RNA sample prep
of formamide.
The mixture was heated to 65°C for 15 min and cooled on ice.
2 µl of 6X sample loading dye and 1 µl of ethidium bromide were added to the samp
The gel was run in 1X MOPS buffer at 60V for 1 hour.
Reverse transcription:
Reverse transcription procedure was used to convert RNA to cDNA sequences as RNA is
highly unstable and prone to degradation. The
downstream procedure was as follows:
11 µl of RNA was taken.

24
The mixture was incubated for 5 min at 65°C
At the end of 5 min sample was immediately placed on ice and centrifuged.
and buffer (Invitrogen) was added.
as added and incubated for 1.5 hrs at 42°C.
Sample at the end of 1.5 hrs was incubated at 72°C for 10 min.
nd incubated at 37°C for 30 min.
erent genes quantitative PCR was used this
pt number in each cycle of logarithmic phase of
py number.
ber of transcripts increase
portional to transcript copy
e)
1 µl (Sense primer)
5 µl of 5x first str
2 µl of 0.1M DTT (Invitrogen) was added.
Sample was incubated for 2 min at 42°C.
1 µl of Superscript (Invitrogen) w
1 µl of RNase was added a
The sample was frozen.
Quantitative PCR (qPCR) or Real time PCR:
To quantify the transcript number of diff
method makes use of the quantification of transcri
PCR. The SYBR green based quantification was used to quantify the transcript co
SYBR green dye can only bind to double stranded DNA and as the num
with each cycle of PCR the fluorescence intensity changes pro
number.
Components: 1 µl (cDNA templat
1 µl (Anti-Sense primer)
7 µl Water
10 µl SYBR green dye
Total 20 µl

25
Gruppe III zipitation (IP) und Western Blot, larvale whole mounts, Immunprä
confocale Mikroskopie (Alice, Mandy)
Immunpräzipitation
isolierung mittels
iner Immunpräzipitation durchgeführt. Präzipitierte Proteine werden einerseits mit einem
roteinisolierung aus adulten Fliegenköpfen mittelsP
Um die Fliegen auf Proteinebene untersuchen zu können, wird eine Protein
e
Western Blot, andererseits mit einer Silberfärbung des Gels nachgewiesen. Wichtig bei allen
Experimenten sind Kontrollen. Diskutieren Sie für die IP die Vor- und Nachteile von Kontrollen
mit Präimmunserum, Weglassen des Antikörpers, und Nullmutanten für das zu präzipitierende
Protein.
Immunpräzipitation

26
Es werden 1ml Fliegen in einem 15ml Falcon abgemessen und in flüssigem Stickstoff
Western Blot
eingefroren. Dann werden sie mittels feinmaschiger Siebe dekapitiert. Die Fliegenköpfe werden
in 800μl gekühltem Lysispuffer homogenisiert, in Eppendorf caps überführt und abzentrifugiert.
Der Überstand wird in ein neues Reaktionsgefäß überführt und für 40 Minuten bei 4°C auf dem
Nutator lysiert. Nach einer 30minütigen Zentrifugation wird der Überstand in ein neues,
gekühltes Reaktionsgefäß überführt. Zu diesem Ansatz werden 300μl von dem Gemisch aus
Lysispuffer mit Proteaseinhibitoren und einer complete Mini Tablette zugegeben. Ebenfalls wird
der Antikörper (nc46) in das Reaktionsgemisch pipettiert und für 10 Minuten auf dem Nutator
inkubiert. Anschließend werden 100μl Protein G-Agarose zugegeben und über Nacht auf dem
Nutator rotiert. Die Protein G-Agarose bindet an die Immunglobuline, an denen das Antigen
gebunden vorliegt. Am nächsten Tag werden die Agarose-Beads dreimal mit Lysispuffer
gewaschen. Zum Schluss wird das Pellet in 40μl 2x Laemmli-Puffer aufgenommen. Zur
Proteindenaturierung wird der Ansatz fünf Minuten bei 96°C aufgekocht, und ein Western Blot
kann durchgeführt werden. Die Proben werden in die Taschen der Fertig-Gele pipettiert und für
1h bei 125 mA laufen gelassen.
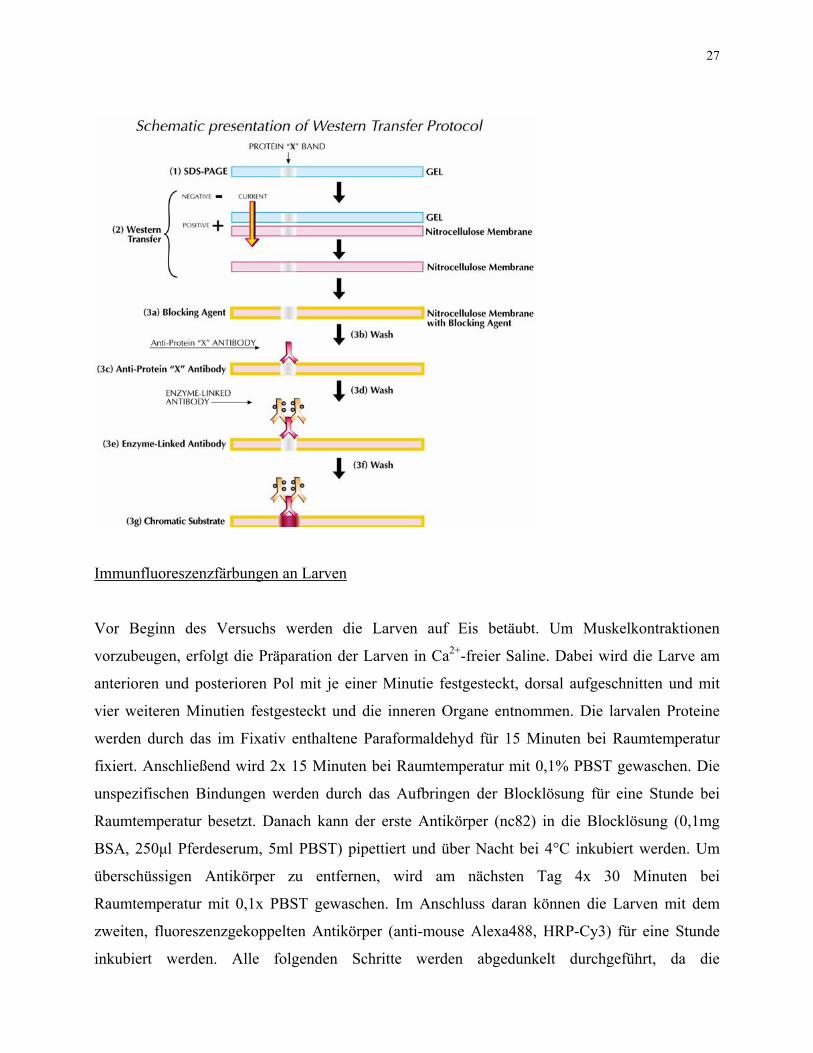
stern Blot werden Proteine auf eine Nitrocellulosemembran transferiert und durch
aufgelegten Röntgenfilm.
Bei einem Weeine Immundetektion visualisiert. Dadurch kann man das Molekulargewicht und die Expressionsstärke eines bestimmten Proteins untersuchen. Für das Blotten wird das Gel aus den Plastikplatten entnommen und mit Whatmanpaper und einer Membran in Transferpuffer befeuchtet. Die Blot-Apparatur wird, wie von BioRad angegeben, zusammengebaut. Das Blotten erfolgt für 1h bei 100V, dadurch werden die Proteine aus dem Gel auf die Membran übertragen. Um Proteine auf der Nitrocellulosemembran nachzuweisen, verwendet man den EnhancedChemiluminescence Nachweis, mit welchem auch kleine Proteinmengen detektiert werden. Nach dem Blotten wird die Membran über Nacht bei 4°C in einer 5%igen Milchpulver-Lösung geblockt, um so noch freie Bindungsstellen abzusättigen. Am nächsten Tag wäscht man die Membran 3x 10 Minuten mit 1x TBST. Nun wird die Membran mit dem ersten Antikörper für eine Stunde inkubiert (3c11 oder nc46). Die unspezifisch gebundenen oder überschüssigen Antikörper werden durch die Waschschritte mit 1x TBST entfernt. Es erfolgt die Inkubation mit dem zweiten Antikörper, der gegen die Fc-Domäne des ersten Antikörpers gerichtet ist (HRP anti-mouse 1:7500). Die ECL-Lösung von Millipore wird im Verhältnis 1:1 zusammenpipettiert, und damit wird die Membran für eine Minute überschichtet. An dem zweiten Antikörper ist das Enzym HRP (horseradish peroxidase) gekoppelt, welches die Umsetzung von Luminol in die oxidierte Form katalysiert. Bei dieser Reaktion wird Licht emittiert, und dieses schwärzt den
27
Immunfluoreszenzfärbungen an Larven
auf Eis betäubt. Um Muskelkontraktionen
orzubeugen, erfolgt die Präparation der Larven in Ca2+-freier Saline. Dabei wird die Larve am
Vor Beginn des Versuchs werden die Larven
v
anterioren und posterioren Pol mit je einer Minutie festgesteckt, dorsal aufgeschnitten und mit
vier weiteren Minutien festgesteckt und die inneren Organe entnommen. Die larvalen Proteine
werden durch das im Fixativ enthaltene Paraformaldehyd für 15 Minuten bei Raumtemperatur
fixiert. Anschließend wird 2x 15 Minuten bei Raumtemperatur mit 0,1% PBST gewaschen. Die
unspezifischen Bindungen werden durch das Aufbringen der Blocklösung für eine Stunde bei
Raumtemperatur besetzt. Danach kann der erste Antikörper (nc82) in die Blocklösung (0,1mg
BSA, 250μl Pferdeserum, 5ml PBST) pipettiert und über Nacht bei 4°C inkubiert werden. Um
überschüssigen Antikörper zu entfernen, wird am nächsten Tag 4x 30 Minuten bei
Raumtemperatur mit 0,1x PBST gewaschen. Im Anschluss daran können die Larven mit dem
zweiten, fluoreszenzgekoppelten Antikörper (anti-mouse Alexa488, HRP-Cy3) für eine Stunde
inkubiert werden. Alle folgenden Schritte werden abgedunkelt durchgeführt, da die

28
Fluoreszenzfarbstoffe lichtempfindlich sind und ausbleichen können. Dann wird 4x 30 Minuten
mit 0,1x PBST gewaschen. Nun können die fertigen Larven in Vectashield eingebettet und bei
4°C aufbewahrt werden. Mit Hilfe eines konfokalen Laserscanning-Mikroskops können die
Präparate angeschaut werden.
Bestimmung der Flugfähigkeit nach Benzer (Benzer, 1973)
Um das Flugvermögen eines Fliegenstamms zu untersuchen, wird der Versuchsaufbau nach
teter 500ml Zylinder als Flugarena
Abbildung: Versuchsaufbau zur Erm ugfähigkeit (Benzer et al., 1973).
Benzer gewählt. Hierbei dient ein mit Paraffinöl beschich
(siehe Abbildung). Es werden jeweils 2x 50 männliche Fliegen jedes Genotyps (Wildtyp und
Nullmutante) getestet. Die Fliegen werden mit Hilfe eines Trichters in den Zylinder geklopft und
bleiben, sobald sie anfangen zu fliegen, an den beschichteten Wänden kleben. Nun kann man die
fixierten Fliegen auszählen. Anschließend wird anhand einer Excel-Tabelle die Flugfähigkeit
bzw. Flugunfähigkeit berechnet.
ittlung der Fl
Präferenz Versuch
Bei diesem Versuch untersuchen wir, ob Fliegen auf bestimmte Alkoholwerte reagieren können.
ierbei wird Mangosaft mit 5% oder 23% Ethanol versetzt. Es werden je 50 männliche Fliegen H
(Wildtyp und Nullmutante) gesammelt und über Nacht ruhen gelassen. Am nächsten Tag werden
diese Fliegen in große Bechergläser geschubst und über Nacht auf Lichttischen belassen. Am
nächsten Morgen wird ausgewertet.

29
2-D-Gelelektrophorese
se dient dazu, Proteingemische aufzutrennen. Für die Auflösung der Die 2D-Gelelektrophore
Proteingemische werden die Proteine durch isoelektrischen Fokussierung getrennt und danach
erfolgt eine SDS-Polyacrylamidgelelektrophorese. Die isolelektrische Fokussierung erfolgt
anhand der Zusammensetzung des Proteins aus sauren und basischen Aminosäuren (1.
Dimension). Wir verwenden einen immobilen pH-Gradienten Streifen als Beladungsmatrix.
Hiermit erfolgt die isoelektrische Fokussierung. In der zweiten Dimension erfolgt eine Trennung
nach der Größe des jeweiligen Proteins. Wir verwenden das ZOOM® 2D PAGE System.
ample Solubilizing Mix:
izer 1 or 2 90.9 μl
hibito Cocktail (Roche®)
μl
μl
ample loading / Strip rehydration mix:
133.5 μl
μl
S
Zoom® 2D Protein Solubil
1 M Tris Base 0.5 μl
100x Protease In r 1 μl
2 M DTT 1
ddH2O 2
S
Zoom® 2D Protein Solubilizer 1 or 2
2 M DTT 1

30
Ampholytes 3-10 (Serva) 2 μl
lume 165 μl
quilibration Solution :
ple Buffer final conc 1x
lkylation Solution:
ample Buffer 400 ml
.0093 g
orgehensweise:
werden gesammelt, in Stickstoff tiefgefroren und dekapitiert.
bei 13000 rpm
atationsmix hergestellt und in die ZOOM® IPGRunner® Kassette geladen.
die Kassette hineingeführt und Luftblasen entfernt. Die Streifen bleiben
unner® Kassette werden Wattepads geklebt und diese werden mit 600 μl
ylierungslösung für 15 min unter Schütteln
ethoden auf RNA-Ebene: RT-PCR
asy Mini-Kits von QIAGEN
ddH2O 5 μl
1% Bromophenolblue trace
Sample homogenate Final vo
E
4x NuPAGE™ LDS Sam
dH2O 4.5 ml
NuPAGE™ Reducing Agent 0.7 ml
A
1x NuPAGE™ LDS S
Iodoacetamide 0
V
Die Wildtypfliegen
Die Köpfe werden in 95 μl Solubilisierungsmix homogenisiert und 2x für 15 min
bei 4°C zentrifugiert.
Dann wird der Rehydr
(Endvolumen 165 μl).
Die Streifen werden in
über Nacht auf 18 °C.
Auf die ZOOM® IPGR
Wasser angefeuchtet. Danach wird die Kassette in die Gelkammer eingespannt und die äußere
Kammer wird mit 600ml Wasser gefüllt. Die Fokussierung erfolgt bei 1500Vh. Danach wird die
Kassette entnommen und bei -80°C tiefgefroren.
Am nächsten Tag werden die Streifen in Alk
behandelt. Danach werden die Streifen in die fertigen 4-12% Bis-Tris NuPAGE™ 2DE Gele
positioniert und mit 400 μl der 0.5% Agarose bedeckt und eine SDS-PAGE wird durchgeführt.
M
Die Isolation der totalen RNA wird mit Hilfe des RNe

31
durchgeführt. Vor Beginn der Arbeit wird der Arbeitsplatz mit Ethanol gereinigt. Des Weiteren
u können, wird eine semiquantitative
n
s wird folgender PCR-Ansatz eingesetzt:
ense
ermix
en
anach werden die Proben mittels einer Gelelektrophorese aufgetrennt.
ruppe IV
werden sterile Filterspitzen verwendet, um eine Kontamination zu vermeiden. Alle
Arbeitsschritte sollten auf Eis erfolgen. Nach der Isolation der totalen RNA wird die genomische
DNA durch Zugabe von 1μl DNase für eine Stunde bei 37°C verdaut. Abgestoppt wird der
Verdau durch zehnminütiges Erhitzen auf 95°C. Die RNA wird nun mittels einer reversen
Transkription in cDNA umgeschrieben. Um eine Primer-Bindung zu erhalten, werden 11μl
mRNA mit je 1μl Oligo-dT-Primer und dNTPs für fünf Minuten bei 65°C inkubiert, danach
sofort auf Eis gestellt und kurz abzentrifugiert. Anschließend werden 4μl 5x First Standard
Buffer und 2μl DTT dazugegeben und der Ansatz wird für zwei Minuten bei 42°C vorinkubiert.
Für die reverse Transkription wird 1μl Super Script II in das Reaktionsgefäß pipettiert, und der
Reaktionsansatz wird für 90 Minuten bei 42°C inkubiert. In dieser Zeit erkennt die reverse
Transkriptase den an die poly A-RNA hybridisierten Oligo-dT-Primer, bindet an ihn und
synthetisiert ausgehend davon den komplementären Strang. Das Abstoppen der Reaktion erfolgt
durch zehnminütiges Erhitzen auf 70°C. Die enthaltene RNA wird durch Zugabe von 1μl RNase
bei einer Inkubation von 30 Minuten bei 37°C verdaut.
Um Transkriptionsunterschiede eines Gens detektieren z
PCR durchgeführt. Man versucht, durch Verwendung verschiedener Zyklenzahlen einen
Unterschied in der Expressionsstärke zu detektieren. Hier werden 25, 28, 31 und 34 Zykle
verwendet.
E
▫ 5μl cDNA
▫ 1μl Primer s
▫ 1μl Primer anti
▫ 8μl H2O
▫ 35μl Mast
50μl Gesamtvolum
D
G

32
Synaptische Proteine: Immunhistochemie, Western Blots, 2D-Gels, ELISA (Partho, Tulip)
In unserer Arbeitsgruppe wurden mit der in der Vorlesung besprochenen Methode „Vom
Antikörper zum Gen“ drei neue Proteine der präsynaptischen Endigung entdeckt, das Cysteine-
String-Protein (CSP), das „Bruchpilot-Prtoein“ (BRP) und das Synapse-Associated-Protein
47kD (SAP47), sowie das Drosophila Homolog zum Synapsin (SYN). Monoklonale Antikörper,
die diese Proteine erkennen, stehen zur Verfügung. Die Antigene für drei weitere monoklonale
Antikörper (MAKs aa2, na21 und nb43) werden gegenwärtig gesucht.
Im Kurs sollen Molekulargewichte und Expressionsstärke (Western Blot, ELISA) bzw.
Expressionsmuster (Immunhistochemie) bzw. Interaktionspartner (Immunpräzipitation) von den
Proteinen ermittelt werden, die durch die zugehörigen Antikörper erkannt werden. In Mutanten
sollen Veränderungen analysiert werden.
Methodik
Antikörper, in Lösung auf einen Gefrierschnitt durch das Gehirn oder auf eine mit Proteinen
beladene Membran (Blot) gebracht, binden nur an die Moleküle, gegen die sie induziert wurden
(Antigene), ungebundener Antikörper wird weggewaschen. Die Verteilung des gebundenen
Antikörpers im Schnitt oder auf dem Blot, und damit die Verteilung des entsprechenden
Antigens, kann man mit Hilfe von Fluoreszenz-Markern oder durch Kopplung von Peroxidase an
die Antikörperbindungsstelle und eine von diesem Enzym katalysierte Farb- oder
Lumineszensreaktion sichtbar machen. In unserem Labor haben sich unterschiedliche Prozeduren
bewährt, an ungeschnittenen Präparaten („wholemounts“) die Inkubation mit Fluoreszenz-
markierten sekundären Antikörpern, die „Enhanced Chemilumineszenz“ (ECL)-Technik am
Western Blot, und die alkaische Phosphatase-Substratreaktion im ELISA.
Bei der Immunhistochemie inkubiert man das fixierte Präparat (Schnitt oder wholemount)
nach Waschen mit dem spezifischen „1.“ Antikörper (z.B. aus der Maus), wäscht wieder und
inkubiert mit dem „2.“ Antikörper, der gegen die konstante Region des 1. Antikörpers gerichtet
ist (z.B. Ziege-anti-Maus IgG). An diesen 2. Antikörper ist ein Fluorophor (fluoreszierendes
Molekül) kovalent gekoppelt, so dass indirekt die Position der Bindungsstelle für den 1.
Antikörper im Gewebe im Fluoreszenzmikroskop lokalisiert werden kann.

33
Beim Western Blot mit ECL-Detektion wird das zu untersuchende Gewebe (statt Gehirn
verwendet man bei Drosophila meist ganze Köpfe) in SDS-Puffer homogenisiert und auf einem
Polyacrylamid-Gel unter denaturierenden Bedingungen elektrophoretisch der Größe nach
aufgetrennt. Aus dem Gel werden die Proteine durch Elektroblotting auf eine
Nitrozellulosemembran transferiert. Nach Absättigung der freien Proteinbindungsstellen der
Membran inkubiert man den Blot mit dem 1. Antikörper, der an „sein“ Antigen auf dem Blot
bindet, und detektiert den 1. Antikörper mit einem HRP-gekoppelten 2. Antikörper, wobei das
Enzym (HRP) eine Lumineszenz-Reaktion katalysiert. Die Lumineszenz (chemische
Lichterzeugung) wird auf dem Blot durch Auflegen eines Röntgenfilms dokumentiert.
Beim Enzyme-linked immunosorbent assay (ELISA) wird das Kopfhomogenat in die
Vertiefungen einer Multititerplatte pipettiert, die Proteine binden dann an die Plastikoberfläche.
Nach Blocken der unspezifischen Bindungsstellen durch Rinderserumalbumin (BSA) wird der
erste Antikörper (Maus monoklonal), dann der Zweitantikörper (anti-Maus-Ig, biotinyliert), dann
der Avidin-alkaische Phosphatase-Komplex, und schließlich das Substrat (pNPP) zugegeben,
jeweils getrennt durch Waschschritte. Die Absorption des gelben Farbniederschlags, der
proportional zur Menge an gebundenem erstem Antikörper (und damit Antigen) entsteht, wird
gemessen.
Der Einfluss von Mutationen oder von der Expression transgener Proteine auf die
synaptische Übertragung kann bei Drosophila am larvalen Nerv-Muskel-Präparat direkt
gemessen werden. Wegen der diffizilen Präparation kann dies im Kurs nur beschrieben werden.
A) Immunoprecipitation
Objective:
1. Immunoprecipitate Synapsin from head homogenates of D.melanogaster.
2. Analyse the immunoprecipitated sample using 2D-PAGE and western blot.
Materials
Lysis buffer (1X) 25 ml Tris 1 M pH 7.5 150 ml NaCl 1 M 2 ml EDTA 1 M
2 ml EGTA 1 M 100 ml Glycerol 1 ml NP-40 Added H2O to make up the volume to 1 l
Aprotinin(100X) 50 μl Complete mini EDTA free

34
protease inhibitor 1 tablet cocktail tablets Leupeptin(1000X) 5 μl Phenylmethylsulfonylfluorid (PMSF)(500X) 10 μl Pepstatin(1000X) 5 μl Protein G-Agarose 50-100 μl • Antibodies
Primary antibody Dilution
3c11 1:50
nc46 1:200 Secondary antibody : Anti Mouse HRP conjugate 1:3000
Procedure
Immunoprecipitation is a technique in which an antigen is isolated by binding to a
specific antibody attached to a sedimentable matrix. The source of antigen in our case for
immunoprecipitation is the homogenized solution of Drosophila heads. Either polyclonal or
monoclonal antibodies from various animal species can be used in immunoprecipitation protocols
but in our case the antibodies used (3c11, nc46 etc) are all mouse monoclonal antibodies.
Prior to performing the actual procedure of immunoprecipitation, the fly heads are
collected. The procedure by which they were collected is as follows: The flies are anaesthetized
using CO2 and then collected in Eppendorf or larger vials depending upon the requirement. The
vials are then dropped into a can of liquid nitrogen and kept immersed for a period of 2-3
minutes, then removed from the can and vortexed for a minute. The contents of the vial are then
dropped onto a clean sheet of paper and fly heads are collected using a soft brush. For larger
number of heads, a sieve is used to collect the heads. The sieve size (500 microns) is such that it
retains the body parts of the fly and the head passes through.

35
The collected heads are put in an eppendorf tube and 20μl of lysis buffer per head is
added to it. The homogenization is done using plastic or glass pestles. The homogenized solution
is kept in the cold room (4°C) for an hour. The sample is centrifuged for an hour at 4°C, the
supernatant is transferred to a fresh eppendorf tube and the pellet is discarded. About 30-40 µl of
the above supernatant is taken in a separate eppendorf and mixed with equal amount of (1:1) 2X
Laemmli buffer and stored at -20°C to be used as a lysate control on an SDS-PAGE.
After the collection and homogenization of the fly heads, the buffers for the
immunoprecipitation are prepared. It is necessary that the buffers with protease inhibitors are
prepared fresh to have maximum efficiency of the inhibitors.
In a separate eppendorf tube the precipitating antibody (400 µl) is mixed with 50 µl of
protein G-Agarose beads and 250 µl of lysis buffer containing protease inhibitors (including
complete mini EDTA free protease inhibitors cocktail tablets). The solution is incubated for 1-2
hours at 4°C on a shaker. The tube is centrifuged for 1-2 minutes at 4°C, the supernatant is
removed and the beads are washed twice with lysis buffer. The sample is then added to the above
antibody bound beads and lysis buffer with protease inhibitors is added to make the final volume
to 750-800 µl for better mixing. The eppendorf tubes are then put on a rotator for 12-15 hours for
better mixing and binding of the antibody antigen complex. Then the sample is centrifuged for 1-
2 minute and the supernatant is discarded, the beads are washed with lysis buffer for 3 times and
the supernatant is completely removed after the final wash. 2X Laemmli buffer is added to the
above beads (1:1) and the sample is boiled at 95°C for 5 minutes and then loaded on an SDS-
PAGE.

36
Immunoprecipitation procedure
The immunprecipitated samples can then be used for 2-D gel elektrophoresis. In particular we are interested to see if the 2D pattern of Synapsin is modified by dephosphorylation of the samples. B) ELISA
Objective
ELISA (Enzyme -linked immunosorbent assay)
1. To quantitatively determine a neuronal protein in head homogenates of different genotypes of D.melanogaster.
2. Detect a possible difference in Synapsin content in SAP47 null mutant flies when compared to wild-type CS.
Materials
PBS (10X) 14.8 g Na2HPO4 4.3 g KH2PO4 72.0 g NaCl Added 1 l H2O and adjusted the pH to 7.4 EDTA 1% • Blocking buffer 1.0 g Bovine Serum Albumin (BSA) (1X) 100 ml of 1X PBS • Detection buffer 12.11 g Tris/HCl 100mM (1X) 0.2 g MgCl2 Added 1 l H2O and adjusted the pH to 9.5 Antibodies
Primary antibody Dilution
3c11 1:50
nc46 1:200

37
Secondary antibody : Anti Mouse Biotin conjugate 1:400
Procedure
ELISA is widely used to detect antibody or antigen quantitatively and/or qualitatively, as
required. In this case the technique will be used to detect the presence of an antigen (protein) .
The various steps involved in the technique are as follows: The antigen is obtained in PBS buffer
by homogenizing the fly heads in PBS buffer using mortar and pestle. About 50 µl of antigen is
coated or loaded onto each well of the ELISA plate. The plate is kept at room temperature on a
shaker for 2 hours. After 2 hours the plate is washed once with water. Blocking solution is added
to each of the wells (300 µl / well). The blocking solution is prepared by making a 1% solution of
BSA in PBS. The plate is then kept on a shaker for about 2 hours at room temperature. On
completion of the blocking step the primary antibody is added to each well (100 µl / well) and
incubated at 4°C on a shaker for 12 hours. The primary antibody solution is prepared by diluting
the antibody in blocking solution.
ELISA procedure
PBS washes (3 times) are performed for 10 minutes duration each. In the next step the
secondary antibody (anti-mouse biotin conjugate) is added to each well (100 µl / well) and the
plate is incubated at room temperature for 2 hours. The secondary antibody solution is prepared
by diluting the antibody in blocking solution (1:400). PBS washes (3 times) are performed for 10
minutes duration each. In the next step, avidin-conjugated alkaline phosphatase (avidin binds
with high affinity to biotin) is added to each of the wells (100 µl / well) and the plate is incubated

38
at room temperature for 1 hour. The solution is prepared by diluting avidin-conjugated alkaline
phosphatase in blocking solution (1:10,000). PBS washes (2 times) are performed for 10 minutes
duration each followed by the third wash using detection buffer (Tris/HCl, MgCl2 pH 9.5) for 10
minutes duration. The substrate (pNPP) is added (100 µl / well) and the plate is kept in the dark
until colour development is observed. The developed colour is measured using the ELISA reader
and the data is analysed using statistical software (Origin Version 7.5). No experiment without
positive (wild type) and negative (null mutant) controls!
C) Protein isolation from fly heads:
Proteins are the most diverse class of all biomolecules and have widely different physical and
chemical properties like solubility, size, charge, specific binding affinities etc. So there is no
single method to isolate the total proteome of any given source. So for total isolation general
procedures are used to try and isolate as many as possible while for specific proteins, specially
designed strategies (based on the properties of the protein) are used to specifically enrich and
purify them from the rest. The first step of proteins isolation is a homogenization of the sample
(cells/tissues), using a ‘lysis buffer’. The main function of the lysis buffer is to provide conditions
suitable to cause the disruption of the tissue, solubilization of the cell membranes to facilitate the
release of proteins, extraction of the released proteins into solution and their maintenance in
stable form. So the basic components of any lysis buffer are:
Buffering agent to maintain pH near physiological conditions,
Lysing agent to solubilize membranes and release proteins,
Salts to maintain physiological conditions or disrupt noncovalent interactions.
For 1-D gels we will use a simpler method of homogenization of the tissue sample in the
Laemmli’s buffer, which is used as a sample loading buffer for SDS-PAGE. It disrupts all non-
covalent interactions and also the covalent disulfide bond, generating linear polypeptides. Its
composition includes:
Laemmli buffer 120mM Tris (1.2 ml of 1M Tris, pH 6.8 stock) (2X) 20% (v/v) Glycerol (2 ml) 4.0% (w/v) SDS (4 ml 10% SDS stock) 0.02% (w/v) Bromophenol blue (0.2 ml of 1% stock) 5.0% β-Mercaptoethanol (0.5 ml added fresh) Final volume made up to 10 ml with dH2O

39
Protocol:
o The required number of wild type (Canton S) flies were collected by anesthetizing the flies
and they were frozen in liq N2.
o All jointed body parts were separated by vortexing vigorously.
o Separated heads were collected in a fresh eppi (for large number of flies, the frozen flies after
vortexing are passed though a 800 µm sieve placed above an 500 µm sieve, which allows
only heads to be collected between the two).
o Required number of heads were then homogenized in 2x Laemmli buffer (10 µl per head).
o The homogenate was spun briefly and then denatured at 90ºC for 5 min.
o The mixture was then again spun briefly and the supernatant was loaded on SDS PAGE gel
and resolved according to their Mr as described below.
1. SDS-PAGE (Na-dodecylsulfate- Polyacrylamide Gel Electrophoresis)
SDS PAGE (24) is based on the principle of differential electrophoretic mobility of
proteins of different sizes (Mr) through a resolving matrix. One such commonly used matrix is a
polymeric mixture of acrylamide and bisacrylamide. Bisacrylamide introduces crosslinks
between linear polyacrylamide chains generating a mesh-like network. It is the ratio of
acrylamide to bisacrylamide with respect to the total volume of the mixture that determines the
'pore size' of the matrix. The higher the ratio of bisacrylamide to acrylamide and the higher the
total acrylamide concentration, the lower will be the pore size and hence the electrophoretic
mobility of proteins. Polymerization of acrylamide and bisacrylamide monomers is initiated by
ammonium persulfate (APS), which generates peroxide free radicals. TEMED is used as a free
radical stabilizer to promote polymerization.

40
(Source: http://www.steve.gb.com)
Sodium dodecyl sulfate (SDS) is an amphipathic detergent with an anionic headgroup and
a lipophilic tail. It binds non-covalently to proteins, with a stoichiometry of around one SDS
molecule per two amino acids, thus masking the intrinsic charge of the proteins and giving them a
uniformly distributed negative charge. This dirupts the non-covalent bonds in proteins. Further
addition of reducing agents like ß-mercaptoethanol or DTT causes reduction of disulfide bridges.
Finally, denaturing this protein mixture by heat causes complete linearization of most proteins
into polypeptide chains.
In an electric field these negatively charged polypeptides migrate toward the anode. The
SDS-treated polypeptides have very similar charge-to-mass ratios, and similar shapes. Thus the
rate of migration of SDS-treated proteins in the gel is effectively determined by the approximate
molecular weight. The relative molecular weight as determined in a gel relative to marker
proteins of known calculated molecular weight Mcalc is denoted as Mr.

41
(Source: http://www.imb-jena.de)
Mini Protean™ (BioRad®) Electrophoresis System:
Resolving gel (Reagents for two mini gels) Gel percentage 12.5% 15%
Reagents (ml) (ml) 30% Acrylamide bisacrylamide(29:1) 3.125 3.75 1.88 M Tris/HCl, pH 8.8 1.5 1.5 dH2O 1.375 0.75 0.5% SDS 1.5 1.5 10% Ammonium persulfate (APS) 0.04 0.04 TEMED 0.006 0.006
5.0% Stacking gel (Reagents for two mini gels)
Reagents (ml) 30%Acrylamide bisacrylamide (29:1) 0.5 0.635 M Tris/HCl, pH 6.8 0.6 dH2O 1.3 0.5% SDS buffer 0.6 10% Ammonium persulfate (APS) 0.03 TEMED 0.004
SDS PAGE Running 125 mM Tris (30.2 g) buffer (10X) 960 mM Glycine (144.2 g)
0.5 % SDS (10 g) pH adjusted to 8.9 and final volume was made to 1l by dH2O (stored at room temperature)
Protocol:
o 0.75 mm thick, 12.5% gel was cast according to the standard recipe.
o Protein samples prepared as lysates in 2x Laemmli buffer were denatured at 90ºC for 5 min.

42
o The mixture was then again spun briefly and the supernatant was loaded on an SDS-
polyacrylamide gel to resolve the proteome over a wide range of Mr.
o Stacking was done at 100 V and resolution was done at 150 V using the Mini Protean™ gel
apparatus (Biorad®).
2. Two dimensional SDS-PAGE
Normal SDS-PAGE involves separation of proteins according to their mass, hence only one
dimensional separation. This is often not enough to resolve complex protein samples as multiple
proteins may have the same Mr. So another dimension of separation based on some other
property of proteins is often used to perform two dimensional electrophoresis (2DE). We will use
isoelectric focusing (IEF) as our new dimension of separation, which separates proteins according
to their isoelectric points (pI). It involves the use of an immobilized pH gradient (IPG) strip, as
the matrix to load samples, which is then subjected to high voltage for isoelectric focusing,
followed by transfer of the proteins (focused as bands on the strip) onto a normal polyacrylamide
gel to run the second dimension as a normal SDS-PAGE. Hence we separate first by pI and then
by Mr. We will use the ZOOM® 2D PAGE setup from Invitrogen.

43
Sample Solubilizing Mix: Zoom® 2D Protein Solubilizer 1 or 2 90.9 µl 1 M Tris Base 0.5 µl 100x Protease Inhibitor Cocktail (Roche®) 1 µl 2 M DTT 1 µl ddH2O 2 µl Sample loading / Strip rehydration mix: Zoom® 2D Protein Solubilizer 1 or 2 133.5 µl 2 M DTT 1 µl Ampholytes 3-10 (Serva) 2 µl ddH2O 5 µl 1% Bromophenolblue trace Sample homogenate Final volume 165 µl Equilibration Solution : 4x NuPAGE™ LDS Sample Buffer final conc 1x dH2O 4.5 ml NuPAGE™ Reducing Agent 0.7 ml

44
Alkylation Solution: 1x NuPAGE™ LDS Sample Buffer 400 ml Iodoacetamide 0.0093 g
Protocol:
o The required number of flies (wildtype and mutant) were collected, frozen and their heads
were separated as described earlier.
o The heads were homogenized in 95 µl of sample solubilizing mix and centrifuged twice for
15 minutes with 13,000 rpm at 4 °C.
o The required amount of the supernatant was then used to prepare the final rehydration mix
and the sample (final volume 165 µl) was loaded in the channel at the curved end of the
ZOOM® IPGRunner® Cassette (see figure below).
o Then the IPG strip(s) was guided gently into each channel having a sample, while holding the
strip at its basic end, with the printed side facing down (see figure below). Air bubbles were
avoided.
o All Sample Loading Wells were sealed using tape after all air bubbles were removed.
o The loaded and sealed ZOOM® IPGRunner® Cassette was put at 18°C overnight to
rehydrate the strips.
o Next day the sealing tape and the two Sample Loading Devices were removed from the
ZOOM® IPGRunner® Cassette.

45
o Two electrode wicks were placed over the exposed ends of the strips on each end of the
cassette and 600 µl of deionized water was added to each. Do not use ultrapure (MilliQ)
water.
o Then the IPGRunner® ‘Cassette + Core sandwich’ was assembled (as shown below) and slid
into the Mini-Cell Chamber of the IPGRunner® and locked.
o The outer chamber was filled with 600 ml of deionized water, while taking care that water
does not spill into the inner chamber.
o Thereafter the setup was connected to the power supply and was focused for 1500-1600 Vh.
o Thereafter the strips were taken out of the cassette and incubated in the equilibration and
alkylation solutions respectively for 15 minutes each with shaking.
o Thereafter the strips were slid into the IPG well of precast 4-12% Bis-Tris NuPAGE™ 2DE
gels, and overlayed with ~400 µl of 0.5% agarose solution prepared in the running buffer (1x
MOPS).
o Once the agarose has solidified, 5 µl of marker was loaded and the second dimension of
electrophoresis (SDS-PAGE) was performed at 100-150V (if required with cooling).
o Finally the second dimension gel was subjected to either staining or western blotting as per
the need.

46
3. Western Blotting
Western blotting is based on the principle of electrophoretic transfer of resolved proteins
from the gel to a porous membrane for adsorption of proteins on its surface in a pattern
resembling their resolution on gel (25). So it involves application of an electric field to SDS-
treated proteins perpendicular to the direction of resolution (along the width of the gel) as shown
below:
It is usually followed by detection of a protein of interest on the membrane using primary (1º)
and secondary (2º) antibodies and a suitable detection chemistry as illustrated below:

47
Transfer buffer 25mM Tris (6.04 g) (1X) 150 mM Glycine (22.52 g) 20% Methanol (400 ml)
pH was adjusted to 8.3 and volume made upto 2000ml with dH2O (stored at 4°C)
Washing buffer 100mM Tris (12.11 g) (10X TBST) 1.5 M NaCl (87.66 g) 0.5% Tween-20 (5.0 ml) pH was adjusted to 7.6 and final volume made upto 1l. Blocking solution 1 g Non fat dry milk powder dissolved in 20 ml (per blot) of
(5% NFDM) washing buffer (1X TBST) and heated to ~60°C and filtered.
Protocol:
o After electrophoresis, the gel was removed from the glass plates and washed in 1x transfer
buffer for 2 mins on shaker. (to remove excess salts that interfere with protein transfer).
o Mean while, nitrocellulose membrane (Protran®, Whatman®) and 3mm Chr paper
(Whatman®) were cut to gel size and equilibrated with blotting pads in 1x transfer buffer.
o Then the layers were assembled in blotting cassettes and placed in Mini Trans-Blot™
(BioRad®) blotting chamber filled with 1x transfer buffer and ice block.
o Blotting was done at 100 V for 60 mins with constant stirring.
o Membrane was stained with Ponceau S to check for success of protein transfer.
o The membrane was blocked in 5% NFDM solution (in 1x TBST) at RT for 1 hr on shaker.
o Membrane was incubated overnight with suitable primary MAb diluted (na21, 1:10) in 1x
TBST at 4ºC with slow shaking.
o Next morning 1º MAb solution was removed and washed thrice (5 min each) with 1x TBST.
o Membrane was incubated in secondary Ab (Goat anti-Mouse-HRP) diluted 1:7500 in 1x
TBST for 45 mins on shaker at RT.
o The 2º Ab was discarded and the membrane were washed thrice (10 min each) with 1xTBST.
o Just before the end of the last wash the detection reagent (ECL™ kit from Amersham GE®)
was prepared by mixing solution A and solution B in equal volumes (1ml each per blot).
o After washing, the membrane was incubated with detection reagent in dark for 1 min.
o Thereafter excess detection reagent was drained off and X-ray films placed on the membrane
for various exposure times in a dark room were developed to detect the antigen as bands.

48
Gruppe V Wholemounts von adulten Gehirnen, confocale Mikroskopie (Kirsa) (Anleitung wird nachgereicht)
Kurzinfo: Gal4-UAS-System
Mithilfe von P-Elementen können Fliegen transformiert werden, d.h. ein fremdes "Trans"-Gen in das genom von Keimbahnzellen inseriert werden. Bei dem Gal4-System ist das inserierte Gen der Transkriptionsaktivator Gal4 aus der Hefe, der unter der Konrrolle eines schwachen Promotors steht und deshalb nur dann exprimiert weren kann, wenn das Gen in der Nähe eines enhancers inseriert. Das exprimierte Gal4-Protein aktiviert die Expression nur solcher Gene, die Gal4-Bindungsstellen, genannt upstream activator sequence (UAS), tragen. Gal4-Gen und UAS-Zielgen werden auf zwei separate, transgene Fliegenlinien aufgeteilt. In der Gal4-Linie („driver line“) wird das Aktivatorprotein Gal4 zellspezifisch exprimiert (durch eine insertion in die Nähe eines zelltypspezifischen enhancers), hat aber kein Zielgen, das es aktivieren könnte. In der UAS-Linie ist das Zielgen, z.B. ein Reporter, zusammen mit der UAS-Sequenz im Genom inseriert, wird aber ohne Gal4-Aktivatorprotein nicht exprimiert. Werden beide Linien gekreuzt, wird in der Nachkommenschaft das Zielgen akiviert. Dadurch erhält man eine große Flexibiltät: Ein Reportergen kann in vielen UAS-Linien mit spezifischen Expressionsmustern exprimiert werden oder verschiedene Reporter können in spezifischen Zellen untersucht werden.

49
Abbildung 5, aus: Brand et al. (1995): Current Opinion in Neurobiology 5: 572-578. Durch zwei getrennte P-Element-Insertionen werden transgene Fliegen hergestellt, die entweder Gal4 zellspezifisch exprimieren, oder ein Gen von Interesse unter UAS-Kontrolle inseriert haben. Kreuzt man beide Linien, kommen beide Transgene in der nächsten Generation zusammen und das Gen von Interesse wird exprimiert.
Kurzinfo: Konfokale Laser-Scanning-Mikroskopie
Reportermoleküle, die eine Visualisierung von Expressionsmustern erlauben, können einerseits Präzipitationsfarbstoffe sein, deren Färbung von enzymatischen Reaktionen abhängt, wie bei der lacZ-Färbung (siehe oben!). Andererseits kann man auch fluoreszierende Moleküle als Reporter verwenden. Ein fluoreszierendes Protein, das natürlich auch genetisch DNA-codiert ist, ist das „green fluorescent protein“ (GFP) aus der Qualle Aequora victoria. Das exprimierte GFP kann mithilfe eines Fluoreszenzmikroskopes bei passender Anregungswellenlänge beobachtet werden. Gegenüber der einfachen konventionellen Fluoreszenzmikroskopie weist die sogenannte konfokale Laser-Scanning-Mikroskopie zwei Unterschiede auf:
1. Das emittierte Licht wird durch eine Blende, ein sogenanntes „pinhole“, geleitet. Dadurch wird hauptsächlich Licht aus einer definierten Ebene, der Fokusebene des Objektivs, detektiert, Licht aus anderen Ebenen wird ausgeblendet (siehe Abbildung 6).
2. Man benötigt zur Anregung des Fluorophors eine monochromatische Lichtquelle, die
relativ hohe Lichtintensität hat, da ja nur das Licht, das durch das pinhole trifft, „verwendet“ wird. Deswegen benutzt man nicht, wie bei der normalen Fluoreszenzmikroskopie, eine Quecksilberdampflampe mit passendem Filter, sondern einen Laser bestimmter Wellenlänge, der das Präparat „abtastet“ (scanning).
Dadurch kann man ein dickeres Präparat, wie z.B. ein Fliegengehirn, auf bestimmten Schnittebenen darstellen. Fluoreszenz, die aus Schnittebenen über oder unter der Fokusebene auftritt, wird ausgeblendet. Diese Schnittebenen können anschließend mithilfe geeigneter Software dreidimensional rekonstruiert werden.

50
Abbildung 6, von Leica Microsystems: Strahlengang eines konfokalen Laser-Scanning Mikroskopes. Hauptsächlich Licht einer definierten Schnittebene (focal plane) wird detektiert.






























