Embed Size (px)
Citation preview

Evidence of oxidative stress response in a mouse model of AA amyloidosis: Immunolocalization of specifie
markers
by
Golnar Katnalvand
Department of Microbiology and Immunology McGill University, Montreal
August 2003
A thesis suhmitted to the Faculty of Graduate Studies and Research in partial fulfillment of the requirements of the degree of Master' s
of Science
© Golnar Kamalvand, 2003

1+1 Library and Archives Canada
Bibliothèque et Archives Canada
Published Heritage Branch
Direction du Patrimoine de l'édition
395 Wellington Street Ottawa ON K1A ON4 Canada
395, rue Wellington Ottawa ON K1A ON4 Canada
NOTICE: The author has granted a nonexclusive license allowing Library and Archives Canada to reproduce, publish, archive, preserve, conserve, communicate to the public by telecommunication or on the Internet, loan, distribute and sell th es es worldwide, for commercial or noncommercial purposes, in microform, paper, electronic and/or any other formats.
The author retains copyright ownership and moral rights in this thesis. Neither the thesis nor substantial extracts from it may be printed or otherwise reproduced without the author's permission.
ln compliance with the Canadian Privacy Act some supporting forms may have been removed from this thesis.
While these forms may be included in the document page count, their removal does not represent any loss of content from the thesis.
• •• Canada
AVIS:
Your file Votre référence ISBN: 0-612-98666-7 Our file Notre référence ISBN: 0-612-98666-7
L'auteur a accordé une licence non exclusive permettant à la Bibliothèque et Archives Canada de reproduire, publier, archiver, sauvegarder, conserver, transmettre au public par télécommunication ou par l'Internet, prêter, distribuer et vendre des thèses partout dans le monde, à des fins commerciales ou autres, sur support microforme, papier, électronique et/ou autres formats.
L'auteur conserve la propriété du droit d'auteur et des droits moraux qui protège cette thèse. Ni la thèse ni des extraits substantiels de celle-ci ne doivent être imprimés ou autrement reproduits sans son autorisation.
Conformément à la loi canadienne sur la protection de la vie privée, quelques formulaires secondaires ont été enlevés de cette thèse.
Bien que ces formulaires aient inclus dans la pagination, il n'y aura aucun contenu manquant.

TABLE OF CONTENTS
ABSTRACT ....................................................................................... .Î
ABRÉGÉ .......................................................................................... iii
ACKNOWLEDGEMENTS ...................................................................... v
CONTRIBUTION OF AUTHORS ............................................................. vi
CHAPTER 1. Literature review, rationale and objectives of the thesis
1. AMYLOIOOSIS - A PROTEIN FOLDING OISOROER ................................ 1
2. CLINICAL ASPECTS OF AA AMYLOIDOSIS ......................................... .4
3. A PARASITE MODEL OF AA AMYLOIOOSIS .......................................... 7
4. PA THOGENESIS OF AA AMYLO IOOSIS ............ '" ........................... '" .. 11
5. PUTATIVE ROLE OF INFLAMMATION-INDUCED OXIDATIVE STRESS IN AMYLOID FORMATION ................................... 14
6. RATIONALE AND OBJECTIVES ......................................................... 17
PREFACE TO CHAPTER 2 ..................................................................... 18
CHAPTER 2. Heme-oxygenase-l response, a marker of oxidative stress, in a mouse model of AA amyloidosis .......................... . 20
PREF ACE TO CHAPTER 3 ..................................................................... 48
CHAPTER 3. Immunohistochemical detection of lipid peroxidation and advanced glycation end products in a mouse model of AA amyloidosis ............................................................... . 49
CHAPTER 4. Concluding remarks ................................................ ... 80
REFERENCES .................................................................................... 82
APPENDIX ........................................................................................ 95

ABSTRACT
M.Sc. Thesis Golnar Kamalvand Microbiology and Immunology
Amyloidosis describes a heterogenous collection of systemic diseases
characterized by the extracellular deposition of proteinaceous amyloid fibrils derived
from normally soluble proteins. With progressive tissue deposition of amyloid, death can
occur through failure of the affected organes). However, the mechanism of amyloid fibril
formation remains obscure. To understand this mechanism, a parasite (alvcolar hydatid
cyst, AHC)-mouse model of inflammation-associated AA amyloidosis, was used. AHC is
a potent inducer of chronic inflammation, serum amyloid A (SAA) synthesis and AA
amyloidosis. It has been proposed that reactive oxygen radicals (ROR) generated by
inflammatory macrophages (m<D) and reticuloendothelial (RE) cells, which are also
intimately involved in SAA clearance, could initiate intra-Iysosomal AA fibril formation.
ROS generated intracellularly could oxidize SAA fragments or other key cellular
proteins, alter them structurally and thus rcnder them resistant to enzymatic degradation
and prone to intralysosomal nascent AA fibril formation.
The objective of this research was to identify oxidative stress (OS) markers
(heme-oxygenase-l, HO-l; 4-hydroxy-2-nonenal, HNE; NE-(Carboxymethyl)lysine,
CML) in peritoneal m<D and splenic/hepatic RE cells obtained from AHC-infected mice
prior to and during AA amyloidosis. Histochemical and peroxidase-immunoperoxidase
methods were used to detect the OS markers. High levels of HO-l, an antioxidant
enzyme; HNE, a product of lipid peroxidation; and CML, an advanced glycation end
product, were found in peritoneal m<D and splenic/hepatic RE cells proximal to AA fibril
deposition. HNE and CML deposits were found in both the tissue interstitium and bound

to AA amyloid deposits, indicating their possible role in the oxidative alteration of
intracellular SAA. OS mediated changes in m<D/RE cells loaded with SAA may prove to
be a prelude to nascent intracellular AA fibril formation.
ii

ABRÉGÉ
Thèse M.Sc. Golnar Kamalvand Microbiologie et Immunologie
L'amylose consiste en la voie finale d'une collection hétérogène de pathologies
systémiques caractérisées par un dépôt amyloïde. Le dépôt protéique extracellulaire
fibrillaire anormal provient d'une source normalement soluble. En progressant. le dépôt
amyloïde entraîne le mauvais fonctionnement des organes affectés ce qui peut provoquer
la mort. Si ce phénomène est déjà amplement traité dans la littérature, le mécanisme
même de la formation de fibrilles amyloïdes demeure obscur. Afin de mieux saisir les
piliers de ce mécanisme, cette étude utilise un modèle de souris de l'amyloidose AA.
infecté de kystes hydatiques alvéolaires (KHA). Le KHA est un puissant catalyseur
d'inflammation chronique et de synthèse du sérum amyloïde A (SAA), lequel le est
précurseur sérique de l'amylose AA.
Les radicaux libres oxygénés (RLO), générés suite à l'activation inflammatoire
des macrophages (m<D) et des cellules réticulo-endothéliales (RE) jouent un rôle
d'importance dans la dégradation de SAA. Aussi cette recherche s'articule-t-elle autour
du prémisse que ces radicaux puissent initier la formation de fibrilles intralysosomale
AA. Les RLO, générés de façon intracellulaire pourraient oxyder les fragments de SAA.
de même que d'autres protéines cellulaires clés. Altérant la structure protéinique, la
présence de RLO se traduirait par une résistance des protéines à la dégradation
enzymatique, d'où la formation initiale de fibrilles intralysosomales AA.
L'objectif de la présente recherche fut d'identifier des marqueurs de stresse
oxydant (SO) (heme-oxygénase-l, HO-l; 4-hydroxy-2-nonénal, HNE; NE
(Carboximéthyl)lysine, CML) dans les m<D inflammatoires péritoneaux et les cellules RE
111

spléniques/hépatiques des souris infectées de KHA avant et pendant le dépôt des fibrilles
AA. Les méthodes histochimiques et la méthode en immunopéroxydase furent utilisées
afin que soient détectés les marqueurs de SO. Conformément à l'hypothèse initialement
formulée, de hauts niveaux d'enzyme anti-oxydant HO-l, du produit de péroxydation
lipidique HNE et d'un produit final de glycolysation avancée CML furent décelés dans
les m<D périt one aux et les cellules RE spléniques/hépatiques à proximité des fibrilles AA.
La protéolysation de dépôt HNE et de CML s'effectua tant dans les tissues
interstitiaux que dans ceux liés aux dépôts amyloïdes AA, d'où la proposition de
l'hypothèse d'un rôle actifs de ces marqueurs de SO dans l'altération oxydative du SAA
intracellulaire. Des changements liés aux SO dans les cellules m<D/RE chargées de SAA
pourraient s'avérer être la preuve du rôle clé du SO dans le prélude de la formation des
fibrilles intracellulaires AA.
IV

ACKNOWLEDGEMENTS
1 wish to thank my supervisor, Dr. Zafer Ali-Khan, for his incessant patience,
support and guidance throughout my master's degree. My work was largely supported by
a research grant (MOP-42526) from the Canadian Institutes of Health Research. 1 would
also like to thank members of my laboratory: Geneviève Pinard, Sheen Ho Hyuk, Hung
Dam and Hannah Phipps-Y onas.
v

CONTRIBUTION OF AUTHORS
This master's of science thesis contains two original papers, one that is published
and one that has recently been submitted, as Chapters 2 and 3 of this thesis. Each of these
chapters contains their own Abstract, Introduction, Materials and Methods, Results,
Discussion, Acknowledgements and References sections. Prefaces that serve as
connecting texts to bridge the published papers are found prior to Chapters 2 and 3. A
general Introduction (Chapter 1) and a section that contains Conclusions and Discussion
(Chapter 4) are also included. References in Chapters 1 and 4 are collated alphabetically
whereas in published papers the y are listed in the order they appear within the text of the
manuscripts. Page numbers of the thesis are found on the bottom center of each page and
should be distinguished from the page numbers of individual manuscripts.
The published papers that comprise the chapters describing the experimental data
of this thesis are listed below. 1 am the principal author in both papers. My research was
conducted under the supervision of Dr. Z. Ali-Khan, who helped in composing the text of
both papers. Sorne experiments in Chapter 2 were performed in collaboration with
Geneviève Pinard who works as a technician in our laboratory.
(1). Kamalvand, G.; Pinard, G.; Ali-Khan, A. Heme-oxygenase-1 response, a
marker of oxidative stress, in a mouse model of AA amyloidosis. Amyloid:
Journal of Protein Folding Disorders. 10: 151-159; 2003 (presented as Chapter 2).
(2). Kamalvand, G.; Ali-Khan, Z. Immunohistochemical detection of lipid
peroxidation end products in a mouse model of AA amyloidosis. Free Radical
Bi%gy and Medicine. (Submitted for publication; presented as Chapter 3).
VI

CHAPTER 1. LITERATURE REVIEW, RATIONALE AND OBJECTIVES OF THE THESIS
1. AMYLOIDOSIS - A PROTEIN FOLDING DISORDER
Amyloidosis is a disorder of protein metabolism. The term 'amyloid' is a generic
term used to describe extracellular, fibrillar prote in deposits associated with disease. The
fibrils are insoluble and generally resist proteolytic digestion. They aggregate to
indefinite lengths in organs and tissue, and through pressure atrophy, destroy the
surrounding normal tissues. Sorne of these disorders are systemic while in other forms the
amyloid deposition is restricted to particular organ systems (Husby, 1992). Clinical
manifestations of amyloidosis vary widely and depend on the organs involved (Stone).
Amyloid deposits exert their pathological effects largely by their physical presence,
distorting tissue architecture and disrupting organ function. Amyloidosis accompanies
and is associated with a number of medical disorders, including adult-onset diabetes,
rheumatoid arthritis (RA), chronic renai dialysis, familial amyloid neuropathy and most
notably Alzheimer's disease (AD), to name a few. Therapy is remarkably deficient and
can mean a fatal outcome, primarily due to organ failure (Tan; Kisilevsky, 1983).
A necessary condition for the formation and deposition of amyloid fibrils is the
presence of an autologous prote in precursor that is abnormal in structure and/or
concentration. The modem classification of amyloidosis is based on the nature of the
precursor plasma protein (Falk). More than 20 forms of amyloid are now recognized
(Westermark); the incidence of these diseases range from being rare to playing central
roles in the pathogenesis of diseases affecting millions of patients (i.e. AD, adult-onset
diabetes). The different amyloid types can be distinguished immunohistochemically with
1

specifie antibody against the precursor and/or amyloid prote in itself. Table 1 lists a few
of the precursors and the diseases the y cause (Bellotti); among the precursors are plasma
constituents (serum amyloid A, Ig light chain, fibrinogen, transthyretin, and
apolipoproteins) or precursors that form localized amyloids (~-protein precursor,
ca1citonin, cystatin C, and atrial natriuretic fàctor).
Although amyloid fibrils are derived from a variety of precursor proteins III
different forms of the disease, they share characteristic tinctorial properties. Amyloid
fibrils have specifie affinity for thioflavin and, when stained with Congo red, they display
an apple-green birefringence under polarized light. Amyloidogenic proteins for different
amyloids are distinct with respect to amino acid sequence, however, aU fibrils are
structurally similar. For example, electron microscopy and X-ray diffraction patterns
reveal that amyloid fibrils have a diameter from 5-13 mn and form rigid, unbranched,
cross ~-pleated sheets (Glenner). Interestingly, this ~-pleated sheet structure is not
normally found in this pure conformation in mammalian tissues, but is found in
invertebrates (Glenner). Their common structural properties imply that amyloid fibrils
have a common mechanism of fibrillization. Consequently, much effort has been directed
towards understanding the fibrillogenesis pathway, with the aim of developing inhibitors
to treat various amyloidoses.
2

Table 1. Nomenclature and classification of amyloidosis
~_!!l~!~l~J:~!"~tejn _____ f~o~einJ~E~~!I!"~_())" ..... AA serum amyloid A (SAA)
AL
AH
ATTR
AIAPP
AFib
AApoI
ACys
A~
AANF
ACal
Ig light chain
Ig heavy chain
transthyretin
islet amyloid polypeptide (amylin)
a-fibrinogen variants
apo A 1 variants
eystatin C variants
~2-mieroglobulin
~-preeursor prote in
atrial natriuretie factor
(pro )ealcitonin
3
- -----"""'-,~""""'--
.. J::~i~i~~L~y~~rome - Reactive (seeondary) amyloidosis
associated with recurrent inflammation (i.e. rheumatoid arthritis)
- Familial Mediterranean fever - Familial amyloid neuropathy
- Idiopathie primary amyloidosis
- Idiopathie primary amyloidosis
- Familial amyloid neuropathy - Familial amyloid eardiomyopathy - Senile systemic amyloidosis
- Type Il diabetes
- Hereditary non-neuropathie renal amyloidosis
- Atherosclerosis - Familial neuropathic and
non-neuropathie amyloidosis
- Hereditary cerebral hemorrhage
- Assoeiated with chronic renal dialysis
- Alzheimer's disease - Down' s syndrome - Hereditary cerebral hemorrhage
- Isolated atrial amyloid
- Medullary eareinomas of the thryroid

2. CLINICAL ASPECTS OF AA AMYLOIDOSIS
Reactive or AA amyloidosis is brought upon by chronic inflammatory disorders
that provoke a sustained acute phase response. The tirst 24-48 ho urs of inflammation are
known as the acute-phase response, characterized by a dramatic increase in hepatic
synthesis of acute phase proteins (Gabay). During chronic inflammation, acute phase
prote in levels, after an initial increase, decrease to steady-state concentrations that are
signiticantly higher than the original baseline levels. Thus, chronic inflammation can
result in continuing tissue damage and, occasionally, complications such as secondary
AA amyloidosis. This form of amyloidosis is characterized by the tissue deposition of
AA tibrils derived from serum amyloid A (SAA) (Glenner).
Many chronic inflammatory disorders of known and unknown etiologies are
known to predispose to AA amyloidosis. Chronic inflammatory conditions may arise
from prolonged microbial or parasitic infections (i.e. leprosy, tuberculosis, leishmaniasis,
malaria, and alveolar hydatid disease), malignant neoplasm (i.e. Hodgkin's disease and
renal carcinoma), and chronic inflammatory disorders including RA, juvenile chronic
arthritis, psoriasis and Crohn's disease (Benson, 1995; Glenner). In developed countries,
where chronic infections such as tuberculosis and leprosy are for the most part contained,
RA is the most frequent predisposing disease, with an estimated 1-10% of incidence of
amyloidosis in RA patients (Glenner, Tan).
While the presence of SAA at high levels is known to be a major predisposing
factor to AA amyloidosis, it is not sufticient on its own to cause amyloid deposits. Only a
limited number of RA patients ranging from 1 to 10% develop AA amyloidoses. Etiology
and pathogenesis of AA amyloidosis, and other forms of amyloidosis, are multifactorial.
4

Genetic susceptibility could be a significant factor; while the frequency of amyloidosis in
RA patients in Caucasians is 1-2%, approximately 10-15% of Japanese RA patients
develop the disease. Yamada et al. have recently shown that certain genetic backgrounds,
particularly SAA polymorphisms, are associated with development of AA amyloidosis
(Yamada, 2003). Their data suggest that a single nucleotide polymorphism within the
SAA 5' flanking region of the gene is related to amyloid development; their data
correlated with differences se en in the frequency of AA amyloidosis in Japanese and
Caucasian ethnic groups (Yamada, 2003). The means by which this allele may regulate
susceptibility has yet to be determined.
Furthermore, there is considerable variability between individual cases with
respect to distribution of amyloid deposits and clinical symptoms. Although the liver and
spleen are the first sites of AA fibril deposition, the kidney is the most common site
leading to clinical signs of disease (Stone). Patients with splenic/hepatic amyloid deposits
can remain asymptomatic. Amyloid deposition in the kidney usually begins in the
glomerular mesangium and around capillary basement membranes, leading to progessive
obliteration of capillary lumina, destruction of glomerular ceUs and, eventually, complete
replacement of glomerulus by a confluent mass of amyloid. Amyloid deposition in the
kidney is characterized by proteinuria that can result in chronic renal failure. The
gastrointestinal tract can also be involved and life-threatening gastrointestinal bleeding
can occur. Amyloid infiltration in blood vessel walls can cause increased risk of
hemorrhage and involvement of other organs, leading to death (Benson, 1995).
Unfortunately, the diagnosis of amyloidosis is usually made when patients already
have advanced disease with resultant compromise in organ function. Hemodialyis or
5

peritoneal dialysis may prolong life. Suppression of the underlying disease proccss by
alkylating agents in rheumatoid arthritis and juvenile chronic arthritis has been shown to
preserve renal function and improve survival in patients. Chronic colchicine
administration has been shown to prevent amyloidosis in patients with familial
Mediterranean fever (Tan). In microbial or parasitic infections, treating the pnmary
inflammatory disease with antibiotics is the key. The length of survival with AA
amyloidosis depends on how early in the course of disease the diagnosis is made.
6

3. A PARASITE MODEL OF AA AMYLOIDOSIS
The discovery that alveolar hydatid cyst (AHC)-infected mlce develop AA
amyloidosis was made in the early 1980s by Ali-Khan et al. (Ali-Khan, 1982). Upon
microscopic observation, susceptible mice infected intraperitoneally with 50-250 ABC,
the larval stage of Echinococcus multilocularis, between 1 to 12 weeks post-infection
(p.i.) depending on the infective inoculum, demonstrated hyaline eosinophilic areas in the
spleen identified as AA amyloid deposits (Alkarmi, 1986). Despite a prompt intlux of
intlammatory cells at the inoculation site and heavy leukocyte infiltration into the tissue
matrix, AHC grows like a tumor in various soft organs and potentially metastasizes
(Treves). The infected mice do not appear to contain the infection (Ali-Khan, 1996). The
proliferating AHC acts as a potent amyloidogen, with the sustained overproduction of
SAA resulting in AA fibril formation.
Echinococcus is a small endoparasitic tlatworm belonging to Class Cestoda. It
exhibits an indirect life cycle in which the adult is hermaphroditic and the larva
proliferates asexually, requiring two mammalian hosts for completion of its life cycle: a
definitive host in which the adult worm develops in the small intestine, and an
intermediate host in which the cysts deve10p in the viscera. The parasite is of pathogenic
and economic significance in intermediate hosts, where the larval parasite develops into a
hydatid cyst. Infection with Echinococcus may be naturally transmitted from the
definitive host to man; ingestion of eggs in contaminated food or drink is the precursor of
alveolar hydatid disease (Rausch).
Echinococcus multilocularis exhibits a mainly holarctic distribution with foxes
primarily as the definitive host, and rodents and man as intermediate hosts; central North
7

America has however become a niche for E. multilacularis since the 1960s. The unique
feature of this species is the multivesicular (alveolar) nature of its metacestode; it is an
infiltrating structure, with no limiting host-tissue barrier, consisting of numerous small
vesicles embedded in a dense stroma of connective tissue, localized initially in the liver
(Rausch). Furthermore, the detachment of germinal ceUs and their subsequent distribution
via the lymph and blood can give rise to the distant metastatic foci characteristic of E.
multi/acularis. Clinical and pathological changes in experimentally infected rodents
include hepato/splenomegaly, increase in body weight due to the proliferating
metacestode, ascites and finally death within five months of infection (Torgerson). In
essential features, the course of AHC-infection in rodents appears to be a wasting disease.
Upon post-mortem examination, infiltration of the liver, peritoneal cavity, other
abdominal organs and the lungs may be evident (Fig. 1).
FIGURE 1. Abdominal view of a post-mortem alveolar hydatid cyst (AHC)-infected mouse
hepatomegaly
AHC mass in the peritoneal cavity
splenomegaly
8

The reactivity of the spleen in response to the AHC-infection appears to play an
important role in the clinical features and the course of the disease. Briet1y, the spleen is a
bi-functional, complex organ that acts as a hematopoietic vascular filter and functions in
the reticular defense system. The splenic parenchyma consists of three parts: the white
pulp, is rich in lymphocytes and the site of germinal centers; the red pulp, typically the
largest part of the spleen, which stores and destroys erythrocytes, is site of a reticular
meshwork; and the perifollicular zone, lying between the white pulp and red pulp, which
contains large venous sinuses, is the first site for antigen and lymphOCyte deposition
(Weiss, 1978a). The reticuloendothelial system is an extensive system of protection and
regulation whose prominent feature is phagocytosis. Macrophages (m<1» are large
capacity phagocytes, as weIl as secretory cells. M<1> synthesize and secrete collagenase,
components of complement, and int1ammatory cytokines. Lysosomes (L Y), upon
secretion, provide a bath of acid hydrolytic enzymes within m<1> that digest phagocytosed
substances. This is notable in the red pulp (Weiss, 1978b).
Elevated SAA level, expansion of the reticuloendothelial cells (monocytoid cells
in the splenic perifollicular zones and Kupffer cells in the liver) and localization of SAA
in the endosomes-Iysosomes of these cells are seen in the pre-amyloidotic phase
(Chronopoulos). The primary targets of amyloid deposition are thus the perifollicular
zone sinuses in the spleen, and portal and central veins and sinus walls in the liver
(Alkarmi, 1984). Progressive amyloid deposition between 1 week and 12 weeks p.i.
significantly distorts the splenic architecture as weil as causes atrophie changes in the
liver, kidneys and GI tract (Ali-Khan, 1983 and 1996).
9

The AHC-infected mouse is a useful in vivo model to investigate the pathogenesis
of AA amyloidosis. The azocasein mouse-model, necessitating daily subcutaneous
injections of the chemical amyloidogen azocasein and producing amyloidosis in
susceptible mice from 9 days to 3 weeks, is the most commonly used model to study the
pathogenesis of AA amyloidosis (Janigan; Sipe, 1993). AHC, however, is a more pote nt
amyloidogen; with a single infective inoculum of 250 AHC, the amyloid induction period
is relatively short - about 1 week p.i. Both the preamyloidotic and the amyloidotic phases
can be studied within a reasonably short time.
In addition, the availability of mouse strains that differ in susceptibility to AA
amyloidosis provides a valuable tool to study factors that are potentially important in AA
amyloidogenesis (Ali-Khan, 1988; Alkarmi, 1984). In susceptible mice, two acute-phase
SAA proteins, SAA 1.1 (previously SAA2) and SAA2.1 (previously SAA 1) are
synthesized; only the SAA1.1 is amyloidogenic (Sipe, 1999). The CE/J mouse strain is
absolutely resistant to AA amyloidosis (Sipe, 1993). The CE/J mouse contains a single
SAA gene, coding for SAA2.2 (previously SAAcEIJ), which seems to be a composite of
SAA1.1 and SAA2.1 genes (Yu). While SAA2.2 differs by only 6 amino acids from
amyloidogenic SAA 1.1, the differences lie in the amino terminal region of SAA 1.1
responsible for amyloidogenicity, thus rende ring the CEl] mouse amyloid resistant (de
Beer). Thus, the CE/J AHC-infected mouse can serve as a valuable tool in studying the
consequences of SAA deposition independent of AA fibril formation.
10

4. P ATHOGENESIS OF AA AMYLOIDOSIS
In AA amyloidosis, high expression and aberrant catabolism of SAA are the main
disposing factors to AA pathogenesis. In response to tissue trauma or an inflammatory
stimulus, activated monocytoid cell-derived cytokines, primarily interleukin (lL)-1, IL-6
and tumor necrosis factor-a (TNF-a), induce the synthesis of acute phase proteins by
liver hepatocytes at the transcriptional level (Benson, 1980; Zahedi). SAA may reach
plasma levels up to 1000-fold greater (l mg/ml) than that found in the non-inflammatory
state. Serum SAA is mainly complexed to high-density lipoproteins (HDL) by displacing
apolipoproteins (apo), apo A-I and A-II, and circulates as acute phase HDL-SAA
complex (Husby, 1994).
SAA, in fact, represents several isoforms encoded by a family of highly conserved
genes found across several mammalian species. While six murine SAA isoforms have
been described, SAA1.1 and SAA2.1 are the prominent acute phase isoforms, produced
in equimolar concentrations during the early phases of inflammation (other SAA
isoforms are constitutive and only ever expressed in minor amounts). While these two
isoforms share 95% structural identity, differing by only six amino acids, SAA 1.1 is more
rapidly cleared from the serum and much more prone to amyloidogenesis (Kluve
Beckerman, 1997; Bell, 1996). Biochemical analysis of purified SAA and AA proteins
has confirmed that the N-terminal two-thirds of SAA1.1 form AA amyloid (Husby,
1994). The pathophysiology of this process, however, is not understood.
Aberrant proteolytic processing of amyloid precursor proteins by activated m<D,
RE cells or microglia has been implicated as a causative factor in many forms of
amyloidosis, with most amyloid fibrils being truncated forms of larger precursors
Il

(Kisilevsky, 1994). Whereas normal unstimulated monocytoid cells successfully degrade
SAA within 4 hours, chronically activated monocytoid cells (i.e. AHC-derived) degrade
SAA differentially, generating N-temlinally intact AA-sized SAA derivatives (Gollaher;
Bell, 1999). Cathepsin B, a major lysosomal cysteine proteinase, has been linked to the
partial degradation of recombinant SAA and to the generation of the most corn mon form
of N-terminally intact AA-sized peptide of ~8 kDa (Yamada, 1995). The plausible link
between these intracellular SAA fragments and extracellular AA fibrils was recently
established by Kluver-Beckerman et al. (Kluve-Beckerman, 1999). They demonstrated
that both native and recombinant murine SAA, co-cultured with m<D, were concentrated
in L Y, generated intracellular N-terminal SAA fragments, and formed both intra-, and
extracellular AA fibrils (Kluve-Beckerman, 1999). In sum, SAA, endocytosed by a yet
unidentified receptor on the surface of m<D/RE cells, is sequestered into the L Y in these
ceIls, where its partial degradation yields amyloidogenic AA-sized fragments.
In AA amyloidosis, the splenic PFZ, populated by a diverse monocytoid cell
population, is invariably the first site of AA deposition, followed shortly after by the
liver, then the kidneys (Du). Fundamental pathogenetic aspects of amyloidosis appear to
rely on local tissue conditions, components or events interacting to create the necessary
conditions for amyloid formation. The extracellular expansion of amyloid fibrils is
generally thought to occur via a nucleation-dependent oligomerization, accelerated by a
'seeding' mechanism (Rochet; Ali-Khan, 2002). In vitro studies show that the addition of
a preformed nucleus or 'seed' to a supersaturated prote in solution accelerates
fibrillization compared with spontaneous self-assembly of fibrils. This phenomenon was
confirmed in vivo in mice undergoing AA amyloidosis; AA fibril deposition was
12

markedly accelerated when the animais were glVen, III addition to an inflammatory
stimulus, an intravenous injection of amyloid enhancing factor (AEF). a concoction of
proteins extracted from AA fibril-laden tissue (Abankwa, Axelrad). Recently, the active
principle of AEF was shown to be the amyloid fibril itself, effective at minute doses of
<1 ng, showing transmittability akin to prion-associated disorders (Lundmark). Further
experiments on murine AA amyloidosis demonstrated cross-nucleation. where fibrils
derived from one form of amyloid was able to serve as a nucleus tor another (Ganowiak,
Johan).
Recently, in an effort to elucidate functions of SAA, a compelling tinding relating
SAA to extracellular AA fibrils was made. Aggregates of SAA, assuming a ~-sheet
conformation, were found to form channels in planar bilayer membranes. and thus could
potentially form such channels in the L Y and plasma membranes (Hirakura). As the se
channels are relatively non-selective, remain open for long periods and are quite large.
they may represent the outlet for nascent AA tibrils into the interstitium, where
nucleation-dependent aggregation can ensue. The precise molecular determinants
necessary for the initial fibrillization remain elusive (Lansbury Jr.). Environmental
factors are thought to be key, including ionic strength, pH and oxidation. in influencing
prote in folding and nucleation (Rochet). Clearly, the elucidation of the fibrillization
pathway is essential to guide ongoing research in amyloidosis research.
13

5. PUTATIVE ROLE OF INFLAMMATION-INDUCED OXIDATIVE STRESS IN AMYLOID FORMATION
The production of intracellular reactive oxygen radicals (ROR) occurs as a
ubiquitous byproduct of both oxidative phosphorylation and the myriad of oxidases
necessary to support aerobic metabolism. The intracellular level of oxidized protein
reflects the balance between the rate of protein oxidation and the rate of oxidized protein
degradation. This balance is a complex function of numerous factors that lead to the
generation of ROR, on the one hand, and of multiple factors that determine the activities
of the prote as es that degrade oxidatively modified proteins (Berlett).
In chronic inflammation, the increased production of cytokines (i.e., IL-l, IL-6,
IL-18, M-CSF, and TNF-a) recruits and activates monocytes that in tum produce high
concentrations of ROR (Gabay, Maury, Migita). In AA amyloidosis, the situation seems
to be exaggerated by SAA and AA fibrils. Recently, Yan et al. showed that SAA1.1 and
AA fibrils bound to murine BV -2 cells through the receptor for advanced glycation end
products (RAGE) and induced expression of several pro-inflammatory cytokines (Yan).
Furthermore, acute phase SAAl.l was shown to enhance the biosynthesis of
cyclooxygenase metabolites, potent mediators of inflammation, in activated monocytes
up to 3-fold (Malle). Thus, inflammation and oxidative stress (OS) act in concert and
perpetuate one another.
The principal damaging intermediates of OS response are the hydroxyl radical,
the superoxide anion and hydrogen peroxide, highly reactive species that alter
macromolecules present at their site of generation (Markesbery). ROR have been
implicated in the modification of lipids, proteins and DNA, lysosome membrane damage
14

and fragmentation/unfolding of cellular proteins. Protein modification, a recognized
manifestation of OS, has been extensively studied in the etiology of AD.
Evidence that ROR production is involved in the pathogenesis of AD IS ever
increasing (Markesbery). Oxidative modification of amyloid P protein (AP), which is a
39- to 42-amino acid peptide cleaved from a longer amyloid precursor prote in (APP),
may be an early event in Ap pathogenesis and may be important in amyloid plaque
formation. As described for AA amyloidosis, microgial cells (monocytes of the brain)
intemalize Ap within lysosomal compartments in their effort to de grade the molecule
(Wegiel). Consequently, while sequestered intracellularly, Ap may be exposed to the
activity of enzymes that together with ROR oxidatively modify Ap. To this end, Head et
al. performed immunohistochemical studies using antibody against oxidized Ap 1-40 in
order to characterize the distribution of oxidized Ap in AD brains. Their results implied a
role for microglial cells in forming oxidatively eross-linked AP, whieh is highly
aggregated and less amenable to de gradation and clearance (Grune). They hypothesized
that oxidatively modified Ap may serve as a seed for the further deposition of
unmodified, soluble Ap into plaques (Head).
The faet that oxidative stress plays an important role in AD pathogenesis seems
cl car, given all the evidence that research has recently provided. The eytopathologie
significance of oxidative damage is seen by the upregulation of antioxidant enzymes.
Heme-oxygenase-l (HO-l) is among the most sensitive indicator of cellular oxidative
stress response and in AD, HO-l response co-localized with amyloid deposits in the brain
(Pappolla; Takeda, 2000a). The net effect of ROR is damaging; upregulations of OS
markers found in AD brains include advanced glycation end-produets (AGE) (Sasaki),
15

nitration (Smith, 1994a), lipid peroxidation adducts (Sayre, 1997a), as weIl as free
carbonyls (Smith, 1996). As in AA amyloidosis, A~ exaggerates the OS response; it has
been shown that neuronal and microgial OS can be induced by A~ by interaction with the
receptor for advanced glycation end products (RAGE) (Sayre, 1997b). It has not been
firmly established whether OS is directly involved in amyloid formation in vivo, be it AA
or A~ amyloid, or if the amyloid fibrils, once formed, trigger an OS reaction. By contrast,
in vitro studies have generated both neurofibrillary tangles (NFT), a hallmark of AD, and
A~ from the precursor exposed to oxidation (Dyrks, Schweers). The protein modulating
properties of ROR, present in amyloid deposits, increase the likelihood that they are
involved in fibril formation.
16

6. RATIONALE AND OBJECTIVES
Activated m<D/RE cell-mediated degradation of SAA is the primary clearance
mechanism of exogenous SAA in vivo. An incessant flow of SAA into these cells,
however, cannot be sustained without causing disturbances in the L Y -mediatcd clearance
of SAA. The resulting retention of parti aIl y degraded, N-terminally intact, SAA
fragments in the L Y can lead to the modification of such fragments. Acid conditions, in
synergy with ROR generated in activated m<D/RE cells, could precipitate intra-LY
nascent AA fibril formation (Ali-Khan, 2002). Such fibrils, after their release, either by
exocytosis or toxic cell death, could act as a nidus in the nucleation-dependent
aggregation of extracellular AA fibril deposits. OS is an established underlying process
during prolonged inflammation. The potential role of ROR in AA fibril formation
remains obscure; oxidative changes in m<D-derived SAA and in amyloidotic tissues need
to be determined.
Using the AHC-infected mouse, a fully characterized model of inflammation
associated AA amyloidosis (Ali-Khan,1996), we show for the first time OS response
prior to and during AA amyloidosis. M<D and splenic/hepatic tissues from pre
amyloidotic and amyloidotic phases were used to immunohistochemically localize
markers of oxidative stress. The expressionlgeneration of three such markers were
monitored in AHC-derived tissues prior to and during AA fibril deposition: heme
oxygenase-l (HO-l), an antioxidant enzyme; 4-hydroxy-2-nonenal (l-INE), a product of
lipid peroxidation; and N€-(carboxymethyl)lysine (CML), an advanced glycation end
product. The following two chapters present our encouraging results and validate our
working hypothesis.
17

PREFACE TO CHAPTER 2
VirtuaIly aIl organisms respond to environmental stress by redirecting their
protein synthetic machinery to pro duce a small set of proteins termed heat shock proteins
(HSP). HO-l, also known as HSP32, is induced in a variety of stressed states including
exposure to heavy metals, UV light, hyperthermia, hypoxia, inflammation and OS
(Maines). HO-l catalyzes the first and rate-limiting step in the degradation of heme, a
ubiquitous iron-containing compound essential for the activity of aIl aerobic ceIls
(Schwartsburd). While HO-l serves as a potent cytoprotective enzyme against OS, a
specific role for HO-l in this capacity has not yet been established. Experimental
evidence stems from observations that cellular resistance to OS correlates positively with
levels of HO-l expression, and cells derived from HO-l-deficient mice are highly
susceptible to the accumulation of ROR and oxidative injury (Poss). The cytoprotective
feature of HO-l may be conferred by bulirubin, the end-pro du ct of heme degradation
known to be an important cellular antioxidant.
lmmnuhistochemical studies of AD brains have shown that intracellular levels of
antioxidant enzymes, namely HO-l, increased several fold in microglial cells adjacent to
NFT and senile plaques (Pappolla; Smith, 1994b). Furthermore, the spatial distribution of
HO-l expression in AD brains was found to be essentially identical to that of the
pathogenic conformational changes of tau protein, the major component of NFT
(Takeda). Similarly, experiments in mice undergoing AA amyloidosis, after injection
with AEF/silver nitrate, demonstrated ~3-fold upregulation ofHO-l expression in splenic
monocytoid cells, resulting from the activation of the NF -KB transcription factor through
engagement of the RAGE receptor (Yan). In this model of AA amyloidosis, AEF/silver
18

nitrate induces amyloid deposition within days of injection and mice are killed after 5
days of treatment (Yan). On the other hand, the AHC-mouse mode!, with mice sacrificed
at different time periods p.i. for up to 12 weeks, allows the immunohistochemical study
of the spatio-temporal distribution of HO-l expression with respect to both SAA and AA
fibril depositions. The results of this study, along with in vitro experiments to ascertain
the possible trigger for HO-l expression in monocytoid ceUs, are presented in the
following chapter.
19

CHAPTER2.
HEME-OXYGENASE-l RESPONSE, A MARKER OF
OXIDATIVE STRESS, IN A MOUSE MODEL OF AA
AMYLOIDOSIS
Golnar Kamalvand, Geneviève Pinard and Zafer Ali-Khan
Department of Microbiology and Immunology, McGill University,
Montreal, QC, Canada
Key words: alveolar hydatid cyst, heme-oxygenase, oxidative stress, AA amyloid, serum
amyloid A, image analysis, western blotting, immunohistochemistry.
Abbreviations: HO-l = heme-oxygenase-l; SAA = serum amyloid A; R-AA = rabbit an ti
mouse AA amyloid IgG; R-HO-l = rabbit anti-mouse HO-l IgG; SF = !!Jplenic follicle;
PFZ = perifollicular zone; RP = red pulp; AHC = alveolar hydatid cyS!: RE ceUs =
reticuloendothelial ceUs; m(j} = macrophages: ROS = reactive oxygen species; ()x-St =
oxidative stress.
CORRESPONDENCE
Dr. Z. Ali-Khan
Department of Microbiology and Immunology, McGill University
Lyman DuffBuilding, 3775 University Street
Montreal, Quebec, H3A 2B4
Tel: (514) 398-3930
Fax: (514) 398-7052
20

ABSTRACT
Expression of heme-oxygenase-l (HO-]), an important marker of oxidative stress,
has been studied extensively in the context of Alzheimer's disease. Evidence of HO-l
expression during AA amyloidosis is, at best, sketchy. Here we present comparative data
on HO-] response in alveolar hydatid cyst (AHC) infected amyloid sensitive (C57BL/6)
and amyloid resistant (CE/J) mou se strains. Histochemical and peroxidase
immunoperoxidase methods were used to monitor serum amyloid A (SAA) and AA fibril
deposition and HO-l expression in hepato-splenic reticuloendothelial (RE) cells of the
AHC-infected mice prior and during AA fibril deposition. Based on the cumulative data,
we conclude that HO-l expression corresponded closely with tissue deposition of SAA,
but was unrelated to AA fibril deposition. To ascertain whether SAA deposition might act
as the trigger for HO-l expression in the RE celIs, macrophages were incubated for up to
72 hr with SAA-containing mouse serum. The SAA-treated macrophages, although
negative for HO-l prote in, demonstrated SAA In the cell extracts and
immunocytochemically in the vacuolar compartments, indicating macrophage-mediated
endocytosis and trafficking of SAA. In sum, these results exclude SAA and AA fibrils as
the primary triggers in the induction of HO-l expression in RE ceIls: the roles of
inflammatory cytokines in this process need to be investigated further.
21

INTRODUCTION
Amyloid-related diseases are characterized by extracellular deposition of non
branching insoluble protein fibrils, called amyloid, in various soft organs u . The in vivo
mechanism of amyloid fibril formation, however, remains obscure. To understand this
mechanism, we are using a parasite (alveolar hydatid cyst, AHC)-mouse model ofreactive
AA amyloidosis 3. AHC grows like a solid tumor in mice, induces chronic inflammation. a
significant increase in serum amyloid A (SAA), the precursor of amyloid A (AA), and
multi-organ AA fibril deposition starting at about 1 week post-infection (p.i.)4.5. Thus,
AHC is a potent inducer of chronic inflammation, SAA synthesis and AA amyloidosis4-6.
Given that chronic inflammation and overproduction of acute-phase SAA are
central to the pathogenesis of AA amyloidosis, we and others have proposed that reactive
oxygen species (ROS) generated by activated macrophages (m<1» and reticuloendothelial
(RE) cells, which are also intimately involved in SAA clearance, could initiate intra
lysosomal nascent AA fibril formation6-9
. Interestingly, oxidative stress (OX-St) and/or
low pH conditions have been ascribed to the generation of amyloid-like fibrils from both
amyloidogenic and non-amyloidogenic proteins IO-13
• More specifically, the role of OX-St
response has been studied extensively in the pathogenesis of Alzheimer's disease (AD)14-
16. As such, heme-oxygenase (HO)-l and several other OX-St markers were localized to
neurofibrillary tangles, around senile plaques, and in neuronal cells l7.18. Takahashi et al.
recently showed intracellular protein-protein interaction in the endoplasmic reticulum
between Alzheimer amyloid precursor protein and HO-2 19• Such an interaction was
22

considered to modulate the protective function of HO, cause exacerbation of oxidative
stress and augmentation of neuronal ceIl death.
HO is a stress prote in and is expressed by different ceIl types including
monocytoid cells20. It functions as the initial and rate-limiting step in toxic heme
de gradation into biliverdin21• Of the three HO isoforms, HO-2 and HO-3 expressions are
constitutive but that of HO-I (aka HSP-32) is inducible at the transcriptionallevel and is
tissue-dependent22. It is found at relatively higher levels in splenic tissues where pro
oxidant heme is degraded21• HO-l expression is also up-regulated in response to heat
shock and inflammatory cytokines and during OX-St by NFKB and AP-I transcription
factors21-23
.
OX-St response is a corollary of chronic inflammation that is considered to be a
key factor in the pathogenesis of AA amyloidosis. Whether an OX-St-related factor plays
any significant role in the disease process, is yet to emerge24-26
. Here we present the
profiles of HO-I expression, a marker of OX-St response, in both splenic and hepatic RE
cells in AA amyloid susceptible (C57BL/6) and AA amyloid resistant (CE/J) mouse
strains and compare the levels of HO-l expression with tissue deposition of SAA and AA
fibrils. Our results indicate that tissue deposition of SAA corresponds with elevated HO-l
expression in the RE cells prior to AA deposition in the AHC-infected C57BL/6 micc;
data from AA amyloid resistant CE/J mice support this conclusion. Furthermore, our
results from in vitro experiments suggest that SAA al one does not induce HO-l response
in m<I> suggesting that other factors including inflammatory cytokines may have a primary
role in oxidative stress response.
23

MATERIALS AND METHODS
Infection
Six-week-old male C57BL/6 and CE/J mice (Jackson Laboratories, Bar Harbor,
Maine) were inoculated intraperitoneally with 250 AHCs, the larval stage of
Echinococcus multilocularis. The methods of inoculum preparation and infection and the
maintenance of AHC in our laboratory have been published elsewhere27. Three to six
mice were sacrificed at various time periods between 3 days and 12 wk p.i. Peritoneal
AHC masses and spleens were harvested from each mouse and weighed. Portions of
spleens and li vers were sectioned (6-8 Ilm thick) using a cryostat and stored at -20°C
until used. Control samples were obtained from non-infected mice of matching strain, sex
and age.
Immuno- and histochemistry
The tissue sections were stained with Congo red28. Skip or adjacent spleen/liver
sections were stained using biotin-strepavidin-peroxidase method29. Rabbit anti mouse
AA amyloid IgO, (R-AA 1.24 mg/mL; working dilution 1 :200 or 1 :400), rabbit anti-HO-
1 IgO (R-HO-l SPA-896; StressOen Biotechnologies Co., Victoria, Canada; working
dilution 1: 1 000) and goat anti-rabbit IgO conjugated to horseradish peroxidase (1:2
dilution, DAKO EnVision+), were used to localize AA fibrils/SAA and HO-l. Following
immunostaining, the sections were stained lightly (10 secs) with Hanis hematoxylin
(Sigma Diagnostics, St. Louis, MO), counterstained with thioflavin S (1 % in distilled
water, 5 min), dehydrated and mounted with permount. SAA-treated m<I> (see below),
24

permeabilized with 2% Triton X-IOO, were immuno-stained for SAA and HO-l, as
described 6.
To determine specificity of immunostaining, control sections were treated either
with the buffer alone or absorbed R-AA/R-HO-l. Purified mouse AA amyloid and HO-I
(SPT-896, Stressgen Biotechnologies Co.) were incubated with the primary antibodies in
a ratio of 20: 1 (w/w) for 30 min at 37°C and then ovemight at 4°C. The treated antibodies
were centrifuged at 4°C for 30 min and the supematants used for immunostaining.
Image analysis
Image analysis was used to quantify concentration of R-HO-I RE cells in the
spleen and liver sections. The sections were digitized using a microscope (Leitz Dialux
20) and a camera (Panasonic WVl550 TV camera), which was connected to a computer
through a video cardo This technique provided grey-scale digitized images where an
image is a two-dimensional array of pixels. Each pixel had a value representing its grey
scale intensity. To tabulate the amount of immunostaining within a given section, a two
level thresholding technique was used on the digital images. This technique yielded a
black and white image, separating stained from non-stained pixels. The user defined
upper and lower threshold values. Pixels with intensity between the upper and lower
thresholds were set to zero (black); these were considered the stained pixels. The rest of
the pixels were set to 1 (white). To normalize the computed numbers, the user had to find
the best upper and lower thresholds with trial and error and by comparing the results with
the original image. Once the image was thresholded, the number of stained pixels was
counted and divided by the total number of image pixels. This provided the user with the
25

percentage of stained pixels. The commands for the computation of the percentage area
of a tissue section occupied by specific staining were written in Matlab®.
Statistical evaluation for the data was performed by the unpaired t-test. A P-value
<0.05 was taken to be statistically significant.
Determination 0/ serum SAA concentration
Retired CDl mice (Charles River, St-Constant, Canada) were injected
subcutaneously with 0.5 ml of 2% AgN03 Serum was collected after 18 hr, pooled, and
its SAA concentration was determined using ELISA, as described 0. Briefly, purified
murine AA amyloid, starting at a concentration of 50ng/50Il1, was used as the standard to
measure SAA concentration. The pooled serum contained 1.06 mg/ml of SAA.
SAA uptake by peritoneal mf/J and RA W264. 7 cells
Peritoneal cells were collected from 6 wk old CD1 mice (Charles River, St
Constant, Canada) and incubated with DMEM for 2 hr to purify peritoneal m<l> by
adherence. Peritoneal m<l> and RA W264.7 cells (kindly provided by Dr A. Descôteaux,
INRS-Institut Armand Frappier, Université du Québec, Laval, Quebec) were cultured at
37°C with 5% CO2 in W-DMEM (DMEM high glucose containing 10% fetal bovine
serum, L-glutamine, and penicillin-streptomycin, Gibco, Burlington, Canada). Three
million cells were plated in a 25 cm2 culture flask and treated with 5 ml W-DMEM
containing 10% SAA serum. At 12, 24 or 72 hr, the cells were trypsinized to remove
SAA bound to the plasma membrane and harvested using a ImM EDTA solution.
Aliquots of m<l>, 500,000 cells, were cytocentrifugated onto glass si ides and
immunostained for the localization of SAA. The remaining ceUs were mixed with ice
cold 2x sample buffer containing 6M urea (50 III of 2x sample buffer / 1 x 106 m<l»,
26

boiled (5 min), centrifuged (10,000 RPM, 30 min), and the prote in extract equivalent to
500,000 cells/lane was used for fractionation and Western blotting.
Western Blotting
Selected C57BL/6, CE/J serum samples and SAA-treated m<D lysates were used
to detect SAA by Western blotting, as described30J1. Briefly, the samples were separated
on 12% SDS-PAGE gels containing 6M ure a, transferred onto a nitrocellulose membrane
and the membrane treated with R-AA (1.24 mg/ml; diluted 1 :3000); enhanced
chemiluminescence kit (Amersham, Baie d'Urfé, Canada) was used for the detection of
SAA band.
27

RESULTS
C57BL/6 mice: Cyst and spleen weights post-infection
As described previously5, splenomegaly correlated with increasing cyst weights
during the course of the infection (Table 1). Between 1 and 12 wk p.i., there was a ~5-
fold increase in the mean spleen weight while the mean cyst weight showed a 48-fold
mcrease.
C57BL/6 mice: Pattern of SAA, AA and HO-I responses
The morphology and the cell composition of the three splenic compartments, i.e.
red pulp (RP), perifollicular zone (PFZ) and splenic follicle (SF), including increased
numbers of megakaryocytes in the RP in the AHC-mice have been described
. 1 52732-34 B' fi SF' hl' d'd f . 1 d prevlOUS y" . ne y, m t e norma mlce were evOl 0 germma centers an
the RP contained rare megakaryocytes34. No R-AA immunoreactivity was observed in
normal spleen and liver sections indicating absence of detectable SAA deposition. By
contrast, significant numbers of R-HO-1 reactive RE cells were detected in the RP of 6 of
6 normal spleen sections (Fig. 2a); HO-1 positive cells occupied approximately 2% of the
splenic tissues (Fig. la). The immunostaining was specific; absorbed R-HO-I-, or TBS-
treated spleen sections failed to show any HO-1 reactivity. Hepatocytes lacked HO-1
reactivity but a few Kupffer cells in the liver sections did stain weakly for HO-I.
At 3 and 5 days p.i., none of the spleen sections displayed Congo red or R-AA
reactive AA fibrils. However, homogeneous R-AA reactivity was localized to the PFZ in
3/5 spleen sections indicating SAA deposition5JL3
2 . Figures 2b and 2c show the high and
low power view, respectively, of global R-AA immunostaining in the PFZ interstitium
28

and within the PFZ cells. Interestingly, R-HO-l reactive RE cell numbers increased in
those spleen sections that demonstrated R-AA reactivity in the PFZ. At 5 days p.L the
RP contained the majority of the R-HO-1 reactive RE cells (Fig. 2d). Figure 1 a depicts
the global profile of R-HO-1 reactive splenic RE cells before and during AA fibril
deposition; the number of R-HO-1 positive cells peaked between 3 and 4 wk p.i., and
then declined due to the replacement of the R-HO-l positive RE cells with increasing
load of AA fibril deposition (see below).
At 1 wk p.i., the spleen sections, although negative for Congo red staining,
displayed stronger R-AA reactivity in the PFZ areas (Table 1), similar to that shown in
figure 2c. In high power view, the PFZ cells displayed punctate cytoplasmic R-AA
reactivity, indicating, as shown previousll3 [, vacuolar localization of SAA32. R-AA
reactivity was also localized around the central arteriole including its endothelium34. At
this time period, R-HO-1 positive RE cells increased in number in both the RP and PFZ
(Fig. la).
At 2 wk p.i., the PFZ sinus walls showed segmental AA fibril deposition Cfable 1;
Fig. 2e), but still contained intact HO-1 positive endothelial cells. The RP had expanded,
contained heavy megakaryocyte cell infiltration and scant focal thiotlavin S (Fig. 2h) or
R-AA positive-AA fibrils (Table 1, Fig. 2f). The RP in the double labeled spleen sections
showed relatively high numbers of R-HO-1 positive RE cells (Fig. 2g), especially around
the thioflavin AA positive foci (Fig. 2h). The liver sections, although negative for AA
deposition at 2 wk p.i., demonstrated strong R-AA reactivity localized to the hepatic
sinuses and perivascularly around the central veins and the peri portal areas. Kupffer cells
labeled strongly with R-HO-l.
29

Between 3 and 4 wk p.i., AA load had inereased in the PFZ and to sorne extent in
the RP (Table 1). This corresponded with redueed numbers of R-HO-l reaetive RE cells
in the splenic parenchyma (Fig. la). As deseribed previously33, liver sections, double
labeled with thioflavin S and R-AA demonstrated seant y thioflavin Sand R-AA reactive
AA fibril in the walls ofhepatie blood vessels and hepatic sinuses. However, the majority
of R-AA reactive hepatic sinuses lacked thioflavin S staining, indieating sequestration of
SAA by the sinus Kupffer cells. Such sinuses also reaeted with R-HO-l (Fig. 2k).
Between 6 and 12 wk p.i., the AA load increased progressively in both the splenic
and hepatic parenchyma33.34 and this eorresponded with a decrease in the number of R
HO-1 positive RE cells (Figs. 1 a,b). At 12 wk p.i., almost aU the PFZ and a major p0l1ion
of the RP were infiltrated by AA fibrils. Consequently sueh an alteration eorresponded
with a precipitous decrease in the number of R-HO-l positive RE eells (Figs. 2iJ).
Since the lag period between SAA and AA fibril deposition is relatively short in
C57BL/6 miee3.5
, it was unclear whether the observed inereased HO-l expression in both
the splenie and hepatie tissues refleeted AHC infeetion-induced inflammatory stress or a
consequence of tissue deposition of SAA or AA fibril. To obtain insights into these
questions, we examined HO-1 response in AHC-infected CE/J mice. These mice do not
develop AA amyloidosis but show elevated circulating non-amyloidogenic SAA in
response to inflammatory stimuli35.36.
AHC-infected CE/J mice: pattern of SM and HO-l response
The CEl J mice examined at different time periods p.i. yielded much greater cyst
masses. The mean cyst weights at 4, 8 and 12 wk p.i. were, respectively, 1.4-fold, 3.5-
fold and 2.4-fold greater than those of the C57BL/6 mice. Regardless of relatively large
30

cyst weights, the CE/J mice did not show AA fibril deposition. However, between 2 and
12 wk p.i., increasing amounts of R-AA reactivity, similar to that shown in Fig. 2c, was
detected in the PFZ including the PFZ cells. Similarly, strong R-AA reactivity was seen
in the Kupffer cells lining the hepatic sinuses (not shown). Figures 1 a and 1 b compare the
splenic and hepatic profiles of R-HO-l reactive RE cells in the AHC-infected C57BL/6
and CE/J mou se strains. Little or no detectable HO-l reactivity was found in tive of five
normal CE/J spleen and liver sections (Fig. 21). In sharp contrast, HO-l expression
peaked at 2 wk p.i., in both the splenic and hepatic tissues in both C57 BL and CE/J mice
( Figs. 1 a,b). As shown in figures 2m and 2n, this reactivity was confined to the RP and
persisted at high levels throughout the course of infection in the CE/J mice. As such, even
at 12 wk p.i., the SF remained intact in the CE/J mice (compare Fig. 2n with 2i). This can
c1early be ascribed to the absence of AA fibril deposition in the CE/J mice.
Effect of acute phase SAA (A-SAA) on HO-l response in cultured macrophages
A-SAA is known to have proinflammatory properties24,37,38. Since the levels of
HO-l expression followed closely with SAA deposition in both splenic and hepatic
tissues in C57BL/6 and CE/J mice, we investigated whether these in vivo findings, as a
manifestation of oxidative stress, could be replicated in vitro. As such, we cultured mouse
resident peritoneal m<D and RA W 264.7 cells for 72 hr in DMEM containing 10% A
SAA serum. The cells were immunostained with R-AA and R-HO-l at 12,24 and 72 hr
(findings for 12 and 24 hr incubations similar, thus not shown). At each time point, each
cell type demonstrated vacuolar R-AA reactivity (Figs. 20,p; only the 72 hr profile shown
here); R-HO-l reactivity was singularly absent. Western blot analysis of the cel1 extracts,
showing ~ 12 kDa R-AA imunoreactive band in the cell lysates, further confirmed
31

intimate interaction between A-SAA and the macrophages, i.e. endocytosis of A-SAA
(Fig. 3).
Detection ofSAA in serafromAHC-infected C57BL/6 and CE/J mice
To confirm tissue (splenic and hepatic) deposition of circulating SAA in the
AHC-infected C57BL/6 and CE/J mice, we immunoblotted selected serum samples using
R-AA. Each C57BL/6 sera (2 ~L/lane), obtained at days 3 and 5 and at 1 and 2 weeks
demonstrated SAA similar in position to that present in the control silver nitrate
stimulated mouse serum (Fig. 4a). Similar, although weaker, SAA reactive bands were
found in the CE/J mou se sera (3 ~L/lane), obtained at 2, 4, 6 and 8 wk p.i. (Fig. 4b).
32

DISCUSSION
The principal finding in this study was that AHC infection induced significantly
increased expression of HO-1 in RE cells of both amyloid-sensitive C57BL/6 and
amyloid-resistant CE/J mice. The factor(s) stimulating the HO-1 response in this model
and its overall implication in AA amyloidosis are under study.
On an immunocytochemical basis, HO-1 positive splenic and hepatic RE cells
increased numerically in both mouse strains (Figs. 1a,b), and this event coincided with
SAA deposition in the spleen (Figs. 2b,c), as well as in the liver. SAA was localized, as
shown previously, both interstitially in the tissues examined and also to the vacuolar
compartments, most likely to the lysosomes, in the RE cells 6. This indicated an intimate
interaction between SAA and the RE cells. Thus, increased HO-1 expression in the RE
cells might tentatively be linked to SAA deposition in the spleniclhepatic tissues.
However, despite a similar tissue SAA deposition pattern in the two mouse strains, their
HO-1 profile levels, as shown in figure la, contrasted significantly, especially between 4
and 12 wk p.i., during the AA fibril deposition phase in C57BL/6 mice. Clearly, this
precipitous decline in the number of splenic HO-l positive cells coincided with tissue
displacement by the increasing AA load (Table 1; Fig. 1 a; Figs. 2i,j). Thus, it is unlikely
that AA fibrils participate directly in increased HO-1 expression in the splenic RE cells.
We found a similar relationship in the hepatic tissues as well (Fig. lb); increased HO-1
expression in the Kupffer cells appeared to be unrelated to AA fibril deposition.
Amyloid-resistant AHC-infected CE/J mice unambiguously confirmed this relationship
(Figs. 1 a,b; Figs. 2m,n); HO-l response in CEl] mice was more pronounced and
33

sustained in the absence of AA fibril deposition. It is aIso worth noting that the pattern of
HO-l response described here (Figs. la,b) was reminiscent of increased ubiquitin (a
stress prote in) expression in m<D/RE ceUs in the AHC-infected C57BL/6 mice. We had
proposed AHC-infection induced inflammatory stress to be the inducing factor for the
b· .. 31 U lqmtm response .
Recently, Yan et al. showed that both AA fibrils and amyloidogenic munne
SAA1.1, but not SAA2.1 or SAA2.2 (CE/J mouse derived), both of which are non-
amyloidogenic, bound to murine BV -2 ceUs, a transformed mouse microglialline. These
ligands bound to the BV -2 ceUs through the receptor for advanced glycation end-products
(RAGE), and induced increased expression of HO-I mRNA, HO-I prote in and several
inflammatory cytokines24. However, studies carried out in vitro and at variance with that
of Yan et al., showed that non-amyloidogenic SAA2.2 binds mouse peritoneal m<D more
avidly than SAA1.1 39. Presumably, the apparent difference in the binding affinities of
SAAI.I and SAA 2.2 to mouse m<l> and BV -2 ceUs could be due to the fact that two
different ceU types were used in these studies; BV -2 ceUs and murine m<D could have
different affinities for different murine SAA isoforms. While the in vivo metabolism of
SAA2.2 in CE/J mice has not been as weU characterized as SAA 1.1/SAA2.1 4o, our data,
nonetheless, clearly show SAA2.2 deposition in both the splenic and hepatic tissues of
AHC-infected CE/J mice. Such a relationship might suggest interaction between SAA2.2
and the RE ceUs in situ, a phenomenon similar to that established in the clearance of
SAA1.1/SAA2.1 from the circulation4o . Thus, the pronounced increase in HO-l
expression in both the splenic and hepatic RE ceUs of C57BL/6 mice closely correlated
with increased tissue deposition of SAA and independent of AA fibril deposition. The
34

validity of this argument is strengthened by the data trom the AHC-infected CE/J mice.
While stress effects are highly complex and multifàctorial, we at this stage are unable to
single out whether SAA alone or in conjunction with inflammatory cytokines, as would
be expected in the AHC-mouse model, effected the induction of HO-l in the RE cells.
SAA is a sensitive indicator of various tissue insults and inflammatory disordcrs
during which its plasma concentration can increase up to 1000-fold41• Clearance of
circulating SAA is known to be mediated through activated m<D/RE cells40 and lysosomes
appear to participate in SAA processing6,8. In view of the fact SAA manifests
proinflammatory properties24,3738, its interaction with monocytoid cells could then
explain the induction of HO-l response. The data derived from the AHC-infected mice
would tend to support this observation (Figs. la,b). However, the in vitro experiment
performed with m<D and SAA-containing mou se serum c1early showed that, despite SAA
endocytosis, the m<D failed to show HO-l response (Figs. 20,p and Fig. 3). Thus, it is
quite likely that multiple factors generated during chronic inflammation in the AHC
infected mice, such as inflammatory cytokines and/or oxidative stress, could have
triggered HO-l expression in the RE cells as a cytoprotective mechanism.
35

REFERENCES
1 Glenner GG (1980). Amyloid deposits and amyloidosis. The beta-fibrillogenesis.
N Engl J Med 302, 1283-1292; 1333-1343
2 Falk RH, Raymond LC and Skinner M (1997). The systemic amyloidosis. N Engl J
Med 337, 898-909
3 Ali-Khan Z, Li W and Chan SL (1996). Animal model for the pathogenesis of reactive
amyloidosis. Parasitol Today 12,297-302
4 Treves S and Ali-Khan Z (1984). Characterization of the inflammatory cells in
progressing tumor-like alveolar hydatid cysts: 1.Kinetics and composition of
inflammatory infiltrates. Tropenmed Parasit 35, 183-188
5 Du T and Ali-Khan Z (1990). Pathogenesis of secondary amyloidosis in an alveolar
hydatid cyst-mouse model: histopathology and immuno/enzyme-histochemical analysis
of splenic marginal zones during amyloidogenesis. J Exp Pathol 71, 313-335
6 Chan SL, Chronopoulos S, Murray J, Laird DW and Ali-Khan Z (1997). Selective
localization ofmurine ApoSAAI/SAA2 in endosomes-Iysosomes of activated
macrophages and their degradation products. Amyloid: Int J Exp Clin Invest 4, 40-48
7 Ali-Khan Z (2002). Searching for an in vivo site tor nascent amyloid fibril formation.
J Alzheimer 's Dis 4, 105-114
8 Kluve-Beckerrnan B, Manaloor J and Liepnicks JJ (1999). Binding, trafficking and
accumulation of serum amyloid A in peritoneal macrophages. Am J Pathol 155, 123-133
9 Shiharima T and Cohen AS (1975). Intralysosomal formation of amyloid fibrils. Am J
Pathol 81, 101-116
36

10 Kelly JW (1996). Alternative confornlations of amyloidogenic proteins govern their
behavior. Curr Opin Struet Biol 6, 11-17
Il Chiti F, Webster P, Toddei N, Clark A, Stefani M, Ramponi 0 and Dobson CM
(1999). Designing conditions for in vitro fonnation of amyloid protofilaments and fibrils.
Proe Natl Aead Sei USA 96, 3590-3594
12 Smith MA, Hirai K, Hsiao K, Pappolla MA, Harris PL, Siedlak SL, Tabaton M and
Perry 0 (1998). Amyloid-beta deposition in Alzheimer transgenic mice is associated with
oxidative stress. J Neuroehem 70, 2212-2215
13 Belloti V and Merlini 0 (1996). CUITent concepts on the pathogenesis of systemic
amyloidosis. Nephrol Dial Transplant 11 (Suppl 9), 53-62
14 Markesbery WR (1997). Oxidative stress in Alzheimer' s disease. Free Radie Biol
Med 23, 134-147
15 Perry 0 and Smith MA (1997). A central role for oxidative damage in the
pathogenesis and therapeutics of Alzheimer's disease. Alzheimer Dis Assoc Disord 2,
319-324.
16 Head E, Oarzon-Rodriguez W, Johnson JK, Lott IT, Cotman CW and Olabe C
(2001). Oxidation of AB and plaque biogenesis in Alzheimer's disease and Down
Syndrome. Neurobiol Dis 8, 792-806.
17 Pappolla MA, Omar RA, Kim KS and Robakis NK (1992). Immunohistochemical
evidence of antioxidant stress in Alzheimer's disease. Am J Pathol 140,621-628
18 Smith MA, Kutty RK, Richey PL, Yan S-D, Stern D, Chader OJ, Wiggert B, Petersen
RB and Perry 0 (1994). Herne oxygenase-l is associated with the neurofibrillary
pathology of Alzheimer's disease. Am J Pathol 145,42-47
37

19 Takahashi M, Dore S, Ferris CD, Tomita T, Sawa A, Wolosker H, Borchelt DR,
Iwatsubo T, Kim SH, Thinakaran G, Sisodia SS and Snyder SH (2000). Amyloid
precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in
Alzheimer's disease. Neuron 28, 461-473
20 Kfunpfer H, Kolb N, Manderscheid M, Wetzler C, Pfeilschifter J and Frank S (2001).
Macrophage-derived heme-oxygenase-l: expression, regulation, and possible functions in
skin repair. Mol Med 7, 488-498
21 Sunderman FW Jr. (1987). Metal induction ofheme oxygenase. Ann N Y Acad Sei
514,65-80
22 Goldbaum ° and Richter-Landsberg C (2001). Stress proteins in oligodendrocytes:
differential effects ofheat shock and oxidative stress. J Neurochem 78, 1233-1242
23 Dong Z, Lavrovsky Y, Venkatachalam MA and Roy AK (2000). Herne oxygenase-l
in tissue pathology. Am J Pathol 156, 1485-1488
24 Yan SD, Zhu H, Zhu A, Golabek A, Du H, Roher A, Yu J, Soto C, Schmidt AM,
Stem D and Kindy MS (2000). Receptor-dependent cell stress and amyloid accumulation
in systemic amyloidosis. Nature 6, 643-651
25 Rysava R, Merta M, Tesar B, Jirsa M and Zima T (1999). Can serum amyloid A or
macrophage colony stimulating factor serve as marker of amyloid formation process?
Biochem Mol Biol Int 47,845-851
26 Ando Y, Nyhlin N, Suhr 0, Holmgren G, Uchida K, El Sahly M, Yamashita T,
Terasaki H, Nakamura M, Uchino M and Ando M (1997). Oxidative stress is found in
amyloid deposits in systemic amyloidosis. Biochem Biophys Res Commun 232,497-502
38

27 Ali-Khan Z (1978). Cellular changes in the lymphoreticular tissue of C57L1J mice
infected with Eehinoeoeeus multiloeularis cysts. Immunology 34, 831-839
28 Puchtler H, Sweet F and Levine M (1962). On the binding of Congo red by amyloid.
J Histoehem Cytoehem 10,355-364.
29 Chronopoulos S, Alizadeh-Khiavi K, Normand J and Ali-Khan Z (1991). Binding of
ubiquitin to experimentally induced murine AA amyloid. J Pathol 163, 199-203
30 Towbin H, Staehelin T and Gordon J (1979). Electrophoretic transfer of proteins
from polyacrylamide gels to nitrocellulose sheets: procedures and sorne applications.
Proe Nat! Aead Sei USA 76,4350-4354
31 Chronopoulos S, Lembo P, Alizadeh-Khiavi K and Ali-Khan Z (1992). Ubiquitin:
Its potential significance in murine AA amyloidogenesis. J Pathol 167,249-259
32 Chronopoulos S, Laird DW and Ali-Khan Z (1994). Immunolocalization of serum
amyloid A and AA amyloid in lysosomes in murine monocytoid cells: confocal and
immunogold electron microscopic studies. J Pathol 173, 361-369
33 Alkarmi T and Ali-Khan Z (1984). Chronic alveolar hydatidosis and secondary
amyloidosis: pathological aspects of the disease in four strains of mice. Br J Exp Pathol
65,405-417
34 Ali-Khan Z, Sipe JD, Du Tao and Riml H (1988). Eehinoeoceus multilocu/aris:
relationship between persistent inflammation, serum amyloid A prote in responses and
amyloidosis in four mouse strains. Exp Parasitol67, 334-345
35 Sipe JD, Carreras 1, Gonnerman W A, Cathcart ES, de Beer MC and de Beer Fe.
(1993). Characterization of the inbred CE/J mouse strain as amyloid resistant. Am .!
PathoI143,1480-1485
39

36 De Beer MC, de Beer FC, McCubbin WD, Kay CM and Kindy MS (1993).
Structural prerequisites for serum amyloid A fibril formation. J Biol Chem 268, 20606-
20612
37 Malle E, Bollmann A, Steinmetz A, Gemsa D, Leis Hl and Sattler W (1997). Serum
amyloid A (SAA) prote in enhances formation of cyclooxygenase metabolites of activated
human monocytes. FEBS Lett 419,215-219
38 Van Lenten Bl, Hama SY, de Beer FC, StatTorini DM, McIntyre TM, Prescott SM,
La Du BN, Fogelman AM and Navab M (1995). Anti-inflammatory HDL becomes pro
inflammatory during the acute phase response. Loss of protective effect of HOL against
LDL oxidation in aortic wall cell cocultures. J Clin lnvest 96, 2758-2767
39 Lian l-S, Elliot-Bryant R, Hajri T, Sipe lD and Cathcart ES (1998). A unique
amyloidogenic apolipoprotein serum amyloid A (apoSAA) isoform expressed by the
amyloid resistant CEll mouse strain exhibits higher affinity for macrophages than
apoSAAI and apoSAA2 expressed by amyloid susceptible CBAI] mice. Biochim
Biophys Acta 1394, 121-126
40 Kluve-Beckerman B, Yamada T, Hardwick 1, Liepnieks 11 and Benson MD (1997).
DifferentiaI plasma clearance of murine acute-phase serum amyloid A proteins SAA 1
and SAA2. Biochem J 322, 663-669
41 Husby G, Marhang G, Dowton G, Sletten K and Sipe D (1994). Serum amyloid A
(SAA): Biochemistry, genetics and the pathogenesis of AA amyloidosis. Amyloid.· Int .J
Exp Clin lnvest 1, 119-187
40

TABLE 1. Mean spleen and cyst weights in normal and alveolar hydatid cyst infected
C57BL/6 mice at various times post-infection (p.i.); also shown here is the pattern of
serum amyloid A (SAA) and amyloid A (AA) fibril deposition in the splenic
perifollicular zones (PFZ) and the red pulp (RP).
Mean weights ± sd AA distribution in spleen SAA deposition Time p.i. Cyst (g) Spleen (mg) AA in PFZ AA in RP in the spleen Normal - 81.3±6 - - -1 wk p.i. 0.1 85±5 - - +"
2 wk p.i. 0.17±0.04 112±7 2+ 1+" ID'
4 wk p.i. 0.46±0.1 147±9 3+ 1+ ID 6 wk p.i. 0.53±0.07 192±5 3+ 3+ ID 8 wk p.i. 1.9±0.2 235±17 4+ 4+ ID 12 wk p.i. 4.8±0.9 390±10 4+ 4+ ID
* +, homogeneous non-fibrillar rabbit anti-mouse AA amyloid IgO (R-AA) reactivity,
indicative of serum amyloid A (SAA) deposition, in the PFZ and the RP.
** Low 1 + to heavy 4+ AA fibril deposition in the PFZ sinuses and the RP
t R-AA immunopositive loci present in the PFZ and the RP but indiscernible (ID)
whether SAA or AA fibrils.
41
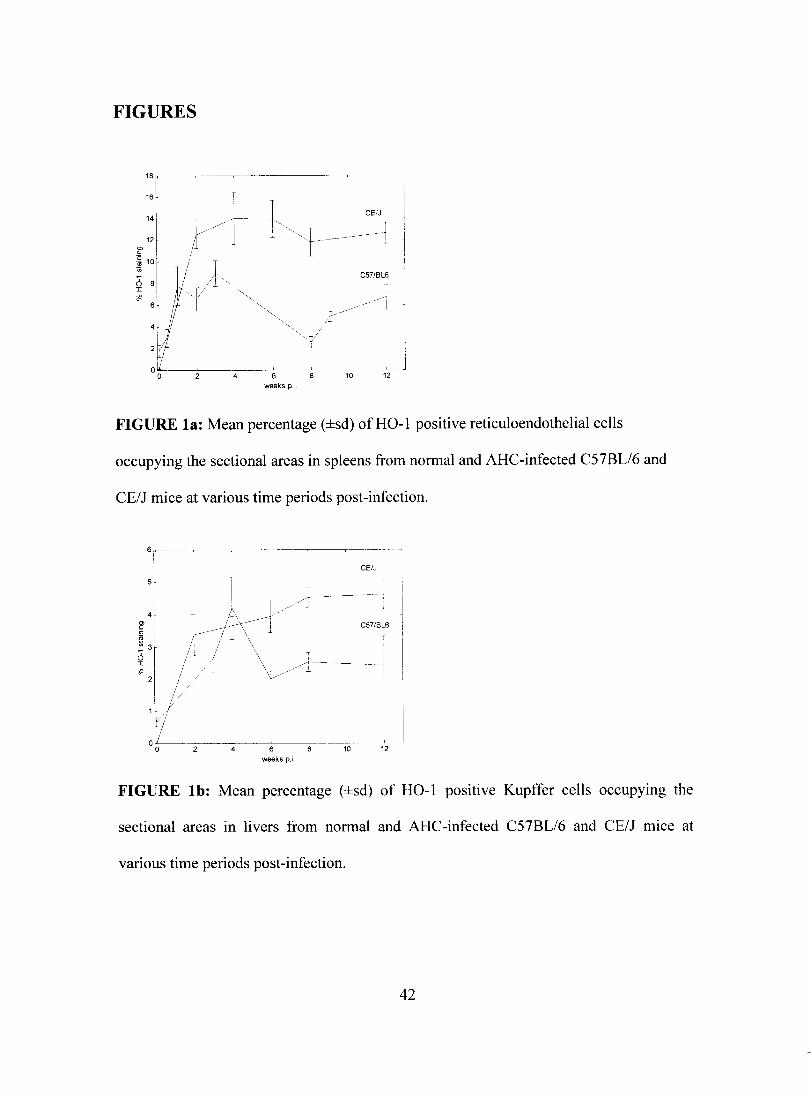
FIGURES
a 1L~----'---~_~L-' ~--'-, ~-----'-- ~~ ______ l __
a 4 6 10 12 weeks p.L
FIGURE la: Mean percentage (±sd) ofHO-l positive reticuloendothelial cells
occupying the sectional are as in spleens from normal and AHC-infected C57BL/6 and
CE/J mice at various time periods post-infection.
6~--~~
1
sr
----r-- 1
CE/J 1
1 CS7/BL6 ~ 4 r l _;t\---i / /~ f3r Ir-j±' " ~'Il/ \/:----1l// L 1
00 ~- ~4--~6~~~ .l __ ~ ___ ~
10 12 weeks p.i
FIGURE lb: Mean percentage (±sd) of HO-l positive Kupffer cells occupymg the
sectional areas in livers from normal and AHC-infected C57BL/6 and CE/J mice at
various time periods post-infection.
42
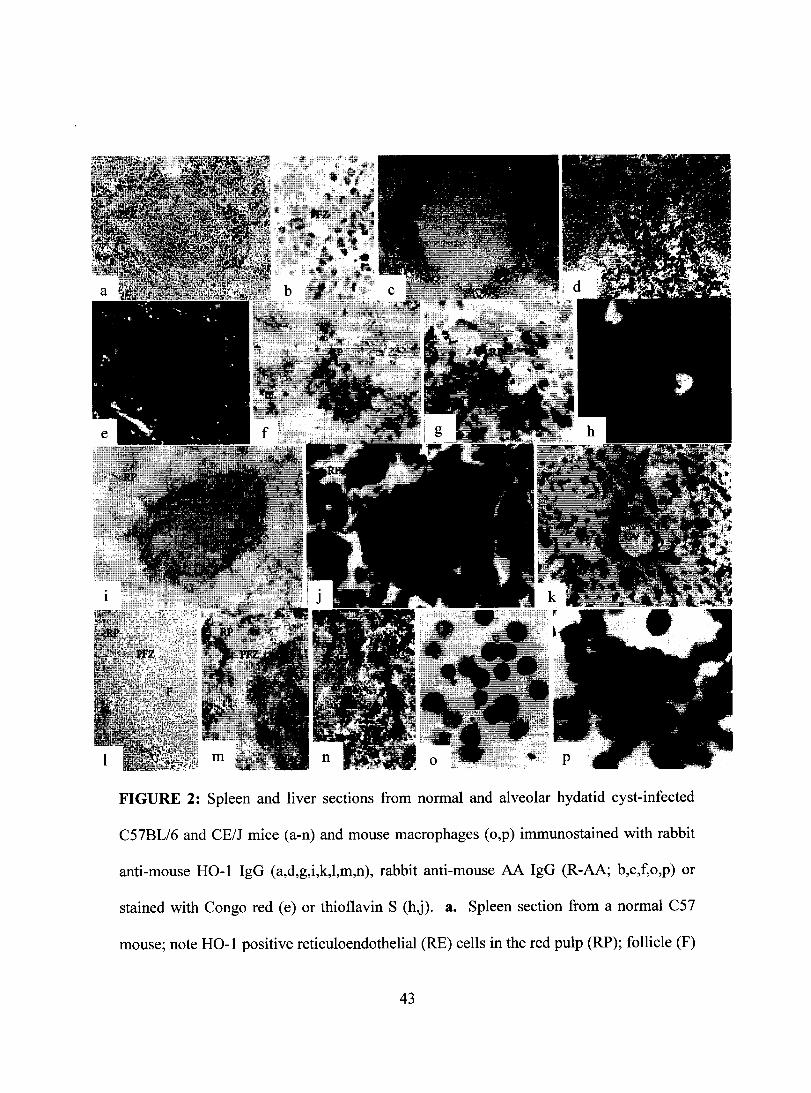
FIGURE 2: Spleen and liver sections from normal and alveolar hydatid cyst-infected
C57BL/6 and CE/J mice (a-n) and mouse macrophages (o,p) immunostained with rabbit
anti-mouse HO-l IgG (a,d,g,i,k,l,m,n), rabbit anti-mouse AA IgG (R-AA; b,c,f,o,p) or
stained with Congo red (e) or thioflavin S (h,j). 3. Spleen section from a normal C57
mouse; note HO-l positive reticuloendothelial (RE) cells in the red pulp (RP); follicle (F)
43

essentially negative for HO-l staining. b. Spleen section from a 3 days post-infection
(p.i.) C57 mouse; note R-AA reactivity, most likely SAA, in the perifollicular (PFZ) cells
and the interstitium. c,d. Spleen sections from a 5 days p.i. C57 mouse; note SAA
immunostaining in the PFZ and the RP (c) and HO-l immunostaining mostly in the RP
(d). e. Spleen section from a 2 wk p.i. C57 mouse; note Congo red positive AA deposits
in the PFZ sinus walls. f-h. Spleen sections from a 2 wk p.i. C57 mouse; note moderate
R-AA immunostaining in the RP (f); double labeling showing HO-l positive RE ce Ils in
the RP (g) around thioflavin S positive AA deposits (h). i,j. A double-Iabeled spleen
section from a 12 wk p.i. C57 mouse; note the decrease in HO-l positive RE cells around
the follicle (i) and the surrounding PFZ and the RP showing massive thioflavin S positive
AA deposits (j). k. Liver section from a 4 wk p.i. C57 mouse; note HO-l positive
hepatic sinuses (S) and central vein endothelium (CV). 1-0. Spleen sections from
normal, 2 wk p.i., and 12 wk p.i. CE/J mice; note residual HO-l immunostaining in the
normal spleen (1), and strong HO-l immunostaining in the morphologically intact RP at
both 2 wk p.i., (m) and 12 wk p.i., (n). o,p. Resident peritoneal macrophages from CD 1
mouse (n) and RAW264.7 cells (0) cultured with 10% SAA-containing mouse serum for
72 hr and reacted with R-AA; note strong vacuolar SAA immunostaining.
Original magnification: a,c,e,i,j ,l,m,n 10x; d,f,k 16x; b,g,h,o,p 60x.
44

Per. m<I> RA W264.7
16.5
-=----~ ,~-'
. r ~ • !il
6.5 kDa -
FIGURE 3: Immunoblot to detect SAA in resident mouse peritoneal macrophages (per.
m<I» and RA W264.7 celllysates. The cells were cultured for 72 hr in DMEM (5% C02,
37°C) containing 10% pooled serum from CDl mice obtained 18 hr after a subcutaneous
injection with 0.5 ml of 2% AgN03. The cells were trysinized, washed with phosphate
buffer, lysed and each lane was loaded with clarified cell extracts equivalent to 500,000
cells; rabbit anti-mouse AA amyloid IgG (1.24 mg/mL, dilution 1 :3000) and ECL kit
were used to deve10p the immunoblot.
45

1 2 3 4 5 1 2 3 4 5
16.5 kDa
a b
FIGURE 4: Immunoblots, developed separate1y, to detect serum amyloid A in sera from
alveolar hydatid cyst-infected C57BL/6 (2 JlL/lane) and CE/J (3 JlL/lane) mice at various
time periods post-infection (p.i.). 3. C57BL/6 mice: lanes 1-4 contain sera at 3 and 5
days, 1 and 2 wk p.i., respectively; lane 5, AgN03-treated CDl mouse (2 JlL/lane). b.
CE/J mice: lane 1, as lane 5 in a.; lane 2-5 contain sera at 2, 4, 6 and 8 wk p.i.,
respectively; rabbit anti-mouse AA amyloid IgG (1.24 mg/mL, dilution 1 :3000) and ECL
kit were used to deve10p the immunoblots.
46

ACKNOLEDGEMENTS
This investigation was supported by a research grant (MOP-42526) from the
Canadian Institute of Health Research. We are thankful to Mr. Joubin Hatamzadeh for
his help with the image analysis set-up.
47

PREFACE TO CHAPTER 3
ln the AHC-mouse model of AA amyloidosis, HO-l expression corresponded
closely with tissue deposition of SAA, but was unrelated to AA fibril deposition. In vitro
studies however, demonstrated that SAA alone was not responsible for the upregulation
in HO-I response in m<I>. Given that chronic inflammation is central to the pathogenesis
of AA amyloidosis, the role of inflammatory cytokines and OS must be clarified in order
to better understand the dynamics of the observed H 0-1 response.
While HO-l serves solely as a marker of antioxidant response, a cytoprotective
response to OS, markers of oxidative modifications explicitly confirm OS and allow for
immunohistochemicallocalization of such modifications. Specifie markers against
different oxidative modifications allow for the determination of the extent and type of
such modifications in tissue, specifically in the fixed and migrating monocytoid cclI
populations. Immunohistochemicallocalization oftwo such oxidative modifications was
performed in spleen sections and peritoneal leukocytes from AHC-infected mice; the
results are presented in the following chapter.
A variety of oxidative modifications is formed from the effect of ROR. Our study
focused on the distribution of markers of lipid peroxidation and advanced glycation end
products, specifically 4-hydroxy-2-nonenal (HNE) and NE-(Carboxymethyl)lysine
(CML), respectively. Both markers have been immunolocalized to chemically diverse
amyloid deposits, including human AA fibrils (Ando). Interestingly, a recent AD study
has shown that a cross-link modification induced by HNE on tau, responsible for the
Alz50 epitope that is prone to NFT formation, is completely coincident with HO-I
expression (Takeda, 2000b).
48

Chapter 3.
Immunolocalization of lipid peroxidation end products in murine AA amyloidosis
Golnar Kamalvand, Zafer Ali-Khan
Department of Microbiology and Immunology, McGill University, Montreal
Quebec, Canada
Running title: Oxidative stress markers in AA amyloidosis
Correspondence: Dr. Zafer Ali-Khan Department of Microbiology and Immunology 3775 University St. McGill University Montreal Quebec, H3A 2B4 Canada Tel: (514) 398-3930 Fax: (514) 398-7052 Email: [email protected]
49

Oxidative stress markers in AA amyloidosis
ABSTRACT
Chronic inflammation, superimposed by amyloid fibril deposition, is believed to
trigger the cascade of oxidative stress response in the affected organs and tissues. We
examined immunohistochemically the distribution oftwo markers of oxidative stress, 4-
hydroxy-2-nonenal (HNE) and NC-(carboxymethyl)lysine (CML), in spleen sections and
peritoneal macrophages (m<D) from mice prior to and during AA amyloidosis. With time,
both HNE and CML immunoreactivities increased significantly, particularly in m<D and
splenic reticuloendothelial cells, known to be associated with the clearance of serum
amyloid A, the precursor of AA fibrils. In addition, the cellular increase in HNE and
CML corresponded with the progression of AA fibril deposition in the splenic tissue.
HNE and CML were localized to the plasma membrane and the cytoplasmic
compartment of m<D and HNE only at the nuc1ear membrane. These mark ers were also
co-Iocalized bound to AA fibril infiltrating the splenic sinus walls. Our results reinforce
the notion that oxidative stress is an integral component of amyloidotic tissues. Both
HNE and CML have been implicated in protein modification and amyloid fibril
formation. The significance of HNE and CML associated with the monocytoid cells,
implicated in SAA clearance and AA fibril formation, is discussed with the pathogenesis
of AA fibrils.
Keywords - AA amyloid, Alveolar hydatid cyst, Oxidative stress, 4-Hydroxy-2-nonenal,
Lipid peroxidation, NE -( carboxymethyl)lysine, Advanced glycation end product,
Immunohistochemistry, Splenic perifollicular zone, Red pulp, Reticuloendothelial cells,
Macrophage.
50

Oxidative stress markers in AA amyloidosis
INTRODUCTION
Amyloid diseases represent a broad spectrum of ailments characterized by the
extracellular deposition of insoluble protein fibrils, originating from a particular
precursor protein, in affected soft organs [1]. Involvement of kidney glomureli, infiltrated
with AA amyloid, in inflammation-associated AA amyloidosis poses a serious c1inical
condition in diseases such as rheumatoid arthritis and chronic bacterial and parasitic
infections such as tuberculosis and leprosy [2,3]. However, the conditions that lead to
transformation of soluble precursor protein, serum amyloid A (SAA), into amyloid A
(AA) fibrils are unknown. AA fibrils are 5 to 13 nm in thickness, have cross ~-pleated
sheet structure, bind Congo red dye and may induce oxidative stress in the tissue
monocytoid cells [4,5]. We have used the alveolar hydatid cyst (AHC)-mouse model to
understand the mechanisms of AA fibril formation [6]; the course of the disease in mice
mimics the course of AA amyloidosis in humans. AHC grows like a solid tumor,
inducing chronic inflammation and a robust SAA response, culminating into multiple
organ AA fibril deposition [3,6].
Chronic inflammation has long been recognized as central to the induction of AA
amyloidosis, although its role in this process has not yet been fully investigated [2,7].
More specificalIy, oxidative stress has recently been implicated as a contributing factor to
the generation of AA fibrils [5,8]. Recently, we showed heme-oxygenase-l (HO-I)
response in the AHC-mice prior to and during AA amyloidosis and we associated this
response to preponderant SAA deposition in the soft organs [9]. In previous studies we
showed that SAA is endocytosed by tissue monocytoid cells, trafticked to lysosomes and
51

Oxidative stress markers in AA amyloidosis
as a result of partial degradation, at least five N-terminally intact SAA derivatives,
similar in molecular masses to that of tissue AA fibril protein, are produced [10, Il]. This
led us to propose that AA fibrils might be generated intracellularly in tissue monocytoid
cells. As such, Kluve-Beckerman et al., in a series of publications, contirmed this finding
[12,13]. However, the conditions that led to AA fibril synthesis were not discussed. Since
multiple mediators of inflammation induce generation of copious amounts of reactive
oxygen radicals (ROR) in the monocytoid cells, it is reasonable to propose that these
processes may affect the metabolism of reticuloendothelial (RE) cell/macrophage (m<D)
associated SAA. We hypothesized that ROR generated in inflammatory m<D/RE cells
could oxidize SAA fragments, render these fragments resistant to enzymatic
degradation/clearance and prone to AA fibril formation [14]. However, the extent that
ROR might affect intracellular nascent AA fibril formation remains to be determined.
Various oxidative modification products are formed from the effect ofROR; protein
modifications that occur under conditions of oxidative stress may result from lipid
peroxidation and/or glycoxidation [15]. Lipid peroxidation, originating in biological
membranes by the c1eavage of oxidized unsaturated fatty acids of cholesterols and
phospholipid esters, generates a complex pattern of aldehydes ofwhich 4-hydroxy-2-
nonenal (HNE) is one of the major reactive products [16]. Glycoxidation ofproteins by
reducing sugars produces advanced glycation end products (AGEs), usually a slow
process, moderated by the presence of oxygen [17]. NE-(Carboxymethyl)lysine (CML), a
glycoxidation product, can also be formed independently through lipid peroxidation [18].
Both markers have been localized to chemically diverse amyloid deposits: HNE
52

Oxidative stress markers in AA amyloidosis
immunoreactivity (IR) was localized to amyloid positive blood vessel walls and m<D
around amyloid deposits in patients with primary and secondary amyloidosis [8], and to
p-amyloid in Alzheimer's disease [19,20]; CML-IR was localized to AA fibrils in kidney
glomeruli [21,22], p2-microglobulin amyloid fibrils in patients with long- term dialysis
[17], and to Alzheimer's p-amyloid [23,24]. These two markers showing HNE-IR and
CML-IR have been co-localized in amyloidotic tissues of patients with uremia [25,26],
and Alzheimer's disease [27]. Modification ofthese proteins through HNE and CML
could render them resistant to proteolytic degradation and present them as tombstone
markers. It has recently been proposed that extensive protein oxidation may render such
aggregates highly resistant to proteolysis [28,29J.
The aim of our investigation was to immunohistochemically monitor the spatio
temporal distribution ofHNE-, and CML-IR in the AHC-mice, prior to and during AA
fibril deposition. HNE and CML were co-Iocalized to peritoneal m<l>, splenic RE cells
and interstitium, and AA fibrils. These data support our previous findings showing stress
response in the AHC-mice undergoing AA amyloidosis [9,30].
53

Oxidative stress markers in AA amyloidosis
MATE RIALS AND METHODS
Infection
In our previous study, we used spleen and liver sections from the AHC-mouse to
examine the profile ofHO-1 response [9]. Tissue sections from the same series of
experiments were used in the present study. Briefly, six-week-old male C57BL/6 mice
(Jackson Laboratories, Bar Harbor, Maine) were inoculated intraperitoneally with 250
alveolar hydatid cysts. Three to six mice were sacrificed betwecn 5 days and 12 wk post
infection (p.i.); cryostat spleen sections (6-8 ~m thick) and cytocentrifuged peritoneal
leukocytes were used for immunostaining. Control samples were obtained from non
infected C57BL/6 mice.
Antibodies
Commercially available rabbit anti-4-hydroxy-2-nonenal serum (R-HNE. Cat #
HNE Il-S, Alpha Diagnostic International, San Antonio, TX) and rabbit anti
carboxymethyllysine serum (R-CML, a generous gift from Dr. George Perry, Institute of
Pathology, Case Western Reserve University, Cleveland, OH) were used in 1:500 and
1 :200 dilutions, respectively, for the detection of lipid peroxidation and advanced
glycation end product formation. The secondary antibody was goat anti-rabbit IgG
conjugated to peroxidase labeled-dextran polymer (K4002, Dako EnVision+, Dako
Corporation, Carpinteria, CA).
Immunohistochemistry
Sorne modifications were applied to the basic method described prcviously [30].
Spleen sections and cytocentrifuged cells were fixed in 4% paraformaldehyde (20 min)
54

Oxidative stress markers in AA amyloidosis
and treated sequentially with the following reagents: 3% H20 2 in methanol for quenching
endogenous peroxidase activity (30 min). washing 3x in phosphate buffered saline (pBS,
pH 7.4), incubation in blocking solution (BS; 0.5% BSA, 0.1 % saponin, 2.5% non-fat dry
milk, 1 % H20 2) containing 5% normal rabbit serum (45 min), incubation in primary
antibody diluted in BS (ovemight), washing 3x in BS, incubation in secondary antibody
diluted in BS containing 5% normal goat serum (l hr), and washing 3x in PBS. Color
reaction was developed with diaminobenzydine (up to 15 min) and the samples were then
lightly counterstained with hematoxylin (Harris' fonnulation, Sigma). Double-labeling of
the immunostained spleen sections were carried out with thioflavin S ( 1 % in distilled
water, 5 min). Specificity of the immunostaining was performed by incubating the
samples in PBS without the primary antibody or in absorbed R-HNE antibody; absorption
was carried out by ovemight incubation of 0.1 ml ofR-HNE with O.lmg ofHNE-KLH
peptide, kindly provided by Dr. George Perry. The absorbed antibody was micro
centrifuged and the supematant used instead ofR-HNE. Digital pictures were taken using
a Nikon 100 camera mounted on a Leitz Dialux 20 microscope.
Between 300 to 400 immunostained cells were examined at each time period,
under 60x (oil immersion); data is presented as the mean of immunopositive cells ± sd.
55

Oxidative stress markers in AA amyloidosis
RESULTS
Cyst and spleen weights and immunohistochemicalloca/ization qfSAA and AA fibri!s in
spleen sections
The data on splenomegaly and peritoneal AHC masses, harvested from mice at
different time post-infection, have been published [9]. Briefly, splenomegaly
corresponded with progressive increase in the peritoneal AHC-biomass and peritonitis
[31]. Between 1 and 12 wk pL there was a 5-fold increase in the mean spleen weight and
a 48-fold increase in the mean AHC weight.
In order to connect the present data with the profiles of SAA and AA tibril
deposition in the splenic tissue, we briefly summarize here our previous findings [9].
Normal spleen sections lacked IR against rabbit anti-mouse AA amyloid antibody (R
AA) which immunoreacts with both SAA and AA amyloid. At 5 days p.i., the spleen
sections were devoid of AA fibril deposition, but they did demonstrate homogeneous R
AA-IR sites at the perifollicular zones (PFZ) indicating commencement of SAA
deposition in the tissue. At 1 wk p.i., spleen sections lacking Congo red staining,
displayed stronger punctate R-AA-IR in RE cells in the PFZ. Between 2 and 12 wk p.i.,
progressive AA fibril deposition caused radical architectural changes in the splenic
parenchyma. Endocytosed SAA with punctate R-AA-IR in the peritoneal mC1l was
detected at 5 days and onwards [32]. In sum, SAA was detected in the splenic tissue
including the tissue m<1> 1 wk prior to AA fibril deposition.
56

Oxidative stress markers in AA amyloidosis
Immunohistochemicallocalization of HNE in peritonealleukocytes
Between 1 to 12 wk p.i., m<D constituted the predominant population. Fig. 1
shows the mean ± sd of m<D with plasma membrane/cytoplasmic-IR. Betwcen 1 and 6 wk
p.i, the number of m<D with HNE-IR increased ~ 3-fold over the normal cells, i.e. from
30% to 80%; the pattern ofHNE-IR in cells, whether membranous or cytoplasmic,
varied. After the peak response at 6 wk p.i., the number of m<D with HNE-IR decrease
gradually to less than 50% by 12 wk p.i. This may indicate freshly recruited m<D to the
peritoneal cavity. Only a fraction of normal m<D displayed light membrane HNE-IR (fig.
1, fig. 2A).
The spatial distribution ofHNE-IR, as shown in fig. 1, varied with time and the
progression of AHC-infection. At 1 wk p.i., ~20% of the m<D displayed a single dot-like
cytoplasmic HNE-IR site (fig. 2B); the IR site was smaller than a nucleus but much larger
than a lysosome (> 1 /-lm). Between 2 and 6 wk p.i., the initial site ofHNE-IR at the
membrane extended to the cytosolic compartment; nuclear HNE-IR was rarely seen at
these time periods. At 8 and 12 wk p.i., while the number ofm<D with HNE-IR decreased
gradually, the positive cells displayed multiple focal HNE-IR in the cytoplasm as weil as
relatively intense HNE-IR in both the plasma and nuclear membranes (fig. 2C,O).
Immunohistochemicallocalization of HNE in spleen sections
The profiles ofHNE-IR localized to three splenic compartments, i.e. splenic
follicle (SF), red pulp (RP) and PFZ, are described here. Normal spleen sections
displayed focal granular interstitial HNE-IR in the RP (fig. 2E,F). As described
57

Oxidative stress markers in AA amyloidosis
previously, the normal spleen sections lacked R-AA-IR, indicating lack of SAA or AA
fibril deposition [9].
At 5 days and 1 wk p.i., spleen sections showed variable HNE-IR, ranging from
light to relatively intense, confined to both the splenic interstitium and the RE cells in the
RP (fig. 20). In high power view, HNE-IR monocytoid cells appeared to abut on the
follicle indicating HNE-IR in the PFZ ceUs (fig. 2H). Occasionally, follicular central
arteriolar endothelium showed intense HNE-IR (fig. 21).
At 2 weeks p.i., the RP had expanded and the PFZ sinus walls were infiltrated
with thioflavin S positive AA fibrils (fig. 2J). Large c1usters of RE ceUs in the RP showed
strong HNE-IR, adjacent to amyloidotic PFZ (fig. 2K). In addition, lumens of both RP
and PFZ sinuses contained diffuse HNE-IR that was also localized to the inner margin of
the infiltrated AA-fibril deposits (fig. 2L,M).
Between 3 and 12 wk p.i., the HNE-IR was global throughout the splenic tissues
including the RE ceUs in the foUicles; HNE-IR to the inner margin of AA-fibrils in the
sinusoids persisted (fig. 2N). By 12 wk p.i., massive infiltration of AA fibrils to both the
RP and the PFZ had diminished the ceU population in the splenic parenchyma; this led to
an overaU diminution ofHNE-IR.
lmmunohistochemicalloca/ization ofCML in peritoneal mc]J
In previous studies, concomitant HNE-, and CML-IR have been localized to
amyloid deposits in patients with diverse amyloidoses [25-27]. Finding of robust HNE-IR
in both peritoneal m<D and splenic tissues led us to examine the incidence of CML.
58

Oxidative stress markers in AA amyloidosis
Figure 3 shows the profile ofm<D with CML-IR; less than 15% of normal m<D
were positive. At 1 wk p.i., there was a ~5-fold increase in the number of m<D with
CML-IR; up to 80% of the cells were positive. Plasma membrane CML-IR \Vas
particularly high (fig. 3, fig. 4A). Notably, at this time period, the number ofm<D with
CML-IR was ~ 3-fold greater than that with HNE-IR.
Between 2 and 6 wk p.i., the number of m<D with CML-IR increased to 90%; up
to 60% of the cells had punctate cytoplasmic CML-IR (fig. 3, fig. 4B). At 8 wk p.L the
number of positive cells decreased to 60% and the majority of such cells displayed
multiple vesicular cytoplasmic CML-IR (fig. 4C). At 12 wk p.i., only~· 30% of cells
displayed CML-IR (fig. 3). Although, with time, the number ofm<D with HNE-IR and
CML-IR had increased, their respective IR pattern differed to sorne extent; CML-IR as
compared to HNE-IR was rarely localized to the nuclear membrane (compare fig. 20 to
fig.4C).
Immunohistochemicallocalization ofCML in spleen sections
Both the pattern and incidence of CML-IR in the splenic tissues were similar to
those described for HNE-IR. Briefly, the normal spleen sections displayed both
interstitial and intracellular CML-IR in the RP (fig. 4D); this reactivity increased at 5
days and sorne ofthe positive RE cells appeared to encroach onto the PFZ (Fig. 4E).
At 2 wk p.i., CML-IR was localized to AA fibril deposits in both the PFZ and the
RP (fig. 4F). Topographically, CML-IR was similar to that ofHNE-IR, that is CML
bound to the inner margin of AA fibrils infiltrating the PFZ sinus walls (fig. 4G,H). The
lumens of such sinuses were devoid ofleukocytes but contained diffuse CML-IR,
59

Oxidative stress markers in AA amyloidosis
indicating release ofthis product by the dying/dead endothelial cells. Between 3 and 12
wk p.i., as indicated previously, AA fibril deposition increased progressively in the
splenic tissue but without any manifest differences in CML-IR (fig. 41). Notably, at these
time periods, both HNE-IR and CML-IR were replica of each other except that the
former appeared segmental while the latter was diffuse and uniform (fig. 2N, 41). Figures
41 and K show co-localization of CML-IR to thiof1avin f1uorescent AA fibrils.
60

Oxidative stress markers in AA amyloidosis
DISCUSSION
The principal findings, based on immunohistochemical studies, were that both
splenic RE cells and peritoneal m<D from mice undergoing AA amyloidosis generated
significantly increased levels of HNE and CML adducts. In addition, each of these
adducts were immunolocalized to the splenie interstitium, eapillary endothelium and the
splenic AA fibril deposits.
Normal m<D/RE cells showed weak HNE-IR and CML-IR but as early as 1 wk
p.i., similar to that ofHO-1 response in this mouse model [9], immunoreactivity against
both the se markers increased remarkably in the monocytoid ceUs. These events, at least
spatio-temporally, appeared to correspond with m<D/RE cell-mediated clearance of
humoral SAA during which humoral SAA is endocytosed, trafficked to lysosomes where
it is either degraded completely or partially, yielding amyloidogenic SAA fragments
[10,30,32,33]. At this point, we do not know whether SAA per se or other mediators of
inflammation triggered the oxidative stress response in the monocytoid cells. In a
previous study, interaction of SAA/ AA fibrils with monocytoid cells, both in vivo and in
vitro, was associated with oxidative stress response [5]. In this context, it is important to
note that marked leukocytosis and chronie inflammation eharacterize AHC infection both
in humans and miee [34,35]; sera from mice undergoing AA amyloidosis show marked
elevation in serum amyloid A, interferon gamma, tumor necrosis tàetor alpha and
granulocyte-macrophage colony stimulating factor (unpublished).
With time and coincident with tissue AA fibril deposition, both HNE-IR and
CML-IR increased in the tissue monoeytoid cells (fig. 1, 2B-D,G-M, 3 and 4A-C,F-I).
61

Oxidative stress markers in AA amyloidosis
However, between 8 and 12 wk p.i., there was a marked numerical reduction in peritoneal
m<l> with HNE-, and CML-IR (fig. 1, fig. 3), while the remaining positive cells displayed
multiple HNE-, and CML-IR loci in the cytoplasmic compartment but only HNE-IR at
the nuclear membrane (fig. 2C,D, fig. 4C). HNE, an amphipatic molecule and a potent
electrophile, is known to enter aU cellular structures without encountering biological
barriers and react with both proteins and DNA [36]. As such, we were able to show HNE
IR at both the plasma and nuclear membranes, in the cytoplasmic compartment and
interstitially bound to the AA fibrils. As to the reason for the precipitous decrease in the
number ofHNE-, and CML-IR positive cells between 8 and 12 wk p.i., we suggest that it
may be due to the ongoing intense int1ammatory reaction and continuo us int1ux of
inflammatory infiltrates at the sites of infection [35,37,38].
Of interest is the finding that both HNE-, and CML-IR were localized to the
endothelium of the central arteriole starting at 5 days p.i., and onwards (fig. 21, CML not
shown). Similar IR was seen in the walls of splenic sinuses infiltrated with AA fibrils
(fig. 2L,M, fig. 4H,I). We suggest that co-Iocalization ofHNE-, and CML-IR at the
endothelium may be related to its dysfunction or degeneration. We have shown flattening
ofhigh endothelial ceUs in the lymphoreticular tissues of the AHC-mice coincident with
marked lymphadenitis, splenomegaly and depletion of T ceUs from the thymus-dependent
areas [31,39]. As to similar changes seen in the endothelium of splenic sinus walls it is
quite likely that they might be amyloid related events. A solid layer of AA fibrils abutting
on the strongly HNE-, and CML-IR endothelium suggests such a possibility (fig. 2L,M,
fig. 4H,I). Indeed, Rocken et al. were able to show a similar pattern of CML-IR in
62

Oxidative stress markcrs in AA amyloidosis
endothelial cells adjacent to AA amyloid deposits found in human and murine kidney.
spleen and liver sections [21]. Furthermore, Yan et al. showed much the same
relationship between oxidative stress response and AA fibrils in murine and human
amyloidotic tissue [5]. They showed both SAA and AA fibrils, by interacting through the
receptor for advanced glycation end products (RAGE), up-reguJated the expression of
HO-l, interleukin-6 and macrophage colony stimulating factor in monocytoid cells [5].
Recently, Miyata et al. coined the term 'carbonyl stress' in relation to P2-
microglobulin amyloid fibril formation and suggested a possible role of increased
accumulation of carbonyl compounds in inducing prote in modification by glycoxidation
and/or by lipid peroxidation reactions [25,26]. As such, simiJar to our findings in the
AHC-mouse model, Myata et al. co-localized both HNE-, and CML-IR to the amyloid
fibrils [25,26]. Evidence shows that in vivo, CML is not only a product of glycoxidation
but also oflipid peroxidation; co-Iocalization ofHNE-, and CML-IR in amyloidotic
tissues manifesting oxidative stress response would be consistent with their common
metabolic origin [15,40]. Thus, the observed elevation in CML-IR in our mouse mode!
undergoing AA fibril deposition could reflect increased lipid peroxidation.
One consequence of prote in oxidation, inc1uding that of glycoxidationllipid
peroxidation, is the formation of intra-, and inter-molecular prote in cross-linking that
confer protease resistance and extreme insolubility to the modified protein [28.29,41,42].
AGE accumulation has been documented in AD [23,24,27] and AGE-modified Ap
protein has been shown to accelerate in vitro the aggregation of soluble Ap peptide into
fibrils [43]. HNE can form Michael adducts with compounds bearing thiol groups (Cys)
63

Oxidative stress markers in AA amyloidosis
and amino groups (His, Lys) at position C3 and can undergo Schiffbase condensation
with Lys residues at the CI carbonyl [15]. When both these reactions occur within one
molecule ofHNE, protein cross-linking results. HNE, generated intracellularly. was
implicated in the inactivation of lysosomal cathepsins via cross-linking and causing
inactivationlfunctional modification [44]. At this stage, our data do not allow us to
directly implicate oxidative stress in amyloid formation. Given that lipid
peroxidationlglycoxidation modify proteins, it is conceivable that such events could
potentially affect m<l>/RE cell-associated SAA rendering it resistant to lysosomal
degradation and prone to AA fibril formation [14].
In sum, with time the tissue IR ofboth HNE and CML intensified and
corresponded with the progression of AA fibril deposition. Our results reinforce the
notion that oxidative stress is an integral component of amyloidotic tissues regardless of
the chemical nature of amyloid deposits. Further investigation is needed to understand the
relationship between oxidative stress-derived factors, defective intracellular clearance of
SAA and nascent AA fibril formation.
64

Oxidative stress markers in AA amyloidosis
ACKNOWLEDGEMENTS
This investigation was supported by a research grant (MOP-42526) t'rom the
Canadian Institutes of Health Research.
65

ABREVIATIONS
AA - amyloid A
AGE - advanced glycation end product
AHC - alveolar hydatid cyst
BS - blocking solution
CML - NE-(carboxymethyl)lysine
HNE - 4-hydroxy-2-nonenal
HO-l - heme-oxygenase-l
IR - immunoreactive/immunoreactivity
M <D - macrophage
PBS - phosphate buffered saline
PFZ - perifollicular zone
SAA - serum amyloid A
SF - splenic follicle
R-AA - rabbit anti-AA amyloid
R-CML - rabbit anti-carboxymethyllysine
RE - reticuloendothelial
R-HNE - rabbit anti-4-hydroxy-2-nonenal
ROR - reactive oxygen radicals
RP - red pulp
Oxidative stress markers in AA amyloidosis
66

Oxidative stress markers in AA amyloidosis
REFERENCES
[1] Glenner, G.G. Amyloid deposits and amyloidosis. The beta-fibrillogenesis. N Engl J
Med. 302:1283-1292,1333-1343; 1980.
[2] Benson, M.D. Amyloidosis. In: Seriver, C.R.; Beaudet A.L.; Sly, W.S.; Valle. D.V ..
eds. The metabalic basis afinherited disease. New York: MeGraw-Hill; 1995:4159-4191.
[3] Ali-Khan, Z.; Li, W.; Chan, S.L. Animal mode! for the pathogenesis ofreaetive
amyloidosis. Parasitai Taday. 12:297-302; 1996.
[4] Schubert, D.; Behl, c.; Lesley, R.; Braek, A.; Darguseh, R.; Sagara, Y.; Kimura. H.
Amyloid peptides are toxie via common oxidative mechanism. Prac Nat! Acad S'ci USA.
92: 1989-93; 1995.
[5] Yan, S.D.; Zhu, H.; Zhu, A.; Golabek, A.; Du, H.; Roher, A.; Yu. J.; Soto, C.;
Schmidt, A.M.; Stem, D.; Kindy, M.S. Receptor-dependent cell stress and amyloid
accumulation in systemic amyloidosis. Nature. 6:643-651; 2000.
[6] Ali-Khan, Z.; Jothy, S.; Alkarmi, T.O. Murine alveolar hydatidosis: A potential
experimental mode! for the study of AA amyloidosis. Br J Exp Pathal. 64:599-611; 1983.
[7] Maury, c.P.J.; Titinen, S.; Laiho, K.; Kaarela, K.; Liljestrom, M. Raised circulating
interleukin-18 levels in reaetive AA amyloidosis. Amylaid: J Prote in Folding Disorcl
9:141-144; 2002.
[8] Ando, Y.; Nyhlin, N.; Suhr, O.; Holmgren, G.; Uchida, K.; El Sahly, M.; Yamashita,
T.; Terasaki, H; Nakamura, M.; Uchino, M.; Ando, M. Oxidative stress is found in
amyloid deposits in systemic amyloidosis. Biochem Biophys Res Commun. 232:497-502;
1997.
67

Oxidative stress markers in AA amyloidosis
[9] Kamalvand, G.; Pinard, G.; Ali-Khan, Z. Heme-oxygenase-l response. a marker of
oxidative stress, in a mouse model of AA amyloidosis. Amyloid: In! J Exp CUn Invest.
10:151-159; 2003.
[10] Chan, S. L.; Chronopoulos, S.; Murray, J.; Laird, D.W.; Ali-Khan, Z. Selective
localization of murine ApoSAA 1 /SAA2 in endosomes-lysosomes of activated
macrophages and their degradation products. Amyloid: Inl J Exp Clin Invesl. 4:40-48;
1997.
[Il] Bell, A. W.; Chan, S.L.; Ali-Khan, Z. N-terminal sequence analysis ofSAA
derivatives purified from murine inflammatory macrophages. Amyloid: Inl.J Exp Clin
Invest. 6:31-36; 1999.
[12] Kluve-Beckerman, B.; Liepnieks, J.1.; Wang. L.; Benson, M.D. A cell culture
system for the study of amyloid pathogenesis: amyloid formation by peritoneal
macrophages cultured with recombinant serum amyloid A. Am .J Pathol. 155: 123-133;
1999.
[13] Kluve-Beckerman, B.; Manaloor, J.1.; Liepnieks, J.1. A pulse-chase study tracking
the conversion of macrophage-endocytosed serum amyloid A into extracellular amyloid.
Arthritis Rheum. 46: 1905-1913; 2002.
[14] Ali-Khan, Z. Searching for an in vivo site for nascent amyloid fibril formation . .J
Alzheimers Dis. 81:105-114; 2002.
[15] Sayre, L.M.; Smith, M.A.; Perry, G. Chemistry and biochemistry of oxidative stress
in neurodegenerative disease. Curr Med Chem. 8:721-738; 2001.
68

Oxidative stress markers in AA amyloidosis
[16] Esterbauer, H.; Schaur, R.G.; Zollner, H. Chemistry and biochemistry of 4-
hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 11 :81-128;
1991.
[17] Miyata, T.; Wada, Y.; Cai, Z.; Iida, Y.; Horie, K.; Yasuda, Y.; Maeda, K; Kurokawa,
K; van Ypersele de Strihou, C. Implication of an increased oxidative stress in the
formation of advanced glycation end products in patients with end-stage rcnal failure.
Kidneylnt. 51:1170-1181; 1997.
[18] Castellani, R.; Perry, G.; Siedlak, S.; Numomura, A.; Shimohama, S.; Zhang, J.:
Montine, T.; Sayre, L.; Smith, M. Hydroxynonenal adducts indicate a role for lipid
peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci Lell.
319:25-28; 2002.
[19] Ando, Y.; Brannstrom, T.; Uchida, K.; Nyhlin, N.; Nasman, B.; Suhr, O.:
Yamashita, T.; Olsson, T.; El Salhy, M.; Uchino, M.; Ando, M. Histochemical detection
of 4-hydroxynonenal protein in Alzheimer's amyloid. J Neurologie Sei. 156: 172-176;
1998.
[20] Takeda, A.; Smith, M.A.; Avila, J.; Nunomura, A.; Siedlak, S.L.; Zhu, X.; Perry, G.;
Sayre, L.M. In Alzheimer's disease, he me oxygenase is coincident with Alz50, an
epitope of T induced by 4-hydroxy-2-nonenal. J Neurochem. 75: 1234-1241; 2000.
[21] Rocken, C; Kientsch-Engel, R.; Mansfeld, S.; Stix, B.; Stubenrauch, K.; Weigle, B.;
Buling, F.; Schwan, M.; Saeger, W. Advanced glycation end products and receptor for
advanced glycation end products in AA amyloidosis. Am J Pathol. 162: 1213-1220; 2003.
69

Oxidative stress markers in AA amyloidosis
[22] Uesugi, N.; Sakata, N.; Nagai, R.; Jono, T.; Horiuchi, S.; Takebayashi, S.
Glycoxidative modification of AA amyloid deposits in renal tissue. Nephrol Dial
Transplant. 15:355-365; 2000.
[23] Smith, M.A.; Taneda, S.; Richey, P.L.; Miyata, S.; Yan, S.D.; Stern, D.; Sayre,
L.M.; Monnier, V.N.; Perry, G. Advanced Maillard reaction end products arc associated
with Alzheimer disease pathology. Proe Nat! Acad Sei USA. 91:571 0-5714; 1994.
[24] Castellani, RJ.; Harris, P.L.R.; Sayre, L.M.; Fujii, J.; Taniguchi, N.; Vitek, M.P.;
Founds, H.; Atwood, C.S.; Perry, G.; Smith, M.A. Active glycation in neurofibrillary
pathology of Alzheimer's disease: Nf.-(carboxymethyl)lysine and hexitol-lysine. Free
Radie Biol Med. 31:175-180; 2001.
[25] Miyata, T.; van Ypersele de Strihou, C.; Kurokawa, K.; Baynes, J.W. Alterations in
non-enzymatic biochemistry in uremia: Origin and significance of 'carbonyl stress' in
long-term uremic complications. Kidney Int. 55:389-399; 1999.
[26] Miyata, T.; Ueda, Y.; Saito, A.; Kurokawa, K. 'Carbonyl stress' and dialysis-related
amyloid. Nephrol Dia! Tramp!ant. 15[Suppll]:25-28; 2000.
[27] Dei, R.; Takeda, A; Niwa, H; Li, M.; Nakagomi, Y.; Watanabe, M.; lnagaki, T;
Washimi, Y.; Yasuda, Y.; Horie, K.; Miyata, T.; Sobue, G. Lipid peroxidation and
advanced glycation end products in the brain in normal aging and in Alzheimer's disease.
Acta Neuropathol. 104: 113-122; 2002.
[28] Grune, T.; Merker, K.; Sandig, G.; Davies, K.J. Selective degradation of oxidatively
modified protein substrates by the proteasome. Biochem Biophys Res Commun. 305:709-
718; 2003.
70

Oxidative stress markers in AA amyloidosis
[29] Grune, T.; Davies, K.1. The proteosomal system and HNE-modified protcins. Mol
Aspects Med. 24: 195-204; 2003.
[30] Chronopoulos, S.; Lembo, P.; Alizadeh-KhiavÏ, K.; Ali-Khan, Z. Ubiquitin: its
potential significance in murine AA amyloidogenesis. J Patho/. 167:249-259; 1992.
[31] Ali-Khan, Z. Cellular changes in the lymphoreticular tissue of C57L1 J mice infected
with Echinococcus multi/ocularis cysts. Immunology. 34:831-839; 1978.
[32] Chronopoulos, S.; Laird, D. W.; Ali-Khan, Z. Immunolocalization of serum amyloid
A and AA amyloid in lysosomes in murine monocytoid cells: confocal and immunogold
electron microscopic studies. J Patho/. 173:361-369; 1994.
[33] Kluve-Beckerman, B.; Yamada, T.; Hardwick, J., Liepnieks, J.1.: Benson, M.D.
DifferentiaI plasma membrane clearance ofmurine acute-phase serum amyloid A
proteins SAA] and SAA2• Biochem J. 332:663-669; 1997.
[34] Ali-Khan, Z.; Rausch, R.L. Demonstration of amyloid and immune complex
deposits in renal and hepatic parenchyma of Alaskan alveolar hydatid disease patients.
Ann Trop Med Parasitol. 81:381-392; 1987.
[35] Du, T.; Ali-Khan, Z. Pathogenesis of secondary amyloidosis in an alveolar hydatid
cyst-mouse model: histopathology and immuno/enzyme-histochemical analysis of splenic
marginal zones during amyloidogenesis. J Exp Pathol. 71:313-335; 1990.
[36] Yamamoto, T.; Shibata, N.; Muramatsu, F.; Sakayori, N.; Kobayashi, M. Oxidative
stress in the human fetal brain: an immunohistochemical study. Pediatr Neurol. 26: 116-
122; 2002.
71

Oxidative stress markers in AA amyloidosis
[37] Devouge, M.; Ali-Khan, Z. Intraperitoneal murine alveolar hydatidosis: relationship
between the size of the larval cyst mass, immigrant inflammatory cells, splenomegaly and
thymus involution. Tropenmed Parasit. 34: 15-20; 1983.
[38] Treves, S.; Ali-Khan, Z. Characterization of the inflammatory cells in progressing
tumor-like alveolar hydatid cysts: 1. kinetics and composition of inflammatory infiltrates.
Tropenmed Parasit. 35: 183-188; 1984.
[39] Ali-Khan, Z.; Sipe, J.O.; Du, T.; Riml, H. Echinoeoccus multilocularis: Relationship
between persistent inflammation, serum amyloid A protein response and amyloidosis in
four mouse strains. Exp Parasitol. 67:334-345; 1988.
[40] Fu, M.; Requena, J.R.; Jenkins, A.J.; Lyons, T.J.; Baynes, J.W.; Thorpe, S.R. The
Advanced glycation end product, NE-(Carboxymethyl)lysine, is a product ofboth lipid
peroxidation and glycoxidation reactions . .J Biol Chem. 271:9982-9986; 1996.
[41] Okada, K.; Wangpoengrakul, C.; Osawa, T.; Toyokunit, S.; Tanaka, K.; Uchida, K.
4-hydroxy-2-nonenal impairrnent of intracellular proteolysis during oxidative stress . .J
Biol Chem. 274:23787-23793; 1999.
[42] Markesbery, W.R. Oxidative stress hypothesis in Alzheimer's disease. Free Radie
Biol Med. 23: 134-147; 1997.
[43] Vitek, M.P.; Bhattacharya, K.; Glendening, M.; Stopa, E.; Vlassara, H.; Bucala, R.;
Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in
Alzheimer's disease. Proc Nat! Acad Sci USA. 91:4766-4770; 1994.
72

Oxidative stress markers in AA amyloidosis
[44] Crabb, J.W.; O'Neil, J.; Miyagi, M.; West, K.; Hoff, H.F. Hydroxynonenal
inactivates cathepsin B by forming Michael adducts with active site residues. Prolein Sci.
Il: 831-840; 2002.
73
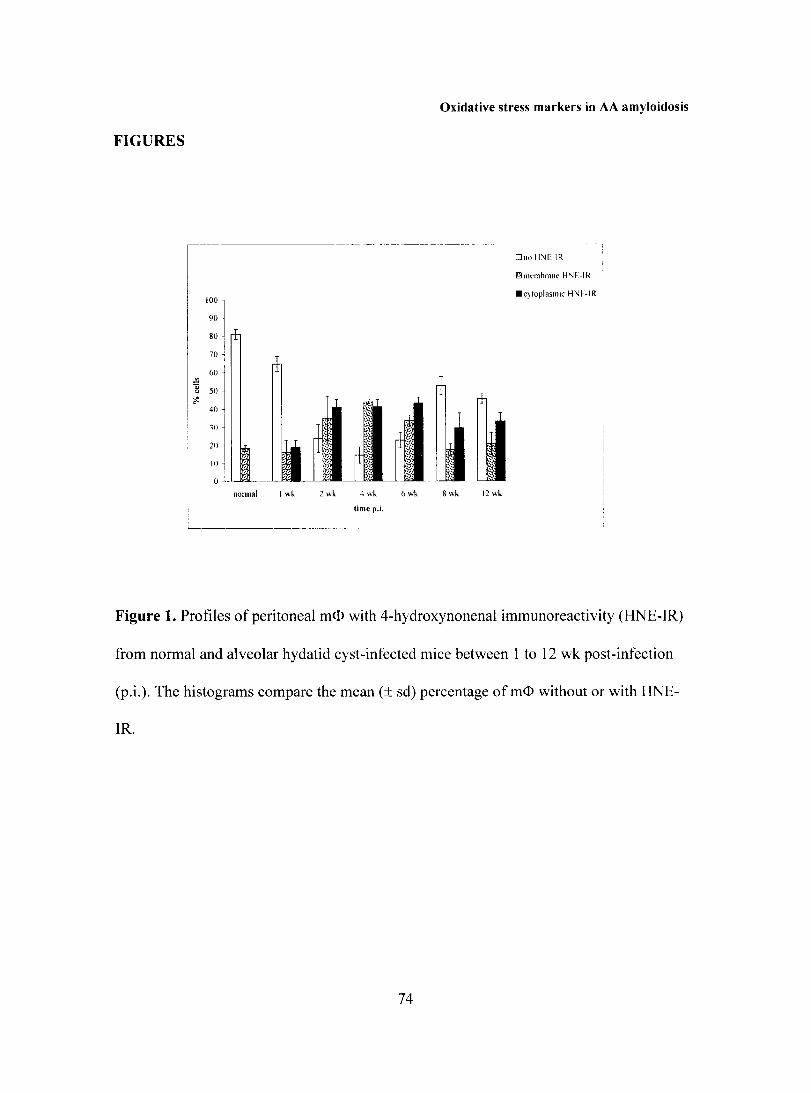
Oxidative stress markers in AA amyloidosis
FIGURES
l '00
1 90 80
Dno Il''E-IR
rn Illcmhrane H'JE-IR.
.c}!oplasmic H:-'E-IR
1 70
60 ~
] 50 ~
40
30
20
10
0
nonnal 1 wh
l timep.i.
Figure 1. Profiles of peritoneal m<D with 4-hydroxynonenal immunoreactivity (HNE-IR)
from normal and alveolar hydatid cyst-infected mice between 1 to 12 wk post-infection
(p.i.). The histograms compare the mean (± sd) percentage of m<D without or with HNE-
IR.
74

• • • .... , .•. ~~ . . '
Oxidative stress markers in AA amyloidosis
Figure 2. Peritoneal macrophages (m<D; A-D) and spleen sections (E-N) from normal and
alveolar hydatid cyst-infected mi ce immunostained with antiserum to 4-hydroxynonenal
(HNE). (A) Normal m<D display residual plasma membrane HNE-immunoreactivity (IR).
(B) At 1 wk post-infection (p.i.), m<l> display focal cytoplasmic HNE-IR. (C,D) At 12 wk
75

Oxidative stress markers in AA amyloidosis
p.i., m<D display HNE-IR at the plasma and nuclear membranes and at multiple cytosolic
sites. (E,F) Spleen sections from a normal mouse; both low power (E) and high power (F)
views showing HNE-IR confined to the red pulp (RP); note both interstitial and
intracellular IR in the reticulendothelial (RE) ceUs. (G-I) At 5 days p.i., note global view
ofHNE-IR in the spleen (G), in the PFZ (H), and in central arteriolar endothelium (1). (1-
M) At 2 wk p.i., note thioflavin S positive AA fibrils in PFZ and RP sinuses (1). a cluster
ofHNE-IR positive RE ceUs in the RP (K), and a double-labeled PFZ sinus showing
segmental HNE-IR (L) co-Iocalized to the inner margin ofthioflavin S positive AA
fibrils (M). (N) At 3 wk p.i., a global view showing segmental pattern of HNE-IR to the
amyloidotic sinus waUs in the PFZ and RP; note lack ofIR in the splenic follicle (SF).
Original magnification: E,G,J,N 16x; B,H,L,M 40x; A,C.I,K 60x; O,F 100x.
76
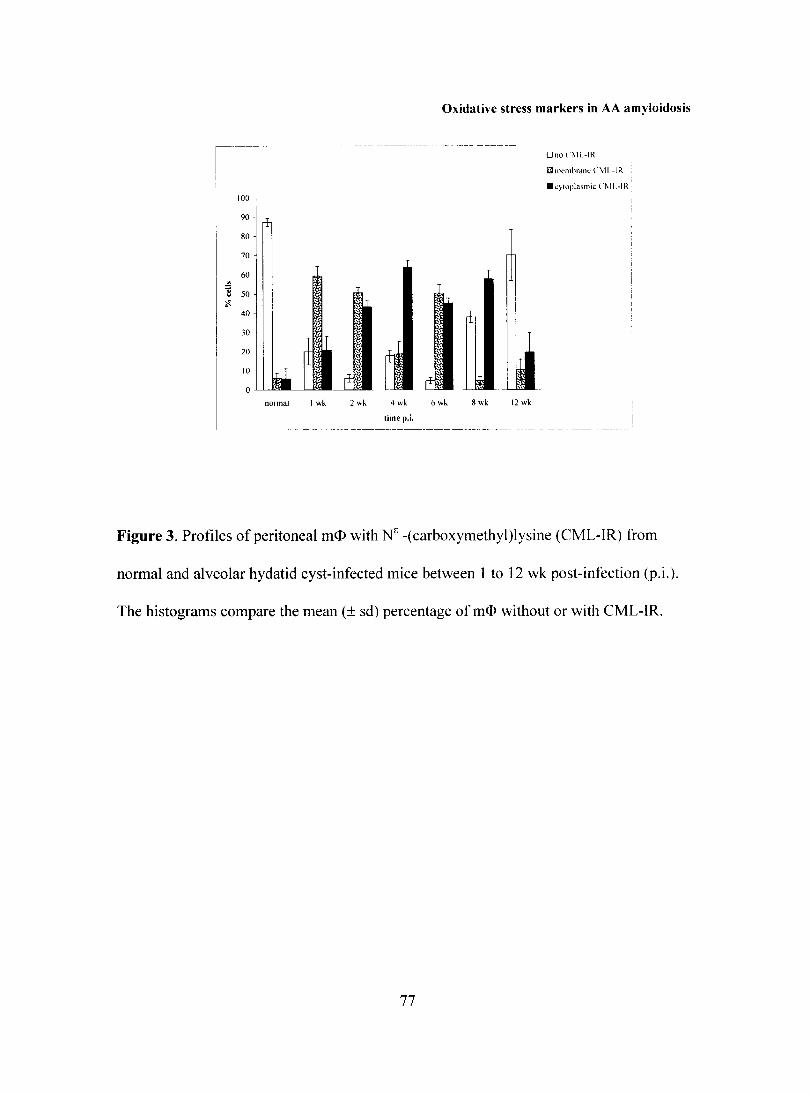
100
90
80
70
60
40
1
30
20
nOlmal 1 wk 2 wk 4 wk
limep.i.
Oxidative stress markers in AA amyloidosis
6 wk 8 wk 12 wk
OnoOIL-IR
~JI1elllbraflt: Ci\1L -IR
.qtoplasmic (,~lI.-IR'
l l:~--,--"
- .. _-_._------------------~---_. -- --- -- - - --
Figure 3. Profiles of peritoneal m<I> with Ne -(carboxymethyl)lysine (CML-IR) from
normal and alveolar hydatid cyst-infected mice between 1 to 12 wk post-infection (p.i.).
The histograms compare the mean (± sd) percentage ofm<D without or with CML-IR.
77
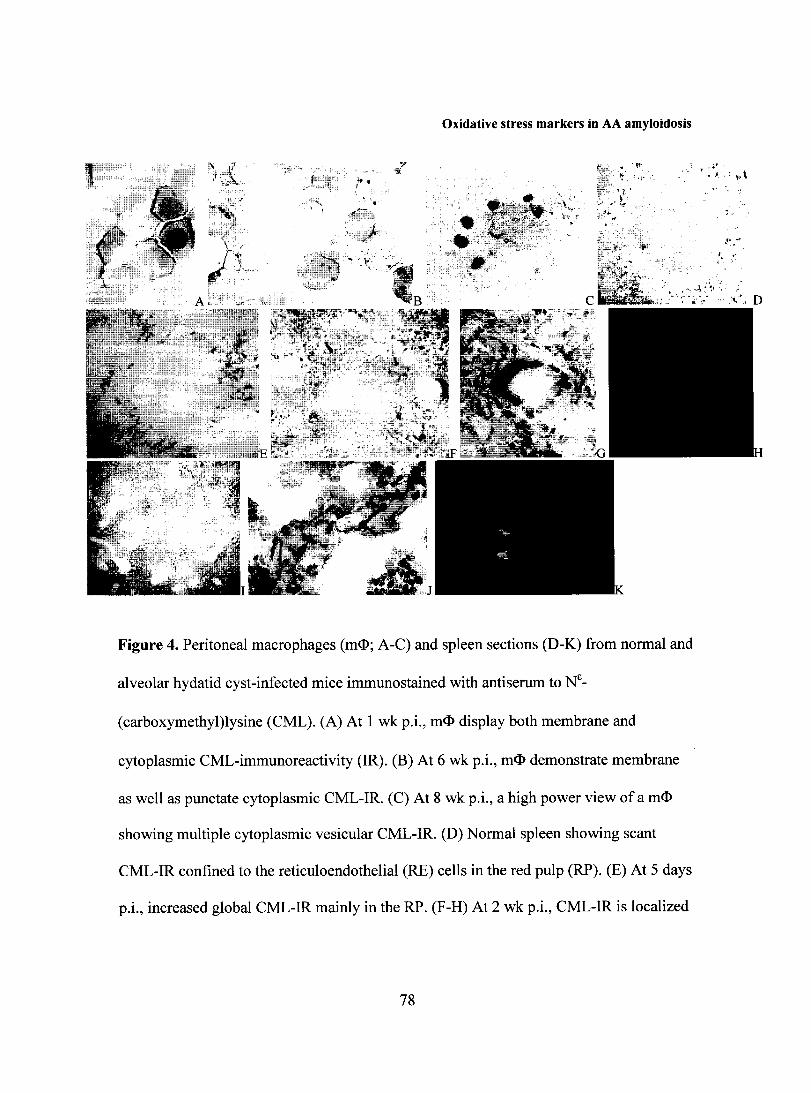
Oxidative stress markers in AA amyloidosis
. . ,~:
f • , r:' ,.; .. , ., ,
~ T~--~ T' --
~<, il", ,.
;".' 1~ .=>J~ ~- - ..,.
Figure 4. Peritoneal macrophages (m<D; A-C) and spleen sections (D-K) from normal and
alveolar hydatid cyst-infected mice immunostained with antiserum to NE_
(carboxymethyl)lysine (CML). (A) At 1 wk p.i., m<l> display both membrane and
cytoplasmic CML-immunoreactivity (IR). (B) At 6 wk p.i., m<l> demonstrate membrane
as well as punctate cytoplasmic CML-IR. (C) At 8 wk p.i., a high power view of a m<D
showing multiple cytoplasmie vesicular CML-IR. (D) Normal spleen showing seant
CML-IR confined to the reticuloendothelial (RE) cells in the red pulp (RP). (E) At 5 days
p.i., inereased global CML-IR mainly in the RP. (F-H) At 2 wk p.i., CML-IR is localized
78

Oxidative stress markcrs in AA amyloidosis
to RE cells in the RP and to AA fibril deposits in the inner margin of PFZ sinus walls (F);
a double-labeled PFZ sinus showing co-localization of CML-IR to the thioflavin S
positive AA fibril deposits (G,H). (1) At 3 wk p.i., note AA fibril-mediated distortion of
splenic architecture and a global view of CML-IR localized to AA fibril infiItrates to the
sinus walls . (J,K) At 12 wk p.i., note uniform CML-IR localized to the margin of
thioflavin S positive AA fibril deposits in the RP. Original magnification: O,E,F,I 16x;
A,B,G,H,J,K 60x; C, 100x.
79

CHAPTER 4. CONCLUDING REMARKS
Chronic inflammation, which is central to the pathogenesis of AA amyloidosis, is
known to instigate OS response. OS-related factors could, in thcir own right cffect the
pathogenesis of AA amyloidosis. Since inflammatory monocytoid cells simultaneously
generate ROR and process SAA, it is plausible that oxidative modification of cell
associated SAA occurs, resulting potentially in its structural alteration and AA fibril
formation. This study represents the groundwork for this hypothesis by providing
unequivocal evidence of OS response in the AHC-infected mouse model of AA
amyloidosis.
This work characterized the immunohistochemical distribution of OS-related
factors generated in vivo in AHC-infected spleenlliver sections and peritoneal m<D. HO-l,
an antioxidant enzyme whose expression implies both inflammation and OS, was
localized to splenic/hepatic SAA deposits, regardless of AA fibril formation. SAA was
not the primary trigger of the observed HO-l response as evidenced by in vitro
experiments. Other groups however, have shown that SAA, through activation of RAGE,
can activate the NFKB pathway, which in tum upregulates HO-l expression (Yan).
A significant increase in levels of HNE, a lipid peroxidation adduct, and CML, an
advanced glycation end-product (AGE), in AHC-mouse derived splenic RE cells and
peritoneal m<D, known to sequester SAA, was demonstrated. Interestingly, HNE and
CML immunoreactivities in peritoneal m<D were demonstrated within cytoplasmic
vesicles, an observation reminiscent of SAA immunoreactivity in lysososmal
compartments of the same cells (Chronopoulos). Furthermore, binding of both HNE and
CML to extracellular AA fibril deposits was evidenced. From these findings, it can only
80

be deduced, and not confirmed, whether SAA or AA fibrils are oxidatively modified
within m<D/RE cells.
Both lipid peroxidation and AGE, through the formation of intra- and inter
molecular protein cross-linkages, have been implicated in protein modification, resistance
to degradation and amyloid fibril formation (Markesbery, Grunne). Thus, defective
catabolism of m<D/RE cell-associated SAA, believed to instigate nascent AA fibril
formation intracellularly, could be associated with oxidation of SAA (Ali-Khan, 2002).
Future work would necessitate structural studies on SAA and oxidized SAA products.
We propose that SAA isolated from AHC-mouse derived m<D/RE cells should be similar
or identical to oxidized recombinant SAA. In the end, the knowledge of the mechanisms
underlying amyloid formation is truly the key in developing compounds capable of
inhibiting fibril formation, in order to retard the progression of amyloid diseases, notably
AD.
81

REFERENCES
Abankwa, G.V.; Ali-Khan, Z. Induction of amyloid enhancing factor and its biological
properties in murine alveolar hydatosis. British Journal of Experimental Pathology.
69: 123-132; 1988.
Ali-Khan, Z. Jothy, S.; Siboo, R. Amyloidosis in experimental murine alveolar
hydatidosis. Trans R Society Tropical Med Hygiene. 76: 1690-1710; 1982.
Ali-Khan, Z.; Jothy, S.; Alkarmi,T. Murine alveolar hydatidosis: A potential
experimental model for the study of AA amyloidosis. British Journal of Experimental
Pathology. 64:599-611; 1983.
Ali-Khan, Z.; Sipe, J.D.; Du, T.; Riml, H. Echinococcus multilocularis: Relationship
between persistent inflammation, serum amyloid A protein response and amyloidosis in
four mouse strains. Experimental Parasitology. 67:334-345; 1988.
Ali-Khan, Z.; Li, W.; Chan, S.L. Animal model for the pathogenesis of reactive
amyloidosis. Parasitology Today. 12:297-302; 1996.
Ali-Khan, Z. Searching for an in vivo site for nascent amyloid fibril formation. Journal
of Alzheimer 's Disease. 4: 1 05-114; 2002.
82

Alkarmi, T.O.; Ali-Khan, Z. Chronic alveolar hydatidosis and secondary amyloidosis:
pathological aspects of the disease in four strains of mice. British Journal of
Experimental Pathology. 65:405-417; 1984.
Alkarmi, T.O.; Ali-Khan, Z.; Zarkada, C.G. Characterization of amyloid protein from
mice infected with alveolar hydatid cyst: Isolation, purification and amino acid
composition. Experimental Mo/ecular Pathology. 45: 142-159; 1986.
Ando, Y.; Nyhlin, N.; Suhr, O.; Holmgren, G.; Uchida, K.; El Sahly, M.; Yamashita,
T.; Terasaki, H.; Nakamura, M.; Uchino, M.; Ando, M. Oxidative stress is found in
amyloid deposits in systemic amyloidosis. Biochemical and Biophysical Research
Communication. 232:497-502; 1997.
Axelrad, M.A. Kisilevski, R.; Willmer, J.; Chen, S.J.; Skinner, M. Further
characterization of amyloid enhancing factor. Laboratory Investigation. 47: 139-146;
1982.
Bell, A. W.; Chan, S.L.; Marcantonio, D.; Ali-Khan, Z. Both murine SAA 1 and SAA2
yield AA amyloid in alveolar hydatid cyst-infected mice. Scandinavian Journal of
Immunology. 43: 173-180; 1996.
83

Bell, A.W.; Chan, S.L.; Ali-Khan, Z. N-terminal sequence analysis of SAA-derivatives
purified from murine inflammatory macrophages. Amyloid: Journal of Protein ràlding
Disorders. 6:31-36; 1999.
Bellotti, V.; Merlini, G. CUITent concepts on the pathogenesis of systemic amyloidosis.
Nephrology Dialysis Transplantation. 11(Suppl 9):53-62; 1996.
Benson, M.D.; Kleiner, E. Synthesis and secretion of serum amyloid prote in A (SAA)
by hepatocytes in mice treated with casein. The Journal oflmmunology. 124:495-499;
1980.
Benson, M.D. Amyloidosis. In: Scriver, C.R.; 8eaudet, A.L.; Sly, W.S.; Valle, D.V., eds.
The Metabolic Rasis of Inherited Disease. New York: McGraw-Hill. 4159-4191; 1995.
Berlett, B.S.; Stadtman, E.R. Prote in oxidation in aging, disease, and oxidative stress.
Journal of Biological Chemistry. 272:20313-20316; 1997.
de Beer, M.C.; de Beer, F.C.; McCubbin, W.D.; Kay, C.M.; Kindy, M.S. Stuctural
prerequisites for serum amyloid A fibril formation. The Journal q( Biological ChemiSl1:V.
268:20606-20612; 1993.
84

Chronopoulos, s.; Laird, D.W.; Ali-Khan, Z. Immunolocalization of serum amyloid A
and AA amyloid in lysosomes in murine monocytoid cells: confocal and immunogold
electron microscopie studies. Journal of Pathology. 173:361-369; 1994.
Du, T.; Ali-Khan, Z. Pathogenesis of secondary amyloidosis in an alveolar hydatid cyst
mouse model: histopathology and immuno/enzyme-histochemical analysis of splcnic
marginal zone ceUs during amyloidogenesis. Journal of Experimental Pathology. 71 :313-
335; 1990.
Dyrks, T.; Dyrks, E.; Hartmann, T.; Masters, C.; Beyreuther, K. Amyloidogenicity
ofbeta A4 and beta A4-bearing amyloid protein precursor fragments by metal-catalyzed
oxidation. Journal of Biological Chemistry. 267: 1821 0-18217; 1992.
Falk, R.H.; Comenzo, R.L.; Skinner, M. The systemic amyloidoses. The l'leM' England
Journal of Medicine. 337:898-909; 1997.
Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to
inflammation. New England Journal of Medicine. 340:448454; 1999.
Ganowiak, K.; HuUman, P.; Engstrom, U.; Gustavsson, A.; Westermark, P. Fibrils
from synthetic amyloid-related peptides enhance development of experimental AA
amyloidosis in mice. Biochemical and Biophysical Research Communication. 199:306-
312; 1994.
85

Glenner, G.G. Amyloid deposits and amyloidosis: The p-fibrilloses. The New England
Journal of Medicine. 302: 1283-1292, 1333-1343; 1980.
Gollaher, c.J.; Bausserman, L.L. Hepatic catabolism of serum amyloid A during an
acute phase response and chronic inflammation. Proceedings of the Society of
Experimental Biology and Medicine. 194:245-250; 1990.
Grune, T.; Reinheckel, T.; Davies, K.J.A. Degradation of oxidized proteins in
mammalian cells. FASEB Journal. 11:526-534; 1997.
Head, E.; Garzon-Rodriguez, W.; Johnson, J.K.; Lott, I.T.; Cotman, c.; Glabe, C.
Oxidation of Ap and plaque biogenesis in Alzheimer's disease and Down syndrome.
Neuobiology of Disease. 8:792-806; 2001.
Hirakura, Y.; Carreras, 1.; Sipe, J.D.; Kagan, B.L. Channel formation by serum
amyloid A: a potential mechanism for amyloid pathogenesis and host defense. Amyloid:
Journal of Pro te in Folding Disorders. 9: 13-23; 2002.
Husby, G. Nomenclature and classification of amyloid and amyloidoses. Journal of'
Internai Medicine. 232:511-512; 1992.
86

Husby, G.; Marhaug, B.; Dowton, K.; Sietten, K.; Sipe, J.O. Serum amyloid A (SAA):
biochemistry, genetics and the pathogenesis of AA amyloidosis. Arnyloid: Journal (~l
Protein Folding Disorders. 1:119-137; 1994.
Janigan, D.T.; Druet, R.L. Experimental amyloidosis: Role of antigenicity and rapid
induction. American Journal of Pathology. 48: 1013-1026; 1966.
Johan, K.; Westermark, G.; Engstrom, U.; Gustavsson, A.; Hultman, P.;
Westermark, P. Acceleration of amyloid prote in A amy1oidosis by amyloid-like
synthetic fibrils. Proceedings of the National Acaderny (if Sciences USA. 95:2558-2563.
Kisilevsky, R. Amy1oidosis: A familiar problem in the Iight of CUITent pathogenetic
developments. Laboratory Investigation. 49:381-390; 1983.
Kisilevsky, R.; Narindrasorasak, S.; Tape, c.; Tan, R.; Boudreau, L. During AA
amyloidogenesis is proteolytic attack on serum amyloid A a pre- or post-fibrillogenic
event? Amyloid: Journal of Prote in Folding Disorders. 1: 174-183; 1994.
Kluve-Beckerman, B.; Yamada, T.; Hardwick, J.; Liepnieks, J.J.; Benson, M.D.
DifferentiaI plasma clearance of murine acute-phase serum amyloid A proteins SAA 1
and SAA2. Biochernical Journal. 332:663-669; 1997.
87

Kluve-Beckerman, B.; Leipnieks, J.J.; Wang, L.; Benson, M.D. A culture system for
the study of amyloid pathogenesis: amyloid formation by peritoneal macrophages
cultured with recombinant serum amyloid A. American Journal of Pathology. 155: 123-
133; 1999.
Lansbury Jr., P.T. Evolution of amyloid: what normal prote in folding may tell us about
fibrillogenesis and disease. Proceedings of the National Academy of Sciences USA.
96:3342-3344; 1999.
Lundmark, K.; Westermark, G.T.; Nystrom, S.; Murphy, c.L.; Solomon, A.;
Westermark, P. Transmissibility of systemic amyloidosis by a prion-like mechanism.
Proceedings of the National Academy of Sciences USA. 99:6979-6984; 2002.
Malle, E.; Bollmann, A.; Steinmetz, A.; Gemsa, D.; Leis, H.-J.; Sattter, W. Serum
amyloid A (SAA) protein enhances formation of cyclooxygenase metabolites of activated
human monocytes. Federation of European Biochemical Societies Lelters. 419:215-219;
1997.
Maines, M.D. Herne oxygenase: function, multiplicity, regulatory mechanisms, and
clinical applications. FASEB Journal. 2:2557-2568; 1988.
Markesbery, W.R. Oxidative stress hypothesis in Alzheimer's disease. Free Radical
Biologyand Medicine. 23:134-147; 1997.
88

Maury, c.P.J.; Tiitinen, S.; Laiho, K.; Kaarela, K.; Liljestrom, M. Raised circulating
interleukin-18 levels in reactive AA amyloidosis. Amyloid.· Journal of Prote in Fofding
Disorders. 9: 141-144; 2002.
Migita, K.; Yamasaki, S.; Shibatomi, K.; Ida, H.; Kita, M., Kawakami, A.; Eguchi,
K. Impaired degradation of serum amyloid A (SAA) prote in by cytokine-stimulated
monocytes. Clinicaf and Experimental Immunology. 123:408-411; 2001.
Pappolla, M.A.; Chyan, Y.-J.; Omar, R.A.; Hsiao, K.; Perry, G.; Smith, M.A.;
Bozner, P. Evidence of oxidative stress and in vivo neurotoxicity of ~-amyloid in a
transgenic mouse model of Alzheimer's disease. American Journal ofPathology.
152:871-877; 1998.
Poss, K.n.; Tonegawa, S. Reduced stress defense in heme-oxygenase-l deficient cells.
Proceedings of the National Academy of Sciences USA. 94: 1 0925-10930; 1997.
Rausch, R.L. Life-cycle patterns and geographic distribution of Echinococcus species.
R.C.A. Thompson, ed. The Biology of Echinococcus and Hydatid Disease. London:
George Allen & Unwin. 44-70; 1986.
Rochet, J.C.; Lansbury Jr., P.T. Amyloid fibrillogenesis: themes and variations.
Current Opinion in Structural Biology. 10:60-68; 2000.
89

Sasaki, N.; Fukatsu, R.; Tsuzuki, K.; Hayashi, Y.; Yoshida, T.; Fujii, N.; Koike, T.;
Wakayama, 1.; Yanagihara, R.; Garruto, R.; Amano, N.; Makita, Z. Advanced
glycation end products in Alzheimer's disease and other neurodegenerative diseases.
American Journal of Pathology. 153: 1149-1155; 1998.
Sayre, L.M.; Zelasko, D.S.; Harris, P.L.R.; Perry,G.; Salomon, R.G.; Smith, M.A.
4-hydroxynonenal-derived advanced lipid peroxidation end products are increased in
Alzheimer's disease. Journal ofNeurochemistry. 68:2092-2097; 1997a.
Sayre, L.M.; Zagorski, M.G.; Surewicz, W.K.; Krafft, G.A.; Perry, G. Mechanisms
of neurotixicity associated with amyloid ~ deposition and the role of free radicais in the
pathogenesis of Alzheimer's disease: a critical appraisal. Chemical Research in
Toxicology. 10:518-526; 1997.
Schweers, O.; Mandelkow, E.M.; Blenart, J.; Mandelkow, E. Oxidation of cysteine-
322 in the repeat domain of microtubule-associated prote in T controis the in vi/ro
assembly ofpaired helical filaments. Proceedings of the National Academy afSciences
USA. 92:8463-8467; 1995.
Shwartsburd, P.M. Self-cytoprotection against stress: feedback regulation of heme
dependent metabolism. Celi Stress & Chaperones. 6:1-5; 2001.
90

Sipe, J.D.; Carreras, 1.; Gonnerman, W.A.; Cathcart, E.S.; de Beer, M.C.; de Beer,
F.C. Characterization ofthe inbred CEIJ mouse strain as amyloid resistant. American
JournalofPathology. 143:1480-1485; 1993.
Sipe, J.D. and Committee. Revised nomenclature for serum amyloid A (SAA). Amyloid:
Journal of Prote in Folding Disorders. 6:67-70; 1999.
Smith, M.A.j Taneda, S.; Richey, P.L. Advanced Maillard reaction products associated
with Alzheimer disease pathology. Proceedings (~{lhe National Academy q{Sciences
USA. 91:5710-5714; 1994a.
Smith, M.A.; Kutty, R.K.; Richey, P.L.; Yan, S.D.; Stern, D.; Chader, G.J.;
Wiggert, B.; Peterson, R.B.; Perry, G. Heme-oxygenase-l is associated with the
neurofibrillary pathology of Alzheimer"s disease. American Journal of Pathology.
145:42-47; 1994b.
Smith, M.A.; Siedlak, S.L.; Richey P.L.; Nagaraj, R.H.; Elhammer, A.; Perry, G.
Quantitative solubilization and analysis of insoluble paired helical filaments from
Alzheimer disease. Brain Research. 717:99-108; 1996.
Stone, M.J. Amyloidosis: A final common pathway for prote in deposition in tissues.
Blood: The Journal of the American Society of Hematology. 75:531-545; 1990.
91

Takeda, A.; Perry, G.; Abraham, N.G. Overexpression of heme-oxygenase-l in
neuronal cells, the possible interaction with tau. Journal of Biological Chemistry.
275:5395-5399; 2000.
Tan, S.Y.; Pepys, M.B. Amyloidosis. Histopathology. 25:403-414; 1994.
Treves, S.; Ali-Khan, Z. Characterization of the inflammatory cells in progressing
tumor-like alveolar hydatid cysts: 1. Kinetics and composition of inflammatory
infiltrates. Tropenmedizin und Parasitologie. 35: 183-188; 1984.
Torgerson, P.R.; Budke, C.M. Echinococcosis - an international public health
challenge. Research in Veterinary Science. 74: 191-202; 2003.
Wegiel, J.; Wang, H.-C.; Imaki, H.; Rubenstein, R.; Wronska, A.; Osuchowski, M.;
Lipniski, W.J.; Walker, L.C.; LeVine H. The role ofmicroglial cells and astrocytes in
fibrillar plaque evolution in transgenic APPsw mice. Neurobilogy ofAging. 22:49-61;
2001.
Weiss, L.P. The spleen; the reticuloendothelial system. Tropical Diseases Research
Series No. 1 : Role of the Spleen in the Immunology of Parasitic Diseases. Geneva:
Schwabe & CO.AG Basel. 7-20; 1978 a.
92

Weiss, L.P. The reticuloendothelial system. Tropical Diseases Research S'eries No. 1 :
Role of the Spleen in the Immunology ofParasitic Diseases. Geneva: Schwabe & CO.AG
Basel. 21-27; 1978 b.
Westermark, P. Classification of amyloid fibril proteins and their precursors: an
ongoing discussion. Amyloid.· Journal of Prote in Folding Disorders. 4:216-218; 1997.
Yamada, T.; Liepnieks, J.J.; Kluve-Beckerman, B.; Benson, M.D. Cathepsin B
generates the most common form of amyloid A (76 residues) as a degradation product
from serum amyloid A. Scandinavian Journal oflmmunology. 41:94-97; 1995.
Yamada, T.; Okuda, Y.; Takasugi, K.; Wang, L.; Marks, D.; Benson, M.D.; Kluve
Beckerman, B. An allele of serum amyloid AI associated with amyloidosis in both
Japanese and Caucasians. Amy/otd: Journal of Protein Folding Disorders. 10:7-11; 2003.
Yan, S.D.; Zhu, H.; Zhu, A.; Golabek, A.; Du, H.; Roher, A.; Yu, J.; Soto, c.;
Schmidt, A.M.; Stern, D.; Kindy, M. Receptor-dependent cell stress and amyloid
accumulation in systemic amyloidosis. Nature. 6:643-651; 2000.
Yu, J.; Zhu, H.; Guo, J.; de Beer, F.C.; Kindy, M.S. Expression ofmouse
apolipoprotein SAAl.l in CE/J mice: isoform-specific effects on amyloidogenesis.
Laboratory Investigation. 80:1797-1806; 2000.
93

Zahedi, K.; Gonnerman, W.A.; de Beer, F.C.; de Beer, M.C.; Steel, D.M .. ; Sipe,
J.D.; Whitehead, A.S. Major acute-phase reactant synthesis during chronic inflammation
in amyloid-susceptible and -resistant mouse strains. Inflammation. 15: 1-14; 1991.
94





























