Embed Size (px)
DESCRIPTION
Émissions de sources fixes Prélèvement et mesurage d'hydrocarbures aromatiques polycycliques à l'émission
Citation preview

© A
FN
OR
200
3 —
Tou
s dr
oits
rés
ervé
sFA124146 ISSN 0335-3931
NF X 43-329Mai 2003
Indice de classement : X 43-329
ICS : 13.040.40
Émissions de sources fixes
Prélèvement et mesurage d'hydrocarbures aromatiques polycycliques à l'émission
E : Stationary source emissions — Sampling and measurement of polycyclic aromatic hydrocarbons and tars at emission
D : Emissionen aus festen Quellen — Probenahme und Messung von polyzyklischen aromatischen Kohlenwassertoffen bei Abgabe
Norme française homologuée par décision du Directeur Général d'AFNOR le 20 avril 2003 pour prendre effetle 20 mai 2003.
Remplace la norme expérimentale XP X 43-329, d’avril 1995.
Correspondance À la date de publication du présent document, il n’existe pas de travaux européensou internationaux traitant du même sujet.
Analyse Le présent document décrit un prélèvement isocinétique et la détermination d’hydro-carbures aromatiques polycycliques (HAP) émis par les sources canalisées. Laméthode s’applique à des effluents gazeux plus ou moins chargés en poussières.
Il est destiné à compléter la série de normes sur la détermination de la concentrationde polluants à l’émission.
Descripteurs Thésaurus International Technique : pollution atmosphérique, air, qualité, émis-sion, effluent gazeux, mesurage, hydrocarbure aromatique, goudron, prélèvementd'échantillon, chromatographie en phase gazeuse, spectrométrie de masse, chro-matographie liquide haute performance, méthode gravimétrique.
Modifications Par rapport au document remplacé, changement de statut.
Corrections
© AFNOR 2003 AFNOR 2003 1er tirage 2003-05-F
Éditée et diffusée par l’Association Française de Normalisation (AFNOR) — 11, avenue Francis de Pressensé — 93571 Saint-Denis La Plaine Cedex Tél. : + 33 (0)1 41 62 80 00 — Fax : + 33 (0)1 49 17 90 00 — www.afnor.fr

Qualité de l'air — Émissions de sources fixes AFNOR X43B
Membres de la commission de normalisation
Président : M PERRET
Secrétariat : MME POTTEVIN — AFNOR
M ARCHENAULT ELYO CYLERGIEM BAILLY MEPACM BARANGER NEREIDES SAM BARRAL ALUMINIUM PECHINEYM BAUJON LHCFM BOEUF SERES SAMME COISY ARELCO ARCMME COL LAB SAMME COMPIANO DIONEXMME COUZINIE ALUMINIUM PECHINEYM DARDIER NORISKO EQUIPEMENTSM DE REYDELLET SAINT GOBAIN ISOVERM DELCHAMBRE OLDHAM FRANCE SAM DEPAUW PROCEDAIR SAM DI RIENZO SICK MAIHAKMME DOCKWILLER TREDIMME FERRE SECAUTOM DZIONSKO ELSAG BAILEY HARTMANN & BRAUNM FIANI ADEME MME GRAND TOTAL FINA ELF FRANCE — CRESM HERAUVILLE VILLE DU HAVRE — LEAM HERMANGE NORISKO EQUIPEMENTSM HUAU DIONEXM JACOB EDF R&DM JANSSENS OLDHAM SAM JULIEN TOTAL FINA ELF FRANCE — CRESMME JUPPEAU ALSESMME KNOCHE STRATENEM LABROSSE SOCIETE ETUDES SECHAUD & METZM LACHENAL LNEMME LAGOUTTE RHODITECHM LEYGUE ECSMME MALLET ETSAM MALNOY SICKM MARIAGE SOCOR SAMME MC CORMACK PECHINEY CRVMME MILHAU ALSTOM POWER BOILERSM MOTSCH ABB AUTOMATIONM MOUGEY CGIAM MOULENE ENVIRONNEMENT SAM NICOL ATOFINA — CRRAMME PAILLIER ADEMEM PELISSIER SAINT GOBAIN RECHERCHEM PERNET AINF SAM PERRET INERISM PEYRICHOU EDF R&DM POULLEAU INERISM PUECH APPAVEMME RAVENTOS INERISM REBER ETSA — LABORATOIRE DE ROUENM REYNAUD CETIATM RICO DION PREVENTION POLLUTIONS RISQUESM RISLER PREFECTURE DE POLICE LABO CENTRALMME ROISSE LECESMME SCHERRER CARSOM TABARIES CNIMM THIEBAUD ATOFINA — CRRAM THYOT AINFM TOURON CONTROLE ET PREVENTIONMME UZZAN BUREAU VERITASM VAUGEOIS CREEDM VICARD STRATENEMLLE VINCENSINI AFNOR CERTIFICATIONM YRIEIX CTBA

— 3 — NF X 43-329
Introduction ......................................................................................................................................................... 4
1 Domaine d'application ....................................................................................................................... 4
2 Références normatives ..................................................................................................................... 5
3 Termes et définitions ......................................................................................................................... 5
4 Principe ............................................................................................................................................... 6
5 Appareillage pour le prélèvement .................................................................................................... 6
6 Prélèvement ....................................................................................................................................... 9
7 Analyse ............................................................................................................................................. 12
8 Expression des résultats ................................................................................................................ 21
9 Vérifications ..................................................................................................................................... 23
10 Rapport d'essai ................................................................................................................................ 24
Annexe A (informative) Quelques caractéristiques des huit HAP dosés ................................................... 26
Annexe B (informative) Caractéristiques de filtres plans utilisables ......................................................... 27
Annexe C (informative) Modes opératoires pour l'essai d'étanchéité ........................................................ 28
Annexe D (informative) Exemple de conditions chromatographiques utilisables en CG/SM .................. 30
Annexe E (informative) Exemple de conditions chromatographiques utilisables en CLHP .................... 31
Annexe F (informative) Purification de l’extrait organique sur microcolonne de silice (méthode CLHP) ............................................................................................................. 32
Annexe G (informative) Ions caractéristiques pour la détection par chromatographie en phase gazeuse/spectrométrie de masse des hydrocarbures aromatiques polycycliques et étalons internes ........................................................................................................................ 33
Annexe H (informative) Valeurs de la pression saturante de la vapeur d’eau ........................................... 34
Bibliographie ..................................................................................................................................................... 35
SommairePage

NF X 43-329 — 4 —
Introduction
a) Avertissement et précautions de sécurité
En raison de la complexité de la méthode, il est souhaitable que les personnels mettant en œuvre ce protocolenormalisé soient particulièrement entraînés, expérimentés et avertis dans le domaine du prélèvement des traces.Il est souhaitable que des installations de préparation des verreries, résines et filtres et de préparation des échan-tillons pour analyse soient dédiées uniquement aux analyses de traces.
L'instabilité physico-chimique des HAP, en présence de O2, NOx, SO2, HCl et de certains métaux lourds d'unepart et en présence de lumière d'autre part, exige que l'opérateur suive scrupuleusement l'ensemble des préco-nisations indiquées dans le présent document lors du prélèvement des échantillons, de leur stockage et de leuranalyse.
b) Manipulation de produits cancérogènes
L'utilisation de HAP certifiés en produits purs ou en mélanges pour préparer des solutions étalons exige quelquesprécautions d'emploi.
Certains HAP étant cancérogènes, éviter autant que possible la manipulation de produits purs solides suscepti-bles de passer dans l'atmosphère, donc d'être inhalés ou fixés sur les surfaces environnantes (utiliser de préfé-rence des solutions étalons prêtes à l'emploi). En cas de manipulation d'HAP à l'état de poudre, il est recommandéde porter un masque facial en coton.
Toute manipulation de produits purs ou en solution nécessite le port de gants de protection : en cas de manipu-lation de HAP à l'état de poudre, il est recommandé d'utiliser des gants à jeter en coton afin d'éviter la dispersionde la poudre par effet électrostatique ; pour les HAP en solution, il est recommandé d'utiliser des gants en latex,à jeter dès contamination, de préférence à des gants en vinyl. Si ceux-ci sont pollués accidentellement par unesolution d'HAP, les changer dès que possible pour éviter un contact avec la peau.
c) Élimination et destruction des déchets
L'élimination de déchets contenant des HAP (solutions, gants, etc.) doit se faire soit par le recours à un organismespécialisé agréé par les pouvoirs publics, soit par destruction chimique effectuée directement au laboratoire.
Dans le deuxième cas, se référer aux méthodes recommandées par le Centre International de Recherche sur leCancer (IARC) décrites dans l'ouvrage suivant : «Laboratory Decontamination and destruction of Carcinogens inLaboratory Wastes — Some Polycyclic Hydrocarbons» — IARC Scientific Publications n° 49 — IARC Lyon 83.
1 Domaine d'application
Le présent document a pour objet la détermination d'hydrocarbures aromatiques polycycliques (HAP) émis parles sources canalisées. Il décrit deux méthodes de prélèvement : la première avec division du débit de prélève-ment, la seconde sans division de débit (voir article 5).
Il décrit en outre deux méthodes d'analyse : la chromatographie en phase liquide haute performance (CLHP) et lachromatographie en phase gazeuse couplée à la spectrométrie de masse (CG/SM).
Les méthodes décrites s'appliquent à des effluents gazeux plus ou moins chargés en poussières.
Parmi les composés de la famille des HAP, les méthodes ont pour objectif de déterminer plus particulièrement lescomposés suivants, réputés représenter la famille des HAP au plan de la cancérogenèse :
— Benzo (a) anthracène ;
— Benzo (k) fluoranthène ;
— Benzo (b) fluoranthène ;
— Benzo (a) pyrène ;
— Dibenzo (a, h) anthracène ;
— Benzo (g, h, i) pérylène ;
— Indéno (1, 2, 3 — c, d) pyrène ;
— Fluoranthène.

— 5 — NF X 43-329
Les méthodes sont également susceptibles de déterminer les HAP possédant au moins trois cycles aromatiques.Pour les HAP à deux cycles des précautions particulières doivent être prises pour éviter les pertes par évaporationet sublimation durant les phases d'extraction et de concentration précédant l'analyse.
Les méthodes de prélèvement et d'analyses retenues dans le présent document permettent de mesurer desteneurs supérieures à 0,5 µg de chacun des composés cités ci-dessus par mètre cube de gaz sec aux conditionsnormales de température et de pression.
NOTE 1 Les conditions normales de température et de pression sont, pour le présent document, de 0 °C (273 K)et 101,3 kPa.
NOTE 2 Au stade actuel des connaissances, les caractéristiques des méthodes n'ont pu être pleinement déterminées.La limite de détection est estimée à 0,1 µg/m3 pour le benzo (a) pyrène. Les limites de détection des composés dépendentde la technique analytique retenue. Elles ne constituent en général pas une limitation à la méthode, cependant, la principaledifficulté est de prendre toutes les précautions possibles pour limiter la valeur des blancs à des niveaux acceptables euégard au niveau de concentration recherché.
2 Références normatives
Le présent document comporte par référence datée ou non datée des dispositions d'autres publications. Ces réfé-rences normatives sont citées aux endroits appropriés dans le texte et les publications sont énumérées ci-après.Pour les références datées, les amendements ou révisions ultérieurs de l'une quelconque de ces publications nes'appliquent à ce document que s'ils y ont été incorporés par amendement ou révision. Pour les références nondatées, la dernière édition de la publication à laquelle il est fait référence s'applique.
A 35-586, Codification des aciers inoxydables français normalisés.
NF X 44-013, Séparateurs aérauliques — Méthode d'essai des filtres à l'aérosol de chlorure de sodium parphotométrie de flamme.
NF EN 13284-1:2001, Émissions de sources fixes — Détermination de la faible concentration en masse depoussières — Partie 1 : Méthode gravimétrique manuelle (indice de classement : X 43-333-1).
3 Termes et définitions
Pour les besoins du présent document, les termes et définitions suivants s'appliquent.
3.1blanc de prélèvementéchantillon prélevé sur le site de façon identique aux échantillons normaux, à l’exception du fait qu’aucun gaz n’estprélevé pendant la durée de l’essai
3.2conditions normalesvaleur de référence sur gaz sec à une pression de 101.325 kPa, arrondis à 101.3 kPa et à une températurede 273.15 K arrondis à 273 K
3.3échantillonnage représentatiféchantillonnage assurant une représentativité des effluents du conduit à caractériser
3.4filtration «dans le conduit»filtration dans le conduit, le filtre inséré dans son logement étant placé immédiatement en aval de la bused’échantillonnage
3.5filtration «hors du conduit»filtration hors du conduit, le filtre inséré dans son logement étant placé en aval de la buse et de la sonded’échantillonnage

NF X 43-329 — 6 —
3.6poussièresparticules de forme, structure ou masse volumique quelconque, dispersées dans la phase gazeuse au point deprélèvement susceptibles d’être recueillies par filtration dans les conditions spécifiées après échantillonnagereprésentatif du gaz à analyser, et qui demeurent en amont du filtre et sur le filtre après séchage dans les condi-tions spécifiées
3.7prélèvement isocinétiqueprélèvement effectué à un débit donné tel que la vitesse et le sens du gaz entrant dans la buse d’échantillonnagesont identiques à la vitesse et au sens du gaz dans le conduit au point de prélèvement
3.8température de filtrationtempérature du gaz échantillonné immédiatement en aval du filtre
NOTE Quelques caractéristiques des composés HAP listés dans ce document figurent en annexe A.
4 Principe
L'échantillon est prélevé de manière isocinétique ; la fraction particulaire est collectée sur un filtre plan dont lematériau doit être choisi en fonction de la température et de la nature physico-chimique des gaz échantillonnés ;la fraction gazeuse est piégée par condensation et par adsorption sur résine XAD2 ou toute autre résine dontl’équivalence de l’efficacité a pu être démontrée.
Les échantillons liquides et solides prélevés sont rapportés en laboratoire et font l'objet d'une extraction/prépara-tion permettant une analyse par chromatographie en phase gazeuse ou chromatographie en phase liquide hauteperformance.
5 Appareillage pour le prélèvement
5.1 Généralités
Deux types de prélèvement peuvent être mis en œuvre : avec ou sans division de débit ; ils sont tous deux enconformité avec la NF EN 13284-1.

— 7 — NF X 43-329
La stabilité physico-chimique des HAP, en présence de O2, NOx, N2, SO2, HCl et de certains métaux lourdscomme le vanadium, est limitée. Elle est d'autant plus faible que la température augmente. C'est la raison pourlaquelle il est nécessaire de :
— maintenir au niveau de la canne de prélèvement et du dispositif de filtration, une température aussi basse quepossible compte-tenu du point de rosée acide des gaz et/ou de l'éventuelle présence de vésicules d'eau, dansle cas d’un prélèvement avec filtre à l’extérieur du conduit. Cette température doit être mentionnée dans le rap-port d'essai ;
— limiter la durée de prélèvement par filtre à 2 h.
Les matériaux utilisés pour la buse, la sonde et le porte-filtre décrits en 5.2 et 5.3, peuvent être le verre, le titane,le PTFE ou l'acier inoxydable ou tout autre matériau n'absorbant pas ou ne réagissant pas avec les composésprésents dans le gaz prélevé.
5.2 Prélèvement avec division du débit
5.2.1 Une sonde en tous points décrite dans la norme NF EN 13284-1, permettant d'amener les gaz à la tem-pérature spécifiée en 5.1.
5.2.2 Un dispositif de filtration dont les caractéristiques sont précisées dans la NF EN 13284-1, fonctionnant àla température spécifiée en 5.1 et équipé d'un filtre plan conforme au 6.2.3.
5.2.3 Un dispositif de mesurage et de réglage du débit permettant l’ajustement et le contrôle de l'isocinétisme.
5.2.4 Un diviseur de débit.
Dans la mesure où celui-ci n’est pas chauffé, il doit être rincé après prélèvement.
5.2.5 Un condenseur en verre, refroidi par circulation d'eau, de façon à ce que la température des gaz à l'entréedu porte-résine (5.2.7) soit inférieure à 20 °C.
5.2.6 Un ballon récupérant les condensats.
5.2.7 Un porte-résine en verre, tel que :
— le temps de séjour des gaz dans la résine soit supérieur à 0.5 s et la vitesse inférieure à 35 cm.s-1 : pour deseffluents chargés, choisir des temps supérieurs à 1 s ;
— le rendement global d’adsorption soit supérieur à 90 % pour des concentrations globales en chacun des HAPsupérieures à 5 µg/m3n.
Le rendement d’adsorption doit être déterminé au moins une fois avec les 8 composés cités en 1. Cette efficacitédoit être déterminée en rajoutant une deuxième cartouche en série.
Pour augmenter le temps de séjour des gaz dans la résine, il est préférable d’augmenter la hauteur de la résineplutôt que d’augmenter le diamètre de la cartouche, ceci afin d’éviter les passages préférentiels.
Les parties en verre (de 5.2.5 à 5.2.7) sont systématiquement protégées de la lumière (en utilisant un habillagede papier aluminium par exemple).
Le porte-résine en verre est positionné :
— verticalement et placé entre le condenseur (5.2.5) et le ballon récupérant les condensats (5.2.6) ;
— horizontalement et raccordé par un té au conduit vertical reliant le condenseur (5.2.5) au ballon récupérant lescondensats (5.2.6).
5.2.8 Un ensemble d'aspiration (avec réglage du débit) et de mesure du débit secondaire de gaz prélevécomportant par exemple :
5.2.8.1 Une pompe étanche avec réglage du débit et permettant de maintenir un débit d'aspiration constantdans les conditions de la mesure. Pour le choix de cette pompe, tenir compte de la dépression importante qu'ilpeut y avoir en aval du filtre utilisé dans le dispositif de filtration.
5.2.8.2 Un indicateur permettant d’ajuster le débit pendant le prélèvement.

NF X 43-329 — 8 —
5.2.8.3 Un compteur à gaz.
Le compteur à gaz peut être humide ou sec ; dans ce dernier cas, il doit être précédé par un dispositif de séchage(dessiccateur chimique ou absorbant physique) assurant une humidité résiduelle inférieure à 10 g.m-3. Dans lamesure du volume de gaz prélevé, l'incertitude totale dépend à la fois de l'incertitude due au compteur à gaz, decelle de la mesure de la température et de la pression, ainsi que de l'étanchéité des dispositifs de mesure etd'extraction.
La mesure du volume doit être réalisée avec une incertitude inférieure à 5 %.
5.2.8.4 Mesures de température et de pression
Une mesure doit être réalisée au niveau du compteur à gaz avec le niveau d’incertitude suivant :
— mesure de la température en K, à ± 1 % ;
— mesure de la pression ± 1 %.
5.3 Prélèvement sans division de débit
5.3.1 Une sonde en tous points décrite dans la norme NF EN 13284-1, permettant d'amener les gaz à la tem-pérature spécifiée en 5.1.
5.3.2 Un dispositif de filtration dont les caractéristiques sont précisées dans la NF EN 13284-1, fonctionnant àla température spécifiée en 5.1 et équipé d'un filtre plan conforme au 6.2.3.
5.3.3 Une jonction entre séparateur et condenseur qui doit être rincée après le prélèvement dans le cas où ellen’est pas chauffée.
5.3.4 Un condenseur en verre, refroidi par circulation d'eau de façon à ce que la température des gaz à l'entréedu porte-résine (5.3.6) soit inférieure à 20 °C.
5.3.5 Un ballon récupérant les condensats.
5.3.6 Un porte-résine en verre, tel que :
— le temps de séjour des gaz dans la résine soit supérieur à 0.5 s et la vitesse inférieure à 35 cm.s-1 : pour deseffluents chargés, choisir des temps supérieurs à 1 s ;
— un rendement global d’adsorption supérieur à 90 % pour des concentrations globales en chacun des HAPsupérieures à 5 µg/m3n.
Le rendement d’adsorption doit être déterminé au moins une fois avec les 8 composés cités en 1. Cette efficacitédoit être déterminée en rajoutant une deuxième cartouche en série.
Pour augmenter le temps de séjour des gaz dans la résine, il est préférable d’augmenter la hauteur de la résineplutôt que d’augmenter le diamètre de la cartouche, ceci afin d’éviter les passages préférentiels.
Les parties en verre (de 5.3.4 à 5.3.6) sont systématiquement protégées de la lumière (en utilisant un habillagede papier aluminium par exemple).
5.3.7 Le porte-résine en verre est positionné :
— verticalement, entre le condenseur (5.3.4) et le ballon récupérant les condensats (5.3.5) ;
— horizontalement et raccordé par un té au conduit vertical reliant le condenseur (5.3.4) au ballon récupérant lescondensats (5.3.5).
5.3.8 Un ensemble d'aspiration et de mesure du débit de gaz prélevé comportant par exemple :
5.3.8.1 Une pompe étanche avec réglage du débit et permettant de maintenir un débit d'aspiration constantdans les conditions de la mesure. Pour le choix de cette pompe, tenir compte de la dépression importante qu'ilpeut y avoir en aval du filtre utilisé dans le dispositif de filtration. À titre d'exemple avec un porte-résine tel quedécrit en 5.3.6, le débit d'échantillonnage est limité à 2 000 l.h-1. Il doit pouvoir être maintenu avec des dépres-sions mesurées derrière le filtre de l'ordre de 40 kPa.

— 9 — NF X 43-329
5.3.8.2 Un indicateur permettant d’ajuster le débit pendant le prélèvement. La valeur du débit peut être vérifiéeau moyen du compteur à gaz (voir 5.3.8.3).
5.3.8.3 Un compteur à gaz.
Le compteur à gaz peut être humide ou sec ; dans ce dernier cas, il doit être précédé par un dispositif de séchage(dessiccateur chimique ou absorbant physique) assurant une humidité résiduelle inférieure à 10 g.m-3. Dans lamesure du volume de gaz prélevé, l'incertitude totale dépend à la fois de l'incertitude due au compteur à gaz, decelle de la mesure de la température et de la pression, ainsi que de l'étanchéité des dispositifs de mesurage etd'extraction.
La mesure du volume au compteur à gaz doit être réalisée avec une incertitude inférieure à ± 5 %.
5.3.8.4 Mesures de température et de pression
Une mesure doit être réalisée au niveau du compteur à gaz avec le niveau d’incertitude suivant :
— mesure de la température en K, à ± 1 % ;
— mesure de la pression ± 1 %.
6 Prélèvement
6.1 Choix de la section de mesurage et nombre de points de prélèvement
Le choix de la section de mesurage et le nombre de points de prélèvements sont précisés par la NF EN 13284-1.
6.2 Préparation du matériel
6.2.1 Généralités
Tout le matériel utilisé dans la chaîne de prélèvement doit être traité de façon à obtenir un blanc de prélèvementcompatible avec les performances désirées (voir 6.3.3).
Après vérification, il doit être stocké sans possibilité de contamination.
Tous les éléments sensibles constituant la chaîne de prélèvement, ayant fait l'objet d'un nettoyage spécial en labo-ratoire, doivent être transportés dans un emballage propre.
6.2.2 Préparation de la sonde de prélèvement et du dispositif de filtration
Laver et brosser avec un écouvillon, éventuellement au détergent.
Rincer à l’eau distillée ou déionisée.
Nettoyer avec un solvant polaire (acétone, méthanol …) puis avec un solvant apolaire (dichlorométhane…).
6.2.3 Choix et préparation du filtre
Le filtre plan doit être choisi en conformité avec les prescriptions du 6.2.7 de la NF EN 13284-1 (voir égalementannexe B).
Il doit par ailleurs être insoluble dans le solvant d'extraction.
En cas de doute sur la propreté du filtre, ce dernier doit être mis au four à 500 °C pendant 5 h ou doit faire l’objetd’un nettoyage par extraction au Soxhlet avec du dichlorométhane pendant 16 h.
Vérifier que le blanc analytique est compatible avec le niveau de concentration à mesurer.

NF X 43-329 — 10 —
6.2.4 Préparation de la verrerie
Toute la verrerie doit être maintenue dans un parfait état de propreté.
Le laboratoire peut suivre toute procédure validée de laboratoire ou l’une des procédures suivantes :
a) Première procédure :
- attention : cette procédure préconise l’emploi d’un mélange sulfo-chromique (H2SO4 concentré saturéen K2Cr2O7). Ce produit, d’un usage extrêmement délicat est interdit dans de nombreux laboratoires enraison de son caractère, corrosif, cancérigène et mutagène ;
- laisser la verrerie séjourner entre 4 h et 12 h dans un mélange sulfo-chromique (H2SO4 concentré saturéen K2Cr2O7) ;
- rincer abondamment avec de l'eau distillée ou déionisée, de qualité reconnue ;
- le séchage final de la verrerie se fait au four ou à l'étuve ;
- maintenir les récipients bouchés après séchage, par une feuille d'aluminium préalablement calcinée à unetempérature comprise entre 400 °C et 500 °C pendant 2 h.
NOTE La pollution de l’aluminium notamment, peut être détectée par un blanc anormal. Dans ce cas-là, il convient decalciner la feuille d’aluminium pendant 2 h à une température comprise entre 400 °C et 500 °C.
b) Seconde procédure :
- nettoyer la verrerie au bac à ultrasons avec un mélange eau — détergent ;
- nettoyer avec un solvant polaire (acétone, méthanol…) puis avec un solvant apolaire (dichlorométhane…) ;
- bouchonner les verreries.
6.2.5 Préparation de la résine XAD2
L’opérateur peut utiliser des résines conditionnées du commerce, ou conditionner lui-même sa résine suivant uneprocédure validée de laboratoire.
À titre d’exemple, la procédure suivante est préconisée :
— conditionner de la résine au soxhlet sur 16 h à l’eau puis 16 h au méthanol et deux fois 16 h au dichloromé-thane (Cf 7.2.2) ;
— sécher les résines sous courant d'azote ;
— remplir le porte-résine nettoyé de résine propre ;
— fermer le porte-résine aux deux extrémités et le stocker à l'abri de la lumière.
6.3 Prélèvement
6.3.1 Généralités
En raison de la complexité de la méthode, il est souhaitable que les personnels soient particulièrement entraînés,expérimentés et avertis dans le domaine du prélèvement des traces.
L'opérateur doit prendre soin d'éviter, lors de la manipulation, toute contamination des parties de l'appareillage quisont ultérieurement en contact avec l'échantillon.
6.3.2 Préparation et vérifications préliminaires
6.3.2.1 Assemblage
Avant mise en place de la sonde dans la veine gazeuse, vérifier le bon assemblage de la chaîne de prélèvement.
6.3.2.2 Contrôle de l'étanchéité
L’opération est à réaliser avant chaque prélèvement (voir 6.3.4.1.1 ou 6.3.4.2.1).
Avant chaque prélèvement, l’équipement complet de prélèvement doit être soumis à ce contrôle. Le rapportd’essai doit décrire la méthode utilisée. En annexe C sont proposés trois modes opératoires.

— 11 — NF X 43-329
6.3.2.3 Calcul du débit d'aspiration et du volume à aspirer
Déterminer le débit d'aspiration de façon à conserver l'isocinétisme au niveau du bec de la buse et choisir levolume à aspirer en fonction de la sensibilité recherchée et de la quantité de polluants présente dans l'effluent.
Le débit sur la résine XAD2 est ajusté de façon à ce que le temps de séjour des gaz dans le porte résine soitsupérieur à 0,5 s et la vitesse inférieure à 35 cms-1.
Simultanément, veiller à ce que le refroidissement du condenseur soit suffisant pour que la température des gazentrant dans le porte-résine contenant la résine XAD2 n'excède pas 20 °C, température qui facilite le piégeagedes composés considérés.
6.3.3 Détermination du blanc de prélèvement
Afin de vérifier la qualité de la mise en œuvre de la procédure, pour chaque campagne de mesure, réaliser unprélèvement appelé «blanc de prélèvement» en suivant toute la procédure hormis qu'aucun effluent n'est extraitde la cheminée ou du conduit.
La valeur du blanc de prélèvement, divisée par le volume d’échantillonnage moyen de la série de mesurages,donne une estimation de la limite de détection (en milligrammes par mètre cube) du processus global de mesu-rage tel qu’il est conduit par les opérateurs. Le blanc de prélèvement inclut les dépôts possibles sur le filtre et surtous les éléments situés en amont du filtre.
Les prélèvements sont considérés comme corrects si la quantité de HAP recueillie pendant le prélèvement est :
— au moins égale à cinq fois (ratio R défini en 10) celle correspondant au blanc de prélèvement lorsque laconcentration mesurée est inférieure à 100 µg.m-3 dans les conditions normales de température et depression ;
— au moins égale à dix fois (ratio R défini en 10.8) celle correspondant au blanc de prélèvement lorsque laconcentration mesurée est supérieure à 100 µg.m-3 dans les conditions normales de température et depression ;
— ou inférieure à une quantité équivalant à une concentration de 0,5 µg/m3.
6.3.4 Exécution du prélèvement
Le mode opératoire décrit ci-dessous peut être adapté en fonction des matériels utilisés. Dans tous les cas veillerà une mise en régime rapide du système de prélèvement.
6.3.4.1 Prélèvement avec division de débit
6.3.4.1.1 Préchauffer la sonde et le dispositif de filtration puis procéder au test d’étanchéité.
6.3.4.1.2 Introduire la sonde dans le conduit et la positionner sur le point de prélèvement.
6.3.4.1.3 Sur la ligne d'aspiration secondaire, relever les valeurs initiales de volume, température et pression.
Maintenir constant à ± 10 % le ratio entre le débit de la ligne principale et de la ligne secondaire.
6.3.4.1.4 Démarrer le prélèvement secondaire approximativement au débit nominal.
6.3.4.1.5 Démarrer le prélèvement principal et ajuster ce débit principal de façon à ce que la somme des débitsremplisse les conditions d'isocinétisme.
6.3.4.1.6 Ajuster le débit secondaire à sa valeur nominale.
6.3.4.1.7 Procéder aux ajustements nécessaires pour maintenir l'isocinétisme et le débit constant sur la lignesecondaire.
6.3.4.1.8 Procéder aux ajustements nécessaires lors de chaque changement de point de mesure.
6.3.4.1.9 Relever périodiquement les valeurs de pression, température et volume sur la ligne secondaire.

NF X 43-329 — 12 —
6.3.4.1.10 En fin de prélèvement, arrêter le prélèvement principal.
6.3.4.1.11 Arrêter le prélèvement secondaire.
6.3.4.1.12 Sur la ligne secondaire, relever les valeurs finales de volume, température et pression.
6.3.4.1.13 Sortir la sonde du conduit.
6.3.4.2 Prélèvement sans division de débit
6.3.4.2.1 Préchauffer la sonde et le dispositif de filtration puis procéder au test d’étanchéité.
6.3.4.2.2 Introduire la sonde dans le conduit et la positionner sur le point de prélèvement.
6.3.4.2.3 Relever les valeurs initiales du volume, de la température et de la pression.
6.3.4.2.4 Démarrer le prélèvement au débit nominal permettant de respecter les conditions d'isocinétisme.
6.3.4.2.5 Procéder aux ajustements nécessaires pour maintenir l'isocinétisme, lors de chaque changement depoint de mesure.
6.3.4.2.6 Relever périodiquement les valeurs du volume, de la pression et de la température.
6.3.4.2.7 En fin de prélèvement, relever les valeurs finales de la pression, de la température et du volume.
6.3.4.2.8 Sortir la sonde du conduit.
6.3.5 Récupération et préparation des échantillons
L'ensemble des échantillons contenant des HAP issus des préparations de 6.3.5.1 à 6.3.5.4 doit être maintenu àl'abri de la lumière et à une température < à 5 °C jusqu'à leur traitement au laboratoire d'analyse. L'ensemble deséchantillons doit faire l'objet d'une extraction dans les 15 j qui suivent le prélèvement. Le délai peut être augmentédans le cas où les échantillons peuvent être maintenus à – 20 °C.
6.3.5.1 Enlever le filtre plan et le mettre dans une boîte en verre fermée.
Effectuer ces opérations à l'abri du vent.
6.3.5.2 Au moins après chaque série d'essais, démonter toutes les parties situées en amont du dispositif defiltration, dispositif de filtration compris. Rincer ces éléments, de préférence sur le lieu du prélèvement, par lavageavec du dichlorométhane. Les produits de rinçage sont conservés dans un récipient bouché.
6.3.5.3 Démonter l'ensemble des verreries situées en aval du dispositif de filtration et procéder au nettoyagede ces verreries «post dispositif de filtration», sur site de préférence, avec du dichlorométhane. Les produits derinçage sont conservés dans un récipient bouché et protégé de la lumière.
6.3.5.4 Isoler le porte-résine en le bouchonnant.
7 Analyse
7.1 Généralités
Les hydrocarbures aromatiques polycycliques peuvent être analysés par chromatographie en phase liquide hauteperformance (CLHP) ou par chromatographie en phase gazeuse (CG/SM) (voir annexes D et E).
Après mise en solution, les extraits organiques obtenus au cours des différentes étapes du protocole peuvent êtreconservés à une température < à 5 °C et à l'abri de la lumière. Le délai maximal est de 15 j entre la mise en solu-tion et l'analyse chromatographique sauf en cas de stockage au congélateur.

— 13 — NF X 43-329
7.2 Produits chimiques utilisés
7.2.1 Généralités
Au cours des prélèvements ou de l'analyse, utiliser uniquement des réactifs de qualité analytique reconnue et del'eau distillée ou dé-ionisée de pureté équivalente de façon à ce que les essais à blanc et les essais qualifiés deblanc de prélèvement conduisent à des valeurs compatibles avec les limites de détection recherchées.
7.2.2 Dichlorométhane
Sa qualité diffère d’un fournisseur à un autre et d’un type à l’autre ; les produits ne sont pas tous d’une qualitésatisfaisante, doser les plus légers de la liste EPA. Prendre du dichlorométhane de qualité Pestipur et éviter laqualité HPLC. Pour la détermination des 8 HAP couverts par le présent document, la qualité du dichlorométhaneimporte peu.
7.2.3 Hexane
7.2.4 Toluène
Le toluène utilisé couramment en laboratoire de chromatographie n’est pas exempt de HAP légers et ne doit enaucun cas être utilisé dans le processus de conditionnement ou d’extraction des HAP des supports de piégeage.Il peut être utilisé dans le cas d’extraction de filtres chargés en poussières carbonées lorsque seuls les HAP lourdssont recherchés (le toluène est dans ce cas plus efficace que le dichlorométhane).
7.2.5 Méthanol ou acétonitrile, qualité CLHP
7.2.6 Diméthylformamide
7.2.7 Chlorure de sodium
7.2.8 Éthylène glycol (utilisé comme keeper pour concentration)
7.2.9 Cyclohexane
7.2.10 Sulfate de magnésium ou sodium anhydre
7.2.11 Solutions étalons
Les HAP étant cancérigènes, l’opérateur doit avoir de préférence recours à des solutions étalons «toutes prêtes»du commerce.
Si l’opérateur prépare lui-même ses solutions, il doit se référer aux règles de sécurité décrites dans le présentdocument (voir introduction) pour la préparation et la manipulation des solutions étalons.
NOTE Des HAP certifiés matériaux de référence peuvent être obtenus auprès du «Standard Measurement and Testing».Direction Générale XII — Commission des Communautés Européennes — 200, rue de la Loi — 1049 Bruxelles (Belgique).
7.2.11.1 Solutions étalons pour CLHP
7.2.11.1.1 Préparation des étalons en solutions mères individuelles (environ 100 µg.ml-1 par composé)
À l'aide d'un entonnoir en verre, introduire environ 10 mg de produit dans une fiole jaugée en verre de 100 ml préa-lablement tarée avec une précision de 0,1 mg. Après pesée précise (à 0,1 mg près), introduire 50 ml d'acétonitrile,boucher et vérifier que la solubilisation est complète. Sinon, accélérer la dissolution en plaçant la solution dansune cuve à ultrasons et en ajoutant quelques millilitres d'un autre solvant (acétone, dichlorométhane). Compléterà 100 ml par de l'acétonitrile.

NF X 43-329 — 14 —
7.2.11.1.2 Préparation du mélange étalon en solution mère (environ à 1 µg.ml-1 par composé)
Un mélange étalon des huit composés à doser est réalisé par dilution des différentes solutions mères dans del'acétonitrile, la concentration de chaque étalon étant approximativement de 1 µg.ml-1.
Les solutions mères doivent être conservées à l’abri de la lumière, à une température < à 5 °C. Elles doivent êtrecontrôlées ou remplacées périodiquement (tous les six mois). Avant utilisation, il est recommandé de laisser repo-ser les solutions quelques heures à température ambiante. Un traitement au bac à ultrasons est conseillé.
7.2.11.1.3 Préparation des mélanges étalons en solution diluée
Ces mélanges étalons sont préparés juste avant chaque série d'analyse par dilution de la solution mère dansl'acétonitrile. Les facteurs de dilution doivent tenir compte du niveau de concentration de l'extrait organique à ana-lyser et du domaine de linéarité du détecteur. Conserver ces mélanges à l'abri de la lumière. La durée de conser-vation est limitée et dépend du niveau de concentration.
7.2.11.2 Solutions étalons pour CG
Les étalons doivent être préparés dans un solvant compatible avec la technique d'injection chromatographiqueutilisée. Les modes «splitless» et «on column» nécessitent l'utilisation d'un solvant de type apolaire (exemple :toluène, heptane, hexane ou cyclohexane).
7.2.11.2.1 Préparation des étalons en solutions mères individuelles (environ 100 µg.ml-1 par composé)
À l'aide d'un entonnoir en verre, introduire environ 10 mg de produit dans une fiole jaugée en verre de 100 ml préa-lablement tarée avec une précision de 0,1 mg. Après pesée précise (à 0,1 mg près), introduire 50 ml de toluène,boucher et vérifier que la solubilisation est complète. Sinon, accélérer la dissolution en plaçant la solution dansune cuve à ultrasons et en ajoutant quelques millilitres d'un autre solvant (acétone, dichlorométhane). Compléterà 100 ml par du toluène.
7.2.11.2.2 Préparation du ou des mélanges étalons (de quelques dixièmes de microgrammes à quelquesmicrogrammes par millilitre par composé).
Ces mélanges étalons sont préparés par dilution des différentes solutions mères dans le cyclohexane de manièreà couvrir la gamme des concentrations à doser.
7.3 Appareillage
AVERTISSEMENT — La verrerie utilisée, pour le prélèvement des échantillons, la préparation des réactifset le dosage, doit être préparée selon 6.2.4.
7.3.1 Matériel courant de laboratoire
7.3.1.1 Microcolonnes garnies de silice prêtes à l'emploi contenant environ 1 ml de silice.
7.3.1.2 Évaporateur rotatif.
7.3.1.3 Pompe à vide.
7.3.1.4 Soxhlet.
7.3.2 Système HPLC comportant en particulier :
7.3.2.1 Une colonne travaillant en phases inversées avec un remplissage de silice greffée C18.
À titre d'exemple, une colonne d'acier de 25 cm de long, de diamètre interne 4,6 mm, garnie d'une phase de silicegreffée C18 (granulométrie 5 µm) convient.
7.3.2.2 Un dispositif d'injection permettant d'introduire une prise d'essai de 5 µl à 40 µl.

— 15 — NF X 43-329
7.3.2.3 Un détecteur fluorimétrique approprié. Des instruments équipés de filtre, ou mieux, de monochroma-teurs à l'excitation et à l'émission peuvent convenir. Le volume de la cellule à circulation doit être réduit, il doit êtreau plus de 30 µl.
7.3.2.4 Un solvant d'élution choisi selon la phase stationnaire utilisée de façon à permettre une séparation opti-male des HAP recherchés.
À titre indicatif, les solvants d'élution tels que acétonitrile-eau ou méthanol-eau sont généralement utilisés.
7.3.3 Système CG/SM
Système d’analyse combinant un chromatographe en phase gazeuse et un spectromètre de masse avec logicield’acquisition de données permettant l’enregistrement et le traitement des chromatogrammes.
Le chromatographe doit être équipé :
— d’un injecteur «On-column» ou un de type chaud sans division de débit (splitless) ;
— d’une alimentation en gaz vecteur Helium de pureté N55 ou équivalent ;
— d’une colonne capillaire haute résolution (Cf 7.5.2.4.1).
7.4 Combinaison des échantillons
Selon le type de résultats recherché, les extraits obtenus par les procédures décrites dans les alinéas suivants,peuvent être regroupés de manière appropriée pour une mesure globale ou des analyses séparées.
L'analyse de la phase HAP particulaire rassemble les échantillons obtenus après le rinçage de la ligne de prélè-vement en amont du dispositif de filtration et après l'extraction du filtre.
L'analyse de la phase gazeuse des HAP rassemble les échantillons obtenus après le nettoyage de la verrerie«post dispositif de filtration», l'extraction des condensats et des résines adsorbantes.
NOTE Les échantillons obtenus après rinçage de la sonde et des verreries peuvent correspondre à plusieurs prélève-ments successifs. Il est admis de les additionner aux échantillons correspondant à chaque prélèvement, au prorata desvolumes de gaz échantillonnés exprimés dans les mêmes conditions de température, de pression et d'humidité.
Dans le cas du prélèvement avec division du débit (5.2), les phases particulaires et gazeuses sont généralementanalysées séparément ; cependant, différents extraits peuvent être mélangés si ce mélange est effectué en tenantcompte du volume total prélevé et du volume prélevé sur le circuit secondaire.
Dans le cas d'un prélèvement sans division de débit (5.3), l'ensemble des échantillons peut être regroupé poureffectuer une analyse unique.
Type de regroupements possibles
HAP particulaires ➞ produit de rinçage de la ligne en amont du dispositif de filtration
filtre
HAP gazeux ➞ produit de rinçage de la ligne en aval du dispositif de filtration
condensats
résine
HAP totaux ➞ produit de rinçage de la ligne en amont du dispositif de filtration
filtre
produit de rinçage de la ligne en aval du dispositif de filtration
produit de rinçage du dispositif de filtration aval
condensats
résine

NF X 43-329 — 16 —
7.5 Extraction et dosage des hydrocarbures aromatiques polycycliques
7.5.1 Méthode chromatographique en phase liquide haute performance (CLHP)
7.5.1.1 Extraction
7.5.1.1.1 Extraction à partir des supports solides
L'extraction des hydrocarbures aromatiques polycycliques des supports solides tels que les résines, filtres ou car-touches, est réalisée à l'abri de la lumière, au Soxhlet ou par technique ASE (Accelerated Solvent Extractor).
a) Extraction au Soxhlet
1) Extraction des filtres
Les filtres sont extraits pendant 16 h au soxhlet par du dichlorométhane de qualité Pestipur.
NOTE Les rendements d’extraction calculés à partir d’essais de dopage se situent entre 69 et 95 % (85 à 95 % pourles 8 HAP considérés dans le présent document).
Le mode opératoire est le suivant :
— introduire le(s) filtre(s) dans un soxhlet de 200 ml ;
— ajouter environ 300 ml de dichlorométhane de qualité Pestipur dans le ballon de 500 ml contenant 2ou 3 pierres ponce à remplacer après 4 cycles de 16 h ;
— placer le réfrigérant sur l’appareillage soxhlet ;
— chauffer à reflux pendant 16 h : le chauffage est réglé de manière à ce que le soxhlet se remplisse de solvant 3à 4 fois par heure ;
— récupérer l’extrait organique et le sécher sur du sulfate anhydre.
Pour des poussières contenant des imbrûlés solides, il peut être nécessaire de substituer le toluène au dichloro-méthane.
b) Extraction des résines
Le mode opératoire est le suivant : extraction au soxhlet : 16 h au dichlorométhane de qualité Pestipur :
— introduire la résine dans la cartouche qui doit être ensuite placée dans le soxhlet, boucher la cartouche avecde la laine de quartz ;
— ajouter le solvant dans le ballon contenant 5 à 6 pierres ponce ;
— placer le réfrigérant sur l’appareillage soxhlet ;
— chauffer à reflux pendant 16 h : le chauffage étant réglé de manière à ce que le soxhlet se remplisse desolvant 3 à 4 fois par heure.
c) Extraction automatique accélérée
Avec l’ASE (Accelerated Solvent Extractor), le processus d’extraction traditionnel est accéléré en maintenant lesolvant à température élevée et la cellule contenant l’échantillon est mise sous pression afin de maintenir le sol-vant chauffé à l’état liquide pendant toute la durée de l’extraction.
7.5.1.1.2 Extraction à partir de phases aqueuses
L'extraction des hydrocarbures aromatiques polycycliques des phases aqueuses est effectuée par extractionliquide-liquide avec du dichlorométhane ou tout autre solvant compatible avec la phase de purification :
— prélever une partie aliquote de l'échantillon. La mettre dans une ampoule à décanter ;
— ajouter 50 ml de dichlorométhane ;
— agiter 5 min ;
— laisser décanter et récupérer la phase organique ;
— effectuer sur le même échantillon deux autres extractions dans les mêmes conditions et rassembler les phasesorganiques ;
— sécher la phase organique sur sulfate de magnésium anhydre.

— 17 — NF X 43-329
7.5.1.2 Concentration
Évaporer l'extrait obtenu à l'aide d'un évaporateur rotatif ou toute autre technique adaptée. L'évaporation est réa-lisée sous vide à une température ne dépassant pas 35 °C. L'évaporation est poursuivie sous gaz inerte aprèstransvasement dans des tubes à concentration. La concentration peut s'opérer dans le tube cônique en évitantd'aller à sec (utiliser par exemple 200 µl du keeper éthylène glycol ; concentrer par exemple jusqu'à 200 µl envi-ron). Le résidu obtenu est ajusté à un volume connu par de l'acétonitrile et/ou du méthanol avant analyse chro-matographique (volume supérieur ou égal à 1 ml) ou par environ 1 ml d'hexane si l'étape de purification estenvisagée.
7.5.1.3 Purification (étape facultative)
Le but de cette purification est d'éliminer les interférences possibles (voir procédure en annexe F).
7.5.1.4 Dosage
Prendre une partie aliquote des extraits préparés précédemment.
Opérer une dilution de la prise d'essai si nécessaire dans de l'acétonitrile et/ou du méthanol.
Le volume de prise d'essai dilué ou non (V), injecté dans la colonne (7.3.1.1) est généralement de 5 µl à 50 µl.
La phase chromatographique (silice greffée en octadécycle), la géométrie de la colonne et la composition de laphase mobile doivent satisfaire à une résolution complète des huit composés retenus.
Les conditions opératoires doivent être sélectionnées de manière à minimiser les risques d'interférences tout enconservant le maximum de sensibilité. Il s'agit de choisir en particulier :
— la nature et la composition de la phase mobile éluante : mélange acétonitrile-eau ou méthanol-eau ;
— les conditions de détection : longueur d'onde à l'excitation et à l'émission.
En fonction des possibilités du matériel, les analyses chromatographiques sont réalisées en régime isocratiqueou gradient, les conditions de détection pouvant être modifiées au cours du temps pour une même injection avecune intervention manuelle d'un opérateur ou automatiquement avec un détecteur programmable. L'extrait peutaussi être injecté plusieurs fois dans des conditions optimales différentes (modification de l'éluant et des condi-tions de détection).
7.5.1.5 Analyses de contrôle
Ces analyses de contrôles doivent être effectuées lors de toute modification du protocole expérimental.
Effectuer parallèlement, dans les conditions d'extraction, concentration et analyse équivalentes (voir de 7.5.1.1à 7.5.1.4), en employant des supports de prélèvement vierges, les mêmes verreries et les mêmes conditions opé-ratoires que celles utilisées pour les échantillons analysés :
— un essai à blanc ;
— un essai après ajout de quantités connues de mélange étalon de HAP, de manière à détecter d'éventuellespertes lors des opérations d'extraction et concentration. Les ajouts d'HAP doivent être du même ordre que lesquantités mesurées lors de l'analyse des échantillons.
Les valeurs de blanc et les taux de récupération des ajouts doivent être mentionnés dans le rapport d'essai.

NF X 43-329 — 18 —
7.5.1.6 Résultats
À partir des chromatogrammes obtenus en 7.5.1.5, les HAP sont identifiés à l'aide des temps de rétention des picscorrespondant aux solutions d'étalonnage (7.2.11).
Calculer la quantité de chaque HAP de l'échantillon, en microgrammes, selon la formule suivante :
... (1)
où :
QHAP est la quantité du HAP considéré de l'extrait injecté, exprimée en microgrammes ;
F est la hauteur de pic relatif au HAP considéré ;
m(s) est la quantité du HAP considéré dans la solution étalon en microgrammes par millilitres ;
F(s) est la hauteur de pic relatif au HAP considéré dans la solution étalon ;
V est le volume de la solution injectée (voir 7.5.1.4), exprimé en millilitres ;
g est le facteur de concentration et de dilution (le cas échéant) de l’extrait (obtenu en 7.5.1.1).
Calculer le taux de récupération de l’étalon interne :
... (2)
où :
C(x,y) est la concentration de l’étalon (x) dans l'extrait injecté ;
V(y) est le volume de l’extrait injecté ;
C(x,add) est la concentration de l’étalon (x) ajoutée à l’échantillon ;
V(x,add) est le volume de la solution étalon (x) ajoutée à l’échantillon.
7.5.2 Méthode chromatographique en phase gazeuse couplée à la spectrométrie de masse (CG/SM)
7.5.2.1 Extraction
7.5.2.1.1 Extraction à partir des supports solides
L’extraction des hydrocarbures aromatiques polycycliques sur supports solides (filtre plan et résines XAD2) estréalisée à l’abri de la lumière au moyen d’un soxhlet ou tout autre moyen équivalent tel que décrit en 7.5.1.1.1.
7.5.2.1.2 Extraction à partir des phases aqueuses
L’extraction des hydrocarbures aromatiques polycycliques dans les phases aqueuses est effectuée par extractionliquide/liquide avec du dichloromethane (ou autre solvant compatible avec le mode opératoire) tel que décriten 7.5.1.1.2.
7.5.2.2 Concentration
La concentration des extraits est réalisée par évaporation au moyen d’un évaporateur rotatif. L’évaporation estréalisée sous vide à une température ne dépassant pas 35 °C. L’extrait est évaporé jusqu’à un volumed’environ 5 ml puis transféré dans un tube concentrateur et concentré sous gaz inerte pour obtenir un extrait quidoit être à 1 ml avec un solvant compatible avec l’analyse par CPG/SM.
Une quantité connue d’étalon interne est ajoutée à chaque extrait avant stockage au refrigérateur.
7.5.2.3 Purification
Si les matrices sont relativement propres, il peut ne pas être nécessaire de procéder à une purification. Les matri-ces complexes peuvent nécessiter une ou plusieurs étapes de purification afin d’éliminer les interférences dues àla présence de composés polaires et autres hydrocarbures, par exemple l’huile. Dans ce cas, la concentration etla purification doivent être conduites tel que décrit ci dessous :
QHAP .= F m s( ) V g⋅ ⋅ ⋅F s( )
-------------------------------------
% recouvrement .= C x y,( ) V y( ) 100⋅ ⋅C x add,( )V x add,( )----------------------------------------------------

— 19 — NF X 43-329
7.5.2.3.1 Concentration
Évaporer l'extrait obtenu à l'aide d'un évaporateur rotatif. L'évaporation est réalisée sous vide à une températurene dépassant pas 35 °C. L'évaporation est poursuivie sous gaz inerte après transvasement dans des tubes àconcentration en évitant d'aller à sec (concentrer jusqu'à 200 µl environ). Le résidu obtenu est ajusté à 50 ml pardu cyclohexane.
7.5.2.3.2 Purification
La purification est réalisée en deux étapes :
a) Extraction liquide-liquide :
Ce procédé permet d'éliminer principalement les hydrocarbures aliphatiques. La phase cyclohexanique (50 ml)est introduite dans une ampoule à décanter, puis soumise à extraction en deux temps successifs (50 mlet 25 ml) par un mélange diméthylformamide-eau (90-10).
Les deux phases diméthylformamide-eau obtenues sont introduites dans une autre ampoule à décanter. Aprèsaddition de 75 ml d'eau distillée, les HAP sont réextraits en deux temps par du cyclohexane (50 ml puis 25 ml).
En cas de besoin, pour favoriser la séparation des phases, ajouter une petite quantité de NaCl à la phaseaqueuse.
Les deux extraits cyclohexaniques sont recombinés et concentrés à l'évaporateur rotatif sous vide à une tem-pérature ne dépassant pas 35 °C. L'évaporation est poursuivie sous gaz inerte après transvasement dans destubes à concentration jusqu'à l'obtention d'un volume de 1 ml environ.
b) Purification sur silice :
L'extrait repris par environ 1 ml de cyclohexane est introduit à l'aide d'une seringue en tête d'une microcolonneprête à l'emploi contenant environ 1 ml de silice. À l'aide d'une seringue du type «Luerlock» ou par soutiragesous vide, les HAP sont élués par 5 ml d'hexane + dichlorométhane (50-50).
La fraction des composés polyaromatiques polaires reste sur la cartouche de purification.
L'éluat est évaporé sous gaz inerte en évitant d'aller à sec (concentrer jusqu'à 200 µl environ).
7.5.2.4 Analyse des échantillons
7.5.2.4.1 Instruments
La séparation chromatographique en phase gazeuse est réalisée au moyen de colonnes capillaires (diamètreinterne 0.25 à 0.32 mm) à film mince (< 0.3 µm) de type méthyl-siloxane ou methyl-phenyl-siloxane. La longueurde la colonne est d’environ 25 m. Les paramètres de températures du chromatographe dépendent des caracté-ristiques de l’instrumentation, de la nature du solvant et de la résolution souhaitée.
Les paramètres types du chromatographe en phase gaz sont les suivants :
— températures initiales de la colonne entre 40 °C et 80 °C pendant 2 min ou 15 °C en dessous du point d’ébul-lition du solvant d’injection dans le cas d’un injecteur «On-column» ;
— rampe de température de la colonne : environ 5 °C à 8 °C/min de manière à obtenir une bonne séparationdes HAP analysés et plus particulièrement le benzo(b)fluoranthène et benzo(k)fluoranthène ;
— température finale de 275 °C à 300 °C pendant plusieurs minutes ;
— température de transfert dans le spectromètre de masse identique à la température finale de la colonne ;
— gaz vecteur : Hélium à environ 30 cm/s à 40 cm/s ;
— température de l’injecteur : 275 °C à 300 °C pour les injecteurs chauds sans division ;
— volume d’injection : de l’ordre de 0,2 µl à 1 µl.
La détection des HAP est réalisée avec un spectromètre de masse d’ionisation par impact d’électron 70-eV fonc-tionnant en mode de contrôle d’ion spécifique (SIM).Pour chaque HAP à analyser choisir un ion primaire pour laquantification (ion cible) et au moins un ion secondaire pour qualifier le composé (ion qualifiant).
L’annexe G montre les ions clés pour les hydrocarbures aromatiques polycycliques et certains composésdeutérés.

NF X 43-329 — 20 —
7.5.2.4.2 Étalon interne
Il convient d’utiliser comme étalons internes des hydrocarbures aromatiques polycycliques deutérés ou aucarbone 13 de pureté supérieure ou égale à 98 %. Il est possible d'utiliser une série complète d’étalons internespour chaque hydrocarbure aromatique polycyclique à analyser ou choisir un certain nombre d’étalons internes,par exemple un par hydrocarbure aromatique polycyclique à 2, 3, 4, 5 cycles.
7.5.2.4.3 Étalonnage des instruments
Les solutions pour étalonnage des hydrocarbures aromatiques polycycliques sont préparées à au moins troisniveaux de concentration pour chaque HAP. En raison des risques associés à l’utilisation des hydrocarbures aro-matiques polycycliques purs, utiliser de préférence des solutions étalons vendues dans le commerce. Les diluerau moyen de fioles jaugées aux plages de concentrations des échantillons. Le National Institute of Standards andTechnology fournit le produit de référence SRM2260 : «PAH in toluène certified». L’étalon NIST est une solutioncertifiée de 23 hydrocarbures aromatiques polycycliques dans le toluène.
Une quantité connue d’étalon interne est rajoutée à chaque solution d’étalonnage avant l’analyse.
Les étalons servent à déterminer les temps de rétention des HAP, leur facteur de réponse relatif par rapport àl’étalon interne correspondant et le rapport entre l’ion «qualifiant»et l’ion «cible».
Le rapport entre l’ion «qualifiant» et l’ion «cible» est calculé selon l’équation suivante :
... (3)
où :
rapport est le rapport entre l’ion «qualifiant» et l’ion «cible» ;
Aqualifiant est la surface de l’ion «qualifiant» (ou secondaire) ;
Acible est la surface de l’ion «cible» (ou primaire).
Le facteur de réponse relatif (RRFi) est calculé pour chaque HAP grâce à l’équation suivante :
... (4)
où :
RRFi est le facteur de réponse relatif du HAP considéré par rapport à l’étalon interne correspondant ;
Si est la surface de l’ion cible du HAP considéré i dans la solution d’étalonnage ;
Se est la surface de l’ion cible de l’étalon interne correspondant à i dans la solution d’étalonnage ;
Qi est la masse du HAP considéré i dans la solution d’étalonnage (µg) ;
Qe est la masse de l’étalon interne correspondant à i dans la solution d’étalonnage (µg).
Si le facteur de réponse relatif est stable sur la plage de fonctionnement, il peut être considéré comme invariantet il est possible d'utiliser le facteur de réponse moyen pour les calculs.
7.5.2.4.4 Analyse
Les extraits d’échantillon sont retirés du réfrigérateur de stockage et mis à température ambiante. Après contrôledes performances du système chromatographe en phase gazeuse par analyse d’une solution d’étalonnage, desinjections d’environ 1 µl de chaque extrait d’échantillon sont effectuées et la réponse du spectromètre de masseest enregistrée. Il est nécessaire d’analyser régulièrement une solution d’étalonnage ainsi qu’un blanc de solvantde manière à vérifier l’absence de contamination entre les injections.
L’identification correcte des HAP dépend :
— du temps de rétention du composé qui doit être compris dans une plage de ± 3 %. Dans le cas où un passeurd’échantillon n'est pas utilisé, il est conseillé d’utiliser les temps de rétention relatif par rapport aux étalonsinternes ;
— de la proportion d’ions «qualifiants» qui ne doit pas différer de la valeur escomptée de plus de ± 30 %.
rapport 100 % Aqualifiant
Acible-----------------------=
RRFi
Si
Se------
Qe
Qi-------⋅=

— 21 — NF X 43-329
La concentration des HAP dans l’extrait est calculée selon l’équation suivante :
... (5)
où :
QHAP est la masse du HAP considéré i dans l’extrait échantillon (µg) ;
Qe est la masse de l’étalon interne correspondant à i ajouté dans l’extrait échantillon (µg) ;
RRFi est le facteur de réponse relatif du HAP considéré par rapport à l’étalon interne correspondant ;
Si est la surface de l’ion cible du HAP considéré i dans l’extrait échantillon ;
Se est la surface de l’ion cible de l’étalon interne correspondant à i dans l’extrait échantillon.
Si la réponse de l’ion cible d’un HAP quelconque sort de la plage de la courbe d’étalonnage initiale du systèmede chromatographie en phase gazeuse/spectromètre de masse, l’extrait doit être dilué et analysé à nouveau.
8 Expression des résultats
8.1 Concentration en poussières
Pour la détermination de la concentration en poussière liée à la mesure isocinétique, exprimer les résultats commeindiqué dans la norme NF EN 13284-1, en tenant compte du débit prélevé dans le circuit secondaire.
8.2 Volume de gaz sec prélevé
Pour la détermination de la concentration en chaque HAP, déterminer tout d'abord des volumes de gaz sec pré-levé Vnor1 et Vnor2 sur les circuits principaux et secondaires, respectivement.
8.2.1 Compteur à gaz sec
... (6)
8.2.2 Compteur à gaz humide
... (7)
où :
Vnor est le volume exprimé en mètres cubes sur gaz sec, aux conditions normales de température et depression
1) ;
Vt,p est le volume mesuré aux conditions réelles de température et de pression, exprimé en mètres cubes ;
t est la température réelle, exprimée en degrés Celsius ;
P est la pression statique totale, exprimée en kilopascals (= pression atmosphérique + pression statiquerelative du compteur à gaz) ;
Ps(H2O) est la pression saturante de la vapeur d'eau à la température du compteur à gaz, exprimée en kilopas-cals (voir annexe H).
1) Les conditions normales de température et de pression sont, pour le présent document, de 0 °C (273 K)et 101,3 kPa.
QHAP
Si
Se------
Qe
RRFi--------------⋅=
Vnor Vt,p 273
273 t+------------------ P
101,3---------------=
Vnor Vt,p 273
273 t+------------------
P Ps H2O –
101,3----------------------------------=

NF X 43-329 — 22 —
8.3 Calcul de la concentration en HAP par rapport au gaz sec
La concentration globale en chaque HAP est ensuite calculée par :
— Pour un prélèvement sans division de débit (5.3) :
... (8)
où :
CHAP est la concentration en HAP, exprimée en microgrammes par mètre cube de gaz sec, dans les conditionsnormales de pression et de température ;
QHAP est la quantité de HAP récupérée, exprimée en microgrammes.
— Pour un prélèvement avec division de débit (5.2) :
... (9)
où :
CHAP est la concentration en HAP, exprimée en microgrammes par mètre cube de gaz sec, dans les conditionsnormales de pression et de température ;
QHAP1 est la quantité de HAP capté dans la ligne de prélèvement principale, exprimée en microgrammes ;
QHAP2 est la quantité de HAP capté dans la ligne de prélèvement secondaire, exprimée en microgrammes ;
Vnor1 est le volume d’air prélevé sur la ligne de prélèvement principale exprimée en mètres cubes sur gaz sec,aux conditions normales de température et de pression ;
Vnor2 est le volume d’air prélevé sur la ligne de prélèvement secondaire, exprimée en mètres cubes sur gazsec, aux conditions normales de température et de pression.
Lorsqu'il est nécessaire d'exprimer le résultat dans un système de référence différent, il y a lieu d'utiliser les rela-tions indiquées ci-après.
8.4 Expression des résultats par rapport au gaz humide dans les conditions normales detempérature et de pression
Les résultats sont obtenus par la relation suivante :
... (10)
où :
CHAPh est la concentration de HAP dans le gaz humide au point de prélèvement, exprimée en microgrammes parmètre cube, dans les conditions normales de pression et de température ;
H est la teneur en vapeur d'eau des gaz au point de prélèvement, exprimée en pourcentage volumique surgaz humide, dans les conditions normales de température et de pression 2).
8.5 Expression des résultats par rapport à une teneur de référence en CO2
Afin de tenir compte d'un éventuel effet de dilution, il est souvent nécessaire d'exprimer la concentration en cha-que HAP par rapport à une teneur de CO2, exprimée sur gaz humide ou sur gaz sec suivant les cas.
À partir de la connaissance de la concentration en chaque HAP au rejet, exprimée en milligrammes de HAP parmètre cube ramenée aux conditions normales de température et de pression, l'eau étant supposée rester sousforme de vapeur, calculer la concentration en milligrammes de HAP, rapportée à une valeur conventionnelle deréférence de concentration en CO2, par l'une des relations :
2) Les conditions normales de température et de pression sont, pour le présent document, de 0 °C (273 K)et 101, 3 kPa.
CHAP QHAP Vnor⁄=
CHAP QHAP1 Vnor1 Vnor2+( ) QHAP2 Vnor2⁄+⁄=
CHAPh CHAP100 H–
100--------------------=

— 23 — NF X 43-329
... (11)
... (12)
où :
(CO2réf)s est la concentration en CO2 dans les conditions de référence, exprimée en pourcentage volumiquesur gaz sec au point de prélèvement ;
(CO2réf)h est la concentration en CO2 dans les conditions de référence, exprimée en pourcentage volumiquesur gaz humide au point de prélèvement ;
(CO2mes)s est la concentration en CO2 dans les points de prélèvement, exprimée en pourcentage volumiquesur gaz sec ;
(CO2mes)h est la concentration en CO2 mesuré au point de prélèvement, exprimée en pourcentage volumiquesur gaz humide.
8.6 Expression des résultats par rapport à une teneur de référence en O2
Afin de tenir compte d'un éventuel effet de dilution, il est souvent nécessaire d'exprimer la concentration en cha-que HAP par rapport à une teneur en O2, généralement exprimée sur gaz sec.
À partir de la connaissance de la concentration en chaque HAP au rejet, exprimée en milligrammes de HAP parmètre cube ramenée aux conditions normales de température et de pression, l'eau étant supposée rester sousforme de vapeur, calculer la concentration en chaque HAP rapportée à une valeur conventionnelle de référencede concentration de O2, par la relation :
... (13)
où :
O2ref est la concentration en oxygène dans les conditions de référence, exprimée en pourcentagevolumique sur gaz sec ;
(O2mes)s est la concentration en oxygène mesuré au point de prélèvement, exprimée en pourcentagevolumique sur gaz sec ;
(O2mes)h est la concentration en oxygène mesuré au point de prélèvement, exprimée en pourcentagevolumique sur gaz humide.
NOTE La concentration en oxygène dans l'air de dilution est pris égal à 21 %.
9 Vérifications
Si la quantité de chaque HAP dosée QHAP n'est pas au moins égale à :
— cinq fois (ratio R défini en 10.o) la quantité dosée lors de l'évaluation du blanc de prélèvement (6.3.3), pour desconcentrations mesurées inférieures à 100 µg.m-3, dans les conditions normales de pression et de température ;
— dix fois (ratio R défini en 10.o) la quantité dosée lors de l'évaluation du blanc de prélèvement (6.3.3), pour desconcentrations mesurées supérieures à 100 µg.m-3, dans les conditions normales de pression et detempérature ;
il est nécessaire d'adapter la durée du prélèvement pour augmenter la quantité d'HAP captée, ou de modifier lemode opératoire pratiqué pour réduire la valeur du blanc de prélèvement.
Il est ainsi possible de limiter l'influence des erreurs résultant de la manipulation, du rinçage du système de pré-lèvement, ainsi que celle des erreurs qui peuvent se produire lors de l'introduction de la sonde dans le conduitpuis de son retrait.
La valeur du blanc de prélèvement évaluée ne doit pas être retranchée du résultat de mesurage.
CHAPCO2s CHAP=CO2ref( )s
CO2mes( )s-------------------------------⋅
CO2ref( )sCO2mes( )h
--------------------------------CHAP=100 H–
100--------------------⋅
CHAPCO2h CHAPh=CO2ref( )h
CO2mes( )s 100 H–( )100
-----------------------------------------------------------------------------------⋅ CHAPh=
CO2ref( )hCO2mes( )h
--------------------------------⋅
CHAPO2s CHAP=21 O2ref–
21 O2mes( )s–----------------------------------------⋅ CHAP=
21 O2ref–
21 O2mes( )h 100100 H–( )
-------------------------–-------------------------------------------------------------------⋅

NF X 43-329 — 24 —
10 Rapport d'essai
Le rapport d'essai doit faire référence au présent document et comporter les points suivants :
a) description du but des essais ;
b) description des conditions de fonctionnement du procédé de production et de leur variation pendant les mesu-res (à défaut mentionner au moins les éléments qui permettent de retrouver ces informations dans les registresd'exploitation) ;
c) caractéristiques de la section de mesure ;
d) nombre de points de mesure et leur position dans la section de mesure ;
e) caractéristiques du matériel de mesure utilisé, en précisant notamment la nature des matériaux utilisés pour laréalisation des différents éléments et la température de filtration et de la canne de prélèvement ;
f) nombre d'essais et leur durée ;
g) méthode analytique mise en œuvre :
1) qualité des réactifs utilisés ;
2) méthode utilisée et conditions analytiques : volume injecté, débit de gaz vecteur, température de l'injecteuret du four pour la CG, etc. ;
3) type de colonne utilisée ;
4) toute modification apportée au mode opératoire ou tout incident susceptible d'avoir agi sur les résultats ;
5) la référence de chaque échantillon et les résultats du dosage pour chaque HAP ;
h) résultat des blancs de prélèvement et analyses de contrôle :
1) résultats du blanc de prélèvement.
Le rapport d'essais doit présenter les résultats obtenus lors de l'évaluation du blanc de prélèvement (QHAP1BFet QHAP2BF) et lors des autres prélèvements. Lorsque plusieurs prélèvements ont été effectués, il faut déter-miner la moyenne et l'écart-type associé à cette moyenne. Les ratios mesure/blanc de prélèvement R, R1 et R2sont calculés de la façon suivante :
R1 = QHAP1 / QHAP1BP ... (14)
R2 = QHAP2 / QHAP2BP ... (15)
... (16)
où :
R1 est le ratio mesure/blanc de prélèvement pour chaque HAP capté dans la ligne de prélèvement principale ;
R2 est le ratio mesure/blanc de prélèvement pour chaque HAP capté dans la ligne de prélèvement secondaire ;
R est le ratio mesure/blanc de prélèvement pour la mesure globale de chaque HAP (correspondant à CHAPcalculé en 8.3) ;
et
QHAP1 est la quantité de HAP capté dans la ligne de prélèvement principale, exprimée en microgrammes ;
QHAP1BP est la quantité de HAP capté dans la ligne de prélèvement principale, exprimée en microgrammes,lors de l'évaluation du blanc de prélèvement (6.3.3) ;
QHAP2 est la quantité de HAP capté dans la ligne de prélèvement secondaire, exprimée en microgrammes ;
QHAP2BP est la quantité de HAP capté dans la ligne de prélèvement secondaire, exprimée en microgrammes,lors de l'évaluation du blanc de prélèvement (6.3.3) ;
CHAP1 est la concentration en chaque HAP capté dans la ligne de prélèvement principale, exprimée enmicrogrammes par mètre cube de gaz sec, dans les conditions normales de pression et detempérature ;
CHAP2 est la concentration en chaque HAP capté dans la ligne de prélèvement secondaire, exprimée enmicrogrammes par mètre cube de gaz sec, dans les conditions normales de pression et detempérature.
2) résultats des analyses de contrôle
RCHAP1 CHAP2+
CHAP1 R1⁄ CHAP2 R2⁄+---------------------------------------------------------------=

— 25 — NF X 43-329
Il est nécessaire de donner la valeur du blanc analytique ainsi que le taux de récupération des ajouts dosés.
i) résultats obtenus
Le rapport d'essai doit en outre indiquer les circonstances particulières et les incidents susceptibles d'avoir agi surles résultats.
Si des contraintes particulières ont imposé une adaptation de la méthode décrite dans le présent document, cesadaptations doivent être décrites et justifiées.

NF X 43-329 — 26 —
Annexe A
(informative)
Quelques caractéristiques des huit HAP dosésInit numérotation des tableaux d’annexe [A]!!!Init numérotation des figures d’annexe [A]!!!Init numérotation des équations d’annexe [Q]!!!
Structure Nomenclature Poids moléculaire
Pointde fusion
Point d'ébullition
Benz[a]anthracène
1,2-Benzanthracène
Tétraphène
2,3-Benzophénanthrène
Naphthanthracène
228.30 162 435 sub
Benzo[k]fluoranthène
8,9-Benzofluoranthène
11,12-Benzofluoranthène
252.32 217 481
Benzo[b]fluoranthène
2,3-Benzofluoranthène
3,4-Benzofluroanthène
Benz[e]acéphénanthrylène
252.32 168 481
Benzo[a]pyrène
1,2-Benzpyrène
3,4-Benzopyrène
Benzo[def]chrysène
252.32 177 496
Dibenz[a,h] anthracène
1,2:5.6-Dibenzanthracène
278.36 270 524
Benzo[ghi]perylène
1,12-Benzopérylène
276.34 278 542
Indeno [1,2,3-cd]pyrène
e-Phénylénépyrène
276.34 164 534
Fluoranthène
Idryl
1,2-Benzacénaphthène
Benzo[jk]fluorène
Benz[a]acénaphthylène
202.26 111 383
—27
—N
F X
43-329
Init numéroInit numéroInit numéro
SARTORIUS MSI
T SM11304 MAGNA
C Nitrate de cellulose Polyamide
É 0,13 ?
C
D ?
N 99,999 ?
T 130 180
C oui ?
P 0,8 0,8
V
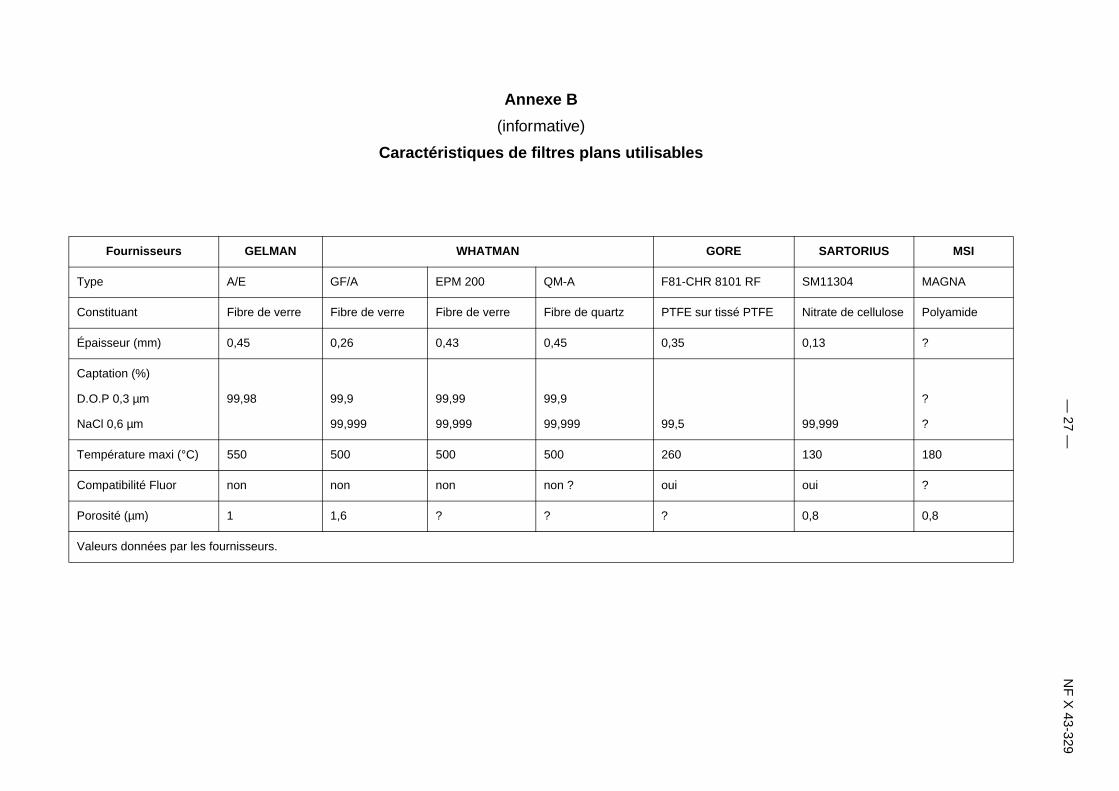
Annexe B
(informative)
Caractéristiques de filtres plans utilisablestation des tableaux d’annexe [B]!!!tation des figures d’annexe [B]!!!tation des équations d’annexe [R]!!!
Fournisseurs GELMAN WHATMAN GORE
ype A/E GF/A EPM 200 QM-A F81-CHR 8101 RF
onstituant Fibre de verre Fibre de verre Fibre de verre Fibre de quartz PTFE sur tissé PTFE
paisseur (mm) 0,45 0,26 0,43 0,45 0,35
aptation (%)
.O.P 0,3 µm 99,98 99,9 99,99 99,9
aCl 0,6 µm 99,999 99,999 99,999 99,5
empérature maxi (°C) 550 500 500 500 260
ompatibilité Fluor non non non non ? oui
orosité (µm) 1 1,6 ? ? ?
aleurs données par les fournisseurs.

NF X 43-329 — 28 —
Annexe C
(informative)
Modes opératoires pour l'essai d'étanchéitéInit numérotation des tableaux d’annexe [C]!!!Init numérotation des figures d’annexe [C]!!!Init numérotation des équations d’annexe [S]!!!
C.1 Méthode A
a) monter complètement l’équipement de prélèvement, y compris le support de filtre et le condenseur/pot àcondensats/cartouche à résine ;
b) laisser l’échantillonneur chauffer jusqu’à obtention de sa température de service ;
c) étanchéifier l’orifice d’entrée de la buse ;
d) mettre en marche la ou les pompe(s) ;
e) après obtention de la pression minimale, relever le débit ; le débit de la fuite ne doit pas dépasser 5 % du débitprévu de gaz prélevé.
C.2 Méthode B
a) monter complètement l’équipement de prélèvement, y compris le support de filtre et le condenseur/pot àcondensats/cartouche à résine ;
b) laisser l’échantillonneur chauffer jusqu’à obtention de sa température de service ;
c) étanchéifier l’orifice d’entrée de la buse ;
d) faire le vide dans l’équipement de prélèvement à une pression basse d’environ 500 hPa à 750 hPa ;
e) isoler le circuit ;
f) relever la pression présente dans le circuit d’échantillonnage au bout de 1 min ;
g) comparer le résultat avec la courbe de la figure C.1 et lire la valeur d’augmentation maximale de pression parunité de temps ou calculer l’augmentation maximale de pression par unité de temps à l’aide de l’équation (C.1).
... (C.1)
où :
∆p est l’augmentation maximale de la pression en kilopascals par minute ;
p0 est la pression à l’intérieur de l’équipement de prélèvement au moment 0, en kilopascals ;
p1 est la pression à l’intérieur de l’équipement de prélèvement au moment 1, en kilopascals ;
pamb est la pression ambiante en kilopascals ;
Vo est le taux de fuite autorisé de l’appareil, soit 5 % ;
V1 est le volume interne de l’équipement de prélèvement en litres ;
v est le débit de prélèvement prévu en l/min.
∆p p1= p0–V0
V1------= v⋅

— 29 — NF X 43-329
Figure C.1 — Augmentation maximale de pression autorisée par unité de temps de 5 % au moment de l'essai d'étanchéité avec une pression faible de 50 kPa
EXEMPLE
Volume interne de l’équipement de prélèvement : Vo = 14 l
Vitesse d’aspiration durant le prélèvement : v = 1,2 m3/h = 20 l/min
Pression minimale durant le prélèvement p0 = 50 kPa
Taux de fuite autorisé 5 %
Pression ambiante : pamb = 102 kPa
Lorsque ces valeurs sont rattachées à l’équation (D.1), on obtient :
∆p = 102 × 0,05/14 × 20 = 7 kPa et p1 = p0 + ∆p = 57 kPa ... (C.2)
C.3 Méthode C
a) monter complètement l’équipement de prélèvement, y compris le support de filtre et le condenseur/pot à con-densats/cartouche à résine ;
b) laisser l’échantillonneur chauffer jusqu’à obtention de sa température de service ;
c) étanchéifier l’orifice d’entrée de la buse ;
d) ouvrir le régulateur de débit de dérivation et s’assurer que le robinet principal est fermé. Mettre en marche lapompe de prélèvement et ouvrir lentement le robinet principal. Lorsque le robinet principal est complètementouvert, fermer lentement le régulateur de débit de dérivation jusqu’à ce que le manomètre indiqueenviron 500 hPa ou la chute de pression prévue en conditions normales de fonctionnement ;
e) laisser le système se vider pendant 1 min ;
f) observer le compteur à gaz et, à l’aide d’un chronomètre, noter tout mouvement du compteur sur une périodesupérieure ou égale à 1 min. Le taux de fuite ne doit pas dépasser 0,6 l/min ou 5 % du débit prélevé prévu sicette valeur est plus élevée ;
g) si la fuite dépasse ce niveau, vérifier le système et localiser la fuite.
Répéter le mode opératoire d’essai d’étanchéité jusqu’à obtention de ce critère.
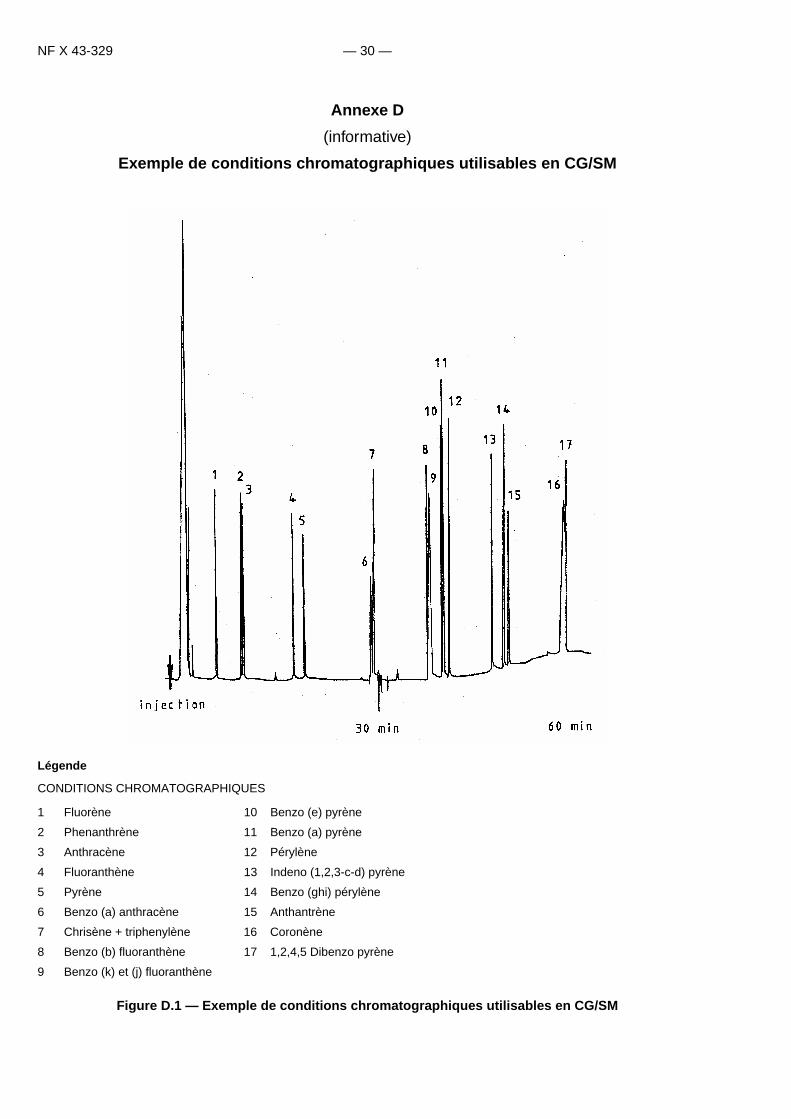
NF X 43-329 — 30 —
Annexe D
(informative)
Exemple de conditions chromatographiques utilisables en CG/SMInit numérotation des tableaux d’annexe [D]!!!Init numérotation des figures d’annexe [D]!!!Init numérotation des équations d’annexe [D]!!!
Légende
CONDITIONS CHROMATOGRAPHIQUES
Figure D.1 — Exemple de conditions chromatographiques utilisables en CG/SM
1 Fluorène 10 Benzo (e) pyrène
2 Phenanthrène 11 Benzo (a) pyrène
3 Anthracène 12 Pérylène
4 Fluoranthène 13 Indeno (1,2,3-c-d) pyrène
5 Pyrène 14 Benzo (ghi) pérylène
6 Benzo (a) anthracène 15 Anthantrène
7 Chrisène + triphenylène 16 Coronène
8 Benzo (b) fluoranthène 17 1,2,4,5 Dibenzo pyrène
9 Benzo (k) et (j) fluoranthène

— 31 — NF X 43-329
Annexe E
(informative)
Exemple de conditions chromatographiques utilisables en CLHPInit numérotation des tableaux d’annexe [E]!!!Init numérotation des figures d’annexe [E]!!!Init numérotation des équations d’annexe [E]!!!
Légende
CONDITIONS CHROMATOGRAPHIQUES
• C18 Vydac 201 TP, 5 µm, L = 25 cm, ∅ int = 4,6 mm
• Éluant acétonitrile — eau ; gradient de 45 % à 100 % acétonitrile en 30 min
• Débit 1,5 ml/min
• Détection UV 254 nm
Figure E.1 — Exemple de conditions chromatographiques utilisables en CLHP
1 Naphtalène 9 Benzo (a) anthracène
2 Acenaphtylène 10 Chrysène
3 Acenaphtène 11 Benzo (b) fluoranthène
4 Fluorène 12 Benzo (k) fluoranthène
5 Phenanthrène 13 Benzo (a) pyrène
6 Anthracène 14 Dibenzo (a,h) anthracène
7 Fluoranthène 15 Benzo (g,h,i) perylène
8 Pyrène 16 Indeno (1,2,3-c,d) pyrène

NF X 43-329 — 32 —
Annexe F
(informative)
Purification de l’extrait organique sur microcolonne de silice (méthode CLHP)
Init numérotation des tableaux d’annexe [F]!!!Init numérotation des figures d’annexe [F]!!!Init numérotation des équations d’annexe [F]!!!
L'extrait repris par environ 1 ml d'hexane (7.5.1.2) est introduit à l'aide d'une seringue en tête d'une microcolonneprête à l'emploi contenant environ 1 ml de silice. À l'aide d'une seringue du type «Luerlock» ou par soutirage sousvide, les HAP sont élués par :
• 5 ml d'hexane + dichlorométhane (50-50).
La fraction des composés polyaromatiques polaires reste sur la cartouche de purification.
L'éluat est évaporé sous gaz inerte en évitant d'aller à sec (concentrer jusqu'à 200 µl environ). Après reprise parun volume connu (de 0,5 ml à 1 ml) d'acétonitrile, l'extrait est injecté en CLHP (voir 7.5.1.4).
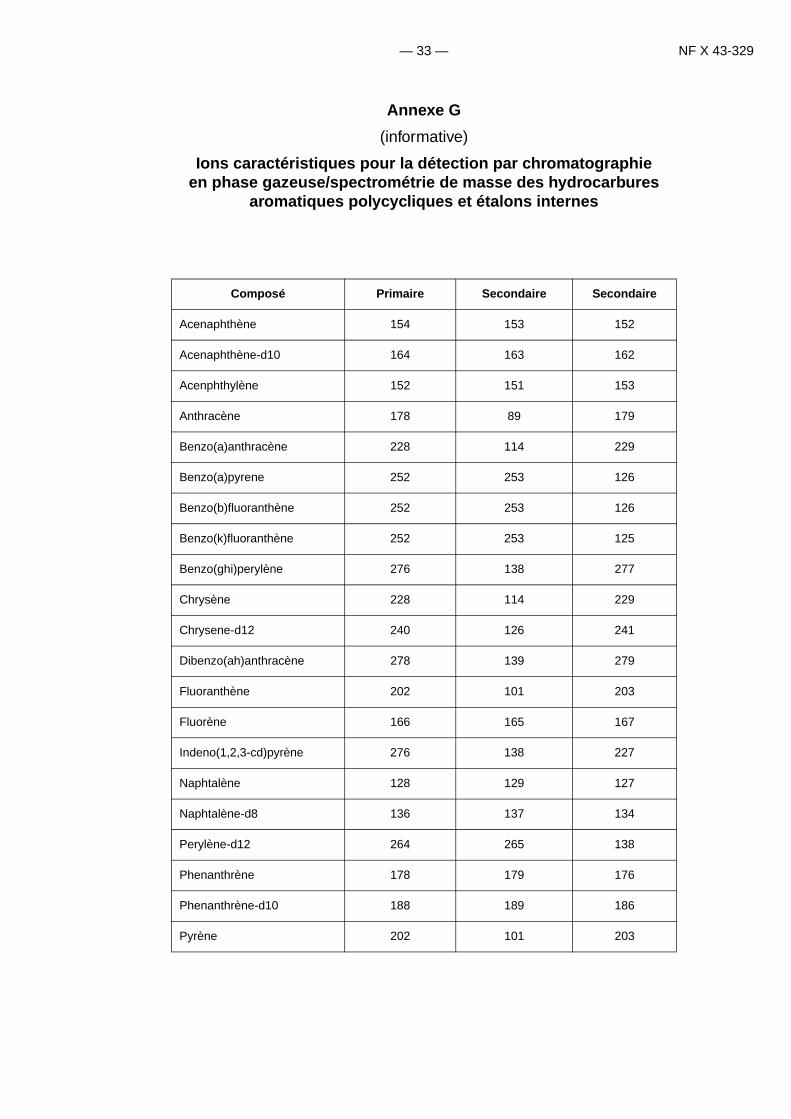
— 33 — NF X 43-329
Annexe G
(informative)
Ions caractéristiques pour la détection par chromatographie en phase gazeuse/spectrométrie de masse des hydrocarbures
aromatiques polycycliques et étalons internes
Init numérotation des tableaux d’annexe [G]!!!Init numérotation des figures d’annexe [G]!!!Init numérotation des équations d’annexe [G]!!!
Composé Primaire Secondaire Secondaire
Acenaphthène 154 153 152
Acenaphthène-d10 164 163 162
Acenphthylène 152 151 153
Anthracène 178 89 179
Benzo(a)anthracène 228 114 229
Benzo(a)pyrene 252 253 126
Benzo(b)fluoranthène 252 253 126
Benzo(k)fluoranthène 252 253 125
Benzo(ghi)perylène 276 138 277
Chrysène 228 114 229
Chrysene-d12 240 126 241
Dibenzo(ah)anthracène 278 139 279
Fluoranthène 202 101 203
Fluorène 166 165 167
Indeno(1,2,3-cd)pyrène 276 138 227
Naphtalène 128 129 127
Naphtalène-d8 136 137 134
Perylène-d12 264 265 138
Phenanthrène 178 179 176
Phenanthrène-d10 188 189 186
Pyrène 202 101 203

NF X 43-329 — 34 —
Annexe H
(informative)
Valeurs de la pression saturante de la vapeur d’eau
Init numérotation des tableaux d’annexe [H]!!!Init numérotation des figures d’annexe [H]!!!Init numérotation des équations d’annexe [H]!!!
La pression atmosphérique de référence est 101,3 kPa.
Température de rosée °C
Pression de vapeur kPa
Température de rosée °C
Pression de vapeur kPa
1 0,66 21 2,50
2 0,71 22 2,66
3 0,76 23 2,82
4 0,82 24 3,00
5 0,88 25 3,18
6 0,94 26 3,38
7 1,01 27 3,58
8 1,08 28 3,80
9 1,15 29 4,03
10 1,23 30 4,27
11 1,32 31 4,52
12 1,41 32 4,78
13 1,50 33 5,06
14 1,61 34 5,35
15 1,71 35 5,66
16 1,83 36 5,98
17 1,95 37 6,32
18 2,07 38 6,67
19 2,21 39 7,04
20 2,35 40 7,43

— 35 — NF X 43-329
Bibliographie
NF X 43-265, Qualité de l'air — Air des lieux de travail — Échantillonnage des hydrocarbures aromatiquespolycycliques (HAP).
ISO 17993, Qualité des eaux — Détermination de 15 hydrocarbures aromatiques polycycliques (HAP) dans l’eaupar HPLC avec détection par fluorescences après extraction liquide-liquide.
NF X 43-025, Qualité de l'air — Air ambiant — Détermination des hydrocarbures aromatiques polycycliques —Dosage par chromatographie liquide haute performance et par chromatographie gazeuse.